

Abstract

This review surveys recent progress in the chemistry of polycyclic heteroaromatic molecules with a focus on structural diversity and synthetic methodology. The article covers literature published during the period of 2016–2020, providing an update to our first review of this topic (Chem. Rev.2017, 117 (4), 3479–3716).
1. Introduction
1.1. Structure and Scope
Since the publication of the first part of this review (denoted CR2017),1 the field of heterocyclic nanographenes and related polycyclic heteroaromatic systems (PHAs) has grown substantially. CR2017 surveyed relevant research published until the end of 2015 (over 1600 references). Soon after publication, it became apparent that, to keep up with the rapid progress of the area, an update might need to be prepared in the next few years. The present review covers relevant literature published since late 2015 until March 2021 and includes close to 800 references (Figure 1). Again, we mainly focus on atomically precise synthetic methods. Accordingly, the present update covers solution syntheses of small molecules and structurally well-defined polymers consisting of extensively fused subunits. Atomically resolved on-surface chemistry is also included.
Figure 1.

Partial citation timeline of CR2017 and the new references included in the present Review.
The scope of the present update retains the selection criteria adopted in CR2017. Briefly, the material is restricted to ring frameworks containing at least (a) one heteroatom, (b) one peri fusion point, (c) five fused rings, and (d) 20 π-conjugated atoms. By way of convention, all tri- and divalent atomic centers are considered to be π-conjugated, and no attempt was generally made to quantify the extent of p-orbital overlap. Curved aromatics are within the scope of the review; however, some highly twisted rings with evidently interrupted π-conjugation were omitted.
The classification of ring frameworks used in this review reflects the decreasing extent of benzenoid (graphene-like) fusion in the PHAs. However, because the review covers a range of research topics and subfields that are interrelated in a complex fashion, it was not always possible to classify such diverse material in a chemically relevant way. Thus, while some of these fields are presented in a single subsection, others may be discussed in more than one place. In such cases, we provide cross references to assist the reader in finding related work.
The present review generally retains the section structure of CR2017. This should help the reader in appreciating the progress in individual areas and in locating background information for the newest results. In some cases, the lowest hierarchy level was modified to better reflect the ongoing research in the field.
To keep the review as concise as possible, we only cite relevant original papers that have not been included in the first part of this review. It should also be noted that the work reviewed herein was often based on earlier (or parallel) developments of related carbocyclic systems or smaller heterocyclic molecules that do not fit into the scope of this review. The corresponding papers are generally not included in this review, but they may be easily identified in the cited references.
1.2. Recent Developments
The most recent developments of PHA chemistry have been motivated by advances in synthetic methodology and by the application potential of heteroatom-doped π-conjugated systems. Representative recent examples of PHA-based materials are highlighted in Chart 1 (for more information, check the corresponding sections). Several bay-annulated perylene diimide (PDI) derivatives, often with nonplanar cores, have emerged as high-performance materials for bulk-heterojunction photovoltaics (69.5,7, C5.4b, and 89.2b) and as effective DSSC dyes (64.2b, cf. Sections 3.2 and 3.3). The use of a π-extended PDI as a functional ligand yielded a highly efficient phosphorescent emitter 83.4. Progress in azaacene chemistry (Sections 2.5.1 and 4.6) provided access to solution-processable nitrogen-doped nanoribbons. Some of these molecules are remarkably long (cf. Scheme 158) and were investigated, e.g., as semiconductors for TFT devices (24.5a–e).
Chart 1. Examples of PHA-Based Materials.
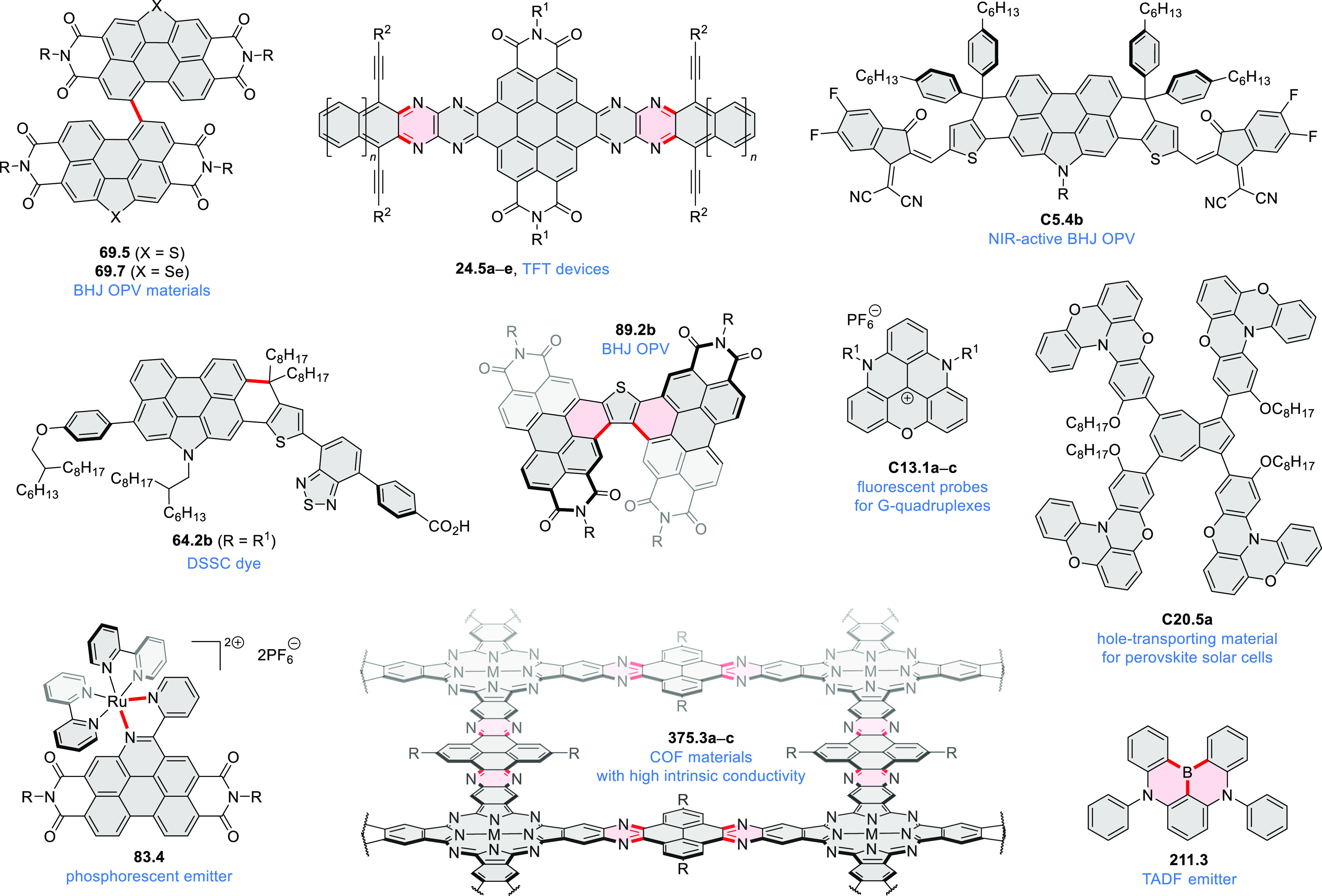
Scheme 158. Iterative Synthesis of Pyrazine-Containing Nanoribbons.
Reagents and conditions: (a)313 CHCl3, AcOH, reflux, 48 h; (b) TFA, water, rt; (c) CHCl3, AcOH, reflux, 4 days; (d) (1) TFA, water, rt, (2) CHCl3, AcOH, reflux, 3 days.
Exploration of boron-doped PHAs has produced many notable methodological advances (Sections 2.3, 3.1, 3.4, 4.1.1, 4.2, 4.3, 4.5, 4.6, 5.1, and5.3.1, 7.3.1). Compound 211.3, an ultrapure-blue emitter based on thermally activated delayed fluorescence, simultaneously highlights the use of B-doping and the utility of “triangular” fusion in designing new materials. C20.5a, combining four oxygen-bridged triphenylamine subunits, contains a similar fusion pattern and was employed as a hole-transporting material for perovskite solar cells. The related heteratriangulenes (Section 4.1), available in a variety of doping patterns, are often emissive and can be used, e.g., for bioimaging applications (C13.1a–c).
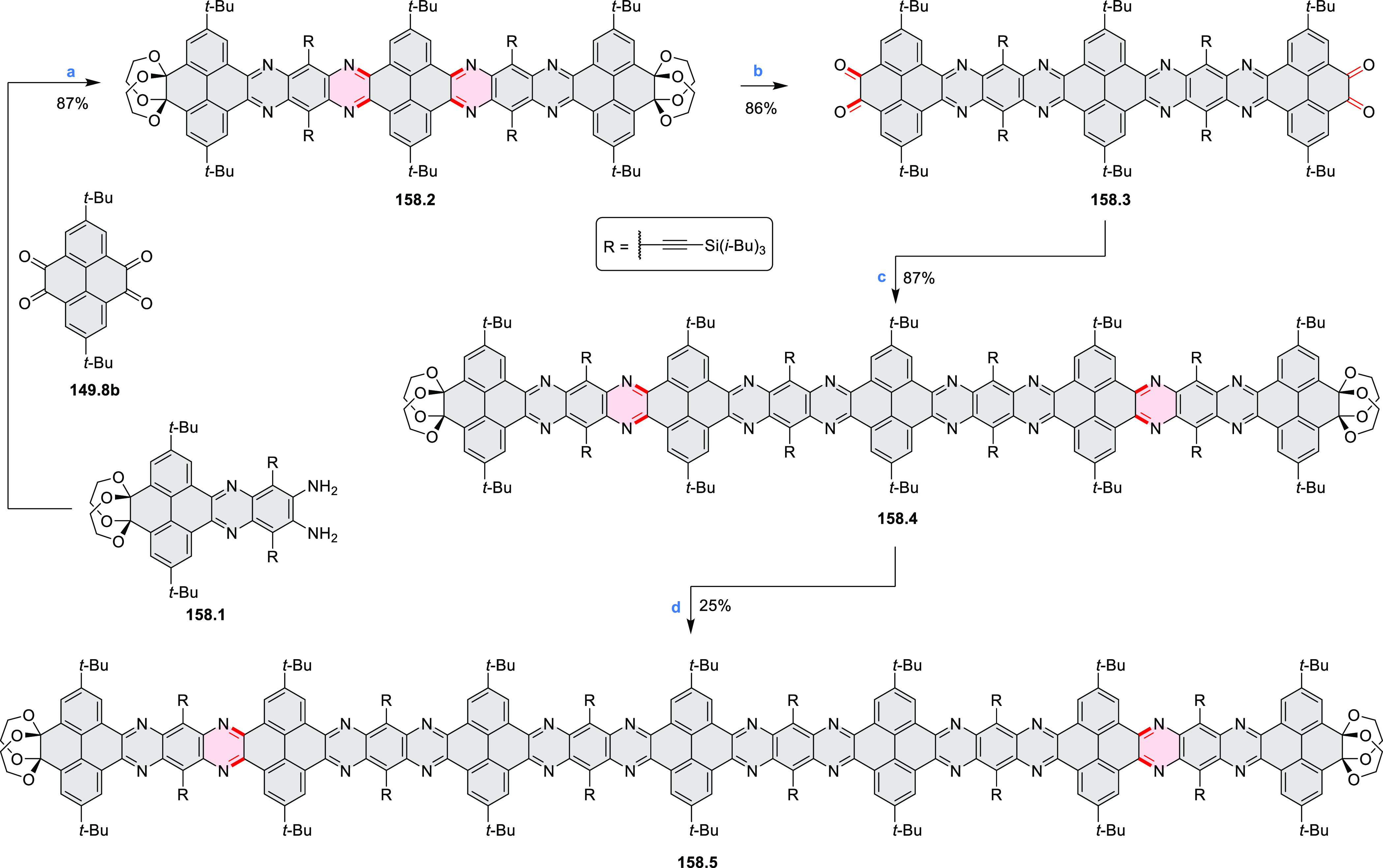
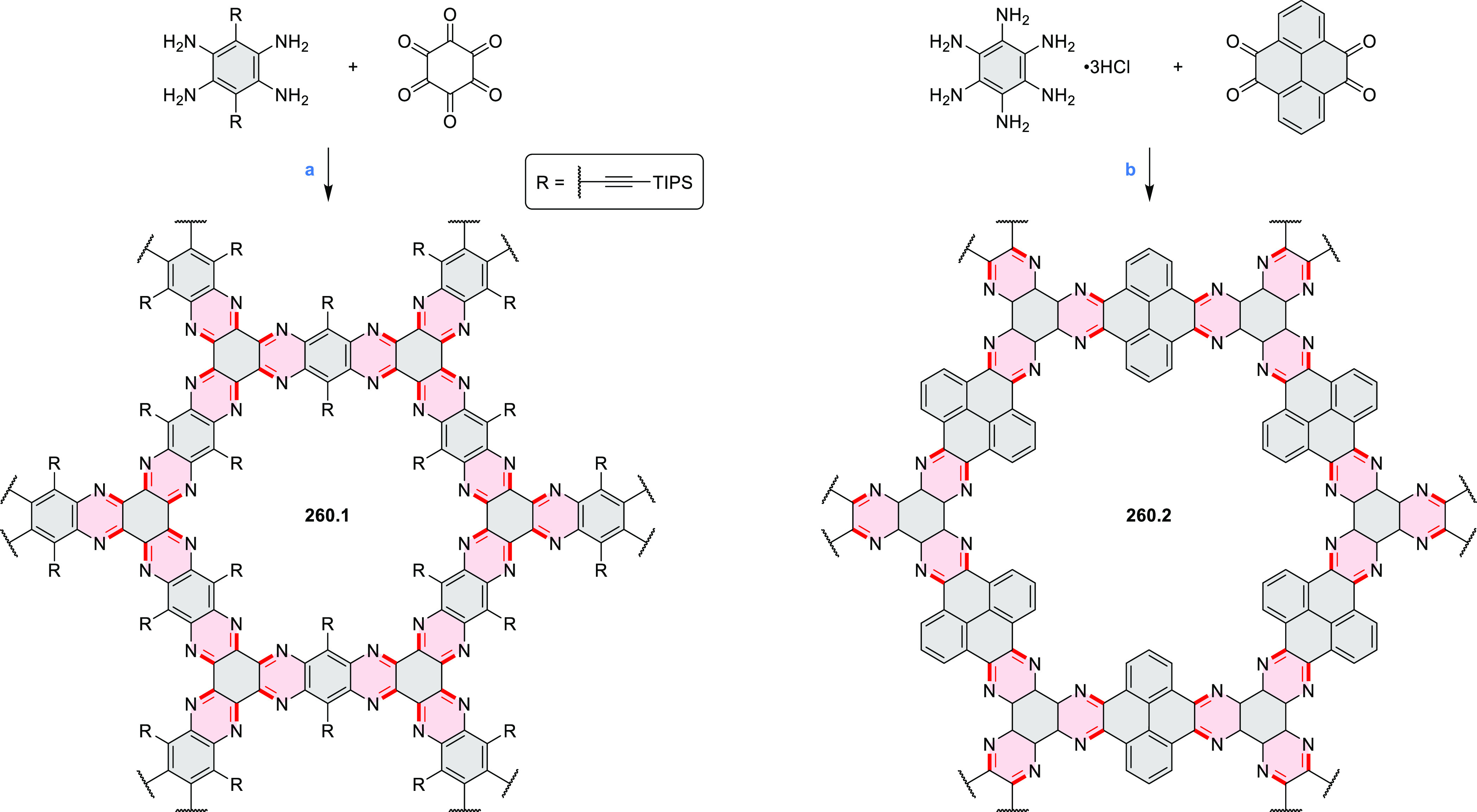
PHA motifs have been incorporated into several types of covalent organic frameworks (COFs, e.g. Scheme 260, Section 6.1.6; Scheme 375, Section 7.5.5; and Scheme 393, Section 7.7.2). COFs 375.2a–c, notable for their high intrinsic conductivity, additionally illustrate the progress in macrocyclic PHAs, which is surveyed in Section 7. This area is notable for the development of several unprecedented fusion motifs and creation of some remarkable 3D-fused structures. A variety of new heteroatom-doped circulenes and coronoids have been developed (Section 6.1), including systems with significant positive or negative curvature.
Scheme 260. Phenazine-Fused Porous Conjugated Frameworks.
Reagents and conditions: (a)484 dioxane/AcOH (1:4), 135 °C, 7 days; (b)485 TfOH, 175 °C.
Scheme 375. Phthalocyanine-Based Pyrazine-Linked Conjugated Two-Dimensional Covalent Organic Frameworks.
Reagents and conditions: (a)739 dimethylacetamide/o-dichlorobenzene (1:1, v/v), H2SO4, 202 °C, 10 days; (b)740 1-methyl-2-pyrrolidone/mesitylene (2:1, v/v), p-TSA (3.5 M), 150 °C, 3 days.
Scheme 393. Shape-Persistent Macrocycles Containing peri-Condensed Pyridine Rings.
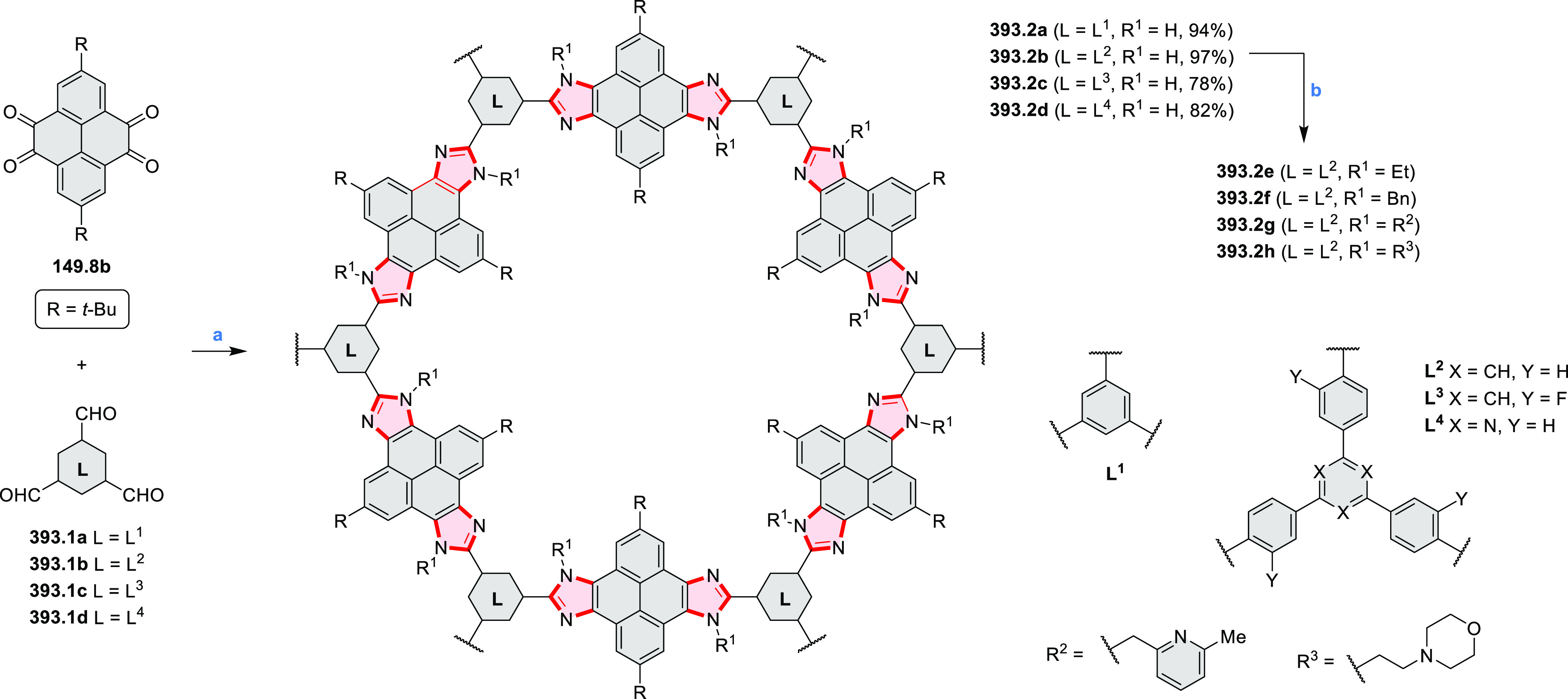
Reagents and conditions: (a)331 NH4OAc, dioxane/mesitylene (1:4, v/v) 150 °C, 5 days; (b) R1-Br, NaH, THF, 65 °C, 2 days.
1.3. Ring-Forming Reactions
The synthetic chemistry of PAHs relies primarily on efficient ring-forming reactions. These transformations are necessary not only to make heterocyclic rings but also to build fused carbocyclic units and create macrocycles. The core methodology of PHA synthesis was summarized in the original review (CR2017, Section 1.4). Below we provide a brief summary of notable recent developments (Schemes 1, 2, and 3).
Scheme 1. Carbocycle Formation via Cyclization and Annulation Reactions.
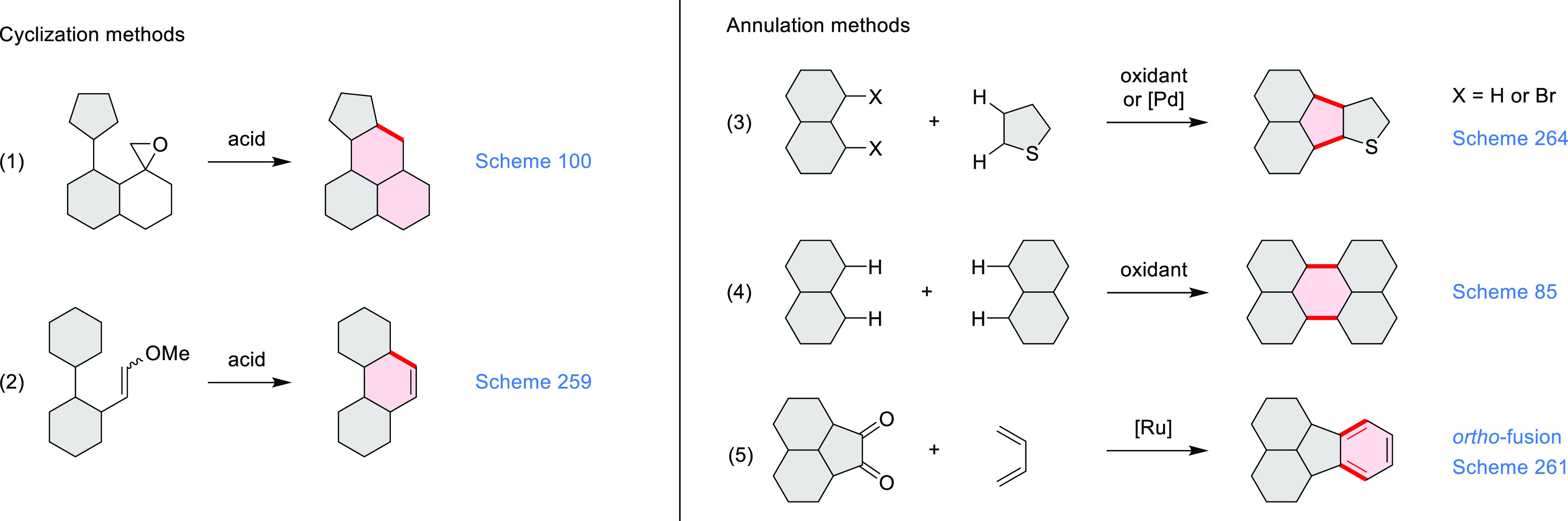
π-Conjugation is implicit in shaded rings. Numbers correspond to representative schemes and charts.
Scheme 2. Heterocycle Formation via Cyclization Reactions.
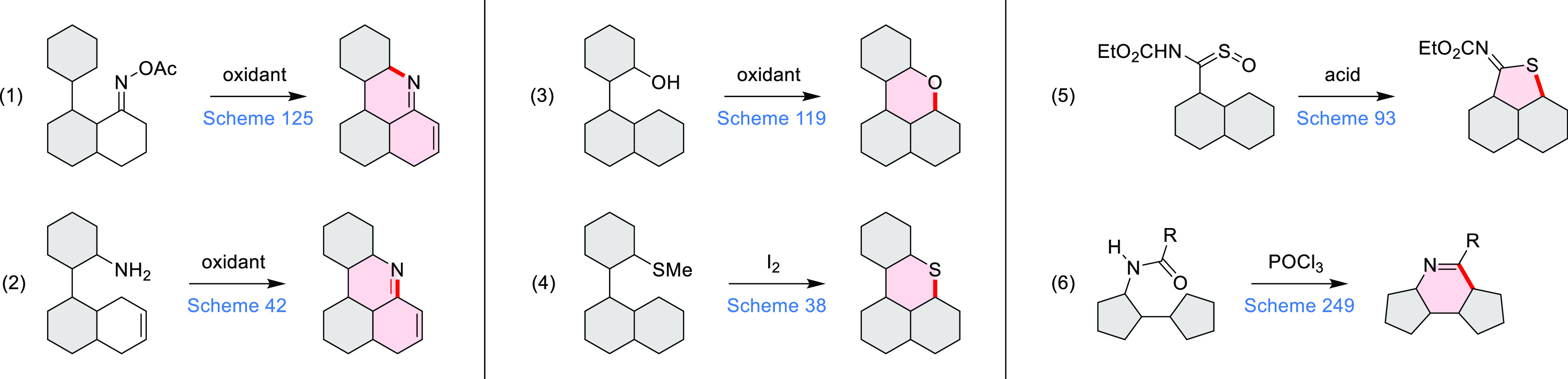
π-Conjugation is implicit in shaded rings. Numbers correspond to representative schemes and charts.
Scheme 3. Heterocycle Formation via Annulation Reactions.
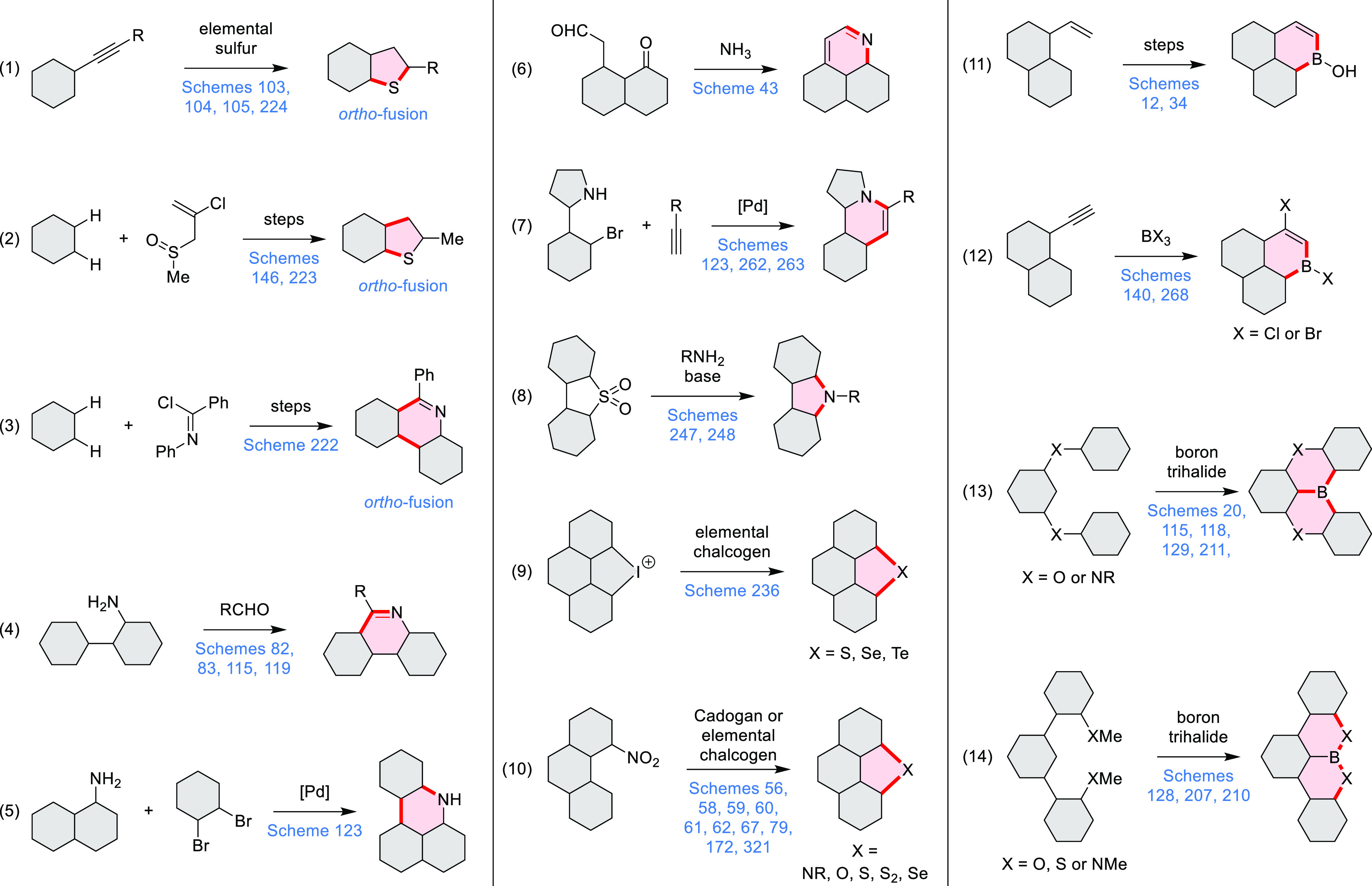
π-Conjugation is implicit in shaded rings. Numbers correspond to representative schemes and charts.
Closures of six- and five-membered carbocyclic rings involve a variety of approaches, both conventional and transition-metal-catalyzed ones. The utility of electrophilic cyclizations has been extended by introduction of new reactive electrophile units, such as the epoxide ring and the 2-methoxyvinyl group (Scheme 1, entries 1 and 2, cf. Schemes 100 and 259). Annulations involving unfunctionalized components, such as the recently reported direct pentannulations of thiophene (cf. Scheme 264) and the oxidative self-dimerization of an electron-rich naphthalene (cf. Scheme 85), can achieve a rapid increase in molecular complexity from readily available starting materials. Ruthenium-catalyzed benzannulations involving 1,3-butadiene, such as the one shown in Scheme 261, provide similar advantages in the construction of ortho-fused benzene rings.
Scheme 100. Syntheses of Pyridine- and Thiophene-Fused Perylenes.
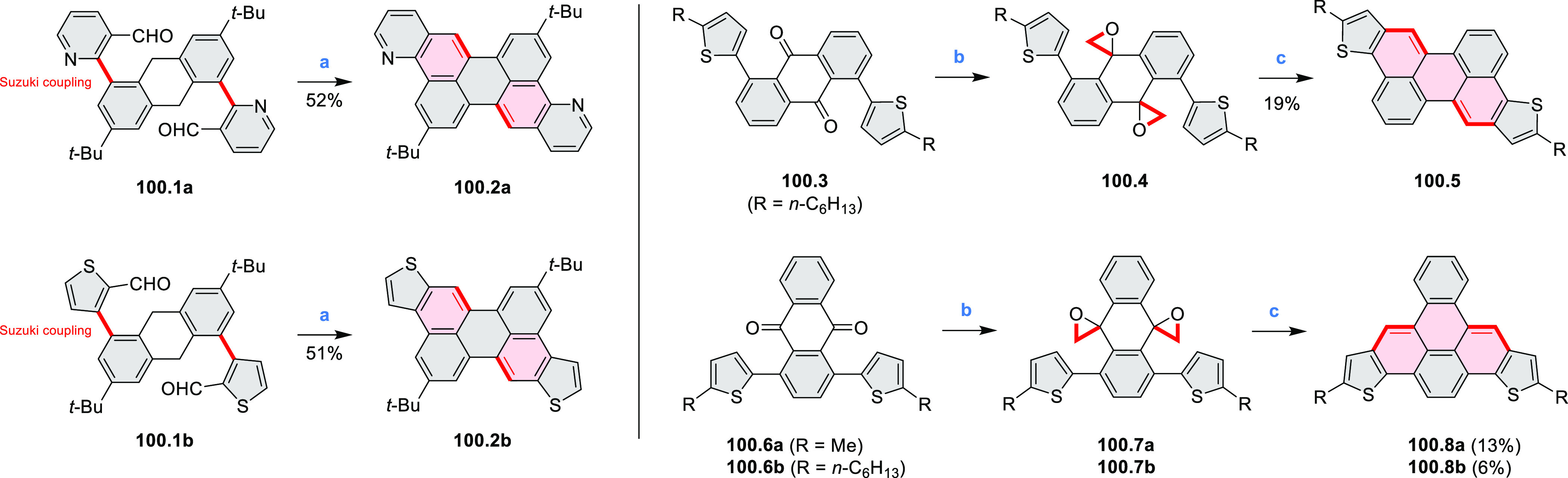
Reagents and conditions: (a)203 KOt-Bu, THF, 60 °C, 16 h; (b)204 Me3SI, sodium hydride, DMSO/THF, 50 °C, 1 h; (c) SnCl2, 1,2-dichloroethane, 60 °C, 12 h.
Scheme 259. Dibenzothiophene–Phenanthrene-Based Macrocycles.
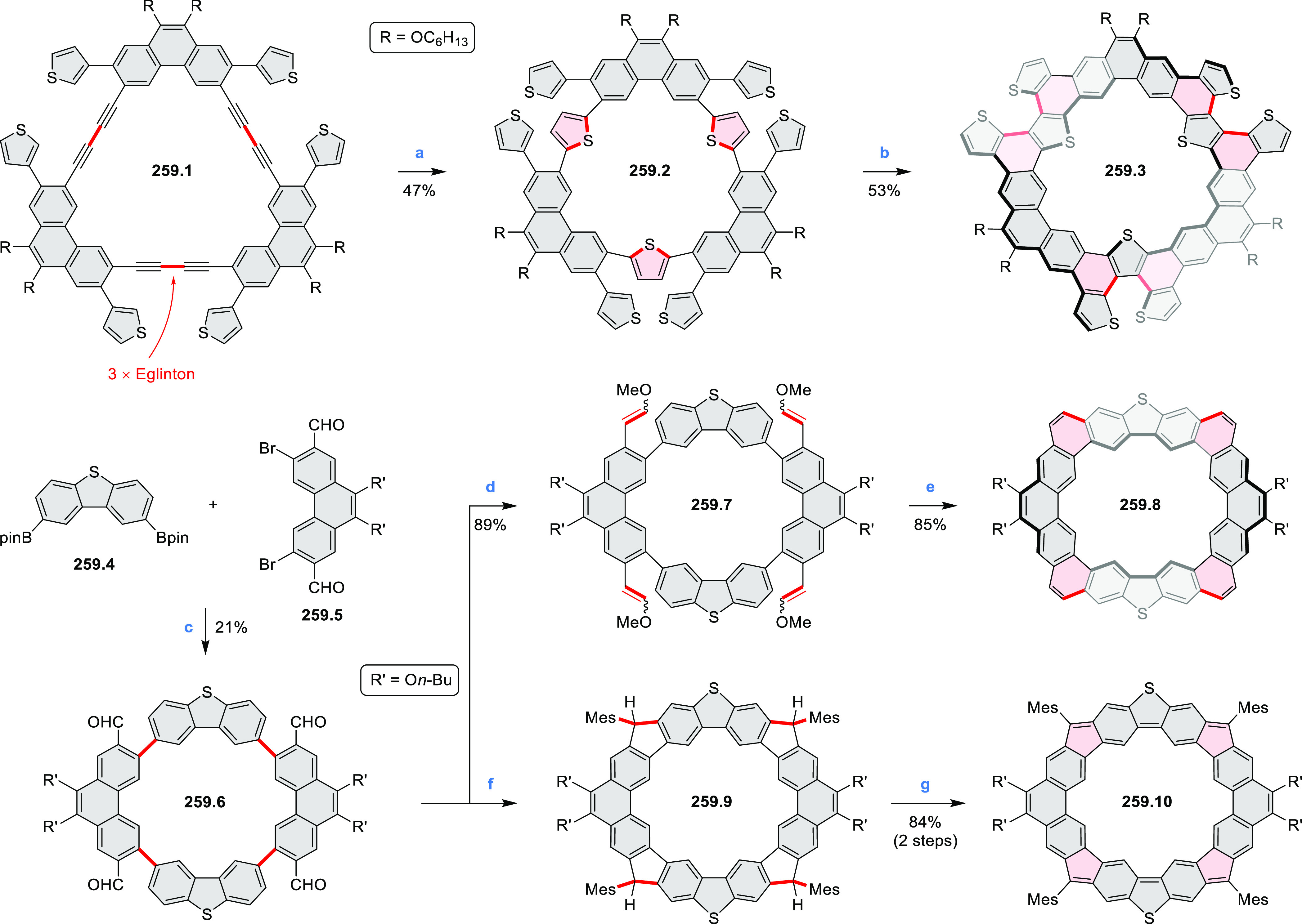
Reagents and conditions: (a)481 Na2S·9H2O, CuI, 1,10-phenanthroline, DMF, 140 °C, N2, 16 h; (b) FeCl3, DCM/MeNO2, bubbling with N2, 0 °C, 10 min; (c)482 Pd2(dba)3, t-Bu3P·HBF4, NaHCO3, THF/H2O, 80 °C, 2 days; (d) premixed [MeOCH2PPh3]+Cl– and t-BuOK, THF, rt, 2 h; (e) Bi(OTf)3, dry 1,2-DCE, rt, 3 h; (f)483 (1) MesMgBr, THF, rt, overnight, (2) BF3·OEt2, DCM, 3 h; (g) DDQ, DCM, rt, 6 h.
Scheme 264. Five-Membered Ring Closures Involving a Thiophene Ring.

Reagents and conditions: (a)494 Pd(OAc)2, t-Bu2PMe·HBF4, K2CO3, DMA, 100 °C, 20 h; (b) (1) 3,5-di-t-butylphenylacetylene, n-BuLi (premixed at −78 °C for 1 h), then rt for 1 h, THF, rt, overnight, (2) SnCl2·H2O, 10% aq. HCl, 50 °C, 2 h; (c)494p-chloranil (1.5 equiv), DCM/TfOH (100/1), rt, 6 h.
Scheme 85. Synthesis of Pyridazine-Fused Perylenes.

Reagents and conditions: (a)167 KMnO4, 1:9 MeOH/H2O, rt, 12 h, 87%; (b) N2H4·H2O, EtOH, rt, overnight, 85%; (c) n-BuBr, NaOH, DMF, 60 °C, overnight, 40%.
Scheme 261. Synthesis of N-Doped Rubicenes.
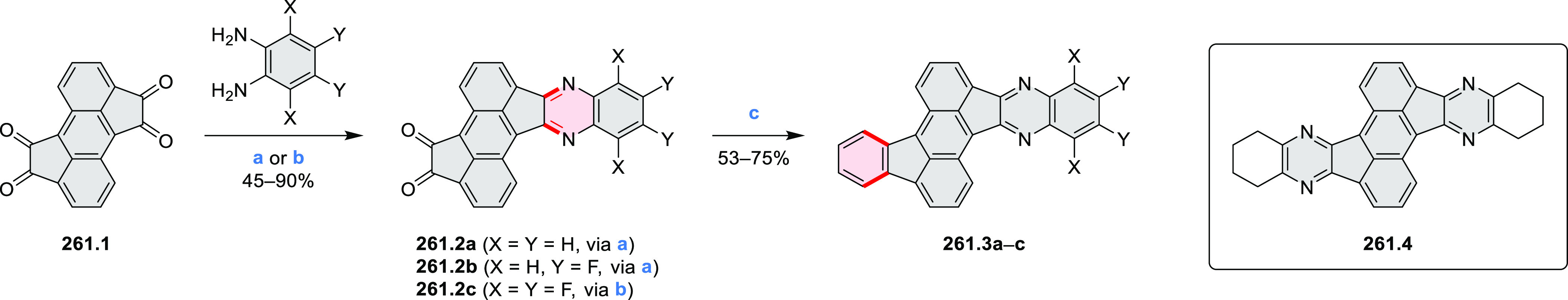
Reagents and conditions: (a)491 pyridine, 90 °C, 16–48 h; (b) Sc(OTf)3, 1,2-DCE, 25 °C, 24 h; (c) (1) 1,3-butadiene, Ru3(CO)12, t-Bu2PMe·HBF4, t-BuOK, i-PrOH, toluene with or without DMA, 140 °C, 48 h, (2) TsOH, toluene, 90 °C, 16 h.
Several oxidative cyclization methods for cove-region heterocycle formation have been recently described (Scheme 2). They include oxidative transformations of oximes and amines (entries 1 and 2, cf. Schemes 125 and 42), phenols (entry 3, cf. Scheme 119), and thioethers (entry 4, cf. Scheme 38), yielding, respectively, N-, O-, and S-doped substructures. Electrophilic heterocyclizations have been used to close both five- and six-membered rings. In one example, the acid-catalyzed dehydrative cyclization of thioamide S-oxide generates a peri-fused sulfur-containing heterocycle (entry 5, cf. Scheme 93). peri-Fused pyridine rings have been closed using an intramolecular variant of the Vilsmeier–Haack reaction (entry 6, cf. Scheme 249).
Scheme 125. Cationic Azapyrenoids.
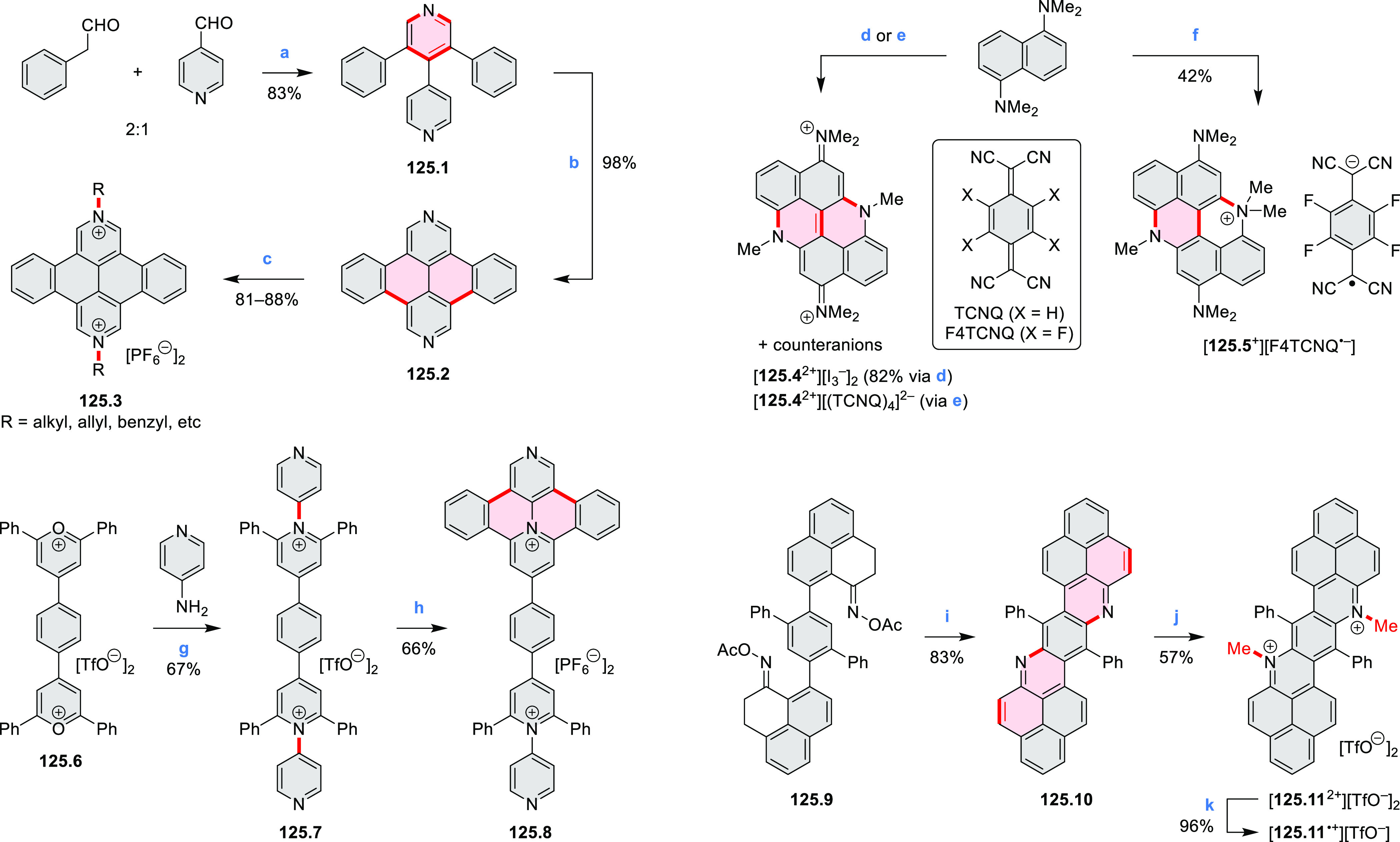
Reagents and conditions: (a)254 (1) KOH, EtOH, 0 °C, rt, 26 h, (2) NH2OH·HCl, reflux, 2 h; (b) 5% HCl, hν (300 nm), air, rt, 24 h; (c) (1) MeI or RBr, DMF, 100 °C, 48 h, (2) KPF4, H2O, rt; (d)255 I2, pyridine/1,4-dioxane, rt, 12 h; (e) TCNQ, MeCN, reflux, 20 h; (f) 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane, MeCN, reflux, overnight; (g)256 (1) DMF, argon, 60 °C, 2 h, (2) toluene, Dean–Stark apparatus, 140 °C, 6 h; (h) (1) MeCN, O2, hν (300–400 nm), 14 h, (2) NH4PF6; (i)257 Fe(acac)3, AcOH, 90 °C, 12 h; (j) MeOTf, 1,2-DCE, 50 °C, 16 h; (k) Et3N, MeCN/DCM, argon, rt, 2 h.
Scheme 42. Synthesis of Diazaperylenoids via Metal-Free C(sp3)–H Aminations with Unprotected Anilines.

Reagents and conditions: (a)71t-BuOK, O2, DMF, 120 °C.
Scheme 119. π-Extended Dioxa- and Diazadioxatriangulene.

Reagents and conditions: (a)77 CuO, nitrobenzene, air, reflux, overnight; (b)241 TFA, toluene, O2, 100 °C, 16 h.
Scheme 38. Synthesis of a Sulfur-Doped Perylenoid.

Reagents and conditions: (a)67 I2, CHCl3, 1 h at 70 °C, 1 h at 80 °C, 22 h at 90 °C.
Scheme 93. Thiophene Imine-Fused Perylene.

Reagents and conditions: (a)192 TfOH, DCM, rt; (b) oxone, 1:1 MeCN/H2O, rt, 1 h.
Scheme 249. Tetrathia[8]circulenes and Their Aza-, Sila-, and Germa-Substituted Derivatives.
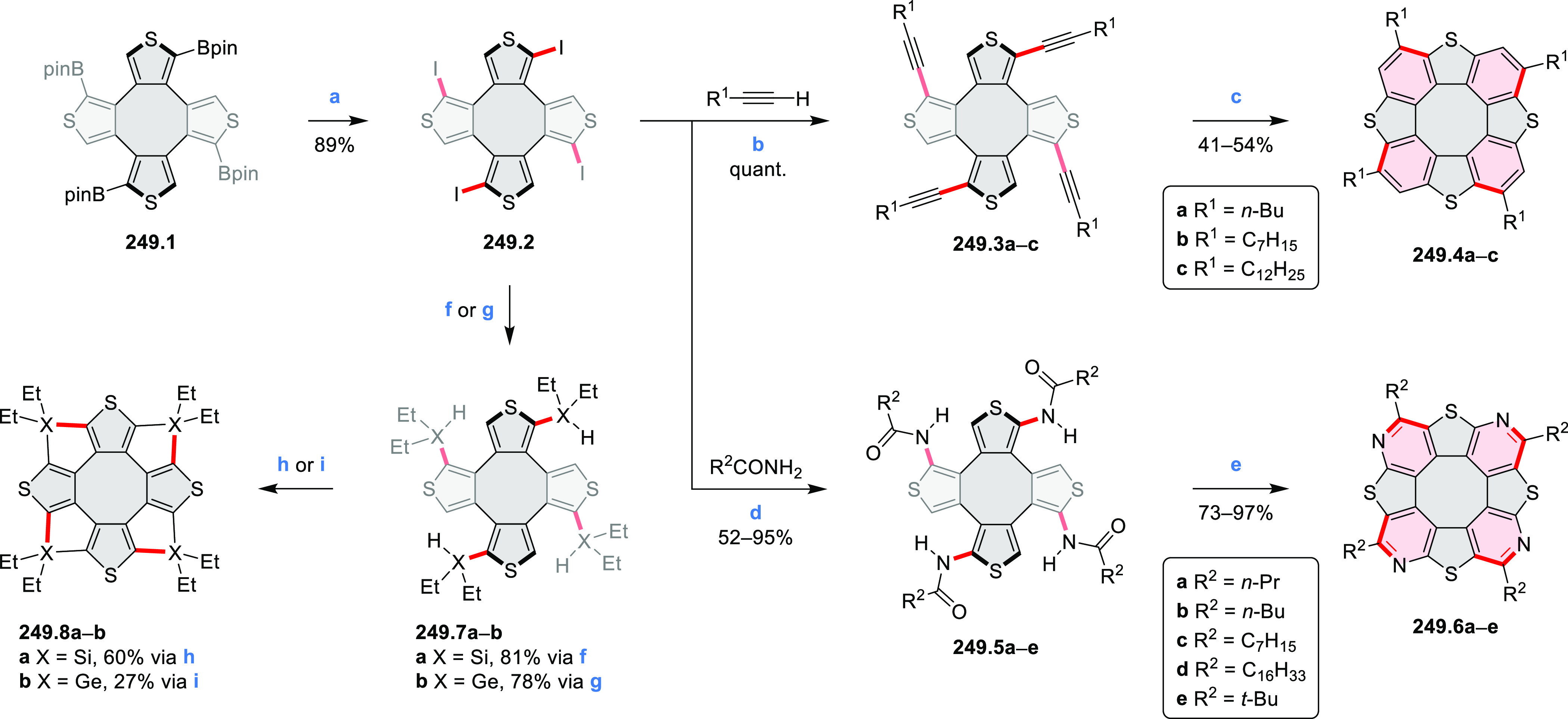
Reagents and conditions: (a)466 NIS, CuI, DMF, 80 °C, 3 h; (b)467 Pd(PPh3)2Cl2, CuI, Et3N, 60 °C, 3 h; (c) DBU, NMP, reflux, 12 h; (d)468 CuSO4·5H2O, K2CO3, N,N′-dimethylethylenediamine, toluene, 80 °C, 20 h; (e) POCl3, toluene, 120 °C, 2 h; (f)466 R2SiH2, Pd(t-Bu3P)2, Et3N, THF, rt, 48 h; (g)469 Et2GeH2, Pd(t-Bu3P)2, i-Pr2NEt, THF, rt, 18 h; (h)466 [RhCl(cod)]2, dppf, toluene, 140 °C, 24 h; (i)469 [RhCl(cod)]2, (C6F5)2PCH2CH2P(C6F5)2, toluene, 140 °C, 20 h.
Examples of heterocycle-forming annulation reactions are collected in Scheme 3. ortho-Thienannulation protocols have been developed for alkynyl-substituted arenes (entries 1 and 2, cf. Schemes 103, 104, 105, and 224) and unfunctionalized arenes (Schemes 146 and 223). Several methods for construction of ortho- or peri-fused pyridines have been reported, some involving functionalized arenes (e.g., entry 3, cf. Scheme 222). In alternative approaches, aminoarenes were condensed with aldehydes (entry 4, cf. Schemes 82, 83, 119, and 242) or with 1,2-dibromobenzene under palladium catalysis (entry 5, cf. Scheme 123). peri-Fused pyridine rings were also formed via the condensation of a 1,5-dicarbonyl-containing arene with ammonia (entry 6, cf. Scheme 43) or via Sonogashira coupling followed by intramolecular hydroamination of alkyne (entry 7, Schemes 262, 123, and 263).
Scheme 103. Heteroannulation of Alkynyl-Substituted Perylene Diimides.
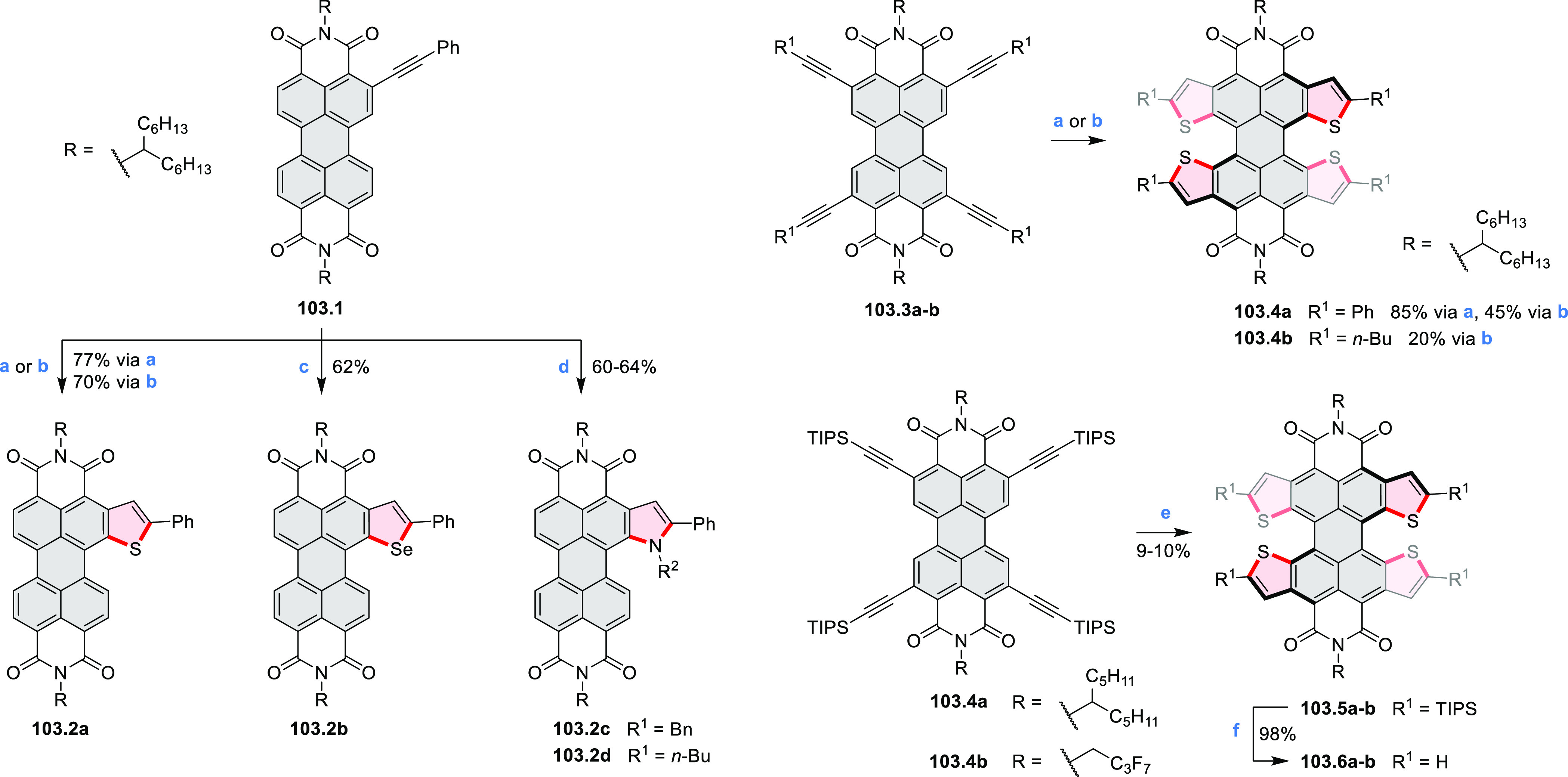
Reagents and conditions: (a)208 sulfur powder, DMA, 140 °C, 20–48 h, 77–85%; (b) K2S, DMF, 80 °C, 20–48 h; (c) selenium powder, DMA, 140 °C, 36 h, 62%; (d) benzylamine or n-butylamine, KOt-Bu, toluene, 4 Å MS, 120 °C, 18 h, 60–64%; (e)210 sulfur powder, 10:1 DMF/H2O, 90 °C, 45 h, 9–10%; (f) TBAF, THF, rt, 1 h, 98%.
Scheme 104. Synthesis of Perylenodithiophenes and Their Dimers.
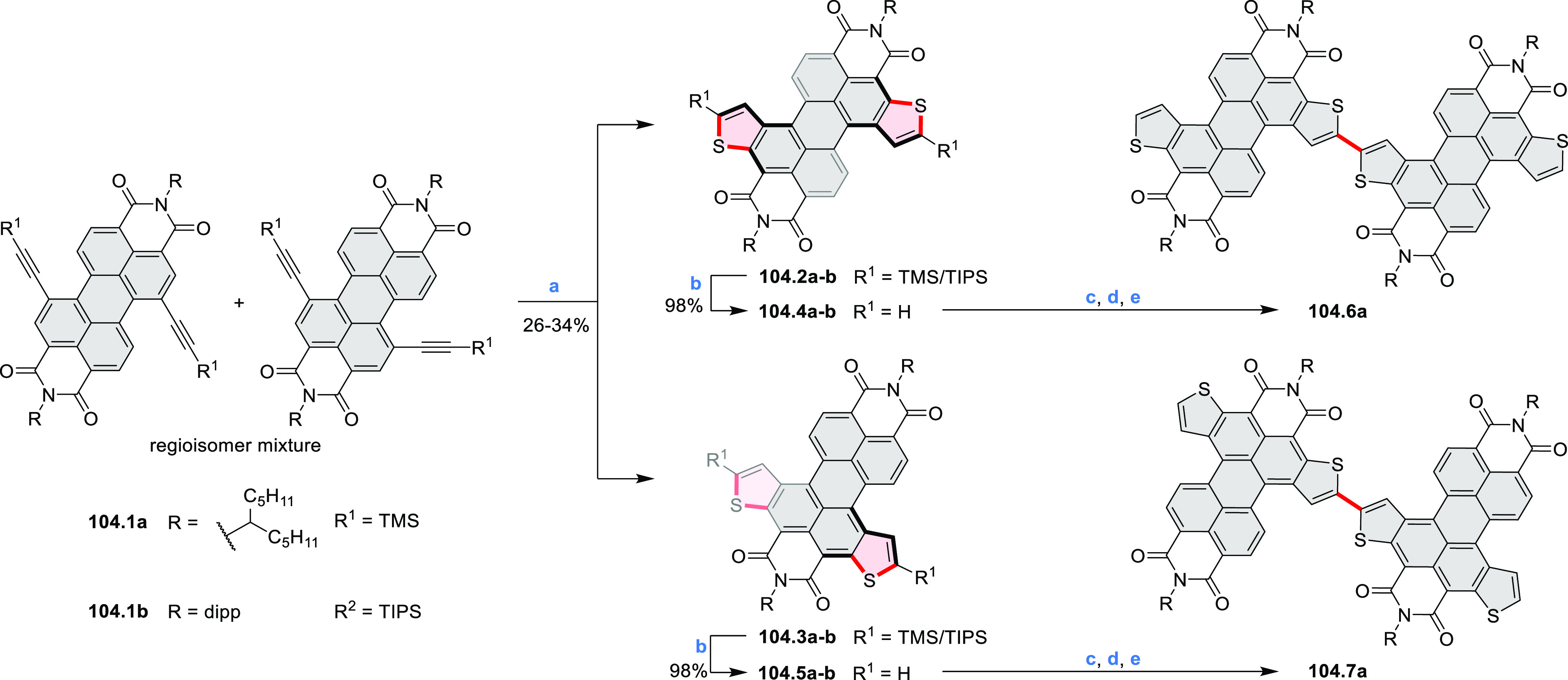
Reagents and conditions: (a)211 sulfur powder, 10:1 DMF/H2O, 90 °C, 12 h, 26–34%; (b) K2CO3, 10:1 THF/MeOH, rt, 3 h, 98%; (c) NBS, 20:1 DCM/AcOH, 55 °C, 79–80%; (d) Cu powder, DMSO, 90 °C, 10 h; (e) PdCl2(dppf), TMEDA, NaBH4, THF, 55 °C, 3 h, 45–66% (two steps).
Scheme 105. Stitching Thienannulations of Alkynyl-Substituted PDIs.
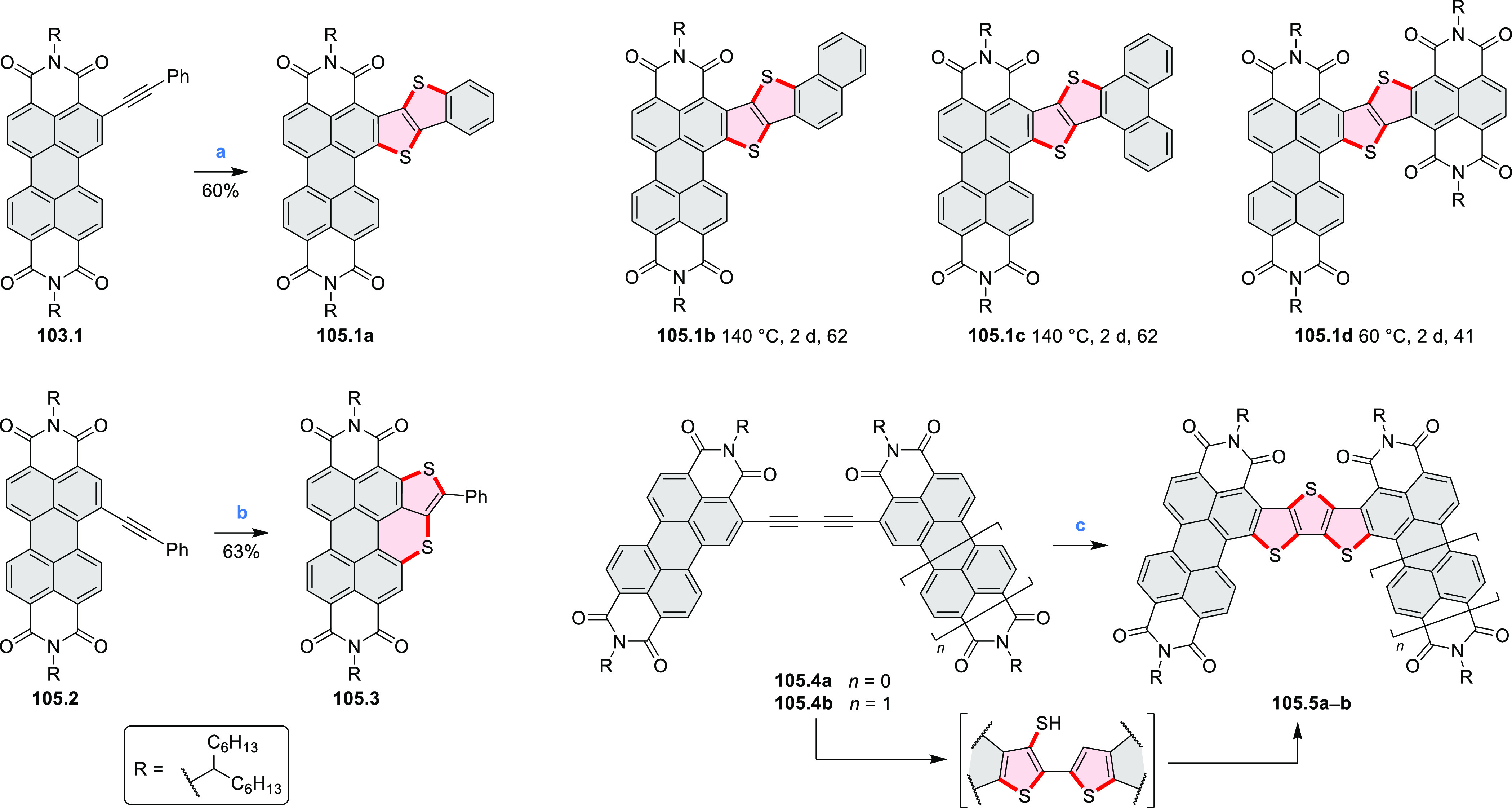
Reagents and conditions: (a)212 K2S, DMF, 170 °C, 12 h, 60%; (b) K2S, DMF, 40 °C, 1 h, 63%; (c) K2S, DMF, 105.5a: 80 °C, 2 days, 61%, 105.5b: 25 °C, 15 h, then 140 °C, 2 days, 79%.
Scheme 224. Thienannulation of Arylethynyl-Substituted Corannulenes with Elemental Sulfur.
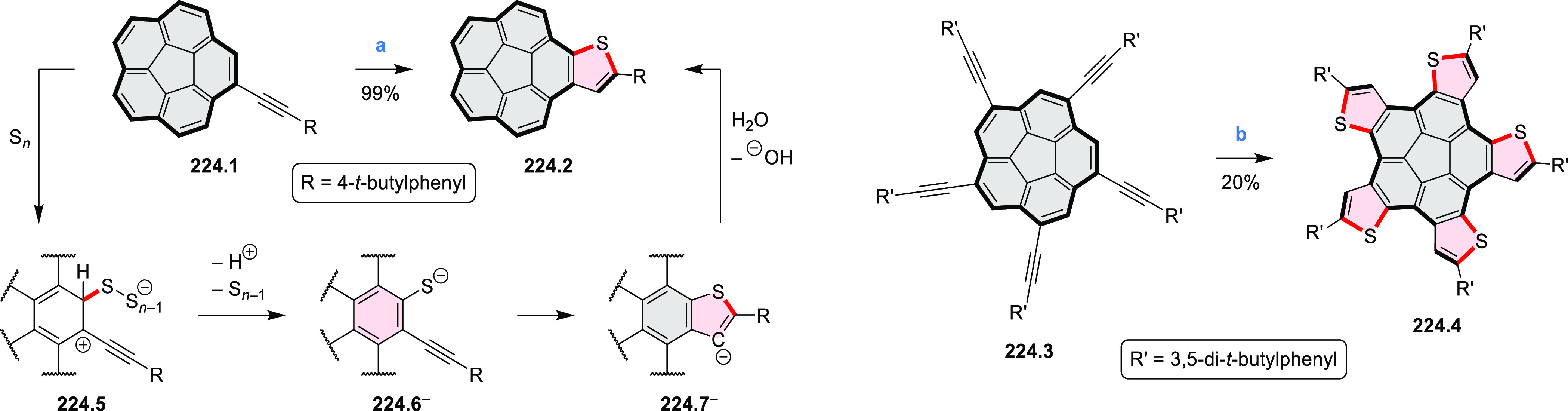
Reagents and conditions: (a)209 S8 (0.5 equiv), 140 °C, 24 h; (b) S8 (5 equiv), DMF, 140 °C, 48 h.
Scheme 146. One-Pot Synthesis of an [a]-Thiophene-Fused Pyrene.

Reagents and conditions: (a)284 (1) 146.5, triflic anhydride, DCE, −30 to 90 °C, MW heating, (2) Et3N, 0 to 50 °C; (b) DDQ, toluene, rt to 80 °C; (c) (1) 146.6, triflic anhydride, DCE, −30 to 90 °C, MW heating, (2) Et3N, 0 to 50 °C.
Scheme 223. Transition-Metal-Free, One-Pot Thienannulation of Corannulene.

Reagents and conditions: (a)284 (1) Tf2O, 1,2-DCE, −30 °C, 20 min, then rt, 1 h, (2) microwave, 90 °C, 12 h, (3) Et3N, rt, 20 min, then 50 °C, 5 h.
Scheme 222. Aza-Annulative π-Extension Reaction of Corannulene.

Reagents and conditions: (a)420 (1) AgPF6, 1,2-DCE, 80 °C, 17 h, (2) p-chloranil, rt, 3 h.
Scheme 82. Azabenzannulation of PDI by Cyclization with an Imine.

Reagents and conditions: (a)162 (1) 3 equiv of R1CHO, TFA, DCM, reflux, 3 h, (2) DCM, rt, hν (white LED), 1 h (12 h for 82.2c), (3) DDQ, DCM, rt, 2 min; (b) (1) 0.33 equiv of dialdehyde, TFA, toluene, reflux, 3 h, (2) DCM, rt, hν (white LED), 12 h, (3) DDQ, DCM, rt, 2 min.
Scheme 83. Coordination Chemistry of a Fused Perylenoid Ligand.

Reagents and conditions: (a)164 TfOH, DMF, 110 °C, 2–3 h, 52–55%; (b) [Cp*IrCl2]2, 40 °C, 18 h, then NH4PF6, EtOH, 67%; (c) Ru(bpy)2Cl2, LiCl, AgClO4, 20:7:1 CHCl3/EtOH/Et3N, 65 °C, 42 h, 69%; (d)165 [(ppy)2IrCl]2, 5:1 DCM/MeOH, 80 °C, 24 h, then NH4PF6, 2 h, 20%.
Scheme 242. [6]Heteracirculenoids Bearing Chalcogenole and Pyridine Rings.

Reagents and conditions: (a)449t-BuONO, DCM, rt, 4 h; (b) Zn, AcOH/EtOH, 0 °C, 10 min, then rt, 3 h; (c) TFA, 120 °C, 12 h.
Scheme 123. Construction of π-Extended Diazapyrenoids by Annulation.
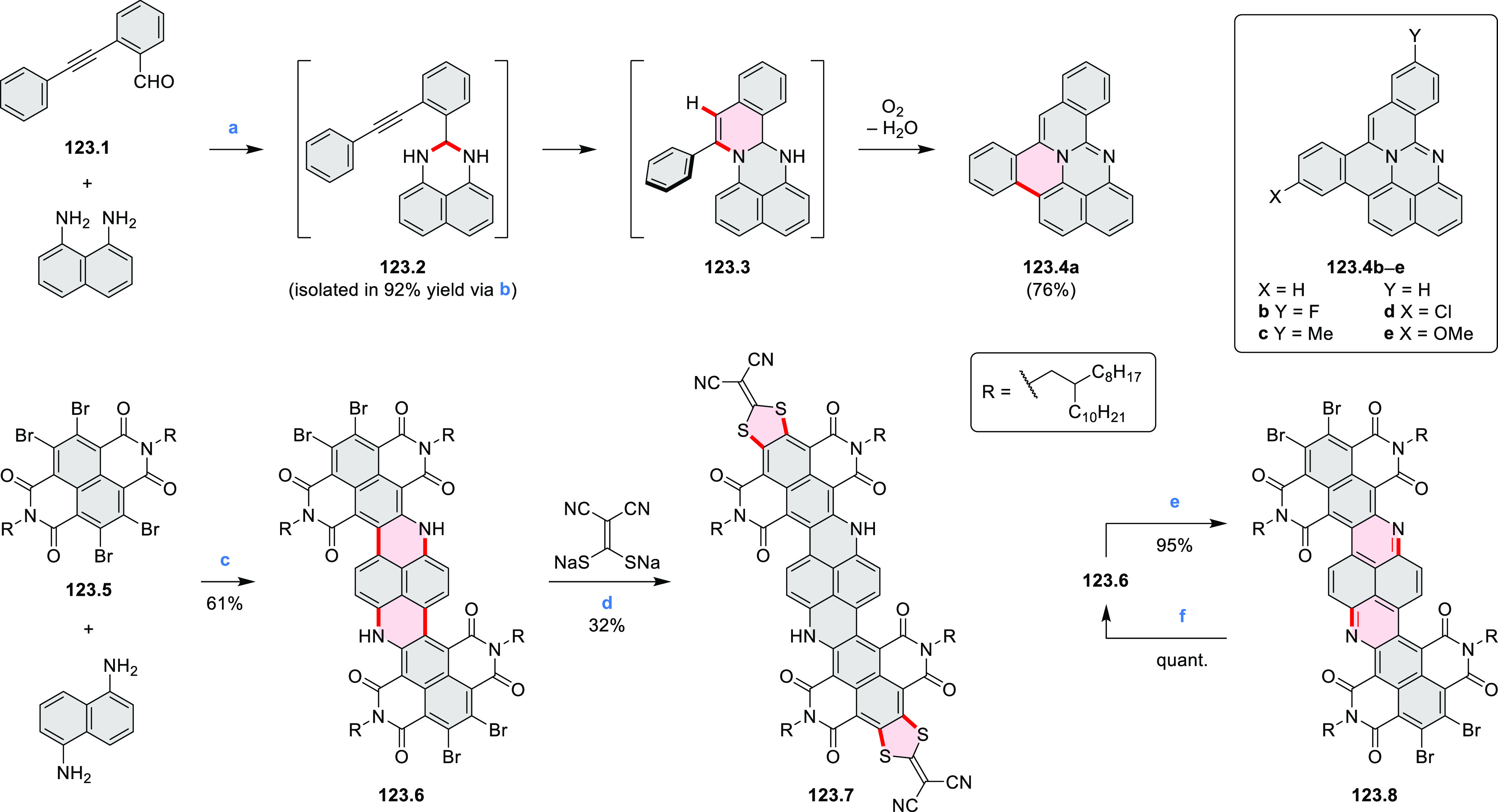
Reagents and conditions: (a)249 Cu(OAc)2 (20 mol %), Cs2CO3 (1.0 equiv), dioxane, O2, 100 °C; (b) dioxane, rt, 6 h; (c)250 K2CO3, THF, 85 °C, 8 h; (d) THF, 80 °C, 36 h; (e) p-phenylenediamine, CHCl3, rt, several seconds; (f) PbO2, CHCl3, 45 °C, 5 h.
Scheme 43. Synthesis of 1,7-Diazaperylene and Its Dialkyl Derivatives.
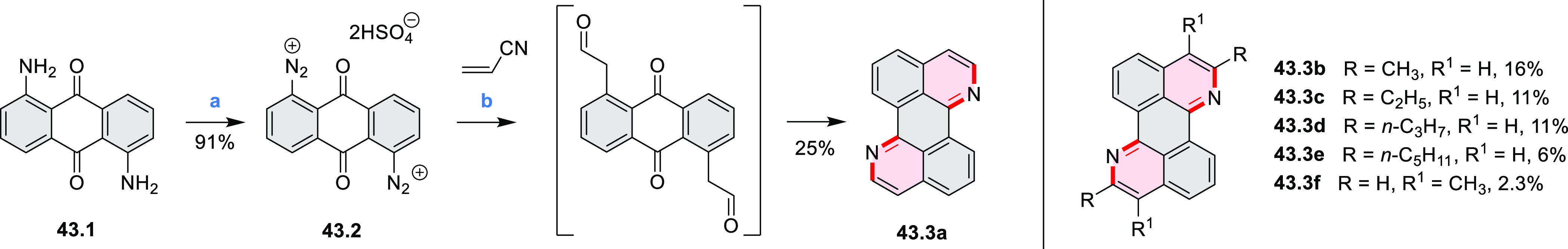
Reagents and conditions: (a)72 NOHSO4, conc. H2SO4, rt, 1 h, 91%; (b) CuCl, H2SO4, t-BuOH, 55–60 °C, then 10:1 25% NH3(aq)/CHCl3, rt, 24 h, 25%.
Scheme 262. Miscellaneous Acenaphthylenes Hetero[a]fused with Nitrogen Heterocycles.
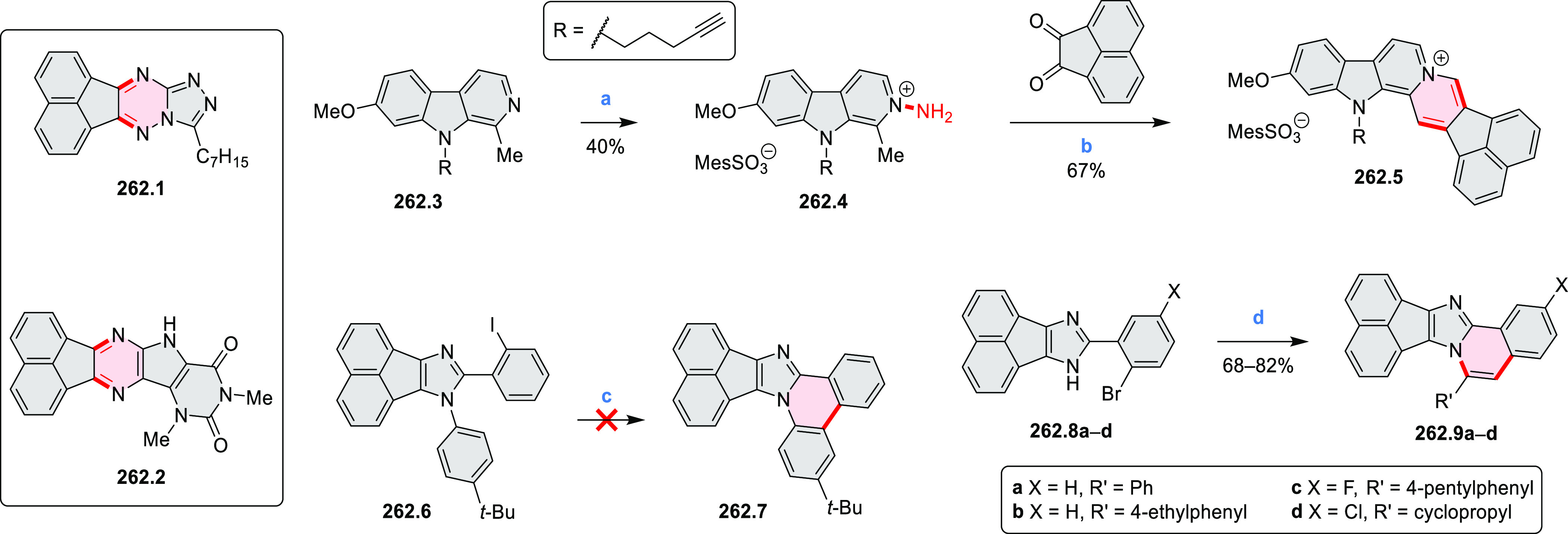
Reagents and conditions: (a)493 MesSO3NH2, DCM, 1 h; (b) acenaphthoquinone, NaOAc, EtOH, reflux, 1 h; (c)333hν (254 nm, 6 × 8 W), 48–72 h; (d)494 R′C≡CH, CuI (10 mol %), 1,10-phenanthroline (20 mol %), Cs2CO3 (1 equiv), dioxane, 100 °C, 26–32 h.
Scheme 263. Formation of an Acenaphtho[1,2-b]benzo[g]indole Skeleton via Cascade Reaction.
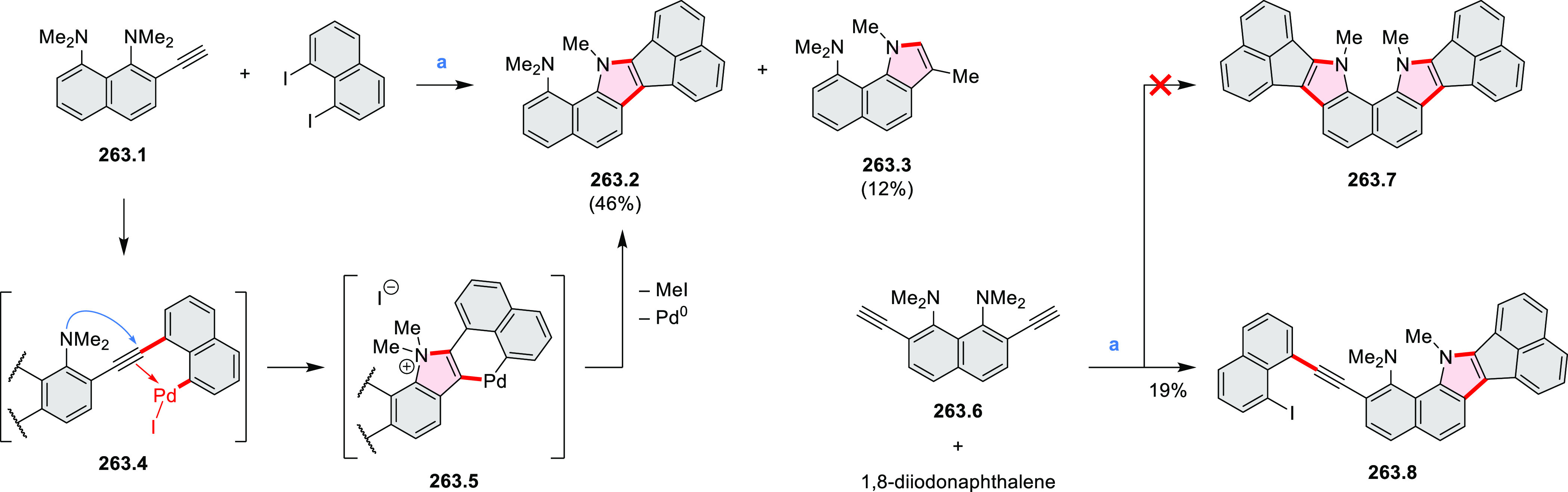
Reagents and conditions: (a)495 Pd2(dba)3, CuI, PPh3, K2CO3, DMF, argon, 60–75 °C, 8–10 h.
Conversions of reactive heterocycles containing heavier heteroelements have found use as a strategy toward nitrogen- and chalcogen-containing rings. In one approach, the dibenzothiophene S,S-dioxide moiety was transformed to the corresponding carbazole moiety via the SNAr mechanism (entry 8, cf. Schemes 247 and 248), whereas the five-membered cyclic iodonium moiety was converted to the fused thiophene, selenophene, and tellurophene rings (entry 9, cf. Scheme 236). Nitroarenes play an important role in the classical Cadogan syntheses of indoles and carbazoles and have also been applied for the formation of oxygen-, sulfur-, and selenium-containing rings in perylenoids (entry 10, cf. Schemes 56, 58, 59, 60, 61, 62, 67, 79, 172, and 321).
Scheme 247. Synthesis of Tetraaza[8]circulene Derivatives via SNAr Reaction.
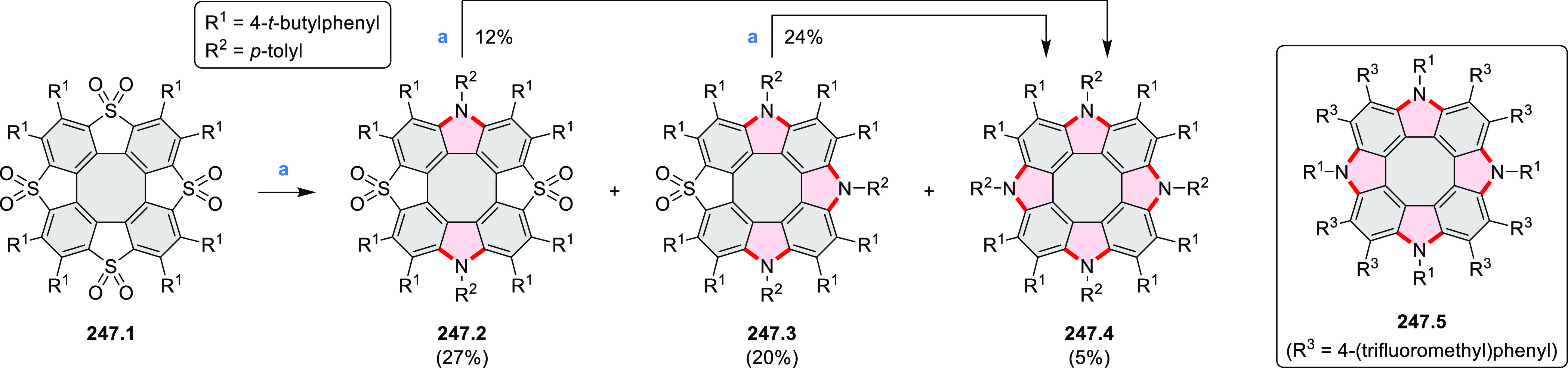
Reagents and conditions: (a)460p-toluidine, KHMDS, 1,4-dioxane/toluene, 85 °C, 24 h.
Scheme 248. Syntheses of Tetrabenzotetraaza[8]circulenes.
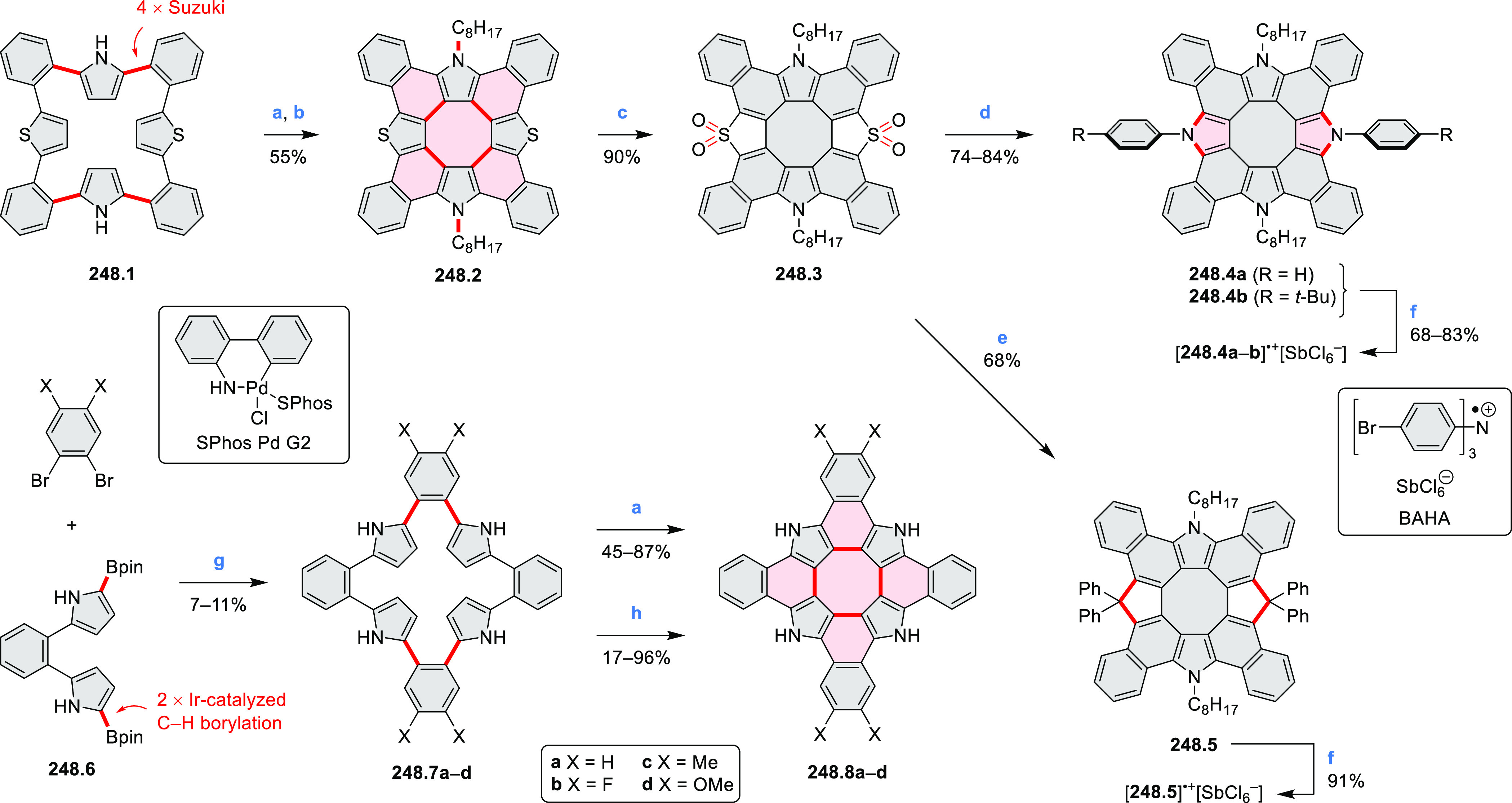
Reagents and conditions: (a)462 DDQ, TfOH, DCM, rt, 10 min–6 h; (b) 1-iodooctane, NaH, DMSO, 50 °C, 38 h; (c) m-CPBA, CHCl3, reflux, 3 h; (d) aniline or 4-tert-butylaniline, KHMDS, dioxane, 90 °C, 24 h; (e)463 diphenylmethane, KHMDS, dioxane, 90 °C, 24 h; (f) BAHA, DCM, rt, 30 min; (g)464 SPhos Pd G2, K3PO4, THF/H2O, 60 °C, 12 h; (h) DDQ, Sc(OTf)3, toluene, darkness, reflux, 2–5 h.
Scheme 236. Syntheses of Unsubstituted Trichalcogenasumanenes from Triphenylene-Based Precursors.
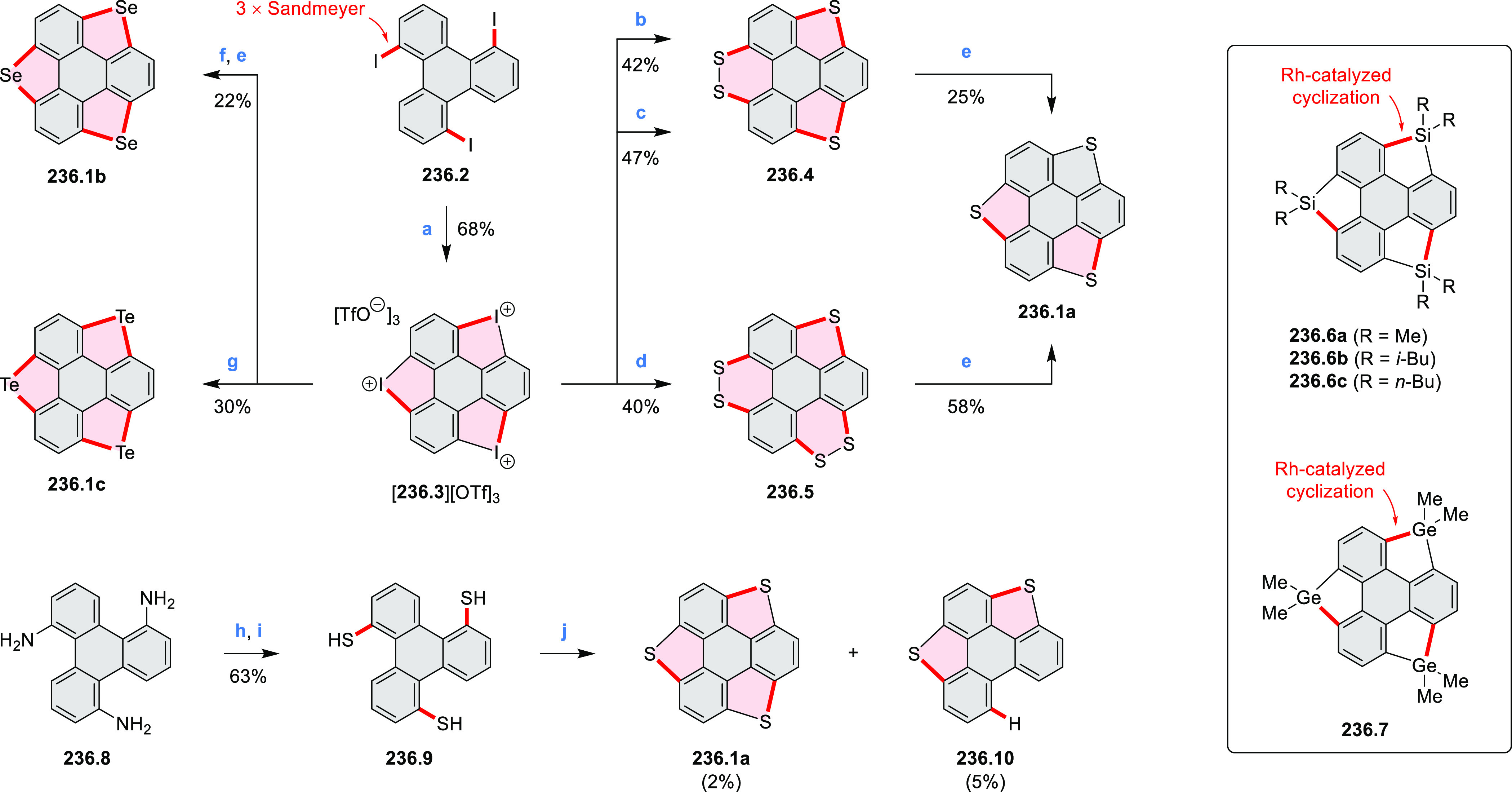
Reagents and conditions: (a)435m-CPBA, TfOH, DCM, 0 °C to rt, 12 h; (b) AcSK, CuCl2, DMSO, 110 °C, 48 h; (c) S8 powder, Cs2CO3, DMSO, 140 °C, 12 h; (d) S8 powder, Cs2CO3, DMSO, 80 °C, 12 h; (e) Cu, tetralin, 200 °C, 1–2 h; (f) Se powder, t-BuOK, DMSO, 70 °C, 24 h; (g)436 Te powder, 2-picoline, DMSO, 125 °C, 12 h; (h)438 (1) HCl, NaNO2, H2O, 0 °C, (2) KSCN, FeCl3, 0 °C, then rt, 1 h; (i) Na2S·9H2O, EtOH/H2O (2/1), reflux, overnight; (j) PdCl2 (90 mol %), DMSO, 120 °C, 24 h.
Scheme 56. Synthesis of Heteroatom-Annulated Perylene Tetraesters.

Reagents and conditions: (a)95 KOH, H2O, 70 °C, 0.5 h, then acidified to pH 8–9 with 1 M HCl, then Aliquat 336, KI, RBr, reflux, 12 h, 69–75%; (b) NaNO2, HNO3, DCM, 0 °C, 1 h, 80–90%; (c)93,95,96 P(OEt)3, reflux, 4 h, 55–62%; (d)94,96 sulfur powder, NMP, 70 °C, 0.5 h, then 180 °C, 17 h, 50–60%; (e)95,96 selenium powder, NMP, 70 °C, 0.5 h, then 180 °C, 17 h, 52–58%; (f) NaH, RBr, THF, reflux, 17 h, 70–80%.
Scheme 58. Synthesis of Unsubstituted N- and S-Annulated Perylenes.

Reagents and conditions: (a)99 sulfur powder, NMP, 70 °C, then 58.1, 180 °C, 10 h, 50%; (b)101 P(OEt)3, reflux, 2 h, 85%; (c) m-CPBA, DCM, −78 to 5 °C, 19%; (d)100 NBS, 1:1 CHCl3/AcOH, rt, 5 h; (e)102 α,α′-dibromo-p-xylene, KOH, KI, THF, reflux, 2 days, 50%; (f) N-methyl-4,4′-bipyridinium hexafluorophosphate, MeCN, reflux, 12 h, 50%.
Scheme 59. Preparation of a NAP Diimide Dimer via Suzuki–Miyaura Coupling Using Nitro and Bromo Derivatives.
Reagents and conditions: (a)103 NaN3, 1:1 DMF/THF, rt, 18 h, 76%; (b) 1-bromooctane, K2CO3, KI, DMF, 90 °C, 18 h, 85%; (c) Br2, DCM, rt, 30 min, quant.; (d) HNO3 (fuming), DCM, rt, 5 min, 66%; (e) 1,4-benzenediboronic acid bis(pinacol) ester, Pd(PPh3)4, K2CO3, 9:1 dioxane/H2O, 110 °C, microwave heating, 2 h.
Scheme 60. O-, S-, and Se-Annulation of Perylene Diimides.

Reagents and conditions: (a)104,105 NMP, O2, 180 °C, 5 h, 30%; (b)106 sulfur powder, NMP, 130 °C, 12 h, 32%; (c)107 sulfur powder, NMP, 70 °C then 60.1b, 190 °C, 3 h, 52%; (d) selenium powder, NMP, 70 °C then 60.1b, 190 °C, 3 h, 48%.
Scheme 61. Synthesis of S- and S2-Annulated Perylene Diimides.
Reagents and conditions: (a)108 HNO3, CAN, DCM, 25 °C, 2 h, 92%; (b)108 HNO3, CAN, DCM, 25 °C, 48 h, 92%; (c)109 HNO3 (fuming), DCM, 0 °C, 20 min then rt, 12 h, 80–86%; (d)108 sulfur powder, DMF, 120 °C, 8 h; (e)108 sulfur powder, DMF, 120 °C, 6 h, 80%; (f)108 sulfur powder, DMF, 80 °C, 2 h, 80%; (g)108 sulfur powder, DMF, 120 °C, 3 h, 85%; (h)109 sulfur powder, NMP, 190 °C, 4 h, 82–86%; (i)110 sublimed sulfur, NMP, 110 °C, 10 h.
Scheme 62. Synthesis of Sulfur-Annulated Fused Perylene Diimides.
Reagents and conditions: (a)111 sulfur powder, NMP, 70 °C then 62.1, 200 °C, 20 min.
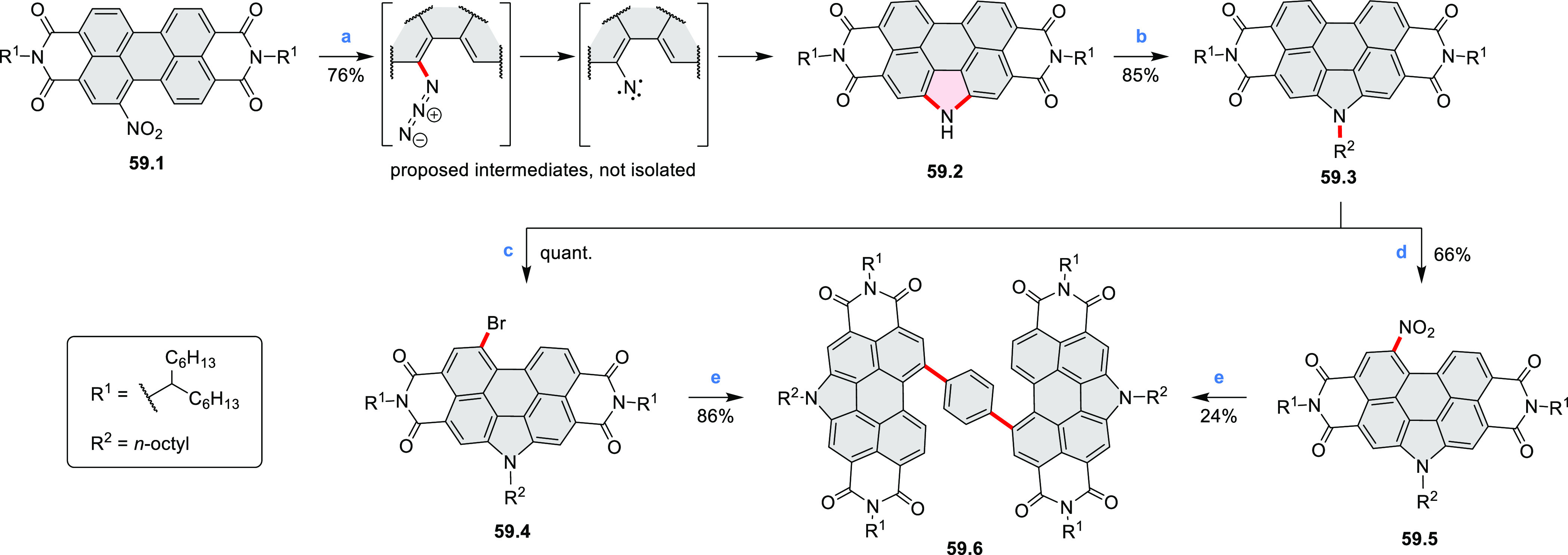
Scheme 67. Synthesis of N-Annulated Perylene Diimide Dimers.
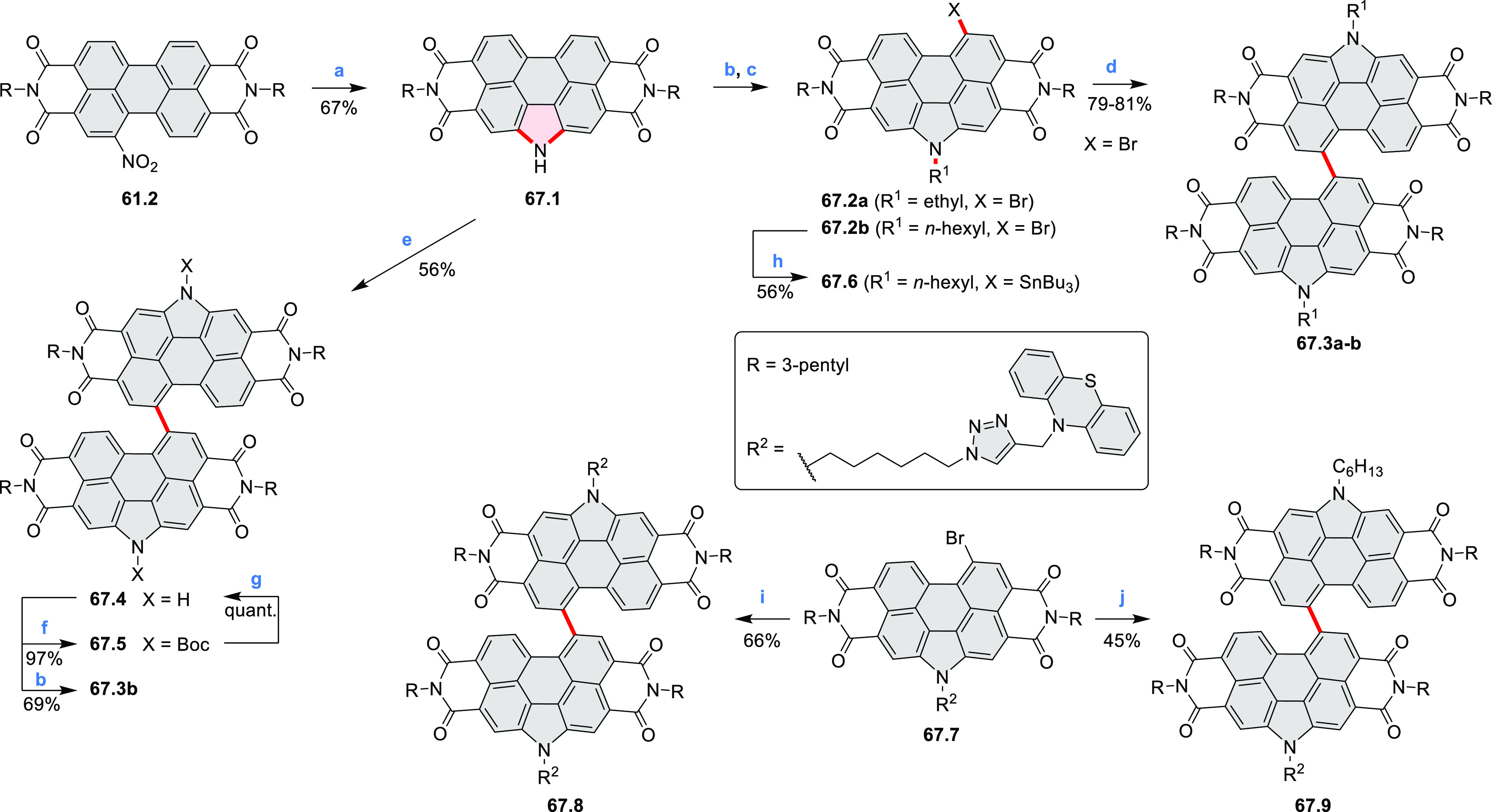
Reagents and conditions: (a)119 PPh3, DMF, 150 °C, 21 h, 67%; (b) n-hexyl bromide, K2CO3, DMF, 120 °C, 18 h, 89–96% for 67.2a–b; (c) Br2, DCM, rt, 2 h, 94–98%; (d) Pd(dba)2, Zn dust, DMF, 100 °C, 3.5 h, 79–81%; (e)120 Br2 (neat), rt, 1 h, 93%, then Pd(dba)2, Zn dust, DMF, 120 °C, 30 min, 56%; (f) Boc2O, DMAP, K2CO3, DMF, 80 °C, 24 h, 97%; (g) 200 °C, 1 h, quant. or 180 °C in thin films; (h)121 hexabutylditin, SiliaCat DPP-Pd, toluene, 100 °C, 1 h, 63%; (i) Zn dust, Pd2(dba)3, DMF, 100 °C, 2.5 h, 66%; (j) 67.6, Pd(PPh3)4, toluene, 180 °C, microwave, 10 min, 45%.
Scheme 79. Chiral Twin N-Annulated Perylene Carboxamides.
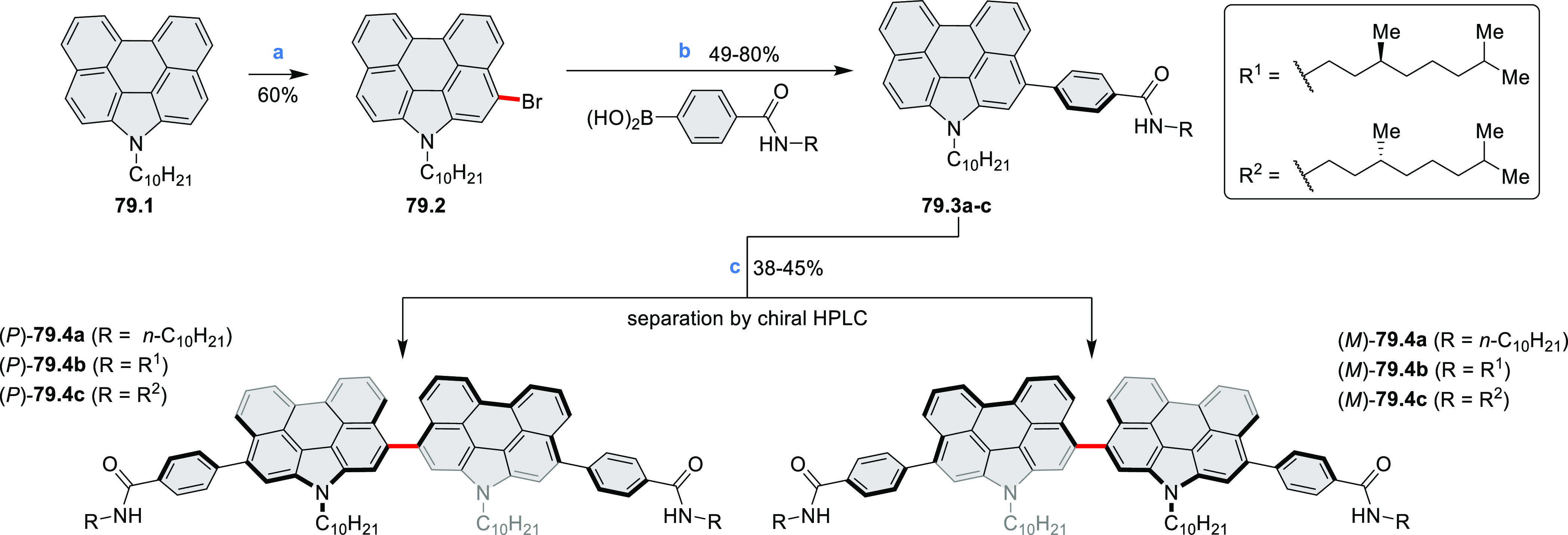
Reagents and conditions: (a)152 NBS, DMF, 0 °C, 30 min, 60%; (b) Pd(PPh3)4, K2CO3, 19:1 THF/water, reflux, 40 h, 49–80%; (c) Sc(OTf)3, DDQ, toluene, rt, 22–48 h, 38–45%.
Scheme 172. Synthesis of Unsymmetrical Azatwistacenes.
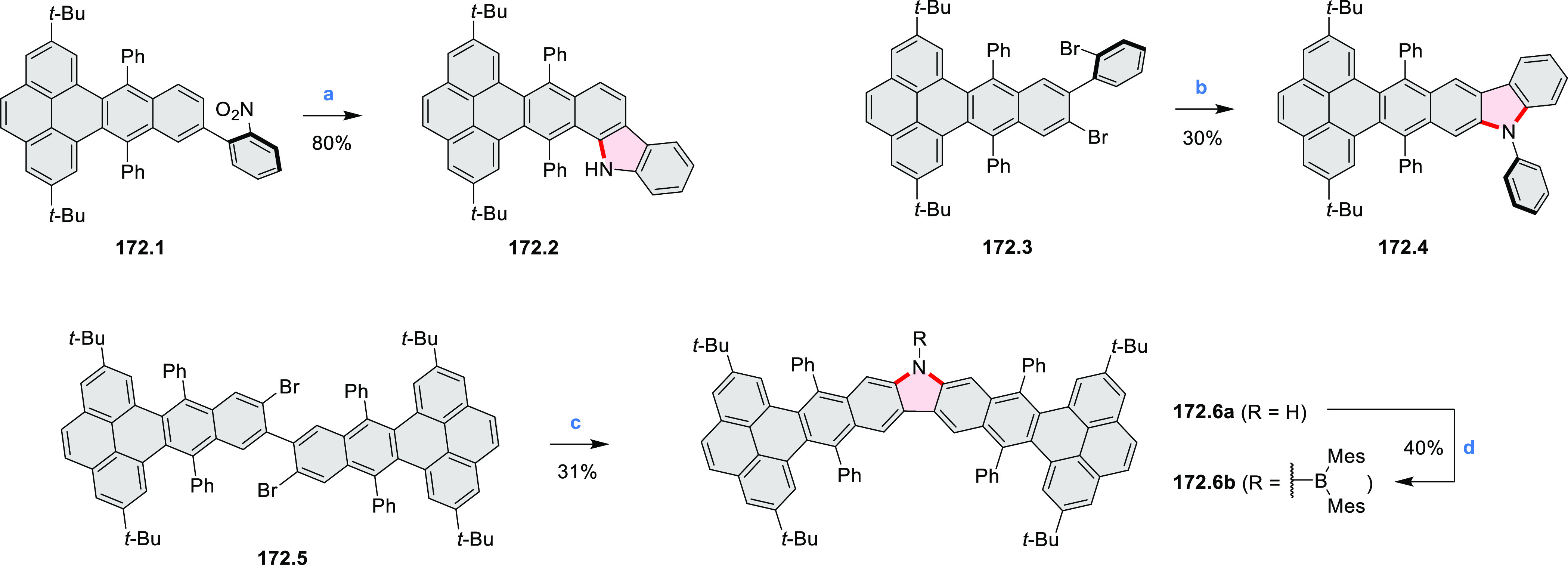
Reagents and conditions: (a)344 P(OEt)3, o-DCB, 160 °C, 24 h; (b) aniline, Pd(OAc)2, PCy3, t-BuOK, toluene, reflux, 48 h; (c)345 NaN3, CuI, N,N′-dimethylethylenediamine, DMSO, 120 °C, 48 h; (d) (1) n-BuLi, THF, −78 °C, 2 h, (2) dimesitylboron fluoride, rt, 24 h.
Scheme 321. Quinolino-Fused Porphyrins.

Reagents and conditions: (a)600 NaBH4, 10% Pd/C, N2, CH3CN/EtOH, rt, 10 min; (b) CH3CN, 1 h, reflux; (c) TFA, conc. H2SO4.
Borylative annulations have emerged as an important method to generate peri-fused boron-containing six-membered rings from olefin-substituted (entry 11, cf. Schemes 138 and 34) and alkyne-substituted precursors (entry 12, Schemes 140 and 268). This strategy was also extended to produce double annulations, leading to concomitant formation of three C–B bonds (entry 13, cf. Schemes 20, 211, 115, 129, and 118) or to borylation of both aromatic carbon atoms and heteroatom substituents (entry 14, cf. Schemes 207, 210, and 128).
Scheme 138. Synthesis of Dithiapyrenoids and Pyrene-Containing Thioxanthene.

Reagents and conditions: (a)277 phenylboronic acid neopentylglycol ester, [Cp*Rh(MeCN)3][SbF6]2, Ag2O, t-AmOH, 120 °C, 48 h; (b) (1) m-CPBA, DCM, rt, (2) TfOH, 1,2-DCE, rt, (3) pyridine, water; (c)67 I2, CHCl3, 1 h at 70 °C, 1 h at 80 °C, 22 h at 90 °C.
Scheme 34. Synthesis of Boron-Doped Perylene.

Reagents and conditions: (a)60,61 (1) HNTf2, chlorobenzene, rt, 90 min then 34.1a added, 110 °C, 5 h or 34.1b added, 160 °C, 24 h, (2) TEMPO, 80 °C, 36 h for 34.1a or 24 h for 34.1b, (3) hydrolytic workup (b) (1) BBr3, DCM, rt, 24 h, (2) MesMgBr, toluene, rt.
Scheme 140. π-Extended Bora- and Diborapyrenes from Alkynes.
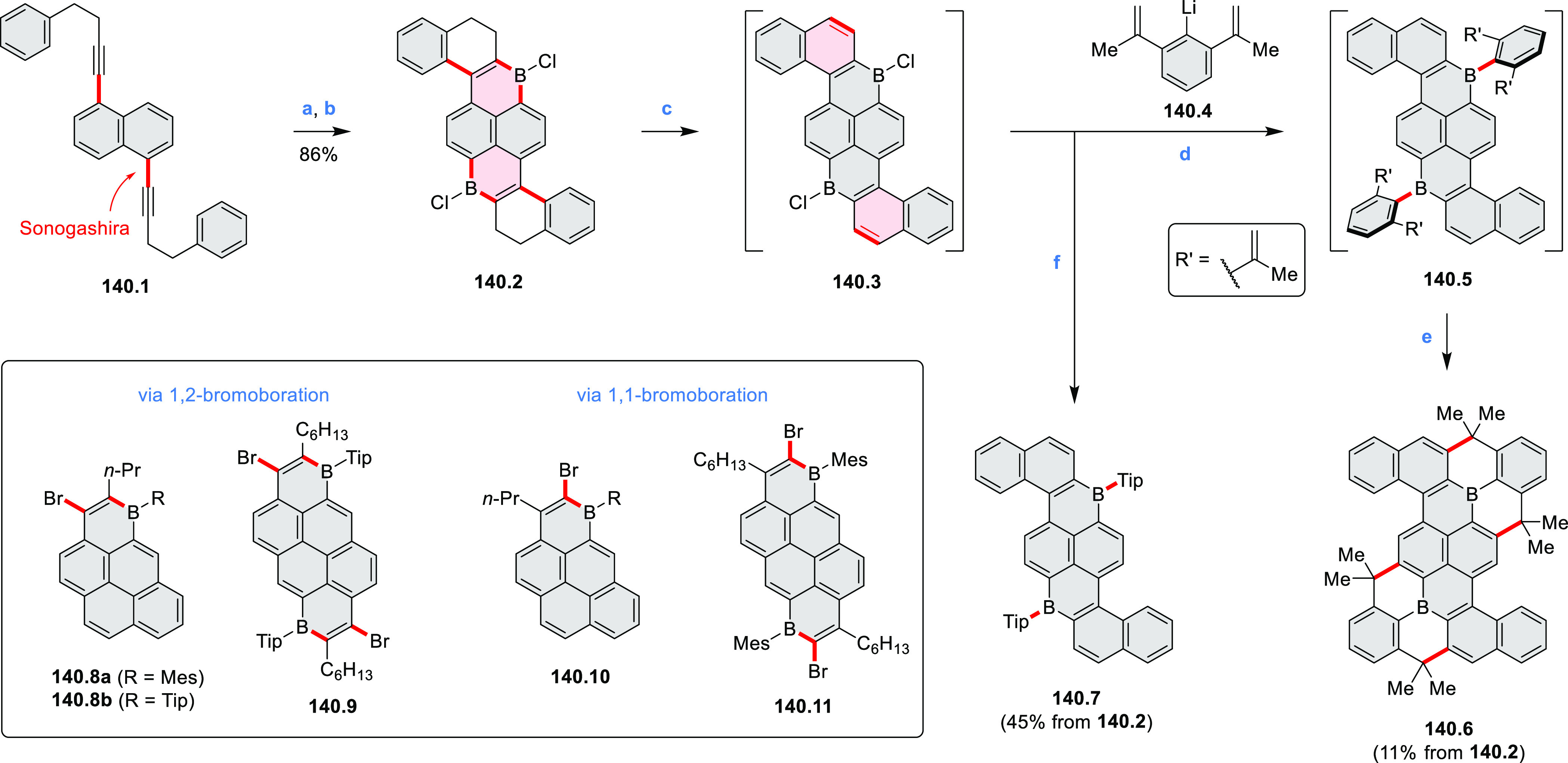
Reagents and conditions: (a)279 (1) BCl3, 2,4,6-tri-tert-butylpyridine, DCM, 5 min, (2) AlCl3, 15 min; (b) AlCl3, 2,6-dichloropyridine, DCM, 18 h; (c) [Ph3C][BF4], 2,4,6-tri-tert-butylpyridine, DCE, 75 °C, 120 h; (d) toluene, rt, overnight; (e) Sc(OTf)3, DCE, 75 °C, 16 h; (f) TipMgBr, toluene, overnight.
Scheme 268. Hetero[e]fused Acenaphthylenes from C–H Annulation Reactions.

Reagents and conditions: (a)506 Pd(PPh3)4 (5 mol %), t-BuOLi (2 equiv), K2CO3 (1 equiv), KOAc (1 equiv), dioxane, N2, 100 °C, 24 h; (b) FeCl3, DCM/MeNO2, 0 °C; (c) FeCl3, DCM/MeNO2, rt; (d) FeCl3, DCM/MeNO2, 0 °C, 15 min, then rt, 2.5 h; (e) (1) DDQ, DCM, 0 °C, 10 min, (2) TfOH, 10 min.
Scheme 20. One-Shot Multiple Borylation toward BN-Doped Nanographenes.

Reagents and conditions: (a)31 BI3, Ph3B, o-DCB, reflux, 20 h; (b) BI3, Ph3B, 1,2,4-trichlorobenzene, 200 °C, 20 h; (c) BI3, o-DCB, reflux, 12 h.
Scheme 211. DABNA and Its Carbazole-Based Analogues.
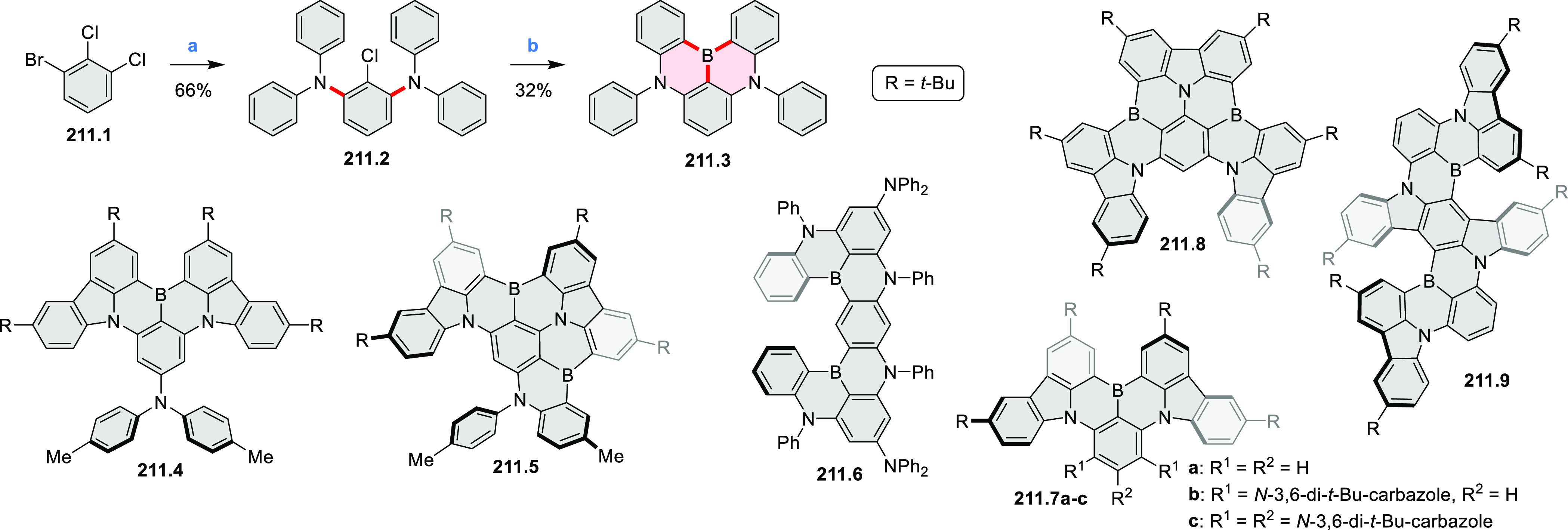
Reagents and conditions: (a)262,397 HNPh2 (2.2 equiv), t-BuOK (2.5 equiv), (AMPHOS)2PdCl2 (1.0 mol %), o-xylene, 80 °C, 2 h then 120 °C, 3 h; (b) (1) t-BuLi (1.2 equiv), t-butylbenzene, 60 °C, 2 h, (2) BBr3 (1.2 equiv), rt, 0.5 h; EtN(i-Pr)2 (2.0 equiv), 120 °C, 3 h.
Scheme 115. Syntheses of Boron-Centered, Oxygen-Containing Heteratriangulenes.
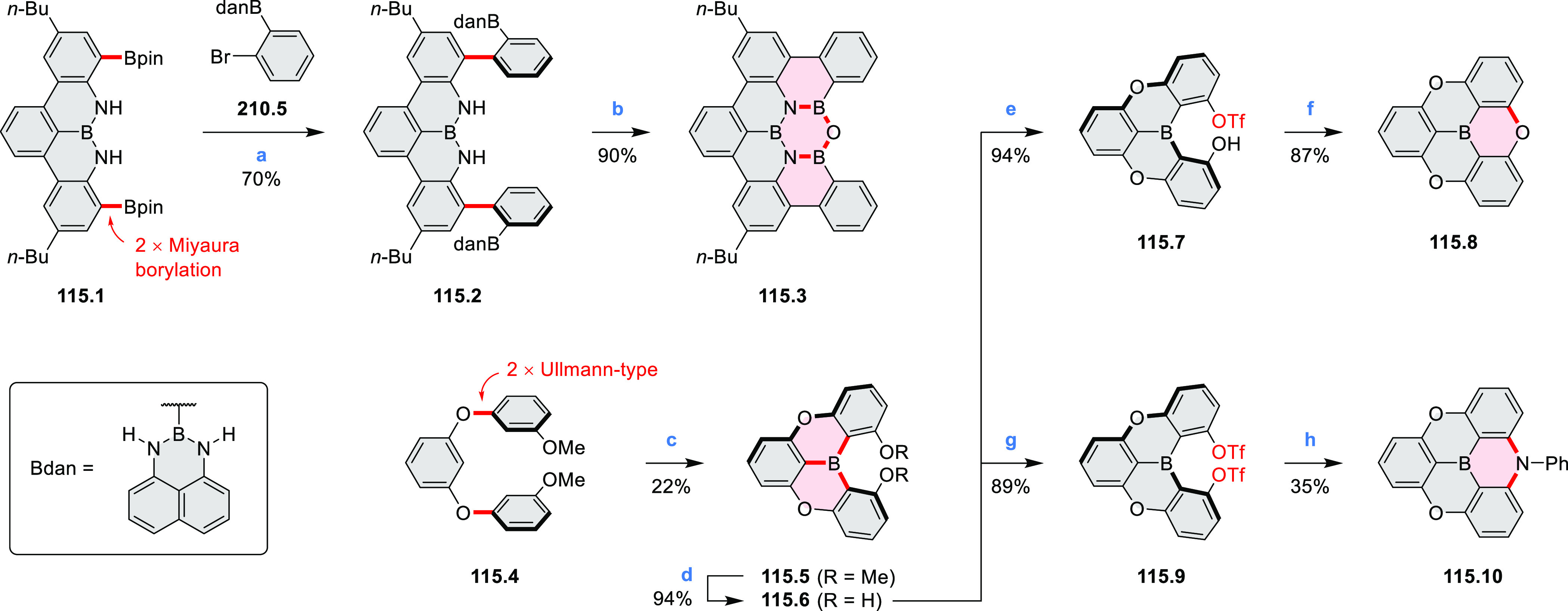
Reagents and conditions: (a)229 Pd(PPh3)4, K2CO3, toluene/EtOH/H2O, reflux, 18 h; (b) aq. H2SO4 (2 M), THF, 50 °C, 3 days; (c)230 (1) n-BuLi, TMEDA, THF, 0 °C, 0.9 h, then rt, 4 h, (2) BF3·Et2O, THF/benzene, −40 °C, 0.4 h, then rt, 2 h, then 87 °C, 21 h; (d) BBr3, DCM, −78 °C, 0.9 h, then rt, 2 h; (e) Tf2O (2.1 equiv), i-PrNEt2 (1.2 equiv), DCM, −78 °C, 1.25 h, then rt, 3 h; (f) DBU, DMF, microwave, 240 °C; (g)231 Tf2O (2.2 equiv), i-PrNEt2 (3.0 equiv), DCM, −78 °C, 1.25 h, then rt, overnight; (h) aniline, LiN(SiMe3)2, 1,4-dioxane, 10 °C, then 150 °C, 20 h.
Scheme 129. Synthesis of BN-Doped Pyrenoids.
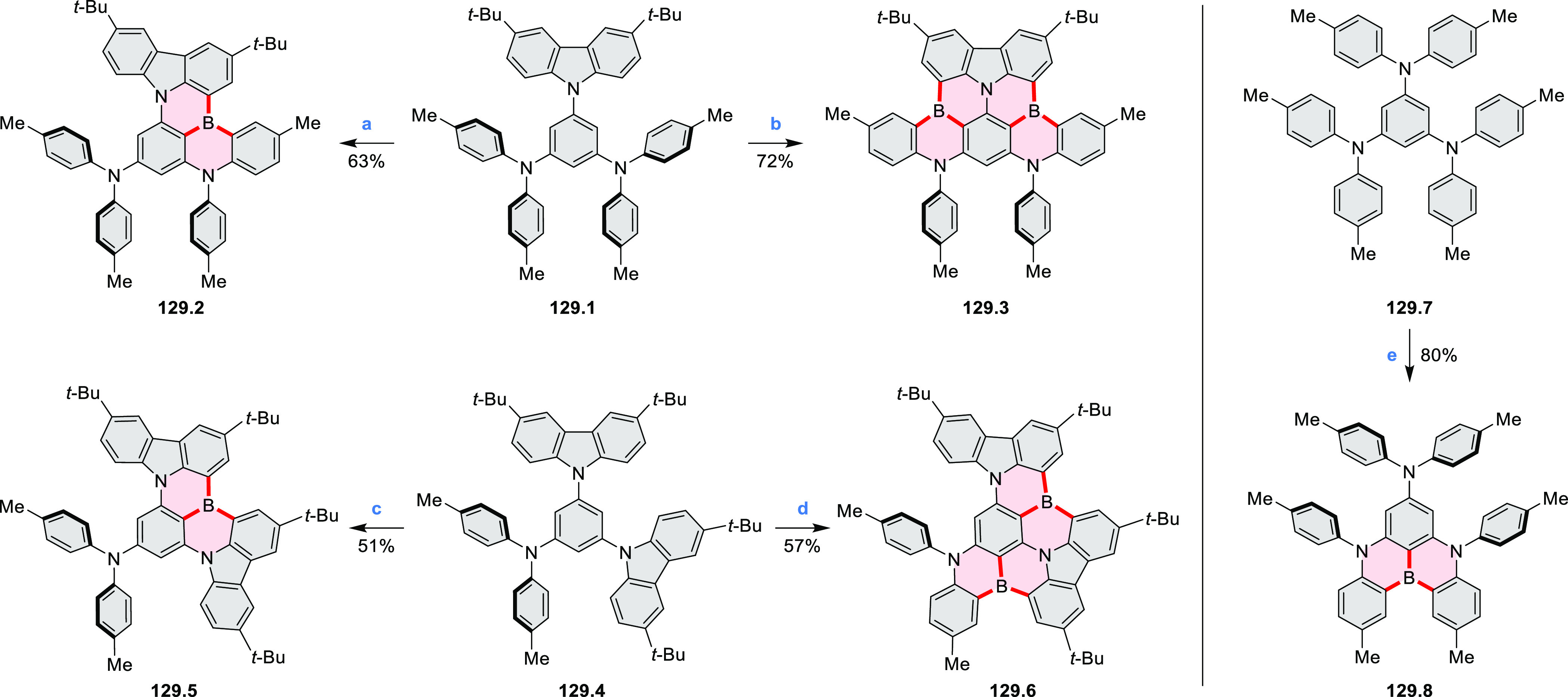
Reagents and conditions: (a)262 2 equiv of BBr3, toluene, reflux, 20 h; (b) 4 equiv of BBr3, chlorobenzene, reflux, 20 h; (c) 12 equiv of BBr3, benzene, reflux, 20 h; (d) 24 equiv of BBr3, chlorobenzene, reflux, 20 h; (e) 1 equiv of BBr3, o-dichlorobenzene, 170 °C, 20 h.
Scheme 118. Divergent Synthesis of the Heteroatom-Centered 4,8,12-Triazatriangulenes.

Reagents and conditions: (a)240t-BuLi, t-butylbenzene, −45 °C, then 50 °C, 30 min to 2 h; (b) BBr3, i-Pr2NEt, −45 °C, 1 h, then 165 °C, 14 h; (c) MeSiCl3, t-butylbenzene, 150 °C, 18 h; (d) (1) PCl3, toluene, 50 °C, 2 h, (2) S8, o-dichlorobenzene, 110 °C, 12 h; (e) m-CPBA, DCM, −30 °C, 1 h; (f) PEt3, o-xylene, 120 °C, 2 days.
Scheme 207. Synthesis of Boronate-Based Benzo[fg]tetracene.
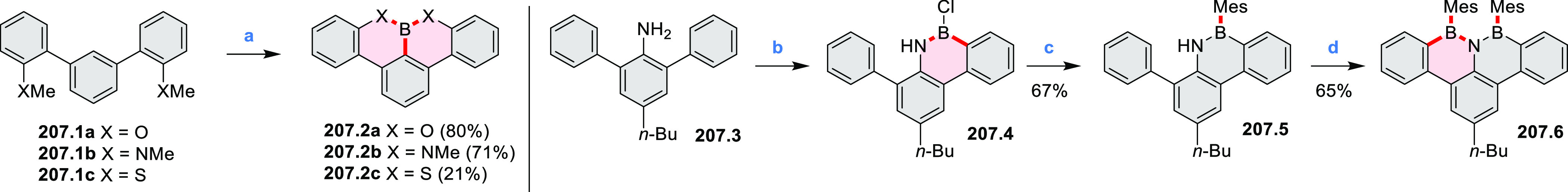
Reagents and conditions: (a)391 BBr3, Brønsted bases, 120 to 200 °C, 18 h; (b)392 (1) BCl3, toluene, 0 °C to reflux, (2) AlCl3, reflux; (c) MesMgBr, benzene, rt; (d) (1) KHMDS, toluene, rt, (2) BCl3, 0 °C to rt, (3) AlCl3, reflux, (4) MesMgBr, benzene, rt.
Scheme 210. Synthesis of a Doubly BN-Doped Dibenzoperylene Derivative.
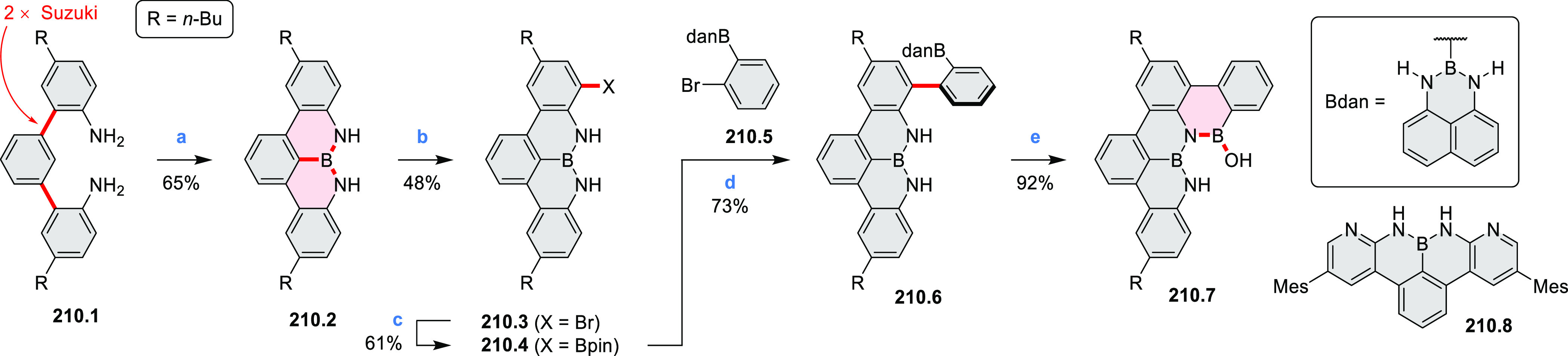
Reagents and conditions: (a)395 BBr3, NaBPh4, toluene, reflux, 18 h; (b) NBS, DCM/CHCl3/MeCN, 0 °C, 2 h; (c) B2pin2, KOAc, Pd(dppf)Cl2, 1,4-dioxane, 90 °C, 18 h; (d) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O, reflux, 18 h; (e) degassed aq. H2SO4 (2 M), THF, 50 °C, 5 days.
Scheme 128. B,N-Doped Pyrenoids.

Reagents and conditions: (a)260 BBr3, i-Pr2EtN, o-DCB, 180 °C; (b)261 PhBCl2, Et3N, 1,2,4-trichlorobenzene, reflux, 36 h; (c) Pd(OAc)2, PCy3, Cs2CO3, o-xylene, reflux, overnight.
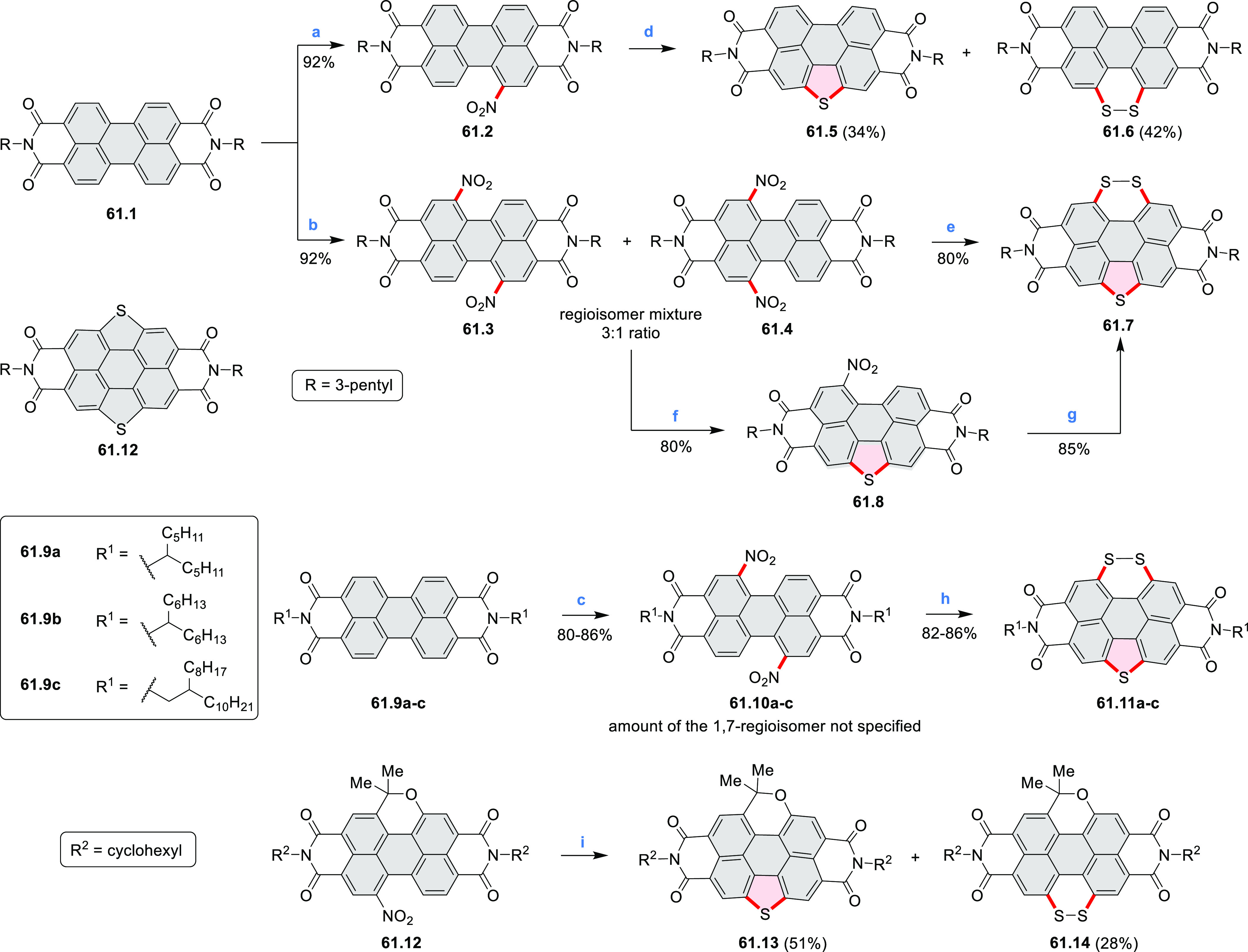
2. Coronenoids
Cyclodehydrogenation of hexaarylbenzenes (HABs) and their heterocyclic analogues remains the most popular route toward hexa-peri-fused coronenes. Complete fusion is occasionally difficult to achieve, and products of partial ring fusion are often reported. For completeness, these species are discussed in Section 2, even though they do not contain a coronene substructure. Likewise, edge-expanded coronenoids containing seven-membered rings are also included below.
2.1. Edge-Doped Aza- and Oxacoronenes
2.1.1. Mono- and Diazacoronenes
In 2017, Jux et al. reported a synthetic route toward pyrrole-containing HBC analogues (Scheme 4).2 The Diels–Alder reactions of each alkyne 4.1a,b at 195–200 °C in a sealed tube in toluene under an argon atmosphere led to the formation of 4.2a,b. Conversion of these compounds into HBC systems was then attempted under dehydrogenative cyclization conditions (DCM/MeNO2/FeCl3), leading to quintuply fused structures 4.3a,b. The fully fused HBC analogues could not be obtained under those conditions. 4.3a showed a helicene-like shape of the molecules in the solid state. The UV–vis spectra of 4.3a,b contain a broad, intense absorption around 360 nm, and due to the lower molecular symmetry, the π → π* transition could be observed as an absorption band around 420 nm. The fluorescence of both [5]helicenes 4.3a,b is similar to HBC derivatives, with two maxima at around 470 and 500 nm. Cyclovoltammetric measurements revealed two reversible oxidation waves at +0.85 V/+0.80 V and +1.29 V/+1.22 V (vs SHE) for 4.3a/4.3b, respectively.
Scheme 4. Synthesis of a HBC-Like System Containing a Pyrrole Ring.

Reagents and conditions: (a)2 2,3,4,5-tetrakis(4-tert-butylphenyl)cyclopenta-2,4-dien-1-one, toluene, 200 °C, 18 h for 4.1a 72 h for 4.1b; (b) 2,3,4,5-tetrakis(4-(t-butyl)phenyl)cyclopenta-2,4-dien-1-one, THF, dioxane, Co2(CO)8, 160 °C, 15 min, microwave, 350 W; (c) FeCl3, DCM, MeNO2, 0 °C.
The Jux group further reported a solution for synthesis of pyridine analogues of hexa-peri-hexabenzocoronene (Scheme 5).3 A key feature of their strategy was the early formation of the C–C bonds at the 3 and 5 positions of the pyridine (5.1a,b, red bonds). These bonds are otherwise unreactive and difficult to close under oxidative conditions. Compounds 5.1a,b and 5.2a,b were obtained via multistep procedures starting from 4-aminopyridine and para-nitroaniline, respectively. Formation of the pseudo-HAB precursors 5.3a,b was achieved via a Suzuki reaction. The final step was carried out under oxidative cyclodehydrogenation conditions with DDQ and triflic acid in DCM, with yields up to 83%. The more soluble product 5.4b was further derivatized via metal coordination, N-alkylation, and N-oxidation. Methylation at the nitrogen atom yielded a pyridinium ion (5.5b), which was isolated as its triflate salt, whereas m-CPBA oxidation produced the corresponding N-oxide 5.6b. 5.4b was shown to act as an apical ligand toward tetrakis(4-tert-butylphenyl)porphyrinato zinc in solution. Formation of the corresponding complex 5.7b was demonstrated by a very large upfield shift of the 2,6-pyridine proton signals from 10.54 to 4.60 ppm, caused by the shielding effect of the aromatic ring current of the porphyrin.
Scheme 5. Synthesis of π-Extended Pyridines.
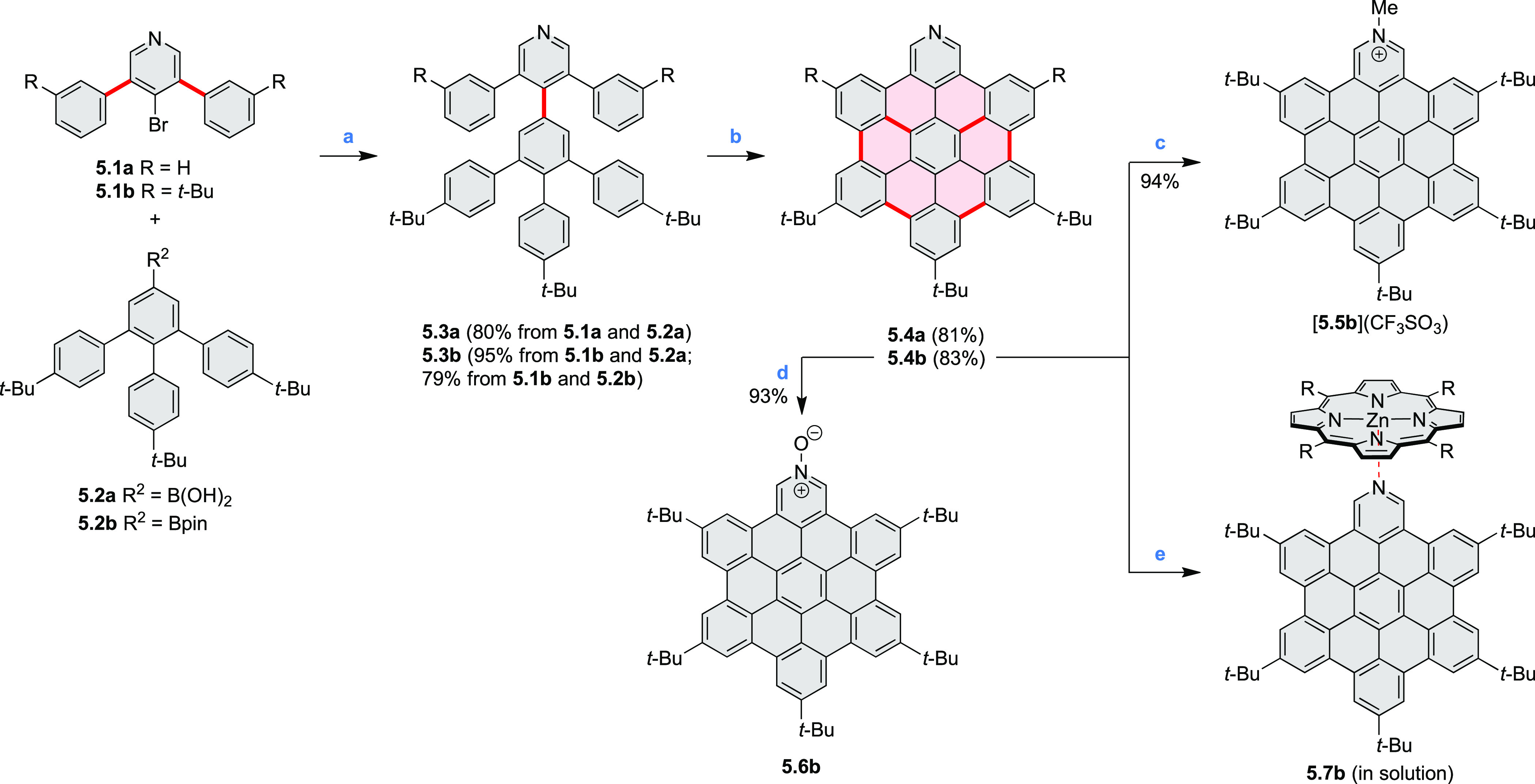
Reagents and conditions: (a)3 Cs2CO3 (2 equiv), 10 mol % of Pd(PPh3)4, THF/H2O (4:1), 17–24 h, 80 °C, N2; (b) 5.4a: DDQ (7 equiv), TfOH (14 equiv), DCM, 1 h, 0 °C, N2; 5.4b: DDQ (7 equiv), TfOH (14 equiv), DCM, 4 h, −50 °C to −20 °C, N2; (c) (1) MeI (excess), CH3CN, 2 h, rt, N2, (2) Ag(OTf) (2.1 equiv), 15 min, rt, N2; (d) m-CPBA (1 equiv), CHCl3, 24 h, 0 °C to rt; (e) tetrakis(4-tert-butylphenyl)porphyrinato zinc (1 equiv), C6D6, rt.
Diimide and tetraester diazacoronenes were synthesized via the Pictet–Spengler reaction of 1,6- and 1,7-diaminoperylenes with picolinaldehyde in 2016 by Würthner et al. (Scheme 6).4 Starting from a 3:2 regioisomer mixture of 1,7- and 1,6-diamino PDIs 6.1–2, azabenzannulated products 6.4–5 were obtained and separated by column chromatography (for singly azabenzannulated analogues, see Scheme 82, Section 3.1.1). Yields of these syntheses were limited by purification problems. Similarly, the bisazabenzannulated perylene tetraester 6.6 was synthesized from the isomerically pure 1,6-diaminoperylene tetraester 6.3 in 37% yield. After hydrolysis of 6.6, the corresponding dianhydride was converted into variously functionalized bisazabenzannulated perylene diimides, such as 6.5 (Scheme 6). The spectroscopic data reveal a hypsochromic shift of the S0–S1 transition band, which is less pronounced in comparison to the parent PDI. The oscillator strengths and the intensities of the S0–S2 transitions strongly increase with core extension because the S0–S2 transition dipole moments are aligned along the laterally elongated molecular axes. Therefore, the spectra of 6.4–6 are not predominated by the lowest-energy S0–S1 transitions but by higher-energy absorption bands.
Scheme 6. Synthetic Routes to Diazacoronene Diimides and Tetraesters.

Reagents and conditions: (a)4 picolinaldehyde, dry DMF, TFA, molecular sieves 3 Å, 110 °C, N2 to O2, 21 h; (b) picolinaldehyde, dry DMF, TFA, molecular sieves 3 Å, 110 °C, N2 to O2, 18 h; (c) (1) H2SO4, AcOH, reflux, 17 h, (2) RNH2, imidazole, pyridine, 120 °C, 6 h.
A soluble amide-containing coronene 7.4 was recently reported by Yamaguchi and Glorius et al.5 The fused framework of 7.4, containing two fused lactam rings, was obtained by 2-fold C–H activation of diazaperylene precursor 7.3 (Scheme 7), which was synthesized in two steps from the commercially available 7.1. 7.4 exhibited a far more red-shifted absorption band (λabs = 673 nm in DCM), in comparison to the diazacoronene 6.4 (λabs = 485 nm in DCM). 7.4 showed a deep green color and an intense NIR emission with the λem of 686 nm and a high fluorescence quantum yield of 0.64 in DCM.
Scheme 7. Synthetic Route to Bis(amide)-Containing Coronene.
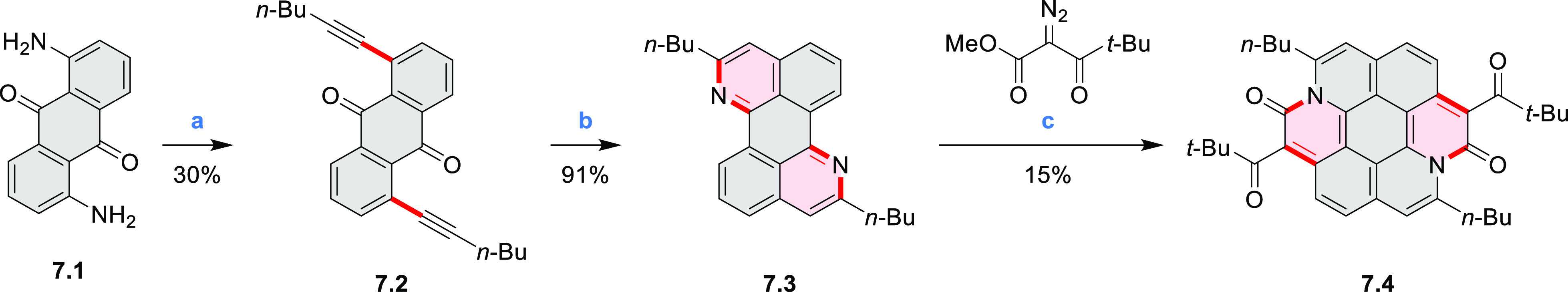
Reagents and conditions: (a)6 (1) NaNO2, H2SO4, (2) KI, water, (3) hex-1-yn-1-yl copper, pyridine; (b) urea, DMF; (c) [RhIIICp·(MeCN)3](SbF6)2, TFE, 120 °C, 14 h, (2) DMAP, TFE, 140 °C, 14 h.
The possibility to induce intramolecular bond formation at the pyrimidine ring of 8.1 by using dibromine was explored by Draper and co-workers (Scheme 8).6 The pyrimidine-containing precursor 8.1 was reacted in neat dibromine at rt to yield different proportions of 8.2, 8.3, and 8.4, depending on the reaction time (5 min to 5 h). Performing the reaction in toluene at 90 °C resulted in the formation of only 8.2 in 85% yield, while refluxing in chloroform was successful in driving the reaction to higher yields of the pentabrominated 8.4. The use of bromine electrophiles was subsequently found to be an efficient method for oxidative coupling of the more electron-rich hexapyrrolylbenzenes (see below).
Scheme 8. Pyrimidine-Containing Nanographenes.

