

Abstract
Purpose:
To provide an updated summary of recent advances in our understanding of the non-canonical roles of apoptotic and DNA double-strand break repair factors in various biological processes, especially in the cellular response to radiotherapy.
Conclusion:
Apoptotic caspases are usually considered as “executioners’’ of unwanted or damaged cells or tissues. However, recent studies indicated they play multiple additional, often counterintuitive roles in many biological processes. Similarly, DNA double-strand break (DSB) repair factors were also found to play unexpected roles beyond repairing damaged DNA. In this review, I will summarize key findings on the non-canonical roles of apoptotic and DSB repair factors in disparate biological and pathological processes such as radiation-induced genetic instability and carcinogenesis, wound healing and tissue regeneration, induced pluripotent stem cell induction, spontaneous and stochastic generation of cancer stem cells, and cancer immunotherapy. I believe these findings will usher in more studies in this exciting and rapidly evolving field.
Keywords: non-canonical roles of apoptotic caspases, Caspase 3, Caspase 6, Caspase 7, Caspase 8, limited MOMP, DNA double-strand breaks, ATM, radiotherapy, immune checkpoint blockade therapy
Apoptosis as a key component of cellular response to stress exposures
Initially proposed in the early 1970s(Kerr et al. 1972), apoptosis refers to a form of programmed cell death in multicellular organisms. It has been recognized as a key biological mechanism to remove unwanted or damaged tissues during development or after stress exposures(Strasser et al. 2000). Apoptosis is characterized by shrinkage of cellular sizes, blebs in the plasma membrane, chromatin condensation, and fragmentation of nuclear DNA. A key function of apoptosis is to facilitate the clearance of dying cells by scavenger cells such as macrophages in a non-inflammatory manner. As such, a distinct feature of apoptotic cells is their disintegration into multiple smaller fragments to facilitate easy phagocytosis by macrophages. The engulfment of apoptotic cells is also facilitated by the flipping of the membrane lipid phosphatidylserine from the inner to outer plasma membrane, which serves as a “eat me” signal(Fadok, Bratton, Frasch, et al. 1998). The engulfment of the apoptotic bodies thus prevents cellular contents from being leaked out into the surrounding tissue microenvironment, which can generate powerful, detrimental inflammatory responses(Fadok, Bratton, Konowal, et al. 1998).
The apoptotic process is very well defined at the molecular level(Elmore 2007). It occurs through either an intrinsic or an extrinsic pathway. In the extrinsic pathway, apoptosis is initiated by the engagement of external ligands to cell surface death receptors such as FAS, TRAIL, or TNFα receptors. The activated receptors subsequently engage downstream factors such as FADD and the “initiator” Caspase 8. Activated Caspase 8 further cleaves and activates “executioner” caspases such as Caspase 3, 6, or 7. The executioner caspases then cleave hundreds of substrates to cause the widely recognized apoptotic phenotype. The intrinsic pathway, on the other hand, usually starts at the mitochondria. Cellular stresses such as radiation-induced DNA damage, high ROS levels, or unfolded protein accumulation can induce the permeabilization of the mitochondrial outer membrane (MOMP). This in turn leads to the migration of SMAC/DIABLO protein into the cytosol and binds inhibitors of apoptosis (IAP) proteins, which pulls them away from caspases and causes their activation. In parallel, cytochrome c, a key component of the electron transport chain in the mitochondria, also leaks into the cytoplasm and forms a complex with Apaf-1 and the “initiator” Caspase 9, which is called the “apoptosome”. Activation of Caspase 9 leads to the cleavage of “executioner” caspases such as Caspase 3, 6, and 7, which leads to full-blown cellular apoptosis.
Genetic instability and bystander effect, two hallmarks of mammalian cells exposed to radiation
Mammalian cells exposed to ionizing radiation are expected to have the following fates: die through apoptosis, necroptosis, mitotic catastrophe, or senescence; or survive with non-lethal DNA damage that is repaired(Eriksson and Stigbrand 2010). Since the early days, the DNA damaging effects of ionizing radiation have long been one of the most important subjects of radiobiology investigations. Since the 1980s, there have been continuous and intensive efforts to study two important phenomena in irradiated mammalian cells for which Dr. John B Little and colleagues at Harvard made seminal contributions: genetic instability and bystander effects(Little 2003).
Radiation-induced genetic instability refers to genomic damage that arises in the progeny of irradiated cells after many cell divisions while radiation-induced bystander effects refer to biological effects that occur in cells not receiving radiation exposure directly but receiving damaging signals from neighboring irradiated cells. Among the earliest examples of radiation-induced genomic instability was the finding of enhanced recombination activities over many cell divisions in irradiated yeast(Fabre 1983) and enhanced frequency of chromosomal aberrations in the somatic cells of full-grown fetuses derived from mouse embryos irradiated at the single-cell stage(Pampfer and Streffer 1989). It was also observed that radiation led to a persistent reduction in cloning efficiency in the progeny of irradiated cells for many generations, which was hypothesized to result from an enhanced frequency of “lethal mutations” in the progeny of irradiated cells(Seymour et al. 1986; Gorgojo and Little 1989; Mendonca et al. 1989; Chang and Little 1991). It was further observed in ethyl methanesulfonate (EMS)-treated CHO cells that elevated mutation frequencies could be observed 10–12 cell generations after the initial exposure. Dr. Little’s group confirmed similar observations in irradiated CHO cells(Little et al. 1990). Since radiation-induced double-strand breaks (DSBs) are not known to persist over 10–12 cell divisions, it was likely the mutations were generated de novo in the progeny cells. Indeed, delayed or persistent mutagenesis of the HPRT(hypoxanthine Phosphoribosyltransferase 1) or the TK1(thymidine kinase 1) gene was observed in at least 10% of clonal populations derived from single cells surviving radiation exposure(Chang and Little 1992; Grosovsky et al. 1996; Little et al. 1997). Persistent genetic instability was also observed at the minisatellite (Li CY et al. 1992) and microsatellite(Li CY et al. 1994; Romney et al. 2001) loci in human cells. Importantly, instability at the minisatellite loci was also observed in x-ray transformed murine cells grown in vivo(Paquette and Little 1994). At the chromosome level, an enhanced frequency of nonclonal chromosomal aberrations was observed in clonal descendants of mouse hematopoietic stem cells examined 12–14 generations after exposure to α radiation(Kadhim et al. 1992). It was also observed in quite a few other cellular systems with low or high LET radiation (Sabatier et al. 1992; Holmberg et al. 1993; Marder and Morgan 1993; Ponnaiya et al. 1997; Limoli et al. 2000; Dugan and Bedford 2003). Mechanistically, it was reported that enhanced oxidative stress was associated with radiation-induced delayed mutagenesis (Limoli et al. 2001; Redpath and Gutierrez 2001). Importantly, a connection between radiation-induced chromosomal instability and malignant transformation was established in mouse mammary epithelial cells in irradiated in vivo(Ullrich and Davis 1999).