Reagents and conditions: (a)6 Br2, toluene, 90 °C, 2 h; (b) Br2, rt, 25 min; (c) Br2, CHCl3, reflux, 3 h.
2.1.2. Dioxacoronenes
New π-extended biscoumarins were reported in 2017 by Gryko and Głowacki et al. (Scheme 9).7 The synthesis, based on a previously developed strategy, involved initial Knoevenagel condensation of 9.1 and 9.2a–c. The resulting 9.3a–c were subjected to Mallory photocyclization, to produce the fused targets 9.4a–c. Compound 9.7 was obtained from 9.5 by using a similar synthetic strategy (Scheme 9).8 The methoxy groups of the intermediate 9.6 were replaced with −OC6H13, and the final ring fusion was achieved by oxidation with FeCl3. Photophysical properties of 9.7 were almost identical to its regioisomers 9.4a–c. These compounds exhibit vibronically resolved absorptions and high fluorescence quantum yields of up to 90%.
Scheme 9. Synthesis of π-Extended Biscoumarins.
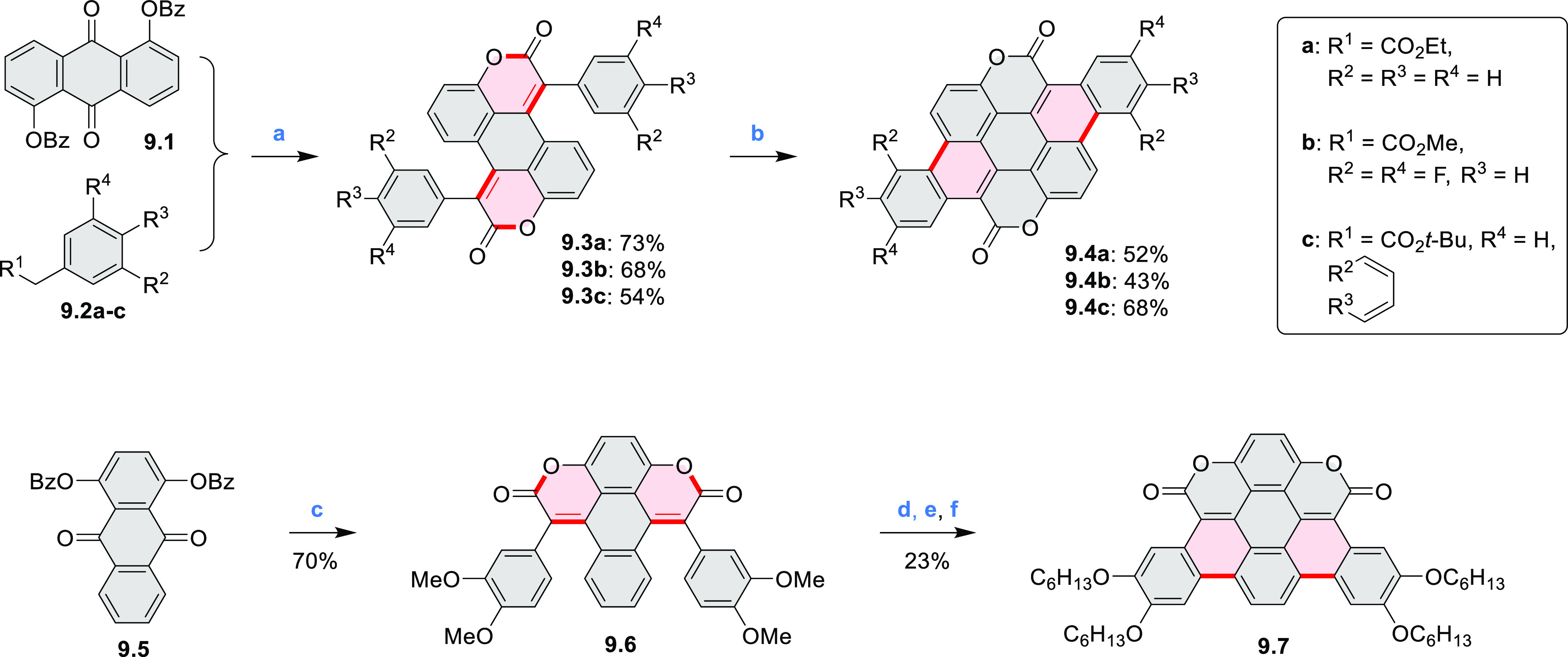
Reagents and conditions: (a)7 K2CO3, DMSO, 100 °C; (b) hν, THF, rt; (c)8 K2CO3, DMSO, 100 °C; (d) BBr3, DCM, −40 °C to rt; (e) C6H13Br, K2CO3, DMF, 60 °C; (f) FeCl3, DCM, rt.
2.1.3. Triazacoronenes
1,5,9-Triazacoronene (TAC) derivatives were explored by Wei et al. as potential materials for electrogenerated chemiluminescence (ECL).9−11 Tris(phenothiazine)-substituted 10.5 was prepared through a triflic-acid-catalyzed 3-fold Pictet–Spengler cyclization and subsequent oxidative aromatization.9 Absorption and fluorescence emission spectra of 10.5 revealed that its electronic properties were affected by intramolecular charge transfer from phenothiazine donors to TAC acceptors in the excited state while showing no charge-transfer interaction in the ground state. π-Extended triazacoronene derivatives 10.4 containing three peri-fused benzopyran units were synthesized in one pot with yields of up to 87%.10 The synthetic approach involved a tandem triflic-acid-catalyzed 3-fold Pictet–Spengler cyclization and a K2CO3-catalyzed ipso-aromatic substitution. In the solid state, 10.4 forms sandwich-type trimeric assemblies stabilized by stacking interactions between the sterically unhindered π surfaces. In 2020, it was reported that the Pictet–Spengler cyclization for synthesizing TACs from the triphenylene triamine and aldehydes proceeds not only under acidic but also under neutral or even alkaline conditions.11 Under optimized conditions (Scheme 10), a wide variety of TAC derivatives (10.2) could be synthesized, with yields reaching 94%.11 Acid-catalyzed conditions were used by Coskun et al. to synthesize conjugated microporous polymers (CMPs) 10.6 from 1,5,9-triaminetriphenylene 10.1 and terephthalaldehyde.12 The optical band gap and surface area of the resulting CMPs were found to correlate with the strength of the acid catalyst.
Scheme 10. 1,5,9-Triazacoronene Derivatives.
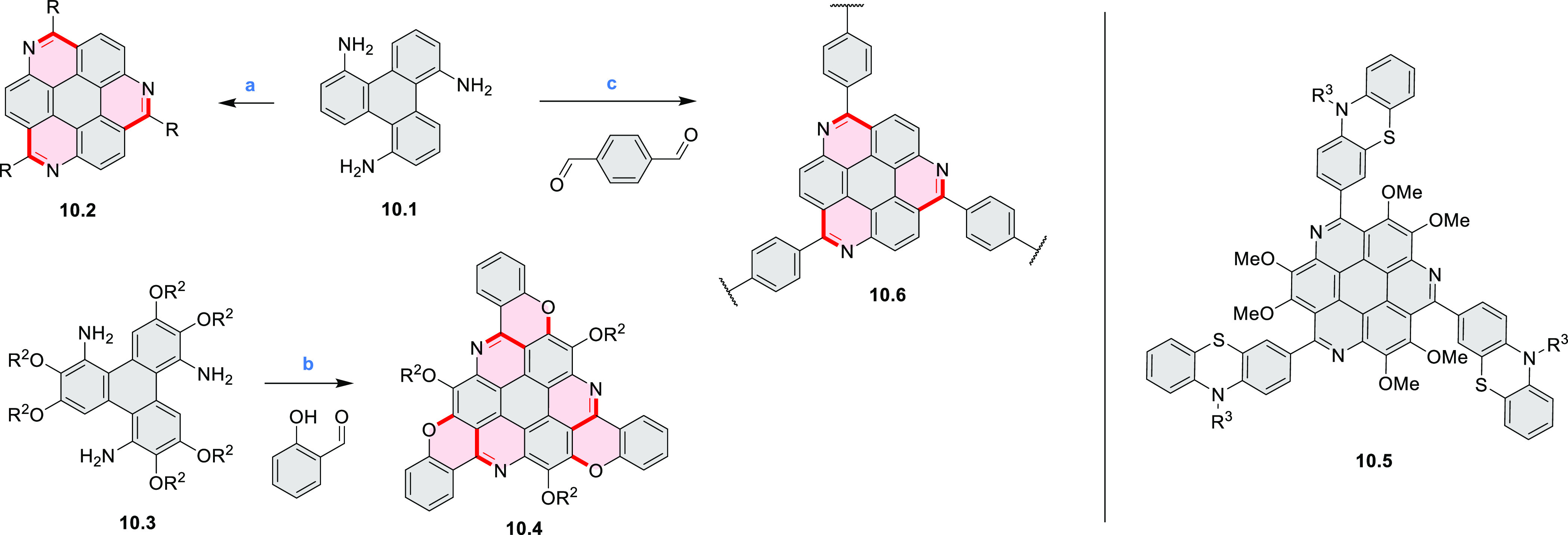
Reagents and conditions: (a)9−12 various aldehydes, DMSO, 130–150 °C, in Ar, then in air; (b)10 (1) DMF or NMP, TfOH, 120 °C, 12 h, (2) 6 equiv of K2CO3, 120 °C, 12 h; (c)12 DMF/dioxane (10:1 v/v), AcOH, TfOH.
2.1.4. Tetra- and Hexaazacoronenes
In 2017, Li et al. reported tetralactam coronenoids 11.3a,b, which were obtained from the commercially available anthraquinone 11.1 via 4-fold Buchwald–Hartwig amination with a 4-alkylaniline followed by microwave-assisted Knoevenagel condensation of the intermediate 11.2 with diethyl malonate (Scheme 11).13 These disclike tetraazacoronenes exhibited optical properties similar to PDIs but had higher LUMO (−3.6 eV) and HOMO (−5.8 eV) levels than those of perylene orange 11.5 (LUMO = −3.8 eV, HOMO = −6.1 eV). Although 11.3 had nearly no fluorescence in solution, it revealed strong photoluminescence in the solid state. 11.3 exhibits high thermal stability (up to 515 °C) and photostability comparable with PDI dyes. Discotic liquid crystals 11.4a–c were designed by introducing wedge-shaped side groups with alkyl tails of different lengths at the periphery of the tetraazacoronene core.14 A high hole mobility μh of 8.84 cm2 V–1 s–1 was determined for 11.4a, whereas 11.4c showed an electron mobility μe of 3.59 cm2 V–1 s–1.
Scheme 11. From Anthraquinone to a Heteracoronene.
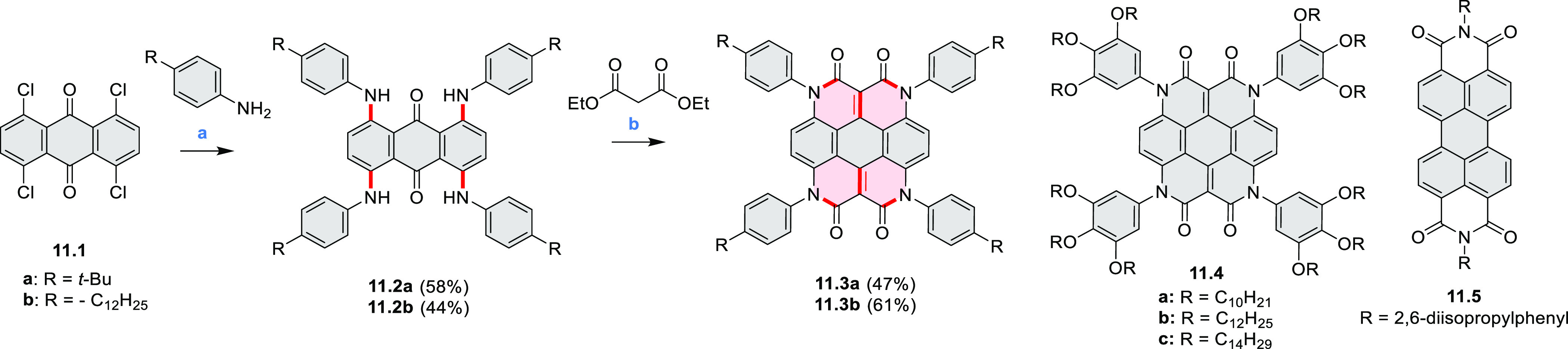
Reagents and conditions: (a)13 Pd2(dba)3, BINAP, Cs2CO3, toluene, 105 °C, 24 h; (b) CH3CO2K, DMF, microwave, 170 °C, 30 min.
The Draper group reported two isomeric types of hexaaza-HBCs, differing in their N-doping pattern (Scheme 12).15 The initial hexaarylbenzenes were obtained as regioisomer mixtures in a dicobalt octacarbonyl-catalyzed cyclotrimerization reaction. For the tert-butyl-substituted precursor 12.2, cyclodehydrogenation using FeCl3 as the oxidant yielded 12.4 and 12.5 in yields of 10% and 20%, respectively, whereas the DDQ/H+-mediated reaction produced 12.5 as the major product. Incomplete ring fusion was similarly observed in the oxidation of 12.3, which yielded 12.6. Fully cyclized products 12.7 and 12.8 were however successfully obtained upon treating the more electron-rich methoxy-substituted HABs with FeCl3.
Scheme 12. Synthesis of Pyrimidine-Containing Hexaaza-HBCs.
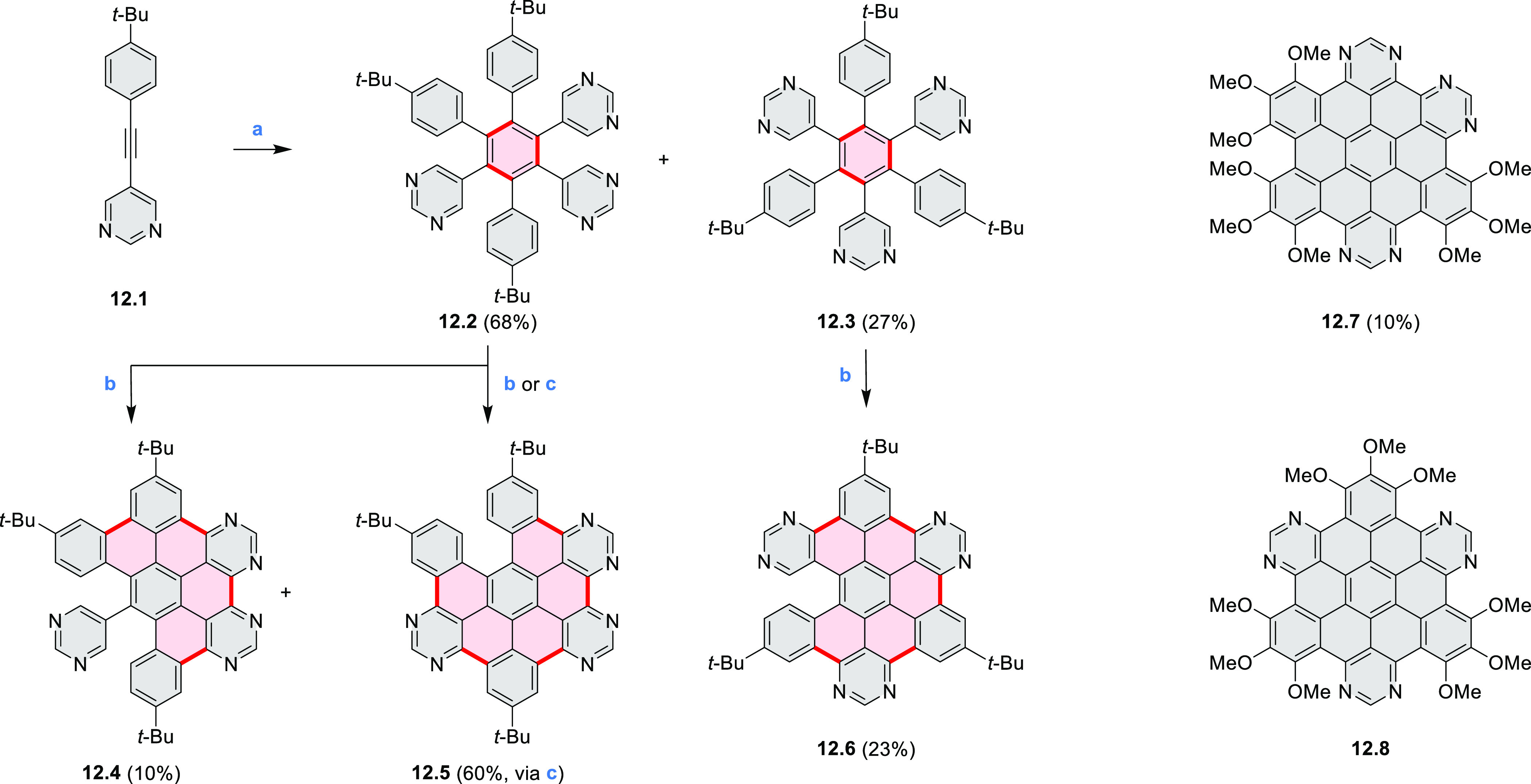
Reagents and conditions: (a)15 Co2(CO)8, dioxane, 115 °C, 24 h, under N2; (b) FeCl3, CH3NO2, 298 K, Ar bubbling, 72 h; (c) DDQ, MeSO3H or CF3SO3H, DCM.
2.2. Internally Doped Azacoronenes
Hexapyrrolohexaazacoronenes (HPHACs) are typically synthesized in two steps, namely, via the SNAr reaction of hexafluorobenzene with the corresponding pyrrole followed by oxidative cyclodehydrogenation of the resulting hexapyrrolylbenzene (Scheme 13; cf. CR2017, section 2.2). Variants of this method provide access to a range of structurally diverse molecules. The use of ethyl substitution introduced by Uno and Takase et al. provides access to more electron-rich and potentially more reactive HPHAC derivatives, such as 13.1,16 which is easily oxidizable to the typical globally aromatic dication 13.12+ containing a 22 π-electron conjugation pathway. According to an MCD analysis and DFT calculations, the NIR absorption band observed for the dication but absent in the spectrum of the parent cyclo[6]pyrrole macrocycle is attributed to a CT transition from the central benzene to the peripheral pyrrole moieties. Oxidation of β-unsubstituted HPHACs 13.2 with silver(I) nitrite did not result in reversible formation of the corresponding dication nor in oxidative dimerization that could be expected on the basis of the known reactivity of corrole or porphyrin chemistry. Instead, the nitrated derivative 13.3 was obtained, displaying redox properties similar to the parent system.17
Scheme 13. Synthesis and Structures of Peripherally Fused Azacoronenes and Their Analogues.
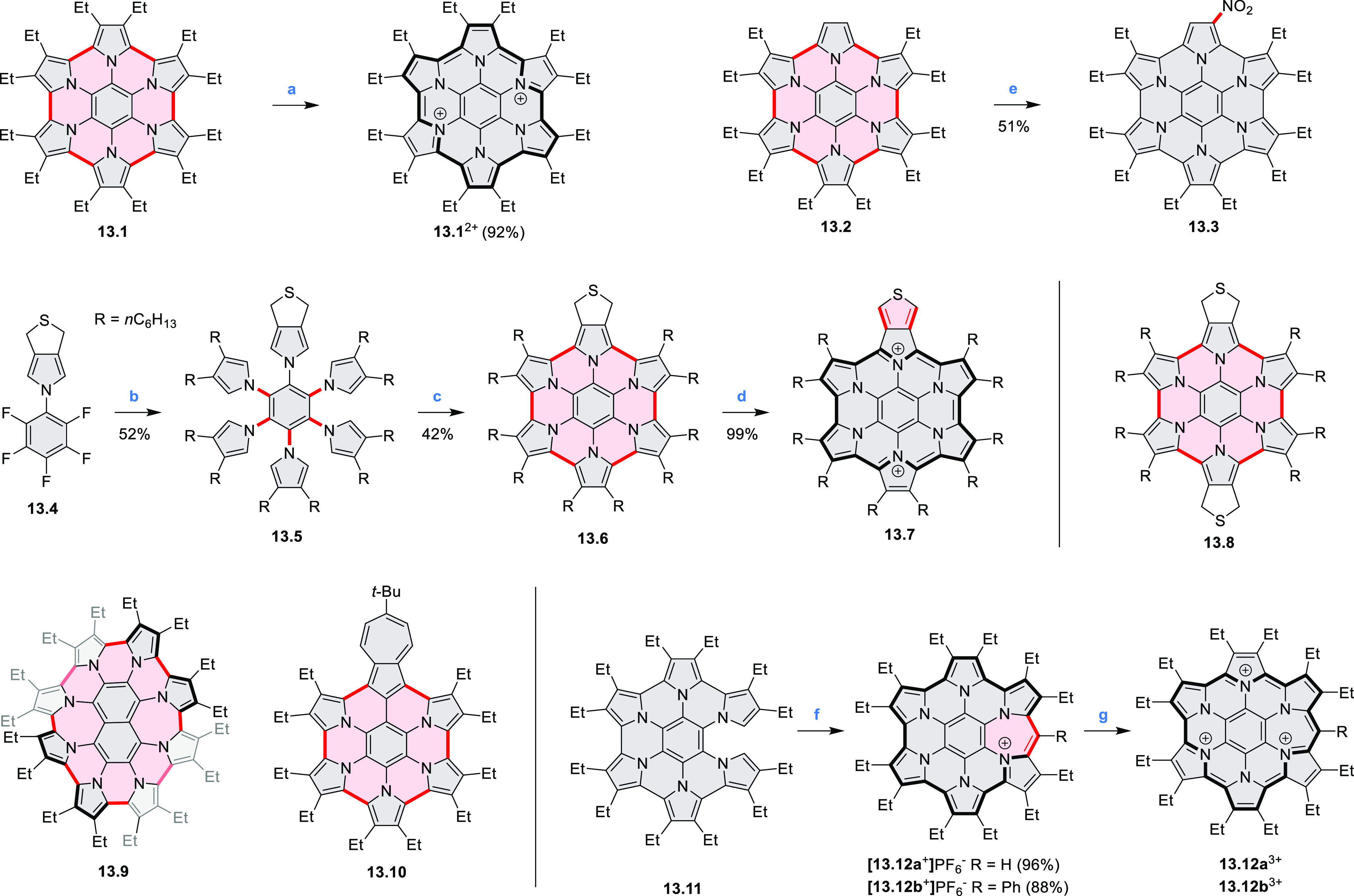
Reagents and conditions: (a)16 NOSbF6 (2.0 equiv) and DCM, rt, 10 min; (b)18 3,4-dihexylpyrrole, NaH, DMF, rt; (c) FeCl3, CH3NO2, rt; (d) I2 (excess), reflux, under N2 flow; (e)17 AgNO2 (9.7 equiv), DCM, rt; (f)21 DMF or N,N-dimethylbenzamide, POCl3, KPF6, DCE; (g) NOSbF6 or BAHA, DCM.
In 2019, Uno and Takase et al. described the preparation of 1,3-dihydrothieno[3,4-a]- and 1,3,8,10-tetrahydrodithieno-[3,4-a;3′,4′-m]-HPHACs (13.6 and 13.8, respectively) by successive SNAr reactions of hexafluorobenzene with (1) 1,3-dihydrothieno[3,4-c]pyrrole and (2) 3,4-dihexylpyrrole, followed by oxidative coupling.18 Upon oxidation with diiodine, 13.6 formed a dehydrogenated dicationic species 13.7, which was not isolated in its neutral form. The dication was stable, and its NMR spectrum was indicative of global diatropicity, consistent with a peripheral aromatic pathway. The formation of a mixed-valence dimer consisting of the neutral 13.6 and its radical cation was observed in a CV measurement performed at high concentration and slow scan speed. 13.10, a HPHAC analogue containing an azulene moiety replacing one of the pyrroles, was synthesized in three steps from a Bpin-substituted azulene derivative, using FeCl3 oxidation in the ultimate step.19 Similarly to the parent HPHAC, 13.10 displayed stable oxidized forms, and its dication was isolated and characterized. Structural and theoretical data demonstrated the existence of a 22π-electron conjugation encompassing the azacoronene core and a tropylium-like conjugation in the outer seven-membered ring.
13.9, a nonplanar core-expanded HPHAC analogue containing two N-doped seven-membered rings, was obtained from the commercially available octafluoronaphthalene and 3,4-diethylpyrrole via SNAr and oxidative coupling reactions.20 X-ray diffraction analyses revealed the distorted structures of both the neutral 13.9 and dication [13.9]2+[PF6–]2 (prepared with AgPF6). In spite of the twisted structure, the nucleus-independent chemical shift (NICS) data of 13.92+ indicated a significant aromaticity increase in the dicationic state, while the anisotropy of the induced current density (ACID) plot demonstrated an amplified current density of the peripheral pathway. These results are consistent with global Hückel aromaticity in the dicationic state, corresponding to peripheral 30 π-electron conjugation.
Peripheral expansion of HPHACs by inserting additional bridges or rings into the rim of the π-conjugated framework leads to systems with significantly modified electronic properties (CR2017, section 2.2). This approach was followed in the synthesis of an antiaromatic expanded azacoronene cation [13.12]+ reported by the Uno group in 2019.21 Synthesis of the partially fused 13.11 was carried out under standard conditions while controlling the amount of oxidant employed. The Vilsmeier–Haack reaction of 13.11 using DMF and POCl3 gave an excellent yield of the intramolecularly cyclized [13.12a]+, rather than α-formyl derivatives of 13.11. The antiaromatic monocation [13.12]+ was readily transformed into the aromatic trication [13.12a]3+.
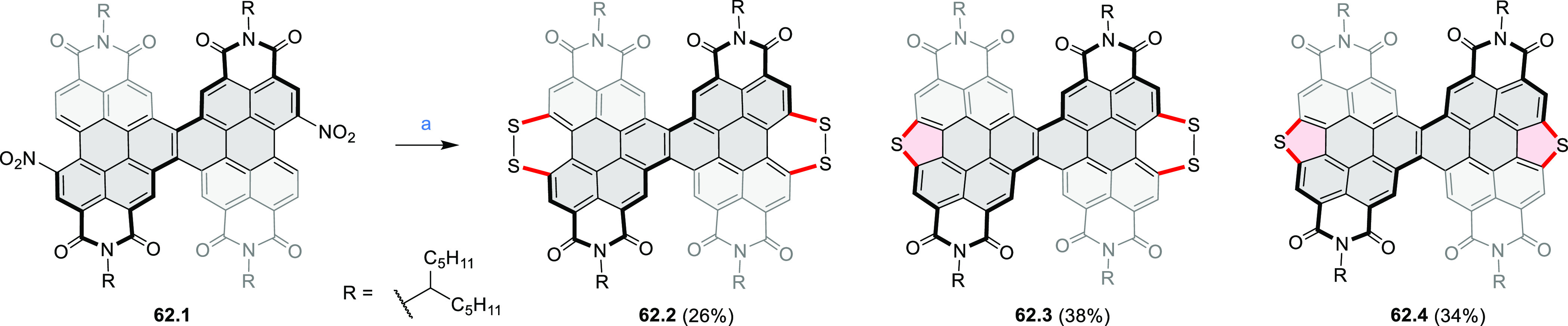
Radial π-extension of HPHACs is conveniently achieved by employing β–β-fused pyrrole building blocks. Our group reported the first such system, the large electron-deficient heterocycle C2.1, which was prepared from the naphthalenemonoimide (NMI)-fused pyrrole 308.1b(22) (Section 7.1) following the standard two-step procedure (Chart 2).23 An XRD analysis of C2.1 revealed a “monkey saddle” conformation, with alternating handedness of the peripheral helicene fragments. The electronic absorption spectrum of C2.1 showed an intense band in the visible region, with a vibronic pattern characteristic of many rylene imide derivatives. C2.1 exhibits considerable solvatochromism in solution, its color changing from purple in toluene, through bluish in DCM, to bluish gray in methanol. While the oxidation behavior of C2.1 was similar to that of its HPHAC parent, the new ring system revealed an exceptional ability to consecutively accept ten electrons at easily accessible potentials, yielding anions with small electronic band gaps and panchromatic UV–vis–NIR absorption. Efficient aerial reoxidation of the reduced nanographenoid indicated its resistance to decomposition.
Chart 2. Radially π-Extended Pyrrole-Fused Azacoronenes.
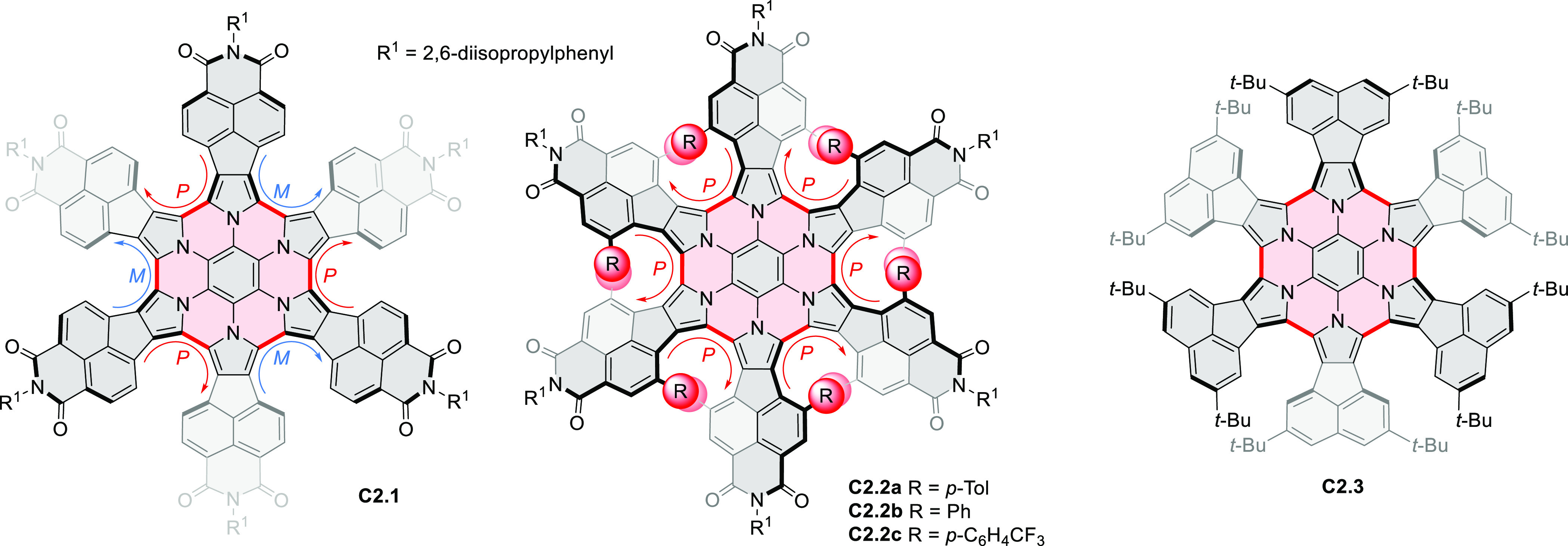
Molecular propellers C2.2a–c, chiral analogues of the snowflake-shaped C2.1, were obtained from sterically hindered hexapyrrolylbenzenes (HPBs), which were treated with bromine electrophiles, N-bromosuccinimide (NBS), and dibromine as oxidative coupling agents.24,25 Ferric chloride, generally an effective oxidant in the syntheses of HPHAC derivatives, applied to the sterically congested precursors of C2.2a–c, produced only mixtures of ill-defined, possibly polymeric products. Subsequent screening of various halogen electrophiles and reaction conditions showed the superior performance of NBS in lactic acid, producing C2.2a–c in 85–88% yields with excellent chemo- and stereoselectivities. Specifically, in contrast to the parent C2.1 the helical sections in C2.2a–c are homochiral. These propeller HPHACs, the first examples of chiral nanographene analogues with deeply embedded nitrogen atoms, possess small band gaps, near panchromatic absorption, and multiredox behavior.
C2.3, an electron-rich analogue of C2.1, with tert-butyl groups replacing the peripheral imide moieties, was synthesized by Uno’s team.26 In that synthesis, CaH2 was found to effectively promote the complete SNAr reaction of C6F6 with the bulky 2,5-di-tert-butyl-8H-acenaphtho[1,2-c]pyrrole. Like other azacoronene derivatives, C2.3 was stable in its oxidized forms, and the NICS calculations demonstrated the existence of global aromaticity in the dication.
A dimeric naphthalimide–azacoronene hybrid linked via a pair of methylene bridges was described by our group in 2020 (Scheme 14).27 Compound 14.2 was formed in a reaction of the partially oxidized HPB 14.1 with paraformaldehyde in the presence of 10-camphorsulfonic acid as the catalyst. In an oxygen-free toluene solution, the 14.2 dimer undergoes photodissociation into a radical monomer. The radical exhibits extensive spin delocalization in its 139-electron π system and spontaneously dimerizes back to a stable σ-dimer and, in the presence of oxygen, is further oxidized to a stable ketone 14.3. Photoinduced switching between the radical and its σ-dimer was found to rely on homolytic cleavage of a weak C(sp3)–C(sp3) bond, but its thermodynamics were controlled by a balance between π-conjugative stabilization, internal strain, and nonbonding interactions. The latter contribution had a decisive influence on the overall energetics of dimer formation and cleavage.
Scheme 14. Synthesis of an Azacoronene Nanosandwich.
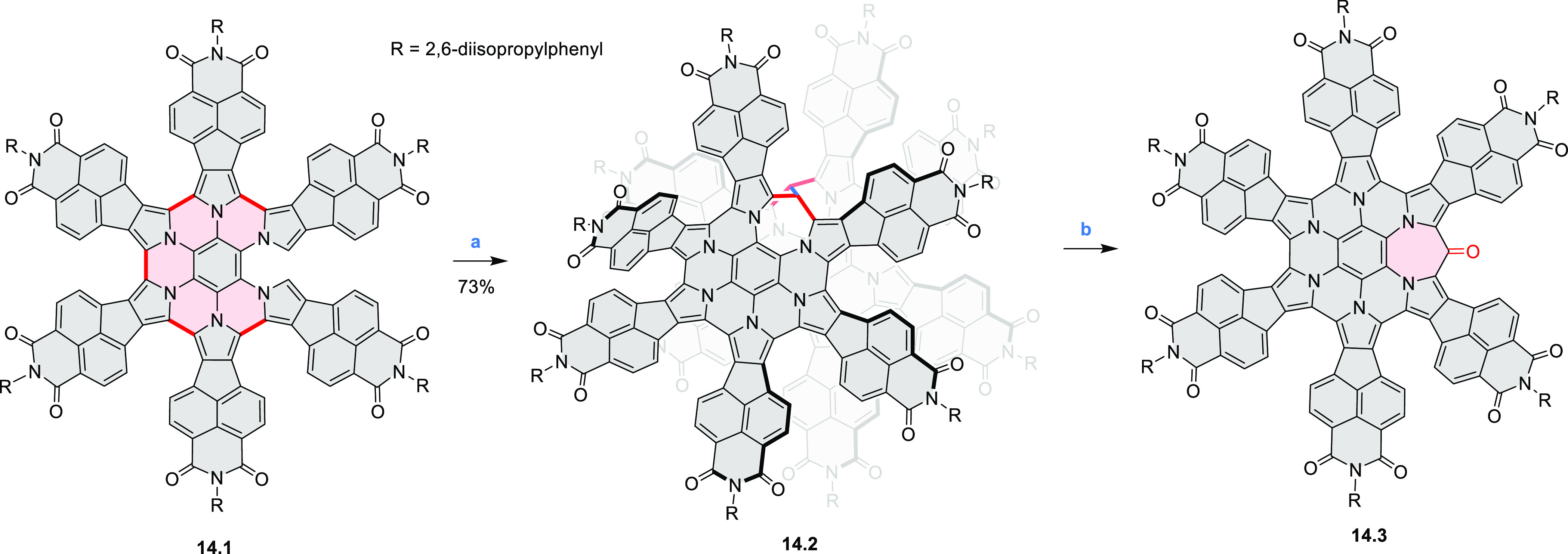
Reagents and conditions: (a)27 6 equiv of 10-camphorsulfonic acid, 4 equiv of paraformaldehyde, CHCl3, pressure tube, 90 °C, 17 h; (b) toluene, 365 nm irradiation, air.
The majority of internally doped azacoronenes is based on the HPHAC design. A different approach to internal doping was proposed by Müllen and co-workers, who obtained pyrazine-containing nanographenes via dimerization of dibenzo-9a-azaphenalene (DBAP) (Scheme 15).28 A DBAP salt 15.1a was dimerized by treatment with a large excess of tributylamine at 190 °C and oxidized with excess DDQ in dry C2D2Cl4, forming the hexabenzoperylene 15.2a. Attempts to oxidize the latter product directly to diaza-HBC derivatives resulted in insoluble solids, which were not characterized. By changing the strategy to on-surface synthesis, small quantities of 15.3b were detected after depositing 15.1 on Ag(111) by molecular beam evaporation and annealing to 270 °C. 15.3b was characterized by STM and FM-AFM; combined scanning probe data and theoretical investigations indicated that 15.3b remained neutral on the surface, retaining the 8π-electron state of the pyrazine ring.
Scheme 15. Synthesis of Diaza-HBP and Internally N-Doped Diaza-HBC.

Reagents and conditions: (a)28 (1) tributylamine, DMSO, 190 °C, Ar, (2) DDQ, C2D2Cl4, 100 °C; (b) in vacuo on the Ag(111) surface, 270 °C.
2.3. B- and BN-Doped Coronenes
2.3.1. Diboracoronenes
A synthesis of stable OBO-doped nanographenes was described in 2016 by Feng and Müllen et al.29 Treatment of hexabromobenzene with 2-methoxyphenylmagnesium bromide in THF provided 16.1a,b, which were then heated in o-dichlorobenzene with BBr3 at 150 °C, to furnish the OBO-doped helical bistetracenes 16.2 with excellent yields (Scheme 16, see also Scheme 51, Section 3.1.2). Bistetracenes 16.2a,b exhibited good stability and strong fluorescence (Φ = 61% for 16.2a and 52% for 16.2b) in comparison to their air-sensitive and nonfluorescent hydrocarbon analogue bistetracene. Single-crystal X-ray analysis revealed a double hetero[5]helicene structure with a highly twisted benzene ring. The cyclodehydrogenation of 16.2a,b in the presence of DDQ/TfOH cleanly transformed the twisted bistetracene analogues into the planar nanographenes 16.3a,b. In contrast to the unstable all-carbon peri-tetracene, the OBO-doped analogues 16.3a,b displayed excellent stability under ambient conditions. Compound 16.3b shows blue fluorescence with a quantum yield of 27%, with the emission spectrum being almost the mirror image of the low-energy absorption band. The Stokes shift was as small as 7 nm, indicating the rigid structure of this nanographene molecule. According to NICS calculations, the A and D rings in OBO-doped peri-tetracenes (Scheme 16) are highly aromatic. Ring B is nonaromatic, and ring C exhibits low aromaticity. These results are consistent with the Clar sextet formulation of 16.3a,b and explain their different properties relative to their hydrocarbon parent.
Scheme 16. Synthesis of OBO-Doped peri-Tetracenes via Bistetracenes.

Reagents and conditions: (a)29 BBr3, o-DCB, rt to 150 °C, 12 h; (b) DDQ, TfOH, DCM, 0 °C to rt.
Scheme 51. Synthesis of BO- and BNO-Doped Perylenoids.

Reagents and conditions: (a)83 (1) t-BuLi, chlorobenzene, −45 °C, 4 h or −45 to 0 °C, 3 h, (2) BBr3, (3) 40 °C, 24 h; (b)29 (1) BBr3, o-dichlorobenzene, 150 °C, 12 h; (c)84 BBr3, o-dichlorobenzene, 180 °C, 24 h; (d)86 6 equiv of BI3, 1,2,4-trichlorobenzene, 180 °C, 20 h.
Atomically precise introduction of group III dopant atoms into bottom-up fabricated semiconducting armchair graphene nanoribbons (AGNRs) was described by Fischer in 201530 and further studied by Garcia-Lekue, Corso, and Pascual et al.31 Nanoribbon 17.3 was obtained in a two-step on-surface reaction of 17.1 (Scheme 17). A clean Au(111) single-crystal surface was precovered with precursor 17.1 and annealed to 200–220 °C for ∼5 min to activate its Ullmann-like coupling and polymerization into structure 17.2. Further annealing to 300–400 °C (∼3 min) induced a cyclodehydrogenation reaction, leading to the completely planarized GNR 17.3. Scanning tunneling microscopy (STM) topography revealed a characteristic modulation of the local density of states along the backbone of 17.3 that is superimposable with the expected position and concentration of dopant B atoms.
Scheme 17. Bottom-Up Synthesis of Substitutionally Boron-Doped Graphene Nanoribbons.

Reagents and conditions: (a)30,31 Au(111), 200–220 °C; (b) Au(111), 300–400 °C.
2.3.2. BN-Containing Systems
Heteracoronene 18.4 containing four embedded NBN fragments was described by Dou, Liu, and Wang (Scheme 18).32 Because of the presence of tetrahedral BF2 fragments, the system is not fully conjugated, yet it can be viewed as an example of potentially useful heterocyclization. Treatment of 18.1 with a mixture of CBr4/PPh3 in DCM afforded the tetra-brominated 6,13-dimethylene-6,13-dihydroquinoxalino[2,3-b]phenazine 18.2 as a yellow solid. 18.2 was then subjected to a Pd-catalyzed alkylamination reaction to give the alkylaminated precursor 18.3. Finally, cyclization of 18.3 with BF3·Et2O/Et3N led to the simultaneous formation of four B–N rings, to produce 18.4 as a bluish violet solid. The compound displayed intense red fluorescence in toluene (Φ = 50%).
Scheme 18. Coronenoid Containing Four NBN Units.
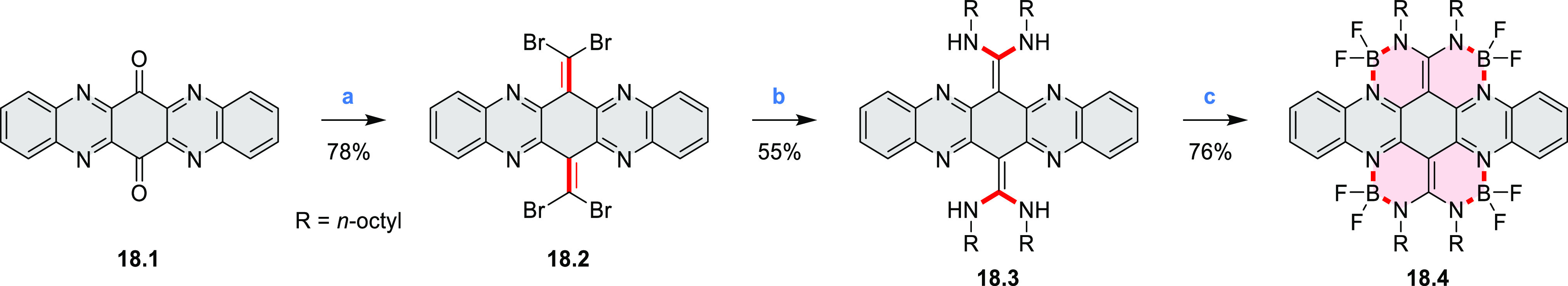
Reagents and conditions: (a)32 CBr4, PPh3, DCM, −50 to 25 °C; (b) R-NH2, t-BuONa, Pd2(dba)3, dppf, toluene, 120 °C; (c) BF3·Et2O, Et3N, DCM, 50 °C.
A solution synthesis of a BN-doped HBC analogue in which the central benzene ring was replaced by a borazine core was described by Bonifazi and co-workers.33 The hexaaryl-substituted borazine precursor 19.3 was obtained in a reaction of 4-xylyl aniline 19.1 with BCl3 followed by addition of (2,6-difluorophenyl)lithium (Scheme 19). Borazine 19.3 was planarized into the target hexa-peri-hexabenzoborazinocoronene 19.4 in a reaction with [i-Pr3Si][CB11H6Cl6] and Me2SiMes2. Along with 19.4, the partially fused BN derivative 19.5 was obtained as the major product (17% yield), suggesting that the ring closures proceed sequentially with the last aryl fusion likely being the rate-determining step. 19.4 exhibited strong blue-violet singlet emission and green phosphorescence.34 Higher cyclization yields were subsequently achieved for precursors containing singly fluorinated aryl groups (Scheme 19).35 In particular, 19.8 afforded borazino-coronene 19.4 in 15% yield, along with an unexpected cleavage product 19.10. The insoluble unsubstituted 19.9, previously reported in an on-surface synthesis (CR2017, section 2.3.2), was also obtained via the same silane-based protocol.35
Scheme 19. Synthesis of a BN-Doped Hexa-peri-hexabenzocoronene.
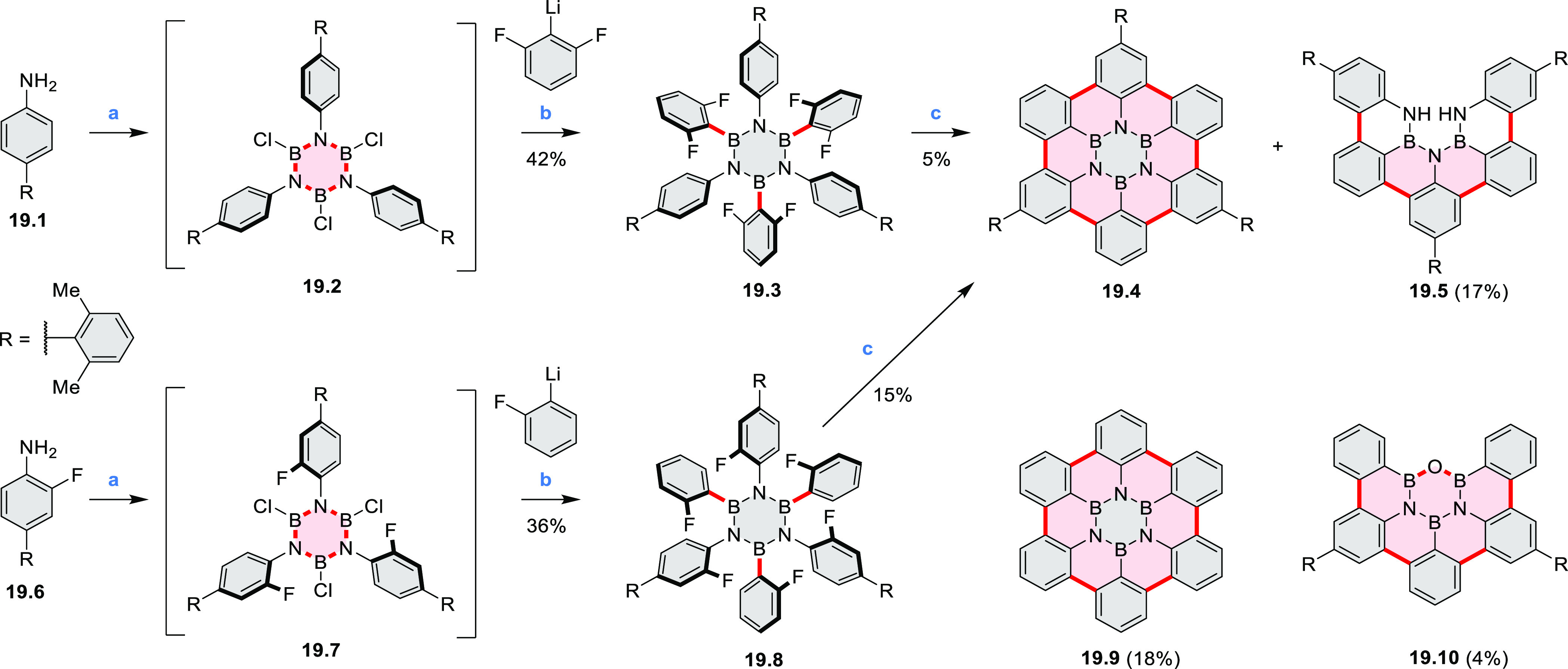
Reagents and conditions: (a)33 BCl3, toluene, reflux; (b) THF, −84 °C to rt; (c) [i-Pr3Si][CB11H6Cl6], Me2SiMes2, PhCl, 110 °C, Schlenk line.
A synthesis of BN-doped hexa-cata-hexabenzocoronene 20.3 was reported by Hatakeyama et al. in 2017 (Scheme 20).3620.1 was obtained via the palladium-catalyzed C–N coupling between 1,3,5-tribromobenzene and di-p-tolylamine and subjected to one-shot quadruple borylation in the presence of 12 equiv of BI3, to afford the quadruply borylated 20.4 in 35% isolated yield and triple borylation compound 20.3 in only 3% yield. Other boron sources, like BCl3 and BBr3, did not give desired borylation products. In the presence of 5.0 equiv of BI3 and 2.0 equiv of Ph3B, selective double borylation took place under reflux in o-DCB to give 20.2 in 76% isolated yield. Triple borylation could be achieved at a more elevated temperature to give 20.3 as the main product (45% isolated yield). 20.2, 20.3, and 20.4 showed deep-blue fluorescence (at 488, 466, and 475 nm, respectively) and small energy differences between the excited singlet and triplet states (0.15–0.18 eV).
2.4. peri-Condensed Coronenes
The propeller-shaped, nitrogen-doped hexapole [7]helicene 21.3 was reported by Wang and co-workers in 2019 (Scheme 21).37 A [2 + 2 + 2] cyclotrimerization of alkyne precursor 21.1, catalyzed by Co2(CO)8 in dioxane at 120 °C, produced the hexaarylbenzene precursor 21.2 containing six dibenzoullazine arms. Extensive screening of cyclodehydrogenation conditions established that photooxidation of 21.2 in chloroform under an aerobic atmosphere was a practical method of preparing 21.3 in modest yield (ca. 8%). Unsuccessful attempts to obtain 21.3 via chemical oxidation indicated that the radical cation of the electron-rich dibenzoullazine unit in 21.2 is probably stable, which prevents it from undergoing further transformation. Although the detailed mechanism of the photochemical conversion of 21.2 into 21.3 was not clarified, control experiments indicated that both oxygen and chloroform were essential. No photoreaction was observed when a solution of 21.2 in chloroform was irradiated under a nitrogen atmosphere or when DCM was used as the solvent.
Scheme 21. Propeller-Shaped Nanographene.
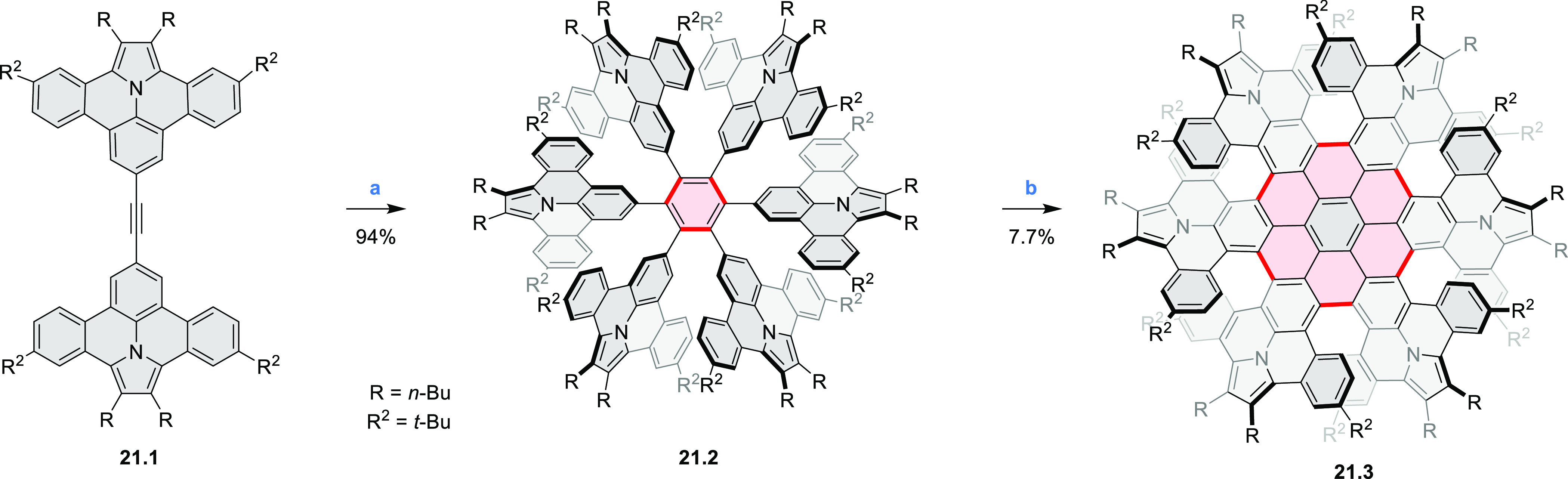
Reagents and conditions: (a)37 Co2(CO)8, dioxane, 120 °C; (b) hν, CHCl3.
A π-extended “superhelicene” containing two HBC units was synthesized by Jux and co-workers (Scheme 22).38 Their four-step synthesis started with diphenyl ether, which was first converted into its 4,4′-dibrominated derivative by aromatic halogenation and subjected to double Sonogashira coupling with 4-tert-butylphenylacetylene to provide 22.1. A Diels–Alder reaction of 22.1 with 2.5 equiv of 2,3,4,5-tetrakis[4-(tert-butyl)phenyl]cyclopenta-2,4-diene-1-one yielded 22.2 which was reacted with DDQ and triflic acid in DCM to produce the helical compound 22.4. The closure of the furan ring is most probably the final step of the cyclodehydrogenation cascade, as shorter reaction times (e.g., 2 h) furnished mixtures of helicene 22.4 and the incompletely cyclized 22.3. Changing the reagents to FeCl3 in nitromethane/DCM allowed us to avoid the closure of the furan ring, thus forming 22.3 selectively. 22.3 was sensitive to light and transformed into helicene 22.4 through photocyclization, causing minor amounts of 22.4 to always be observed during photophysical measurements. 22.4 showed an almost 103-fold amplification of photoluminescence dissymmetry factors gPL of a π-extended superhelicene when embedded in an achiral conjugated polymer matrix, from approximately 3 × 10–4 in solution to 0.15 in a blend film in the solid state.39
Scheme 22. Synthesis of Oxa[7]superhelicene.
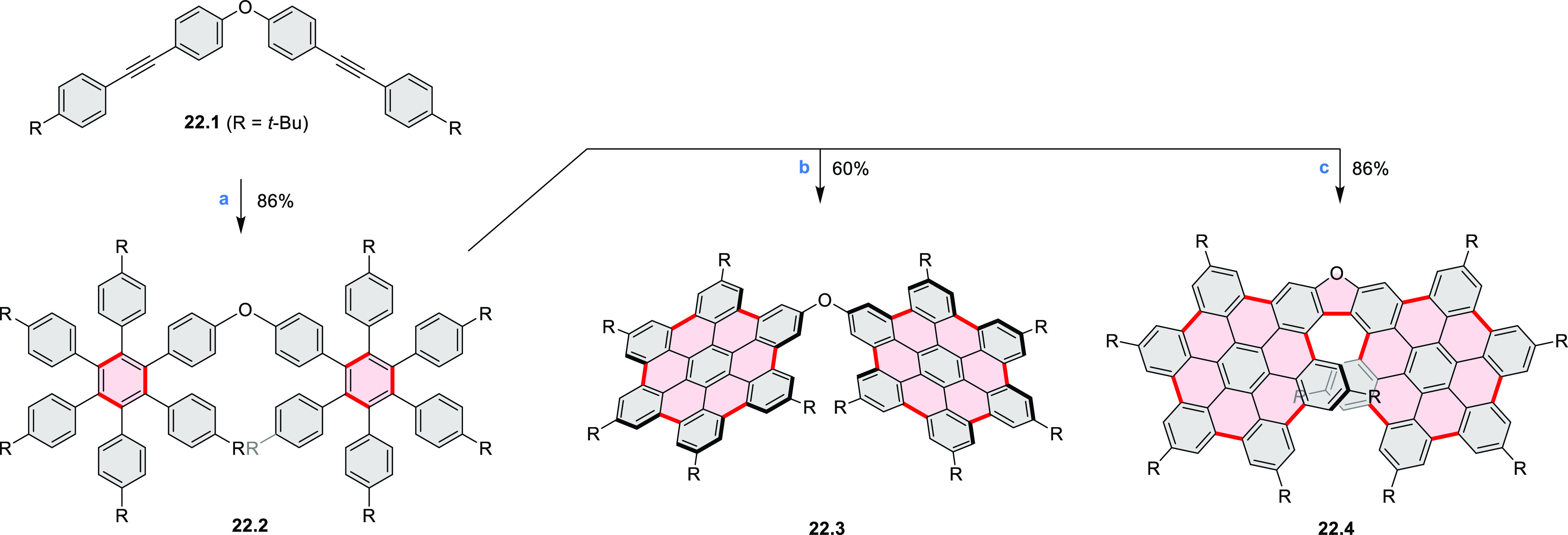
Reagents and conditions: (a)38 2,3,4,5-tetrakis(4-tert-butylphenyl)cyclopenta-2,4-dien-1-one (2.5 equiv), toluene, N2, 23 h, 220 °C (pressure flask); (b) anhydrous FeCl3 (35 equiv), MeNO2, DCM, N2, 25 min at 0 °C, 20 h at rt; (c) DDQ (15 equiv), triflic acid (30 equiv), DCM, N2, 25 min at 0 °C, 20 h at rt.
A persulfurated coronene “sunflower” 23.3 was reported in 2017 by Feng and Müllen et al. (Scheme 23).40 Its synthesis was carried out starting from coronene, which was chlorinated to the dodecachloro derivative 23.1. Nucleophilic replacement of all peripheral chloro substituents was achieved using lithium benzylthiolate at rt, to afford dodecakis(benzylthio)coronene 23.2 as a red powder in 62% yield. After reductive cleavage of the protective benzyl groups under Birch conditions, the resulting dodecalithium species was treated with aqueous hydrogen chloride and hydrogen peroxide to afford the desired product 23.3 as a dark-red solid. Because of the low solubility of 23.3, its characterization by NMR or XRD was not possible. The structure was studied by mass spectrometry, IR, and Raman spectroscopy and by scanning tunneling microscopy (STM). Compound 23.3 could be reduced with sodium borohydride to afford the more readily soluble perthiolated coronene 23.4.
Scheme 23. Persulfurated Coronene.

Reagents and conditions: (a)40 phenylmethanethiol, NaH, DMI, 0 °C to rt, 16 h; (b) (1) Li, THF, MeOH, NH3, −78 °C to rt, 4 h, (2) HCl/H2O2/water, rt; (c) NaBH4.
Thiophene-fused extended HBCs with the proposed structures C3.1 and C3.2 were reported by Jin et al. (Chart 3).41 These structures were prepared by FeCl3 oxidation of the corresponding hexaarylbenzenes, which were assembled using either the cobalt-catalyzed cyclotrimerization or the Diels–Alder cycloaddition route. The formation of C3.1 and C3.2 was validated only by mass spectrometry.
Chart 3. Peripherally Fused HBC Systems41,42.

A well-defined nanographene–Re complex C3.3 was synthesized by Li and co-workers.42 The ligand was obtained via condensation reaction of 1,10-phenanthroline-5,6-diamine with the corresponding coronene-diketone derivative (new bonds indicated in red, Chart 3). The ligand was then treated with an excess of Re(CO)5Cl in hot toluene to yield C3.3. The nanographene-containing complex showed a significantly less negative potential for electrocatalytic CO2 reduction as well as visible-light-driven photocatalytic CO2 reduction without the need for a photosensitizer.
2.5. ortho-Condensed Coronenoids
2.5.1. Coronenoids Fused to Azaheterocycles
Three N-heteroarenes with azaacene units fused to a coronene nucleus were described by Bunz et al. (Scheme 24).43 Green light irradiation of a chloroform solution of 24.1 with a catalytic amount of iodine yielded the key coronene precursor 24.2. Palladium-catalyzed coupling of this compound with o-diaminoarenes 24.3a–e produced 24.4a–e in good yields (up to 83%). Finally, the dihydropyrazine species 24.4a–e were oxidized with MnO2 at rt. The resulting 24.5a–c showed decreased solubility, as a consequence of increased π-stacking. The most soluble 24.5e derivative was used for fabrication of a proof-of-concept thin-film transistor, yielding electron mobilities of 8 × 10–4 cm2 V–1 s–1 in polycrystalline films. This relatively low charge carrier mobility was attributed to the small domain size and polycrystalline nature of the fabricated films. A related phenothiazine-fused PDI 24.6 was synthesized by Aratani and Yamada et al.44 The latter system was prepared via DDQ/TfOH-mediated oxidation of the corresponding phenothiazine-linked PDI.
Scheme 24. Coronene-Containing N-Heteroarenes.
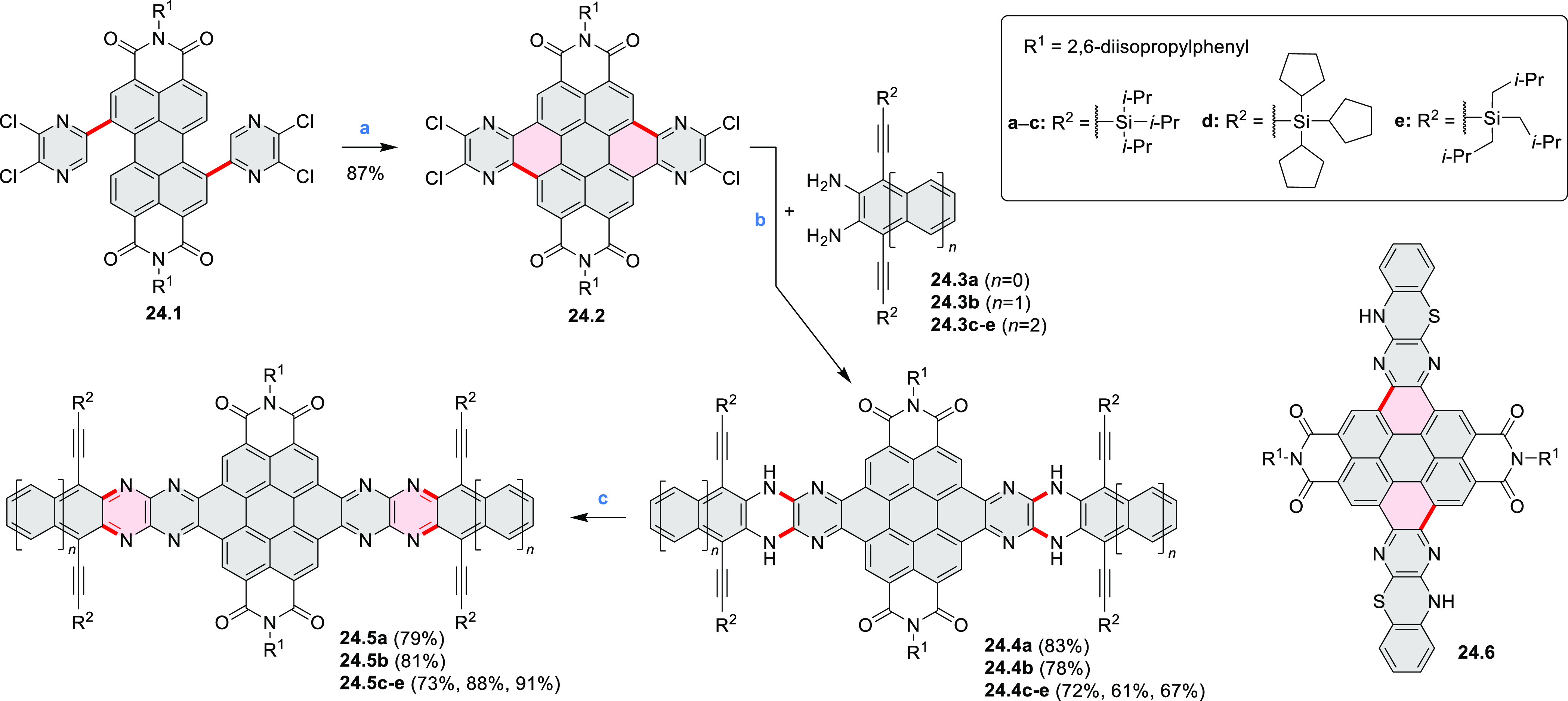
Reagents and conditions: (a)43 I2, hν, rt; (b) Pd(dba)2, RuPhos, DIPEA, CHCl3, 60 °C; (c) MnO2, CHCl3, rt.
2.5.2. Thieno-Fused Coronenoids
A strategy toward tetraheteracoronenes derived from soluble diarenoperylenes was presented by Mastalerz et al. in 2018 (Scheme 25).4525.1a,b were selectively brominated at the 8,16 positions by treating 2.2 equiv of NBS in DCM at rt for 1 h. The aryl bromides 25.2a,b were then appended with thienyl units via Suzuki–Miyaura cross-coupling, leading to 25.3a,b. Finally, photocyclization of these compounds in the presence of catalytic I2 and propylene oxide as an acid scavenger provided the extended coronenes 25.4a,b in 61–65% yields. Compound 25.4c was obtained in the same manner, starting from a thiophene-substituted perylene.
Scheme 25. cata-Condensed Heteroannulated Coronenes.
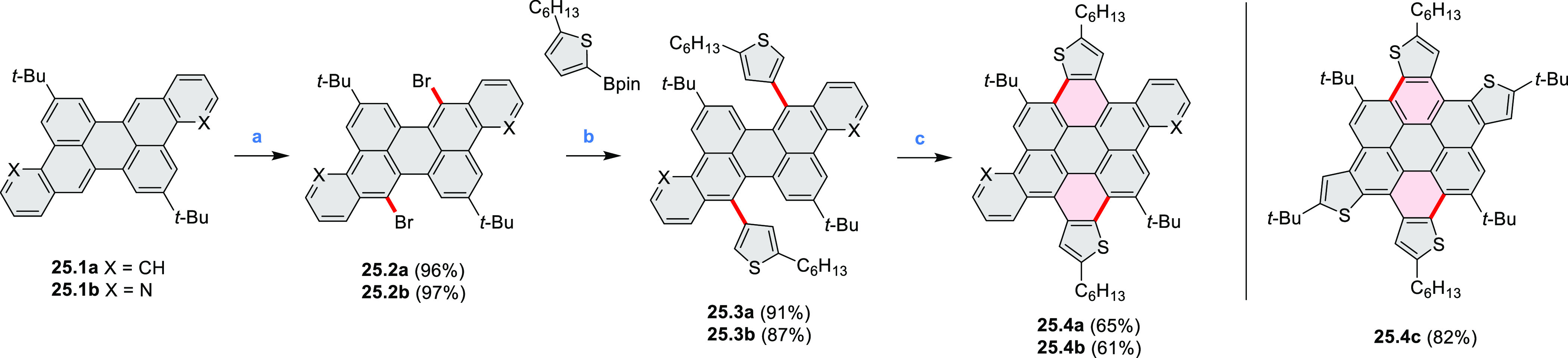
Reagents and conditions: (a)45 2.2 equiv of NBS, DCM, rt, 1 h; (b) 5 mol % of Pd2(dba)3, 7.5 mol % of t-Bu3P·HBF4, THF, K2CO3, 80 °C 16 h; (c) I2, hν, propylene oxide, cyclohexane, 4 h.
Benzofuran- and benzothiophene-fused analogues of c-HBC 26.4a,b were synthesized by Loo et al. in 2015 (Scheme 26).4626.1 was obtained via Corey–Fuchs reaction using isopropyl phosphite with tetrabromomethane and dibenzoanthraquinone. In the Suzuki–Miyaura coupling step, N-methyliminodiacetic acid (MIDA) boronates 26.2a,b were used instead of boronic acid derivatives. Their gradual conversion to the boronic acid counterparts during the reaction allowed the coupling to compete favorably with boronic acid decomposition, thus leading to higher synthetic yields, particularly in the case of the 2-benzofuranyl derivative 26.2a. Intermediates 26.3a,b were then subjected to ring closing via photocyclization in toluene, leading to 26.4a,b. Unlike in the syntheses of many c-HBC derivatives, chemical oxidation was not necessary to effect complete ring closure of the precursors to yield 26.4a,b, likely because peripheral congestion was reduced by introduction of five-membered rings. Such benzofuran- and benzothiophene-containing c-HBC derivatives showed stronger visible-light absorption, in comparison with c-HBC, which was attributed to a simultaneous decrease in molecular symmetry and an increase in conjugation relative to the parent c-HBC compound.
Scheme 26. Synthesis of O- and S-Doped Contorted Coronenes.

Reagents and conditions: (a)46 Pd(OAc)2, SPhos, K3PO4, dioxane/water, 60 °C; (b) hν, I2, toluene, 2-methyloxirane.
Three-dimensional S-doped nanographenes featuring a cyclooctatetraene core 27.3 were reported by the groups of Molina-Ontoria, Guldi, and Martín (Scheme 27).47 The initial tetraalkyne 27.1 was prepared via Sonogashira coupling from a tetrabrominated cyclic tetrathiophene, which was obtained via an oxidative dimerization of 2,2′-dibromo[3,3′]bithiophene. Compound 27.1 was subjected to a microwave-assisted [4 + 2] cycloaddition with 2,3,4,5-tetrakis[4-(1,1-dimethylethyl)phenyl]2,4-cyclopentadien-1-one to give rise to 27.2. Finally, the treatment of the latter species with FeCl3 produced the corresponding fully cyclodehydrogenated 27.3 in moderate yield. Two different crystal polymorphs of 27.3 were crystallographically characterized. 27.3 underwent triplet energy transfer to C60, but in contrast to its all-benzene analogue, it showed no electron transfer to TCNE.
Scheme 27. Sulfur-Doped Three-Dimensional Nanographenes.
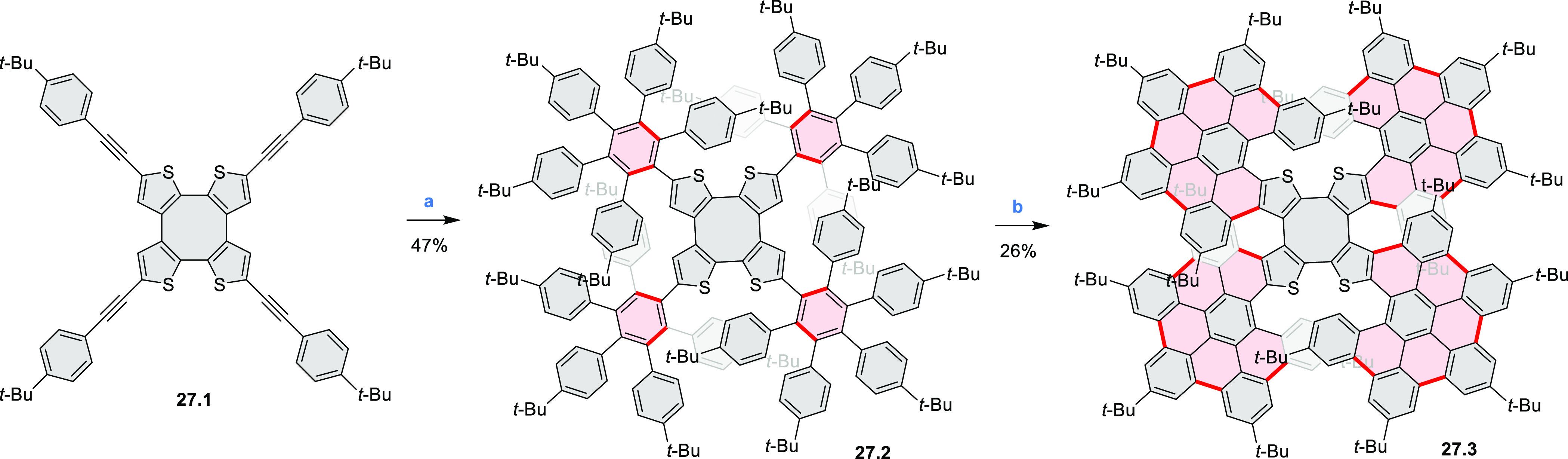
Reagents and conditions: (a)47 2,3,4,5-tetrakis(4-tert-butylphenyl)cyclopenta-2,4-dien-1-one, MW, 300 °C; (b) FeCl3, MeNO2, DCM, 0 °C.
A series of trisbenzothieno[1,2:7,8:13,14]hexa-peri-hexabenzocoronenes were synthesized by Pisula and Feng et al. (Scheme 28).48 Compounds 28.2a–f bearing alkyl or alkoxy substituents were synthesized via an intramolecular oxidative cyclodehydrogenation reaction of triarylbenzenes 28.1a–f. The products were well soluble in common organic solvents, including DCM, toluene, and THF. Unlike its alkoxy analogues, the alkyl-substituted 28.2a could be selectively oxidized with m-CPBA to produce the triple sulfone 28.3, which was obtained as a red powder. The HOMO level of a methoxy analogue of 28.2b–c increased to −4.98 eV relative to the alkyl-substituted 28.2a (−5.06 eV), while the LUMO level also increased from −1.80 eV (28.2a) to −1.74 eV (28.2b,c). The HOMO/LUMO levels of 28.3a (−5.58/–2.34 eV) decreased compared with those of 28.2a (−5.06/–1.80 eV), reflecting the conversion of the electron-rich benzothiophene ring into an electron-poor thiophene-S,S-dioxide unit.
Scheme 28. Tris(benzothiophene)-Fused Hexa-peri-hexabenzocoronenes.

Reagents and conditions: (a)48 FeCl3, DCM, MeNO2, rt, 1 h; (b) m-CPBA, THF, rt, 1 h.
Highly π-extended donor–acceptor hybrids consisting of a perylene or naphthalene diimide fused to a hexabenzocoronene core (e.g., C4.1–2, Chart 4) were described in 2020 by Guldi and Hirsch et al.49 The hexaphenylbenzenes were obtained from the corresponding brominated 1,2-ryleneimidebenzimidazole in a sequence consisting of palladium-catalyzed Sonogashira cross-coupling, Diels–Alder reaction, and FeCl3-mediated oxidative coupling. Linearly fused systems such as C4.2 showed enhanced π conjugation, leading to red-shifted absorption and fluorescence. All conjugates revealed a CT character in the ground and excited states. For (PDI–HBC)s, the CT character of the ground state was reflected in a slight hypsochromic shift of the PDI absorption.
Chart 4. Ryleneimide–HBC Hybrids.

3. Perylenoids
3.1. Heteraperylenoids
3.1.1. Monoheteraperylenoids
Azaperylene 29.2 was obtained by Tang et al. via highly regioselective 3-fold photocyclodehydrogenation of the tetraphenylethylene derivative 29.1 (Scheme 29).50 In contrast to most PAHs, azaperylene 29.2 did not suffer from strong fluorescence quenching in the solid state and exhibited high emission quantum yields of 49% in solution and 21% as a solid. Dimers bound through π–π interactions were revealed in the solid-state structure of 29.2, but further π-stacking was prevented by steric crowding. The dimeric nature of 29.2 was proposed to be responsible for the prominent red-shift of its emission in the solid state (λem = 566 and 609 nm) compared to that in solution (λem = 476 nm). Methylation of the pyridine units in 29.1 with iodomethane followed by ion exchange with potassium hexafluorophosphate led to 29.3 in 95% yield. This compound underwent photocyclodehydrogenation to 29.4 upon UV irradiation in the presence of oxygen. 29.4 could also be synthesized by dimethylation of 29.2. While 29.3 was weakly emissive in solution, 29.4 displayed emission at 580 nm with a quantum yield of 18% in a PBS-buffered aqueous medium. The transformation from 29.3 to 29.4 was performed in HeLa cells upon UV irradiation, which resulted in a strong increase in fluorescence that could be observed through yellow or red channels.
Scheme 29. Synthesis of N-Doped Benzo[ghi]perylene and Its Dication.

Reagents and conditions: (a)50 I2, propylene oxide, hν (500 W high-pressure mercury vapor lamp), rt, 2 h, 64%; (b) (1) CH3I, MeCN, reflux, overnight, (2) KPF6, acetone, 2 h, 95%; (c) MeOH, hν (500 W high-pressure mercury vapor lamp), O2, 10%; (d)51,52 iodine, propylene oxide, toluene, hν.
A similar synthetic approach was employed by Raouafi and Aloui in their synthesis of N-doped benzo[ghi]perylenes 29.7a,b (Scheme 29). Compounds 29.7a,b were obtained via double oxidative photocyclization of π-extended stilbene derivatives 29.5a,b. In the course of this reaction, the intermediate helicenes 29.6a,b were not isolated. Target perylenes 29.7a,b were well-soluble in common organic solvents and exhibited absorption and emission in the visible region.
Würthner and co-workers reported the synthesis of 3-azaperylenes through a cross-coupling methodology. In the reaction of 5-quinolineboronic ester 30.1 with 1,8-dibromonaphthalene at high temperature, the initial Suzuki coupling was followed by cyclization through C–H activation to provide the 3-azaperylene 30.2 (Scheme 30).53 This represents the first synthesis of unsubstituted 3-azaperylene, which was previously only isolated from natural sources. A different set of optimized conditions allowed us to perform this coupling with the more electron-rich 5,6-dibromoacenaphthalene to give 30.3. Several nonheterocyclic analogues were prepared along with these examples. The N-doping was observed to cause a red-shift of emission (main peak at 457 nm in 30.2 and 488 nm in 30.3) and an increase in Stokes shifts.
Scheme 30. Synthesis of Azaperylenes Through Intramolecular C–H Activation.

Reagents and conditions: (a)53 [Pd2(dba)3]·CHCl3, P(m-tolyl)3, Cs2CO3, 1-chloronaphthalene, 160 °C, 16 h, 50%; (b) Pd(PPh3)2Cl2, Cs2CO3, mesitylene, 120 °C, 16 h, 25%.
Dehydrogenative cyclization of 4-naphthylcoumarins via a variant of the Scholl reaction54 was used by the group of Zhang to synthesize perylenoid structures 31.3 and 31.7 (Scheme 31).55 These compounds were then selectively brominated and derivatized under optimized Suzuki coupling conditions. Irradiation of unprocessed Suzuki reaction mixtures with blue LED light under air resulted in an efficient electrocyclization and dehydrogenation, leading to the fused products 31.5a–p. The final compounds were intensely fluorescent, with emission color tunable from blue to red depending on the substitution pattern. Peak emission wavelength varied from 424 nm in 31.5b,c to 570–605 nm in the electron-rich compounds 31.5k–n. These analogues also had the highest fluorescence quantum yields (31.5l: 67.9%, 31.5m: 75.2%, 31.5l: 53.0%).
Scheme 31. Synthesis of Extended Perylenoids Containing a Coumarin Substructure.
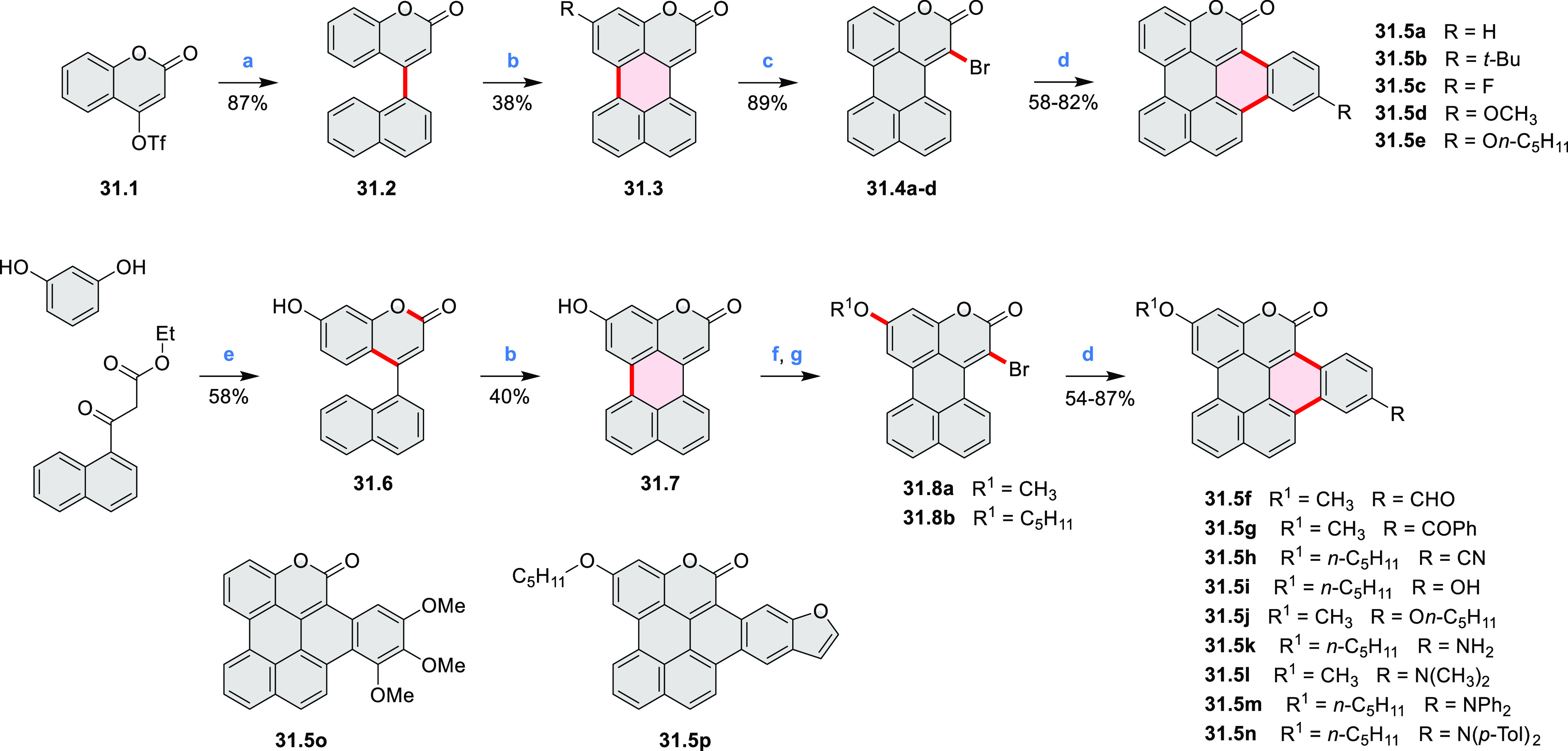
Reagents and conditions: (a)55 1-naphthylboronic acid, Pd(PPh3)4, K2CO3, 4:1 toluene/H2O, 90 °C, 2 h, 87%; (b) AlCl3, NaCl, 140 °C, 4 h, 38%; (c) NBS, Bz2O2, DCM, 30 °C, 12 h, 89%; (d) RB(OH)2, Pd(PPh3)2Cl2, K3PO4, 5:1 EtOH/H2O, 90 °C, 8 h, then 5:1 EtOH/H2O, air, hν (blue LED), rt, 3 h, 54–87%; (e) MeSO3H, rt, 16 h, 58%; (f) MeI, K2CO3, DMF, rt, 5 h, 85% or 1-bromopentane, Na2CO3, KI, DMF, reflux, 16 h, 48%; (g) NBS, Bz2O2, CHCl3, rt, 1 h, 82–84%.
Diheteratriangulenium salts with a π-extension resulting in a monoheteraperylenoid substructure were evaluated as fluorescent dyes by Laursen and co-workers (Scheme 32).56 These compounds are extended analogues of a larger series of triangulenium fluorophores (see Chart 13 and Scheme 122, Section 4.1). Different synthetic pathways were suitable for dioxa and diaza variants. Addition of lithiated o-dimethoxybenzene to the dichlorobenzanthrone 32.2, followed by a one-step dehydration, demethylation, and nucleophilic aromatic substitution, provided 32.4. Unlike other dioxatriangulenium examples, 32.4 could not be transformed into its diaza analogues in reactions with primary amines. In contrast, condensation of carbenium salts 32.8 and 32.10 with methylamine gave the diazahelicenes 32.9 and 32.11, respectively. Both compounds were cyclized to 32.12 via heating in polyphosphoric acid. Emission spectra of the resulting heteraperylenoids were red-shifted relative to their triangulenium analogues lacking the π extension. Compound 32.4 had its main emission peak at 595 nm (Φ = 78%), while for 32.12 a weaker red luminescence at 652 nm (Φ = 30%) was observed. While the solubility of these compounds prevented their use in cell imaging, the morpholine-appended compound 32.13 was successfully introduced into cells. It displayed emission enhancement upon DNA binding, which was attributed primarily to deaggregation.57
Scheme 32. Synthesis of π-Extended Triangulenium Dyes.
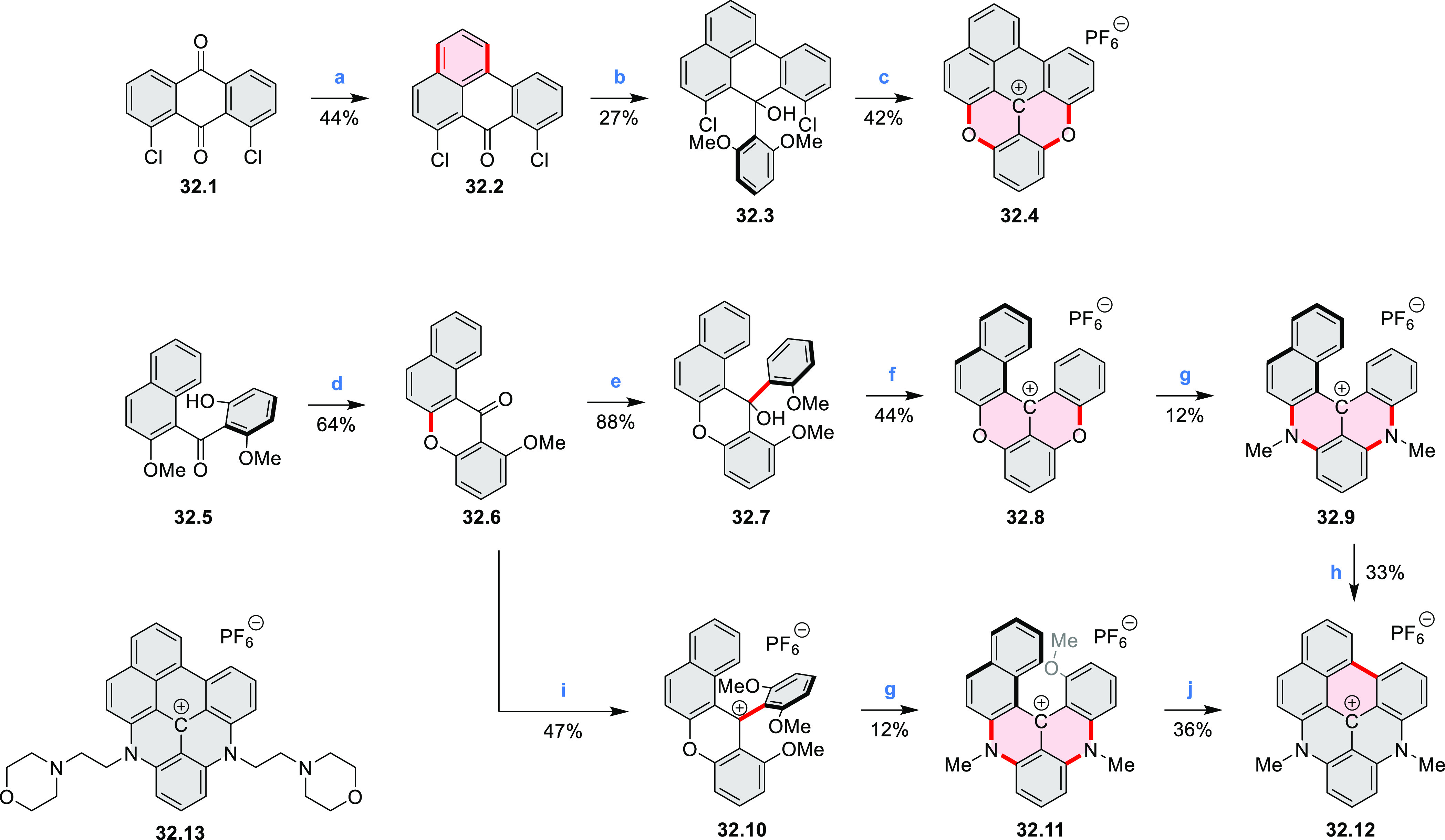
Reagents and conditions: (a)56 (1) Al, H2SO4, 25 °C, 18 h, (2) glycerol, H2SO4, 125 °C, 3.5 h; (b) m-dimethoxybenzene, TMEDA, n-BuLi, 1:1 benzene/Et2O, rt, 1 h; (c) (1) 48% HBr(aq), AcOH, reflux, 48 h, (2) 0.2 M KPF6(aq); (d) neat, 225 °C, 5 h; (e) Li, o-bromoanisole, benzene/Et2O, reflux, 30 min; (f) pyridine hydrochloride, 190 °C, 5 min; (g) methylamine, PhCO2H, NMP, 90–95 °C, 18 h; (h) PPA, 110 °C, 30 h; (i) (1) m-dimethoxybenzene, TMEDA, n-BuLi, 1:1 benzene/Et2O, reflux, 2 days, (2) 6 M HCl(aq), 0.2 M KPF6(aq); (j) PPA, 110 °C, 1 h.
Chart 13. Diazaoxa- and Azadioxatriangulenium Salts.
Scheme 122. Diheteratriangulenium Dyes with an Isopropylene Bridge.
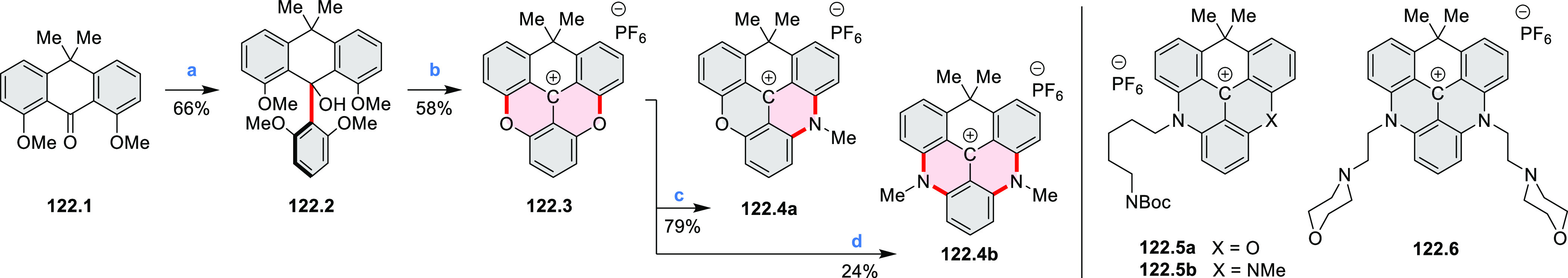
Reagents and conditions: (a)248m-dimethoxybenzene, TMEDA, n-BuLi, 1:1 benzene/Et2O, 0 °C to rt, 3 h; (b) pyridine hydrochloride, 190–200 °C; (c) 2 equiv of methylamine, PhCO2H, NMP, 70 °C, 3 days; (d) 60 equiv of methylamine, PhCO2H, 1:1 EtOH/NMP, reflux, 5 days.
An electrophilic borylation reaction was used to introduce boron as a fusion point in the indole-fused boraperylene 33.3 (Scheme 33).58 This was done through a two-step, one-pot sequence from 33.1, where the 3-position of indole was borylated first, followed by further intramolecular borylation. Highly electrophilic borenium salts derived from chloroboranes and dichloropyridine in the presence of AlCl3 are thought to be the electrophilic species responsible for the second step of this process.59 To prevent indole oligomerization caused by strong acid, an excess of 2,4,6-tri(tert-butyl)pyridine was added as a noncoordinating base. Compound 33.3 was obtained in 30% yield, partly because of its vulnerability to protodeborylation, which caused significant product loss during chromatographic purification.
Scheme 33. Indole-Fused Boraperylene Prepared by Electrophilic Borylation.

Reagents and conditions: (a)58 BCl3, 2,4,6-tri(tert-butyl)pyridine, AlCl3, DCM, 1 h; (b) 2,4,6-tri(tert-butyl)pyridine, 2,6-dichloropyridine, AlCl3, DCM, rt, 18 h.
3.1.2. Diheteraperylenoids
In 2017, Würthner and co-workers used a new one-pot strategy in the synthesis of a stable 3,9-diboraperylene as its corresponding borinic acid 34.2a (Scheme 34).60 The sequence consists of alkene hydroboration followed by C–H borylation with an NHC-borenium ion. The doubly boron-doped 34.2a exhibits absorbance in the visible region, with the lowest-energy maximum at 561 nm and bright fluorescence in CHCl3 solution (λmax = 603 nm, Φ = 63%). The electron-deficient analogue 34.2b showed an even higher fluorescence quantum yield (λmax = 603 nm, Φ = 0.95). Cyclic voltammetry studies performed on 34.2a showed two reversible one-electron reductions at moderate potentials of −1.30 and −1.64 eV vs Fc+/Fc in DMSO. These two reduction potentials were anodically shifted relative to those of perylene. The B-hydroxyl groups in 34.2a were subsequently replaced with B-mesityl groups, yielding 34.3a.61 The B–C bonds in 34.2a were also used as reactive handles for Suzuki coupling, giving a saddle-shaped hydrocarbon with two seven-membered rings.6234.3a was implemented in organic thin-film transistors (OTFTs), exhibiting n-type charge-carrier mobilities of 3 × 10–3 cm2 V–1 s–1. It was also used as an acceptor in combination with donor polymers in bulk-heterojunction solar cells with power conversion efficiencies of up to 3%.61 Moreover, this C–H borylation method was expanded to other polyaromatic boronic acids including triangulene (Chart 12, Section 4.1) and pyrenoid systems (Scheme 140, Section 4.3).
Chart 12. Diboratriangulenes.

Wagner and co-workers reported the synthesis of a boron-doped tetrabenzopentacene 35.3 and oxadiborepin 35.4 from a single starting material (Scheme 35).63 First, a single lithium–halogen exchange of 1,8-dibromonaphthalene followed by condensation with 35.1 led to 35.2, which was obtained as a mixture of atropisomers. Intramolecular Yamamoto coupling of 35.2 in pyridine led to the expected product 35.3. However, performing the same reaction in THF provided the oxadiborepin 35.4 in high yield. The authors proposed that 35.3 undergoes a Ni-catalyzed transformation to 35.4 upon introduction of oxygen and moisture while quenching the reaction with a stream of air. The absence of this transformation in pyridine was attributed to its interaction with the Lewis-acidic boron atoms.
Scheme 35. Synthesis of a Boron-Doped Tetrabenzopentacene.

Reagents and conditions: (a)63 1,8-dibromonaphthalene, 1 equiv of n-BuLi, Et2O, 0 °C – rt, 45 min, then 35.1, rt, overnight, 86%; (b) Ni(cod)2, cod, 2,2′-bipyridyl, pyridine, rt, 24 h, 79%; (c) Ni(cod)2, cod, 2,2′-bipyridyl, THF, rt, 24 h, 81%.
A two-step procedure developed by Takata and co-workers provided access to two types of helical polymers containing either a dioxaperylene or dioxapyrene substructure (cf. Scheme 137, Section 4.3).64 Polycondensation of 137.1 with the anthraquinone spacer 36.1 efficiently gave the polymeric precursor 36.2 in 86% yield (Scheme 36). A subsequent intramolecular cyclization reaction of 36.2 upon treatment with H2SO4 quantitatively afforded a screw-shaped helical polymer 36.3. The product was partially sulfonated, bearing an average of 1.9 sulfonic acid groups per monomer on its fluorene moieties. This caused 36.3 to be soluble in water and DMSO.
Scheme 137. Synthesis of a Screw-Shaped Polymer.
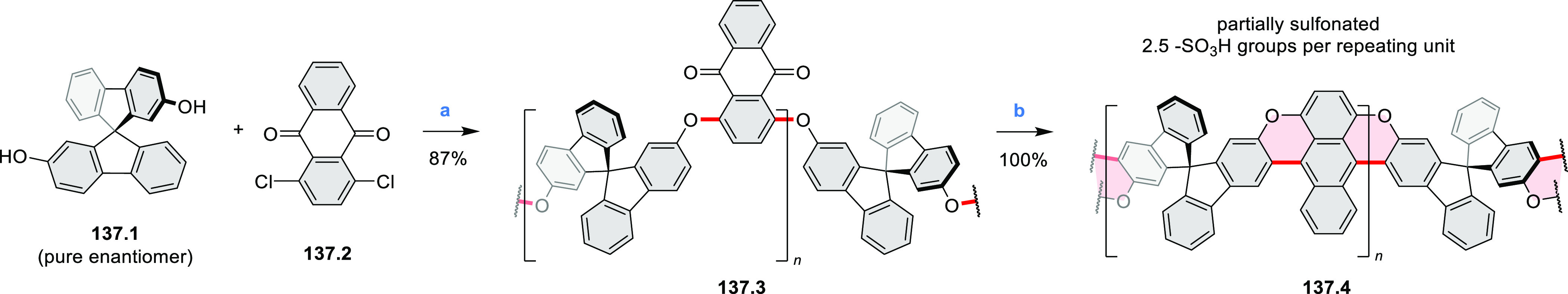
Reagents and conditions: (a)64 K2CO3, diphenyl sulfone, 210 °C, 5 h; (b) (1) H2SO4, 160 °C, 6 h, (2) tetrabutylammonium iodide, 120 °C, 6 h.
Scheme 36. Synthesis of a Coil-Shaped Dioxaperylenoid Polymer.
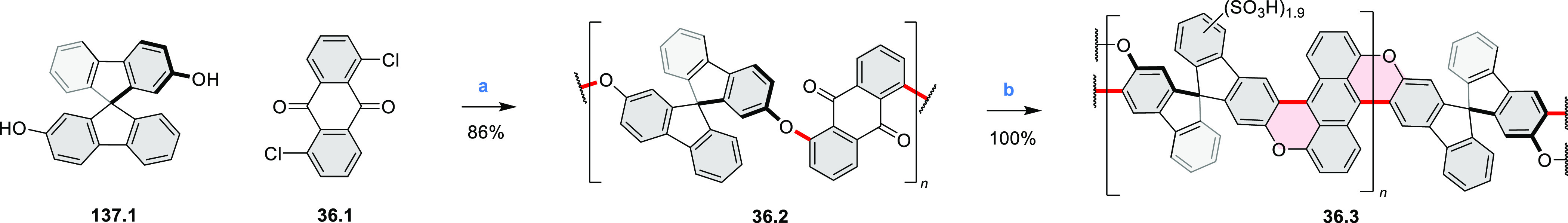
Reagents and conditions: (a)64 K2CO3, diphenyl sulfone, 210 °C, 5 h; (b) (1) H2SO4, 160 °C, 6 h, (2) tetrabutylammonium iodide, 120 °C, 6 h.
A 6π electrocyclization and oxidation upon prolonged irradiation with visible light converted the methylated diazaxanthilidene 37.1 into compound 37.2 that contained a dioxaperylene substructure (Scheme 37).65,66 This compound had red-shifted absorption and emission peaks (λabs = 499 nm, λem = 597 nm in H2O) compared to 37.1 (λabs = 410 nm, λem = 536 nm). Compound 37.1 and its dimethylated analogue showed good cell permeability and low cytotoxicity and were studied as dyes for bioimaging. Both dyes were observed to localize in lysosomes. Photocyclization of 37.1 to 37.2 was performed in live HeLa cells upon irradiation with 405 nm laser light. This transformation could be conveniently detected because of the significant difference in emission wavelengths of 37.1 to 37.2. These features allowed labeling of individual cells in densely populated samples.
Scheme 37. Photocyclization of Monomethylated Diazaxanthilidene.

Reagents and conditions: (a)65 H2O, hν (fluorescent light bulb), 3 days, 64%.
In 2017, Liu and Fang developed an efficient five-step route toward a series of π-extended S-doped perylene (Scheme 38) and pyrene derivatives (Scheme 138, Section 4.3).67 The bis(o-methylthio)-substituted precursor for the final annulation step exists as a pair of atropisomers 38.1a and 38.1b, which can be cleanly separated using normal silica gel chromatography. When treated with I2 under the same conditions, 38.1a and 38.1b were converted into the same product 38.2 in 27% and 36% yield, respectively, along with a trace amount of the half-closed 38.3. Crystal structures of 38.2 showed that the two terminal phenyl groups were bent symmetrically to the opposite sides of the pyrene plane, resulting in a helical structure with C2 symmetry. The dihedral angle between the helicene blades was 50.3° in the monoclinic polymorph and 62.4° in the triclinic one.
Embedding of two pyridinium rings in the hexabenzoperylene framework of 39.3 was achieved by Sato and co-workers in a simple two-step procedure (Scheme 39).68 First, condensation of a triphenylpyrylium salt 39.1 with p-phenylenediamine gave the dipyridinium product 39.2, which was photocyclized by UV irradiation in MeOH/DCM solution in the presence of air, providing 39.3 in 15% yield. This compound had a twisted double helicene structure and was determined to exist as a mixture of enantiomers (meso form was not found). 39.3 exhibited yellow luminescence in solution (λmax = 548 nm, Φ = 22%), while the solid-state emission was much weaker (Φ = 0.4%) and red-shifted. 39.3 was also found to undergo four reversible reductions in voltammetric experiments.
Scheme 39. Synthesis of a Diazonia Derivative of Hexabenzoperylene.

Reagents and conditions: (a)68p-phenylenediamine, DMF, 150 °C, 20 h, 39%; (b) 2:5 MeOH/DCM, air, hν (450 W high-pressure mercury vapor lamp), 14 h, 15%.
The preparation of previously unknown N-containing tetrabenzopentacenes 40.2 and 40.4 was reported by Gorodetsky and co-workers (Scheme 40).69 The final cyclization steps involved a base-mediated cyclodehydrohalogenation to form C–C bonds between the acene core and the pendant naphthyls or quinolines of 40.1 and 40.3, respectively. Moreover, when precursor 40.3 or 40.5 was reacted under Heck-type coupling conditions, N-doped rubicene derivatives 40.7 and 40.6 were obtained, respectively. 40.2 displayed absorption peaks at 555, 600, and 650 nm, while the absorption features of 40.4 were slightly blue-shifted (543, 588, and 638 nm). Rubicene derivatives 40.6 and 40.7 showed blue-shifted absorption compared to tetrabenzopentacene derivatives. Within the N-doped derivatives, tetrabenzopentacenes, or rubicenes, the electronic properties were found be influenced by the precise location of the nitrogen dopants; in particular, 40.4 and 40.7 featured a lower-lying LUMO and HOMO than, respectively, 40.2 and 40.6. From electrochemical measurements, HOMO and LUMO energies were calculated to be −5.23, −3.58 and −4.94, −3.50 eV, for 40.4 and 40.2, respectively, in agreement with a computational study (−5.13, −3.11 vs −4.89, −2.87 eV).
Scheme 40. Synthesis of N-Doped Tetrabenzopentacenes and N-Doped Rubicenes.

Reagents and conditions: (a)69 KOH, o-xylene, reflux; (b) Pd(OAc)2, PCy3, DBU, DMA, 150 °C; (c) KOH, quinoline, 180 °C.
Two planar N-doped benzo[ghi]perylene derivatives, namely, 7,8-diazabenzo[ghi]peryleneimide 41.2 and 1,2-diazonia-7,8-diazabenzo[ghi]peryleneimide 41.3, were synthesized to study the effects of nitrogen lone pairs on the electronic structure of N-doped aromatics (Scheme 41).7041.2 was obtained in 14% yield from 1,12-diazaperylene 41.1, which was subjected to a Diels–Alder reaction with maleic anhydride followed by imidization with 4-heptylamine. 41.3 was similarly prepared via the Diels–Alder reaction of 41.1 but with 4-phenyl-1,2,4-triazoline-3,5-dione. Nitrogen atoms connected to the perylene core in 41.3 significantly increased its HOMO level, resulting in a DFT HOMO–LUMO gap of 2.63 eV, smaller than the value of 3.45 eV determined for 41.2. This change caused a significant red-shift of the spectral features of 41.3: in particular, its emission band was in the 570–780 nm range, while 41.2 emitted at 420–550 nm.
Scheme 41. Synthesis of 7,8-Diazabenzo[ghi]peryleneimide and 1,2-Diazonia-7,8-diazabenzo[ghi]peryleneimide.

Reagents and conditions: (a)70 (1) maleic anhydride, p-chloranil, 220 °C, (2) 4-heptylamine, 150 °C; (b) p-chloranil, 150 °C.
In 2017, Alabugin and co-workers developed a direct method for intramolecular C–H amination under mild conditions in the presence of KOt-Bu and molecular oxygen in DMF.71 The scope of this methodology was demonstrated by the synthesis of several quinolines and 9-azaphenanthrenes, as well as the dibenzo- and dinaphthoperylenes 42.2a–d (Scheme 42). The reaction is thought to proceed via radical anion intermediates formed by deprotonation of the NH2 group followed by hydrogen atom abstraction from the adjacent methylene bridge by the DMF radical.
A scalable synthesis of 1,7-diazaperylene and its alkyl derivatives (43.3a–f, Scheme 43) was reported by Langhals and co-workers.72 Diazotization of 1,5-diaminoanthraquinone 43.1 provided the isolable and stable double diazonium salt 43.2. The subsequent copper-catalyzed Meerwein arylation reaction with acrylonitrile provided the corresponding dialdehyde, which was used without purification in a condensation with NH3. The use of t-BuOH as the main solvent combined with H2O and H2SO4 was important to avoid the reduction of 43.2 to anthraquinone through hydrogen abstraction from a solvent molecule. Using acrylonitrile derivatives with a terminal alkene function furnished dialkylated 1,7-diazaperylenes, albeit in decreased yields. A very low yield was also obtained with an internal alkene (crotononitrile), leading to 43.3f.
The synthesis of 2,7-diazaperylene diimides was recently described by Okamoto and co-workers (Scheme 44).73 The diazaperylene core in 44.3 was formed through condensation of the nitrile-functionalized anthraquinone 44.2. The hydroxyl groups were then removed to afford the diester 44.4. A challenging functionalization of this highly electron-deficient compound through bromination under harsh conditions and Pd-catalyzed carbonylation led to the dianhydride 44.5. Finally, diimides 44.6a–c were prepared through condensation with amines. In the solid state, C–H···N hydrogen-bonding contacts were observed, which, along with π-stacking interactions, governed the aggregation patterns. These compounds were successfully applied as n-type organic semiconductors with high electron mobility.73−75 Notably, their high chemical stability allowed the use of photolithography for patterning.73
Scheme 44. Synthesis of 1,7-Diazaperylene Diimides.

Reagents and conditions: (a)73 methyl cyanoacetate, KOt-Bu, DMSO, 50 °C, 3 h, 38%; (b) H2SO4, rt, 30 min, 87%; (c) Tf2NPh, DMAP, Et3N, DCM, −20 °C to rt, 2 h, 22%; (d) Pd(PPh3)4, HCOOH, Et3N, DMF, 80 °C, 2 h, 83%; (e) NBS, H2SO4, 50 °C, 2.5 h, 47%; (f) 2,4,6-trichlorophenyl formate, Pd(OAc)2, Xantphos, Et3N, toluene, 100 °C, 12 h, 22%; (g) TsOH·H2O, o-dichlorobenzene, 120 °C, 24 h, 89%; (h) RNH2, EtCOOH, o-dichlorobenzene, 150 °C, 11–38 h.
In 2016, Li and co-workers developed an efficient N–H/C–H one-pot coupling method for the preparation of benzo[kl]acridines, including the N,S- and N,O-containing benzoperylenes 45.3 and 45.4, respectively (Scheme 45).76 This strategy is based on the reaction of 1,8-dibromonaphthalene 45.1a or 1,8-diiodonaphtalene 45.1b with a secondary aromatic amine. The reaction was proposed to proceed via Buchwald–Hartwig amination followed by intramolecular C–H arylation. The catalyst system used here was essentially a mixture of Pd sources and ligands known to be effective for these two transformations (Pd2(dba)3/(t-Bu)3P for the amination and Pd(OAc)2/Cy3P for the C–H arylation). The same palladium-catalyzed domino approach gave easy access to other benzo[kl]acridine derivatives (Scheme 183, Section 5.1.1; Scheme 294, Section 6.4.2). High yields were obtained using 45.2a,b and other cyclic and acyclic diarylamines; carbazoles however were not suitable substrates.
Scheme 45. Synthesis of N,S- and N,O-Containing Benzoperylene Derivatives.

Reagents and conditions: (a)76t-BuONa, Pd(OAc)2, Pd2(dba)3, Cy3P, (t-Bu)3P, toluene, 90 °C, 10 h.
Scheme 183. Syntheses of Azabenzophenalenoids.

Reagents and conditions: (a)76t-BuONa, Pd(OAc)2, Pd2(dba)3, Cy3P, (t-Bu)3P, toluene, 90 °C, 10 h.
Scheme 294. Syntheses of [de] and [kl]Fused Dibenzoazepines via Palladium-Catalyzed Annulation.

Reagents and conditions: (a)76t-BuONa, Pd(OAc)2 (3 mol %), Pd2(dba)3 (3 mol %), PCy3 (7 mol %), t-Bu3P (7 mol %), dry toluene, 90 °C, 10 h; (b)558 Pd(OAc)2 (10 mol %), PPh3 (20 mol %), norbornene (1 equiv), Cs2CO3 (4 equiv), toluene, 95 °C, 6 h; (c)559 PdCl(C3H5)(dppb) (2 mol %), KOAc (2 equiv), DMA, 150 °C, 72 h.
In 2020, Bonifazi et al. reported the synthesis of oxygen-doped nanographenes, including the π-extended dioxaperylenoid 46.3 (Scheme 46).77 The important precursor 46.1 was obtained from the cross-coupling of 1,8-diiodopyrene and the appropriate arylboronic acid. Compound 46.1 was demethylated using BBr3 to give 46.2. Subsequently, a 2-fold oxidative Pummerer cyclization using CuO in nitrobenzene at 200 °C furnished the target nanographene 46.3 in a high overall yield. Mixed-valence crystals containing the oxidized form of 46.3 were obtained using electrocrystallization with n-Bu4ClO4/THF as the electrolyte. The asymmetric unit [(46.3)3(ClO4)2·THF·0.5H2O] of the mixed-valence complex contains three independent molecules of 46.3 and two perchlorate anions. The molecules of 46.3 stack at distances of 3.21–3.42 Å, and they have similar bond lengths, indicating charge delocalization over the stacks. The authors also reported the construction of other oxygen-doped polyaromatic skeletons using the oxidative Pummerer reaction (see Scheme 119, Section 4.1, and Scheme 141, Section 4.3).
Scheme 46. π-Extended Dioxaperylenoid.

Reagents and conditions: (a)77 BBr3, DCM, 0 °C to rt, overnight; (b) CuO, nitrobenzene, air, 200 °C, overnight.
Scheme 141. Oxygen-Doped Pyrenoids via Oxidative Pummerer Cyclization.

Reagents and conditions: (a)159 BBr3, DCM, 0 °C to rt, overnight; (b) CuO, nitrobenzene, 240 °C, overnight.
New stable derivatives of Thiele’s hydrocarbon, possessing a bridged tetraaryl-p-quinodimethane structure, were reported by Tanioka, Muranaka, Uchiyama, and co-workers.7847.2 was obtained in 17% yield by treatment of iso-aminobenzopyranoxanthene 47.1 with concentrated sulfuric acid at high temperature (Scheme 47). Analogues bearing shorter N-alkyl chains 47.5a–c were synthesized via a one-step reaction from the corresponding 2-(4-dialkylamino-2-hydroxybenzoyl)benzoic acids 47.3a–c and p-dimethoxybenzene 47.4. A tert-butyl-substituted derivative 47.5d and chloro-substituted 47.5e were also prepared in a similar manner, with the yields of 10% and 1%, respectively. Despite their quinodimethane substructure, 47.2 and 47.5a–c showed no evidence of diradical character. Their closed-shell nature was attributed to the presence of a strong electron donor and acceptor moieties, favoring zwitterionic resonance forms over the diradical ones. The chemical oxidation of 47.5c proceeded smoothly with an equivalent amount of silver hexafluoroantimonate in DCM, providing the radical cation salt 47.6a in 93% yield after recrystallization (Scheme 47).79 The oxidation of 47.5c was also achieved using DDQ in DCM. The resulting radical cation salts were stable in air and showed no evidence of dimerization in the solid state. 47.6a had an intense NIR absorption with peaks at 927, 1007, 1094, and 1303 nm. Thin films of 47.6b displayed high electric conductivity of 7.7 × 10–3 S cm–1.
Scheme 47. Synthesis of Bridged Tetraaryl-p-quinodimethanes.

Reagents and conditions: (a)78 conc. H2SO4, 140 °C, 3 h; (b) conc. H2SO4, 75 to 100 °C, 48 h, then 160 °C, 2 h (6 h for 47.5a); (c) conc. H2SO4, 95 to 130 (150) °C, 78 (75) h (47.5e); (d)79 AgSbF6, DCM, rt; (e) DDQ, DCM, rt.
Cyclizations of naphthyl-, azanaphthyl-, or 2,7-naphthyridinyl-substituted perylenes 48.1a–c were reported by Hasobe and co-workers to produce terrylenes 48.2a–c (Scheme 48).80 Oxidative coupling conditions were used for the naphthyl-containing substrate 48.1a; this approach is however known to fail with electron-deficient N-doped compounds. Thus, cyclization of 48.1b–c was performed under the radical anion coupling conditions (CR2017, Section 3.1), i.e., via treatment with potassium in xylene followed by air bubbling (cf. Scheme 84, Section 3.3). Electrochemical measurements revealed that the first one-electron reduction and oxidation potentials became positively shifted with the increasing number of nitrogens in the terrylene core (Ered1 = −1.45, −1.28, and −1.12 V and Eox1 = 0.54, 0.66, and 0.75 V, respectively, in the series 48.2a–c). The HOMO–LUMO gaps remained similar (2.39, 2.37, and 2.35 eV for 48.2a–c, respectively), corresponding to an ca. 40 nm red-shift of absorption and emission features in 48.2c compared to 48.2a. Protonation of the N-doped terrylenes with TFA in DCM solution produced new NIR absorption bands: a broad peak between 600 and 820 nm for 48.2b and a sharp peak at 800 nm for 48.2c.
Scheme 48. Synthesis of Azaterrylene Derivatives.

Reagents and conditions: (a)80 (1) Sc(OTf)3, DDQ, xylene, 165 °C, 1 day, (2) N2H2·H2O, rt, 1 h; (b) (1) potassium, xylene, 130 °C, 16–17 h, (2) air, 4 h.
Scheme 84. Mechanochemical Cyclodehydrogenation of Polyarylpyridines.

Reagents and conditions: (a)166 CoI2, 1,3-bis(diphenylphosphino)propane, Zn, NMP, or DMA, 80 °C, 24 h; (b) K metal, ball milling (30 Hz), rt, 2 h.
3.1.3. Tri-, Tetra-, and Hexaheteraperylenoids
Electrochemical and photophysical properties of azaperylenes containing from 1 to 4 nitrogen atoms were compared in a study by Sato, Hasobe, and co-workers.81 The new diazaperylene 49.2b and triazaperylene 49.2e were prepared along with previously known compounds (Scheme 49). A common synthetic methodology based on radical anion coupling was used in all cases. Precursors 49.1a–f were obtained via Suzuki or Negishi-type coupling reactions using derivatives of naphthalene, quinoline, and 2,7-naphthyridine as building blocks. The final cyclization was performed by treatment with K metal followed by exposure to air, providing 49.2a–f in low yields. It was found that increasing the number of embedded nitrogens resulted in lower HOMO as well as LUMO energies, with only a small increase of the band gap. The emission quantum yield was significantly lowered in the triazaperylene 49.2e (32%) and tetraazaperylene 49.2f (0.013%) compared to 49.2a–d (69–81%). This was explained by stabilization of energy levels of the nonemissive excited states corresponding to an n−π* transition. In the series of azaperylenes, compound 49.2d had the highest proton affinity, which was attributed to intramolecular proton exchange between N atoms positioned in the bay region. Introduction of further N atoms in 49.2e–f led to a lower proton affinity.
Scheme 49. Synthesis of Azaperylenes via Radical Anion Coupling.

Reagents and conditions: (a) (1) 10–12 equiv of K, DME, rt, or DME, 100 °C for 49.2f, or toluene, 95 °C for 49.2e, ca. 1 day, (2) air, 4 h.
In 2016, Bonifazi and co-workers described the preparation of O-doped benzo-fused rylenes by using a stepwise planarization strategy involving the formation of C–O bonds through an intramolecular oxidative coupling (Scheme 50).82 Intramolecular etherification of the quaternaphthalenes 50.4a and 50.4b by treatment with CuI and PivOH in DMSO afforded 50.5a,b as mixtures of atropisomers. Demethylation of methoxy groups with BBr3 followed by the second ring-closure reaction led to the formation of the hexaoxa derivatives 50.6a and 50.6b in 29% and 36% yield, respectively. A similar sequence of transformations applied to 50.1a,b yielded 50.3a,b. Single-crystal X-ray diffraction analysis showed that the tetraoxa derivative 50.3b forms face-to-face π–π stacks in the solid state.
Scheme 50. Synthesis of O-Doped Benzorylenes.

Reagents and conditions: (a)82 CuI, pivalic acid, DMSO, 130–145 °C; (b) BBr3, DCM, 0 °C.
In 2016, Hatakeyama et al. reported a two-step synthesis of double [5]helicenes possessing two boronate substructures at the ring junction (Scheme 51).83 The annulation synthetic step consisted of lithium–halogen exchange of 51.1a,b followed by trapping of the resulting aryllithium with boron tribromide to give the diborylated intermediates 51.2a,b. These readily underwent boron-assisted demethylative cyclization at 40 °C providing 16.2a and 16.2b in 55% and 60% yield, respectively (Scheme 51). Shortly afterward, a more efficient synthesis of the same compounds was presented by Feng, Müllen, and co-workers (Scheme 51).29 Heating a solution of 16.1a,b in o-dichlorobenzene at 150 °C in the presence of BBr3 gave the corresponding OBO-doped bistetracenes 16.2a,b in 92% and 90% yield, respectively. The reaction was thought to proceed via demethylation to form the intermediates 51.3a,b followed by intramolecular C–H borylation. Moreover, compounds 16.2a,b were converted into OBO-doped peritetracenes by cyclodehydrogenation (Scheme 16, Section 2.3). The same methodology was also used to prepare the double [7]heterohelicene 51.5.84 The low yield obtained in this example was attributed to the significant increase in steric strain upon cyclization. Electron-withdrawing character of the OBO fragments incorporated into 16.2a allowed us to obtain its dianionic form upon treatment with Na or K in the presence of 18-crown-6.85 A related synthesis described by Hatakeyama and co-workers employed double borylation of the electron-rich compound 51.6 with BI3 to provide the fully fused product 51.7 through the formation of four new rings. Of the three [4]helicenes in this compound, the lateral ones had dihedral angles of 21.0° and 24.8° in the crystal structure, while the central helicene had a greater twist of 41.4°.
Blue luminescence of the products (16.2a: 405, 430 nm, Φ = 68%; 16.2b: 411, 436 nm, Φ = 65%) was a very good match for pure RGB blue which is desirable in OLEDs.83 Compound 16.2b had an increased barrier to epimerization compared to 16.2a and was separated into enantiomers, allowing the detection of CPL with a dissymmetry factor of 1.7 × 10–3 in the optically pure material. Emission of the double [7]helicene 51.5 was weaker and red-shifted relative to the [5]helicene analogues, with a peak at 487 nm and a quantum yield of 26%.84 OLED devices prepared from 51.7 combined with charge-transporting polymers displayed emission at 505 nm, which was close to a pure green color.
The BNB-doped perylenoid 52.5 (Scheme 52) was reported by Wagner and co-workers together with smaller phenalenoid analogs with BNB or BOB doping.87 Reduction of the 1,8-naphthalenediboronic anhydride 52.1 provided the diborane 52.2, which was then condensed with the aniline 52.3, giving 52.4. Treatment of this compound with Mg or K metal as a reducing agent resulted in B–C coupling, providing 52.5. This compound had a twisted geometry and was a rare example of a cryptoracemate (a racemic mixture that forms chiral crystals). The same authors also reported a synthesis of bis-BO- and bis-BN-doped perylenoids (Scheme 52).88 The key transformation was the Au-catalyzed cyclization of alkynes with adjacent OH or NH functions. In this manner, borinic acids 52.7a,b were converted into the tetraheteraperylenoids 52.8a,b in 89% yield for both analogues. Aminoboranes 52.9a–c, obtained via condensation of 52.7a–c with TMS2NMe, were also cyclized by this methodology. Somewhat harsher conditions were needed, but the products 52.10a–c were obtained in similarly high yields as 52.8a,b.
Scheme 52. Synthesis of BN- and BO-Doped Perylenoids.
Reagents and conditions: (a)87 LiAlH4, Et2O, 0 °C to rt, 12 h, then Me3SiCl, −78 °C to rt, 12 h; (b) C6H6, 50 °C, 4.5 h; (c) Mg, THF, 50 °C, 2.5 h; (d)88 R–CC–SnR′3, Pd(Pt-Bu3)2, toluene, 80 °C, 4 h, then rt, 14 h; (e) 10–20 mol % of (Ph3P)Au(NTf2), DCM, 60 °C, 4 h; (f) TMS2NMe, C6D6, 120 °C, 2 days; (g) 25 mol % of (Ph3P)Au(NTf2), DCM, 60 °C, 2 days.
Thermal annealing of tetraazaanthraquinodimethanes 53.1a–c on the Au(111) surface caused a rearrangement with elimination of ethylene to form 53.2a–c (Scheme 53).89 This process formed four new pyridine rings while breaking two pyrazine rings, a process named “heterocyclic segregation” by the authors. The transformation was proposed to proceed with a transfer of hydrogen atoms from the lateral phenyl rings to the pyrazine vinylene bridge. This transfer could plausibly be intramolecular (via intermediate a1) or mediated by the Au surface (via intermediate a2). Both types of intermediates formed from 53.1a were detected after annealing at decreased temperatures (370–470 K). The products and intermediates were analyzed on the surface using STM, and no preparative synthesis was attempted.
Scheme 53. Formation of Tetraazaperylenoids on the Gold Surface.
Reagents and conditions: (a)89 Thermal deposition on the Au(111) surface in a vacuum and then annealing at 470–550 K.
An N,S-doped dibenzoperylene derivative with a double hetero[4]helicene structure was synthesized via dimerization of phenothiazine (Scheme 54).90 The synthetic route started with oxidative homocoupling of 54.1 using DDQ to provide 54.2. Then, oxidation of 54.2 by a combination of DDQ and scandium triflate resulted in intramolecular C–N bond formation and gave the fused phenothiazine dimer 54.3. These two steps could also be performed in one pot. The enantiomers of 54.3 were much more stable toward racemization than other double [4]helicenes, presumably as a consequence of the highly bent structure of phenothiazine. Upon oxidation with BAHA, 54.3 was converted into a crystalline radical cation salt, which was stable under ambient atmosphere and lighting. The radical cation possessed a NIR absorption band at 1500 nm.
Scheme 54. Synthesis of Phenothiazine Dimers.

Reagents and conditions: (a)90 DDQ, CHCl3, rt, 7 h; (b) DDQ, Sc(OTf)3, CHCl3, reflux, 23 h; (c) (1) DDQ, DCM, rt, 2 h, (2) DDQ, Sc(OTf)3, reflux, 18 h; (d)91 Ni(cod)2, cod, 2,2′-bipyridyl, THF, 60 °C, 24 h; (e) DDQ 3.1 equiv, CHCl3, rt, 24 h, then N2H4·H2O; (f) m-CPBA, CHCl3, rt, 40 min.
Another kind of phenothiazine dimer was obtained by Yamamoto homocoupling of the dibromophenothiazine 54.4 followed by DDQ oxidation to form the central N–N bond, providing 54.6.91 This compound was further oxidized to the disulfoxide 54.7 upon treatment with m-CPBA, resulting in a cis/trans mixture due to the sulfoxide geometry. The respective disulfone could also be formed using an excess of m-CPBA but was not characterized due to its low solubility. 54.6–7 had a “butterfly” structure with a ridge formed by the hydrazine substructure. 54.6 was nonemissive, while the two isomers of 54.7 displayed emission close to 540 nm. Compound cis-54.7 had a stronger emission (Φ = 15%) than its trans isomer (Φ = 5%).
In 2021, Zhang et al. described the synthesis of the thiazine-fused buckybowl 55.3 and its derivatives (Scheme 55).92 The synthesis began with the copper(I)-catalyzed cyclodimerization of the dibromophenothiazine 55.1. The same transformation under Buchwald–Hartwig conditions only led to a complex product mixture. The resultant compound 55.2 was then cyclized under palladium catalysis to give the target buckybowl 55.3 in 49% yield. Upon oxidation by H2O2, the corresponding bis(S,S-dioxide) 55.4 was obtained in 70% yield. Chemical oxidation of 55.3 could take place using 1 or 2 equiv of AgSbF6 to give the radical cation 55.3•+ and the dication 55.32+, respectively. According to single-crystal XRD data, the bowl depth of the neutral compound 55.3 (0.59 Å) was deeper than that of the radical cation (0.37 Å), respectively, while the dication assumed a planar geometry.
Scheme 55. Synthesis of Phenothiazine-Containing Buckybowls.
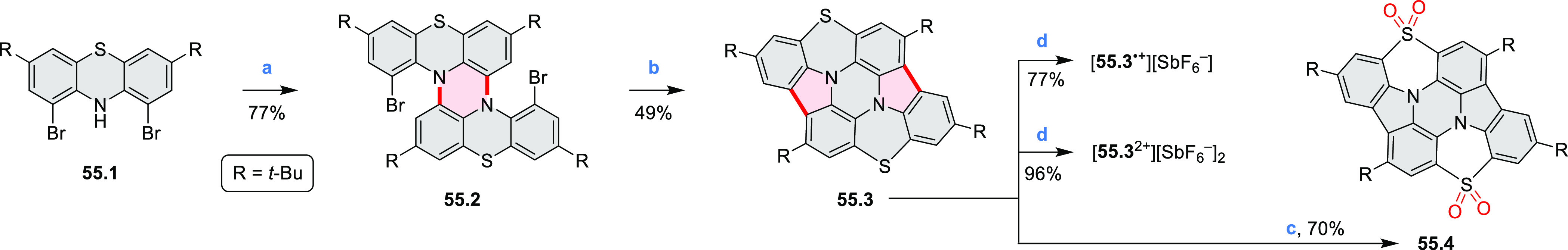
Reagents and conditions: (a)92 CuI, K2CO3, 18-crown-6, o-dichlorobenzene, 180 °C, 48 h; Pd(OAc)2, PCy3·HBF4, K2CO3, DMA, 170 °C, 48 h; (c) 30% H2O2, AcOH, CHCl3, 60 °C, 12 h; (d) AgSbF6 (1 or 2 equiv), dry DCM, rt, 20 min.
3.2. [ghi]Heteroannulated Perylenoids: Five-Membered Rings
In a series of articles by Achalkumar and co-workers, the commercially available perylene dianhydride 56.1 was derivatized into tetraesters 56.2a–e via hydrolysis followed by SN2 esterification (Scheme 56). NaNO2-mediated nitration of these intermediates afforded the 6-nitro derivatives 56.3a–e in high yields, in contrast to the low-yielding nitration of unsubstituted perylene. The nitro group was then used as a reactive handle for various annulations (see CR2017, Section 3.2 for earlier related work). The Cadogan reaction with triethyl phosphite provided N-annulated perylenes (NAPs) 56.4a–e, which were further derivatized via N-alkylation to afford 56.7b–d.93 Treatment of 56.3a–e with elemental S or Se in NMP afforded, respectively, S-annulated 56.5a–e94 and Se-annulated 56.5a–e.95 Moderate yields of 50–60% were generally observed in all annulation reactions. The products were luminescent, having green emission in THF solutions of 56.4b–d and 56.7b–d (492–495 nm), light blue in 56.5a–d (461–464 and 481–482 nm), and bluish green in 56.6a–d (weak and broad emission peaking at 476 nm). The heteroannulated compounds were generally capable of self-assembly into π-stacked columns, giving rise to columnar hexagonal liquid crystalline phases. This behavior was disrupted in the N-alkylated 56.7b–d, where the additional alkyl chain was thought to disfavor self-assembly. In 56.4e, 56.5e, and 56.5e, which were functionalized with branched tetrahydrogeranyl chains, the temperature range of the hexagonal columnar mesophase was especially broad (from −50 to 285 °C for 56.4e).96 These liquid crystalline materials were used as electroluminescent layers in OLED devices either in neat form or as dopants in a polyvinyl carbazole matrix. The latter OLED variant provided a higher emission intensity.
Achalkumar and co-workers also investigated the conversion of heteroannulated perylene tetraesters into mono- and diimides (Scheme 57).97,98 Both transformations were effected by microwave heating in imidazole in the presence of 1.1 or 2.05 equiv of the respective amine. Diimides 57.3a–c were prepared via the anhydride intermediates 57.2a–c, while monoimides 57.4a–c were made directly from the tetraesters 57.1a–c. A possibility of elaborating monoimides into unsymmetrically substituted diimides was discussed but only realized experimentally with a nonannulated perylene core. Self-assembly into a hexagonal columnar mesophase as seen in tetraester analogues (Scheme 56) was also observed in the diimides 57.3a and 57.3b. In 57.3c an additional oblique columnar phase was detected. Solution luminescence of the monoimides and diimides was red-shifted in comparison to the tetraesters, with two main emission peaks in the 500–600 nm range.
Scheme 57. Synthesis of N-Annulated Perylene Monoimides and Diimides.

Reagents and conditions: (a) p-toluenesulfonic acid monohydrate, toluene, 100 °C, 30 h, 90%; (b)98 tris(n-dodecyloxy)benzylamine (2.05 equiv), zinc acetate, imidazole, 165 °C, microwave, 30 min, 80–90%; (c) n-octadecylamine (1.1 equiv), imidazole, 165 °C, microwave, 35 min, 55%.
Simple examples of introducing [ghi]heteroannulation into unsubstituted perylenes include bay nitration followed by Cadogan reaction to provide N-annulated 58.3 or treatment with elemental sulfur, leading to S-annulated 58.2 (Scheme 58). In particular, the Cadogan reaction is significantly more efficient with the otherwise unsubstituted 1-nitroperylene 58.1 than with perylene tetraesters or perylene diimides (PDIs). The resulting compounds can be derivatized by 3,10-dihalogenation as shown in the example of 58.5. The S-annulated 58.2 could also be oxidized to sulfoxide upon treatment with m-CPBA.99 The sulfoxide 58.4 undergoes deoxygenation upon irradiation with UV or violet light (420 nm). While photodeoxygenation of related sulfoxides is known to generate atomic oxygen, the mechanism of this process in the case of 58.5 is currently unclear. The dibrominated 58.5 was observed to self-assemble on the Ag(111) surface, forming three distinct networks, all of which contained pores flanked by six molecules of 58.5.100 This ordering was driven by σ-hole interactions, with Br···Br halogen bonding contacts as well as a weaker S···Br chalcogen bonding.
The simple N-annulated perylene 58.3 formed a complex with diiodine, which was crystallized out of a benzene solution.101 The crystal structure revealed polymeric chains of I atoms adjacent to columnar stacks of 58.3. These chains were nearly linear with distances of 3.054–3.174 Å between adjacent I atoms. These values are significantly larger than the I–I covalent bond length (2.70 Å) and were thought to imply that the I atoms have a partial negative charge. A similar polyiodide is believed to be present in the iodine–starch complex, as implied by the similarities in low-wavenumber Raman signals. Alkylation on the pyrrolic N atom of 58.3 led to a conjugate with a positively charged viologen unit.102 This compound was nonemissive, while blue emission with a 435 nm maximum was present in its N-benzyl counterpart. This effect was attributed to photoinduced electron transfer from the N-annulated perylene to the viologen segment. This charge-separated state recombined in approximately 20 ps.
N-Annulation of bay-nitrated PDIs can be accomplished by a simple treatment with NaN3 at rt (Scheme 59),103 which presents a valuable alternative to the Cadogan reaction using triethyl phosphite or PPh3 under much harsher conditions. Indeed, 59.2 was obtained in 76% yield, which is an improvement over 50–70% efficiencies generally seen using the Cadogan approach with PDIs or perylene tetraesters. The authors propose a displacement of the nitro group followed by spontaneous decomposition of the resulting azido derivative to nitrene as the reaction mechanism. As no bay-azido-substituted PDIs are known, this mechanistic hypothesis is likely to be correct. Another unusual transformation demonstrated in this work is the use of the bay-NO2 substituent as a reactive handle for Suzuki cross-coupling. While this reaction was inefficient with 59.5, providing the bis-PDI 59.6 in only 24% yield, much better results were obtained with nonannulated PDIs. This difference is attributed to the reactivity of the NO2 group being decreased by the additional electron density from the pyrrolic ring.
As an alternative to the synthetic sequence shown in Scheme 57, the heteroatom annulation can be done after diimide formation (Scheme 60). Bay-nitrated PDI 60.1a was converted into the O-annulated derivative 60.2 in 30% yield by heating in NMP under an oxygen atmosphere.104,105 Additionally, the S-annulated analogue 60.3 was prepared using a similar methodology.104,106 Spectroscopic and electrochemical data of this compound were reported and compared with several nonannulated analogues.106 The O-annulated compound 60.2 displayed slightly red-shifted absorption and emission (λabs = 510 nm, λem = 522 nm) in comparison to 60.3 (λabs = 500 nm, λem = 515 nm). Self-assembly into a needle-like morphology was reported for 60.2, while 60.3 formed nanosheets.104
S- and Se-annulation was performed on the diimide 60.1b functionalized with optically pure (R)-tetrahydrogeranyl chains in similar yields as shown above for perylene tetraesters.107 Aggregation of these compounds occurred in 2:3 CHCl3/MeOH, and the resulting CD features had opposite signs in 60.5 and 60.4. This chiroptical difference translated into macroscopic features of dried samples, where 60.4 formed 200 nm wide ribbons with a left-handed twist, while 60.5 produced 100 nm wide right-handed ribbons. Conductivity of these fibers was sensitive to the presence of NH3 gas via formation of donor–acceptor complexes, resulting in up to 340-fold current response for 60.4 in the presence of 100 ppm of NH3.
Perylene diimides such as 61.1 and 61.9a–c can be nitrated at the bay positions to mono- or dinitro derivatives (Scheme 61). In the latter case, a mixture of 1,6- and 1,7-regioisomers is obtained, as demonstrated by 61.3 and 61.4, which formed in a 3:1 ratio.108 S-Annulation of the dinitro derivatives resulted in the formation of 61.7 and 61.11a–c,109 each with a sulfide and a disulfide bridge across the two perylene bays. A disulfide-bridged compound could also be formed from the mononitro derivative 61.2 by performing the S-annulation under relatively gentle conditions (120 °C rather than 180–190 °C), resulting in a separable mixture of 61.5 and S2-annulated 61.6. An even lower reaction temperature allowed us to obtain the singly annulated nitro derivative 61.8. Additionally, 61.12, annulated with two five-membered rings, was prepared according to a previously reported method.
Compared to 61.12, 61.7 and 61.11c have an additional broad absorption band at 500–700 nm, which was tentatively assigned to a charge-transfer transition involving the electron-rich disulfide. Similar spectroscopic differences were seen between compounds 61.13 and 61.14, where the absorption maximum of 61.14 was red-shifted to 654 nm, relative to 548 nm in 61.13.110 Compound 61.14 was additionally nonemissive, while 61.13 displayed an orange fluorescence. A lack of luminescence was also reported in the case of the S/S2-annulated 61.7.
A dinitro derivative of a fused perylene diimide dimer, 62.1, was efficiently converted into a mixture of S- and S2-annulated products under similar conditions as shown above (Scheme 62).111 At longer reaction times, product mixtures enriched in 62.4 were obtained. As in the compounds with a single perylene core, introduction of S2 bridges caused a decrease of the optical band gap, giving rise to new absorption features in the 600–700 nm range. DFT calculations showed that the highest HOMO amplitude is present on the S2 bridges in 62.2 and 62.3, in contrast to a very low HOMO amplitude on the S bridges in 62.4. This observation implies that the aromaticity of S-annulated rings is absent in S2-annulated analogues, where the sulfur atoms simply act as electron-rich substituents.
1,4-Addition of Grignard reagents was demonstrated by Wudl and Zheng et al. as an effective catalyst-free method to directly functionalize the 2,5,8,11-positions of bay-annulated perylene diimides with aryl substituents (Scheme 63).112 The S-heterocyclic annulated 63.2a was prepared in 70% yield via a Stille-type coupling reaction between 63.1 and bis(tributyltin)sulfide. The reaction of 63.2a with the corresponding Grignard reagent, 2-thienylmagnesium or 2-mesitylmagnesium bromide, afforded tetrasubstituted S-containing perylene diimides, 63.2b and 63.2c, respectively, in reasonable yields. Preliminary studies on OSCs utilizing these molecules as nonfullerene acceptors showed PCE values up to 5.07% for the thienyl-substituted 63.2b.
Scheme 63. Synthesis of Dithiophene-Annulated Tetraarylperylenes.

Reagents and conditions: (a)112 Pd(PPh3)4, bis(tributyltin)sulfide, toluene, reflux, 10 h; (b) 2-thienylmagnesium bromide, THF, 0 °C to rt, 12 h; (c) mesitylmagnesium bromide, THF, 0 °C to rt, 12 h.
In 2015, Wang and co-workers designed and synthesized two D–A organic dyes consisting of electron-rich N-annulated 6H-thieno[3′,2’:5,4]-benzo[cd]perylene or its isomer 13H-thieno[2′,3′:3,4]cyclopenta[b]perylene, linked to an electron acceptor, 4-(benzo[c]-[1,2,5]thiadiazol-7-yl)benzoic acid (Scheme 64).113 Thiophene-substituted 64.1b underwent intramolecular Friedel–Crafts cyclization in the presence of Amberlyst 15 acting as a solid acid catalyst to concurrently afford 64.2a and 64.3a in 30% and 60% yield, respectively. The electron-rich compounds 64.2a and 64.3a were treated with tert-butyllithium and chlorotrimethylstannane to generate the corresponding stannanes, which were cross coupled with butyl 4-(7-bromobenzo[c]-[1,2,5]thiadiazol-4-yl)benzoate to yield 64.2b and 64.3b after the KOH-mediated hydrolysis. The final compounds were tested as dyes for DSSCs. The best results were obtained with 64.2b, with a PCE of up to 12% when irradiated with 100 mW cm–2, AM 1.5G simulated sunlight. This was the first example of a metal-free organic dye reaching such a high PCE without any coadsorbate.
Scheme 64. Synthesis of N-Annulated Thienoperylene Dyes.
Reagents and conditions: (a)113 Amberlyst 15, toluene, reflux, 12 h; (b) t-BuLi, THF, −78 °C, 1 h, then chlorotrimethylstannane, −78 °C to rt, 24 h; (c) butyl 4-(7-bromobenzo[c][1,2,5]thiadiazol-4-yl)benzoate, Pd(PPh3)2Cl2, toluene, reflux, 12 h; (d) KOH, 3:1 THF/H2O, reflux, 8 h, then HCl. *64.2a and 64.3a were obtained in 1:2 ratio.
In 2017, Wang et al. developed another D–A dye based on a NAP decorated with 4-(benzo[c]-[1,2,5]thiadiazol-7-yl)benzoic acid.114 As presented in Scheme 65, N-annulated perylene 65.1 was subjected to the addition reaction with (4-hexylphenyl)magnesium bromide, generating a tertiary alcohol, which was cyclized into 65.2a in 98% yield in the presence of Amberlyst 15 sulfonic acid resin. 65.2a was brominated and derivatized in a Sonogashira coupling with butyl 4-(7-ethnylbenzo[c]thiadiazol-4-yl)benzoate. Finally, this precursor underwent hydrolysis in the presence of potassium hydroxide followed by acidification with H3PO4 to afford the target product 65.2b. Compound 65.4b, acting as a control dye, was synthesized by an analogous approach. DSSCs containing 65.2b adsorbed on TiO2 particles reached a PCE of up to 12.6% with no coadsorbent. A slightly lower efficiency (11.7%) was achieved with 65.4b, which was attributed to the inhibition of charge carrier recombination by the additional 4-hexylphenyl groups in 65.2b.
Scheme 65. Synthesis of N-Annulated Perylene Dyes.

Reagents and conditions: (a)114 4-hexylphenylmagnesium bromide, THF, reflux, overnight; (b) Amberlyst 15, toluene, reflux, overnight; (c) NBS, THF, 0 °C, 30 min; (d) butyl 4-(7-ethynylbenzo[c][1,2,5]thiadiazol-4-yl)benzoate, Pd2(dba)3, P(t-Bu)3, Cs2CO3, dioxane, reflux, 5 h; (e) (1) KOH, 3:1 THF/H2O, reflux, 8 h, (2) phosphoric acid.
In their further research on donor–acceptor organic dyes, Wang and co-workers reported two more molecules with a NAP-based unit as the central module of the electron donor and ethynylbenzothiadiazole-benzoic acid as the electron acceptor, C5.2a and C5.2b,115 along with the previously reported model dye C5.1 (Chart 5). The thiophene-fused compounds C5.2a and C5.2b were more electron rich, corresponding to a higher HOMO energy and decreased HOMO–LUMO gap compared to C5.1. An additional alkoxy substituent in C5.2b resulted in a further decrease in the energy gap attributed to conformational rigidification by an O···S chalcogen bonding interaction. DSSC devices containing these dyes in combination with TiO2 particles displayed high PCE values of 11.5% with C5.2a and 12.4% with C5.2b under AM 1.5G irradiation. The same group also described compounds C5.3a and C5.3b where the benzothiadiazole acceptor unit was attached to the fused thiophene.116 The additional ethynyl linker in C5.3b resulted in a decreased HOMO–LUMO gap and red-shifted absorption and NIR emission peaks. These dyes were incorporated into transparent DSSCs that partially transmitted visible light in the 600–750 nm range. The devices achieved PCE values of 10.1% with C5.3a and 10.3% with C5.3b, which were considered high for transparent cells.
Chart 5. N-Annulated Perylene Dyes.

The synthetic approach shown in Scheme 65 was further extended to prepare symmetrically derivatized NAPs C5.4a and C5.4b.117 In this case, very narrow HOMO–LUMO gaps of 1.74 eV for C5.4a and 1.70 eV for C5.4b were achieved, which allowed the absorption bands to extend into the NIR region. Bulk heterojunction organic photovoltaic cells were prepared using these compounds and the PTB7-Th polymer as the donor material. Remarkably, the cells were able to capture NIR radiation with wavelengths of up to 1000 nm. Compound C5.4b afforded much higher electron mobility and external quantum efficiency values, allowing us to achieve a PCE of 10.21% under a simulated solar irradiation of 100 mW/cm2.
A star-shaped molecule with a central electron-deficient triazine ring connected to three electron-rich NAP subunits was designed as a prospective emissive layer in OLEDs.118 To obtain the target 66.3, the NAP tetraester 66.1 was treated with cyanuric chloride 66.2 in the presence of n-BuLi as a base (Scheme 66). The star-shaped 66.3 exhibits bright green fluorescence with a high quantum yield. When applied in green OLED devices, 66.3 displayed an external quantum efficiency of 1.76%, which was improved to 2.57% by embedding it in a polyvinylcarbazole matrix.
Scheme 66. Synthesis of a NAP–Triazine Hybrid.

Reagents and conditions: (a)118n-BuLi, THF, reflux, 5 h.
N-Annulated perylene diimide dimers such as 67.3a,b were obtained by Negishi-type homocoupling of aryl bromide precursors (Scheme 67). An alternative Cadogan annulation procedure using PPh3 in DMF rather than neat P(OEt)3 was used in the synthetic sequence.119 Dimerization could also be performed using 67.1 to provide 67.4 with free pyrrolic NH groups.120 While the low solubility of 67.4 in organic solvents impaired its processability, it could be Boc protected to 67.5, which was highly soluble, similarly to the N-alkylated analogues. This compound was then deprotected upon thermal annealing of a thin film. The unprotected 67.4 interacted with bases; with DBU it was deprotonated to a dianion with a strongly red-shifted absorption. Apart from homodimers, a perylene dimer 67.9 with unsymmetric N-substitution was prepared via a Stille coupling of the stannylated 67.6 with 67.7.121 These donor–acceptor conjugates were characterized by quenching of the PDI emission (67.3b: 588 nm in solution, 633 nm in film),122 attributed to charge transfer.
Compared with their monomeric analogues, the perylene diimide dimers showed decreased aggregation in thin films. This led to an improved film morphology which corresponded to higher PCE values of bulk heterojunction OSC devices that used these materials as electron acceptors.122 PCE values in the range of 5–7% were reported for compounds 67.3a,b in blends with PTB7-Th and TTFQx-T1 donor polymers.119,120 Additionally, N-annulation decreased the electron affinity of the perylene core, leading to higher open-circuit voltages in OSCs.122
A similar Negishi-type homocoupling of aryl bromides was used in the final step toward the tetrameric N-annulated PDIs C6.1–2 (Chart 6).123 The reaction was performed using Pd2(dba)3 and Zn dust at 100 °C in DMA, providing C6.1 and C6.2 in 81% and 32% yield, respectively. The triazole linkages were formed via CuAAC prior to the final homocoupling step. While the spectral features of C6.2 were essentially a sum of its building blocks (monomeric and dimeric N-annulated PDI), compound C6.1 displayed signs of folding. OPV devices including C6.1–2 as coacceptors alongside PC61BM achieved a PCE of 7.0–7.2% under simulated sunlight and up to 13.7% under LED light irradiation, demonstrating their utility in powering small devices via indoor light recycling.
Chart 6. N-Annulated PDI Tetramers with Dimeric Cores.

N-Annulated PDI dimer 68.4 was evaluated for OSC applications alongside compounds 68.5 and 68.6 containing electron-rich core subunits (Scheme 68).124 The N-annulation was carried out similarly as shown in Scheme 67, and the final compounds were prepared via Suzuki or Stille cross-coupling reactions. In particular, the homocoupling of 68.1 to provide 68.4 in 49% yield is notable as an alternative to the Negishi-type approach shown above. OSC devices containing these compounds in blends with a PTB7-Th donor polymer had PCEs of up to 5.29% for 68.4, which was in agreement with the results observed with the structurally similar 67.3a,b. Compounds 68.5 and 68.6 displayed lower PCE values but higher open-circuit voltage (up to 1.14 V for 68.5) due to the higher LUMO energy.
Scheme 68. Preparation of N-Annulated PDI Conjugates via Cross-Coupling Reactions.
Reagents and conditions: (a)124 B2pin2, Pd(dppf)Cl2, NaOAc, 10:3/1 DMF/toluene/H2O, 70 °C, overnight, 49%; (b) Pd(PPh3)4, toluene, 100 °C, 24 h, 72%; (c) Pd(PPh3)4, K2CO3, 2:1 toluene/H2O, reflux, 2 days, 41%.
Synthesis of an S-annulated perylene diimide dimer was accomplished via an unusual synthetic route with no recourse to bay nitration. Partial dehalogenation of the tetrachloro-PDI 69.1 followed by bromination provided 69.3 (Scheme 69).125 This compound underwent Ullmann-type homocoupling under surprisingly mild conditions to the dimeric 69.4. Finally, the S-annulation was performed via a Stille-type cross-coupling with bis(tributyltin)sulfide, giving the target 69.5. A Se-annulated analogue 69.7(126) was also prepared using the same annulation approach as shown above in Scheme 56, with a final reductive dimerization step similar to that in Scheme 67. Compounds 69.5 and 69.7 displayed similar spectroscopic properties with visible absorption in the 400–600 nm range in thin films. Bulk heterojunction OSCs formulated with 69.5 and 69.7 in tandem with the PDBT-T1 donor polymer and 1,8-diiodooctane additive achieved a PCE of 7.2% and 8.4%, respectively. With 69.7, the OSCs possessed remarkably high fill factors approaching 70%.
Scheme 69. Synthesis of S- and Se-Annulated Perylene Diimide Dimers.

Reagents and conditions: (a)125 CuI, l-proline, 110 °C, 24 h, 33%; (b) Br2, H2SO4, rt, 2 days, 90%; (c) Cu, DMSO, 65 °C, 12 h, 76%; (d) (Bu3Sn)2S, Pd(PPh3)4, toluene, reflux, 12 h, 42%; (e)126 Zn dust, Pd2(dba)3, DMF, 55 °C, 1 h, 88%.
Acetylene linkers can be introduced into N-annulated PDI dimers and PDI–dye conjugates using Sonogashira coupling (Scheme 70).127,128 In the preparation of 70.1 via cross-coupling with TMS–acetylene, thorough removal of Pd residues with a dimercaptotriazine (DMT) resin was important to achieve high yields. 70.1 was subsequently used as a building block in the synthesis of the homodimer 70.3 via oxidative alkyne homocoupling and in Sonogashira syntheses of 70.2(127) and 70.5a–c.128 Compared to 67.3b, the acetylene-spaced dimers 70.2 and 70.3 have a decreased steric strain, which allows them to adopt a conformation with coplanar PDI units. This greatly strengthens their aggregation and reduces solubility. Such a behavior is detrimental to the morphology of their blends with donor polymers for OSC applications; as a result, compounds such as 67.3b perform much better. Increased freedom of intramolecular rotation, which is facilitated by the presence of acetylene spacers, was also present in the donor–acceptor conjugates 70.5a–c. In this case, the accessibility of coplanar conformations results in intense charge–transfer absorption bands which extend into the NIR range (up to approximately 900 nm in 70.5c). Overall panchromatic absorption across the visible spectrum is thus achieved.
Scheme 70. Synthesis of N-Annulated Perylene Diimide Dimers with Acetylene Linkers.
Reagents and conditions: (a)127 TMS-acetylene, Pd(PPh3)4, CuI, (i-Pr)2NH, 40 °C, 24 h, then SiliaMetS DMT, DCM, rt, 4 h, 98%; (b) K2CO3, 1:1 CHCl3/MeOH, rt, 10 min, 99%; (c) Pd(PPh3)4, CuI, 1:10 (i-Pr)2NH/toluene, 60 °C, 0.3–5 h; (d) Pd(PPh3)4, CuI, K2CO3, 1:1 CHCl3/MeOH, rt, air, 3 h, 71%.
Palladium-catalyzed C–H arylation of thiophenes in the presence of catalytic pivalate was extensively used by Welch and co-workers to prepare conjugates of N-annulated PDI 67.2b with various thiophene-containing units (Scheme 71).129−136 This methodology provided symmetrically substituted 71.1a–g in a single step in moderate yields. Additionally, unsymmetrical 71.4(129) and 71.5(133) could be prepared via two consecutive steps. A product of a single arylation (71.3b) was also elaborated into the dimeric 71.6 via thiophene homocoupling.135 As the borane functionality was reactive under the direct arylation conditions, Stille coupling was used instead to prepare 71.2h.136 In contrast to the analogues with ethynyl spacers (Scheme 70), these compounds adopt twisted conformations wherein the PDI is out of the plane of the adjacent thiophene ring. As a result, the low-energy charge transfer absorption is much weaker in 71.2b(130) than in its ethynyl-linked counterpart 70.4a. Further conformational control was realized using fused (71.2f)134 or twisted (71.2h)136 core subunits. Additionally, solvent vapor annealing of thin films could cause molecular reorganization, causing a remarkable enhancement of low-energy absorption of the unsymmetrical 71.4.129 Strong absorptions seen in the 600–800 nm range for 71.1c(130) and 71.6(135) were presumably due to the planarity of their bithiophene units, resulting in greater π-electron delocalization. When tested as acceptor materials in bulk heterojunction OSC devices, the best results were obtained with 71.2a (PCE of 5.6%).131 However, the conjugates shown here generally did not exceed the performance of the structurally simpler N-annulated PDI dimers (Scheme 67).
Scheme 71. Preparation of Thiophene-Linked Dimeric N-Annulated PDIs via Direct Heteroarylation.
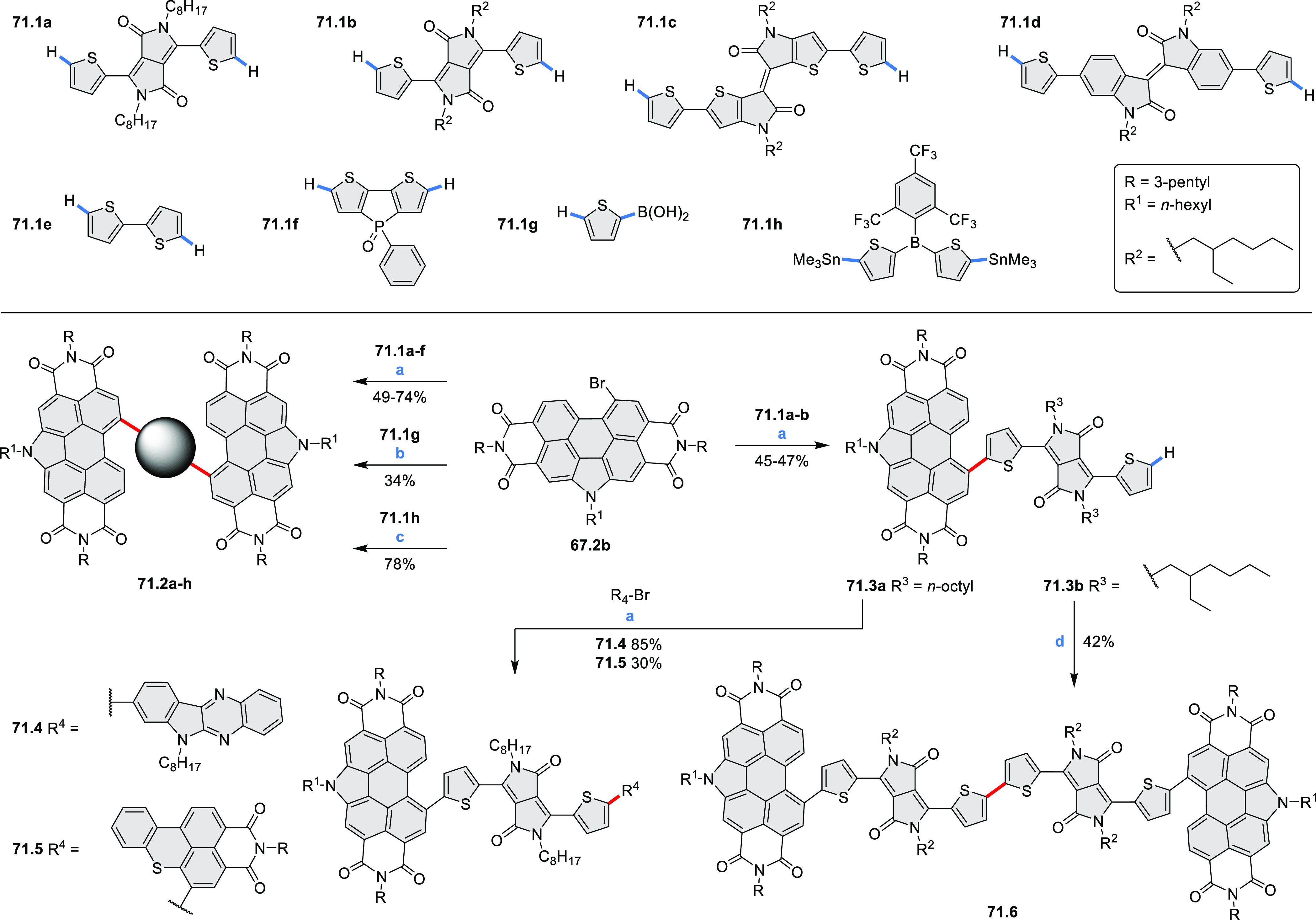
Reagents and conditions: (a)129 SiliaCat DPP-Pd, PivOH, K2CO3, DMA, 80–90 °C, 24 h (or microwave, 80 °C, 4 h for 71f), 49–74%; (b)136 SiliaCat DPP-Pd, PivOH, Cs2CO3, DMA, 80 °C, 18 h, 34%; (c) Pd2(dba)3, P(o-Tol)3, toluene, 80 °C, 36 h, 78%; (d)135 Pd(OAc)2, pivalic acid, K2CO3, N,N’-dimethylacetamide, 80 °C, 24 h.
Compound 71.2 g was converted into the fused product 72.1 in a fast and efficient oxidation with FeCl3 (Scheme 72).137 Short reaction time was in this case essential for good results, as impurities were formed after longer periods of up to 30 min. OSCs prepared with 72.1 blended with PTB7-Th possessed unusually high open-circuit voltage in the range of 1.05–1.15 V. However, PCE values of up to only 2.78% could be obtained.
Scheme 72. Preparation of an N-Annulated PDI Dimer with a Fused Thiophene Bridge.

Reagents and conditions: (e)137 FeCl3, MeCN, toluene, 50 °C, 1 min, quant.
When the thienothiophene-based building blocks 73.1a and 73.1b were subjected to C–H arylation, their terminal thiophene rings reacted at both the α and β positions, yielding the highly sterically congested compounds 73.2a(138) and 73.2b,138,139 each bearing four N-annulated PDI units (Scheme 73). Preparation of these compounds was chromatography-free and selective: 73.2b was obtained in 70% yield with only the trisubstituted compound as a major byproduct. DFT calculation showed that the PDIs are orthogonal to the core subunits of 73.2b, which adopts a butterfly-shaped structure. A low-energy emission (650–900 nm) was observed, indicating an unusually large Stokes shift. When applied as an acceptor material in OSCs, 73.2b reached moderate PCE values of up to 3.41%, reflecting the low electron mobility of this material in comparison with the hole mobility.
Scheme 73. Direct Heteroarylation Leading to Tetrameric N-Annulated PDI Conjugates.
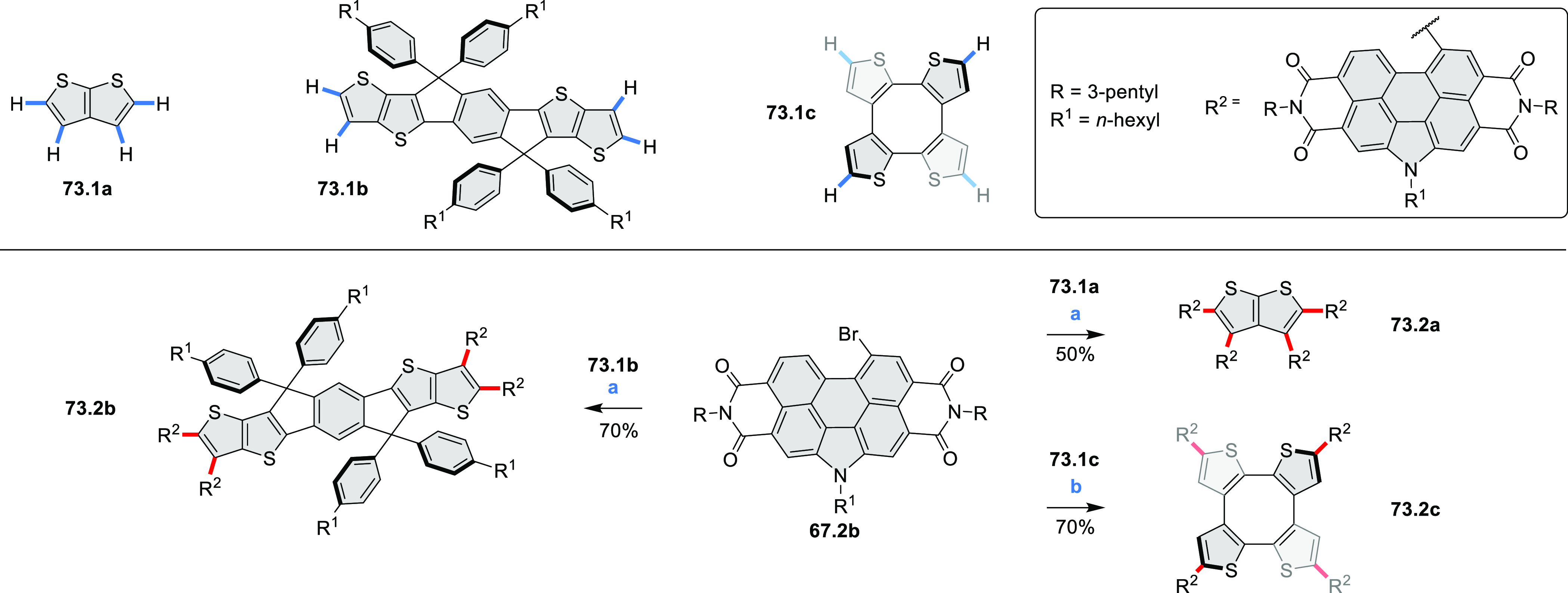
Reagents and conditions: (a)139 SiliaCat DPP-Pd, PivOH, K2CO3, DMA, 120 °C, 24 h, 70%; (b)140 SiliaCat DPP-Pd, AcOH, Cs2CO3, DMA, 120 °C, 24 h, 70%.
Another sterically congested tetrakis PDI) conjugate prepared by this methodology was 73.2c, where a saddle-shaped structure was achieved through introduction of a cyclooctatetrathiophene core.140 In this case, using catalytic acetate instead of pivalate allowed us to achieve a higher yield. This improvement was attributed to decreased steric strain in the concerted palladation–deprotonation step, which is responsible for the C–H activation of thiophenes. OSC devices formulated with 73.2c, a PTB7-Th donor polymer, and a 1-chloronaphthalene additive displayed PCEs of up to 4.26%, with improved fill factor and short-circuit current in comparison to 73.2b.
Similarly to N-annulated perylene diimides, their S- and Se-annulated analogues can be efficiently brominated to 6-bromo derivatives126 such as 74.1a–c and further derivatized via cross-coupling reactions. A series of compounds with four S- or Se-annulated PDI units located on different core synthons were prepared in this manner via Suzuki coupling in the final step (Scheme 74).141−143 In all cases, the S- or Se-annulation was introduced in earlier steps similarly as shown above in Scheme 60. The sterically constrained structures of these molecules were intended to prevent aggregation via π-stacking and lead to improved morphology and phase separation in blends with donor polymers. OSCs built using 74.3 combined with the PBDT-TS1 polymer and diphenyl ether additive reached a PCE of 6.17%.141 Importantly, 74.3 greatly outperformed its analogue lacking the S-annulation (3.62%). A similar trend in PCE was observed, with 74.4 performing better than a nonannulated counterpart (8.28% vs 7.16%).142 In both cases, the improvement was attributed to balanced carrier mobilities as well as to the increase of LUMO energies caused by the electron-donating character of the S atom. Among dyes derived from tetraphenylethylene, the Se-containing 74.5b displayed a higher maximum PCE of 7.63% compared to 6.85% for 74.5a.143 Compound 74.5b performed better across the full range of parameters, showing remarkably high open-circuit voltage (1.078 V) and fill factor (68.8%) as well as well-balanced carrier mobility (μh = 3.21 × 10–4 cm2 V–1 s–1, μe = 1.37 × 10–4 cm2 V–1 s–1).
Scheme 74. Synthesis of Three-Dimensional Thiophene-Annulated PDI Dyes.
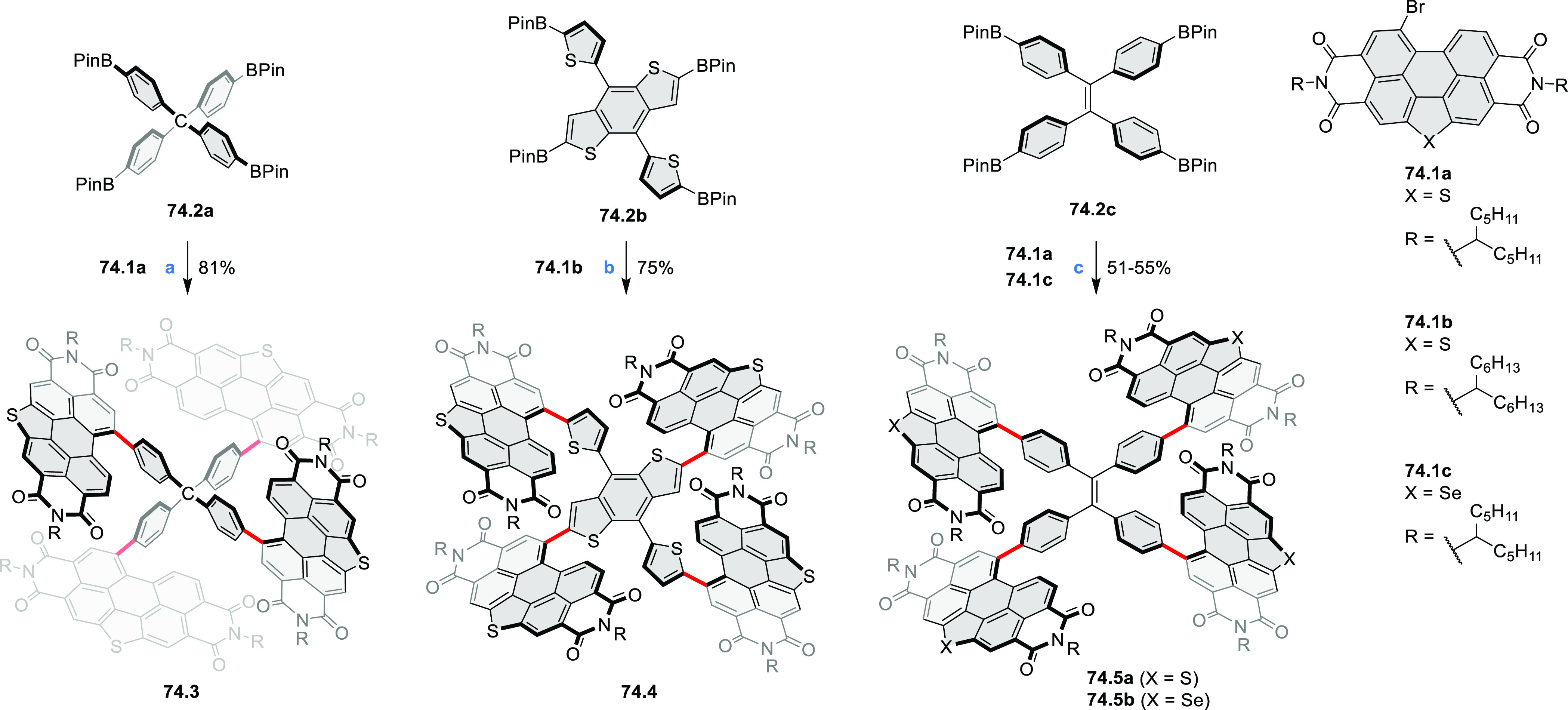
Reagents and conditions: (a)141 Pd(PPh3)4, K2CO3, THF, reflux, 2 days, 81%; (b)142 Pd2(dba)3, P(p-C6H4OMe)3, K2CO3, THF/H2O, 16 h, reflux, 75%; (c)143 Pd(PPh3)4, K2CO3, 2:1 THF/H2O, 80 °C, 2 days, 51–55%.
Chen, He, and co-workers used Stille coupling to efficiently append Se-annulated peryleneimide units onto thiophene-based cores to provide 75.3–5 (Scheme 75).144 Compound 75.4 was found by DFT calculations to have the greatest steric constraints, preferentially adopting a conformation where two of its PDI units are nearly orthogonal. This feature was thought to disfavor aggregation and was found to be beneficial for the performance of organic photovoltaics based on combinations of these materials with the PBDB-T thiophene-based donor polymer. Electron mobility, hole mobility, and short-circuit current were the highest with 75.4, resulting in the best PCE value (75.3: 4.10%, 75.4: 5.82%, 75.5: 5.10%). A related compound, 75.2, likewise had a twisted geometry, with an ca. 119° angle between PDI planes according to molecular modeling.145 Although not directly comparable to 75.3–5 because of the use of different donor polymers, OSCs based on 75.2 displayed a lower performance, with PCE of up to 2.53%.
Scheme 75. Synthesis of Acceptors with Two, Three, and Four Se-Annulated Peryleneimide Units.
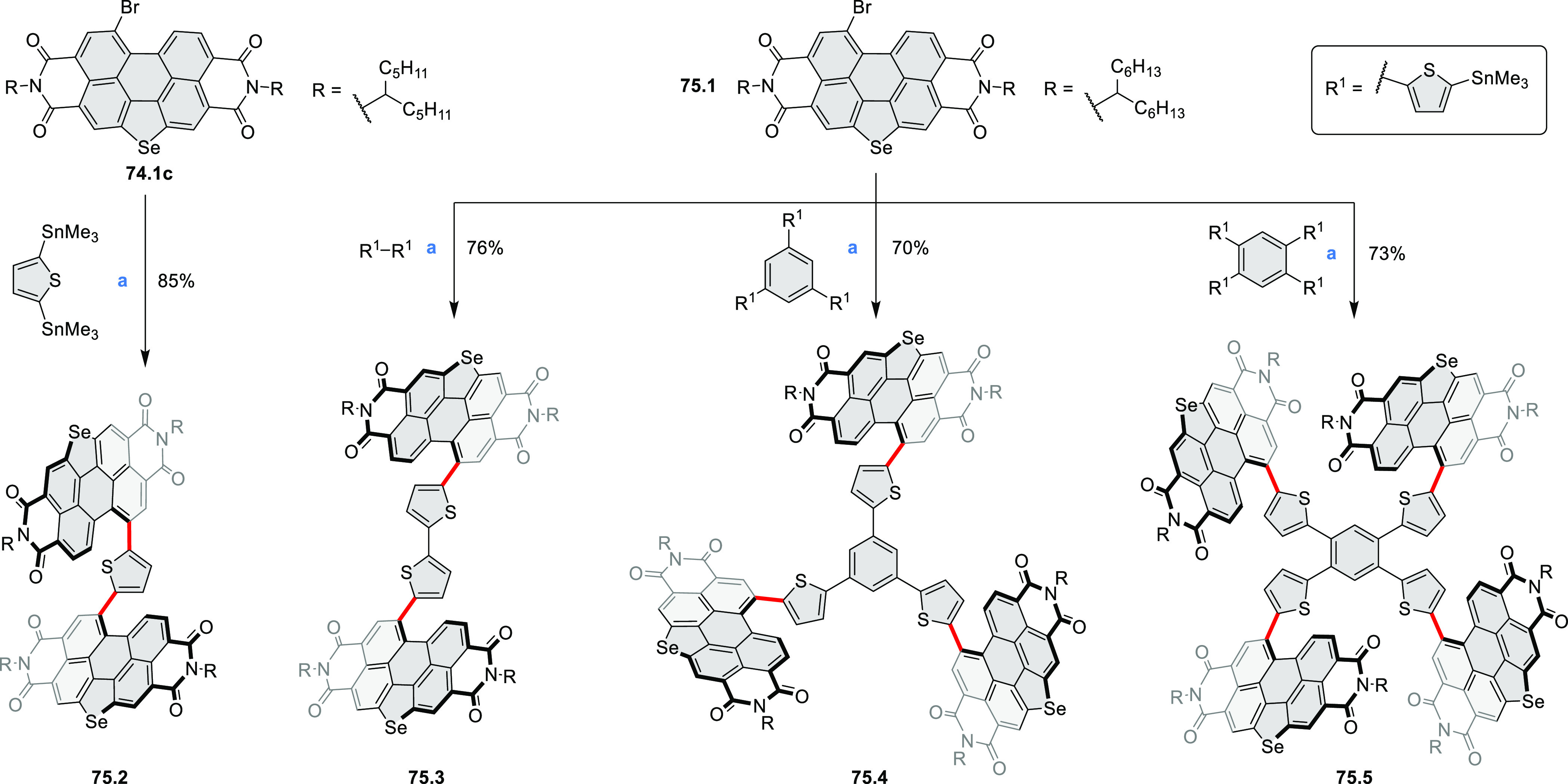
Reagents and conditions: (a)144,145 Pd2(dba)3, P(o-tolyl)3, toluene, reflux, 24 h, 70–85%.
Double S- or Se-annulated PDIs linked via a vinyl or a thiophene bridge were prepared using a similar approach based on Stille coupling (Scheme 76). Likewise, 76.1a,b and 76.2 adopt a twisted structure which prevents aggregation and helps to form smooth films upon spin coating.146 These compounds were used as interfacial materials in perovskite solar cells, positioned as an interlayer between the perovskite absorbing layer and the fullerene-based electron transport layer. The presence of the interlayer improved charge transport and inhibited charge recombination, resulting in better device performance (no interlayer: PCE 17.53%, FF 77.50%; 76.2 interlayer: PCE 20.41%, FF 83.86%). The Se-containing 76.1b was further converted into the fused dimer 76.3 via UV irradiation in the presence of catalytic I2.147 This fused product was predicted by DFT modeling to adopt a highly twisted geometry with a nearly 35° angle between the PDI planes. The absorption spectrum of 76.3 has maxima at 471 and 509 nm, which are blue-shifted relative to the precursor 76.1b (483 nm, 517 nm). HOMO and LUMO levels were estimated to be similar in both compounds. When used in OSC devices alongside the PBDB-T donor polymer, 76.3 enabled higher and more balanced carrier mobilities compared to 76.1b. This corresponded to higher PCE values of up to 7.41% for 76.3, achieved with a 1,8-diiodooctane additive.
Scheme 76. Synthesis of Vinylene- and Thiophene-Bridged S- and Se-Annulated PDI Dimers.