Drs. Hatsumi Nagasawa and John B. Little were the first to report radiation-induced bystander effect, where genetic changes were observed in mixed irradiated and non-irradiated cell populations even in cells that had not received any radiation exposure(Nagasawa and Little 1992). Strikingly, 20–40% of the cells were observed to possess sister chromosome exchange when only 0.1–1.0% of nuclei were traversed by an α particle. These observations were later confirmed by other investigators(Deshpande et al. 1996). Soon afterward, it was shown that enhanced mutagenesis at the gene levels could also be observed in bystander cells(Nagasawa and Little 1999; Huo et al. 2001). At the mechanistic level, evidence was shown that reactive oxygen species(ROS) were involved in α particle-induced chromosomal changes(Narayanan et al. 1997). However, based on the inability to suppress enhanced bystander mutagenesis by DMSO after microbeam irradiation, it was also suggested that ROS were not involved in bystander mutagenesis(Zhou H et al. 2000). On the other hand, Azzam et al presented evidence that oxidative stress mediated by flavin-containing oxidase-generated superoxide and hydrogen peroxide were involved in micronucleus formation in bystander cells(Azzam et al. 2002). At the genetic level, molecular mechanisms proposed to be involved in regulating bystander effects include the p53/p21Waf1 pathway, gap junctions, and MAPK kinases (Azzam et al. 1998; Bishayee et al. 1999; Azzam et al. 2001; Azzam et al. 2002; Howell and Bishayee 2002). In addition, cells deficient in the NHEJ pathway were also found to have elevated frequencies of bystander effect(Little et al. 2003; Nagasawa et al. 2003). Importantly, radiation-induced bystander effect was also observed in vivo in mice(Watson et al. 2000; Xue et al. 2002). Taken together, for both radiation-induced genomic instability and bystander effects, ROS and RNS (reactive nitrogen species) were suggested to be responsible for radiation-induced delayed cellular injury(Azzam et al. 2012) with the caveat of conflicting reports from different labs using different cellular and assay systems as outlined above. However, the upstream molecular mechanisms involved in the prolonged induction of reactive oxygen or nitrogen species by radiation are not clearly defined.
Sublethal caspase activation as a key mechanism mediating radiation-induced genetic instability and carcinogenesis
Despite numerous studies that validated the initial observations of radiation-induced delayed genetic instability in mammalian cells, the molecular mechanisms involved were not understood well. Theoretically, radiation-induced mutagenesis of key DNA repair genes is one plausible mechanism for the widespread and transgenerational upregulation of mutation rates in the progeny of irradiated cells. However, such a scenario is highly unlikely given the high prevalence of the delayed genetic instability phenotype (10–30% of survivor cells) that would require radiation-induced mutation rates to be in the range of 10−2–10−1 per gene per cell division for the putative DNA repair genes. However, under most circumstances, mutation rates in most mammalian genes are usually in the range of 10−5–10−7 per gene per cell division, including the so-called “mutator” cell lines with mismatch repair deficiencies (DeMars and Held 1972; Kat et al. 1993; Bhattacharyya et al. 1994). Therefore, the delayed genetic instability observed in irradiated cells is more likely caused by non-genetic processes or factors induced by radiation that can be transmitted into the progeny of irradiated cells at relatively high frequencies (e.g. 10–30% of irradiated cells).
We believe that the recently identified phenomenon of limited MOMP (mitochondrial outer membrane permeability) and sublethal caspase activation play critical roles in mediating both radiation-induced genetic instability and carcinogenesis(Ichim et al. 2015; Liu et al. 2015). Using a novel fluorescent reporter system that allowed us to monitor the activation of apoptotic caspases using live imaging, we showed that ionizing radiation-induced a persistent, transgenerational activation of apoptotic caspase 3 and 7, two executioner caspases canonically associated with certain death of host cells from apoptosis, in a subpopulation of irradiated mammary epithelial cell line MCF10A. More importantly, we showed that a significant fraction of the irradiated cells with apoptotic caspase activation could survive it(Liu et al. 2015). Furthermore, those cells had ongoing induction of DNA DSBs, as evidenced by the continued presence of γH2AX foci in the progeny of irradiated cells for weeks after initial radiation exposure. They also formed soft agar colonies, which measured their abilities to grow in an anchorage-independent manner, a distinct property of cancer cells over normal cells(Cifone and Fidler 1980). Consistently, these cells also form tumors in nude mice at much higher frequencies than those without ongoing DSB induction. Mechanistically, we identified endonuclease G (EndoG) is a key downstream factor for radiation-induced persistent genomic instability. EndoG normally resides within the mitochondria. During apoptosis, EndoG migrates to the nucleus to cleave the DNA in between nucleosomes to cause DNA fragmentation that is a hallmark of apoptosis(Li LY et al. 2001). However, we discovered that after cellular exposure to radiation, the mitochondria became leaky and EndoG migrated to the nucleus to cause DNA double-strand breaks and nuclear fragmentation without killing host cells. We established the importance of EndoG by showing that its down-regulation in irradiated cells prevented soft agar colony and tumor formation in nude mice in irradiated MCF10A cells. The importance of Casp3 was also confirmed in the well-recognized two-stage DMBA+TPA chemical carcinogenesis model. Casp3 genetic deficiency significantly reduced DMBA+TPA induced skin carcinogenesis in C57BL/6 mice. Similar results were also obtained independently by another group which established the roles of limited MOMP/sublethal caspase 3 activation in stress-induced carcinogenesis(Ichim et al. 2015). Therefore, the above studies provided clear evidence that radiation can induce persistent genetic instability and oncogenic transformation in mammalian cells and both depend on limited MOMP and sublethal activation of apoptotic caspases and endonucleases (Fig. 1).
Figure 1.