Reagents and conditions: (a)146 Pd(PPh3)2Cl2, toluene, reflux, 24 h, 77–80%; (b)147 I2, toluene, hν (Hg lamp), 36 h, 55%.
UV irradiation in the presence of catalytic I2 was also used to achieve the fusion of three PDI blocks to a common benzene core, leading to the propeller-shaped 77.3 (Scheme 77).148 This compound was then subjected to late-stage nitration, providing a mixture of bay-nitrated isomers, followed by Se annulation to provide 77.4. These molecules are highly twisted because of steric crowding, especially in 77.4 where the Se annulation further rigidifies the PDI. In the solid-state structure of 77.4, pairs of Se···O chalcogen bonding contacts with a distance close to 3.0 Å were observed between adjacent molecules. OSCs built using 77.3 or 77.4 blended with the PDBT-T1 donor polymer displayed higher open-circuit voltage for 77.4 (1.00–1.01 V vs 0.96–0.97 V for 77.3), reflecting the higher LUMO level of 77.4. Blend films with 77.4 also displayed higher carrier mobilities, which were otherwise well-balanced with both compounds (μe/μh = 1.3–1.5). These favorable properties resulted in PCE values reaching 8.28% for 77.3 and 9.28% for 77.4 in the presence of a 1,8-diiodooctane additive. A similar approach was also used in the preparation of dimeric benzene-fused PDIs 77.8a,b.149 In this case, 77.6 was exposed to sunlight in the presence of I2, providing the fused 77.7 regioselectively despite its steric congestion. Perylene tetraesters were then converted into PDIs in the final steps. In films of 77.8a blended with PBDB-T, μe was higher by an order of magnitude than for 77.8b, implying that long alkyl chains of 77.8b impeded charge transport. This difference was reflected in a much better OSC performance of 77.8a (PCE up to 7.41%, JSC = 11.34 mA/cm2) compared to 77.8b (PCE up to 4.57%, JSC = 8.28 mA/cm2).
Scheme 77. Synthesis of Benzene-Fused Se-Annulated PDI Dimers and Trimers.
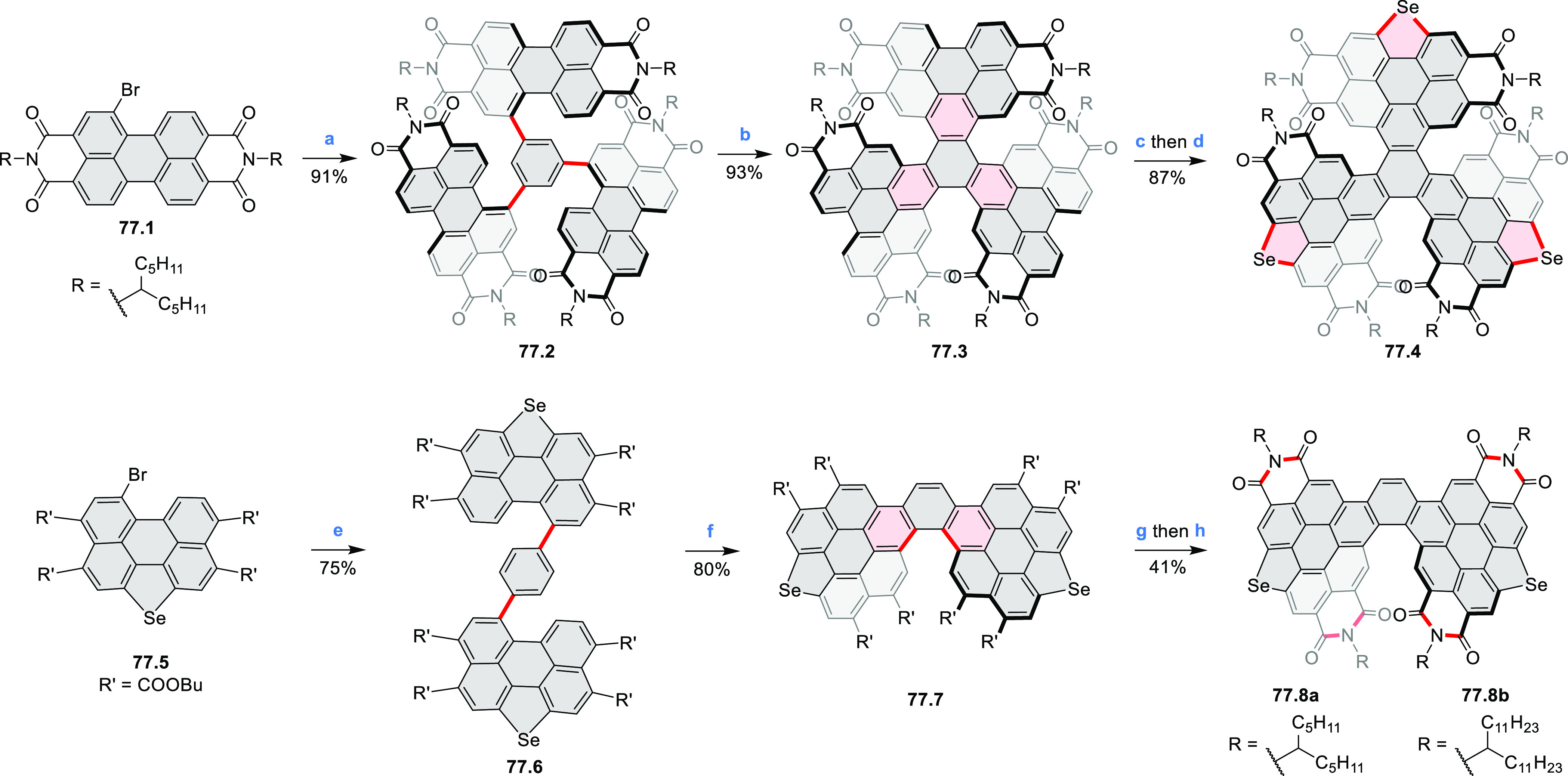
Reagents and conditions: (a)148 1,3,5-benzenetriboronic acid tris(pinacol)ester, Pd(PPh3)4, Na2CO3, 2:1 THF/H2O, reflux, 48 h, 91%; (b) I2, toluene, hν (Hg lamp), rt, 24 h, 93%; (c) HNO3 (fuming), DCM, rt, 12 h, quant.; (d) selenium powder, NMP, 190 °C, 12 h, 87%; (e)149 Pd(PPh3)4, K2CO3, 2:1 THF/H2O, 85 °C, 13 h, 75%; (f) I2, DCM, hν (sunlight), rt, 8 h, 80%; (g) ClSO3H, rt, 4 h, quant.; (h) 6-undecylamine or 12-tricosylamine, imidazole, 150 °C, 4 h, 41%.
N-Annulated PDI chromophores were appended to a phenylene spacer via Buchwald–Hartwig amination (Scheme 78).150 The isomeric products 78.2–3 displayed nearly identical UV–vis spectra and electrochemical behavior. In the solid state, however, the slightly convex PDI units were nearly parallel in 78.2 but close to orthogonal in 78.3. Additive-free OSCs containing blends of these compounds with the PTB7-Th donor polymer performed better for 78.3, in line with several reports of superior properties of PDI-based acceptor materials with a twisted structure. The use of a 1-chloronaphthalene additive enabled greater optimization for 78.2, leading to a PCE of 5.01%, compared to 4.15% for 78.3 with a 1,8-diiodooctane additive. These results were explained by demonstrating a significantly faster symmetry-breaking charge separation in 78.2 than in 78.3, attributed to better conjugation through the para-phenylene bridge.151 This process was shown to be particularly favorable in polar solvents (THF and acetone), where charge separation became exergonic.
Scheme 78. Synthesis of Phenylene-Bridged Isomeric N-Annulated Perylene Diimide Dimers.

Reagents and conditions: (a)150 Pd(OAc)2, t-BuOK, P(t-Bu)3, toluene, 110 °C, o/n, 47–54%.
N-Annulated perylene 79.1 (prepared via the Cadogan reaction similarly as shown in Scheme 56) was selectively monobrominated to the 3-bromo derivative 79.2, which was then derivatized via Suzuki coupling to 79.3a–c (Scheme 79).152 These fragments were oxidatively homocoupled at the 10 position in a process enabled by the electron-donating character of the pyrrolic ring. In the dimers 79.4a–c, rotation around the central C–C bond was sufficiently restricted to permit resolution of atropisomers by HPLC. These compounds underwent self-assembly via a combination of π-stacking and hydrogen-bonding interactions between the amide groups. The aggregation occurred upon addition of cyclohexane to a DCM solution and was complete in 19:1 cyclohexane/DCM. A preference for homochiral aggregation was observed, which was determined by the axial chirality of the biperylenyl core, overriding the point chirality of the tetrahydrogeranyl chains.
Compounds C7.1a–d (Chart 7), obtained using similar synthetic methods, formed aggregates in nonpolar solvent, analogous to those observed for 79.4a–c. In the case of C7.1a, intramolecular hydrogen bonding between adjacent amide groups caused aggregation in toluene to proceed with a lag period of 10–40 min, followed by the formation of a distinct intermediate and final aggregates.153 The lag period could be eliminated upon seeding with the intermediate aggregate, indicating that it possessed active sites for supramolecular polymerization. The formation of fibrillar aggregates in methylcyclohexane was also reported for compounds C7.1b–d.154 Columnar assembly with NAP units oriented in the same direction in adjacent monomers was identified. A mixture of M- and P-type aggregates was always obtained, regardless of point chirality in the alkyl side chains. The helicity of these structures was postulated to spontaneously interconvert via intrastack stereomutation. This flexible chirality was used to produce CD response in solution upon mechanical agitation, with opposite CD spectra being obtained with clockwise and counterclockwise stirring. Aggregation properties were also studied for the related C7.2a,b, functionalized with trialkoxyphenyl groups.155 Analog C7.2a with ester-appended lateral groups formed a single type of fibrillar aggregate with an absorption spectrum red-shifted relatively to the monomeric species. The presence of amide linkers in C7.2b enabled intra- and intermolecular hydrogen bonding, resulting in a more complex behavior reminiscent of that outlined above for C7.1a. Overall, four types of fibrillar and nanosheet aggregates were formed by C7.2b under different conditions.
Chart 7. N-Annulated Perylenes with Amide-Bound Side Chains.

N-Annulated perylenes were incorporated as electron-rich donor units into push–pull copolymers (Chart 8). These systems were evaluated as dyes for organic photovoltaics in combination with the fullerene-based acceptor PC71BM (C8.1a,b and C8.2a,b) or for green-selective photodiodes (C8.3). The polymers were prepared via Pd-catalyzed Suzuki or Stille polymerization reactions from the respective diboronic acid or dibromide precursors. In materials C8.1a and C8.1b, the presence of a branched alkyl chain resulted in a slight broadening (ca. 20 nm) of the visible absorption band in the 400–600 nm range.156 The photovoltaic performance of these polymers was inferior to an analogue containing carbazole donor units, with PCEs of 2.4–2.5% (5.8% for the carbazole-based material). This decrease was attributed mainly to a lower yield of charge-separated excited states.
Chart 8. Polymers Containing N-Annulated Perylene Units.

In copolymers C8.2a,b, the longer alkyl chain caused C8.2b to adopt a more planar conformation in the solid state, resulting in slightly red-shifted absorption and emission spectra.157 In photovoltaic devices with films composed of polymer and PC71BM in a 1:4 weight ratio, C8.2b afforded a higher PCE of 4.80%, compared to 3.63% for C8.2a. A planar heterojunction photodiode was prepared with spin-coated films of ZnO and polymer C8.3.158 The ICT absorption of C8.3 in the amorphous solid film (538 nm with 138 nm fwhm) produced a green-selective response of the device. At a reverse bias voltage (−5 V), the photodiode was characterized by a low dark current of 0.68 nA/cm2 and a strong response to 550 nm light with a detectivity of 1.04 × 1012 Jones.
3.3. [ghi]Heteroannulated Perylenoids: Six- and Seven-Membered Rings
[ghi]Benzoxepine-fused perylene diimide 80.2 was obtained via Cu-mediated ring closure of the phenol-substituted PDI precursor 80.1 (Scheme 80).159 Smaller ring fusion was also possible by this method, leading to analogues with five- and six-membered rings (Scheme 141, Section 4.3). Fusion of the benzoxepine ring in 80.2 led to a hypsochromic shift and a quenching of the emission quantum yield by 16% compared to the parent PDI. Another method to access the benzoxepine-fused PDI framework is the Pd-catalyzed intramolecular cyclization to effect the fusion of a pendant phenyl ether substituent (Scheme 80).160 C–H activation at the ortho position of this substituent enables its coupling to a bay-brominated PDI core. This method was applied on PDIs 80.3a–d and was reported to be unsuccessful when the respective perylene tetraesters were used instead. While the cyclized compounds 80.4a,b were obtained in relatively low yields, a greater efficiency was attained with the brominated substrates 80.3c,d, leading to 80.4c,d. The Br substituents on the electron-rich phenyl ether groups were not affected under the reaction conditions. Subsequent dehalogenation of 80.4c was thus a more efficient way of preparing 80.4a than its direct synthesis from 80.3a. The cyclized products had a curved geometry with a 22° twist angle in the perylene core. This resulted in good solubility in organic media even in the absence of large imide-bound solubilizing substituents.
Scheme 80. Synthesis of Benzoxepine-Fused Perylene Diimides.

Reagents and conditions: (a)159 Cu(OAc)2, Cs2CO3, PivOH, DMSO, 140 °C, overnight, 94%; (b)160 Pd(OAc)2, PCy3·HBF4, K2CO3, DMA, 100–120 °C, 2–4 h (c) Pd(PPh3)4, Cs2CO3, paraformaldehyde, 1:1 DMF/toluene, 80 °C, 17 h, 97%.
Diels–Alder cycloaddition with benzynes was used to prepare PDIs fused with various aromatics, including the indole-fused molecule 81.1 (Scheme 81).161 Benzyne intermediates were obtained by diazotization of anthranilic acid derivatives or by desilylation of o-trimethylsilyl aryl triflates. The indole-fused 81.1 was characterized by higher HOMO and LUMO levels and much lower extinction coefficients compared to analogues fused with carbocyclic units.
Scheme 81. Benzyne Cycloaddition to Perylene Diimide.

Reagents and conditions: (a)161 CsF, 1:1 toluene/MeCN, 80 °C, 36 h, 64%.
Goujon, Hudhomme, and co-workers described the preparation of azabenzannulated PDI derivatives in a one-pot condensation–photocyclization–oxidation sequence from 1-aminoPDI 82.1 and various aryl aldehydes (Scheme 82).162 In the first step, an imine was formed from these starting materials in the presence of TFA. After concentrating the reaction mixture under vacuum, the crude imine was redissolved in DCM and subjected to visible-light irradiation, resulting in photocyclization to a secondary amine intermediate. This species was rapidly oxidized with DDQ, providing the azabenzannulated products 82.2a–e in high yields. The dimeric products 82.3–4 were prepared in the same manner from the respective dialdehydes. The resulting compounds were emissive, with moderate to high quantum yields. Electron-rich groups on the nitrogen-containing ring resulted in increased HOMO levels and narrower band gaps. Thus, 82.2b, 82.2e, and the dimeric 82.4 were notable for broad and red-shifted emission bands (552, 541, and 572 nm, respectively). On the other hand, electron-deficient substituents in 82.2c,d had little effect on the spectral properties. An earlier report by Würthner and co-workers included a similar methodology used to prepare 82.5a–f, although with lower yields compared to the protocol described above.4 The synthesis of related perylene dianhydrides, anhydride imides, and tetraesters was also presented, as well as double azabenzannulations to provide diazacoronenes (see Scheme 6, Section 2.1.1).
Dehydrogenative condensation of amino-substituted PDIs with 2-formylpyridine according to a previously reported method163 led to ligands 83.2a–c (Scheme 83). These compounds were then used to prepare the Ir(III) and Ru(II) complexes 83.3–5. Phosphorescence in the NIR region (700–1100 nm) was observed with 83.3 and 83.4.164 The ruthenium complex 83.4 had a particularly high phosphorescence quantum yield of 11% with a 4.2 μs lifetime. Low quantum yield (<1%) with a 33 μs lifetime was recorded for 83.3. For compound 83.5 a strong aggregation-induced dependence of the emission spectrum on concentration was reported.165 Strong NIR emission with peaks at 736 and 824 nm was seen at high concentration (10–2 M) but not in dilute solution (10–5 M). Emission at 758 nm was also seen in films prepared from neat 83.5 as well as in a blend with PMMA.
Wang, Yoshikai, and co-workers reported the synthesis of pentasubstituted pyridines via Co-catalyzed [2 + 2 + 2] cyclotrimerization between two alkynes and a nitrile (Scheme 84).166 The reaction was proposed to proceed via an initial coupling of two alkyne molecules into a cobaltacyclopentadiene, followed by the insertion of nitrile. This process required cobalt(0) complexes, which formed in situ in the presence of Zn dust. Among the compounds prepared by this methodology, the tetra- and pentaarylpyridines 84.1a–e were subsequently cyclized into the N-doped perylenoid PAHs 84.2a–e. This cyclodehydrogenation was achieved via an unusual method of solvent-free ball milling of 84.1a–e with K metal. Attempts to effect the same transformation under oxidative coupling conditions (e.g., with FeCl3 in MeNO2/DCM) failed, apparently because of the electron-deficient and basic character of pyridine. In the case of 84.1d, an unexpected demethylation occurred, providing 84.2d in 12% yield.
A pyridazine-fused perylene 85.3 was prepared in a short and efficient sequence from dihydroxynaphthalene 85.1 (Scheme 85).167 First, oxidation in a dilute KMnO4 solution formed the perylene ring system in 85.2. This reaction was proposed to proceed via a diradical intermediate. While the trans tautomer of 85.2 was predicted to be more stable than the cis form, their interconversion was fast at rt, and thus the condensation product 85.3 was efficiently formed from the cis tautomer and hydrazine. The product was further derivatized by O-alkylation to provide 85.4. The crystal structure of 85.3 revealed a twisted geometry of the perylene core with a dihedral angle close to 30°. The twist arises due to the steric clash between the alkoxy substituents. Compound 85.3 displayed a broad red emission peaking at 628 nm, while a narrower emission blue-shifted to 521 nm was seen in 85.4. Large Stokes shifts were observed in both cases.
Inverse electrons demand Diels–Alder cycloaddition between cyclopentadienone 86.1 and alkynes 86.2a,b, providing the sterically crowded products 86.3 and 86.5 (Scheme 86).168 Multiple fusions of the closely positioned phenyl groups in these molecules were then accomplished via oxidative coupling with DDQ and TfOH. These conditions were chosen as optimal, as incomplete dehydrogenation was observed when using FeCl3 in MeNO2. The fused product 86.6 was thus obtained from 86.5 in 91% yield. Compound 86.3 was converted into a mixture of diastereomers 86.4a,b in 72% overall yield, which is an excellent result considering that 27 C–C bonds were formed in a single step. The diastereomers were successfully separated via silica gel chromatography; enantiomers of 86.4a were also resolved by semipreparative chiral HPLC. While the planar compound 86.6 was highly insoluble, 86.4a,b had good solubility in several organic solvents because their twisted structures prevented aggregation. The propeller-shaped D3-symmetric 86.4a had a higher solubility than the C2-symmetric 86.4b, which was observed to aggregate in DCM at micromolar concentrations. In the CD spectra of the pure enantiomers of 86.4a, a strong Cotton effect was present with a peak Δε of 762 and −768 M–1 cm–1, respectively, which was attributed to the high rigidity of the extended chiral structure.
Scheme 86. Synthesis of Twisted N-Doped Nanographenes.
Reagents and conditions: (a)168o-xylene, 80 °C, 30 min, then reflux, 12 h; (b) DDQ, TfOH, DCM (high dilution), 0 °C to rt, 4 h.
Oxidative coupling was successfully employed in the cyclization of compounds 87.1a,b, which were readily assembled via cross-coupling reactions, into 87.2a,b (Scheme 87).169 When FeCl3 was used as the oxidant, yields close to 70% were achieved for the formation of three C–C bonds. These cyclized products were then elaborated into acceptor-fused donor–acceptor triads through cross-coupling with diketopyrrolopyrrole units. The resulting products 87.3a,b had an intense absorption band in the 500–800 nm range, which was absent in 87.2a,b. The triazole-containing 87.3b was more electron rich and had a slightly larger band gap than 87.3a. Compound 87.3b also had nearly two times higher extinction coefficients in the 550–650 nm range. OSCs built using 87.3a,b blended with the PC71BM fullerene-based acceptor achieved PCEs of up to 2.88% with 87.3b.
Scheme 87. Synthesis of Thiadiazole- and Triazole-Fused Perylenes.

Reagents and conditions: (a)169 FeCl3, MeNO2, 0 °C, 3 h, 69–72%; (b) ArB(OH)2, Pd(PPh3)4, K2CO3, 2:1 toluene/H2O, 85 °C, overnight, 78–80%.
π-Extended acridinium dyes with a perylenoid substructure were prepared, as shown in Scheme 88.170 Condensation of 2-naphthol with an aldehyde led to the dibenzoxanthene 88.1. This compound was oxidized to the xanthenium salt 88.2. The pyrylium oxygen atom in this compound could be displaced by an aniline, leading to the dibenzoacridinium 88.3. This product readily cyclized to 88.4 upon exposure to sunlight without any catalyst or oxidant.170 A similar cyclization of 88.2 was also reported to be possible, allowing us to switch the order of steps. Finally, donor–acceptor compounds 88.5 and 88.6(171) were obtained via cross-coupling reactions. Charge transfer upon photoexcitation was reported for these compounds with the expanded acridinium subunit acting as an acceptor.
Scheme 88. Synthesis of Expanded Acridinium Cations.
Reagents and conditions: (a)170 1:25 conc. HCl(aq)/AcOH, reflux, 36 h, 78%; (b) PbO2, AcOH, reflux, 6.5 h, 77%; (c) HBF4, 1:2 AcOH/MeCN, rt, 24 h, 93%; (d) 3,4,5-trimethoxyaniline, NMP, molecular sieves, reflux, 8 h, 38%; (e) MnO2, HCl(g), Ac2O, reflux, 1 h, 66%; (f) 1:1 MeCN/H2O, hν (sunlight), rt, 91%; (g) 4-(dimethylamino)phenylboronic acid, Pd(PPh3)4, Na2CO3, 5:8 DME/DMF, 95 °C, 24 h, 19%; (h)171 alkyne, Pd(PPh3)4, CuI, Et3N, DMF, 80 °C, 30 h, 19%.
Perylene diimides containing S2-annulated bays108,110,111 were obtained and studied along with their analogues containing five-membered thiophene annulations and are discussed in Section 3.2 (Schemes 61 and 62).
Fusion of two PDI units to a common heterocyclic ring was reported by Jen and co-workers (Scheme 89).172 Oxidation with FeCl3 was used to effect double bay cyclization, to produce products 89.2a–c with a furan, thiophene, or selenophene core, respectively. A tellurophene analogue 89.8 and its singly fused analogue 89.9 could also be prepared using the same method, with a selectivity dependent on reaction conditions.173 Alternatively, bay cyclizations could be achieved by visible or near-UV irradiation in the presence of I2. The latter approach was used for the postpolymerization modification to afford 89.6,174 as well as to prepare 89.4 with a thienothiophene core.175 Compound 89.2b was also obtained by photocyclization in 88% yield, comparable to that achieved with FeCl3 oxidation. The resulting compounds 89.2a–c and 89.8 adopt a twisted structure, where the angle between the two PDI planes is predicted to increase with the bulk of the central heteroatom.172,173 Similar twist angles were reported for the DFT-optimized structures of 89.2b (23°)172 and the polymer 89.6 (24.4°).174 The fused compounds had higher LUMO levels and larger band gaps than their heterocycle-bridged precursors, which corresponded to shorter wavelengths of absorption onset. Similar HOMO and LUMO energies were reported for compounds with different heterocyclic cores (89.2a–c and 89.8). Ring fusion was generally observed to increase electron mobility in donor–acceptor blend films, which translated to the better performance of OSC devices. PCE significantly improved with ring fusion in the tellurophene series (89.7: 1.45%, 89.9: 3.26%, 89.8: 7.52%), where 89.8 notably outperformed the fullerene-based acceptor PC61BM.173
Scheme 89. Synthesis of Perylene Diimides Fused to Five-Membered Heterocycles.
Reagents and conditions: (a)172 FeCl3, MeNO2, reflux, 3 h; (b)175 I2, toluene, hν (incandescent lamp), air, rt, 24 h, 84%; (c)174 I2, DCM, hν (xenon lamp), rt, 10 h, quant.; (d)173 FeCl3, MeNO2/chlorobenzene, 130 °C, 3 days, 66%; (e) FeCl3, MeNO2/toluene, 90 °C, 8 h, 37%.
Irradiation in the presence of oxygen with catalytic I2 provided access to further examples of thienothiophene-fused dimeric PDIs C9.1–2 and a dimeric perylene tetraester C9.3 (Chart 9).176 The planarity of these molecules facilitated the formation of self-assembled monolayers on the HOPG surface. C9.1 formed rows of molecules with contacts along the unsubstituted perylene edges. Its short alkylamine chains occupied the space between the rows. Longer chains in C9.2 entirely occupied the areas of contact between adjacent molecules arranged in parallel rows with greater intermolecular distances than in C9.1. The dimeric perylene tetraester C9.3 formed a honeycomb pattern with 4 nm voids surrounded by six molecules.
Chart 9. Thienothiophene-Fused Perylene Diimides and Perylene Tetraesters.

Methods of introducing ring fusion summarized in Scheme 89 were used to synthesize several dimeric fused PDIs with extended thiophene-based cores (Chart 10). In most of these examples, the [ghi] cyclization was performed in the last synthetic step. The only exception was the polymer C10.3, where a fused monomer was prepared prior to polymerization. The respective method of annulation (oxidative cyclization with FeCl3 or irradiation in the presence of catalytic I2) is indicated for each molecule. In the C10.1a–c series, compound C10.1b was predicted by DFT to be nearly planar.177 In contrast, C10.1a and C10.1c were expected to be twisted because of a steric clash between the PDIs and core-bound alkyl groups. Extension of the electron-rich core resulted in increased HOMO and LUMO energies and a decreased band gap. In C10.1b and C10.1c, an absorption band at 600–650 nm was present, which was assigned to intramolecular charge transfer, highlighting their acceptor–donor–acceptor architecture. Core extension also resulted in weaker and red-shifted emission bands and in a strong increase of a two-photon absorption cross section. OSCs prepared with C10.1a–c blended with PTB7-Th displayed higher VOC and PCE values than their nonfused analogues.178 A slight increase of VOC with core extension was also observed. The highest PCE of 6.06% was reached for the planar compound C10.1b.
Chart 10. Further Examples of Dimeric Thiophene-Fused Perylene Diimides.
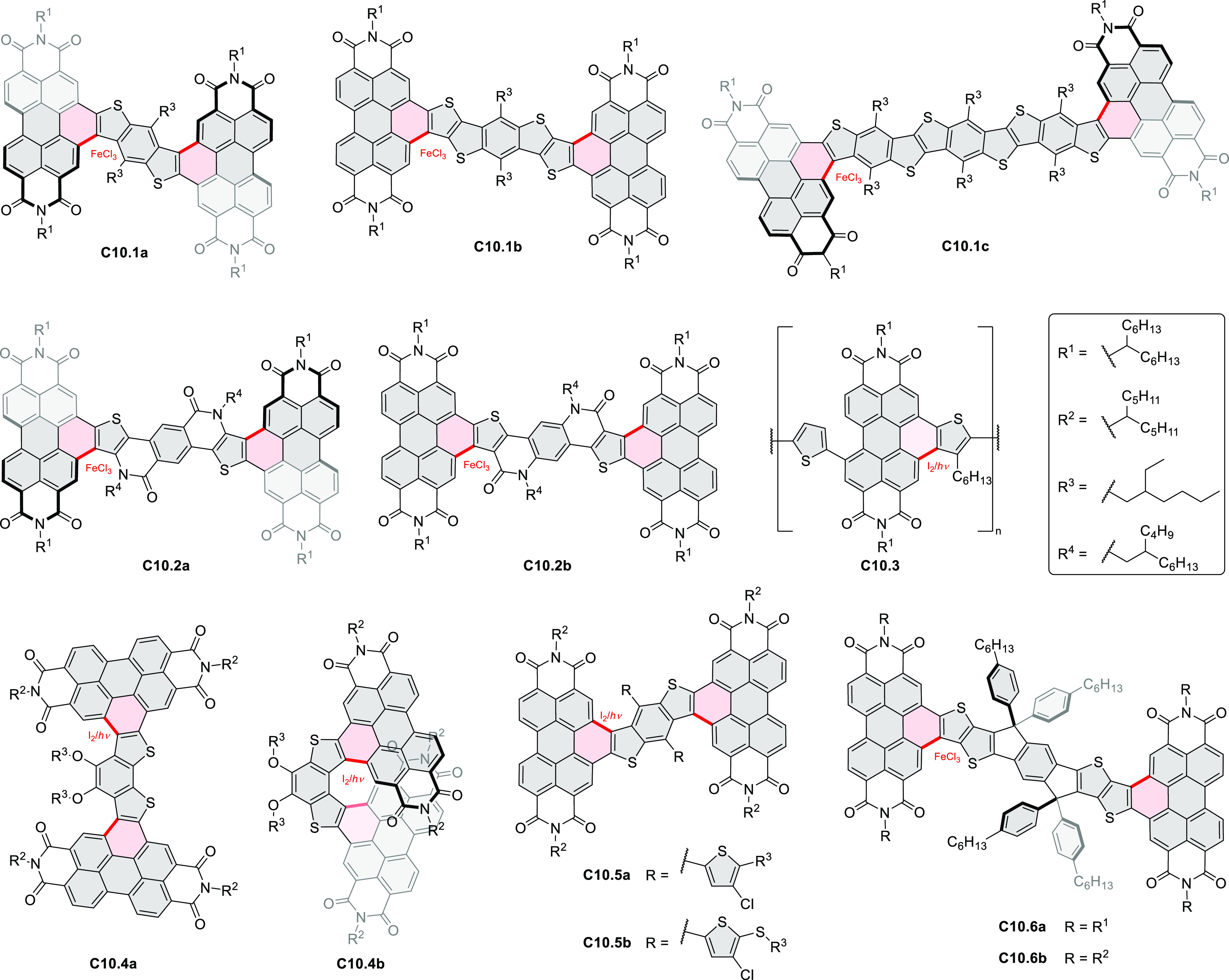
Similarly, the optimized structure of C10.2a was twisted, while its isomer C10.2b was predicted to be planar.179C10.2a had a strongly red-shifted low-energy absorption band compared to C10.2b. Its emission was also red-shifted and much weaker (λem = 647 nm, ΦF = 0.042), in contrast to the bright emission of the planar isomer C10.2b (λem = 547 nm, ΦF = 0.52). A slightly better performance in OSCs was observed for C10.2b (PCE = 4.97%) compared to C10.2a (PCE = 4.79%). Significant differences in geometry were also evident between the isomeric compounds C10.4a and C10.4b.180 The more sterically congested C10.4b had partially overlapping PDI units with a 42.6° dihedral angle between them. Conversely, a smaller tilt of 12.7° was predicted for C10.4a, which displayed a higher band gap and lower HOMO energy than C10.4b. OSCs built using blends of these compounds with PTB7-Th and a 1-chloronaphthalene additive achieved PCEs of up to 4.34% for C10.4b and 2.89% for C10.4a. The main improvement observed for the twisted isomer C10.4b was the short-circuit current (JSC = 10.27 mA cm–2 and JSC = 6.79 mA cm–2 for C10.4a). Compounds C10.5a,b had planar conjugated backbones, but their pendant thiophene groups decreased the tendency to aggregate.181C10.5b contained additional thioether functions, which resulted in a higher HOMO level and a decreased band gap as determined by cyclic voltammetry. Absorption spectra of the two analogues were broadly similar. OSCs using these compounds as acceptors had high open-circuit voltage values of up to 1.14 V, with a better overall performance recorded for C10.5a.
Polymer C10.3 proved to be a more efficient acceptor in OSCs compared to its analogue with nonfused PDI units as well as the one in which both PDI bays were fused.182 Open-circuit voltages of up to 1.03 V were obtained with the PBDB-T donor, while the highest PCE (4.60%) was achieved with PTB7-Th. Compounds C10.6a,b were reported in OSC applications by several groups.183,184 A good photovoltaic performance was achieved with a PCE of up to 7.33%, a VOC close to 1 V, and a high JSC of 13.24 mA cm–2. A later study of the photophysical properties of C10.6b in blends with PTB7-Th revealed delayed photoluminescence indicative of nongeminate charge recombination.184 This effect was proposed to be an indicator for fast screening of photovoltaic blends.
The above method of preparing thiophene-fused PDIs was further extended to sterically congested targets with four PDIs connected to thiophene-based cores (Scheme 90).185−187 The requisite precursors, 90.1a–c, were prepared via cross-coupling reactions. Conformational freedom in this series of compounds decreases from 90.1a, through 90.1b with a partially fused core, to 90.1c, in which a fully rigid spirocyclic core was introduced. Fused final products 90.2a–c were then obtained upon cyclodehydrogenation with FeCl3, which led to significant changes in molecular geometry. While in 90.1a adjacent PDI units were nearly orthogonal, compound 90.2a adopted a “double-decker” structure, characterized by staggered though largely coplanar PDIs and a tilted central benzene ring. In 90.2b, two PDIs were integrated into the fully fused section of the molecule, to which the remaining fused PDIs were orthogonal. Compound 90.2c had a very rigid x-shaped structure with two orthogonal fully conjugated π-systems.
Scheme 90. Synthesis of Tetrameric Thiophene-Fused PDIs.
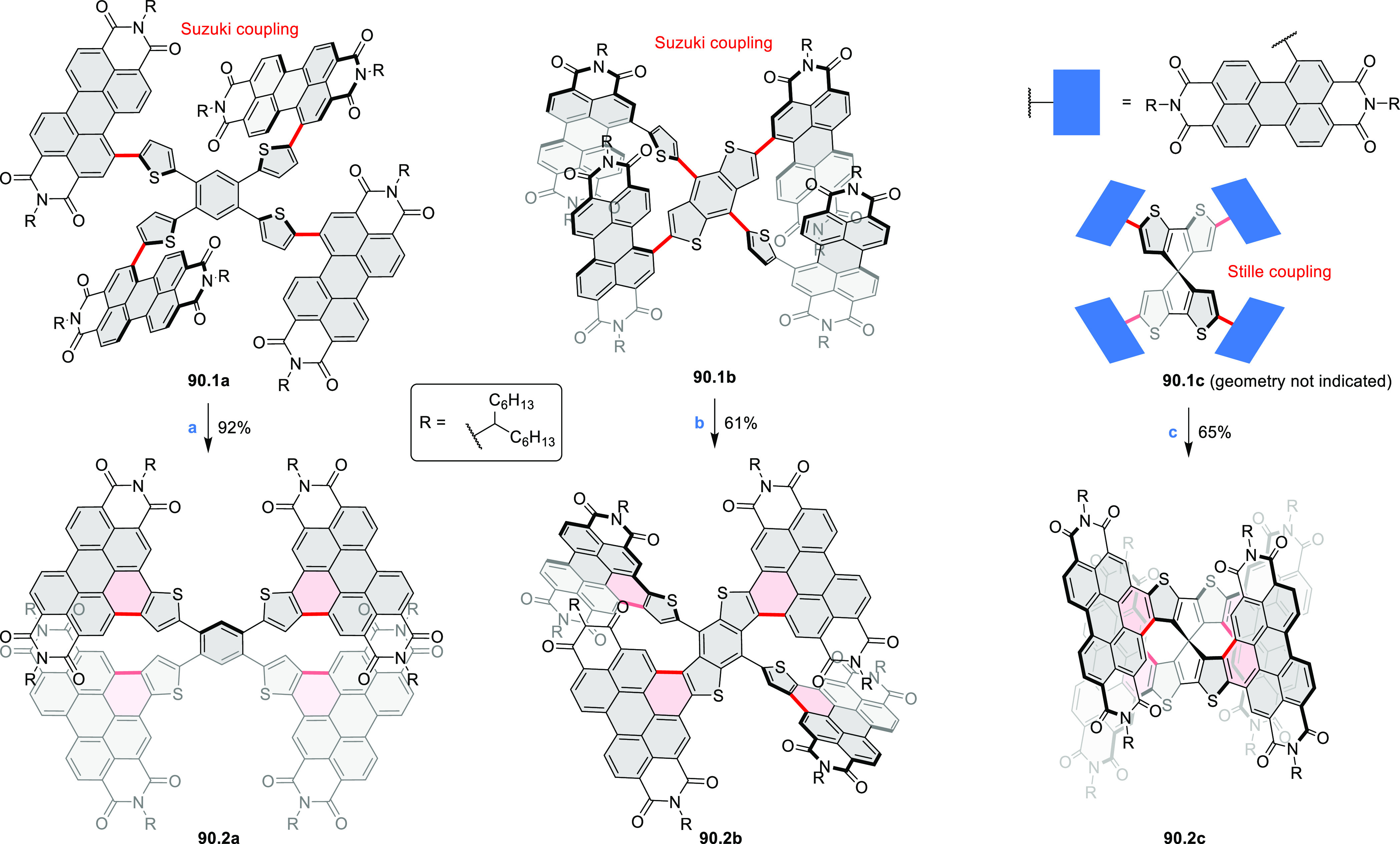
Reagents and conditions: (a)185 FeCl3, 25:1 toluene/MeNO2, rt, 2 h, 92%; (b)186 FeCl3, 2:1 toluene/MeNO2, 0 °C to rt, 5 h, 61%; (c)187 FeCl3, 2:1 toluene/MeNO2, 110 °C, 5 h, 65%.
Compared to 90.1a, its fused analogue 90.2a had higher HOMO and LUMO levels, decreased band gap, and higher extinction coefficients across the entire visible spectrum.185 OSCs containing blends of 90.2a with a P3TEA donor polymer achieved a remarkably high PCE of 10.37% and open-circuit voltage of 1.13 V, significantly improving upon the performance of the PC71BM fullerene-based acceptor (PCE = 7.71%, VOC = 0.90 V). A much lower performance (PCE = 6.67%, VOC = 1.05 V) was also recorded for the nonfused precursor 90.1a. Using the finely tuned donor material P3TAE188 enabled further increase of VOC to 1.20 V, albeit with a lower PCE of 7.83%. Different changes in energy levels were observed between 90.1b and 90.2b, where the latter had a greater band gap.186 This resulted in a shorter wavelength of absorption onset but was compensated by higher absorption in the 300–500 nm range. As in the previous case, the fused compound 90.2b produced more efficient OSCs in blends with the PTB7-Th donor (90.1b: PCE 5.16%, VOC 0.85 V; 90.2b: PCE 7.56%, VOC 0.92 V). In contrast to the other fused tetrameric compounds, 90.2c had a much lower band gap of 1.87 eV, compared to 2.16 eV in 90.2a and 2.46 eV in 90.2b.187 Combined with PTB7-Th in OSCs, 90.2c achieved a PCE up to 8.75%, with a VOC of 0.90 V and a very high short-circuit current (JSC) of 16.6 mA cm–2.
A thiazole-fused perylene diimide was prepared via Stille coupling of the brominated PDI 91.1 followed by UV irradiation of the Stille reaction mixture in the presence of I2 with no prior purification or workup (Scheme 91).189 The product 91.2a was further functionalized through C–H arylation of its thiazole ring under Pd/Cu-catalyzed conditions. A particularly high-yielding cross-coupling occurred with 2-bromothiophene to provide 91.3f. This methodology was extended to the synthesis of bridged dimers, providing the thiophene-bridged 91.4a in 75% yield and thienothiophene-bridged 91.5 with a lower efficiency. The synthesis of analogues with dithienothiophene and 2,2′-bithiophene cores was not successful. Compound 91.2a also underwent oxidative homocoupling in the presence of Ag2O and Cu(I) catalyst to form 91.6 in 90% yield. Additionally, a free radical iodination initiated by KOt-Bu with pentafluoroiodobenzene as the source of iodine was developed.190 This method was used to make 91.3 g, which was elaborated via Stille coupling into the thiophene-bridged dimer 91.4b.
Scheme 91. Synthesis and C–H Arylation of Thiazole-Annulated PDI.
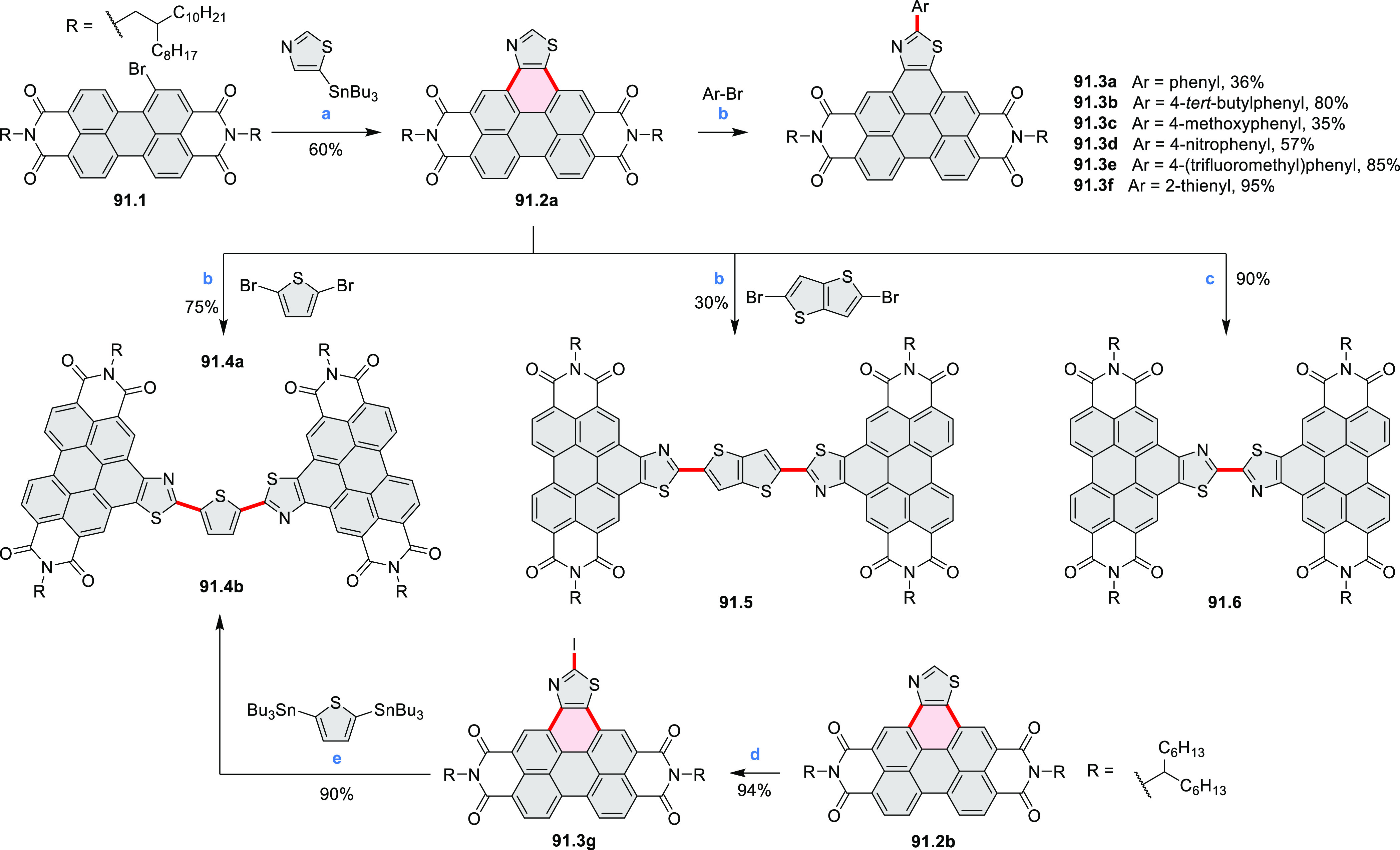
Reagents and conditions: (a)189 Pd(PPh3)4, CuI, 90 °C, 4 h, then I2, hν (254 nm), rt, 3 h; (b) bis(chloro-di-tert-butylphosphine)palladium dichloride, Cu(Xantphos)I, K2CO3, DMF, 135 °C, overnight; (c) Ag2O, Cu(Xantphos)I, K2CO3, DMF, 135 °C, overnight, 90%; (d)190 KOt-Bu, toluene, rt, 30 min, 94%; (e) Pd(PPh3)4, toluene, 110 °C, overnight, 90%.
Photocyclization in the presence of I2 was also used to prepare isatin- and isoindigo-fused PDIs (Scheme 92).191 Exposure to sunlight was used in the synthesis of 92.1, whereas the preparation of 92.2 was effected under blue (450 nm) LED irradiation in a flow reactor. In contrast to the heterocycle-fused compounds in Scheme 89, oxidation with FeCl3 or DDQ was not successful in the case of 92.2. Compound 92.1 was further elaborated into dimeric structures via condensation reactions with the isatin ketone group. The resulting compounds were used to build bottom-gate/bottom-contact OFET devices that displayed on/off current ratios up to 2 × 106.
Scheme 92. Synthesis of Isatin- and Isoindigo-Fused Perylene Diimides.

Reagents and conditions: (a)191 Pd2(dba)3, P(t-Bu)3·HBF4, K3PO4, THF, 80 °C, 12 h; (b) I2, CHCl3, air, hν (sunlight), 25 °C, 24 h; (c) I2, toluene, air, hν (blue LED), 90 °C, 12 h (flow reactor).
3.4. [cd]Heteroannulated Perylenoids
Perylene was selectively derivatized in a 3-position in an SEAr reaction with ethoxycarbonyl isothiocyanate in the presence of triflic acid to provide 93.1 (Scheme 93, see Scheme 142, Section 4.4, for related fused pyrenes).192 Oxidation to thioamide S-oxide 93.2 and subsequent cyclization under acidic conditions provided 93.3 in very high yield (95%). The thiophene-imine-fused perylene 93.3 was strongly emissive in DCM solution, with the emission maximum at 606 nm and a quantum yield of 54%.
Scheme 142. Triflic-Acid-Promoted Cyclization of Thioamide S-Oxides.

Reagents and conditions: (a)192 TfOH, DCM, rt.
Condensation of perylenetetracarboxylic acid anhydride imides with 1,2-diaminoarenes leads to benzimidazole-fused peryleneimides. Yuksel et al. reported the dinitrile derivative 94.2 (Scheme 94, see also Scheme 130, Section 4.2).193 The compound was fluorescent, with emission peaks at 535, 582, and 629 nm in CHCl3. A peryleneimide–tetrathiafulvalene conjugate with a fused benzimidazole linker 94.3 was prepared by Guldi and Liu et al. through condensation in pyridine in the presence of imidazole.194 Compound 94.3 underwent binding to graphene sheets and was used to improve the ultrasonic exfoliation of graphite, providing dispersions with higher optical density and greater stability. Signs of graphene p-doping were present, reflecting the electron-deficient character of peryleneimide. Grozema and Jager et al. reported the condensation between 94.4 and 1,2-diaminobenzene in propionic acid.195 A significantly higher yield of 87% was obtained, implying that Brønsted acid catalysis may be more effective for this reaction. The product was a 2:1 mixture of regioisomers 94.5a,b.
Scheme 94. Synthesis of Benzimidazole-Fused Peryleneimides.
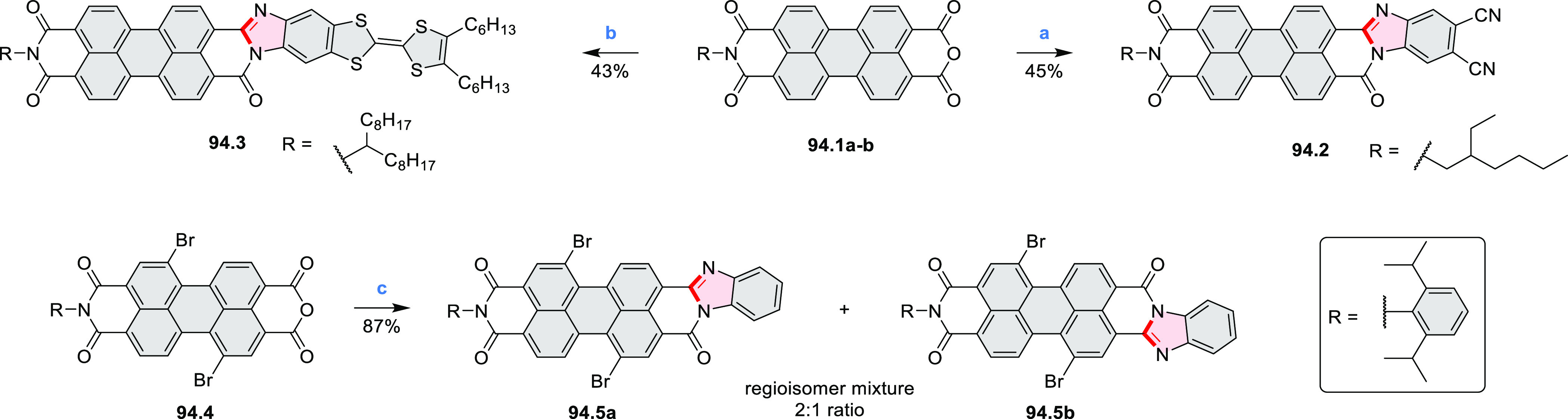
Reagents and conditions: (a)193 4,5-diaminophthalonitrile, Zn(OAc)2, quinoline, 150 °C, 18 h, 45%; (b)194 1,2-diaminoarene, imidazole, pyridine, 130 °C, 23 h, 43%; (c)195o-phenylenediamine, propionic acid, reflux, 3 h, 87%.
Scheme 130. Benzimidazole-Fused π-Systems Derived from Naphthalene Monoimide Monoanhydride.
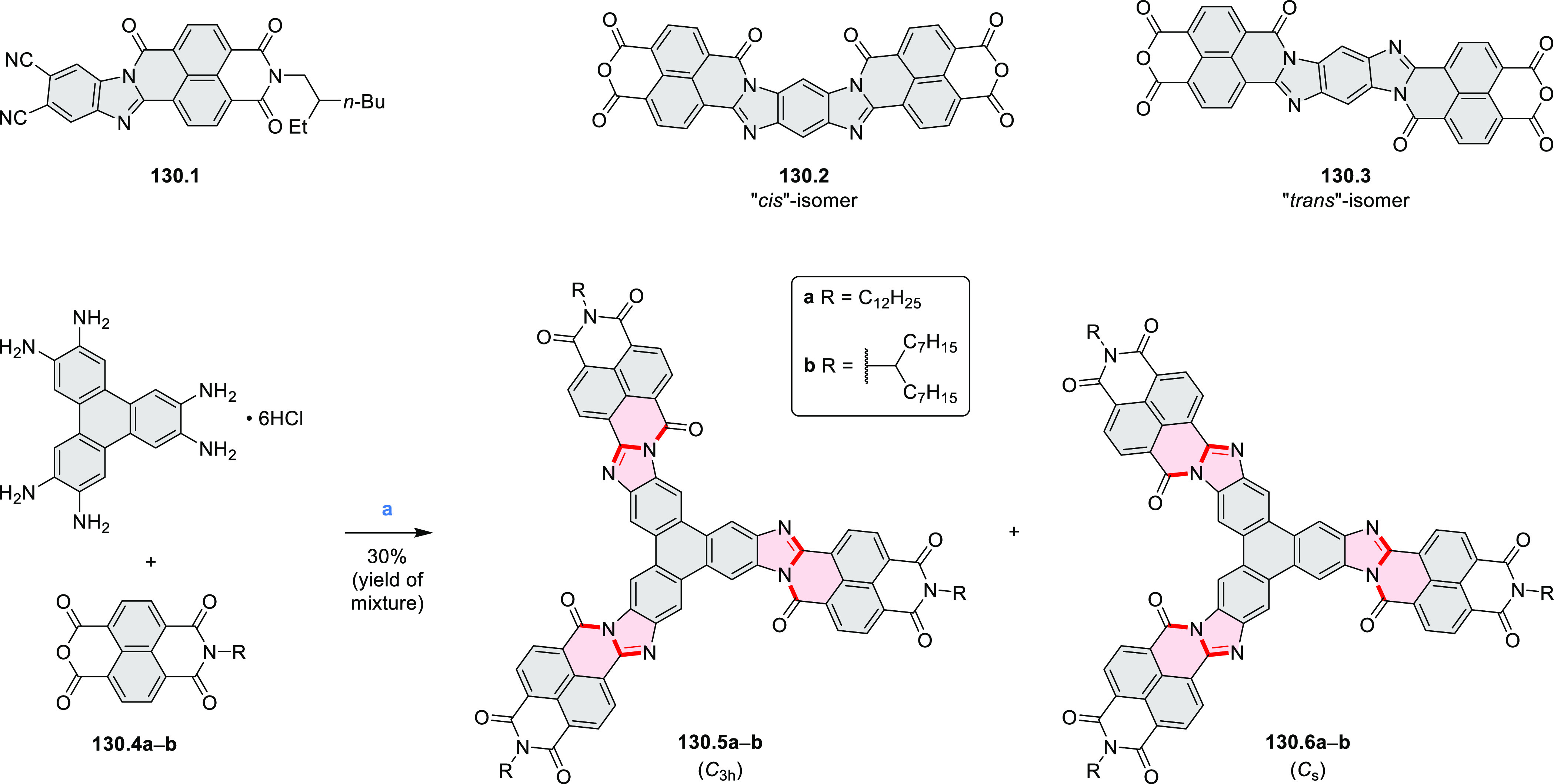
Reagents and conditions: (a)263,264 (1) Et3N, quinoline, rt, 30 min, (2) 130.2a/b, Zn(OAc)2, 180 °C, overnight.
Condensations of 1,2,4,5-benzenetetramine with perylene anhydrides in 1:2 stoichiometry provide access to benzodiimidazole-fused PDI dyads (Scheme 95). This approach was applicable to monoanhydride 95.1 as well as the dianhydride 95.3, with subsequent steps converting the diester function into anhydrides to provide 95.2(196) and anhydrides into imides to produce 95.4a,b.197 Compound 95.2 was prepared along with the naphthalene-based analogue 130.2 (Scheme 130, Section 4.2). In both condensations with benzenetetramine, mixtures of regioisomers were obtained, which were not separated.
Scheme 95. Synthesis of Dimeric Benzimidazole-Fused Perylenes.
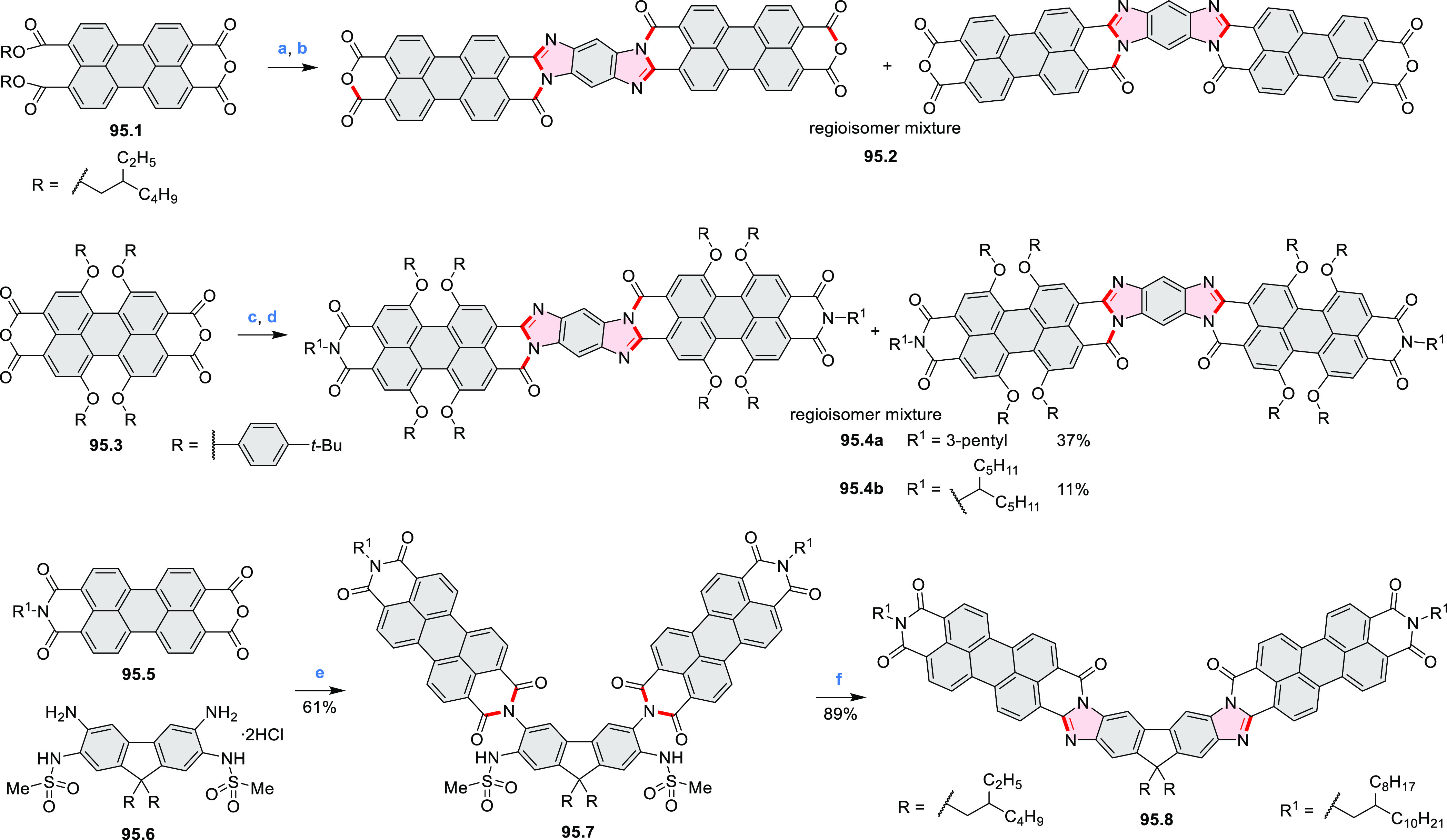
Reagents and conditions: (a)196 1,2,4,5-tetraaminobenzene tetrahydrochloride, pyridine, reflux, overnight, 41%; (b) TsOH·H2O, toluene, 110 °C, 24 h, 95%; (c)197 1,2,4,5-tetraaminobenzene, ZnCl2, quinoline, 170 °C, 18 h, 21%; (d) R1NH2, Zn(OAc)2, imidazole, 140 °C, 2 h; (e)198 Zn(OAc)2, quinoline, 145 °C, 14 h, 61%; (f) 1:1 H2SO4/propionic acid, 150 °C, 5 h, 89%.
A single regioisomer 95.8 could however be prepared via a stepwise condensation as reported by Schönamsgruber and Hirsch.198 The fluorene-2,3,6,7-tetramine derivative 95.6 with the amino groups on C2 and C7 protected as sulfonamides was used as the key starting material. Initial condensation involving the free amino groups on 95.6 formed the diimide 95.7. Further condensation could proceed upon cleavage of sulfonamide groups under acidic conditions, providing 95.8. Aside from enabling regioselectivity, this method had a higher overall yield than direct condensations with 1,2,4,5-benzenetetramine. Compound 95.8 displayed an absorption maximum at 614 nm and a NIR emission peak at 708 nm, strongly red-shifted relative to monomeric perylene diimides.198 A strong NIR two-photon absorption of up to 1110 GM was measured in the 1200–1400 nm range, suggesting an application in optical power-limiting materials. In the case of 95.4a, an absorption peak at 650 nm was observed, which was further red-shifted to 773 nm upon protonation of its basic imidazole rings.197 Its broad NIR emission with maxima at 744 and 915 nm was significantly quenched by protonation.
Condensation of the air-stable tetraamine 96.1 with naphthalene-1,4,5,8-tetracarboxylic acid dianhydride was used to obtain the conjugated polymer 96.2 (Scheme 96).199 The same approach also gave access to an NDI analogue 126.4 (Scheme 126, Section 4.2). The absorption spectrum of 96.2 in DCM solution covers the entire visible region and extends into the NIR, with two intense peaks at 697 and 758 nm and at 711 and 803 nm in the thin film. In a blend with the P3HT donor polymer, 96.2 was used to construct bulk heterojunction photodetectors, showing high detectivity in the NIR range (1.6 × 1010 Jones at 800 nm).
Scheme 96. Polycondensation of 96.1.

Reagents and conditions: (a)199 naphthalene-1,4,5,8-tetracarboxylic acid dianhydride, m-cresol, 5 h at 80 °C then 32 h at 200 °C.
Scheme 126. Benzimidazole-Fused Carbonyl-Containing Pyrenoids.
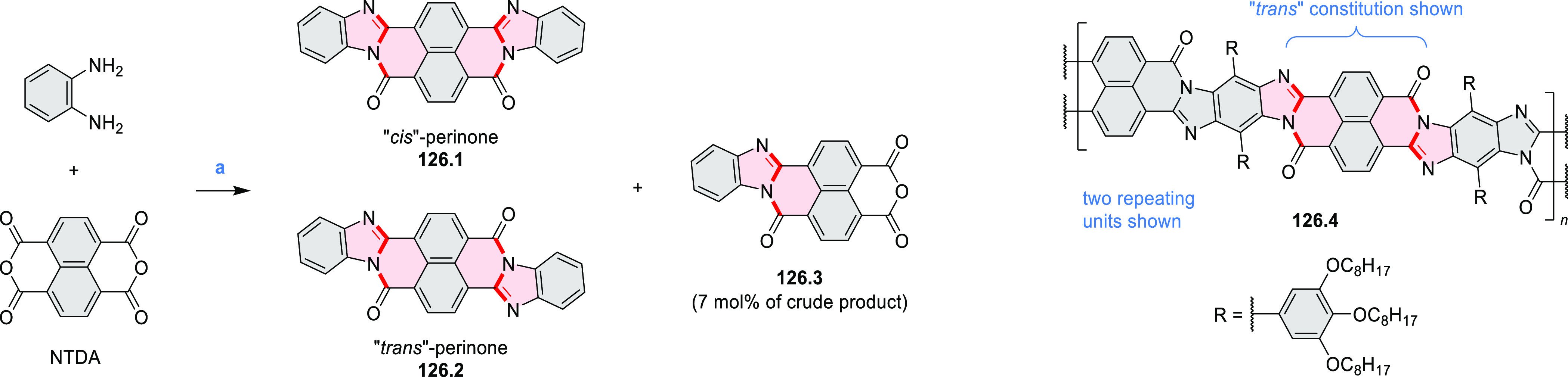
Reagents and conditions: (a)258 H2O, 180–250 °C, autoclave or microwave reactor.
Bonifazi and co-workers reported syntheses of furan- and pyranopyran-fused perylenes from common precursors.200 The Cu-catalyzed aerobic dimerization of perylenol 97.1 provided 97.3, while the cross-coupling of 97.1 with 2-naphthol gave 97.2 along with the homocoupled products 97.3 and binaphthol (Scheme 97). Upon treatment with TsOH, compounds 97.2–3 were condensed into the furan-fused derivatives 97.4a and 97.5a, respectively. It was also possible to cyclize 97.2 and 97.3 into the corresponding pyranopyran-fused derivatives 97.4b and 97.5b. This oxidative etherification was effected in the presence of CuI and pivalic acid in DMSO, according to the methodology previously developed in the group (see Scheme 50).82 The furan-fused compounds had nonplanar structures with interplanar angles close to 17°, while the pyranopyran-fused analogues were planar. All compounds 97.1–5 were strongly emissive, with quantum yields of 50–88%. The wavelengths of absorption and emission peaks increased on going from the nonfused precursors 97.2–3, through furan-fused, to pyran-fused compounds, a change attributed mainly to higher HOMO energies caused by O-annulation. Additionally, spectral features of biperylenol-derived systems were red-shifted relative to the respective perylenol–naphthol analogues. Tuning of the emission wavelength in the range between 463 nm in 97.1 and 649 nm in 97.5b was thus demonstrated.
Scheme 97. Synthesis of Thiophene-Fused Perylenoids Through Dehydrogenative Photocyclizations.
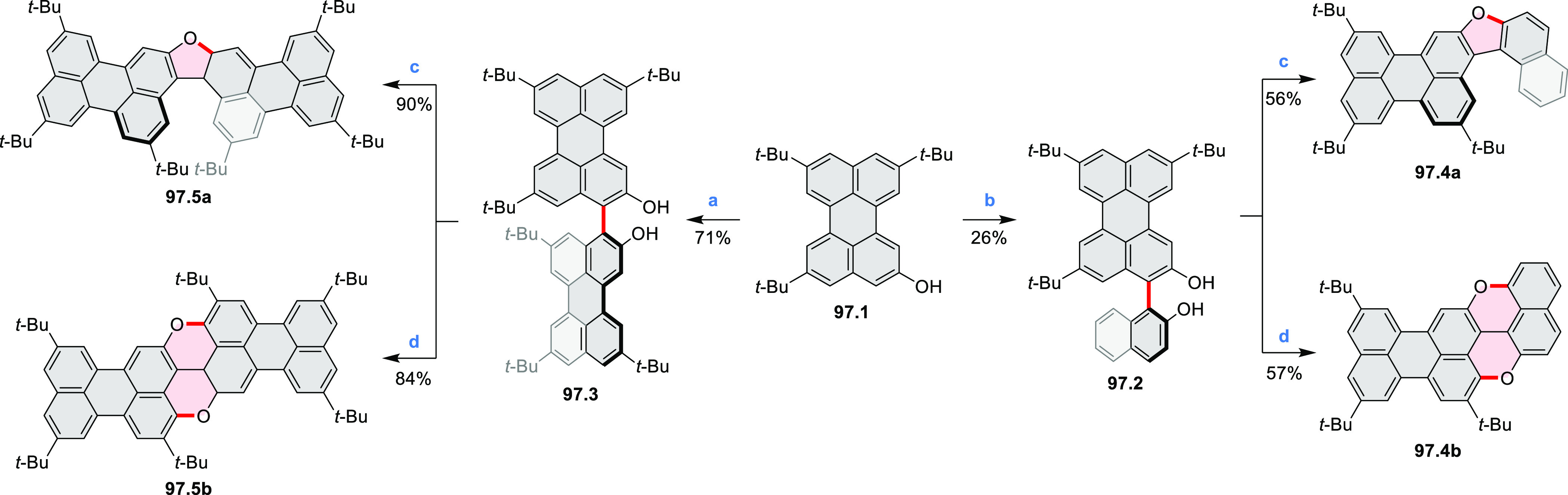
Reagents and conditions: (a) [Cu(OH)(Cl)(TMEDA)], DCM, air, rt, 1 h, 71%; (b) 2-naphthol, [Cu(OH)(Cl)(TMEDA)], DCM, air, rt, 2 h, 26%; (c) TsOH, toluene, reflux, 4 h; (d) CuI, PivOH, DMSO, 140 °C, 2 h.200
Peryleneimides linked to a thiophene or terthiophene through a fused pyrazine ring were prepared in a condensation reaction from acenaphthene-derived diketones 98.2a,b (Scheme 98).201 The terthiophene-containing 98.3c displayed panchromatic absorption with a broad low-energy band peaking at 668 nm and extending up to ca. 900 nm. This band, which was absent in 98.3a,b, was assigned to an intramolecular charge transfer absorption. Compounds 98.3a–c were studied as n-type semiconductors for OFET devices, with 98.3a showing an electron mobility (μe) of up to 1.5 × 10–4 cm2 V–1 s–1.
Scheme 98. Synthesis of Pyridazine-Bridged Peryleneimide-thiophene Assemblies.

Reagents and conditions: (a)201 AlCl3, chlorobenzene, reflux, 6 h, 80% from 98.1a or 20% from 98.1b; (b) benzeneseleninic anhydride, chlorobenzene, 130 °C, overnight, 78–95%; (c) TsOH, CHCl3, 50 °C, overnight.
In 2020, Ingleson and Zysman-Colman et al. reported a bromoboration–electrophilic borylation sequence for the preparation of the boron-containing perylenoids 99.2–3 (Scheme 99).202 For instance, 3-(p-tolylethynyl)perylene (99.1) was treated with BBr3 and 2,4,6-tri-tert-butylpyridine to bring about the 1,1-bromoboration of the alkyne unit, followed by borylative cyclization with C4 of the perylene core. When the resulting compound 99.2a was stirred in untreated DCM under air, the B–Br bond was hydrolyzed to give 99.2b in 88% yield. Alternatively, 99.2a was reacted with 2,4,6-triisopropylphenylmagnesium bromide (TipMgBr) to give 99.2c in 50% yield. Similarly, the doubly peri-fused perylenoids 99.3 were obtained from the corresponding 3,9-dialkynyl-substituted perylene precursor.
Scheme 99. peri-Fused Perylenoids Bearing One or Two Boron Atoms.

Reagents and conditions: (a)202 BBr3, 2,4,6-tri-tert-butylpyridine, o-dichlorobenzene, heptane, rt, 24 h; (b) DCM, air, 1 h, then aq. HCl; (c) TipMgBr, o-dichlorobenzene, 48 h.
3.5. ortho-Heteroannulated Perylenoids
Mastalerz et al. reported a base-promoted double cyclization of dihydroanthracenes bearing o-formylaryl substituents to yield the corresponding doubly fused perylenes, including the pyridine- and thiophene-fused examples 100.2a,b (Scheme 100).203 Several nonheterocyclic analogues were reported along with the compounds shown here, with yields of 45–79%. The transformation was accomplished by treatment of 100.1a,b with KOt-Bu in THF solution, giving 100.2a,b in satisfactory yields. The products were soluble in organic solvents despite the large and planar π-surfaces. Compound 100.2a was strongly luminescent (λem = 446 nm, Φ = 67%), while 100.2b had a weaker and red-shifted emission (λem = 477 nm, Φ = 24%).
Related thieno-fused perylene structures were accessed via a procedure consisting of a Corey–Chaykovsky reaction followed by dehydrative cycloaromatization that was developed by Kakiuchi et al. (Scheme 100).204 The reaction of anthraquinone 100.3 with trimethylsulfonium iodide and sodium hydride at 50 °C gave the corresponding diepoxide 100.4. The use of SnCl2 as a catalyst for the dehydrative cycloaromatization was found to be successful, and this two-step protocol gave the desired dithiophene-fused perylene 100.5 in 19% yield. The synthesis of thiophene-containing pyrenoids 100.8a,b was performed in a similar fashion.
A perylene extended through fusion with four benzothiophenes was prepared via an oxidative cyclization of the precursor 101.1 bearing o-(methylmercapto)phenyl groups (Scheme 101).205 Based on a previously known methodology, the reaction was initiated by oxidation of thioethers to sulfoxides upon treatment with I2. The sulfoxide underwent an acid-mediated cyclization and condensation to form a methylsulfonium intermediate. Finally, this species was demethylated by a base (AcO– or I–) to complete the process. Propylene oxide was added to the reaction medium to scavenge excess HI formed during thioether oxidation, thus preventing the cleavage of butyl ethers. Under optimized conditions, the 4-fold cyclization to 101.2 proceeded in a high yield of 89%. Compound 101.2 was strongly twisted, with interplanar angles of 46.7° and 57.0° between its terminal benzene rings observed in the solid-state structure. A green emission at 504 nm with a quantum yield of 68% was seen in DCM solution of 101.2. Enantiomers of this compound were separated using chiral HPLC, leading to the observation of CPL with a dissymmetry factor of 1.09 × 10–3 in the optically pure samples.
Scheme 101. Synthesis of a Benzothiophene-Fused Perylene via Oxidative Cyclization.

Reagents and conditions: (a)205 I2, 4:3:4 CHCl3/AcOH/propylene oxide, 55 °C, 12 h, 89%.
The synthesis of π-extended dithia[6]helicene 102.2 reported by Itami et al.206 demonstrates the utility of ortho-phenylene-linked precursors for the synthesis of sterically encumbered nanographenes (Scheme 102). When the quadruply substituted naphthalene 102.1 was treated with an excess of MoCl5 (20 equiv), the tetrachlorinated 102.2a was produced in 26% yield. Lowering the proportion of the oxidant (12 to 16 equiv) did not afford well-defined products. It was assumed that chlorination took place immediately after cyclization, and the introduced chlorine atoms served to cap the reactive sites of intermediate 102.2, corresponding to the most nucleophilic positions of perylene. A subsequent palladium-catalyzed dechlorination of 102.2 quantitatively furnished the halogen-free product 102.2b. A single-crystal X-ray diffraction analysis revealed that the configuration of the double helicene corresponds to the pair of enantiomers (P,P)-102.2b and (M,M)-102.2b. The meso isomers, (P,M)-102.2a or (P,M)-102.2b, were not detected. It was shown in subsequent work that the formation of the more extensively fused product 102.3a was possible under somewhat more forced conditions.207 The additional formation of a seven-membered ring in 102.3a occurred when the reaction mixture was additionally purged with O2 during MoCl5 oxidation.
Scheme 102. Synthesis of π-Extended S-Doped Double [6]Helicenes.

Reagents and conditions: (a)206 MoCl5 (20 equiv), DCM (5 mM), rt, 40 min; (b) Pd/C, HCO2H, Et3N, pyridine, 130 °C, 25–72 h; (c)207 MoCl5, O2-purged dry DCM, rt, 27 h.
Perylene diimides [a]-fused with thiophenes, selenophenes, and pyrroles were prepared using an ethynylarene heteroannulation approach (Scheme 103).208 The phenylethynyl-substituted PDI 103.1 was converted into the thiophene-fused 103.2a via treatment with elemental sulfur at 140 °C, based on an earlier methodology.209 Alternatively, K2S, Na2S·9H2O, or KSAc were used as the source of sulfur to decrease the reaction temperature to 80 °C while retaining yields above 50%. Similarly, heating 103.1 with Se in DMA provided the selenophene-fused product 103.2b, and treatment with primary amines in the presence of a base resulted in the pyrrole-fused 103.2c,d. All annulations are thought to proceed via nucleophilic attack on the distal carbon atom of the alkyne, assisted by its conjugation with the electron-deficient PDI core. Analogous conditions were also used for the cyclization of tetraalkynes 103.3a,b. While the synthesis of 103.4a was efficient using elemental sulfur, the n-butyl-substituted analogue 103.4b could only be obtained upon treatment with K2S at a lower temperature (80 °C). A relatively low reaction temperature of 90 °C was also employed when using sulfur in 10:1 DMF/H2O medium, although low yields were observed for the TIPS-functionalized quadruply cyclized products 103.5a,b.210 These derivatives were then desilylated to afford 103.6a,b. These fused PDIs were determined by X-ray crystallography to be strongly twisted, with an ca. 53° interplanar angle between the blades of the [5]helicene substructure. Despite the large twist angle, enantiomers of 103.6a were observed to interconvert on the time scale of minutes at rt.
The same method was used for the cyclization of PDIs 104.1a,b bearing two alkynyl substituents in the bay positions (Scheme 104).211 The starting materials were used as mixtures of 1,6- and 1,7-disubstituted regioisomers derived from mixed 1,6- and 1,7-dibromo PDIs. Isomeric fused products 104.2–3 were thus obtained in modest yield and separated by chromatography. These were further processed by desilylation to 104.4–5. Finally, dimers 104.6a–7a were prepared via double α-bromination of 104.4–5 at the fused thiophene moieties, dimerization through Ullmann coupling, and dehalogenation to remove unreacted thienyl bromides. In the solid-state structures of both 104.4b and 104.5b, slightly twisted geometries were observed, with interplanar angles of 23–26°. Dimers 104.6a–7a had narrower band gaps and red-shifted absorption spectra, with the low-energy π–π* absorption at 705 and 708 nm, respectively, in contrast to those of the monomeric 104.4a–5a (respectively, 638 and 654 nm).
Treating ortho-(arylethynyl)-substituted PDIs with K2S in DMF at even higher temperatures (140–170 °C) than shown above in Scheme 103 led to a stitching thienannulation, providing the thienothiophene-fused structures 105.1a–c (Scheme 105),212 rather than products of a single annulation such as 103.2a. The same reaction proceeded at a lower temperature for a more electrophilic substrate (alkyne-bridged PDI–NDI conjugate), providing 105.1d. This process is thought to begin with a nucleophilic addition of S3•– to alkynes. The formation of S3•– upon addition of K2S to DMF was confirmed by its characteristic absorption at 617 nm. The bay-phenylethynyl substrate 105.2 led instead to the thienobenzothiopyran 105.3, apparently because of the more electrophilic nature of PDI compared to a phenyl group. Triple thienannulations of butadiyne-bridged substrates were also demonstrated, providing the dithienothiophene-fused 105.5a,b. Lateral thiophene rings in these products readily formed at ambient or slightly elevated temperatures, while closure of the central ring required harsher conditions. The 2-thieno-3-mercaptothiophene intermediates were isolated by running the reaction at 25–40 °C. Apart from the perylenoid products shown here, several NDI-derived analogues were reported, demonstrating the versatility of this methodology for electron-deficient aromatics.
A perylene derivative 106.3 fused with two benzothiophene motifs was prepared via dehydrogenative cyclization of 106.2 upon irradiation in the presence of I2 (Scheme 106).213 The diester functions of 106.3 were then converted into imides via treatment with primary amines and imidazoles in refluxing o-dichlorobenzene. These compounds were able to form liquid crystalline mesophases thanks to their tendency to self-assemble. The use of doubly branched alkyl chains in 106.4b led to a much wider temperature window for the mesophase. A cyclodehydrochlorination reaction upon irradiation under basic conditions was used to prepare 106.6 by forming four C–C bonds in a single step.214 Several nonheterocyclic PAHs as well as 2-azatriphenylene were prepared by the same method, achieving higher overall yields and yields per cyclization than in the case of 106.6. The relatively low efficiency with a thiophene-containing substrate was attributed to a greater likelihood of rearrangement reactions.
Scheme 106. Synthesis of Thiophene-Fused Perylenoids Through Dehydrogenative Photocyclizations.
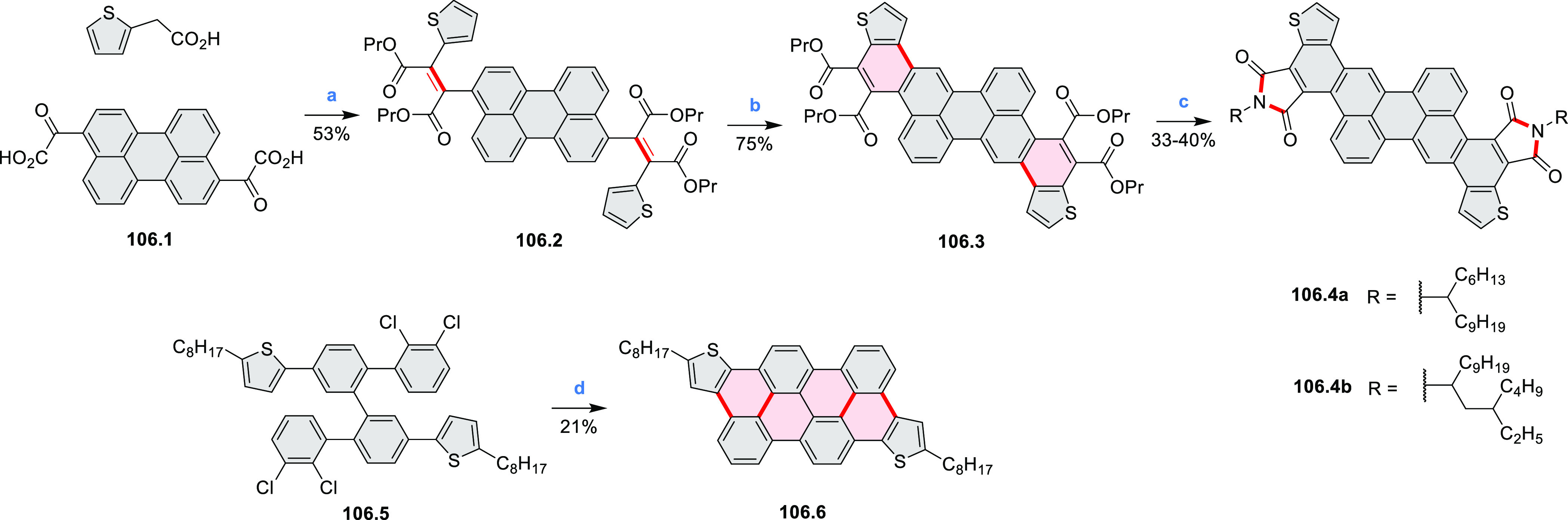
Reagents and conditions: (a)213 Ac2O, Et3N, THF, reflux, 16 h, then 1-bromopropane, DBU, 1:10 PrOH/THF, reflux, 10 h, 53%; (b) I2, 1:13 dioxane/toluene, hν (incandescent lamp), reflux, 6 days, 75%; (c) amine hydrochloride, imidazole, o-dichlorobenzene, reflux, 16 h, 33–40%; (d)214 Na2CO3, 77:1 acetone/H2O, rt, hν (medium pressure mercury lamp), 5 h, 21%.
PAHs with a picene substructure, including the perylenoid 107.4, were prepared via an oxidative coupling cyclization (Scheme 107).215 Sterically congested precursors 107.3 and 107.6 were prepared in a rhodium-catalyzed double annulation reaction from the corresponding dialkynes (107.2 and 107.5) and the biphenyl boronic acid 107.1. The subsequent oxidation of 107.3 with FeCl3 as an oxidant provided the quadruply cyclized, C2-symmetrical product 107.4 in 90% yield. Compound 107.6 upon treatment with DDQ and MsOH led to the triply cyclized, desymmetrized product 107.7 in 49% yield, in which one five-membered ring was formed. The other side of the molecule did not cyclize in the same manner due to steric repulsion. Crystal structures of 107.4 and 107.7 revealed a twisted geometry in both products, with 37–39° interplanar angles between the thiophene-containing blades.
Scheme 107. Synthesis of Thiophene-Fused Perylenoids.
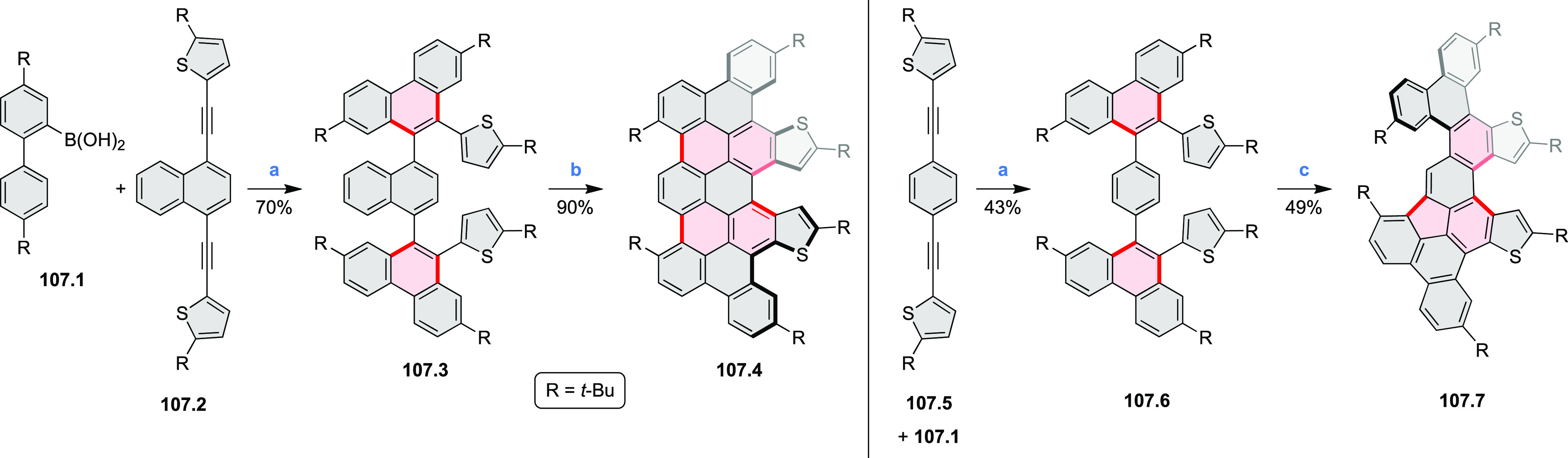
R = t-Bu. Reagents and conditions: (a)215 [Rh(Cp*)Cl2]2, AgOAc, DMF, 100 °C, 16 h, 70%; (b) FeCl3, 1:2 MeNO2/DCM, rt, 1 h, 90%; (c) MeSO3H, DDQ, DCM, 0 °C, 2 h, 49%.
A similar cyclodehydrogenation was used to prepare thiophene-fused nanographenes 108.3a,b that contained multiple instances of [4]- and [5]helicene substructures (Scheme 108). The bi(thienophenanthryl) precursors were assembled in two steps, starting with a Pt-catalyzed cyclization of the diyne 108.1 to provide 108.2a. This compound was then converted into the cyclodehydrogenation precursor 108.2b by dehydrogenation and into 108c,d by Suzuki coupling. The final cyclodehydrogenation was conducted using the DDQ oxidant in the presence of TfOH, giving 108.3a–c in moderate to high yields, which corresponded to 90–94% yield per cyclization.
Scheme 108. Cyclodehydrogenation toward Thiophene-Fused Nanographenes.
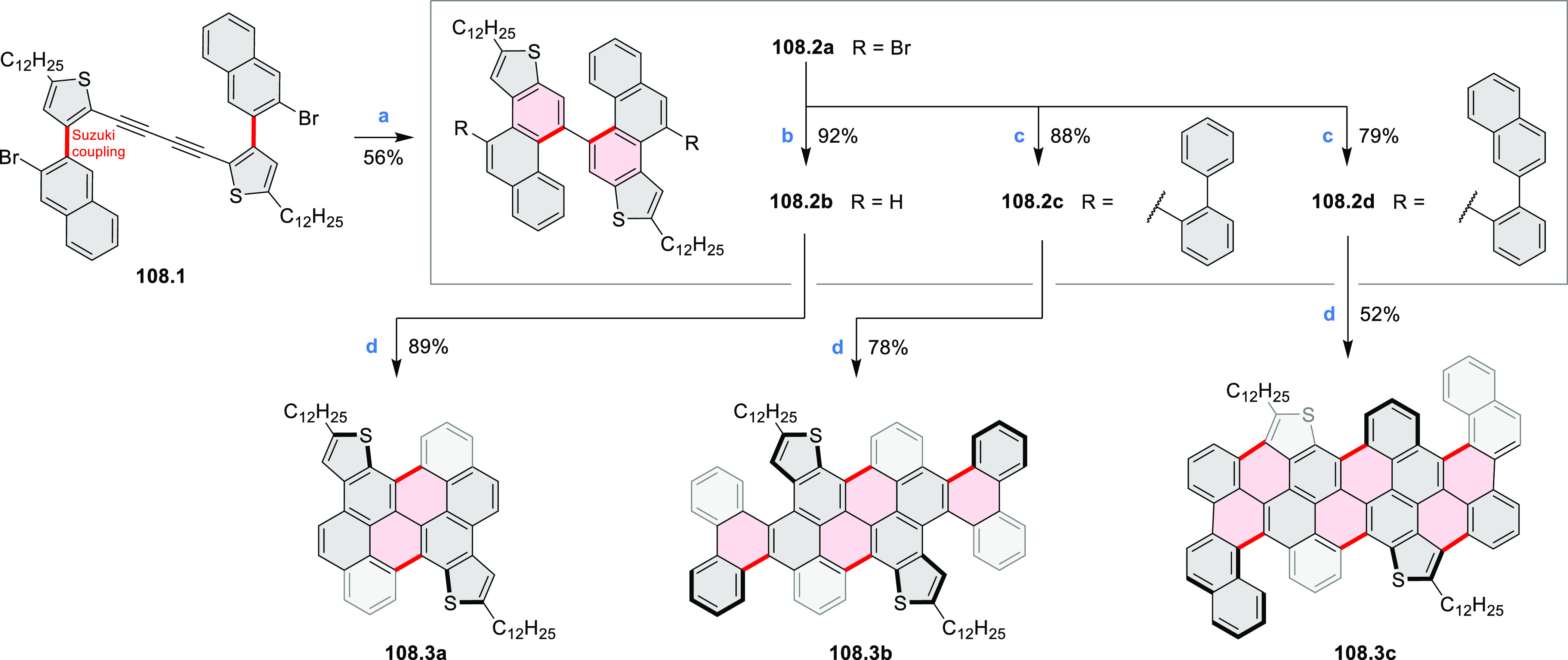
Reagents and conditions: (a) PtCl2, toluene, 100 °C, 20 h; (b) n-BuLi, THF, −78 °C, 1 h; (c) RB(pin), Pd(PPh3)4, K2CO3, toluene/EtOH/H2O, 90–100 °C, 24 h; (d) DDQ, TfOH, DCM, −35 °C for 108.3a or 0 °C for 108.3b,c, 30–90 min.
Dihedral angles in 108.3a–c were estimated by DFT calculations to be 15–19° in thieno[4]helicenes, 24.1° in the carbo[4]helicenes of 108.3b, and 33–34° in thieno- and carbo[5]helicenes of 108.3c. All [4]helicene substructures readily underwent inversion at rt, while the helicity of [5]thienohelicenes in 108.3b was reported to be configurationally stable. Greater π-extension corresponded to decreased HOMO–LUMO gaps and red-shifted spectral features (108.3a: λmax = 460 nm, Φ = 4.5%; 108.3b: λmax = 505 nm, Φ = 13.8%; 108.3c: λmax = 569 nm, Φ = 3.8%).
Reaction of perylene diimides with DBU was reported to produce fused products 109.2a,b, containing a 5–6–7 ring system (Scheme 109).216 This transformation was proposed to proceed through a nucleophilic attack of the imine nitrogen of DBU on the ortho position of the PDI followed by dehydrogenation. Another nucleophilic addition of the enediamine carbon at the bay position with further loss of hydrogen was proposed to complete the process. Subjecting the bay-alkynyl-substituted PDI 109.3 to similar reaction conditions provided the pyrrole-annulated 109.5 in 14% yield.217 The DBU-derived caprolactam ring remained appended to the pyrrolic N atom of 109.5. The benzannulated PDI 109.4 was isolated alongside 109.5.
Scheme 109. Reactions of PDIs and bay-Alkynyl PDI with DBU.
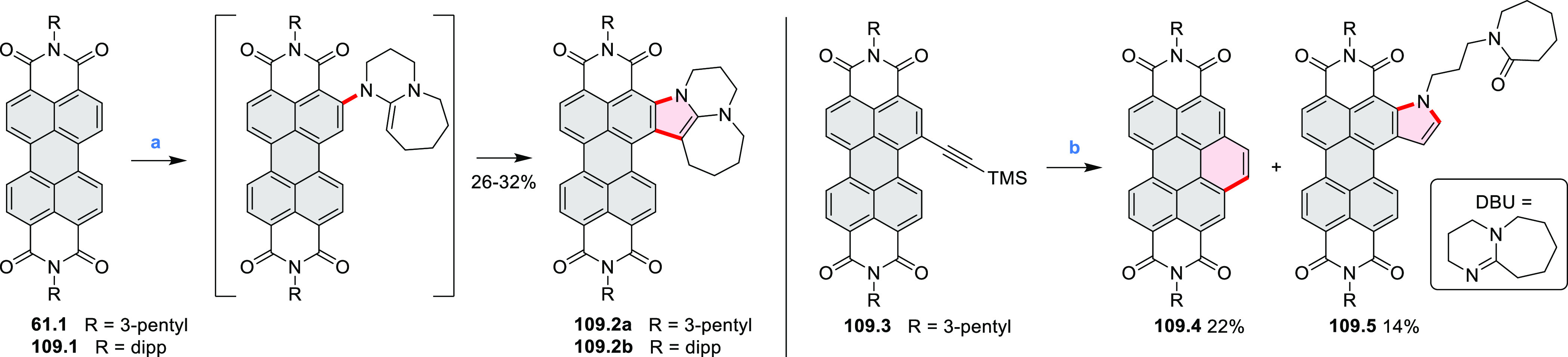
Reagents and conditions: (a)216 DBU, toluene, 140 °C, 24 h, 26–32%; (b)217 DBU, toluene, 120 °C, overnight.
Compound 109.2a had a broad absorption band extending into the NIR region (580–950 nm), which was thought to include π–π* as well as charge transfer transitions.216 Stabilization of the charge-transfer excited state in polar solvents caused a bathochromic shift of this absorption band (ca. 70 nm difference between toluene and DMSO). Quenching of this low-energy absorption and strong enhancement of the emission peaks at 532 and 569 nm occurred upon protonation. In 109.5, the electron-donating character of the fused pyrrole caused a strong red-shift of the PDI absorption features to a 625 nm maximum.217 A red emission (λem = 654 nm, Φ = 25%) was also observed. Overall, pyrrole fusion of PDIs provides a way of achieving panchromatic absorption.
Imidazole-fused PDIs were accessed via the reaction of 109.1 with NaNH2 and benzonitriles which were used as both reagent and solvent (Scheme 110).218 Using unsubstituted benzonitrile provided a mixture of single and double annulation products 110.1a and 110.2a, whereas with chlorinated benzonitriles only the singly annulated 110.1b,c were obtained. In analogy to the pyrrole-annulated systems (Scheme 109), PDIs extended with imidazole rings had strongly red-shifted spectral features. Further bathochromic shifts occurred upon deprotonation of the imidazoles with a strong base (tetraoctylammonium hydroxide) in THF solution. Unlike the weakly emissive pyrrole-fused PDIs, 110.1a–c displayed luminescence quantum yields of 98–99% (λem = 584–597 nm). 110.2a had a slightly weaker deep red emission (λem = 584–597 nm, Φ = 85%). Imidazolide anions had weak emission peaks red-shifted into the NIR region, with the longest wavelength observed for the dianion of 110.2a (λem = 802 nm, Φ = 5%). These properties were used to demonstrate colorimetric and fluorescent sensing of pH and CO2.
Scheme 110. Synthesis of Imidazole-Fused PDIs.

Reagents and conditions: (a)218 NaNH2, respective benzonitrile used as solvent, 150–170 °C, aeration, 0.5–3 h.
Double helicenes 111.4–5 were obtained via fusion of four indole or benzothiophene moieties to a PDI core (Scheme 111).219 First, the tetraiodo PDI 111.1 (accessible from unsubstituted PDI)220 was functionalized with o-bromoaniline via an SNAr reaction or with o-aminothiophenol via Cu-catalyzed cross-coupling to provide 111.2–3. Compound 111.2 was then cyclized using Pd-catalyzed intramolecular C–H activation, while for 111.3 the cyclization was effected by diazotization and subsequent reduction with hydroquinone. The products 111.4–5 had a high barrier to epimerization (ca. 65 kcal mol−1 as determined by DFT) and were separated into their P and M enantiomers by chiral HPLC.
Scheme 111. Synthesis of Tetraindole- and Tetra(benzothiophene)-Fused PDIs.
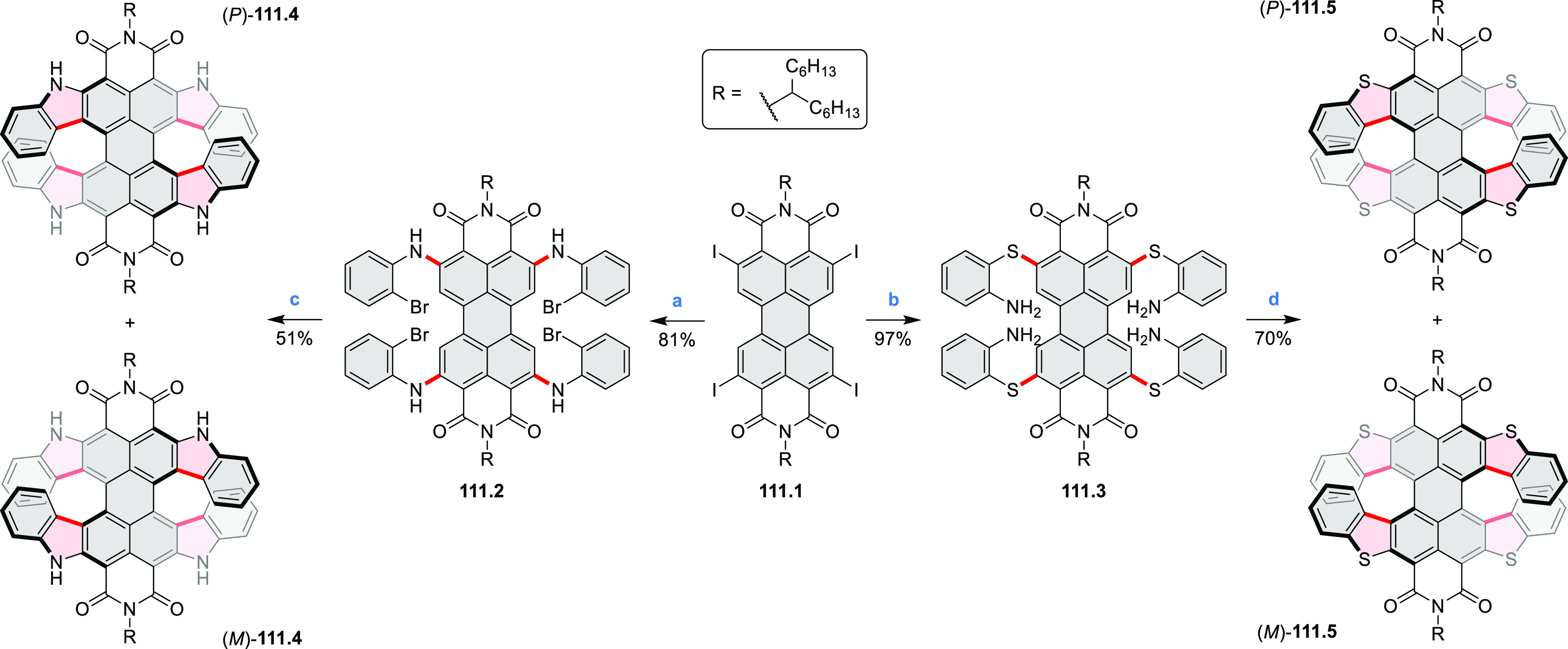
Reagents and conditions: (a)219o-bromoaniline, KOt-Bu, neat, 120 °C, 24 h, 81%; (b) o-aminothiophenol, CuI, l-proline, Cs2CO3, CTAB, 3:1 toluene/H2O, reflux, overnight, 97%; (c) Pd(OAc)2, P(t-Bu)3·HBF4, Cs2CO3, DMF, 150 °C, 24 h, 51%; (d) isopentyl nitrite, 1:1 DCM/AcOH, 0 °C, 1 h, then hydroquinone, rt, 30 min, 70%.
Absorption features of 111.4–5 were heavily red-shifted relative to the parent PDIs, with the lowest-energy absorption band at 737 nm in 111.4. Notably, the CD features of pure enantiomers extended into the NIR region. Consequently, OFET devices prepared using (P)-111.4 and (M)-111.4 displayed differences in photocurrent when illuminated by left- or right-handed circularly polarized light at 635 or 730 nm.
Another example of a double [7]heterohelicene is 112.2 with four benzofurans fused to a dibenzoperylene core (Scheme 112).221 This compound was prepared in 34% yield from the tetrakis(benzofuranyl)terphenyl 112.1 upon cyclodehydrogenation with DDQ in the presence of TfOH. The product 112.2 had ca. 30° interplanar angles between helicene blades in the solid state. It was configurationally stable, with a 50.5 kcal mol−1 barrier to interconversion estimated by DFT calculations, and was separated into (P,P) and (M,M) enantiomers. Compound 112.2 had a lowest-energy absorption band at 488 nm and a bright emission (λmax = 511 nm, Φ = 71%) in DCM solution. Upon treatment with AgBF4, it was reversibly converted into a radical cation, which displayed panchromatic absorption with a broad NIR band at 770–1200 nm.
Scheme 112. Synthesis of a Double Tetraoxa[7]helicene.

Reagents and conditions: (a)221 DDQ, TfOH, DCM, 0 °C, 2 h.
4. Pyrenoids
4.1. Triangulenes
Heteroatom-doped triangulenes can be conveniently classified as either heteroatom- or carbon-centered (Sections 4.1.1 and 4.1.2, respectively), depending on the element located at the center of the fused framework. Other heteroatoms are usually located at some of the three central positions along the zigzag edges of the triangulene parent. In the absence of additional fusion, pyrene is the largest leading substructure present in the triangulene core. However, several peri-fused triangulenes have been discussed in Sections 2 and 3, e.g., 4.3a,b, 12.4–6, 19.10, 20.4, 27.3, 32.12, and 84.2e.
4.1.1. Heteroatom-Centered Heteratriangulenes
In 2020, Hamzehpoor and Perepichka reported a series of emissive azatriangulenetrione derivatives 113.2a–e (Scheme 113).222 Compounds 113.2a–e were obtainable from the 3-fold intramolecular Friedel–Crafts acylation of the corresponding triester precursors 113.1a–e. In the solid state, the unsubstituted azatriangulenetrione 113.2a exhibits phosphorescence at 538 nm with a lifetime of 28.6 ms and a high photoluminescence quantum yield (PLQY) of 42%. The solids of the trihalo-substituted congeners 113.2b–d also show phosphorescence within 560–589 nm, albeit with shorter lifetimes and lower PLQYs. This trend observed from the four emitters was ascribed to the heavy atom effect. In stark contrast, the tri-t-butyl-substituted derivative 113.2e fluoresces at 495 nm with a lifetime of 10.0 ns with PLQY of 11% in the solid state. The single-crystal structures of all five compounds indicate the formation of π-stacks. Contrary to the coaligned, stacking molecules observed for 113.2a–d, the molecules of 113.2e are rotated by 60° relative to the neighbors along the stack to accommodate the bulk of t-butyl groups. This work demonstrated that the solid-state emission properties could be tuned by the crystal packing structures, which in turn could be controlled by the substituents on the π-skeleton.
Scheme 113. Azatriangulenetriones from Triarylamine Precursors.

Reagents and conditions: (a)222 (1) KOH, MeOH/H2O, (2) SOCl2, DMF, then SnCl4, DCM.
Enantiomeric azatriangulenetriones C11.1a,b, reported in 2016 by Meijer and Schmidt et al., possess trialkoxy-substituted benzamide side chains, with each alkoxy group in either the R or S configuration (Chart 11).223 The self-assembly process of each enantiopure sample dissolved in o-dichlorobenzene was monitored by CD spectroscopy at an initial temperature of 80 °C, and two different aggregation states could be obtained upon cooling. The kinetically trapped state A was achieved by direct cooling to 7 °C, whereas the thermodynamic state B was reached by undercooling to −5 °C and then returning to 7 °C. The two states were stable for hours. States A and B were proposed to arise via isodesmic and nucleation–elongation processes, respectively. Notably, mixing the aggregates A and B of the same enantiomer resulted in the complete conversion from A into B, as revealed by CD and UV–vis spectroscopy. On the other hand, when C11.1a in state A was mixed with C11.1b in state B, the CD signal stayed midway between those of the parent solutions, indicating the absence of state conversion. This work demonstrated the enantioselective nature of the self-assembling processes for the reported molecules.
Chart 11. Benzamide-Functionalized Azatriangulenetrione Derivatives.
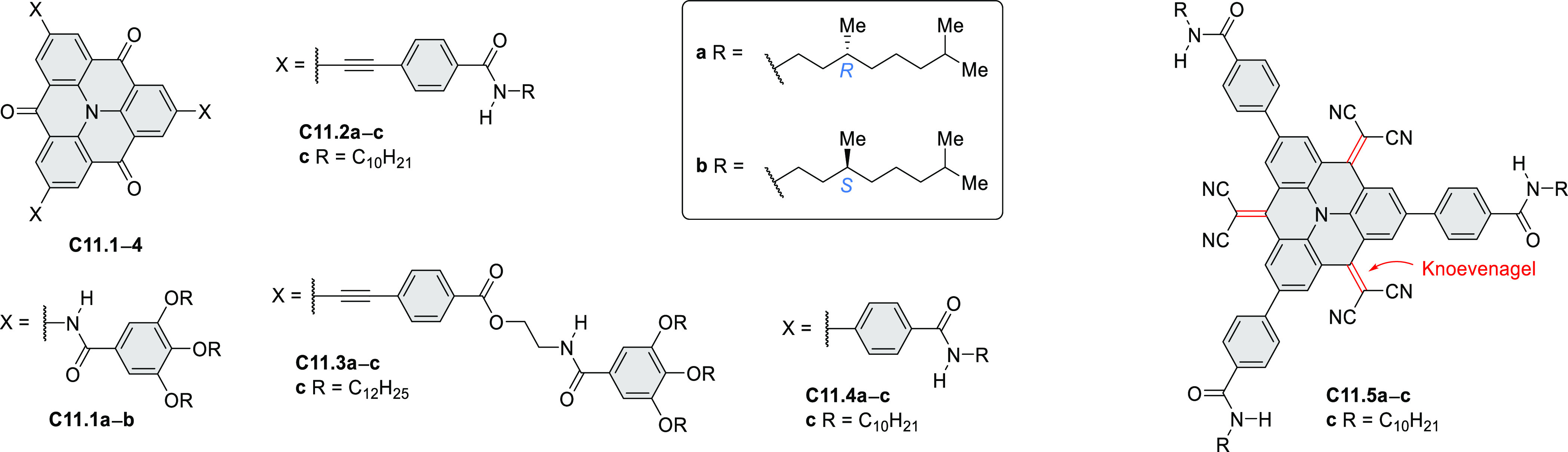
Chiral and achiral trisubstituted azatriangulenetriones prepared in subsequent years unveiled the pathway complexity of their supramolecular assembling processes. The trialkynyl derivatives C11.2a–c224 and C11.3a–c225 were synthesized by Sánchez and Casado et al., while the triaryl derivatives C11.4a–c were explored by Sánchez and Ortí et al.226 On the basis of CD spectroscopic evidence, all these azatriangulenetriones were proposed to form self-assembled helical stacks in the solution phase. Tris(dicyanovinylidene)-substituted azatriangulenes C11.5a–c were obtained by Knoevenagel condensation of C11.4a–c with malononitrile in the presence of TiCl4 and pyridine.794 DFT calculations suggested the presence of C1- and C3-symmetrical conformations for compounds C11.5a–c, with the C1 conformation being less stable by 2.5 kcal mol−1 and accessible through a barrier of 5.4 kcal mol−1. As shown spectroscopically, the monomeric species with different molecular symmetries were found to self-assemble into different aggregates, which coexisted in freshly prepared solutions. The interconversion between the two aggregates is unfavorable at rt because of the steric effects of the dicyanovinylidene groups within the assembly. Equilibration to the C3-based assembly could be achieved by heating the solution followed by cooling. The overall process involves deaggregation and the conformation flipping of monomeric C11.5 from C1 to C3 symmetry at a high temperature, followed by reassembly upon cooling.
In 2020, Kato et al. reported the preparation of the sulfur-containing azatriangulene analogues 114.4–5 (Scheme 114).227 The intermediate 114.2 was obtainable from the Ullmann-type reaction between dimethyl 2-iodoisophthalate (114.1) and phenothiazine. Compound 114.2 was then treated with MeMgBr to give the diol 114.3. Subsequent H3PO4-promoted cyclization yielded the target molecule 114.4 (62% yield) and the side product 114.5 (2%). Upon one-electron oxidation by BAHA, compound 114.4 could be converted to its radical cation 114.4•+ as the SbCl6– salt, which was characterized by the UV–vis–NIR and ESR spectroscopy. The three-line splitting observed in the ESR spectrum of [114•+][SbCl6–] results from the hyperfine coupling of the unpaired electron with the 14N nucleus, with a coupling strength of 0.72 mT.
Scheme 114. Azatriangulene Derivatives Bearing Peripheral sp3-Hybridized Carbon Atoms.
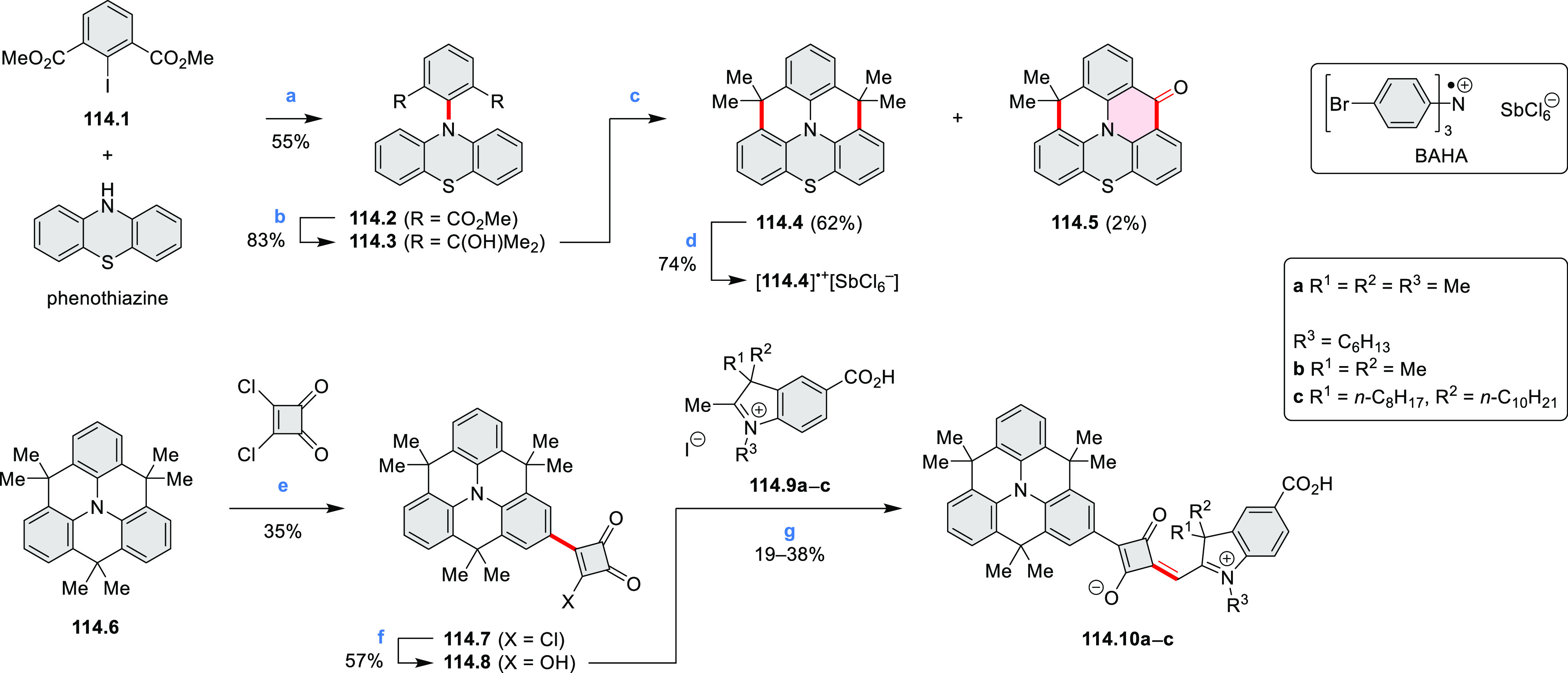
Reagents and conditions: (a)227 Cu, CuI, K2CO3, n-Bu2O, 150 °C, 48 h; (b) MeMgBr, toluene, 110 °C, 15 h; (c) 85% H3PO4, rt, 4 h; (d) tris(4-bromophenyl)ammoniumyl hexachloroantimonate (BAHA), DCM, N2, rt, 1 h; (e)228 benzene, 80 °C, 24 h; (f) AcOH, H2O, HCl, 100 °C, 12 h; (g) benzene/n-BuOH/pyridine, Dean–Stark apparatus, reflux, 5 h.
In 2016, Nithyanandhan et al. reported the zwitterionic dyes 114.10a–c comprising an azatriangulene donor moiety (Scheme 114). The synthesis involved the condensation of the known azatriangulene 114.6 with squaryl dichloride, followed by hydrolysis of the C–Cl bond to afford compound 114.8. Subsequent condensation with the indolinium salts 114.9a–c provided the target sensitizers 114.10a–c. Upon evaluation of the dye-sensitized solar cell performance, compound 114.10c displayed the highest power conversion efficiency (PCE) of 6.73% and an open-circuit voltage (VOC) of 0.53 V without any coadsorbent.
In 2020, Fingerle and Bettinger reported the synthesis of the nanographene 115.3 incorporating a boroxadizine (B3N2O) unit (Scheme 115).229 The diboronate ester 115.1 could be prepared from the known compound 210.2 in two steps. Using similar procedures outlined in Scheme 210 (Section 5.3.1), the target molecule 115.3 could be obtained. The UV–vis spectrum of 115.3 contains three broad absorption maxima within 330–370 nm. These peaks are blue-shifted relative to the lowest-energy absorption of the all-carbon analogue (dibenzo[fg,ij]phenanthro[9,10,1,2,3-pqrst]pentaphene). The small Stokes shift (221 cm–1) of 115.3 suggested molecular rigidity in the excited state. The NICS(1) value calculated for the boroxadizine ring in 115.3 is −1.0 ppm, reflecting the nonaromatic character of the B3N2O unit.
In two reports, Kitamoto, Oi et al. described syntheses of boron-centered heteratriangulenes 115.8 and 115.10 (Scheme 115).230,231 Among the three attempted synthetic routes, the successful one involved the precursor 115.4, which was derived from 2-fold Ullmann-type coupling between resorcinol and 3-iodoanisole. 3-Fold ortho-directed lithiation followed by treatment with boron trifluoride etherate afforded the annulated product 115.5 in 22% yield. This strategy ensured that the two methoxy groups would remain intact during borylation. Subsequent double demethylation with boron tribromide furnished the diol 115.6. Controlled esterification with Tf2O led to singly (115.7) and doubly (115.9) triflated products in good yield. For the monotriflate 115.7, an intramolecular SNAr reaction using DBU as the base smoothly produced the boratriangulene 115.8 bearing three peripheral oxygen atoms. For the bis(triflate) 115.9, a double SNAr reaction with aniline in the presence of LiHMDS produced the boratriangulene 115.10 bearing two peripheral oxygen atoms and one phenylamino group in 35% yield. Both 115.8 and 115.10 were shown by single-crystal XRD analysis to possess a flat triangulene skeleton in the solid state.
Yamamura and Nabeshima reported the X-ray structures and computational results on six “phosphangulene” derivatives bearing various axial substitutions (Scheme 116).232 In addition to the known phosphangulene oxide (116.2a) and sulfide (116.2b), the authors prepared the selenide (116.2c), P-methylphosphonium iodide [116.3][I], borane adduct (116.4), and phosphine tungsten pentacarbonyl complex (116.5) from phosphangulene (116.1). According to the XRD and DFT data of the six derivatives, the phosphine cone angle of the tungsten complex 116.5 was the smallest, while that of the phosphonium cation 116.3+ was the largest. Natural bond orbital (NBO) analysis on the optimized structures showed that the bowl depth increased with the s-orbital character of the phosphorus atom. The heteronuclear coupling constants (1JP–Se, 1JP–C, and 1JP–W) for 116.2c, [116.3][I], and 116.5 were noticeably larger than those of the corresponding triphenylphosphine adducts, i.e., Ph3PS, Ph3PMe+I–, and Ph3PW(CO)5. This difference was consistent with the increased s-orbital character in these phosphangulene adducts.
Scheme 116. Phosphangulene Derivatives.
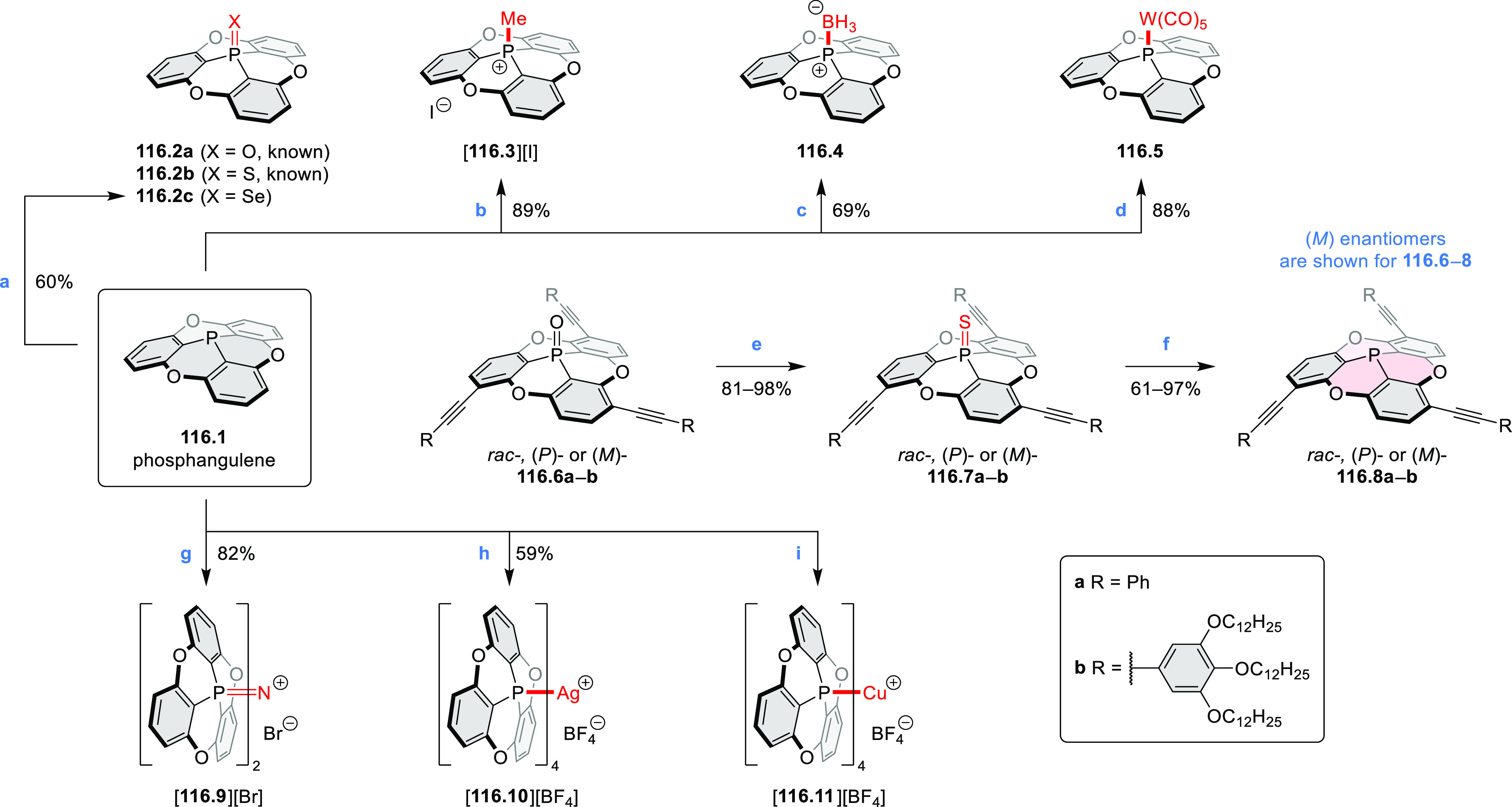
Reagents and conditions: (a)232 Se, CDCl3, rt, 25 days; (b) MeI, CDCl3, 55 °C, 54 h; (c) BH3·Me2S, CDCl3, rt, 10 min; (d) W(CO)6, THF, preirradiated with a high-pressure mercury lamp (400 W) at rt for 30 min, then rt, 10 min; (e)233,234 Lawesson’s reagent, toluene, reflux. 12–16 h; (f) P(NMe2)3, toluene, reflux, 15–24 h; (g)237 (1) Br2, 1,1,2,2-tetrachloroethane, 25 °C, 2 h, (2) NH2OH·HCl, reflux, 20 h; (h)238 AgBF4, DCM, 25 °C, 1 h; (i) [Cu(MeCN)4]BF4, DCM/MeCN, 25 °C, 12 h.
In 2015, Yamamura, Sukegawa, and Nabeshima explored the synthesis and supramolecular properties of the tris(arylethynyl)-substituted phosphangulenes 116.8a,b.233 The known phosphangulene oxide 116.6a was treated with Lawesson’s reagent to give the corresponding sulfide 116.7a in good yield. Desulfurization with tris(dimethylamino)phosphine furnished the target molecule 116.8a. This reaction sequence was performed starting with the racemic sample and with both enantiopure samples of 116.6a with retention of configuration. In the cocrystal of rac-116.8a with C60, the formation of a 2:1 complex was observed. Specifically, the fullerene was encapsulated by one (P)-116.8a molecule and one (M)-116.8a, resulting in an achiral cavity. In contrast, the oxide 116.6a and sulfide 116.7a each formed a complex with C60 in 4:1 ratio, to give a capsule-like structure. This difference in host–guest behavior was attributed to the deeper bowl depth of the 116.8a relative to the corresponding oxide and sulfide. In 2016, the same group reported a similar phosphangulene analogue 116.8b bearing nine long alkoxy chains in racemic and enantiopure forms.234 The formation of a columnar liquid crystalline phase for rac-, (P)-, and (M)-116.8b was supported by DSC and XRD analyses and by CD spectroscopy for the enantiopure samples.
Wuest et al. reported X-ray crystal structures of various polymorphs of phosphangulene chalcogenides 116.2a–c235 and those of the various solvates originating from the cocrystallization of 116.2a–c with C60 or C70.236 Furthermore, they characterized the crystal structures of the bis(phosphangulene)iminium salts (including [116.9][Br])237 and the silver(I) (116.10+) and copper(I) (116.11+) complexes featuring a metal center that is tetracoordinated by phosphangulene (Scheme 116).238
In 2016, Yamamura, Hasegawa, and Nabeshima reported three heteratriangulenes 117.1–3 that are formed by conceptually replacing one, two, or three oxygen atoms in phosphangulene (116.1) with sulfur atoms (Scheme 117).239 The synthesis begins with the treatment of the known triarylphosphine oxide 117.4 with dimethylthiocarbamoyl chloride using DABCO as the base. The product 117.5 containing one new O-thiocarbamate group and one cyclized phosphaxanthene unit was isolated in 92% yield. Newman–Kwart rearrangement of 117.5 followed by alkoxide-promoted cyclization gave the doubly cyclized product 117.7 in 68% yield over two steps. Finally, the phosphine oxide 117.7 could be reduced using trichlorosilane to the target phosphine 117.1 in 46% yield. The other two target molecules 117.2–3 were obtained using similar reactions. X-ray crystal structures of 117.1–3 revealed a progressive decrease of their bowl depths (1.88, 1.68, and 1.46 Å, respectively), reflecting the increasing circumference of these molecules caused by the introduction of sulfur atoms.
Scheme 117. Synthesis of Phosphorus-Centered, Sulfur-Containing Heteratriangulenes.
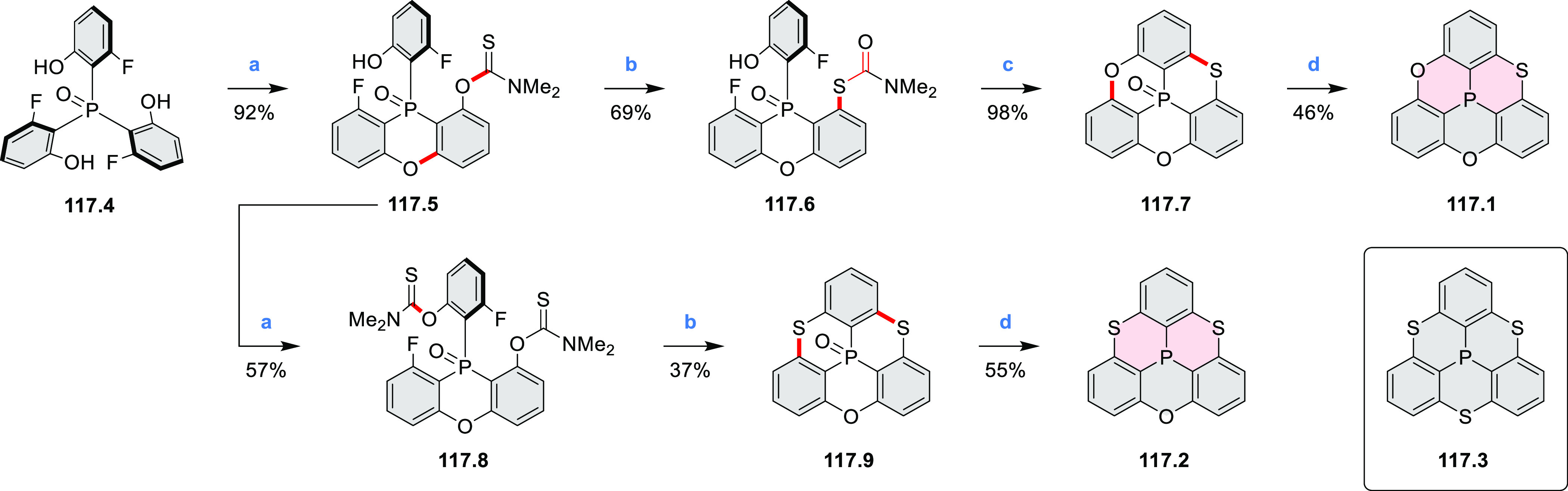
Reagents and conditions: (a)239 dimethylthiocarbamoyl chloride, DABCO, DMF, rt, 30 min, then 75 °C, 2–17 h; (b) 240 °C, argon, 2–5 h; (c) t-BuOK, DMF, 140 °C, 21 h; (d) SiHCl3, Et3N, toluene, 120 °C, 61–72 h.
In 2017, the Hatakeyama group developed synthetic routes to boron-, phosphorus-, and silicon-centered 4,8,12-triazatriangulenes.240 These syntheses relied on efficient incorporation of heteroatoms into the macrocyclic precursor through electrophilic C–Li and C–H substitution (Scheme 118). The starting macrocycle 118.1 was in turn obtained in four steps starting from 1-bromo-2,3-dichlorobenzene. Postsynthetic modification of 118.4 by desulfurization with PEt3 gave the corresponding phosphine 118.6 in 92% yield, whereas oxidation with m-CPBA led to the corresponding phosphine oxide 118.5 in 90% yield. Unlike the bowl-shaped structures of 118.3–6, the boron-centered 118.2 adopts a planar geometry. The fluorescence spectrum of 118.2 showed a sharp and strong emission band at λ = 399 nm with Φ = 0.54. Notably, the fwhm of this peak (26 nm) is one of the smallest values observed for organic light-emitting materials. This fwhm value in combination with a small energy difference between the S1 and T1 states (ΔEST = 0.21 eV) make 118.2 a promising candidate as an OLED material.
4.1.2. Carbon-Centered Heteratriangulenes
Diboratriangulenes C12.1a,b were obtained by employing the C–H borylation strategy reported by Würthner et al. (Chart 12, cf. Scheme 34, Section 3.1.2).61 The borinic acid C12.1a was synthesized from its diolefin precursor, and upon stepwise treatment with BBr3 and MesMgBr, it was converted into the dimesityl-substituted derivative C12.1b. Both compounds were stable under ambient conditions. The related diboratriangulenes C12.2a,b were prepared according to the bromoboration–electrophilic C–H borylation protocol reported by Ingleson and Zysman-Colman et al. (cf. Scheme 99, Section 3.4).202 The authors attributed the lower photoluminescence quantum yield of C12.2b (5.9%) relative to that of C12.1b (34%) to the heavy atom effect in the bromine-containing compound.
The oxidative Pummerer cyclization employed by Bonifazi et al. for the synthesis of an oxygen-doped perylenoid (see Scheme 46, Section 3.1.2) was also used to prepare the π-extended dioxatriangulene 119.2 (Scheme 119).77 A series of π-extended N,O-doped systems, including the triangulene 119.6, was reported in 2018 by Mastalerz et al.241 The synthesis involved the Pictet–Spengler reaction of biphenyldiamine 119.3 with the substituted salicylaldehyde 119.4 in 1:2 stoichiometry in the presence of TFA and O2. The intermediate 119.5 was not isolated but spontaneously underwent a thermally induced ipso-substitution to give the desired tetraheteratriangulene 119.6 in 80% yield. In contrast, the same ipso-substitution applied to other starting materials required higher reaction temperatures (>180 °C, see Scheme 205, Section 5.2.2). Compound 119.6 emits at 538 nm when dissolved in chloroform and at 433 nm when dissolved in THF, with the quantum yields of 8% and 30%, respectively. The effect was caused by protonation of 119.6 in the slightly acidic chloroform. Addition of K2CO3 to the chloroform solution gave an emission spectrum which resembled that obtained in THF.
Scheme 205. Synthesis of Oxygen- and Nitrogen-Doped Hydrocarbons.

Reagents and conditions: (a)241 3,5-di-tert-butyl-2-hydroxybenzaldehyde, toluene, TFA, O2, 100 °C, 16 h; (b) neat, 180 °C, 24 h.
In 2015, Matsuda et al. reported photochemical cleavage of triazatriangulene derivatives 120.2a–c substituted with an apical arylethynyl group at the central carbon (Scheme 120).242 These derivatives were prepared from the reaction between the known triazatriangulenium salt [120.1][BF4] and acetylide anions generated via deprotonation or desilylation. Upon irradiation (365 nm) of 120.2a–c dissolved in ethanol, the photoinduced C–C bond cleavage could be detected by the yellow fluorescence at 562 nm and the red color of the resulting cation [120.1]+. This transformation was also corroborated by the emergence of 1H resonances corresponding to [120.1]+, when 120.2a–c dissolved in ethanol-d6 was irradiated. Under these conditions, the acetylenic resonance of the expected arylacetylene product was not observable presumably because of H/D exchange.
Scheme 120. Synthesis of the Triazatriangulene Derivatives Bearing Different Axial Groups at the Central Carbon Atom.

Reagents and conditions: (a)242n-BuLi, THF, 0 °C, 1 h then rt, 1 h, (2) [120.1][BF4], rt, overnight; (b) (1) KOH powder, THF, 0 °C, 30 min, (2) [120.1][BF4], 0 °C.
Lewis-acid reactivity of the known trioxatriangulenium cation 121.1+ toward phosphines and an N-heterocyclic carbene as Lewis bases was explored by Gianetti et al. (Scheme 121).243 When [121.1][BF4] was treated with PMe3, P(n-Pr)3, or P(n-Bu)3, the corresponding stable Lewis adducts 121.2a–c could be isolated in good yield. The 31P NMR spectrum of 121.2a shows a resonance at 39.7 ppm, indicative of a phosphonium species. In addition, the structure of 121.2a was confirmed by single-crystal XRD analysis. In contrast, when [121.1][BAr′4] was exposed to the weakly basic PPh3 or the bulky P(t-Bu)3 in a deuterated solvent, no evidence for the formation of Lewis adducts was observed by 1H and 31P NMR spectroscopy. In the UV–vis absorption spectrum of an equimolar mixture of [121.1][BAr′4] and P(t-Bu)3 in DCM, an extra weak absorption band emerged, with maxima at 576 and 626 nm, besides the major bands of [121.1]+ at 458 and 483 nm. On the basis of absorption spectroscopy and DOSY data, the authors postulated the dynamic existence of the frustrated encounter complexes 121.2d,e in the solution state. Furthermore, when [121.1][BAr′4] was mixed with 1,3-di-t-butylimidazol-2-ylidene, single-electron transfer occurred, to generate the trioxatriangulenyl radical 121.1•, observable by EPR spectroscopy. The C4-adduct 121.3 and C2-adduct 121.4 were isolated as products of this reaction. The former regioisomer was the major product at rt, and the regioselectivity was reversed when the reaction was performed at −78 °C.
Scheme 121. Lewis Acid–Base Chemistry of a Trioxatriangulenium Cation.

Reagents and conditions: (a)243 MeCN (for a–c), CDCl3 or CD3CN (for d), CD2Cl2 or CD3CN (for e), rt, 5 min; (b) 1,3-di-t-butylimidazol-2-ylidene, toluene, rt, 1 h; (c) 1,3-di-t-butylimidazol-2-ylidene, toluene, −78 °C, 30 min.
Vilar and Kuimova et al. studied heteratriangulenium cations C13.1–2 as optical probes for G-quadruplexes (Chart 13). The introduction of 2-(4-morpholinyl)ethyl and propyl groups at the nitrogen atoms in C13.1–2 was achieved using established methods. In the presence of DNA, the emission of C13.1a was enhanced and bathochromically shifted, whereas the emission of C13.2 was quenched.244 The former compound was then shown to bind with single- and double-stranded DNAs, as well as G-quadruplex DNA, with log Ka of 5.7–6.1 determined using emission titration experiments. The fluorescence lifetime of C13.2 observed in vitro is longer for G-quadruplexes than for double- and single-stranded DNAs. Compound C13.2 was found to permeate into cells and localize mainly in the nucleus. Further photophysical and computational investigations suggested the importance of the morpholine moiety for the fluorescence enhancement upon binding with DNA.245 In particular, the propyl-substituted derivative C13.1b displayed a reversed fluorescence selectivity for double-stranded DNA over G-quadruplexes.
Lacour, Gruenberg, Vauthey, and Bakker et al. reported a series of diazaoxatriangulenium cations [C13.3a–c]+ as the hexafluorophosphate and trifluoroacetate salts (Chart 13).246 The free NH group rendered these three cations pH-sensitive (pKa = 7.3–8.4). In particular, the hexadecyl-substituted cation C13.3a+ and its deprotonated form C13.4 emit fluorescence in the red part of the visible spectrum with quantum yields of 14–16% and lifetimes of 7.7–7.8 ns. Cellular studies with the neutral C13.4 demonstrated selective imaging of late endosomes in HeLa cells. The lipophilic dodecyl side chain was found to be crucial for achieving such selectivity.
In 2018, Lacour et al. reported nine cationic diazaoxatriangulenium-based dyes C13.5–13 substituted with one electron-donating or electron-withdrawing group at one of the peripheral carbons (Chart 13).247 The nitro and formyl species (C13.5 and C13.8, respectively) were derived directly from the known N,N′-unfunctionalized precursor via appropriate substitution reactions and further used as starting materials for the other seven derivatives. The substituents not only influenced the redox potentials but also tuned the optical properties relative to the unfunctionalized triangulenium. The absorption maxima of C13.5–13 span a range of 528–640 nm in acetonitrile, exhibiting variable fluorescence in the yellow–red range of the visible spectrum. For instance, the emission maxima of the 1,2,3-triazole C13.7 and the nitrile C13.12 were observed at 607 and 556 nm, respectively.
Laursen and co-workers reported further analogues of the above compounds, in which one of the peripheral heteroatoms was replaced with an sp3 carbon bridge (Scheme 122).248 The triangulenium structure was assembled through an addition of ortho-lithiated m-dimethoxybenzene to 122.1, followed by condensation in molten pyridine hydrochloride. The resulting dioxatriangulenium salt 122.3 could be further transformed into the azaoxa and diaza analogues 122.4a,b by treatment with methylamine. Other primary amines such as N-Boc-cadaverine were incorporated in a similar manner, leading to compounds 122.5a,b and 122.6.57 Introduction of nitrogen atoms caused a bathochromic shift of emission peaks, while the quantum yields increased slightly (122.3: λmax = 535 nm, Φ = 54%; 122.4a: λmax = 584 nm, Φ = 57%; 122.4b: λmax = 624 nm, Φ = 61%). Additionally, emission of 122.4b was red-shifted by ca. 50 nm compared to its closest oxygen-bridged analogue C13.1c. A further red-shift could be achieved by π-extension (see Scheme 32, Section 3.1.1). The triangulenium salts were introduced into cells for fluorescence imaging, where 122.4b localized in the nucleus and displayed good photostability. The appended morpholine units in 122.6 resulted in emission quenching due to photoinduced electron transfer. Emission enhancement occurred in an acidic environment or upon binding to DNA.
4.2. Azapyrenoids
In 2017, Feng, Liu, and Wang reported a copper-catalyzed domino tricyclization reaction for the construction of the heptacyclic dibenzo[1,2:7,8]quinolizino[3,4,5,6-kla]perimidine (123.4a) from 2-(phenylethynyl)benzaldehyde (123.1) and naphthalene-1,8-diamine (Scheme 123).249 The optimized conditions involved heating a dioxane solution of the two reactants in the presence of Cu(OAc)2, Cs2CO3, and O2 gas, giving 123.4a in 76% yield. Stirring the two reactants in the absence of any reagents at rt afforded a high yield of the aminal 123.2, which was thus proposed as an intermediate for the domino reaction. Presumably, 123.2 undergoes intramolecular hydroamination to produce 123.3, which is then dehydrogenated to give the target molecule. The functional group tolerance of this transformation was examined, and a library of substituted derivatives including 123.4b–e were isolated in 67–81% yield.
In 2015, Zhang and Pei et al. reported the diazapyrenoids 123.6–8 that are fused with two naphthalene diimide units (Scheme 123).250 The authors found that 1,5-naphthalenediamine reacted with the tetrabromo NMI derivative 123.5 in a 1:2 stoichiometry to yield the doubly annulated product 123.6. Notably, this conversion did not necessitate palladium catalysis, and only a weak base (K2CO3) was required. 123.6 was further annulated with sodium 1,1-dicyanoethylene-2,2-dithiolate to form compound 123.7. Besides, the two NH groups in 123.6 could be oxidized by p-phenylenediamine to give 123.8. In the UV–vis–NIR absorption spectra, compound 123.6 showed a broad band between 600 and 1200 nm, while the corresponding band for compound 123.7 is bathochromically shifted by roughly 100 nm (log ε ≈ 5). The oxidized derivative 123.8 absorbs in the NIR range much less intensely (log ε ≈ 3). The narrower band gap of 123.7 in comparison with 123.6 was ascribed to a lower LUMO level (−4.72 eV vs −4.35 eV, respectively), as determined by CV measurements.
In 2016, Monkman and Bujak et al. reported flavanthrone-derived dyes 124.3a–e for application in organic electronics (Scheme 124).251 Flavanthrone 124.2 (see CR2017, Section 4.2 for historical developments of its chemistry) was obtained through the acid-mediated 2-fold cyclization of compound 124.1, which in turn was obtained via Ullmann reaction of the bromo-substituted precursor. Next, 124.2 was reduced by Na2S2O4 in the presence of NaOH, followed by alkylation with alkyl bromides under phase-transfer conditions in one pot. The five dialkoxy-substituted products 124.3a–e were isolated in 59–69% yield. STM images showed that the dioctyloxy-substituted derivative 124.3c self-assembled in monolayers deposited on a highly oriented pyrolytic graphite (HOPG) surface. Among compounds 124.3a,c,e, which show electroluminescence in a guest/host-type OLED, 124.3c displayed a luminance value approaching 1900 cd m–2 and the highest luminous efficiency (>3 cd A–1). Later, the dithienyl-substituted analogue 124.4 was studied by Bujak et al. as a potential luminophore.252
Scheme 124. Diaza- and Tetraazapyrenoids from Condensation of Ketone Precursors.
Reagents and conditions: (a)251 aq. HCl, reflux, 24 h; (b) (1) Na2S2O4, NaOH, H2O, 60 °C, 1 h, (2) CnH2n+1Br, Aliquat 336, toluene, 90 °C, 24 h; (c)252 (1) 2-octylthiophene and n-BuLi (premixed in Et2O at −78 °C for 1.5 h), −78 °C, then rt, overnight, (2) SnCl2·H2O, rt, 3 h; (d)253 MsOH, 150 °C, 16 h; (e) (1) NaH, DMF, rt, 2 h, (2) EtI, rt, 24 h; (f) N2H4·H2O, EtOH, 130 °C, 72 h; (g) toluene, 120 °C, 48 h.
A selective synthesis of the fused phthalazine derivative 124.8 (Scheme 124) was developed by Zhu and Zhang.253 Upon heating the known diester 124.5 in methanesulfonic acid, the 2-fold Friedel–Crafts acylation took place regioselectively to give the angularly fused product 124.6 rather than the isomeric, linearly fused quinacridone skeleton. Compound 124.6 was ethylated at both NH groups and condensed with hydrazine to yield the tetraazapyrenoid 124.8 in 36% over two steps. A [4 + 2] cycloaddition–elimination sequence involving the pyridazine ring in 124.8 and dimethyl acetylenedicarboxylate was envisaged to provide compound 124.10. However, only the [2 + 2 + 2] cycloadduct 124.9 was isolated in this reaction.
In 2017, Wei and Li et al. reported a family of viologens (N,N′-disubstituted 4,4′-bipyridinium salts) in which the pyridyl rings are connected by two ortho-phenylene linkages (Scheme 125).254 First, phenylacetaldehyde and pyridine-4-carboxaldehyde underwent base-catalyzed condensation in a 2:1 stoichiometry to give a 1,5-dialdehyde intermediate, which was then condensed with hydroxylamine to generate the new pyridine ring in 125.1. Owing to the electron-poor nature of 125.1, subsequent cyclodehydrogenation did not take place when DDQ was used as the oxidant. Instead, the Mallory photocyclization worked smoothly to furnish the doubly cyclized product 125.2 in 98% yield. Reactions of 125.2 with various alkyl halides, followed by salt metathesis, gave ten viologens 125.3 in the form of hexafluorophosphate salts in 81–88% yield. All new viologens showed intense blue emission in the range of 421–453 nm.
In 2020, Wallis et al. showed that the diiminium species 125.42+ can be obtained by oxidative dimerization of 1,5-bis(dimethylamino)naphthalene (Scheme 125).255 This transformation is comprised of two types of bond formation, namely, the intermolecular C–C coupling of the carbon atoms para to the dimethylamino groups and the intramolecular C–N coupling that generates the new hexagonal rings in 125.42+. The bis(triiodide) salt [125.42+][I3–]2 was obtained with diiodine as the oxidant at rt, whereas [125.42+][(TCNQ)4]2– was obtained using tetracyanoquinodimethane (TCNQ) as the oxidant in refluxing acetonitrile. Interestingly, 1,5-bis(dimethylamino)naphthalene was oxidized by 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) to give the monocationic quaternary ammonium salt [125.5+][F4TCNQ•–]. All three salts were unambiguously confirmed by X-ray crystallography. In particular, the negative charge carried by TCNQ in the solid state of [125.42+][(TCNQ)4]2– was estimated from bond length measurements. Additionally, the IR spectrum of this salt exhibits two C≡N stretching frequencies at 2185 and 2154 cm–1.
Hromadová, Kolivoška, and Valášek et al. reported the cationic diazapyrenoid 125.8 in which one positively charged nitrogen center occupies an interior position of the π-skeleton (Scheme 125).256 The known bis(pyrylium) salt 125.6 was doubly condensed with 4-aminopyridine in DMF, followed by azeotropic removal of water. The resulting bis(pyridinium) salt 125.7 was then subjected to Mallory photocyclization, yielding the doubly cyclized product 125.8 with one diazapyrenoid unit. Further ring closures of compound 125.8 could not be achieved either by altering the Mallory reaction conditions or by FeCl3-mediated oxidation. Both 125.7 and 125.8 show a reversible two-electron reduction wave centered at −1.0 V, implying a limited effect on the LUMO levels imposed by the double cyclization. This observation was reflected in DFT data obtained for 125.7 and 125.8 (ELUMO = −3.78 and −3.75 eV, respectively).
Chen et al. reported the radical cation 125.11•+ consisting of two azapyrenoid moieties (Scheme 125, cf. Scheme 136).257 The key step in the synthesis involves the electrophilic cyclization and dehydrogenative aromatization of the bis(O-acetyl oxime) 125.9 in the presence of iron(III) acetylacetonate. The resulting polycyclic arene 125.10 was isolated in 83% yield. 2-Fold methylation of 125.10 with methyl triflate gave the dicationic species 125.112+ as the triflate salt. Single-electron reduction of this dication with triethylamine quantitatively afforded the radical cation salt [125.11•+][TfO−] which possessed appreciable air stability (>7 days) in the solid form. This salt was characterized by X-ray crystallography and EPR spectroscopy in the solution and solid states. On photoexcitation, solutions of [125.11•+][TfO−] showed red fluorescence with a quantum yield of 9.3%. Upon dispersion in a PMMA film, [125.11•+][TfO−] fluoresces with a higher quantum yield (19.3%), suggesting an inhibition of the radiationless decay of the excited state. The salt dissolved in DCM showed excellent photostability, with a half-life of 9.5 × 104 s under irradiation at 350 nm.
Scheme 136. Synthesis of π-Radical Cation [136.4]•+.

Reagents and conditions: (a) s-BuLi, TMEDA, THF, −78 °C, 45 min, then phenalenone, −78 °C to rt, overnight;273 (b) TfOH, 1,1,2,2-tetrachloroethane, 120 °C, 12 h; (c) Na2S2O4, MeCN, rt, 2 h.
In 2018, Unterlass et al. reported a green hydrothermal synthesis of perinones 126.1–2 (Scheme 126, cf. CR2017, Section 4.2).258 In their protocol, o-phenylenediamine and NTDA in a molar ratio of 2:1 were suspended in deionized water in a nonstirred batch autoclave and heated at 200 °C for 16 h. Purification and NMR analysis confirmed the formation both the “cis”- and “trans”-isomers of perinone (126.1–2) in a 2:3 ratio. The singly condensed side product 126.3 was also identified, and its emergence could not be avoided under various screened conditions. The reaction time could be shortened to 12 min when the mixture was stirred under microwave heating at 200 °C. The perinone-based conjugated ladder polymer 126.4 was prepared by Wang and Qiao et al. in addition to its perylene-derived congener (see Scheme 96, Section 3.4) and evaluated in bulk heterojunction photodetectors.199
Tetraaryl-substituted imidazole 127.1 bearing electron-donating groups was used by the Gryko group as a precursor to the fully conjugated benzo[e]pyrene scaffold 127.2 (Scheme 127).259 For this transformation, (bis(trifluoroacetoxy)iodo)benzene was used as the oxidant, which was shown to work well for a range of ortho-fused imidazole targets. In the synthesis of 127.2, 2.5 equiv of the oxidant was sufficient to achieve complete reaction, and no partially fused products were observed, indicating that the first oxidative coupling facilitated the ultimate fusion. The emission spectrum recorded for 127.2 shows three maxima at 400, 422, and 444 nm. The fluorescence quantum yield in acetonitrile was determined to be 32%.
Scheme 127. Imidazole-Fused Pyrene via Oxidative Coupling.

Reagents and conditions: (a)259 PIFA, BF3·Et2O, toluene, rt.
An NBN-doped pyrene 128.2 was obtained via electrophilic C–H borylation by Wakamiya and Wang in 2018 (Scheme 128).260 This double annulation was also used for construction of related phenalenoid systems (see Scheme 209, Section 5.3.1). Irreversible oxidation peaks were observed for 128.2 in cyclic voltammetry; however, the attempts to construct an NBN-containing seven-membered ring from 128.2 were unsuccessful, which was ascribed to the instability of the anthracene moiety toward oxidants. 128.2 has small band gap of ca. 2.5 eV resulting from a high-lying HOMO and a low-lying LUMO, making the compound potentially suitable for OPV applications.
Scheme 209. Synthesis of NBN-Dibenzophenalene Derivatives.
Reagents and conditions: (a)394 Cs2CO3, Pd(PPh3)4, toluene/EtOH; (b) BCl3, Et3N, o-DCB.
In 2020, Park and Shin et al. reported the preparation of B2N2-ixene (128.5, Scheme 128).261 The key precursor, 2,2′-(buta-1,3-diyne-1,4-diyl)dianiline (128.3), was obtained from the Glaser coupling of 2-ethynylaniline. Treatment of 128.3 with PhBCl2 and Et3N brought about the chloroboration of alkyne followed by B–N bond formation to yield compound 128.4 in 28% yield. Next, palladium-catalyzed cyclization involving the phenyl groups and the C–Cl bonds afforded the target B2N2-ixene (128.5) in 40% yield. In the solid state, 128.5 showed a contorted structure, in which the four peripheral benzene rings deviated from planarity. The molecules packed into a 1D columnar pattern with a short intermolecular distance of 3.685 Å, which may have resulted from favorable dipolar interactions.
Electrophilic borylation with BBr3 was used by Hatakeyama and co-workers to prepare various BN-doped polycyclic aromatics (Scheme 129).262 In 129.1, boron was selectively introduced at the central benzene, ortho to the carbazolyl substituent, where the highest HOMO amplitude was predicted by DFT. The product of single borylation (129.2) was obtained with 2 equiv of BBr3 in refluxing toluene. With a greater excess of BBr3 and higher temperature, double borylation to 129.3 was also accomplished. The starting material 129.4 bearing two carbazolyl groups reacted in a similar manner, providing 129.5–6, respectively. For compound 129.7 with three diarylamine substituents, relatively harsh conditions (170 °C) were needed to establish a single peri fusion through borylation, giving 129.8 in 80% yield. All fused products shown here, as well as their differently substituted variants, were characterized by strong, narrowband photoluminescence, with quantum yields of 80–88% and fwhm of 26–39 nm in the PMMA matrix. Emission maxima were found in the 453–504 nm range, making these borylated derivatives suitable for fabrication of deep blue, sky blue, and green OLEDs. Products of double borylation (129.3 and 129.6) displayed red-shifted emission relative to their singly borylated counterparts, which was attributed to π-extension.
Condensations of NTDA and related monoimides with aromatic oligoamines provided access to a variety of perinone analogues, such as the dicyano-substituted 130.1(193) or the isomeric dianhydrides 130.2–3196 (Scheme 130, for syntheses of the corresponding perylene-based analogues 94.2 and 95.2; see Section 3.4). Liu, Ma, and Navarro et al. explored the disk-shaped triphenylene-tris(naphthaleneimidazole)s 130.5–6 as n-type semiconductors (Scheme 130).263,264 These compounds were synthesized through the condensation of 2,3,6,7,10,11-triphenylenehexaamine with the naphthalene monoimide monoanhydrides 130.4a,b. In each case, the C3h-symmetrical (130.5) and the Cs-symmetrical (130.6) products were obtained as a mixture. In OFET devices, the average and maximum electron mobilities for the annealed thin films of the dodecyl analogues (130.5a,6a) were 1.0 × 10–3 and 1.3 × 10–3 cm2 V–1 s–1, respectively. Both values are at least 1 order of magnitude larger than those (8.5 × 10–5 and 1.3 × 10–4 cm2 V–1 s–1) of the branched alkyl analogues (130.5b,6b). The linear alkyl side chains were believed to facilitate molecular organization, thereby enhancing electron transport in the thin films.
Mastalerz and Vaynzof et al. reported the design and synthesis of triptycene-triaroylenimidazoles (TTAIs) 131.6–7, which constitute a class of three-dimensional acceptor molecules (Scheme 131).265,266 The synthetic pathway is based on the C3v-symmetrical triaminotrinitro-substituted triptycene 131.1 and its Cs-symmetrical isomer 131.2. Both molecules were obtained from the corresponding triptycenetriamines via an acetylation–nitration–deacetylation sequence. Compounds 131.1–2 underwent 3-fold condensation with 131.3a–c in quinoline at 180 °C in the presence of Zn(OAc)2 to give 131.4a–c (C3v) and 131.5a–c (Cs) in 39–66% yield. The nitro groups in these compounds were then reduced by SnCl2 to amino groups, and then the crude products were subjected to intramolecular condensation to furnish the target TTAIs 131.6a–c (C3v) and 131.7a–c (Cs) with different symmetries. This stepwise condensation approach does not result in the formation of isomers (compare with Scheme 126). Organic solar cells were fabricated using each TTAI and the donor polymer PTB7 in 1:1 ratio. The photovoltaic performance of the TTAIs was found to be insensitive to molecular symmetry, but it was dependent on the solubilizing groups. For the C3v-symmetrical TTAIs (131.6a−c), the maximum PCEs were 0.90%, 2.97%, and 2.76%, respectively. The Cs-symmetrical series (131.7a–c) exhibited the same trend. The high open-circuit voltage values (VOC) determined for these photovoltaic devices were 0.15–0.2 V higher than those for PTB7–fullerene-based devices.
Scheme 131. Synthesis of Cs- and C3v-Symmetric π-Extended Triptycene Derivatives.
Reagents and conditions: (a)265,266 Zn(OAc)2·2H2O, quinoline, 180 °C, 24 h; (b) SnCl2·2H2O, EtOAc, 85 °C, 12 h; (c) DMF (for a) or Zn(OAc)2·2H2O, quinoline (for b and c), 140 °C, 72 h.
4.3. Other Heterapyrenoids
Fluorinated biphenyls can be converted into dibenzofurans and fused xanthenes using an alumina-promoted oxodefluorination reported by Akhmetov et al. in 2020 (Scheme 132).267 The reaction occurred on the surface of activated γ-Al2O3 and did not require other reagents. An attempt to convert 132.1 into 132.4 using an analogous approach showed low selectivity with pentannulation products being formed alongside the target xanthene derivative. However, 132.5 with two fluorine atoms in the cove region was efficiently transformed into 132.6, providing access to the elusive naphtho[2,1,8,7-klmn]xanthene core.
Scheme 132. Fused Xanthenes via Alumina-Promoted Oxodefluorination.

Reagents and conditions: (a)267 γ-Al2O3, 190 °C, 12 h.
A single-step triple annulation was used to prepare pyrylium-containing oxapyrenoids 133.3a–j from simple precursors (Scheme 133).268 The products were obtained in moderate to high yields from 4-formyl-1-naphthols 133.1a,b and symmetric diarylalkynes 133.2a–h in the presence of a rhodium catalyst and Cu(OAc)2 acting as a stoichiometric oxidant. The multistep mechanism proposed for this transformation consists of three distinct cyclizations. Each of these stages involves an O-directed C–H activation by the rhodium catalyst, followed by insertion of alkyne into the resulting rhodacycle. Remarkably, different products were obtained by simply using NH4BF4 as an additive instead of NaBF4. The ammonium nitrogen was incorporated into the products in the place of the aldehyde-derived oxygen, leading to pyridinium-fused oxaphenalenes (Scheme 196, Section 5.1.3). Compounds 133.3a–j as well as phenalenoids 196.1a,b and 196.2a–d were emissive, with luminescence quantum yields of 35–69%. Emission wavelength in this series of compounds varied from 470 nm in 196.1a to 623 nm in the most electron-rich pyrenoid 133.3j. A selection of pyrylium and pyridinium products were tested as fluorescent probes for use in cell cultures. These compounds permeated into cells and localized in the mitochondria of HepG2 cells. Compound 133.3a displayed the best performance in terms of high photostability and low cytotoxicity.
Scheme 133. Synthesis of Pyrylium-Fused Oxapyrenes.

Reagents and conditions: (a)268 [Cp*RhCl2]2, Cu(OAc)2, NaBF4, THF, 150 °C, 24 h.
Scheme 196. Synthesis of Pyridinium-Fused Oxaphenalenes.

Reagents and conditions: (a)268 [Cp*RhCl2]2, Cu(OAc)2, THF, 100 °C, 12 h; (b) [Cp*RhCl2]2, Cu(OAc)2, NH4BF4, THF, 150 °C, 24 h.
In 2018, Bonifazi reported the first synthesis of a chemically and thermally stable, O-doped zigzag-edged nanoribbon 134.7 (Scheme 134).269 A similar strategy was employed as in the synthesis of O-doped benzorylenes such as 134.4a (see Scheme 50, Section 3.1.3).82 The synthetic procedure started with a C–C oxidative heterodimerization of 134.1b and 134.2, which gave the racemic binaphthol 134.3b in 35% yield. Surprisingly, oxidative O-annulation of 134.3b following a procedure successfully applied in the synthesis of 134.4a(82) failed, yielding the desired product in only 7% yield along with extensive degradation. However, under alternative conditions with a catalytic amount of CuCl, K2CO3, and N-methylimidazole in m-xylene at 120 °C, the peri-xanthenoxanthene (PXX) 134.4b was obtained in 64% yield and was subsequently demethylated using n-dodecanethiol to provide 134.5. This compound was then homodimerized into 134.6 in the presence of a Cu–TMEDA catalyst. The final oxidative ring-closure reaction gave the desired zigzag nanoribbon 134.7 in 17% yield. The Cu(I)-mediated cyclization methodology shown here is an alternative to the earlier approach using a Hg(II) oxidant.270 Photophysical and electrochemical data showed that the extension of the 134.4 PXX core into the molecular ribbon 134.7 reduced the band gap. This change was caused by an increase of the HOMO energy from −5.14 eV in 134.4 to −4.74 eV in 134.7, which is a desirable feature for prospective p-type organic semiconductors. Compound 134.7 had a yellow emission (λmax = 540 nm, Φ = 38%), which was red-shifted relative to 134.4b (λmax = 457 nm, Φ = 48%).
Scheme 134. Synthesis of Zig-Zag O-Doped peri-Acenoacenes.

Reagents and conditions: (a)269 CuCl2, 1-phenylethan-1-amine, MeOH, DCM, 0 °C to rt, 2 h; (b) CuI, pivalic acid, DMSO, 130 °C, 2 h; (c)82 CuCl, K2CO3, N-methylimidazole, 120 °C, 20 h, 64%; (d) n-dodecanethiol, NaOH, NMP, 130 °C, 4 h, 93%; (e) Cu-TMEDA, DCM, rt, 5 min; (f) CuCl, K2CO3, N-methylimidazole, 120 °C, 20 h.
Frontier orbital energy levels of the O-doped anthranthrene core (134.4a, Scheme 134) can be tuned by introducing electron-withdrawing imide groups (Scheme 135).271 Regioselective bromination of the 1,8-naphthalic anhydride hydroxyl derivative 135.1 (95%) followed by imidization (97%) and methylation (100%) gave 135.2 in an overall excellent yield. 135.2 was then subjected to in situ Miyaura borylation and Suzuki coupling, providing 135.3 after demethylation. The final ring closure was achieved by a Cu-promoted oxidative etherification, which gave good yields despite the presence of electron-withdrawing imide groups. The same approach was utilized in the synthesis of 135.8a bearing one N-octylimide functionality. Excited-state redox potentials of 135.4a and 135.8a were comparable to those of organometallic photocatalysts such as [Ru(bpy)3]2+ and [Ir(ppy)3]. These compounds were thus successfully used to promote a photoredox dehalogenation of several alkyl and aryl halides in the presence of DIPEA as a stoichiometric reducing agent.
Scheme 135. Synthetic Strategies Toward the Preparation of peri-Xanthenoxanthene Imides.
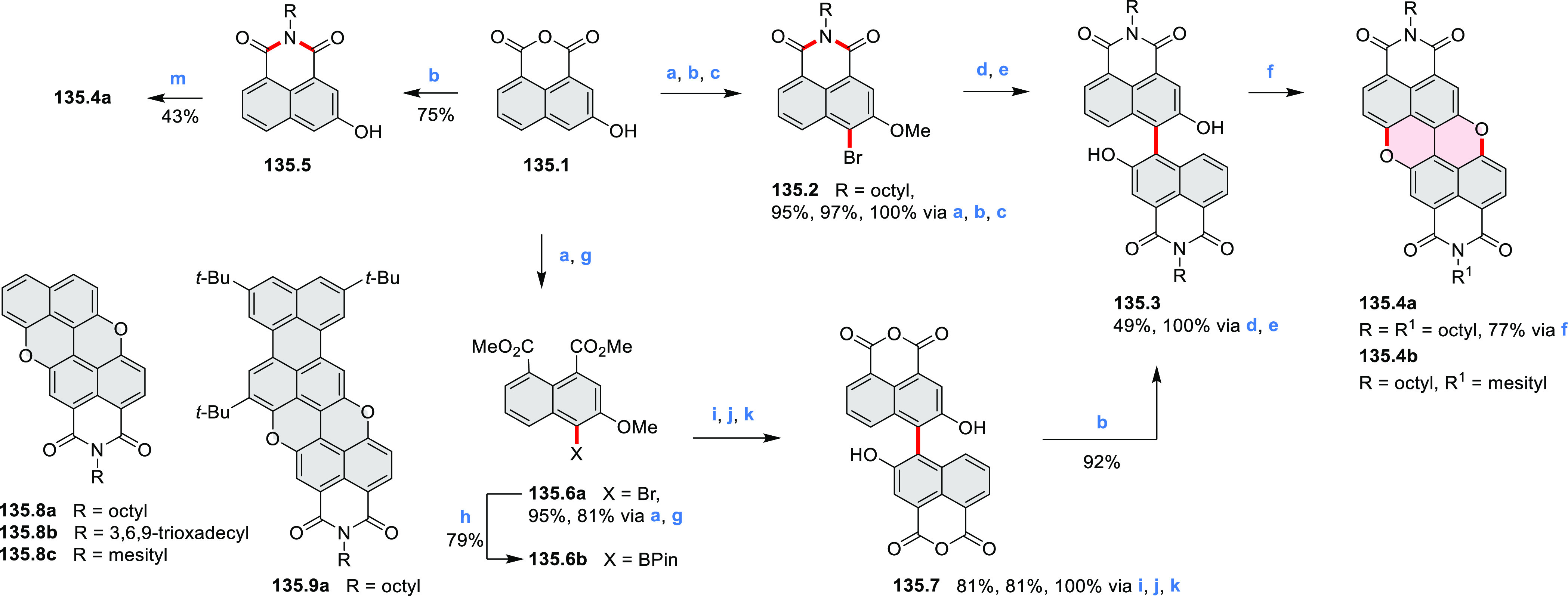
Reagents and conditions: (a)271,272 Br2, dioxane, reflux, 2.5 h; (b) i-Pr2NEt, n-octylamine, dioxane, reflux, 16–24 h; (c) K2CO3, CH3I, MeCN, reflux, 4 h; (d) Cs2CO3, B2Pin2, Pd(dba)2, SPhos, dioxane, reflux, 18 h; (e) BBr3, DCM, 0 °C to rt, 16 h; (f) pivalic acid, CuI, DMSO, 120 °C, 5 h; (g) DBU, MeI, MeOH, reflux, 18 h; (h) B2Pin2, KOAc, Pd(PPh3)2Cl2, 1,4-dioxane, reflux, 16 h; (i) 135.6a, 135.6b, K3PO4, Pd(dba)2, SPhos, 1,4-dioxane, H2O, reflux; (j) (1) KOH, i-PrOH, reflux, 13 h, (2) HCl(aq.), AcOH, reflux, 24 h; (k) HBr(aq.), AcOH, 126 °C, 24 h; (m) CuCl, DMSO, 120 °C, 24 h, air.
To further investigate the synthetic methods toward the peri-xanthenoxanthene diimide 135.4a, a direct oxidative Pummerer dimerization was examined (Scheme 135).272 The homocoupling of 135.5 was done in up to 43% yield in the presence of CuCl in DMSO under air. An alternative pathway toward 135.4a and related compounds was also described in the same study. In this approach, imidization was performed in the penultimate step (such as from the dimeric anhydride 135.3), allowing for convenient introduction of various N-substituents. Compound 135.4b with differentially substituted imides was also prepared. Moreover, monoimides 135.8a–c and the peri-π-extended analogue 135.9a were obtained by the same approach.
In 2019, Chen and co-workers reported the synthesis of a bisphenalenyl radical cation [136.3]•+ (Scheme 136).273 Treatment with triflic acid caused demethylation of the methoxy groups in 136.2, which was followed by cyclization to generate the dipyrylium salt [136.3]OTf2 after stirring in tetrachloroethane at 120 °C for 12 h. Reduction of the dication [136.3]2+ in the presence of either strong reducing reagents (i.e., Na, K, and Zn) or mild ones (e.g., sodium dithionite) provided the π-radical cation [136.3]•+, while the corresponding neutral species could not be obtained. The electronically stabilized π-radical cation [136.3]•+ shows multiple short intermolecular contacts (<3.4 Å) in its X-ray crystal structure. DFT simulations revealed that these close π–π interactions enabled intermolecular spin–spin coupling, implying that [136.3]•+ achieved electrostatically enhanced intermolecular covalent bonding interactions in two dimensions. Single-crystal devices fabricated from [136.3]•+ demonstrated average electrical conductivities of 1.31 × 10–2 S/cm.
The natural product xylindein C14.1 from the wood-staining fungi, Chlorociboria aeruginosa (Chart 14, see CR2017, Section 4.3), is of potential interest as a fluorescent probe as well as an organic semiconductor. Its problematic purification was addressed by Remcho, who developed a centrifugal partition chromatography (CPC) method utilizing a new biphasic solvent system for the preparative-scale isolation and purification of C14.1.274 Electronic properties of C14.1 for potential use in optoelectronic devices were explored by Ostroverkhova and co-workers (Chart 14).275,276 Aggregation of xylindein in THF was detected in the 8–180 μM concentration range by the emergence of a NIR absorption band at 720 nm. This band was thought to arise from a combination of hydrogen bonding and π-stacking interactions. Photoresponsive conductive films were prepared from xylindein and from its blends with PMMA or crystalline nanocellulose.
Chart 14. Xylindein.

In 2015, Takata and co-workers developed a homochiral helical polymer consisting of alternating spirobifluorene and dioxapyrenoid moieties (Scheme 137).64 First, the poly(aryl ether) precursor 137.3 was synthesized through polycondensation of the optically pure spirobifluorene 137.1 and the anthraquinone spacer 137.2. Next, a postpolymerization cyclization was done using concentrated H2SO4 followed by addition of an iodide salt as a reducing agent. In contrast to related polymers with rigid structures, the resulting 137.4 was highly soluble in water because of incidental introduction of sulfonyl groups into the polymer backbone. The sulfonation ratio was estimated to be 2.5 per repeating according to elemental analysis. The isomeric polymer 36.3 with a dioxaperylene substructure was synthesized using the same approach (see Scheme 36, Section 3.1.2).
Miura and co-workers developed a new rhodium-catalyzed C–H activation method for a direct arylation of naphthalene derivatives bearing thioether groups.277 The coupling products 138.2a,b were readily transformed into the corresponding S-doped polyaromatics 138.3a,b (Scheme 138). This process involved oxidation of the thioethers to sulfoxides upon treatment with m-CPBA, followed by cyclization in the presence of acid. The same approach was used to prepare sulfur-containing π-extended phenalenoids (Scheme 197, Section 5.1.3). In a related example, the pyrene-containing thioxanthene 138.5 was obtained via a new strategy of extending the pyrene core from its K region (Scheme 138).67 The final ring annulation was achieved by treating the o-methylthio precursor 138.4 with an excess of iodine in chloroform at an elevated temperature. A product of double cyclization was similarly obtained (Scheme 38, Section 3.1.2). 138.5 was almost perfectly planar in the solid state, with a torsion angle of only 1.43° between the pyrene and benzene fragments. The molecules of 138.5 were arranged into columnar stacks with a π–π distance of 3.554 Å.
Scheme 197. π-Extended Thiaphenalenoids.

Reagents and conditions: (a)376 [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), 3 equiv of Ag2O, 1 equiv of PivOH, HFIP, 100 °C, N2 atmosphere, 24 h.
A series of boron-containing polycyclic aromatic hydrocarbons were synthesized through the ruthenium-catalyzed annulation of arylborane and -silane enynes as the key step (Scheme 139).278 A single cyclization was performed to furnish the silicon-containing 7H-benzo[de]anthracene derivatives 139.2a,b (see also Scheme 193, Section 5.4.1). Double cyclization of an enediyne was also possible, providing the boron-doped 6H-benzo[cd]pyrene 139.6. Si/B exchange was achieved by treating 139.2a,b with excess neat BBr3 at rt followed by mesitylation. 139.3b readily undergoes a light-induced conversion to the fully planarized 139.4b upon irradiation with UV light in the presence of iodine. All the B-doped products proved to be benchtop-stable and showed deep blue photoluminescence with quantum yields of 79–91%.
Scheme 139. Synthesis of Polycyclic Conjugated Arylboranes.

Reagents and conditions: (a)278 PPh3Ru(cymene)Cl2, NH4PF6, 1,2-DCE, 75 °C; (b) (1) neat BBr3, rt, (2) MesMgBr, THF, rt; (c) I2, hν, toluene, rt; (d) PPh3Ru(cymene)Cl2, NH4PF6, 1,2-DCE, 80 °C.
Scheme 193. Synthetic Routes to Boron-, Nitrogen-, and Sulfur-Doped PAHs.
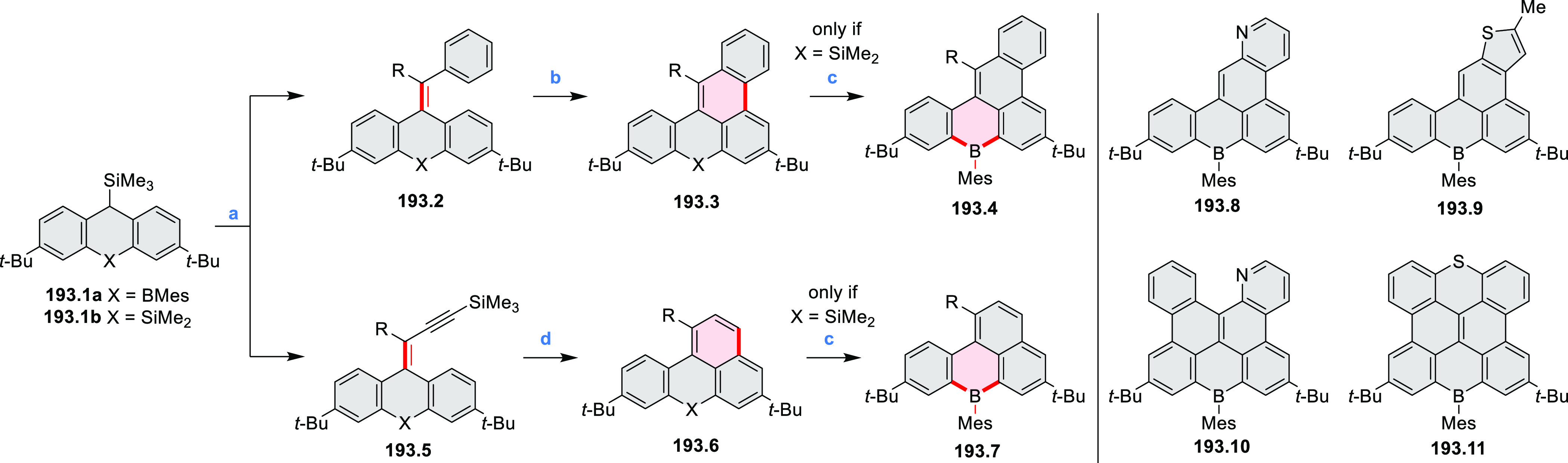
Reagents and conditions: (a)373n-BuLi, Et2O; (b) UV irradiation (Hg medium pressure lamp), I2, propylene oxide, cyclohexane; (c) (1) excess neat BBr3, (2) MesMgBr, toluene/THF; (d) (1) [n-Bu4N]F, THF, (2) (PPh3)Ru(cymene)Cl2, NH4PF6, (CH2Cl)2.
In 2017, Ingleson et al. described the preparation of π-extended diborapyrene derivatives 140.5–6 (Scheme 140).279 In the synthesis, the dialkynyl-substituted naphthalene 140.1 was treated with BCl3 and 2,4,6-tri-tert-butylpyridine to initiate electrophilic cyclization involving the phenyl groups. The resulting BCl2-containing intermediate was reacted with stoichiometric amounts of AlCl3 and 2,6-dichloropyridine to induce the C–H borylative cyclization with the naphthalene core to give the diborapyrene derivative 140.2 in 86% yield. Subsequent dehydrogenative aromatization with trityl tetrafluoroborate yielded compound 140.3, which then reacted with either the aryllithium 140.4 or 2,4,6-triisopropylphenylmagnesium bromide (TipMgBr) to give 140.5 and 140.7, respectively. The former product 140.5 could undergo 4-fold electrophilic cyclization to give 140.6. In subsequent work, Ingleson and Zysman-Colman et al. reported the boron-doped π-extended pyrenes 140.8–11 through a bromoboration–electrophilic borylation sequence on the corresponding monoalkynyl- and dialkynyl-substituted pyrenes (for details, see Scheme 99, Section 3.4).202 A bromine-free analogue of compound 140.9 was also prepared using an alternative approach from an olefinic precursor, as reported by Würthner et al. (cf. Scheme 34, Section 3.1.2).61
The Pummerer oxidative annulation reaction was utilized to extend PAHs through the formation of an intramolecular C–O bond with a suitable phenol substituent (Scheme 141).159 The pyrane-containing 141.3 was synthesized by treatment of 141.2 with copper(II) oxide in boiling nitrobenzene. Depending on the topology of the phenol-substituted precursor, five- and seven-membered rings could be obtained (furans and oxepines, respectively, cf. 141.4 and Scheme 80, Section 3.3). The annulations led to substantial changes in the photophysical and electrochemical properties relative to the nonfused precursors. Fusion with the pyrane ring in 141.3 caused a strong bathochromic shift and enhancement of quantum yields (141.1: λem = 384 nm, Φ = 25%; 141.3: λem = 450 nm, Φ = 100%). Conversely, the oxepin annulation had a detrimental effect on the emission properties of 80.2 (Scheme 80, Section 3.3). The same cyclization approach was used for the preparation of the π-extended pyrenoid 141.5 bearing two oxygen atoms (for its perylenoid isomer 46.3, see Scheme 46, Section 3.1.2).77
4.4. [cd]-Heterofused Pyrenoids
In 2018, Zakrzewski and co-workers developed a TfOH-promoted cyclization method of polycyclic aromatic N-ethoxycarbonylthioamide S-oxides, which provides pyrenes with a [cd]fused thiophene imine unit (Scheme 142, see Scheme 93, Section 3.4, for perylene analogues).192 The proposed reaction mechanism involves the intermediacy of an electrophilic sulfur species, either a protonated iminosulfenic acid or an iminosulfenium cation. These species may attack either the peri- or ipso-position of the arene, leading to regioisomeric products like 142.2b and 142.2b′. Generation of sulfenium-type electrophiles in this reaction was also supported by isolation in a small amount of an intermolecular coupling product. In contrast to 142.2a and 142.2b′, which exhibit very weak emission (Φf < 0.01), 142.2b fluoresces with a quantum yield of 64% in a DCM solution.
4.5. [a]-Heterofused Pyrenoids
In 2021, Nagahora et al. reported a synthesis of thiopyrylium-fused aromatics, including the pyrene derivative 143.4 (Scheme 143).280 Diaryl thioether 143.3 with a cyclic-acetal-protected aldehyde function was prepared in an SNAr-type reaction in the presence of Cu2O in trimethylpyridine. Treatment of this compound with TfOH resulted in acetal cleavage and condensation, leading to the thiopyrylium-containing 143.4. Compared to analogues that contained smaller aromatic systems, 143.4 had a much smaller HOMO–LUMO gap, resulting in an additional weak and broad NIR absorption band with a maximum at 686 nm.
Scheme 143. Synthesis of Thiopyrylium-Fused Pyrene.

Reagents and conditions: (a)280 Cu2O, 2,4,6-trimethylpyridine, 150 °C, 48 h, 59%; (b) TfOH, DCM, rt, 2 h, 64%.
A vicinal electrophilic diborylation reaction reported by the Wagner group provides access to doubly boron-doped PAHs such as the benzopyrene-containing 144.4 (Scheme 144).281 The latter system was obtained by treating 4,5-dichloro-1,2-bis(trimethylsilyl)benzene 144.1 with BBr3 in the presence of benzo[a]pyrene, followed by addition of mesitylmagnesium bromide. The transformation was proposed to proceed through a highly electrophilic diborylated intermediate 144.2 formed from 144.1 in a reaction with BBr3. Other B-doped aromatics were synthesized in this manner, including the fluoranthene-fused 144.5. Compound 144.4 exhibited intense photoluminescence with a maximum at 503 nm and a quantum yield of 82% in cyclohexane solution. The emission maximum position depended on solvent polarity and was bathochromically shifted by about 100 nm in acetonitrile.
Scheme 144. Vicinal Diborylation Reaction toward Doubly Boron-Doped Polycyclic Arenes.
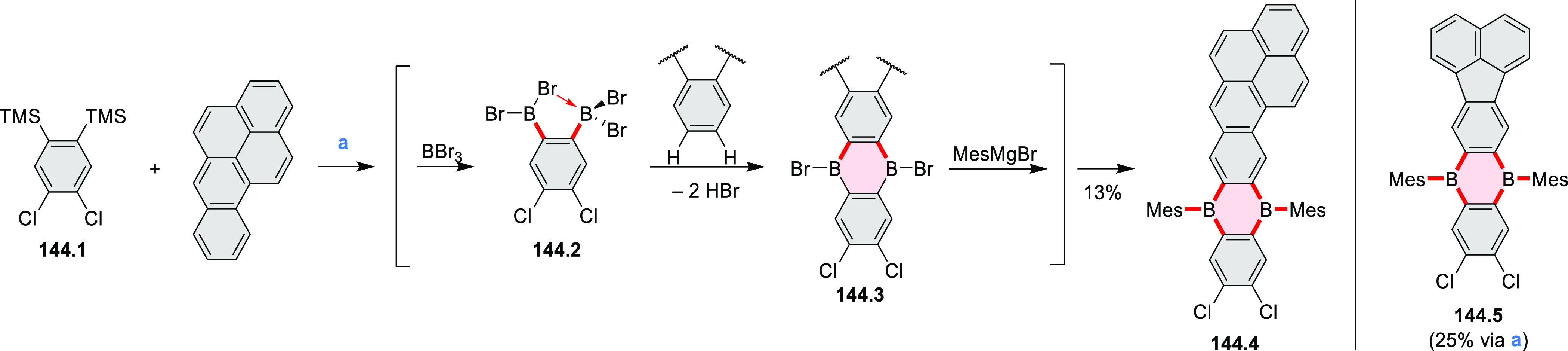
Reagents and conditions: (a)281 (1) BBr3, n-hexane, ampule flame-sealed under vacuum, 120 °C, 2.5 days, (2) MesMgBr, toluene/THF, 0 °C, then rt, overnight.
Synthesis of heterofused pyrene derivative 145.3 was achieved by an iridium-catalyzed direct fusion of pyrene-1-oxime methyl ether 145.1 and benzothiophene 145.2, which is a successful example of a cascade C–H/C–H cross-coupling/cyclization strategy (Scheme 145).282 The reaction was proposed to begin with an oxime-directed C–H activation of 145.1 followed by C–H arylation of benzothiophene in an [IrII]–[IrIV] catalytic cycle with Ag2O as the terminal oxidant. The resulting cross-coupled intermediate underwent cyclization via two consecutive one-electron oxidations by Ag2O.
Scheme 145. Synthesis of a Benzo[4,5]thieno[3,2-b]pyridine-Fused Pyrene.

Reagents and conditions: (a)282 [Cp*IrCl2]2, AgSbF6, Ag2O, Zn(OTf)2, hexafluoroisopropanol, 140 °C, 24 h.
The majority of the recently reported [a]hetero-fused pyrenoids contains a thiophene ring (see Scheme 141 for related furan derivatives and ref (283) for systems containing phosphole and silole rings). In 2019, Procter and co-workers developed a one-pot approach for the synthesis of dihydrothiophene-fused arenes proceeding through a sequence of an intermolecular interrupted Pummerer reaction between an activated sulfoxide 146.5 and nonprefunctionalized pyrene 146.1, followed by Claisen-type [3,3]-sigmatropic rearrangement and cyclization (Scheme 146).284 The oxidation/aromatization of the 2,3-dihydrobenzothiophene 146.2 by treatment with DDQ in hot toluene led to the thiophene-fused 146.3. Alternatively, using the chlorinated starting material 146.6 produced 146.3 in 62% yield without the final oxidative step. In that case, intermediate 146.4 was formed, which was converted to 146.3 by demethylation and HCl elimination. This transition-metal-free thienannulation was also utilized to obtain various thiophene-fused systems, e.g., ones containing fluoranthene (146.7) and corannulene (Scheme 223, Section 6.1.1).
In 2018, Sun and Chen reported a synthesis of the two anthanthrene derivatives fused with thiophene and benzothiophene, 147.3 and 147.4, respectively (Scheme 147).285 Their synthetic route started with intramolecular Friedel–Crafts cyclization of 1,6-bisthienylopyrene 147.1, providing the key intermediate 147.2 in high yield. Treatment of this compound with lithium alkynide prepared from TIPS-acetylene resulted in 1,4- and 1,2-addition to its α,β-unsaturated ketone substructures. After subsequent treatment with SnCl2, the tetrasubstituted product 147.3 was obtained in 19% yield. 147.4 bearing two additional benzene rings was synthesized using a similar approach. Both products exhibited a red emission at 662–663 nm. An additional vibronic NIR emission band was observed at 715 nm in 147.3 and 723 nm in 147.4. Likewise, two isomeric pyrenodithiophenediones 147.6 and 147.9 were converted into their TIPS-ethynyl functionalized derivatives 147.7a–c and 147.10a–c. In this case, mixtures of derivatives with two, three, and four TIPS-ethynyl substituents were obtained.286 Increasing the number of appended alkynes lowered the LUMO energy levels while having little impact on the HOMO energy. These changes were reflected in bathochromic shifts of absorption bands.
Scheme 147. Synthesis of TIPS-Substituted Thieno-Fused Anthanthrenes and Pyrenes.
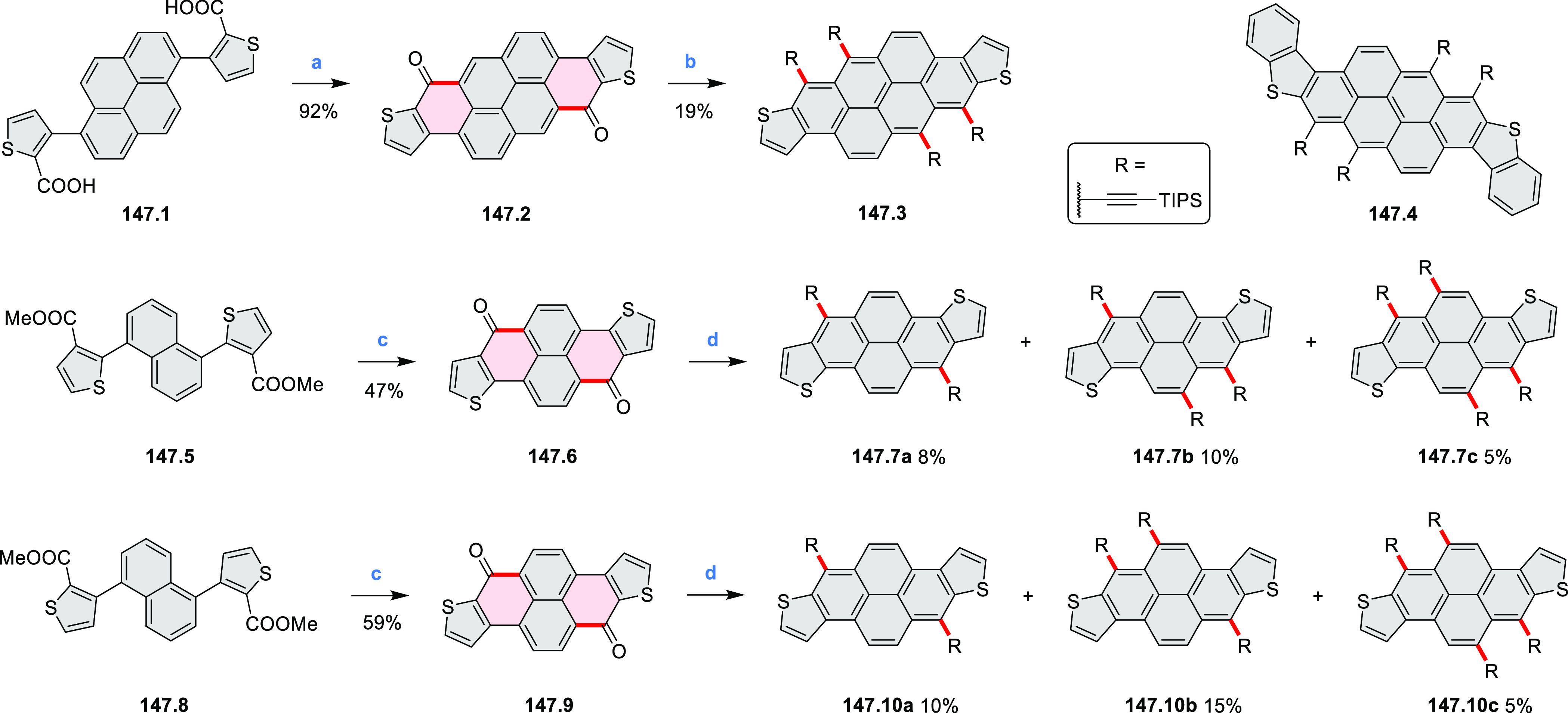
Reagents and conditions: (a)285 polyphosphoric acid, 90–120 °C, 24 h; (b) (1) triisopropylsilylacetylene, n-BuLi, THF, 0 °C, 1 h then 147.2, rt, 6 h, (2) SnCl2·2H2O, HCl, rt, 3 h; (c)286 TfOH, MsOH, 140 °C, 4 h; (d) (1) triisopropylsilylacetylene, n-BuLi, Et2O, 0 °C, 1 h, then 147.6 or 147.9 in THF, rt, overnight, (2) SnCl2·2H2O, HCl, rt, 1 h.
Oxidative coupling with FeCl3 was used to convert two isomeric bis(pyren-2-yl)thiophenes 148.1a,b into the fully fused compounds 148.2a,b (Scheme 148).287 Attempted cyclization on thiophenes carrying three and four pyren-2-yl groups was not successful. While compound 148.2b with a [b]fused thiophene was obtainable in high yield, its [c]fused isomer 148.2a was unstable and could only be obtained in 5% yield. The stable isomer 148.2b was analyzed by single-crystal X-ray diffraction, revealing a 39° angle between its [5]helicene blades.
Scheme 148. Synthesis of a Dithiophene-Fused Benzopyrene.

Reagents and conditions: (a)287 FeCl3, MeNO2, 0 °C, 10 min for 148.2a or 1 h for 148.2b.
4.6. Pyrazine-Fused Systems (Pyrazaacenes)
Condensation of pyrene diones and tetraones, such as 149.1 and 149.8 (Scheme 149), with aromatic amines provides a very general approach toward large N-doped “pyrazaacenes”, usually with ribbon-like structures (cf. CR2017, Section 4.6, for earlier developments and Scheme 260, Section 6.1.6, for related 2D materials). Pyrazaacenes can be tailored to perform a variety of functions; for instance, they can be elaborated into ligands. Poyatos and Peris described the preparation of NHC ligands containing fused phenanthro[4,5-abc]-phenazine units (for acenaphtho analogues see Chart 25, Section 6.2.1).288 First, azolium salts 149.2a,b were prepared by condensation of diketone 149.1a,b and 1,3-dibutyl-5,6-diaminobenzimidazolium iodide 149.1 in yields of 78 and 79%, respectively. The coordination of 149.2a–d to iridium was then achieved by deprotonation with potassium tert-butoxide and subsequent addition of [IrCl(cod)]2. Furthermore, treatment of 149.3c,d with carbon monoxide in methylene chloride afforded the related carbonyl complexes 149.4c,d in excellent yields. Addition of π-stacking additives such as pyrene and hexafluorobenzene was investigated for its possible effect on the electron-donating character of the ligands.
Scheme 149. Synthesis of Iridium Complexes Containing Phenanthro[4,5-abc]phenazino[11,12-d]imidazol-2-ylidene.
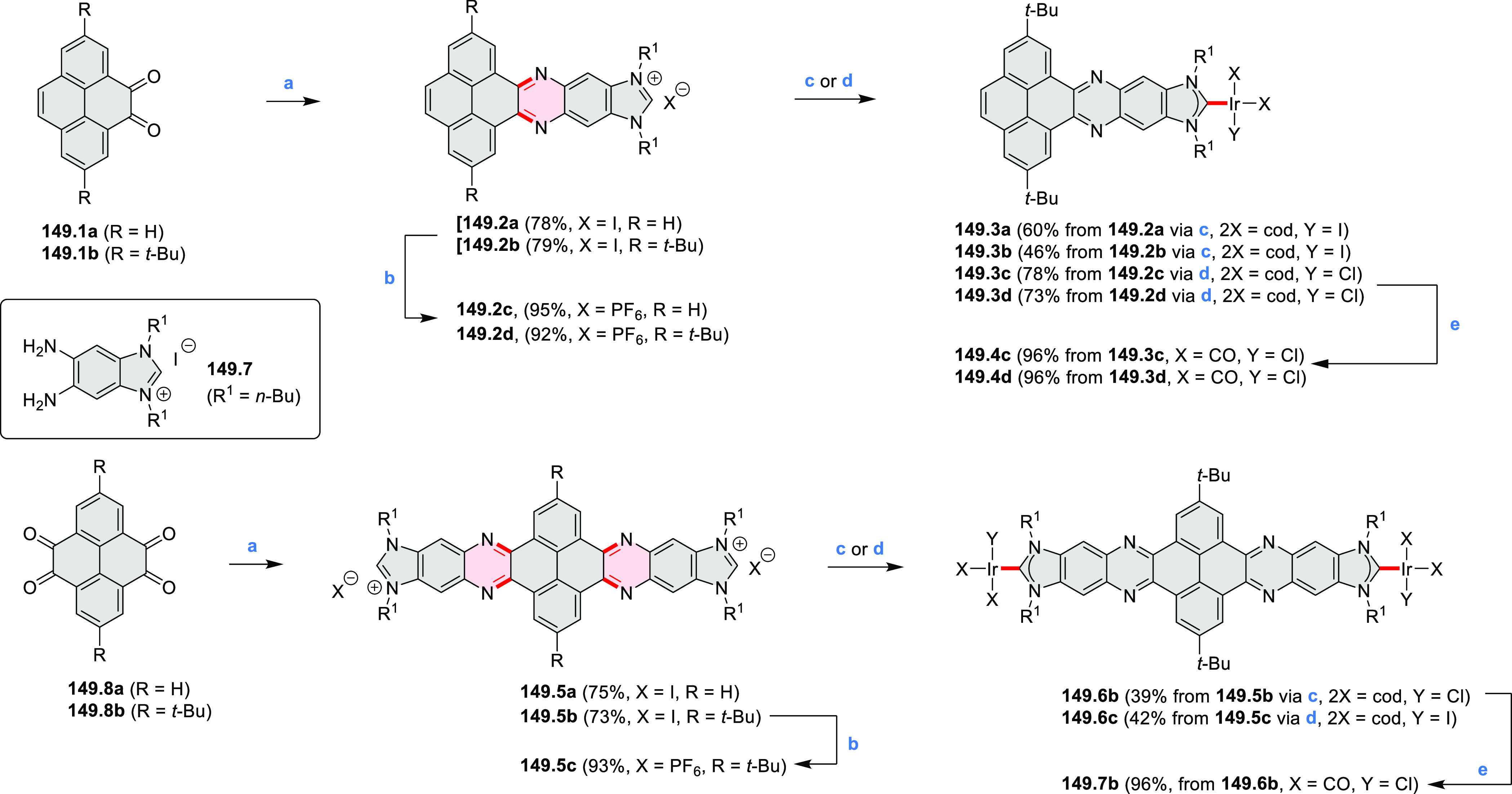
Reagents and conditions: (a)288,289149.7, MeOH, reflux, overnight; (b) NH4PF6, MeOH/DCM (3:1), rt, overnight; (c) potassium tert-butoxide, NaI (in the case of 149.3a,b), [IrCl(cod)]2, THF, rt, overnight; (e) CO, DCM, 0 °C, 20 min; (d) potassium tert-butoxide, [IrCl(cod)]2, KI (in case of 149.6c), THF, rt, overnight.
Chart 25. Acenaphthylene-Based Quinoxalines.
Janus-like bis-N-heterocyclic carbene (bis-NHC) ligands 149.5a,b were obtained by the same group by following a similar direct condensation approach with the corresponding pyrene-4,5,9,10-tetraone 149.8a,b.289 Anion metathesis of 149.5b with ammonium hexafluorophosphate allowed the preparation of the corresponding PF6 salt, 149.5c, in 93% yield. Due to their higher solubility, the di-tert-butyl-substituted bisazoliums 149.5b and 149.5c were chosen for coordination experiments. The reaction of the bis-azolium salt 149.5c with [IrCl(cod)]2 produced the di-iridium complex 149.6c in 42% yield. In the absence of the KI additive, the reaction of 149.5b afforded the chlorine analogue complex 149.6b. Treatment of 149.6b with carbon monoxide in DCM gave the tetra-carbonyl species 149.7b. The X-ray crystal structure of 149.6c revealed a metal-to-metal distance of 22.4 Å.
In 2017, Zhu and Zhang successfully synthesized a pyrene-containing nitrogen-rich nanoribbon with 15 six-membered rings linearly annulated in one row.290 Compound 150.3 was obtained in a good yield of 70% through the reaction between diamine 150.2 and an excess amount of tetraketone 150.1 (Scheme 150). Both substrates were substituted by triisopropylsilyl groups to increase their solubility and crystallizability. Next, the condensation of 150.3 and 150.4 followed by reductive ring opening was performed yielding 150.6. Finally, the target compound 150.7 was prepared by condensing diketone 150.3 with diamine 150.6 in a mixture of chloroform and AcOH under reflux conditions. The single X-ray crystal structure of 150.7 shows a slightly twisted structure at the two pyrene units which might stabilize this large azaacene system by releasing the strain resulting from structure packing. The optical band gap of 150.7 was found to be 1.85 eV, and this compound exhibits emission maxima at 661 nm with a fluorescence quantum yield of 3%. The low Φf value might be attributed to the self-absorption effect and intersystem crossing.
Scheme 150. Synthesis of a Pyrene-Fused N-Heteroacene.
Reagents and conditions: (a)290 (1) CHCl3/AcOH (2:1), 80 °C, 36 h, (2) MnO2, DCM; (b) CHCl3/AcOH (2:1), 80 °C, 30 h; (c) LiAlH4, THF, 0 °C to rt, 14 h; (d) CHCl3/AcOH (4:1), 80 °C, 48 h.
In their search for solid-state electron acceptors, the Mastalerz group revisited four previously known di- and tetracyano-substituted pyrene-fused pyrazaacenes and synthesized a new derivative 151.2.291 Except 151.1, single crystals from all known compounds were grown for the first time. 151.1 and 151.2 were prepared by condensing pyrenedione 149.1a with 2,3-diaminomaleonitrile and 4,5-diaminophthalonitrile, respectively, under acidic conditions (Scheme 151). Tetracarbonitriles 151.3a,b and 151.4 were obtained similarly from the corresponding pyrenetetraones 149.8a,b. The electron-deficient nature of these compounds and low-lying LUMOs (up to −3.9 eV) makes them suitable as electron-transport materials. X-ray crystallographic analyses revealed that all the structures formed face-to-face columnar stacks with small π–π distances (ca. 3.29–3.51 Å; average: 3.37 Å). Calculated charge transfer integrals for electron transport (69–76 meV for 151.3b, 101–172 meV for 151.2, 108 meV for 151.4, and 113–252 meV for 151.1) and small reorganization energies (91–256 meV) indicated that those four compounds might be useful as n-type semiconductors.
Scheme 151. Synthesis of Di- and Tetracyanopyrazines.

Reagents and conditions: (a)291 2,3-diaminomaleonitrile, EtOH/AcOH (1:1), 80 °C, 11–16 h; (b) 4,5-diaminophthalonitrile, EtOH/AcOH (1:1), 80 °C, 11–16 h.
Asymmetric pyrazine–pyrene–imidazole systems with a D−π–A structure were similarly synthesized (Scheme 152).292 In the second step, the Debus–Radziszewski condensation was employed, furnishing the target imidazoles 152.3a and 152.3b in moderate yields. The color of 152.3a and 152.3b changed from light yellow to brown upon protonation with trifluoroacetic acid in DCM solution. Thin films of 152.3b showed reversible electrochromic behavior accompanied by color alteration from yellow to green under different bias voltages, observable by in situ UV–vis and NIR spectroscopy.
Scheme 152. Synthesis of Unsymmetrically Fused Pyrazaacenes.

Reagents and conditions: (a)292 1,2-diamino-4,5-didodecyloxybenzene, AcOH, CHCl3, reflux; (b) AcOH, reflux.
Stepwise annulation protocols were employed in the syntheses of thienyl-substituted pyrazaacenes, such as the [1,2,5]thiadiazolo[3,4-g]quinoxalines, reported by Zhang et al.293 Here, the key annulation step was performed between pyrene-4,5-dione 149.1a or 2,7-di-tert-butylpyrene-4,5-dione 149.1b and thiadiazole-fused diamine 153.1, providing the target compound 153.2a and 153.2b, respectively (Scheme 153). In OFET investigations, 153.2a and 153.2b showed typical p-type characteristics with mobilities of up to 0.05 and 0.0055 cm2 V–1 s–1 and on/off current ratios of 1 × 106 and 1 × 104, respectively. Because of bulkier substitution, compound 153.2b adopted looser packing in the solid state, and the larger π– π distances reduced the transport performance. A related thiadiazole-based charge-transfer system, 153.3, showed an excellent nonvolatile tristate memory behavior, with a ternary device yield as high as 78% and retention stability longer than 104 s.294 The ribbon-like 153.4 with 12 linearly fused aromatic six-membered rings was well soluble in chlorinated solvents but could also be easily crystallized, showing a zigzag packing motif in the solid state.295 Single-crystal organic field effect transistors fabricated from 153.4 through an organic ribbon mask technique exhibited hole mobility of up to μh = 8.1 × 10–3 cm2 V–1 s–1. The related nanoribbon 153.5 was shown by Lee and co-workers to form organogels with hydrocarbon solvents and select halogenated solvents, with the lowest critical gelation concentration of 1.2 mM in hexadecane.296153.5 showed a relatively stabilized ELUMO of −3.74 eV due to the electron-deficient nature of the core and reduced band gap of 1.55 eV as a result of intramolecular charge transfer between the core and axial thiophene.
Scheme 153. Synthesis of Thiophene-Substituted Pyrazine-Fused Pyrenoids.
Reagents and conditions: (a)293 AcOH, 100 °C, 24 h.
Thiadiazole-capped pyrazaacenes were obtained in several laboratories via stepwise condensations summarized in Scheme 154 and investigated as organic semiconductors.297−299 In particular, 154.3a and 154.3b were used as LED emitters, showing red–NIR emission bands, centered at 658 and 721 nm, respectively.297154.3a was also explored as an active material in resistive memory devices.298 Charge carrier transport properties in the homologous series consisting of 154.3a, 154.5a, and 154.7 were evaluated in “top contact bottom gate” FETs.299 In particular, devices based on a 154.3a single crystal exhibited an average electron mobility of about 6 × 10–4 cm2 V–1 s–1, with an ON/OFF ratio of 2000 and a threshold voltage (Vth) of 25 V. For a single crystal of 154.7, the average mobility was higher than that of 154.3a, with an electron mobility of approximately 5 × 10–3 cm2 V–1 s–1, an ON/OFF ratio of 500, and a threshold voltage (Vth) of 20 V. The highly soluble variant 154.5b substituted with tert-butyl and TIBS groups was used for OFET device fabrication.300 Thin films of 154.5b deposited from solution show an n-type behavior and a maximum electron mobility of μemax = 1.4 × 10–4 cm2 V–1 s–1 without any device optimization.
Scheme 154. Synthesis of Thiadiazoloquinoxaline-Containing Long Pyrene-Fused N-Heteroacenes.
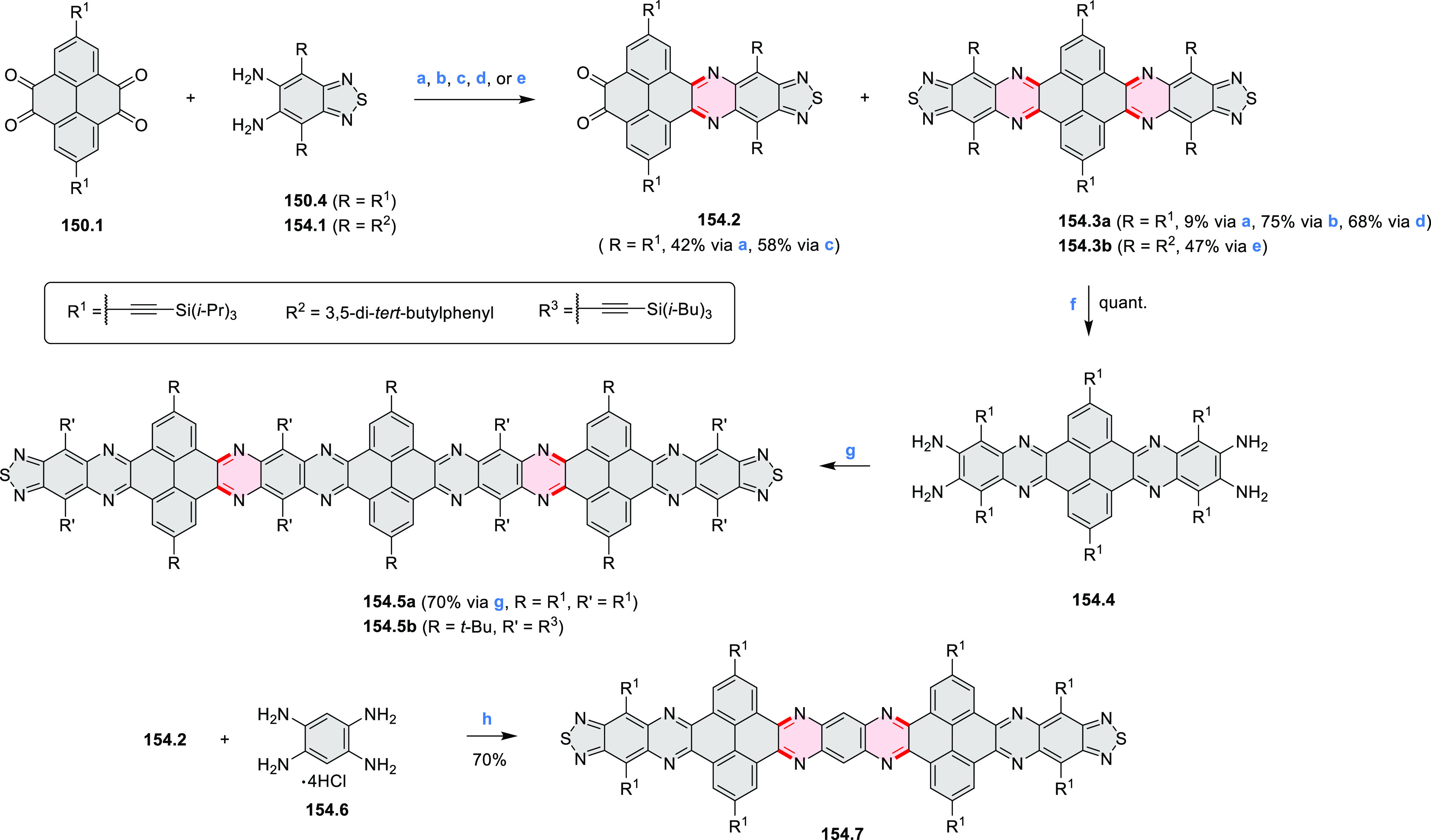
Reagents and conditions: (a)299150.1/150.4 (1:1), CHCl3/AcOH (2:1), reflux, 1 day; (b)298150.1/150.4 (1:2), CHCl3/AcOH (1:1), 80 °C, 1 day; (c)298150.1/150.4 (1:1), CHCl3/AcOH (1:1), reflux, 1 day; (d)297150.1/150.4 (1:2.9), AcOH, 132 °C, overnight; (e)297150.1/154.1 (1:3), AcOH, reflux, overnight; (f)299 LiAlH4, THF, 0 °C to rt, overnight; (g)299154.2, CHCl3/AcOH (1:1), reflux, 1 day; (h)299 CHCl3/AcOH (1:1), reflux, 1 day.
A mechanochemical synthesis under solvent-free ball-milling conditions was reported as an efficient route to C15.1a–e, C15.2a–e, C15.3a–c, and C15.4a–c (Chart 15).301 Di- or tetraketone pyrenes were used as precursors along with the appropriate 1,2-diaminoarenes, and the reactions took 3 to 4 h under ambient laboratory conditions with p-toluenesulfonic acid as the catalyst. In 2018, thin films of two pyrazaacenes, C15.4c and its dodecyloxy-substituted derivative C15.5, on silicon/native silica (Si/SiO2)native and fused silica substrates were analyzed with respect to their microstructural and anisotropic optical properties (Chart 15).302 Because of solubility differences, thin films of C15.4c were obtained by organic molecular beam deposition in ultrahigh vacuum conditions, while C15.5 layers were produced from solution by spin coating. The two films exhibited different textures, each with strongly uniaxial anisotropy. The molecules of C15.4c were arranged parallel to the surface, while the cores of C15.5 were cofacially packed and tilted with respect to the surface normal. In another report, Tegeder et al. investigated the adsorption geometry and electronic properties of C15.3a and its 2,11-di-tert-butyl analogue C15.4a as a function of coverage on Au(111).303 Both molecules adopted a planar geometry with respect to the substrate in both a single monolayer and thin films (up to 10 monolayers thick). In contrast, in the crystal structures, a tilt of up to 82° was observed between molecular planes in neighboring stacks.
Chart 15. Pyrene-Fused Azaacenes.

Further work on pyrazaacene–triptycene hybrids was reported by Mastalerz and co-workers. Triptycenylene end caps were, for instance, introduced to enhance the solubility of quinoxalinophenanthrophenazines 155.2a,b (Scheme 155).304 In this way, the formation of LC phases was avoided, a behavior often caused by functionalization with long alkyl and alkoxy chains. Compounds 155.2a,b were synthesized in 60% and 93% yield, respectively, by condensing diammonium triptycene dichloride 155.1 with pyrene tetraketones 149.8a,b. In 2015, Mastalerz described four polymorphs of triptycene-based 155.3b, an organic molecule of intrinsic microporosity (OMIM), which showed an unusual gas sorption behavior with a very large hysteresis (Scheme 155).305 The material could not be activated by solvent exchange; however, by thermal treatment, high specific surface areas of up to 350 m2 g–1 could be generated reproducibly. Quinoxalinophenanthrophenazines 155.6a,b with triptycenylene units were used as the model compounds to get a deeper impact of the dispersion interaction of the tert-butyl groups and the triptycenylene units on the formation of the unusual packing motif of 155.3a.306 The monotriptycenylene-substituted 155.6a and 155.6b were obtained from the annulation reactions between either 155.7a or 155.5 with the corresponding o-phenylene diamine in 67% and 63% yield, respectively. Well-balanced dispersion contributions of tert-butyl groups and one triptycenylene unit at the periphery of the π plane both seemed to affect packing motifs, leading 155.6a and 155.6b to adopt a brick-wall arrangement of π planes in the solid state.
Scheme 155. Synthesis of Triptycene End-Capped Quinoxalinophenanthrophenazines.
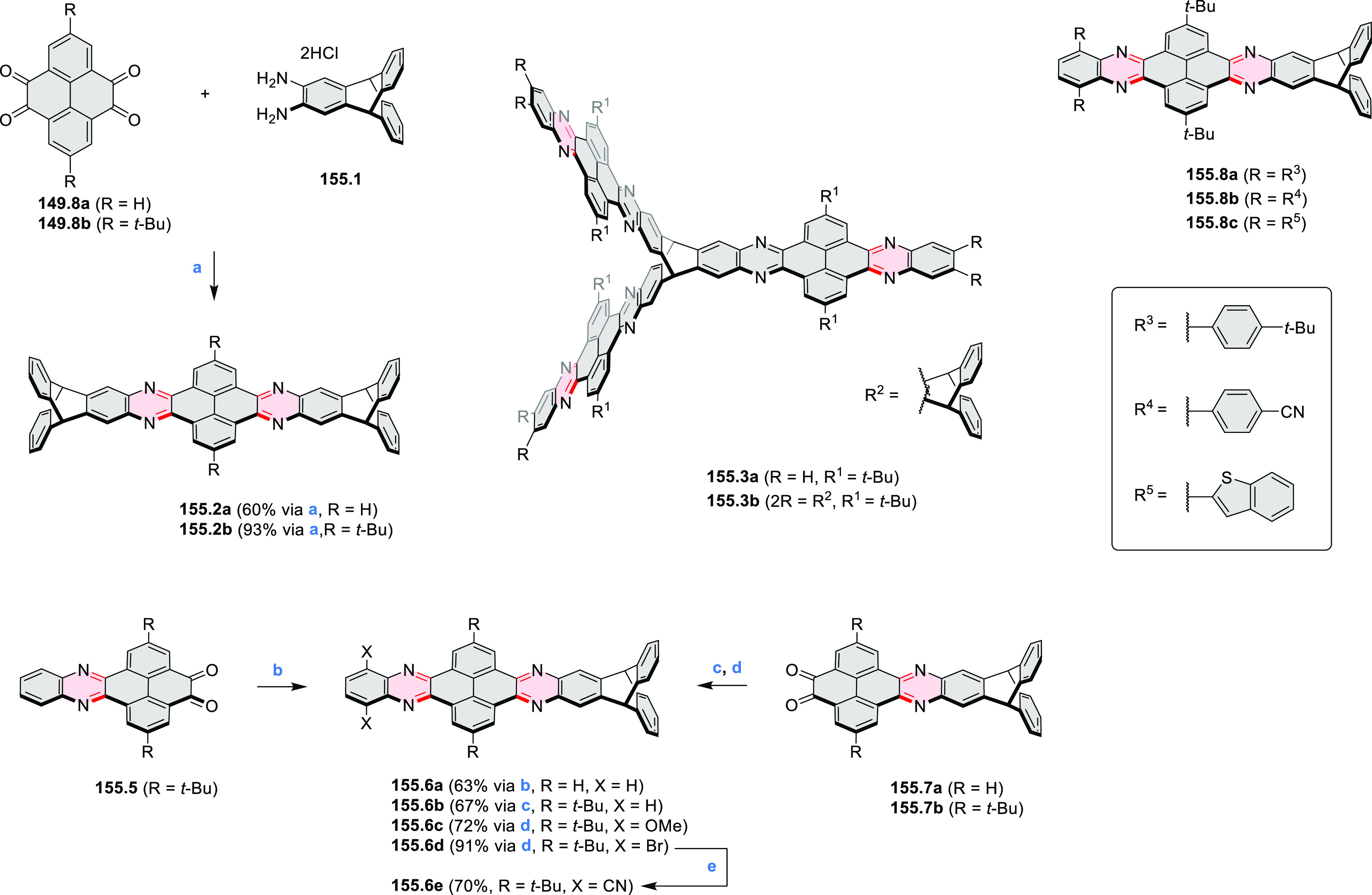
Reagents and conditions: (a)304 KOAc, CHCl3, AcOH, 70 °C, 16 h; (b)306155.1, KOAc, CHCl3, AcOH, 70 °C, 16 h; (c)306o-phenylenediamine, CHCl3, AcOH, 70 °C, 16 h; (d)307 appropriate o-phenylenediamine, CHCl3, AcOH, 70 °C, 16 h; (e) NMP, CuCN, 180 °C 15 h.
To study the effect of substitution on crystal packing, the Mastalerz group obtained 17 single-crystal solvates of four QPP structures (155.6c–e and 155.6b).306,307 All crystal structures formed the cofacial π–π dimers and the degree of overlap, and the arrangement of these dimers to each other depended on solvents, substituents, and crystallization conditions. For some crystalline structures of 155.6b, a high degree of LUMO–LUMO overlap and high transfer integrals were found. Related investigations were performed on three triptycene-end-capped QPP derivatives 155.8a–c bearing aromatic substituents at the peripheral phenylene units.308 Absorption spectra of these films indicated increased π stacking tendency of 155.8b and 155.8c relative to 155.8a. The formation of π-dimers occurred to a different degree in all crystal structures, showing that the directing capability of the triptycene end capping was also pronounced in the presence of larger aromatic substituents that could disturb the packing by competing π-stacking interactions (e.g., in 155.8c). The group also reported charge-transfer (CT) complexes based on 155.6c with six small electron-deficient molecules.309
In 2021, Mastalerz et al. reported the synthesis and properties of the π-extended tribenzotriquinacene (TBTQ) derivatives C16.1–2 bearing one and three quinoxalinophenanthrophenazine units (Chart 16).310 These two species were obtained in two steps from the corresponding diamino- and hexamino-substituted TBTQs, respectively. Both compounds C16.1–2 had similar absorption and emission profiles. In particular, compound C16.2 exhibited solvatofluorochromism, with the emission maximum becoming increasingly red-shifted in the order of hexane, toluene, THF, chloroform, and DCM.
Chart 16. Quinoxalino–Phenanthrophenazine-Fused Tribenzotriquinacenes.

In 2019, Hu and Baumgarten reported triptycene-based three-dimensionally extended pyrazaacenes (156.3–6) obtained by an iterative approach (Scheme 156).311156.3a was synthesized by the condensation of the intermediate diketone 156.2 and hexamine triptycene hydrochloride salt [156.1][6HCl]. To further increase the length of nanoribbons to 156.4–6, the thiadiazole units were reduced by a large excess of LiAlH4 in THF to the corresponding diamine moieties, which were then condensed with 156.2. The diameters of 156.3–6 were calculated to be 3.66, 6.06, 8.48, and 10.88 nm, respectively. Compared to their linear counterparts (154.5a, 18 rings, only soluble in hot o-dichlorobenzene, Scheme 154),299156.3–6 (156.6 with 22 rings) all showed good solubility in DCM, THF, chlorobenzene, and tetrachloroethane.
Scheme 156. Synthesis of Triptycene-Based Three-Dimensional Pyrene-Fused N-Heteroacenes.
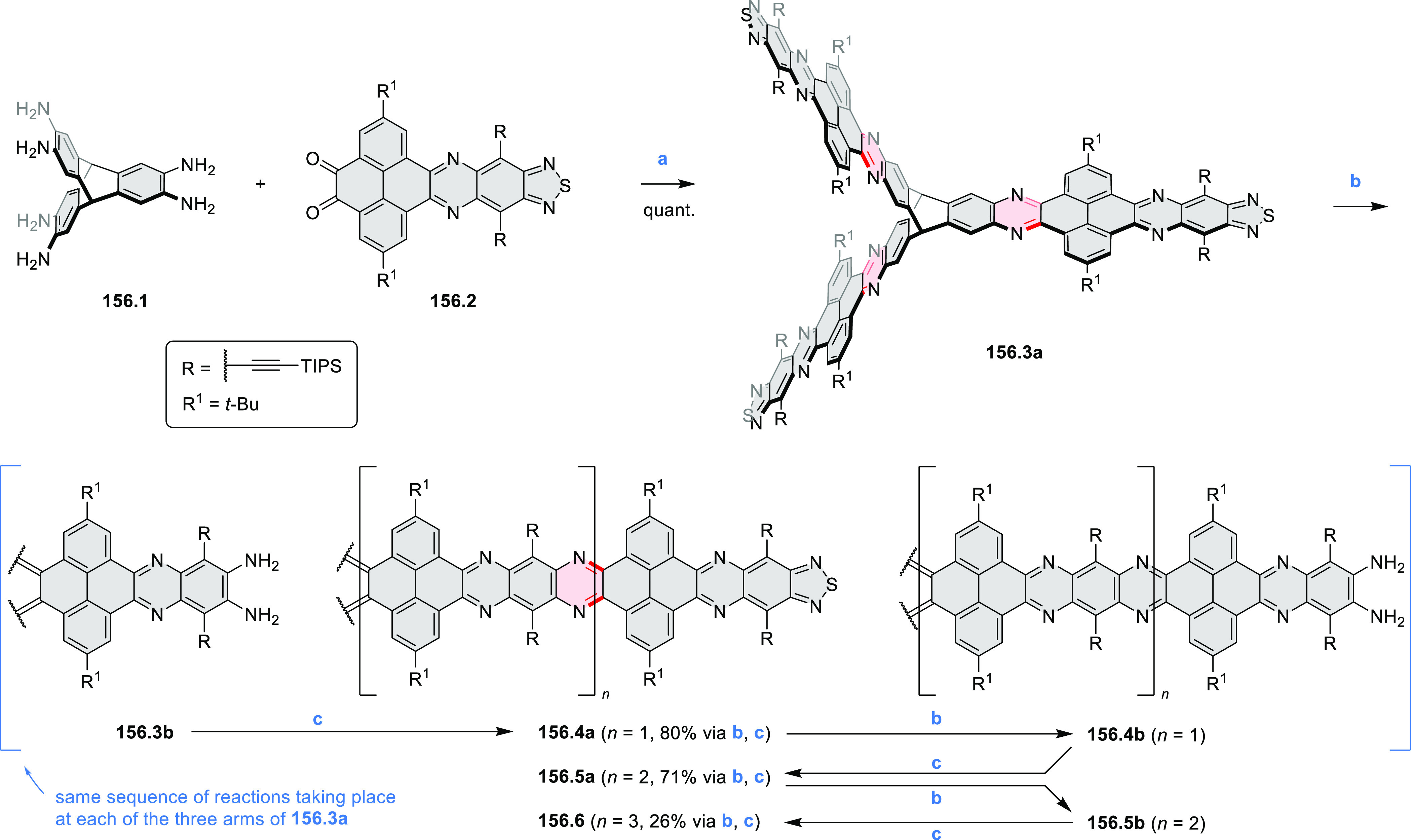
Reagents and conditions: (a)311 KOAc, AcOH, chlorobenzene, reflux, 2 days; (b) LiAlH4, THF, 0 °C to rt; (c) 156.2, KOAc, AcOH, chlorobenzene, reflux, 2 days; (d) (1) LiAlH4, THF, 0 °C to rt, (2) 156.2, KOAc, AcOH, chlorobenzene, reflux, 2 days.
In 2021, Sun et al. reported the synthesis of three [10]CPP dimers 157.1a–c with a rigid pyrazaacene linker (Scheme 157).312 The combined VT-NMR spectroscopic analysis and theoretical analysis indicated rapid interconversion of cis and trans conformers at rt, with trans conformers favored by 2.3 kcal mol–1 and an energy barrier of 9.0 kcal mol–1 for the cis–trans interconversion. Compound 157.1c in DCM exhibited three absorption maxima at 284, 340, and 429 nm and a broad absorption in the range of 440–550 nm. This compound exhibited a large effective Stokes shift of 276 nm, emitting at 616 nm with a quantum yield of up to 80%. A host–guest binding study of 157.1c revealed that it could bind two C60 molecules with K1 = 4.15 ± 0.58 × 105 M–1 and K2 = 3.63 ± 0.44 × 103 M–1.
Scheme 157. Dimeric Cycloparaphenylenes with a Rigid Aromatic Linker.

Conformations interconvert at rt.
In 2018, Melle-Franco and Mateo-Alonso reported on an iterative assembly of a small molecular building block into pyrazaacene nanoribbons containing up to 30 conjugated linearly fused rings (Scheme 158).313 The selectivity of individual iterations was achieved using diacetal-protected o-dione functionalities, which were unmasked before each consecutive condensation step. In the last completed iteration, performed without isolating the intermediate tetraone, the 7.7 nm long tetradecabenzodotriacontacene 158.5 was obtained in a yield of 25%. The solubility of these nanoribbons could be ensured by introduction of relatively small tert-butyl and tri-isobutylsilyl substituents. Electronic spectra showed the expected bathochromic shift and an increase of absorptivity in the series 158.2, 158.4, and 158.5. The photoluminescence spectra in chloroform were almost superimposable over the whole series of nanoribbons; however, the quantum yields of 158.2 (0.27), 158.4 (0.14), and 158.5 (0.11) decreased with increasing lengths. Pseudophotoconductivities were found to be nearly length-invariable for this family of N-doped graphene nanoribbons.
Electronic properties of pyrazaacenes can be fine-tuned by ring fusion and introduction of substituents (Scheme 159). In one recent example, pyrene–phenazine imides 159.1a,b and 159.2a,b were synthesized by annulation of an appropriate diaminophthalimide and the corresponding di- or tetraketone (Scheme 159).314 These systems self-assemble into high aspect ratio crystalline tapes and might be usable as n-channel semiconductors with charge carrier mobilities ranging from 0.18 up to 4.1 cm2 V–1 s–1 for 159.2b and 159.1a, respectively. The luminescence of the N-alkylated imides, 159.1b and 159.2b, was quenched in the presence of acid. Pyrene-fused azahexacene 159.4a and azaheptacene 159.4b containing boron–nitrogen units were synthesized by treatment of either 159.3a or 159.3b with BF3·Et2O and triethylamine in DCM at 50 °C (Scheme 159).315159.4a and 159.4b exhibited low-lying LUMO energy levels and high electron affinities, thus demonstrating their n-type character. As a result, solution-processed OFET devices based on 159.4b exhibited unipolar n-type characteristics with an electron mobility of up to 0.21 cm2 V–1 s–1. A lowering of LUMO energies correlating with the electron-withdrawing character of substituents was also demonstrated for perylene-functionalized acenes 159.5–7 and 159.7a–c.316
Scheme 159. Synthesis of Azaacenes Containing BN Units.

Reagents and conditions: (a)315 BF3·Et2O, Et3N, DCM, 50 °C; (b)316 malononitrile, Al2O3, toluene, reflux, 12 h.
Three isomeric pyrazaacene dimers that undergo singlet fission with triplet quantum yields as high as 125% were developed by Franko, Guldi, and Mateo-Alonso (Scheme 160).317 Cyclocondensation of dione 160.1 and anthracenediamine 160.2 yielded the asymmetric dibenzodiazahexacene 160.3b with a boronate ester that was further engaged in cross-coupling reactions (Scheme 160). The dimers were obtained via microwave-assisted Suzuki reaction between the precursor 160.3b with ortho-, meta-, and para-diiodobenzene, affording, respectively, the desired 160.4, 160.5, and 160.6. Pyrazaacene monomer 160.3a and dimers 160.4–6 were all found to fluoresce in the red to NIR region with the quantum yields of 25%, 19%, 20%, and 13%, respectively, recorded in toluene. The optical HOMO–LUMO gaps (Egap ≈ 1.8 eV) and electrochemical HOMO–LUMO gaps (Egap ≈ 1.8 eV) were found to be nearly identical for all the pyrazaacene derivatives and, according to computational results, met the energetic requirements (2T1=S1) for singlet fission (SF) in dimers. A detailed investigation of the excited-state dynamics using a kinetic model fit, from femtosecond (fs-TAS) and nanosecond transient absorption (ns-TAS) data, showed that 160.6 underwent SF with 1(T1T1) triplet quantum yields (TQYs) of 125% in toluene. The lower TQYs of 92% found in benzonitrile indicated that a change in solvent polarity affected the energetic levels of excited-state species. TQYs of 1(T1T1) were found to be lower in value for 160.4 and 160.5 with 82% and 70% in toluene. The observed high TQYs in the o-dimer (160.6) were ascribed to the close spatial proximity between the two dibenzoazahexacenes, leading to an enhanced electronic communication in comparison with the m-dimer (160.5) or p-dimer (160.4).
Scheme 160. Synthesis of Pyrene-Fused Azaacene Dimers.
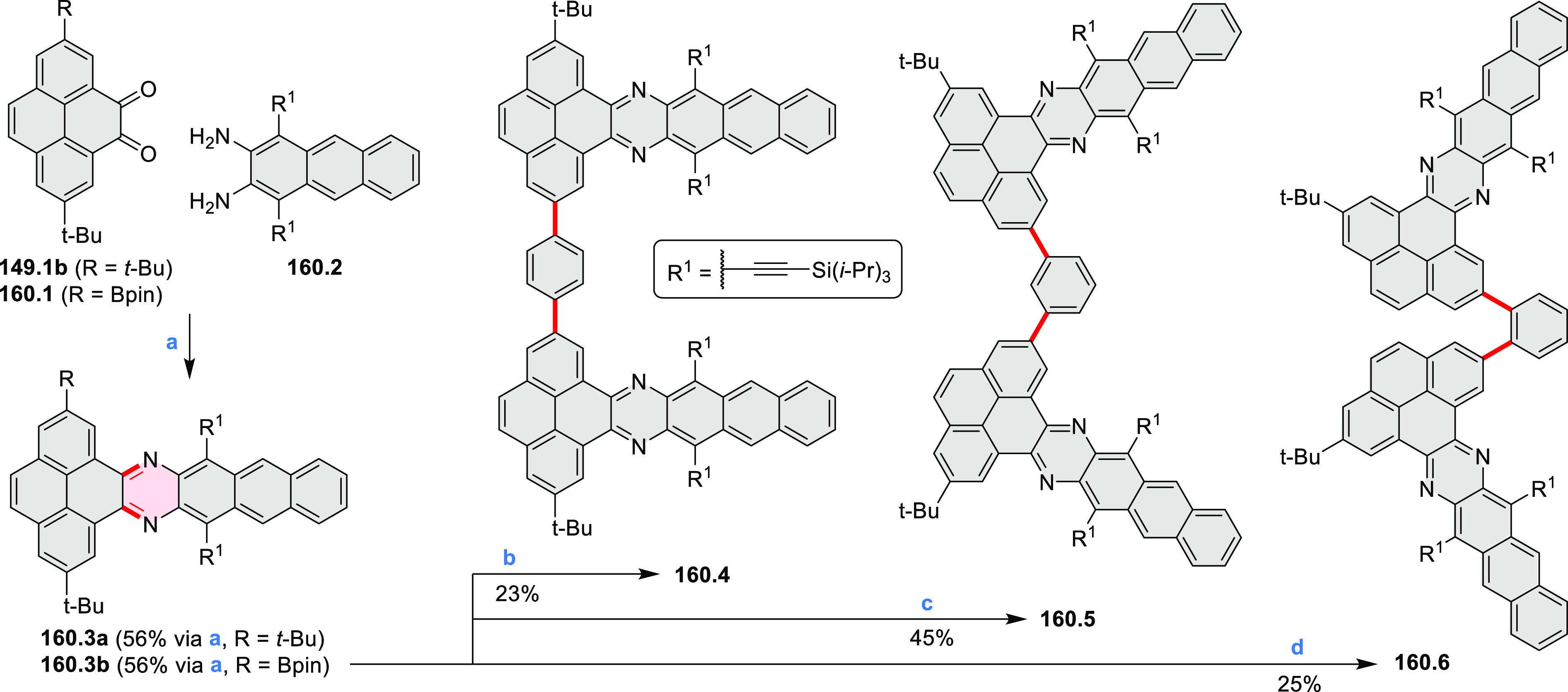
Reagents and conditions: (a)317 CHCl3, AcOH, reflux; (b) p-diiodobenzene, PdCl2(dppf), K3PO4, THF, H2O, 80 °C, 11 min, microwave heating; (c) m-diiodobenzene, PdCl2(dppf), K3PO4, THF, H2O, 80 °C, 11 min, microwave heating; (d) o-diiodobenzene, PdCl2(dppf), K3PO4, THF, H2O, 80 °C, 11 min, microwave heating.
Homopolymerization of an alkoxy-substituted pyrazaacene was reported by Ling and Mo et al. (Scheme 161).318 The poly(2,11-diquinoxalinopyrene) was obtained by subjecting the bromo derivative 161.1b to Yamamoto reaction conditions. By varying the reaction time, it was possible to obtain oligomers with variable lengths and molar weights of up to 7300 Da. The oligomers showed limited band gap dependence on their length, indicating that the conjugation between subunits was relatively weak.
Scheme 161. Synthesis of Poly(2,11-diquinoxalinopyrene).

Reagents and conditions: (a)318 (1) bis(1,5-cyclooctadiene) nickel(0), 1,5-cyclooctadiene, 2,2′-bipyridine, DMF, 85 °C, 30 min, (2) 161.1b in toluene, 12–48 h, (3) bromobenzene, 4 h.
Electrochromic copolymers with the general structures 162.5–6 were obtained by Stille cross-coupling of the pyrazaacene 162.2 with dibromothiophene 162.4 and one of the distannanes 162.1 or 162.3 in varying molar ratios (Scheme 162).319 All three polymers 162.5a–c had band gaps smaller than 1.85 eV and satisfactory thermal stability. The polymers showed electrochromic switching with high optical contrast and fast response time in both the visible and near-infrared regions. For 162.5c, the optical contrasts were 53.35% at 540 nm and 66.49% at 1325 nm, with coloration efficiencies of 163 cm2 C–1 at 540 nm and 205 cm2 C–1 at 1325 nm. A similar performance was obtained for 162.6a–c, which had somewhat larger band gaps of ca. 2.0 eV.
Scheme 162. Synthesis of Thiophene- and Pyrazaacene-Based Oligomers.
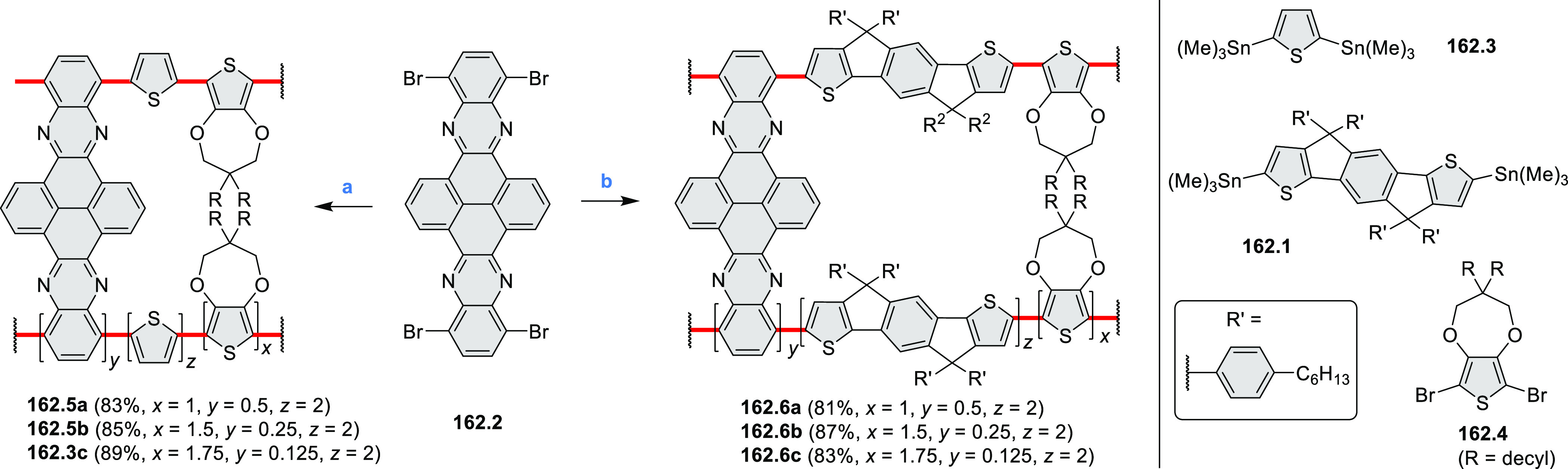
Reagents and conditions: (a)319 Pd(PPh3)2Cl2, toluene, 100 °C, 48 h, 162.2/162.3/162.4 were used in molar ratios of 0.5:2:1, 0.25:2:1.5, and 0.125:2:1.75 for 162.5a, 162.5b, and 162.5c, respectively; (b)320 Pd(PPh3)2Cl2, toluene, 100 °C, 48 h, 162.2/162.1/162.4 were used in molar ratios of 0.5:2:1, 0.25:2:1.5, and 0.125:2:1.75 for 162.6a, 162.6b, and 162.6c, respectively.
Papageorgiou, Reichert, and Mateo-Alonso investigated the reaction of tetraketone 149.8b and the tetraamine 163.1 for the in situ formation of pyrene-fused pyrazaacene-based oligomers on three close-packed coinage metal surfaces (Au, Ag, and Cu) under ultrahigh vacuum conditions (Scheme 163).321 They found that, in contrast to the reaction on Ag(111), the reactants desorb from the Au(111) surface before reacting, whereas they decompose on the Cu(111) surface during thermal treatment. To promote cyclocondensation, 149.8b and 163.1 were deposited onto the Ag(111) surface at rt and subsequently annealed gradually to 510 K. The simplified classification of the occurring oligomers includes straight 163.2 and bent 163.3 dimers and diketone-terminated trimers 163.4–6 (Scheme 163). Formation of tetramers and longer oligomers was also observed. To investigate the effect of stoichiometry, the tetraketone to tetraamine ratio was tuned from ca. 1:5 to 4:1. For every investigated stoichiometry and a given oligomer length, the straight oligomer type was preferred to the corresponding bent type.
Scheme 163. Synthesis of Pyrene-Fused Pyrazaacenes on Metal Surfaces.
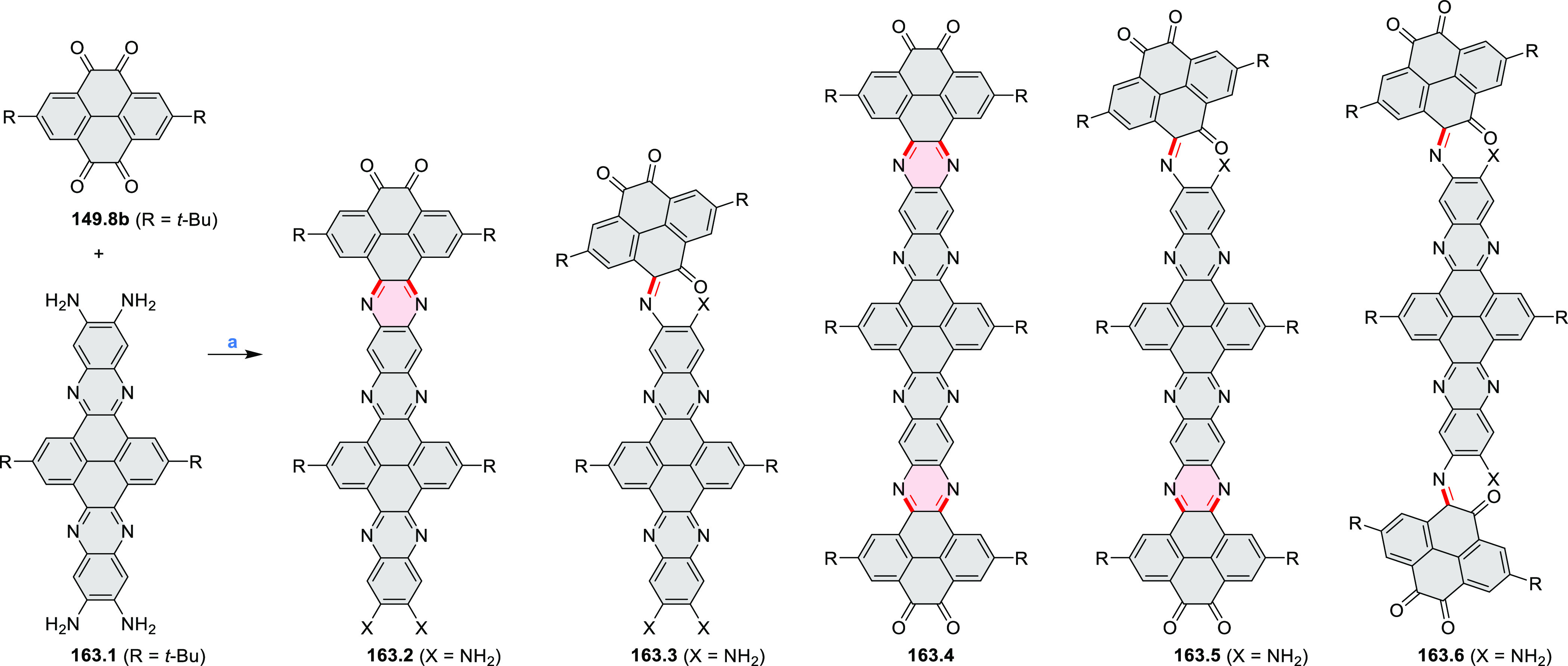
Reagents and conditions: (a)321 Ag(111), TSTM = rt to 510 K, Us = 2.22 V, It = 0.09 nA.
In 2019, Li and co-workers synthesized tetraazatetraoxadecacene C17.1a and tetraazatetrathiadecacene C17.1b, heteroacenes with ten linearly fused rings containing, respectively, embedded O/N or S/N atoms (Chart 17).322 Crystal structures showed that the O-containing C17.1a exhibited linear conformations, while the S-containing C17.1b was noticeably bent. C17.1a showed a fluorescence quantum yield of 0.55 in DCM, which was more than 10 times higher than that of C17.1b (Φf = 0.05). Upon simple reprecipitation, C17.1a and C17.1b self-assembled into microwires and microrods, respectively, which demonstrated distinctive nonlinear optical properties depending on the orientation of transition dipoles. The synthesis of pyrene-fused tetraazaheptacene C17.2 consisting of two terminal pyrene units and a central tetraazaanthracene core substituted by two solubilizing triisopropylsilylethynyl (TIPS) groups was described by Hueso and Mateo-Alonso et al.323C17.2 exhibited a strong absorption in the UV–vis region with the lowest-energy transition at 576 nm and an orange to red photoluminescence, which is centered at 620 nm and exhibits a shoulder at 666 nm. The high stability of C17.2 allowed the preparation of thin films via sublimation. Charge transport studies on thin films of C17.2 show a p-type semiconducting behavior for heptacene C17.2. Hole mobilities are in the range of 0.2 × 10–6 cm2 V–1 s–1. Symmetric donor–acceptor N-heteroacene C17.3 possessing electron-donating decyloxy substituents and pyrazine rings as acceptor units were investigated by Li, Lu, and Zhang as materials for memory devices.298
Chart 17. Symmetric Heteroacenes and Pyrazine-Fused Pyrenoids.

As shown above, synthetic methods available in pyrazaacene chemistry provide access to various nanoribbon structures with nonequivalent termini. This possibility was also explored by Xiao and co-workers, who reported a family of π-extended systems C18.1–5 with the two ends capped, respectively, with a pyrene moiety and a variety of ring systems (Chart 18).324 In particular, compound C18.1a was obtained by reacting the appropriate diamine derivative with thionyl chloride in the presence of triethylamine in DCM, while compound C18.1b was prepared through the reaction between the same diamine and selenium oxide in hot ethanol. All these compounds exhibit weak fluorescence (Φf = 0.024–0.13), with emissions ranging from the yellow through orange up to red region. Spectral features of derivatives containing pyrazine rings, i.e., C18.2–5, were significantly affected by addition of a strong protic acid. Further examples of similar designs include hexacenes C18.6a,b,325 the thiophene-fused system C18.7,326 and pyrazaacenes C18.8–10, which were successfully employed in memory devices.298,327
Chart 18. Unsymmetrical Pyrazaacenes.
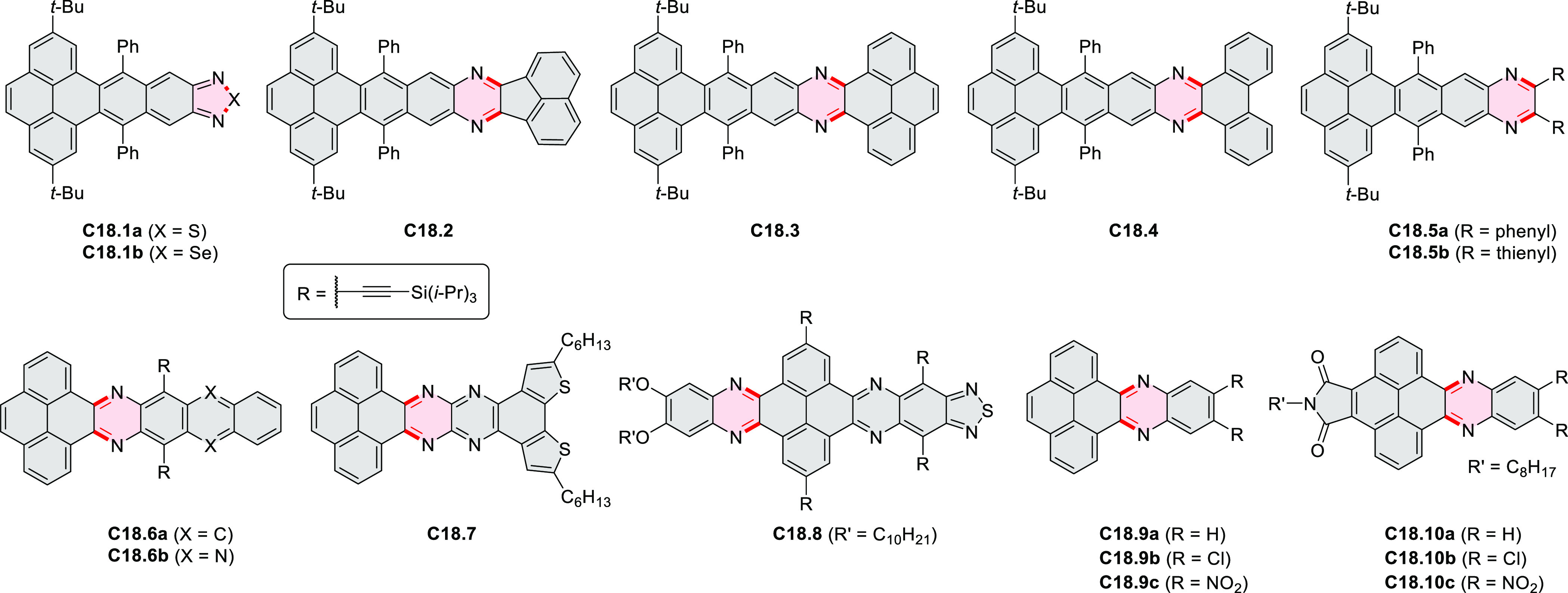
In 2020, Mateo-Alonso et al. reported a series of rotaxanes C19.1a–c consisting of a nitrogenated nanographene and a tetraamide-based macrocycle (Chart 19).328 The synthesis was accomplished using the clipping strategy, in which the macrocycle formation was assisted by hydrogen bonding between the sp2-hybridized nitrogen atoms on the nanographene and amide groups. The structure of C19.1b was confirmed by X-ray crystallography. All three mechanically interlocked systems showed improved photostability under UV irradiation relative to the isolated nanographene.
Chart 19. Mechanically Interlocked Nitrogenated Nanographenes.
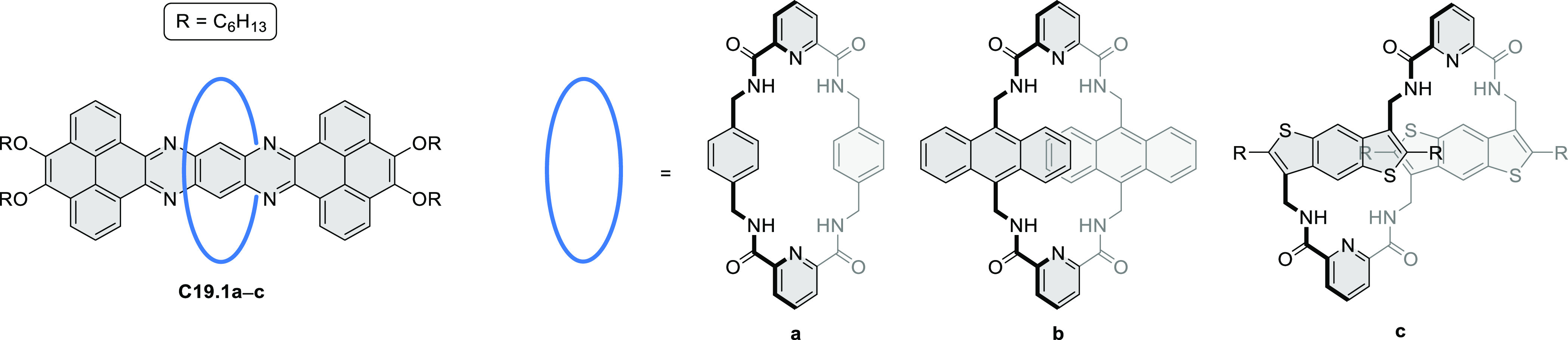
4.7. Other [e]Fused Pyrenoids
4.7.1. Pyrrole- and Indole-Containing Systems
Benzoylpyrenes 164.1 and 164.5, obtained via an oxidative ring opening of cyclopentadienone units, were used by Mastalerz et al. for the synthesis of hetero-[c]-annulated pyrenes (Scheme 164; for related systems, see Schemes 170, 173, and 177, in subsequent subsections).329 Pyrrole rings, in particular, were obtained in a two-step approach consisting of condensation with hydrazine hydrate followed by reductive ring contraction with zinc dust, yielding 164.3 and 164.4. These two compounds exhibit blue emission with quantum yields of 37 and 40%, respectively. An alternative method, due to Liu and Zhang, involved heating with molten methylammonium formate, yielding the N-methylated derivative 164.6 in moderate yield (45%, Scheme 164; cf. Schemes 173 and 177 below).330 Fluorescence emission 164.6 was blue-shifted relative to the starting dibenzoylpyrene 164.5 as well as the chalcogen-containing analogues (173.3, Scheme 173, and 177.3–4, Scheme 177).
Scheme 164. Synthesis of the Pyridazine- and Pyrrole-Extended Pyrenes.

Reagents and conditions: (a)329 N2H5OH, pyridine, water, reflux, 3 h; (b) Zn, DCM, AcOH, rt, 3 h; (c)330 HCOONH3CH3, 145 °C, 24 h.
Scheme 170. Synthesis of the Pyridazine-Extended Pyrenes.

Reagents and conditions: (a)329 N2H5OH, pyridine, water, reflux, 3 h; (b)341 N2H5OH, MeOH, 70 °C, 72 h.
Scheme 173. Synthesis and Reactivity of Furan-Extended Pyrenes.
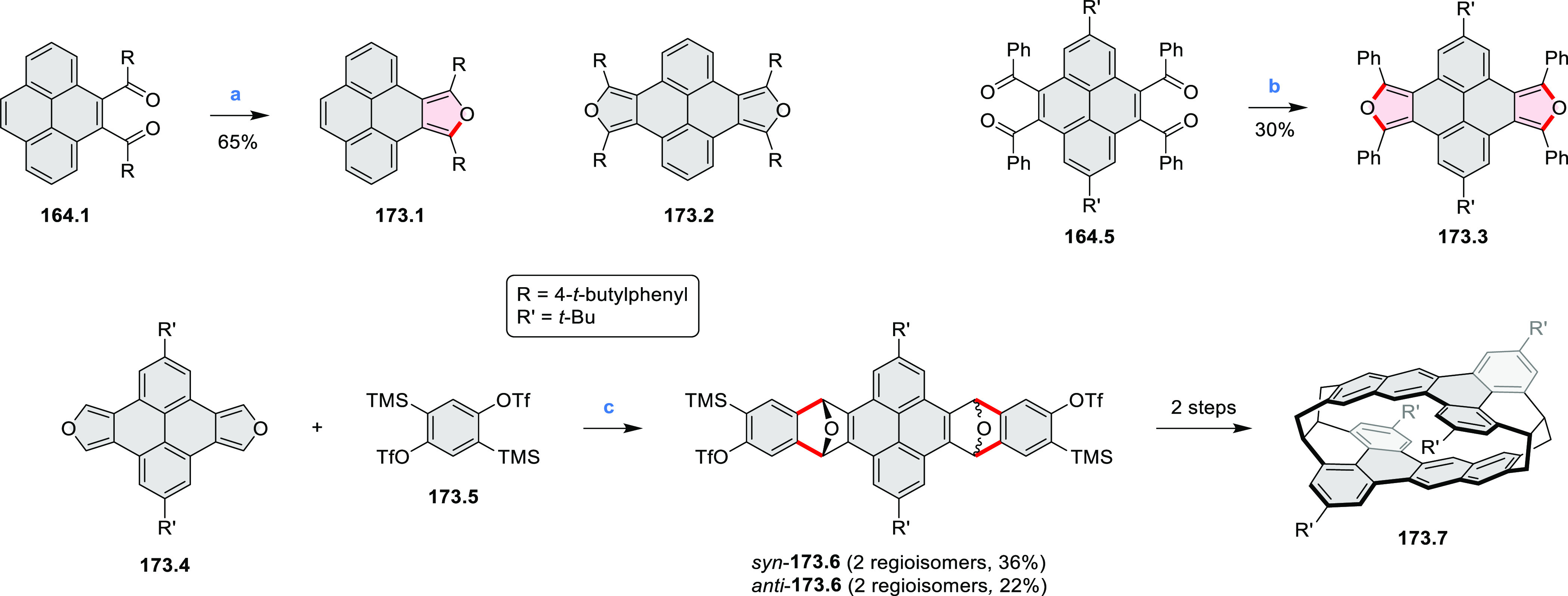
Reagents and conditions: (a)329 LiAlH4, Et2O, rt, 30 min; (b)330 NaBH4, MeOH, rt, overnight; (c)347 CsF, MeCN/THF, 40 °C, overnight.
Scheme 177. Synthesis of Thiophene and Selenophene Extended Pyrenes.

Reagents and conditions: (a)329 P4S10, NaHCO3, MeCN, reflux, 20 h; (b)330 Lawesson’s reagent, reflux, overnight, in the dark; (c) Woollins’ reagent, reflux, 2 h, in the dark.
4.7.2. Imidazole-Containing Systems
Pyrene diones and tetraones such as 149.1a and 165.7 provide straightforward access to pyreno-fused mono- and diimidazoles, which can be used as handles for further ring fusion or metal coordination. The Debus–Radziszewski reaction used in these syntheses (Scheme 165) is efficient enough to be used in COF syntheses (cf. Scheme 393, Section 7.7.1),331 but it will produce regioisomers for N-substituted diimidazoles (e.g., anti-165.9 and syn-165.9).332 π-Extension of such imidazoles via light-induced direct arylation in the solid state was reported in 2016 by Skonieczny and Gryko.333 Photochemical direct arylation was then performed via evaporation of a solution of 165.5 on a quartz glass followed by irradiation with a UV lamp, to produce 165.6. The same approach was also applied to acenaphthylene-1,2-dione (Scheme 262, Section 6.2.1). Fusion of the additional rings in 165.6 led to a dramatic increase of fluorescence quantum yield from less than 0.0001 for 165.5 to 0.75 for 165.6. Oxidative annulation of imidazoles 165.1a–f with diphenylacetylene in the presence of [RuCl2(p-cymene)]2 as a catalyst was found to produce the isoquinolino-fused products 165.2a–f.334 The same reaction sequence was employed to obtain naphthalene and thiophene analogues 165.3 and 165.4, respectively. 165.2e showed distinct solvatofluorochromism, which was attributed to intramolecular charge transfer due to the electron-withdrawing NO2 group.
Scheme 165. Syntheses of Pyrenoimidazole Derivatives.
Reagents and conditions: (a)334 4-R-benzaldehyde (R = H, OCH3, CN, CF3, NO2, or CO2CH3), NH4OAc, AcOH, reflux, 24 h; (b) [RuCl2(p-cymene)]2, 1-adamantanecarboxylic acid, Cu(OAc)2, K2CO3, diphenylacetylene, 120 °C, 16 h; (c)333 4-tert-butylaniline, 2-iodobenzaldehyde, NH4OAc, AcOH, 110 °C, 5 h; (d) hν, 48–72 h; (e)331 benzaldehyde, NH4OAc, dioxane, 120 °C, 3 days; (f)332 aniline, benzaldehyde, NH4OAc, AcOH, reflux, 2 h.
One- or two-step alkylation of 166.1 was used by Poyatos and Peris et al. to obtain the tetraalkyldiimidazolium salts 166.3–4 (Scheme 166).335 Intermediates 166.2a–e were obtained as mixtures of the anti and syn regioisomers, which could usually be separated and converted into the corresponding tetraalkyl targets. The reaction between [{Pt(ppy)(μ-Cl)2}2] and [syn-166.4b][I2] performed in the presence of sodium acetate and sodium iodide produced a mixture of the corresponding bis-N-heterocyclic carbene complexes 166.5a and 166.6a, as two isomers were obtained in 6:4 molar ratio. Iodide abstraction with a silver salt and subsequent reaction with NaCN afforded a mixture of the isomeric complexes 166.5b and 166.6b, which contain strong-field CN– auxiliary ligands. All the compounds 166.2–6 showed emissions in the range of 379–423 and 370–420 nm for the neutral bisazole 166.2a–e and dicationic bisazolium compounds 166.3–6, respectively, with quantum yields in the range of Φf = 0.20–0.55. Both the emissions and the quantum yields were only slightly sensitive to the nature of the bisazoles and their substituents.
Scheme 166. Pyrene-Fused Imidazolium Cations and Their NHC Complexes.
Reagents and conditions: (a)335 (1) NaOH, DMSO, rt, 2 h, (2) RBr or RI (2 equiv), rt for 30 min, then 37 °C, overnight; (b) (1) NaOH, DMSO, rt, 2 h, (2) RBr or RI (excess), rt for 30 min, then 37 °C, overnight; (c) MeI (excess), 100 °C, 24 h; (d) [Pt(ppy)(μ-Cl)]2, (ppy = 2-phenylpyridinate), NaOAc, NaI, DMSO, 100 °C, overnight; (e) (1) AgBF4, DCM, 1 h, (2) NaCN, rt, overnight.
For 166.2eanti and syn isomers were obtained as a mixture in 1:1 ratio and were not separated. Total yield is given.
Isomers 166.5a and 166.6a were obtained as a mixture in 6:4 ratio and were not separated. Total yield is given.
166.5b and 166.6b were obtained as a mixture in 1:1 ratio and were not separated. Total yield is given.
Two mononuclear ruthenium(II) and osmium(II) complexes containing a pyrene-fused biimidazole ligand were designed by Baitalik et al. as cyanide ion sensors for aqueous media.336 The monometallic complexes 167.2a,b were synthesized by reacting 167.1 with the appropriate metal precursor (Scheme 167). Both complexes showed high selectivity toward cyanide ions in aqueous media in the presence of an excess of other anions, with measurable responses in absorption and emission spectra as well as in voltammetric measurements. Analogous binuclear complexes 167.4a,b were found to interact with CT-DNA via an intercalative binding mode with intrinsic binding constants on the order of 105 M–1.337,338
Scheme 167. Synthesis of the Pyrene-Biimidazole-Based Ru(II) and Os(II) Complexes.

Reagents and conditions: (a)336 (1) cis-[Ru(bpy)2Cl2]·2H2O, AgClO4, EtOH, reflux, 1 h, (2) 167.1, reflux, 10 h; (b) (1) cis-Os(bpy)2Cl2, EtOH, water, reflux, 40 h, (2) NaClO4, rt; (c)338 (1) cis-[Ru(bpy)2Cl2]·2H2O, ethylene glycol, reflux, overnight, (2) NaClO4, water, rt; (d) (1) cis-Os(bpy)2Cl2, ethylene glycol, reflux, overnight, (2) NaClO4, water, rt.
Pyrenodiimidazole 149.8a was copolymerized with vinylene 168.2 and thiophene 168.4 by Stille coupling reaction to furnish two conjugated polymers, 168.3 and 168.5 (Scheme 168).339 The two polymers exhibited coplanar backbones, similar LUMO energy levels at −3.7 eV, and uniformly delocalized HOMOs with low energies of ca. −5.7 eV. X-ray diffraction and grazing-incident X-ray diffraction measurements demonstrated that 168.5 adopted a highly ordered structure, reflected in an enhanced hole mobility of 0.015 cm2 V–1 s–1 in organic thin-film transistors. In contrast, the thin film of 168.3 was disordered with a hole mobility of up to 2.22 × 10–3 cm2 V–1 s–1.
Scheme 168. Synthesis of Pyrenodiimidazole Polymers.

Reagents and conditions: (a)339 5-Bromothiophene-2-carbaldehyde, NH4OAc, AcOH, reflux, 10 h; (b) 1-bromooctadecane, DMF, 100 °C, 12 h; (c) Pd(PPh3)4, THF, 65 °C, 16–20 h.
When a mixture of pyrene-4,5-dione 149.1a, ammonium acetate, and glacial acetic acid was heated at 100 °C for 10 h, self-condensation was observed, resulting in the formation of 169.2a, 6H-phenanthro[4,5-cde]pyreno[4′,5′:4,5]imidazo[1,2-a]azepin-6-one in 25% yield (Scheme 169).340 To address the solubility problem of 169.2a, tert-butyl-substituted pyrene-4,5-diones 149.1b and 169.1c were subjected to the same conditions to afford analogous products with both improved solubility and yields. 169.2b exhibited solvato-, mechano-, and acidofluorochromic properties, presumed to originate from excimer/monomer equilibria.
Scheme 169. Self-Condensation of Pyrene-4,5-dione.

Reagents and conditions: (a)340 NH4OAc, AcOH, 100 °C.
4.7.3. Other Nitrogen-Containing Systems
Pyridazines 170.2a,b were obtained by direct condensation of corresponding benzoylpyrenes with hydrazine hydrate (Scheme 170).329 Both 170.2a and 170.3a underwent reductive contraction to yield the corresponding pyrrole analogues (Scheme 164, Section 4.7.1). 170.2b and 170.3b exhibited moderate two-photon absorption cross-section values, reaching 454 GM in the case of 170.3b.341
Structural variety can be created by condensing pyrene di- and tetraones with untypical aromatic diamines. A recent example is provided by condensations with 3,4-diamino-5-heptyl-1,2,4-triazole 171.5, which yielded the triazolo–triazines 171.6–7 (Scheme 171, cf. Scheme 262, Section 6.2.1, for an acenaphthylene analogue).342 LUMO energy levels were calculated to be −3.77 eV and −3.95 eV for 171.6 and 171.7, respectively, indicating their potential utility as n-type semiconductors. Condensations with pure enantiomers of 2,2′-diamino-1,1′-binaphthalene 171.1 furnished the chiral systems 171.2–3 containing twisted 1,4-diazocine rings.343 The CD spectra of 171.2–3 demonstrated chiral induction between the chiral binaphthalene group(s) and the pyrene chromophore.
Scheme 171. Synthesis of Chiral Pyrene-Based Compounds.
Reagents and conditions: (a)343 (R)- or (S)-(±)-2,2′-diamino-1,1′-binaphthalene, CF3COOH, toluene, 60 °C, 3 h; (b)342 AcOH, reflux, overnight.
Azatwistacenes 172.2 and 172.4 containing fused carbazole moieties were synthesized using respectively the Cadogan reaction and a Pd-catalyzed annulative amination (Scheme 172, for a furan derivative see Scheme 175, Section 4.7.4).344 The former reaction was regioselective, producing exclusively the angularly fused isomer 172.2. The related bent twistacene 172.6a was synthesized from the respective dibromo precursor using a Cu-catalyzed amination reaction.345 Compound 172.2 displayed blue fluorescence in DCM solution with maxima peaks at 455 and 475 nm and a quantum yield of 0.29, while 172.4 showed the bathochromic shift to 481 and 510 nm, resulting in green emission (Φf = 0.32). Twistacenes 172.2 and 172.4 were tested as dopants in multilayered OLED devices and exhibited promising electroluminescent performance. 172.6a was found to be solvatofluorochromic, its fluorescence changing from green to yellow with increasing solvent polarity. In contrast, the dimesitylboryl derivative 172.6b emitted blue light with no significant solvent dependence.
Scheme 175. Synthesis of Oxatwistacene.

Reagents and conditions: (a)351 (1) I2, DCM, (2) n-BuLi, THF; (b) I2, DCM; (c)344 Pd(OAc)2, K3PO4, 2-di-t-butylphosphino-2′-methylbiphenyl, toluene, 100 °C, 24 h.
4.7.4. Oxygen-Containing Systems
Pyrenoids with K-region-fused furans 173.1–3 were recently accessed via LiAlH4 or NaBH4 reduction of the corresponding benzoyl derivatives followed by aqueous acidic workup (Scheme 173).329,330 This method complements other synthetic transformations of pyrene ketones leading to [c]heteroannulated products (Schemes 164, 170, and 177). 173.1 and 173.2 show blue emission with the quantum yields of 0.16 and 0.24, respectively. The furan rings in such furan-fused pyrenes can act as dienes in Diels–Alder reactions. For example, as reported by Miao et al., pyrenodifuran 173.4 reacted with the benzyne generated in situ from 173.5 in a 1:2 stoichiometry to give 173.6 as an isomeric mixture. The regioisomeric mixture of syn-173.6 could be isolated and was then converted to the hydrogenated zigzag nanobelt 173.7 in two steps. The first fully conjugated zigzag nanobelt, reported in 2021 by Itami and Segawa et al., was synthesized using a similar synthetic approach.346
Laterally π-extended twistacenes (also known as twistarenes) consisting of 9,14-diphenyldibenzo[de,qr]tetracene and a fused heterocyclic subunit have been explored as functional dyes for various applications. A pyran-containing system 174.2 was synthesized in 42% yield via a one-step Pd-catalyzed annulation between 174.1 and 1-naphthol (Scheme 174).348 Under controlled conditions, 174.2 assembled into various morphologies (nanowires or nanospheres), as evidenced by SEM, UV–vis absorption, and emission data. The fabricated electroluminescent devices based on 174.2 had a maximum brightness of 4355 cd m–2 (bias voltage at 9.0 V) with CIE coordinates of (0.14, 0.53). 174.3a and 174.3b were obtained by annulative nucleophilic substitution of 174.1b with catechol or 2-aminophenol, respectively (Scheme 174).349 The same approach proved to be effective in the syntheses of their sulfur-containing analogues (Scheme 180, Section 4.7.5). Photodetector devices fabricated by using compound 174.3b and single-walled carbon nanotubes as the donor and acceptor, respectively, produced a steady photocurrent upon irradiation of a halogen lamp, displaying good stability and photosensitivity.
Scheme 174. Synthesis of Oxygen- and Nitrogen-Doped Twistacenes.

Reagents and conditions: (a)348 1-naphthol, PPh3, Cs2CO3, Pd(OAc)2, DMF, 140 °C, 24 h; (b)349 catechol, NaH, NMP, 205 °C, 12 h; (c) 2-aminophenol, NaH, NMP, 205 °C, 12 h; (d)350 K2CO3, DMF, 110 °C.
Scheme 180. Synthesis of Sulfur-, Selenium-, and Nitrogen-Doped Twistacenes.

Reagents and conditions: (a)349 toluene-3,4-dithiol, K2CO3, DMF, 100 °C, 1 h; (b) 2-aminobenzenethiol, K2CO3, DMF, 130 °C, 1 h; (c)358 Na2S·9H2O, NMP, 195 °C, overnight; (d) (1) Se, sodium borohydride, EtOH, 0 °C, 40 min, (2) 180.3 in NMP, then 190 °C, overnight.
Dioxa derivative 174.5 and its more π-extended analogue 174.7 were also obtained using nucleophilic annulations, this time with the twistarene building block 174.4 acting as a nucleophile (Scheme 174).350174.7 adopted a reclining chair conformation in the solid state, rather than the usual twisted structure. Self-assembly of 174.5 and 174.7 was investigated via the surfactant-assisted reprecipitation method. It was shown that 174.5 could self-assemble into nanobelts, and 174.7 formed nanowires in the presence of cetyltrimethylammonium bromide.
Benzofuran-fused twistacene 175.5 was synthesized by the intramolecular O-arylation of 175.4 (Scheme 175, for its nitrogen analogues, see Scheme 172, Section 4.7.3).344 Compound 175.5 exhibited blue fluorescence in DCM solution (λmaxem = 456 and 479 nm, QY = 38%). The related twistacenes 175.2a,b were synthesized by diiodine-induced cyclization of the corresponding precursors 175.1a,b followed by dehalogenation with n-BuLi (Scheme 175).351 When the triphenylamine-substituted 175.1c was reacted under the same I2-mediated conditions, the dimeric 175.3 formed in 65% yield. Twistacenes 175.2a,b and 175.3 as well as their precursors 175.1a–c emitted blue light (QY = 0.26–0.49). Additionally, 175.2a,b and dimeric 175.3 showed an increase of two-photon absorption cross sections relative to the starting twistacenes 175.1a–c, which was attributed to the introduction of electron-rich furan units.
A donor–acceptor twistarene 176.3 containing 12 linearly fused rings was reported by Xiao and Zhang et al. (Scheme 176).352 In the final step, 176.1 was treated with 4,5,9,10-tetrabromo-2,7-didodecylbenzo[lmn][3,8]phenanthroline-1,3,6,8-tetraone 176.2 in the presence of potassium carbonate to afford the desired 176.3 as a dark red solid. 176.3 was thermally stable up to 436 °C, had absorption maxima at 510 and 538 nm, and emitted weak red fluorescence (Φf up to 1.9% in toluene). A solution-processed memory device based on 176.3 had an ON/OFF current ratio of 103.46:1 and a threshold voltage of −2.44 V.
Scheme 176. Synthesis of a Twelve-Ring Twistheteroacene.

Reagents and conditions: (a)352 K2CO3, DMF.
4.7.5. Sulfur-Containing Systems
Many of the sulfur-containing [e]fused derivatives discussed below were obtained as S-doped analogues of systems discussed in preceding subsections. For instance, the thiophene-fused pyrenes 177.1 and 177.2 were obtained by the Mastalerz group from the previously mentioned benzoylpyrenes in a reaction with sodium bicarbonate and phosphorus pentasulfide (Scheme 177).329 The longest absorption wavelength of the monofused system 177.1 was bathochromically shifted by 30 nm in comparison with 177.2. This effect was explained by the lower π-conjugation in 177.2, on the basis of additional XRD evidence. Liu and Zhang obtained the related 177.3 and its selenium analogue 177.4 using, respectively, Lawesson’s and Woollins’ reagents (Scheme 177).330177.3, 177.4, and the oxygen analogue 173.3 all exhibited the same HOMO energy level of −5.29 eV, lower than the HOMO of −5.06 eV for the nitrogen analogue 164.6.
Several thieno-fused pyrenoids were obtained via cyclization of thienyl-containing substituents. Verbitskiy et al. used oxidative photocyclization to obtain 178.2a–c in moderate yields (Scheme 178).353 The reactivity of precursors depended on substitution (R, t-Bu < H < CF3), correlating with the electron-withdrawing character of the R group. Compounds 178.2a–c were somewhat more fluorescent (Φ = 0.1–0.13) than their pyrimidine parents 178.1a–c (Φ = 0.05–0.07). Further examples include the mesomorphic fused triphenylene 178.4 obtained by FeCl3-mediated cyclodehydrogenation354 and bishelicenes 178.6a–e, prepared using Mallory photocyclization.355,356178.6a,b were investigated as active layers in p-type OFET top contact devices, exhibiting unusual electronic stability.
Scheme 178. Synthesis of Thiophene- and Benzothiophene-Fused [e]-Pyrene.

Reagents and conditions: (a)353 I2, MeCN, UV light irradiation, rt, 130 h (178.1a), 30 h (178.1b), 150 h (178.1c); (b)354 iron(III) chloride, nitromethane, 0 °C, 1.5 h; (c)355,356 I2, benzene, hν.
In 2020, Murakami and Itami reported a synthetic route to thiophene-fused PAHs by a palladium-catalyzed annulative dimerization of phenylene triflate through 2-fold inter- and intramolecular C–H activation.357 Optimized conditions utilized palladium chloride, tributylphosphonium tetrafluoroborate, K2CO3, and pivalic acid in cyclopentyl methyl ether at 140 °C. The benzothiophene-substituted phenylene triflate 179.1 was converted into 179.2a and 179.2b in 88% yield as a 4:1 mixture of regioisomers (Scheme 179). The reaction of 179.2b resulted in a complex mixture containing 179.3b, whereas the regioisomer 179.2a reacted smoothly to give the bisbenzothiophene extended pyrene derivative 179.3a in 79% yield.
Scheme 179. Annulative Dimerization of Phenylene Triflate.

Reagents and conditions: (a)357 PdCl2, P(n-Bu)3·HBF4, K2CO3, pivalic acid, cyclopentyl methyl ether, 140 °C, 20 h; (b) FeCl3, DCM, 0 °C, 2.5 h. * Obtained as a mixture of two regioisomers 179.2a and 179.2b in 4:1 ratio (overall yield). ** n.d. = not determined. Obtained as a complex mixture.
π-Extension of twistacenes to include sulfur-containing rings was achieved using methods described in preceding sections. Specifically, the S- and N-doped 180.2a and 180.2b were obtained by nucleophilic cyclization of the dibromo building block 180.1 (Scheme 180).349180.2a had the expected twisted structure in the solid state, with an additional bend at the 1,4-dithiine ring. 180.2b deposited on single-wall carbon nanotubes produced a steady photocurrent upon irradiation of a halogen lamp. Terminal thiophene and selenophene rings in 180.4a,b were synthesized through cyclization of the alkyne precursor 180.3 with Na2S·9H2O or selenium and sodium borohydride, respectively.358180.4a and 180.4b emitted blue fluorescence with quantum yields of 0.39 and 0.04, respectively, and self-assembled into nanoparticles upon reprecipitation from a mixture of THF and water.
Condensation of 2,7-di-tert-butyl-4,5,9,10-tetrabromopyrene 181.1 with 1,2-benzenedithiol in the presence of potassium carbonate was used by Wang et al. to prepare the S-doped hexacene 181.2 (Scheme 181).359 Chemical oxidation of 181.2 was carried out with NO{Al[OC(CF3)3]4} at different molar ratios, affording the yellow-green radical cation salt 181.2•+{Al[OC(CF3)3]4}− and green diradical dication salt 181.22+••·2{Al[OC(CF3)3]4}−, respectively. Both salts were isolated as crystals that were thermally stable under anaerobic conditions at rt. The spin density in 181.2•+ is delocalized on one sulfur-doped ring and the central pyrene unit, while that in 181.22+•• is delocalized on the whole molecule. The diradical dication 181.22+•• featured a robust triplet ground state, representing the first example of a high-spin sulfur-containing hydrocarbon diradical (with no nitrogen atoms incorporated).
Scheme 181. S-Doped Hexacene via Nucleophilic Substitution.

Reagents and conditions: (a)359 1,2-benzenedithiol, K2CO3, DMF, reflux, 12 h; (b) NO{Al[OC(CF3)3]4} (1 equiv), toluene, rt, 12 h; (c) NO{Al[OC(CF3)3]4} (2 equiv), DCM, rt, 12 h.
5. Phenalenoids
Most systems discussed in this section contain at least one heteroatom incorporated into the phenalene substructure (see CR2017, Section 5 for scope definition and earlier examples). A unique example of an extended phenalene containing an ortho-fused heterocycle is the pro-aromatic bisphenaleno-thieno[3,2-b]thiophene (182.3) reported by Kim and Chi et al. in 2015 (Scheme 182).360 It was synthesized from diacid 182.1, which was converted into the corresponding acyl chloride and subjected to double Friedel–Crafts acylation, to afford the desired diketone 182.2 in 85% yield. Addition of triisopropylsilyl ethynyl lithium to 182.2, followed by SnCl2 reduction, gave the target 182.3 in 45% yield. Transient absorption spectra of 182.3 exhibited a ground-state bleach signal at around 690 nm and a weak excited-state absorption band in the 450–600 nm region. 182.3 displayed amphoteric redox behavior with two reversible oxidation waves (E1/2ox = −0.03, +0.44 V vs Fc+/Fc) and two reversible reduction waves (E1/2 = −1.73, −1.38 V vs Fc+/Fc).
Scheme 182. Synthetic Route of Bisphenaleno-thieno[3,2-b]thiophene.

Reagents and conditions: (a)360 (1) SOCl2, dry DCM, reflux, (2) AlCl3, dry DCM, 0 °C to rt, overnight; (b) (1) (triisopropylsilyl)ethynyllithium, THF, 0 °C to rt, (2) SnCl2, toluene, rt, 12 h.
5.1. Monoheteraphenalenes
5.1.1. Azaphenalenes
The 1-azaphenalene motif, which also appears in some larger fused frameworks (cf. Schemes 45, Section 3.1.2, and 294, Section 6.4.2), can be assembled in several ways, e.g., using the previously discussed tandem cross-coupling annulation reported by Li et al. (183.2, Scheme 183).76 In this synthesis, Buchwald–Hartwig amination was presumed to precede the arylation step. An alternative approach, employing nucleophilic aromatic substitution in the ring-forming step, was used by Zeng et al. to prepare the NIR fluorescent rhodamine dye 183.4.361183.4 showed a near-infrared emission at 830 nm, significantly red-shifted in comparison with conventional rhodamine dyes.
The reactivity of 4-amino-5-hydrazineyl-4h-1,2,4-triazole-3-thiol 184.1 toward cyclic anhydrides was studied by El-Shaieb et al. in 2019 (Scheme 184).362 Depending on the anhydride, several types of heterocyclic structures were obtained, including cyclic hydrazides and triazoloterazine derivatives. The most complex ring system, the pentacyclic 184.3, was obtained in the reaction with 1,8-naphthalic anhydride 184.2.
Scheme 184. Reactivity of 4-Amino-5-hydrazineyl-4h-1,2,4-triazole-3-thiol.

Reagents and conditions: (a)362 glacial acetic acid, reflux, 4 h.
An unexpected formation of the blue azaphenalenoid 185.4 was observed by Furuta and Shimizu et al. during the synthesis of meso-2,6-dichlorophenyl-substituted tribenzosubporphyrin (185.3) in a reaction of phthalimide (185.1) and 2,6-dichlorophenylacetic acid (185.2, Scheme 185).363 No similar product was isolated in the reaction using phenylacetic acid, indicating potential relevance of the chloro substituents in 185.2. The mechanism of this unusual transformation remains to be elucidated.
Scheme 185. Byproduct Formation in the Synthesis of a Tribenzosubporphyrin.

Reagents and conditions: (a)363 B(OH)3, 360 °C.
Several nitrogen-containing phenalenoids were obtained using a rhodium-catalyzed oxidative annulation of β-enamino esters with internal alkynes, described by Sun et al. (Scheme 186).364 For instance, diphenylacetylene and the unprotected β-enamino ester 186.1 could be reacted in the presence of [Cp*RhCl2]2 and Cu(OAc)2 to afford 186.2 in 73% yield. When 4-aminocoumarins were used as substrates in the same reaction, extended systems, such as 186.4, were formed. This double annulation was shown to work with a variety of alkynes, β-enamino esters, and 4-aminocoumarins.
Scheme 186. Synthesis of Nitrogen-Containing Phenalenoids via Rhodium-Catalyzed Multiple C–H Bond Activation and Oxidative Annulation.

Reagents and conditions: (a)364 [Cp*RhCl2]2, Cu(OAc)2, DMF, 100 °C, Ar, 24 h.
Assembly of 1-azaphenalene fragments was the key step in the synthesis of N-doped heptazethrene and octazethrene diradicaloids reported by Wu and co-workers in 2019 (Scheme 187).365 Suzuki coupling between 1-acetamido-8-bromonaphthalene (187.1) and diboronic acids 187.2a,b gave the corresponding intermediates 187.3a,b. Demethylation of 187.3a,b with boron tribromide afforded diols 187.4a,b, which were further subjected to hydrazine-mediated deprotection and cyclization to give precursors 187.5a,b. Chemical oxidation of 187.5a,b by lead(IV) oxide in dry DCM gave the respective dehydrogenated products 187.6a,b, which were, however, too unstable to be isolated. 187.6a exhibited a major absorption band with maximum (λmax) at 598 nm and a weak shoulder peak at 660 nm. 187.6b displayed a similar band structure to 187.6a with λmax = 644 nm together with a shoulder peak at 728 nm.
Scheme 187. N-Doped Heptazethrene and Octazethrene.

Reagents and conditions: (a)365 Pd(PPh3)4, 2 M Na2CO3, 1,2-dimethoxyethane, 115 °C, 48 h; (b) BBr3, dry DCM, rt, 48 h; (c) hydrazine monohydrate (80%), DMSO/i-PrOH, 140 °C, 72 h; (d) PbO2, dry DCM, rt, 30 min.
In 2019, Arikawa, Shimizu, and Shintani et al. reported the synthesis of a polycyclic zwitterion azoniadibenzo[a,j]phenalenide 188.6.366 The synthesis of 188.6 was accomplished in six steps from 2-bromo-5-(tert-butyl)-1,3-bis(methoxycarbonyl)benzene 188.1 (Scheme 188). The amination of 188.1 with bis(4-tert-butylphenyl)amine gave 188.2, and hydrolysis of its methyl esters followed by 2-fold intramolecular Friedel–Crafts acylation resulted in 188.3. Reaction of 188.3 with mesityllithium gave the diol 188.4, which was then reduced by Et3SiH/HBF4·Et2O to give the cation 188.5. Deprotonation of 188.5 using NaH in THF/hexane in a degassed sealed tube gave 188.6, which was isolated by recrystallization under an inert atmosphere. 188.6 is stable at ambient temperature in a degassed solution or in the solid state under argon. The negative charge of 188.6 delocalized over the periphery of the main core, and a positive charge localized near the nitrogen center. 188.6 has an open-shell singlet ground state, which was demonstrated using NMR, ESR studies, and DFT calculations.
Scheme 188. Polycyclic Heterahelicenes.
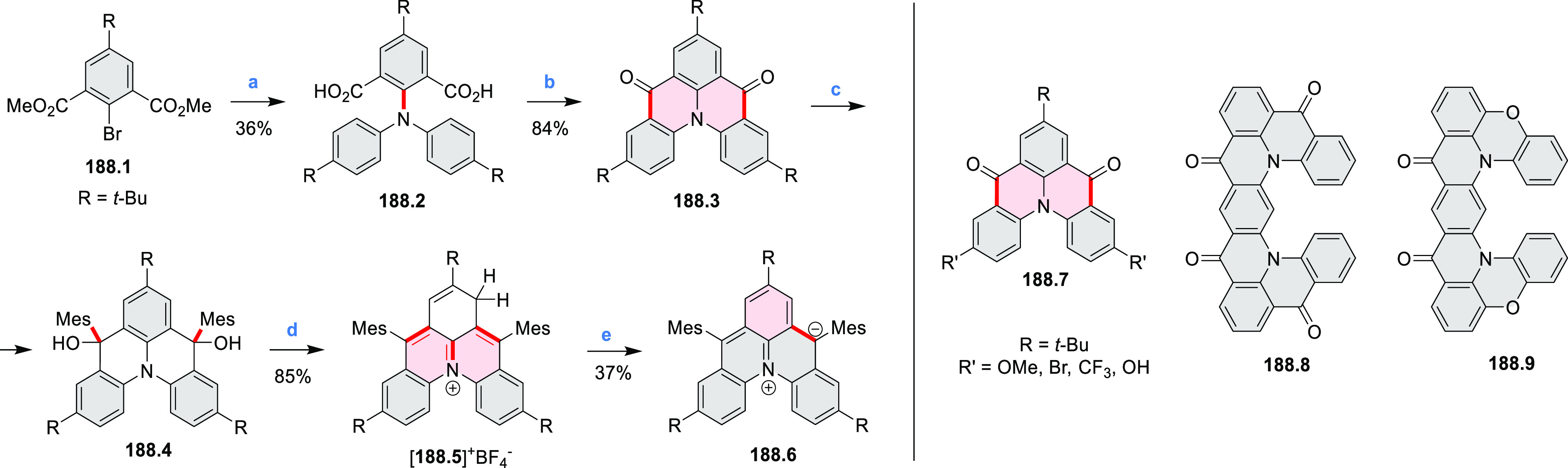
Reagents and conditions: (a)366 (1) bis(4-tert-butylphenyl)amine, Cu, 18-crown-6, K2CO3, 200 °C, 24 h, (2) NaOH, EtOH, 100 °C, 9 h; (b) (1) (COCl)2, DMF, DCM, 45 °C, 1 h, (2) SnCl4, 45 °C, 12 h; (c) MesLi, THF, −15 °C to rt, 18 h; (d) Et3SiH, HBF4·Et2O, DCM, rt; (e) NaH, THF/hexane, rt, 4 h.
Methoxy-, bromo-, CF3-, and hydroxy-substituted bridged triarylamine helicenes (188.7), analogous to 188.3, were synthesized by Shang and Yamamoto et al.367 Triarylamine helicenes 188.7 were synthesized from 188.1 in a similar to 188.3 manner (Scheme 188). The final cyclizations were carried out in neat sulfuric acid under ambient conditions, providing the products in 65–98% yield. Among the four derivatives, methoxy 188.7 showed the highest photoluminescence quantum yield (38%) and longest lifetime (13.98 ns), while the hydroxy-substituted 188.7 showed solvent- and pH-dependent luminescence in alkaline conditions. All four derivatives had low oxidation potentials ranging from −1.37 to −1.00 V.
cis-Quinacridone derivatives structurally related to 188.7 were reported as delayed fluorescence luminogens by Yasuda et al. in 2021.368188.8–9 were synthesized in two or three steps, starting with diethyl 4,6-dibromoisophthalate and Buchwald–Hartwig amination. The resulting arylamine-functionalized isophthalates were then subjected to intramolecular Friedel–Crafts acylation. Deep-blue OLEDs based on 188.8 achieved a high maximum external electroluminescence (EL) quantum efficiency ηext of 19.0% with a narrow fwhm of 37 nm.
A synthesis of acridines from o-aminoaryl ketones and arylboronic acids was developed by Zhang and Zhang et al., who used copper(II)-mediated relay reactions that involved intermolecular Chan–Lam cross-coupling and subsequent intramolecular cyclization.369 In particular, the fused ceramidonine 189.2 was synthesized from 1-aminoanthracene-9,10-dione 189.1 and 1-naphthylboronic with Cu(OTf)2 (1.2 equiv) as the activator (Scheme 189).
Scheme 189. Synthesis of a Ceramidonine via a Copper Trifluoroacetate-Mediated Relay Reaction.

Reagents and conditions: (a)369 Cu(OTf)2, 1,1,2,2-tetrachloroethane (TCE), 100 °C, 60 h.
A transformation of iptycene 190.2 into planar acridinones 190.3–4 was reported by Chuang et al. in 2017 (Scheme 190).370 The starting 190.2 was obtained in a reaction between 190.1 and p-benzoquinone in the presence of Cu(OAc)2 as the oxidant. The inverse electron-demand Diels–Alder reaction between 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine and 190.2 yielded the fully planarized compound 190.3. The cycloaddition of 190.2 and the electron-rich 1,3-diphenylisobenzofuran occurred at the outer double bond and, after subsequent acid-catalyzed aromatization, produced the π-extended product 190.4. A competition experiment between methyl acrylate and the iminoquinone moiety in 190.5 carried out in the presence of 10 mol % of Pd(OAc)2 and Cu(OAc)2/O2 as the catalyst and the oxidant, respectively, resulted in ortho C–H activation and insertion of methyl acrylate at position 3 of the furan moiety, followed by [4 + 2] cycloaddition of the iminoquinone, to yield 190.6.
Scheme 190. Synthesis of Planar Acridinone Heterocyclics.

Reagents and conditions: (a)370 Cu(OAc)2 (2 equiv), AcOH (1.8 equiv), o-DCB, 140 °C, under air, 24 h; (b) DCM, 80 °C, reflux, overnight; (c) (1) toluene, 180 °C, 16 h, (2) p-TsOH, 8 h, 180 °C; (d) 10 mol % of Pd(AcO)2, Cu(AcO)2, O2, TFE, 120 °C, 24 h.
As part of their research on anthraquinone imine dyes for DSSCs, Sundermeyer and co-workers reported in 2016 a four-step synthesis of benzoacridinone 191.3 (Scheme 191).371 The requisite precursor 191.1 had to be obtained via a monoacetal intermediate, to avoid selectivity problems. 191.1 was then photolyzed in a Mallory-type reaction, yielding 191.3. In the latter reaction boron trifluoride was used as a Lewis-acid activator to promote the 6π electrocyclization of the imide.
Scheme 191. π-Extended Acridones.

Reagents and conditions: (a)371 BF3·Et2O, hν, rt, 1 h.
5.1.2. Boraphenalenes
A route to 8-borabenzo[gh]tetraphenes was developed by Yamaguchi and Wagner et al. (Scheme 192).372 Compound 192.1 undergoes smooth Si/B exchange with BBr3 at rt to afford the highly reactive bromoborane 192.2. This intermediate was not isolated but immediately converted into the stable triarylboranes 192.3 and 192.4 by addition of an appropriate aryllithium reagent. An intramolecular, Sc(OTf)3-mediated Friedel–Crafts cyclization carried out on 192.4 gave the planarized boron-doped PAH 192.5. 192.3 and 192.5 had similar electronic properties; in particular, both were strongly fluorescent, with quantum yields of 85% and 89%, respectively.
Scheme 192. Synthetic Routes to 8-Borabenzo[gh]tetraphenes.
Reagents and conditions: (a)372 excess neat BBr3, 25 °C; (b) 1.5 equiv of MesLi, toluene/THF, 0 °C; (c) 1.1 equiv of [2,6-bis(propen-2-yl)phenyl]lithium, toluene/THF, 25 °C; (d) 1 equiv of Sc(OTf)3, 1,2-DCE, reflux temperature.
A synthetic approach to B-doped PAHs containing additional N and S centers was described by Wagner’s group in 2016 (Scheme 193).373 The strategy generally relied on the tricyclic starting material 193.1a to circumvent destructive side reactions between BBr3 and the Lewis basic heteroatoms during the Si/B exchange. For similar reasons, the stilbene-type photocyclization was performed to set up the polycyclic framework and omitted the Ru-catalyzed enyne benzannulation approach. Using 193.1a and pyridine-2-aldehyde, the preparation of the monopyridine derivative 193.8 was achieved in 2 steps and 35% yield. Similarly, the use of phenyl(pyridin-2-yl)methanone yielded the extended congener 193.10. Photocyclization of a thienyl substituent was also possible, with the formation of PAH 193.9, while pyrrole- and furan-containing precursors proved to be inert under UV–vis irradiation. Thioxanthen-9-one was sufficiently electrophilic toward the lithiated 193.1a to furnish the Peterson olefination product, which could be photocyclized to give 193.11. A similar synthetic approach to form 193.7 through the Ru-catalyzed cyclization of aryl enynes was also described by the Wagner group.278
Liu and Feng et al. reported a one-pot synthesis of B-doped PAHs from branched aryl-substituted alkynes (Scheme 194).374 In particular, 194.2 was obtained by heating 194.1 with boron tribromide (BBr3) and 2,4,6-tri(tert-butyl)pyridine and subsequent reaction with mesitylmagnesium bromide at rt. It was found that the bulky Brønsted base played an important role in the formation of 194.2. In the proposed mechanism, the intermediate 194.3 was formed via a 6-endo-dig borylative cyclization of alkyne 194.1 in the presence of BBr3. Subsequently, 194.3 underwent 1,4-boron migration to afford intermediate 194.4, followed by an electrophilic borylation. The resulting six-membered boracycle intermediate was then converted into the final 194.2 after workup with MesMgBr.
Scheme 194. One-Pot Synthetic Routes to B-Doped PAHs.

Reagents and conditions: (a)374 (1) BBr3, TBP, TCB, 200 °C, 24 h, (2) MesMgBr, rt, 1 h.
5.1.3. Oxa- and Thiaphenalenes
The photochemistry of benzannulated derivatives of 2-phenylphenol was investigated by Lukeman and Wang et al. (Scheme 195).375 Preparation of derivatives 195.1 and 195.4 was achieved by Suzuki coupling of an appropriate methoxyaryl boronic acid and aryl bromide, followed by demethylation with BBr3. Under dioxygen-free conditions, 195.1 was found to undergo photocyclization to dihydrobenzoxanthene 195.2. Treatment of the latter intermediate with carbon-supported palladium in oxygenated toluene resulted in an over 90% conversion into benzo[k,l]xanthene 195.3. Irradiation of 195.4 gave rise to a single photoproduct, which was identified as naphtho[1,2,3-kl]xanthene 195.6. The primary photocyclization product 195.5 could be identified as an intermediate using NMR spectroscopy.
Scheme 195. Photocyclization Reactions of Arylphenols.

Reagents and conditions: (a)375hν, deoxygenated H2O/CH3CN; (b) Pd/C, O2, toluene.
Pyridinium-fused oxaphenalenes 196.2a–d were obtained from 4-formyl-1-naphthols 133.1a,b and alkynes 133.2a,b in a rhodium-catalyzed triple cyclization (Scheme 196).268 As the pyridinium nitrogen atoms in these products were derived from the NH4BF4 additive, pyrylium-containing pyrenoid products 133.3a–j were obtained when NaBF4 was used instead (Scheme 133, Section 4.3). Additionally, omitting the additive and using 133.1a and alkyne in a 1:1 stoichiometry provided products of a single cyclization 196.1a,b in high yields. These oxaphenalenes were thought to be intermediates in the triple cyclizations toward 196.2a–d as well as 133.3a–j (see also Scheme 133, Section 4.3 for a discussion of photophysical properties of these products).
A one-step synthesis of benzo[de]thioacenes via a Rh-catalyzed peri-selective heteroarylation/Ag-mediated SET intramolecular cyclization sequence of 1-(methylthio)naphthalene 197.1 and its analogues was developed by You et al. in 2019 (Scheme 197).376 Specifically, 197.1 underwent one-shot Rh-catalyzed annulation with either benzofuran or benzothiophene in the presence of [Cp*RhCl2]2, AgSbF6, Ag2O, PivOH, and HFIP as a solvent. The reaction is applicable to the synthesis of benzo[de]thioacenes and showed that the benzothiophenes and naphthalenes with a variety of functional groups, as well as other carbo- and heterocyclic precursors, yielded products such as 197.4–7. Related fused benzothioxanthenes were obtained by oxidative cyclization of ammonium salts (197.8)377 and via acid-mediated cyclization of sulfoxides (197.9–11, cf. Scheme 138, Section 4.3).277 Variously substituted 197.8 derivatives displayed large Stokes shifts and efficient singlet oxygen generation in the absence of heavy atoms in their structures (quantum yields of up to 0.82).
The synthesis and characterization of sulfur-doped dibenzohepta- and dibenzooctazethrene (Scheme 198) were reported by Zhang and Wu et al. in 2020.378 The intermediates 198.3a,b were first prepared via Suzuki coupling between 2-methylnaphthaleneboronic acid 198.1 and 198.2a,b (Scheme 198). Oxidation of 198.3a,b with hydrogen peroxide afforded sulfoxides 198.4a,b. The two-step sequential intramolecular cyclization of 198.4a,b in the presence of Eaton’s reagent followed by refluxing in pyridine gave 198.5a,b. Chemical oxidation of the neutral 198.5a,b with NO2SbF6 produced radical cations 198.6a,b•+, while the oxidation of neutral 198.7a,b with either 1 and 2 equiv of NOSbF6 gave the corresponding radical cations 198.7a,b•+ and dication 198.7a,b2+, respectively.
Scheme 198. Sulfur-Doped Dibenzohepta- and Dibenzooctazethrene.

Reagents and conditions: (a)378 Pd2(dba)3, Na2CO3, SPhos, toluene/ethanol/H2O (10:1:1), 110 °C; (b) 30% H2O2, acetic acid/chloroform (1:1), rt; (c) (1) Eaton’s reagent, rt, (2) pyridine/H2O, reflux; (d) NO2SbF6, DCM/MeCN, rt.
5.2. Diheteraphenalenes
5.2.1. Pyridoacridines and Other 1,6-Diheteraphenalenoids
The azaoxoaporphine alkaloid deazaascididemin 199.4 as well as various ring-A-modified analogues of ascididemin-type pyridoacridine alkaloids were reported by the Bracher group in 2015 (Scheme 199).379 Their approach involved a Suzuki or Negishi cross-coupling reaction, followed by directed remote ring metalation and intramolecular trapping of the adjacent ester group. The protocol made it possible to introduce structural variation at a critical part of the pentacyclic ring system, which is of special pharmacological interest. The same group later reported a related strategy, which relied on the biaryl 199.5 obtained from 4-bromobenzo[c][2,7]naphthyridine via Negishi cross-coupling.380199.5 was converted into the respective organomagnesium product by bromine–magnesium exchange with i-PrMgCl·LiCl, followed by intramolecular addition to the ester group which produced the expected pyridoacridone 199.6, isomeric to 199.4, in 28% yield.
Scheme 199. Synthesis of Ascididemin-Type Analogues (Deazaascididemin).
Reagents and conditions: (a)379 corresponding boronic acid, KF, Pd2(dba)3, (t-Bu)3P, THF, 80 °C, 30 min; (b) TMPMgCl·LiCl, THF, 2 h at 0 °C, then 16 h, rt; (c)380i-PrMgCl·LiCl, THF, 0 °C, 2 h.
An improved synthesis of styelsamine D (200.4), deoxyamphimedine (200.5), and amphimedine (200.6) was developed by Copp et al. (Scheme 200).381 Coupling of kynuramine 200.3b, which was obtained using an improved procedure, with N-Boc-protected 4-(2-aminoethyl)benzene-1,2-diol using a two-step sequence, afforded styelsamine D 200.4 in 34% yield. Reaction of 200.4 with paraformaldehyde afforded the two pentacyclic natural products deoxyamphimedine (200.5) in 48% yield and demethyldeoxyamphimedine in 52% yield. The yield of 200.5 could be improved up to 66% by increasing the amount of added paraformaldehyde. The versatility of the approach was demonstrated by the synthesis of non-natural analogues of 200.6 and 200.5.
Scheme 200. Synthesis of Amphimedine.
Reagents and conditions: (a)381 AcOH, DMSO/HCl (conc.); (b) O2, 1 N NaOH; (c) HBr, AcOH, 80 °C; (d) (1) Ag2O, CeCl3·7H2O, MeOH/AcOH (1:1), 40 °C, (2) 6 N HCl, 90 °C; (e) (CH2O)n, AcOH, 60 °C, 5 h; (f) K3[Fe(CN)6], aq. NaOH, 0 °C.
Regioselective postfunctionalization of racemic and enantiopure cationic diaza[4]helicenes (201.4+) was described by Lacour et al. in 2016 (Scheme 201).382,383 Overall, more than 20 new chromophores were prepared from [201.1]+BF4– in moderate to excellent yields (55–99%). These systems showed tunable redox and optical characteristics dependent on the nature of the Y group, with electron-donating substituents shifting the low-energy absorption band toward the far-red and NIR regions. The water-soluble zwitterionic dye with pH-dependent absorption and emission properties (201.2) was obtained via the Vilsmeier–Haack and Pinnick reactions of [201.1]+BF4–.383 Depending on its protonation state, compound 201.2 showed an efficient and reversible turn-on of electronic circular dichroism at 300 nm and of circularly polarized luminescence in the red domain.
Scheme 201. Polycyclic Heterahelicenes.

Reagents and conditions: (a)382,383 (1) DMF, POCl3, 90 °C, 8 h, (2) NaH2PO4, NaClO2, H2O2, MeCN, 60 °C, 1 h; (b) 1 M NaOH; (c) 1 M HCl.
Sequential exchange of oxygen atoms in dioxa[6]helicene [202.1]+BF4– with nitrogen bridges was reported in 2019 by Lacour and Poblador-Bahamonde et al. (Scheme 202).384 With a smaller amount of amine (3 equiv) and benzoic acid additive (1.5 equiv), it was possible to achieve preferential formation of azaoxa species [202.2a]+. Upon increasing the stoichiometry of reagents (25 equiv of amine and 12.5 equiv of benzoic acid) along with a prolonged reaction time (7 h) at 70 °C, the diaza [202.2b]+ species could be obtained from either [202.1]+BF4– or [202.2a]+. Amines with n-octyl, i-propyl, alcohol, carboxylic acid, and ester functional groups were generally compatible with these reaction conditions, and two different R groups could be introduced using the sequential protocol.
Scheme 202. Cationic Helicenes.

Reagents and conditions: (a)384 RNH2 (3 equiv), PhCOOH, NMP, 60 °C, 3 h; (b) RNH2 (25 equiv), PhCOOH, NMP, 70 °C, 7 h; (c)385 1,3,3-trimethyl-2-methyleneindoline, benzophenone, 80 °C, 21 h; (d) 1,3,3-trimethyl-2-methyleneindoline, (i-PrO)3Al, benzophenone, 80 °C, 24 h; (e) i-Pr2NEt (3.0 equiv) at 90 °C, NMP.
In 2020, Bosson, Jacquemin, and Lacour et al. showed that tertiary alkyl amines can be oxidized to enamines by cationic dioxa[6]helicene, which further reacts as an electrophile and oxidant to form mono or bis donor−π–acceptor coupling products.385 Cationic dioxa[6]helicene 202.1 readily oxidized tertiary alkyl amines such as diisopropylethylamine into the corresponding enamine. Then, nucleophilic oxidative addition of such an in situ generated intermediate to 202.1 led to the formation of merocyanine-like products, such as 202.3c. With 1,3,3-trimethyl-2-methyleneindoline as a nucleophile, mono- and bis-addition products of oxidative coupling 202.3a and 202.3b were selectively prepared. Because of the extended π-delocalization, optical properties of 202.3 displayed strong hyper- and bathochromic shifts relative to 202.1. These salts absorbed and emitted in the far-red and NIR domains (λmaxabs of up to 791 nm, λmaxem of up to 887 nm) and had relatively strong ECD signals in the NIR range (up to |Δε| = 60 M–1 cm–1 at ca. 800 nm for the most red-shifted derivative).
V-Shaped dioxaphenalene fluorescent dyes were described by Tsubaki et al. (Scheme 203).386,387 Initially, precursor 203.1 was obtained in a reaction between MOM-protected 1,3,6-trihydroxy-9H-xanthen-9-one and an appropriate naphthyllithium. The acid-mediated removal of the five MOM groups and intramolecular cyclization under basic conditions produced the quinoidal dioxaphenalene 203.2 in 65% yield. Subsequent treatment of 203.2 with acetic anhydride afforded the corresponding diacetate compound 203.3. Derivatives of 203.2 containing piperidinyl groups387 were shown to be promising bioimaging fluorescent dyes in experiments using HeLa cells.
Scheme 203. V-Shaped Dioxaphenalene Dyes.

Reagents and conditions: (a)386,387 (1) H2SO4, (2) K2CO3; (b) Ac2O, DMAP.
A one-step synthesis of azathiaphenalenes was reported by Mongin et al. in 2020, as part of their research on thioxanthone chemistry (Scheme 204).388 The amine 204.1, obtained from the corresponding iodo derivative via CuI-catalyzed amination, was reacted with 2-iodobenzothiophene and 2-iodobenzofuran in the presence of activated copper (0.2 equiv) and potassium carbonate (2 equiv), yielding the corresponding hexacyclic products 204.3a,b. Both compounds fluoresced in the green region of the visible spectrum, with quantum yields of ca. 0.5.
Scheme 204. N-Arylation of 1-Amino-9-thioxanthone by Heteroaryl Iodides.

Reagents and conditions: (a)388 0.2 equiv of activated Cu, 2 equiv of K2CO3, Bu2O, reflux, 24 h.
5.2.2. Other Diheteraphenalenoids
A xanthene-containing phenalenoid 205.3 was reported by Mastalerz et al., who found that 205.2, the product of the Pictet–Spengler reaction between 205.1 and a substituted salicylaldehyde, cyclized spontaneously to give 205.3, when heated without solvent at 180 °C (Scheme 205).241 This efficient transformation could also be used to make 205.4 from the corresponding diamine. In the final step, however, significantly higher reaction temperatures (300 °C) were needed. The fused systems were fluorescent in solution with a significant solvatochromism and quantum yields ranging up to 72%. An alternative multicomponent synthesis of similar chromophores, 205.5a,b, was reported by Bornadiego, Díaz, and Marcos.389
A route to helical heteroatom-doped PAHs consisting of double ortho-C–H activation of benzamides and subsequent N,O-double annulations with aryl alkynes was reported by Lan and You et al. in 2019 (Scheme 206).390 To suppress the formation of the alternative N,N-double annulation product (206.1), the reaction between benzamide and diphenylacetylene had to be optimized toward preferential formation of the desired N,O-annulated target 206.2. The best result was obtained in the catalyst system of [Cp*RhCl2]2/CF3CO2Ag, in the presence of several additives. A wide scope of alkynes and para-substituted benzamides was demonstrated, giving the desired products in moderate to good yields. Upon treatment of one of these products, 206.3, with PIFA and BF3·Et2O, the π-extended phenalenoid 206.4 was obtained in 60% yield. 206.4 possessed two cove regions and exhibited strong blue emission both in solution and in a PS film.
Scheme 206. Double C–H Activation/Annulation of Benzamides with Aryl Alkynes.

Reagents and conditions: (a)390 [Cp*RhCl2]2 (5 mol %), CF3CO2Ag (30 mol %), Ag2O, Mg(OAc)2·4H2O, PivOH, DCE, 85 °C, under N2, 12 h; (b) phenyliodine(III) bis(trifluoroacetate), BF3·Et2O, DCM, N2, −40 °C, 2 h.
5.3. Higher Heteraphenalenes
5.3.1. N-Doped Systems
A direct demethylative borylation applied to the synthesis of benzo[fg]tetracenes containing boronate ester, amide, and thioester substructures was reported by Hatakeyama et al. in 2016 (Scheme 207).391 The demethylative direct borylation of a teraryl with two methoxy groups (207.1a) provided the boronate-based benzo[fg]tetracene (207.2a) in one step. The key to successful synthesis was the proper choice of the boron source and Brønsted base, which facilitated the boron-mediated demethylation and successive electrophilic arene borylation. The versatile direct borylation was applied to the synthesis of benzo[fg]tetracenes containing boronate amide or thioester substructures (207.2b,c). The synthesis of a π-extended, hexacene analogue of 207.2a was performed in a similar manner. The best yield (80%) for the formation of 207.2a was obtained by using 1 equiv of BBr3 and 2 equiv of 2,2,6,6-tetramethylpiperidine (TMP). NaBPh4, acting as a noncoordinative Brønsted base, was used in the synthesis of 207.2b, providing an improved yield of 71%. The synthesis of 207.2c was the least efficient, despite using the harshest reaction conditions (200 °C in an autoclave). 207.2c was observed to be air-stable but immediately decomposed in protic solvents, whereas 207.2a,b showed substantial stability.
A four-step synthesis of the BNB-doped zigzag-edged benzo[fg]tetracene 207.6 was reported by Bettinger et al. in 2017.392 Preliminary experiments showed that the 2-fold addition of boron atoms to the aniline with BCl3 or BBr3 was not successful. The electrophilic borylation of one phenyl group could however be achieved using boron trichloride and aluminum trichloride. The resulting 1,2-dihydro-1,2-azaborine derivative 207.4 was too unstable to be isolated and was transformed into the mesityl derivative 207.5 using MesMgBr in diethyl ether. After deprotonation of the nitrogen center in 207.5 with KHMDS, borylation of the phenyl ring proceeded upon addition of boron trichloride and aluminum trichloride. The crude reaction product was directly transformed into the dimesityl compound 207.6 by treatment with MesMgBr. Incorporation of the BNB moiety at the zigzag periphery of the benzo[fg]tetracene resulted in significant bathochromic shifts of the absorption spectrum and reasonably strong fluorescence (Φf = 0.21) with a large Stokes shift (3100 cm–1).
A structurally related framework, 208.3, was obtained by Zeng et al., who used a similar sequential borylation approach (Scheme 208).393208.3 was chemically reduced with an excess of metallic potassium in the presence of [18]-crown-6 in anhydrous THF. The resulting salt of dianion [208.3]2–, isolated as a deep blue solid, was stable in an inert atmosphere but sensitive to moisture. Comproportionation of [208.3]2– with an equivalent amount of neutral 208.3 produced, instead of the expected radical anion, the diamagnetic rearrangement product [208.4]2–, which was crystallographically characterized. [208.4]2– was found to contain a completely planar BNB-doped benzo[cd]fluoranthene skeleton.
Scheme 208. Preparation of 1,9-Dibora-9a-azaphenalenyl (DBAP) Anionic Species.

Reagents and conditions: (a)393 dichlorophenylborane, toluene, 0 °C to reflux; (b) (1) LiHMDS, toluene, 0 °C to rt, (2) dichlorophenylborane, 0 °C to reflux; (c) 18-crown-6, K, THF; (d) 208.3.
Dibenzo-fused 1,9-diaza-9a-boraphenalenes featuring zigzag edges with a nitrogen–boron–nitrogen bonding pattern were synthesized by Feng’s group in 2016 (Scheme 209).394 Quinquephenyl 209.3, prepared via Suzuki coupling from appropriate building blocks, was treated with BCl3 and triethylamine at 180 °C to afford 209.4 as a yellow crystalline solid. 209.4 exhibited split emission bands at 448 nm and a high fluorescence quantum yield (ΦPL = 0.83). NBN-doped phenalenoids 209.6–9 were synthesized by Wakamiya and Wang et al. in 2018 using a similar synthetic approach.260 Their syntheses consisted of a palladium-catalyzed double Suzuki coupling reaction, followed by a 2-fold electrophilic aromatic borylation. The oxidative coupling of 209.7 using DDQ led to the formation of a fully fused planar compound 209.8 in 50% yield.
In 2019, Bettinger et al. reported a series of BN-doped systems containing phenalenoid and perylenoid cores (Scheme 210).395 The synthetic pathway involved the diamino-substituted m-terphenyl 210.1, which was subjected to electrophilic borylation with BBr3 in the presence of NaBPh4 as a noncoordinating base. Instead of the commonly used o-dichlorobenzene, the use of the lower-boiling toluene as the solvent was found to work well for this transformation. The resulting benzo[f,g]tetracene derivative 210.2 was singly brominated and subjected to Miyaura borylation, to give the boronate ester 210.4, which was coupled with aryl bromide 210.5, bearing a 1,8-naphthalenediamine-protected boryl group (Bdan). The resulting 210.6 was treated with excess degassed aqueous sulfuric acid, and the target molecule 210.7 was isolated in 92% yield. The synthesis of an analogue of 210.2 possessing two additional nitrogens 210.8 was reported by Bonifazi in 2020.396
Ultrapure-blue TADF emitters, benefiting from the multiple resonance effect, were developed by Hatakeyama in 2016.397 The key compound 211.3 (DABNA-1) was synthesized in two steps from a commercially available starting material, 1-bromo-2,3-dichlorobenzene 211.1 (Scheme 211). The boron atom was introduced in one pot via a lithium–chloride exchange reaction of 211.2 with tert-butyllithium, electrophilic trapping with boron tribromide, and tandem electrophilic arene borylation to give 211.3 in 32% yield. In OLED devices, a substituted variant of 211.3 (denoted DABNA-2) exhibited pure blue emission at 467 nm with a narrow fwhm of 28 nm, CIE coordinates of (0.12, 0.13), and an IQE of ≈100%. Carbazole-based DABNA analogues 211.4–5 were subsequently developed by the same group,262 whereas the related compounds 211.6–8 were reported by the groups of Hatakeyama et al. and Yasuda et al.398 All these compounds exhibit narrowband thermally activated delayed fluorescence with emission spectra ranging from blue to red. The organic light-emitting diode devices employing these products as emitters exhibited emission with high external quantum efficiencies up to 31.8% for 211.6c.
Benzo[4]helicenes 212.3a–d, with a “reversed” BNB pattern were reported by Hatakeyama et al. and by Zysman-Colman and Wang et al. (Scheme 212).399 The borinic acid 212.2 was prepared from readily available 212.1 and converted into 212.3a–d via borylation followed by the treatment with the corresponding phenyllithium. Small ΔES–T values and emission enhancement at higher temperatures supported the TADF behavior of these derivatives.
Scheme 212. Intramolecular Borylation via Sequential B–Mes Bond Cleavage.

Reagents and conditions: (a)399t-BuLi and MesB(OMe)2, THF, −78 °C; (b) (1) BBr3, toluene, 110 °C, 24 h, (2) R–Li (2.4 equiv), THF or Et2O, −78 °C to rt, 12 h.
Wakamiya and Murata et al. explored semiconducting properties of various structures based on oxygen-bridged triphenylamine subunits (Chart 20).400−402 The isomeric dimers C20.1 and C20.2 showed, respectively, 1D and 2D stacking patterns in the crystalline state, which influenced their charge-transporting behavior. While C20.1 showed strong field dependency in the bulk charge mobilities, C20.2 exhibited ambipolar charge-transporting properties with comparable hole and electron mobilities (ca. 10–3 cm2 V–1 s–1). The fluorescence spectra of amorphous films of C20.1 and C20.2 suggested the coexistence of a monomer-like random orientation and a π-stacking orientation. Hole-transporting materials, in which the two identical (C20.3) or different subunits (C20.4) were connected via a 1,3-phenylene linker, were reported in 2016.401 These compounds showed excellent amorphous stability and did not crystallize even after annealing at 160 °C. Vacuum-deposited amorphous films of C20.3 and C20.4 combined transparency and good hole mobility. C20.5a–c and C20.6 were designed as hole-transporting materials for perovskite solar cells, showing better performance (PCE of 16.5% for C20.5a) than the commonly used Spiro-OMeTAD.400
Chart 20. Oxygen-Bridged Triphenylamine Skeletons.

Phosphorescent iridium and platinum complexes with N,B,O-doped polycyclic ligands were described by Yuan et al. (Chart 21).403,404 Single-crystal structures indicated that the B-containing structures were more planar than their N-doped analogues, leading to different π–π-stacking and electronic characteristics. Solid-state packing could be further modified by coordination of pyridine ligands to the boron centers (C21.5a,b). More importantly, by controlling the number of embedded boron or nitrogen atoms (C21.3a,b vs C21.4a,b), solution-processed OLED devices incorporating these materials as emitting layers could achieve a phosphorescence color variation from green to deep red and showed low-efficiency roll-off and turn-on voltage. In particular, the boron derivative C21.4a showed good color purity with a narrow full width at half-maximum (1211 cm–1) and CIE coordinates (0.67, 0.31) in the deep-red region. Platinum complexes C21.1–2 showed a significant dependence of intermolecular interactions in the solid state on the nature of the X heteroatom (B vs N), attributed to both steric and electronic effects. Under a hypoxic atmosphere, the N-containing neutral complex C21.1b displayed dual emission with both fluorescence and phosphorescence, whereas only fluorescence was observed in air. In contrast, the boron analogue C21.1a showed only phosphorescence at all dioxygen levels.
Chart 21. Metal Complexes with NBO-Doped Polycyclic Ligands.

5.3.2. Tricycloquinazolines and Related Systems
The century-old tricycloquinazoline motif 213.2 (see CR2017, Section 5.3.15.3.1) was revisited by Ruoff et al. in 2018 (Scheme 213).405 Using an ionothermal protocol, 213.2 was prepared in excellent yields by heating a mixture of 213.1 and 1 equiv of ZnCl2 at 300–350 °C in flame-sealed ampules. In the same way, heat treatment of the diamine 213.3 in the presence of 1 equiv of ZnCl2 at 300 to 400 °C produced covalent networks with the idealized structure 213.4. 213.4 showed high chemical and thermal stability, a surface area greater than 1800 m2 g–1, and large pore volumes of up to 0.93 cm3 g–1. Excellent CO2 uptake capacity was demonstrated for these materials, reaching 7.16 mmol g–1 at 273 K and 1 bar.
Scheme 213. Synthesis of Porous Covalent Quinazoline Networks.

Reagents and conditions: (a)405 ZnCl2, 300–400 °C.
Tricycloquinazoline was found to use a bridging ligand in a series of iridium complexes 213.5–7 reported by Shensky et al.406 Transient absorption bands observed for all three complexes in the 475–900 nm range implied the potential for reverse-saturable absorption (RSA) at those wavelengths. The presence of RSA behavior was further confirmed for all three compounds using Z-scan measurements. The enhanced triplet cross section and ratio of triplet cross section to ground-state cross section for 213.5 suggested that it might be more beneficial for RSA applications than 213.6–7.
5.4. Phenalenoids with Nonbenzenoid peri-Fusion
5.4.1. Cyclopenta[cd]phenalenes
Fused and heteroatom-doped derivatives of ullazines have been explored by several research groups (see also Scheme 21, Section 2.4, and CR2017, Section 5.4.1 for earlier developments). An efficient synthesis of 6-aza-ullazines was described by Langer and co-workers in 2017 (214.2, Scheme 214).407 The procedure involved metal-free cyclization promoted by p-toluenesulfonic acid as the key ring-forming step. These aza derivatives showed similar absorption and emission spectra but higher oxidation potentials than the parent ullazine. Three naphtho-fused derivatives 215.3a–c were synthesized by Berger and Feng et al. from the known dione 215.1 (Scheme 215).408 Nucleophilic addition of lithium triisopropylsilylacetylenide, phenyllithium, and benzo[b]thiophen-2-yllithium to 215.1 gave diols 215.2a–c, which were subsequently reduced with anhydrous SnCl2 to yield 215.3a–c. These targets had easily accessible radical cationic and anionic states, which were generated using in situ spectroelectrochemistry.
Scheme 214. Synthesis of Azaullazines.

Reagents and conditions: (a)407p-TSA, xylene, 120 °C, 24 h.
Scheme 215. π-Extended Ullazine with a Benzoisoindole Core.

Reagents and conditions: (a)408 for 215.2a: n-BuLi, triisopropylsilylacetylene, THF, 0 °C to rt, 3 days, 215.2b: phenyllithium, THF, rt, 3 days, 215.2c: n-BuLi, 2-bromobenzothiophene, THF, −78 °C to rt, 3 days; (b) SnCl2, DCM, rt, 3 days.
In 2019, Bunz, Feng, and Berger et al. reported ullazine derivatives (216.3–7) obtained through cycloaddition reactions of the previously reported polycyclic aromatic azomethine ylides (PAMYs, Scheme 216).409,410 Intermediates 216.2a,b were initially obtained via 1,3-dipolar cycloaddition of PAMYs, generated in situ from the corresponding salts 216.1a,b by addition of triethylamine (TEA), followed by dehydrogenation with DDQ. 216.2a,b were then condensed with hydrazine to yield the final compounds 216.3a,b as yellow solids.409 The cycloaddition reaction of PAMY 216.1b with ortho-1,2-naphthoquinone and the following oxidation gave the intermediate 216.4, which provided the helicene-like 216.5 upon condensation with ortho-benzenediamine. The same synthetic method was used to obtain larger fused frameworks by cycloaddition of PAMY to anthracene-1,2,6,7-tetraketone, acting as a single or double dipolarophile. Subsequent condensation with ortho-benzenediamine provided 216.6–7 with two and four [5]helicene motifs, respectively.410
Scheme 216. Synthetic Routes of the Ullazine Derivatives.
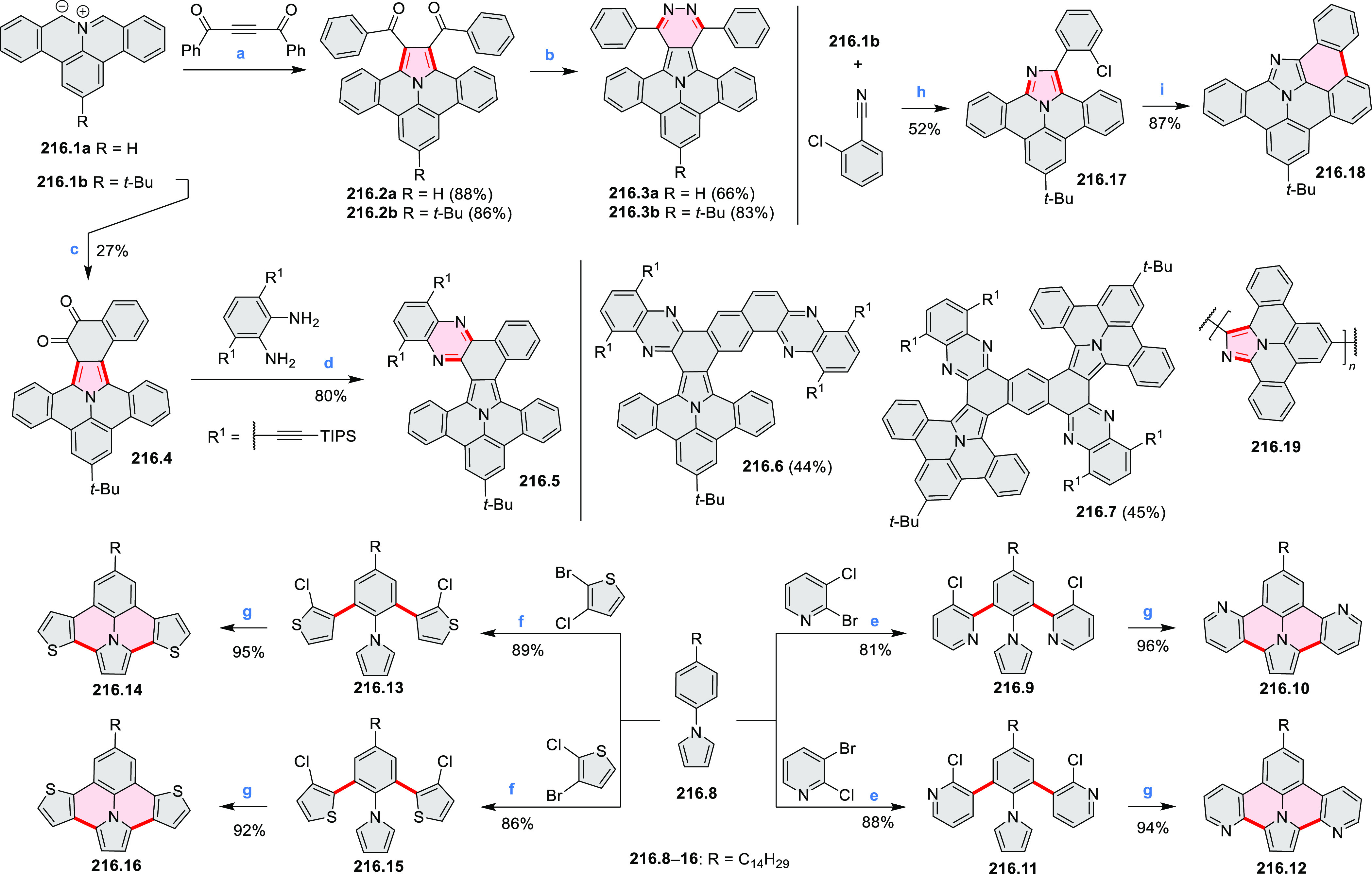
Reagents and conditions: (a)409 (1) 1,4-diphenylbut-2-yne-1,4-dione, DCM, 5 min, rt, (2) DDQ, toluene, rt, 3 h; (b) hydrazine, ethanol, overnight, 80 °C; (c)410 (1) 1,2-naphthoquinone, DCM, TEA, rt, 5 min, (2) DDQ, toluene, rt, 3 h; (d) HOAc, toluene, 100 °C, 72 h; (e)413 Pd(dppf)Cl2, Na2CO3, DMSO, 100 °C, 16 h; (f) PdCl2(PPh3)2, PPh3, K2CO3, DMF/H2O, 90 °C, 24 h; (g) hν (254 nm), decalin, 100 °C, 2 h; (h)411 (1) i-Pr2NEt, CsF, DMSO, 160 °C, 12 h, (2) DDQ, DCM, rt, 20 min; (i) Pd(AcO)2, P(t-Bu)2Me·HBF4, DBU, DMA, 160 °C, 12 h.
Ito et al. reported the 1,3-dipolar cycloaddition between the PAMY derived from 216.1b with nitriles to produce highly fused imidazole derivatives such as 216.17.411 This transformation had a broad substrate scope and good functional group compatibility. Subsequent palladium-catalyzed intramolecular cyclization provided access to even more π-extended imidazoles such as 216.18. Analogous cycloaddition chemistry was used by Palma et al. for on-surface oligomerization of cyano-substituted PAMYs into dibenzoullazine chains 216.19 on Au(111), Ag(111), and h-BN/Cu(111) surfaces.412
A metal-free, photochemical cyclodehydrochlorination reaction was developed by Morin et al. as an alternative route to π-extended ullazine derivatives annulated with electron-poor pyridine (216.10 and 216.12) and electron-rich thiophene units (216.14 and 216.16).413 The common starting material 216.8 was coupled to 2-bromo-3-chloropyridine and 3-bromo-2-chloropyridine to give the pyridyl-substituted precursors 216.9 and 216.11. Likewise, coupling with 2-bromo-3-chlorothiophene and 3-bromo-2-chlorothiophene gave compounds 216.13 and 216.15, respectively. When subjected to the photochemical cyclodehydrochlorination reaction, all four precursors produced the ullazine analogues 216.10, 216.12, 216.14, and 216.16 in excellent yields.
A series of π-extended cycl[3,3,2]azines (217.2–4), structurally related to ullazines, were reported by Klauk, Müllen, and Li et al. in 2018 (Scheme 217).414 Following an established route, the initial aldol condensation was carried out on 217.1 in imidazole solution, giving 217.2 in good yields. Malononitrile was reacted with 217.2 under acidic conditions, to produce the singly and doubly condensed products, 217.3 and 217.4. These two systems were found to be strong electron acceptors with low-lying LUMO energy levels of −3.99 and −3.95 eV, respectively. Organic thin-film transistors based on 217.4 were fabricated by vapor deposition, exhibiting extraordinarily stable n-type semiconductor character under ambient conditions, with the highest electron mobility of 0.06 cm2 V–1 s–1 retained consistently for more than 30 months.
Scheme 217. Synthesis of π-Extended Cycl[3,3,2]azines.

Reagents and conditions: (a)414 imidazole, 120 °C, 2 h; (b) AcOH, Ac2O, malononitrile, reflux.
In 2019, Moresco et al. reported the on-surface synthesis of a nitrogen-doped nanographene bearing five- and seven-membered rings (Scheme 218).415 The extended ullazine 218.2 was first synthesized in four steps from 1,4-naphthoquinone and the iminium salt 218.1 in 90% overall yield (for related cycloaddition reactions, see Scheme 221, Section 6.1.1). Afterward, compound 218.2 was deposited on an atomically clean Au(111) surface. The sample was annealed starting at 160 °C, and structural changes began to emerge after annealing to 250 °C as observed by STM. After annealing at 300 °C, three new species were observed and identified at high resolution with a CO-functionalized tip to be dephenylated, doubly cyclized species 218.3, the triply cyclized species 218.4, and the quadruply cyclized species 218.5 in relative abundances of 25%, 65%, and 10%, respectively.
Scheme 218. On-Surface Synthesis of Fused Azananographenes.
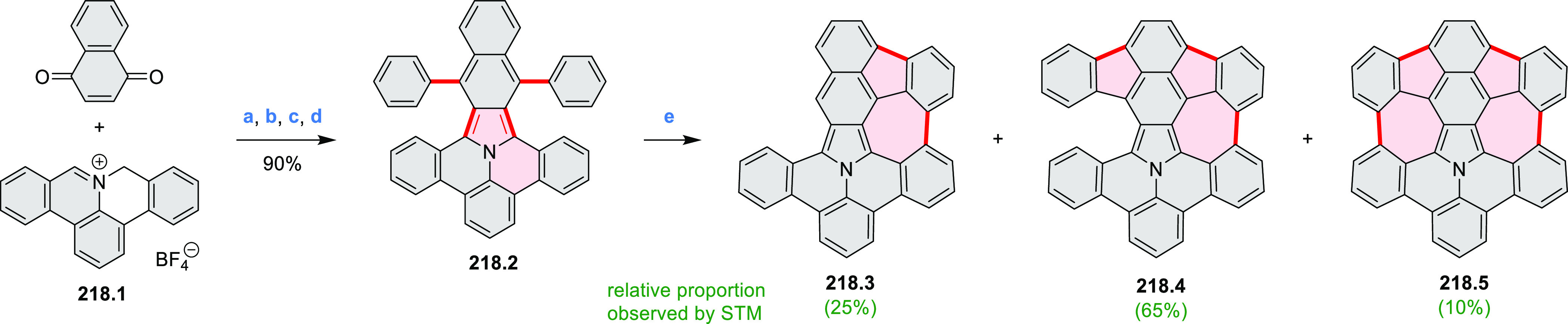
Reagents and conditions: (a)415 Et3N, DCM, rt, 5 min; (b) DDQ, toluene, rt, 3 h; (c) PhLi (excess), THF, rt, 3 days; (d) SnCl2, DCM, rt, 2 h; (e) 300 °C on Au(111).
Scheme 221. Azabuckybowl Synthesis via 1,3-Dipolar Cycloaddition of Corannulene.
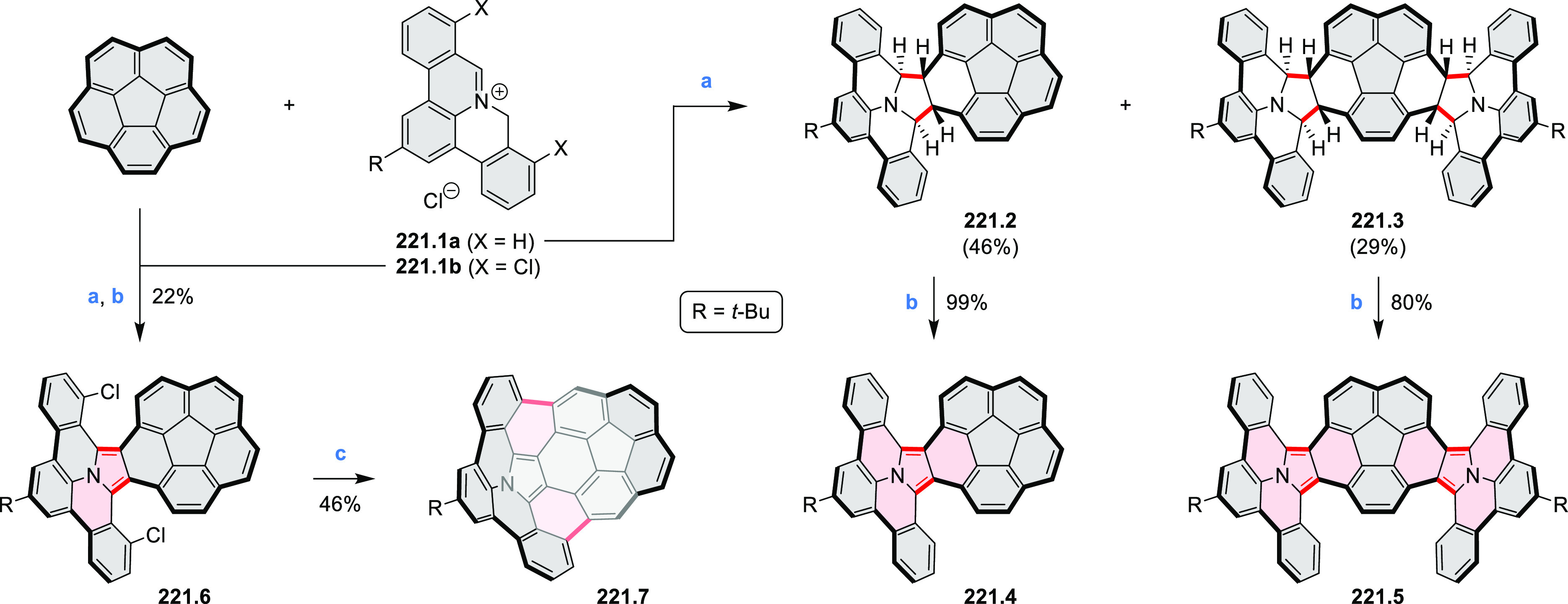
Reagents and conditions: (a)418,419i-Pr2NEt, DMSO, 120–140 °C, 1–20 h; (b) DDQ (1.8–4.2 equiv), DCM, rt, 10 min–14 h; (c) Pd(OAc)2, t-Bu2PMe·HBF4, DBU, DMA, 150 °C, 24 h.
Gryko and co-workers reported the synthesis of butterfly-shaped pyrrolo[3,2-b]pyrroles containing dual [6]helicene substructures (Scheme 219).416 Specifically, 219.4–5 were obtained in a three-step procedure, involving efficient intramolecular oxidative aromatic coupling in the final ring-forming step. These nanographenoids had nonplanar, twisted structures and were moderately emissive in solution (QY up to 32%) with observable solvatochromism.
Scheme 219. π-Extended Pyrrolopyrroles with a Double-Helicene Structure.
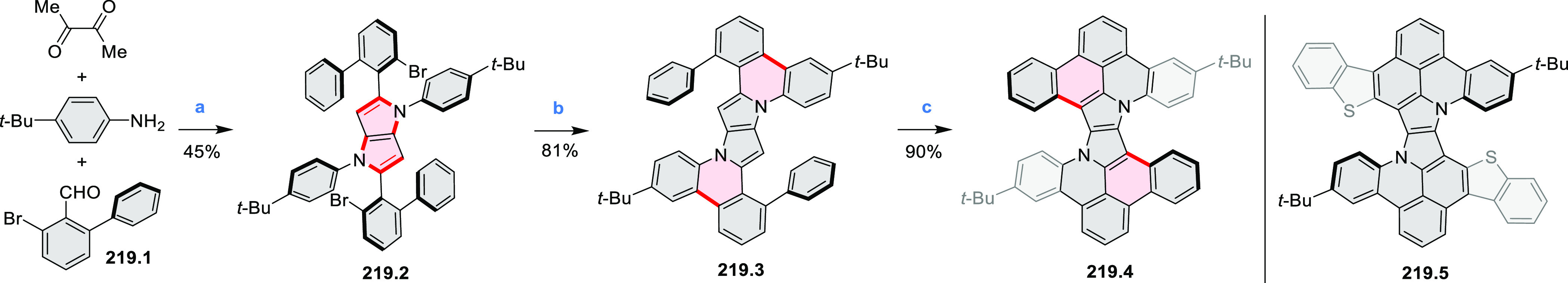
Reagents and conditions: (a)416p-TsOH, AcOH, 90 °C, 3 h; (b) Pd(AcO)2, PPh3, Cs2CO3, toluene, 120 °C, 3 h; (c) FeCl3, DCM, MeNO2, rt, 0.5 h.
The groups of Jiao, Wang, and Pei reported in 2020 the synthesis of azadipyrromethene-based polycyclic aromatics containing 13 fused rings (Scheme 220).417 The installation of phenyl rings at 2,6-positions of the tetraaryl azadipyrromethenes 220.2 was achieved using Suzuki coupling, and the resulting intermediates were converted into BF2 complexes and subjected to oxidative ring-fusion reaction. The latter step used DDQ/TfOH as the oxidant to give the target 220.4. In comparison with 220.3, the fully annulated 220.4 showed a sharp absorption between 800 and 880 nm (red-shifted by more than 160 nm relative to 220.3) and emission with a very small Stokes shift.
Scheme 220. Synthesis of π-Extended Aza-BODIPYs.