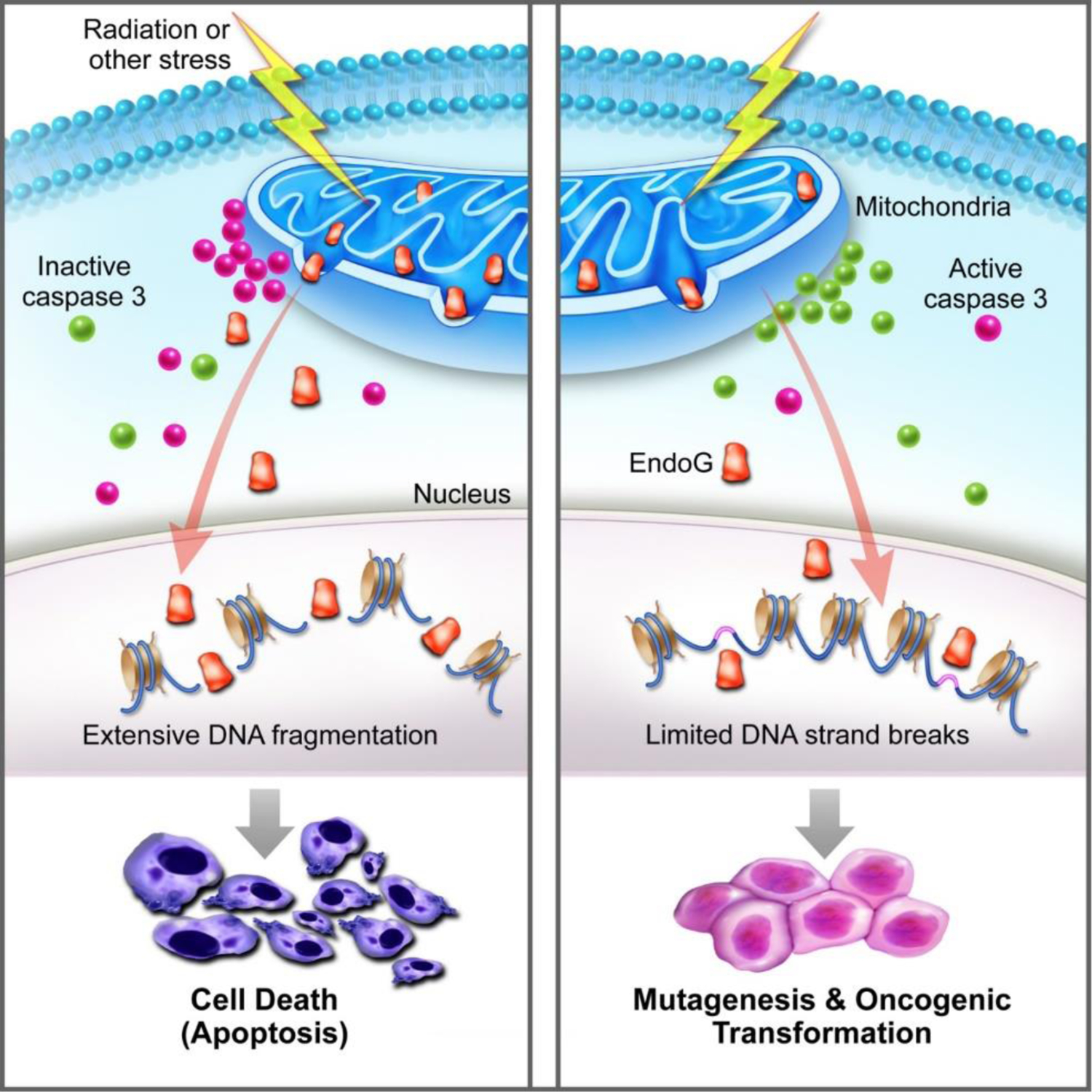
A schematic diagram illustrating how sublethal caspase activation facilitates stress-induced genetic instability and oncogenic transformation. The left panel shows the conventional scenario where mitochondrial permeability changes lead to activation of Casp3 and leakage of endonuclease G that kills the host cells. The right panel, on the other hand, shows partial leakage and survival of the cells with secondary genetic damage and oncogenic transformation. From Liu et al, Molecular Cell, 2015, PMID:25866249.
Related to the above studies, Cartwright et al examined the role of Caspase 3 in Myc-induced oncogenic transformation of MCF10A human mammary epithelial cells(Cartwright et al. 2017). By use of the CRISPR/Cas9 gene-editing technique, we generated Casp3 gene knockout MCF10A cells and transduced the Myc gene into the cells. In contrast to parental MCF10A cells, which were readily transformed after Myc transduction, MCF10A cells with Casp3KO were resistant to Myc-induced transformation. Myc was found to induce Casp3 activation, consistent with its long-established ability to induce apoptosis(Evan et al. 1992). However, the fact that Casp3 deficiency attenuated Myc-induced transformation was very surprising since apoptosis was in general considered a tumor-suppressive process. We further discovered that Myc transduction was accompanied by significant induction of DSBs mediated by Casp3 and downstream apoptotic DNA fragmentation factor EndoG. Genetic deletion of EndoG, similar to Casp3 knockout, prevented Myc-induced transformation of MCF10A cells. These results, therefore, indicate that limited MOMP plays a similar role in facilitating oncogene-induced transformation as it does in radiation and chemically-induced oncogenesis(Liu et al. 2015).
Unexpected roles of apoptotic caspases in stimulating wound healing, tissue regeneration, tumor repopulation after radiotherapy, and tumor cell metastasis
In addition to perpetuating radiation-induced persistent genetic instability and malignant transformation, apoptosis was found to play unexpected roles in several important biological processes. Li et al reported a surprising finding where Caspase 3 plays a key role in facilitating mouse skin wound healing and liver regeneration(Li F, Huang, et al. 2010), which was referred to as the “Phoenix Rising” pathway (Fig. 2). Mechanistically, activated Caspase 3 in dying cells in damaged tissues could activate calcium-independent phospholipase A2 (iPLA2) by cleaving it, which led to the production of more arachidonic acid, which in turn led to more production of PGE2 that was a potent stimulator of tissue regeneration. The same pathway was also found to function in irradiated tumor cells. Dying tumor cells utilized the same “Phoenix Rising” pathway to promote tumor cell repopulation after radiotherapy and caspase 3 inhibition could potently reduce tumor repopulation after radiotherapy(Huang et al. 2011; Donato et al. 2014). Our findings thus provide mechanistic insights for the long-established phenomenon of tumor repopulation, one of the 4 “Rs” in radiobiology teachings(Hall and Giaccia 2011).
Figure 2.