Reagents and conditions: (a)417 CH3CO2NH4, EtOH, reflux; (b) (1) 2 equiv of Br2, DCM, rt, (2) 4-alkylphenylboronic acid, Pd(PPh3)4, Na2CO3, (3) Et3N, BF3·Et2O; (c) TfOH, DDQ, DCM, rt.
6. Nonbenzenoid Fusion
6.1. Circulenoids and Related Systems
6.1.1. Heterofused Circulenes
In 2017, Tokimaru, Ito, and Nozaki reported the first 1,3-dipolar cycloaddition between corannulene and a polyaromatic azomethine ylide (Scheme 221).418 In the presence of Hünig’s base, the iminium salt 221.1a was deprotonated to give the corresponding azomethine ylide (the 1,3-dipole), which then reacted with corannulene (the dipolarophile). The single and double cycloadducts 221.2 and 221.3 were isolated in 46% and 29% yield, respectively. The X-ray structures of both compounds indicated the 1,3-dipole approached the rim C=C bonds of corannulene from the convex face in an exo manner. On DDQ-mediated dehydrogenation of 221.2–3, the corresponding pyrrole-fused corannulenes 221.4–5 were obtained in high yields. In 2018, the same authors reported an analogous reaction sequence with the iminium salt 221.1b bearing two chlorine atoms.419 The palladium-catalyzed ring closures of 221.6 afforded compound 221.7 with two new six-membered rings in 46% yield. The highly curved structure of 221.7 was evident from the large bowl depth of 4.19 Å determined from the X-ray structure. Compared with the HOMO (−5.12 eV) and LUMO (−2.52 eV) levels of the noncyclized analogue 221.4, the additional π-conjugation in 221.7 lowered the LUMO level (−3.04 eV) but did not alter the HOMO level (−5.11 eV).
In 2020, an aza-annulative π-extension (aza-APEX) reaction, applicable to unfunctionalized aromatics, was described by Itami and Ito et al. (Scheme 222).420 In one of the trials, a solution of corannulene, N-phenylbenzimidoyl chloride (222.1), and AgPF6 in 1,2-dichloroethane was heated at 80 °C, followed by addition of p-chloranil. The quinoline-fused corannulene derivative 222.2 was isolated in 41% yield. The authors proposed the following mechanism based on extensive DFT calculations. First, the imidoyl chloride 222.1 was converted to the nitrilium salt 222.3 upon the action of AgPF6. An initial π-complex formed between the nitrilium ion and the arene gave rise to the carbocationic intermediate [222.4]+. Afterward, an intramolecular C–C bond formation between the cationic center and the N-phenyl group generated a new six-membered ring in [222.5]+. Proton transfers assisted by PF6– gave the rearomatized species [222.6]+, which was subsequently dehydrogenated by p-chloranil to produce the target product.
Procter et al. reported a transition-metal-free, one-pot protocol for the thienannulation of arene substrates via 2-fold C–H functionalization (Scheme 223).284 In a representative example, the overall reaction between corannulene and 2-chloro-3-(methylsulfinyl)prop-1-ene (223.1) gave rise to the corannulene derivative 223.2 bearing an ortho-fused methylthiophene unit in 18% yield. Mechanistically, the sulfoxide 223.1 was first activated by triflic anhydride to become the salt 223.3, which participated in an intermolecular interrupted Pummerer reaction with corannulene. The resulting sulfonium ion 223.4+, upon microwave heating, underwent a [3,3]-sigmatropic rearrangement into the neutral intermediate 223.5, which then spontaneously cyclized in the presence of acid to give the sulfonium species 223.6+. Eventually, addition of triethylamine followed by heating effected the elimination of HCl and removal of the sulfur-bonded methyl group, thereby providing the desired thienannulation product 223.2.
Another transition-metal-free thienannulation of arylethynyl-substituted arenes using elemental sulfur was reported by Itami and Segawa et al. (Scheme 224).209 In the typical procedure, a solution of (4-tert-butylphenyl)ethynyl-substituted corannulene 224.1 in DMF was heated in the presence of elemental sulfur for 24 h. The cyclization product 224.2 bearing an ortho-fused thiophene unit was obtained in 99% yield. Likewise, the 5-fold thienannulation of the C5-symmetrical pentakis(arylethynyl)-substituted corannulene 224.3 furnished the corresponding pentathieno-fused derivative 224.4 in 20% yield. Based on a series of experiments, the authors proposed the following mechanism. First, the electrophilic Sn forms a C–S bond with the arene to form the zwitterion 224.5, in which the positive charge is stabilized by both the neighboring aromatic ring and the alkyne unit. Subsequently, the elimination of elemental sulfur (Sn–1) and a proton generates the rearomatized thiolate intermediate 224.6–. Then, the nucleophilic sulfur anion attacks the alkyne unit, forming the carbanionic species 224.7–, which eventually leads to the target product upon protonation by residual water. The authors showed that this simple procedure could be carried out on a decagram scale and could allow multiple thienannulations.
In 2018, Bao et al. prepared the corannulene imide derivative 225.2 bearing two ortho-fused indole units via the triphenylphosphine-mediated 2-fold reductive cyclization of the bis(2-nitrophenyl)-substituted precursor 225.1 (Scheme 225).421 They found that the diindole-fused derivative 225.2 had an absorption onset at 588 nm, significantly red-shifted from that of the corannulene imide 225.3 without any indole fusions (437 nm). Using DFT calculations, they also determined that the diindole-fused derivative 225.2 possessed a bowl-to-bowl inversion barrier of 7.2 kcal mol−1, which was lower than that of 225.3 (9.8 kcal mol−1).
Scheme 225. Synthesis of Diindole-Fused Corannulene Imide by Reductive Ring Closure.

Reagents and conditions: (a)421 PPh3, dry o-dichlorobenzene, 180 °C, 36 h.
In 2016, Rajeshkumar and Stuparu reported a two-step synthesis of π-extended corannulenes (Scheme 226).422 First, the corannulene-based phosphonium ylide 226.1 underwent the Wittig reaction with 2-thiophenecarboxaldehyde to furnish an E/Z isomeric mixture of the stilbenoid product 226.2 in 95% yield. The Mallory reaction of 226.2 in the presence of iodine and propylene oxide gave the benzothiophene-fused corannulene derivative 226.3 in good yield. If benzo[b]thiophene-2-carboxaldehyde (226.4) was used in the first step, another corannulene derivative 226.5 bearing a fused dibenzothiophene unit could be obtained. According to the authors, this work was the first example of π-extension of the bowl-shaped corannulene nucleus via photocyclization, which served as an alternative to known methods such as metal-catalyzed cyclization and flash vacuum pyrolysis.
Scheme 226. π-Extension of Corannulene via Photocyclization.
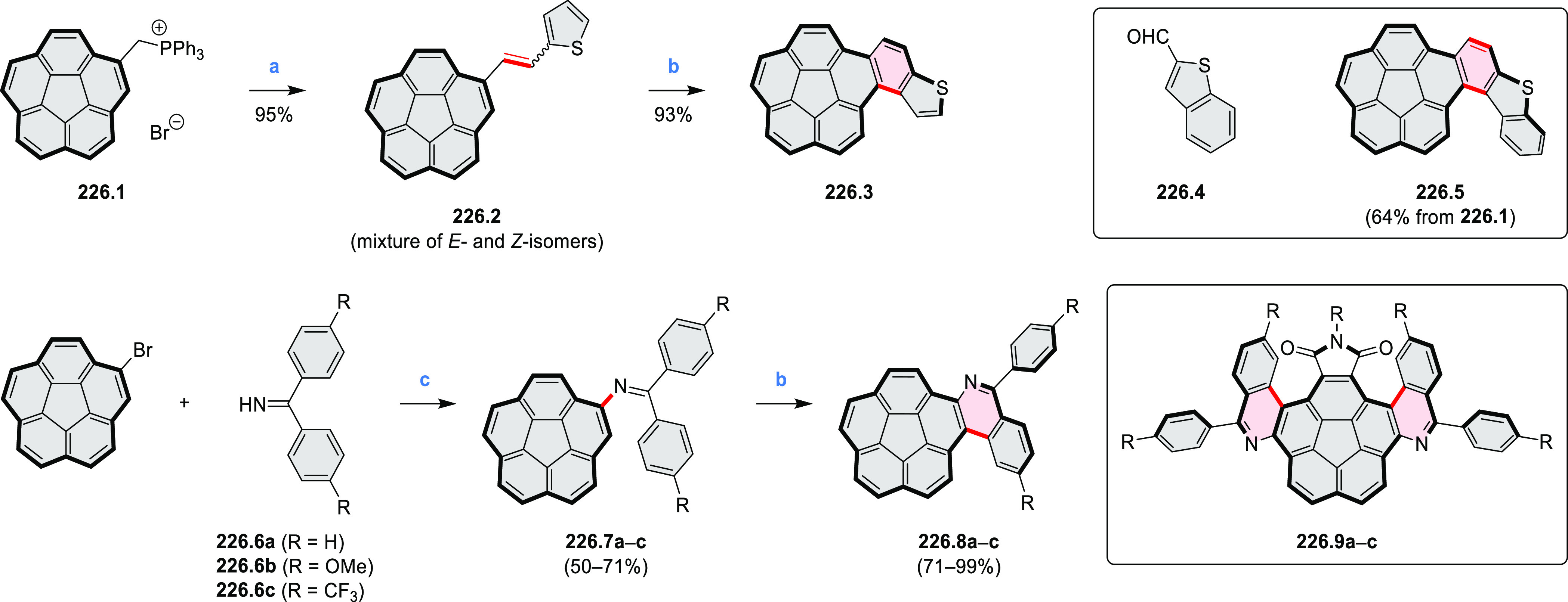
Reagents and conditions: (a)422 (1) n-BuLi, THF, 0 °C, 30 min, (2) 2-thiophenecarboxaldehyde, rt, 2 h; (b) I2, propylene oxide, medium-pressure Hg lamp (125 W), toluene; (c)423 Pd2(dba)3, rac-BINAP, t-BuONa, toluene, 110 °C, 12 h.
In 2021, Stuparu et al. reported the analogous photocyclization for the imine-containing corannulenes 226.7a–c.423 These substrates were prepared from the Buchwald–Hartwig amination of bromocorannulene with the respective benzophenone imines 226.6a–c. Subsequent photochemical cyclization of 226.7a–c provided the isoquinoline-fused corannulenes 226.8a–c in 71–99% yield. Both the electron-rich methoxy and electron-poor trifluoromethyl groups were tolerated in this reaction. Doubly fused corannulenes 226.9a–c could also be formed from the appropriate precursors via the presumably more demanding 2-fold photocyclization in 44–99% yield. This result demonstrated the uniqueness of corannulene reactivity since the Mallory reaction is typically not applicable to N-aryl imines.
In 2019, a simple synthetic route to corannulene-based electron acceptors containing the sulfone functionality was presented by Stuparu et al.424 As shown in Scheme 227, 1,2,5,6-tetrabromocorannulene (227.1) was subjected to nucleophilic aromatic substitution by 1,2-benzenedithiol in the presence of a strong base. The corannulene derivative 227.2 bearing two ortho-fused benzodithiine units was formed in 80% yield. Upon oxidation by m-CPBA, compound 227.2 was converted to the corresponding tetrasulfone 227.3 in 58% yield. The reduction potentials of 227.3 and of the benchmark electron acceptor, PC61BM, were determined by square-wave and cyclic voltammetry. The electron affinities of the corannulene derivative 227.3 and PC61BM were found to be similar (0.9–1 V). The significant increase in electron-accepting capability of 227.3 relative to corannulene was attributed to the electron-withdrawing sulfone groups positioned in a rigid fused polycyclic framework.
Scheme 227. Synthesis of Doubly Fused Corannulene Tetrasulfone.

Reagents and conditions: (a)424 (1) benzene-1,2-dithiol, t-BuOK, DMF, rt, 1 h, (2) 60 °C, 21 h; (b) m-CPBA, DCM, rt, 48 h.
In 2018, Siegel and Baldridge et al. reported a synthetic approach toward azaindenocorannulene analogues bearing a pyridine unit.425 As shown in Scheme 228, 2-fluoro-3-pyridylcorannulene (228.1a) was subjected to silyl cation C–F activation/coupling to furnish the corresponding peri-fused corannulene 228.2 in 26% yield. The same product was obtainable, in 60% yield, from the palladium-catalyzed C–Cl activation/coupling of another substrate 228.1b. In related work by the same group, three cyclization substrates 228.5a,b and 228.6 were prepared from the enantiopure dibromocorannulene imide 228.3 by Suzuki–Miyaura cross-coupling.426 Their palladium-catalyzed ring closures successfully afforded 228.7a,b and 228.8 in 30–45% yield. Each of these azaindenocorannulenes existed as two diastereomers with opposite bowl configurations. HPLC analysis of time-sequenced aliquots of 228.7b revealed an inversion half-life of over 100 days (in diphenyl ether at 218 °C). The corresponding bowl inversion barrier exceeded 190 kcal mol−1, implying exceptional configurational stability.
Scheme 228. Synthesis of Azaindenocorannulenes.
Reagents and conditions: (a)425 [i-Pr3Si]+[CB11H6Cl6]−, (MeS)2SiMe2, chlorobenzene, microwave (225 W), 110 °C, 2 h; (b) Pd(PCy3)2Cl2, DBU, DMA, microwave (300 W), 160 °C, 0.5 h; (c)426 2-chloropyridine-3-boronic acid (4 equiv), Pd(PPh3)4, K2CO3, THF/H2O (9/1), 100 °C, 1.5 h; (d) 2-chloropyridine-3-boronic acid (8 equiv), Pd(PPh3)4, K2CO3, THF/H2O (9/1), 100 °C, 1.5 h; (e) 2-chlorophenylboronic acid, Pd(PPh3)4, K2CO3, THF/H2O (9/1), 100 °C, 1.5 h; (f) Pd(PCy3)2Cl2, DBU, DMA, 160–180 °C, 6–7 h.
In 2017, Miao et al. reported the synthesis and properties of tetrabenzo[7]circulene.427 The authors demonstrated the flexibility of their synthetic route for accessing heterofused analogues with four and two benzo fusions replaced with thieno fusions, i.e., compounds 229.1 and 229.2, respectively (Scheme 229). The synthesis began with the intramolecular Friedel–Crafts acylation of 229.3 to yield the diketone 229.4 bearing a seven-membered ring. On treatment with CBr4 and PPh3, one of the ketone groups in 229.4 was converted to the dibromovinylidene group in compound 229.5 in 75% yield. Surprisingly, the ketone group in 229.5 was inert to excess CBr4 and PPh3, presumably because of the steric blocking caused by the bromine atoms on the convex side of the molecule. Suzuki–Miyaura cross-coupling of the dibromide 229.5 allowed introduction of two 3-bromo-2-thienyl groups, leading to compound 229.6. Finally, palladium-catalyzed C–H activation of 229.6 gave the doubly cyclized product 229.7 in 75% yield. The ketone group in 229.7 could smoothly react with CBr4 and PPh3 to yield the corresponding dibromovinylidene derivative 229.8 in 90% yield. A repetition of the previous cross-coupling–cyclization sequence on 229.8 offered tetrathieno[7]circulene 229.1 (17% yield over two steps). The same two-step sequence performed on 229.8, using 2-bromophenylboronic acid as the alternative coupling partner, provided the unsymmetrical [7]circulene derivative 229.2 possessing two thieno and two benzo fusions (42% yield over two steps).
Scheme 229. Preparation of 4-Fold Fused [7]Circulene Derivatives Bearing Four or Two Thieno Fusions.

Reagents and conditions: (a)427 NaCl, AlCl3, 150 °C, 0.5 h; (b) CBr4, PPh3, toluene, 80 °C, 24 h; (c) (3-bromothiophen-2-yl)boronic acid pinacol ester, Pd(PPh3)4, K2CO3, THF/H2O (10/1), reflux, 24 h; (d) Pd(PPh3)4, Cs2CO3, toluene, reflux, 24 h; (e) 2-bromophenylboronic acid, Pd(PPh3)4, K2CO3, THF/H2O (10/1), reflux, 24 h.
6.1.2. [5]Heteracirculenoids
In 2017, Scott et al. reported a synthesis of the benzannulated azacorannulene derivative 230.4 (Scheme 230).428 They first prepared the bis(2-halophenyl)-substituted indenoisoquinoline derivatives 230.1a,b using known synthetic methods. When the first substrate 230.1a was heated with Pd(PCy3)2Cl2 and a strong base, single cyclization and C–Cl bond cleavage took place to give the product 230.2 in 15% yield. The cyclization was confirmed by 1H NMR spectroscopy to involve the pyridine nucleus. In other words, the singly cyclized isomer 230.3 was not found. For the second substrate 230.1b, flash vacuum pyrolysis (FVP) at 1000 °C produced the desired doubly cyclized product, i.e., 5-azadibenzo[a,g]corannulene (230.4) in 28% yield. The nitrile 230.5 was observed as a minor product which presumably originated from the thermal opening of 230.4. Furthermore, compound 230.4 underwent rapid hydrolysis in the presence of silica gel to yield the amino aldehyde 230.6. Hence, the nitrogen atom in 230.4 was assumed to provide a reactive site for ring-opening reactions to release the inherent ring strain.
Scheme 230. Attempted Synthesis of 5-Azadibenzo[a,g]corannulene by Solution-Phase Synthesis and Flash Vacuum Pyrolysis.
Reagents and conditions: (a)428 Pd(PCy3)2Cl2, DBU, DMA, 150 °C, 3 days; (b) FVP, 1000 °C; (c) silica gel, dry DCM, 24 h.
A four-step synthesis reported in 2018 by Hatakeyama et al. enabled the preparation of a corannulene derivative 231.5 possessing B–N units on two of the “spoke” bonds (Scheme 231).429 First, the commercially available 4,7-dibromobenzo[c][1,2,5]thiadiazole (231.1) was doubly coupled with phenylboronic acid to give compound 231.2 in 96% yield. Second, sulfur extrusion of the thiadiazole ring in 231.3 using zinc metal provided the diamine 231.3 in 86% yield. Third, condensation of 231.3 with diphenylacetic acid furnished compound 231.4 bearing a new imidazole ring. In the final step, heating an o-dichlorobenzene solution of 231.4 with BBr3 at 200 °C effected the electrophilic C–H borylation to produce B2N2-containing corannulene 231.5 in 32% yield. The final product could be obtained in multigram quantities (3.6 g). According to X-ray data, 231.5 possessed a much shallower bowl depth (0.15 Å) than corannulene (0.87 Å). The authors ascribed this observation to the longer rim C–B bonds (1.54–1.58 Å) in 231.5 than the rim C–C bonds (1.45–1.47 Å) in corannulene. NICS calculations suggested the nonaromatic character of the central C3N2 ring and of all four C4BN rings, with NICS(0) values ranging from −2.4 to −0.4 ppm. Moreover, the BN units rendered compound 231.5 strongly blue fluorescent at 424 nm, with a 69% quantum yield. Using compound 231.5, the authors reported the first fabrication of a corannulene-based OLED.
Scheme 231. Gram-Scale, Four-Step Synthesis of B2N2 Corannulene.

Reagents and conditions: (a)429 phenylboronic acid, Pd(PPh3)4, K2CO3, THF/H2O (1:1), reflux, 3 days; (b) Zn, acetic acid, 40 °C, 22 h; (c) 2,2-diphenylacetic acid, 180 °C, 62 h; (d) BBr3, o-dichlorobenzene, reflux, 16 h.
In 2018, Nozaki and Ito et al. reported the preparation and liquid-crystalline (LC) properties of 5-fold functionalized azapentabenzocorannulene derivatives.430 Using the methodology discussed earlier (see Scheme 221), compound 232.3 could be obtained in 34% yield. Compound 232.3 was then subjected to palladium-catalyzed 3-fold cyclization to give the unsubstituted azabuckybowl 232.4 (Scheme 232). Subsequently, the iridium-catalyzed 5-fold direct borylation of 232.4 furnished the 2,5,8,11,14-pentaborylated derivative 232.5 in 70% yield over two steps. Finally, compound 232.5 was submitted to Suzuki–Miyaura coupling with aryl bromides bearing long alkoxy substituents to yield the three pentaarylated products 232.6a–c in 55–82% yield. XRD analyses of 232.6a–c at 180–200 °C on cooling showed that these compounds form LC mesophases with hexagonally arranged columnar structures. The stability of these mesophases was attributed to the strong π–π stacking between the azapentabenzocorannulene cores. In the same year, Tokimaru, Ito, and Nozaki reported a conjugated azapentabenzocorannulene–corannulene hybrid 221.7 (see Scheme 221, Section 6.1.1).419
Scheme 232. Regioselective Five-Fold C–H Functionalization of Azabuckybowl.
Reagents and conditions: (a)430 (a) i-Pr2NEt, DMSO, 100 °C, 9 h; (b) DDQ, DCM, rt, 5 min; (c) Pd(OAc)2, t-Bu2PMe·HBF4, DBU, DMA, 140 °C, 18 h; (d) B2pin2, [Ir(OMe)(cod)]2, 4,4′-di-t-butyl-2,2′-bipyridine, t-BuOK, THF, 85 °C, 10 days; (e) 1,2,3-trialkoxy-5-bromobenzene, Pd2(dba)3·CHCl3, SPhos, Cs2CO3, toluene/H2O (2/1), 80–85 °C, 3–5 days.
In 2019, Petrukhina, Nozaki, and Ito et al. reported the chemical reduction of the known tert-butyl-substituted azapentabenzocorannulene 233.1 with sodium and cesium in THF in the presence of 18-crown-6 (18c6) (Scheme 233).431 The two-electron reduction of 233.1 with sodium metal gave the dianion 233.12– which existed with [Na+(18c6)(THF)2] and [Na+(18c6)(THF)] in 2:3:1 stoichiometry in the crystal structure. The analogous reduction of 233.1 with varied amounts of cesium allowed isolation of both the radical anion and dianion salts. In the former salt [Cs+(18c6)](233.1•–), the 18c6-capped cesium ion was bound to the concave side of 233.1•–. In the latter salt [Cs+(18c6)]2(233.12–), two cesium ions coordinated to the concave and convex faces of 233.12–. In both cases, the binding of cesium ions took place asymmetrically at the six-membered rings of the azabuckybowl.
Scheme 233. Chemical Reduction of an Azapentabenzocorannulene Derivative.

Reagents and conditions: (a)431 Na (8 equiv), 18-crown-6, THF, Ar, 25 °C, 48 h; (b) Cs (2 equiv), 18-crown-6, THF, Ar, 25 °C, 5 min; (c) Cs (5 equiv), 18-crown-6, THF, argon, 25 °C, 30 min.
In 2017, Shinokubo and Hiroto et al. reported the properties of azapentabenzocorannulene dimer 234.2 in which the two buckybowl units are linked through a C–C bond (Scheme 234).432 The synthesis involved subjecting the known polyaromatic precursor 234.1 to palladium-catalyzed intra- and intermolecular C–C bond formation using the catalyst system of Pd(OAc)2 (0.2 equiv) and PCy3·HBF4 (0.4 equiv). Compound 234.2 was formed in 31% yield and cocrystallized with C60 in 1:2 ratio. As revealed by XRD analysis, the two azabuckybowl units of 234.2 are oriented in opposite directions relative to the central C–C bond, while the concave face of each unit was coordinated to one C60 molecule. In solution, however, molecules of 234.2 were found to form a 1:1 complex with C60, while precipitation was observed at higher C60 concentrations. SEM images of the precipitate revealed the presence of fiber-like aggregates, resulting from the formation of one-dimensional chain-like supramolecular assemblies. The 1:1 binding ratio of 234.2 and C60 in the precipitate was established by variable-temperature 1H NMR spectroscopy using toluene-d8 as the solvent.
Scheme 234. Bis(azapentabenzocorannulene)s.

Reagents and conditions: (a)432 Pd(OAc)2 (0.2 equiv), PCy3·HBF4 (0.4 equiv), K2CO3 (8 equiv), DMA, 130 °C, 29 h; (b)433 3,6-dibromo-9H-carbazole, Pd(PPh3)4, Cs2CO3, THF/H2O (4/1), reflux, 4 h; (c) 3,6-diiodophenanthrene, Pd(PPh3)4, Cs2CO3, dioxane/H2O (20/3), 80 °C, 1 h.
Shinokubo, Hiroto, and Kim et al. reported the synthesis and the host–guest chemistry of molecular tweezers bearing two azabuckybowl units connected by a carbazole or phenanthrene linker.433 The azabuckybowl derivative 234.3 bearing one boronate ester group was cross-coupled, in 1:2 ratio, with 3,6-dibromo-9H-carbazole or 3,6-diiodophenanthrene to furnish the tweezers 234.4 and 234.5, respectively. Titration experiments with C60 and C70 as the guests were performed using fluorescence spectroscopy. For the carbazole-based tweezer 234.4 in toluene, the 1:1 association constant with C60 and C70 was 4.4 × 107 M–1 and 7.0 × 108 M–1, respectively. For the phenanthrene-based tweezer 234.5, the corresponding values were 3.0 × 108 (with C60) and 6.3 × 107 M–1 (with C70), indicating an opposite binding preference. The cocrystal structure of 234.5 with C60 or with C70 indicated that, in each case, the fullerene molecule interacted with both azabuckybowl units of the same host molecule in a concave–convex manner.
In 2018, Yokoi, Hiroto, and Shinokubo described the reversible π- and σ-dimerization of azabuckybowl radical cations 235.1•+ and 235.2•+ (Scheme 235).434 Their synthesis involved subjecting the polyaromatic precursor 234.1 to the coupling conditions outlined in Scheme 234, but with excess amounts of the catalyst Pd(OAc)2 (2 equiv) and ligand source PCy3·HBF4 (4 equiv). In this way, the singly cyclized product 235.1 with two reductively cleaved C–Br bonds was obtained in 5% yield, in addition to the known azabuckybowl 235.2 (37%). Compound 235.1 was revealed by X-ray crystallography to adopt an essentially planar conformation. Both the planar and bowl-shaped compounds were subjected to one-electron oxidation by BAHA, generating the corresponding radical cations 235.1•+ and 235.2•+, which were characterized by X-ray crystallography as their SbCl6– salts. In the former salt, a dimeric structure [235.1]22+ was observed, in which two planar motifs were stacked face-to-face. The closest interplanar distance found between two α-carbon atoms of the individual pyrrole units was 3.14 Å. In the other salt, a dimeric structure [235.2]22+ was observed, in which two bowl-shaped motifs were linked in a convex–convex fashion. A uniquely long C–C bond (1.64 Å) connected the two subunits via α-positions of their respective pyrrole rings. The reversible dimerization behavior of 235.1•+ and 235.2•+ was also probed by variable-temperature NMR, ESR, and UV–vis–NIR absorption experiments. The bowl-shaped radical cation 235.2•+ could react with excess methanol to yield the methoxylated cation 235.3+. However, no such conversion was observable with the planar radical cation 235.1•+.
Scheme 235. Synthesis and Reversible Dimerization of Azabuckybowl Derivatives.
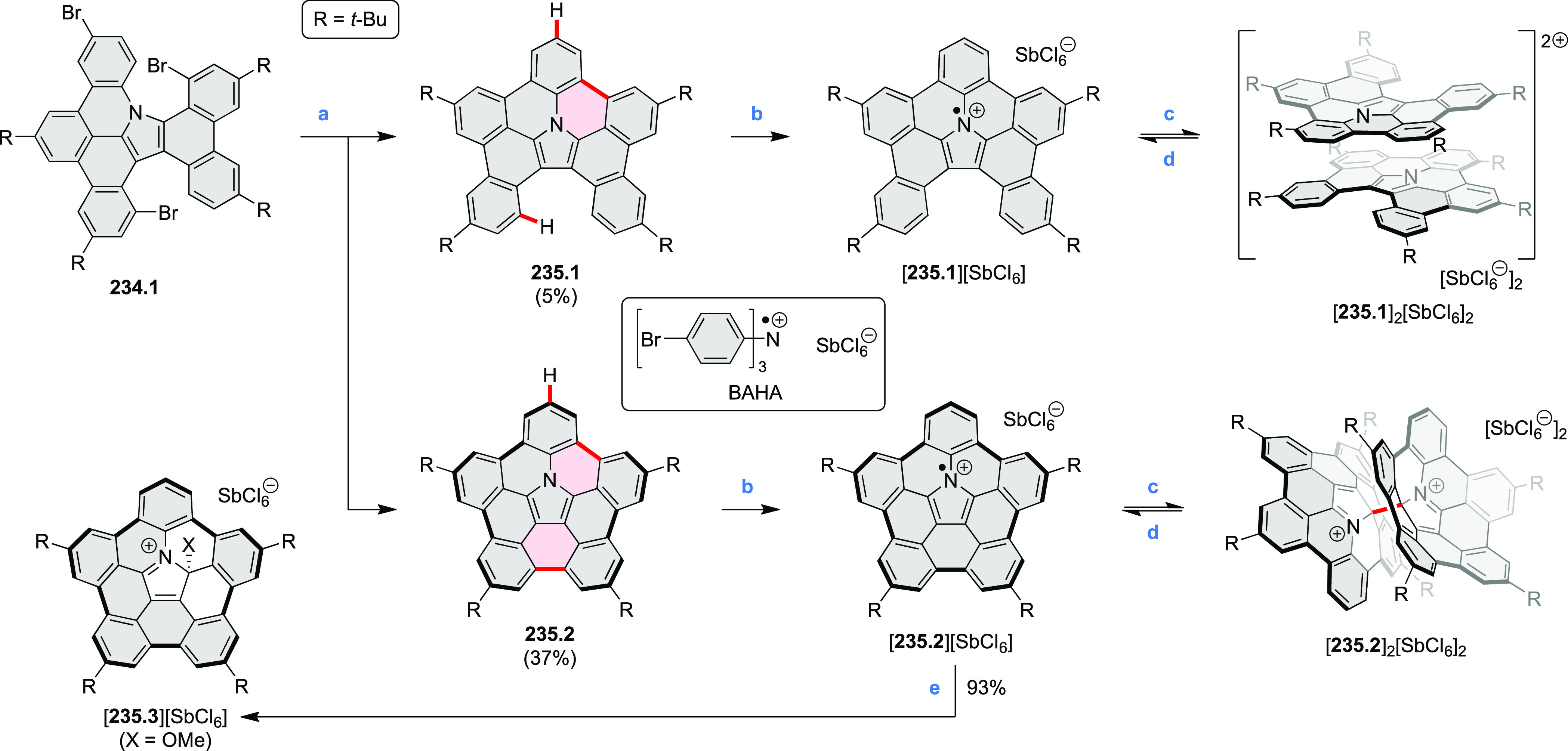
Reagents and conditions: (a)434 Pd(OAc)2 (2 equiv), PCy3·HBF4 (4 equiv), K2CO3 (8 equiv), DMA, 130 °C, 32 h; (b) BAHA (1 equiv); (c) crystallization or cooling in solution; (d) heating in solution; (e) MeOH, DCM/toluene/Et2O (3:2:1) (for crystallization) or CDCl3 (for NMR).
6.1.3. [6]Heteracirculenoids
In two papers, Xu and Tan et al. reported the synthesis of unsubstituted trichalcogenasumanenes (236.1a–c) from 1,5,9-trisubstituted triphenylene precursors. Their approach used the triiodonium cation [236.3]3+ as the common precursor. Oxidation of 1,5,9-triiodotriphenylene (236.2) by m-CPBA in the presence of triflic acid generated the triiodonium salt [236.3][OTf]3 in 68% yield. The identity of the salt was substantiated by 1H, 13C, and 19F NMR spectroscopy and by MALDI–TOF mass spectrometry.435 Using potassium thioacetate or sulfur powder as the sulfur source and appropriate conditions, [236.3][OTf]3 could afford the sulfur-containing derivatives 236.4 or 236.5 in fair yields. Both these compounds could then be desulfurized by heating with copper powder in tetralin at 200 °C to furnish trithiasumanene (236.1a). Likewise, [236.3][OTf]3 was subjected to selenation with selenium powder followed by copper-mediated deselenation to give triselenasumanene (236.1b) in 22% overall yield. In subsequent work, tritellurasumanene (236.1c) was directly formed from [236.3][OTf]3, by heating with tellurium powder in the presence of 2-picoline as the base.436 Additionally, the synthetic versatility of the triiodonium salt was demonstrated by Xu and Tan et al. in the syntheses of the trisilasumanenes 236.6a–c and the trigermasumanene 236.7 involving rhodium-catalyzed cyclodehydrogenation of Si/Ge–H and C–H bonds.437
In 2018, Xu and Tan et al. described the oxidative cyclization of 2-biphenylthiols into the corresponding dibenzothiophenes, which was achieved with PdCl2 as the catalyst and DMSO as both the solvent and oxidant.438 This set of conditions was also applicable to the 3-fold cyclization of triphenylene-1,5,9-trithiol (236.9) to yield trithiasumanene (236.1a, 2%) alongside the incompletely fused 236.10 (5%, Scheme 236). The authors ascribed the low yield of 236.1a to the instability of the molecule under the reaction conditions. Despite the low yield, this preparation represented the first trithiasumanene synthesis using a direct “stitching” method, as opposed to desulfurative ring contractions (Schemes 236 and 240).
Scheme 240. Synthesis and Reactivity of Trichalcogenasumanene Derivatives Bearing Two Different Chalcogens.
Reagents and conditions: (a)447n-BuLi (4, 5, or 10 equiv), TMEDA, hexane, 60 °C, 3 h; (b) Se powder (10 equiv), THF, −78 °C to rt, 8 h; (c) Cu nanopowder, 215 °C, 160 min; (d) Te powder (10 equiv), ultrasound, rt, 12 h; (e) oxone, THF/H2O (4/1), rt, 12 h; (f) 30% aq. H2O2, DCM/AcOH (5/1), 24 h; (g) Br2, DCM, rt, 15 min.
Hexabromination of trithiasumanene with elemental bromine in the presence of iron powder was shown by Xie and Tan et al. to give 237.1 in high yield (Scheme 237).439 Subsequent 6-fold nucleophilic aromatic substitution of this compound with aryl thiolates furnished the sulfur-rich products 237.2a–c in 35–40% yield. The hexakis(phenylthiol)-substituted derivative 237.2a formed cocrystals with C60 and with C70, which were analyzed by X-ray crystallography. In the solid state, both fullerenes coordinated to the concave face of the buckybowl 237.2a in a 1:1 stoichiometry. The six phenyl groups in 237.2a were found to direct away from C60 and C70, lacking any interaction with the fullerene molecule in both crystal structures. The bowl depths observed in both cocrystals were 0.76 Å, in contrast to the deeper bowl depth (0.83 Å) found in the crystal of pure 237.2a. The bowl shallowing upon complexation with fullerenes was believed to strengthen the intermolecular interaction.
Scheme 237. Six-Fold Functionalized Trithiasumanenes.

Reagents and conditions: (a)439 Br2, Fe, nitrobenzene, 80 °C, 2 days; (b) RSNa, dry 1,3-dimethyl-2-imidazolidinone, rt, 8 h; (c)440 BBr3, DCM, 0 °C to rt, then H2O; (d) R′Br, K2CO3, DMF, 75 °C.
In a recent report, Furukawa, Akutagawa, and Saito et al. demonstrated a new method for generation of ferroelectricity based on bowl-to-bowl inversion of trithiasumanene.440 The attendant dipole inversion was envisioned to induce ferroelectric response in the solid state because of the relatively low inversion barrier compared to corannulene and sumanene. Hexaalkoxytrithiasumanenes 237.4a–d were prepared by applying a dealkylation–realkylation procedure to the ethoxy-substituted precursor 237.3. The alkoxy chains were installed to enhance the flexibility and internal thermal energy of the columnar assemblies upon heating. The temperature and frequency dependence of the dielectric constants of 237.4a–d was examined, alongside their ferroelectric polarization–electric field (P–E) hysteresis curves. The π-stacked assemblies of 237.4a–d underwent polarization upon application of a pulse voltage, followed by a dipole relaxation pathway through bowl-to-bowl inversion in bulk.
In 2017, Saito and Fuji et al. reported the formation of the phosphole sulfides 238.5 from the triphenylene precursor 238.4b.441 Based on this method, Shao et al. proposed a general route to replace one chalcogen atom in the hexabutoxy-substituted heterasumanenes 238.1a–c with a phosphorus(V) group (Scheme 238).442 For instance, the opening of one thiophene ring in the trithiasumanene (238.1a) with 2.2 equiv of n-BuLi generated the dilithiated intermediate 238.2a. Sequential treatment of 238.2a with excess PhPCl2 and excess sulfur powder gave rise to compound 238.3a bearing one PhP=S group in 25% yield. This one-pot protocol was also feasible for the syntheses of the selenium-based and tellurium-based congeners, i.e., 238.3b and 238.3c, in 41% and 90% yield, respectively. For all three phosphorus(V)-containing sumanenes 238.3a–c, the cocrystal structures with AgNO3 indicated coordination between Ag+ and the sulfur atom on the P=S functionality. The thiophene-containing derivative 238.3a was shown to be a potential sensitive fluorescence sensor for Ag+, with a limit of detection as low as 0.21 μM, which is superior to the World Health Organization standard for drinking water (0.5 μM).
Scheme 238. Ring-Modified Trichalcogenasumanenes.
Reagents and conditions: (a)441,442n-BuLi (2.2 equiv), TMEDA, hexane, −30, 60 °C or reflux, 30 min–3 h; (b) (1) PhPCl2, THF, −78 °C to rt, 3–4 h, (2) S powder, rt, 12.5–14 h; (c)443 THF, −78 °C to rt, then reflux, 12 h; (d)444 30% aq. H2O2, DCM/AcOH (1/2), 60 °C, 24 h.
In 2018, Furukawa and Saito et al. reported the incorporation of group 14 elements (silicon, germanium, and tin) into the dithiasumanene structure.443 Compounds 238.6a,b were initially treated with n-BuLi, and the resultant dilithiated species 238.2a,d were reacted with SiHCl3, SiCl4, GeCl4, or SnCl4 to furnish the corresponding spirocyclic structures 238.7aa–bc with Si, Ge, or Sn located at the spiro junction. Alternatively, the reaction between 238.2a,d and Ph2ZCl2 (Z = Si, Ge, or Sn) afforded the corresponding heterasumanenes 238.8aa–bc. In the solid state, the dihedral angle between sumanene subunits in 238.7ba–bc ranged from 84.7° to 88.9°. As shown by CV measurements, the monomeric heterasumanenes 238.8ba–bc displayed only one quasi-reversible anodic wave at E1/2 = 0.40–0.41 V, while the spirocyclic analogues 238.7ba–bc showed two reversible anodic waves at E1/2 = 0.40–0.41 and 0.54–0.55 V. The inferred stability of the radical cations of 238.7ba–bc was attributed to the stabilizing spiroconjugation between the two subunits. Such spiroconjugative effects were corroborated by the orbital shapes of HOMO and HOMO–1 obtained from DFT calculations.
In 2017, Shao et al. reported the oxidation of hexabutoxytrithiasumanene (238.1a) to the corresponding tris(S,S-dioxide) 238.9 (Scheme 238).444 The reaction worked when excess hydrogen peroxide was used as the oxidant, whereas the use of m-CPBA for the same conversion proved ineffective. Compound 238.9 displayed strong indigo fluorescence in DCM solution (λem = 463 nm) and in the solid state (λem = 478 nm). The 1:1 cocrystal of 238.9 with 2,3,6,7,10,11-hexabutoxytriphenylene (238.4a) showed strong yellow fluorescence in the range of 500–700 nm.
The preparation of the hexabutoxy-substituted tritellurasumanene 238.1c mentioned above, as well as another analogue 239.1, was described by Shao et al. earlier in 2016 (Scheme 239).445 Hexaalkoxytriphenylenes 238.4a,b were separately hexalithiated using n-BuLi and subjected to ultrasound-assisted reaction with tellurium powder to afford the triply tellurated products 238.1c and 239.1, and the doubly tellurated products 239.2a,b. These successful conversions confirmed that ultrasound could promote the surface contact between tellurium powder, which dissolves poorly in hexane, with the solvated hexalithiated intermediate. Moreover, both tritellurasumanenes could react with elemental bromine to yield the tris(Te,Te-dibromide)s 239.3a,b quantitatively. Likewise, the reaction with iodine generated the tris(Te,Te-diiodide)s 239.4a,b as supported by 1H and 13C NMR spectroscopy. In a later paper by the same group, the reaction of 238.1c with FeCl3 in DCM/MeNO2 produced the tris(Te,Te-dichloride) 239.5 in 84% yield.446 Similar halogenations were not feasible with the trithia- and triselenasumanenes 238.1a,b. Besides, the known oxone-mediated oxidative ring opening of the benzene unit in 238.1a,b did not take place for the tritellurasumanene 238.1c as inferred from the absence of any carbonyl stretch in the IR spectrum of the product mixture.
Scheme 239. Gram-Scale, One-Pot Synthesis of a Tritellurasumanene Derivative and Its Subsequent Halogenation.

Reagents and conditions: (a)445 (1) n-BuLi, TMEDA, hexane, 60 °C, 3 h, (2) Te powder, ultrasound, rt, 12 h; (b) Br2, DCM, rt, 15 min; (c) I2 (yield not given); (d)446 FeCl3, DCM, MeNO2, rt, 2 h.
Shao et al. reported the systematic preparation of a series of six trichalcogenasumanenes 223.1a–3b, each bearing two different kinds of chalcogen atoms (Scheme 240).447 For the preparation of the two monothia analogues, compound 240.4a was deprotonated by excess n-BuLi to generate the tetralithiated species 240.5a. A subsequent selenation–deselenation sequence successfully produced the S1Se2-sumanene derivative 240.1a. Similarly, the ultrasound-assisted reaction between 240.5a and tellurium powder directly afforded the S1Te2-sumanene derivative 240.1b in 49% yield. The preparation of the monoselena (240.2a,b) and monotellura (240.3a,b) analogues employed 240.2b and 239.2a as the respective starting materials. According to X-ray diffraction data, the bowl depths of the S1Se2- (240.1a, 0.49 Å) and S2Se1-analogues (240.2a, 0.42 Å) were shallower than that of the S3-analogue (238.1a, 0.77 Å), whereas the Se1Te2-analogue (240.2b) assumed a planar geometry. Using either oxone or 30% H2O2 as the oxidant, the oxidative C–C bond cleavage of the S1Se2- and S2Se1-analogues occurred regioselectively at the benzene ring between thiophene and selenophene to yield the ring-opened diesters 240.8–9. Exposure of both the S2Te- and Se2Te-analogues to elemental bromine resulted in 2-fold bromination at the tellurium atom, furnishing compounds 240.10a,b (see Scheme 239).
In 2020, Wang and Shi et al. reported the copper-catalyzed dechalcogenization of aryl dichalcogenides to the corresponding aryl chalcogenides.448 This reaction required substoichiometric amounts of CuI as the catalyst and 1,10-phenanthroline as the ligand and involved heating in a solvent mixture of DMSO and water at 160 °C. The reaction had a broad substrate scope and was applied to the preparation of trithia- and triselenasumanene derivatives (Scheme 241). First, compound 241.1 containing two cyclic disulfide units was converted into the singly and doubly desulfurized products, i.e., 241.2 and 238.1a, in 20% and 40% yield, respectively. Similarly, the single cyclic diselenide unit in compound 241.3 was smoothly deselenated to give the 238.1b in 95% yield. These conditions were significantly milder than those used previously (see Schemes 236 and 240).
Scheme 241. Trithiasumanene and Triselenasumanene Derivatives via Copper-Catalyzed Dechalcogenization.

Reagents and conditions: (a)448 CuI (40 mol %), 1,10-phenanthroline (80 mol %), K2CO3 (6 equiv), DMSO/H2O (20:1), air, 160 °C; (b) CuI (20 mol %), 1,10-phenanthroline (40 mol %), K2CO3 (3 equiv), DMSO/H2O (20:1), air, 160 °C.
In 2018, Shao et al. described the [6]heteracirculenoids 242.2aa–bb possessing two chalcogenole rings and one pyridine ring that are fused with the central benzene ring (Scheme 242).449 These compounds represent hybrids of trichalcogenasumanene and triazacoronene. The triphenylenodithiophene 238.6a and triphenylenodiselenophene 240.4b were separately subjected to a nitration–reduction sequence to give the amines 242.1a,b in 73–80% yield. Subsequent Pictet–Spengler reaction with benzaldehyde or picolinaldehyde afforded the four target molecules 242.2aa–bb in 70–80% yield. The protonation-dependent optical properties of the two sulfur-containing compounds 242.2aa–ab could be modulated by addition of trifluoroacetic acid and triethylamine. The molecular frameworks of the two pyridyl-substituted analogues 242.2ab,bb were planar in the solid state. However, the structures became bowl-shaped in the complexes 242.3a,b, in which the 2,2′-bipyridine moiety was bound to a Zn2+ ion.
In 2017, Shao et al. obtained a series of trichalcogenasumanene-derived amidine-containing polycycles 243.1–3 (Scheme 243).450 In the synthetic route, the trithia- and triselenasumanenes 238.1a,b first underwent oxone-induced benzene ring opening to give the corresponding diesters, followed by base-promoted hydrolysis and dehydration reactions, yielding the seven-membered cyclic anhydrides 243.4a,b. These intermediates were then condensed with various aromatic diamines in the presence of DCC to give the target polycycles. For instance, the double condensation with o-phenylenediamine led to the [7–5–6]-fused polycycles 243.1a,b. Analogously, the [7–5–6]-fused (243.2a,b) and [7–7–6]-fused polycycles (243.3a,b) could be obtained using the suitable diamines. Among these three fusion types, only the last two displayed twisted molecular geometries in their X-ray crystal structures. The enantiomers of the highly twisted compound 243.3b were separated by chiral HPLC and were found to be configurationally stable. Thin films of 243.2a,b exhibited hole mobility (μh) of 6 × 10–4 cm2 V–1 s–1, while those of 243.1a,b and 243.3a,b were poor semiconductors. Single crystals of 243.2a could be fabricated into an OFET device with a p-type semiconducting behavior. In 2018, Shao and Sun et al. reported the use of tert-butyl hydroperoxide for the oxidative ring opening of two benzene rings in trithia- and triselenasumanenes (238.1a,b) to give the tetraesters 243.6a,b, which could be converted to the cyclic diimides 243.7a,b.451 Based on open-aperture Z-scan measurements, the authors concluded that 243.7a,b were potentially superior to C60 as optical-limiting materials.
Scheme 243. Oxidative Ring Opening of Trichalcogenasumanenes.
Reagents and conditions: (a)450 oxone, THF/H2O (4:1), rt, 2 h; (b) (1) NaOH, EtOH/H2O (10:1), reflux, 2 h, (2) aq. HCl (3 N); (c) Ac2O, reflux; (d) o-phenylenediamine, DCC, THF, reflux, 8 h; (e)451t-BuOOH (6 equiv), DCM, 40 °C, 5 h; (f) (1) p-toluidine, DCC, THF, reflux, 12 h.
The same group reported the synthesis and reactivity of a trichalcogenasumanene-derived ortho-quinone 244.1a,b (Scheme 244).446 On treatment of the trithia- and triselenasumanenes 238.1a,b with FeCl3 in a mixed solvent of DCM/MeNO2, one of the dibutoxybenzene moieties was converted to an ortho-quinone unit. According to the authors, this was the first reported case of such a direct dealkylative oxidation. (The different reactivity of the tritellurasumanene 238.1c toward FeCl3 has been addressed in Scheme 239.) The ortho-quinones 244.1a,b were then condensed with 1,2-diphenylethane-1,2-diamine to afford the pyrazine-fused trichalcogenasumanene derivatives 244.2a,b in 30–33% yield. Similarly, the use of aromatic ortho-diamines in the same reaction gave rise to a series of π-extended derivatives 244.3aa–bc. Thin films of 244.3aa and 244.3ba behaved as a p-type semiconductor, with respective hole mobilities of 1.7 × 10–3 and 2.6 × 10–3 cm2 V– s–1. Surprisingly, treatment of the ortho-quinone with 1,8-naphthalenediamine produced the aforementioned [7–6–6]-fused polycycles 243.2a,b in 70–76% yield. The mechanism of this transformation was postulated to involve a ring rearrangement of the strained aziridine intermediate 244.5a,b. The properties of 244.2a,b452 and 244.3ba(453) as materials were explored in subsequent work.
Scheme 244. Synthesis and Reactivity of Trichalcogenasumanene-Derived ortho-Quinone.
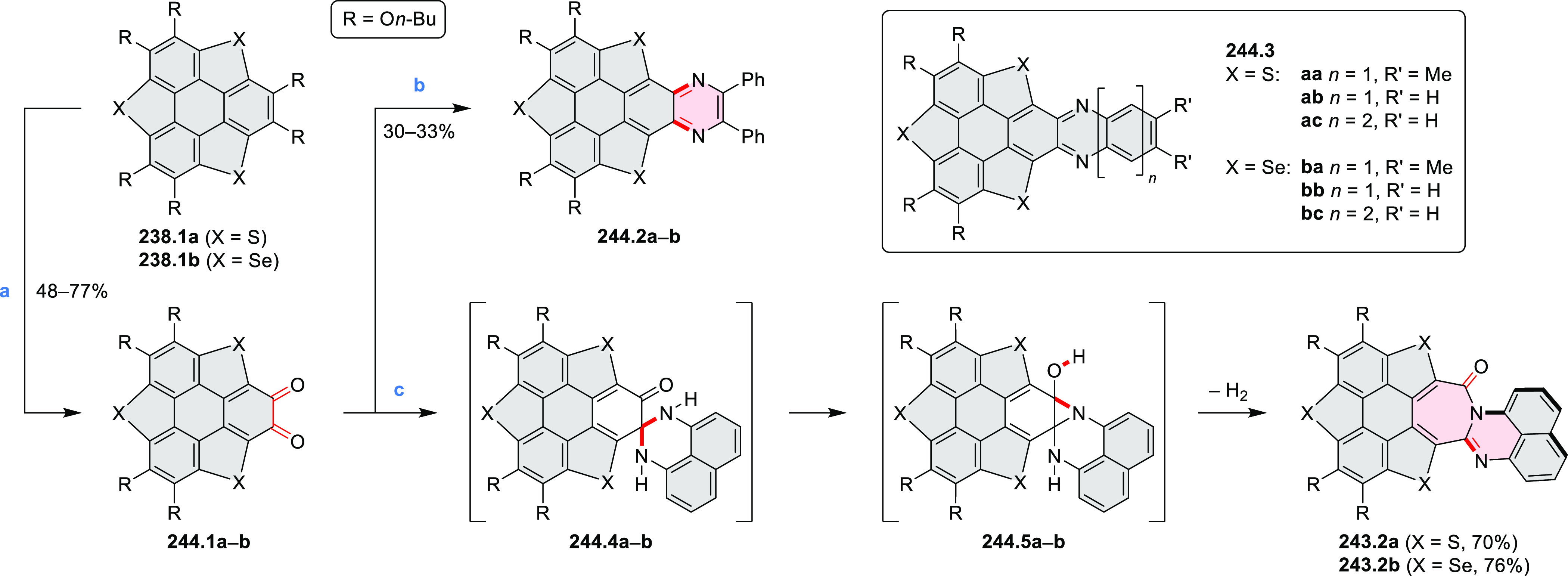
Reagents and conditions: (a)446 FeCl3, DCM, MeNO2, rt, 2 h; (b) 1,2-diphenylethane-1,2-diamine, AcOH, reflux, 12 h; (c) 1,8-naphthalenediamine, AcOH, reflux, 4 h.
Applying their synthetic strategy toward the pyrazine-fused trichalcogenasumanenes 244.2, Shao et al. prepared a series of new D1–A–D2 π-systems C22.1aa–bb consisting of the electron-donating tetrathiafulvalene (D1) and trichalcogenasumanene (D2) units connected through an electron-accepting pyrazine unit (A) (Chart 22).454 These compounds displayed a broad absorption band (λ = 450–720 nm) as a consequence of intramolecular charge transfer (ICT) transition contributed by both D1 and D2. Femtosecond transient absorption (fs-TA) measurements were carried out under photoexcitation at λ = 385 nm. Two transient states with charge separation, CS1 followed by CS2, emerged with absorption maxima at 889 and 1105 nm, respectively. In the first state (CS1), the tetrathiafulvalene unit (D1) was in the radical cation state. Afterward, a transition from CS1 to the second state (CS2) took place via the ICT from D2 to D1•+. Contrary to these D1–A–D2 systems, compounds C22.2 and 244.3ab, possessing D–A scaffolds, exhibited only one transient charge-separated state.
Chart 22. Tetrathiafulvalene-Fused Compounds with a D1–A–D2 or D–A Structure.

In 2017, Sakurai et al. reported a series of C3-symmetrical triaryl-substituted triazasumanenes (C)-245.2a–f. These enantiopure systems were obtained by Liebeskind–Srogl arylation of the corresponding triazasumanene (C)-245.1 (Scheme 245).455 The 3-fold reaction occurred in good yields with various 2- and 4-substituted arylboronic acids but failed with 2,6-dimethylphenylboronic acid. Based on the CD spectra of both enantiomers of 245.2f, as well as the X-ray crystal structure of (C)-245.2f, the authors concluded that the coupling reaction was not accompanied by any racemization. Later, the same group reported the enantiopure tris(2-hydroxyphenyl)-substituted analogue of (A)-245.2, which showed dual emission in the crystalline state, caused by excited-state intramolecular proton transfer (ESIPT).456 The emission at 631 nm was not observable in solution. In 2018, Sakurai and Higashibayashi et al. reported the use of the same catalyst system, but with organosilanes instead of boronic acids, to induce the desulfurization of (C)-245.1.457 Thus, the triply desulfurized product (C)-245.4 was obtained in 50% yield, accompanied by traces of the doubly desulfurized (C)-245.3. The authors found that the triazasumanene (C)-245.4 was acid labile. Based on 1H NMR and mass spectral data, the imine cleavage was proposed to take place at a flank bond rather than a rim bond to give the hydrolysis product 245.5.
Scheme 245. Dativizations of Enantiopure Tris(methylthio)-Substituted Triazasumanene.

Reagents and conditions: (a)455 RB(OH)2, Pd2(dba)3, tri(2-furyl)phosphine, copper(I) thiophene-2-carboxylate (CuTC), THF, 50 °C, 12 h; (b)457 Et2MeSiH or poly(methylhydrosiloxane) (PMHS), Pd2(dba)3, tri(2-furyl)phosphine, CuTC, THF, 50 °C, 30 min; (c) MeOH/H2O, pH = 2.01; (d) DCl, CD3OD/D2O.
In contrast to all the trichalcogenasumanenes and the C=N-containing triazasumanenes presented above, the pyrrole-containing triazasumanene skeleton is known to be highly strained. In 2017, Chen, Tanaka, and Osuka explored the construction of tribenzo-fused triazasumanenes 246.1a,b via the fold-in synthesis (Scheme 246).458 They proposed subjecting the ortho-phenylene-bridged cyclic tripyrroles 246.2a,b to 3-fold C–C bond coupling at the pyrrolic β-carbons to create the central six-membered ring. It was found that the combination of FeCl3 and AgOTf in DCM/MeNO2 transformed 246.2a into the unexpected product 246.3 in 7% yield, instead of the desired 246.1a. Under the same conditions, the N,N′,N″-trimethylated macrocycle 246.2b produced only a complex mixture of products. The structure of 246.3 was established by X-ray crystallography to result from (i) β–β coupling of rings A and B, (ii) α–β′ addition between ring C and each of the rings A and B, and (iii) 1,2-hydrogen shift of the NH group in rings A and B. The alternative reductive approach using the hexabrominated cyclic tripyrrole 246.4 produced only debromination under standard Yamamoto conditions, whereas the use of Pd[P(t-Bu)3]2 and K3PO4 in refluxing toluene yielded the singly cyclized product 246.5 (36% yield) and the fully debrominated product 246.2b (10% yield). The authors speculated that, after the first cyclization of 246.4, subsequent transmetalation of the C–PdII–Br to generate a further cyclized C–PdII–C species was energetically disfavored due to the narrower interior space and higher rigidity of the molecule.
Scheme 246. Attempted Oxidative and Reductive Fold-In Syntheses of Strained Triazasumanene Derivatives.

Reagents and conditions: (a)458 FeCl3, AgOTf, DCM/MeNO2 (10:1), rt, 24 h; (b) NBS, DMF, 0 °C, 6 h; (c) MeI, NaH, DMF, rt, 3 h; (d) Pd[P(t-Bu)3]2, K3PO4, toluene, 110 °C, 24 h.
6.1.4. [7]Heteracirculenes
The stilbenoid substrate C23.1 bearing two 2-naphthyl groups connected to the carbazole core via olefin linkages was explored as a precursor to aza[9]helicenes by Bedekar et al. (Chart 23).459 Mallory photocyclization of C23.1, performed using a high-pressure mercury vapor lamp in the presence of I2 and THF, yielded a mixture of mainly ortho-fused products C23.2–4 containing linear (L) or angular (A) fused subsections. They were accompanied by the quasi-circulene system C23.5, containing a seven-membered ring fused with one pyrrole and five benzene rings. This ALF compound was proposed to originate from C23.4 via an additional ring closure.
Chart 23. Aza[n]helicenes via Photochemical Cyclization.

6.1.5. [8]Heteracirculenes
In 2017, Miyake et al. reported a two-step synthesis of tetraaza[8]circulene derivatives from the corresponding tetrathia[8]circulene-based precursors (Scheme 247).460 The first step involved the formation of tetrathia[8]circulene octoxide, such as 247.1, through the oxidation of sulfur atoms by m-CPBA. Such octoxides are negatively curved and were recently shown to exhibit AIE behavior in the solid state.461 In the second step, 247.1 was heated with p-toluidine in the presence of excess KHMDS to generate a separable mixture of the partially substituted products, 247.2 (27% yield) and 247.3 (20% yield), accompanied by the desired tetraaza[8]circulene 247.4 (5% yield). Another analogue 247.5 bearing a different substitution pattern could be formed in 25% yield without any partially substituted products.
In 2020, Tanaka et al. reported the synthesis and single-electron oxidation of the tetraazatetrabenzo[8]circulenes 248.4a,b bearing two aryl groups on opposite pyrrole units (Scheme 248).462 The synthesis started with oxidative cyclization of the macrocyclic precursor 248.1, followed by N,N′-dialkylation to introduce solubilizing groups. The resulting diazadithia[8]circulene 248.2 was subjected to m-CPBA oxidation to furnish the bis(S,S-dioxide) 248.3 in 90% yield. Subsequent SNAr reaction with aromatic amines in the presence of KHMDS (see Scheme 247) provided the target diarylated tetraazatetrabenzo[8]circulenes 248.4a,b in 74–84% yield. Chemical oxidation of these compounds with BAHA gave the corresponding radical cation salts [248.4a,b]•+[SbCl6–], both structurally confirmed by X-ray crystallography. The ESR spectra of both radical cations measured in DCM/toluene at 20 K show sharp signals with g-factors of 2.0155 and 2.0154, respectively. Unrestricted DFT calculation on 248.4a•+ predicted spin delocalization over the entire [8]circulene skeleton, but not on the N-aryl substituents. Notably, the NIR absorptions of 248.4a•+ extended to 2000 nm. An SNAr reaction of 248.3 was also possible with the diphenylmethyl anion, leading ultimately to the diazatetrabenzo[8]circulene analogue 248.5 bearing two sp3-hybridized carbon atoms in the polycyclic framework.463
In 2021, the same group presented an improved synthesis of the o-phenylene-spaced cyclic tetrapyrroles 248.7a–d.464 Hence, the boronate-containing dipyrrolylbenzene 248.6 underwent Suzuki–Miyaura cross-coupling with the appropriate ortho-dibromobenzenes to give the desired macrocycles 248.7a–d in 7–11% yield. In comparison, the previous “reverse” coupling strategy suffered from lower yields attributed to the low stability of the corresponding dibromo-substituted dipyrrolylbenzene. Subsequently, two different sets of oxidative aromatic coupling conditions allowed eight-membered ring closures to afford the tetraazatetrabenzo[8]circulenes 248.8a–d.
Controlled N-alkylation of the known tetrabenzotetraaza[8]circulene C24.1 (Chart 24), performed by reacting C24.1 with 2 or 3 equiv of 1-iodobutane and excess NaH, produced the corresponding singly, doubly, and triply N-butylated products C24.2–4.465 It was noted that double butylation occurred regioselectively on opposite pyrrole rings to yield C24.3. Attempts to oxidize C24.3 using MnO2, NiO2, and FeCl3 to give compound C24.5 were unsuccessful.
Chart 24. Structures of Tetraaza[8]circulene Derivatives.

In a series of papers, Miyake et al. demonstrated the synthetic versatility of the tetraiodo-substituted β,β-cyclooctatetrathiophene (β,β-COTh) 249.2 (Scheme 249). This compound was derived from the known tetraborate 249.1 by treatment with NIS and CuI.466 The 4-fold Sonogashira coupling of the tetraiodide 249.2 with aliphatic terminal alkynes gave the tetrasubstituted 249.3a–c in quantitative yields.467 Subsequent 4-fold cycloisomerization mediated by DBU gave rise to the desired tetrathia[8]circulene derivatives 249.4a–c in 41–54% yield. Alternatively, the tetraiodide was coupled with primary amides to give the tetraamides 249.5a–e, which were then subjected to the Vilsmeier–Haack conditions to give the tetraazatetrathia[8]circulenes 249.6a–e.468 Besides, the silicon- (249.8a)466 and germanium-containing (249.8b)469 hetera[8]circulenes could be obtained in two steps from the tetraiodide 249.2 by employing palladium and rhodium catalysis. The X-ray structures of 249.4a,6a,8a,b showed that these polycyclic frameworks were essentially planar. Based on an isodesmic model, the association constants for the heptyl-substituted compounds 249.4b and 249.6c in CDCl3 were experimentally determined to be 58.9 and 115 M–1, respectively. For the tetragermatetrathia[8]circulene 249.8b, phosphorescence at 524 nm was observable upon irradiation (λex = 310 nm) of a frozen toluene sample at 77 K.
Synthetic routes toward hetera[8]circulenes 250.1–2 bearing two pyrrole and two chalcogenole units were explored by Wong et al. (Scheme 250).470ortho-Directed lithiation of the cyclic carbazole dimer 250.3 with n-BuLi, followed by addition of either water or TMSCl, furnished 250.4 and 250.5, respectively, in fair yields. The former tetrabromide 250.4 was subjected to the known reaction sequence consisting of (i) lithiation, (ii) chalcogenization with sulfur or selenium powder, and (iii) dechalcogenization using copper powder (cf. Scheme 240). However, the desired products 250.1a,b were not obtained. For the TMS-substituted tetrabromide 250.5, however, a simple lithiation–chalcogenization sequence performed at low temperatures smoothly produced the target diazadithia- and diazadiselena[8]circulenes 250.2a,b in 75–78% yield. The structures of both 250.2a,b were elucidated by X-ray crystallography. In particular, the mean interior angle in the eight-membered ring was 135.0° for 250.2a and 134.8° for 250.2b, each almost identical to that of a regular octagon (135°).
Scheme 250. Synthesis of Diazadithia- and Diazadiselena[8]circulenes under Mild Conditions.

Reagents and conditions: (a)470 (1) n-BuLi, THF, −78 °C, (2) H2O, −78 °C, 1 h, then rt, 2 h; (b) (1) n-BuLi, THF, −78 °C, (2) TMSCl, −78 °C, 1 h, then rt, 2 h; (c) n-BuLi, THF, −78 °C, 3 h, (2) S powder or Se powder, −78 °C, 2 h, then rt, overnight; (d) Cu powder, 250 °C.
In 2020, Pittelkow et al. reported the preparation of 251.8–9, the first hetero[8]circulenes containing three different heterocycles (Scheme 251).471 The first key step in their synthesis was the thermal Newman–Kwart rearrangement of the 4,4′-bicarbazole derivative 251.2 bearing two O-thiocarbamate functionalities at C-3 and C-3′. The resulting diazathia[7]helicene 251.4 was debenzylated to give 251.5 with free pyrrolic NH. Intramolecular oxidative coupling of 251.5 using chloranil and BF3·OEt2 produced the eight-membered ring closure product 251.6 in 93% yield. The same cyclization could also take place after sulfone formation to give 251.7 in 94% over two steps. More remarkably, a demethylation–oxidative coupling sequence on 251.5 directly generated diazaoxathia[8]circulene 251.8 in 77% yield. The NICS(0) values calculated for the central eight-membered ring in the planar [8]circulenes 251.8 and 251.9 were respectively +7.73 and +8.50 ppm, i.e., higher than those determined for the nonplanar counterparts 251.6 and 251.7 (+7.63 and +7.41 ppm, respectively).
Scheme 251. Synthesis of a Planar Diazaoxathia[8]circulene Skeleton by Compression of a Nonplanar [7]Helicene Precursor.
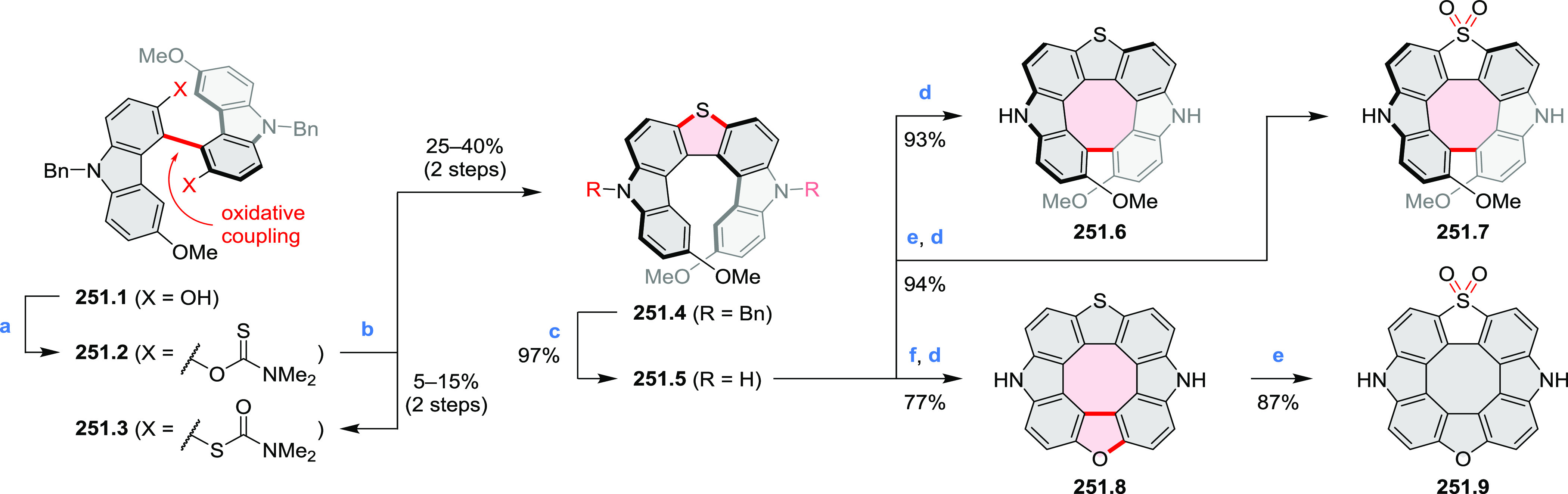
Reagents and conditions: (a)471 (1) NaH, DMF, 0–25 °C, 40–45 min, (2) dimethylthiocarbamoyl chloride, 85 °C, 2 h; (b) 300 °C, 25 min; (c) t-BuOK, O2, DMSO, 25 °C, 1 h; (d) chloranil, BF3·OEt2, MeCN/DCM, 25 °C, 40–45 min; (e) m-CPBA, THF/DCM, 25 °C, 30–45 min; (f) n-Bu4NI, BCl3, MeCN/DCM, 25 °C, 1 h.
In 2018, Tanaka and Osuka et al. showed that compounds 252.1a–c underwent intramolecular oxidative coupling to different extents under two different sets of conditions (Scheme 252).472 Conducting the cyclization at low temperatures with PIFA as the oxidant, the three triaza[7]helicene derivatives 252.2a–c resulting from a double cyclization were isolated in 40–71% yield. In contrast, the Sc(OTf)3-mediated cyclization of 252.1a,b at a high temperature yielded the triply cyclized compounds 252.3a,b bearing an additional eight-membered ring in 82–90% yield. Subsequently, the same group examined the reactivity of two additional substrates, 252.1d–e, toward PIFA.473 The dichloro-substituted 252.1d underwent a similar double cyclization to give the corresponding triaza[7]helicene 252.2d in high yield. However, the dimethoxy derivative 252.1e furnished only the triply cyclized 252.3e (97% yield) without any sign of the double cyclization product 252.2e. Importantly, direct formation of the triazaoxa[8]circulene derivative 252.4 was achieved by simply treating 252.3e with BBr3. The diol intermediate 252.3f was not detected even at lower reaction temperatures. This observation was attributed to the rapid cyclodehydration of 252.3f driven by strain release.
Scheme 252. Oxidative and Dehydrative Cyclization Reactions between Pyrrole and Indole Units.
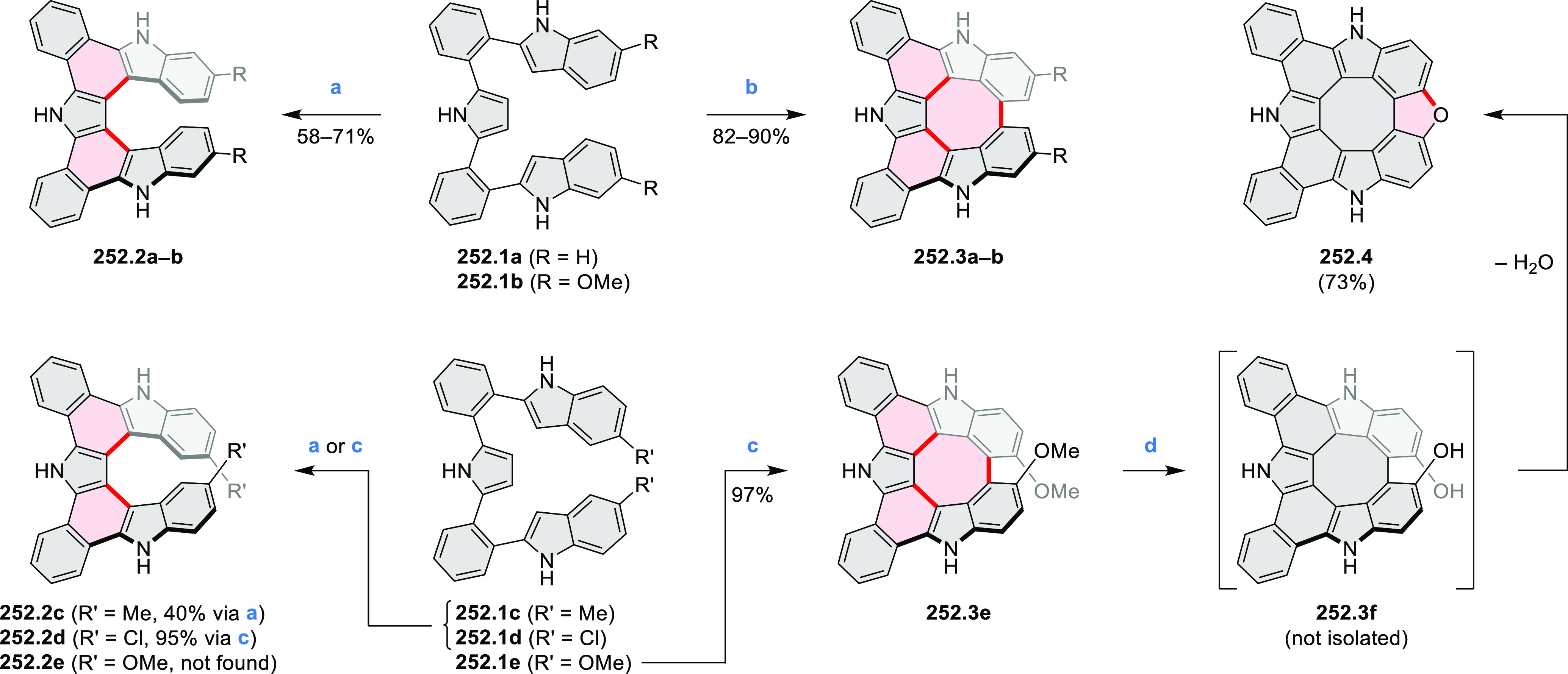
Reagents and conditions: (a)472 (1) PIFA (2.5 equiv), DCM, −78 °C, 30 min, (2) rt, 1 h; (b) DDQ (5 equiv), Sc(OTf)3 (5 equiv), toluene, reflux, 2–3 h; (c)473 PIFA (3.0–3.5 equiv), DCM, −78 °C, 3 h, (2) rt, 45 min; (d) BBr3, DCM, rt, 12 h.
In 2016, Wang and Rajca et al. described the synthesis and properties of a series of thiophene-based double helices rac-253.3–5 of different lengths (Scheme 253).474 Following their earlier results on the regioselective deprotonation of β,β-cyclooctatetrathiophene (β,β-COTh), the racemic tetrasubstituted β,β-COTh derivative rac-253.1 was exposed to n-BuLi (2 equiv) in THF at −78 °C for 2 h, and then heated at 60 °C for 2 h. As a result of the excellent ipsilateral selectivity, the generated dilithiated species rac-253.2 had both lithium centers occupying the same bay region. Then, two different transformations were performed on rac-253.2. First, the oxidative self-coupling promoted by CuBr2 afforded the double helix rac-253.5 in 42% yield. Second, the conversion of rac-253.2 to the corresponding organozinc chloride species followed by Negishi coupling with the dibromide 253.6 furnished the shorter double helix rac-253.3a in 33% yield. This racemate was thereby submitted to ipsilateral deprotonation on the β,β-COTh moiety followed by treatment with CuCl2, to generate, besides the dichlorination side product rac-253.3b, two intermolecularly coupled products. The first one was the target double helix rac-253.4 resulting from the formation of two C–C bonds between two reactant molecules of the same chirality. The second one, meso-253.7, originated from the C–C bond formation between a pair of enantiomers. In the latter case, chlorination occurred at the remaining lithiated sites because the second C–C bond formation was impossible. Since the yield of rac-253.4 (26%) was approximately twice that of meso-253.7 (11%), the first C–C bond formation was thought the be highly diastereoselective. A similar diastereoselectivity also accounted for the formation rac-253.5 in reasonable yields.
Scheme 253. Iterative Deprotonation–Coupling Approach toward Thiophene-Based Double Helices,
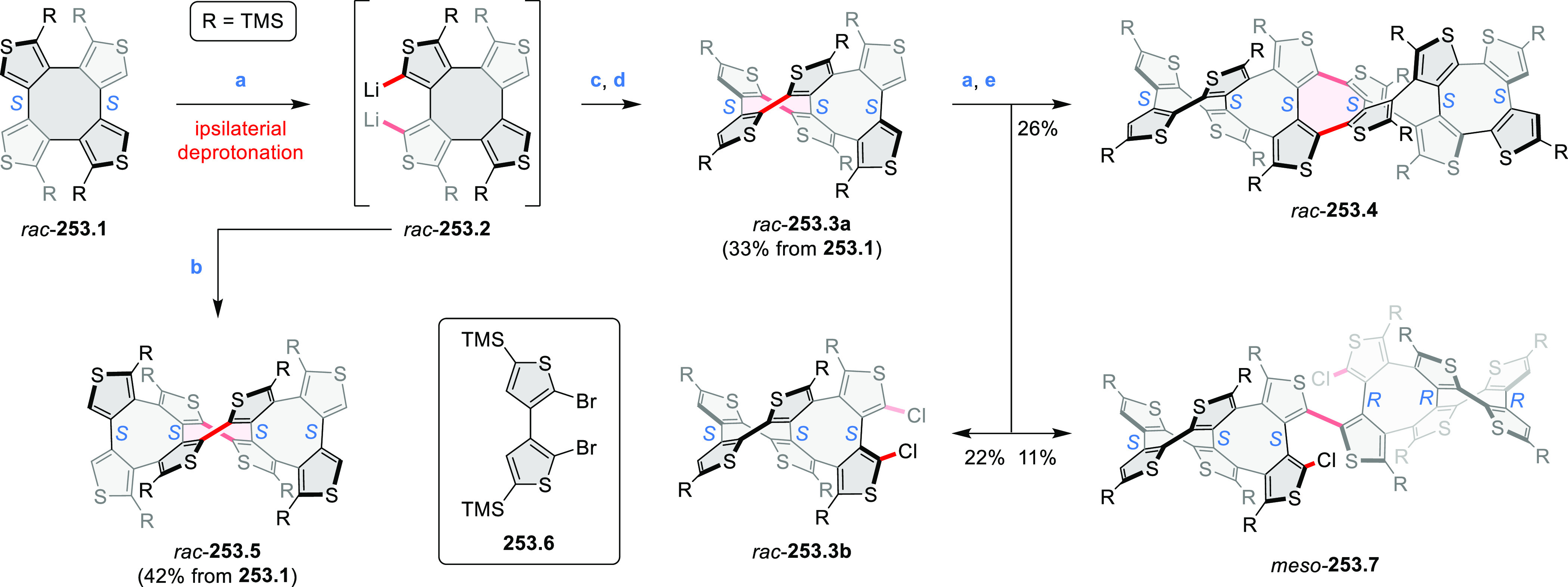
For all the racemates, the absolute configurations of the chiral axes of depicted enantiomers are shown to facilitate discussion.
Reagents and conditions: (a)474n-BuLi (2.1 equiv), THF, −78 °C, 2 h, then 60 °C, 2 h; (b) CuBr2, Et2O, −78 °C, 1 h, then rt, overnight; (c) ZnCl2, −78 °C, then rt; (d) 253.6, Pd(PPh3)4, Et2O, 120 °C, 48 h; (d) CuCl2, Et2O, −78 °C, 2 h, then rt, 24 h.
Subsequently, Wang and Li et al. reported an attempted synthesis of compound 254.6 featuring a cross-dimensional double-helical structure based on the β,β-COTh core (Scheme 254).475 The common starting material rac-254.1 was prepared via the Negishi coupling reaction between the 3,3′-bithiophene derivative 253.6 or 254.7 with the appropriate β,β-COTh precursor in 2:1 ratio in a way similar to that shown in Scheme 253. The treatment of rac-254.1 with n-BuLi generated the tetralithiated species rac-254.2, which could be transmetalated with ZnCl2 and coupled with 3-bromothiophene to yield the 4-fold arylated product rac-254.4a in 26% overall yield. The tetralithiated species could also be reacted with 1,2-dibromo-1,1,2,2-tetrachloroethane to afford, in 80% overall yield, the tetrabromide 254.3, which was then cross-coupled with phenylboronic acid to give rac-254.4b in 34% yield. However, when either rac-254.5 or rac-254.3 was reacted with the respective 3,3′-bithiophene 253.6 or 254.7, the expected Negishi coupling did not take place to yield 254.6. The authors reasoned that the formidable torsional strain in the double helical skeleton might account for this unsuccessful transformation.
Scheme 254. Arylation and Attempted Vertical Extension of a Thiophene-Based Double Helix.
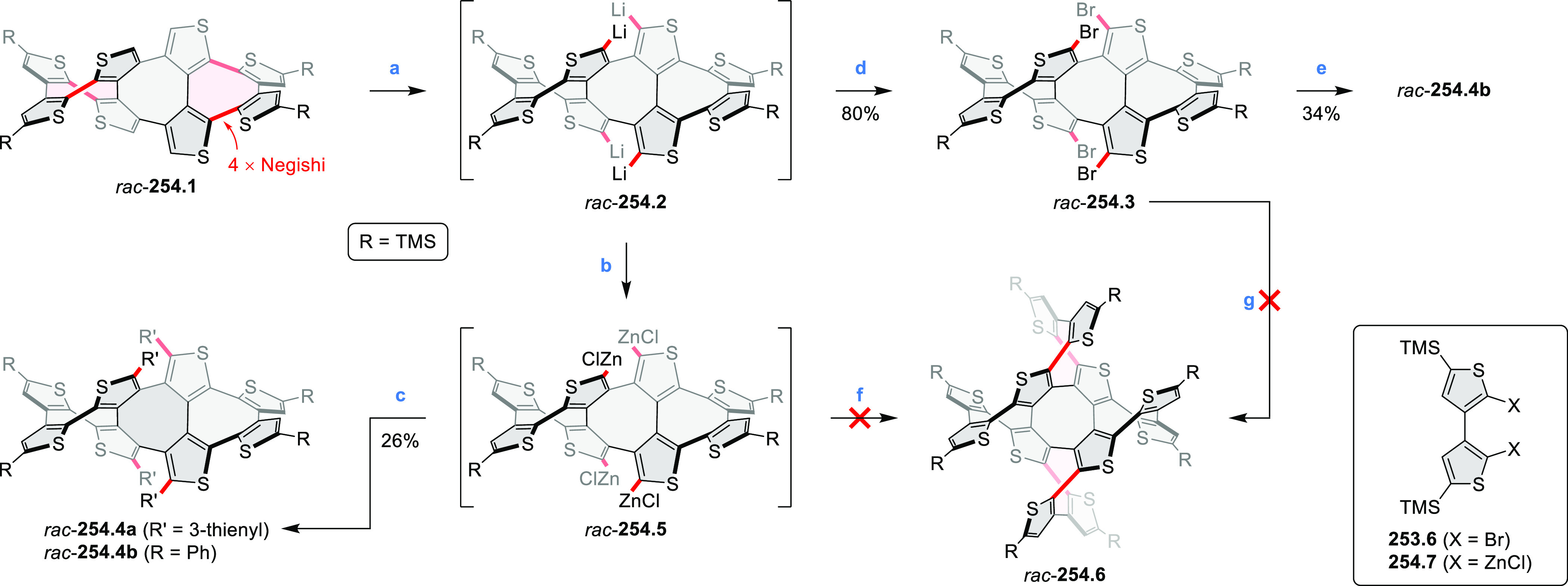
Reagents and conditions: (a)475n-BuLi, THF, −78 °C, then 60 °C, 2 h; (b) ZnCl2, −78 °C, then rt; (c) 3-bromothiophene, Pd(PPh3)4, THF, 120 °C, 48 h; (d) C2Br2Cl4, −78 °C, then rt, overnight; (e) phenylboronic acid, Cs2CO3, THF/H2O (50:1), 100 °C; (f) 253.6, Pd(PPh3)4, THF, 120 °C; (g) 254.7, Pd(PPh3)4, THF, 120 °C.
In 2018, Wang and Shi et al. reported an alternative strategy toward the known octathia[8]circulene 255.6 (“sulflower”, cf. CR2017, Section 6.1.5) using β,β-COTh-based starting materials (Scheme 255).476 The tetrakis(trimethylsilyl)-substituted β,β-COTh derivative 253.1 was quadruply deprotonated by n-BuLi and then reacted with bis(phenylsulfonyl)sulfide to give the doubly annulated product 255.1 in 31% yield. The four TMS groups were quantitatively acidolyzed by TFA, and the resultant compound 255.3 underwent quadruple deprotonation by LDA, followed by double annulation, to yield the “sulflower” 255.6 in 60% yield. This acidolysis–deprotonation–annulation sequence could also be performed stepwise, involving the intermediacy of compounds 255.2,4–5. The OFET device performances of compounds 255.3,5,6 were examined. The hole mobilities of 255.5 and 255.6 (6.8 × 10–4 and 2.6 × 10–3 cm2 V–1 s–1, respectively, at 80 °C) were both higher than that of “sulflower” (6.4 × 10–4 cm2 V–1 s–1 at the same temperature).
Scheme 255. Synthesis of Sulflower and Related Systems.

Reagents and conditions: (a)476n-BuLi, Et2O, −78 °C, then 60 °C, 2 h; (b) (PhSO2)2S, −78 °C, then rt, overnight; (c) TFA, DCM, rt, monitor by TLC until complete reaction; (d) LDA (4.1 or 2.1 equiv), THF, 0 °C, 2 h; (e) TFA, DCM, rt, then aq. NaHCO3 right after addition of TFA.
Ito, Kawai, and Foster et al. reported a combined in-solution and on-surface synthesis of a π-extended diaza[8]circulene 256.4 (Scheme 256).477 In their approach, based on the PAMY chemistry discussed earlier (see Schemes 221 and 232), dibenzo[a,e]cyclooctatetraene (256.1) reacted in a stepwise manner with the iminium salts 232.2 and 221.1a to give the products 256.3a,b with two newly fused pyrrole units. Notably, direct 2-fold cycloaddition of 256.1 proved unachievable. X-ray diffraction data showed that 256.3b adopted a tub-shaped geometry. Conversion of 256.3a into the diaza[8]circulene 256.4 could not be achieved in solution but was successfully realized via on-surface synthesis. Compound 256.3a was deposited on Au(111) at rt under ultrahigh vacuum and annealed at ca. 700 K to promote cyclodehydrogenation. STM images recorded before and after the reaction showed that some of the adsorbed molecules changed from the tub conformation to a flat structure, whereas some remained unchanged. The high-resolution CO-STM images confirmed the planarity of the Au(111)-adsorbed molecule. Bond lengths in the eight-membered ring were 1.42, 1.56, and 1.69 Å, indicating a pronounced bond order variation. The eight-membered ring was believed to be highly strained judging from its distortion from the regular octagonal shape. DFT calculations on 256.4 predicted that, in the gas phase, the planar and saddle conformations were 12.7 and 2.7 kcal mol−1 above the most stable twisted conformation.
Scheme 256. Combined In-Solution/On-Surface Approach toward a π-Extended Diaza[8]circulene Derivative,
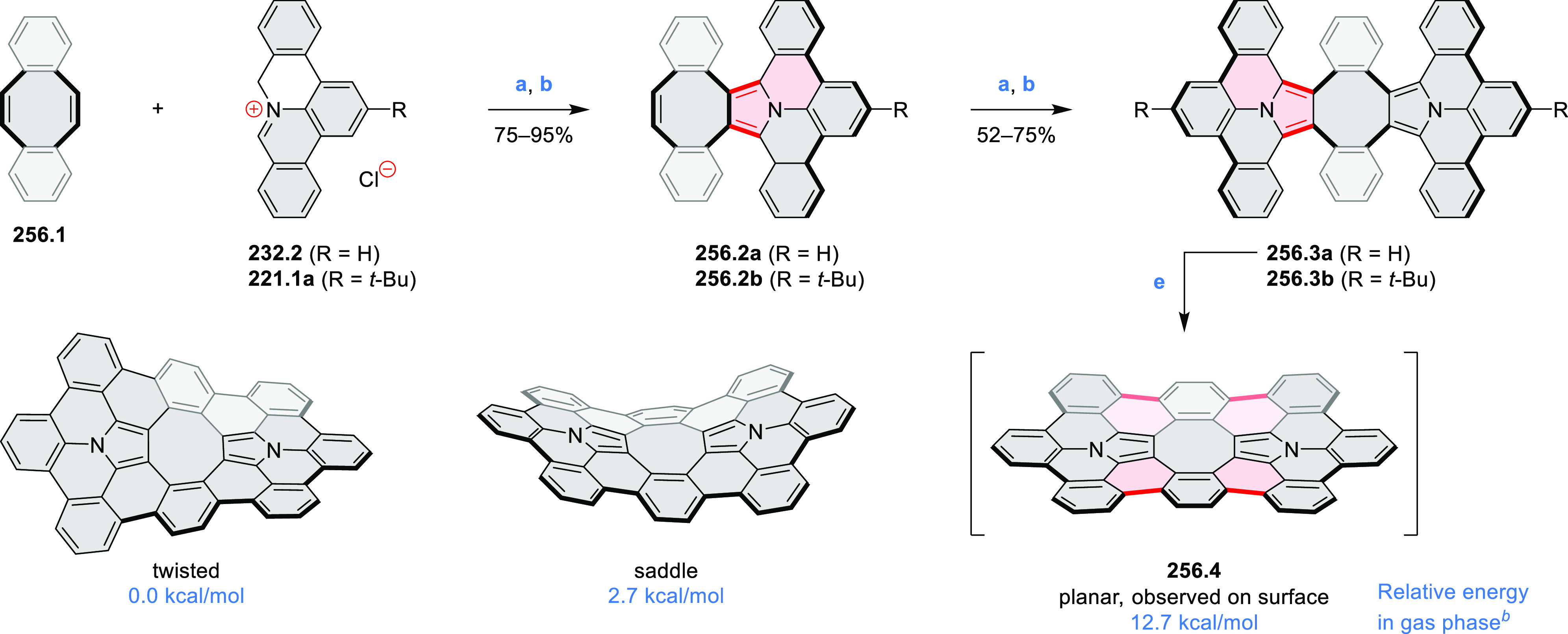
Reagents and conditions: (a)477i-Pr2NEt, DMSO, 140 °C, 6 h–18 h; (b) DDQ, DCM, rt, 10–1 h; (e) >430 °C on Au(111).
Level of theory: B3LYP/6-31G(d).
6.1.6. Larger Circulenes and Coronoids
In 2020, Pittelkow and Baryshnikov et al. reported the isolation of dihydrodiazatrioxa[9]circulene 257.2, which represented the first [n]circulene system with n > 8 (Scheme 257).478 Compound 257.2 was synthesized via a high-yielding oxidation of the known diazatrioxa[10]helicene 257.1 by DDQ, and was characterized by X-ray crystallography. The same group later reported the fully conjugated diazatrioxa[9]circulene.479 The synthesis began with the acid-catalyzed condensation of the carbazolediol 257.3 with 0.5 equiv of glyoxal to give compound 257.4 in 89% yield. Oxidation with chloranil/BF3·Et2O (cf. Scheme 251, Section 6.1.5) furnished the incompletely dehydrogenated 257.5, which could be converted into the fully conjugated hetera[9]circulene 257.6 by subsequent quantitative oxidation with DDQ. The tetrahydro[10]circulene 257.7 could also be formed from further condensation of compound 257.4 with glyoxal. The XRD data of compound 257.6 show a planar conformation, with a significant contribution from a [9]radialene character for the nine-membered ring. Both compounds 257.6 and 257.7 show well-defined blue fluorescence at 400–500 nm. The fully conjugated hetera[9]circulene 257.6 is photolabile in aerated solvents, while the tetrahydrohetera[10]circulene is stable under similar conditions. The instability of the former compound was attributed to the intersystem crossing from the S1 state to the reactive T1 state with spin density that is localized on the α-carbon atom of the furofuran moiety.
Scheme 257. Syntheses of Derivatives of Diazatrioxa[9]circulene and Diazatetraoxa[10]circulene.
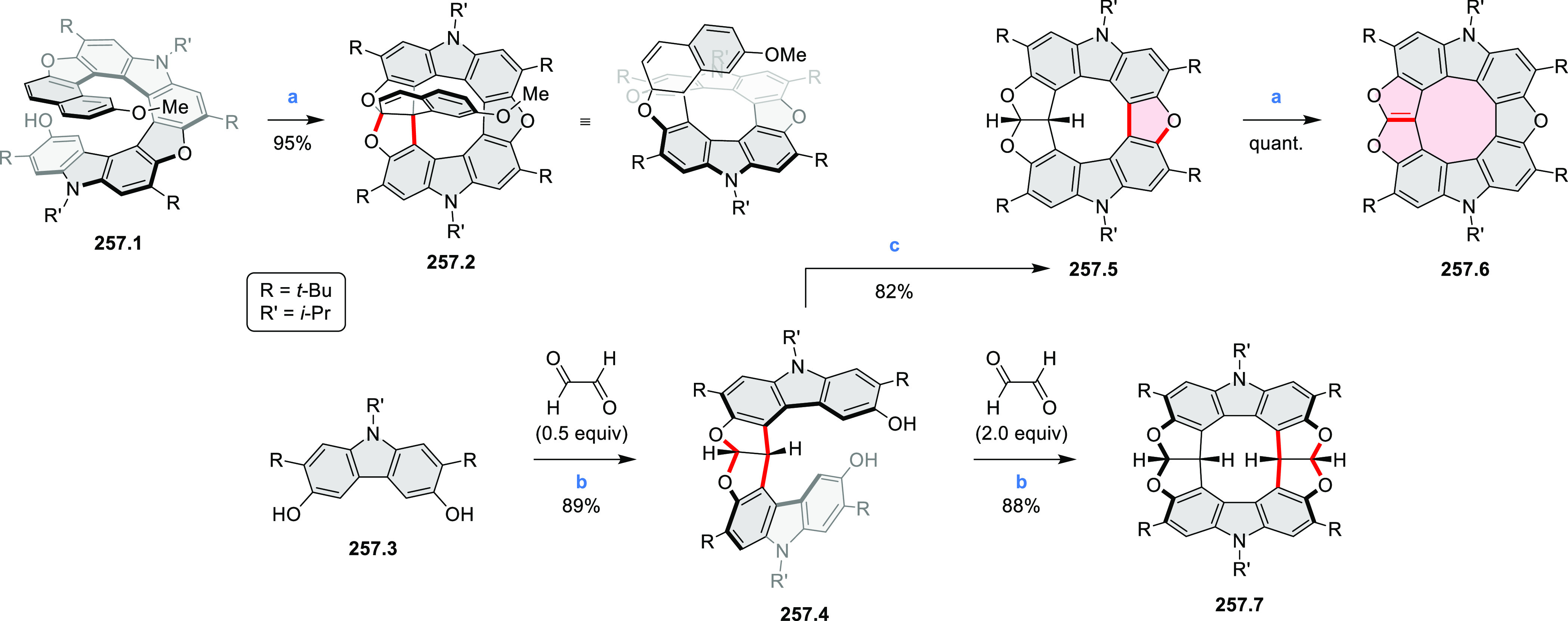
Reagents and conditions: (a)478 DDQ, DCM, rt; (b)479 H2SO4, AcOH, rt, 5–16 h; (c) chloranil, BF3·Et2O, DCM, rt, 45 min.
In 2016, Wu, Casanova, Ding, and Casado et al. reported the macrocyclic tetraradicaloid 258.1a and hexaradicaloid 258.1b, each consisting of alternating quinoidal and aromatic carbazole units (Scheme 258).480 The Suzuki–Miyaura cross-coupling of the known carbazole monomers 258.2 and 258.3 followed by recycling GPC allowed the isolation of 258.4a (2% yield) and 258.4b (29% yield) containing four and six carbazole units, respectively. A conventional mesitylation–cyclization sequence on 258.4a and 258.4b led to the respective 4-fold (25% yield) and 6-fold cyclized products (41% yield) as stereoisomeric mixtures, which were then individually oxidized by DDQ to generate the target macrocycles 258.1a,b in quantitative yields. Magnetic properties of 258.1a,b were investigated by superconducting quantum interfering device (SQUID) measurements. The results suggested that both molecules possessed a singlet ground state, in agreement with computational predictions. The singlet–triplet energy gaps (ΔES–T) were estimated to be −0.25 (for 258.1a) and −0.30 kcal mol−1 (for 258.1b), confirming the accessibility of the paramagnetic triplet state upon thermal excitation.
Scheme 258. Synthesis of Carbazole-Based Macrocyclic Tetraradicaloid and Hexaradicaloid.
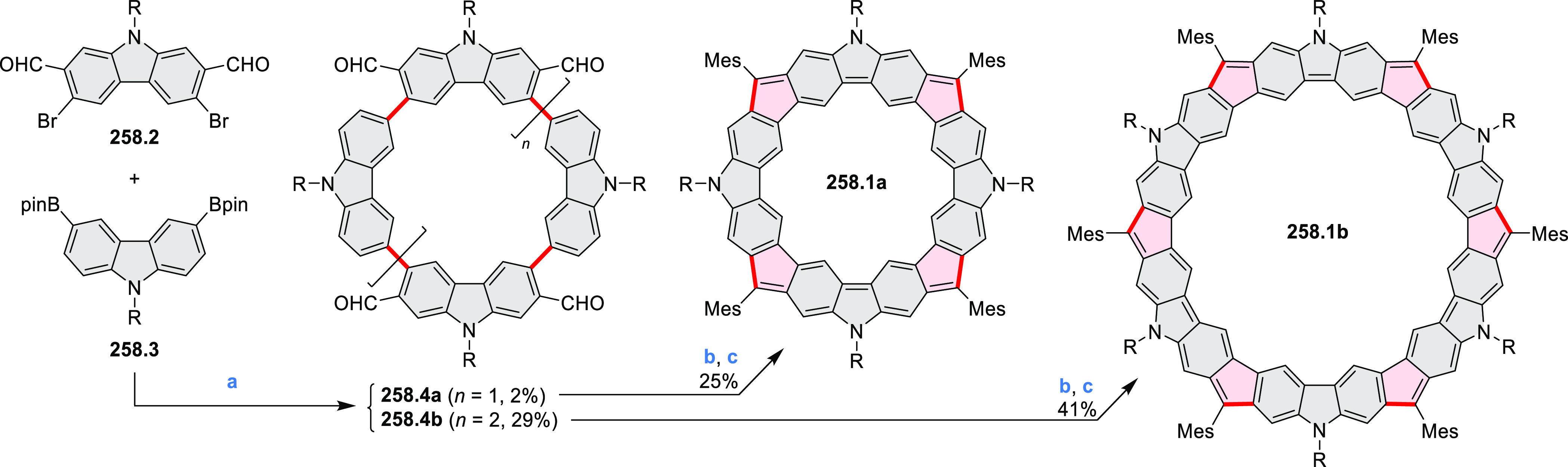
Reagents and conditions: (a)480 Pd2(dba)3, t-Bu3P·HBF4, NaHCO3, THF/H2O, 85 °C, 72 h; (b) (1) MesMgBr, THF, rt, 24 h, (2) BF3·OEt2, DCM; (c) DDQ, DCM, rt, 10–30 min.
Three examples of thiophene-containing coronoids (cycloarenes) have been recently reported (Scheme 259). In 2018, Miao et al. reported the preparation of the heterocycloarene 259.3.481 The first macrocyclic intermediate, 259.1, was obtained via the Eglinton reaction of the corresponding diyne precursor. The 1,3-diyne units in 259.1 were converted into thiophene units in 259.2 using Na2S, CuI, and 1,10-phenanthroline. Eventually, the action of FeCl3/MeNO2 induced the 6-fold oxidative cyclization among the thiophene moieties of 259.2, to produce the target heterocycloarene 259.3, which was isolated in 53% yield. This compound displayed a lowest-energy absorption maximum at 455 and a rather small Stokes shift of 0.20 eV in the emission spectrum. The broad aromatic resonances in the 1H NMR spectrum of 259.3 were attributed to the hindered inversion of the trithia[5]helicene units in the solution state.
The thiophene-containing analogue of octulene, 259.8, was reported in 2020 by Wu, Lu, and Zhao.482 The dibenzothiophene- and phenanthrene-based building blocks (259.4 and 259.5) were reacted in a 1:1 ratio under Suzuki–Miyaura cross-coupling conditions. The macrocyclic product 259.6 bearing four aldehyde groups was isolated in 21% yield after recycling GPC. Next, the Wittig reaction between 259.6 and (methoxymethylene)triphenylphosphane Ph3P=CH(OMe) was used to install four 2-methoxyvinyl groups in high yield. The resultant compound 259.7 was submitted to Bi(OTf)3-catalyzed electrophilic ring closures to afford the target 259.8 in 85% yield. This molecule was predicted by DFT calculation to assume a saddle-shaped conformation. Upon complexation with C60 or C70, the fluorescence emissions of 259.8 at 466 and 495 nm were quenched. The fluorescence titration data showed a higher association constant of 259.8 with C60 (1.25 × 106 M–1) than with C70 (9.49 × 105 M–1) in toluene. Subsequently, Lu and Wu et al. reported the preparation of the tetraradicaloid 259.10 from the macrocycle 259.6.483 Here, 259.6 was treated with mesitylmagnesium bromide followed by BF3·Et2O to give 259.9 as a stereoisomeric mixture. Dehydrogenation by DDQ provided the fully conjugated compound 259.10 in 84% yield over two steps. From the single-crystal XRD data of 259.10, the two benzenoid rings in the dibenzothiophene unit show significant bong length alternation, indicating the dominance of the quinoidal form as shown in Scheme 259. Based on VT-EPR data, the singlet–triplet energy gap of 259.10 was determined to be −3.47 kcal mol−1.
Heteroatom-doped coronoid substructures can be found in some of the recently reported 2D polymers obtained by condensation of aromatic components. In 2017, Mateo-Alonso et al. reported the first solvothermal synthesis of 260.1, a conjugated mesoporous polymer (CMP) bearing fused pyrazine units (Scheme 260).484 This CMP was prepared from the condensation of bis(TIPS-ethynyl)-substituted benzenetetramine and hexaketocyclohexane in dioxane/acetic acid at 135 °C. According to the authors, the known, alternative ionothermal synthesis employing harsher conditions is incompatible with the TIPS groups. Using semiempirical calculations, 260.1 was predicted to assume a highly twisted framework with the bulky TIPS-ethynyl substituents being congested in the node regions. Upon sonication, the CMP 260.1 could form homogeneous dispersions in TFA, DMF, and EtOH/H2O (1:1). The EtOH/H2O dispersion was examined by atomic force microscopy (AFM) and found to consist of exfoliated layers with a mean diameter of 200 nm. In 2021, Baek, Oh et al. reported another fused aromatic framework 260.2 via the condensation of benzenehexamine trihydrochloride and 4,5,9,10-pyrenetetraone in triflic acid at 175 °C.485 The structure of 260.2 was supported by FT-IR spectroscopy, X-ray photoelectron spectroscopy, and elemental analysis. The isolated thin flakes of 260.2 showed remarkable electron and hole mobilities of 996 and 501 cm2 V–1 s–1, respectively.
6.2. Fused Acenaphthylene Derivatives
6.2.1. Hetero[a]fused Acenaphthylenes
The acenaphtho[1,2-b]quinoxaline structure is generally prepared via the condensation between acenaphthoquinone and aromatic ortho-diamines (cf. CR2017, Section 6.2.1). Recent additions to this class of heteroaromatics are summarized in Chart 25. The thiadiazole-fused compound C25.1 was shown by Baumgarten et al. to form heat-to-head dimers with intermolecular N–S and weak N–N interactions, and stack at a distance of 3.37–3.39 Å in the solid state.486 Compound C25.2 was an unexpected product characterized by Bunz et al., and was proposed to arise from the ring rearrangement of the penultimate spirocyclic species C25.3.487 In 2018, Bunz, Dreuw, Freudenburg et al. reported a series of acenaphthylene-based quinoxalines C25.4a–c.488 In particular, among C25.4a,c and three other pyracylene-based quinoxalines C26.3a–c tested for organic light-emitting diode (OLED) performance (see Chart 26, section 6.2.7), the green-emitter C25.4c had the highest luminance (5.8 × 103 cd m–2), maximum efficiency (2.88 cd A–1) and efficacy (0.83 lm W–1). Compound C25.5 reported in 2017 by Kothavale and Sekar consists of the electron-donating morpholine group and the electron-accepting pyrazine and phenanthroline units, and was found to exhibit negative acidochromism, i.e., a blue-shifted absorption band upon protonation of the morpholine group.489
Chart 26. Pyracylene-Based Quinoxalines.

The acenaphthylene–quinoxaline motif has also been introduced in ligand design (Chart 25). In 2015, Valdes, Poyatos, and Eduardo reported the iridium(I) complexes C25.6a–c bearing an N-heterocyclic carbene (NHC) ligand containing fused acenaphtho[1,2-b]quinoxaline (see also Scheme 149 in Section 4.6).288 In the crystal structure of C25.6a, the NHC ligand was positioned quasi-orthogonally to the coordination plane around the iridium center, with stacking distances of 3.47 Å between adjacent molecules. In 2016, Liu, Xing et al. reported a series of ruthenium(II) polypyridyl complexes C25.7a–d.490 All four complexes featured the same new bidentate ligand containing an acenaphthylene–quinoxaline terminus. The complexes were demonstrated to induce A549 cell apoptosis via a reactive oxygen species (ROS)-mediated mitochondrial dysfunction pathway.
In 2021, Krische et al. reported a two-step synthesis of the π-extended diazarubicenes 261.3a–c (Scheme 261).491 Quinoxalines 261.2a–c were initially obtained by selective condensation of one diketone moiety of 261.1 with an appropriate ortho-phenylenediamine (the formation of the tetrafluoro congener 261.2c required the presence of Sc(OTf)3). Compounds 261.2a–c were then subjected to ruthenium-catalyzed transfer hydrogenative benzannulation with 1,3-butadiene to yield the target 261.3a–c. In addition, the tetraaza-substituted rubicene 261.4 was prepared from 261.1 in two steps. The UV–vis spectra of 261.2–4 in chloroform were similar and resembled that of the parent rubicene. The absorption and emission peaks were rather insensitive to the variation of solvent (cyclohexane and acetonitrile), indicating the absence of charge-transfer character despite the presence of nitrogen- and/or fluorine-rich regions in these molecules.
Examples of acenaphthylenes [a]fused to nitrogen-containing heterocycles are shown in Scheme 262. Fusco and Centore et al. reported the synthesis of compound 262.1 bearing a fused triazolotriazine unit.342 This nitrogen-rich polyaromatic skeleton showed a reversible reduction wave in cyclic voltammetry, corresponding to an estimated LUMO level of −3.59 eV (see Scheme 171, Section 4.7.3). In 2017, the preparation of the nitrogen-rich 262.2 from a pyrrolo[3,2-d]pyrimidine-based diamine was reported by Popov et al.492 Bellón and Alajarín et al. reported the preparation of the new acenaphtho- and indolo-fused quinolizinium salt 262.5 using the base-induced condensation of the N-aminopyridinium salt 262.4 with acenaphthoquinone.493 This transformation relied on the CH acidity of the vicinal methyl group in 262.4.
Gang, Wang, and Liu reported a palladium-free Sonogashira coupling reaction for the efficient synthesis of a family of imidazo[2,1-a]isoquinolines, such as 262.9a–d, from the corresponding arylated acenaphtho[1,2-d]imidazole precursors 262.8a–d (Scheme 262).494 The proposed mechanism involved a cascade process of Sonogashira coupling followed by intramolecular hydroamination of the new internal alkyne unit. In another attempt to build a similar π extension on acenaphtho[1,2-d]imidazole, the diaryl precursor 262.6 failed to undergo the crystalline-state photochemical direct arylation to generate the fused target 262.7 (for successful examples, see Scheme 165, Section 4.7.2).333
In 2018, Gulevskaya et al. reported a cascade reaction between the ethynyl-substituted derivatives of the proton sponge and 1,8-diiodonaphthalene, which generated the acenaphtho[1,2-b]benzo[g]indole skeleton (Scheme 263).495 For instance, compound 263.1 reacted with 1,8-diiodonaphthalene under Sonogashira coupling conditions to give compound 263.2 in 46% yield. The proposed mechanism involved the intermediate 263.4, which resulted from the initial diarylacetylene product with the remaining C–I bond further activated by palladium. The nucleophilic dimethylamino group induced a bicyclization process, resulting in the palladacycle 263.5, which led to the final product upon reductive elimination and elimination of methyl iodide. Interestingly, when the diyne 263.6 was submitted to the same reaction with 1,8-diiodonaphthalene, the double fusion product 263.7 was not found, but compound 263.8 was obtained in 19% yield.
In 2017, Li, Hartl, and Yang et al. reported optimized conditions for a palladium-catalyzed C–H activation reaction of benzo[b]thiophene with 1,8-dibromonaphthalene, which gave the acenaphthylene product 264.1 in 69% yield (Scheme 264).496 By applying this protocol, it was also possible to react fused dithiophenes with 1,8-dibromonaphthalene in a 1:2 ratio, to obtain compounds 264.2a–c and 264.3 in 26–90% yield. Additionally, the para-benzoquinone 264.3 could successfully be converted to the diyne 264.2d in 22%. When an anodic potential was applied to concentrated solutions, the neutral molecules of 264.2a,c–d were found to interact with their own radical cations, to form dimers such as [264.2a]2•+
In 2020, Murata et al. reported a cross-dehydrogenative coupling reaction between tetracene and benzene, using p-chloranil as the oxidant in the presence of triflic acid, which was found to produce indeno[1,2,3-fg]tetracene.497 This method also worked when benzene is replaced with t-butylbenzene, fluorobenzene, anisole and 2-methylthiophene. In the last case, compound 264.4 was formed in 29% yield (Scheme 264). Notably, a solution of 264.4, when illuminated with artificial light under air, showed negligible photodegradation as monitored by absorption spectroscopy. This contrasted with the behavior of rubrene, which is much more photolabile. In 2017, Murata and Murata et al. reported the synthesis of the doubly fused tetracene 281.3 (for synthesis, see Scheme 281, section 6.2.7),498 which is a π-extended analogue of 264.4.
Scheme 281. π-Extended Heterocycle-Fused Pyracylenes.
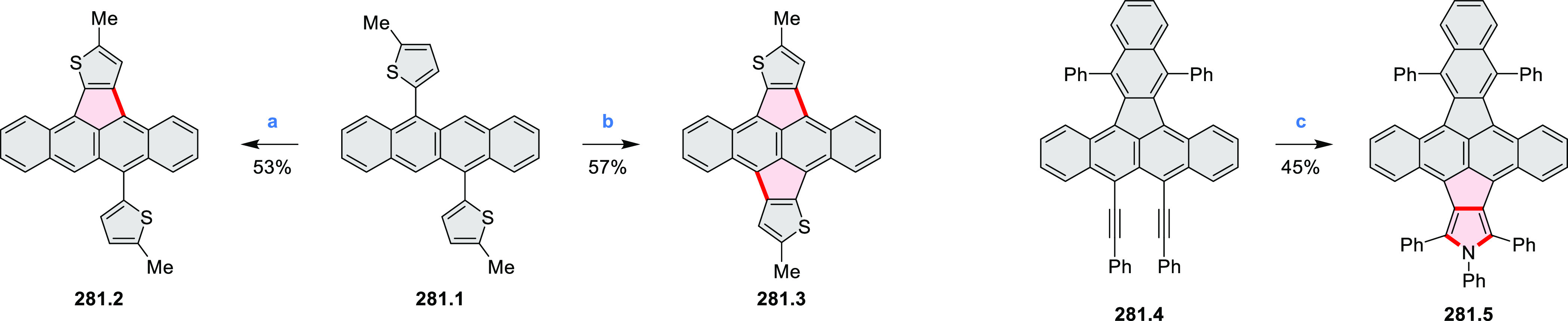
Reagents and conditions: (a)498p-chloranil (1.0 equiv), DCM/TfOH (100/1), 0 °C, 10 min; (b) p-chloranil (2.0 equiv), DCM/TfOH (100:1), 0 °C, 10 min; (c)525 PhNH2, PdCl2, Et3N, DMSO, 90 °C, 8 h.
In 2017, Jeng et al. reported the preparation of conjugated polymers 265.3–4 based on the emeraldicene core499−501 and examined their optoelectronic and photovoltaic properties (Scheme 265).502 To synthesize the monomer, compound 265.1 was subjected to palladium-catalyzed cyclopentannulation followed by dibromination to give 265.2 in an overall yield of 59%. Subsequent copolymerization via Suzuki–Miyaura coupling with 265.5 or via Stille coupling with 265.6 furnished the corresponding polymers 265.3–4 in 82% and 28% yield, respectively. The visible-light-transparent emeraldicene–fluorene copolymer 265.3 absorbed mostly in the UV and NIR regions. For the polymer solar cell device constructed using 265.3, a power conversion efficiency (PCE) of 2.5% was demonstrated by the authors. When fabricated with a cross-linker additive, the device showed a greatly enhanced thermal stability.
Scheme 265. Thiophene-Fused Rubicene-Based Conjugated Polymers,
Reagents and conditions: (a)502 Pd(OAc)2 K2CO3, n-Bu4NHSO4, MeCN, reflux, 24 h; (b) NBS, THF, 0–35 °C, 12 h; (c) 265.5, Pd(PPh3)4, K2CO3, toluene, 110 °C, microwave, 12 h; (d) 265.6, Pd(PPh3)2Cl2, toluene, 110 °C, microwave, 12 h; (e)503 FeCl3, DCM/MeNO3, argon, rt, overnight
Only one out of three possible isomeric constitutions of the repeat unit is shown for polymers 265.7a and 265.8a–c. Calculation was performed on the repeat unit of the structures shown (B3LYP/6-31G**). Alternative regioisomeric repeat units were also calculated.
In 2017, Plunkett et al. reported the preparation of the conjugated ladder polymers 265.8a–c featuring singly or doubly thieno-fused rubicene units (Scheme 265).503 They were prepared from the oxidative six-membered ring closure of the corresponding copolymers (such as 265.7a) using FeCl3/MeNO2 in 89–93% yield. Because the cyclopentannulation was not regioselective, the precursor polymer 265.7a, as well as the cyclized product 265.8a, contained regioisomeric repeat units. Polymer 265.8a was essentially insoluble, except when heated in high-boiling chlorinated solvents. According to DFT calculations, the repeating units of 265.8a are noticeably flatter than those of 265.8b,c, as quantified by the smaller splay angles of the thiahelicene units in 265.8a.
In 2017, Takasu et al. reported a series of π-extended fluoranthenes through a KHMDS-promoted anionic-radical cascade.504 As exemplified by the thiophene-fused congener 266.2, the precursor 266.1 was refluxed in diglyme in the presence of KHMDS (3 equiv) and cis-1,2-cyclohexanediol (20 mol %) to afford the doubly cyclized product in 41% yield (Scheme 266). It was suggested that KHMDS plays two roles in this cascade reaction. First, the strong base deprotonates 266.1 to give the corresponding enolate, which undergoes intramolecular [2 + 2] cycloaddition followed by nucleophilic displacement to yield the key intermediate 266.3. Second, KHMDS functions as a one-electron reductant. It provides an electron to 266.3 via single electron transfer (SET), thereby inducing subsequent ring rearrangement of 266.3•–.
Scheme 266. Thieno[j]fluoranthenes.

Reagents and conditions: (a)504 KHMDS (3 equiv), cis-1,2-cyclohexanediol (20 mol %), diglyme, reflux, 24 h.
6.2.2. Hetero[e]fused Acenaphthylenes
A series of fused pentacyclic compounds such as 267.3a–f were prepared in an intermolecular domino C–H annulation reaction between the picolinamide 267.1 and 2 equiv of a 1,4-diaryl-1,3-butadiyne 267.2a–f (Scheme 267).505 The reaction showed high regioselectivity, producing five new C–C bonds and four fused rings in a single operation. To proceed with improved selectivity, the transformation required [RhCp*Cl2]2 (20 mol %), AgSbF6 (40 mol %), and excess Cu(OAc)2 (4.0 equiv). This RhIII-catalyzed CuII-assisted annulation pathway was studied using DFT calculations, and the proposed sequence of C–C bond formation is shown in Scheme 267.
Scheme 267. Hetero[e]fused Acenaphthylenes from C–H Annulation Reactions,

Reagents and conditions: (a)505 [RhCp*Cl2]2 (20 mol %), AgSbF6 (40 mol %), Cu(OAc)2 (4.0 equiv), dioxane, 130 °C, 3 h; (b)284 (1) Tf2O, 1,2-DCE, −30 °C, 20 min, then rt, 1 h, (2) microwave, 90 °C, 12 h, (3) Et3N, rt, 20 min, then 50 °C, 5 h.
The sequence of formation of the five new C–C bonds determined by DFT calculation for 267.3a is shown in red.
Shi, Huang, and Blakey et al. recently explored the reactivity of heterocycle-substituted diolefins 268.3a–c in oxidative coupling reactions (Scheme 268).506 These three compounds were synthesized via the known palladium-catalyzed cross-coupling of the dibromides 268.2 with the corresponding N-tosylhydrazones 268.1 in a 1:2 stoichiometry (76–95% yield). Compound 268.3a underwent 4-fold cyclodehydrogenation when treated with FeCl3/MeNO2 at 0 °C, to give the dithieno-fused rubicene 268.4 in 64% yield. When the same reaction took place at rt, further aromatic chlorination occurred to give the dichloride 268.5 in 60% yield. The tert-butyl-substituted precursors 268.3b,c were cyclized without chlorination, to furnish to the products 268.6 (using FeCl3/MeNO2) and 268.7 (using DDQ/TfOH). In the absorption spectra, a red-shift of 13 nm was observed for the selenophene-fused 268.7 relative to the thiophene-fused congener 268.6. This phenomenon was ascribed to the greater quinoidal character of the π-system containing the selenium atom. Based on femtosecond transient absorption experiments, the accelerated intersystem crossing from the S1 state to the T1 state was confirmed for the Se-containing 268.7 in comparison to 268.6.
In 2016, Mastalerz et al. reported the preparation of a series of π-extended truxene derivatives, including two heterocycle-fused analogues 269.1a,b (Scheme 269).507 The first key step in their work was the 3-fold iridium-catalyzed direct borylation of the tri-tert-butyl-substituted truxene 269.2, which took place selectively at the least hindered aromatic sites to give 269.3 in 96% yield. Subsequent Suzuki–Miyaura cross-coupling of 269.3 with 3-bromothiophene-2-carboxaldehyde led to the trialdehyde 269.4a in excellent yield. The second key step involved the condensation between the aldehyde and methylene groups in the presence of t-BuOK to give the product of 3-fold cyclization 269.1a in 92% yield. Likewise, the pyridine-fused analogue 269.1b was prepared from the triboronate 269.3 in 64% yield over two steps. The α-positions of the thiophene rings in 269.1a were easily functionalizable via bromination and borylation.
Scheme 269. Synthesis of Heterocycle-Fused π-Extended Truxenes.
Reagents and conditions: (a)507 B2pin2, [Ir(OMe)(cod)]2, 4,4′-di-t-butyl-2,2′-bipyridine, t-BuOK, THF, 80 °C, 24 h; (b) 3-bromothiophene-2-carboxaldehyde, Pd2(dba)3 (10 mol %), t-Bu2PMe·HBF4 (17 mol %), K2CO3, THF/H2O (4:1), 80 °C, 16 h; (c) t-BuOK, THF, 60 °C, 16 h.
6.2.3. Hetero[d]fused Acenaphthylenes
In 2018, Plunkett et al. reported the two cyclopentannulated anthracenedithiophenes 270.3 and 270.4, the latter being the cyclodehydrogenation product of the former (Scheme 270).508 The five-membered rings in compound 270.3 were constructed through the 2-fold palladium-catalyzed cyclopentannulation of the dibromide 270.1 with the diarylacetylene 270.2 in 39% yield. The DFT-optimized geometry of 270.3 had a contorted structure with large splay angles of roughly 40° around the (thia)[5]helicene moieties (cf. Scheme 265, Section 6.2.1). The absorption edge of 270.4 at about 830 nm was significantly bathochromically shifted relative to that of 270.3 (670 nm). Moreover, the LUMO energy of the π-system was estimated by cyclic voltammetry to drop from −3.44 to −3.70 eV upon six-membered ring closure.
Scheme 270. Synthesis of a Contorted Polyarene in a Cyclopentannulation–Cyclodehydrogenation Sequence,

Reagents and conditions: (a)508 Pd2(dba)3, tri(o-tolyl)phosphine, KOAc, LiCl, DMF/toluene, 130 °C; (b) FeCl3, DCM/MeNO2.
Level of theory: B3LYP/6-311G(d,p).
6.2.4. Carbazole-Based and Related Systems
Cyclocondensation of carbazole with diethyl malonate in a 1:2 ratio was reported to yield compound 271.1 (Scheme 271).509 When 271.1 was heated with NaOH in ethylene glycol, the lactone ring opening followed by decarboxylation gave rise to compound 271.2, which could be derivatized into the three compounds 271.3–5 in subsequent steps. Compound 271.3 underwent cyclodehydration in the presence of Eaton’s reagent to give the benzofuran-fused product 271.6 (19% yield), while the azides 271.4 and 271.5 could undergo thermal electrocyclization to give, respectively, the furazan N-oxide 271.7 (66% yield) and the isoxazole 271.8 (78% yield).
Scheme 271. Lactam-Fused Carbazole Derivatives,
Reagents and conditions: (a)509 diethyl malonate (3 equiv), diphenyl ether, 250–260 °C, 7 h; (b) NaOH, ethylene glycol, 180 °C, 3 h; (c) P2O5, MsOH, 150 °C, 20 min; (d) DMF, reflux, 2 h; (e) bromobenzene, reflux, 3 h; (f)510 AcOH, reflux, air, 30–40 h.
The chains of atoms in 271.1 that originate from the first and second reacting diethyl malonate molecules are shown in blue and green, respectively.
In 2017, Garazd et al. reported the formation of the peri-fused carbazole analogues 271.12a,b, when a solution of l-tryptophan and 2-formylbenzoic acid (271.9a) or opianic acid (271.9b) in acetic acid was heated to reflux for 30–40 h (Scheme 271).510 The initial Pictet–Spengler reaction product 271.10a,b was proposed to further cyclize to give the lactam 271.11a,b, which furnished the observed products upon decarboxylative ring rearrangement in the presence of atmospheric oxygen. Compounds 271.12a,b represent benzannulated analogues of the natural product canthin-6-one.
In 2016, Huang, Yu, and Xie et al. reported the preparation of the diindole-fused azapentacenone 272.3 using an improved procedure (Scheme 272).511 First, the commercially available 272.1 was subjected to the copper(I)-catalyzed Ullmann-type reaction with carbazole to give 272.2 in 76% yield. Afterward, 272.2 was hydrolyzed and activated by SOCl2. Subsequent 2-fold intramolecular Friedel–Crafts acylation led to the ring-closed compound 272.3 in 72% yield. Based on various data, the performance of 272.3 as an organic cathode material for lithium-ion battery was concluded to be superior to the parent azapentacenone without the two peri-fusions.
Scheme 272. Synthesis of a Diindole-Fused Azapentacenone.

Reagents and conditions: (a)511 CuI, K2CO3, 18-crown-6, o-dichlorobenzene, 180 °C, N2, 24 h; (b) (1) NaOH, MeOH, reflux, 10 h, then aq. HCl; (c) DMF (cat.), SOCl2, reflux, 1 h; (d) AlCl3, DCM, rt, 12 h.
In 2015, Higashibayashi et al. reported two new pyridazinodicarbazoles 273.1a,b, in addition to the previously known 273.1c (Scheme 273).512 Compounds 273.1a,b were obtained through oxidative N–N bond formation in 1,1′-bicarbazoles 273.2a,b, induced using [n-Bu4N][MnO4]. Interestingly, when 9,9′-bicarbazole 273.3 was subjected to the Yamamoto coupling conditions, the cyclized product 273.1b was not obtained, but the 1,1′-bicarbazole 273.2b was isolated in 20% yield, implying the occurrence of a reductive N–N bond cleavage. In 2016, the same group reported the synthesis and redox chemistry of the hydrazinobuckybowl 273.4.513 The known compound 273.5 was reductively desulfurized using naphthalene and metallic lithium to give [2]cyclo-1,8-carbazolylene 273.6 and the further C–C bond cleavage side product 273.2c in 62% and 12% yield, respectively. The former product underwent a DDQ-mediated N–N bond formation, followed by reductive quenching with hydrazine, to yield the hydrazinobuckybowl 273.4 almost quantitatively (for an alternative approach, see Scheme 388, Section 7.7.2). The chemical oxidation of 273.4 with 1.0 or 2.1 equiv of NOSbF6 led to the formation of the SbF6– salts of the radical cation [273.4]•+ and the dication [273.4]2+, respectively. In the crystalline state, the skeletons of 273.4, [273.4]•+, and [273.4]2+ adopted a twisted, bowl-shaped, and planar geometry, respectively.
Scheme 273. Fused Bicarbazoles.
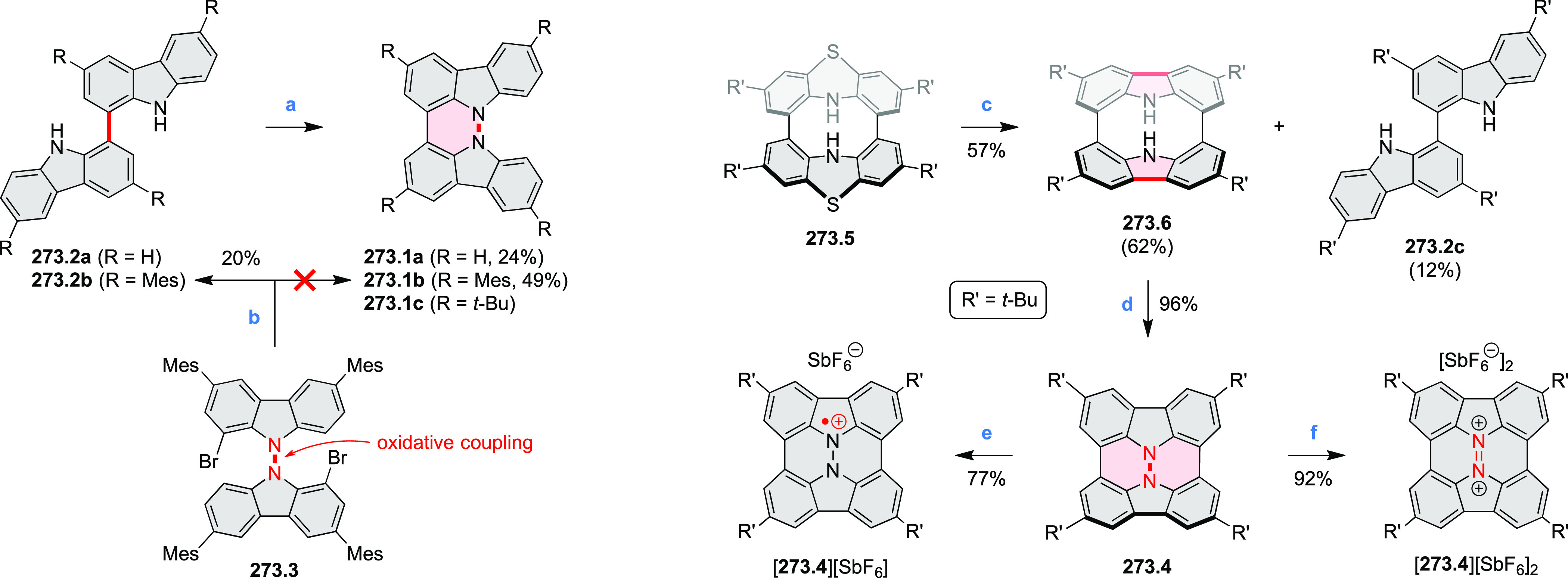
Reagents and conditions: (a)512 [n-Bu4N][MnO4], pyridine, 70 °C, 24 h; (b) Ni(cod)2 (150 mol %), cod (150 mol %), 2,2′-bipyridyl (150 mol %), THF, 55 °C, 24 h; (c)513 naphthalene, Li, THF, 0 °C, 1 h; (d) (1) DDQ, DCM, 0 °C, then rt, 2 h, (2) N2H4·H2O, 0 °C; (e) NOSbF6 (1.0 equiv), DCM/MeCN, rt, 1 h; (f) NOSbF6 (2.1 equiv), DCM/MeCN, rt, 30 min.
Scheme 388. Synthesis and Oxidation of [n]Cyclo-1,8-carbazolylenes.
Reagents and conditions: (a)759 slow addition of 388.2 over 1 h to Ni(cod)2 (250 mol %), cod (250 mol %), 2,2′-bipyridyl (150 mol %), THF, 80 °C, then, 80 °C, 1 h; (b) n-BuLi, toluene, −80 °C, then 0 °C, 30 min, (2) BBr3, −80 °C, then rt, 3 h; (c) PhSiCl3, Et3N, 1,2-DCE, 90 °C, 12 h; (d) PCl3, Et3N, 1,2-DCE, 90 °C, 16 h; (e) m-CPBA, DCM, 30 min; (f)760 DDQ (1.5 equiv), CHCl3 stabilized by 0.3–1.0% EtOH, rt; (g) DDQ (2.0 equiv), EtOH-free CHCl3, rt, 95%.
6.2.5. Carbonyl-Free Azafluoranthenes
In 2015, Lipson et al. reported a three-component synthesis of extended pyrazoles of the general structure 274.3 (R = aryl), obtained via cyclocondensation of 5-amino-3-methylpyrazole (274.1), an arylglyoxal hydrate (274.2), and 1,3-indanedione (Scheme 274).514 Subsequent condensation with hydrazine hydrate performed for the p-methoxyphenyl derivative was shown to provide access to the pyrazole-fused triazafluoranthene 274.4 in 52% yield.
Scheme 274. Carbonyl-Free Azafluoranthene Derivatives.

Reagents and conditions: (a)514 EtOH, reflux, 1 h; (b) N2H4·H2O, EtOH, reflux, 8 h; (c)515 Cu(OTf)2 (100 mol %), MeCN, 80 °C, 12 h; (d) Cu(OTf)2 (10 mol %), Selectfluor (1.0 equiv), MeCN, 80 °C, 12 h; (e)517ortho-phenylenediamine (2.5 equiv), Na2CO3 (3 equiv), Cu(OAc)2 (0.1 equiv), i-PrOH/ethylene glycol (9:1), argon, reflux, 16 h; (f) TsOH·H2O (0.1 equiv), EtOH, reflux.
In 2018, Patil et al. developed a library of cationic nitrogen-doped polycyclic arenes via copper-promoted intramolecular [4 + 2] cycloaddition cascade.515 Upon heating with an equimolar amount of Cu(OTf)2 in acetonitrile, 274.5 was transformed to the doubly cyclized quaternary ammonium salt [274.6][OTf] in 60% yield (Scheme 274). When an external oxidant (Selectfluor) was added, the use of a catalytic amount of Cu(OTf)2 sufficed to mediate the same bicyclization to give [274.6][BF4] in a similar yield. Substituted and fused analogues of [274.6][OTf] were also synthesized and were found to exhibit tunable emission wavelengths. It was later found that the outcome of the reaction depends on the oxidant; in particular, the use of a hypervalent iodine reagent led to spirocyclized products.516
In 2019, Wen et al. reported the double amination of heterocyclic iodonium cations with primary amines as a general method to construct polycyclic heteroaromatic fragments which might be useful in drug discovery.517 For instance, the reaction of the chromone-fused iodonium cation 274.7+ with ortho-phenylenediamine in the presence of Cu(OAc)2 and Na2CO3 resulted in the fused indole derivative 274.8 in 68% yield (Scheme 274). Further acid-catalyzed cyclization of 274.8 produced the peri-fused heteroarene 274.9 in 95% yield. (See also Scheme 295, Section 6.4.2)
Scheme 295. [de]Fused Dibenzoazepines and Analogues via Direct Cyclization.
Reagents and conditions: (a)560 AuCl(PPh3) (10 mol %), AgSbF6 (10 mol %), 1,2-DCE, 80 °C, 24 h; (b) AuCl[P(C6F5)3] (10 mol %), AgOTf (10 mol %), 1,2-DCE, rt, 3 h; (c)518 Cs2CO3, CuI, DMF, microwave, 150 °C, 10 min; (d)517 AcCl (1.2 equiv), Et3N (2.0 equiv), DCM, rt, 4 h; (e) polyphosphoric acid, POCl3 (10 equiv) 120 °C, 3 h.
6.2.6. Miscellaneous Azaacenaphthylenes and Azafluoranthenes
In 2016, Xu and Chen et al. reported a one-pot, four-component Ugi procedure for the synthesis of a series of benzimidazoisoquinolines including 275.1a,b (Scheme 275).518 The ortho-halophenyl groups in these two compounds could further react with the NH group of the benzimidazole unit in an Ullmann-type reaction. Consequently, the quinoxaline-fused products 275.2a,b were isolated in 80–87% yield (for a related product with a seven-membered ring, see Scheme 295, Section 6.4.2).
Scheme 275. Synthesis of a Fused Benzimidazoisoquinoline–Quinoxaline.

Reagents and conditions: (a)518 Cs2CO3, CuI, DMF, microwave, 150 °C, 10 min.
Another three-component cascade reaction, developed by Xu and Li et al. for the synthesis of pyridodiindoles, involved a condensation of the 1-Boc-protected 3-aminoindole (276.1), a substituted benzaldehyde, and benzyl isocyanide (276.3) (Scheme 276).519 The reaction required 10% perchloric acid in methanol under microwave heating at 100 °C. When methyl 2-formylbenzoate (276.2) was used as the aldehyde component, the expected aryl-substituted pyridodiindole 276.4 was found to further cyclize to give the lactam 276.5 in 78% yield. In the mechanism proposed by the authors, the imine 276.6 formed from 276.1 and 276.2 is attacked by the nucleophilic carbon atom of benzyl isocyanide to give 276.7, which undergoes a dehydrogenative imine formation and a pseudo-Neber rearrangement to give the azirine 276.8. Cleavage of the C–N single bond yields the intermediate 276.9. The latter species rearranges to 276.10, which further oxidatively cyclizes to give the penultimate intermediate 276.4.
Scheme 276. Three-Component Cascade Condensation.
Reagents and conditions: (a)519 10% HClO4, MeOH, microwave, 100 °C, 10 min.
In 2019, Xi et al. reported the MeOTf-induced annulation of N-(2-cyanoaryl)indoles to give the corresponding indolo[1,2-a]indol-10-imines.520 When applied to the substrate N-(2-cyanophenyl)carbazole (277.1), the same protocol produced the indole-fused acridinimine 277.2 nearly quantitatively (Scheme 277). The proposed mechanism involves an initial activation of the nitrile group upon methylation. The resultant intermediate 277.3+ undergoes electrophilic cyclization to give 277.4+, which eventually gives the target molecule upon deprotonation.
Scheme 277. MeOTf-Induced Cyclization of N-(2-Cyanophenyl)carbazole.

Reagents and conditions: (a)520 MeOTf (1.2 equiv), 1,2-DCE, air, 90 °C, 24 h.
In 2019, Tilve et al. reported the cyclization of compound 278.1 in the presence of NaHMDS to give the known compound 278.2 (45% yield), which is an analogue of the natural product alpkinidine (Scheme 278).521 In an attempt to increase the cyclization yield by introducing an alkyl chain, 278.1 was subjected to cyclization using sodium ethoxide following by treatment with allyl bromide. The combined yield of the O- and C-allylated polycycles 278.3–4 was 88%.
Scheme 278. Base-Promoted Cyclization toward an Alpkinidine Analogue.

Reagents and conditions: (a)521 NaHMDS, THF, −95 °C, 10 min, then rt, 4 h; (b) (1) NaOEt, EtOH, rt, 2 h, (2) allyl bromide, 6 h.
In 2021, Mastalerz et al. reported the synthesis of two indigo derivatives 279.1–2, both end-capped with triptycene units, in an attempt to solubilize the otherwise insoluble indigo pigment (Scheme 279).522 In the synthetic route, 2-aminotriptycene (279.3) was subjected to the sequential treatments of chloral hydrate, hydrochloric acid, and hydroxylamine. The resulting compound 279.4 underwent cyclization in MsOH to give the isatin 279.5 in 85% yield. Next, 279.5 was heated with PCl5 in toluene at 100 °C, followed by reduction by thiophenol in one pot to furnish the indigo derivative 279.1 in 57% yield. Further annulation was accomplished with the use of 2-thiopheneacetyl chloride, yielding the Cibalackrot analogue 279.2 in 30% yield. The solubilities of compounds 279.1 and 279.2 in DCM were determined to be 10 and 38 mmol/L. The former compound was thus 66 times more soluble than the parent indigo (0.15 mmol/L) without the end-caps.
Scheme 279. Synthesis of Triptycene-Capped Indigo Derivatives.
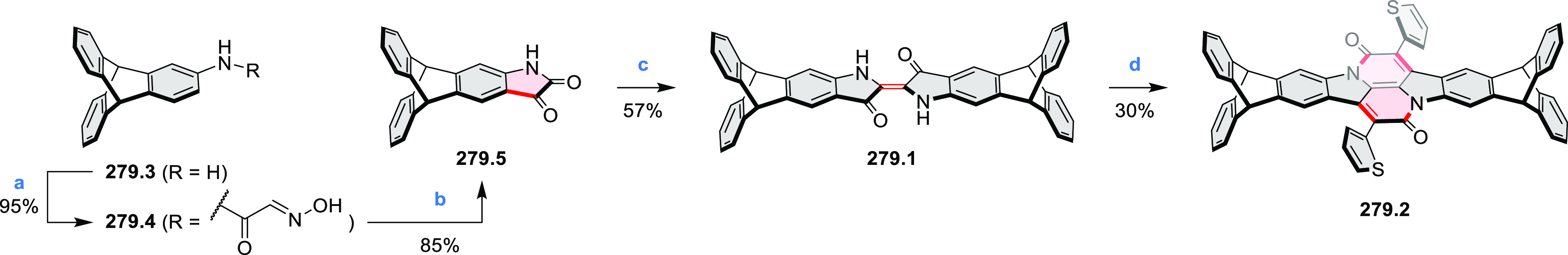
Reagents and conditions: (a)522 Cl3CCH(OH)2, Na2SO4, H2O, 50 °C, then conc. HCl, 5 min, (2) aq. NH2OH·HCl, 80 °C, 20 h; (b) MsOH, 50 °C, 4 h; (c) (1) PCl5, toluene, 100 °C, 4 h, (2) PhSH, 50 °C, 16 h; (d) 2-thiopheneacetyl chloride, o-xylene, 155 °C, 48 h.
Koutentis and co-workers showed that, when heated with excess S4N4 in DMF, benzotriazinones 280.1a,b produced the corresponding thiadiazole derivatives 280.2a,b, accompanied by an unusual peri-fused product 280.3a,b and a small quantity of the amine 280.4a,b (Scheme 280).523 Thermolysis of the 1,4-thiazino-fused 280.3a,b efficiently produced the ring-contracted triazafluoranthenones 280.5a,b.
Scheme 280. Preparation of Thiadiazolo-Fused Triazafluoranthenones.

Reagents and conditions: (a)523 S4N4, DMF, 153 °C, 7 h; (b) m-terphenyl, 270 °C, 3 h.
6.2.7. Pyracylene-Based Systems
In analogy to acenaphthylene-fused quinoxalines, their pyracylene-fused analogues are prepared via diketone–diamine condensation (Chart 26). In 2017, the three polymorphs of compound C26.1 were found by Zhang, Li et al. to exhibit different stacking modes and interlayer distances, which correlated with the trends in solid state fluorescence quantum yields.524 Shown in the chart are also the pyracylene-based quinoxalines C26.2a–c and C26.3a–e explored by Bunz, Dreuw, Freudenburg et al. as potential OLED emitters.488
In 2017, Murata, Murata et al. explored the reactivity of 5,11-bis(5-methyl-3-thienyl)tetracene (281.1) in oxidative pentannulation reactions (Scheme 281.1).498 Using one or two equivalents of p-chloranil in the presence of triflic acid, the singly cyclized (281.2) and doubly cyclized (281.3) products were isolated in 53% and 57% yield, respectively. Both ring closures took place regioselectively with the tetracene backbone. According to the authors, this cyclization was the first example of fusing a thiophene unit into a fully unsaturated five-membered ring using oxidative coupling. Such transformations had previously been accomplished only by palladium-catalyzed protocols (such as Schemes 264 and 265, Section 6.2.1). Experimental and theoretical data implied that the electronic structure of 281.3 had a strong antiaromatic contribution associated with the newly formed five-membered rings.
In 2019, Hamura et al. reported a new method for construction of the benzo[5,6]indeno[1,2,3-fg]tetracene skeleton involving intramolecular benzoallene–alkyne cycloaddition.525 The resulting diacetylenes, such as the derivative 281.4, were used as precursors to pyracylene derivatives (Scheme 281). In particular, the palladium-catalyzed bicyclization of 281.4 with aniline under previously known conditions afforded the pyrrole-fused pyracylene derivative 281.5 in 45% yield.
6.2.8. Extended Thiaacenaphthylenes
The known sulfur-rich acenes, tetrathiotetracene (C27.1) and hexathiopentacene (C27.2), are potential candidate cathodes in lithium-ion batteries (Chart 27, cf. CR2017, Section 6.2.8). In 2019, Long, Hu et al. developed a zone-melting chemical-vapor-transport (ZM-CVT) apparatus, which enabled the large-scale synthesis of single crystals of C27.1–2 in a solvent-free, continuous manner.526 The lithium-ion battery performance and cycling stability of the single crystals of both acenes were compared, with hexathiopentacene C27.2 being a better material in this respect. A two-step, three-electron lithiation mechanism was proposed for the hexathiopentacene battery instead of the typically assumed two-electron mechanism.
Chart 27. Edge-Fused Organosulfur Acenes.

In 2016, Hissler, Nyulászi et al. reported the optical and electrochemical properties of the thiophene- and phosphole-fused polycyclic arene 282.1.527 As shown in Scheme 282, the synthesis involved the Fagan–Nugent reaction of the diyne 282.2 with Cp2ZrCl2 and n-BuLi to give the zirconacycle 282.3, followed by stepwise treatment with CuI and PhPCl2, to give the crude phosphole 282.4. Subsequent oxidation with elemental sulfur gave the corresponding phosphole sulfide 282.5 in 48% overall yield. Photochemical cyclization effected the single six-membered ring closure of 282.5 to give 282.1 in 50% yield, while the doubly cyclized product was not detected. An analogue of 282.5 containing juxtaposed benzene and thiophene rings was similarly obtained.
Scheme 282. Thiophene- and Phosphole-Fused Polyarene.

Reagents and conditions: (a)527 Cp2ZrCl2, THF, −78 °C, then n-BuLi, overnight; (b) (1) CuI, 0 °C, 20 min, (2) PhPCl2, −78 °C, 15 h; (c) S8, DCM, rt, 5 h; (d) I2, propylene oxide, toluene, hν (mercury vapor lamp), 20 h; (e) FeCl3, DCM.
A synthesis of the phenanthro[9,8-bc:10,1-b′c′]dithiophene derivative 283.1 substituted with two bulky TBDMS groups was developed by Ooyama, Oshita et al. (Scheme 283).528 In the synthesis, the penultimate intermediate 283.4 originated from the sequential treatments of the bis(sulfoxide) 283.3 with TMEDA, n-BuLi and tert-butyldimethylsilyl chloride (TBDMSCl). The cyclodehydrogenation of 283.4 using FeCl3/MeNO2 gave compound 283.1 in 50% yield. This fused polyarene exhibited an intense vibronically structured absorption (λmax = 598 nm) and fluorescence (λem = 613 nm), which were red-shifted relative to those the noncyclized precursor 283.4. Notably, the Stokes shift of 283.1 (409 cm–1) was significantly smaller than that of 283.4 (3513 cm–1).
Scheme 283. Synthesis of a Phenanthrodithiophene Derivative.

Reagents and conditions: (a)528 NaIO4, THF/H2O, rt, 12 h; (b) (1) TMEDA, THF, −80 °C, 30 min, (2) n-BuLi, THF, 30 min, (3) TBDMSCl, 20 min, then rt, 12 h; (c) FeCl3, toluene/MeNO2, rt,.
In 2019, Huang, Zhao et al. reported the reactivity of a series of 2,2′-diaryl-substituted 3,3′-bithiophene derivatives toward FeCl3-mediated oxidative coupling (Scheme 284).529 It was found that the double oxidative cyclization of 284.1a–b was indeed successful, but it furnished monobromides 284.3a–b, rather than the expected dibromides 284.2a–b. When the phenyl-substituted compounds 284.4a–b were subjected to identical oxidative conditions, they produced unexpected rearrangement–cyclization products 284.5a–b. Their identities were deduced from NMR data and further confirmed by independent synthesis. The emergence of the unexpected product was rationalized under the radical cation mechanism for oxidative coupling. Specifically, the radical cation 284.4•+ was proposed to undergo C–C bond formation to give the distonic radical cation 284.7, which then isomerized to 284.8. Subsequent C–C bond cleavage generated 284.9, completing an overall 1,4-shift of the 3,4-dialkoxyphenyl group. Subsequent ring closures would then lead to the observed polyaromatic skeleton of 284.5.
Scheme 284. Synthesis of a Sulfur-Hybridized Pyracylene Derivative and an Unexpected Aryl Shift during Oxidative Cyclization,
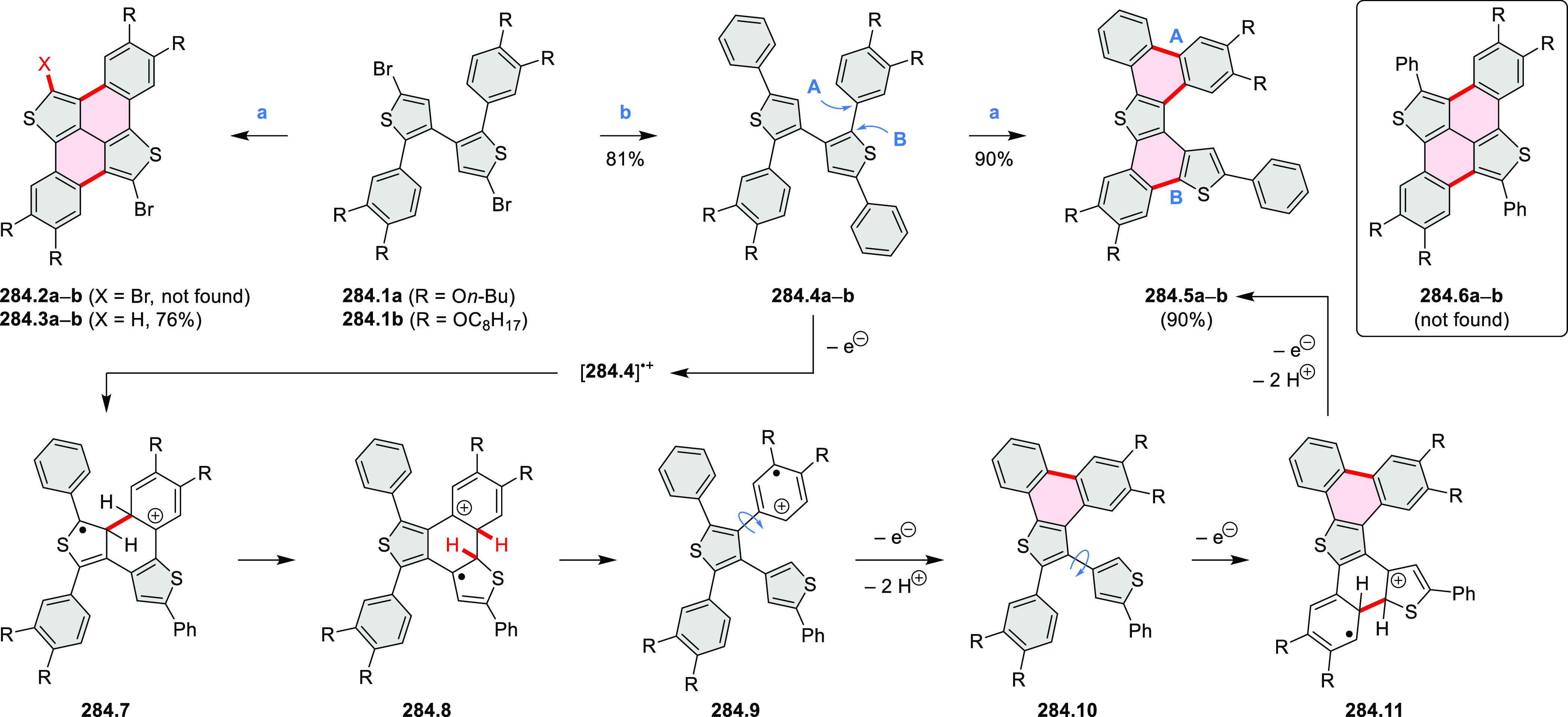
Reagents and conditions: (a)529 FeCl3, DCM/MeNO2, 5 min; (b) phenylboronic acid, K2CO3, Pd(PPh3)4, THF/H2O, N2, dark, 70 °C, overnight.
Two corresponding carbon atoms (A and B) of compounds 284.4 and 284.5 are labeled in blue for a better understanding.
6.2.9. Fused Oxaacenaphthylenes
In 2015, Wang, Han et al. reported a palladium-catalyzed C–H annulation reaction of mono- and disubstituted coumarin derivatives using diaryliodonium cations as coupling partners.530 In particular, when benzo[g]coumarin (285.1) was heated with diphenyliodonium triflate 285.2 in the presence of Pd(OAc)2 in DMF at 100 °C, the diarylation product 285.3 was formed in 70% yield (Scheme 285). Preliminary mechanistic studies suggested the involvement of double Pd(II)–Pd(IV) catalytic cycles.
Scheme 285. Palladium-Catalyzed Diarylation with Diaryliodonium Salts.

Reagents and conditions: (a)530 Pd(OAc)2 (10 mol %), DMF, 100 °C, 24 h.
6.3. Cyclopenta[cd]indene Systems
6.3.1. Fused Pyrrolo[3,2,1-hi]indoles
The pyrrolo[3,2,1-hi]indole structure is most prevalent in its [a,d] dibenzannulated form, i.e., as the indolo[3,2,1-jk]carbazole structure. Two commonly used strategies to construct this extended skeleton include intramolecular direct arylation or oxidative coupling, both under palladium catalysis. Recent examples employing the former strategy are shown in Scheme 286. As reported by Lee et al., compounds 286.3,531286.5,532 and 286.6,533 which are sky blue to violet emitters, were synthesized from the carbazole precursors possessing ortho-bromoaryl groups attached to the nitrogen atoms.
Scheme 286. Pentannulated Carbazoles via Palladium-Catalyzed Direct Arylation.
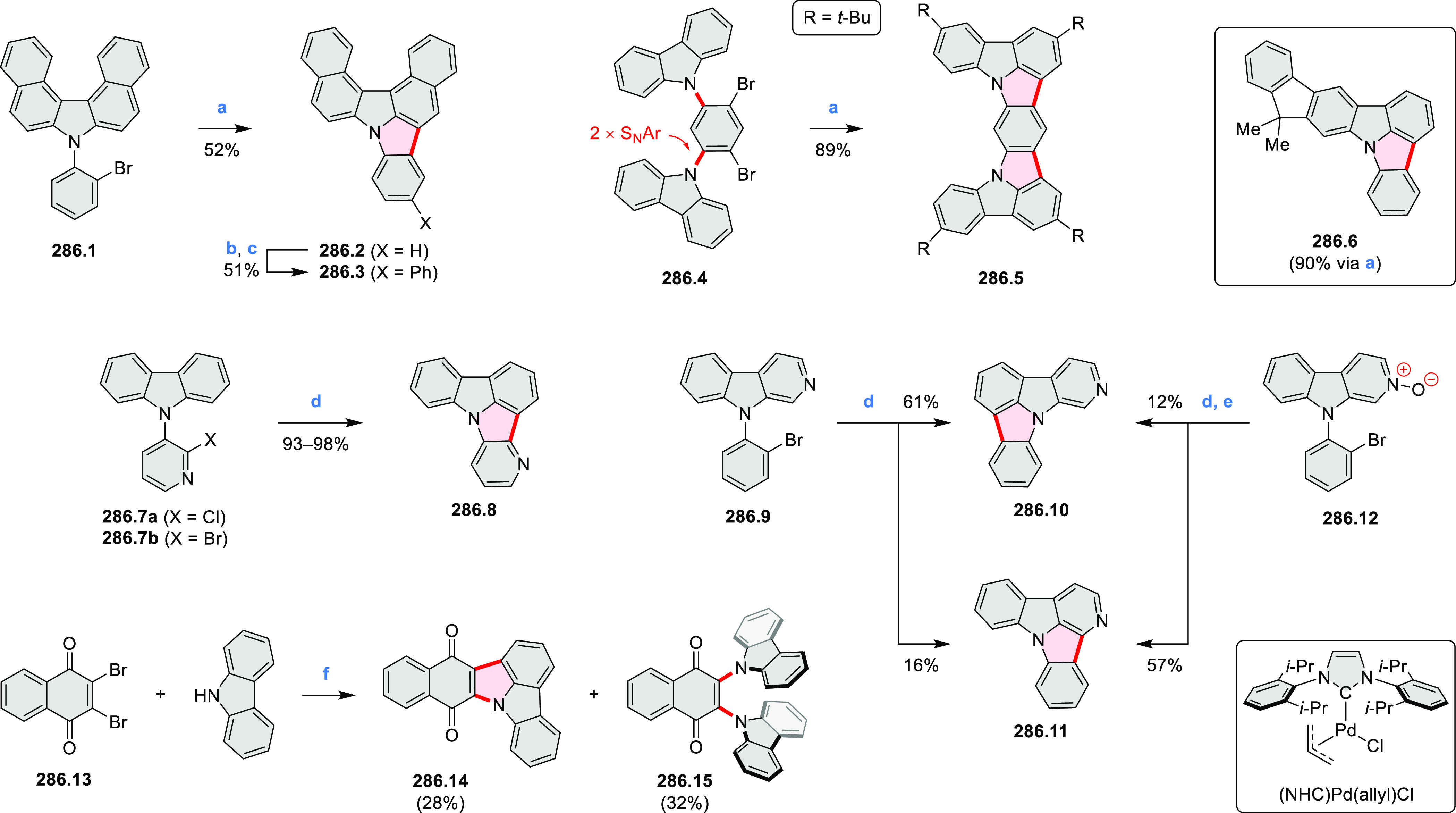
Reagents and conditions: (a)531−533 Pd(OAc)2, PPh3, [BnNEt3]Cl, K2CO3, DMA, reflux, 6 h; (b) NBS, DMF, rt, 12 h; (c) diphenylamine, Pd(dba)2, t-Bu3P, t-BuONa, xylene, reflux, 12 h; (d)534 (NHC)Pd(allyl)Cl (5 mol %), K2CO3 (2 equiv), DMA, argon, 130 °C, 4–6 h; (e) Fe powder, AcOH, 60 °C, argon, 2 h; (f)535 Pd(OAc)2, PPh3, t-BuONa, toluene, 90 °C, 24 h.
Kautny showed that the use of (NHC)Pd(allyl)Cl as the catalyst granted access to a series of aza-substituted indolocarbazoles.534 For example, 286.8 was obtained in good yield from the 9-(2-halo-3-pyridyl)carbazoles 286.7a–b using the optimized conditions. For the unsymmetrical carbazole 286.9, the C–Br bond preferentially reacted with the benzo nucleus to give 286.10 (61% yield), with the regioisomer 286.11 (16% yield) being the minor product. Interestingly, when the N-oxide 286.12 was subjected to the same cyclization followed by reduction, the yields of 286.10 and 286.11 became 12% and 57%, respectively, indicating an electronic control during cyclization.
Langer et al. reported the palladium-catalyzed reaction of 2,3-dibromo-1,4-naphthoquinone (286.13) with carbazole to give the annulation product 286.14 in 28% yield, and the 2-fold C–N coupling product 286.15 in 32% yield.535
A route to the doubly pentannulated acridone derivative 287.5, developed by Deng and Zhang in 2019, started with the singly fused acridone 287.2, which was available from an intramolecular Friedel–Crafts acylation step (Scheme 287).536 Attempted direct conversion of 287.2 into the target molecule 287.5 was unsuccessful because of the rigidity of the fused molecular skeleton. Thus, the ketone functionality in 287.2 was quantitatively reduced by borane to furnish the more flexible molecule 287.3, which proved susceptible to the subsequent palladium-catalyzed cyclization, yielding 287.4. Finally, dichromate oxidation of 287.4 yielded the target ketone 287.5 in 95% yield. The bowl-shaped geometry of 287.5 was confirmed by X-ray crystallography.
Scheme 287. Stepwise Cyclization Approach toward Highly Fused Acridones.
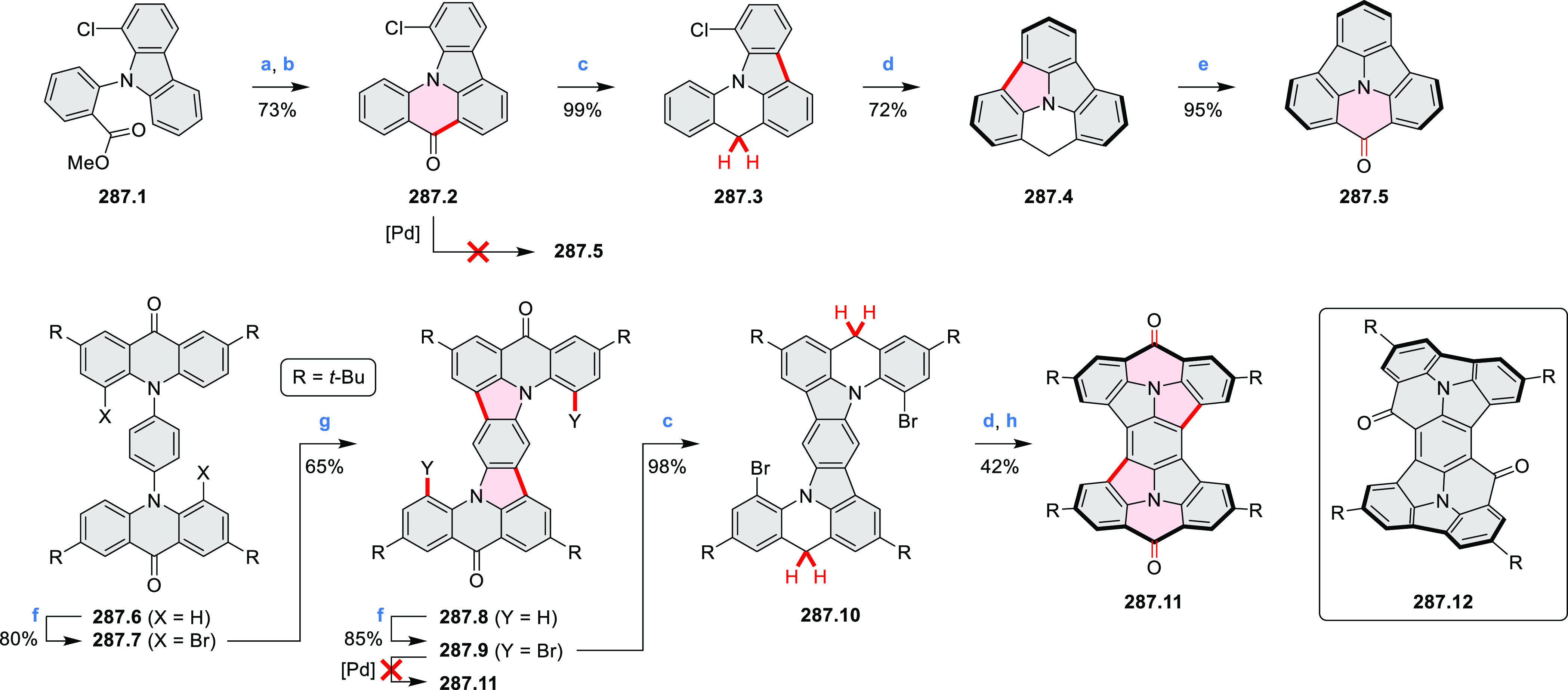
Reagents and conditions: (a)536 NaOH, EtOH/H2O, reflux, 3 h; (b) polyphosphoric acid, 180 °C, 4 h; (c) BH3·THF, THF, reflux, 4 h; (d) Pd(OAc)2, PCy3·HBF4, K2CO3, DMA, 170 °C, 48 h; (e) Na2Cr2O7, AcOH, reflux, 1 h; (f)537 Br2, DCM, rt, 48 h; (g) Pd(OAc)2, PCy3·HBF4, K2CO3, DMA, 130 °C, 24 h; (h) DDQ, DCM/dioxane/H2O, 0 °C to rt, 16 h.
In 2020, Zhou and Zhang reported an analogous approach toward the highly fused bisacridone 287.11 (Scheme 287).537 The synthesis began with selective 2-fold bromination of the bisacridone 287.6 to give 287.7, followed by double cyclization. The resulting 287.8 was subjected to another dibromination step, furnishing the dibromide 287.9. Again, the rigid molecule 287.9 could not directly cyclize to give 287.11. Instead, the reduction–cyclization–oxidation sequence allowed isolation of the target diketone 287.11 in 41% yield over three steps. In 2021, Song and Zhang reported the isomeric nanoboat 287.12 with a lower-symmetry fusion mode.538 The curved, boat-like structures of both 287.11 and 287.12 were established by X-ray crystallography. The absorption profile of the latter nanoboat is bathochromically shifted from that of the former one. Based on DFT results, the HOMO levels of both molecules do not differ significantly, but the less symmetrical molecule 287.12 possesses a lower LUMO level (−2.94 eV) than does 287.11 (−2.50 eV). This difference in HOMO–LUMO gap is also reflected in the red-shifted fluorescence wavelength of 287.12 (524 nm) relative to that of 287.11 (475 nm).
In 2015, Patureau et al. reported optimized conditions for the oxidative cyclization of 9-arylcarbazoles such as 288.1a–c to give the corresponding indolocarbazoles 288.2a–c in 38–70% yield (Scheme 288).539 In addition to palladium(II) pivalate, the presence of Ag2O, CuO, and an oxygen atmosphere was found essential to achieve the conversion. The trisubstituted products 288.3a–c were also obtained from their respective precursors. However, the dimethoxy-substituted indolocarbazole 288.3d could not be obtained under those conditions. Subsequently, the same group developed a synthesis of N-aryl-substituted π-extended carbazole derivatives, such as 288.5a–d, directly from the corresponding aryl(2-naphthyl)amines (Scheme 288).540 The authors showed that a modified set of reaction conditions, with Pd(OAc)2 in place of palladium(II) pivalate, successfully induced pentannulation of 288.5a–d to give 286.2 and 288.6b–d in 17–62% isolated yield.
Scheme 288. Pentannulated Carbazoles via Palladium-Catalyzed Oxidative Cyclization.
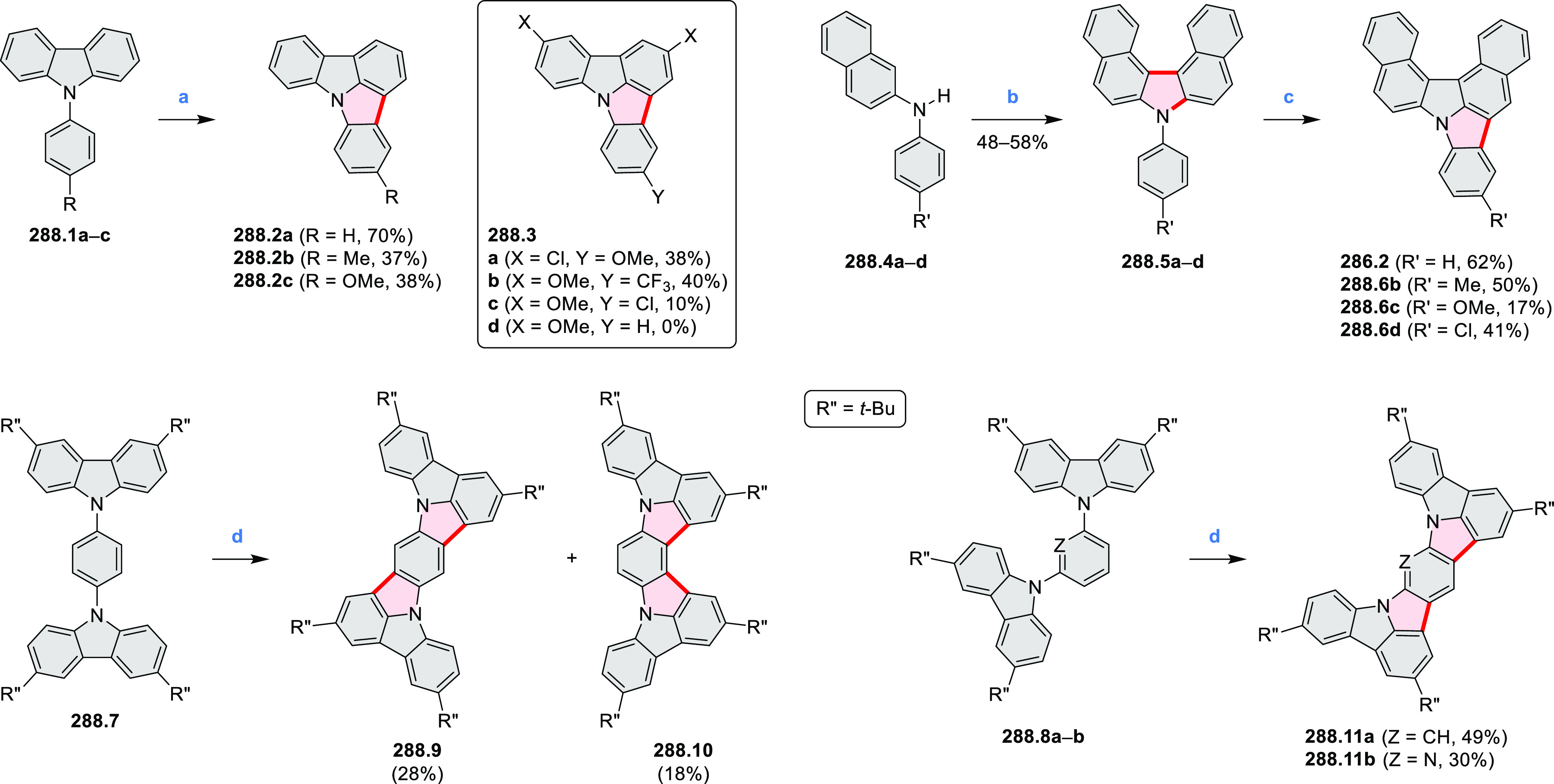
Reagents and conditions: (a)539 palladium(II) pivalate (10 mol %), Ag2O (1.2 equiv), CuO (1.2 equiv), pivalic acid, O2 (1 atm), 130 °C, 24 h; (b)540 Ag2O (1 equiv), toluene/cumene/AcOH, air, 60 °C, 2 h; (c) Pd(OAc)2 (10 mol %), Ag2O (1.2 equiv), CuO (1.2 equiv), pivalic acid, air, 130 °C, 3 days; (d)541 palladium(II) trifluoroacetate (30 mol %), AgOAc (6.0 equiv), pivalic acid, 160 °C, 48 h.
For oxidative cyclization of di-9-carbazolyl-substituted derivatives 288.7–8, Miura et al. employed palladium(II) trifluoroacetate as the catalyst and silver(I) acetate as the oxidant (Scheme 288).541 The para-dicarbazolyl substrate 288.7 provided the isomeric doubly cyclized products 288.9 and 288.10 in 28% and 18% yield, respectively. The meta-dicarbazolyl-substituted benzene 288.8a and pyridine 288.8b afforded the corresponding double fusion products 288.11a–b in 49% and 30% yield, respectively. Interestingly, the corresponding ortho-dicarbazolylbenzene produced a seven-membered ring under these conditions (Scheme 301, Section 6.4.4).
Scheme 301. Syntheses of Fused 1,4- and 1,5-Diazocines.