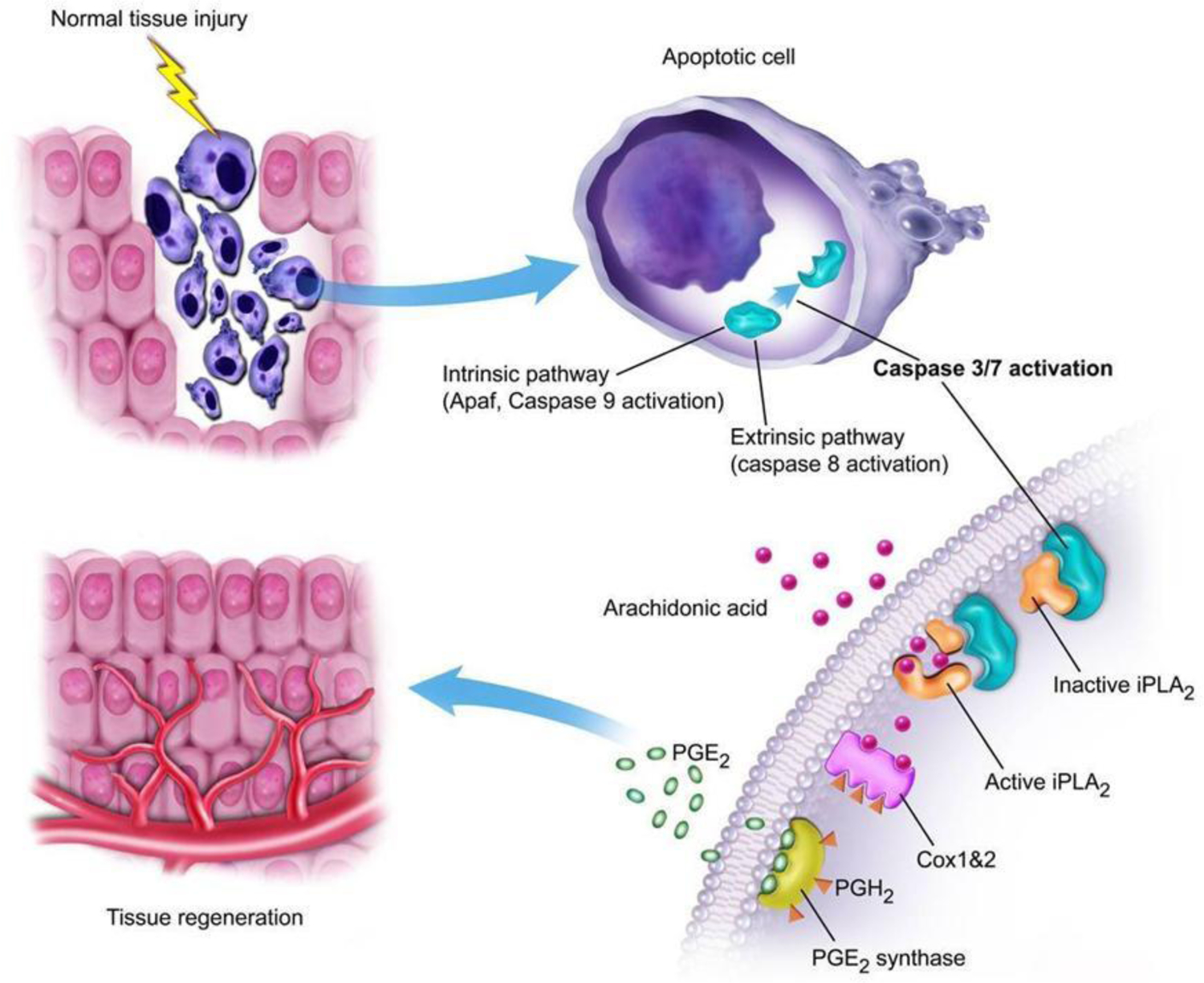
Illustration of the “Phoenix Rising” pathway of wound healing and tissue regeneration. Damage in tissues leads to the activation of Caspase 3/7, which cleaves and activates iPLA2 and increased production of arachidonic acid, a precursor for the production of PGE2, a known stimulating signal for stem cell growth and tissue regeneration. From Li et al, Science Signaling, 2010, PMID: 20179271.
Besides stimulating tumor repopulation after radiotherapy, Caspase 3 was found to play important roles in regulating the migratory behaviors of colon cancer cells(Zhou M et al. 2018). Genetic knockout of Casp3 in colon cancer cells was found to make them less clonogenic in soft agar assays and more sensitive to DNA damaging agents such as radiation and mitomycin C. In addition, Casp3KO colon cancer cells metastasized significantly less than parental control cells, and this phenotype was correlated with significantly increased expression of E-cadherin and decreased expression of N-cadherin, Snail, and ZEB1, biomarkers for the epithelial to mesenchymal (EMT) phenotype characteristic of metastatic cancer cells.
Key roles of apoptotic caspases in mediating epigenetic reprogramming in iPSC induction
Another unexpected finding concerning apoptotic caspases is their role in the epigenetic reprogramming of induced pluripotent stem cells (iPSCs) from fibroblasts(Li F, He, et al. 2010). The establishment of iPSCs from various differentiated cells is a landmark finding made by Shinya Yamanaka in 2006(Takahashi and Yamanaka 2006). It fundamentally changed our view on the plasticity of epigenetic control in differentiated cells and ushered in a new era in stem cell biology and regenerative medicine. Many new factors and mechanisms have since been identified that regulate the epigenetic states of mammalian cells. Li et al discovered that apoptotic caspases 3 & 8 played critical roles in iPSC derivation from human fibroblasts using Yamanaka factors(Li F, He, et al. 2010). Yamanaka et al observed substantial apoptosis in their initial reprogramming experiments(Takahashi and Yamanaka 2006). Based on conventional wisdom, one would expect that inhibiting caspases and reducing the number of apoptotic cells would boost the rate of iPSCs derived from the reprogramming process. Unexpectedly, Li et al discovered that inhibition of apoptotic caspases significantly attenuated, and in some cases completely prevented the formation of iPSCs(Li F, He, et al. 2010). Mechanistically, Li et al showed that many human fibroblast cells underwent significant, steadily increasing Caspase 3&8 activation in the 2–3 weeks following transduction of iPSC induction factors. Interestingly, many of the cells with Caspase 3&8 activation survived the process, reminiscent of the limited MOMP phenomenon observed in radiation and oncogene-induced carcinogenesis discussed earlier(Liu et al. 2015; Cartwright et al. 2017). Li et al further showed that Caspases 3&8 were required to cleave and deactivate the Rb protein, a gatekeeper for the cellular epigenetic state, to facilitate reprogramming (Fig. 3). A key role for Rb in preventing epigenetic plasticity has also been shown by others in mouse MEF cells. For example, Rb inactivation was shown to facilitate the reprogramming of MEF cells into an embryonic stem (ES) cell-like state by a cell cycle independent mechanism(Kareta et al. 2015).
Figure 3.
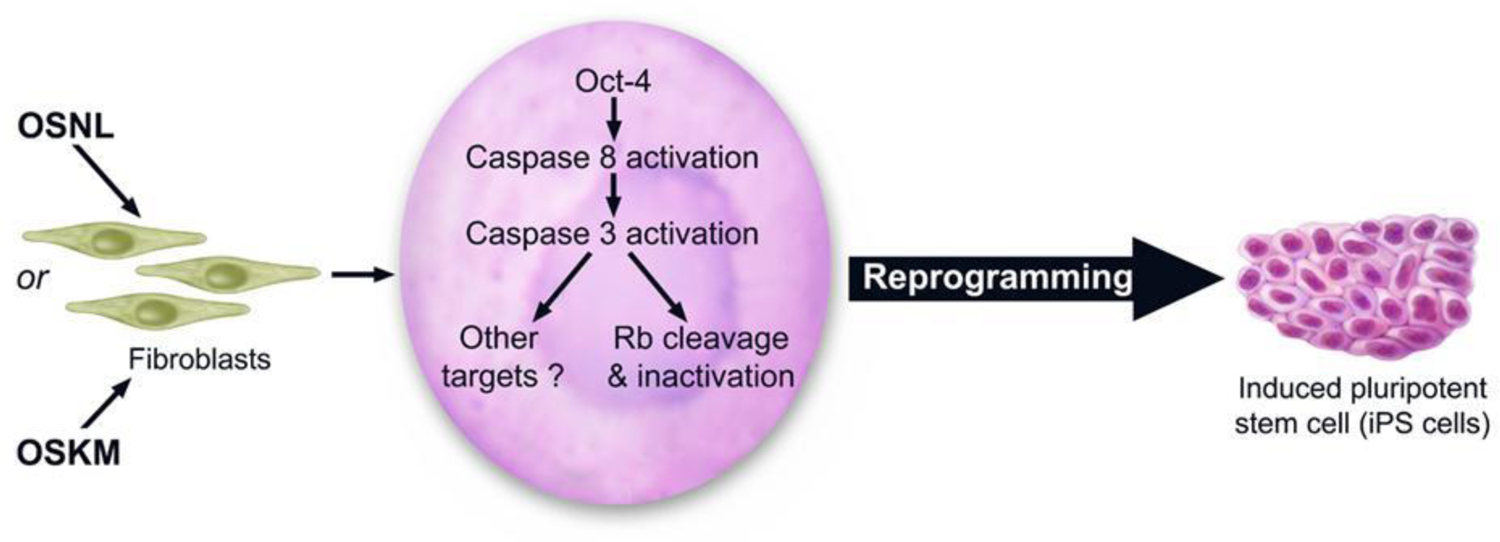
An illustration on the stem cell factors, in particular Oct4, activates Caspases 3 & 8 to deactivate Rb to facilitate reprogramming of differentiated cells into induced pluripotent stem cells (iPSCs). From Li et al, Cell Stem Cell, 2010, PMID: 20887956.
Paradoxical role of ATM in promoting oncogenic transformation
The fact that it was possible to use Yamanaka factors to reprogram terminally differentiated cells into ES-like iPSCs in a matter of 2–3 weeks is illuminating from many perspectives. What is remarkable is that given the right combination of transcriptional factors, it is possible to induce profound changes in the transcriptional levels of thousands of genes, which involve the activation and silencing of thousands of genes by 4–5 transcription factors, to achieve the reprogramming of terminally differentiated cells. Using the same line of reasoning, our laboratory developed a similar protocol to transform primary human cells into cancer cells using a combination of oncogenes. Earlier, the transformation of primary human cells had been a tedious, months-long process that required sequential transduction of 4–5 oncogenes into targeted primary cells(Hahn et al. 1999). Inspired by the Yamanaka protocol for iPSC induction, we developed an oncogenic cocktail that included the stem cell transcriptional factors/oncogenes Oct4 and oncogenes Myc, dominant-negative p53 (p53DD), Ras, cyclin D, and CDK4. Our data indicated that similar to iPSC induction by the Yamanaka factors, Oct4 in combination with the oncogenes could potently transform primary cells such as astrocytes into cancer cells in a matter of 2–3 weeks(Li F et al. 2016). The transformed cells were very tumorigenic, with only 100 cells required to form tumors in nude mice. The feasibility and ease with which we could transform tumor cells make it possible to study the whole transformation process in vitro in 2–3 weeks. Indeed, we also used the system to study the role of the DNA double-strand break repair factor ATM in oncogenesis. We discovered that simultaneous transduction of 4 transgenic factors (Oct4, Myc, Ras, and p53DD) into human fibroblast cells generated an increasing amount of DNA DSBs over 1–3 weeks(Liu et al. 2020). Consistently, increased amounts of γH2AX and phosphorylated ATM were observed. What is the role of ATM in this transformation process? On the one hand, ATM was a key sensor of DNA double-strand breaks and a central coordinator of the DNA damage response that is responsible for sensing and repairing the strand breaks. In addition, ATM was traditionally recognized as a tumor suppressor. Biallelic ATM mutation carriers were at significantly higher risk for leukemias and lymphomas(Taylor et al. 1996). On the other hand, the roles of ATM in solid tumors were less clear. No evidence exists for its involvement in solid tumors except for earlier reports on its potential role in breast cancer(Swift et al. 1987). However, such involvement remains controversial because of conflicting reports(Renwick et al. 2006). To determine the role of ATM in solid tumor carcinogenesis, Liu et al generated ATM knockout in human fibroblasts. These cells were then transduced with the oncogenic cocktail (Oct4, Myc, Ras, p53DD). In contrast to parental control of human fibroblast cells, ATMKO cells become quite resistant to oncogenic transformation, contrary to conventional wisdom. Even the few morphologically transformed colonies that emerge failed to form soft agar colonies. They also failed to form tumors in nude mice when injected with 1 million or more cells. This is in sharp contrast to transformed parental cells, which could form tumors with less than 100 cells injected(Liu et al. 2020). It was further shown that mechanistically DNA double-strand breaks generated during the transformation process activated the DNA damage response (DDR) and ATM, as indicated by the appearance of γH2AX foci and phosphorylated ATM. ATM activation, instead of suppressing transformation, facilitated it by phosphorylating and deactivating KAP1, which led to chromatin relaxation that allowed for transcriptional reprogramming that was key for transformation.
Collaboration of apoptotic caspases and ATM in promoting the generation of cancer stem cells and their maintenance
So far it is clear that both apoptotic caspases and DNA double-strand repair factors can play non-canonical, facilitative roles in carcinogenesis and tumor response to cytotoxic treatments such as radiotherapy. Are there any interactions between the two? Liu et al conducted a study where they attempted to answer this question(Liu et al. 2017). In multiple human cancer cell lines, they found evidence for spontaneous mitochondrial cytochrome c leakage and caspase activation, clear evidence for limited MOMP. Correspondingly, they discover constitutive induction of γH2AX foci, indicative of DNA double-strand breaks. Theoretically, these DSBs could also be explained by the previously described phenomenon of oncogenic stress(Bartkova et al. 2005; Gorgoulis et al. 2005; Odoux et al. 2008), where oncogene activation disrupts cell cycle and DNA replication regulation, which can lead to slow or stalled replication progression, leading to DNA damage. To establish a causal relationship between the observed, spontaneously arising DSBs and limited MOMP, Liu et al knocked out Casp3, 6, 7 and downstream apoptotic DNA fragmentation nucleases EndoG and CAD/DFF40. Importantly, in the knockout cell, the incidence of spontaneous DSBs was reduced by more than 50%, indicating that limited MOMP accounted for at least 50% of the spontaneous arising DSBs. What are the consequences of spontaneous limited MOMP and DSB induction in tumor cells? Liu et al demonstrated that cells with limited MOMP were more potent in forming tumors because they were rendered more stem cell-like by DNA damage response induced by the DSBs. Mechanistically, activated ATM leads to activation of stem cell factors such as Oct4, Stat3, or NF-kB, which endowed tumor cells with stem cell-like properties (Fig. 4). This previously unexplored mechanism also provided an explanation for the stochastic nature of the emergence and disappearance of cancer stem cells (Odoux et al. 2008), which had not been clearly understood previously. Furthermore, the authors demonstrated that inhibition of caspases or ATM made tumors grow significantly slower in vivo, thereby providing potential therapeutic targets for future studies (Liu et al. 2017).
Figure 4.