Reagents and conditions: (a)541 palladium(II) trifluoroacetate (30 mol %), AgOAc (6.0 equiv), pivalic acid, 160 °C, 48 h; (b)567 neat, 145–150 °C.
The pyridodiindole 289.1 bearing a 2-bromophenyl substituent was obtained in 85% yield during substrate screening for three-component condensation developed by Xu, Li, et al. (Scheme 289; for synthesis and mechanism, see Scheme 276, Section 6.2.6).519 Compound 289.1 could be cyclized in an Ullmann-type reaction under microwave heating to give the peri-fused compound 289.2 in 63% yield. In contrast to syntheses described earlier in this section, the formation of the 5–5–6 fusion is completed with a C–N bond formation rather than with a C–C coupling.
Scheme 289. Formation of [5,5,6] Fusion via Ullmann-Type Reaction.

Reagents and conditions: (a)519 Cs2CO3, CuI, DMF, microwave, 120 °C, 10 min.
A range of dyes designed for optoelectronic applications have been prepared from the multistep functionalization of unsubstituted indolo[3,2,1-jk]carbazole (Chart 28). In 2015, Lumpi, Baumgartner et al. reported a series of P-substituted dithienophosphole oxides, including the indolocarbazole-containing analogues C28.1a–b.542 Among the reported molecules, only C28.1a displayed vibronically well-resolved phosphorescence, and possessed a high triplet energy (2.87 eV), making it suitable as a potential host material for phosphorescent OLED (PhOLED). Similarly, compounds C28.2a,3 reported by Lumpi, Chen et al. in 2016 were also studied in terms of PhOLED applications.543 Compounds C28.2b, C28.4a–b,544 and C28.5a–b545 bearing various donor units (i.e., carbazole, carbazolylcarbazole, acridine and diphenylamino groups) were developed by Lee et al. as sky blue to deep blue TADF emitters, respectively. The same group reported that the derivative C28.6 bearing a 2-cyanoindolo[3,2,1-jk]carbazole acceptor and a 9-carbazolyl donor group was a green TADF emitter,546 while the dicarbazolyl analogues C28.7a–b were deep blue TADF emitters.547 Related designs include C28.8, a greenish blue TADF emitter, reported by Lee, Hong et al.,548 and the blue phosphorescent indolocarbazole–benzothiophene conjugates C28.9a–b.549 The use of indolocarbazoles 231.10a–b in perovskite solar cells was explored in 2019 by Wang, Jiang et al.550
Chart 28. Substituted Indolocarbazoles.
Indolocarbazole dyads connected either via a direct C–C bond or through an aromatic linker are shown in Chart 29. The dimer C29.1 was prepared by Lumpi, Chen et al. as part of their PhOLED study mentioned above.543 In 2017, Bintinger, Horkel et al. reported the use of thiophene-based linkers to construct indolocarbazole dyads such as C29.2a–b and C29.3, for potential application in OLED devices.551 Compound C29.4 incorporating the m-phenylene linker, obtained by Lee et al., was used as a host for green PhOLED devices.552
Chart 29. Covalently Linked Bis(indolocarbazole)s.

6.3.2. Fused Pyrrolo[2,1,5-cd]indolizines and related systems
In 2017, Charette et al. reported a new general synthesis of imidazo[2,1,5-c,d]indolizine fluorophores, e.g. the naphtho[a]-fused derivative 290.6 (Scheme 290).553 The synthesis started with treating the commercially available 6-bromopicolinaldehyde (290.1) with hydroxylammonium sulfate to provide the oxime 290.2, followed by reduction by zinc powder to give the ammonium salt 290.3·AcOH quantitatively over two steps. The final three steps, i.e., acylation, electrophilic cyclization, and palladium-catalyzed cyclization, provided the naphtho[a]-fused compound 290.6 and a range of its analogues with or without ring fusions on the [a] side. The last three steps had been reported earlier by the same group for the preparation of the benzo[a]-fused analogue. This improved method avoided the use of the expensive amine 290.3 as the starting material. Emission properties of these fluorophores could be tuned over the entire visible range by changes of substitution.
Scheme 290. π-Extended Imidazole- and 1,3-Azaphosphole-Fused Indolizine Derivatives.
Reagents and conditions: (a)553 (HONH3)2SO4, NaOAc, EtOH, rt, 16 h; (b) Zn, AcOH, rt, 10 min; (c) 1-naphthylacetyl chloride, Et3N, DCM, 0 °C, then rt, 16 h; (d) Tf2O, 2-methoxypyridine; DCM, 50 °C, 16 h; (e) Pd2(dba)3 (cat.) t-Bu3P·HBF4 (cat.), K2CO3, DMF, 110 °C, 16 h; (f)554n-BuLi, TMEDA, THF, 0 °C, 1 h; (g) PhPCl2, 0 °C, 10 min, then rt, 30 min; (h) aq. H2O2 (30%), 30 min; (i) S8, 30 min.
In 2018, Mathey, Duan, Chen et al. described a series of 1,3-azaphosphole oxides and phosphides, including the π-extended derivatives 290.10a–b, which were prepared in a one-pot procedure from the pyrrolo[1,2-a]quinoline precursor 290.7.554 Specifically, the bay region of the pyrrolo[1,2-a]quinoline skeleton was dilithiated with n-BuLi in the presence of TMEDA to give 290.8. Subsequent treatment with PhPCl2 generated the 1,3-azaphosphole unit in the intermediate 290.9. Eventual addition of H2O2 or elemental sulfur afforded the target oxide 290.10a (66% yield) and sulfide 290.10b (55% yield), respectively. Compound 290.10a was shown to be useful as an emitting dopant in OLEDs and as a bioimaging dye.
Unexpected formation of a dibenzo-fused triazino[2,1,6-cd]pyrrolizine ring system was reported by Li et al. (Scheme 291).555 When the known compound 291.2a, obtained by trimerization of phthalonitrile (291.1a), was heated in a ceramic crucible at 230 °C, the “phthalorubine” 291.3a was isolated in 47% yield. Likewise, using 4-methoxyphthlonitrile (291.1b) or 4-(tert-butylthio)phthalonitrile (291.1c) as the starting materials yielded phthalorubines 291.3b–c of unspecified regiochemistry. Besides, the cyclization of 291.2a–c could proceed in a refluxing mixture of acetic acid and ethanol, affording the amidinium salts 291.7a–c in 40–51% yield. When 4,5-di(9-carbazolyl)phthalonitrile was used as the starting material, only the latter milder conditions could mediate the cyclization to give the amidinium salt 291.6. The study was then extended to metal coordination chemistry. First, the two cyano groups in 291.3a were reductively removed using triisopropylsilane and a rhenium(I) catalyst. The resultant compound 291.4 acted as a C,N bidentate ligand toward iridium(III), forming the mononuclear complex 291.5. Second, 291.7a was annulated with acetylacetone to give the pyrimidinyl-substituted derivative 291.8, which was then reacted with K2PtCl4 to give the inherently chiral complex 291.9.
Scheme 291. Dibenzannulated Derivatives of Triazino[2,1,6-cd]pyrrolizine.
Reagents and conditions: (a)555t-BuOK (1 equiv), DMF, 0 °C, 1 h; (b) neat, N2, 230 °C, 3 h; (c) [Rh(cod)Cl]2 (5 mol %), P(Oi-Pr)3 (0.1 equiv), i-Pr3SiH (4 equiv), ethylcyclohexane, reflux, 15 h; (d) IrCl3, dry ethylene glycol, N2, reflux, 24 h; (e) EtOH/AcOH (1:1), reflux, 24 h; (f) acetylacetone, pyridine, reflux, 24 h; (g) K2PtCl4, 2-ethoxyethanol/H2O (3:1), N2, 80 °C, 24 h.
6.4. peri-Fused Seven- and Eight-Membered Rings
6.4.1. peri-Fused Cycloheptatrienes
In 2017, Kwong et al. reported a palladium-catalyzed three-component reaction of aryl iodide, 8-bromo-1-naphthoic acid and norbornadiene, which provided access to carbo- and heterocyclic frameworks containing seven-membered rings (Scheme 292).556 For example, 3-iodobenzo[b]thiophene could be converted into compound 292.1 in 95% yield under optimized conditions. Likewise, 4-iododibenzofuran could be transformed into 292.2 in 85% yield. On the basis of DFT calculations, the authors proposed a Pd(0)–Pd(II)–Pd(IV) catalytic cycle for the reaction.
Scheme 292. Three-Component Synthesis of Fused Cycloheptatrienes.

Reagents and conditions: (a)556 Pd(OAc)2 (5 mol %), PCy3 (15 mol %), Cs2CO3, 1,4-dioxane, 130 °C, 18 h.
In 2016, Samineni, Srihari, and Mehta reported the application of a ring-expansion strategy to the total synthesis of the natural product radermachol (293.5, Scheme 293, for earlier work on radermachol, see CR2017, Section 6.4.1).557 The aryne precursor 293.1 was synthesized in four steps from 1,4-dimethoxynaphthalene. The naphthyne generated from 293.1 successfully inserted into 1,3-indanedione to give 293.2 in 78% yield. Subsequent C-acylation with 3-methylcrotonoyl chloride furnished 293.3. Next, a demethylation–cyclodehydration sequence generated the furo fusion in radermachol, which was obtained in 65% yield over two steps.
Scheme 293. Total Synthesis of Radermachol.

Reagents and conditions: (a)557 1,3-indanedione, CsF, MeCN, 75–80 °C, 1.5 h; (b) 3-methylcrotonoyl chloride, AlCl3, 55 °C, 2 h; (c) Me3SiI, CDCl3, 25 °C, 27 h; (d) TsOH, benzene, reflux, 5 h.
6.4.2. Fused Azepines and Diazepines
The one-pot N–H/C–H coupling reaction reported by Li et al. (for details, see Scheme 45, Section 3.1.2) was also applicable to the annulation of 5H-dibenzo[b,f]azepine (294.1).76 As shown in Scheme 294, the use of either 1,8-dibromo- or 1,8-diiodonaphthalene as the coupling partner under the optimized reaction conditions could lead to the [de]annulated dibenzoazepine 294.2 in 79–83% yield. The nonplanar geometry of 294.2 was established by X-ray crystallography. In 2016, Jiao et al. reported the palladium-catalyzed annulation of the dibenzoazepine-based carbamic chloride 294.3 with 2-iodotoluene to give 294.4 in 41% yield.558 In addition to the precatalyst, the phosphine ligand and the base, the addition of norbornene was necessary. DFT calculations suggested a Pd(0)–Pd(II)–Pd(IV) catalytic cycle enabled by the participation of norbornene. The two new C–C bonds were formed by C3 acylation of 2-iodotoluene, followed by C4 arylation of dibenzoazepine. As shown by Doucet, Soulé et al., dibenzo[b,f]azepines can be pentannulated using the palladium-catalyzed direct C10-arylation with aryl bromides (Scheme 294).559 In a particular example using the optimized conditions, the diazepine 294.5 and 1,2-dibromobenzene were heated in DMA in the presence of PdCl(C3H5)(dppb) and KOAc. The indeno-fused product 294.6 was isolated in 50% yield. The reaction proceeded via two successive C–H bond activations.
In 2016, Shibata et al. reported the gold(I)-catalyzed cycloisomerization of 9-(2-alkynylphenyl)carbazoles, such as 295.1–g, to give the a series of carbazole-fused azepine derivatives including 295.2a–g (Scheme 295).560 In the case 295.1a, the use of AuCl(PPh3) and AgSbF6 at a high temperature led to the cyclized product 295.2a in 20% yield. A much higher yield (99%) could be achieved with the use of AuCl[P(C6F5)3] and AgOTf at rt. The authors examined the regioselectivity of this reaction with several unsymmetrical carbazoles. For example, the cycloisomerization of 295.3 occurred preferentially at the electron-rich aromatic ring to give the azepine 295.4, while the regioisomer 295.5 could not be detected.
A different type of direct cyclization employed precursors 295.5a–e obtained in the already mentioned four-component condensation reported by Xu, Chen et al. (Scheme 295, cf. Scheme 275, section 6.2.6).518 An Ullmann-type cyclization of the pendant 2-bromophenyl substituent led to the corresponding fused benzimidazoisoquinolines 295.6a–e in 86–95% yield. Similarly, compound 295.7 (Scheme 274, Section 6.2.5) was converted into a fused diazepine derivative 295.8 in a two-step acylation sequence, as shown in Scheme 295.517
Phenanthroimidazole 296.1, synthesized by Skonieczny and Gryko in a three-component condensation discussed in Section 4.7.2 (Scheme 165) was found to be susceptible to photoinduced intramolecular direct arylation of the 4-tert-butylphenyl ring, generating the azepine-fused product 296.2 in 47% yield (Scheme 296).561 This protocol worked successfully without using any sensitizer or base.
Scheme 296. peri-Naphtho-Fused Azepine Derivatives via Heptannulation.
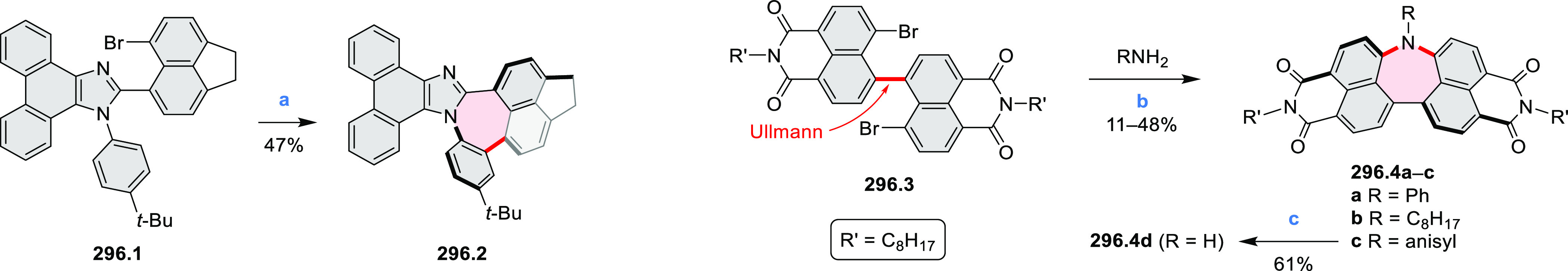
Reagents and conditions: (a)561hν (254 nm, 2 × 8 W), DCM, rt; (b)562 Pd2(dba)3·CHCl3, BrettPhos, t-BuONa, toluene, 80 °C, 9 h; (c) anisole, TFA, toluene, 70 °C, 4 h.
In 2019, Shinokubo, Akutagawa, Kim et al. reported a series of dinaphthoazepine diimides, such as 296.4a–d, which can be considered ring-expanded analogues of perylene diimides (Scheme 296).562 The precursor 296.3 was prepared from the intermolecular Ullmann-type coupling of the corresponding bromoiodonaphthalene monoimide. Buchwald–Hartwig amination of 296.3 with various primary amines yielded the fused azepines 296.4a–c in 11–48% yield. In particular, the 4-methoxybenzyl (anisyl) group in 296.4c could be cleaved by excess anisole and trifluoroacetic acid to give 296.4d in 61% yield. These fused azepines possessed flexible, nonplanar conformations, and exhibited ambipolar redox activity.
In 2016, Anand et al. reported the oxidative cyclodimerization of the indolylpyrrolylmethane 297.1 upon successive additions of DDQ and Cu(OAc)2 to give the diazaheptalene derivative 297.2 in 1% yield (Scheme 297). While low-yielding, this transformation is notable for creating a sequence of six linearly fused rings of different sizes in just one synthetic step. Under identical conditions, the isomer of 297.1 containing a 2-pyrrolyl group yielded structurally different products. In the solid state, the molecules of 297.2 were planar and stacked at a distance of ca. 3.3 Å.
Scheme 297. Fused Diazaheptalene via Copper-Assisted Oxidative Cyclodimerization.

Reagents and conditions: (a)563 (1) DDQ, THF, 1 h, (2) Cu(OAc)2, overnight.
6.4.3. Oxa- and Thia- 7-Membered Rings
In 2017, Khlebnikov et al. reported the construction of the indole-fused dibenzoxazepine derivative 298.3 (Scheme 298).564 The aziridine 298.1 reported by the same group was subjected to cycloaddition with dimethyl maleate in refluxing toluene to give the pyrrolidine 298.2 stereoselectively in 91% yield. Exposure of 298.2 to AIBN and tributyltin hydride at a high temperature brought about the free radical cyclization that led to the indole-fused dibenzoxazepine 298.3 in 56% yield. When the pyrrolidine 298.2 was replaced by the previously reported pyrroline 298.4 or pyrrole 298.5, the same radical cyclization could proceed to give the same product 298.3 in 68–72% yield using a lesser amount of AIBN and shorter reaction time.
Scheme 298. Synthesis of Indole-Fused Dibenzoxazepine via Radical Cyclization.
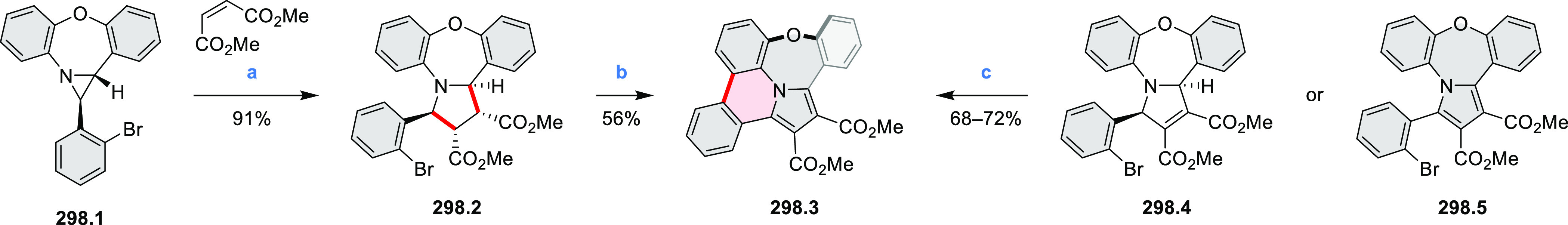
Reagents and conditions: (a)564 toluene, reflux, 8 h; (b) AIBN (2.2 equiv), Bu3SnH (0.5 equiv), toluene, 80 °C, 7 h; (c) AIBN (1.1 equiv), Bu3SnH (0.5 equiv), toluene, 80 °C, 4 h.
In 2020, Shinokubo, Fukui, Yamada et al. reported the synthesis and properties of the dinaphthothiepine diimides 299.2a–b (Scheme 299),565 analogous to the azepines 296.4a–d mentioned above. The construction of the fused thiepine ring was accomplished by the SNAr reaction of the dibromides 296.3 and 299.1 by sodium sulfide using DMF as the solvent. 299.2a–b could be converted into the corresponding sulfoxides 299.3a–b or sulfones 299.4a–b depending on the oxidation conditions. Both 299.2a and the sulfoxide 299.3a could undergo sulfur extrusion upon electron injection by decamethylcobaltocene followed by quenching with chloranil to give the perylene diimide 299.5. Besides, the same extrusion reactions could take place upon electrochemical reduction and photoirradiation, as substantiated by voltammetric and spectroscopic measurements. In contrast, photoirradiation of the sulfone 299.4a did not lead to the PDI product 299.5.
Scheme 299. Synthesis and Reactivity of Dinaphthothiepine Diimides.
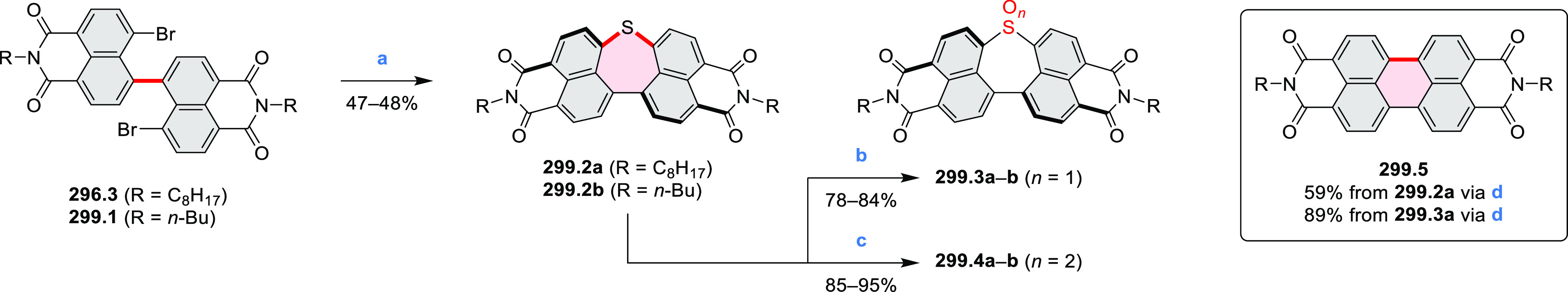
Reagents and conditions: (a)565 Na2S, DMF, 60 °C; 3 h; (b) m-CPBA, DCM, −80 °C, 30 min, then rt, 1 h; (c) aq. H2O2, Na2WO4·2H2O, MeN(n-C8H17)3·HSO4, phenylphosphonic acid, toluene/H2O, 50 °C; (d) (1) Co(Cp*)2, DCM, rt, 20 min, (2) chloranil.
6.4.4. Aza-, Oxa- and Thia-8-Membered Rings
In 2020, Mastalerz et al. reported the synthesis and of the chiral monkey saddle 300.3 bearing three fused azocine rings (Scheme 300).566 The key intermediate in the synthesis is the tribromotruxenone 300.1, which was first triply cross-coupled with 2-aminophenylboronic acid hydrochloride (300.2), followed by imine bond formation to give the triply cyclized product 300.3 in 45% yield. In the racemic single crystal, the chiral monkey saddle conformation was confirmed for both the (Ra,Ra,Ra)- and (Sa,Sa,Sa)-enantiomers of 300.3. The authors managed to separate the enantiomers by chiral HPLC, and the inversion barrier and half-life of enantiopure 300.3 were found to be 113 ± 6 kJ mol−1 and 7.7 ± 0.1 h, respectively.
Scheme 300. Synthesis of a Triaza Monkey Saddle.

Reagents and conditions: (a)566300.2, Pd2(dba)3, t-Bu3P·HBF4, K2CO3, THF/H2O (1:1), 80 °C, 48 h; (b) AcOH/CHCl3 (1:10), 80 °C, 18 h.
In addition to the meta- and para-dicarbazolylbenzenes discussed above (see Scheme 288section 6.3.1), Miura et al. also tested the reactivity of the ortho isomer 301.1 toward the same oxidative cyclization (Scheme 301).541 In the latter reaction, the fused 1,4-diazocine 301.2 was isolated in 13% yield, and its twisted molecular structure was unequivocally established by X-ray crystallography. The feasibility of eight-membered ring closure was attributed to the proximity of the two carbazole units in 301.1. A different approach to peri-fused diazocines was described by Deeb et al., who showed that heating an equimolar mixture of 1,8-naphthalenediamine and the ester-containing coumarin 301.3 could furnish the annulation product 301.4 which exists in equilibrium with the 1,5-diazocine tautomer 301.4′.567
In 2017, Igarashi, Tobisu, and Chatani reported a general, gram-scale synthesis of a series of cyclic diarylborinic acids, including the cyano-substituted analogue 302.1 (Scheme 302).568 Under Pd-catalyzed cross-coupling conditions, compound 302.1 underwent an unusual ring-opening annulation with 1,8-dibromonaphthalene, yielding the fused oxocine derivative 302.2 in 48% yield. Kumar et al. showed the formation of a fused oxocine ring in an iodonium-based annulation reaction (Scheme 302).569 When diphenyleneiodonium triflate (302.3) and quinolin-4(1H)-one (302.4) were heated in the presence of Pd(OAc)2, the C–H arylation product 302.5 and the annulation product 302.6 were isolated in yields of 20% and 70%, respectively. The identity of the oxocine 302.6 was supported by NMR and mass spectral data, and this product was believed to originate from the C–O bond formation of the minor product 302.5.
Scheme 302. Syntheses of peri-Fused Oxocines via Annulative Coupling.

Reagents and conditions: (a)568 Pd2(dba)3, t-Bu3P·HBF4, Cs2CO3, H2O/t-amyl alcohol, 100 °C, 48 h; (b)569 Pd(OAc)2 (5 mol %), AcOH, 100 °C, 12 h.
In 2021, Shinokubo, Fukui et al. reported the preparation of the dinaphthodithiocine diimide 303.2 (Scheme 303),570 a larger expanded congener of thiepine diimides 299.2a–b. The synthesis used the known dibromonaphthalene imide 303.1, which was treated with Na2S in DMF to give the annulation product 303.2 in moderate yield. As revealed by X-ray crystallography, molecules of 303.2 assumed a nonplanar V-shaped geometry with a dihedral angle of 67° between the two naphthalene units. This conformation was shown by VT-NMR experiments and DFT calculations to be sufficiently rigid, with an inversion barrier greater than 30 kcal mol−1.
Scheme 303. Synthesis of Dinaphthodithiocine Diimide.

Reagents and conditions: (a)570 Na2S (1.2 equiv), DMF, rt, 3 h.
7. Macrocyclic Systems
7.1. [b]-Fused (β–β-Fused) Porphyrinoids
Fusion along the β–β bonds of pyrrole subunits is one of the most frequently used methods of peripheral extension used in porphyrin chemistry. Below we summarize the most recent examples of such systems containing peri-fused moieties (for earlier developments, see CR2017, Section 7.1). Variously substituted acenaphthylene-fused dithiaporphyrins were reported by Hogan and Sutherland (Scheme 304).571 Thiophene building blocks were prepared by using the Hinsberg condensation between dimethyl 2,2′-thiodiacetate and acenaphthoquinone 304.1 to give the diester 304.2, which was transformed into 304.5a–c in three steps. Condensation of these dicarbinols with pyrrole and the subsequent oxidation by DDQ afforded acenaphthylene-fused dithiaporphyrins, 304.6a–c. Compounds 304.6a and 304.6b displayed broad visible light spectrum absorptions extending into the near-IR range with the onsets of absorption at 1000 and 1100 nm, respectively. The curvature and low oxidation potentials of these dithiaporphyrins were reflected in their interactions with [60]fullerene.
Scheme 304. Acenaphthylene-Fused Dithiaporphyrins.
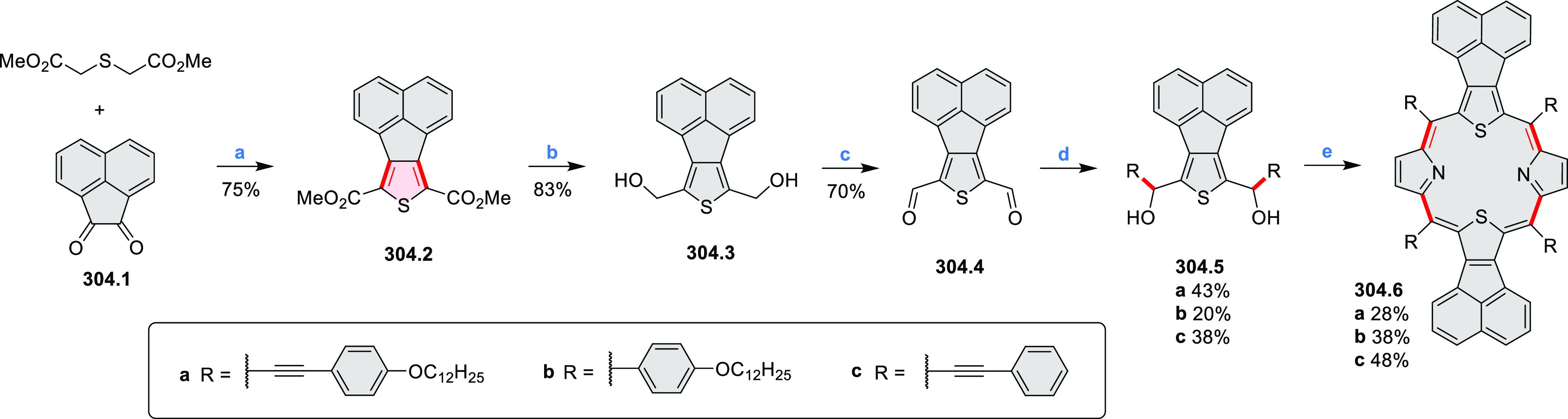
Reagents and conditions: (a)571 (1) NaOMe, THF, (2) MeOH, H2SO4; (b) LiAlH4, diethyl ether; (c) MnO2, DCM; (d) n-BuLi, TMEDA, 304.5a from 1-ethynyl-4-dodecyloxybenzene; 304.5b from 1-bromo-4-dodecyloxybenzene; 304.5c from ethynylbenzene; (e) (1) BF3·Et2O, pyrrole, (2) DDQ.
In 2020, Lash and co-workers reported the incorporation of dihydropyracylene-fused subunits into extended porphyrin chromophores.572 The synthesis of 305.3a was performed using the typical MacDonald condensation with tripyrrane 305.1 and dialdehyde pyrrole 305.2 (Scheme 305). Subsequent metalation afforded the nickel(II) complex 305.3b. In addition, tetraphenyl-tetrakis(dihydropyracylo)porphyrin 305.5a was also prepared by reacting dihydropyracylopyrrole 305.4 with benzaldehyde in the presence of BF3·Et2O, followed by oxidation with DDQ, which gave the desired product in 41% yield. The UV–vis absorption spectrum of 305.5a did not show any significant shift in the Soret band peak compared to the tetraacenaphthoporphyrin derivative (309.4a, R = H, meso-aryl = phenyl), however, the doubly protonated 305.5a produced a red-shift of 26 nm in the Soret band, apparently caused by the additional five membered rings.
Scheme 305. Dihydropyracyloporphyrins.

Reagents and conditions: (a)572 (1) TFA, DCM, 2 h, (2) DDQ, 1 h; (b) Ni(OAc)2, 16 h, reflux; (c) benzaldehyde, BF3·Et2O, CHCl3, N2, 20 min, (2) DDQ, 20 min.
In 2016, Okujima, Mack, and Kobayashi reported a family of porphyrins containing one fused benzofluoranthene moiety accompanied by a variety of other fused subunits (Scheme 306).573 The synthesis was done via 3 + 1 condensation approach fusing a tripyrrane, e.g. 306.1, and a bicyclo[2.2.2]octadiene (BCOD) pyrrole, such as 306.2, which afforded the corresponding BCOD-fused porphyrin, e.g. 306.3. The π-extended porphyrins 306.5–11 were then obtained from the corresponding BCOD precursors via retro-Diels–Alder reactions in near-quantitative yields. The introduction of additional fused-ring moieties at β positions of pyrrole rings and variation of fusion patterns along the x or y axes of the macrocycle significantly affected the electronic structure of these porphyrins. Among the derivatives, the phenanthroline and acenaphthylene-fused porphyrins 306.7 and 306.10 had the narrowest HOMO–LUMO gaps and their lowest absorption bands (Q bands) were shifted into the red/NIR region of the spectrum (close to 700 nm).
Scheme 306. [b]-Condensed Porphyrins Containing “Remote” peri-Fusion Points.
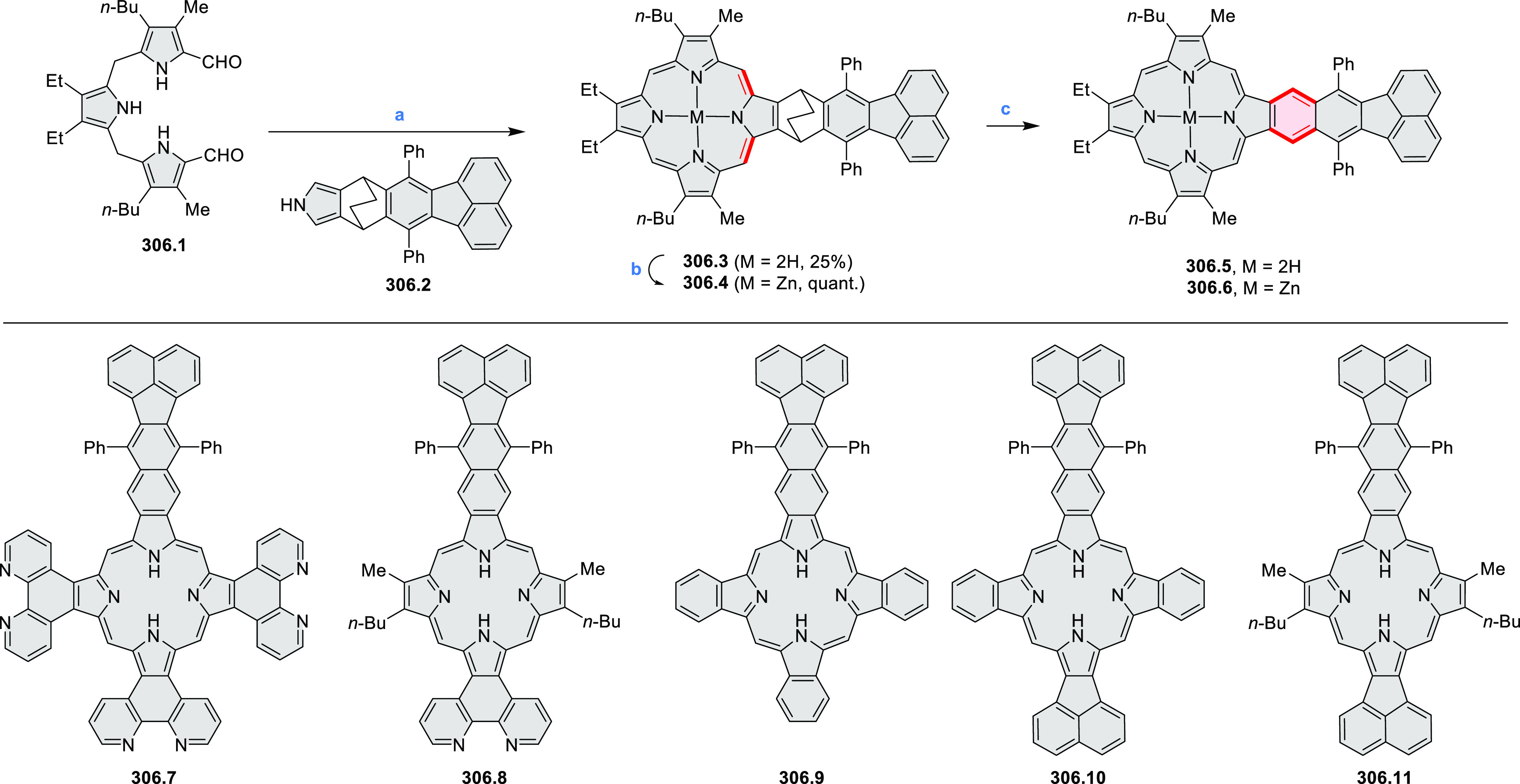
Reagents and conditions: (a)573 (1) 306.2, TFA, CHCl3, (2) Et3N, DDQ; (b) Zn(OAc)2·2H2O, CHCl3/MeOH; (c) Δ, 350 °C, reduced pressure, 0.5 to 3 h, quantitative yields.
The “3 + 1” condensation strategy was further used by Lash and co-workers in the first syntheses of porphyrin analogues incorporating pyrene units (Scheme 307).574 Specifically, 307.3 was obtained via acid-catalyzed condensation of the pyrene dialdehyde 307.1 with the tripyrrane 307.2, whereas thiapyreniporphyrin 307.7 was prepared via Lewis acid-catalyzed condensation of the thiophene dicarbinol 307.6 with the pyrene tripyrrane 307.5. In the free base form, 307.3 and 307.7 were found to be nonaromatic, but showed significant diatropicity upon protonation, as also shown by NICS calculations and ACID plots.
Scheme 307. Synthesis of [b]-Fused Porphyrins Containing Pyrene Subunits.

Reagents and conditions: (a)574 (1) TFA, DCM, N2, 5 h, (2) DDQ, 3 h; (b) Pd(OAc)2, CHCl3/CH3CN, reflux, 2 h; (c) (1) BF3·Et2O, DCM, N2, 3 h, (2) DDQ, 5 h.
In 2016, our group reported a family of band gap-tunable pyrroles structurally related to rylene dyes which were used as building blocks for the synthesis of electron-deficient porphyrin–ryleneimide hybrids (Scheme 308).22 The naphthalenediamide-substituted (NDA) macrocycle, 308.2a was synthesized from the monopyrrole 308.1a under modified Lindsey conditions, and the porphyrin product was isolated as the trifluoroacetate salt in 42% yield. The naphthalene diimide-fused (NMI) analogue, 308.3a could be obtained either via direct condensation of the monopyrrole 308.1b or, more efficiently, by hydrolysis and imidization of 308.2a. The corresponding metalloporphyrins, 308.2b–c and 308.3b were obtained from the free-base porphyrins 308.2a and 308.3a, respectively. The presence of electron deficient moieties in 308.3b provided an ability to accept up to 8 electrons via electrochemical reduction and revealed reversible formation of eight charged states, characterized by remarkably red-shifted optical absorptions, extending beyond 2200 nm. The π-extended NDA-fused porphyrins were subsequently explored as potential photosensitizing agents for antimicrobial photodynamic therapy (aPDT).575 Their aPDT efficiency could be easily tuned by metal coordination, showing that this family of dyes might be used for treatment of infections caused by Gram-positive bacteria. NMI porphyrins were also incorporated into energy-transfer cassettes containing peripheral BODIPY units.576 The free base porphyrin cassette 308.5a was obtained by condensing the octaacid 308.2d with the amino-functionalized BODIPY 308.7, and was converted into metal complexes 308.5b and 308.5c (Scheme 308). The Br-functionalized NMI metalloporphyrins 308.4a and 308.4b were subjected to Suzuki coupling with the catecholate-protected BODIPY 308.8 to afford the larger analogues 308.6a and 308.6b. The femtosecond transient absorption measurements and theoretical predications demonstrated efficient excitation energy transfer pathways in 308.5b and 308.5c.
Scheme 308. Synthesis of Donor–Acceptor π-Extended Porphyrins.
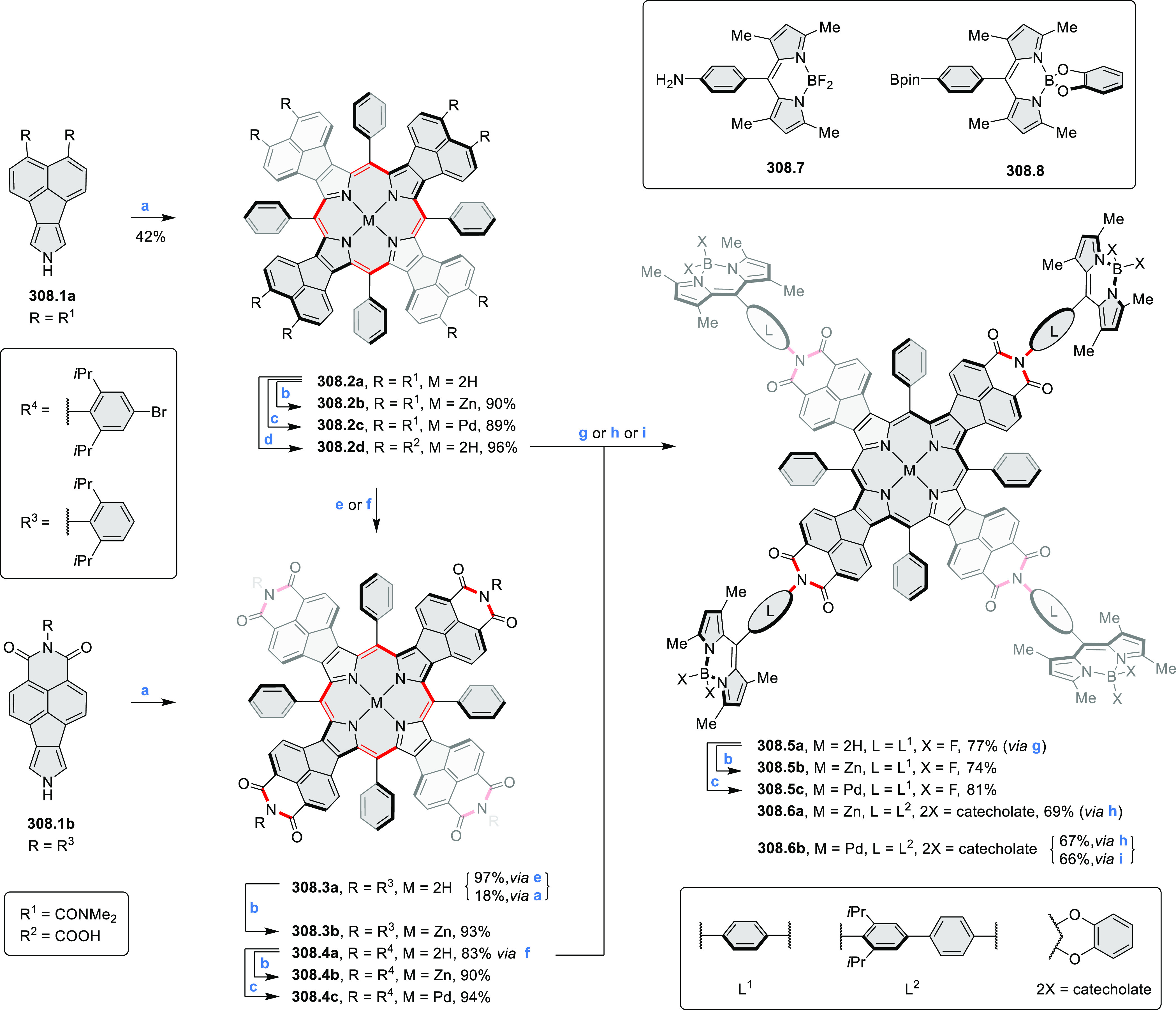
Reagents and conditions: (a)22 (1) benzaldehyde, p-TsOH·H2O, CHCl3/MeOH (100:1 v/v), (2) DDQ, rt; (b)22,576 Zn(OAc)2, CHCl3/MeOH; (c)575,576 Pd(OAc)2, CHCl3/MeOH; (d)22 HClconc, reflux; (e)22 2,6-diisopropylaniline, AcOH, 160 °C; (f)576 4-bromo-2,6-diisopropylaniline, AcOH, 160 °C; (g)576308.7, AcOH, 160 °C; (h)576308.8, Pd(dppf)Cl2 (0.5 equiv), K2CO3, 120 °C, toluene/H2O (4:1 v/v); (i)576 Pd(dppf)Cl2 (1.5 equiv), K2CO3, 120 °C, toluene/H2O (4:1, v/v).
Later in 2020, we developed synthetic routes to mixed donor–acceptor porphyrins containing NDA/NMI pyrrole subunits.577 The approach shown in Scheme 309 was utilized to circumvent the differential reactivity of electron-rich and electron-poor pyrroles and minimizing the undesired possibilities of reaction products. 309.3a was prepared in 12% yield by condensing the acenaphthopyrrole dicarbinol 309.2 with one equivalent of diamide pyrrole 308.1a. Similarly, 309.4a was obtained from a cross condensation of 308.1a, the unsubstituted 309.1 and 309.2 in a 1:1:2 molar ratio, to afford the product in 10% yield. Subjecting the two porphyrins to acid hydrolysis followed by imidization with 2,6-diisopropylaniline gave the corresponding imides 309.5a and 309.6a. Free bases and zinc complexes of these porphyrins displayed distinct optical properties resulting from desymmetrization, notably more extended NIR bands, with the absorption onset approaching 1000 nm. Interestingly, despite the smaller number of electron-withdrawing NMI moieties, the HOMO–LUMO gap of all these mixed D–A porphyrins were reduced relative to those of the fully symmetrical 308.3a–b.
Scheme 309. Synthesis of Donor–Acceptor Porphyrins with Mixed Ring Fusion Patterns.

Reagents and conditions: (a)577 (1) 308.1a and 309.2 (+ 309.1, for 309.4a), p-TSA, CHCl3/MeOH (100:1 v/v), 1 h, (2) DDQ, 2 h; (b) Zn(OAc)2·2H2O, CHCl3/MeOH (3:1 v/v); (c) HCl, reflux, 24 h; (d) 2,6-diisopropylaniline, acetic acid, 20 h, reflux.
In 2018, Jiang and Wang reported the synthesis of a rylene-annulated phthalocyanine by integrating electron deficient PDI subunits with the comparatively electron-rich metallophthalocyanine core.578 The three-step synthetic route started with the Suzuki–Miyaura cross-coupling of 77.1 with pinacol (3,4-dicyanophenyl)boronate to give 310.1 in 95% yield (Scheme 310). 310.1 was then further irradiated in toluene solution with a catalytic amount of I2, to afford 310.2 and its regioisomer. In the final step, zinc(II) phthalocyanine 310.3 was obtained in a microwave-assisted condensation reaction of 310.2 in the presence of zinc acetate and DBU. The four imide-bound branched alkyl chains provided good solubility for 310.3 in chloroform, THF, and o-dichlorobenzene solvents. 310.3 exhibited a nearly planar conformation and showed cage-like packing motif in the solid state. The UV–vis–NIR absorption spectrum of 310.3 displayed an absorption maximum at 785 nm and a high molar extinction coefficient of ca. 300,000 M–1 cm–1. A significant red-shift of the Q-band relative to the parent nonfused phthalocyanine was attributed to the greater π-electron delocalization in 310.3.
Scheme 310. Synthesis of a Rylene-Annulated Phthalocyanine.
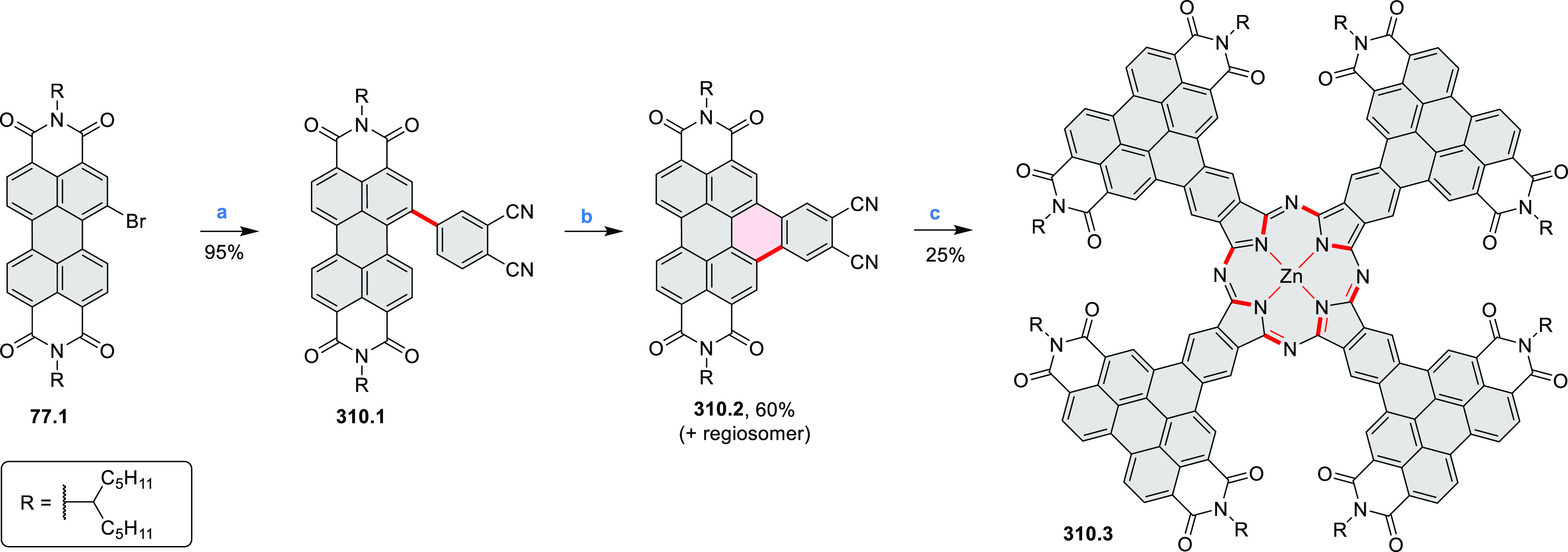
Reagents and conditions: (a)578 3,4-dicyanobenzeneboronate pinacol ester, Pd(PPh3)4, K2CO3, THF/H2O, reflux, 12 h; (b) hν, I2, toluene, 95 °C, 48 h, Zn(OAc)2, DBU, n-hexanol, 160 °C, MW, 1 h.
7.2. Benzo[cd]-fused porphyrinoids
7.2.1. Benzo[cd]-Fusion via meso-Substituent Coupling
In 2020, Zhuang and Wang reported a method of controlling delocalized magnetism in metal-free porphyrins by peripheral fusion of ortho-dimethylphenyl substituents.579 The three metal-free porphyrins 311.3–5 with different π-electron topologies were obtained from the precursor 5,10,15,20-tetrakis(2,6-dimethylphenyl)porphyrin 311.1 using on-surface synthesis on Au(111) (Scheme 311). 311.3 resulted from double methyl group cleavage followed by formation of five-membered rings, whereas 311.5 was a product of incomplete dehydrogenation. On the basis of high-resolution nc-AFM imaging, 311.3 was assigned a closed-shell structure, whereas compounds 311.4–5 were described as open-shell species, with two and one unpaired electrons, respectively. The π-electron magnetism in these porphyrins could be switched on/off via scanning tunneling microscope manipulation by tuning the interfacial charge transfer from the Au(111) substrate. In the same year, Fasel and co-workers examined the structural and electronic properties the three zinc porphyrins 311.6, 311.9, and 311.10 bearing respectively zero, two, and four meso-fused phenalenyl moieties.580 Combined experimental and theoretical data revealed a triplet ground state for 311.9 and a charge-transfer-induced open-shell character for the intrinsically closed-shell 311.10. Formation of intramolecular- and intermolecular-fused products on Cu(111), Au(111) or Ag(110) surfaces was also demonstrated for other free-base or cobalt(II) porphyrins.581−585
Scheme 311. Precise Control of π-Electron Magnetism in Metal-Free Porphyrins.
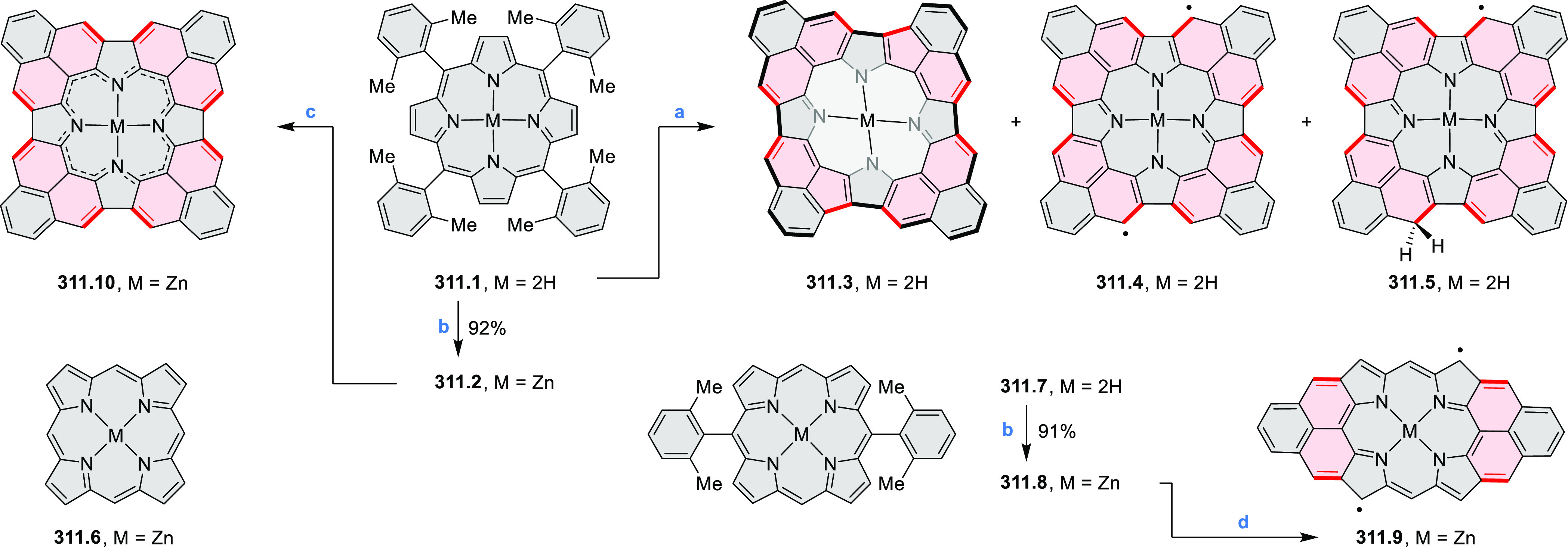
Reagents and conditions: (a)579 Au(111), 295 °C; (b)580 Zn(OAc)2, THF, reflux; (c)580 Au(111), 300 °C; (d)580 Au(111), 330 °C.
A related benzannulation was observed by Peruncheralathan, Srinivasan et al. (Scheme 312). They reported the formation of meso-fused β–β′ carbaporphyrin dimer 312.2 and meso-fused monomer 312.3, upon treating carbatriphyrin(3.1.1) 312.1 with PtCl2 under aerial conditions.586 The molecular structure of 312.2 was confirmed by single-crystal X-ray diffraction, which revealed the fusion of one meso-Mes group resulting in the formation of a 1H-benzo[f]indole unit. The UV–vis spectrum of 312.2 in DCM showed an intense absorption at 355 nm and a prominent band at 505 nm, while 312.3 showed relatively weaker bands between 400 and 1000 nm.
Scheme 312. Synthesis of a β–β′-Linked meso-Fused Carbatriphyrin(3.1.1) Dimer.

Reagents and conditions: (a)586 PtCl2 (6.5 equiv), chlorobenzene, reflux.
In 2020, Lungerich, Jux, Drewello, and co-workers reported a mass spectrometry-based gas-phase study of a porphyrin-based conical graphene fragment 313.2a–d formed by means of 8-fold fjord region-selective cyclodehydrofluorination (Scheme 313).587 The γ–ortho cyclization and was found to depend on the choice of metal and functionalized fluorinated meso-aryl porphyrins. The coupling mechanism was presumed to consist of C–C nucleophilic addition followed by 1,2-elimination of HF. Attempts to perform an analogous in-solution dehydrochlorination of 313.1e to produce the fully cyclized product 313.2e resulted only in the formation of inseparable mixtures of partially coupled products.
Scheme 313. Porphyrin-Based Conical Nanocarbons.

Reagents and conditions: (a)587 gas phase, ΔE, N2; (b) Pd(PCy3)2Cl2 (8 equiv), DMAc/DBU (v/v 4:1), MW, N2, 180 °C, 4 h.
In 2019, Holten, Lindsey et al. reported NIR-absorbing bacteriochlorins annulated with aromatic rings (Scheme 314).588 Suzuki–Miyaura cross-coupling of 314.1 with (8-bromonaphthalen-1-yl)boronic acid and (2-bromophenyl)boronic acid afforded the arylated precursors 314.2 and 314.4 as mixtures of atropisomers which were used as the mixture in the next step. Palladium-catalyzed intramolecular direct arylation afforded the target β,meso-annulated bacteriochlorins 314.3 and 314.5 in 37% and 80% yield, respectively. The UV–vis–NIR absorption spectrum of 314.5 was significantly red-shifted compared to that of 314.3, with the lowest-energy Q bands lying at 1033 and 913 nm, respectively. These bacteriochlorins were characterized by extremely short lowest excited singlet state lifetimes (7 ps for 314.5 and 150 ps for 314.3) and vanishingly small fluorescence quantum yields.
Scheme 314. Synthesis of β,meso-Annulated Bacteriochlorins.

Reagents and conditions: (a)588 (2-bromophenyl)boronic acid or (8-bromonaphthalen-1-yl)boronic acid, Pd(PPh3)4, K2CO3, toluene, DMF, 100 °C; (b) Pd(PPh3)4, SPhos, K2CO3, DMF, 80 to 100 °C, 6 h; (c) Pd(PPh3)4, PCy3·HBF4, K2CO3, DMF, 110 °C, 2 h.
Examples of naphthalene-fused porphyrins C30.3(589) and C30.4–5590 obtained using intramolecular oxidative aromatic coupling were reported by Gryko et al. (Chart 30). Prasanthkumar, Giribabu and co-workers developed a related design based on a doubly fused anthracene linker C30.6 (Chart 30).591 The formation a 1:1 charge transfer complex between C30.6 and PDI was observed spectroscopically in CHCl3. The face-to-face π–π stacking responsible for self-assembly of the CT complex led to further aggregation into spherical and nanorod-shaped aggregates, investigated by TEM and AFM microscopy.
Chart 30. meso-β-Pentannulated and Benzannulated Porphyrins.
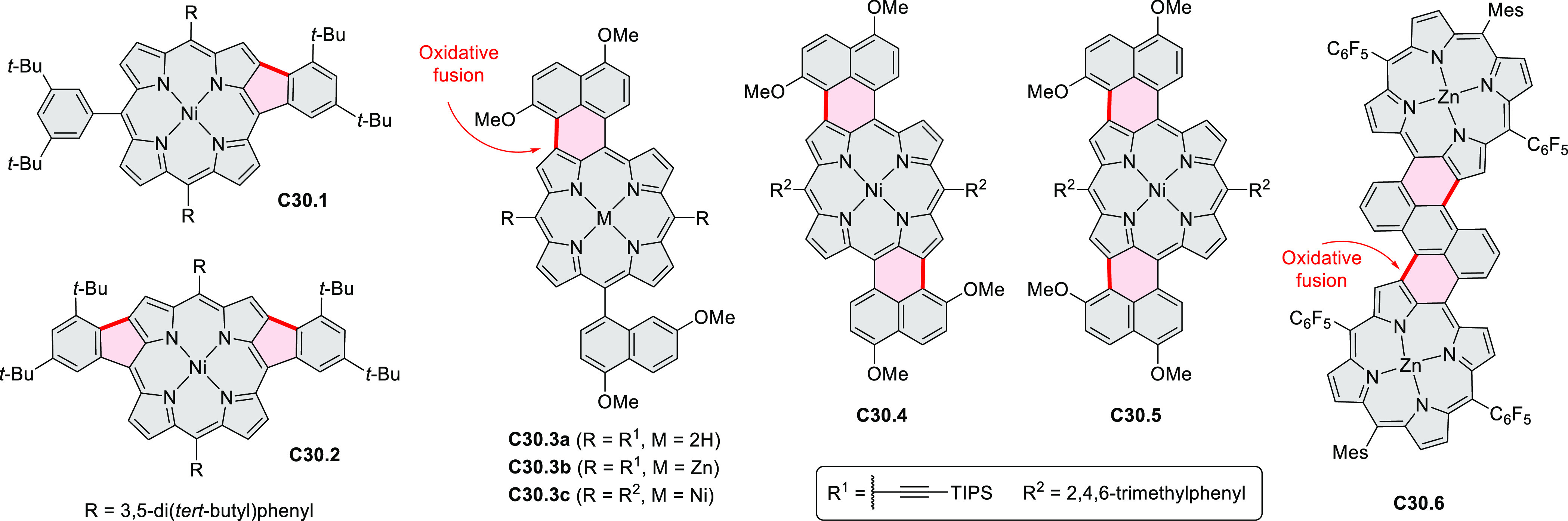
7.2.2. Other Benzo-Fused Systems
In 2020, Sankar and Kadish electrochemical and chemical syntheses of cyano-substituted naphtho-fused π-extended tetraphenylporphyrins from the corresponding tetracyano precursors (Scheme 315).592 The electrosynthetic method involved an application of a controlled reducing potential of −1.6 V or −1.7 V at a platinum electrode, followed by an oxidizing potential of 0.0 V in DCM solution, while the chemical synthesis proceeded via a cyanide anion induced electron transfer. Both approaches were found to produce the same decyanated products 315.2a–d in 86–94% yields (Scheme 315). The UV–vis–NIR absorption of 315.2a–d in DCM showed a significant red-shift of the lowest-energy Q bands in comparison with the parent 315.1a–d.
Scheme 315. Synthesis of Cyano-Substituted π-Extended Porphyrins.

Reagents and conditions: (a)592 TBAP, DCM, glassy carbon or Pt cathode, controlled applied potential; (b) TBACN, CHCl3, stir, 30 min.
7.2.3. Pyrido[cd]fused Systems
In 2016, Pawlicki et al. reported an indolizinone-fused porphyrin obtained via condensation between a meso-N-pyrrolyl-substituted porphyrin and an aldehyde.593 A transmetalation of 316.1b yielded 316.1a, which was condensed with p-tolualdehyde and oxidized with DDQ to give the lactam 316.2a (Scheme 316). 316.2b was obtained by a transmetalation because direct condensation was ineffective for 316.1b. Compounds 316.2a or 316.2b reacted efficiently with sodium ethoxide in the presence of phosphonium (Ph3P+CH2PhCl–) or tetraethylammonium salt (Et4N+Cl–) to give 316.3a or 316.3b, respectively. Compound 316.2a or 316.2b showed red-shifted absorption reaching the wavelength of about 680 nm and tailing up to 700 nm. Deprotonation of 316.3b upon titration with n-Bu4N+F– formed 316.4a, containing a peri-fused pyridine ring.
Scheme 316. Synthesis of meso-Pyrrole Porphyrins.
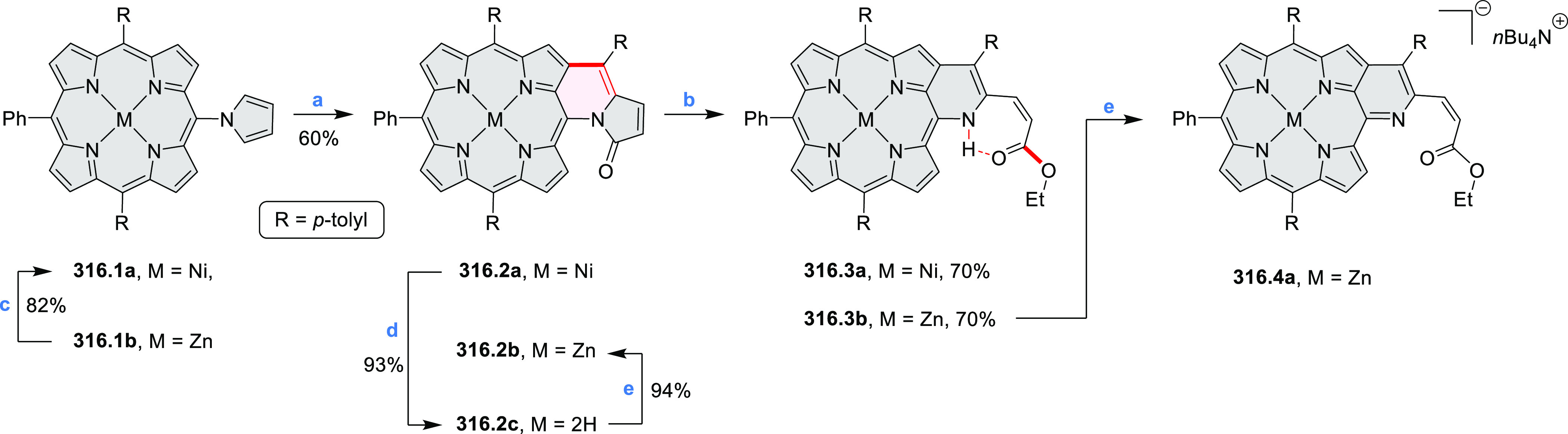
Reagents and conditions: (a)593 (1) DCM, TFA, p-tolualdehyde, rt, 16 h, (2) DDQ (2 equiv); (b) EtONa (10 equiv), Ph3P+CH2PhCl– or Et4N+Cl– (10 equiv), EtOH, then addition of 316.2a or 316.2b, 24 h; (c) (1) TFA (10%) followed by neutralization with Et3N, (2) Ni(OAc)2, DMF, reflux, 1 h; (d) TFA/H2SO4 neutralization with Na2CO3, 2 h; (e) CHCl3/MeOH, Zn(OAc)2, 1 h; (e) THF, nBu4N+F–, N2.
The doubly fused porphyrin 317.4a was synthesized by Osuka et al. via stepwise oxidation of the meso-phenoxazinyl Ni(II) porphyrin 317.1 and was subsequently converted into the corresponding free base porphyrin 317.4b and its zinc complex 317.4c (Scheme 317).594 In contrast, the oxidation of 317.1 with DDQ and FeCl3 at rt resulted in the fusion of two aryl groups, yielding the doubly phenylene-fused porphyrin 317.2. The authors also transformed the previously reported doubly diphenylamine-fused nickel(II) complex 317.4d into the free base 317.4e and zinc complex 317.4f. The doubly phenoxazine-fused porphyrins exhibited features attributed to their highly planar structures, such as higher fluorescence quantum yields, formation of an offset face-to-face dimer both in solution and the solid state, and generation of a mixed-valence π-radical cation dimer upon electrochemical oxidation. The one-electron oxidation of 317.7a and 317.7d using BAHA gave the corresponding dichlorinated radical cations 317.8a and 317.8d were found to be much more stable than 317.5a which was slowly chlorinated at the reactive β-positions.
Scheme 317. Synthesis of Diarylamine-Fused Porphyrins.
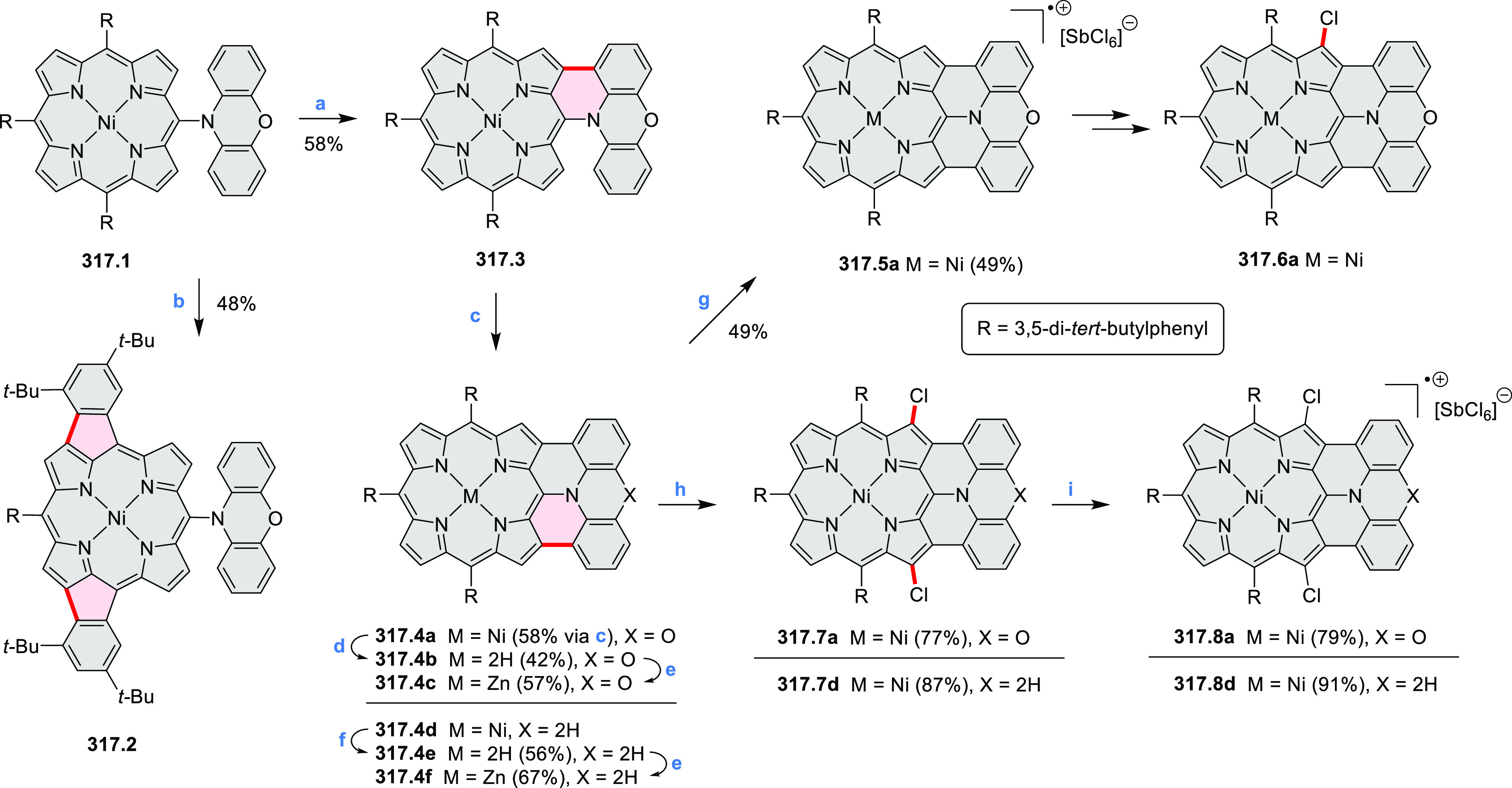
Reagents and conditions: (a)594 DDQ (10 equiv), Sc(OTf)3 (10 equiv), ClCH2CH2Cl/MeNO2 70 °C, 1 h; (b) DDQ (10 equiv), FeCl3 (10 equiv), DCM/MeNO2, rt; (c) (1) FeCl3 (10 equiv), DCM/MeNO2, 70 °C, 1 h, (2) HCOOH, NEt3, Pd2(dba)3, SPhos, toluene, 120 °C, 1 h; (d) TFA/conc. H2SO4; (e) Zn(OAc)2·2H2O, DCM; (f) (1) p-tolylmagnesium bromide, toluene, (2) 3 M HClaq; (g) BAHA (1.1 equiv), DCM, rt, 10 min; (h) 2-chloro-1,3-bis(methoxycarbonyl)guanidine (2.2 equiv), CHCl3, 0 °C (10 min) to rt, 2.5 h for 317.7a and 7 h for 317.7a; (i) BAHA (1.1 equiv), DCM, rt, 10 min for 317.7a and 5 min for 317.7a.
Structurally related diarylamine-fused subporphyrins 318.4a–b were synthesized by the Osuka group through a one-pot procedure involving nucleophilic aromatic substitution followed by electron-transfer-mediated intramolecular C–H/C–I coupling (Scheme 318).595 Of these two compounds, 318.4b showed a more red-shifted absorption attributed to the effective conjugation between the electron-donating dimethylamino group and the subporphyrin core. The fluorescence quantum yields (Φf) of 318.4a and 318.4b were found to be 0.21 and 0.18, respectively. In another work by Yorimitsu and Osuka, a similar synthetic strategy led to the formation of meso–meso-linked diphenylamine-fused porphyrin dimers.596318.6a and 318.6c were prepared from 318.5 using a similar procedure. Upon oxidation with BAHA, 318.6a and 318.6c showed different reactivity, yielding respectively a dicationic closed-shell quinoidal dimer 318.7a and the doubly linked porphyrin dimer 318.8a.
Scheme 318. Synthesis of Diarylamine-Fused Subporphyrins and Porphyrin Dimers.
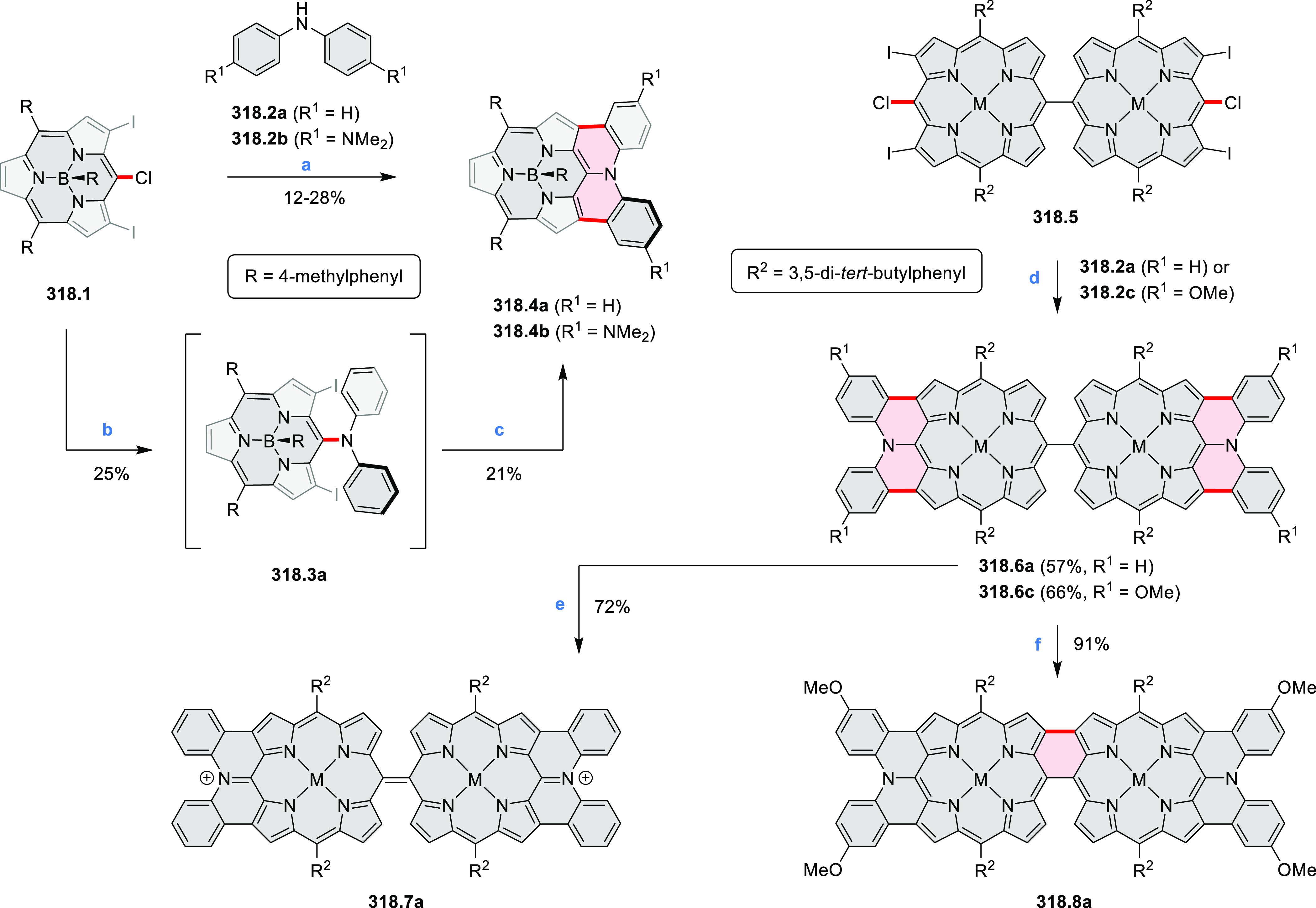
Reagents and conditions: (a)595318.2a or 318.2b (3 equiv), NaOt-Bu (12 equiv), DMF, 100 °C, 15 min; (b)595318.2a (3 equiv), DMF, rt, 30 min; (c)595 NaOt-Bu (12 equiv), DMF, 100 °C, 10 min; (d)596318.2a or 318.2c (2.5 equiv), NaOt-Bu (10 equiv), DMF, 100 °C; (e)596 BAHA (6.0 equiv), DCM, rt, 10 min; (f)596 BAHA (6.0 equiv), DCM, rt, 15 min, then H2NNH2·H2O.
Osuka et al. reported meso-substituted porphyrin dimers 319.3 and 319.5, which were synthesized in a SNAr reaction of 319.1 with the corresponding phenylene-diamine 319.2a or 319.2b (Scheme 319).597 Then, palladium-catalyzed 4-fold C–H arylation conditions on 319.3 and 319.5 led to the formation of fully fused, nonplanar systems 319.4 and 319.6. The X-ray crystal structure of 319.4 revealed that the two [4]helicene-like segments had opposite helicities, whereas in 319.6, the corresponding two segments were homochiral. Both fused-porphyrin dimers revealed a highly reversible electrochemical oxidation and produced dication diradicals upon chemical oxidation. A related synthesis, reported later by the same authors, produced an inseparable mixture of 319.7 and its helically twisted isomer 319.8, which could not be separated.598 A helically twisted nitrogen-doped fused porphyrin dimer 319.9 was however synthesized using benzene-1,3,5-triamine via t-BuONa-promoted one-pot reaction. Zinc complex 319.10, prepared from 319.9, showed fluorescence at around 650–700 nm in DCM with a quantum yield (Φf) of 0.04.
Scheme 319. Synthesis of Diarylamine-Fused Porphyrin Dimers.
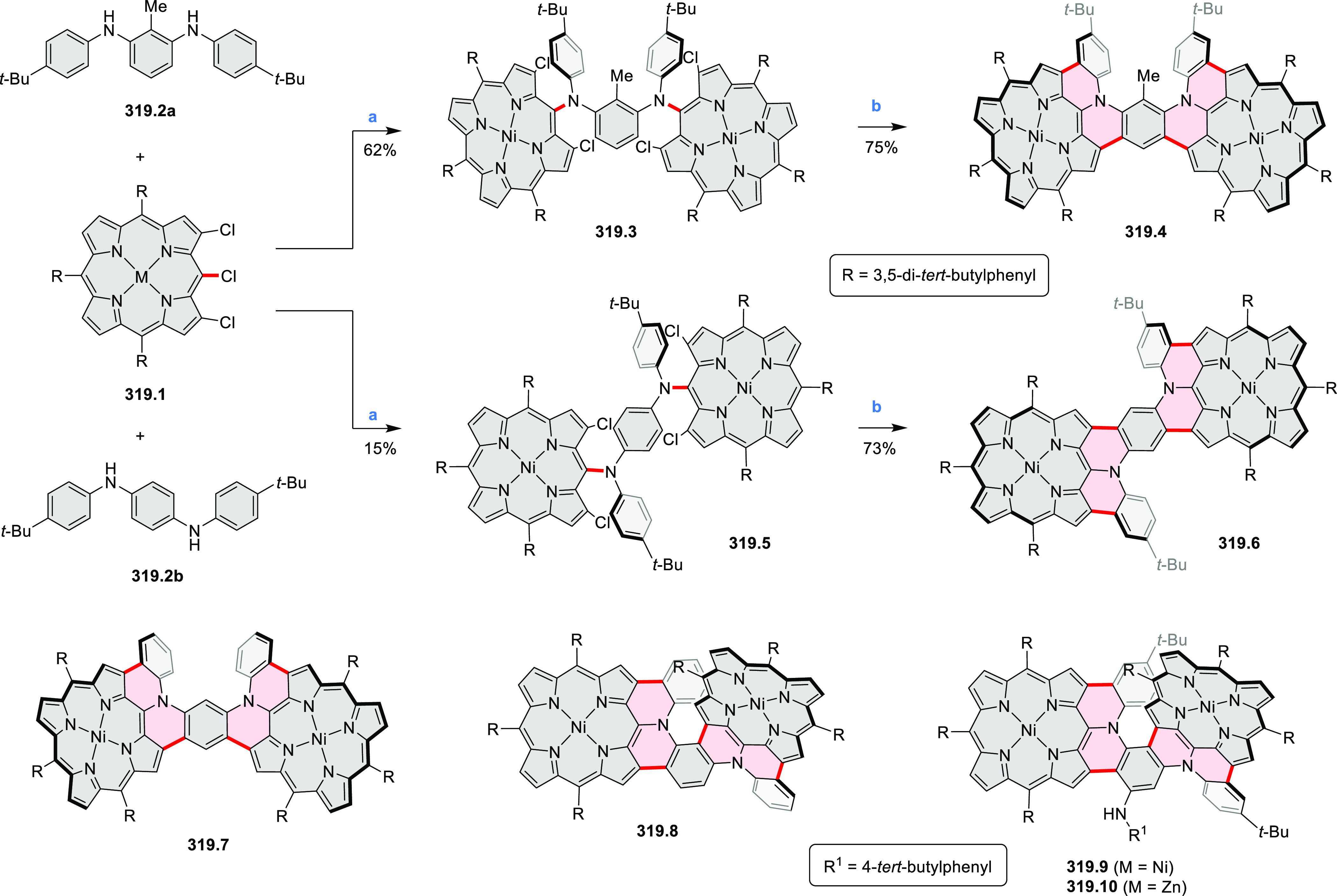
Reagents and conditions: (a)597t-BuONa (11 equiv), DMF, 60 °C, 10 h; (b) Pd(OAc)2, PCy3·HBF4, pivalic acid, K2CO3 (10 equiv), DMA, 140 °C, 13 h.
A synthesis of 1,5-naphthyridine-fused porphyrin dimer 320.2 was developed in 2018 by Osuka and co-workers (Scheme 320).599 The platinum catalyzed intramolecular cyclization of β-to-β ethynylene-bridged meso-amino Ni(II) porphyrin dimer 320.1 resulted in the formation of a six-membered ring via a 6-endo-dig cyclization and the subsequent oxidation with PbO2 afforded 320.2 in 84% yield. The redox active 1,4-diazabutadiene unit in 320.2 was found to be interconvertible with its reduced 1,2-diaminoethene linkage in 320.3 via reduction with NaBH4 and oxidation with PbO2. The UV–vis absorption of 320.2 in DCM showed an intense absorption band at 1011 nm in the NIR region which is drastically different than that of 320.3, suggesting a substantial difference in their electronic interaction between the two porphyrin units via fused 1,4-diazabutadiene or 1,2-diaminoethene linkage. The electrochemical HOMO–LUMO gap 320.2 and 320.3 was found to be 0.99 and 1.8 eV, respectively.
Scheme 320. Synthesis of a 1,5-Naphthyridine-Fused Porphyrin Dimer.
Reagents and conditions: (a)599 PtCl2, toluene, rt, 3 h, and then, PbO2, DCM, rt, 1 h; (b) NaBH4, DCM/MeOH, rt, 15 min; (c) PbO2, DCM, rt, 5 min.
In 2020, Brückner and co-workers described a route to quinoline-annulated metalloporphyrins involving an intramolecular SNAr displacement of an o-fluorine atom on a meso-pentafluorophenyl group by the neighboring β-amino substituent (Scheme 321).600 The reduction of the nitro group in 321.1a–c led to the formation of a β-aminoporphyrin, which were then directly used in the next step involving an intramolecular SNAr-type annulation reaction to afford the desired products 321.2a–c in 50% isolated yields (over two steps). The free base 321.2d was also obtained by demetalation of the copper complex. The quinoline-annulated porphyrins 321.3a–c, obtained by Boechat, Neves, Cavaleiro and co-workers via oxidative cyclization of corresponding β-aminoporphyrins, were used as potential photodynamic photosensitizers.601 Among these, 321.3a was found to most efficiently inactivate Gram-positive bacteria S. aureus, because of efficient singlet oxygen generation and the less hydrophobic character, which enhanced its affinity for bacteria. The quinoline-fused nickel(II) complex 321.4a, obtainable via Cadogan-type cyclization, was derivatized in a variety of ways (e.g., 321.4b–c602). An isocyanide monomer containing N-appended 321.4a was copolymerized with poly(3-hexylthiophene) to afford a photovoltaic donor material.603 Furthermore, isatin–porphyrin conjugates were obtained via peripheral Buchwald–Hartwig amination of 321.4a.604
The porphyrin dimer 322.2 was obtained by Ruppert et al. upon subjecting 322.1 to various monoelectronic oxidants, with best results obtained when using 0.55 equiv of [Fe(phen)3]3+ in the presence of base (Scheme 322).605 Another porphyrin dimer, 322.4 was obtained in 18% yield upon slow and simultaneous addition of equimolar solution of 322.1 and PIFA to a suspension of sodium carbonate in DCM. Intramolecular cyclization was preferred under these conditions, leading to the formation of an intermediate 322.3, which subsequently afforded the N–N linked dimeric product 322.4. The UV–vis absorption spectrum of 322.4 had the lowest-energy Q-band at 675 nm, somewhat red-shifted relative to that of 322.2.
Scheme 322. Oxidative Coupling of Enaminoporphyrin.
Reagents and conditions: (a)605 Fe(phen)3(PF6)3, Na2CO3, DCM, rt; (b) PIFA, Na2CO3, DCM, rt.
In 2015, Pawlicki and Latos-Grażyński reported a NIR chromophore based on fused meso-aminoporphyrin, switchable using a redox process or protonation of the meso-nitrogen.606 The synthesis consisted of the Buchwald–Hartwig amination of 323.1 followed by attempted oxidative cyclization of the resulting 323.2a–c (Scheme 323). The latter step was relatively more effective for 323.2c, yielding ultimately the fused product 323.4c (with 323.3c as an intermediate), whereas 323.2a and 323.2b were only oxidized to produce the corresponding imine cations 323.3a,b. 323.4c and its reduced form 323.5c were mutually convertible via a two-electron redox process. In addition, 323.6c could be obtained by deprotonation of 323.5c. Each of these species showed different electronic properties, reflected by significant changes in the absorption and fluorescence spectra.
Scheme 323. Synthesis of Fused meso-Aminoporphyrins.

Reagents and conditions: (a)606 arylamine (5 equiv), Pd(OAc)2, DPEPhos, Cs2CO3, THF, 65 °C, 3 h; (b) DDQ, DCM; (c) DDQ, DCM, TFA; (d) NaBH4, THF; (e) HCl; (g) DCM; (f) TBAF, THF.
Brückner and Zhu demonstrated the use of quinoline-annulated porphyrin derivatives as potential contrast agents for photoacoustic imaging (PAI), however, their insolubility in aqueous solutions prevented the in vivo assessment.607 To obtain water-soluble products that derivatives be useful for tumor tomography, the group employed the hydroxy-substituted 324.2b, which was PEGylated with methyl-capped PEG-mesylates to provide 324.2c with tetraethylene glycol chains, as well as 324.2d with longer chains (n ∼ 12), or substituted with a fluorescent BODIPY tag (324.2e–f, Scheme 324).608324.2c was found to be only slightly soluble in pure water whereas 324.2d was freely soluble in alcohols, water, serum, and PBS buffer. 324.2d absorbed strongly within the spectroscopic window and was very weakly fluorescent. It had low acute toxicity, accumulated in the tumor site, and was excreted unaltered via renal pathways.
Scheme 324. Quinoline-Annulated Porphyrins.

Reagents and conditions: (a)608 pyridine, reflux, 48 h; (b) BBr3, DCM; (c) Me(OCH2CH2)4OMs or Me(OCH2CH2)12OMs, Cs2CO3, DMF, 90 °C; (d) (1) Zn(OAc)2·2H2O, DCM/MeOH, (2) 324.3 (1 equiv), Na2CO3, copper(I) thiophene-2-carboxylate, CH3CN, 50 °C, (3) HCl.
In 2016, Brückner described the OsO4-mediated dihydroxylation of quinoline-annulated porphyrins generating a quinoline-annulated dihydroxychlorin in a regioselective fashion and highlighted its importance as an effective strategy to red-shift the absorption spectra of these annulated chlorin analogs.609 The osmate ester 325.2 was obtained in 84% yield by reacting the quinoline-annulated porphyrin 325.1 with stoichiometric amounts of OsO4. 325.2 was then treated with H2S to afford the quinoline-annulated dihydroxychlorin 325.3 in 54% yield as a racemic mixture (Scheme 325). The oxidation of 325.3 using Dess–Martin periodinane (DMP) resulted in the formation of dione product 325.4. The direct conversion of a pyrrole β, β′bond to a lactone moiety was made by treating 325.3 with cetyltrimethylammonium permanganate (CTAP) which yielded 325.5 as regioisomers (2-oxa-3-oxo-quinoline annulated porphyrin or 3-oxa-2-oxoquinoline annulated porphyrin). Compounds 325.6a–b were obtained from the oxidative cleavage of diol 325.3 using sodium periodate. The UV–vis absorption spectrum of 325.6a were found to be much more red-shifted than the other derivatives reaching up to 1000 nm, while 325.3 showed the lowest energy Q like band at 842 nm.
Scheme 325. Synthesis of Quinoline-Annulated Chlorin and Derivatives.

Reagents and conditions: (a)609 OsO4, pyridine, CHCl3, rt; (b) H2S, CHCl3, rt; (c) DMP, DCM, rt; (d) DCM, CTAP; (e) NaIO4/silica, EtOH, or MeOH.
7.2.4. Pyrano- and Thiopyrano[cd]fused Systems
In 2018, Devillers described chemical and electrochemical syntheses of C–N fused pyridinium-containing porphyrins from precursors bearing a pyridin-2-ylthio meso-substituent (Scheme 326).610 The chemical oxidation of nickel(II) porphyrins 326.1a–c using 1.2 equiv of PIFA produced fused cationic derivatives, which were isolated as hexafluorophosphate salts 326.2a, 326.2b, and 326.2c in 98%, 81%, and 88% yield, respectively. Furthermore, the chemical oxidation of 326.2c with 1 equiv of PIFA followed by anion exchange step led to the regioselective formation of doubly C–N fused dicationic pyridinium-based porphyrin 326.3. These singly and doubly C–N-fused products were also obtained via exhaustive electrolysis under mild conditions.
Scheme 326. Intramolecular Oxidative C–N Coupling of Pyridin-2-ylthio-meso-substituted Porphyrins.
Reagents and conditions: (a)610 (1) PIFA (1 equiv), DCM, rt, (2) anion exchange: X = CF3COO to PF6.
In 2019, D. Wu and J. You et al. described a divergent synthesis of various meso-N/O-heteroarene-fused porphyrins via rhodium-catalyzed [4 + 2] annulation (Scheme 327).611 The reaction of 327.1 and diphenylacetylene in the presence of [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), and Ag2O (2 equiv) afforded the doubly pyridine-fused anti-quinoidal porphyrin 327.6a as a major product. In the same reaction, a high polarity product was also obtained which was identified as the pyridinium-fused 327.9a. With the optimized reaction conditions, the scope of this reaction was extended to a variety of alkynes with either electron-donating or electron-withdrawing groups, affording the desired products 327.6–10 with controllable chemoselectivity and complete anti-selectivity.
Scheme 327. Synthesis of meso-N/O-Heteroarene-Fused Porphyrins via a [4 + 2] Oxidative Annulation Strategy.
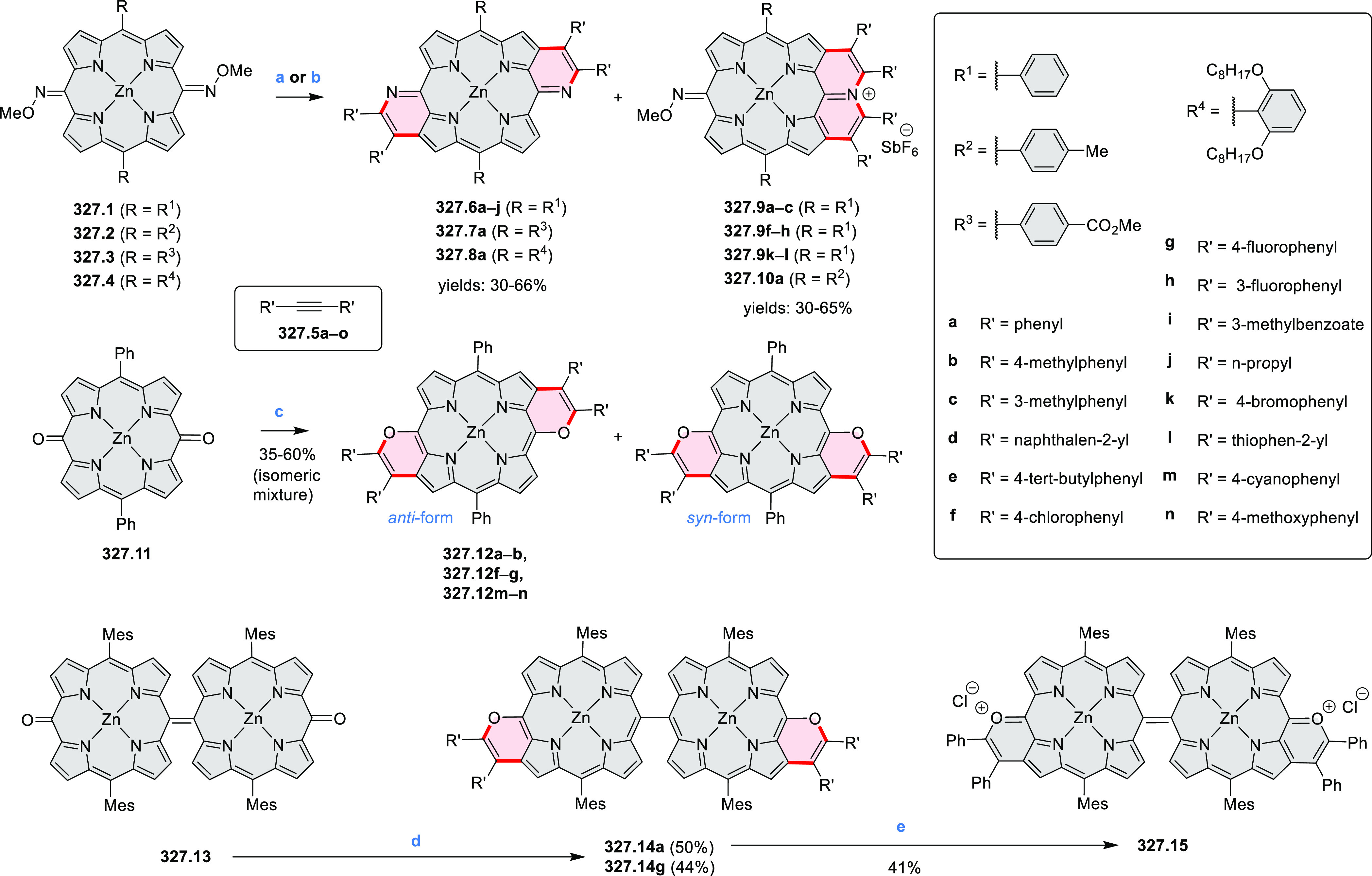
Reagents and conditions: (a) Standard conditions for 348.6–8: 348.5 (0.2 mmol), [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), THF (0.5 mL), 100 °C, N2, 24 h; (b) standard conditions for 348.9–10: 348.5 (0.2 mmol), [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), Ag2O (2.0 equiv), NaSbF6 (2.0 equiv), DCE (0.5 mL), 120 °C, N2, 24 h; (c) 348.5 (0.2 mmol), [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), Ag2O (2.0 equiv), DCE, or 1,4-dioxane (0.5 mL), 120 °C, N2, 24 h; (d) [Cp*RhCl2]2 (5 mol %), AgSbF6 (20 mol %), Ag2O (2.0 equiv), DCE, or 1,4-dioxane (0.5 mL), 120 °C, N2; (e) FeCl3 (10 equiv), DDQ (10 equiv), DCM/MeNO2.
The synthesis of meso-O-containing heteroarene-fused porphyrins 327.12 was carried out under similar conditions using the 5,15-dioxoporphyrin 327.11 and diphenylacetylene as the reaction substrates. The doubly pyran-fused porphyrin 327.12a was obtained as a mixture of syn- and anti-isomers. The scope of this reaction was extended to a variety of alkynes, which typically produced regioisomeric mixtures, which could occasionally be separated chromatographically. Using this annulation approach, the doubly pyran-fused porphyrin dimers 327.14a and 327.14g were also prepared in 50% and 44% yields, respectively. Upon oxidation of 327.14a with FeCl3 and DDQ, the doubly pyrylium-fused porphyrin dimer 327.15 was obtained, which displayed intense near-infrared (NIR) Q bands reaching up to 1300 nm in DCM. ACID and NICS(1) analyses suggested the presence of 22π aromatic conjugation in syn/anti-327.14a, reflecting the involvement of the pyrylium rings in π conjugation.
7.2.5. Benzo-Fused Porphyrin Oligomers
The preparation of a fused subporphyrin dimer 328.2 was reported in 2016 by Kim, Osuka and co-workers.612 The bromination of 328.1 followed by intramolecular Yamamoto coupling led exclusively to the anti diastereomer of fused subporphyrin dimer 328.2 (Scheme 328). The unobserved syn isomer was predicted by DFT calculations to have a higher energy than the anti form. The UV–vis absorption spectrum of 328.2 in DCM exhibited a broad Soret-like band and Q-like band at 641 nm with a tail reaching ca. 850 nm, considerably red-shifted relative to 328.1, and showed a significantly smaller electrochemical HOMO–LUMO gap of 1.72 eV. The same authors reported the synthesis of triply linked subporphyrin dimer 328.6 by stepwise reductive elimination of the doubly PtII-bridged subporphyrin dimer 328.3 (Scheme 328).613 Attempted reductive elimination of 328.3 resulted in the ligand-exchanged product 328.4 and a trace amount (<1%) of fused dimer. In contrast, treatment of 328.3 with BAHA gave the doubly fused PtII-bridged dimer 328.5 in 67% yield and subsequently, further reductive elimination resulted in the formation of the desired triply linked subporphyrin dimer 328.6 in 78% yield. 328.6 had a domed structure with a positive Gaussian curvature and a bowl-depth of 1.65 Å. The UV–vis spectrum of 328.6 in DCM showed the lowest-energy absorption band at 942 nm and a small electrochemical HOMO–LUMO gap of 1.35 eV, suggesting the effective π-conjugation extending over the whole molecule.
Scheme 328. Synthesis of Doubly and Triply Fused Subporphyrin Dimers.

Reagents and conditions: (a)612 (1) NBS, CHCl3, 0 °C, 3 h, (2) Ni(cod)2, 1,5-cod, DMF, 80 °C, 3 h; (b)613 PPh3 (10 equiv), toluene, 120 °C, 3 h; (c)613 BAHA (4.0 equiv), DCM, rt, 1 h, then H2NNH2·H2O; (d)613 dppp (5 equiv), toluene, 120 °C, 1 h.
In 2018, Furukawa, Osuka et al. reported the synthesis of a trimethylenemethane (TMM) diradical fused with three nickel(II) meso-triarylporphyrins (Scheme 329).614 Dialdehyde 329.1 was reacted with two equivalents of a porphyrinylmagnesium reagent derived from 329.1, to afford the diol 329.3 as a 4:1 mixture of diastereomers. The acid-mediated cyclization of 329.3 followed by DDQ oxidation resulted in a highly polar product which was identified as the corresponding radical cation, which was further reduced with ascorbic acid to yield the TMM diradical 329.4. The diradical 329.4 was found be quite stable under ambient conditions because of the extensively delocalized spin density. The X-ray crystal structure of 329.4 revealed a pseudo-C3-symmetric propeller-like conformation, in which the three hetero[5]helicene moieties adopted a twisted helical conformation with the same twist direction. The lower limit of ΔEST from SQUID measurements was estimated for 329.4 to be +2.8 kcal mol–1 (J1/kB ≈ 715 K), consistent with a theoretical value of ΔEST ≈ +3.0 kcal mol–1 (J1/kB ≈ 750 K). These results indicated a triplet ground state, reflecting a strong ferromagnetic interaction between the spins. 329.4 displayed strong absorptions in the NIR region (700–1350 nm) and four-step redox interconversion with a narrow electrochemical gap of 0.48 V.
Scheme 329. Synthesis of a Trimethylenemethane Diradical Fused with Three Porphyrins.
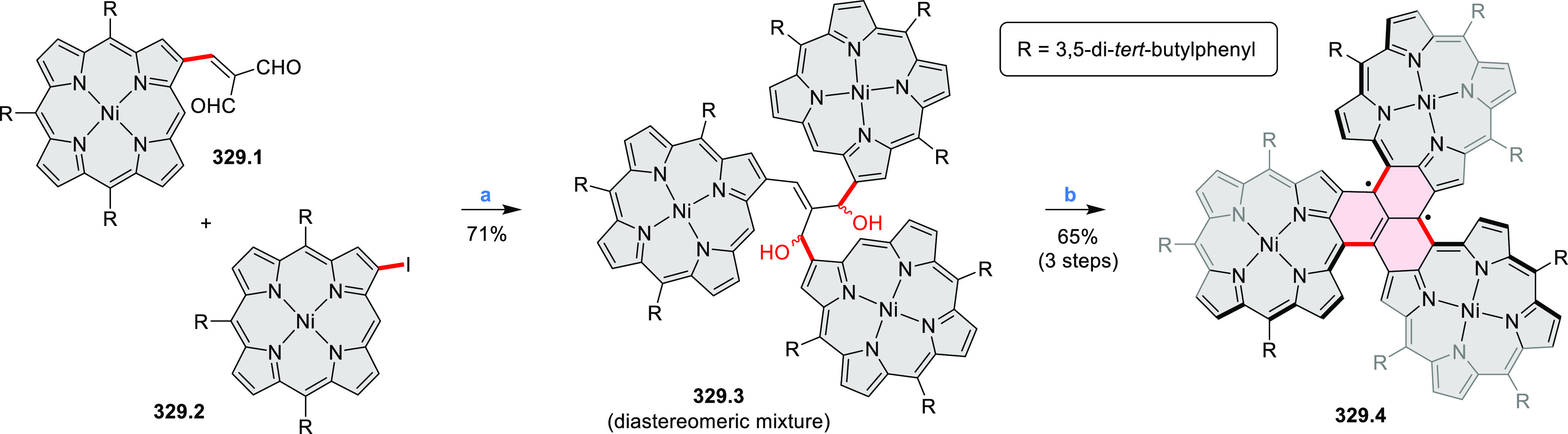
Reagents and conditions: (a)614i-PrMgCl·LiCl (1.1 equiv), THF, −40 °C, then 329.1 (0.4 equiv), −40 °C to rt, 2 h; (b) (1) 25% H2SO4, 1,2-dichloroethane, 60 °C, 11 h, (2) DDQ (10 equiv), DCM, rt, 15 min, (3) ascorbic acid (10 equiv), DCM/MeOH.
2,6-Naphthoquinodimethane- and 1,5-naphthoquinodimethane-bridged porphyrin dimers were synthesized in 2019 by Wu et al. (Scheme 330).615330.1 and 330.2 were treated with 2-mesitylmagnesium bromide to give the corresponding diols, which were then subjected to BF3·OEt2 mediated Friedel–Crafts alkylation reaction to give the dihydro precursors 330.3 and 330.4, respectively. In the subsequent step, the oxidative dehydrogenation of 330.3 and 330.4 with DDQ afforded the target product 330.5 and 330.6, both in 45% yield over three steps. The compound 330.5 showed greater stability, which facilitated its isolation via silica gel column chromatography. 330.6 exhibited a larger diradical character, a smaller singlet–triplet energy gap (ΔEST = −2.60 kcal mol–1) and a smaller two-photon absorption (TPA) cross section value compared to the 330.5 isomer.
Scheme 330. Synthesis of Naphthoquinodimethane-Bridged Porphyrin Dimers.

Reagents and conditions: (a)615 (1) 2-mesitylmagnesium bromide, dry THF, 8 h, rt, (2) BF3·OEt2, dry DCM, 5 min, rt; (b) DDQ, dry toluene, rt.
The syntheses of doubly naphthalene-fused porphyrins and syn- and anti-fused-anthracene-bridged porphyrin dimers were reported in 2021 by Song et al. (Scheme 331).616 In a representative example, 331.5 was reacted with 2-mesitylmagnesium bromide to provide corresponding carbinol, and intramolecular Friedel–Crafts alkylation and subsequent oxidation with DDQ afforded 331.6 in 62% yield. Similarly, other derivatives 331.2, 331.4, 331.8, and 331.10, were obtained from corresponding aldehyde containing porphyrin precursors over three steps. In the case of 331.10, the final oxidation step was carried out using t-BuOK/O2. The monoradical 331.6 and the open-shell syn dimer 331.8 were found to be very stable: the half-life of 331.8 in 1,2-dichlorobenzene under ambient air and at 80 °C was about 28 days, despite its high-spin triplet ground-state carbon diradical character. The electrochemical HOMO–LUMO gaps of 331.6 and 331.8 were estimated to be 0.62 and 0.97 V, respectively.
Scheme 331. Synthesis of Singly and Doubly Naphthalene-Fused Porphyrins and Anthracene-Bridged Porphyrin Dimers.
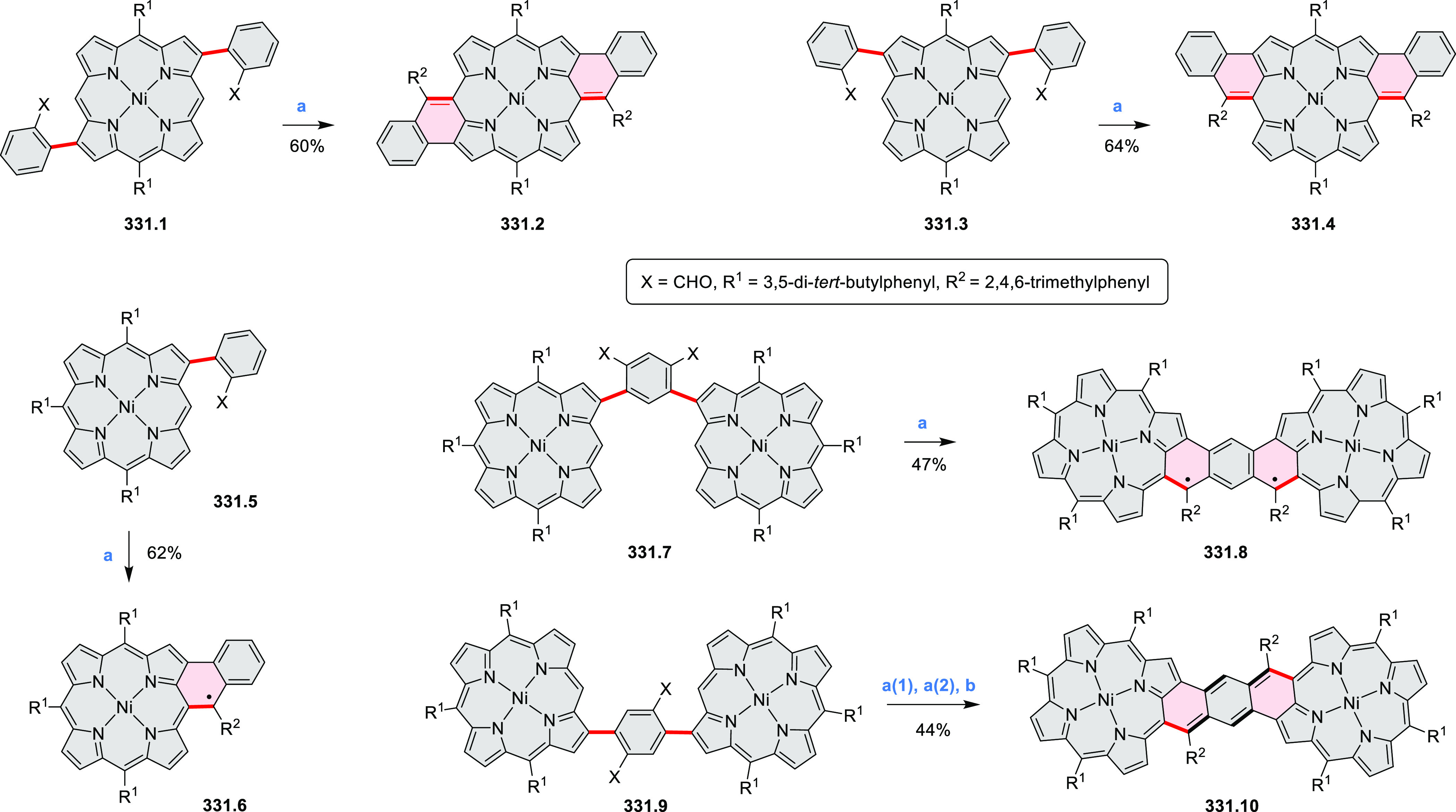
Reagents and conditions: (a)616 (1) 2-mesitylmagnesium bromide, dry THF, rt, (2) BF3·OEt2, dry DCM, (3) DDQ, DCM; (b) t-BuOK, THF.
7.2.6. Oxobenzo- and Oxonaphtho-Fused Porphyrinoids
Bacteriochlorophylls and their derivatives such as 332.1 possess a highly reactive β-ketoester moiety in ring E, which has been used to develop π-extended derivatives with various fused ring systems (Scheme 332).617,618 In 2012, Kozyrev et al. reported the synthesis of bacteriochlorins bearing fused quinoxaline, benzimidazole, and perimidine aromatic rings.619 The synthetic design aimed to induce large bathochromic shifts of Qy absorption bands by simultaneously extending the conjugation on pyrrole C and introducing electron-withdrawing substituents on the opposite pyrrolic unit A. An in situ autoxidation of 332.1 using aqueous LiOH in THF for 24 h followed by an acidic workup and re-esterification with diazomethane, afforded the bacteriochlorin α-diketone 332.3 in 68% yield. Using 332.3 as the starting material, various highly conjugated annulated bacteriochlorins 332.4–6 were synthesized by condensing with diamines in the presence of pyridine/TFA. Additionally, the annulated cyclohexenenone ring systems 332.7–9 were also synthesized using the diazomethane ring-enlargement approach. These derivatives showed the largest bathochromic shifts, with Qy absorptions in the range of 870–890 nm. More recently, radionuclide-containing analogues of 332.7 were investigated as PET imaging agents.620
Scheme 332. Synthesis of Bacteriochlorins Bearing Fused Quinoxaline, Benzimidazole, and Perimidine Aromatic Rings.
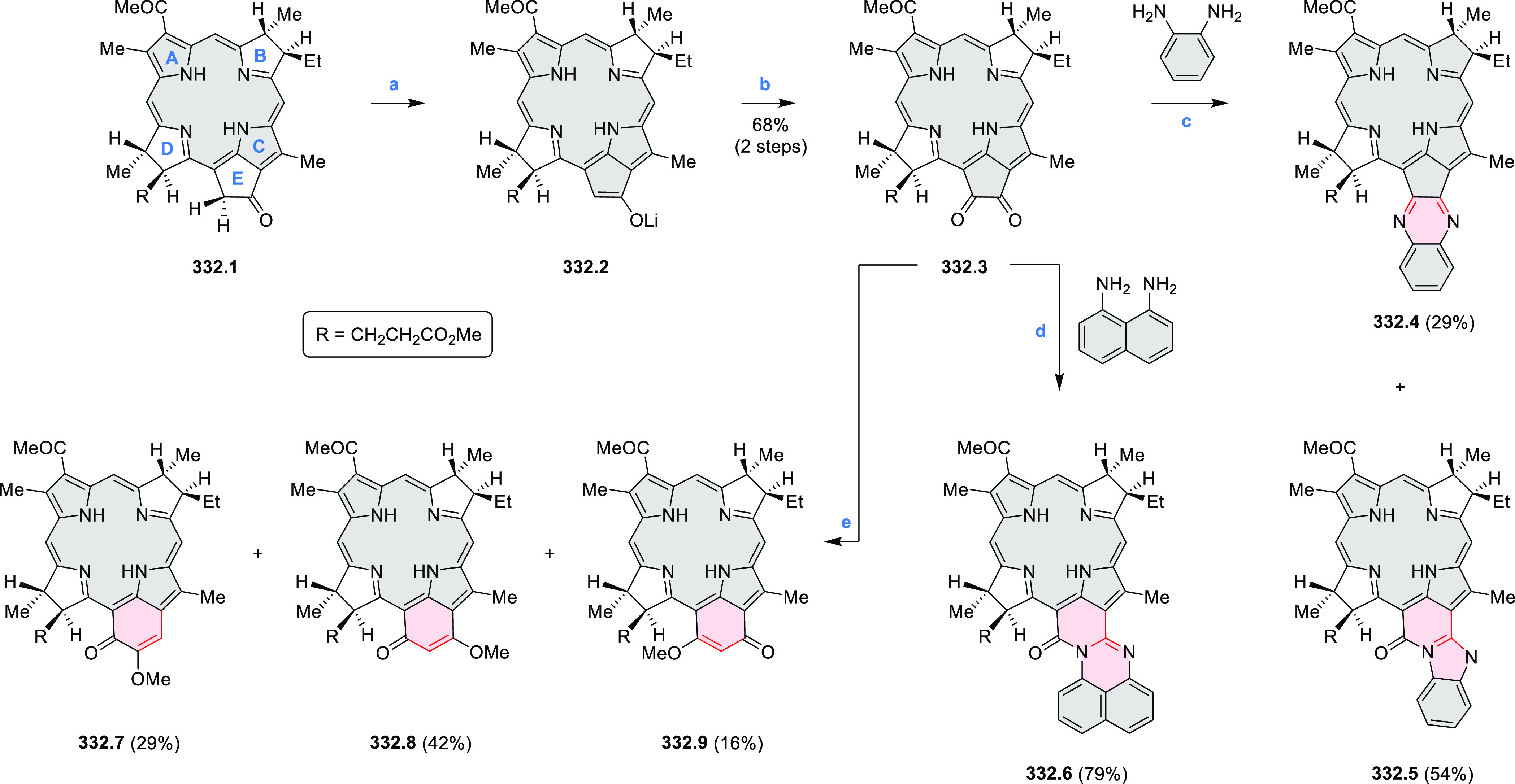
Reagents and conditions: (a)619 LiOH/H2O, THF, 24 h; (b) O2; (c) 1,2-phenylenediamine hydrochloride, pyridine, TFA, reflux; (d) 1,8-diaminonaphthalene hydrochloride, pyridine, TFA, reflux; (e) CH2N2, DCM, N-methyl-N-nitroso-p-toluenesulfonamide, rt, overnight.
In a recent report, Ruppert, Harvey et al. investigated electronic communication in platinum(II)-bridged porphyrin dyads C31.2–3 (Chart 31, for earlier work, see CR2017, Section 7.2.6).621,622 These systems revealed an extremely fast rate of S1 singlet energy transfer with very small S1 lifetimes of 2.1 ps for C31.2b, 0.8 ps for C31.2a, and 105 fs for C31.2c. The rate of singlet energy transfer, kET(S1), for dyad C31.2c was found to be 8.3 × 1012 s–1 which was 5.5-fold faster than that of palladium(II) linked dyad C31.2d. In the enaminothioketone dyad C31.3c the S1 energy transfer process occurred faster than 49 fs, i.e., more rapidly than in C31.2c (M= Pt, X = O, 105 fs) and C31.2d (M= Pd, X = O, 650 fs). The rate of energy transfer for C31.3c (kET(S1)≥ ∼ 15 × 1012 s–1) was found to be higher than ever reported for both natural systems and laboratory model compounds and this enhanced rate was ascribed to a significant increase in MO coupling and predominance of the Dexter mechanism.
Chart 31. Porphyrin Oligomers Linked by Metal Ions.
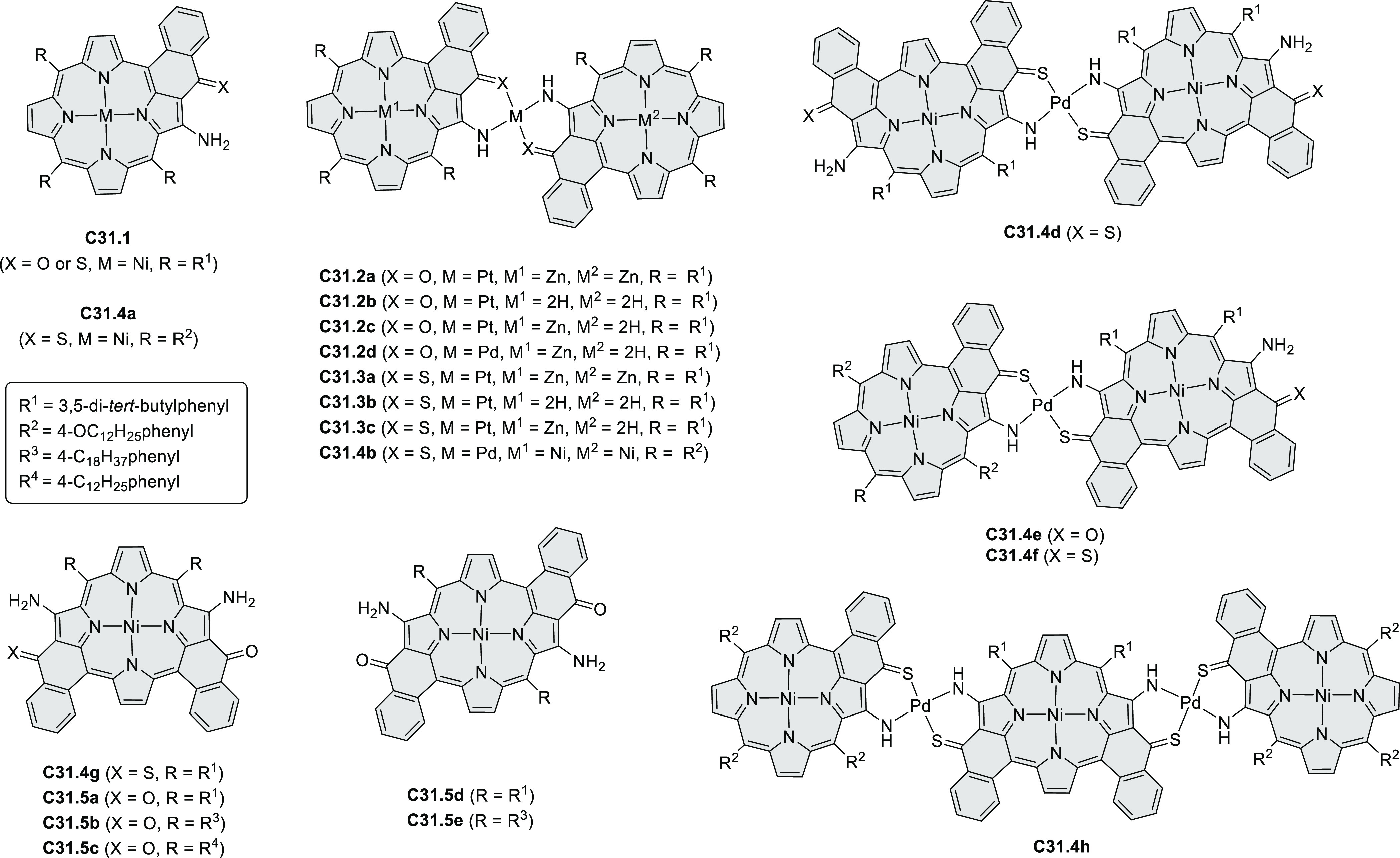
Self-assembly of enaminoketone and enaminothioketone monomers, dimers, and trimers at a highly oriented pyrolytic graphite (HOPG)/liquid interface was explored by Kikkawa and Ruppert (Chart 31).623 Among all the compounds tested, the dodecyloxyphenyl-substituted monomer C31.4a, and dimers C31.4b and C31.4f showed the formation of highly ordered two-dimensional self-assembled structures. In a later report, nanoribbons were built from symmetrical porphyrins C31.5a–c or C31.5d–e stabilized by either hydrogen bonding between the enaminoketone fragments or by coordination of nickel(II).624
7.2.7. Indole- and Carbazole-Based Porphyrinoids
In 2019, Kobayashi and Ng reported a series of air-stable boron(III) carbazosubphthalocyanines, which can be seen as core-expanded subphthalocyanine (SubPc) analogues.625 The synthesis of 333.3a–d was achieved by the condensation of 1,3-diiminoisoindolines 333.1a–d and diamine 333.2, followed by boron-induced complexation and cyclization (Scheme 333). The choice of BF3·Et2O instead of other Lewis acidic boron sources such as BCl3 and BBr3 was needed to obtain the desired product, stabilized by the axial fluoride ligand. These analogues represented the first examples of antiaromatic SubPc derivatives and the smallest antiaromatic azaporphyrinoids.
Scheme 333. Synthesis of Boron(III) Carbazosubphthalocyanines.

Reagents and conditions: (a)625 (1) EtOH, reflux, (2) BF3·Et2O, DBU, 1-chloronaphthalene, 150 °C.
In 2019, Maeda Ema et al. reported π-extended carbazole-based porphyrins containing peripherally fused benzene rings (Scheme 334).626 Ethynyl-substituted isophlorins 334.2a–d were prepared via bromination of 334.1 with NBS followed by Stille coupling. In the next step, Pt-catalyzed intramolecular cyclization of the ethynyl substituent in 334.2a–d afforded the benzo-fused isophlorins 334.3a–d, which were subsequently oxidized to the porphyrin-like 334.4a–d. ortho-Fused analogues 334.10 were synthesized via Glaser coupling of diethynylbenzocarbazole 334.5 and diethynylcarbazole 334.6, which yielded both the unsymmetrical 334.7 and symmetrical carbazole dimer 334.8. In the next step, the annulation reaction of 334.7a with Na2S·9H2O provided 334.9a, which upon further oxidation afforded 334.10a. 334.10b was also synthesized in the same manner. The oxidation of compounds 334.3 and 334.9 showed drastic changes in their electronic properties. The UV–vis–NIR absorption displayed intense broad NIR absorption bands at 963 and 1180 nm for 334.3a and at 910 and 1100 nm 334.9a. Their absorption onset reached over 1400 nm. Compounds 334.3b–d displayed similar spectra while 334.9b showed slightly red-shifted NIR bands revealing the additional fusion moieties clearly perturbed the π-electron systems of 334.3 and 334.9.
Scheme 334. Synthesis of π-Extended Carbazole-Based Porphyrins.
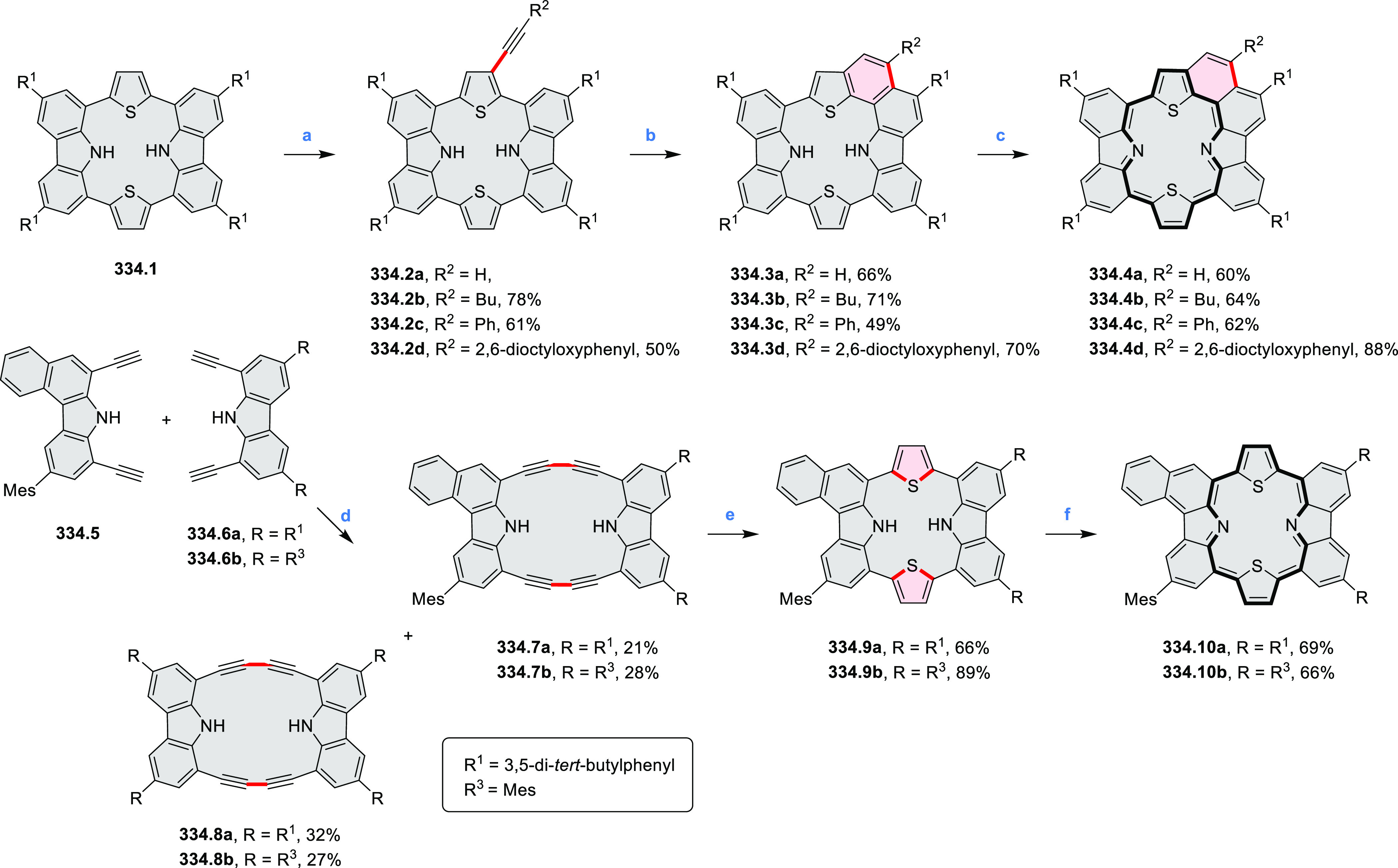
Reagents and conditions: (a)626 (1) NBS, CHCl3, 0 °C, (2) Bu3SnC≡CR2, Pd(PPh3)4, toluene, reflux; (b) PtCl2, toluene, reflux; (c) MnO2, DCM, rt; (d) Cu(OAc)2·2H2O, pyridine, toluene, air, rt; (e) Na2S·9H2O, toluene, 2-methoxyethanol, reflux; (f) PbO2, DCM, rt.
Substituent effects of cyano and ethynyl groups on the photophysical properties of carbazole-based porphyrins 335.1a–c were investigated by Maeda, Yoshioka et al. (Scheme 335).627 Additionally, thia- and selenaporphyrins 335.1d–e bearing electron-donating ethylenedioxy groups were also prepared for the comparison. The introduction of the ethynyl groups into the conjugated macrocycle induced the red-shifts while that of cyano groups induced blue shifts to the lowest-energy bands. Selenaporphyrin 335.2e displayed stronger and red-shifted absorption in the NIR region, with the lowest-energy band at 1178 nm. A chiral analogue 335.2f displayed a CD spectrum with Cotton effects in the NIR region.628 Palladium complexes 335.3a–c of the carbazole-based macrocycles displayed a weak antiaromatic character derived from the 20π isophlorin framework.629 These metal complexes exhibited weak near-infrared (NIR) absorption at 700–1000 nm.
Scheme 335. Carbazole-Based Porphyrins.

Related porphyrinoids containing a single carbazole subunit were reported in 2019 by Kim, Song et al. (Scheme 336).630 The palladium-catalyzed Suzuki–Miyaura cross-coupling reaction between the dibromocarbazole 336.4 and diboryltripyrrane 336.5 followed by MnO2 oxidation afforded the macrocycle 336.3d along with its palladium(II) complex 336.3e (Scheme 336). By extension of this strategy, the directly linked oligomers 336.8 and 336.9 were also synthesized from 336.6a and 336.7a–b. Later in 2020, Gokulnath and co-workers reported an alternative, higher-yielding route to 336.3a–c, based on the [3 + 1] acid-catalyzed condensation,.631 The macrocycles 336.3a and 336.3b exhibited red emission with two characteristic bands from 600 to 850 nm with the fluorescence quantum yield (ΦF) of 7.6% and 12.8%, respectively. The furan-containing macrocycle 336.3a showed a sensing capability toward mercury(II) ions.
Scheme 336. Synthesis of Asymmetric Carbazole-Based Porphyrins and Their Oligomers.
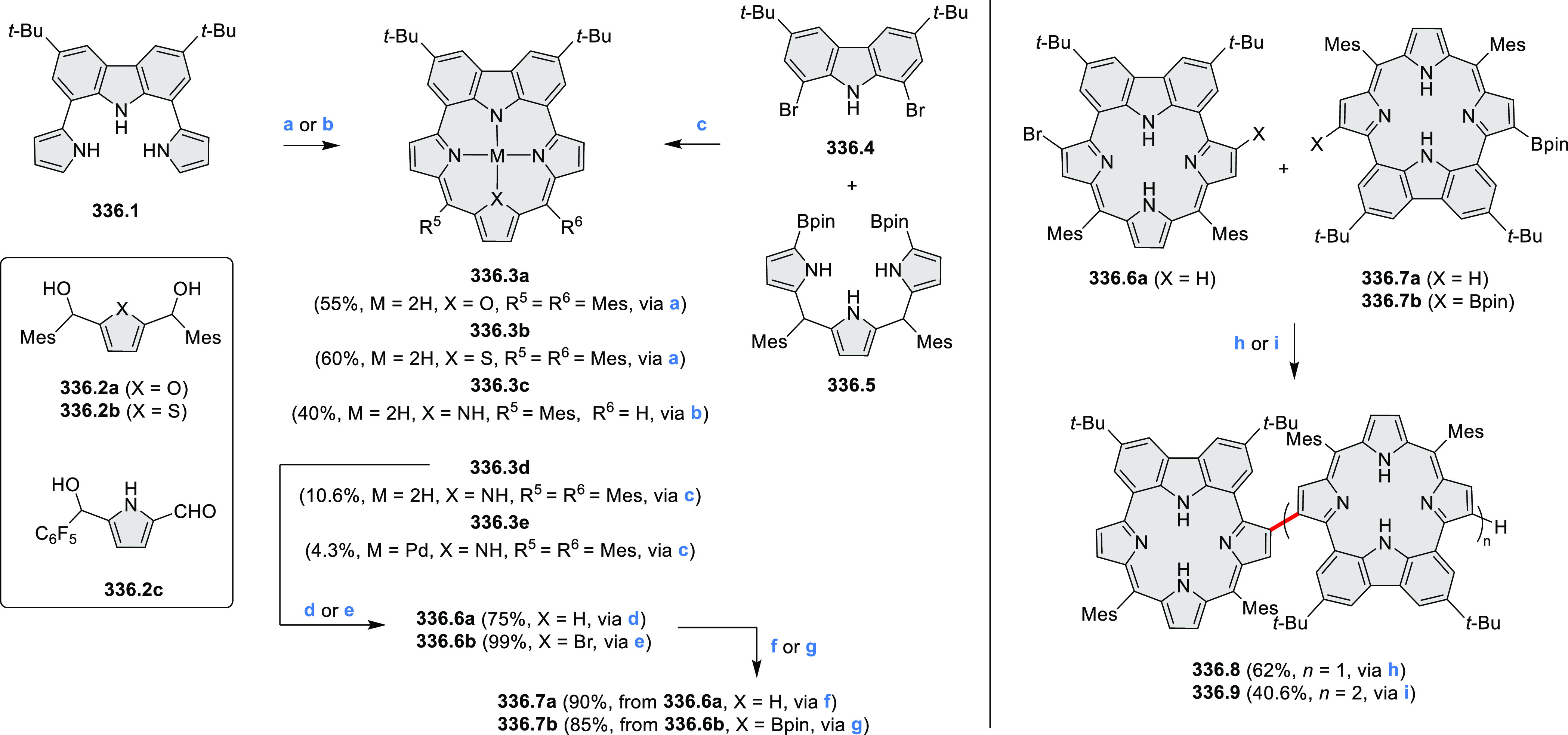
Reagents and conditions: (a)631 (1) 336.2a or 336.2b, p-TSA (0.2 equiv), DCM, rt, dark, 1 h, (2) DDQ (1 equiv), 10 min; (b)631 (1) 336.2c, CF3COOH (1 equiv), DCM, rt, dark, 90 min, (2) DDQ (2 equiv), 10 min; (c)630 (1) Pd2(dba)3, SPhos, CsF, Cs2CO3, reflux, 48 h, (2) MnO2; (d)630 NBS (0.9 equiv), CHCl3; (e)630 NBS (3.7 equiv), CHCl3; (f)630 (Bpin)2 (2.9 equiv), Pd(dppf)Cl2, KOAc, 1,4-dioxane, reflux; (g)630 (Bpin)2 (9.8 equiv), Pd(dppf)Cl2, KOAc, 1,4-dioxane, reflux; (h)630336.6a/336.7a (1:1), Pd2(dba)3, PPh3, CsF, Cs2CO3, toluene/DMF, reflux, 48 h; (i)630336.6a/336.7b (1:0.5), Pd2(dba)3, PPh3, CsF, Cs2CO3, toluene/DMF, reflux, 48 h.
The synthetic route to thiophene-bridged carbazole-based porphyrin dimers, reported by Maeda, Ema, and co-workers, started with the Glaser coupling reaction of ethynyl-substituted carbazole-based isophlorins 334.2a to afford the butadiyne-bridged dimers 337.1 (Scheme 337).632 In the next step, the annulation reaction on 337.1 produced the thiophene-bridged dimers 337.3 in 35% yield. Finally, the oxidation of 337.1 and 337.3 with PbO2 afforded the carbazole-based diporphyrins 337.2 and 337.4, respectively. Diporphyrins 337.5 and 337.6 linked at 3,3′-positions of carbazole moiety were similarly prepared. Absorption spectra indicated that the diporphyrins linked at carbazole moiety 337.5 and 337.6 had stronger intramolecular electronic interactions within the diporphyrins than those of 337.2 and 337.4. Electrochemical HOMO–LUMO gaps of 337.5 and 337.6 were found to be 0.761, and 0.739 eV, respectively.
Scheme 337. Thiophene-Bridged Carbazole-Based Diporphyrins.
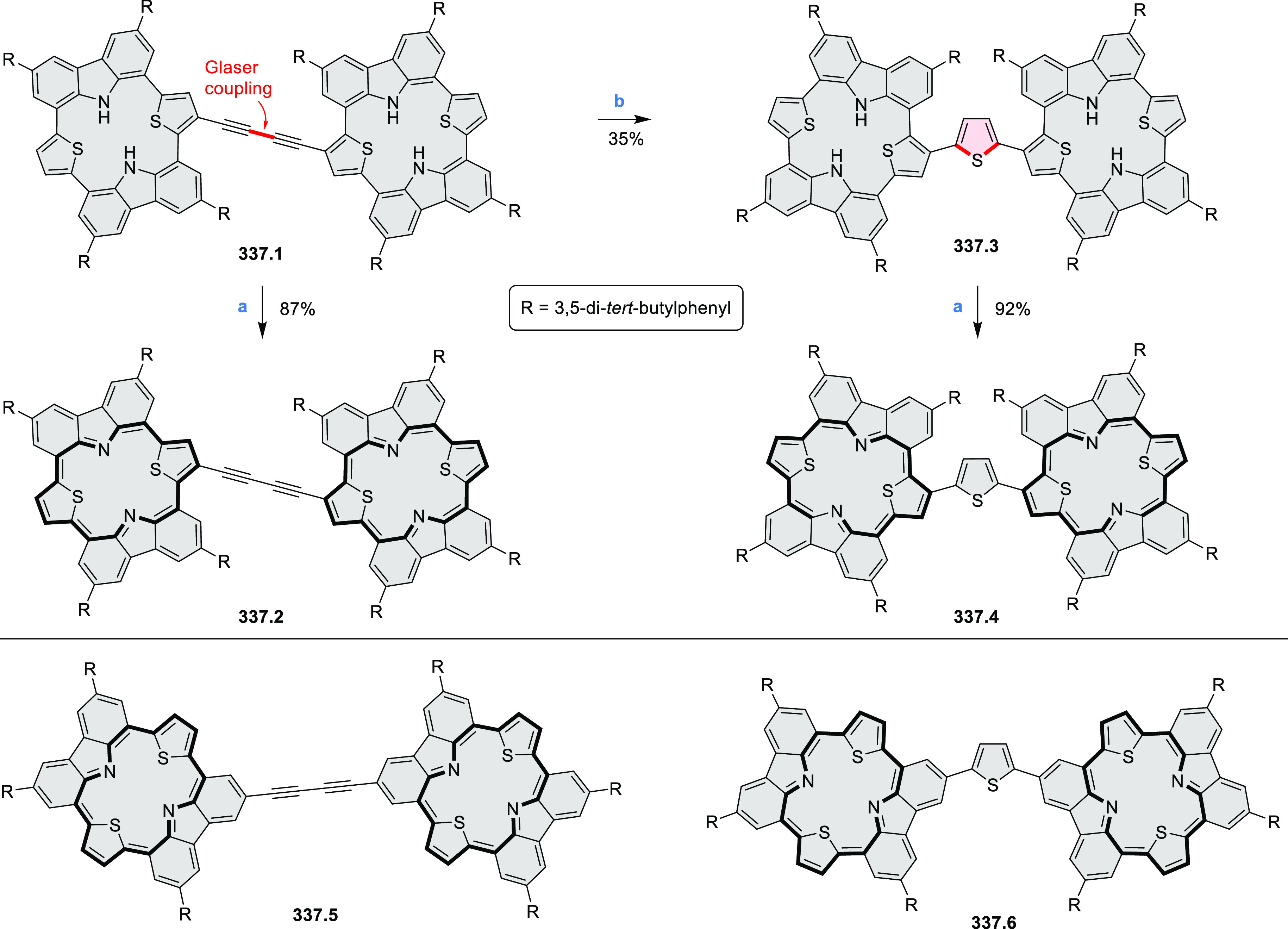
Reagents and conditions: (a)632 PbO2, DCM; (b) Na2S·9H2O, p-xylene, 2-methoxyethanol, reflux.
Directly linked carbazole-based diporphyrin 338.2 and fused diporphyrin 338.4 were obtained using a similar approach, consisting of (i) Glaser coupling, (ii) annulation, and (iii) oxidation of the isophlorin porphyrin (Scheme 338).633 In these macrocycles, the 2,6-dioctyloxyphenyl groups were used to improve the solubility. The UV–vis–NIR absorption spectrum of 338.4 in DCM showed NIR bands at 948, 1085, and 1269 nm, bathochromically shifted in comparison with 338.2 (887, 932, 969, and 1077 nm). This red-shift was ascribed to the effective π-delocalization over the directly fused linkage between the porphyrin-like subunits in 338.4. Cyclic voltammograms of 338.4 showed one oxidation wave at −0.012 V and two reductions at −0.535 and −0.837 V, corresponding to a narrow electrochemical gap of 0.523 eV.
Scheme 338. Directly Linked and Fused Carbazole-Based Diporphyrins.
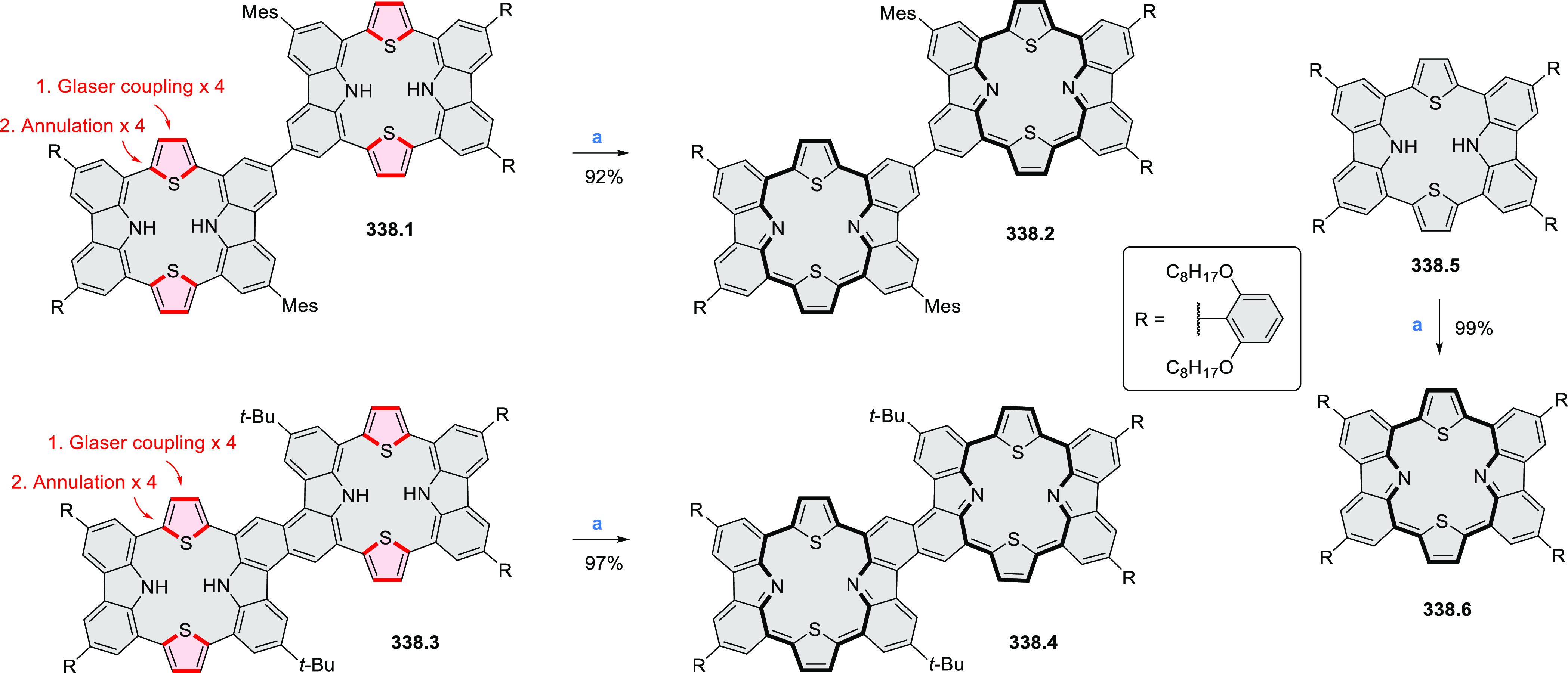
Reagents and conditions: (a)633 PbO2, DCM.
Maulbetsch and Kunz reported a synthesis of “carbenaporphyrins” via 1,3-dipolar cycloaddition (Scheme 339).634 The azide precursor 339.2 was obtained from a Sandmeyer-type reaction and subjected to copper-catalyzed 1,3-dipolar cycloaddition with 339.1, to produce the macrocycle 339.3a. The latter system had a nonplanar geometry in the solid state, with the carbazole and triazole planes tilted in opposite directions to avoid steric congestion of C–H and N–H bonds. 339.3a was alkylated by Meerwein’s salt to yield 339.3b in nearly quantitative yield. Deprotonation of 339.3b with LiHMDS led to the lithium complex 339.4a. Then, transmetalation with ScCl3 resulted in the desired scandium complex 339.4b, which exhibited orange fluorescence. The latter species could be further converted into the Cp derivative 339.4c via ligand exchange.
Scheme 339. Synthesis of Carbenaporphyrins.
Reagents and conditions: (a)634 CuSO4·5H2O, tris((1-benzyl-4-triazolyl)methyl)amine, sodium ascorbate, Et3N, THF, Ar; (b) 2Me3OBF4, DCM; (c) LiHMDS (6.6 equiv), THF-d8; (d) ScCl3(THF)3, THF-d8; (e) LiCp, THF-d8; (f) LiHMDS, THF, LiCp.
7.3. Naphtho[2,1,8,7-cdef]-fused porphyrinoids
7.3.1. Monoporphyrinoids
In 2020, Mai reported a doubly anthracene-fused porphyrin 340.4 as a photocatalyst visible light-induced aerobic oxidation of amines to imines under ambient conditions (Scheme 340).635 In the final step of their synthesis, cyclodehydrogenation of the anthryl-substituted porphyrin 340.3 using DDQ and a catalytic amount of triflic acid produced 340.4 in a high yield of 97%. These cyclodehydrogenation conditions were more efficient than employing FeCl3 and catalytic AgOTf/DDQ. UV–vis–NIR absorption spectrum of 340.4 in THF displayed a large red-shift in the lowest-energy wavelength band compared to 340.3, with Soret-band absorption at 565 nm and Q-bands in the NIR region (850 to 1150 nm).
Scheme 340. Synthesis of Bisanthracene-Fused Porphyrins.

Reagents and conditions: (a)635 NBS, DCM/MeOH (10:1, v/v); (b) 9-anthraceneboronic acid, Pd(PPh3)4, K2CO3, toluene/DMF (5:1, v/v), 120 °C; (c) TfOH, DDQ, DCM.
Diphenylmethane-fused porphyrin 341.3a, reported in 2016 by Osuka et al., was obtained in a two-step reaction sequence beginning with a SNAr reaction of 341.1 with diphenylmethyllithium which produced 341.2 in 65% yield (Scheme 341).636 Then, a 2-fold palladium-catalyzed intramolecular C–H arylation of 341.2 afforded 341.3a with a doubly fused structure with a loss of one hydrogen atom at the diphenylmethane segment. Based on spectroscopic evidence, compound 341.3a was identified as the neutral porphyrin radical, which was found to be very stable under ambient conditions. Demetalation of 341.3a resulted in the freebase radical 341.3b, which was likewise air- and moisture-stable. The UV–vis–NIR absorption spectra of these compounds in DCM displayed a relatively weak Soret-like band at 481 nm and Q-like bands at 582 and 620 nm for 341.3a, and a broadened Soret-like band at 475 nm with Q-like bands at 564 and 633 nm for 341.3b, along with several weak and broad bands reaching up to 1500 nm. The observed low-energy broad absorption bands in the UV–vis–NIR were indicative of the presence of radical character in these porphyrins. Excited-state dynamics of the radical porphyrins were studied by femtosecond transient absorption (TA) measurements, which showed two decay components with lifetimes of 0.5 and 8 ps for 341.3a, and 0.3 and 9 ps for 341.3b, suggesting the lowest excited state relaxation to the open-shell ground state. In 1H NMR titration experiments, chemical oxidation and reduction of 341.3a (and 341.3b) led to the generation of cationic 341.4a–b and anionic species 341.5a–b with perturbed electronic properties.
Scheme 341. Synthesis of Diphenylmethane-Fused Porphyrins.
Reagents and conditions: (a)636,637 (2-R1-C6H4)2CHLi (2.0 equiv), THF, −98 °C to rt, 2 h; (b)636,637 Pd(OAc)2, PCy3·HBF4, K2CO3 (5.0 equiv), toluene, reflux, overnight; (c)636 H2SO4/CF3CO2H, 0 °C to rt, 45 min; (d)636 BAHA, CDCl3, rt; (e)636 cobaltocene, [D8]THF, rt.
Later in 2017, Furukawa, Osuka et al. reported helically chiral analogues of these diphenylmethane-fused porphyrins.637 The preparation of rac-341.6b–c and rac-341.7a–b from more sterically hindered diarylmethane derivatives, containing either ortho substituents, or additional benzene rings. The formation of rac-341.6b–c was accompanied by partial rearrangement (type B products, Scheme 341). The crystallographic analysis of rac-341.7a revealed a fully conjugated framework containing a [6]helicene unit. Experimental bond lengths as well as DFT calculations indicated that the radical was stabilized by full delocalization over the whole fused π-system.
In 2016, Tanaka, Kim, Osuka et al. developed a synthesis of di-peri-dinaphthoporphyrin (Scheme 342).638 Initially, 342.2a was obtained by the Ramirez olefination of 342.1 with tetrabromomethane in the presence of triphenylphosphine, which was then metalated with a nickel salt to give the 342.2b complex. Then, 342.2b was subjected to Stille coupling with 8 equiv of tributyl(phenylethynyl)tin to yield the quinodimethane-type porphyrin 342.3b. The treatment of 342.3b with 0.6 equiv of PtCl2 afforded a 2-fold cyclized product 342.4b in 21% yield, whereas when 2 equiv of PtCl2 was used, the 4-fold cyclization product 342.5b was formed in low yield along with many side products. This complex reaction mixture hampered the isolation of 342.5b in a pure form. Compound 342.5b could instead be obtained in up to 11% yield using 8 equiv of a different platinum reagent, Pt(MeCN)2Cl2.
Scheme 342. Synthesis of Di-peri-dinaphthoporphyrin Porphyrins.
Reagents and conditions: (a)638 CBr4, PPh3, toluene, 80 °C, 12 h; (b) M = Ni: Ni(acac)2, toluene, reflux, 3 h; M = Zn: Zn(OAc)2·2H2O, DCM, MeOH, rt, 1 h; (c) tributyl(phenylethynyl)tin, Pd2(dba)3, P(2-furyl)3, toluene, 80 °C, 1 h; (d) PtCl2 or Pt(MeCN)2Cl2, toluene, 90 °C, 12 h; (e) HCl, DCM, quantitative yield.
Synthesis of the free base porphyrin 342.5a was instead achieved starting from the 342.2c precursor by first converting to 342.3c, then cycloisomerization with 2 equiv of the platinum reagent and subsequent demetalation of 342.5c. The UV–vis spectrum of 342.5b exhibited a sharp Soret-like band at 522 nm and a red-shifted band at 897 nm with a very weak absorption tail reaching to around 1300 nm. Such weak absorption tails were referred to the presence of NIR dark states, supporting the 24 π antiaromatic circuit as the dominant resonance contributor in 342.6. This conclusion was also supported by the combined spectroscopic investigations from 1H NMR, cyclic voltammetry, and DFT calculations. An alkyl-substituted di-peri-dinaphthoporphyrin 342.8 was obtained following a similar synthetic pathway.639342.8 exhibited slightly shorter intermolecular packing distance in the solid state (ca. 3.402 Å) and showed blue-shifted absorption relative to its phenyl-substituted analogue.
Mono- and bis(diphenylborane)-fused porphyrins were obtained through a sequence of (i) Si–B exchange reaction, (ii) intramolecular bora-Friedel–Crafts reaction, and (iii) ring-closing Si–B exchange (Scheme 343).640 The mono- (diphenylborane)-fused nickel(II) porphyrin, 343.2a underwent demetalation under strongly acidic conditions to afford 343.2b, which was further metalated with Pd, Cu, and Zn salts, yielding 343.2a–e. The bis(diphenylborane)-fused porphyrin 343.4 was synthesized from 343.3 in a similar manner. Absorption spectra of mono and bis(diphenylborane)-fused porphyrins 343.2a–e and 343.4 showed significant red-shifts compared to the reference porphyrin with no boron-fusion. This effect was ascribed to extended electronic interactions between the empty p-orbital of the boron atom of the fused diphenylborane group and the porphyrin π-network. The addition of pyridine at the boron center in the porphyrin disrupted of the electronic interaction of the boron atom as indicated by UV–vis titration experiments. Compound 343.2e interacted with Lewis bases, displaying regioselective binding of DMAP at the boron atom and 3,5-difluoropyridine at the zinc center.
Scheme 343. Synthesis of Diphenylborane-Fused Porphyrins.