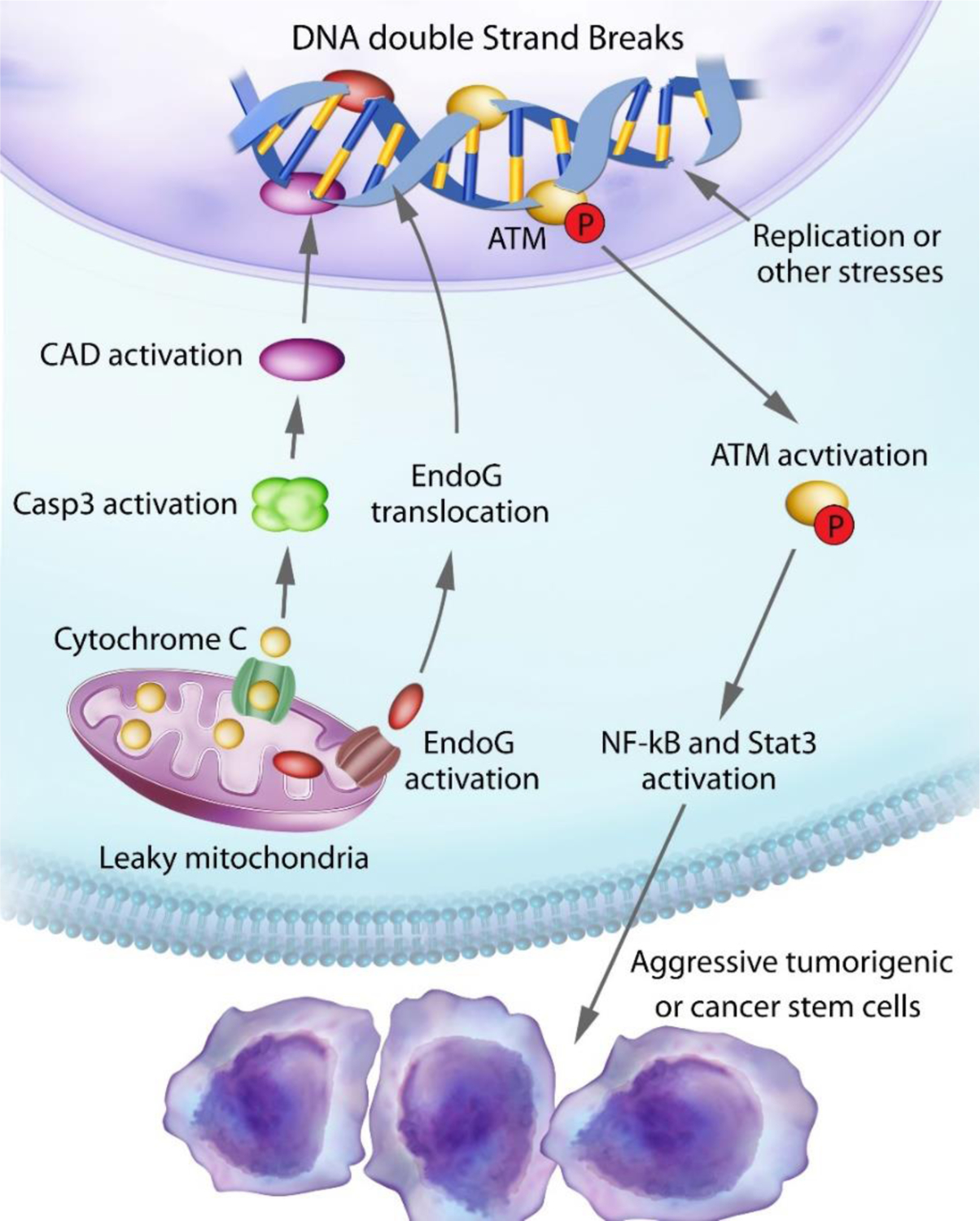
An illustration of our findings on spontaneous DNA double-strand breaks induction and their roles in maintaining their stemness and tumorigenicity of cancer cells. Leaky mitochondria lead to migration of EndoG and CAD from the mitochondria to the nucleus, which generates DNA DSBs and activation of ATM, which in turn activates NF-κB and Stat3 that confers stem-like properties to cancer cells. From Liu et al, Cell Research, 2017, PMID:28227983.
ATM inhibition as a strategy to boost the efficacy of immune checkpoint blockade therapy.
Cellular innate immunity is a key determining factor of the tumor microenvironment and tumor response to immunotherapy. It is usually involved in the mammalian cellular response to viral infections. Some of the key players in cellular innate immunity include the cGAS/STING pathway for sensing cytosolic double-stranded DNA (dsDNA)(Li XD et al. 2013; Cai et al. 2014) and MAVS/MDA5 for cytosolic dsRNA(Hou et al. 2011). Studies have shown that the cGAS/STING pathway was critical for ICB therapy and radiotherapy in preclinical models(Deng et al. 2014; Demaria et al. 2015; Wang et al. 2017). DNA methyltransferase inhibitors or radiotherapy were shown to activate endogenous retroviruses, which could activate the RIG-I/MDA5 pathway and synergize with immunotherapy and enhance radiotherapy(Chiappinelli et al. 2015; Roulois et al. 2015; Lee et al. 2020). Therefore, there were efforts aimed at developing STING agonists to enhance cancer immunotherapy with promising results in preclinical models (Corrales et al. 2015; Demaria et al. 2015). However, because most STING agonists are synthetic homologs of 2’3’-cGAMP and have to be delivered intratumorally due to poor bioavailability, efforts are being made to develop STING agonists that can be delivered systemically(Ramanjulu et al. 2018; Chin et al. 2020; Gajewski and Higgs 2020; Pan et al. 2020). Interestingly, recent studies suggest that ATM inhibitors may work as potent activators of the cGAS/STING pathway. Kinase deficient ATM in Drosophila ATM could trigger an innate immune response(Petersen et al. 2012). Furthermore, unrepaired DNA lesions in ATM knockout mice induce STING pathway activation and promote antiviral and anti-bacterial response(Hartlova et al. 2015). It was further reported that ATM inhibition could activate a type I interferon response in pancreatic tumor cells that could enhance ICB therapy(Zhang et al. 2019). However, it was not clear what are the molecular mechanisms involved in triggering spontaneous type I interferon response activation in ATM-deficient cells. Hu et al showed that increased type I interferon response was dependent on the cGAS/STING pathway in multiple murine and human tumor cells. ATM inhibition led to the down-regulation of TFAM, a key factor in mtDNA replication and mitochondrial biogenesis, which led to cytoplasmic leakage of mtDNA and activation of the cGAS/STING pathway(Hu et al. 2021). Importantly, they provided strong evidence that leaked mtDNA, rather than nuclear DNA was responsible for the high basal levels of cGAS/STING activation. The authors also show that ATM inhibition, either through genetic knockout or chemical inhibition, could synergize with ICB therapy. Since ATM inhibitors are already in clinical trials to enhance radiotherapy, these results therefore could potentially be translated into human cancer patients rather quickly. Furthermore, the authors also showed from analyzing a large cohort of cancer patients treated at Memorial Sloan Kettering Cancer Institute that human tumors with ATM mutations responded significantly better to ICB therapy, indicating ATM mutations could be used as a predictive biomarker for ICB treatment(Hu et al. 2021).
Conclusion
Recent studies have demonstrated that apoptotic and DNA double-strand break repair factors play many non-canonical roles in diverse biological processes. However, despite the increasing number of studies and novel revelations, we are only touching the tip of the iceberg. It is all but certain that more fruits will be borne in this fertile area of research. While “to live or to die” is the key issue that we associate apoptotic caspases with, they certainly do plenty of other things in between.
Acknowledgment
The study is supported in part by grants from US National Institutes of Health grants CA208852, CA216876, and a grant from the Emerson Collective.
Footnotes
Declaration of Interest:
The author declares no conflict of interest.
References
- Azzam EI, de Toledo SM, Gooding T, Little JB. 1998. Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat Res 150(5):497–504. [PubMed] [Google Scholar]
- Azzam EI, de Toledo SM, Little JB. 2001. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha-particle irradiated to nonirradiated cells. Proc Natl Acad Sci U S A 98(2):473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam EI, De Toledo SM, Spitz DR, Little JB. 2002. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from alpha-particle-irradiated normal human fibroblast cultures. Cancer Res 62(19):5436–5442. [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C et al. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434(7035):864–870. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya NP, Skandalis A, Ganesh A, Groden J, Meuth M. 1994. Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci U S A 91(14):6319–6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishayee A, Rao DV, Howell RW. 1999. Evidence for pronounced bystander effects caused by nonuniform distributions of radioactivity using a novel three-dimensional tissue culture model. Radiat Res 152(1):88–97. [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54(2):289–296. [DOI] [PubMed] [Google Scholar]
- Cartwright IM, Liu X, Zhou M, Li F, Li CY. 2017. Essential roles of Caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Little JB. 1991. Delayed reproductive death in X-irradiated Chinese hamster ovary cells. Int J Radiat Biol 60(3):483–496. [DOI] [PubMed] [Google Scholar]
- Chang WP, Little JB. 1992. Persistently elevated frequency of spontaneous mutations in progeny of CHO clones surviving X-irradiation: association with delayed reproductive death phenotype. Mutat Res 270(2):191–199. eng. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A et al. 2015. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162(5):974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, Vernier W, Ali SH, Kissai M, Lazar DC et al. 2020. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369(6506):993–999. [DOI] [PubMed] [Google Scholar]
- Cifone MA, Fidler IJ. 1980. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc Natl Acad Sci U S A 77(2):1039–1043. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ et al. 2015. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11(7):1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV et al. 2015. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 112(50):15408–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMars R, Held KR. 1972. The spontaneous azaguanine-resistant mutants of diploid human fibroblasts. Humangenetik 16(1):87–110. [DOI] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T et al. 2014. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41(5):843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Goodwin EH, Bailey SM, Marrone BL, Lehnert BE. 1996. Alpha-particle-induced sister chromatid exchange in normal human lung fibroblasts: evidence for an extranuclear target. Radiat Res 145(3):260–267. [PubMed] [Google Scholar]
- Donato AL, Huang Q, Liu X, Li F, Zimmerman MA, Li CY. 2014. Caspase 3 Promotes Surviving Melanoma Tumor Cell Growth after Cytotoxic Therapy. J Invest Dermatol [DOI] [PMC free article] [PubMed]
- Dugan LC, Bedford JS. 2003. Are chromosomal instabilities induced by exposure of cultured normal human cells to low- or high-LET radiation? Radiat Res 159(3):301–311. [DOI] [PubMed] [Google Scholar]
- Elmore S 2007. Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson D, Stigbrand T. 2010. Radiation-induced cell death mechanisms. Tumour Biol 31(4):363–372. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. 1992. Induction of apoptosis in fibroblasts by c-myc protein. Cell 69(1):119–128. eng. [DOI] [PubMed] [Google Scholar]
- Fabre F 1983. Mitotic transmission of indued recombinational activity in yeast. In: Friedberg EC, Bridges BA, editors. Cellular Response to DNA Damage Alan R. Liss; p. 379–384. [Google Scholar]
- Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. 1998. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ 5(7):551–562. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101(4):890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Higgs EF. 2020. Immunotherapy with a sting. Science 369(6506):921–922. [DOI] [PubMed] [Google Scholar]
- Gorgojo L, Little JB. 1989. Expression of lethal mutations in progeny of irradiated mammalian cells. Int J Radiat Biol 55(4):619–630. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr., Kastrinakis NG, Levy B et al. 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434(7035):907–913. [DOI] [PubMed] [Google Scholar]
- Grosovsky AJ, Parks KK, Giver CR, Nelson SL. 1996. Clonal analysis of delayed karyotypic abnormalities and gene mutations in radiation-induced genetic instability. Mol Cell Biol 16(11):6252–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. 1999. Creation of human tumour cells with defined genetic elements. Nature 400(6743):464–468. [DOI] [PubMed] [Google Scholar]
- Hall E, Giaccia A. 2011. Radiobiology for the Radiologist Sixth ed. Philadelphia: Lippincott Williams & Wilkins. [Google Scholar]
- Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA et al. 2015. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42(2):332–343. [DOI] [PubMed] [Google Scholar]
- Holmberg K, Falt S, Johansson A, Lambert B. 1993. Clonal chromosome aberrations and genomic instability in X-irradiated human T-lymphocyte cultures. Mutat Res 286(2):321–330. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. 2011. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146(3):448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell RW, Bishayee A. 2002. Bystander effects caused by nonuniform distributions of DNA-incorporated (125)I. Micron 33(2):127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, Li F, Li CY. 2021. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Invest 131(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC et al. 2011. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17(7):860–866. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L, Nagasawa H, Little JB. 2001. HPRT mutants induced in bystander cells by very low fluences of alpha particles result primarily from point mutations. Radiat Res 156(5 Pt 1):521–525. [DOI] [PubMed] [Google Scholar]
- Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D et al. 2015. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57(5):860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadhim MA, Macdonald DA, Goodhead DT, Lorimore SA, Marsden SJ, Wright EG. 1992. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 355(6362):738–740. [DOI] [PubMed] [Google Scholar]
- Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, Cecchini MJ, Spacek D, Batista LF, O’Brien M et al. 2015. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P. 1993. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci U S A 90(14):6424–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Pan D, Bao X, Hu M, Li F, Li CY. 2020. Endogenous Retrovirus Activation as a Key Mechanism of Anti-Tumor Immune Response in Radiotherapy. Radiat Res [DOI] [PMC free article] [PubMed]
- Li CY, Yandell DW, Little JB. 1992. Evidence for coincident mutations in human lymphoblast clones selected for functional loss of a thymidine kinase gene. Mol Carcinog 5(4):270–277. [DOI] [PubMed] [Google Scholar]