Reagents and conditions: (a)640 BBr3, o-dichlorobenzene, 80 °C; (b) TFA/H2SO4/DCM, 0 °C; (c) PdCl2(PhCN)2, PhCN, 140 °C; (d) Cu(OAc)2, DCM/MeOH, rt; (e) Zn(OAc)2·2H2O, DCM/MeOH, rt.
A triphenylsilane-fused Ni(II) porphyrin in which the meso-linked silicon atom was embedded in a triply linked skeleton was subsequently reported by the same group.641 The synthesis of phosphorus analogues, summarized in Scheme 344, involved a sequence of nucleophilic aromatic substitution, oxidation, and palladium-catalyzed intramolecular cyclization.642 In the latter step, double cyclization via intramolecular C–H arylation of 344.3 using palladium–pivalic acid catalytic system gave the doubly cyclized product 344.4a in 45% yield. The success of this cyclization could be attributed to the electron-deficient nature of the phenyl groups at the diphenylphosphine oxide moiety. 344.5a was obtained by the reduction of 344.4a with HSiCl3 in 85% yield. The zinc(II) complex of the diphenylphosphine-fused porphyrin 344.5a could not be obtained because of its unstable nature. In the solid state, 344.4a and 344.5a showed planar structures. 344.5a formed antiparallel face-to-face dimer stabilized by P–Ni bonds, while 344.4b formed an analogous dimer stabilized by O–Zn bonds. The embedded P = O and P centers acted as an electron-accepting and an electron-donating unit, respectively, which perturbed the optical and electronic properties of nickel(II) porphyrins. According to an NMR spectroscopic investigation in CDCl3, the dimerization was entropy-driven, with a large entropy gain of ΔSD = 207 J K–1 mol–1. Later in 2017, the same group synthesized a mesityl-substituted derivative 344.6 and heterocycle-containing analogues 344.7–13 with a goal of stabilizing a planar phosphorus(III) center embedded in a fused aromatic skeleton.643
Scheme 344. Synthesis of Diphenylphosphine-Fused Porphyrins.
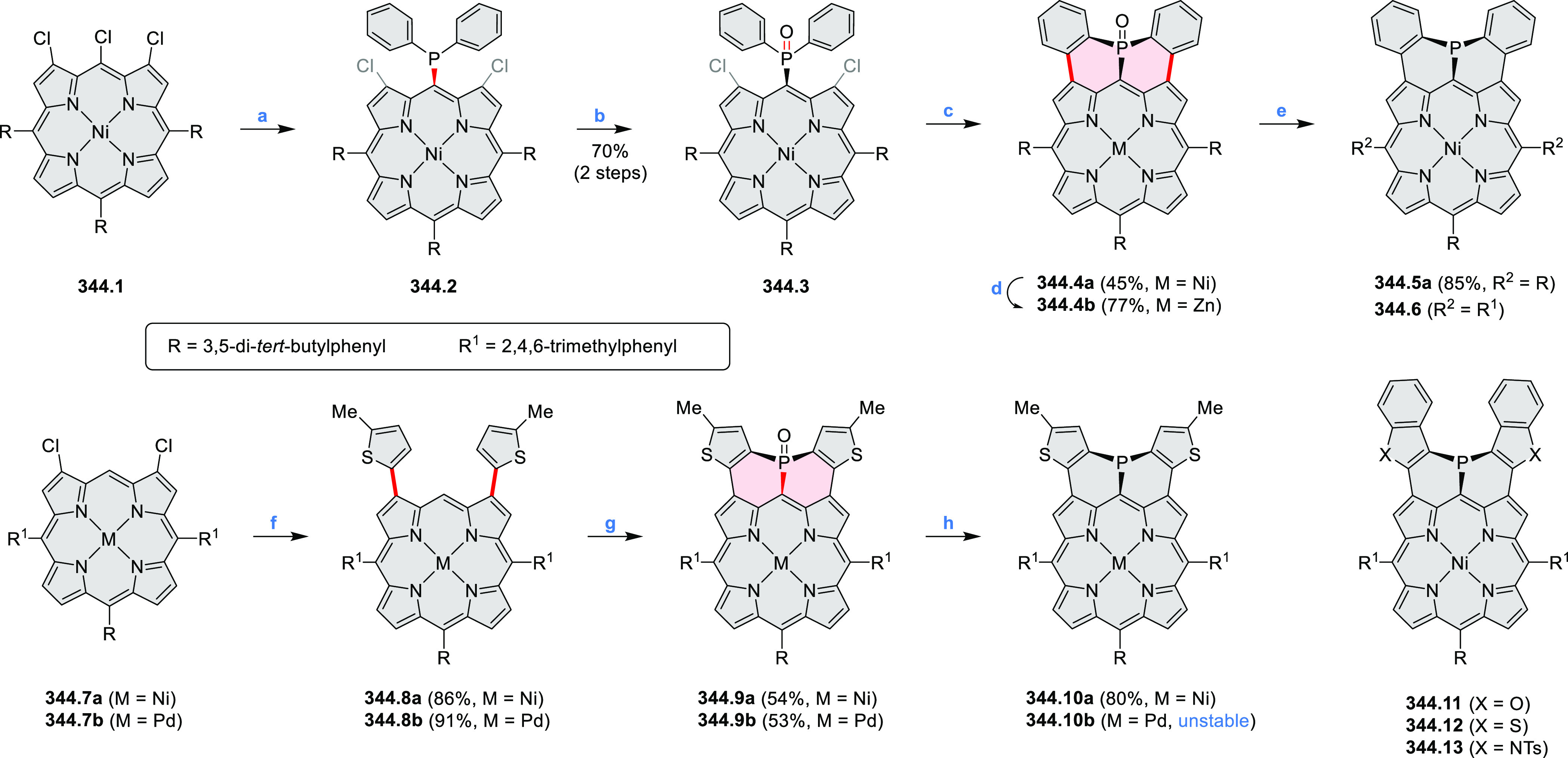
Reagents and conditions: (a)642 LiPPh2, THF, rt; (b)642 H2O2, DCM, rt; (c)642 Pd(OAc)2, PCy3·HBF4, t-BuCO2H, K2CO3, DMA, 120 °C; (d)642 conc. H2SO4, TFA, 0 °C, and then, Zn(OAc)2·2H2O, DCM/MeOH, rt; (e)642,643 HSiCl3, toluene, reflux; (f)643 5-methyl-2-thienylzinc chloride lithium chloride, Pd2(dba)3, RuPhos, THF, reflux; (g)643 PBr3, ZnI2, o-dichlorobenzene, 180 °C; then, H2O2; (h)643 HSiCl3, toluene, rt.
In 2018, Müllen, Narita et al. reported the synthesis of the β-, meso-, β-triply fused porphyrin-nanographene conjugates 345.5b and 345.6b by fusing porphyrin with π-extended HBCs having two extra K-regions.644 These π-extended electronic networks were expected to facilitate the electron delocalization over the porphyrin and HBC units, thus narrowing the HOMO–LUMO gap and producing large red-shifts in the UV–vis–NIR absorption spectra. Boronate 345.1 was subjected to a Suzuki coupling reaction with 345.2–b, which afforded the corresponding one- and 2-fold coupling products 345.3a–b and 345.4a–b (Scheme 345). The mesityl-substituted 345.3b and 345.4b underwent oxidative cyclodehydrogenation with DDQ/TfOH to provide the triply fused porphyrin-nanographene conjugates 345.5b (90%) and 345.6b (97%), respectively. The attempts to obtain the corresponding free bases using trifluoroacetic acid and sulfuric acid resulted in decomposition. The UV–vis–NIR absorption of 345.5b in THF showed the maximum absorption peak at 866 nm with the tail extended up to around 1000 nm, while 345.6b showed largely red-shifted absorption with the tail extending to approximately 1400 nm upon fusion of another nanographene unit to the porphyrin core. The planar structure of 345.6b was validated by STM measurements which also revealed a self-assembled network at the interface of 1,2,4-trichlorobenzene (TCB) and highly oriented pyrolytic graphite (HOPG).
Scheme 345. Synthesis of Triply Fused Porphyrin–Nanographene Conjugates.
Reagents and conditions: (a)644 (1) Pd(PPh3)4, K2CO3, toluene/DMF (1:1, v/v), 110 °C, 36 h, (2) Pd(PPh3)4, triethylamine, formic acid, toluene, 100 °C, 2 h; (b) DDQ, DCM/triflic acid (100:1, v/v), rt, 10 h.
7.3.2. Porphyrin Tapes
In 2016, Yorimitsu, Kim, Osuka et al. described a regioselective oxidative fusion of 10,15,20-triaryl nickel(II)-porphyrins bearing electron-withdrawing substituents at the 5-position (Scheme 346).645 Upon oxidation with DDQ and FeCl3, fusion of the 10- and 20-aryl groups at the 12- and 18-positions was observed, with selectivities dependent on the character of the meso substituents. The meso–meso linked diporphyrin 346.5 was obtained by Ni(0)-mediated reductive homocoupling of 346.2c. The zinc complex 346.6 prepared via demetalation of 346.5 was subjected to the oxidative fusion reaction with DDQ and Sc(OTf)3 which afforded 346.7 in 60% yield. This quadruply phenylene-fused diporphyrin displayed red-shifted absorption bands in the near-infrared region, smaller optical and electrochemical HOMO–LUMO gap, and a larger TPA cross section value than the diporphyrin analogues lacking phenylene fusion. In 2020, Jux et al. reported similar cyclizations on the symmetrically substituted nickel(II) meso-tetra(3,5-di(tert-butyl)phenyl)porphyrin.646 The singly or doubly fused products, C30.1 or C30.2, could be obtained by varying the amount of the FeCl3 oxidant and reaction time (Chart 30).
Scheme 346. Synthesis of Doubly and Quadruply Phenylene-Fused Porphyrins.
Reagents and conditions: (a)645 DDQ, FeCl3, DCM/MeNO2, rt; (b) DDQ, FeCl3, DCM/MeNO2, rt, 4 h; (c) NCS, CuCl, DMF/toluene, 80 °C, 1 h; (d) NEt3/HCOOH, Pd2(dba)3, SPhos, toluene, 120 °C, 13 h; (e) Ni(cod)2, DMF, 100 °C, 9 h; (f) (1) TFA/conc. H2SO4, 0 °C, 30 min, (2) Zn(OAc)2·2H2O, DCM, MeOH, rt, 1 h; (g) DDQ, Sc(OTf)3, toluene, 70 °C, 1 h.
A fully π-conjugated macrocycle containing triply linked porphyrin tape units was reported by Anderson and co-workers.647 A K-shaped tetrapyridyl template was employed to synthesize the cyclic porphyrin octamer C32.1 containing six butadiyne links and two β,meso,β-fused connections (Chart 32). The formation of the nanoring C32.1 with 2:2 complex of porphyrin tetramer precursor and the template was confirmed by 1H diffusion NMR spectroscopy of the complex. The UV–vis–NIR spectra of C32.1 nanoring showed the lowest absorption energy band at 1304 nm indicating the π-conjugation is extended around the whole macrocycle; with a dominant contribution of S0 → S2 transitions, as predicted by TD-DFT calculations. NMR-monitored titrations with oxidants revealed that C32.1 exhibited global antiaromatic and aromatic ring currents, in the [C32.1]4+ and [C32.1]6+ oxidation states, respectively.
Chart 32. Porphyrin Nanobelt from Triply Linked Porphyrin Tapea.

a meso-Aryl rings and double bonds are omitted for clarity (meso-aryl = 3,5-bis(trihexylsilyl)phenyl).
Triply linked corrole dimers were described in 2016 by Tanaka, Kim, Osuka et al.612 The synthesis was achieved by intramolecular oxidative fusion reaction of 347.1a–c with DDQ, which gave the target dimers 347.3a–c in 39% yield (Scheme 347). High-dilution conditions were essential to avoid undesirable intermolecular oxidative couplings. The UV–vis–NIR spectrum of the fused dimer 347.3a in DCM exhibited split Soret-like bands at 520 and 546 nm and a broad band at 653 nm along with a weak absorption tail extending into the NIR region. It was found that the molar absorption coefficient of 347.3a was distinctly weaker than that of the meso-meso linked corrole dimer 347.1a, presumably because of the nonaromatic nature of the former species. Metalation of 347.3a,c gave the corresponding zinc(II) fused dimers 347.5a,c which were also characterized as nonaromatic complexes. 347.3c was reduced with NaBH4 to give 347.4c, characterized by corrole-like oxidation level, which however reverted to 347.3c under aerobic conditions. Spectroscopic investigations involving 1H NMR, UV–vis absorption spectroscopy and DFT calculations indicated the recovery of moderate aromaticity in 347.4c upon reduction.
Scheme 347. Synthesis of Triply Linked Corrole Dimers.
Reagents and conditions: (a)612 DDQ (3.5 equiv), CHCl3 (ca. 0.03 mM), reflux, 30 min; (b)648p-chloranil (1 equiv), CHCl3, reflux, 15 min; (c)648 DDQ (2 equiv), CHCl3, reflux, 30 min; (d) NaBH4, THF/MeOH; (e) air; (f)612,649 M = Zn: Zn(OAc)2·2H2O, CHCl3, reflux, 20 h; M = Cu: Cu(acac)2 (30 equiv), toluene/MeOH, reflux, 20 h; (g)649 AgBF4 (2 equiv), DCM/EtNO2, rt, 5 min; (h)648 Co(OAc)2·2H2O, pyridine, 100 °C, 15 min; (i)648 DDQ (3.5 equiv), CHCl3, reflux, 30 min; (j) p-chloranil (1 equiv), CHCl3, reflux, 3.5 h.
It was subsequently found that the radical dimer 347.2a was formed from 347.1a upon using mild oxidant, p-chloranil, in refluxing CHCl3 with a short reaction time (Scheme 347).648 This diradical dimer could be further oxidized with DDQ, producing 347.3a in 83% yield. 347.2a was found be stable under ambient conditions, which was ascribed largely to the strong intramolecular antiferromagnetic interaction of the two spins, with the experimental spin-exchange integral JS–T value of −2.19 kcal·mol–1. The UV–vis absorption spectrum of 347.2a in DCM exhibited a Soret-like band at 386 nm and a broad band around 520 nm along with a broad NIR band with a maximum at 981 nm, in agreement with the diradical structure.
The copper complex 347.6a was subsequently obtained from 347.3a in a reaction with Cu(acac)2 in toluene under reflux conditions (Scheme 347).649 Oxidation of 347.6a with 2 equiv of AgBF4 afforded 347.7a, which was characterized as a dicationic bis-copper(II) species. Oxidation of the meso–meso linked cobalt corrole dimer 347.8a containing two axially coordinated pyridines with DDQ resulted in the formation of two products 347.9a and 347.10a.648 The major product 317 was found to be a meso–meso-linked cobalt corrole dimer and the minor one 347.10a was a fused dimer bearing pyridine and cyanide as axial ligands. An attempt to obtain the triply fused cobalt corrole dimer 347.11a from 347.10a was not successful. The authors proposed that the cyanide anion could be derived from the decomposed DDQ, which was later supported by a ligand exchange reaction with aqueous NaCN. A SQUID measurement on 347.9a revealed a strong antiferromagnetic interaction with JS–T = −1.76 kcal·mol–1, suggesting a similar singlet diradical character as in 347.2a. The UV–vis–NIR absorption spectrum of 347.9a in DCM(containing 1% pyridine) was found to be similar to that of 347.2a, with a broad NIR band at 1115 nm, whereas the fused corrole dimer 347.10a displayed panchromatic absorption with a weak absorption tail reaching up to 1500 nm.
Gallium(III) complexes of these triply linked corrole dimers were later found to assemble into unique face-to-face π-dimers (Scheme 348).650348.1b–c were obtained from 347.3b–c in two steps, via initial reduction with NaBH4 followed by metalation with GaCl3 in pyridine. In the solid state, 348.1b was found to be a dimeric, with two Ga(III) corrole dimers bridged by apically coordinated hydroxy groups. The 1H NMR spectra of these dimers showed very broad peaks, indicating their open-shell character, which was further confirmed by ESR spectroscopy. SQUID measurements indicated the structures 348.1b and 348.1c to be singlet diradicals with spin-exchange integral values (JS-T) of −475 cm–1 (−684 K) and −447 cm–1 (−643 K), respectively. The two radicals were antiferromagnetically coupled by through-space interaction between the two corrole tapes, which was apparently reflected in the short face-to-face distance (3.24 Å). In line with its diradical character, 348.1b showed a strong Soret-like band at 611 nm along with weak and broad bands with peaks at 1900 nm and exhibited a narrow electrochemical band gap of 0.37 V. Dimer 348.1b underwent nucleophilic addition to form the diamagnetic monomers 348.2b and 348.3b upon reacting with para-ethoxycarbonylphenyl zinc iodide lithium chloride complex.
Scheme 348. Synthesis of Face-to-Face Dimers and Monomeric Gallium(III) Corrole Tapes.

Reagents and conditions: (a)650 (1) NaBH4, THF/MeOH, (2) GaCl3 (50 equiv), pyridine, reflux, workup; (b) R1ZnI·LiCl (20 equiv), TMSCl (40 equiv), THF, reflux.
In 2019, Ogawa reported the synthesis of dinuclear triple-decker metal complexes with a fused diporphyrin and two tetraphenylporphyrin ligands in their neutral, dianionic, and diprotonated forms (Scheme 349).651349.2, obtained via lithiation of 349.1, was reacted with 349.3a to yield the terbium(III) complex 349.4a along with a mixture of quadruple-decker and quintuple-decker complexes. The Yb(III) complex 349.4b was obtained via a similar synthetic protocol. The neutral forms of these metal complexes had large open shell biradical characters, with the experimental spin-exchange integral J = −1.4 kcal mol–1 determined for 349.3b using the magnetic susceptibility measurements. These neutral complexes possessed very small HOMO–LUMO gaps (0.33–0.36 eV) and showed multiple redox states.
Scheme 349. Synthesis of Triple Decker Complexes Containing a Triply Linked Diporphyrin.
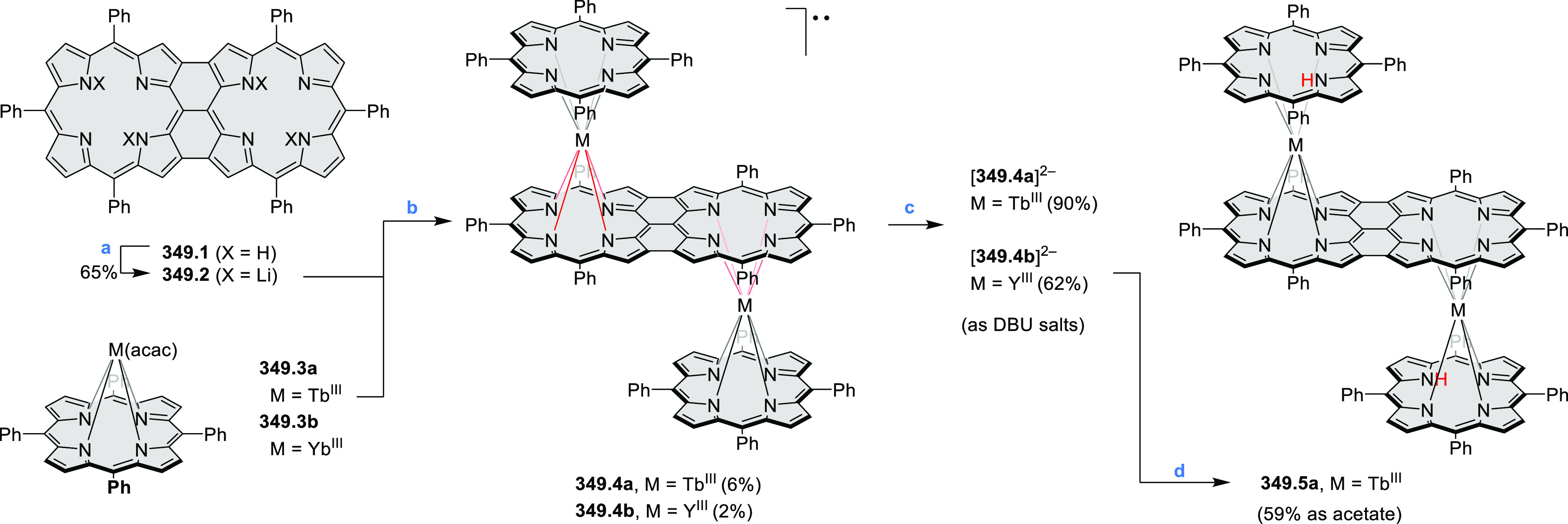
Reagents and conditions: (a)651 LiHMDS (6.6 equiv), 1,2,4-trichlorobenzene 120 °C, 1 h; (b) 349.3a (2 equiv) or 349.3a (2 equiv), 1,2,4-trichlorobenzene 120 °C, 2 h; (c) DBU (116 equiv), DCM, rt, 1 h; (d) CH3COOH (excess), DCM, rt, N2, 1 h
The preparation of electron-deficient hybrid porphyrin tapes, ADA and AAA comprising meso-3,5-di-tert-butylphenyl-substituted Zn(II)-porphyrins (D) and meso-pentafluorophenyl-substituted Zn(II)-porphyrins (A) was described by Kim, Osuka et al.652 The syntheses were achieved via cross-condensation of meso-formyl porphyrins 350.1–2 with oligopyrromethanes 350.3a–b (Scheme 350). For instance, acid-catalyzed condensation of mono-formyl porphyrin 350.1 and 5-(pentafluorophenyl) dipyrromethane 350.3a followed by the DDQ oxidation afforded the trimeric nickel(II) porphyrin 350.4a, which was subsequently converted into the zinc(II) complex 350.4b. In the subsequent step, the oxidative fusion of 350.4b using the combination of DDQ and Sc(OTf)3 resulted in the formation of the desired AAA-type porphyrin tape 350.6a in 48% yield. ADA and ADDA porphyrin tapes, 350.6b and 350.9a, were similarly prepared. The UV–vis–NIR spectrum of 350.9a displayed a significant bathochromic shift (Q-like bands at 1400 and 1657 nm) and a large value of TPA cross-section of 1400 GM.
Scheme 350. Synthesis of Porphyrin Tapes.
Reagents and conditions: (a)652 (1) 350.3a, CH3SO3H (15 mol %), (2) DDQ (3 equiv), DCM, 0 °C, 2 h, then rt 15 min; (b) (1) 350.3b, p-TsOH·H2O (1 equiv), (2) DDQ (6 equiv), DCM, 0 °C, 2 h, then rt 15 min; (c) (1) H2SO4, TFA, rt, 5 days, (2) Zn(OAc)2·2H2O (60 equiv), CHCl3/MeOH, 50 °C, o/n; (d) (1) H2SO4, TFA, 0 °C, 30 min, (2) Zn(OAc)2·2H2O (60 equiv), CHCl3/MeOH, 50 °C, o/n; (e) DDQ (8 equiv), Sc(OTf)3 (8 equiv), toluene, 100 °C, 5 h; (f) DDQ (8 equiv), Sc(OTf)3 (8 equiv), toluene, 80 °C, 4 h; (g) H2SO4, TFA, 0 °C, 30 min, (2) Zn(OAc)2·2H2O (40 equiv), CHCl3/MeOH, 50 °C, o/n; (h) DDQ (5 equiv), Sc(OTf)3 (5 equiv), toluene, rt, 3 h; (i) DDQ (12 equiv), Sc(OTf)3 (12 equiv), toluene, 80 °C, 3 h.
The singly fused diporphyrin quinone 350.10 was reported by Yamashita, Sugiura et al.653 The compound was obtained in low yield in a tandem PIFA-induced oxidation of a 5,15-diarylporphyrin along with the expected monomeric dioxoporphodimethane. 350.10 displayed strong panchromatic absorption between 300 and 1000 nm, with a large maximum at 724 nm, and possessed a significantly reduced electrochemical HOMO–LUMO gap relative to the monomeric quinone.653
Osuka et al. introduced seven membered rings into porphyrin tapes to produce significant curvature of the π system.654 Carbonyl-containing arch-tapes were obtained from the bridged porphyrin dimers 351.1 or trimers 351.5 and 351.8 via an intramolecular oxidative fusion using DDQ and Sc(OTf)3 (Scheme 351). Subsequently, the Luche reduction with NaBH4 and CeCl3 produced the carbinol 351.3a, which was directly subjected to ionic hydrogenation reaction with HBF4·Et2O and BH3·NEt3 to afford the methylene-containing dimer 351.4a in 88% yield. The same two-step procedure was applied in the syntheses of 351.7 and 351.10. Because of their curved shape, these arch-porphyrin tapes were found to be highly soluble. In comparison with the planar trimers 351.11a–b, containing bulky meso-substituents to prevent aggregation, UV–vis–NIR absorption spectra of carbonyl-containing arch tapes were most red-shifted, with lowest-energy Q bands at 1260 and 1254 nm and electrochemical HOMO–LUMO gaps of 0.87 and 0.85 eV for 351.5 and 351.8, respectively. The methylene-bridged arch tape 351.7 exhibited strong association with C60 in toluene, with an association constant of (1.5 ± 0.4) × 107 M–1 at 25 °C.
Scheme 351. Synthesis of Carbonyl- and Methylene-Containing Porphyrin Arch Tapes.
Reagents and conditions: (a)654 DDQ/Sc(OTf)3 (5 equiv), toluene, 60 C, 2–3 h; (b) NaBH4, CeCl3, DCM/MeOH, −80 °C; (c) (1) HBF4·OEt2, DCM, rt, 10 min, (2) BH3·NEt3, rt, 10 min; (d) DDQ/Sc(OTf)3 (15 equiv), toluene, 60 °C, 5 h.
Later in 2018, Osuka reported the synthesis of singly and doubly 1,2-phenylene-expanded porphyrin arch-tape dimers 352.2–5 (Scheme 352).655 The oxidative approach described above was not successful with the ortho-phenylene precursor 352.1a, which produced a rearranged product 352.4 when treated with DDQ and Sc(OTf)3. The structure of this product was consistent with an oxidation-induced 1,2-migration of the phenylene bridge to the neighboring β-position. Nickel(0)-mediated reductive coupling of the chlorinated precursor 352.1b produced 352.2, which upon oxidation with DDQ and Sc(OTf)3 in the subsequent step furnished the desired arch-tape dimer 352.3. Interestingly the doubly 1,2-phenylene-expanded arch-tape dimer 352.5 could be obtained using the oxidative method at slightly higher temperature. X-ray crystallographic investigations revealed that the embedded eight-membered rings in the phenylene-containing dimers produced even more contorted structures than the carbonyl-containing porphyrin tapes bearing seven-membered rings. The Q-band of the absorption spectrum became more red-shifted with the increasing molecular contortion and the electrochemical HOMO–LUMO gaps became smaller, reaching respectively 1294 nm and 0.77 eV in 352.3. In subsequent work, the same authors prepared sulfone-containing arch-tape dimers which also exhibited large molecular contortion.656 The synthesis of 352.3 was achieved by first converting the bridging sulfur unit in 352.6a into the sulfone. The resulting 352.6b was then subjected to the oxidative fusion reaction using AuCl3 and AgOTf instead of DDQ and Sc(OTf)3, to provide the target 352.7 in 58% yield.
Scheme 352. Synthesis of 1,2-Phenylene- and Sulfone-Containing Porphyrin Arch-Tape Dimers.
Reagents and conditions: (a)655 Palau′Chlor, CHCl3, rt, 6 h; (b)655 Ni(cod)2, 1,5-cyclooctadiene, DMF, 100 °C, 9 h; (c)655 DDQ, Sc(OTf)3, toluene, 80 °C, 2 h; (d)655 DDQ, Sc(OTf)3, toluene, 60 °C, 1 h; (e)656 H2O2, NaWO4·2H2O, MeN(nOct)3·HSO4, PhP(O)(OH)2, toluene, 50 °C, 13 h; (f)656 AuCl3, AgOTf, (CHCl2)2, 80 °C, 30 min.
meso-Unsubstituted porphyrins can be oxidatively coupled using electrochemical methods to produce benzo-fused dimers.657 Two-step electropolymerization unsubstituted magnesium porphine 353.1 in acetonitrile solutions produced films, which were proposed to contain triply linked porphine tapes 353.3a (Scheme 353).658 The addition of water or a strong proton accepting additive such as lutidine to the magnesium porphyrin solution during the course of its electrooxidation was found to strongly accelerate the rate of film growth. This lutidine addition increased the efficiency of this process without modifying the redox properties and electric conductivity of the obtained polyporphine films. The intermediate magnesium(II) polyporphine 353.2a could be transmetalated to the corresponding cobalt(II) species 353.2b,659 which was electrochemically converted into the fused polymer 353.3b.660 In comparison to 353.2b, 353.3b was found to be more stable toward potential cycling in an aqueous matrix, and was thus employed for sensing of sulfite in aqueous solutions. Structurally related polymeric porphyrin tapes were also obtained by using oxidative chemical vapor deposition (oCVD)661−666 and were utilized in photocatalysis667 and ammonia sensing.668
Scheme 353. Electropolymerization of Metalloporphyrins.
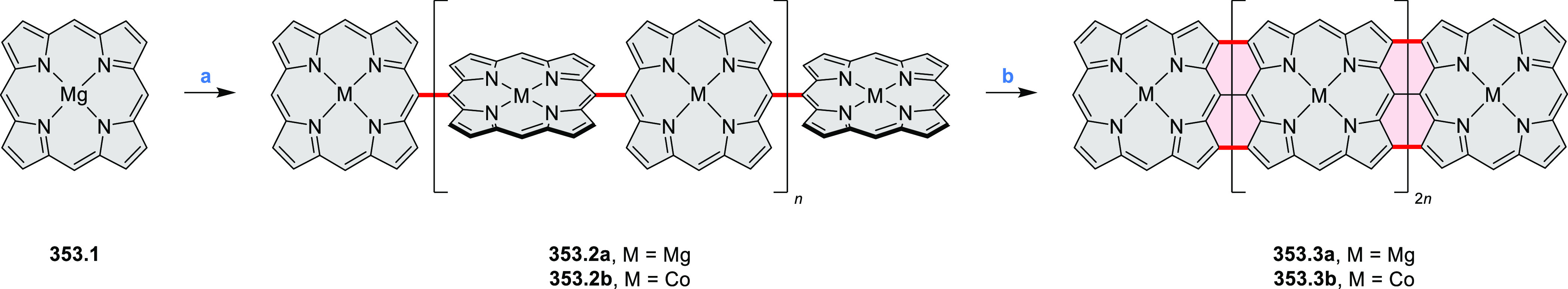
Reagents and conditions: (a)658 electrochemical potential, Eapp = 0.35 V, CH3CN; (b)658,660Eapp > 0.65 V, in TBAPF6/CH3CN.
In 2020, Bottari, Fasel, Torres et al. reported an on-surface synthesis of a structurally precise porphyrin–GNR hybrid 354.4a. The two-step annealing process used by the authors consisted of Ullmann-type coupling and subsequent cyclodehydrogenation of the free-base porphyrin precursor 354.2 bearing a bromodianthryl moiety (Scheme 354).669 The atomically precise structure of the triply fused hybrid was characterized by STM and nc-AFM methods. The electronic properties were investigated by scanning tunneling spectroscopy (STS) in combination with DFT calculations, which revealed a low electronic gap of 0.4 eV. Later in 2021, the same authors reported on-surface synthesis of GNRs with increased lengths fused at either one or both of their termini with a porphyrin macrocycle (354.3a–e, 354.4b–c).670 Combined high-resolution STM, STS and DFT data revealed weak hybridization of the electronic states of the GNR and the porphyrin moieties despite the high degree of conjugation in these hybrids.
Scheme 354. On-Surface Synthesis of Singly and Doubly Porphyrin-Capped Graphene Nanoribbon Segments.
Reagents and conditions: (a)669 200–350 °C on the Au(111) surface; (b)670 10,10′-dibromo-9,9′-bianthracene, 200–350 °C, Au(111).
7.4. [cd]-Fused porphyrinoids with Five- and Seven-membered rings
7.4.1. Indeno[1,2,3-cd]porphyrins
Indeno[1,2,3-cd]-fused porphyrins have been typically obtained by either CDA-based cyclizations or on-surface synthesis (cf. CR2017, Section 7.4.2). CDA type reactions may occasionally occur spontaneously, as evidenced by a recent report by Cavaleiro, de Souza, Neves et al., who observed formation of 355.2 under Heck-type conditions (Scheme 355).671 The UV–vis spectrum of 355.2 exhibited a broad and split Soret band, which was ascribed to the high deformation of the macrocycle caused by its five-membered ring.
Scheme 355. Synthesis of β-Substituted Dithiaporphyrins by a Heck Reaction.

Reagents and conditions: (a)671 styrene, Pd(OAc)2, K2CO3, PPh3, DMF/toluene (1:1, v/v), reflux, 1 h.
On-surface synthesis of stable corrole radicals on Ag(111) was achieved via site-specific dehydrogenation of a pyrrole N–H bond in 5,10,15-tris(pentafluorophenyl)corrole 356.1 (Scheme 356).672 The process was triggered by annealing at 330 K under ultrahigh-vacuum conditions. It was found that surface-adsorbed corrole radicals were stable up to 430 K. At higher temperatures radical-cascade reactions caused site-selective ring closure in 356.2, which produced an extended π-conjugated corrole system 356.3. Above 550 K, formation of covalently coupled corroles on Ag(111) was revealed.
Scheme 356. On-Surface Site-Selective Cyclization of Corrole Radicals.

Reagents and conditions: (a)672 Ag(111), 330 K; (b) Ag(111), 430 K.
Free bases of the multiply fused porphyrins 357.1–3 were studied in 2017, Ishizuka, Kojima et al., who found a significant difference in the first and second protonation constants, which was ascribed to the structural rigidity of these macrocycles (Scheme 357).673 In particular, the first protonation of the quadruply fused porphyrin 357.1 (QFP) proceeded smoothly with TFA as the acid source, whereas the second protonation required a large excess of the acid, with the corresponding equilibrium constants of K1 = (1.3 ± 0.1) × 105 M–1 and K2 = 7.3 ± 0.3 M–1, respectively. Br- and Ph-functionalized QFP derivatives 357.5a and 357.6 were reported show red-shifts in their absorption spectra, caused by substitution effects.674 Additionally 357.5a underwent unusual ligand-induced dimerization process, which involved dissociation of one of the nonfused pyrroles. This dimerization caused hypsochromic shifts of absorption bands, resulting from H-type aggregation of the two porphyrins. NH tautomerization behavior was studied for the unsymmetrically substituted free base 357.8, which was obtained in three steps from 357.4.675 ΔG‡298 for tautomerization of 357.8 was found to be larger than that of free base tetraphenylporphyrin, and the difference was ascribed to the more severe steric congestion of the NH protons in the deformed QFP core. On the basis of kinetic and thermodynamic analyses, NH tautomerism in 357.8 was proposed to involve dissociation of a π-stacked dimer stabilized by dipole–dipole interactions.
Scheme 357. Synthesis of Quadruply Ring-Fused Porphyrinsa.
Reagents and conditions: (a)673 TFA, CHCl3; (b)674 NBS (5 equiv), CHCl3; (c)675 NBS (1.9 equiv), CHCl3; (d)674 (1) phenylboronic acid, Pd(PPh3)4, K2CO3, (2) TFA, NaHCO3, (3) Zn(OAc)2·2H2O; (e)675 2,4,6-trimethylphenylboronic acid, Pd(PPh3)4, K2CO3; (f)675 TFA, CHCl3.
7.4.2. Other Cyclopenta-Fused Systems
π-Extended cyclopenta-fused systems can be accessed by chemical modification of chlorophyll derivatives. In a recent example, pyridine-fused chlorins such as 358.2 were elaborated via conventional condensation chemistry (Scheme 358).676 In the final step, 358.1 was reacted with ammonium acetate in acetic acid to close the heterocyclic ring. These pyridine fused chlorophylls showed significant bathochromic shifted absorption in the near-infrared region (748–766 nm) with unusually broadened Q bands, as well as small electrochemical HOMO–LUMO gaps (1.89–2.02 eV).
Scheme 358. Synthesis of π-Extended Derivatives from Enone-Substituted Chlorins.

Reagents and conditions: (a)676 NH4OAc, acetic acid, N2, reflux.
In 2019, Higashino and Imahori described a synthesis of phosphole-fused dehydropurpurins via titanium-mediated [2 + 2+1] cyclization.677 The reaction of bis(alkynyl)porphyrin 359.1a with excess Ti(Oi-Pr)4 and i-PrMgCl produced the phosphole-fused product 359.2a in 62% yield (Scheme 359). The latter species was converted into a range of derivatives, including the trivalent 359.4a–c, which were obtained by reacting 359.3a–c with P(NMe2)3 in toluene solution. These phospholes were gradually reoxidized under air to the respective oxides 359.2a–c, probably owing to the electron-rich nature of the porphyrin core. Dehydropurpurins 359.4a–c showed diminished absorption (and slightly red-shifted Q-bands) and smaller fluorescence quantum yields (Φf) relative to the P = O derivatives, a difference potentially caused by the 24π antiaromatic character of the phosphole-containing π-system of the porphyrin. Later in 2020, the same group synthesized the pyrrole-fused dehydropurpurins 359.5a–c via similar [2 + 2+1] cyclization strategy.678 The antiaromatic 24π-electron conjugation in these pyrrole-fused 7,8-dehydropurpurins appeared to be more significant than in their phosphole counterparts.
Scheme 359. Phosphole-Fused and Pyrrole-Fused Dehydropurpurins.
Reagents and conditions: (a)677 (1) Ti(Oi-Pr)4−i-PrMgCl, (2) PhPCl2, (3) H2O2 or S8, Et2O; (b) conc. H2SO4, CF3COOH; (c) Zn(OAc)2·2H2O, DCM/MeOH; (d) Lawesson’s reagent, toluene; (e) P(NMe2)3, toluene; (f) air; (g)678 aniline, PdCl2, Et3N, toluene/DMSO.
In 2018, Shinokubo et al. attempted the synthesis of 360.2a using the nickel-mediated tandem double cyclization of ethynylene-linked dibromodiporphyrins 360.1a (Scheme 360).679 However, presumably because of the high reactivity of 360.2a, it underwent a thermal [2 + 2] cycloaddition at the fused C–C double bond to afford the X-shaped cyclobutane-linked tetraporphyrin 360.3a as a mixture of syn and anti isomers. Spectroscopic investigations indicated that the formation of these X-shaped tetraporphyrins might involve a thermally activated triplet state of 360.2a in a thermal [2 + 2] cycloaddition reaction. It was later found that a differently substituted tetraporphyrin 360.3c, obtained regioselectively in the syn form, underwent two-step four-electron oxidation with BAHA.680 Subsequent spontaneous ring opening produced two molecules of the etheno-fused diporphyrin dication [360.2c]2+, which was found to be nearly planar in the solid state. Reduction of the latter species with excess of cobaltocene regenerated the cyclobutane-linked tetraporphyrin 360.3c. This redox-mediated cyclobutane cycloreversion could also be induced electrochemically and produced large hysteresis in the cyclic voltammogram. The unique reactivity of the etheno-fused diporphyrins 360.2a–c was attributed to the contribution of antiaromaticity to their macrocyclic conjugation.
Scheme 360. Synthesis of Etheno-Fused Diporphyrin Dications.
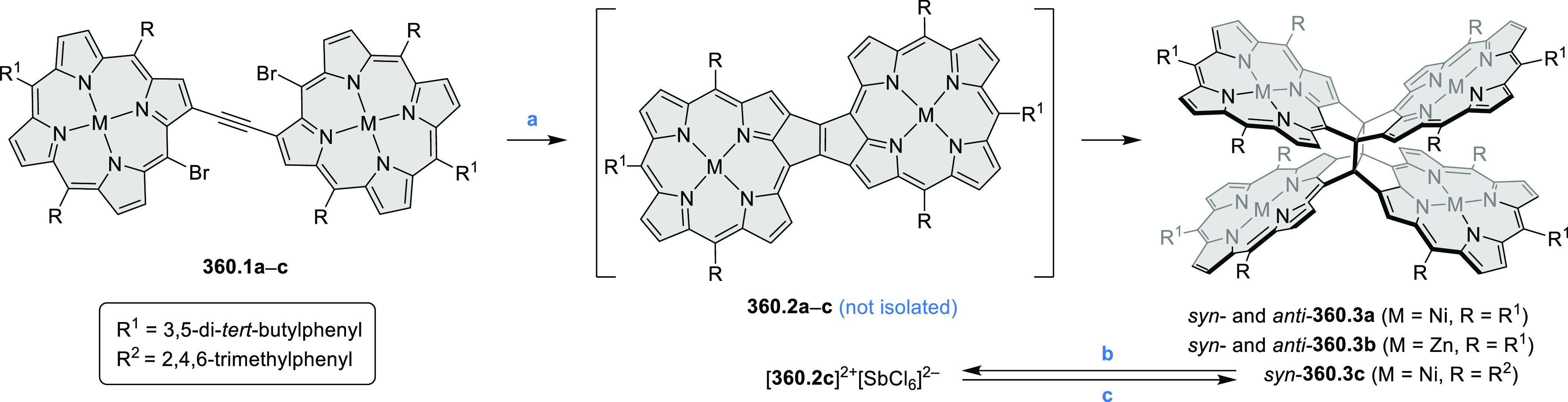
Reagents and conditions: (a)679 Ni(cod)2, 2,2′-bipyridyl, THF; (b)680 BAHA (4 equiv), DCM, rt; (c)680 CoCp2 (4 equiv), DCM, rt.
7.4.3. Fused 7-Membered Rings
An oxidative fusion reaction of the pyrrole-substituted norrole 361.1, reported in 2016 by Furuta, Xie et al., produced in the N–Cmeso-fused pyrrolyl isonorrole 361.2 accompanied by pentaphyrin 361.3 (Scheme 361).681 Upon refluxing in toluene for 10 h, 361.2 underwent skeletal rearrangement to the azepine-fused system 361.4, again accompanied by 361.3. Subsequent azepine fusion, yielding the doubly annulated product 361.5, occurred when 361.4 was stirred in DMF for 4 h at 30 °C. This reaction was facilitated by the basic nature of DMF solvent, which induced deprotonation of the NH group of 361.4 followed by its nucleophilic attack at the C6F5 moiety. Bathochromic shifts of Q-bands observed for the peripherally annulated products 361.4 and 361.5 relative to 361.2 were explained by the recovery of conjugation in the two former compounds. Dimeric copper complexes based on the extended frameworks of 361.4–5 were subsequently reported by the same authors.682
Scheme 361. Synthesis of Singly and Doubly N-CAr-Fused Confused Corroles.

Reagents and conditions: (a)681 DDQ (1 equiv), DCM; (b) NBS, DCM; (c) toluene, reflux; (d) DMF, 30 °C.
Osuka et al. reported the synthesis of an azepine-fused nickel(II) porphyrin dimer via oxidative amination of β–β linked Ni(II) porphyrin dimer (Scheme 362).683 The synthesis of the amine precursor 362.3c was achieved in 3 steps starting from the β-iodoporphyrin 362.1. Oxidative fusion of 362.3c with PbO2, followed by treatment with SnCl2, afforded 362.2a in 75% yield. 362.2b was obtained through a different route by performing a 2-fold Buchwald–Hartwig amination on 362.3d with 4-tert-butylaniline. Oxidation of 362.2a gave successively a stable neutral aminyl radical and a nitrenium ion, whereas 362.2b afforded a formal nitrenium dication in a single-step, with the two-electron oxidation mainly taking place on the bridging nitrogen.
Scheme 362. Synthesis of β -to- β-Linked meso-Aminoporphyrin Dimers.

Reagents and conditions: (a)683 Ni(cod)2, DMF, 100 °C; (b)683,684 I2, AgNO2, DCM/MeCN, rt; (c)683,684 NaBH4, Pd/C, DCM/MeOH, rt; (d)683 PhICl2, CHCl3, 0 °C; (e)684 PbO2, DCM, rt; (f)683 PbO2, DCM, rt, and then, SnCl2, DCM, rt; (g)683 4-tert-butylaniline, Pd-PEPPSI-IPr, NaOt-Bu, toluene, 120 °C.
The same authors later showed that the oxidation of the dimer 362.3f carrying two meso-amino groups gave a hybrid product containing a helical tetrapyrrin fused to an intact porphyrin macrocycle.684 The oxidation was proposed to involve a meso-aminyl radical intermediate, in which the α-carbon of the cleaved macrocycle formed a N–C(α) bond with the nitrogen atom of the other porphyrin segment. Similarly, a sulfur-bridged analogue 362.6 was also synthesized from 362.4c. 362.5 and 362.6 displayed intense NIR absorption bands at 1200–1400 nm and reversible redox processes. The more red-shifted absorption and the greater ease of first reduction suggested a more effective π-conjugation through the direct β–β linkage in 362.5 in comparison with 362.6. The helical structure of the tetrapyrrin-fused Ni(II) porphyrins enabled chiral resolution into stable enantiomers.
Oxidation of the β–meso-linked porphyrin dimer 363.1 resulted in the formation of three differently fused porphyrin dimers 363.2, 363.3, and 363.4 (Scheme 363).685 Specifically, the use of PbO2 in DCM at rt gave 363.2 and 363.3 in yields of 68 and 19%, respectively, whereas oxidation with MnO2 produced 363.4 was obtained in 1% yield along with 363.2 (58%) and 363.3 (9%). The UV–vis–NIR absorption spectra showed perturbed optical properties with a large red-shift in the lowest-energy Q-like band (937 nm) for 363.3. The electrochemical gaps were found to be in the order of 363.4 > 363.2 > 363.3.
Scheme 363. Oxidative Reactivity of meso-Amino meso-to-β-Linked Nickel(II) Aminoporphyrin.
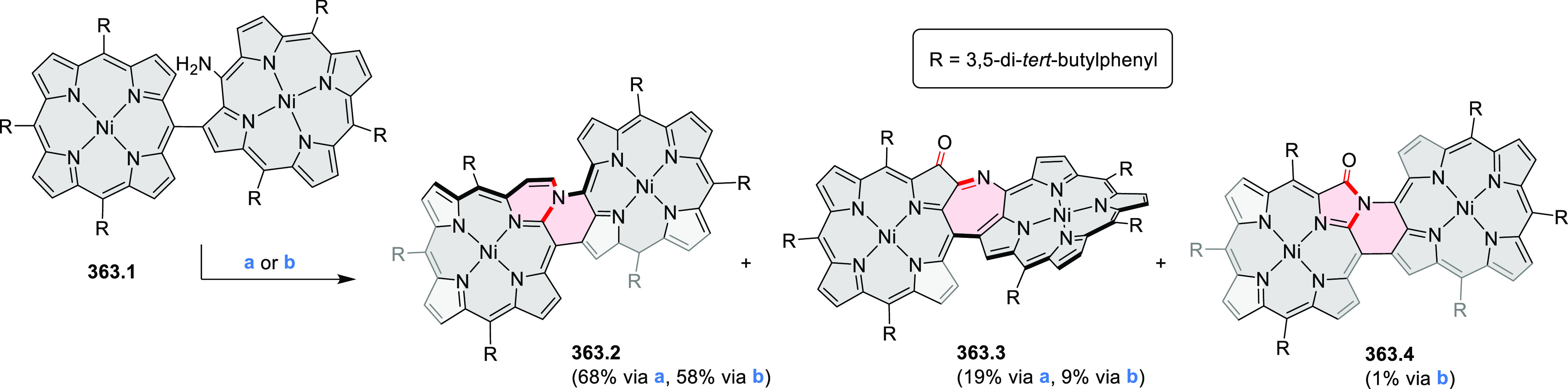
Reagents and conditions: (a)685 PbO2 (100 equiv), DCM, rt, 12 h; (b) MnO2 (100 equiv), DCM, rt, 16 h.
7.5. Porphyrinoids with Polycyclic Subunits
7.5.1. Porphyrinoids with Benzannulated Bipyrrole Units
Benzo and naphthobipyrrole fragments have been introduced into porphyrinoid frameworks as a means of rigidifying the macrocycle (for earlier work, see CR2017, Section 7.5.1). The effect of this rigidification is most apparent in expanded macrocycles, e.g. sapphyrins (364.2a–e),686 rosarins,687 or rubyrins.688 Peripherally substituted antiaromatic naphthorosarins 364.2a–c were obtained by Byon, Sessler, Lee et al. from the corresponding naphthobipyrroles 364.1 (Scheme 364).687364.2c, bearing both peripheral fluorine and meso-2,6-dichlorophenyl substituents, was reduced to a stable 25 π-electron radical species upon treating with TFA. Subsequent formation of the 26 π-electron trication, [364.2c-H3]+, was found to be slower than in the case of [364.2a-H3]+ or [364.2b-H3]+. This behavior of naphthorosarins is contrasted with the chemistry of the analogous naphthorubyrin, which prefers 26 π-electron conjugation and could not be oxidized to the corresponding 24 π-electron state.688 The properties of naphthorosarins can be further tuned by substitution: the electron-deficient 364.3 was found to form a cofacially stacked noncovalent dimer,689 whereas 364.2a was found to undergo nucleophilic substitution with catechol to yield 364.4.690
Scheme 364. Naphthobipyrrole Macrocycles.
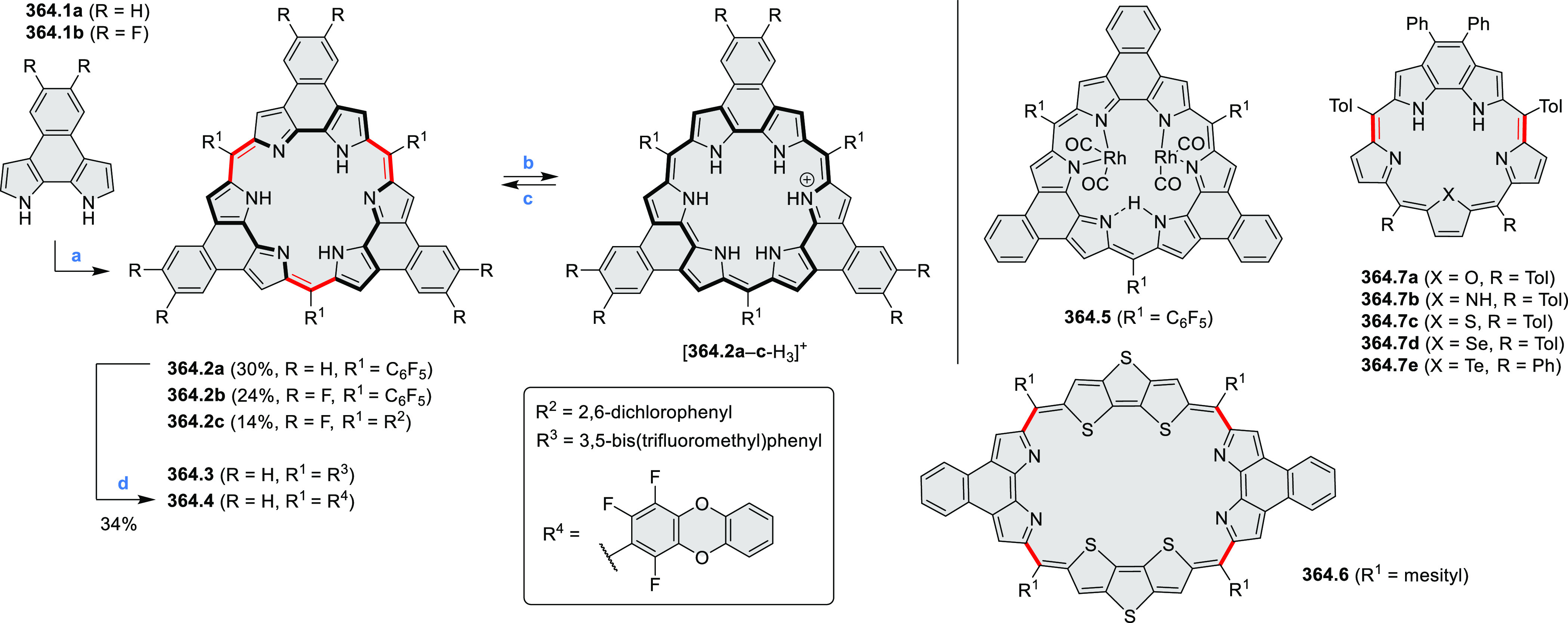
Reagents and conditions: (a)687 appropriate aldehyde, TFA, DCM, (2) DDQ, TEA; (b)687 TFA, cobaltocene, THF-d8; (c)687 TEA; (d)690 catechol, K2CO3, NMP, 100 °C, 6 h.
In a naphthorosarin rhodium complex 364.5, prepared by Sessler, Lee and co-workers, the macrocycle maintained its planarity, but the two rhodium ions reside on opposite sides of the molecular plane, resulting in inherent chirality.691 The rhodium(I)–rhodium(I) distance was found to be 3.143 Å. The UV–vis absorption spectrum of 364.5 showed multiple absorption bands between 416 and 496 nm, along with bands at 623 and 653 nm, which was distinct to that of the free base 364.2a. Cyclic voltammogram measurements revealed that upon metalation, the tendency to undergo reduction or oxidation was less pronounced than in the free base naphthorosarin and consequently, reduction to the corresponding 26 π-aromatic form became more difficult.
A core-modified rigid [32]octaphyrin(1.0.1.0.1.0.1.0) 364.6 derived from rigid naphthobipyrrole and dithienothiophene (DTT) precursors were reported by Zhang, Kim, Sessler et al.692 The X-ray crystallographic analysis of revealed the macrocycle was essentially a planar system and further spectroscopic investigations indicated a weakly 32 π-electron antiaromatic or nonaromatic character of 364.6. 364.6 underwent proton-coupled two-electron reduction to produce the aromatic 34 π-electron state, in the presence of HCl and other hydrogen halides. The intermediate 33 π-electron radical species were observed upon addition of acids (such as TFA) that are less redox active than HCl to solutions of 364.6.
7.5.2. Cyclooctatetraene-Fused Systems
A modified synthetic procedure to obtain cyclooctatetraene (COT) fused corrole dimer was reported by Z. Gross et al. (Scheme 365).693365.1 was heated in 1,2,4-trichlorobenzene at 200 °C for 24 h in air which allowed the isolation of corrole dimer 365.2 in 18% yield. Upon treating 365.2 with GaCl3 in pyridine under inert conditions, 365.3 was obtained, wherein one pyridine ligand was coordinated to each metal center. Upon recrystallization in pyridine, the tetrapyridine complex 365.4 was obtained in quantitative yield. The fusion of the two corrole units in 365.2–4 was found to produce significant distortion of bond lengths in the COT ring. Extensive π delocalization through the COT bridge in these systems was reflected in their absorption spectra, which contained low-energy bands reaching into the near-infrared (λmax at 720–724 nm).
Scheme 365. Synthesis of a Cyclooctatetraene-Bridged Gallium(III) Corrole Dimer.

Reagents and conditions: (a) 1,2,4-trichlorobenzene, reflux, 24 h; (b) (1) GaCl3, pyridine, N2, (2) DCM, n-heptane; (c) pyridine.
7.5.3. Thiophene-Fused Systems
In 2016, Chandrashekar and co-workers reported Möbius-aromatic [32]π heptaphyrins C33.1a–b, which were obtained via acid-catalyzed cross-condensation of two modified tripyrranes with pentafluorobenzaldehyde (Chart 33).694 Proton NMR studies indicated weak Möbius aromaticity at 298 K; however, upon lowering the temperature, the molecule adopted a [4n]π Möbius conformation with a strong diatropic ring current. This aromaticity was further supported by the ACID plots obtained from DFT calculations. Similarly designed, planar [30]π heptaphyrin C33.3 and [34]π octaphyrins C33.4a–b were later reported to be aromatic in freebase and protonated forms.695 The more expanded dithienothiophene-containing [40]π nonaphyrin was found to adopt a twisted figure-eight conformation.696
Chart 33. Porphyrinoids Containing Polycyclic Thiophene-Based Subunits.
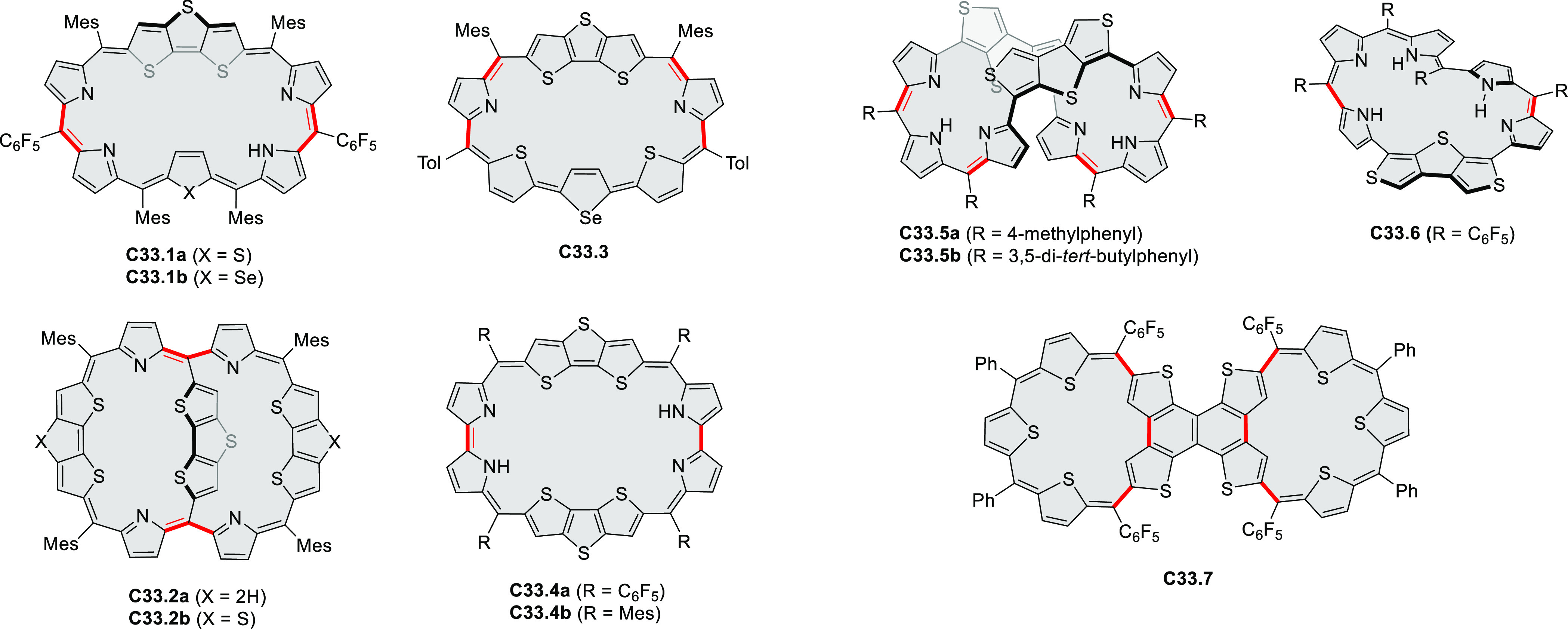
Bicyclic dithienothiophene-bridged [34]octaphyrins C33.2a–b were reported to form Baird-type aromatic triplet states.697 The synthetic route to these macrocycles involved an acid-catalyzed condensation reaction involving a diformyl dithienothiophene as the source of the bridging unit. The absorption spectrum of C33.2a in DCM showed two B-like bands at 501 and 605 nm, which suggested two competing π-conjugation pathways, containing [26]π and [34]π electronic-circuits, within the same nonplanar framework. Two-electron electrooxidation of C33.2a led to the generation of a 40π-electron triplet species, showing features consistent with global Baird aromaticity. Later in 2019, Park, Kim and co-workers reported a detailed study on the significant changes in the macrocyclic aromaticity of C33.2b upon C60 complexation.698 The binding ability of the bowl-shaped macrocycle with C60 was monitored with spectroscopic titration, which confirmed 1:1 binding stoichiometry in a toluene solution with an association constant of about 1.8 × 103 M–1. 1H NMR spectroscopy also indicated a host–guest complexation occurred by effective π–π interactions between the bowl-shaped macrocycle and fullerene. Furthermore, femtosecond transient absorption measurements revealed that formation of the photoinduced charge-separated state and the triplet excited-state populations of the bowl-shaped and rigid expanded porphyrin could be controlled by a simple complexation with C60.
In 2017, Higashino, Imahori et al. reported the synthesis of thiophene-fused dithiaoctaphyrins C33.5a–b.699 These systems were characterized by coexistence of a macrocyclic [36]π-electron circuit and noncyclic cross-conjugation. In a related [28]hexaphyrin C33.6, switching between these two types of conjugation was realized via a change in the topology of the π system.700 A naphthalene-fused dimer C33.7 of an antiaromatic expanded isophlorin was reported by Kögerler, Anand et al. in 2017.701C33.7 was obtained from atypical [3 + 2] acid-catalyzed condensation followed by a rare β–β oxidative dehydrogenation which occurred between the adjacent two ring-inverted thiophene rings. Fused dimer C33.7 exhibited marginal peripheral aromaticity rather than strong global diatropicity or paratropicity and weak electronic communication as a result of cross-conjugation.
7.5.4. Systems with Acene and Heteroacene Subunits
A bis-“dicarbacorrole” 366.2 obtained by incorporating a dibenzo[g,p]chrysene moiety into a macrocyclic structure was reported in 2017 by Kim, Sessler, and co-workers (Scheme 366).702 This ligand system possesses two contracted corrole-like cores and stabilized higher oxidation states of metal ions. 366.2 was obtained in 8% yield in an acid catalyzed condensation reaction between 366.1 and pentafluorobenzaldehyde, followed by oxidation with DDQ. The trianionic cores in 366.2 were used for copper and palladium coordination to obtain mono- and bis-metal complexes, 366.3–7.702,703 Compounds 366.2 and 366.4 were both antiaromatic, each containing two formally 16 π-electron dicarbacorrole subunits, whereas the mixed Cu/Pd complex 366.5 was an organic π radical, consisting of a fused 15 π-electron nonaromatic subunit and a 16 π-electron antiaromatic subunit. Formation of these organic radicals was ascribed to one-electron transfer from the ligand backbone to the Pd center. The UV–vis–NIR absorption spectra of bis-Pd 366.7 in toluene displayed an intense NIR absorption band at ca. 1420 nm, whereas 366.2 and 366.4 were characterized by very weak or negligible NIR absorptions over 1000 nm. A detailed spectroscopic analysis revealed a closed-shell ground state for the bis-Pd complex 366.7, whereas the mono-Pd complex 366.6 existed as stable monoradical. The electronic structure of 366.7 was described as quinoidal with zwitterionic contributions such as 366.7’.703
Scheme 366. Synthesis of Dibenzo[g,p]chrysene-Fused Bisdicarbacorrole.
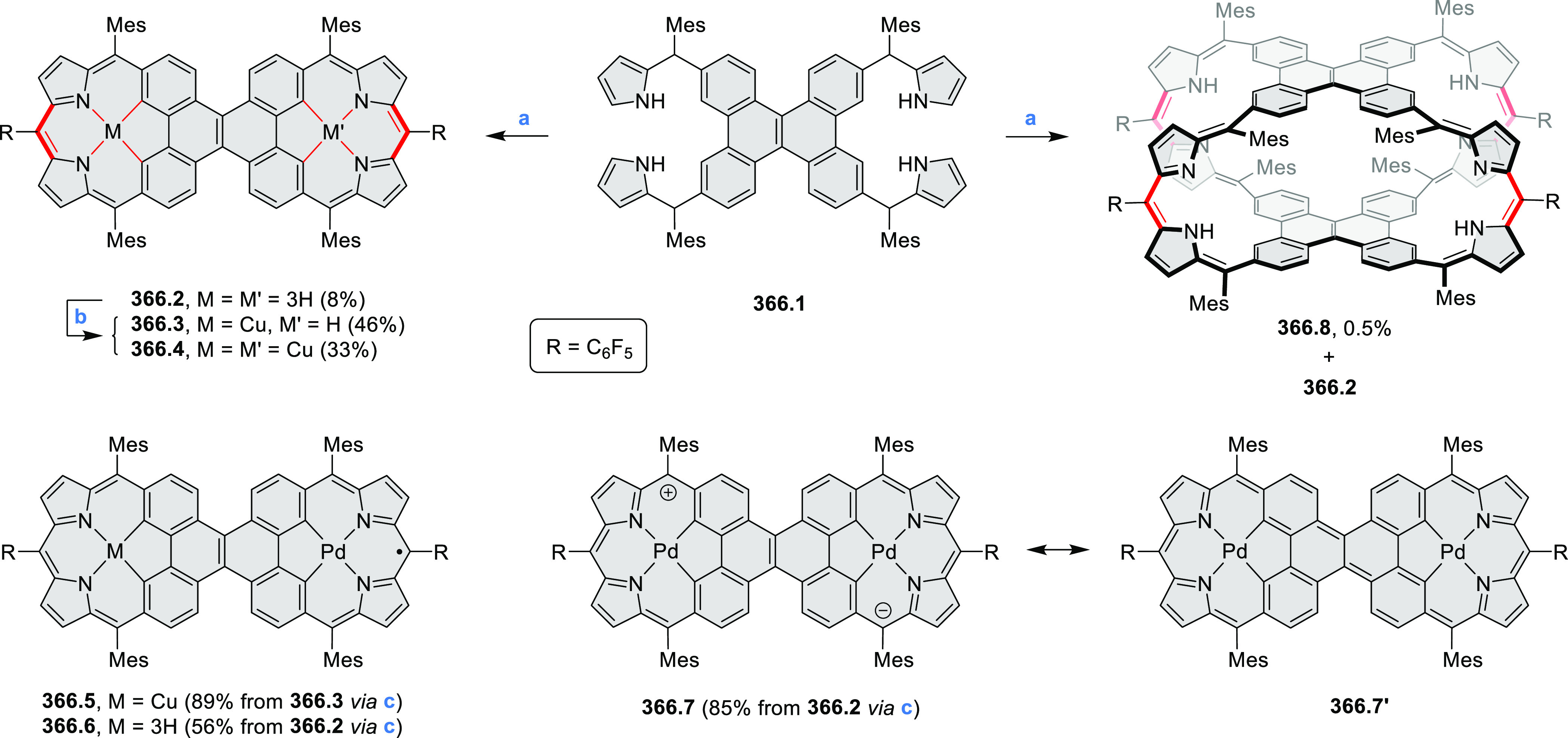
Reagents and conditions: (a)702,704 (1) pentafluorobenzaldehyde, BF3·OEt2, DCM, rt, (2) DDQ, rt; (b) Cu(OAc)2·H2O (73.8 equiv), CHCl3, reflux, 48 h; (c)702,703 Pd(PhCN)2Cl2 (3 equiv for 366.5; 2 equiv for 366.6; 15 equiv for 366.7), PhCN, 180 °C.
In parallel work, the authors utilized the 366.1 to prepare a fully conjugated three-dimensional expanded carbaporphyrin.704 An acid catalyzed [2 + 4] condensation reaction with pentafluorobenzaldehyde in DCM followed by DDQ oxidation resulted in the formation of the target product 366.8 in 0.5% yield along with the more favored product 366.2. The neutral form of this cage 366.8 was nonaromatic, but it gained a global aromatic character upon protonation, which was inferred from the large negative NICS value (−11.63) and diatropic ring current observed in an ACID plot. In addition, the size of the cavity in 366.8 increased upon protonation to ca. 143 Å3.
The phenanthrene-containing porphyrinoid 367.3 was obtained by Szyszko, Latos-Grażyński, and co-workers as an intermediate during the acid-catalyzed condensation reaction between 367.1 and 367.2 (Scheme 367).705367.3 underwent subsequent intramolecular oxidative ring fusion to yield the “helicenophyrin” 367.4, which was shown to adopt a figure-of-eight conformation in the solid state. 367.4 appeared to be relatively difficult to protonate, but it formed a monoanion upon treatment with TBAF. When treated with BF3·OEt2, 367.4 gave the doubly fused macrocyclic product 367.5 in 60% yield, instead of an expected boron complex. The X-ray structure revealed that this macrocycle 367.5 incorporated an ortho-fused system of seven aromatic rings, forming an S-shaped helicene ribbon. Compounds 367.4–5 were both chiral and could be separated into enantiomers using chiral HPLC.
Scheme 367. Expanded Carbaporphyrins Incorporating Aza-[5]helicene Motifs.

Reagents and conditions: (a)705 (1) p-nitrobenzaldehyde, BF3·OEt2, CHCl3, 2 h, (2) DDQ, 10 min; (b) BF3·OEt2, toluene, triethylamine, reflux, 90 min, (2) triethylamine.
Later in 2018, Latos-Grażyński and co-workers explored the reactivity of phenanthrene-containing porphyrinoids 368.1 and 368.5 toward protic and Lewis acids (Scheme 368).706 When titrated with HBF4·Et2O, 368.1 underwent two-step protonation, yielding ultimately the meso-protonated dication 368.3. Reaction of 368.1 with sulfuric acid (85%) at 40 °C produced the intermediate 368.4, which underwent spontaneous oxidation during workup to give 368.5 in 70% yield. The latter species formed two well-defined species, a monocation and a trication, when treated with HBF4·Et2O. Upon long exposure to tetrafluoroboric acid, 368.5 underwent borylation at the carbonyl oxygen atoms, forming the aromatic BF2 derivative 368.6, which was identified by XRD analysis. These phenanthrene-containing macrocycles acted as trianionic ligands and could be transformed into copper(III) complexes 368.9–10.707 The dione 368.5 was subsequently transformed into π-extended phenanthriporphyrins 369.2a–h via condensation with aliphatic and aromatic amines (Scheme 369).708
Scheme 368. Phenanthriporphyrin.
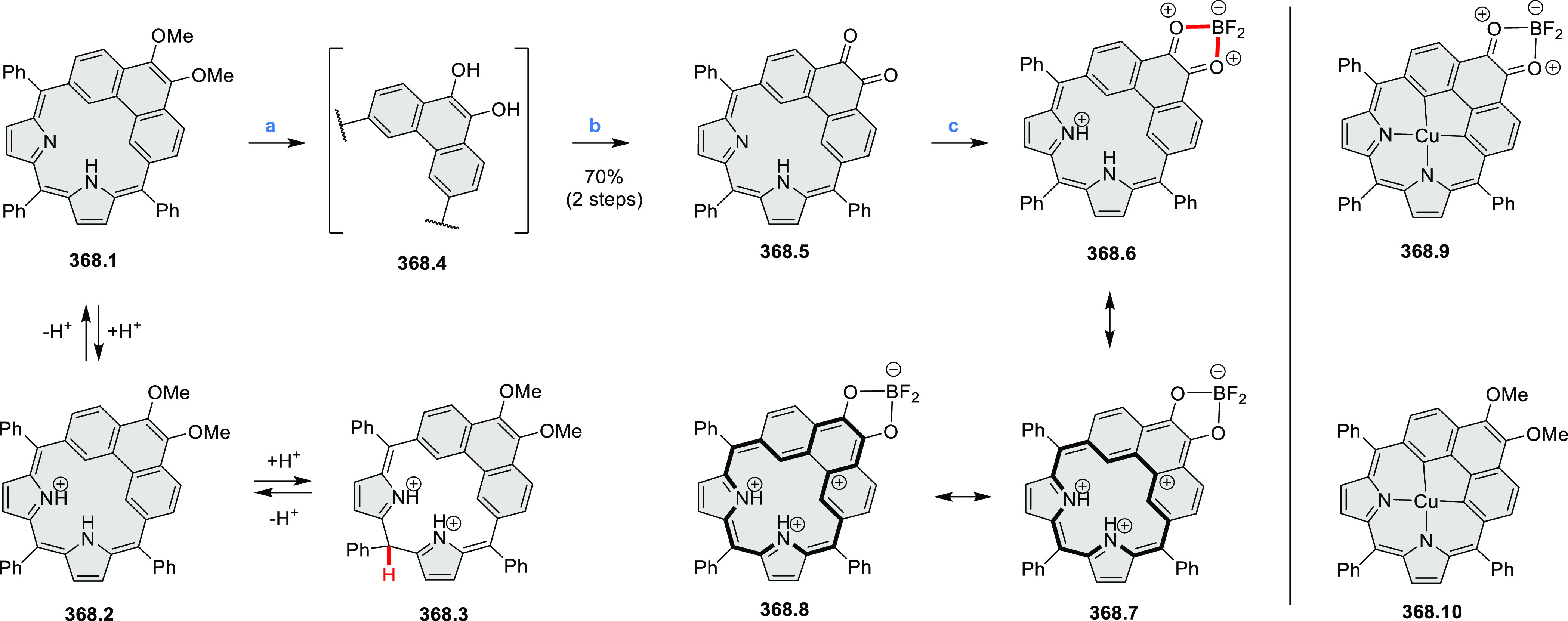
Reagents and conditions: (a)706 H2SO4, 0 to 40 °C, 28 h; (b) air oxidation; (c) HBF4·Et2O.
Scheme 369. Phenanthrene- and Naphthalene-Containing Porphyrinoid Macrocycles.
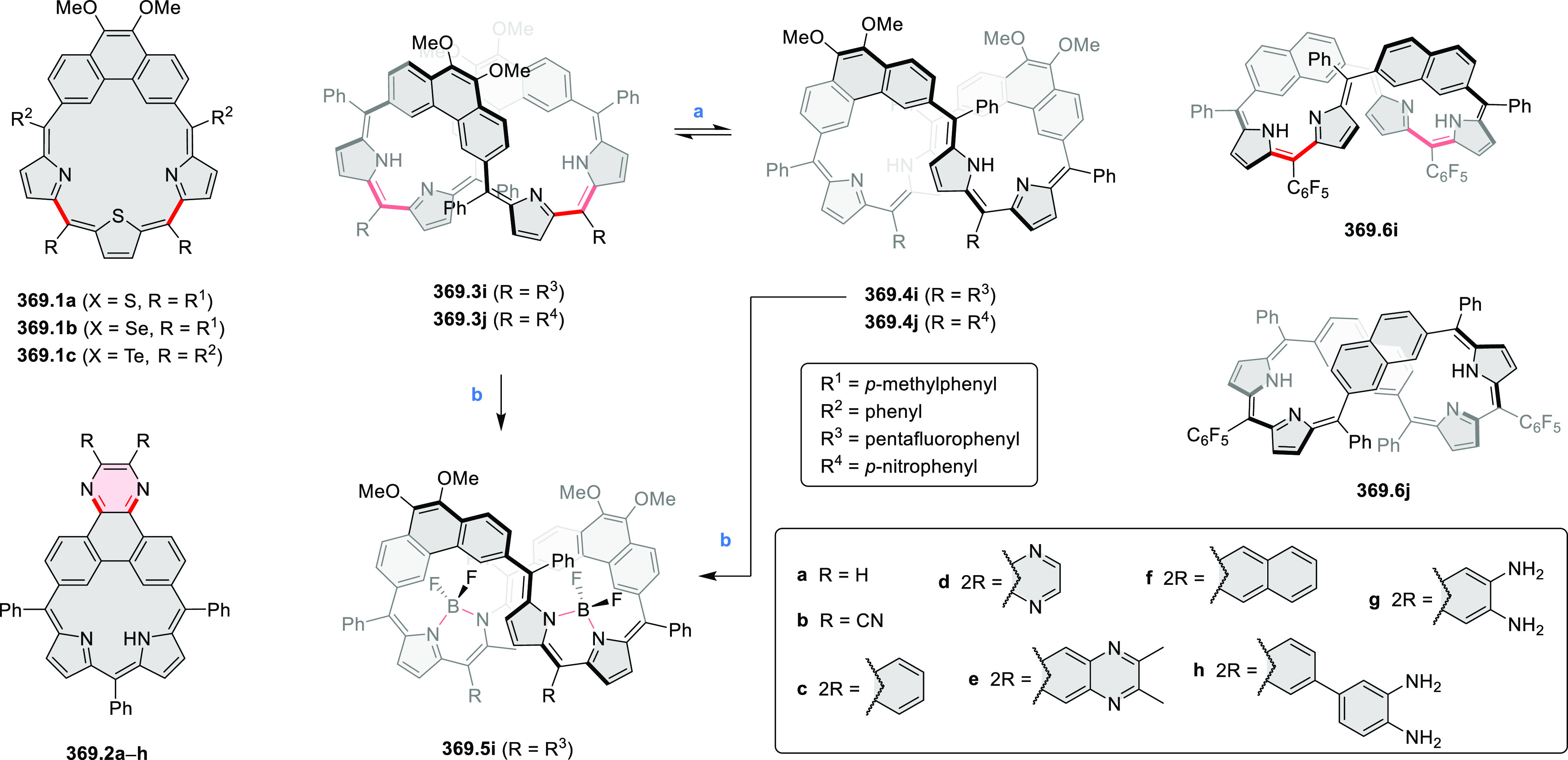
Reagents and conditions: (a) Δ, amine; (b) (1) BF3·OEt2, TEA, toluene, reflux, 3 h, (2) TEA.
Phenanthrene and naphthalene building blocks was similarly used for the synthesis of phenanthrisapphyrins 369.1a–c709 and larger macrocycles710−712 (Scheme 369). Diphenanthrioctaphyrins 369.3i and 369.4i were obtained as conformationally locked macrocycles in a single macrocyclization reaction.710 The UV–vis absorption of 369.3i exhibited three intense bands at 255, 374, and 458 nm and a weaker and broad absorption extending up to 900 nm, while the low-energy bands for 369.4i were slightly blue-shifted. Both 369.3i and 369.4i adopted a figure-eight shape, differing in the relative alignment of the phenanthrene moieties. DFT calculations showed that the 369.4i was more stable than 369.3i by 6.6 kcal mol−1. Interconversion between these two species could be induced by hydrogen bond acceptors such as amines. Both 369.3i and 369.4i produced the same bisboron(III) complex 369.5i with a conformation corresponding to that found in 369.4i. A similar conformational dichotomy was subsequently observed in a di-2,7-naphthihexaphyrin(1.1.1.1.1.1), which formed two conformationally locked stereoisomers 369.6i and 369.6j, which could be interconverted using acid–base chemistry under either kinetic or thermodynamic control.712
Anthriporphyrin 370.3a was synthesized by Cho and co-workers from the anthracene–thiophene dicarbinol 370.2 and dipyrromethane 370.1 (Scheme 370).713 The anthracene unit in 370.3a reacted with dimethyl acetylenedicarboxylate to produce the Diels–Alder phlorin-like adduct 370.4a. For each macrocycle, the corresponding palladium complex, 370.3b or 370.4b, was obtained under standard conditions. Naphthalene-containing analogues 370.5a,b714 and the bismacrocyclic systems 370.7a(715) were obtained in a similar way. In each case, the corresponding palladium(II) complex could be obtained.
Scheme 370. Anthracene- and Naphthalene-Based Carbaporphyrins.
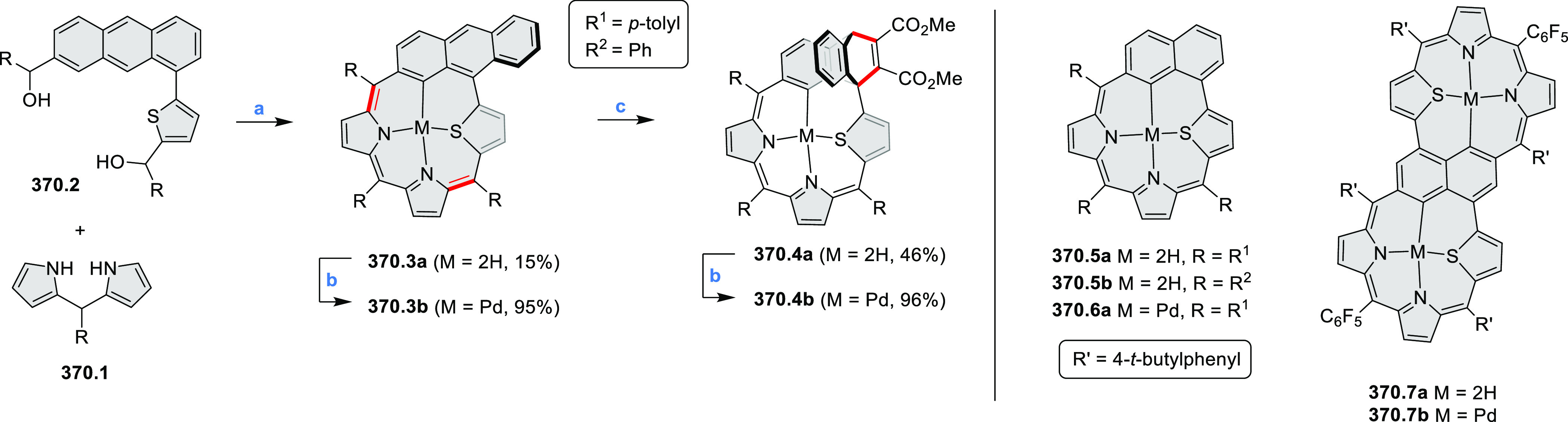
Reagents and conditions: (a)713 (1) BF3·OEt2, DCM, 1 h, (2) p-chloranil, 20 min; (b) Pd(OAc)2, DCM/CH3CN; (c) dimethyl acetylenedicarboxylate, o-DCB, 145 °C.
In 2019, Ravikanth et al. reported meso-fused carbatriphyrins(2.1.1) based on a fused fluorene motif (Scheme 371).716 Condensation of pentafluorobenzaldehyde and the fluorene-based tripyrranes 371.1a,b produced the phlorin-like 371.2a,b rather than the expected fully conjugated 371.3a. However, the phosphorus(V) complex 371.4a was obtained in 48% yield by treating the phlorin 371.2a with PCl3 in toluene/triethylamine. 371.4a showed features consistent with macrocyclic aromaticity, with the dominant 18-electron conjugation pathway. In a similar way, meso-fused heterabenziporphyrins 371.5a–c and their palladium complexes 371.6a,b were obtained by the same group.717 These systems showed weak absorptions in the NIR range, with a progressive red-shift observed in analogues containing heavier chalcogen heteroatoms. Related designs from the Ravikanth group include the strained fluorenophyrins 371.7a,b,718 dibenzofuran/dibenzothiophene-based macrocycles 371.8a–c,719 and their contracted benzofuran/benzothiophene analogues 371.9a,b.720
Scheme 371. Hybrid Porphyrinoid Macrocycles.
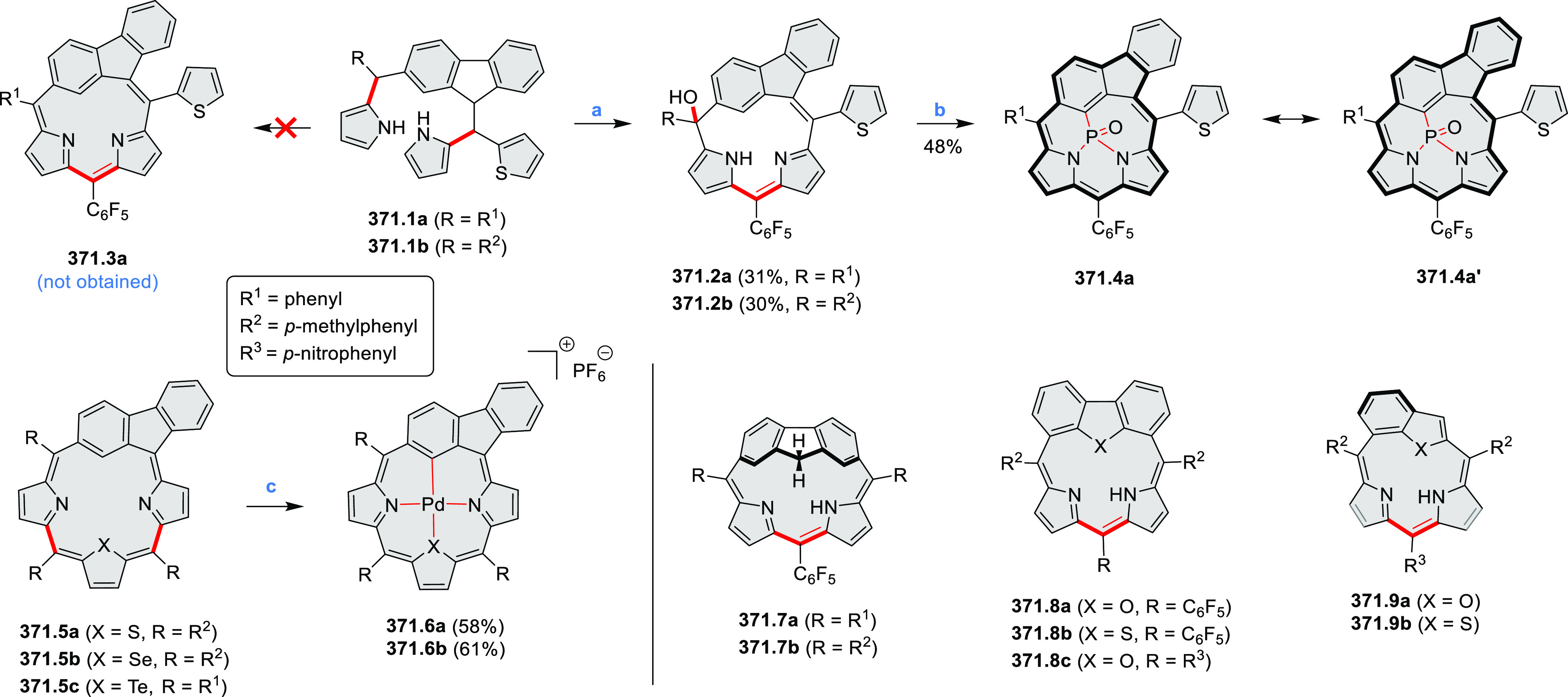
Reagents and conditions: (a)716 (1) BF3·OEt2, C6F5CHO, (2) DDQ; (b)716 PCl3, TEA, toluene, reflux, 1 h; (c)717 PdCl2, CHCl3/CH3CN, reflux.
2,7-Naphthiporphyrins 372.5a–c containing various chalcogens in the coordination core were obtained by Szyszko, Latos-Grażyński, and co-workers from the tripyrrane analogue 372.3 (Scheme 372).721 These porphyrinoids had a larger coordination cavity than m-benziporphyrins and showed an ability to form organophosphorus(V) complexes 372.7a,b when treated with excess phosphorus(III) trichloride in the presence of triethylamine. In these complexes the macrocycle was formally reduced relative to its oxidation state in 372.5a–c. The reduced free base 372.6b could be reversibly generated from the Se analogue 372.5b.
Scheme 372. Synthesis of 28-Hetero-2,7-naphthiporphyrins.
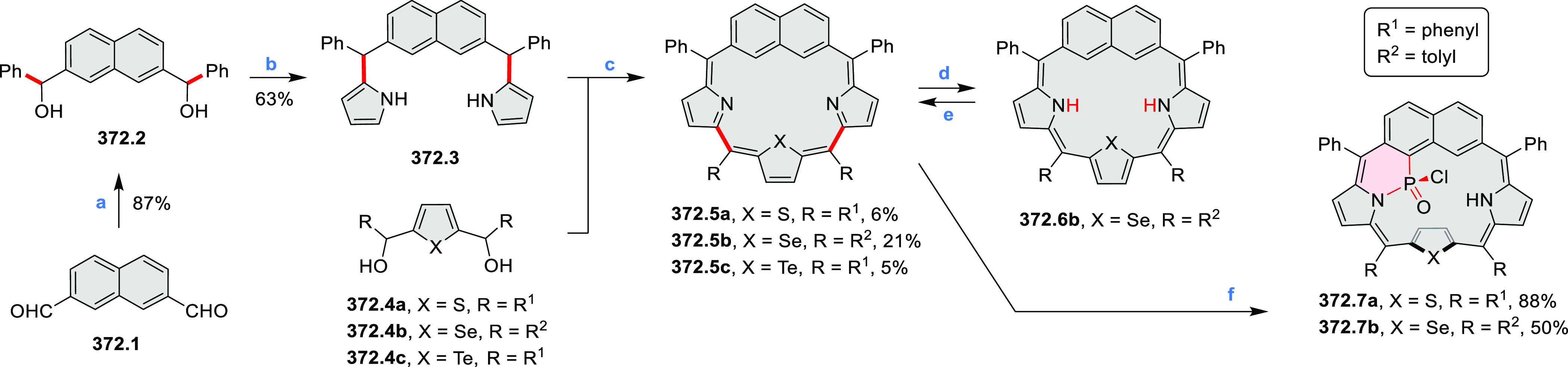
Reagents and conditions: (a)721 (1) phenylmagnesium bromide, THF, (2) H3O+; (b) (1) pyrrole, BF3·OEt2, CHCl3, N2, reflux, 48 h, (2) triethylamine; (c) (1) BF3·OEt2, CHCl3, (2) DDQ, 15 min; (d) Zn(Hg), toluene, N2; (e) O2; (f) PCl3, TEA, toluene, reflux, 80 min.
7.5.5. Systems with Macrocyclic Subunits
Large supermacrocycles based on porphyrin subunits are typically highly nonplanar and show limited π-conjugation. A relatively high degree of coplanarity between subunits is displayed by 2,5-pyrrolylene-linked cyclic porphyrin dimers C34.1a,b and trimers C34.1d–c, obtained by Song et al. via Suzuki–Miyaura coupling of 2,5-diborylpyrrole with appropriate 3,7-dibromoporphyrins (Chart 34).722C34.1a adopted a bent geometry, in which the pyrrole nitrogens were pointing inward, whereas C34.1d showed a more coplanar triangular molecular shape with a reverse alignment of pyrrole bridges. Zinc complexes C34.1c and C34.1f were also prepared from the corresponding free base oligomers. The UV–vis absorption spectra showed a split Soret band at λmax = 419 and 496 nm for C34.1c and at λmax = 416 and 510 nm for C34.1f and a long tailing Q-band indicating strong electronic interactions in the cyclic oligomers. The cyclic trimer C34.1f displayed ultrafast excitation energy transfer (690 fs) mediated by through-bond electronic interaction via the pyrrole linkage. A similar synthetic approach was further utilized to make several new analogues of cyclic porphyrin oligomers incorporating 6,6″-terpyridylene bridging units.723
Chart 34. Porphyrin–Pyrrolylene Supermacrocycles.

The synthesis of radially π-conjugated porphyrinylene/phenylene nanohoops C35.1–4 was reported by Meyer, Delius, and co-workers (Chart 35).724 The X-ray structure of C35.1 exhibited an oval-shaped nanohoop structure with an average diameter of ca. 13.2 Å. DFT calculations predicted the ring strain of 54 kcal mol–1 for C35.1. The UV–vis absorption spectrum of C35.1 showed bathochromic shifts of Soret and Q bands relative to immediate unstrained precursors. The observed red-shifts were ascribed to the narrowing of the HOMO/LUMO gap with increasing ring strain. For the π-extended analogues C35.2–4, a slight blue-shift of absorption features was indeed observed. The small nanohoop C35.1 was found to accommodate C60 and C70 with binding constants of ca. 3 × 108 M–1 and ca. 2 × 107 M–1, respectively. The naphthalene-containing nanohoop C35.3 was found to bind more strongly with C70 by a factor of 5.
Chart 35. Porphyrin Nanohoops and Nanorings.

In 2019, the Wu group reported a template-free synthesis of porphyrin-based macrocycles containing aromatic porphyrins strapped with quinoidal oligothiophene units (Chart 35).725 The cyclic porphyrin dimer C35.5 and trimer C35.6 were obtained via Yamamoto coupling reaction followed by DDQ oxidation. The solid-state structure of dimer C35.5 showed a cyclophane-like distorted geometry. Theoretical calculations, including ACID, NICS, and ICSS, indicated the local aromatic character of the porphyrin and nearby thiophene rings. The electrochemical HOMO–LUMO gaps of C35.5 and C35.6 were estimated to be 1.52 and 1.35 eV, respectively. The solid-state structure of the dication of C35.5 exhibited a chair-shaped conformation facilitating p-orbital overlap between the porphyrin, thiophene, and bithiophene units. The dications of C35.5 and C35.6 were both found to show global aromaticity with a dominant 54 π and 82 π conjugation pathway, respectively.
Bismacrocyclic structures, constructed by strapping a large porphyrinoid ring with a π-conjugated subunit, can exhibit unusual aromaticity resulting from the coexistence of multiple conjugation pathways (for earlier work, see CR2017, Section 7.5.5). Internally 1,3-phenylene-strapped [26]π- and [28]π-hexaphyrins, 373.1 and 373.2, exhibit Hückel aromaticity and antiaromaticity, respectively, in their ground states (Scheme 373).726 The excited-singlet-state aromaticity of these hexaphyrins was evaluated based on femtosecond transient absorption measurements, and it was found that the aromaticity reversal takes place in the lowest singlet excited states of [26]-and [28]hexaphyrins where 373.1 becomes antiaromatic and 373.2 could be assumed to be aromatic. Subsequently, this excited-state aromaticity reversal study was investigated by using time-resolved IR spectroscopy, and the results were rationalized in terms of the aromaticity-driven change in the molecular structures and IR spectral features.727 An even larger system, dodecaphyrin 373.7, was reported by the Osuka group.728 This 56 π-electron macrocycle could be reversibly oxidized to its 54- and 52-electron congeners in a stepwise manner. The 52 π-electron macrocycle was metalated with palladium(II), yielding a quadruply twisted structure. The same group reported a range of bismacrocyclic [46]decaphyrins 373.8a–e strapped with a variety of aromatic subunits.729,730 A structurally related three-dimensional cage structure consisting of thiophene units was reported by the Wu group.731
Scheme 373. Internally Strapped Porphyrinoid Macrocycles.
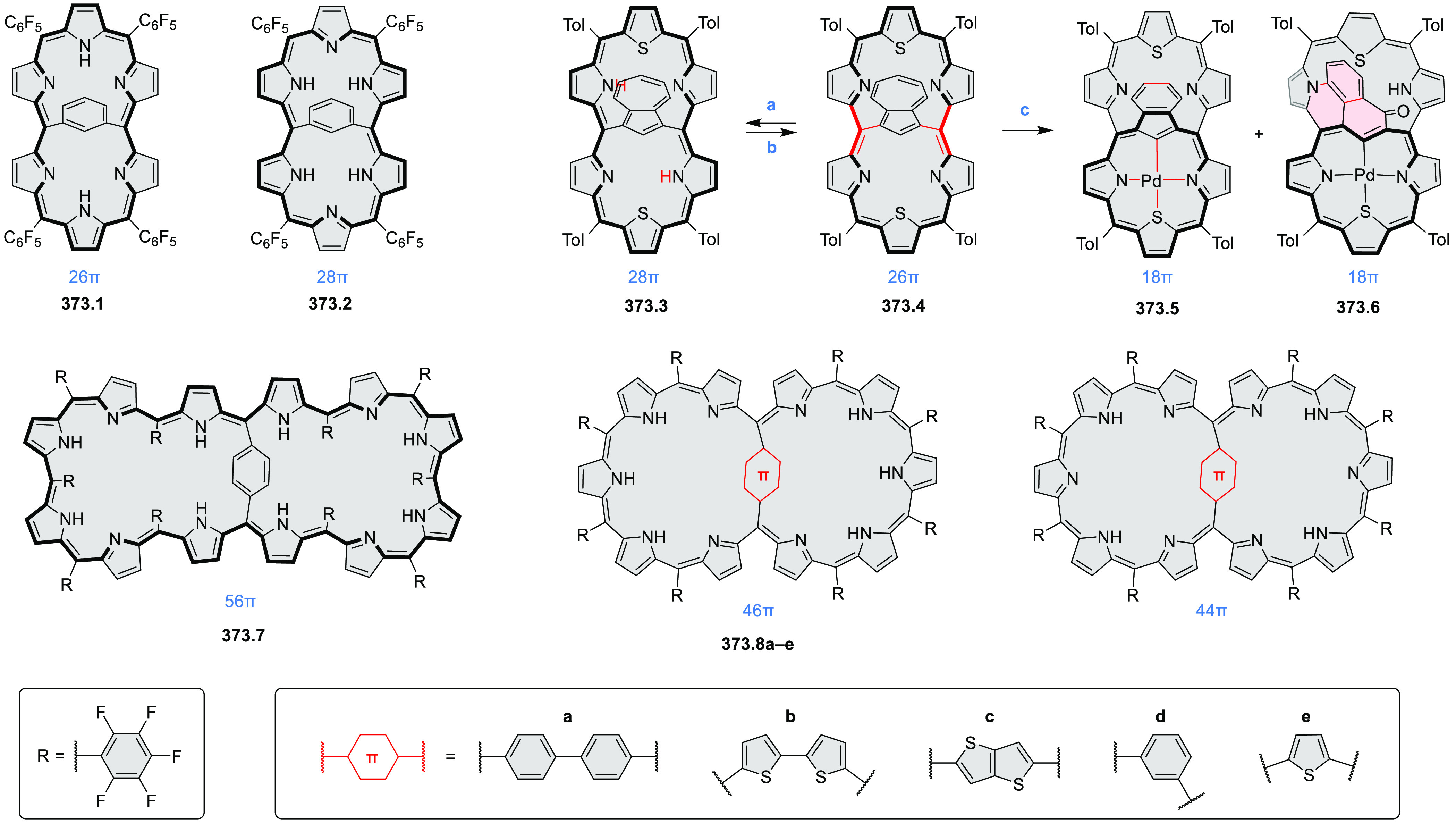
Reagents and conditions: (a)732 Zn/H+; (b) O2; (c) Pd(OAc)2, DMF.
An azulene-strapped hexaphyrin 373.4, reported by the Latos-Grażyński group, is an aromatic 26 π-electron system, and its absorption spectrum showed intense bands at 455 and 565 nm along with a NIR band at 1038 nm.732 The chemical reduction of 373.4 with zinc powder in the presence of gaseous HCl produced the highly antiaromatic 28π-electron macrocycle 373.3. A mononuclear palladium(II) complex obtained from 373.4 upon insertion of palladium(II) in DCM/MeCN retained the aromaticity of the original macrocycle. In DMF, however, palladium insertion produced a range of products, including the two rearranged species 373.5 and 373.6. Each of these systems showed a predominance of an 18π-electron pathway restricted to the Pd-containing macrocycle.
“Earring” porphyrins C36.1, C36.2, and C36.6, containing tripyrrin loops fused to the macrocyclic core, were developed by Song and Kim et al. (Chart 36).733,734 The synthetic strategy, based on Suzuki–Miyaura coupling, was subsequently extended to develop the heterole-fused derivatives C36.3–5.735 These porphyrinoids possess multiple coordination cavities and can accommodate two or three metal ions per molecule, with meso carbons acting as organometallic donors in the earring loops. The UV–vis–NIR absorption spectra of the metalated complexes C36.1c, C36.2b, and C36.6b were significantly red-shifted, reaching up to 1400–1500 nm in DCM. Even larger red-shifts, reaching up to 2200 nm, were observed for the metal complexes C36.3a and C36.3c. In line with these observations, the electrochemical HOMO–LUMO gaps of C36.1c, C36.2b, C36.3a, and C36.3c were 0.99, 0.88, 0.80, and 0.78 eV, respectively.
Chart 36. Edge-Sharing π-Extension in Porphyrinoids with Multiple Cavities.
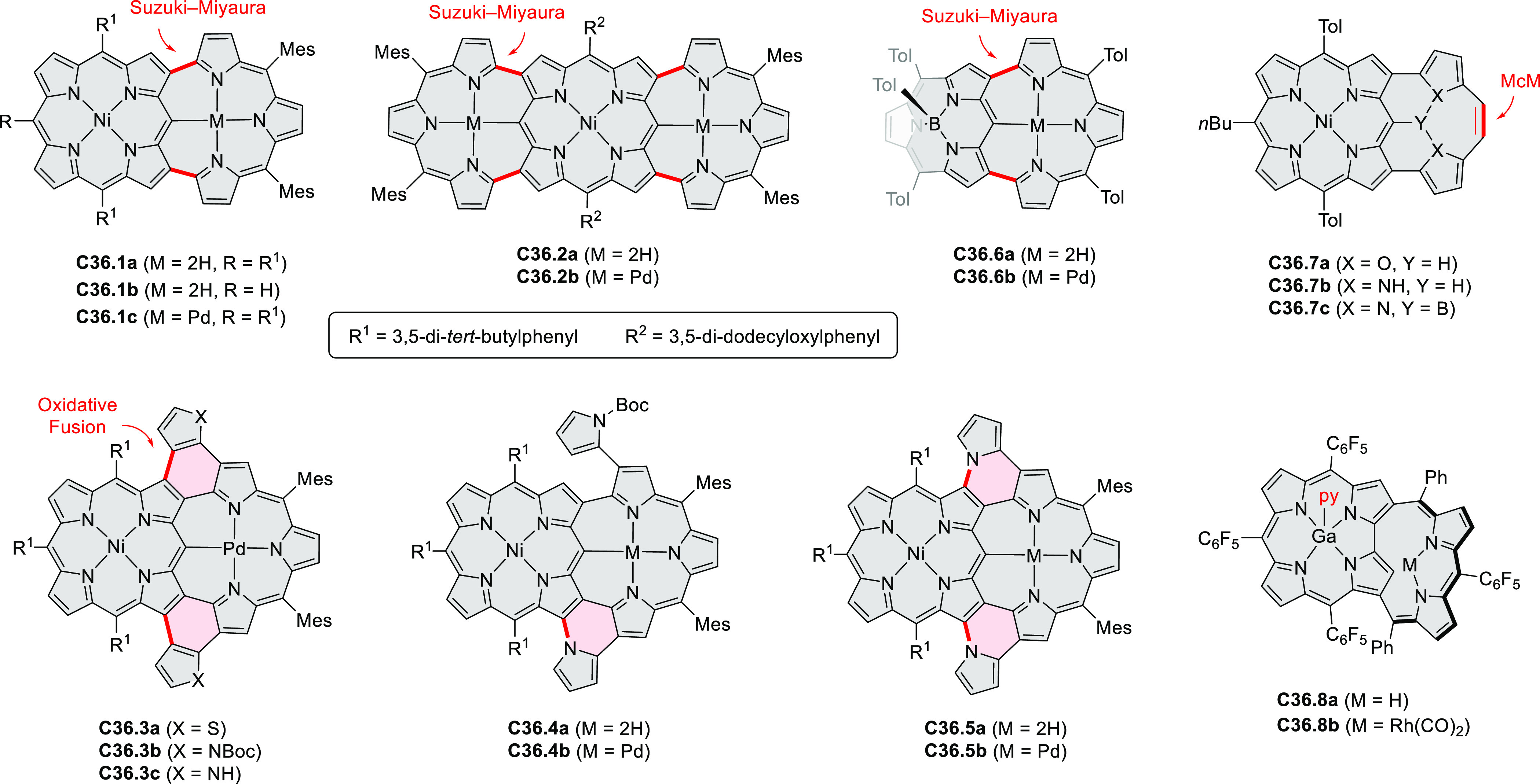
A similar design was employed in the synthesis of hybrid macrocycles C36.7a,b, reported by Pawlicki et al., in which the triphyrin(2.1.1)-like loop was closed via the McMurry reaction.736 The loop in C36.7b accommodated boron(III), yielding the boron(III) complex C36.7c with a direct B–C bond. Boron coordination strongly affected the optical properties of C36.7c, which showed a strong absorption at 807 nm. Lateral π-extension of corrole by attaching a dipyrrin loop produced the hybrid bismacrocycle C36.8a containing a nonplanar corrorin-like subunit.737 The electronic absorption spectra of C36.8a and its rhodium complex C36.8b showed weak broad Q-like bands in the 500–800 nm region. C36.8a was also found to be emissive, with a low quantum yield of 1.3%.
In 2019, Yamada and co-workers reported the synthesis of porphyrin(2.1.2.1)-based nanobelt 374.4 via condensation reaction of 1,2,4,5-tetra(pyrrol-2-yl)benzene 374.1 and arylaldehyde (Scheme 374).738 This condensation reaction with multiple-reactive site containing precursor 374.1 resulted in the formation of different products 374.2–4, depending upon the amount of acid and reaction time. The solid-state structure of 374.4 showed a belt-shaped structure with C3h symmetry, in which the porphyrin moieties maintained the arch-shaped structures similar to those in 374.2. The absorption spectrum of 374.4 in DCM showed a maximum at 512 nm, while 374.3 showed red-shifted absorption at 558 nm. Cyclic voltammetry revealed that 374.4 had an ability to both release and accept five electrons, indicating that the shape of the nanobelt stabilized multicationic and multianionic states. The concave-shaped cavity of 374.4 acted as a receptor capable of cooperative binding of two C60 molecules, yielding a 1:2 supramolecular complex.
Scheme 374. Synthesis of a Porphyrin(2.1.2.1)-Based Nanobelt.

Reagents and conditions: (a)738 (1) RCHO, BF3·Et2O (5–70 mol %), (2) DDQ, (3) Ni(OAc)2.
Well-ordered two-dimensional polymers with porous frameworks can be obtained by dione–diamine condensations (cf. Scheme 260, Section 6.1.6, and Scheme 393, Section 7.7.2). This approach was successfully applied for the synthesis of phthalocyanine-based COFs 375.2a–c by Mirica et al. (Scheme 375).739 and by Dong and Feng et al.740 The synthetic procedure of Mirica et al. involved the reaction of 375.1a and 149.8a in a DMAc/o-DCB mixture in the presence of sulfuric acid for 10 days, which afforded the desired COF material 375.2a. This synthesis was also achieved by using microwave heating, which significantly reduced the reaction time, albeit at the expense of slightly diminished crystallinity. The structure of 375.2a featured square apertures with a side length of 2.2 nm and showed high chemical and thermal stability. The intrinsic bulk conductivity of the 375.2a reached 2.51 × 10–3 S/m and increased by 3 orders of magnitude with iodine doping. Upon integration into chemiresistive devices, this conductive COF showed excellent responses to various reducing and oxidizing gases, including NH3, H2S, NO, and NO2 with limits of detection at the ppb level. The synthetic procedure described by Dong and Feng et al.740 involved condensation of the reactants with p-TSA as the catalyst in a heated NMP/mesitylene mixture. The resultant compounds 375.2b–c were found to be p-type semiconductors, both with a band gap of ∼1.2 eV and relatively similar electronic properties. It was subsequently found that 375.2b exhibited enhanced conductivity upon molecular iodine doping, with the carrier mobility reaching ∼22 cm2 V–1 s–1.741
7.6. Internally Fused Porphyrinoids
Doubly N-confused porphyrins containing an internally fused benzene ring were reported in 2020 by Uno, Mack, and co-workers (Scheme 376).742 When the bicyclo-fused bis(dipyrromethane) 376.2 was condensed with methylal under acidic conditions, a mixture of 376.3a and 376.3b was obtained in moderate yields. The proposed mechanism involved acid-induced skeletal opening of the bicyclo[2.2.1]heptadiene ring to give an intermediate with a formyl group attached to the sp3 carbon. 1H NMR spectra, X-ray structures, and theoretical calculations indicated the coexistence of both macrocyclic 18π and local 6π aromaticity in these systems, in spite of the presence of the formally antiaromatic 2,6-diaza-s-indacene subunit in the macrocycle.
Scheme 376. Benzene-Fused Doubly N-Confused Porphyrins.

Reagents and conditions: (a)742tert-butyl 5-acetoxymethyl-3,4-diethylpyrrole-2-carboxylate, catalytic p-TsOH·H2O, AcOH, rt, 3 h; (b) TFA, N2, CH2(OMe)2, DCM, p-chloranil.
In 2017, Yoneda and Neya reported the synthesis of [22]pentaphyrins 377.3a,b containing β,β-disubstituted pyrroles flanking a single free meso position (Scheme 377).743 The substitution of an electron-withdrawing group at the periphery in 377.3b suppressed its oxidative decomposition relative to 377.3a. 377.3a and 377.3b showed a roughly planar nonfused macrocyclic structure in the solid state and displayed strong 22 π-electron Hückel aromaticity. In methanol solution, compound 377.3b underwent an N-fusion reaction to give N-fused pentaphyrin 377.4, which exhibited solvent-dependent polymorphism, yielding either Hückel- or Möbius-type conformations in the solid state. Spectroscopic and theoretical evidence suggested that the Hückel conformer 377.4 was nonaromatic, whereas the Möbius conformer 377.4′ had a Möbius aromatic character.
Scheme 377. N-Fused Pentaphyrins and Their Reactivity.

Reagents and conditions: (a)743 (1) p-TsOH·H2O, DCM, N2, 90 min, (2) DDQ, 1 h; (b) CH3OH, 25 °C, 4 h.
Osuka et al. reported the synthesis of doubly N-fused [24]pentaphyrin 378.2 and its silicon complex 378.3, which were obtained in 6% and 38% yield, respectively, upon treating the singly fused 378.1 with MeSiCl3 and N,N-diisopropylethylamine in DMF at 80 °C (Scheme 378).744 The X-ray crystallographic analyses of these doubly fused structures revealed twisted geometries with a Möbius structure exhibiting effective macrocyclic π-conjugation. Upon addition of a fluoride ion, the silicon complex 378.3 afforded a monoanionic hexacoordinate SiIV complex 378.6, which could be converted back into 378.3 when treated with acid. Additionally, 378.3 was found to undergo Tamao–Fleming oxidation, which resulted in the formation of the nonaromatic β-keto doubly N-fused pentaphyrin 378.4 and Hückel-antiaromatic triply fused [24]pentaphyrin 378.5 in moderate yields.
Scheme 378. Synthesis of Doubly N-Fused [24]Pentaphyrin and Its Reactivity.

Reagents and conditions: (a)744 MeSiCl3 (100 equiv), DIPEA (300 equiv), DMF, 80 °C, 14 h; (b) KHCO3 (4 equiv), excess aq. H2O2, THF, rt, 45 min; (c) TBAF, CD2Cl2; (d) H3O+.
Metalation of the N-fused [22]pentaphyrin(1.1.1.1.1) 379.1 with palladium(II) trifluoroacetate in refluxing THF under aerobic conditions afforded two complexes of N-fused [22]pentaphyrin(1.1.1.1.0) 379.2 and 379.3, formed by extrusion of one of the meso carbons (Scheme 379).745 X-ray crystallographic analyses revealed fairly planar structures for both species, and the structural features indicated that the carbonyl moiety in 379.3 was conjugated to the sapphyrin core. Oxidation of 379.2 with DDQ and Sc(OTf)3 led to the formation of the heterodimer 379.4, in which two sapphyrin units were linked via a C–N bond. The UV–vis absorption spectrum of 379.4 in DCM exhibited Soret-like bands at 457 and 521 nm and Q-like bands at 638, 697, and 799 nm. Cyclic voltammograms showed two pseudoreversible oxidations and two reversible reduction waves for 379.4 with an electrochemical HOMO–LUMO gap of 1.63 eV.
Scheme 379. Palladium Insertion-Triggered Reactivity of N-Fused Pentaphyrin.

Reagents and conditions: (a)745 Pd(CF3CO2)2 (5 equiv), THF, reflux; (b) DDQ (1.2 equiv), Sc(OTf)3 (0.7 equiv), toluene, rt.
In 2019, Furuta and Xie et al. reported the synthesis of a thianorhexaphyrin 380.2, which showed unprecedented reactivity with oxidants accompanied by multiple N–C fusions and skeletally rearranged products (Scheme 380).746380.2 was prepared in 46% yield from 380.1 upon reacting it with 4 equiv of DDQ under high dilution conditions. Upon further addition of DDQ (2 equiv), three different multiply fused products were obtained, including the extensively fused 380.5–6. The unusual pyrrolo[1,2]isothiazole moiety in 380.6 could be desulfurized under a variety of conditions, to produce pyrrolizine species 380.3. The formally 26 π-electron macrocycle 380.2 had a nonaromatic character, while 380.3, 380.5, and 380.6 showed apparent globally aromatic features according to calculations.
Scheme 380. Oxidative Reactivity and Ring Fusion in Twisted Thianorhexaphyrin.

Reagents and conditions: (a)746 DDQ, DCM; (b) p-chloranil, DCM; (c) O2, Et3N/MeCN; (d) H2O2/AcOH, THF; (e) m-CPBA, DCM; (f) PPh3, DCM.
The N-fused heptaphyrin 381.2 was isolated as a byproduct in a macrocyclization reaction between the benzo-tetrathiafulvalene-fused pyrrole 381.1 and pentafluorobenzaldehyde (Scheme 381).747 The X-ray crystal structure revealed that the central macrocyclic core exists in a highly strained, nonplanar figure-of-eight conformation. The electronic spectrum of 381.2 extended up to 1600 nm, revealing the intrinsic intramolecular charge transfer (CT) character of the chromophore. A fused thiaheptaphyrin(1.1.1.1.1.1.0) 381.3a, structurally related to 380.2, was obtained by sequential dehydrogenation of a heptapyrromethane analogue (Scheme 381).748 In the solid state, the brominated derivative 381.3b was found to contain a pyrrolo[3,2-b]pyrrolizine fragment and to adopt a figure-eight-like conformation. The UV–vis–NIR spectrum of 381.3a in DCM displayed absorption maxima at 428, 653, and 808 nm and a broad low-energy band at 950–1350 nm.
Scheme 381. N-Fused Heptaphyrins.

Reagents and conditions: (a)747 (1) pentafluorobenzaldehyde, BF3·H2O, DCM, 3 h, rt, (2) DDQ, 1 h, rt.
An internally bridged, chiral corrole 382.2a was obtained by Goldberg and Gross et al. upon treating 382.1a with phosgene (Scheme 382).749 The XRD analysis showed that the corrole ring in 382.2a was severely twisted to minimize steric repulsion between the carbonyl group and the N-pyrrole proton. The same group later reported the rhodium(I) complexes 382.3a and 382.4a, which were utilized as cyclopropanation catalysts, with 382.4a showing a higher catalytic reactivity than 382.3a.750 In 2017, Biswal, Kar, and co-workers reported modified synthetic protocol to synthesize further N21,N22-carbamide-corrole derivatives 382.2b–h.751 The latter compounds were obtained by treating 382.1b–h with ammonium carbonate in the presence of pyridine, which was found to act as both a solvent and a weak base.
Scheme 382. Synthesis of N21,N22-Carbamide-corroles.
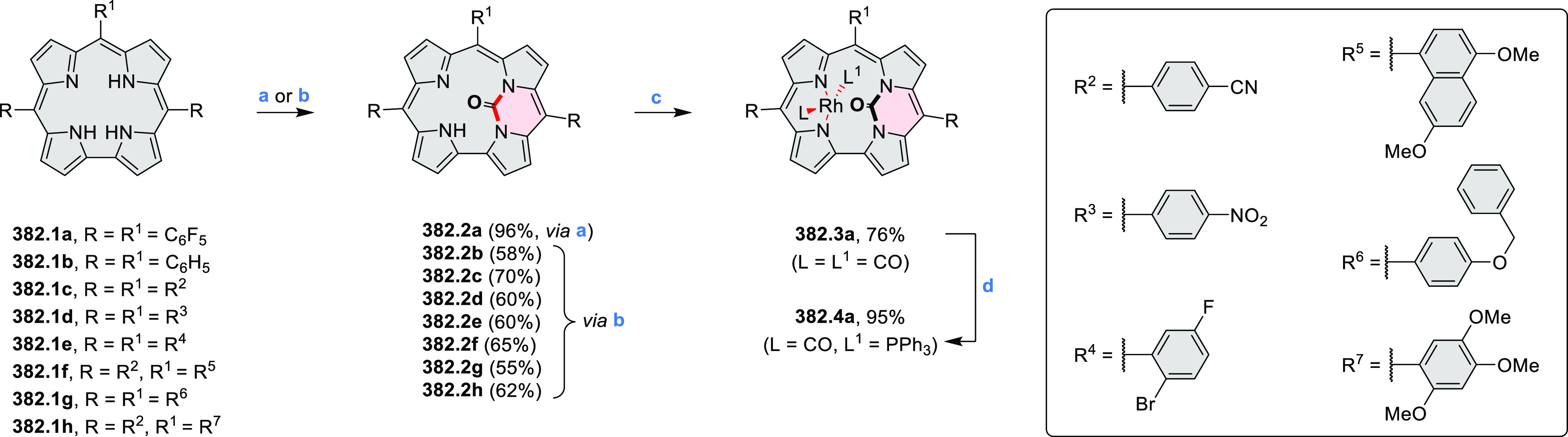
Reagents and conditions: (a)749 COCl2, toluene, 0 °C (10 min), then pyridine, toluene, N2, reflux (15 min); (b)751 (NH4)2CO3 (excess), CHCl2, pyridine, 110 °C, 3 h; (c)750 Rh2(CO)4Cl2, LiHMDS, benzene, reflux; (d)750382.3a, PPh3, benzene.
Internal N-fusion was induced inside para-benziporphyrin upon insertion of boron(III) and phosphorus(V).752 Reactions of 383.1 with PCl3 or PhBCl2 in a boiling toluene/Et3N mixture afforded, respectively, 383.2 and two stereoisomers of 383.3 (Scheme 383). Protonation of 383.2 occurred at one of the meso positions, also leading to stereoisomerism. Further, the oxidation of 383.2 and 383.3 with an excess of bromine in strictly anhydrous conditions led to two-electron oxidation and the formation of aromatic complexes 383.4 and 383.5, respectively.
Scheme 383. Synthesis of Boron(III) and Phosphorus(V) N-Fused p-Benziporphyrin.

Reagents and conditions: (a)752 PCl3, Et3N, H2O, toluene, 3 h; (b) PhBCl2, Et3N, toluene, 5 h; (c) Br2, CHCl3; (d) Br2, DCM, or DDQ/MeOH
7.7. peri-Fused Cyclophanes
7.7.1. Pyridine-Fused Cyclophanes
Coordination chemistry of the previously reported bisphenanthroline macrocycle 384.2 (cf. CR2017, Section 7.7.1) was explored by Surendranath et al. (Scheme 384).753 The poorly soluble iron(III) complex 384.3, obtained by metalation with anhydrous FeCl3, contained an apical chloride ligand and retained the deprotonated peripheral nitrogens characteristic of the free-base ligand. 384.3 and the corresponding μ-oxo complex, 384.4, were found to be good models of Fe–N–C electrocatalysts. In particular, the electrochemical and catalytic properties of 384.3 showed a high selectivity for the four-electron reduction of O2 in both acidic and alkaline media. In parallel work, Nabae et al.754 reported a related blue-colored complex 384.5, which was proposed to contain iron(II), a peripherally protonated bisphenanthroline ligand, and two axially coordinated DMF molecules. Upon heating this product in water at 85 °C, the triple-decker complex 384.6 was isolated and characterized crystallographically. The fourteen-membered macrocycle in 384.6 had relatively strong Fe–N bonds with an average bond distance of 1.90 Å, which is markedly shorter than that in porphyrin complexes (2.0 Å).
Scheme 384. Bisphenanthroline peri-Fused Cyclophanes and Their Complexes.

Reagents and conditions: (a)753 NH3, 280–300 °C, 36 h, (b)753 FeCl3, N(n-Bu)3, DMF, 170 °C, 36 h, (c)753 EtOH, 48 h, Soxhlet, (d)754 FeBr2, DMF; (e)754 H2O under air, rt.
In 2019, Kumagai et al. reported the synthesis of “TriQuinoline”, a macrocycle consisting of three quinoline units concatenated at the 2- and 8-positions in a head-to-tail fashion (Scheme 385).755 In an initial synthetic approach, 385.8 was employed to promote nucleophilic cyclization, followed by spontaneous oxidative aromatization which afforded the TriQuinoline 385.5b in rather poor yield (5.7%). In an alternative strategy, 2-fold benzylic radical bromination on 385.1 resulted in the dibromide product 385.2, which upon exposure to aqueous Na2CO3 at 80 °C afforded the aldehyde 385.3. In the subsequent step, a key intermediate product was obtained upon removal of the N-Boc group with trifluoroacetic acid, which afforded the TFA salt of cyclic imine 385.4. The final step involved an unusual uninterrupted Povarov reaction, which furnished the desired 385.5a. 385.5a was found to be an efficient DNA intercalator for the inhibition of topoisomerase I activity. 385.5a formed supramolecular binary and ternary complexes with [12]cycloparaphenylene (CPP) and coronene, stabilized by π stacking and CH···π interactions.
Scheme 385. Shape-Persistent Macrocycles Containing peri-Condensed Pyridine Rings.

Reagents and conditions: (a)755 AIBN, NBS (2.5 equiv), CCl4, 77 °C, 5 h; (b) Na2CO3, 1,4-dioxane, 80 °C, 14 h; (c) TFA (10 equiv), dichloroethane, 70 °C, 18 h; (d) n-butyl vinyl ether (10 equiv), MeCN, 45 °C, 25 h; (e) n-butyl vinyl ether (10 equiv), degassed CH3CN, rt; (f) air; (g) 2,3-dihydrofuran (50 equiv), MeOH, 25 °C, 24 h; (h) nBuLi, THF, −78 to 25 °C, 16 h.
Synthesis of a shape-persistent m-diethynylene-phenylene bridged macrocycle incorporating two quinoline moieties was described by Shimasaki and Shibata et al.756386.2 was obtained in 23% yield from 386.1 by deprotection of the TMS group with KOH followed by cyclodimerization in a high-dilution Sonogashira cross-coupling reaction (Scheme 386). X-ray crystal structure revealed that 386.2 adopts an almost planar C2h symmetrical parallelogram structure. Fluorescence of 386.2 had a maximum at 420 nm with a quantum yield of 47%.
Scheme 386. Shape-Persistent Macrocycles Containing peri-Condensed Pyridine Rings.

Reagents and conditions: (a)756 (1) KOH, MeOH/THF (2:1 v/v), rt, 24 h, (2) Pd(PPh3)4, CuI, NEt3/THF (1:2, v/v), 50 °C, 24 h.
In 2020, Isobe and co-workers reported the bottom-up synthesis of a nitrogen-doped phenine nanotube with a discrete, rigid structure by combining eight 2,4,6-trisubstituted pyridine units with thirty-two 1,3,5-trisubstituted benzene units (Chart 37).757 The target nanotube fragment C37.1 was obtained by forming eight aryl–aryl bonds along the edges of the tube in a Yamamoto-type coupling reaction. C37.1 has a porous structure based on the graphitic lattice of (12,12)-carbon nanotube with a length index of tf = 7.0. The UV–vis absorption spectrum of N-doped macromolecule showed absorptions reaching up to 378 nm, corresponding to an optical gap of 3.28 eV, and the fluorescence quantum yield of ca. 16%.
Chart 37. Nitrogen-Doped Phenine Nanotubes.

7.7.2. Pyrrole-Fused and Related Cyclophanes
Fold-in syntheses of macrocyclic tetrabenzopentaaza[9]helicene and pentabenzohexathia[9]/[5]helicene were reported in 2017 by Tanaka, Kim, and Osuka et al.758 Oxidative fusion reaction of the 1,2-phenylene-bridged cyclic hexapyrrole 387.1a with 5 equiv of DDQ and Sc(OTf)3 gave a pale-yellow compound 387.2a in 4% yield (Scheme 387). Regioselectivity in this oxidation was high, producing only the 4-fold fusion product 387.2a even upon increasing temperature or increasing oxidant equivalents. Similarly, by oxidation of 387.1b with 8 equiv of DDQ and Sc(OTf)3, a thiophene-based system 387.2b was obtained in 66% yield. On the basis of X-ray crystallographic analysis and spectroscopic studies, the structure was identified as the double helicene product 387.2b resulting from a 5-fold oxidative fusion along with an unexpected 1,2-aryl shift. 387.2a exhibited well-defined emission at 427 and 453 nm with a high fluorescence quantum yield, Φf = 0.31. In contrast, 387.2b emitted very weakly with Φf = 0.018. Both macrocycles were chiral and could be separated into enantiomers.
Scheme 387. Oxidative Fusion Reactions of ortho-Phenylene-Bridged Cyclic Hexapyrrole and Hexathiophene.

Reagents and conditions: (a)758 DDQ, Sc(OTf)3, toluene, reflux.
In 2017, Higashibayashi et al. reported the isolation of the members in the [n]cyclo-1,8-carbozolylene family 388.1aa–bc (Scheme 388).759 In their procedure, each of the 2,8-dibromo carbazole derivatives 388.2a,b was slowly added to a THF solution of Ni(cod)2, cod, and 2,2′-bipyridyl at 80 °C. The reductive cyclooligomerization of 388.2a produced the macrocyclic trimer (11%) and tetramer (8.0%), whereas 388.2b yielded the macrocyclic trimer, tetramer, and hexamer (16%, 7.9%, and 1.8%, respectively). The absence of the cyclic hexamer in the former case was ascribed to the lack of solubilizing substituents. The corresponding hydrazinobuckybowls 388.3a and 273.4 were isolated in each case as a minor side product. [3]Cyclo-1,8-carbazolylene 388.1ba was further used as a tridentate ligand for boron, silicon, and phosphorus, affording the corresponding flake-shaped complexes 388.4a–d. In 2017, the same group attempted to prepare the macrocycle 388.5 via the DDQ-mediated dehydrogenation of the [3]cyclo-1,8-carbozolylene 388.1ba (Scheme 388).760 When ethanol-stabilized chloroform was used as the solvent, the α-ethoxyimine structure 388.6 (87% yield) was isolated. In contrast, the use of ethanol-free chloroform gave the ring-rearranged ketone 388.7 (95% yield), which was shown by DFT calculations to be 6.6 kcal mol−1 more stable than the presumed α-hydroxyimine intermediate. These dearomatized products were proposed to arise from the desired 388.5, which was too unstable to be isolated.
In 2016, Suzuki et al. described the synthesis of three N-substituted tetracyclo(2,7-carbazoles) to investigate macrocyclic ring currents inside nanohoops.761C38.1a, C38.1c, and C38.1d were obtained using a platinum-mediated cyclooligomerization strategy (Chart 38). The X-ray crystal structure of C38.1a revealed the alternant conformation of the macrocycle, which was also predicted to be most stable by DFT calculations. C38.1d contained a 5,5-dimethylnonane bridge between two neighboring anti-carbazoles, which acted as a covalently bound probe for determination of NMR shielding inside the macrocycle. Accordingly, the methyl groups of the bridge were found to resonate at −2.70 ppm. Cyclocarbazole C38.1b was also employed in an OFET device by Poriel et al. and was found to produce a hole mobility μFE of 1.1 × 10–5 cm2 V–1 s–1.762 Analogous tetramers of 3,7-dibenzo[b,d]thiophene (C38.2) and 3,7-dibenzo[b,d]thiophene-5,5-dioxide (C38.3) were similarly obtained by Yamago et al.763C38.3 was characterized by the single-crystal X-ray structure which revealed the anti-orientations with respect to the adjacent DBTO unit. The fluorescence spectra of C38.2 and C38.3 showed emission maxima at 510 nm (ΦF = 0.21) and at 429 and 529 nm (Φf = 0.41), respectively.
Chart 38. [4]Cyclo(2,7)carbazoles and Related Systems.

In 2018, Shimasaki and Shibata reported the synthesis of fully conjugated macrocycles composed of two m-diethynylene-phenylene-bridged dibenzofuran, dibenzothiophene, and carbazole units.764 The Sonogashira cross-coupling reaction between the diiodide precursors 389.2a–c and the deprotected diethynyl derivatives 389.1a–c under high dilution conditions afforded the desired macrocycles 389.3a–c in moderate yields (Scheme 389). The three compounds showed UV absorptions with onsets below 400 nm and were fluorescent, with ΦF of up to 0.57 for 389.3a.
Scheme 389. peri-Fused Dibenzofuran-, Dibenzothiophene-, and Carbazole-acetylene Macrocycles.

Reagents and conditions: (a)764 (1) TBAF, THF, rt, 1 h, (2) 389.2a, 389.2b, or 389.2c, Pd(PPh3)4, CuI, NEt3/THF (1:1 v/v), rt, 24 h.
Hybrid cyclophanes consisting of carbazole and pyridine units were reported in 2019 by Pawlicki et al.765 The dialdehyde precursor 390.1 was subjected to the low valent titanium coupling in 1,4-dioxane which resulted in products of both intramolecular and intermolecular cyclization (Scheme 390). 390.2a and 390.2b were obtained in 15% and 12% yield. In this reaction, cyclodimers 390.3a–c were also isolated as minor products. Upon UV irradiation of 390.3c in air-free CDCl3 for 72 h, an intramolecular cyclobutane-linked product 390.4 was obtained as a result of close proximity of the two vinylene bridges, which allowed the [2 + 2] photodimerization. In contrast to the helically twisted 390.3c, compound 390.4 was found to be silent in the CD experiments in line with its achiral structure.
Scheme 390. Synthesis of a Carbazole-Based Cyclophane.

Reagents and conditions: (a)765 TiCl4, Zn, pyridine, dioxane, reflux; (b) CHCl3 or CDCl3, 365 nm, 72 h.
In 2015, Tanaka et al. reported the synthesis of mesogenic macrocycles composed of four bis(salicylidene)-o-phenylenediamine (salphen) moieties alternating with four carbazoles.766391.4e was synthesized in 22% yield in a 4:4 condensation reaction between the carbazole 391.1b and o-phenylenediamine 391.2e (Scheme 391). A similar cyclization approach was used to synthesize the other macrocycles 391.4a–d. An X-ray crystallographic analysis of 391.4a revealed a highly symmetric structure where the inner cavity size was ca. 9 Å. Metalated derivatives 391.5–6 self-assembled into columnar liquid-crystalline phases showing high fluidity over a wide range of temperatures. Later in 2017, the same authors reported the synthesis of expanded macrocycles containing diindolocarbazole units, which were design to retain the geometry of their carbazole-based congeners.767 The structure of the giant macrocycle 391.7b was unambiguously confirmed using STM, which showed a hollow square structure with a diagonal of 2.5 nm. 391.7a formed a Colr thermotropic phase stable over a range of 16–213 °C.
Scheme 391. Synthesis of an Imine-Bridged Carbazole-Based Cyclophane.
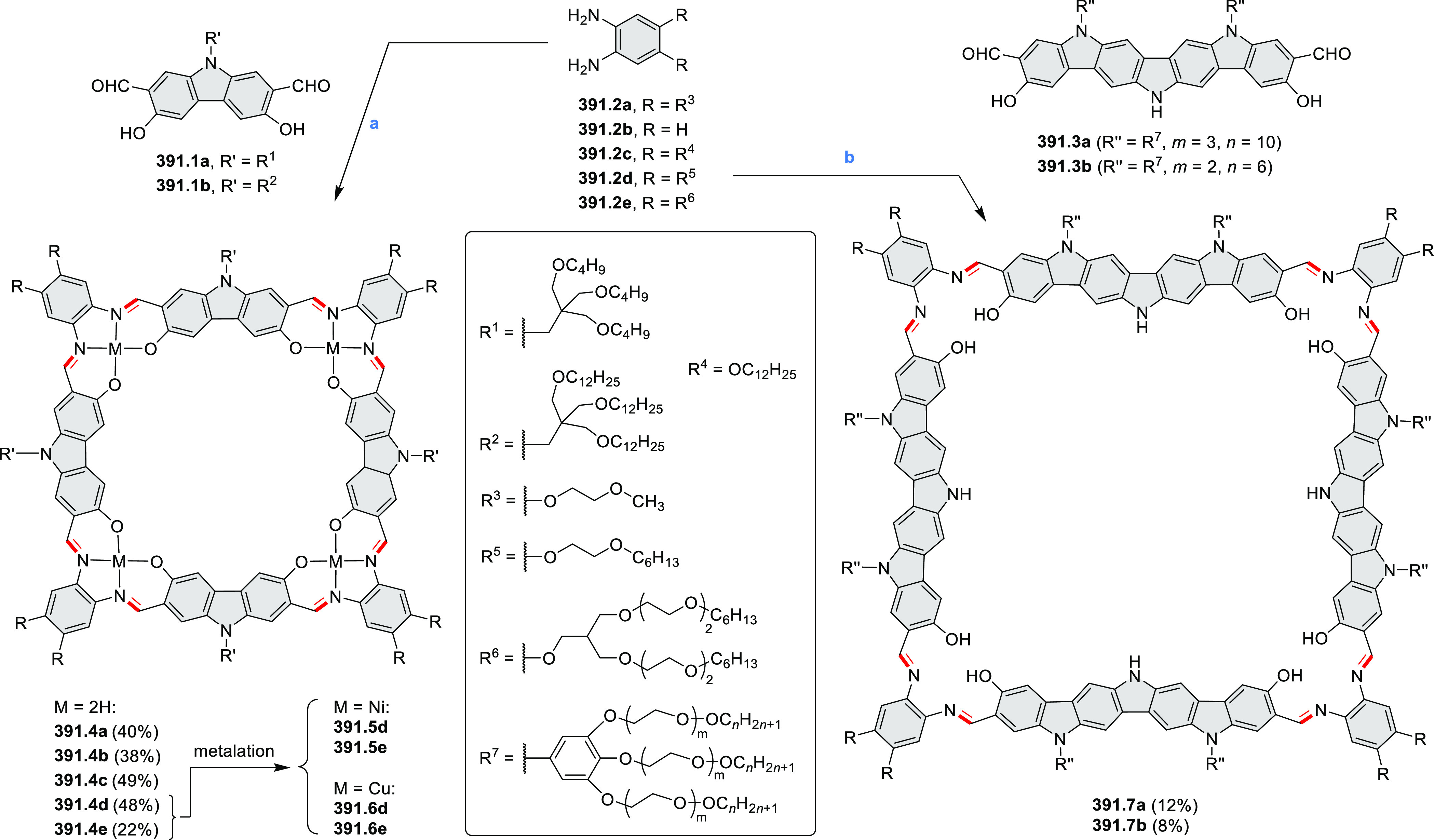
Reagents and conditions: (a)766 EtOH, reflux, 48 h; (b)767 EtOH, CH3CN, reflux, 36.5 to 38 h.
Trimeric macrocycles with alternating carbazole and triazole units can be formed via CuAAC cyclooligomerization (Scheme 392). Following the first description of these structures by Flood and co-workers (see CR2017, Section 7.7.2), a recent development was the study of their self-assembly in the presence of anionic guests.768 A combination of anion binding by triazoles acting as C–H hydrogen bond donors and macrocycle stacking caused the formation of a 3:2 host–guest complex of 392.2b and a bisulfate anion in CHCl3. A hydrogen-bonded bisulfate dimer was thus bound inside a stack of three 392.2b molecules. Addition of MeCN to the medium weakened host–host interaction and allowed the 2:2 complex to form instead. A solid-state structure of 392.2a cocrystallized with NBu4HSO4 revealed 2:2:2 assemblies of 392.2a with HSO4– and NBu4+.
Scheme 392. Synthesis of Tricarbazolotriazolophane Macrocycles.
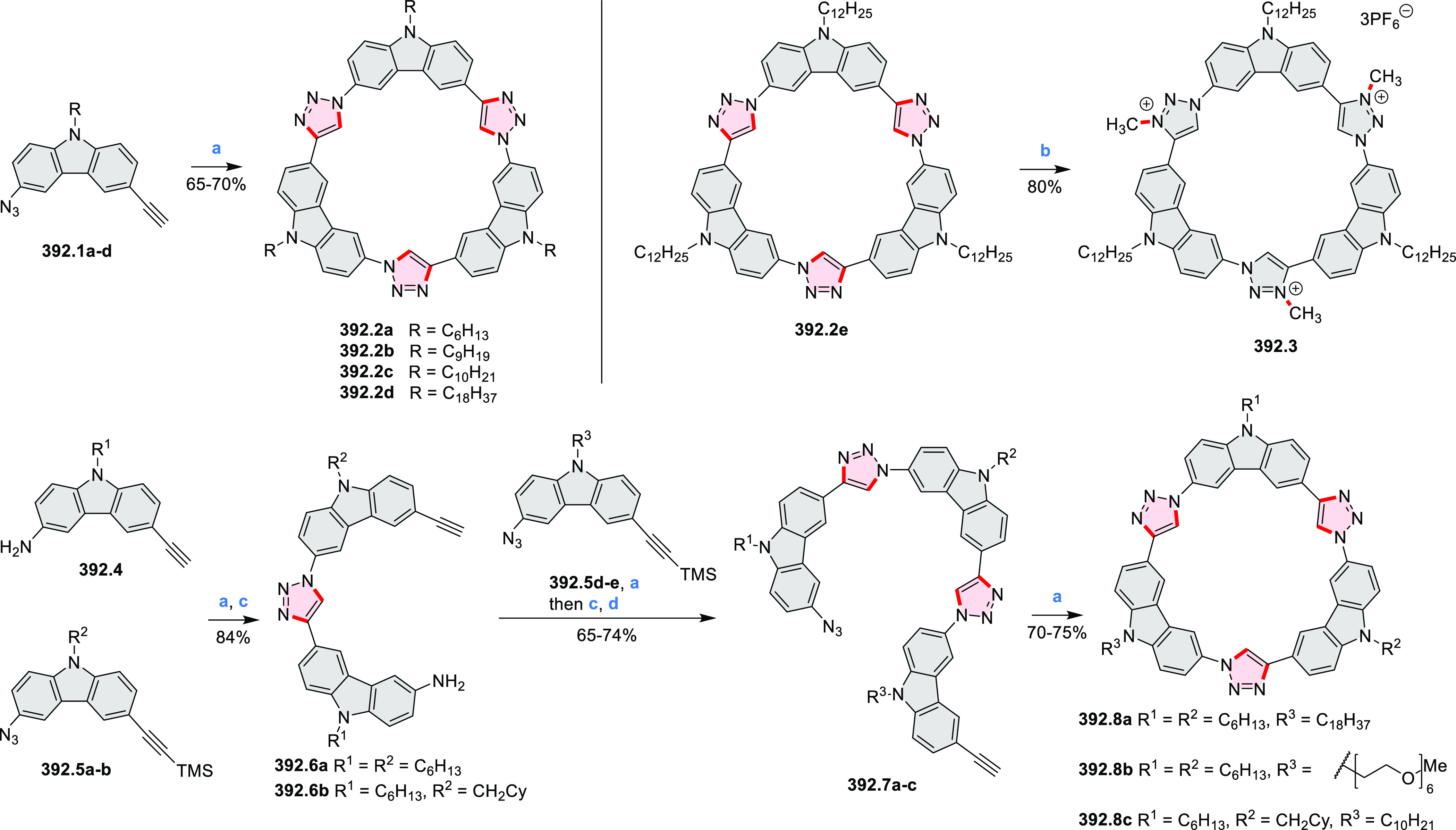
Reagents and conditions: (a)768,769 CuSO4, sodium ascorbate, TBTA, 2:1:1 THF/EtOH/H2O, 55–70 °C, 6–7 h; (b)772 (1) Me3OBF4, DCM, rt, 4 d, (2) NH4PF6, H2O/acetone, rt, 2 h; (c)769 K2CO3, 2:1 THF/MeOH, rt, 1 h; (d) (1) NaNO2, TsOH, THF/H2O, 0 °C, 30 min, (2) NaN3, 0 °C to rt, 1.5 h.
A stepwise synthesis of tricarbazolotriazolophanes was also performed, which allowed us to prepare compounds bearing dissimilar N-substituents on the carbazole groups.769 The low reactivity of TMS-capped alkynes in the CuAAC reaction and the use of arylamine as a precursor to the triazole reactive handle enabled the synthesis of dimers 392.6a,b followed by open trimers 392.7a–c. These precursors were cyclized in the final step, providing the macrocycles 392.8a–c with two or three different N-substituents. On the graphite surface, the products tended to assemble into rings of six molecules each, with different substitution patterns resulting in different 2D polymorphs. These assemblies were stabilized by hydrogen bonding interactions between the triazole N atoms and the carbazole C–H units on the outside of the macrocycle, and their self-organization behavior was further studied using STM imaging and computational methods.770,771
Nakamura and co-workers demonstrated enhancement of the anion affinity of a tricarbazolotriazolophane by N-alkylation of its triazoles.772 This was performed via treatment with a trimethyloxonium salt, providing the tricationic product 392.3 in 80% yield. Halide binding in DCM was consistent with 1:1 association and was enhanced by 2 orders of magnitude in the alkylated receptor 392.3 compared to 392.2. Anion affinity order was I– > Br– ≈ Cl–, reflecting the better size match of I– with the macrocycle cavity.
Ding and Wang reported pyrenodiimidazole-based COFs 393.2a–d, which were obtained using the Debus–Radziszewski reaction under solvothermal conditions from the tetraone 149.8b, ammonium acetate, and the corresponding trialdehydes 393.1a–d (Scheme 393, cf. Scheme 165, Section 4.7.2).331 Their structures were proposed to contain hexagonal lattices of cyclophane-like pores. These imidazole-linked COFs were found be insoluble in common solvents and showed high thermal stability (up to 400 °C) and chemical inertness toward strong acids and bases. These imidazole-linked COFs were further postmodified via N-alkylation to yield 393.2e–h which could be used as functional materials. BET surface areas determined for these systems using nitrogen adsorption–desorption measurements were in the rage of ca. 500 to 800 m2 g–1.
7.7.3. Sulfur-Containing peri-Fused Cyclophanes
In 2018, the Nuckolls group described two large, isomeric macrocycles C39.1 (“cis”) and C39.2 (“trans”) consisting of helical PDI ribbons connected via phenylene–thiophene linkers (Chart 39).773 These systems were synthesized from regioisomerically pure PDI building blocks, which were assembled into cyclodimers using the platinacycle method. The macrocycles were explored as n-type charge-transport materials in OFET devices. The cis isomer C39.1 exhibited electron mobility of ∼4.1 × 10–3 cm2 V–1 s–1, which was four times greater than that of the trans-isomer C39.2 (∼9.9 × 10–4 cm2 V–1 s–1). This difference in mobility was ascribed to the greater flexibility of the former species, which enhanced intermolecular contacts in thin films.
Chart 39. Sulfur-Containing peri-Fused Macrocycles.

Phenanthrylene–thienylene macrocycles 394.10–11 were obtained by Mazal and co-workers via sulfide-mediated annulation of the corresponding butadiynylene precursors 394.8–9 (Scheme 394).774 The cyclotetramer 394.11f was more easily oxidizable than the corresponding cyclotrimer and formed a stable radical cation upon treatment with DDQ. Self-association of these macrocycles due to π–π stacking was quantified using 1H NMR; the association constant was determined to be 291 ± 33 M–1 for 394.11f. Later in 2018, the same group reported another series of fully conjugated donor–acceptor (D–A) phenanthrylene–thienylene structures as shape-persistent macrocycles (Scheme 394).775 The open-chain diiodo compound 394.1 was used as the precursor for Sonogashira coupling reaction with acetylene gas, which afforded two macrocyclic products in a combined yield of 48%. The resulting inseparable products 394.2a and 394.2b were directly used as a mixture in the next step, in which the butadiynylene linker of 394.2b was selectively transformed into the 2,5-thienylene moiety. To obtain the D–A macrocycles, the acetonide protection of the quinone functionality was removed by trifluoroacetic acid (TFA) in a two-phase DCM/water system, yielding the quinone 394.3c. The latter quinone was subjected to condensation reactions with diamines to produce peripherally extended macrocycles 394.4c–7c.
Scheme 394. Sulfur-Containing peri-Fused Macrocycles.
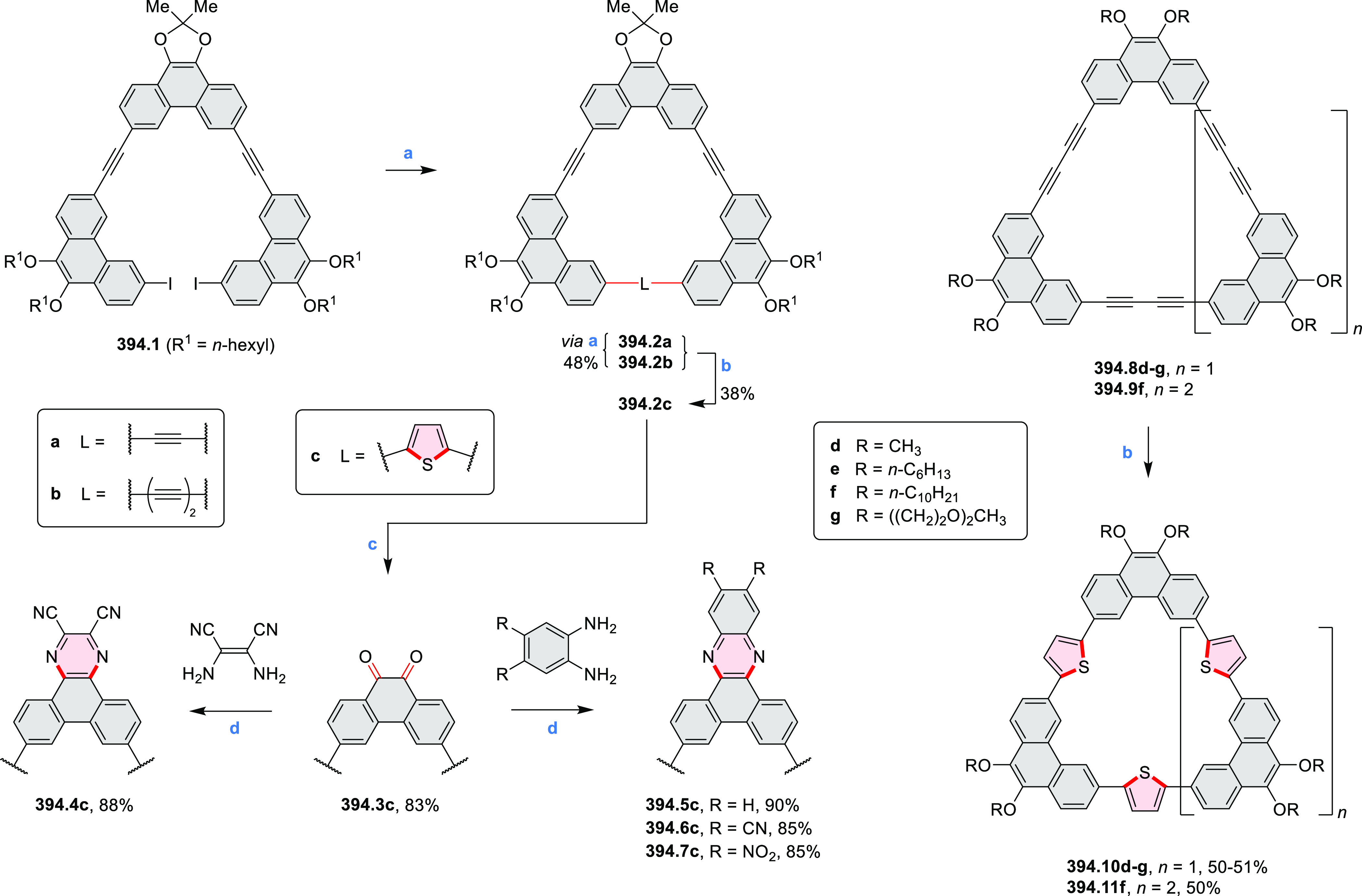
Reagents and conditions: (a)775 acetylene, Pd(PPh3)4, CuI, Et3N, THF, 70 °C, 3 h; (b)774,775 Na2S·H2O, toluene, 2-methoxyethanol, 150 °C, 12 h; (c)775 TFA, DCM, H2O, rt, 12 h; (d)775p-TsOH, AcOH, EtOH, DCM, 110 °C, 12 h.
Cyclophanes containing phenothiazine and related subunits were previously reported by several groups (cf. CR2017, Section 7.7.3). More recently the known system 395.8 was explored as an antimicrobial agent, capable of inhibiting both bacterial and fungal pathogens (Scheme 395).776 In 2020, Jiang, Zhu, and co-workers reported the synthesis of phenoxathiin-based molecular belts.777 The biphenolic building blocks 395.2 and 395.5 were obtained over a series of steps starting from 395.1a. Ullmann-type cyclization of 395.2 or 395.5 with the dibromoarene 395.1b followed by an intramolecular Friedel–Crafts reaction resulted in the formation of cyclo[8]phenoxathiins 395.4 and 395.7, respectively. 395.4 had a bowl-like geometry and was found to form a capsule-like 2:1 complex with C60, stabilized by C–H···S hydrogen bonds. The complex was stable not only in the solid state but also in solution, with a high formation constant β2 = 3.6 × 109 M–2. In the crystal, 395.7 formed a ring-in-ring supramolecular complex with [2,2]paracyclophane, characterized by a distorted octagonal conformation.
Scheme 395. Phenoxathiin- and Phenazine-Containing Cyclophanes.
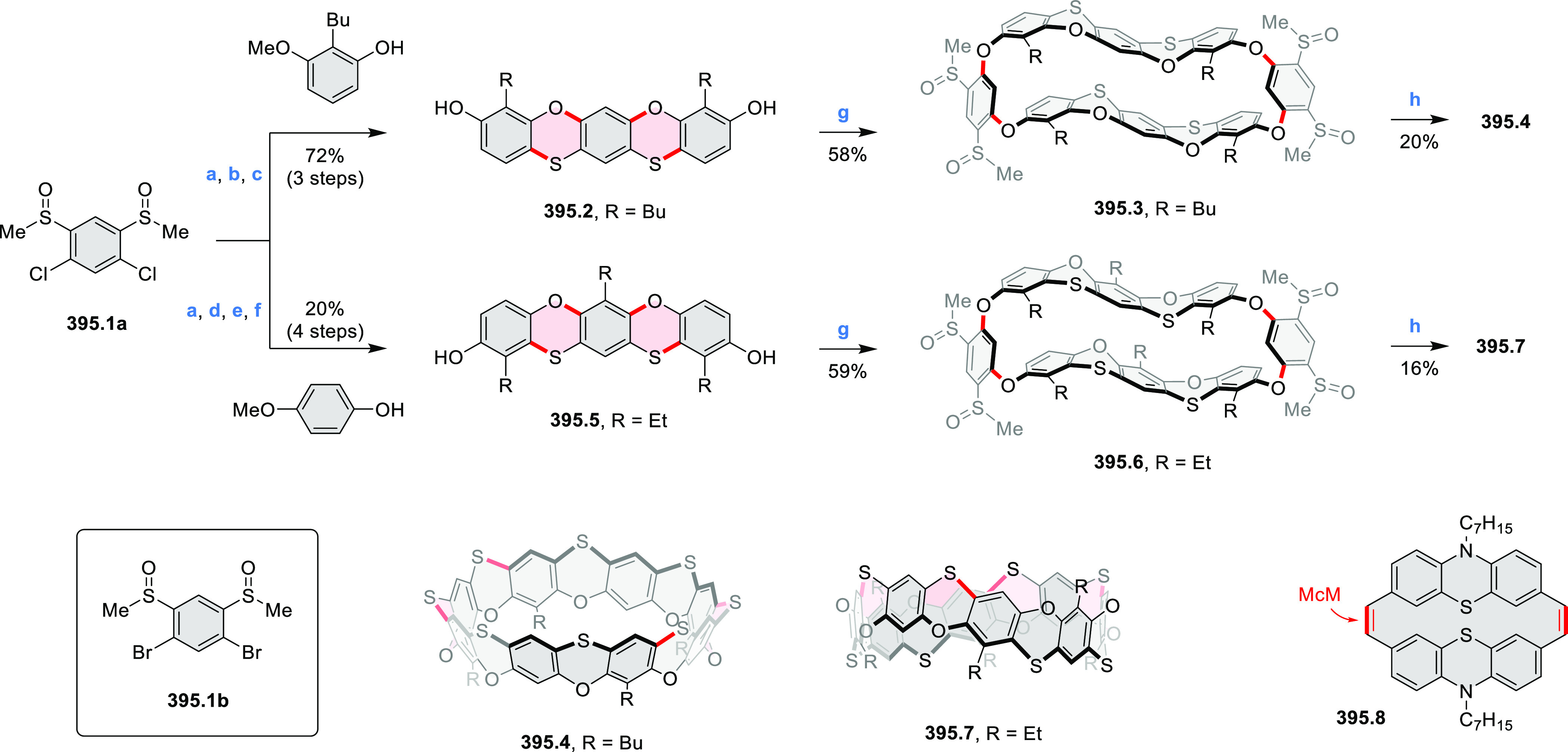
Reagents and conditions: (a)777 Cs2CO3, DMSO, 120 °C, 26 h; (b) (1) Tf2O, dichloroethane, rt, 1 h, (2) pyridine, rt, 10 h; (c) BBr3, DCM, −15 °C to rt, 20 h; (d) (1) CF3SO3H, P2O5, rt, 26 h, (2) pyridine/H2O (1:1, v/v), 105 °C, 18 h; (e) (1) nBuLi, THF, −5 °C, 1 h, (2) CH3CH2I, rt, 24 h; (f) BBr3, DCM, −10 °C to rt, 20 h; (g) 395.1b, CuI, Cs2CO3, N,N-dimethylglycine, DMAc, 150 °C, 48 h; (h) (1) CF3SO3H, 80 °C, 48 h, (2) pyridine/H2O (1:1, v/v), 105 °C, 15 h.
peri-Fused macrocycles, cyclotrimer 396.3a and cyclotetramer 396.3b, were reported by the Wu group (Scheme 396).778 These macrocycles were synthesized via Yamamoto coupling of the dibromo monomer 396.1 followed by the reduction with SnCl2 to afford the fully conjugated products. These macrocycle products exhibited good stability under ambient conditions, and the attachment of bulky 4-tert-butyl-2,6-dimethylphenyl groups improved the solubility. NMR investigations indicated the closed-shell ground-state electronic structure for both macrocycles, with 36 π- and 48 π-electron antiaromatic conjugation pathways in 396.3a and 396.3b, respectively. The dications of 396.3a and 396.3b obtained by oxidation with NO[SbF6] were characterized as globally aromatic open-shell singlet diradical dications. The singlet–triplet energy gaps of [396.3a]2+ and [396.3b]2+ were estimated to be −2.90 and −2.60 kcal mol–1, respectively.
Scheme 396. Sulfur-Containing peri-Fused Macrocycles.

Reagents and conditions: (a)778 Ni(cod)2, THF, 65 °C; (b)778 SnCl2, DCM, rt; (c)779p-chloranil, rt, 2 h.
In another report by the Wu group, the dihydro-expanded porphycene 396.4, obtained via McMurry coupling, was subjected to further oxidative dehydrogenation to produce the cyclopentabithiophene-bridged macrocycle 396.5.779 ESR and SQUID measurements revealed that 396.5 had an open-shell singlet ground state with a small tetraradicaloid contribution and a small singlet–triplet energy gap (ΔES–T) of −0.92 kcal mol–1. The cyclic voltammogram of 396.5 displayed four oxidation waves at −0.64, −0.54, −0.09, and +0.22 V and one reduction at −1.45 V (vs Fc/Fc+) with a small electrochemical HOMO–LUMO energy gap of 0.81 eV. Despite these features, the macrocycle exhibited very good stability under ambient conditions.
7.7.4. Oxygen-Containing Cyclophanes
Preparation of the furan-fused [12]cyclo-p-phenylene macrocycle 397.2 was reported in 2017 by Tsubaki et al.780 The target cyclic compound 397.2 consisting of dinaphthofuran units and biphenylene units was synthesized from 397.1 using the platinacycle method (Scheme 397). The UV–vis spectrum of 397.2 in DCM exhibited a maximum at 361 nm and was similar to that of 397.1, reflecting the weak electronic communication between the dinaphthofuran fragments. The fluorescence quantum yield of compound 397.2 was 7% with the emission maximum at 439 nm. In 2016, Higashibayashi et al. reported the synthesis of [3]cyclo-4,6-dibenzofuranylene 397.3 by Ni(0)-mediated reductive coupling of 4,6-dibromodibenzofuran.781 The flake-shaped C2 symmetric structure for this cyclotrimer 397.3 was found to be the most stable conformer as predicted by the DFT calculations.
Scheme 397. Oxygen-Containing peri-Fused Macrocycles.

Reagents and conditions: (a)780 (1) PtCl2(cod), THF, reflux, 2 days, (2) PPh3, toluene, reflux, 3 h.
7.7.5. Heteroatom-Bridged Cyclophanes
In 2017, Ito and co-workers investigated redox properties of the known tetraazacyclophanes C40.1 and newly synthesized tetraazacyclophanes C40.2a,b containing 9,10-anthracenylene moieties (Chart 40).782C40.2a and C40.2b were obtained in 14% and 8% yield from a Buchwald–Hartwig reaction. The dication of C40.2a was prepared upon addition of 2 equiv of AgSbF6 and was isolated as an air-stable salt, while the dication of C40.2b was too unstable to be isolated. X-ray crystallographic analysis of the dications of C40.1 and C40.2a revealed different types of structural deformation caused by steric demand of constituent aromatic moieties, which led to different spin density distributions. The singlet–triplet energy gap (ΔES–T) values for the dications of C40.1 and C40.2a were determined to be 0.3 kcal mol–1 and −1.0 kcal mol–1, respectively, by SQUID measurements, indicating, respectively, a ground-state triplet and singlet for these two species. Later in the same year, Sakamaki, Iwanaga, and Ito reported two larger macrocyclic derivatives hexaaza[16]paracyclophane, C40.3 and C40.4.783 The authors were able to isolate the stable trication salt of C40.4a and found that the three segmented phenylenediamine-based radical spins were found to be mutually antiferromagnetically coupled with J/kB ≃ −74 K, leading to a typical spin-frustrated three-spin system.
Chart 40. Polycyclic Arylamine Cyclophanes.

A related diradical dication salt 398.2 was prepared by Wang and Rajca et al. via oxidation of tetraazacyclophane 398.1 with an Ag(I) aluminate reagent (Scheme 398).784 The product proved to be thermally stable up to 180 °C and could be stored under ambient conditions. It had a triplet ground state with a singlet–triplet energy gap of approximately 0.5 kcal mol–1. In the solid state, 398.2 formed 1D chains with contacts between the peripheral p-tolyl groups. Neighboring chains were largely separated by the aluminate anions. These assemblies were found to possess intrachain antiferromagnetic coupling of J′/k = −5.4 K, which was associated with the intermolecular C···C contacts, including π–π interactions.
Scheme 398. Oxidation of a Tetraazacyclophane to a Diradical Dication.

Reagents and conditions: (a)784 2 equiv of Ag[Al(OC(CF3)2CH3)], DCM, rt, 1 day.
In 2019, Chi and co-workers reported the synthesis of a hybrid macrocycle 399.4, containing alternating para-quinodimethane and triphenylamine subunits (Scheme 399). The initial precursor 399.3 was obtained in a Lewis acid-mediated intermolecular Friedel–Crafts alkylation between the triarylamine 399.1 and the diol 399.2 in a dilute DCM solution (Scheme 399). Subsequently, oxidative dehydrogenation of 399.3 with DDQ in toluene afforded 399.4 in 73% yield. This macrocycle showed moderate 32 π-electron global antiaromaticity, reflecting the participation of the bridging nitrogen atoms in π-conjugation. The dication and tetracation of 399.4 showed an open-shell diradical character with mall singlet–triplet gaps of −1.07 and −2.96 kcal mol–1, respectively. These two species were globally aromatic and antiaromatic, respectively, with [30]- and [28]annulene structures.
Scheme 399. Polycyclic Arylamine Cyclophanes.

Reagents and conditions: (a)785 BF3·OEt2, DCM; (b) DDQ, toluene.
Macrocycles consisting of two hexabenzocoronene-based units connected by benzidine linkers were described by Isobe, Wu, and co-workers. The macrocyclization step was a double Yamamoto coupling of aryl bromide precursors in the presence of Ni(cod)2, which provided 400.1 and related molecules in 7–15% yields. Conjugated final products 400.2–4 were obtained from these precursors in quantitative yields upon oxidation with PbO2 (Scheme 400).786 Steric crowding in the dimethyl-substituted bay regions of 400.1–2 induced chirality in their hexabenzocoronene blocks. Only the homochiral dimers of 400.1 were obtained in the Yamamoto cyclodimerization from a racemic starting material. Compound 400.1 was separated into enantiomers by chiral HPLC and used to prepare optically pure samples of 400.2. Compounds 400.2–4 possessed oligoradicaloid characters due to the quinoidal structure of the oxidized benzidine linkers. Weak and broad low-energy absorption bands characteristic of such structures emerged in the final oxidative step. Their ESR signals were weak at rt but increased upon heating, revealing ΔES-T close to 7 kcal mol−1 in all cases. The hydrogenated precursors of 400.3–4 selectively encapsulated fullerene C70 via interactions with their concave surfaces.
Scheme 400. Conjugated Macrocyclic Hexabenzocoronene Dimers.

Reagents and conditions: (a)786 PbO2, CDCl2CDCl2, rt, 5 min.
The synthesis of helically twisted macrocycles, including the nitrogen-bridged derivative 401.4, was reported by Itami and co-workers (Scheme 401).787 Based on a previously reported method, condensation of p-phenylenediacetaldehyde 401.1 with 1-bromo-4-ethynylbenzene 401.2 led to the sterically congested phenanthrene 401.3. This compound was then coupled with aniline under Buchwald–Hartwig amination conditions to provide the cyclic dimer 401.4 in 12% yield. The nitrogen linker in 401.4 was the smallest linker in a series of compounds prepared via a similar approach. This resulted in a particularly high configurational stability with a racemization barrier of 31.7 kcal mol−1 according to computational analysis. Emission of 401.4 (λem = 466 nm, Φ = 41%) was bathochromically shifted relative to the analogues with aromatic linkers.
Scheme 401. Twisted Macrocycle with a Nitrogen Bridge.

Reagents and conditions: (a)787 BF3·Et2O, MS4 Å, 10:1 DCM/HFIP, rt, 3 h; (b) aniline, t-BuONa, Pd2(dba)3, P(t-Bu)3, toluene, 90 °C, 16 h.
The formation of oxygen- and sulfur-bridged cyclopyrenylene macrocycles was reported in 2019 by Aratani and Yamada et al. (Scheme 402).788 While performing emission studies of the cyclopyrenylene trimer 402.1, the authors observed a spectral change under ambient light in DCM, with a clear isosbestic point and a new peak emerging at 480 nm. The change was found to result in quantitative formation of the cyclic ether 402.2, containing an oxygen bridge between two pyrene units. Excitation of the cyclopyrenylene trimer at 515 nm resulted in singlet dioxygen 1O2* (1Δg) sensitization with a quantum yield of 0.33 at 298 K, yielding ultimately the oxygen insertion product. When treated with Na2S·9H2O, 402.1 was also transformed into the cyclic thioether 402.3, providing the latter product in 67% conversion yield. The fluorescence quantum yields (Φf) for 402.2 and 402.3 were found to be 0.54 and 0.44, respectively.
Scheme 402. Oxygen-Containing peri-Fused Macrocycles.

Reagents and conditions: (a) O2, hν; (b) Na2S·9H2O, DMF, 80 °C.
Synthesis of a series of thiacalix[n]dithienothiophenes C41.1a–g (n = 4–10) and selenacalix[4]dithienothiophene C41.2 were reported by Hasegawa and co-workers (Chart 41).789,790 The sulfur-containing macrocycles were prepared through a Pd-catalyzed coupling reaction of an appropriate dibromo precursor with tributylstannyl sulfide. The combined yield of the cyclic homologues C41.1a–g was as high as 75% when toluene solvent was used. Formation of larger macrocycles was found to be suppressed in DMF, whereas the highest conversion was found for reactions performed in a 1:1 mixture of DMF/toluene. An XRD analysis showed that C41.1a formed a puckered quadrilateral conformation with a large square cavity with a diagonal distance of 12.8 Å, and the shortest distance between facing walls was found to be 8.6 Å. Compound C41.1b showed an envelope-shaped pentagonal geometry with diagonal S···S distances in the range of 13.2–15.6 Å. UV–vis absorption spectra showed a red-shifted band for the larger macrocycles, which is attributed to the conjugation between the DTT units of each macrocycle through the sulfide linkers. Cyclic voltammograms showed reversible multielectron redox processes with low oxidation potentials, reflecting efficient electronic delocalization in these systems. Binding studies of these macrocycles with C60 revealed that C41.1a formed a Janus-head complex with two molecules of C60, whereas C41.1b and C41.1c formed stable 1:1 complexes with C60. The related selenium derivative C41.2 was prepared in 29% yield from the palladium-catalyzed coupling of 2,5-dibromodithienothiophene derivatives with (nBu3Sn)2Se. In this reaction only the tetrameric macrocycle was obtained, in contrast to the synthesis of thiacalix[n]dithienothiophenes. The cyclic tetramer C41.2 underwent similar complexation with C60 to produce 1:2 adducts in the solid state.
Chart 41. Miscellaneous Heteroatom-Bridged Cyclophanes.