- Li CY, Yandell DW, Little JB. 1994. Elevated frequency of microsatellite mutations in TK6 human lymphoblast clones selected for mutations at the thymidine kinase locus. Mol Cell Biol 14(7):4373–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, He Z, Shen J, Huang Q, Li W, Liu X, He Y, Wolf F, Li CY. 2010. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 7(4):508–520. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY. 2010. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal 3(110):ra13. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Liu X, Sampson JH, Bigner DD, Li CY. 2016. Rapid Reprogramming of Primary Human Astrocytes into Potent Tumor-Initiating Cells with Defined Genetic Factors. Cancer Res 76(17):5143–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. 2001. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412(6842):95–99. eng. [DOI] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341(6152):1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli CL, Kaplan MI, Giedzinski E, Morgan WF. 2001. Attenuation of radiation-induced genomic instability by free radical scavengers and cellular proliferation. Free Radic Biol Med 31(1):10–19. [DOI] [PubMed] [Google Scholar]
- Limoli CL, Ponnaiya B, Corcoran JJ, Giedzinski E, Kaplan MI, Hartmann A, Morgan WF. 2000. Genomic instability induced by high and low LET ionizing radiation. Adv Space Res 25(10):2107–2117. [DOI] [PubMed] [Google Scholar]
- Little JB. 2003. Genomic instability and bystander effects: a historical perspective. Oncogene 22(45):6978–6987. [DOI] [PubMed] [Google Scholar]
- Little JB, Gorgojo L, Vetrovs H. 1990. Delayed appearance of lethal and specific gene mutations in irradiated mammalian cells. Int J Radiat Oncol Biol Phys 19(6):1425–1429. [DOI] [PubMed] [Google Scholar]
- Little JB, Nagasawa H, Li GC, Chen DJ. 2003. Involvement of the nonhomologous end joining DNA repair pathway in the bystander effect for chromosomal aberrations. Radiat Res 159(2):262–267. [DOI] [PubMed] [Google Scholar]
- Little JB, Nagasawa H, Pfenning T, Vetrovs H. 1997. Radiation-induced genomic instability: delayed mutagenic and cytogenetic effects of X rays and alpha particles. Radiat Res 148(4):299–307. eng. [PubMed] [Google Scholar]
- Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, Li CY. 2015. Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell 58(2):284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hu M, Liu P, Jiao M, Zhou M, Lee AK, Li F, Li CY. 2020. ATM Paradoxically Promotes Oncogenic Transformation via Transcriptional Reprogramming. Cancer Res 80(8):1669–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li F, Huang Q, Zhang Z, Zhou L, Deng Y, Zhou M, Fleenor DE, Wang H, Kastan MB et al. 2017. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res 27(6):764–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder BA, Morgan WF. 1993. Delayed chromosomal instability induced by DNA damage. Mol Cell Biol 13(11):6667–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonca MS, Kurohara W, Antoniono R, Redpath JL. 1989. Plating efficiency as a function of time postirradiation: evidence for the delayed expression of lethal mutations. Radiat Res 119(2):387–393. [PubMed] [Google Scholar]
- Nagasawa H, Huo L, Little JB. 2003. Increased bystander mutagenic effect in DNA double-strand break repair-deficient mammalian cells. Int J Radiat Biol 79(1):35–41. [PubMed] [Google Scholar]
- Nagasawa H, Little JB. 1992. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res 52(22):6394–6396. [PubMed] [Google Scholar]
- Nagasawa H, Little JB. 1999. Unexpected sensitivity to the induction of mutations by very low doses of alpha-particle radiation: evidence for a bystander effect. Radiat Res 152(5):552–557. [PubMed] [Google Scholar]
- Narayanan PK, Goodwin EH, Lehnert BE. 1997. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res 57(18):3963–3971. [PubMed] [Google Scholar]
- Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA, Lagasse E. 2008. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res 68(17):6932–6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampfer S, Streffer C. 1989. Increased chromosome aberration levels in cells from mouse fetuses after zygote X-irradiation. Int J Radiat Biol 55(1):85–92. [DOI] [PubMed] [Google Scholar]
- Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, Trotter BW, Altman MD, Buevich AV, Cash B et al. 2020. An orally available non-nucleotide STING agonist with antitumor activity. Science 369(6506). [DOI] [PubMed] [Google Scholar]
- Paquette B, Little JB. 1994. In vivo enhancement of genomic instability in minisatellite sequences of mouse C3H/10T1/2 cells transformed in vitro by X-rays. Cancer Res 54(12):3173–3178. [PubMed] [Google Scholar]
- Petersen AJ, Rimkus SA, Wassarman DA. 2012. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc Natl Acad Sci U S A 109(11):E656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnaiya B, Cornforth MN, Ullrich RL. 1997. Radiation-induced chromosomal instability in BALB/c and C57BL/6 mice: the difference is as clear as black and white. Radiat Res 147(2):121–125. [PubMed] [Google Scholar]
- Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, Tran JL, Moore P, Lehmann S, Eberl HC et al. 2018. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564(7736):439–443. [DOI] [PubMed] [Google Scholar]
- Redpath JL, Gutierrez M. 2001. Kinetics of induction of reactive oxygen species during the post-irradiation expression of neoplastic transformation in vitro. Int J Radiat Biol 77(11):1081–1085. [DOI] [PubMed] [Google Scholar]
- Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K et al. 2006. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38(8):873–875. [DOI] [PubMed] [Google Scholar]
- Romney CA, Paulauskis JD, Little JB. 2001. X-ray induction of microsatellite instability at autosomal loci in human lymphoblastoid WTK1 cells. Mutat Res 478(1–2):97–106. [DOI] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ et al. 2015. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162(5):961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier L, Dutrillaux B, Martin MB. 1992. Chromosomal instability. Nature 357(6379):548. [DOI] [PubMed] [Google Scholar]
- Seymour CB, Mothersill C, Alper T. 1986. High yields of lethal mutations in somatic mammalian cells that survive ionizing radiation. Int J Radiat Biol Relat Stud Phys Chem Med 50(1):167–179. [DOI] [PubMed] [Google Scholar]
- Strasser A, O’Connor L, Dixit VM. 2000. Apoptosis signaling. Annu Rev Biochem 69:217–245. eng. [DOI] [PubMed] [Google Scholar]
- Swift M, Reitnauer PJ, Morrell D, Chase CL. 1987. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med 316(21):1289–1294. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. eng. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Metcalfe JA, Thick J, Mak YF. 1996. Leukemia and lymphoma in ataxia telangiectasia. Blood 87(2):423–438. [PubMed] [Google Scholar]
- Ullrich RL, Davis CM. 1999. Radiation-induced cytogenetic instability in vivo. Radiat Res 152(2):170–173. [PubMed] [Google Scholar]
- Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. 2017. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A 114(7):1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GE, Lorimore SA, Macdonald DA, Wright EG. 2000. Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Res 60(20):5608–5611. [PubMed] [Google Scholar]
- Xue LY, Butler NJ, Makrigiorgos GM, Adelstein SJ, Kassis AI. 2002. Bystander effect produced by radiolabeled tumor cells in vivo. Proc Natl Acad Sci U S A 99(21):13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, Parsels LA, Shi J, Ramnath N, Wahl DR et al. 2019. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res 79(15):3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Randers-Pehrson G, Waldren CA, Vannais D, Hall EJ, Hei TK. 2000. Induction of a bystander mutagenic effect of alpha particles in mammalian cells. Proc Natl Acad Sci U S A 97(5):2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Liu X, Li Z, Huang Q, Li F, Li CY. 2018. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int J Cancer 143(4):921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]