Lhoták and co-workers reported the phenoxathiin-based thiacalix[4]arene C41.3a, which was accessed via an acid-catalyzed rearrangement of a spirodienone derivative, obtained by oxidation of thia[4]calixarene with chloramine-T.791−793 The inherently chiral C41.3a was alkylated, which led to the configurationally stable derivatives C41.3b,c with stereoselective formation of a partial cone conformer, which was determined using NMR and XRD analyses.
8. Conclusions
The field of polycyclic heteroaromatics covers a wide array of diverse yet interrelated topics. The research presented in this review is unified by a shared interest in two-dimensionally extended π-conjugation as a means of creating new properties in heterocyclic systems. Developments of the last five years, summarized herein, have expanded the repertoire of synthetic methods and spawned a plethora of new molecules. These achievements are of high relevance from the fundamental viewpoint, contributing to our understanding of physical properties of aromatic compounds and providing access to previously unknown structural motifs. These new structures often display unusual features, such as intrinsic curvature, three-dimensional aromaticity, or open-shell character. At the same time, by altering ring fusion and heteroatom placement in PHA molecules, it is possible to tune very precisely their electronic properties and solid-state behavior. In consequence, practically useful characteristics can be engendered even in structurally simple motifs, with clear benefits for materials science.
Future developments in the field will likely be driven by a continuing search for novel structural motifs. Here, introduction of seven-membered and larger rings may be seen as a particularly promising strategy toward such new structures. Unusual two-dimensional fusion patterns based on enlarged rings are easily accessible in pyrrole-based chemistry, notably among porphyrinoids, but can also be elaborated by modifications of benzenoid structures. Apart from changing the topology of the fused framework, large rings can induce out-of-plane distortions of the π-electron surface and may yield systems with pronounced negative curvature. Likewise, one can anticipate increasing interest in positively and neutrally curved heteroaromatics, which may be obtained using methods developed for carbocyclic targets. While much of the chemistry of heterocyclic nanographene analogues has been based on nitrogen, exploration of other heteroatoms, notably boron and oxygen but also some of the less-studied elements such as selenium or tellurium, is likely to have a significant impact on further evolution of the field, especially as a route toward functional materials.
Elaboration of new PHA motifs is often challenged by synthetic difficulties, and further progress will thus undoubtedly depend on the availability of efficient ring-forming reactions. Cyclizations based on oxidative, reductive, and transition-metal-catalyzed protocols are among the most fundamental methods available in the synthetic toolbox, and their further refinements will have a direct impact not only on the scope of structures that can be made but also on their large-scale availability. Annulative approaches, which enable multicomponent assembly of complicated targets, are an important alternative to direct cyclizations and will play an increasing role in heteroaromatic chemistry. Unique preparative opportunities have been offered by the remarkable development of on-surface synthesis that occurred over the past decade. While these methods currently do not give access to bulk quantities of products, they can provide unique chemical selectivities. Given the possibility of structural analysis on the single-molecule level, on-surface synthesis will surely remain an important tool of chemical discovery.
With the growing availability of polycyclic heteroaromatics, their use in diverse branches of organic chemistry and materials science will continue to expand. Because of their anisotropic shapes and structural rigidity, PHA motifs are suitable components for 1D and 2D polymers, framework materials, and supramolecular assemblies, such as molecular cages. They can also serve as functional platforms for the development of novel fluorophores, NIR dyes, charge-transfer assemblies, open-shell organics, and other systems relying on extensive π-electron conjugation. This broad range of applications has been one of the driving forces of ongoing research and will likely shape the field for years to come.
Acknowledgments
Financial support from the Foundation for Polish Science (TEAM POIR.04.04.00-00-5BF1/17-00 to M.S.) and the National Science Center of Poland (grants UMO-2018/29/B/ST5/01842 and UMO-2019/35/B/ST4/00401 to M.S. and 2017/27/N/ST5/00613 to L.M.) is gratefully acknowledged.
Glossary
Abbreviations
- 18c6
18-crown-6
- Ac
acetyl
- acac
acetylacetonate
- ACID
anisotropy of induced current density
- AFM
atomic force microscopy
- AIBN
azobis(isobutyronitrile)
- AIE
aggregation-induced emission
- AM
air mass (coefficient)
- BAHA
tris(4-bromophenyl)aminium hexachloroantimonate
- BHJ
bulk heterojunction
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- BODIPY
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
- Bpin
4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl
- bpy
2,2′-bipyridyl
- BrettPhos
2-(dicyclohexylphosphino)-3,6-dimethoxy-2′,4′,6′-triisopropyl-1,1′-biphenyl
- CAN
ceric ammonium nitrate
- CD
circular dichroism
- CDA
catalytic direct arylation
- CIE
Commission International de l’Eclairage
- CMP
conjugated mesoporous polymer
- cod
1,5-cyclooctadiene
- COF
covalent organic framework
- Colh
columnar hexagonal (phase)
- COT
cyclooctatetraene
- COTh
cyclooctatetrathiophene
- m-CPBA
m-chloroperoxybenzoic acid
- CPP
cycloparaphenylene
- CR2017
Part 1 of this Review1
- CT
charge transfer
- CTAB
cetyltrimethylammonium bromide
- CTAP
cetyltrimethylammonium permanganate
- CuAAC
copper-catalyzed azide–alkyne cycloaddition
- CV
cyclic voltammetry
- DABCO
1,4-diazabicyclo[2.2.2]octane
- dba
trans,trans-dibenzylideneacetone
- DBAP
dibenzo-9a-azaphenalene
- (o-)DCB
(ortho-)dichlorobenzene
- DCC
dicyclohexylcarbodiimide
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DFT
density functional theory
- DIC
dichloroisocyanuric acid
- dipp
2,6-di(isopropyl)phenyl
- DIPEA
N,N-diisopropylethylamine
- DMA, DMAc
N,N-dimethylacetamide
- DMAP
4-dimethylaminopyridine
- DME
dimethoxyethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DMT
dimercaptotriazine
- DOSY
diffusion-ordered spectroscopy
- DPP
diphenylphosphine
- dppb
1,1-bis(diphenylphosphino)butane
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- dppp
1,1-bis(diphenylphosphino)propane
- DSC
differential scanning calorimetry
- ECL
electrogenerated chemiluminescence
- ESIPT
excited-state intramolecular proton transfer
- ESR
electron spin resonance
- Fc
ferrocene
- FET
field-effect transistor
- FF
fill factor
- FT-IR
Fourier-transform infrared (spectroscopy)
- FVP
flash vacuum pyrolysis
- fwhm
full width at half-maximum
- GNR
graphene nanoribbon
- GPC
gel permeation chromatography
- HBC
hexa-peri-hexabenzocoronene
- c-HBC
hexa-cata-hexabenzocoronene
- HFIP
1,1,1,3,3,3-hexafluoro-2-propanol
- HOMO
highest occupied molecular orbital
- HOPG
highly ordered pyrolytic graphite
- HPHAC
hexapyrrolohexaazacoronene
- HPLC
high-performance liquid chromatography
- ICT
intramolecular charge transfer
- Ir(ppy)3
iridium(III) fac-tris(2-phenylpyridine)
- ITO
indium tin oxide
- KHMDS
potassium bis(trimethylsilyl)amide
- LB
Langmuir–Blodgett (films)
- LC
liquid crystal
- LiHMDS
lithium bis(trimethylsilyl)amide
- LUMO
lowest unoccupied molecular orbital
- MALDI
matrix-assisted laser desorption ionization
- MCD
magnetic circular dichroism
- MIDA
N-methyliminodiacetic acid boronate
- MO
molecular orbital
- MOM
methoxymethyl
- MW
microwave (heating)
- NaHMDS
sodium bis(trimethylsilyl)amide
- NAP
N-annulated perylene
- NBS
N-bromosuccinimide
- nc-AFM
noncontact atomic force microscopy
- NDI
naphthalene diimide
- NHC
N-heterocyclic carbene
- NICS
nucleus-independent chemical shift
- NIR
near-infrared
- NIS
N-iodosuccinimide
- NMI
naphthalene monoimide
- NMP
N-methylpyrrolidone
- NMR
nuclear magnetic resonance
- NTDA
naphthalenetetracarboxylic dianhydride
- OFET
organic field-effect transistor
- OLED
organic light-emitting diode
- OPA
one-photon absorption
- OPV
organic photovoltaic(s)
- OSC
organic solar cell
- OTFT
organic thin-film transistor
- PAH
polycyclic aromatic hydrocarbon
- Palau′Chlor
2-chloro-1,3-bis(methoxycarbonyl)guanidine
- PAMY
polycyclic aromatic azomethine ylide
- PC(61)BM
[6,6]-phenyl-C61-butyric acid methyl ester
- PC71BM
[6,6]-phenyl-C71-butyric acid methyl ester
- PCE
(photovoltaic) power conversion efficiency
- Pd-PEPPSI-IPr
(1,3-bis(2,6-diisopropylphenyl)imidazolidene)(3-chloropyridyl) palladium(II) dichloride
- PDBT-T1
poly[[5,10-bis(5-octyl-2-thienyl)dithieno[2,3-d:2′,3′-d′]benzo[1,2-b:4,5-b′]dithiophene-2,7-diyl]-2,5-thiophenediyl[5,7-bis(2-ethylhexyl)-4,8-dioxo-4H,8H-benzo[1,2-c:4,5-c′]dithiophene-1,3-diyl]-2,5-thiophenediyl]
- PDI
perylene diimide
- PDT
photodynamic therapy
- PET
photoinduced electron transfer
- PHA
polycyclic heteroaromatic (system)
- phen
1,10-phenanthroline
- PhOLED
phosphorescent organic light-emitting diode
- PIFA
phenyliodine bis(trifluoroacetate)
- PLED
polymer light-emitting diode
- PLQY
photoluminescence quantum yield
- PMMA
poly(methyl methacrylate)
- PPA
polyphosphoric acid
- Ppy
2-phenylpyridine
- PS
polystyrene
- PTB7
poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]]
- PTB7-Th
poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-)-2-carboxylate-2–6-diyl)]
- QFP
quadruply fused porphyrin
- RE
reductive elimination
- room temperature
rt
- RuPhos
2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl
- SCE
standard calomel electrode
- SF
singlet fission
- SPhos
2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl
- Spiro-OMeTAD
2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene
- SQUID
superconducting quantum interference device
- STM
scanning tunneling microscopy
- STS
scanning tunneling spectroscopy
- SubPc
subphthalocyanine
- TADF
thermally activated delayed fluorescence
- TAT
7,8,15,16-tetraazaterrylene
- TBA
tetrabutylammonium (cation)
- TBAP
tetrabutylammonium perchlorate
- TBDMS
tert-butyldimethylsilyl
- TBP
2,4,6-tri(tert-butyl)pyridine
- TBTA
tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine
- TBTQ
tribenzotriquinacene
- TCB
1,2,4-trichlorobenzene
- TCNE
tetracyanoethylene
- TCNQ
tetracyanoquinodimethane
- TEA
triethylamine
- TEM
transition electron microscopy
- Tf
trifluoromethanesulfonyl
- TFE
2,2,2-trifluoroethanol
- TFT
thin-film transistor
- THF
tetrahydrofuran
- TIBS
tri-iso-butylsilyl
- Tip
2,4,6-triisopropylphenyl
- TIPS
triisopropylsilyl
- TFA
trifluoroacetic acid
- TMEDA
tetramethylethylenediamine
- TMP
2,2,6,6-tetramethylpiperidinyl-magnesium chloride lithium chloride complex (Knochel–Hauser base)
- TMSCl
trimethylsilyl chloride
- TOF
time-of-flight
- Tol
p-Tol, para-tolyl
- TPA
two-photon absorption
- TTF
tetrathiafulvalene
- p-TSA
p-toluenesulfonic acid
- XRD
X-ray diffraction
Biographies
Arseni Borissov is originally from Novosibirsk, Russia. He obtained an MChem degree (2014) at the University of Edinburgh. He then proceeded to complete a doctoral degree (2019) at the University of Oxford, cosupervised by Prof. Paul D. Beer and Prof. Martin D. Smith and working on anion receptor synthesis. After remaining as a research associate in the same department in the group of Prof. Stephen P. Fletcher, he joined the group of Professor Marcin Stępień at the University of Wrocław in 2020. His research interests include host–guest chemistry, organic synthesis methodology, and polycyclic aromatic chromophores.
Yogesh Kumar Maurya was born in 1992 in India. He received his M.Sc. degree (2015) in chemistry from National Institute of Science Education and Research, Bhubaneswar, India. He earned his Ph.D. degree in 2018 from Kyushu University, Japan, for his research work on N-confused/N-linked metallocorroles under the guidance of Professor Hiroyuki Furuta. In 2018, he joined the group of Professor Marcin Stępień at the University of Wrocław as a postdoctoral researcher. His research interests include the synthesis and design of near-infrared absorbing materials, notably the π-extended conjugated systems based on pyrrole chemistry.
Liliia Moshniaha was born in 1995 in Vinnytsia, Ukraine. She received her M.Sc. degree (2018) in organic chemistry from the University of Wrocław. Currently, she is a doctoral candidate at the same University under the guidance of Professor Marcin Stępień. Her research interests are focused on the synthesis of “impossibly strained” and fluorescent oligopyrrolic aromatic compounds. In 2018, she received a PRELUDIUM fellowship from the National Science Center of Poland.
Wai-Shing Wong was born in 1990 in Hong Kong. He obtained his Ph.D. degree (2018) in Prof. H.-F. Chow’s group at the Chinese University of Hong Kong for his work on fenestrindane chemistry. He then continued to work as a research fellow in the same group. In 2020, he started his postdoctoral research in Prof. M. Stępień’s group at the University of Wrocław. His current research interests are polyaromatic macrocycles and oligoradicals.
Marika Żyła-Karwowska was born in 1990 in Nowa Ruda, Poland. She received her M.Sc. (2014) and Ph.D. degrees in chemistry (2018) from the University of Wrocław. Currently, she is an FNP TEAM postdoctoral researcher in the Stępień Group at the same University.
Marcin Stępień was born in 1977 in Wrocław, Poland. He received his Ph.D. (2003) from the University of Wrocław under the guidance of Prof. L. Latos-Grażyński. Following a period of postdoctoral research in the laboratory of Prof. J. L. Sessler (University of Texas), he continued to work at the University of Wrocław, where he completed his habilitation (2010) and became a titular professor (2017). His research team explores the synthetic chemistry of nanocarbons and their heterocyclic analogues, with a focus on unusual chromophores, curved aromatics, and open-shell organic systems.
Author Contributions
§ A.B., Y.K.M., L.M., W.-S.W., and M.Z.-K. contributed equally.
The authors declare no competing financial interest.
This paper was originally published ASAP on December 1, 2021. References 399 and 400, and the accompanying text were corrected, and the revised version was reposted on December 17, 2021.
References
- Stępień M.; Gońka E.; Żyła M.; Sprutta N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. 10.1021/acs.chemrev.6b00076. [DOI] [PubMed] [Google Scholar]
- Ammon F.; Sauer S. T.; Lippert R.; Lungerich D.; Reger D.; Hampel F.; Jux N. Unexpected Formation of [5]Helicenes from Hexaarylbenzenes Containing Pyrrole Moieties. Org. Chem. Front. 2017, 4, 861–870. 10.1039/C7QO00112F. [DOI] [Google Scholar]
- Reger D.; Schöll K.; Hampel F.; Maid H.; Jux N. Pyridinic Nanographenes by Novel Precursor Design. Chem. - Eur. J. 2021, 27, 1984–1989. 10.1002/chem.202004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze M.; Philipp M.; Waigel W.; Schmidt D.; Würthner F. Library of Azabenz-Annulated Core-Extended Perylene Derivatives with Diverse Substitution Patterns and Tunable Electronic and Optical Properties. J. Org. Chem. 2016, 81, 8394–8405. 10.1021/acs.joc.6b01573. [DOI] [PubMed] [Google Scholar]
- Greßies S.; Ito M.; Sakai M.; Osaki H.; Kim J. H.; Gensch T.; Daniliuc C.; Ando N.; Yamaguchi S.; Glorius F. Twofold C-H Activation Enables Synthesis of a Diazacoronene-Type Fluorophore with Near Infrared Emission Through Isosteric Replacement. Chem. - Eur. J. 2021, 27, 2753–2759. 10.1002/chem.202004080. [DOI] [PubMed] [Google Scholar]
- Delaney C.; Ó Máille G. M.; Twamley B.; Draper S. M. One-Pot, High-Yielding, Oxidative Cyclodehydrogenation Route for N-Doped Nanographene Synthesis. Org. Lett. 2016, 18, 88–91. 10.1021/acs.orglett.5b03312. [DOI] [PubMed] [Google Scholar]
- Wecławski M. K.; Jakešová M.; Charyton M.; Demitri N.; Koszarna B.; Oppelt K.; Sariciftci S.; Gryko D. T.; Głowacki E. D. Biscoumarin-Containing Acenes as Stable Organic Semiconductors for Photocatalytic Oxygen Reduction to Hydrogen Peroxide. J. Mater. Chem. A 2017, 5, 20780–20788. 10.1039/C7TA05882A. [DOI] [Google Scholar]
- Wecławski M. K.; Deperasińska I.; Banasiewicz M.; Young D. C.; Leniak A.; Gryko D. T. Building Molecular Complexity from Quinizarin: Conjoined Coumarins and Coronene Analogs. Chem. - Asian J. 2019, 14, 1763–1770. 10.1002/asia.201800757. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zhang Q.; Chen K.; Gao H.; Qi H.; Shi X.; Han Y.; Wei J.; Zhang C. Triphenothiazinyl Triazacoronenes: Donor-Acceptor Molecular Graphene Exhibiting Multiple Fluorescence and Electrogenerated Chemiluminescence Emissions. J. Mater. Chem. C 2017, 5, 4293–4301. 10.1039/C7TC00314E. [DOI] [Google Scholar]
- Liu B.; Shi D.; Yang Y.; Liu D.; Li M.; Liu E.; Wang X.; Zhang Q.; Yang M.; Li J.; et al. Triazacoronene Derivatives with Three Peri -Benzopyrano Extensions: Synthesis, Structure, and Properties: Triazacoronene Derivatives with Three Peri -Benzopyrano Extensions: Synthesis, Structure, and Properties. Eur. J. Org. Chem. 2018, 2018, 869–873. 10.1002/ejoc.201701386. [DOI] [Google Scholar]
- Sun Y.-X.; Wang X.-G.; Shen G.-D.; Yang T.; Yang Y.-H.; Li J.; Yang M.-Y.; Sun H.-M.; Wei J.-F. A 6π Azaelectrocyclization Strategy towards the 1,5,9-Triazacoronenes. Adv. Synth. Catal. 2020, 362, 1651–1656. 10.1002/adsc.201901433. [DOI] [Google Scholar]
- Lee J.; Buyukcakir O.; Kwon T.; Coskun A. Energy Band-Gap Engineering of Conjugated Microporous Polymers via Acidity-Dependent in Situ Cyclization. J. Am. Chem. Soc. 2018, 140, 10937–10940. 10.1021/jacs.8b05978. [DOI] [PubMed] [Google Scholar]
- Duan R.; Schollmeyer D.; Müllen K.; Li C. From Anthraquinone to Heterocoronene as Stable Red Chromophore. J. Mater. Chem. C 2018, 6, 1334–1337. 10.1039/C7TC05073A. [DOI] [Google Scholar]
- De J.; Bala I.; Gupta S. P.; Pandey U. K.; Pal S. K. High Hole Mobility and Efficient Ambipolar Charge Transport in Heterocoronene-Based Ordered Columnar Discotics. J. Am. Chem. Soc. 2019, 141, 18799–18805. 10.1021/jacs.9b09126. [DOI] [PubMed] [Google Scholar]
- Wijesinghe L. P.; Perera S. D.; Larkin E.; O Maille G. M.; Conway-Kenny R.; Lankage B. S.; Wang L.; Draper S. M. [2 + 2 + 2] Cyclotrimerisation as a Convenient Route to 6N-Doped Nanographenes: A Synthetic Introduction to Hexaazasuperbenzenes. RSC Adv. 2017, 7, 24163–24167. 10.1039/C7RA02648J. [DOI] [Google Scholar]
- Oki K.; Takase M.; Kobayashi N.; Uno H. Synthesis and Characterization of Peralkylated Pyrrole-Fused Azacoronene. J. Org. Chem. 2021, 86, 5102–5109. 10.1021/acs.joc.0c03042. [DOI] [PubMed] [Google Scholar]
- Sasaki Y.; Takase M.; Mori S.; Uno H. Synthesis and Properties of NitroHPHAC: The First Example of Substitution Reaction on HPHAC. Molecules 2020, 25, 2486. 10.3390/molecules25112486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno H.; Ishiwata M.; Muramatsu K.; Takase M.; Mori S.; Okujima T. Oxidation Behavior of 1,3-Dihydrothieno[3,4-a]HPHAC. Bull. Chem. Soc. Jpn. 2019, 92, 973–981. 10.1246/bcsj.20190022. [DOI] [Google Scholar]
- Sasaki Y.; Takase M.; Okujima T.; Mori S.; Uno H. Synthesis and Redox Properties of Pyrrole- and Azulene-Fused Azacoronene. Org. Lett. 2019, 21, 1900–1903. 10.1021/acs.orglett.9b00515. [DOI] [PubMed] [Google Scholar]
- Oki K.; Takase M.; Mori S.; Shiotari A.; Sugimoto Y.; Ohara K.; Okujima T.; Uno H. Synthesis, Structures, and Properties of Core-Expanded Azacoronene Analogue: A Twisted π-System with Two N-Doped Heptagons. J. Am. Chem. Soc. 2018, 140, 10430–10434. 10.1021/jacs.8b06079. [DOI] [PubMed] [Google Scholar]
- Oki K.; Takase M.; Mori S.; Uno H. Synthesis and Isolation of Antiaromatic Expanded Azacoronene via Intramolecular Vilsmeier-Type Reaction. J. Am. Chem. Soc. 2019, 141, 16255–16259. 10.1021/jacs.9b09260. [DOI] [PubMed] [Google Scholar]
- Zhylitskaya H.; Cybińska J.; Chmielewski P.; Lis T.; Stępień M. Bandgap Engineering in π-Extended Pyrroles. a Modular Approach to Electron-Deficient Chromophores with Multi-Redox Activity. J. Am. Chem. Soc. 2016, 138, 11390–11398. 10.1021/jacs.6b07826. [DOI] [PubMed] [Google Scholar]
- Żyła-Karwowska M.; Zhylitskaya H.; Cybińska J.; Lis T.; Chmielewski P. J.; Stepień M. An Electron-Deficient Azacoronene Obtained by Radial π Extension. Angew. Chem., Int. Ed. 2016, 55, 14658–14662. 10.1002/anie.201608400. [DOI] [PubMed] [Google Scholar]
- Navakouski M.; Zhylitskaya H.; Chmielewski P. J.; Lis T.; Cybińska J.; Stepień M. Stereocontrolled Synthesis of Chiral Heteroaromatic Propellers with Small Optical Bandgaps. Angew. Chem., Int. Ed. 2019, 58, 4929–4933. 10.1002/anie.201900175. [DOI] [PubMed] [Google Scholar]
- Navakouski M.; Zhylitskaya H.; Chmielewski P. J.; Żła-Karwowska M.; Stępień M. Electrophilic Aromatic Coupling of Hexapyrrolylbenzenes. A Mechanistic Analysis. J. Org. Chem. 2020, 85, 187–194. 10.1021/acs.joc.9b02556. [DOI] [PubMed] [Google Scholar]
- Sasaki Y.; Takase M.; Kobayashi N.; Mori S.; Ohara K.; Okujima T.; Uno H. Radially π-Extended Pyrrole-Fused Azacoronene: A Series of Crystal Structures of HPHAC with Various Oxidation States. J. Org. Chem. 2021, 86, 4290–4295. 10.1021/acs.joc.0c02825. [DOI] [PubMed] [Google Scholar]
- Moshniaha L.; Żyła-Karwowska M.; Chmielewski P. J.; Lis T.; Cybińska J.; Gońka E.; Oschwald J.; Drewello T.; Rivero S. M.; Casado J.; et al. Aromatic Nanosandwich Obtained by σ-Dimerization of a Nanographenoid π-Radical. J. Am. Chem. Soc. 2020, 142, 3626–3635. 10.1021/jacs.9b13942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Y.; Richter M.; He Y.; Björk J.; Riss A.; Rajesh R.; Garnica M.; Hennersdorf F.; Weigand J. J.; Narita A.; et al. Exploration of Pyrazine-Embedded Antiaromatic Polycyclic Hydrocarbons Generated by Solution and on-Surface Azomethine Ylide Homocoupling. Nat. Commun. 2017, 8, 1948. 10.1038/s41467-017-01934-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Y.; Narita A.; Zhang W.; Feng X.; Müllen K. Synthesis of Stable Nanographenes with OBO-Doped Zigzag Edges Based on Tandem Demethylation-Electrophilic Borylation. J. Am. Chem. Soc. 2016, 138, 9021–9024. 10.1021/jacs.6b04092. [DOI] [PubMed] [Google Scholar]
- Cloke R. R.; Marangoni T.; Nguyen G. D.; Joshi T.; Rizzo D. J.; Bronner C.; Cao T.; Louie S. G.; Crommie M. F.; Fischer F. R. Site-Specific Substitutional Boron Doping of Semiconducting Armchair Graphene Nanoribbons. J. Am. Chem. Soc. 2015, 137, 8872–8875. 10.1021/jacs.5b02523. [DOI] [PubMed] [Google Scholar]
- Carbonell-Sanromà E.; Garcia-Lekue A.; Corso M.; Vasseur G.; Brandimarte P.; Lobo-Checa J.; de Oteyza D. G.; Li J.; Kawai S.; Saito S.; et al. Electronic Properties of Substitutionally Boron-Doped Graphene Nanoribbons on a Au(111) Surface. J. Phys. Chem. C 2018, 122, 16092–16099. 10.1021/acs.jpcc.8b03748. [DOI] [Google Scholar]
- Min Y.; Dou C.; Tian H.; Liu J.; Wang L. A Disk-Type Polyarene Containing Four B-N Units. Chem. Commun. 2019, 55, 3638–3641. 10.1039/C9CC00769E. [DOI] [PubMed] [Google Scholar]
- Dosso J.; Tasseroul J.; Fasano F.; Marinelli D.; Biot N.; Fermi A.; Bonifazi D. Synthesis and Optoelectronic Properties of Hexa-Peri-Hexabenzoborazinocoronene. Angew. Chem., Int. Ed. 2017, 56, 4483–4487. 10.1002/anie.201700907. [DOI] [PubMed] [Google Scholar]
- Fresta E.; Dosso J.; Cabanillas-González J.; Bonifazi D.; Costa R. D. Origin of the Exclusive Ternary Electroluminescent Behavior of BN-Doped Nanographenes in Efficient Single-Component White Light-Emitting Electrochemical Cells. Adv. Funct. Mater. 2020, 30, 1906830. 10.1002/adfm.201906830. [DOI] [Google Scholar]
- Dosso J.; Battisti T.; Ward B. D.; Demitri N.; Hughes C. E.; Williams P. A.; Harris K. D. M.; Bonifazi D. Boron-Nitrogen-Doped Nanographenes: A Synthetic Tale from Borazine Precursors. Chem. - Eur. J. 2020, 26, 6608–6621. 10.1002/chem.201905794. [DOI] [PubMed] [Google Scholar]
- Matsui K.; Oda S.; Yoshiura K.; Nakajima K.; Yasuda N.; Hatakeyama T. One-Shot Multiple Borylation toward BN-Doped Nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. 10.1021/jacs.7b10578. [DOI] [PubMed] [Google Scholar]
- Guo X.; Yuan Z.; Zhu Y.; Li Z.; Huang R.; Xia Z.; Zhang W.; Li Y.; Wang J. A Nitrogen-Doped Hexapole [7]Helicene versus Its All-Carbon Analogue. Angew. Chem., Int. Ed. 2019, 58, 16966–16972. 10.1002/anie.201907972. [DOI] [PubMed] [Google Scholar]
- Reger D.; Haines P.; Heinemann F. W.; Guldi D. M.; Jux N. Oxa[7]Superhelicene: A π-Extended Helical Chromophore Based on Hexa-Peri-Hexabenzocoronenes. Angew. Chem., Int. Ed. 2018, 57, 5938–5942. 10.1002/anie.201800585. [DOI] [PubMed] [Google Scholar]
- Wade J.; Brandt J. R.; Reger D.; Zinna F.; Amsharov K. Y.; Jux N.; Andrews D. L.; Fuchter M. J. 500-Fold Amplification of Small Molecule Circularly Polarised Luminescence through Circularly Polarised FRET. Angew. Chem., Int. Ed. 2021, 60, 222–227. 10.1002/anie.202011745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R.; Pfeffermann M.; Skidin D.; Wang F.; Fu Y.; Narita A.; Tommasini M.; Moresco F.; Cuniberti G.; Berger R.; et al. Persulfurated Coronene: A New Generation of “Sulflower. J. Am. Chem. Soc. 2017, 139, 2168–2171. 10.1021/jacs.6b12630. [DOI] [PubMed] [Google Scholar]
- Yin J.; Hu Y.; Zhang D.; Li X.; Jin W. Synthesis and Characterization of Symmetrical Sulfur-Fused Polycyclic Aromatic Hydrocarbons with Controlled Shapes. Tetrahedron 2017, 73, 5794–5799. 10.1016/j.tet.2017.08.020. [DOI] [Google Scholar]
- Qiao X.; Li Q.; Schaugaard R. N.; Noffke B. W.; Liu Y.; Li D.; Liu L.; Raghavachari K.; Li L. Well-Defined Nanographene-Rhenium Complex as an Efficient Electrocatalyst and Photocatalyst for Selective CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 3934–3937. 10.1021/jacs.6b12530. [DOI] [PubMed] [Google Scholar]
- Endres A. H.; Schaffroth M.; Paulus F.; Reiss H.; Wadepohl H.; Rominger F.; Krämer R.; Bunz U. H. F. Coronene-Containing N-Heteroarenes: 13 Rings in a Row. J. Am. Chem. Soc. 2016, 138, 1792–1795. 10.1021/jacs.5b12642. [DOI] [PubMed] [Google Scholar]
- Kojima M.; Tamoto A.; Aratani N.; Yamada H. Rearrangement of an Aniline Linked Perylene Bisimide under Acidic Conditions and Visible to Near-Infrared Emission from the Intramolecular Charge-Transfer State of Its Fused Derivatives. Chem. Commun. 2017, 53, 5698–5701. 10.1039/C7CC01520H. [DOI] [PubMed] [Google Scholar]
- Yang X.; Rominger F.; Mastalerz M. Cata-Condensed Heteroannulated Coronenes via Selective Bromination of Diarenoperylenes as the Key Step. Org. Lett. 2018, 20, 7270–7273. 10.1021/acs.orglett.8b03181. [DOI] [PubMed] [Google Scholar]
- Davy N. C.; Man G.; Kerner R. A.; Fusella M. A.; Purdum G. E.; Sezen M.; Rand B. P.; Kahn A.; Loo Y.-L. Contorted Hexabenzocoronenes with Extended Heterocyclic Moieties Improve Visible-Light Absorption and Performance in Organic Solar Cells. Chem. Mater. 2016, 28, 673–681. 10.1021/acs.chemmater.5b04503. [DOI] [Google Scholar]
- Urieta-Mora J.; Krug M.; Alex W.; Perles J.; Fernández I.; Molina-Ontoria A.; Guldi D. M.; Martín N. Homo and Hetero Molecular 3D Nanographenes Employing a Cyclooctatetraene Scaffold. J. Am. Chem. Soc. 2020, 142, 4162–4172. 10.1021/jacs.9b10203. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Marszalek T.; Müllen K.; Pisula W.; Feng X. Derivatizing Tribenzothiophene-Fused Hexa-Peri-Hexabenzocoronenes with Tunable Optoelectronic Properties. Chem. - Asian J. 2016, 11, 2107–2112. 10.1002/asia.201600753. [DOI] [PubMed] [Google Scholar]
- Dusold C.; Haines P.; Platzer B.; Guldi D. M.; Hirsch A. Helically and Linearly Fused Rylenediimide-Hexabenzocoronenes. Chem. - Eur. J. 2021, 27, 6511–6521. 10.1002/chem.202005235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Wang H.; Roose J.; He Z.; Zhou Y.; Yan Y.; Cai Y.; Shi H.; Zhang Y.; Sung H. H. Y.; et al. A Luminescent Nitrogen-Containing Polycyclic Aromatic Hydrocarbon Synthesized by Photocyclodehydrogenation with Unprecedented Regioselectivity. Chem. - Eur. J. 2015, 21, 17973–17980. 10.1002/chem.201503147. [DOI] [PubMed] [Google Scholar]
- Raouafi S.; Aloui F. New Functional Benzo[Ghi]Perylene Derivatives: Synthesis and Photophysical Properties. J. Mol. Struct. 2019, 1196, 685–690. 10.1016/j.molstruc.2019.07.022. [DOI] [Google Scholar]
- Raouafi S.; Aloui F. Synthesis and Photophysical Properties of New Nitrile Grafted Benzo[Ghi]Perylene Derivatives. J. Mol. Struct. 2019, 1195, 153–160. 10.1016/j.molstruc.2019.05.100. [DOI] [Google Scholar]
- Pigulski B.; Ximenis M.; Shoyama K.; Würthner F. Synthesis of Polycyclic Aromatic Hydrocarbons by Palladium-Catalysed [3 + 3] Annulation. Org. Chem. Front. 2020, 7, 2925–2930. 10.1039/D0QO00968G. [DOI] [Google Scholar]
- Tasior M.; Deperasińska I.; Morawska K.; Banasiewicz M.; Vakuliuk O.; Kozankiewicz B.; Gryko D. T. Vertically π-Expanded Coumarin - Synthesis via the Scholl Reaction and Photophysical Properties. Phys. Chem. Chem. Phys. 2014, 16, 18268–18275. 10.1039/C4CP02003K. [DOI] [PubMed] [Google Scholar]
- Xue W.; Wang D.; Li C.; Zhai Z.; Wang T.; Liang Y.; Zhang Z. π-Expanded Coumarins: One-Pot Photo Synthesis of 5 H -Benzo[12,1]Tetrapheno[7,6,5- Cde ]Chromen-5-Ones and Photophysical Properties. J. Org. Chem. 2020, 85, 3689–3698. 10.1021/acs.joc.9b03327. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.; Santella M.; Bogh S. A.; Muñoz A. V.; Andersen H. O. B.; Hammerich O.; Bora I.; Lincke K.; Laursen B. W. Extended Triangulenium Ions: Syntheses and Characterization of Benzo-Bridged Dioxa- and Diazatriangulenium Dyes. J. Org. Chem. 2019, 84, 2556–2567. 10.1021/acs.joc.8b02978. [DOI] [PubMed] [Google Scholar]
- Lewis B. W.; Bisballe N.; Santella M.; Summers P. A.; Vannier J.-B.; Kuimova M. K.; Laursen B. W.; Vilar R. Assessing The Key Photophysical Properties of Triangulenium Dyes for DNA Binding by Alteration of the Fluorescent Core. Chem. - Eur. J. 2021, 27, 2523–2536. 10.1002/chem.202003875. [DOI] [PubMed] [Google Scholar]
- Escande A.; Crossley D. L.; Cid J.; Cade I. A.; Vitorica-Yrezabal I.; Ingleson M. J. Inter- and Intra-Molecular C-H Borylation for the Formation of PAHs Containing Triarylborane and Indole Units. Dalton Trans. 2016, 45, 17160–17167. 10.1039/C6DT03526D. [DOI] [PubMed] [Google Scholar]
- Del Grosso A.; Ayuso Carrillo J.; Ingleson M. J. Regioselective Electrophilic Borylation of Haloarenes. Chem. Commun. 2015, 51, 2878–2881. 10.1039/C4CC10153G. [DOI] [PubMed] [Google Scholar]
- Farrell J. M.; Schmidt D.; Grande V.; Würthner F. Synthesis of a Doubly Boron-Doped Perylene through NHC-Borenium Hydroboration/C-H Borylation/Dehydrogenation. Angew. Chem., Int. Ed. 2017, 56, 11846–11850. 10.1002/anie.201706346. [DOI] [PubMed] [Google Scholar]
- Farrell J. M.; Mützel C.; Bialas D.; Rudolf M.; Menekse K.; Krause A.-M.; Stolte M.; Würthner F. Tunable Low-LUMO Boron-Doped Polycyclic Aromatic Hydrocarbons by General One-Pot C-H Borylations. J. Am. Chem. Soc. 2019, 141, 9096–9104. 10.1021/jacs.9b04675. [DOI] [PubMed] [Google Scholar]
- Farrell J. M.; Grande V.; Schmidt D.; Würthner F. A Highly Warped Heptagon-Containing Sp2 Carbon Scaffold via Vinylnaphthyl Π-Extension. Angew. Chem., Int. Ed. 2019, 58, 16504–16507. 10.1002/anie.201909975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke J.; Schickedanz K.; Bamberg M.; Menduti L.; Schollmeyer D.; Bolte M.; Lerner H.-W.; Wagner M. Selective Access to Either a Doubly Boron-Doped Tetrabenzopentacene or an Oxadiborepin from the Same Precursor. Chem. Sci. 2019, 10, 9017–9027. 10.1039/C9SC03115D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z.; Okuda H.; Koyama Y.; Seto R.; Uchida S.; Sogawa H.; Kuwata S.; Takata T. Exact Helical Polymer Synthesis by a Two-Point-Covalent-Linking Protocol between C2-Chiral Spirobifluorene and C2- or Cs-Symmetric Anthraquinone Monomers. Chem. Commun. 2015, 51, 10423–10426. 10.1039/C5CC02086G. [DOI] [PubMed] [Google Scholar]
- Tran M. N.; Rarig R.-A. F.; Chenoweth D. M. Synthesis and Properties of Lysosome-Specific Photoactivatable Probes for Live-Cell Imaging. Chem. Sci. 2015, 6, 4508–4512. 10.1039/C5SC01601K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M. N.; Chenoweth D. M. Photoelectrocyclization as an Activation Mechanism for Organelle-Specific Live-Cell Imaging Probes. Angew. Chem., Int. Ed. 2015, 54, 6442–6446. 10.1002/anie.201502403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Liu Z.; Fang Q. Synthesis, Structures, and Optoelectronic Properties of Pyrene-Fused Thioxanthenes. Org. Lett. 2017, 19, 1382–1385. 10.1021/acs.orglett.7b00276. [DOI] [PubMed] [Google Scholar]
- Sato K.; Seki Y.; Suga S.; Ikeda Y.; Yamaguchi M. Reinvestigation of the Photoreaction of 1,4-Bis(2,4,6-Triphenylpyridinio)Benzene: Synthesis of a Diazonia Derivative of Hexabenzoperylene by Multiple Photocyclization. J. Photochem. Photobiol., A 2016, 331, 8–16. 10.1016/j.jphotochem.2016.06.022. [DOI] [Google Scholar]
- Park Y. S.; Dibble D. J.; Kim J.; Lopez R. C.; Vargas E.; Gorodetsky A. A. Synthesis of Nitrogen-Containing Rubicene and Tetrabenzopentacene Derivatives. Angew. Chem., Int. Ed. 2016, 55, 3352–3355. 10.1002/anie.201510320. [DOI] [PubMed] [Google Scholar]
- Tokuo K.; Sakai H.; Sakanoue T.; Takenobu T.; Araki Y.; Wada T.; Hasobe T. Control of the Electrochemical and Photophysical Properties of N-Substituted Benzo[Ghi]Perylene Derivatives. Mater. Chem. Front. 2017, 1, 2299–2308. 10.1039/C7QM00301C. [DOI] [Google Scholar]
- Evoniuk C. J.; Gomes G.; dos P.; Hill S. P.; Fujita S.; Hanson K.; Alabugin I. V. Coupling N-H Deprotonation, C-H Activation, and Oxidation: Metal-Free C(Sp3)-H Aminations with Unprotected Anilines. J. Am. Chem. Soc. 2017, 139, 16210–16221. 10.1021/jacs.7b07519. [DOI] [PubMed] [Google Scholar]
- Langhals H.; Reichherzer S.; Mayer P.; Polborn K. A Three-Step Synthesis of 1,7-Diazaperylene and Derivatives. Synthesis 2021, 53, 713–722. 10.1055/s-0040-1707293. [DOI] [Google Scholar]
- Okamoto T.; Kumagai S.; Fukuzaki E.; Ishii H.; Watanabe G.; Niitsu N.; Annaka T.; Yamagishi M.; Tani Y.; Sugiura H.; et al. Robust, High-Performance n-Type Organic Semiconductors. Sci. Adv. 2020, 6, eaaz0632 10.1126/sciadv.aaz0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai S.; Ishii H.; Watanabe G.; Annaka T.; Fukuzaki E.; Tani Y.; Sugiura H.; Watanabe T.; Kurosawa T.; Takeya J.; et al. Cooperative Aggregations of Nitrogen-Containing Perylene Diimides Driven by Rigid and Flexible Functional Groups. Chem. Mater. 2020, 32, 9115–9125. 10.1021/acs.chemmater.0c01888. [DOI] [Google Scholar]
- Sawada T.; Makita T.; Yamamura A.; Sasaki M.; Yoshimura Y.; Hayakawa T.; Okamoto T.; Watanabe S.; Kumagai S.; Takeya J. Low-Voltage Complementary Inverters Using Solution-Processed, High-Mobility Organic Single-Crystal Transistors Fabricated by Polymer-Blend Printing. Appl. Phys. Lett. 2020, 117, 033301. 10.1063/5.0006651. [DOI] [Google Scholar]
- Zagranyarski Y.; Skabeev A.; Ma Y.; Müllen K.; Li C. Facile Synthesis of Annulated Heterocyclic Benzo[Kl]Acridine Derivatives via One-Pot N-H/C-H Coupling. Org. Chem. Front. 2016, 3, 1520–1523. 10.1039/C6QO00371K. [DOI] [Google Scholar]
- Dorđevic L.; Valentini C.; Demitri N.; Meziere C.; Allain M.; Salle M.; Folli A.; Murphy D.; Manas-Valero S.; Coronado E.; Bonifazi D.; et al. O-Doped Nanographenes: A Pyrano/Pyrylium Route Towards Semiconducting Cationic Mixed-Valence Complexes. Angew. Chem., Int. Ed. 2020, 59, 4106–4114. 10.1002/anie.201914025. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Tanioka M.; Muranaka A.; Miyamoto K.; Aoyama T.; Ouyang X.; Kamino S.; Sawada D.; Uchiyama M. Stable Thiele’s Hydrocarbon Derivatives Exhibiting Near-Infrared Absorption/Emission and Two-Step Electrochromism. J. Am. Chem. Soc. 2018, 140, 17857–17861. 10.1021/jacs.8b11092. [DOI] [PubMed] [Google Scholar]
- Harada M.; Tanioka M.; Muranaka A.; Aoyama T.; Kamino S.; Uchiyama M. A Remarkably Air-Stable Quinodimethane Radical Cation. Chem. Commun. 2020, 56, 9565–9568. 10.1039/D0CC04025H. [DOI] [PubMed] [Google Scholar]
- Hirono A.; Sakai H.; Hasobe T. Synthesis and Electrochemical and Photophysical Properties of Azaterrylene Derivatives. Chem. - Asian J. 2019, 14, 1754–1762. 10.1002/asia.201801410. [DOI] [PubMed] [Google Scholar]
- Hirono A.; Sakai H.; Kochi S.; Sato T.; Hasobe T. Electrochemical Properties and Excited-State Dynamics of Azaperylene Derivatives. J. Phys. Chem. B 2020, 124, 9921–9930. 10.1021/acs.jpcb.0c07532. [DOI] [PubMed] [Google Scholar]
- Stassen D.; Demitri N.; Bonifazi D. Extended O-Doped Polycyclic Aromatic Hydrocarbons. Angew. Chem., Int. Ed. 2016, 55, 5947–5951. 10.1002/anie.201509517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T.; Nakatsuka S.; Hirai H.; Yasuda N.; Kumar J.; Kawai T.; Hatakeyama T. Two-Step Synthesis of Boron-Fused Double Helicenes. J. Am. Chem. Soc. 2016, 138, 5210–5213. 10.1021/jacs.6b01674. [DOI] [PubMed] [Google Scholar]
- Wang X.-Y.; Wang X.-C.; Narita A.; Wagner M.; Cao X.-Y.; Feng X.; Müllen K. Synthesis, Structure, and Chiroptical Properties of a Double [7]Heterohelicene. J. Am. Chem. Soc. 2016, 138, 12783–12786. 10.1021/jacs.6b08664. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Wang X.-Y.; Wei Z.; Müllen K.; Petrukhina M. A. Charging OBO-Fused Double [5]Helicene with Electrons. Angew. Chem., Int. Ed. 2019, 58, 14969–14973. 10.1002/anie.201908658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda N.; Oda S.; Matsumoto R.; Yoshioka M.; Fukushima D.; Yoshiura K.; Yasuda N.; Hatakeyama T. Solution-Processable Pure Green Thermally Activated Delayed Fluorescence Emitter Based on the Multiple Resonance Effect. Adv. Mater. 2020, 32, 2004072. 10.1002/adma.202004072. [DOI] [PubMed] [Google Scholar]
- Scholz A. S.; Massoth J. G.; Bursch M.; Mewes J.-M.; Hetzke T.; Wolf B.; Bolte M.; Lerner H.-W.; Grimme S.; Wagner M. BNB-Doped Phenalenyls: Modular Synthesis, Optoelectronic Properties, and One-Electron Reduction. J. Am. Chem. Soc. 2020, 142, 11072–11083. 10.1021/jacs.0c03118. [DOI] [PubMed] [Google Scholar]
- Kaehler T.; Bolte M.; Lerner H.-W.; Wagner M. Introducing Perylene as a New Member to the Azaborine Family. Angew. Chem., Int. Ed. 2019, 58, 11379–11384. 10.1002/anie.201905823. [DOI] [PubMed] [Google Scholar]
- Sun K.; Sugawara K.; Lyalin A.; Ishigaki Y.; Uosaki K.; Taketsugu T.; Suzuki T.; Kawai S. Heterocyclic Ring-Opening of Nanographene on Au(111). Angew. Chem., Int. Ed. 2021, 60, 9427–9432. 10.1002/anie.202017137. [DOI] [PubMed] [Google Scholar]
- Sakamaki D.; Kumano D.; Yashima E.; Seki S. A Double Hetero[4]Helicene Composed of Two Phenothiazines: Synthesis, Structural Properties, and Cationic States. Chem. Commun. 2015, 51, 17237–17240. 10.1039/C5CC07680C. [DOI] [PubMed] [Google Scholar]
- Shindo Y.; Nomura S.; Saikawa Y.; Nakata M.; Tanaka K.; Hanaya K.; Sugai T.; Higashibayashi S. Synthesis and Properties of Hydrazine-Embedded Biphenothiazines and Application of Hydrazine-Embedded Heterocyclic Compounds to Fluorescence Cell Imaging. Asian J. Org. Chem. 2018, 7, 1797–1801. 10.1002/ajoc.201800364. [DOI] [Google Scholar]
- Zhu G.; Song Y.; Zhang Q.; Ding W.; Chen X.; Wang Y.; Zhang G. Modulating the Properties of Buckybowls Containing Multiple Heteroatoms. Org. Chem. Front. 2021, 8, 727–735. 10.1039/D0QO01452D. [DOI] [Google Scholar]
- Gupta R. K.; Pathak S. K.; Pradhan B.; Rao D. S. S.; Prasad S. K.; Achalkumar A. S. Self-Assembly of Luminescent N-Annulated Perylene Tetraesters into Fluid Columnar Phases. Soft Matter 2015, 11, 3629–3636. 10.1039/C5SM00463B. [DOI] [PubMed] [Google Scholar]
- Gupta R. K.; Pradhan B.; Pathak S. K.; Gupta M.; Pal S. K.; Ammathnadu Sudhakar A. Perylo[1,12- b, c, d ] Thiophene Tetraesters: A New Class of Luminescent Columnar Liquid Crystals. Langmuir 2015, 31, 8092–8100. 10.1021/acs.langmuir.5b01187. [DOI] [PubMed] [Google Scholar]
- Gupta R. K.; Pathak S. K.; Pradhan B.; Gupta M.; Pal S. K.; Sudhakar A. A. Bay-Annulated Perylene Tetraesters: A New Class of Discotic Liquid Crystals. ChemPhysChem 2016, 17, 859–872. 10.1002/cphc.201501028. [DOI] [PubMed] [Google Scholar]
- Gupta R. K.; Das D.; Gupta M.; Pal S. K.; Iyer P. K.; Achalkumar A. S. Electroluminescent Room Temperature Columnar Liquid Crystals Based on Bay-Annulated Perylene Tetraesters. J. Mater. Chem. C 2017, 5, 1767–1781. 10.1039/C6TC04166C. [DOI] [Google Scholar]
- Gupta R. K.; Shankar Rao D. S.; Prasad S. K.; Achalkumar A. S. Columnar Self-Assembly of Electron-Deficient Dendronized Bay-Annulated Perylene Bisimides. Chem. - Eur. J. 2018, 24, 3566–3575. 10.1002/chem.201705290. [DOI] [PubMed] [Google Scholar]
- Gupta R. K.; Achalkumar A. S. Microwave-Assisted Method for the Synthesis of Perylene Ester Imides as a Gateway Toward Unsymmetrical Perylene Bisimides. J. Org. Chem. 2018, 83, 6290–6300. 10.1021/acs.joc.8b00161. [DOI] [PubMed] [Google Scholar]
- Chintala S. M.; Petroff II J. T.; Barnes A.; McCulla R. D. Photodeoxygenation of Phenanthro[4,5-Bcd]Thiophene S-Oxide, Triphenyleno[1,12-Bcd]Thiophene S-Oxide and Perylo[1,12-Bcd]Thiophene S-Oxide. J. Sulfur Chem. 2019, 40, 503–515. 10.1080/17415993.2019.1615065. [DOI] [Google Scholar]
- Xing L.; Jiang W.; Huang Z.; Liu J.; Song H.; Zhao W.; Dai J.; Zhu H.; Wang Z.; Weiss P. S.; et al. Steering Two-Dimensional Porous Networks with σ-Hole Interactions of Br···S and Br···Br. Chem. Mater. 2019, 31, 3041–3048. 10.1021/acs.chemmater.9b01126. [DOI] [Google Scholar]
- Madhu S.; Evans H. A.; Doan-Nguyen V. V. T.; Labram J. G.; Wu G.; Chabinyc M. L.; Seshadri R.; Wudl F. Infinite Polyiodide Chains in the Pyrroloperylene-Iodine Complex: Insights into the Starch-Iodine and Perylene-Iodine Complexes. Angew. Chem., Int. Ed. 2016, 55, 8032–8035. 10.1002/anie.201601585. [DOI] [PubMed] [Google Scholar]
- Santoni M.-P.; Santoro A.; Salerno T. M. G.; Puntoriero F.; Nastasi F.; Di Pietro M. L.; Galletta M.; Campagna S. Photoinduced Charge Separation in a Donor-Spacer-Acceptor Dyad with N-Annulated Perylene Donor and Methylviologen Acceptor. ChemPhysChem 2015, 16, 3147–3150. 10.1002/cphc.201500615. [DOI] [PubMed] [Google Scholar]
- Rocard L.; Hatych D.; Chartier T.; Cauchy T.; Hudhomme P. Original Suzuki-Miyaura Coupling Using Nitro Derivatives for the Synthesis of Perylenediimide-Based Multimers. Eur. J. Org. Chem. 2019, 2019, 7635–7643. 10.1002/ejoc.201901319. [DOI] [Google Scholar]
- Ma Y.; Shi Z.; Zhang A.; Li J.; Wei X.; Jiang T.; Li Y.; Wang X. Self-Assembly, Optical and Electrical Properties of Five Membered O- or S-Heterocyclic Annulated Perylene Diimides. Dyes Pigm. 2016, 135, 41–48. 10.1016/j.dyepig.2016.06.027. [DOI] [Google Scholar]
- Li W.; Wang Q.; Ma Y.; Jiang T.; Zhu Y.; Shao Y.; Sun C.; Wu J. Photophysical Property, Electronic Structure and Solid-State Packing of O-Heterocyclic Annulated Perylene Diimide. Pigm. Resin Technol. 2019, 48, 256–262. 10.1108/PRT-04-2018-0034. [DOI] [Google Scholar]
- Zhang F.; Huang X.; Wei X.; Ren H.; Jiang T.; Li X.; Wu J.; Ma Y. Synthesis and Properties of Bay Unilaterally Extended and Mono-Substituted Perylene Diimides. J. Chem. Res. 2020, 44, 60–66. 10.1177/1747519819886502. [DOI] [Google Scholar]
- Deng Q.; Zhou E.; Huang Y.; Qing W.; Zhai H.; Liu Z.; Wei Z. Chalcogen-Substitution Modulated Supramolecular Chirality and Gas Sensing Properties in Perylenediimides. Chem. Commun. 2019, 55, 4379–4382. 10.1039/C9CC01443H. [DOI] [PubMed] [Google Scholar]
- Chen L.; Xia P.; Du T.; Deng Y.; Xiao Y. Catalyst-Free One-Pot Synthesis of Unsymmetrical Five- and Six-Membered Sulfur-Annulated Heterocyclic Perylene Diimides for Electron-Transporting Property. Org. Lett. 2019, 21, 5529–5532. 10.1021/acs.orglett.9b01847. [DOI] [PubMed] [Google Scholar]
- Li G.; Li D.; Liu X.; Xu H.; Zhang J.; Wang S.; Liu Z.; Tang B. Novel Dithiano-Thieno Fused Perylene Diimides: Synthesis, Characterization and Application in Organic Thin-Film Transistors (OTFTs). Chem. Commun. 2019, 55, 9661–9664. 10.1039/C9CC04133H. [DOI] [PubMed] [Google Scholar]
- Wang R.; Li G.; Zhou Y.; Hao P.; Shang Q.; Wang S.; Zhang Y.; Li D.; Yang S.; Zhang Q.; et al. Facile Syntheses, Characterization, and Physical Properties of Sulfur-Decorated Pyran-Annulated Perylene Diimides. Asian J. Org. Chem. 2018, 7, 702–706. 10.1002/ajoc.201800011. [DOI] [Google Scholar]
- Li X.; Wang H.; Nakayama H.; Wei Z.; Schneider J. A.; Clark K.; Lai W.-Y.; Huang W.; Labram J. G.; de Alaniz J. R.; et al. Multi-Sulfur-Annulated Fused Perylene Diimides for Organic Solar Cells with Low Open-Circuit Voltage Loss. ACS Appl. Energy Mater. 2019, 2, 3805–3814. 10.1021/acsaem.9b00492. [DOI] [Google Scholar]
- Li X.; Wang H.; Schneider J. A.; Wei Z.; Lai W.-Y.; Huang W.; Wudl F.; Zheng Y. Catalyst-Free One-Step Synthesis of Ortho-Tetraaryl Perylene Diimides for Efficient OPV Non-Fullerene Acceptors. J. Mater. Chem. C 2017, 5, 2781–2785. 10.1039/C7TC00263G. [DOI] [Google Scholar]
- Yao Z.; Zhang M.; Li R.; Yang L.; Qiao Y.; Wang P. A Metal-Free N-Annulated Thienocyclopentaperylene Dye: Power Conversion Efficiency of 12% for Dye-Sensitized Solar Cells. Angew. Chem., Int. Ed. 2015, 54, 5994–5998. 10.1002/anie.201501195. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wu H.; Jin L.; Zhang J.; Yuan Y.; Wang P. A Perylene-Based Polycyclic Aromatic Hydrocarbon Electron Donor for a Highly Efficient Solar Cell Dye. ChemSusChem 2017, 10, 2962–2967. 10.1002/cssc.201700916. [DOI] [PubMed] [Google Scholar]
- Wang J.; Xie X.; Weng G.; Yuan Y.; Zhang J.; Wang P. A Low-Energy-Gap Thienochrysenocarbazole Dye for Highly Efficient Mesoscopic Titania Solar Cells: Understanding the Excited State and Charge Carrier Dynamics. ChemSusChem 2018, 11, 1460–1466. 10.1002/cssc.201800186. [DOI] [PubMed] [Google Scholar]
- Xie X.; Sun D.; Wei Y.; Yuan Y.; Zhang J.; Ren Y.; Wang P. Thienochrysenocarbazole Based Organic Dyes for Transparent Solar Cells with over 10% Efficiency. J. Mater. Chem. A 2019, 7, 11338–11346. 10.1039/C9TA03115D. [DOI] [Google Scholar]
- Yao Z.; Liao X.; Gao K.; Lin F.; Xu X.; Shi X.; Zuo L.; Liu F.; Chen Y.; Jen A. K.-Y. Dithienopicenocarbazole-Based Acceptors for Efficient Organic Solar Cells with Optoelectronic Response Over 1000 Nm and an Extremely Low Energy Loss. J. Am. Chem. Soc. 2018, 140, 2054–2057. 10.1021/jacs.7b13239. [DOI] [PubMed] [Google Scholar]
- Gupta R. K.; Ulla H.; Satyanarayan M. N.; Sudhakar A. A. A Perylene-Triazine-Based Star-Shaped Green Light Emitter for Organic Light Emitting Diodes: A Perylene-Triazine-Based Star-Shaped Green Light Emitter for Organic Light Emitting Diodes. Eur. J. Org. Chem. 2018, 2018, 1608–1613. 10.1002/ejoc.201800161. [DOI] [Google Scholar]
- Hendsbee A. D.; Sun J.-P.; Law W. K.; Yan H.; Hill I. G.; Spasyuk D. M.; Welch G. C. Synthesis, Self-Assembly, and Solar Cell Performance of N-Annulated Perylene Diimide Non-Fullerene Acceptors. Chem. Mater. 2016, 28, 7098–7109. 10.1021/acs.chemmater.6b03292. [DOI] [Google Scholar]
- Vespa M.; Cann J. R.; Dayneko S. V.; Melville O. A.; Hendsbee A. D.; Zou Y.; Lessard B. H.; Welch G. C. Synthesis of a Perylene Diimide Dimer with Pyrrolic N-H Bonds and N-Functionalized Derivatives for Organic Field-Effect Transistors and Organic Solar Cells. Eur. J. Org. Chem. 2018, 2018, 4592–4599. 10.1002/ejoc.201801055. [DOI] [Google Scholar]
- Cann J. R.; Cabanetos C.; Welch G. C. Synthesis of Molecular Dyads and Triads Based Upon N-Annulated Perylene Diimide Monomers and Dimers. Eur. J. Org. Chem. 2018, 2018, 6933–6943. 10.1002/ejoc.201801383. [DOI] [Google Scholar]
- Nazari M.; Cieplechowicz E.; Welsh T. A.; Welch G. C. A Direct Comparison of Monomeric vs. Dimeric and Non-Annulated vs. N-Annulated Perylene Diimide Electron Acceptors for Organic Photovoltaics. New J. Chem. 2019, 43, 5187–5195. 10.1039/C8NJ06491A. [DOI] [Google Scholar]
- Cann J.; Farahat M. E.; Welch G. C. Hybrid Tetrameric Perylene Diimide Assemblies. ChemSusChem 2021, 14, 3511. 10.1002/cssc.202002784. [DOI] [PubMed] [Google Scholar]
- You F.; Zhou X.; Huang H.; Liu Y.; Liu S.; Shao J.; Zhao B.; Qin T.; Huang W. N-Annulated Perylene Diimide Derivatives as Non-Fullerene Acceptors for Solution-Processed Solar Cells with an Open-Circuit Voltage of up to 1.14 V. New J. Chem. 2018, 42, 15079–15087. 10.1039/C8NJ02566E. [DOI] [Google Scholar]
- Sun D.; Meng D.; Cai Y.; Fan B.; Li Y.; Jiang W.; Huo L.; Sun Y.; Wang Z. Non-Fullerene-Acceptor-Based Bulk-Heterojunction Organic Solar Cells with Efficiency over 7%. J. Am. Chem. Soc. 2015, 137, 11156–11162. 10.1021/jacs.5b06414. [DOI] [PubMed] [Google Scholar]
- Meng D.; Sun D.; Zhong C.; Liu T.; Fan B.; Huo L.; Li Y.; Jiang W.; Choi H.; Kim T.; et al. High-Performance Solution-Processed Non-Fullerene Organic Solar Cells Based on Selenophene-Containing Perylene Bisimide Acceptor. J. Am. Chem. Soc. 2016, 138, 375–380. 10.1021/jacs.5b11149. [DOI] [PubMed] [Google Scholar]
- Cann J.; Dayneko S.; Sun J.-P.; Hendsbee A. D.; Hill I. G.; Welch G. C. N-Annulated Perylene Diimide Dimers: Acetylene Linkers as a Strategy for Controlling Structural Conformation and the Impact on Physical, Electronic, Optical and Photovoltaic Properties. J. Mater. Chem. C 2017, 5, 2074–2083. 10.1039/C6TC05107C. [DOI] [Google Scholar]
- Cann J. R.; Cabanetos C.; Welch G. C. Spectroscopic Engineering toward Near-Infrared Absorption of Materials Containing Perylene Diimide. ChemPlusChem 2017, 82, 1359–1364. 10.1002/cplu.201700502. [DOI] [PubMed] [Google Scholar]
- Payne A.-J.; Li S.; Dayneko S. V.; Risko C.; Welch G. C. An Unsymmetrical Non-Fullerene Acceptor: Synthesis via Direct Heteroarylation, Self-Assembly, and Utility as a Low Energy Absorber in Organic Photovoltaic Cells. Chem. Commun. 2017, 53, 10168–10171. 10.1039/C7CC05836E. [DOI] [PubMed] [Google Scholar]
- McAfee S. M.; Dayneko S. V.; Hendsbee A. D.; Josse P.; Blanchard P.; Cabanetos C.; Welch G. C. Applying Direct Heteroarylation Synthesis to Evaluate Organic Dyes as the Core Component in PDI-Based Molecular Materials for Fullerene-Free Organic Solar Cells. J. Mater. Chem. A 2017, 5, 11623–11633. 10.1039/C7TA00318H. [DOI] [Google Scholar]
- McAfee S. M.; Dayneko S. V.; Josse P.; Blanchard P.; Cabanetos C.; Welch G. C. Simply Complex: The Efficient Synthesis of an Intricate Molecular Acceptor for High-Performance Air-Processed and Air-Tested Fullerene-Free Organic Solar Cells. Chem. Mater. 2017, 29, 1309–1314. 10.1021/acs.chemmater.6b04862. [DOI] [Google Scholar]
- Hendsbee A. D.; Dayneko S. V.; Pells J. A.; Cann J. R.; Welch G. C. N-Annulated Perylene Diimide Dimers: The Effect of Thiophene Bridges on Physical, Electronic, Optical, and Photovoltaic Properties. Sustain. Energy Fuels 2017, 1, 1137–1147. 10.1039/C7SE00056A. [DOI] [Google Scholar]
- Payne A.-J.; Rice N. A.; McAfee S. M.; Li S.; Josse P.; Cabanetos C.; Risko C.; Lessard B. H.; Welch G. C. Donor or Acceptor? How Selection of the Rylene Imide End Cap Impacts the Polarity of π-Conjugated Molecules for Organic Electronics. ACS Appl. Energy Mater. 2018, 1, 4906–4916. 10.1021/acsaem.8b00929. [DOI] [Google Scholar]
- Welsh T. A.; Laventure A.; Baumgartner T.; Welch G. C. Dithienophosphole-Based Molecular Electron Acceptors Constructed Using Direct (Hetero)Arylation Cross-Coupling Methods. J. Mater. Chem. C 2018, 6, 2148–2154. 10.1039/C7TC05631A. [DOI] [Google Scholar]
- Breukelaar W. B.; McAfee S. M.; Welch G. C. Exploiting Direct Heteroarylation Polymerization Homocoupling Defects for the Synthesis of a Molecular Dimer. New J. Chem. 2018, 42, 1617–1621. 10.1039/C7NJ04285J. [DOI] [Google Scholar]
- Welsh T. A.; Laventure A.; Alahmadi A. F.; Zhang G.; Baumgartner T.; Zou Y.; Jäkle F.; Welch G. C. Borane Incorporation in a Non-Fullerene Acceptor To Tune Steric and Electronic Properties and Improve Organic Solar Cell Performance. ACS Appl. Energy Mater. 2019, 2, 1229–1240. 10.1021/acsaem.8b01793. [DOI] [Google Scholar]
- Laventure A.; Stanzel S.; Payne A.-J.; Lessard B. H.; Welch G. C. A Ring Fused N-Annulated PDI Non-Fullerene Acceptor for High Open Circuit Voltage Solar Cells Processed from Non-Halogenated Solvents. Synth. Met. 2019, 250, 55–62. 10.1016/j.synthmet.2019.02.010. [DOI] [Google Scholar]
- Welsh T. A.; Payne A.-J.; Welch G. C. Synthesis of Aromatic Imide Tetramers Relevant to Organic Electronics by Direct (Hetero)Arylation. New J. Chem. 2019, 43, 9333–9337. 10.1039/C9NJ01553A. [DOI] [Google Scholar]
- Payne A.-J.; Song J.; Sun Y.; Welch G. C. A Tetrameric Perylene Diimide Non-Fullerene Acceptor via Unprecedented Direct (Hetero)Arylation Cross-Coupling Reactions. Chem. Commun. 2018, 54, 11443–11446. 10.1039/C8CC06446F. [DOI] [PubMed] [Google Scholar]
- Koenig J. D. B.; Laventure A.; Welch G. C. Harnessing Direct (Hetero)Arylation in Pursuit of a Saddle-Shaped Perylene Diimide Tetramer. ACS Appl. Energy Mater. 2019, 2, 8939–8945. 10.1021/acsaem.9b01978. [DOI] [Google Scholar]
- Fan W.; Liang N.; Meng D.; Feng J.; Li Y.; Hou J.; Wang Z. A High Performance Three-Dimensional Thiophene-Annulated Perylene Dye as an Acceptor for Organic Solar Cells. Chem. Commun. 2016, 52, 11500–11503. 10.1039/C6CC05810H. [DOI] [PubMed] [Google Scholar]
- Luo Z.; Liu T.; Cheng W.; Wu K.; Xie D.; Huo L.; Sun Y.; Yang C. A Three-Dimensional Thiophene-Annulated Perylene Bisimide as a Fullerene-Free Acceptor for a High Performance Polymer Solar Cell with the Highest PCE of 8.28% and a VOC over 1.0 V. J. Mater. Chem. C 2018, 6, 1136–1142. 10.1039/C7TC05261H. [DOI] [Google Scholar]
- Li G.; Wang S.; Liu T.; Hao P.; Liu Z.; Li F.; Yang L.-M.; Zhang Y.; Li D.; Yang S.; et al. Non-Fullerene Acceptor Engineering with Three-Dimensional Thiophene/Selenophene-Annulated Perylene Diimides for High Performance Polymer Solar Cells. J. Mater. Chem. C 2018, 6, 12601–12607. 10.1039/C8TC04926B. [DOI] [Google Scholar]
- Qu J.; Mu Z.; Lai H.; Xie M.; Liu L.; Lu W.; Chen W.; He F. Effect of the Molecular Configuration of Perylene Diimide Acceptors on Charge Transfer and Device Performance. ACS Appl. Energy Mater. 2018, 1, 833–840. 10.1021/acsaem.7b00277. [DOI] [Google Scholar]
- Liang Y.; Deng P.; Wang Z.; Guo Z.; Lei Y. Novel Perylene Diimide Acceptor for Nonfullerene Organic Solar Cells. Funct. Mater. Lett. 2019, 12, 1950022. 10.1142/S179360471950022X. [DOI] [Google Scholar]
- Wu F.; Luo Z.; Zhu L.; Chen C.; Lu H.; Chen Z.; Tang J.; Yang C. Sulfur-Annulated Perylenediimide as an Interfacial Material Enabling Inverted Perovskite Solar Cells with over 20% Efficiency and High Fill Factors Exceeding 83%. J. Mater. Chem. A 2019, 7, 21176–21181. 10.1039/C9TA07349C. [DOI] [Google Scholar]
- Li M.; Wang H.; Liu Y.; Zhou Y.; Lu H.; Song J.; Bo Z. Perylene Diimide Acceptor with Two Planar Arms and a Twisted Core for High Efficiency Polymer Solar Cells. Dyes Pigm. 2020, 175, 108186. 10.1016/j.dyepig.2020.108186. [DOI] [Google Scholar]
- Meng D.; Fu H.; Xiao C.; Meng X.; Winands T.; Ma W.; Wei W.; Fan B.; Huo L.; Doltsinis N. L.; et al. Three-Bladed Rylene Propellers with Three-Dimensional Network Assembly for Organic Electronics. J. Am. Chem. Soc. 2016, 138, 10184–10190. 10.1021/jacs.6b04368. [DOI] [PubMed] [Google Scholar]
- Hu M.; Zhao X.; Zhu G.; Zhang Y.; Yuan Z.; Wang L.; Huang X.; Hu Y.; Chen Y. Seleno Twisted Benzodiperylenediimides: Facile Synthesis and Excellent Electron Acceptors for Additive-Free Organic Solar Cells. Chem. Commun. 2019, 55, 703–706. 10.1039/C8CC09367A. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Fu H.; Meng D.; Jiang W.; Sun Y.; Wang Z. Isomeric N-Annulated Perylene Diimide Dimers for Organic Solar Cells. Chem. - Asian J. 2018, 13, 918–923. 10.1002/asia.201800058. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Ma Z.; Niu X.; Zhang W.; Tao M.; Guo Q.; Wang Z.; Xia A. Bridge-Mediated Charge Separation in Isomeric N-Annulated Perylene Diimide Dimers. J. Am. Chem. Soc. 2019, 141, 12789–12796. 10.1021/jacs.9b05723. [DOI] [PubMed] [Google Scholar]
- Buendıa J.; Greciano E. E.; Sánchez L. Influence of Axial and Point Chirality in the Chiral Self-Assembly of Twin N -Annulated Perylenecarboxamides. J. Org. Chem. 2015, 80, 12444–12452. 10.1021/acs.joc.5b02309. [DOI] [PubMed] [Google Scholar]
- Greciano E. E.; Sánchez L. Seeded Supramolecular Polymerization in a Three-Domain Self-Assembly of an N-Annulated Perylenetetracarboxamide. Chem. - Eur. J. 2016, 22, 13724–13730. 10.1002/chem.201602344. [DOI] [PubMed] [Google Scholar]
- Buendia J.; Calbo J.; Orti E.; Sanchez L. Flexible Chirality in Self-Assembled N-Annulated Perylenedicarboxamides. Small 2017, 13, 1603880. 10.1002/smll.201603880. [DOI] [PubMed] [Google Scholar]
- Greciano E. E.; Calbo J.; Ortí E.; Sánchez L. N-Annulated Perylene Bisimides to Bias the Differentiation of Metastable Supramolecular Assemblies into J- and H-Aggregates. Angew. Chem., Int. Ed. 2020, 59, 17517–17524. 10.1002/anie.202005837. [DOI] [PubMed] [Google Scholar]
- Qin R.; Guo D.; Ma H.; Yang J.; Jiang Y.; Liu H.; Liu Z.; Song J.; Qin C. Effect of Molecular Structures of Donor Monomers of Polymers on Photovoltaic Properties. ACS Omega 2019, 4, 19177–19182. 10.1021/acsomega.9b02476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Kim Y. J.; Ha Y. H.; Zhao Q.; Park C. E.; Kim Y.-H. Effects of Alkyl Chain Length on the Optoelectronic Properties and Performance of Pyrrolo-Perylene Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 8859–8867. 10.1021/acsami.5b01444. [DOI] [PubMed] [Google Scholar]
- Sung M. J.; Yoon S.; Kwon S.-K.; Kim Y.-H.; Chung D. S. Synthesis of Phenanthro[1,10,9,8- Cdefg ]Carbazole-Based Conjugated Polymers for Green-Selective Organic Photodiodes. ACS Appl. Mater. Interfaces 2016, 8, 31172–31178. 10.1021/acsami.6b12410. [DOI] [PubMed] [Google Scholar]
- Đorđević L.; Milano D.; Demitri N.; Bonifazi D. O-Annulation to Polycyclic Aromatic Hydrocarbons: A Tale of Optoelectronic Properties from Five- to Seven-Membered Rings. Org. Lett. 2020, 22, 4283–4288. 10.1021/acs.orglett.0c01331. [DOI] [PubMed] [Google Scholar]
- Fujimoto K.; Izawa S.; Takahashi A.; Inuzuka T.; Sanada K.; Sakamoto M.; Nakayama Y.; Hiramoto M.; Takahashi M. Curved Perylene Diimides Fused with Seven-Membered Rings. Chem. - Asian J. 2021, 16, 690–695. 10.1002/asia.202100066. [DOI] [PubMed] [Google Scholar]
- Zink-Lorre N.; Doncel-Giménez A.; Font-Sanchis E.; Calbo J.; Sastre-Santos Á.; Ortí E.; Fernández-Lázaro F. Diels-Alder Reaction on Perylenediimides: Synthesis and Theoretical Study of Core-Expanded Diimides. Org. Chem. Front. 2019, 6, 2860–2871. 10.1039/C9QO00682F. [DOI] [Google Scholar]
- Goujon A.; Rocard L.; Cauchy T.; Hudhomme P. An Imine Photocyclization as an Alternative to the Pictet-Spengler Reaction for the Synthesis of AzaBenzannulated Perylenediimide Dyes. J. Org. Chem. 2020, 85, 7218–7224. 10.1021/acs.joc.0c00600. [DOI] [PubMed] [Google Scholar]
- Hao L.; Jiang W.; Wang Z. Integration of Nitrogen into Coronene Bisimides. Tetrahedron 2012, 68, 9234–9239. 10.1016/j.tet.2012.08.084. [DOI] [Google Scholar]
- Schulze M.; Steffen A.; Würthner F. Near-IR Phosphorescent Ruthenium(II) and Iridium(III) Perylene Bisimide Metal Complexes. Angew. Chem., Int. Ed. 2015, 54, 1570–1573. 10.1002/anie.201410437. [DOI] [PubMed] [Google Scholar]
- Shi J.; Fan J.; Qu Z.; Wang S.; Wang Y. Solution Concentration-Dependent Tunable Emission in Cyclometalated Iridium Complex Bearing Perylene Diimide (PDI) Ligand: From Visible to near-Infrared Emission. Dyes Pigm. 2018, 154, 263–268. 10.1016/j.dyepig.2018.03.011. [DOI] [Google Scholar]
- Wang C.-S.; Sun Q.; García F.; Wang C.; Yoshikai N. Robust Cobalt Catalyst for Nitrile/Alkyne [2 + 2+2] Cycloaddition: Synthesis of Polyarylpyridines and Their Mechanochemical Cyclodehydrogenation to Nitrogen-Containing Polyaromatics**. Angew. Chem., Int. Ed. 2021, 60, 9627–9634. 10.1002/anie.202017220. [DOI] [PubMed] [Google Scholar]
- Wei H.; Qiu T.; Huang X.; Zhou J.; Guo J.; Jiang C.; Luo S.; Zeng Z.; Wu J. A Facile Approach toward 1,2-Diazabenzo[Ghi]Perylene Derivatives: Structures and Electronic Properties. Chem. Commun. 2017, 53, 6740–6743. 10.1039/C7CC03362A. [DOI] [PubMed] [Google Scholar]
- Hahn U.; Maisonhaute E.; Nierengarten J.-F. Twisted N-Doped Nano-Graphenes: Synthesis, Characterization, and Resolution. Angew. Chem., Int. Ed. 2018, 57, 10635–10639. 10.1002/anie.201805852. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Li J.; Li M.; Chen C.; Xu S.; Tang X.; Chen L.; Chen R.; Huang W. Synthesis and Application of Perylene-Embedded Benzoazoles for Small-Molecule Organic Solar Cells. Org. Lett. 2018, 20, 6376–6379. 10.1021/acs.orglett.8b02650. [DOI] [PubMed] [Google Scholar]
- Benniston A. C.; He X.; Lemmetyinen H.; Tkachenko N. V. Charge Transfer Properties of a Donor-Acceptor Dyad Based on an Expanded Acridinium Cation. RSC Adv. 2013, 3, 4995. 10.1039/c3ra22813d. [DOI] [Google Scholar]
- He X.; Benniston A. C.; Lemmetyinen H.; Tkachenko N. V. Charge Shift/Recombination and Triplet Formation in a Molecular Dyad Based on a Borondipyrromethene (Bodipy) and an Expanded Acridinium Cation. ChemPhotoChem. 2018, 2, 277–282. 10.1002/cptc.201700184. [DOI] [Google Scholar]
- Zhong H.; Wu C.-H.; Li C.-Z.; Carpenter J.; Chueh C.-C.; Chen J.-Y.; Ade H.; Jen A. K.-Y. Rigidifying Nonplanar Perylene Diimides by Ring Fusion Toward Geometry-Tunable Acceptors for High-Performance Fullerene-Free Solar Cells. Adv. Mater. 2016, 28, 951–958. 10.1002/adma.201504120. [DOI] [PubMed] [Google Scholar]
- Yang L.; Gu W.; Lv L.; Chen Y.; Yang Y.; Ye P.; Wu J.; Hong L.; Peng A.; Huang H. Triplet Tellurophene-Based Acceptors for Organic Solar Cells. Angew. Chem., Int. Ed. 2018, 57, 1096–1102. 10.1002/anie.201712011. [DOI] [PubMed] [Google Scholar]
- Wang L.; Hu M.; Zhang Y.; Yuan Z.; Hu Y.; Zhao X.; Chen Y. Single-Strand and Ladder-Type Polymeric Acceptors Based on Regioisomerically-Pure Perylene Diimides towards All-Polymer Solar Cells. Polymer 2019, 162, 108–115. 10.1016/j.polymer.2018.12.041. [DOI] [Google Scholar]
- Hartnett P. E.; Matte H. S. S. R.; Eastham N. D.; Jackson N. E.; Wu Y.; Chen L. X.; Ratner M. A.; Chang R. P. H.; Hersam M. C.; Wasielewski M. R.; et al. Ring-Fusion as a Perylenediimide Dimer Design Concept for High-Performance Non-Fullerene Organic Photovoltaic Acceptors. Chem. Sci. 2016, 7, 3543–3555. 10.1039/C5SC04956C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.-P.; Xue L.-W.; Wei Q.; Liang M.; Deng K.; Zhang Z.-J.; Jiang P. Seeing Modulability Self-Assembled Monolayers of π-Conjugated Perylene Derivatives by Scanning Tunneling Microscopy. J. Phys. Chem. C 2016, 120, 18607–18615. 10.1021/acs.jpcc.6b04975. [DOI] [Google Scholar]
- Cai Z.; Vázquez R. J.; Zhao D.; Li L.; Lo W.; Zhang N.; Wu Q.; Keller B.; Eshun A.; Abeyasinghe N.; et al. Two Photon Absorption Study of Low-Bandgap, Fully Conjugated Perylene Diimide-Thienoacene-Perylene Diimide Ladder-Type Molecules. Chem. Mater. 2017, 29, 6726–6732. 10.1021/acs.chemmater.7b01512. [DOI] [Google Scholar]
- Cai Z.; Zhao D.; Sharapov V.; Awais M. A.; Zhang N.; Chen W.; Yu L. Enhancement in Open-Circuit Voltage in Organic Solar Cells by Using Ladder-Type Nonfullerene Acceptors. ACS Appl. Mater. Interfaces 2018, 10, 13528–13533. 10.1021/acsami.8b01308. [DOI] [PubMed] [Google Scholar]
- Carlotti B.; Cai Z.; Kim H.; Sharapov V.; Madu I. K.; Zhao D.; Chen W.; Zimmerman P. M.; Yu L.; Goodson T. Charge Transfer and Aggregation Effects on the Performance of Planar vs Twisted Nonfullerene Acceptor Isomers for Organic Solar Cells. Chem. Mater. 2018, 30, 4263–4276. 10.1021/acs.chemmater.8b01047. [DOI] [Google Scholar]
- Li X.; Wu K.; Zheng L.; Deng Y.; Tan S.; Chen H. Synthesis and Characterization of Novel Benzodithiophene-Fused Perylene Diimide Acceptors: Regulate Photovoltaic Performance via Structural Isomerism. Dyes Pigm. 2019, 168, 59–67. 10.1016/j.dyepig.2019.04.027. [DOI] [Google Scholar]
- Tian Z.; Guo Y.; Li J.; Li C.; Li W. Benzodithiophene-Fused Perylene Bisimides as Electron Acceptors for Non-Fullerene Organic Solar Cells with High Open-Circuit Voltage. ChemPhysChem 2019, 20, 2696–2701. 10.1002/cphc.201900309. [DOI] [PubMed] [Google Scholar]
- Yang J.; Chen F.; Ran H.; Hu J.-Y.; Xiao B.; Tang A.; Wang X.; Zhou E. Design and Synthesis of a Novel N-Type Polymer Based on Asymmetric Rylene Diimide for the Application in All-Polymer Solar Cells. Macromol. Rapid Commun. 2018, 39, 1700715. 10.1002/marc.201700715. [DOI] [PubMed] [Google Scholar]
- Li S.; Liu W.; Li C.-Z.; Lau T.-K.; Lu X.; Shi M.; Chen H. A Non-Fullerene Acceptor with a Fully Fused Backbone for Efficient Polymer Solar Cells with a High Open-Circuit Voltage. J. Mater. Chem. A 2016, 4, 14983–14987. 10.1039/C6TA07368A. [DOI] [Google Scholar]
- Ziffer M. E.; Jo S. B.; Zhong H.; Ye L.; Liu H.; Lin F.; Zhang J.; Li X.; Ade H. W.; Jen A. K.-Y.; et al. Long-Lived, Non-Geminate, Radiative Recombination of Photogenerated Charges in a Polymer/Small-Molecule Acceptor Photovoltaic Blend. J. Am. Chem. Soc. 2018, 140, 9996–10008. 10.1021/jacs.8b05834. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Li Y.; Huang J.; Hu H.; Zhang G.; Ma T.; Chow P. C. Y.; Ade H.; Pan D.; Yan H. Ring-Fusion of Perylene Diimide Acceptor Enabling Efficient Nonfullerene Organic Solar Cells with a Small Voltage Loss. J. Am. Chem. Soc. 2017, 139, 16092–16095. 10.1021/jacs.7b09998. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Zhao D.; Yang J.; Sharapov V.; Cai Z.; Li L.; Zhang N.; Neshchadin A.; Chen W.; Yu L. Propeller-Shaped Acceptors for High-Performance Non-Fullerene Solar Cells: Importance of the Rigidity of Molecular Geometry. Chem. Mater. 2017, 29, 1127–1133. 10.1021/acs.chemmater.6b04287. [DOI] [Google Scholar]
- Sun H.; Song X.; Xie J.; Sun P.; Gu P.; Liu C.; Chen F.; Zhang Q.; Chen Z.-K.; Huang W. PDI Derivative through Fine-Tuning the Molecular Structure for Fullerene-Free Organic Solar Cells. ACS Appl. Mater. Interfaces 2017, 9, 29924–29931. 10.1021/acsami.7b08282. [DOI] [PubMed] [Google Scholar]
- Liu J.; Ma L.-K.; Sheong F. K.; Zhang L.; Hu H.; Zhang J.-X.; Zhang J.; Li Z.; Ma C.; Han X.; et al. Carboxylate Substitution Position Influencing Polymer Properties and Enabling Non-Fullerene Organic Solar Cells with High Open Circuit Voltage and Low Voltage Loss. J. Mater. Chem. A 2018, 6, 16874–16881. 10.1039/C8TA04935A. [DOI] [Google Scholar]
- Shi Q.; Andreansky E. S.; Marder S. R.; Blakey S. B. Synthesis and C-H Functionalization Chemistry of Thiazole-Semicoronenediimides (TsCDIs) and -Coronenediimides (TCDIs). J. Org. Chem. 2017, 82, 10139–10148. 10.1021/acs.joc.7b01604. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Zhang S.; Zhang J.; Oswald V. F.; Amassian A.; Marder S. R.; Blakey S. B. KOtBu-Initiated Aryl C-H Iodination: A Powerful Tool for the Synthesis of High Electron Affinity Compounds. J. Am. Chem. Soc. 2016, 138, 3946–3949. 10.1021/jacs.5b12259. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Xue N.; Xiao C.; Ravva M. K.; Guo Y.; Wu L.; Zhang L.; Li Z.; Yue W.; Wang Z. Effect of Conjugation Length on the Properties of Fused Perylene Diimides with Variable Isoindigos. J. Mater. Chem. C 2019, 7, 12263–12269. 10.1039/C9TC04078A. [DOI] [Google Scholar]
- Witalewska M.; Wrona-Piotrowicz A.; Makal A.; Zakrzewski J. Polycyclic Aromatic N -Ethoxycarbonyl Thioamide S -Oxides and Their Triflic Acid Promoted Cyclization to Fluorescent Thiophene Imine-Fused Arenes. J. Org. Chem. 2018, 83, 1933–1939. 10.1021/acs.joc.7b02867. [DOI] [PubMed] [Google Scholar]
- Aksakal N. E.; Bayar M.; Dumrul H.; Atilla D.; Chumakov Y.; Yuksel F. Structural and Optical Properties of New Naphthalene and Perylene Imide Imidazoles. Polycyclic Aromat. Compd. 2019, 39, 363–373. 10.1080/10406638.2017.1327871. [DOI] [Google Scholar]
- Volland M.; Zhou P.; Wibmer L.; Häner R.; Decurtins S.; Liu S.-X.; Guldi D. M. Nanographene Favors Electronic Interactions with an Electron Acceptor Rather than an Electron Donor in a Planar Fused Push-Pull Conjugate. Nanoscale 2019, 11, 1437–1441. 10.1039/C8NR06961A. [DOI] [PubMed] [Google Scholar]
- Inan D.; Dubey R. K.; Westerveld N.; Bleeker J.; Jager W. F.; Grozema F. C. Substitution Effects on the Photoinduced Charge-Transfer Properties of Novel Perylene-3,4,9,10-Tetracarboxylic Acid Derivatives. J. Phys. Chem. A 2017, 121, 4633–4644. 10.1021/acs.jpca.7b03806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Chen W.; Wang Z.; Jie K. C. W.; Liu M.; Zhang Q. Synthesis and Exploration of Ladder-Structured Large Aromatic Dianhydrides as Organic Cathodes for Rechargeable Lithium-Ion Batteries. Chem. - Asian J. 2017, 12, 868–876. 10.1002/asia.201700070. [DOI] [PubMed] [Google Scholar]
- Schönamsgruber J.; Hirsch A. Benz-Bisimidazole-Bridged Perylenes - Linearly Expanded Chromophores: Benz-Bisimidazole-Bridged Perylenes. Eur. J. Org. Chem. 2015, 2015, 2167–2174. 10.1002/ejoc.201403561. [DOI] [Google Scholar]
- Chen L.; Zhang K.; Tang C.; Zheng Q.; Xiao Y. Controllable and Stepwise Synthesis of Soluble Ladder-Conjugated Bis(Perylene Imide) Fluorenebisimidazole as a Multifunctional Optoelectronic Material. J. Org. Chem. 2015, 80, 1871–1877. 10.1021/jo5028529. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Qi J.; Qiao W.; Wang Z. Y. Soluble Ladder Conjugated Polypyrrones: Synthesis, Characterization and Application in Photodetectors. Dyes Pigm. 2015, 113, 160–164. 10.1016/j.dyepig.2014.08.010. [DOI] [Google Scholar]
- Miletić T.; Fermi A.; Orfanos I.; Avramopoulos A.; De Leo F.; Demitri N.; Bergamini G.; Ceroni P.; Papadopoulos M. G.; Couris S.; et al. Tailoring Colors by O Annulation of Polycyclic Aromatic Hydrocarbons. Chem. - Eur. J. 2017, 23, 2363–2378. 10.1002/chem.201604866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Echegaray P.; Mancheño M. J.; Arrechea-Marcos I.; Juárez R.; López-Espejo G.; López Navarrete J. T.; Ramos M. M.; Seoane C.; Ortiz R. P.; Segura J. L. Synthesis of Perylene Imide Diones as Platforms for the Development of Pyrazine Based Organic Semiconductors. J. Org. Chem. 2016, 81, 11256–11267. 10.1021/acs.joc.6b02214. [DOI] [PubMed] [Google Scholar]
- Yuan K.; Kahan R. J.; Si C.; Williams A.; Kirschner S.; Uzelac M.; Zysman-Colman E.; Ingleson M. J. The Synthesis of Brominated-Boron-Doped PAHs by Alkyne 1,1-Bromoboration: Mechanistic and Functionalisation Studies. Chem. Sci. 2020, 11, 3258–3267. 10.1039/C9SC05404A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Rominger F.; Zschieschang U.; Klauk H.; Mastalerz M. Facile Synthetic Approach to a Large Variety of Soluble Diarenoperylenes. Chem. - Eur. J. 2016, 22, 14840–14845. 10.1002/chem.201603336. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Yamada K.; Watanabe K.; Kochi T.; Ie Y.; Aso Y.; Kakiuchi F. Synthesis of Dibenzo[ h, Rst ]Pentaphenes and Dibenzo[ Fg, Qr ]Pentacenes by the Chemoselective C-O Arylation of Dimethoxyanthraquinones. Org. Lett. 2017, 19, 3791–3794. 10.1021/acs.orglett.7b01666. [DOI] [PubMed] [Google Scholar]
- Li B.; Peng W.; Luo S.; Jiang C.; Guo J.; Xie S.; Hu Y.; Zhang Y.; Zeng Z. Diagonally π-Extended Perylene-Based Bis(Heteroacene) for Chiroptical Activity and Integrating Luminescence with Carrier-Transporting Capability. Org. Lett. 2019, 21, 1417–1421. 10.1021/acs.orglett.9b00152. [DOI] [PubMed] [Google Scholar]
- Fujikawa T.; Mitoma N.; Wakamiya A.; Saeki A.; Segawa Y.; Itami K. Synthesis, Properties, and Crystal Structures of π-Extended Double [6]Helicenes: Contorted Multi-Dimensional Stacking Lattice. Org. Biomol. Chem. 2017, 15, 4697–4703. 10.1039/C7OB00987A. [DOI] [PubMed] [Google Scholar]
- Fujikawa T.; Segawa Y.; Itami K. Laterally π-Extended Dithia[6]Helicenes with Heptagons: Saddle-Helix Hybrid Molecules. J. Org. Chem. 2017, 82, 7745–7749. 10.1021/acs.joc.7b01540. [DOI] [PubMed] [Google Scholar]
- Wu J.; He D.; Wang Y.; Su F.; Guo Z.; Lin J.; Zhang H.-J. Selective Ortho-π-Extension of Perylene Diimides for Rylene Dyes. Org. Lett. 2018, 20, 6117–6120. 10.1021/acs.orglett.8b02557. [DOI] [PubMed] [Google Scholar]
- Meng L.; Fujikawa T.; Kuwayama M.; Segawa Y.; Itami K. Thiophene-Fused π-Systems from Diarylacetylenes and Elemental Sulfur. J. Am. Chem. Soc. 2016, 138, 10351–10355. 10.1021/jacs.6b06486. [DOI] [PubMed] [Google Scholar]
- Zeng C.; Xiao C.; Feng X.; Zhang L.; Jiang W.; Wang Z. Electron-Transporting Bis(Heterotetracenes) with Tunable Helical Packing. Angew. Chem., Int. Ed. 2018, 57, 10933–10937. 10.1002/anie.201805614. [DOI] [PubMed] [Google Scholar]
- Zeng C.; Meng D.; Jiang W.; Wang Z. Synthesis of Isomeric Perylenodithiophene Diimides. Org. Lett. 2018, 20, 6606–6609. 10.1021/acs.orglett.8b02983. [DOI] [PubMed] [Google Scholar]
- Su F.; Chen S.; Mo X.; Wu K.; Wu J.; Lin W.; Lin Z.; Lin J.; Zhang H.-J.; Wen T.-B. Trisulfur Radical Anion-Triggered Stitching Thienannulation: Rapid Access to Largely π-Extended Thienoacenes. Chem. Sci. 2020, 11, 1503–1509. 10.1039/C9SC05332H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral M. G. B.; Pereira de Oliveira Santos D. M.; Bentaleb A.; Hillard E. A.; Cristiano R.; Gallardo H.; Durola F.; Bock H. Columnar Liquid-Crystalline Dibenzopentacenodithiophenes by Photocyclization. Chem. - Eur. J. 2016, 22, 8043–8047. 10.1002/chem.201600773. [DOI] [PubMed] [Google Scholar]
- Daigle M.; Picard-Lafond A.; Soligo E.; Morin J.-F. Regioselective Synthesis of Nanographenes by Photochemical Cyclodehydrochlorination. Angew. Chem., Int. Ed. 2016, 55, 2042–2047. 10.1002/anie.201509130. [DOI] [PubMed] [Google Scholar]
- Tokoro Y.; Oishi A.; Fukuzawa S. Twisted Polycyclic Aromatic Systems Prepared by Annulation of Bis(Arylethynyl)Arenes with Biphenylboronic Acids. Chem. - Eur. J. 2016, 22, 13908–13915. 10.1002/chem.201602474. [DOI] [PubMed] [Google Scholar]
- Regar R.; Mishra R.; Mondal P. K.; Sankar J. Metal-Free Annulation at the Ortho - and Bay -Positions of Perylene Bisimide Leading to Lateral π-Extension with Strong NIR Absorption. J. Org. Chem. 2018, 83, 9547–9552. 10.1021/acs.joc.8b01303. [DOI] [PubMed] [Google Scholar]
- Regar R.; Sekhar A. R.; Mishra R.; Sankar J. Bay- and Ortho- Ring Annulated Perylenediimides: Synthesis and Their Panchromatic Absorption. Indian J. Chem. Sect. B Org. Chem. Med. Chem. 2018, 57B, 308–313. [Google Scholar]
- Pfeifer D.; Klimant I.; Borisov S. M. Ultrabright Red-Emitting Photostable Perylene Bisimide Dyes: New Indicators for Ratiometric Sensing of High PH or Carbon Dioxide. Chem. - Eur. J. 2018, 24, 10711–10720. 10.1002/chem.201800867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Song I.; Ahn J.; Han M.; Linares M.; Surin M.; Zhang H.-J.; Oh J. H.; Lin J. π-Extended Perylene Diimide Double-Heterohelicenes as Ambipolar Organic Semiconductors for Broadband Circularly Polarized Light Detection. Nat. Commun. 2021, 12, 142. 10.1038/s41467-020-20390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; He D.; Zhang L.; Liu Y.; Mo X.; Lin J.; Zhang H. Direct Synthesis of Large-Scale Ortho-Iodinated Perylene Diimides: Key Precursors for Functional Dyes. Org. Lett. 2017, 19, 5438–5441. 10.1021/acs.orglett.7b02718. [DOI] [PubMed] [Google Scholar]
- Chang H.; Liu H.; Dmitrieva E.; Chen Q.; Ma J.; He P.; Liu P.; Popov A. A.; Cao X.-Y.; Wang X.-Y.; et al. Furan-Containing Double Tetraoxa[7]Helicene and Its Radical Cation. Chem. Commun. 2020, 56, 15181–15184. 10.1039/D0CC06970A. [DOI] [PubMed] [Google Scholar]
- Hamzehpoor E.; Perepichka D. F. Crystal Engineering of Room Temperature Phosphorescence in Organic Solids. Angew. Chem., Int. Ed. 2020, 59, 9977–9981. 10.1002/anie.201913393. [DOI] [PubMed] [Google Scholar]
- Haedler A. T.; Meskers S. C. J.; Zha R. H.; Kivala M.; Schmidt H.-W.; Meijer E. W. Pathway Complexity in the Enantioselective Self-Assembly of Functional Carbonyl-Bridged Triarylamine Trisamides. J. Am. Chem. Soc. 2016, 138, 10539–10545. 10.1021/jacs.6b05184. [DOI] [PubMed] [Google Scholar]
- Valera J. S.; Sánchez-Naya R.; Ramírez F. J.; Zafra J. L.; Gómez R.; Casado J.; Sánchez L. Solvent-Directed Helical Stereomutation Discloses Pathway Complexity on N-Heterotriangulene-Based Organogelators. Chem. - Eur. J. 2017, 23, 11141–11146. 10.1002/chem.201702391. [DOI] [PubMed] [Google Scholar]
- Valera J. S.; Gómez R.; Sánchez L. Tunable Energy Landscapes to Control Pathway Complexity in Self-Assembled N-Heterotriangulenes: Living and Seeded Supramolecular Polymerization. Small 2018, 14, 1702437. 10.1002/smll.201702437. [DOI] [PubMed] [Google Scholar]
- Dorca Y.; Valera J. S.; Cerdá J.; Aragó J.; Gómez R.; Ortí E.; Sánchez L. Synergy of Axial and Point Chirality to Construct Helical N-Heterotriangulene-Based Supramolecular Polymers. ChemNanoMat 2018, 4, 781–784. 10.1002/cnma.201800186. [DOI] [Google Scholar]
- Dorca Y.; Cerdá J.; Aragó J.; Orti E.; Sánchez L. Flipping Motion To Bias the Organized Supramolecular Polymerization of N-Heterotriangulenes. Chem. Mater. 2019, 31, 7024–7032. 10.1021/acs.chemmater.9b01653. [DOI] [Google Scholar]
- Kato S.; Matsuoka T.; Suzuki S.; Asano M. S.; Yoshihara T.; Tobita S.; Matsumoto T.; Kitamura C. Synthesis, Structures, and Properties of Neutral and Radical Cationic S,C,C-Bridged Triphenylamines. Org. Lett. 2020, 22, 734–738. 10.1021/acs.orglett.9b04575. [DOI] [PubMed] [Google Scholar]
- Karjule N.; Munavvar F. M.; Nithyanandhan J. Heterotriangulene-Based Unsymmetrical Squaraine Dyes: Synergistic Effects of Donor Moieties and out-of-Plane Branched Alkyl Chains on Dye Cell Performance. J. Mater. Chem. A 2016, 4, 18910–18921. 10.1039/C6TA08651A. [DOI] [Google Scholar]
- Fingerle M.; Bettinger H. F. Embedding a Boroxazine Ring into a Nanographene Scaffold by a Concise Bottom-up Synthetic Strategy. Chem. Commun. 2020, 56, 3847–3850. 10.1039/D0CC00471E. [DOI] [PubMed] [Google Scholar]
- Kitamoto Y.; Suzuki T.; Miyata Y.; Kita H.; Funaki K.; Oi S. The First Synthesis and X-Ray Crystallographic Analysis of an Oxygen-Bridged Planarized Triphenylborane. Chem. Commun. 2016, 52, 7098–7101. 10.1039/C6CC02440H. [DOI] [PubMed] [Google Scholar]
- Kitamoto Y.; Oda K.; Ogino K.; Hiyama K.; Kita H.; Hattori T.; Oi S. Synthesis of an Azadioxa-Planar Triphenylborane and Investigation of Its Structural and Photophysical Properties. Chem. Commun. 2021, 57, 2297–2300. 10.1039/D0CC08331C. [DOI] [PubMed] [Google Scholar]
- Yamamura M.; Nabeshima T. Relationship between the Bowl-Shaped Geometry of Phosphangulene and an Axial Group on the Phosphorus Atom. Bull. Chem. Soc. Jpn. 2016, 89, 42–49. 10.1246/bcsj.20150288. [DOI] [Google Scholar]
- Yamamura M.; Sukegawa K.; Nabeshima T. Tuning the Depth of Bowl-Shaped Phosphine Hosts: Capsule and Pseudo-Cage Architectures in Host-Guest Complexes with C60 Fullerene. Chem. Commun. 2015, 51, 12080–12083. 10.1039/C5CC04194E. [DOI] [PubMed] [Google Scholar]
- Yamamura M.; Sukegawa K.; Okada D.; Yamamoto Y.; Nabeshima T. Chiroptical Switching Caused by Crystalline/Liquid Crystalline Phase Transition of a Chiral Bowl-Shaped Molecule. Chem. Commun. 2016, 52, 4585–4588. 10.1039/C6CC00995F. [DOI] [PubMed] [Google Scholar]
- Heskia A.; Maris T.; Wuest J. D. Foiling Normal Patterns of Crystallization by Design. Polymorphism of Phosphangulene Chalcogenides. Cryst. Growth Des. 2019, 19, 5390–5406. 10.1021/acs.cgd.9b00907. [DOI] [Google Scholar]
- Heskia A.; Maris T.; Wuest J. D. Putting Fullerenes in Their Place: Cocrystallizing C60 and C70 with Phosphangulene Chalcogenides. Cryst. Growth Des. 2019, 19, 5418–5428. 10.1021/acs.cgd.9b00954. [DOI] [Google Scholar]
- Heskia A.; Maris T.; Wuest J. D. Bis(Phosphangulene)Iminium Salts. Holding on to Fullerenes with Phangs. Cryst. Growth Des. 2020, 20, 1319–1327. 10.1021/acs.cgd.9b01568. [DOI] [Google Scholar]
- Heskia A.; Maris T.; Aguiar P. M.; Wuest J. D. Building Large Structures with Curved Aromatic Surfaces by Complexing Metals with Phosphangulene. J. Am. Chem. Soc. 2019, 141, 18740–18753. 10.1021/jacs.9b08179. [DOI] [PubMed] [Google Scholar]
- Yamamura M.; Hasegawa T.; Nabeshima T. Synthesis of Phosphorus-Centered and Chalcogen-Bridged Concave Molecules: Modulation of Bowl Geometries and Packing Structures by Changing Bridging Atoms. Org. Lett. 2016, 18, 816–819. 10.1021/acs.orglett.6b00105. [DOI] [PubMed] [Google Scholar]
- Nakatsuka S.; Gotoh H.; Kinoshita K.; Yasuda N.; Hatakeyama T. Divergent Synthesis of Heteroatom-Centered 4,8,12-Triazatriangulenes. Angew. Chem., Int. Ed. 2017, 56, 5087–5090. 10.1002/anie.201701246. [DOI] [PubMed] [Google Scholar]
- Elbert S. M.; Reinschmidt M.; Baumgärtner K.; Rominger F.; Mastalerz M. Benzopyrano-Fused N-Heterocyclic Polyaromatics: Benzopyrano-Fused N-Heterocyclic Polyaromatics. Eur. J. Org. Chem. 2018, 2018, 532–536. 10.1002/ejoc.201701796. [DOI] [Google Scholar]
- Yokoyama S.; Hirose T.; Matsuda K. Photochemical Cleavage of the Axial Group Attached to the Central Carbon Atom of Triazatriangulene. Chem. Lett. 2015, 44, 76–78. 10.1246/cl.140897. [DOI] [Google Scholar]
- Shaikh A. C.; Veleta J. M.; Moutet J.; Gianetti T. L. Trioxatriangulenium (TOTA+) as a Robust Carbon-Based Lewis Acid in Frustrated Lewis Pair Chemistry. Chem. Sci. 2021, 12, 4841–4849. 10.1039/D0SC05893A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivalingam A.; Izquierdo M. A.; Marois A. L.; Vyšniauskas A.; Suhling K.; Kuimova M. K.; Vilar R. The Interactions between a Small Molecule and G-Quadruplexes Are Visualized by Fluorescence Lifetime Imaging Microscopy. Nat. Commun. 2015, 6, 8178. 10.1038/ncomms9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivalingam A.; Vyšniauskas A.; Albrecht T.; White A. J. P.; Kuimova M. K.; Vilar R. Trianguleniums as Optical Probes for G-Quadruplexes: A Photophysical, Electrochemical, and Computational Study. Chem. - Eur. J. 2016, 22, 4129–4139. 10.1002/chem.201504099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallabregue A.; Moreau D.; Sherin P.; Moneva Lorente P.; Jarolímová Z.; Bakker E.; Vauthey E.; Gruenberg J.; Lacour J. Selective Imaging of Late Endosomes with a PH-Sensitive Diazaoxatriangulene Fluorescent Probe. J. Am. Chem. Soc. 2016, 138, 1752–1755. 10.1021/jacs.5b09972. [DOI] [PubMed] [Google Scholar]
- Delgado I. H.; Pascal S.; Besnard C.; Voci S.; Bouffier L.; Sojic N.; Lacour J. C-Functionalized Cationic Diazaoxatriangulenes: Late-Stage Synthesis and Tuning of Physicochemical Properties. Chem. - Eur. J. 2018, 24, 10186–10195. 10.1002/chem.201801486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M.; Rostgaard K. R.; Liao Z.; Madsen A. Ø.; Martinez K. L.; Vosch T.; Laursen B. W. Design, Synthesis, and Time-Gated Cell Imaging of Carbon-Bridged Triangulenium Dyes with Long Fluorescence Lifetime and Red Emission. Chem. Sci. 2018, 9, 3122–3130. 10.1039/C8SC00089A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B.-B.; Liu J.-Q.; Wang X.-S. Cu(OAc)2-Catalyzed Aerobic Oxidative Dehydrogenation Coupling: Synthesis of Heptacyclic Quinolizino[3,4,5,6-Kla]Perimidines. J. Org. Chem. 2017, 82, 1817–1822. 10.1021/acs.joc.6b02644. [DOI] [PubMed] [Google Scholar]
- Xie J.; Shi K.; Cai K.; Zhang D.; Wang J.-Y.; Pei J.; Zhao D. A NIR Dye with High-Performance n-Type Semiconducting Properties. Chem. Sci. 2016, 7, 499–504. 10.1039/C5SC03045E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotwica K.; Bujak P.; Data P.; Krzywiec W.; Wamil D.; Gunka P. A.; Skorka L.; Jaroch T.; Nowakowski R.; Pron A.; et al. Soluble Flavanthrone Derivatives: Synthesis, Characterization, and Application to Organic Light-Emitting Diodes. Chem. - Eur. J. 2016, 22, 7978–7986. 10.1002/chem.201600513. [DOI] [PubMed] [Google Scholar]
- Kotwica K.; Bujak P.; Skorka L.; Jaroch T.; Nowakowski R. Luminophore from Forgotten Dye: Di(Alkylthiophene) Derivative of Benzo[h]Benz[5,6]Acridino[2,1,9,8-Klmna]Acridine. Synth. Met. 2017, 232, 117–122. 10.1016/j.synthmet.2017.08.002. [DOI] [Google Scholar]
- Zhu G.; Zhang G. Access to a Phthalazine Derivative Through an Angular Cis-Quinacridone. J. Org. Chem. 2021, 86, 1198–1203. 10.1021/acs.joc.0c02344. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Liu D.; Song M.; Shi D.; Liu B.; Cheng K.; Lu Y.; Liu H.; Yang M.; Wang W.; et al. Facile Synthesis of π-Extended Viologens: Electron-Deficient Polycyclic Aza-Aromatics. Chem. - Eur. J. 2017, 23, 7409–7413. 10.1002/chem.201700018. [DOI] [PubMed] [Google Scholar]
- Yang S.; Bristow J. C.; Addicoat M. A.; Wallis J. D. One Step Conversion of 1,5-Bis(Dimethylamino)Naphthalene to Salts of “Back to Back” Bis-Acridine Derivatives. New J. Chem. 2020, 44, 9621–9625. 10.1039/D0NJ00757A. [DOI] [Google Scholar]
- Šebera J.; Sebechlebská T.; Nováková Lachmanová Š.; Gasior J.; Moreno Garcia P.; Mészáros G.; Valášek M.; Kolivoška V.; Hromadová M. Investigation of the Charge Transport in Model Single Molecule Junctions Based on Expanded Bipyridinium Molecular Conductors. Electrochim. Acta 2019, 301, 267–273. 10.1016/j.electacta.2019.01.132. [DOI] [Google Scholar]
- Imran M.; Wehrmann C. M.; Chen M. S. Open-Shell Effects on Optoelectronic Properties: Antiambipolar Charge Transport and Anti-Kasha Doublet Emission from a N -Substituted Bisphenalenyl. J. Am. Chem. Soc. 2020, 142, 38–43. 10.1021/jacs.9b10677. [DOI] [PubMed] [Google Scholar]
- Taublaender M. J.; Glöcklhofer F.; Marchetti-Deschmann M.; Unterlass M. M. Green and Rapid Hydrothermal Crystallization and Synthesis of Fully Conjugated Aromatic Compounds. Angew. Chem., Int. Ed. 2018, 57, 12270–12274. 10.1002/anie.201801277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skonieczny K.; Jażwiński J.; Gryko D. T. The Synthesis of Imidazo[1,2-f]Phenanthridines, Phenanthro-[9,10-d]Imidazoles, and Phenanthro[9’,10’:4,5]Imidazo[1,2-f]-Phenanthridines via Intramolecular Oxidative Aromatic Coupling. Synthesis 2017, 49, 4651–4662. 10.1055/s-0036-1589053. [DOI] [Google Scholar]
- Yang D.-T.; Nakamura T.; He Z.; Wang X.; Wakamiya A.; Peng T.; Wang S. Doping Polycyclic Arenes with Nitrogen-Boron-Nitrogen (NBN) Units. Org. Lett. 2018, 20, 6741–6745. 10.1021/acs.orglett.8b02850. [DOI] [PubMed] [Google Scholar]
- Pati P. B.; Jin E.; Kim Y.; Kim Y.; Mun J.; Kim S. J.; Kang S. J.; Choe W.; Lee G.; Shin H.-J.; et al. Unveiling 79-Year-Old Ixene and Its BN-Doped Derivative. Angew. Chem., Int. Ed. 2020, 59, 14891–14895. 10.1002/anie.202004049. [DOI] [PubMed] [Google Scholar]
- Oda S.; Kumano W.; Hama T.; Kawasumi R.; Yoshiura K.; Hatakeyama T. Carbazole-Based DABNA Analogues as Highly Efficient Thermally Activated Delayed Fluorescence Materials for Narrowband Organic Light-Emitting Diodes. Angew. Chem., Int. Ed. 2021, 60, 2882–2886. 10.1002/anie.202012891. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Hanifi D.; Alvarez S.; Antonio F.; Pun A.; Klivansky L. M.; Hexemer A.; Ma B.; Liu Y. Charge Transport Anisotropy in n -Type Disk-Shaped Triphenylene-Tris(Aroyleneimidazole)s. Org. Lett. 2011, 13, 6528–6531. 10.1021/ol202814y. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Hanifi D. A.; Fernández-Liencres M. P.; Klivansky L. M.; Ma B.; Navarro A.; Liu Y. Understanding Electron Transport in Disk-Shaped Triphenylene-Tris(Naphthaleneimidazole)s through Structural Modification and Theoretical Investigation. ACS Appl. Mater. Interfaces 2017, 9, 20010–20019. 10.1021/acsami.7b03795. [DOI] [PubMed] [Google Scholar]
- Menke E. H.; Lami V.; Vaynzof Y.; Mastalerz M. π-Extended Rigid Triptycene-Trisaroylenimidazoles as Electron Acceptors. Chem. Commun. 2016, 52, 1048–1051. 10.1039/C5CC07238G. [DOI] [PubMed] [Google Scholar]
- Menke E. H.; Leibold D.; Lami V.; Hofstetter Y. J.; Mastalerz M.; Vaynzof Y. Triptycene-Trisaroyleneimidazoles as Non-Fullerene Acceptors - Influence of Side-Chains on Solubility, Device Morphology and Performance. Org. Electron. 2017, 47, 211–219. 10.1016/j.orgel.2017.05.004. [DOI] [Google Scholar]
- Vladimir A.; Mikhail F.; Amsharov K. Alumina-Promoted Oxodefluorination. RSC Adv. 2020, 10, 10879–10882. 10.1039/D0RA01369B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Yan L.; Liu Y.; Yang Y.; You J. Switchable Cascade C-H Annulation to Polycyclic Pyryliums and Pyridiniums: Discovering Mitochondria-Targeting Fluorescent Probes. Chem. Commun. 2020, 56, 15080–15083. 10.1039/D0CC06997C. [DOI] [PubMed] [Google Scholar]
- Berezin A.; Biot N.; Battisti T.; Bonifazi D. Oxygen-Doped Zig-Zag Molecular Ribbons. Angew. Chem., Int. Ed. 2018, 57, 8942–8946. 10.1002/anie.201803282. [DOI] [PubMed] [Google Scholar]
- Wetherby A. E.; Benson S. D.; Weinert C. S. Reaction of Bis(Bis(Trimethylsilyl)Amido)Mercury(II) with 3,3′-Disubstituted Binaphthols: Cyclization via an Intramolecular Electrophilic Aromatic Substitution Reaction. Inorg. Chim. Acta 2007, 360, 1977–1986. 10.1016/j.ica.2006.10.005. [DOI] [Google Scholar]
- Sciutto A.; Fermi A.; Folli A.; Battisti T.; Beames J. M.; Murphy D. M.; Bonifazi D. Customizing Photoredox Properties of PXX-Based Dyes through Energy Level Rigid Shifts of Frontier Molecular Orbitals. Chem. - Eur. J. 2018, 24, 4382–4389. 10.1002/chem.201705620. [DOI] [PubMed] [Google Scholar]
- Sciutto A.; Berezin A.; Lo Cicero M.; Miletić T.; Stopin A.; Bonifazi D. Tailored Synthesis of N-Substituted Peri-Xanthenoxanthene Diimide (PXXDI) and Monoimide (PXXMI) Scaffolds. J. Org. Chem. 2018, 83, 13787–13798. 10.1021/acs.joc.8b02076. [DOI] [PubMed] [Google Scholar]
- Wehrmann C. M.; Charlton R. T.; Chen M. S. A Concise Synthetic Strategy for Accessing Ambient Stable Bisphenalenyls toward Achieving Electroactive Open-Shell π-Conjugated Materials. J. Am. Chem. Soc. 2019, 141, 3240–3248. 10.1021/jacs.8b13300. [DOI] [PubMed] [Google Scholar]
- Boonloed A.; Weber G. L.; Ramzy K. M.; Dias V. R.; Remcho V. T. Centrifugal Partition Chromatography: A Preparative Tool for Isolation and Purification of Xylindein from Chlorociboria Aeruginosa. J. Chromatogr. A 2016, 1478, 19–25. 10.1016/j.chroma.2016.11.026. [DOI] [PubMed] [Google Scholar]
- Giesbers G.; Van Schenck J.; Gutierrez S. V.; Robinson S.; Ostroverkhova O. Fungi-Derived Pigments for Sustainable Organic (Opto)Electronics. MRS Adv. 2018, 3, 3459–3464. 10.1557/adv.2018.446. [DOI] [Google Scholar]
- Giesbers G.; Van Schenck J.; Quinn A.; Van Court R.; Vega Gutierrez S. M.; Robinson S. C.; Ostroverkhova O. Xylindein: Naturally Produced Fungal Compound for Sustainable (Opto)Electronics. ACS Omega 2019, 4, 13309–13318. 10.1021/acsomega.9b01490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S.; Nishii Y.; Miura M. Thioether-Directed Peri-Selective C-H Arylation under Rhodium Catalysis: Synthesis of Arene-Fused Thioxanthenes. Org. Lett. 2019, 21, 233–236. 10.1021/acs.orglett.8b03675. [DOI] [PubMed] [Google Scholar]
- Hertz V. M.; Lerner H.-W.; Wagner M. Ru-Catalyzed Benzannulation Leads to Luminescent Boron-Containing Polycyclic Aromatic Hydrocarbons. Org. Lett. 2015, 17, 5240–5243. 10.1021/acs.orglett.5b02604. [DOI] [PubMed] [Google Scholar]
- Crossley D. L.; Kahan R. J.; Endres S.; Warner A. J.; Smith R. A.; Cid J.; Dunsford J. J.; Jones J. E.; Vitorica-Yrezabal I.; Ingleson M. J. A Modular Route to Boron Doped PAHs by Combining Borylative Cyclisation and Electrophilic C-H Borylation. Chem. Sci. 2017, 8, 7969–7977. 10.1039/C7SC02793A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahora N.; Hirano K.; Yasuda A.; Sasamori T.; Shioji K.; Okuma K. The Electronic Structure of Thioxanthylium Scaffolds. Heterocycles 2021, 102, 451–464. 10.3987/COM-20-14372. [DOI] [Google Scholar]
- John A.; Bolte M.; Lerner H.-W.; Wagner M. A Vicinal Electrophilic Diborylation Reaction Furnishes Doubly Boron-Doped Polycyclic Aromatic Hydrocarbons. Angew. Chem., Int. Ed. 2017, 56, 5588–5592. 10.1002/anie.201701591. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Zhang Y.; Tang J.; Zhang L.; Liu Q.; Li Q.; Gao G.; You J. Ir-Catalyzed Cascade C-H Fusion of Aldoxime Ethers and Heteroarenes: Scope and Mechanisms. ACS Catal. 2020, 10, 203–209. 10.1021/acscatal.9b04274. [DOI] [Google Scholar]
- Mocanu A.; Szücs R.; Caytan E.; Roisnel T.; Dorcet V.; Bouit P.-A.; Nyulászi L.; Hissler M. Synthesis, Optical, and Redox Properties of Regioisomeric Benzoheterocycles-Fused Pyrene. J. Org. Chem. 2019, 84, 957–962. 10.1021/acs.joc.8b02884. [DOI] [PubMed] [Google Scholar]
- Yan J.; Pulis A. P.; Perry G. J. P.; Procter D. J. Metal-Free Synthesis of Benzothiophenes by Twofold C-H Functionalization: Direct Access to Materials-Oriented Heteroaromatics. Angew. Chem., Int. Ed. 2019, 58, 15675–15679. 10.1002/anie.201908319. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Li J.; Zhang S.; Wang Q.; Dai G.; Liu B.; Zhu X.; Li Z.; Kolodziej C.; McCleese C.; et al. Stable 2D Bisthienoacenes: Synthesis, Crystal Packing, and Photophysical Properties. Chem. - Eur. J. 2018, 24, 14442–14447. 10.1002/chem.201802411. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Gu W.; Zhang G.; Liu L.; Lv A.; Zhang L. Isomeric Pyrenodithiophenediones and Their Derivatives: Synthesis, Reactivity, and Device Performance. J. Org. Chem. 2019, 84, 5936–5942. 10.1021/acs.joc.9b00579. [DOI] [PubMed] [Google Scholar]
- Hossain M. A.; Yamashita K.; Hirabayashi K.; Shimizu T.; Goto K.; Sugiura K. Thiophene-Fused Dinaphthopentaphenes: Versatile Applications of 1,2-Bis(Pyren-2-Yl)Aromatics in the Synthesis of π-Expanded Molecules. ChemistrySelect 2017, 2, 4343–4348. 10.1002/slct.201700152. [DOI] [Google Scholar]
- Valdés H.; Poyatos M.; Peris E. Postmodification of the Electronic Properties by Addition of π-Stacking Additives in N-Heterocyclic Carbene Complexes with Extended Polyaromatic Systems. Inorg. Chem. 2015, 54, 3654–3659. 10.1021/acs.inorgchem.5b00250. [DOI] [PubMed] [Google Scholar]
- Valdés H.; Poyatos M.; Peris E. A Nanosized Janus Bis-N-Heterocyclic Carbene Ligand Based on a Quinoxalinophenanthrophenazine Core, and Its Coordination to Iridium. Organometallics 2015, 34, 1725–1729. 10.1021/acs.organomet.5b00208. [DOI] [Google Scholar]
- Wang Z.; Gu P.; Liu G.; Yao H.; Wu Y.; Li Y.; Rakesh G.; Zhu J.; Fu H.; Zhang Q. A Large Pyrene-Fused N-Heteroacene: Fifteen Aromatic Six-Membered Rings Annulated in One Row. Chem. Commun. 2017, 53, 7772–7775. 10.1039/C7CC03898D. [DOI] [PubMed] [Google Scholar]
- Mastalerz M.; Ueberricke L.; Ciubotaru I.; Ghalami F.; Mildner F.; Rominger F.; Elstner M. Di- and Tetracyano Substituted Pyrene-Fused Pyrazaacenes -Aggregation in the Solid State. Chem. - Eur. J. 2020, 26, 11634–11642. 10.1002/chem.202002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.-B.; Feng G.-F.; Zhao P.-C.; Chang F.-F.; Huang W. Asymmetric Imidazole/Pyrene/Pyrazine Based D-π-A Compounds Showing Visualized Acidichromism and near-Infrared Electrochromism. Tetrahedron Lett. 2015, 56, 6912–6914. 10.1016/j.tetlet.2015.10.100. [DOI] [Google Scholar]
- Gu P.-Y.; Zhang J.; Long G.; Wang Z.; Zhang Q. Solution-Processable Thiadiazoloquinoxaline-Based Donor-Acceptor Small Molecules for Thin-Film Transistors. J. Mater. Chem. C 2016, 4, 3809–3814. 10.1039/C5TC03222A. [DOI] [Google Scholar]
- Li Y.; Zhang C.; Gu P.; Wang Z.; Li Z.; Li H.; Lu J.; Zhang Q. Nonvolatile Tri-State Resistive Memory Behavior of a Stable Pyrene-Fused N-Heteroacene with Ten Linearly-Annulated Rings. Chem. - Eur. J. 2018, 24, 7845–7851. 10.1002/chem.201801146. [DOI] [PubMed] [Google Scholar]
- Gu P.-Y.; Wang Z.; Liu G.; Yao H.; Wang Z.; Li Y.; Zhu J.; Li S.; Zhang Q. Synthesis, Full Characterization, and Field Effect Transistor Behavior of a Stable Pyrene-Fused N-Heteroacene with Twelve Linearly Annulated Six-Membered Rings. Chem. Mater. 2017, 29, 4172–4175. 10.1021/acs.chemmater.7b01318. [DOI] [Google Scholar]
- Lee S. H.; Paredes M. S. V.; Rappenecker T. J.; Robins K. A.; Lee D.-C. Optimized Synthesis of Thermally Stable Axially Modified Pyrazine-Acene Nanoribbon with Gelation Properties. New J. Chem. 2020, 44, 3604–3611. 10.1039/C9NJ06303J. [DOI] [Google Scholar]
- Cortizo-Lacalle D.; Pertegás A.; Melle-Franco M.; Bolink H. J.; Mateo-Alonso A. Pyrene-Fused Bisphenazinothiadiazoles with Red to NIR Electroluminescence. Org. Chem. Front. 2017, 4, 876–881. 10.1039/C7QO00227K. [DOI] [Google Scholar]
- Li Y.; Wang Z.; Zhang C.; Gu P.; Chen W.; Li H.; Lu J.; Zhang Q. Thiadizoloquinoxaline-Based N-Heteroacenes as Active Elements for High-Density Data-Storage Device. ACS Appl. Mater. Interfaces 2018, 10, 15971–15979. 10.1021/acsami.8b05178. [DOI] [PubMed] [Google Scholar]
- Hu B.-L.; Zhang K.; An C.; Schollmeyer D.; Pisula W.; Baumgarten M. Layered Thiadiazoloquinoxaline-Containing Long Pyrene-Fused N-Heteroacenes. Angew. Chem., Int. Ed. 2018, 57, 12375–12379. 10.1002/anie.201803230. [DOI] [PubMed] [Google Scholar]
- Cortizo-Lacalle D.; Gozalvez C.; Melle-Franco M.; Mateo-Alonso A. A Thiadiazole-Capped Nanoribbon with 18 Linearly Fused Rings. Nanoscale 2018, 10, 11297–11301. 10.1039/C8NR03516D. [DOI] [PubMed] [Google Scholar]
- Sahoo P. K.; Giri C.; Haldar T. S.; Puttreddy R.; Rissanen K.; Mal P. Mechanochemical Synthesis, Photophysical Properties, and X-Ray Structures of N-Heteroacenes. Eur. J. Org. Chem. 2016, 2016, 1283–1291. 10.1002/ejoc.201600005. [DOI] [Google Scholar]
- Fratczak E. Z.; Makowski T.; Moustafa R. M.; El-Assaad T. H.; Moneta M. E.; Uznanski P.; Kaafarani B. R. Spectroscopic Characterization of the Structural Properties of Quinoxalinophenanthrophenazine Thin Films. J. Mater. Chem. C 2018, 6, 781–789. 10.1039/C7TC04757F. [DOI] [Google Scholar]
- Maass F.; Stein A.; Kohl B.; Hahn L.; Gade L. H.; Mastalerz M.; Tegeder P. Substrate-Directed Growth of N-Heteropolycyclic Molecules on a Metal Surface. J. Phys. Chem. C 2016, 120, 2866–2873. 10.1021/acs.jpcc.5b12080. [DOI] [Google Scholar]
- Kohl B.; Baumgaertner K.; Rominger F.; Mastalerz M. Quinoxalinophenanthrophenazines (QPPs) and Hexabenzoovalenes (HBOs) - Proving the Solubility Enhancement by Triptycene End-Capping. Eur. J. Org. Chem. 2019, 2019, 4891–4896. 10.1002/ejoc.201900819. [DOI] [Google Scholar]
- Kohl B.; Rominger F.; Mastalerz M. Crystal Structures of a Molecule Designed Not To Pack Tightly. Chem. - Eur. J. 2015, 21, 17308–17313. 10.1002/chem.201502847. [DOI] [PubMed] [Google Scholar]
- Kohl B.; Bohnwagner M. V.; Rominger F.; Wadepohl H.; Dreuw A.; Mastalerz M. Attractive Dispersion Interactions Versus Steric Repulsion of Tert-Butyl Groups in the Crystal Packing of a D3h-Symmetric Tris(Quinoxalinophenanthrophenazine). Chem. - Eur. J. 2016, 22, 646–655. 10.1002/chem.201503863. [DOI] [PubMed] [Google Scholar]
- Ueberricke L.; Holub D.; Kranz J.; Rominger F.; Elstner M.; Mastalerz M. Triptycene End-Capped Quinoxalinophenanthrophenazines (QPPs): Influence of Substituents and Conditions on Aggregation in the Solid State. Chem. - Eur. J. 2019, 25, 11121–11134. 10.1002/chem.201902002. [DOI] [PubMed] [Google Scholar]
- Ueberricke L.; Wieland S.; Rominger F.; Mastalerz M. Triptycene End-Capped Quinoxalinophenanthrophenazines with Aromatic Substituents - Synthesis, Characterization, and Single-Crystal Structure Analysis. Org. Mater. 2019, 01, 050–062. 10.1055/s-0039-1700847. [DOI] [Google Scholar]
- Ueberricke L.; Ghalami F.; Zhang W.-S.; Rao V.; Rominger F.; Schröder R. R.; Elstner M.; Mastalerz M. Isostructural Charge-Transfer Cocrystals Based on Triptycene End-Capped Quinoxalinophenanthrophenazine. Cryst. Growth Des. 2021, 21, 1329–1341. 10.1021/acs.cgd.0c01619. [DOI] [Google Scholar]
- Ueberricke L.; Benke B. P.; Kirschbaum T.; Hahn S.; Rominger F.; Bunz U. H. F.; Mastalerz M. Synthesis and Optoelectronic Properties of a Quinoxalino-Phenanthrophenazine (QPP) Extended Tribenzotriquinacene (TBTQ). Chem. - Eur. J. 2021, 27, 2043–2049. 10.1002/chem.202003666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.-L.; An C.; Wagner M.; Ivanova G.; Ivanova A.; Baumgarten M. Three-Dimensional Pyrene-Fused N-Heteroacenes. J. Am. Chem. Soc. 2019, 141, 5130–5134. 10.1021/jacs.9b01082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Xu Z.; Deng H.; Zhou Z.; Dang Y.; Sun Z. Dimeric Cycloparaphenylenes with a Rigid Aromatic Linker. Angew. Chem., Int. Ed. 2021, 60, 7649–7653. 10.1002/anie.202016995. [DOI] [PubMed] [Google Scholar]
- Cortizo-Lacalle D.; Mora-Fuentes J. P.; Strutyński K.; Saeki A.; Melle-Franco M.; Mateo-Alonso A. Monodisperse N-Doped Graphene Nanoribbons Reaching 7.7 Nanometers in Length. Angew. Chem., Int. Ed. 2018, 57, 703–708. 10.1002/anie.201710467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X.; Zhao J.; Zhang W.; Chen L. Novel N-Channel Organic Semiconductor Based on Pyrene-Phenazine Fused Monoimide and Bisimides. Chin. Chem. Lett. 2018, 29, 331–335. 10.1016/j.cclet.2017.09.015. [DOI] [Google Scholar]
- Min Y.; Dou C.; Tian H.; Geng Y.; Liu J.; Wang L. N-Type Azaacenes Containing B-N Units. Angew. Chem., Int. Ed. 2018, 57, 2000–2004. 10.1002/anie.201712986. [DOI] [PubMed] [Google Scholar]
- Wang C.-Z.; Feng X.; Elsegood M. R. J.; Warwick T. G.; Teat S. J.; Redshaw C.; Bi Y.-S.; Yamato T. Pyrene-Fused Pyrazaacenes with Eight Rectilinearly Arranged Aromatic Rings. Asian J. Org. Chem. 2019, 8, 155–160. 10.1002/ajoc.201800608. [DOI] [Google Scholar]
- Mora-Fuentes J. P.; Papadopoulos I.; Thiel D.; Álvarez-Boto R.; Cortizo-Lacalle D.; Clark T.; Melle-Franco M.; Guldi D. M.; Mateo-Alonso A. Singlet Fission in Pyrene-Fused Azaacene Dimers. Angew. Chem., Int. Ed. 2020, 59, 1113–1117. 10.1002/anie.201911529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M.; Li X.; Sun L.; Niu X.; Liang Q.; Cai X.; Huang J.; Ling J.; Mo Y. Special Photophysical Properties of Poly(2,11-Diquinoxalinopyrene)s. Chin. J. Polym. Sci. 2017, 35, 1097–1109. 10.1007/s10118-017-1961-2. [DOI] [Google Scholar]
- Chu T.; Ju X.; Han X.; Du H.; Zhang Y.; Zhao J.; Zhang J. Synthesis and Electrochromic Properties of Cross-Linked and Soluble Conjugated Polymers Based on 5, 8, 14, 17-Tetrabromoquinoxaline[2’, 3′:9,10]Phenanthro[4,5-Abc]Phenazine as the Multifunctionalized Acceptor Unit. Org. Electron. 2019, 73, 43–54. 10.1016/j.orgel.2019.06.004. [DOI] [Google Scholar]
- Chu T.; Yue H.; Zhao Y.; Du H.; Zhang Y.; Han X.; Zhao J.; Zhang J. Synthesis and Characterization of D-A Type Conjugated Electrochromic Polymers with Cross-Linked Structure Employing a Novel and Multi-Functionalized Molecular as the Acceptor Unit. J. Electroanal. Chem. 2019, 848, 113276. 10.1016/j.jelechem.2019.113276. [DOI] [Google Scholar]
- Jiang L.; Papageorgiou A. C.; Oh S. C.; Saglam Ö.; Reichert J.; Duncan D. A.; Zhang Y.-Q.; Klappenberger F.; Guo Y.; Allegretti F.; et al. Synthesis of Pyrene-Fused Pyrazaacenes on Metal Surfaces: Toward One-Dimensional Conjugated Nanostructures. ACS Nano 2016, 10, 1033–1041. 10.1021/acsnano.5b06340. [DOI] [PubMed] [Google Scholar]
- Yu X.; Wan J.; Yuan C.; Guo N.; Shen Y.; Li J. Tetraazatetraoxodecacene and Tetraazatetrathiodecacene: Synthesis, Crystal Structures, Linear and Third-Order Nonlinear Optical Properties. Dyes Pigm. 2019, 161, 130–136. 10.1016/j.dyepig.2018.07.038. [DOI] [Google Scholar]
- Marco A. B.; Gozalvez C.; Olano M.; Sun X.; Atxabal A.; Melle-Franco M.; Hueso L. E.; Mateo-Alonso A. Bis(Triisopropylsilylethynyl)-Substituted Pyrene-Fused Tetraazaheptacene: Synthesis and Properties. Phys. Chem. Chem. Phys. 2016, 18, 11616–11619. 10.1039/C5CP07656K. [DOI] [PubMed] [Google Scholar]
- Pan H.; Song T.; Yin X.; Jin P.; Xiao J. Synthesis, Crystal Analysis, and Optoelectronic Properties of Diazole-Functionalized Acenes and Azaacenes. Chem. - Eur. J. 2018, 24, 6572–6579. 10.1002/chem.201705657. [DOI] [PubMed] [Google Scholar]
- Antonicelli G.; Gozalvez C.; Atxabal A.; Melle-Franco M.; Hueso L. E.; Mateo-Alonso A. K-Conjugated Dibenzoazahexacenes. Org. Lett. 2016, 18, 4694–4697. 10.1021/acs.orglett.6b02332. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Lo K.-C.; Szeto L.; Chan W.-K. Synthesis of Pyrazinopyrazine-Fused Azaacenes through Direct Condensation Reactions between Quinoxalinediamine and Diketones. J. Org. Chem. 2020, 85, 6372–6379. 10.1021/acs.joc.9b03504. [DOI] [PubMed] [Google Scholar]
- Wu Z.-H.; Sun W.-J.; Tian H.-H.; Yu Z.-F.; Guo R.-X.; Shao X.; Zhang H.-L. 9,10-Imide-Pyrene-Fused Pyrazaacenes (IPPA) as N-Type Doping Materials for High-Performance Nonvolatile Organic Field Effect Transistor Memory Devices. Adv. Electron. Mater. 2019, 5, 1800598. 10.1002/aelm.201800598. [DOI] [Google Scholar]
- Riaño A.; Carini M.; Melle-Franco M.; Mateo-Alonso A. Mechanically Interlocked Nitrogenated Nanographenes. J. Am. Chem. Soc. 2020, 142, 20481–20488. 10.1021/jacs.0c10345. [DOI] [PubMed] [Google Scholar]
- Baumgärtner K.; Kirschbaum T.; Krutzek F.; Dreuw A.; Rominger F.; Mastalerz M. K-Region-Extended [ c ]-Heteroannulated Pyrenes. Chem. - Eur. J. 2017, 23, 17817–17822. 10.1002/chem.201703988. [DOI] [PubMed] [Google Scholar]
- Chen W.; Long G.; Kanehira K.; Zhang M.; Michinobu T.; Liu M.; Zhang Q. A Direct Method to Access Substituted Pyreno[4,5-c:9,10-c’] Difuran and Its Analogues. Asian J. Org. Chem. 2018, 7, 2213–2217. 10.1002/ajoc.201800122. [DOI] [Google Scholar]
- Wang P.-L.; Ding S.-Y.; Zhang Z.-C.; Wang Z.-P.; Wang W. Constructing Robust Covalent Organic Frameworks via Multicomponent Reactions. J. Am. Chem. Soc. 2019, 141, 18004–18008. 10.1021/jacs.9b10625. [DOI] [PubMed] [Google Scholar]
- Liu Y.-L.; Yang L.; Guo Y.-Q.; Xu G.-Q.; Qu B.; Fu Y. Synthesis and Configurational Character Study of Novel Structural Isomers Based on Pyrene-Imidazole. Molecules 2019, 24, 2293. 10.3390/molecules24122293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skonieczny K.; Gryko D. T. Light-Induced Direct Arylation in the Solid Crystalline State as a Strategy Towards π-Expanded Imidazoles. Chem. - Asian J. 2016, 11, 2513–2517. 10.1002/asia.201600752. [DOI] [PubMed] [Google Scholar]
- Karthik S.; Ajantha J.; Nagaraja C. M.; Easwaramoorthi S.; Gandhi T. Synthesis and Photophysics of Extended π-Conjugated Systems of Substituted 10-Aryl-Pyrenoimidazoles. Org. Biomol. Chem. 2016, 14, 10255–10266. 10.1039/C6OB01760F. [DOI] [PubMed] [Google Scholar]
- Ibáñez S.; Guerrero A.; Poyatos M.; Peris E. Fluorescent Pyrene-Based Bis-Azole Compounds: Synthesis and Photophysical Analysis. Chem. - Eur. J. 2015, 21, 10566–10575. 10.1002/chem.201501179. [DOI] [PubMed] [Google Scholar]
- Mardanya S.; Karmakar S.; Bar M.; Baitalik S. Pyrene-Biimidazole Based Ru(II) and Os(II) Complexes as Highly Efficient Probes for the Visible and near-Infrared Detection of Cyanide in Aqueous Media. Dalton Trans. 2015, 44, 21053–21072. 10.1039/C5DT03766B. [DOI] [PubMed] [Google Scholar]
- Mardanya S.; Karmakar S.; Mondal D.; Baitalik S. Homo- and Heterobimetallic Ruthenium(II) and Osmium(II) Complexes Based on a Pyrene-Biimidazolate Spacer as Efficient DNA-Binding Probes in the Near-Infrared Domain. Inorg. Chem. 2016, 55, 3475–3489. 10.1021/acs.inorgchem.5b02912. [DOI] [PubMed] [Google Scholar]
- Mardanya S.; Mondal D.; Baitalik S. Bimetallic Ru(II) and Os(II) Complexes Based on a Pyrene-Bisimidazole Spacer: Synthesis, Photophysics, Electrochemistry and Multisignalling DNA Binding Studies in the near Infrared Region. Dalton Trans. 2017, 46, 17010–17024. 10.1039/C7DT03355A. [DOI] [PubMed] [Google Scholar]
- Wen H.; Gong X.; Jia Z.; Han P.; Lin B.; Ye S.; Sun Y.; Zhang X.; Yang H. Conjugated Polymers Constructed by a Novel Pyrene-Fused Polycyclic Building Block and Their Applications as Organic Electronic Materials. Dyes Pigm. 2016, 130, 16–23. 10.1016/j.dyepig.2016.03.009. [DOI] [Google Scholar]
- Shahrokhi F.; Zhao Y. Self-Condensation of Pyrene-4,5-Dione: An Approach To Generate Functional Organic Fluorophores. Org. Lett. 2019, 21, 9306–9310. 10.1021/acs.orglett.9b03297. [DOI] [PubMed] [Google Scholar]
- Fan M.; Shen Y.; Zheng Y.; Yu X.; Li M.; Xu L.; Wang J.; Yang L.; Li J. Pyridazine-Containing Diazatwistanthracene and Tetraazatwisttetracene: Synthesis, Crystal Structures and Third Order Non-linear Optical Properties. ChemistrySelect 2019, 4, 2810–2814. 10.1002/slct.201804065. [DOI] [Google Scholar]
- Fusco S.; Maglione C.; Velardo A.; Piccialli V.; Liguori R.; Peluso A.; Rubino A.; Centore R. N-Rich Fused Heterocyclic Systems: Synthesis, Structure, Optical and Electrochemical Characterization. Eur. J. Org. Chem. 2016, 2016, 1772–1780. 10.1002/ejoc.201501283. [DOI] [Google Scholar]
- Zheng Y.; Zhao L.; Zhang Y.; Wang C.; Wang K.; Qi D.; Jiang J. Novel Chiral Binaphthalene-Linked Pyrenes. Synthesis, Structure, and Spectroscopy. Dyes Pigm. 2017, 141, 245–250. 10.1016/j.dyepig.2017.02.031. [DOI] [Google Scholar]
- Lv B.; Xiao J.; Zhou J.; Zhang X.; Duan J.; Su W.; Zhao J. Synthesis, Crystal Analyses, Physical Properties, and Electroluminescent Behavior of Unsymmetrical Heterotwistacenes. ACS Appl. Mater. Interfaces 2016, 8, 18998–19003. 10.1021/acsami.6b07304. [DOI] [PubMed] [Google Scholar]
- Jin P.; Tian F.; Han Y.; Wang L.; Zhao X.; Xiao J. Dimesitylboryl-Decorated Azaarene: Synthesis, Enhanced Stability and Optoelectronic Property. Chem. - Asian J. 2019, 14, 4395–4399. 10.1002/asia.201901349. [DOI] [PubMed] [Google Scholar]
- Cheung K. Y.; Watanabe K.; Segawa Y.; Itami K. Synthesis of a Zigzag Carbon Nanobelt. Nat. Chem. 2021, 13, 255–259. 10.1038/s41557-020-00627-5. [DOI] [PubMed] [Google Scholar]
- Chen H.; Gui S.; Zhang Y.; Liu Z.; Miao Q. Synthesis of a Hydrogenated Zigzag Carbon Nanobelt. CCS Chem. 2021, 3, 613. 10.31635/ccschem.020.202000189. [DOI] [Google Scholar]
- Tian F.; Song T.; Wang T.; Xiao J.; Zhao X. 11,16-Di-Tert-Butyl-9,18-Diphenylbenzo[Kl]Benzo[8,9]Triphenyleno [2,3-b]Xanthene: Synthesis, Photophysics, Self-Assembly and Electroluminescent Properties. Asian J. Org. Chem. 2019, 8, 399–403. 10.1002/ajoc.201900048. [DOI] [Google Scholar]
- Liu Z.; Wang W.; Xu W.; Chen H.; Zhang X.; Ren T.; Wang X.; Zhao J.; Xiao J. Synthesis, Characterization and Photocurrent Behavior of Asymmetrical Heterotwistacenes. Dyes Pigm. 2015, 115, 143–148. 10.1016/j.dyepig.2014.12.025. [DOI] [Google Scholar]
- Pan H.; Duan J.; Zhai G.; Jin P.; Zhao X.; Jiang L.; Xiao J. Synthesis, Optoelectronic and Self-Assembly Properties of Diazadioxaacene Derivatives. Chem. - Asian J. 2017, 12, 2121–2126. 10.1002/asia.201700700. [DOI] [PubMed] [Google Scholar]
- Song T.; Han Y.; Jin P.; Li X.; Song Y.; Xiao J. The Enhanced Two-Photon Absorption Behavior of Twistfuranacenes to Phenylacetylene-Functionalized Twistacenes. J. Mater. Chem. C 2019, 7, 6344–6351. 10.1039/C9TC01568J. [DOI] [Google Scholar]
- Duan J.; Gu P.; Xiao J.; Shen X.; Liu X.; Yi Y.; Zhang Q. Synthesis, Physical Properties and Memory Device Application of a Twelve-Ring Fused Twistheteroacene. Chem. - Asian J. 2017, 12, 638–642. 10.1002/asia.201700048. [DOI] [PubMed] [Google Scholar]
- Verbitskiy E. V.; Cheprakova E. M.; Makarova N. I.; Dorogan I. V.; Metelitsa A. V.; Minkin V. I.; Slepukhin P. A.; Svalova T. S.; Ivanova A. V.; Kozitsina A. N.; et al. Heteroacenes Bearing the Pyrimidine Scaffold: Synthesis, Photophysical and Electrochemical Properties. Eur. J. Org. Chem. 2016, 2016, 1420–1428. 10.1002/ejoc.201501450. [DOI] [Google Scholar]
- Zhao K.-Q.; Jing M.; An L.-L.; Du J.-Q.; Wang Y.-H.; Hu P.; Wang B.-Q.; Monobe H.; Heinrich B.; Donnio B. Facile Transformation of 1-Aryltriphenylenes into Dibenzo[Fg,Op]Tetracenes by Intramolecular Scholl Cyclodehydrogenation: Synthesis, Self-Assembly, and Charge Carrier Mobility of Large π-Extended Discogens. J. Mater. Chem. C 2017, 5, 669–682. 10.1039/C6TC04530H. [DOI] [Google Scholar]
- Moriguchi T.; Higashi M.; Yakeya D.; Jalli V.; Tsuge A.; Okauchi T.; Nagamatsu S.; Takashima W. Synthesis, Characterization and Air Stable Semiconductor Properties of Thiophene-Condensed Pyrene Derivatives. J. Mol. Struct. 2017, 1127, 413–418. 10.1016/j.molstruc.2016.07.104. [DOI] [Google Scholar]
- Moriguchi T.; Yakeya D.; Tsuge A.; Jalli V. Synthesis of Three New Thiophene Condensed Pyrene Derivatives, Crystal Structure and Evaluation of Their Photophysical Properties. J. Mol. Struct. 2018, 1157, 348–354. 10.1016/j.molstruc.2017.12.081. [DOI] [Google Scholar]
- Uryu M.; Hiraga T.; Koga Y.; Saito Y.; Murakami K.; Itami K. Synthesis of Polybenzoacenes: Annulative Dimerization of Phenylene Triflate by Twofold C-H Activation. Angew. Chem., Int. Ed. 2020, 59, 6551–6554. 10.1002/anie.202001211. [DOI] [PubMed] [Google Scholar]
- Lv B.; Shen X.; Xiao J.; Duan J.; Wang X.; Yi Y. Synthesis, Single Crystal, and Physical Properties of Asymmetrical Thiophene/Selenophene-Fused Twistacenes. Chem. - Asian J. 2015, 10, 2677–2682. 10.1002/asia.201500733. [DOI] [PubMed] [Google Scholar]
- Tang S.; Zhang L.; Ruan H.; Zhao Y.; Wang X. A Magnetically Robust Triplet Ground State Sulfur-Hydrocarbon Diradical Dication. J. Am. Chem. Soc. 2020, 142, 7340–7344. 10.1021/jacs.0c02141. [DOI] [PubMed] [Google Scholar]
- Shi X.; Lee S.; Son M.; Zheng B.; Chang J.; Jing L.; Huang K.-W.; Kim D.; Chi C. Pro-Aromatic Bisphenaleno-Thieno[3,2-b]Thiophene versus Anti-Aromatic Bisindeno-Thieno[3,2-b]Thiophene: Different Ground-State Properties and Applications in Field-Effect Transistors. Chem. Commun. 2015, 51, 13178–13180. 10.1039/C5CC04243G. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Huang K.; Cai S.; Liu C.; Jiao X.; He S.; Zhao L.; Zeng X. Synthesis of Near-Infrared Fluorescent Rhodamines via an SNArH Reaction and Their Biological Applications. Org. Biomol. Chem. 2018, 16, 7163–7169. 10.1039/C8OB01701H. [DOI] [PubMed] [Google Scholar]
- El-Shaieb K. M.; Mohamed A. H.; Abdel-Latif F. F. Investigation of the Reactivity of 4-Amino-5-Hydrazineyl-4H-1,2, 4-Triazole-3-Thiol towards Some Selected Carbonyl Compounds: Synthesis of Novel Triazolotriazine-, Triazolotetrazine-, and Triazolopthalazine Derivatives. Z. Naturforsch., B: J. Chem. Sci. 2019, 74, 847–855. 10.1515/znb-2019-0140. [DOI] [Google Scholar]
- Shiina Y.; Karasaki H.; Mori S.; Kobayashi N.; Furuta H.; Shimizu S. A Novel Isoindole-Containing Polyaromatic Hydrocarbon Unexpectedly Formed during the Synthesis of Meso-2,6-Dichlorophenyl-Substituted Tribenzosubporphyrin. J. Porphyrins Phthalocyanines 2016, 20, 1049–1054. 10.1142/S1088424616500541. [DOI] [Google Scholar]
- Peng S.; Liu S.; Zhang S.; Cao S.; Sun J. Synthesis of Polyheteroaromatic Compounds via Rhodium-Catalyzed Multiple C-H Bond Activation and Oxidative Annulation. Org. Lett. 2015, 17, 5032–5035. 10.1021/acs.orglett.5b02510. [DOI] [PubMed] [Google Scholar]
- Chow C. H. E.; Han Y.; Phan H.; Wu J. Nitrogen-Doped Heptazethrene and Octazethrene Diradicaloids. Chem. Commun. 2019, 55, 9100–9103. 10.1039/C9CC04564C. [DOI] [PubMed] [Google Scholar]
- Arikawa S.; Shimizu A.; Shintani R. Azoniadibenzo[a, j]Phenalenide: A Polycyclic Zwitterion with Singlet Biradical Character. Angew. Chem., Int. Ed. 2019, 58, 6415–6419. 10.1002/anie.201902006. [DOI] [PubMed] [Google Scholar]
- Yan C.; Shang R.; Nakamoto M.; Yamamoto Y.; Adachi Y. The Substituent Effect of Bridged Triarylamine Helicenes on Light-Emitting and Charge Transfer Properties. Chem. Lett. 2020, 49, 457–460. 10.1246/cl.200089. [DOI] [Google Scholar]
- Min H.; Park I. S.; Yasuda T. Cis-Quinacridone-Based Delayed Fluorescence Emitters: Seemingly Old but Renewed Functional Luminogens. Angew. Chem., Int. Ed. 2021, 60, 7643–7648. 10.1002/anie.202016914. [DOI] [PubMed] [Google Scholar]
- Wu H.; Zhang Z.; Ma N.; Liu Q.; Liu T.; Zhang G. Synthesis of Acridines from o -Aminoaryl Ketones and Arylboronic Acids by Copper Trifluoroacetate-Mediated Relay Reactions. J. Org. Chem. 2018, 83, 12880–12886. 10.1021/acs.joc.8b01828. [DOI] [PubMed] [Google Scholar]
- Raju S.; Annamalai P.; Chen P.-L.; Liu Y.-H.; Chuang S.-C. Iptycenes with an Acridinone Motif Developed through [4 + 2] Cycloaddition of Tethered Naphthalene and Iminoquinone via a Radical Reaction. Chem. Commun. 2017, 53, 6247–6250. 10.1039/C7CC03030D. [DOI] [PubMed] [Google Scholar]
- Prinzisky C.; Meyenburg I.; Jacob A.; Heidelmeier B.; Schröder F.; Heimbrodt W.; Sundermeyer J. Optical and Electrochemical Properties of Anthraquinone Imine Based Dyes for Dye-Sensitized Solar Cells. Eur. J. Org. Chem. 2016, 2016, 756–767. 10.1002/ejoc.201501309. [DOI] [Google Scholar]
- Hertz V. M.; Ando N.; Hirai M.; Bolte M.; Lerner H.-W.; Yamaguchi S.; Wagner M. Steric Shielding vs Structural Constraint in a Boron-Containing Polycyclic Aromatic Hydrocarbon. Organometallics 2017, 36, 2512–2519. 10.1021/acs.organomet.6b00800. [DOI] [Google Scholar]
- Hertz V. M.; Massoth J. G.; Bolte M.; Lerner H.-W.; Wagner M. En Route to Stimuli-Responsive Boron-, Nitrogen-, and Sulfur-Doped Polycyclic Aromatic Hydrocarbons. Chem. - Eur. J. 2016, 22, 13181–13188. 10.1002/chem.201602406. [DOI] [PubMed] [Google Scholar]
- Zhang J.-J.; Tang M.-C.; Fu Y.; Low K.-H.; Ma J.; Yang L.; Weigand J. J.; Liu J.; Yam V. W.-W.; Feng X. One-Pot Synthesis of Boron-Doped Polycyclic Aromatic Hydrocarbons via 1,4-Boron Migration. Angew. Chem., Int. Ed. 2021, 60, 2833–2838. 10.1002/anie.202011237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukeman M.; Simon H.; Wan P.; Wang Y.-H. Photocyclization and Photoaddition Reactions of Arylphenols via Intermediate Quinone Methides. J. Org. Chem. 2015, 80, 11281–11293. 10.1021/acs.joc.5b01580. [DOI] [PubMed] [Google Scholar]
- Yang S.; Cheng R.; Zhang M.; Bin Z.; You J. Rh/Ag-Mediated Peri -Selective Heteroarylation/Single Electron Transfer Annulation Cascade of 1-(Methylthio)Naphthalenes and Analogues: Road Less Traveled to Benzo[ de ]Thioacenes. ACS Catal. 2019, 9, 6188–6193. 10.1021/acscatal.9b01426. [DOI] [Google Scholar]
- Zhang L.; Huang Z.; Dai D.; Xiao Y.; Lei K.; Tan S.; Cheng J.; Xu Y.; Liu J.; Qian X. Thio-Bisnaphthalimides as Heavy-Atom-Free Photosensitizers with Efficient Singlet Oxygen Generation and Large Stokes Shifts: Synthesis and Properties. Org. Lett. 2016, 18, 5664–5667. 10.1021/acs.orglett.6b02902. [DOI] [PubMed] [Google Scholar]
- Chow C. H. E.; Phan H.; Zhang X.; Wu J. Sulfur-Doped (Dibenzo)Heptazethrene and (Dibenzo)Octazethrene Diradicaloids. J. Org. Chem. 2020, 85, 234–240. 10.1021/acs.joc.9b02796. [DOI] [PubMed] [Google Scholar]
- Plodek A.; König M.; Bracher F. Synthesis of the Azaoxoaporphine Alkaloid Sampangine and Ascididemin-Type Pyridoacridines through TMPMgCl·LiCl-Mediated Ring Closure. Eur. J. Org. Chem. 2015, 2015, 1302–1308. 10.1002/ejoc.201403502. [DOI] [Google Scholar]
- Melzer B. C.; Plodek A.; Bracher F. Functionalization of 4-Bromobenzo[c][2,7]Naphthyridine via Regioselective Direct Ring Metalation. A Novel Approach to Analogues of Pyridoacridine Alkaloids. Beilstein J. Org. Chem. 2019, 15, 2304–2310. 10.3762/bjoc.15.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil I. M.; Barker D.; Copp B. R. Bioinspired Syntheses of the Pyridoacridine Marine Alkaloids Demethyldeoxyamphimedine, Deoxyamphimedine, and Amphimedine. J. Org. Chem. 2016, 81, 282–289. 10.1021/acs.joc.5b02312. [DOI] [PubMed] [Google Scholar]
- Delgado I. H.; Pascal S.; Wallabregue A.; Duwald R.; Besnard C.; Guénée L.; Nançoz C.; Vauthey E.; Tovar R. C.; Lunkley J. L.; et al. Functionalized Cationic [4]Helicenes with Unique Tuning of Absorption, Fluorescence and Chiroptical Properties up to the Far-Red Range. Chem. Sci. 2016, 7, 4685–4693. 10.1039/C6SC00614K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal S.; Besnard C.; Zinna F.; Di Bari L.; Le Guennic B.; Jacquemin D.; Lacour J. Zwitterionic [4]Helicene: A Water-Soluble and Reversible PH-Triggered ECD/CPL Chiroptical Switch in the UV and Red Spectral Regions. Org. Biomol. Chem. 2016, 14, 4590–4594. 10.1039/C6OB00752J. [DOI] [PubMed] [Google Scholar]
- Labrador G. M.; Besnard C.; Bürgi T.; Poblador-Bahamonde A. I.; Bosson J.; Lacour J. Stereochemical Significance of O to N Atom Interchanges within Cationic Helicenes: Experimental and Computational Evidence of near Racemization to Remarkable Enantiospecificity. Chem. Sci. 2019, 10, 7059–7067. 10.1039/C9SC02127B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson J.; Labrador G. M.; Besnard C.; Jacquemin D.; Lacour J. Chiral Near-Infrared Fluorophores by Self-Promoted Oxidative Coupling of Cationic Helicenes with Amines/Enamines. Angew. Chem., Int. Ed. 2021, 60, 8733–8738. 10.1002/anie.202016643. [DOI] [PubMed] [Google Scholar]
- Yamagami A.; Ishimura H.; Katori A.; Kuramochi K.; Tsubaki K. Syntheses and Properties of the V-Shaped Dimeric Xanthene Dyes. Org. Biomol. Chem. 2016, 14, 10963–10972. 10.1039/C6OB01967F. [DOI] [PubMed] [Google Scholar]
- Yamagami A.; Kawano K.; Futaki S.; Kuramochi K.; Tsubaki K. Syntheses and Properties of Second-Generation V-Shaped Xanthene Dyes with Piperidino Groups. Tetrahedron 2017, 73, 7061–7066. 10.1016/j.tet.2017.10.064. [DOI] [Google Scholar]
- Mokhtari Brikci-Nigassa N.; Nauton L.; Moreau P.; Mongin O.; Duval R. E.; Picot L.; Thiéry V.; Souab M.; Baratte B.; Ruchaud S.; et al. Functionalization of 9-Thioxanthone at the 1-Position: From Arylamino Derivatives to [1]Benzo(Thio)Pyrano[4,3,2-de]Benzothieno[2,3-b]Quinolines of Biological Interest. Bioorg. Chem. 2020, 94, 103347. 10.1016/j.bioorg.2019.103347. [DOI] [PubMed] [Google Scholar]
- Bornadiego A.; Díaz J.; Marcos C. F. Tandem Synthesis of 4-Aminoxanthones Is Controlled by a Water-Assisted Tautomerization: A General Straightforward Reaction. Org. Biomol. Chem. 2019, 17, 1410–1422. 10.1039/C8OB02527D. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu J.; Yin J.; Zhang Y.; Han W.; Lan J.; Wu D.; Bin Z.; You J. Double Ortho-C-H Activation/Annulation of Benzamides with Aryl Alkynes: A Route to Double-Helical Polycyclic Heteroaromatics. J. Org. Chem. 2019, 84, 15697–15705. 10.1021/acs.joc.9b02339. [DOI] [PubMed] [Google Scholar]
- Numano M.; Nagami N.; Nakatsuka S.; Katayama T.; Nakajima K.; Tatsumi S.; Yasuda N.; Hatakeyama T. Synthesis of Boronate-Based Benzo[ Fg ]Tetracene and Benzo[ Hi ]Hexacene via Demethylative Direct Borylation. Chem. - Eur. J. 2016, 22, 11574–11577. 10.1002/chem.201602753. [DOI] [PubMed] [Google Scholar]
- Fingerle M.; Maichle-Mössmer C.; Schundelmeier S.; Speiser B.; Bettinger H. F. Synthesis and Characterization of a Boron-Nitrogen-Boron Zigzag-Edged Benzo[Fg]Tetracene Motif. Org. Lett. 2017, 19, 4428–4431. 10.1021/acs.orglett.7b01873. [DOI] [PubMed] [Google Scholar]
- Wei H.; Liu Y.; Gopalakrishna T. Y.; Phan H.; Huang X.; Bao L.; Guo J.; Zhou J.; Luo S.; Wu J.; et al. B-N-B Bond Embedded Phenalenyl and Its Anions. J. Am. Chem. Soc. 2017, 139, 15760–15767. 10.1021/jacs.7b07375. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhang F.; Schellhammer K. S.; Machata P.; Ortmann F.; Cuniberti G.; Fu Y.; Hunger J.; Tang R.; Popov A. A.; et al. Synthesis of NBN-Type Zigzag-Edged Polycyclic Aromatic Hydrocarbons: 1,9-Diaza-9a-Boraphenalene as a Structural Motif. J. Am. Chem. Soc. 2016, 138, 11606–11615. 10.1021/jacs.6b04445. [DOI] [PubMed] [Google Scholar]
- Fingerle M.; Stocker S.; Bettinger H. F. New Synthesis of a Dibenzoperylene Motif Featuring a Doubly Boron-Nitrogen-Doped Bay Region. Synthesis 2019, 51, 4147–4152. 10.1055/s-0039-1690687. [DOI] [Google Scholar]
- Tasseroul J.; Lorenzo-Garcia M. M.; Dosso J.; Simon F.; Velari S.; De Vita A.; Tecilla P.; Bonifazi D. Probing Peripheral H-Bonding Functionalities in BN-Doped Polycyclic Aromatic Hydrocarbons. J. Org. Chem. 2020, 85, 3454–3464. 10.1021/acs.joc.9b03202. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Shiren K.; Nakajima K.; Nomura S.; Nakatsuka S.; Kinoshita K.; Ni J.; Ono Y.; Ikuta T. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect. Adv. Mater. 2016, 28, 2777–2781. 10.1002/adma.201505491. [DOI] [PubMed] [Google Scholar]
- a Kondo Y.; Yoshiura K.; Kitera S.; Nishi H.; Oda S.; Gotoh H.; Sasada Y.; Yanai M.; Hatakeyama T. Narrowband Deep-Blue Organic Light-Emitting Diode Featuring an Organoboron-Based Emitter. Nature Photonics 2019, 13 (10), 678–682. 10.1038/s41566-019-0476-5. [DOI] [Google Scholar]; b Yang M.; Park I. S.; Yasuda T. Full-Color, Narrowband, and High-Efficiency Electroluminescence from Boron and Carbazole Embedded Polycyclic Heteroaromatics. J. Am. Chem. Soc. 2020, 142, 19468–19472. 10.1021/jacs.0c10081. [DOI] [PubMed] [Google Scholar]
- a Oda S.; Kawakami B.; Kawasumi R.; Okita R.; Hatakeyama T. Multiple Resonance Effect-Induced Sky-Blue Thermally Activated Delayed Fluorescence with a Narrow Emission Band. Org. Lett. 2019, 21 (23), 9311–9314. 10.1021/acs.orglett.9b03342. [DOI] [PubMed] [Google Scholar]; b Knöller J. A.; Meng G.; Wang X.; Hall D.; Pershin A.; Beljonne D.; Olivier Y.; Laschat S.; Zysman-Colman E.; Wang S. Intramolecular Borylation via Sequential B-Mes Bond Cleavage for the Divergent Synthesis of B,N,B-Doped Benzo[4]Helicenes. Angew. Chem., Int. Ed. 2020, 59, 3156–3160. 10.1002/anie.201912340. [DOI] [PubMed] [Google Scholar]
- Nishimura H.; Ishida N.; Shimazaki A.; Wakamiya A.; Saeki A.; Scott L. T.; Murata Y. Hole-Transporting Materials with a Two-Dimensionally Expanded π-System around an Azulene Core for Efficient Perovskite Solar Cells. J. Am. Chem. Soc. 2015, 137, 15656–15659. 10.1021/jacs.5b11008. [DOI] [PubMed] [Google Scholar]
- Nishimura H.; Hasegawa Y.; Wakamiya A.; Murata Y. Development of Transparent Organic Hole-Transporting Materials Using Partially Oxygen-Bridged Triphenylamine Skeletons. Chem. Lett. 2017, 46, 817–820. 10.1246/cl.170164. [DOI] [Google Scholar]
- Nishimura H.; Fukushima T.; Wakamiya A.; Murata Y.; Kaji H. The Influence of Quasiplanar Structures of Partially Oxygen-Bridged Triphenylamine Dimers on the Properties of Their Bulk Films. Bull. Chem. Soc. Jpn. 2016, 89, 726–732. 10.1246/bcsj.20160031. [DOI] [Google Scholar]
- Li Q.; Shi C.; Huang M.; Wei X.; Yan H.; Yang C.; Yuan A. B- and N-Embedded Color-Tunable Phosphorescent Iridium Complexes and B-N Lewis Adducts with Intriguing Structural and Optical Changes. Chem. Sci. 2019, 10, 3257–3263. 10.1039/C8SC04252G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.; Li F.; Li Q.; Zhao W.; Cao Y.; Zhao Q.; Yuan A. B- and N-Embedded π-Conjugation Units Tuning Intermolecular Interactions and Optical Properties of Platinum(II) Complexes. Inorg. Chem. 2021, 60, 525–534. 10.1021/acs.inorgchem.0c03078. [DOI] [PubMed] [Google Scholar]
- Buyukcakir O.; Yuksel R.; Jiang Y.; Lee S. H.; Seong W. K.; Chen X.; Ruoff R. S. Synthesis of Porous Covalent Quinazoline Networks (CQNs) and Their Gas Sorption Properties. Angew. Chem., Int. Ed. 2019, 58, 872–876. 10.1002/anie.201813075. [DOI] [PubMed] [Google Scholar]
- Shensky W. M.; Ferry M. J.; O’Donnell R. M.; Ensley T. R.; Shi J. Nonlinear Optical Characterization of Multinuclear Iridium Compounds Containing Tricycloquinazoline. Appl. Opt. 2017, 56, B179–B183. 10.1364/AO.56.00B179. [DOI] [PubMed] [Google Scholar]
- Boldt S.; Parpart S.; Villinger A.; Ehlers P.; Langer P. Synthesis and Properties of Aza-Ullazines. Angew. Chem., Int. Ed. 2017, 56, 4575–4578. 10.1002/anie.201701347. [DOI] [PubMed] [Google Scholar]
- Richter M.; Schellhammer K. S.; Machata P.; Cuniberti G.; Popov A.; Ortmann F.; Berger R.; Müllen K.; Feng X. Polycyclic Heteroaromatic Hydrocarbons Containing a Benzoisoindole Core. Org. Chem. Front. 2017, 4, 847–852. 10.1039/C7QO00180K. [DOI] [Google Scholar]
- Richter M.; Fu Y.; Dmitrieva E.; Weigand J. J.; Popov A.; Berger R.; Liu J.; Feng X. Polycyclic Aromatic Hydrocarbons Containing A Pyrrolopyridazine Core. ChemPlusChem 2019, 84, 613–618. 10.1002/cplu.201900031. [DOI] [PubMed] [Google Scholar]
- Richter M.; Hahn S.; Dmitrieva E.; Rominger F.; Popov A.; Bunz U. H. F.; Feng X.; Berger R. Helical Ullazine-Quinoxaline-Based Polycyclic Aromatic Hydrocarbons. Chem. - Eur. J. 2019, 25, 1345–1352. 10.1002/chem.201804751. [DOI] [PubMed] [Google Scholar]
- Li Q.-Q.; Ochiai K.; Lee C.-A.; Ito S. Synthesis of π-Extended Imidazoles by 1,3-Dipolar Cycloaddition of Polycyclic Aromatic Azomethine Ylides with Nitriles. Org. Lett. 2020, 22, 6132–6137. 10.1021/acs.orglett.0c02203. [DOI] [PubMed] [Google Scholar]
- Riss A.; Richter M.; Paz A. P.; Wang X.-Y.; Raju R.; He Y.; Ducke J.; Corral E.; Wuttke M.; Seufert K.; et al. Polycyclic Aromatic Chains on Metals and Insulating Layers by Repetitive [3 + 2] Cycloadditions. Nat. Commun. 2020, 11, 1–8. 10.1038/s41467-020-15210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D.; Aumaitre C.; Morin J.-F. Photochemical Synthesis of π-Extended Ullazine Derivatives as New Electron Donors for Efficient Conjugated D-A Polymers. J. Mater. Chem. C 2019, 7, 3015–3024. 10.1039/C8TC05288C. [DOI] [Google Scholar]
- Skabeev A.; Zschieschang U.; Zagranyarski Y.; Klauk H.; Müllen K.; Li C. Carbonyl-Functionalized Cyclazines as Colorants and Air-Stable n-Type Semiconductors. Org. Lett. 2018, 20, 1409–1412. 10.1021/acs.orglett.8b00183. [DOI] [PubMed] [Google Scholar]
- Skidin D.; Eisenhut F.; Richter M.; Nikipar S.; Krüger J.; Ryndyk D. A.; Berger R.; Cuniberti G.; Feng X.; Moresco F. On-Surface Synthesis of Nitrogen-Doped Nanographenes with 5–7 Membered Rings. Chem. Commun. 2019, 55, 4731–4734. 10.1039/C9CC00276F. [DOI] [PubMed] [Google Scholar]
- Krzeszewski M.; Kodama T.; Espinoza E. M.; Vullev V. I.; Kubo T.; Gryko D. T. Nonplanar Butterfly-Shaped π-Expanded Pyrrolopyrroles. Chem. - Eur. J. 2016, 22, 16478–16488. 10.1002/chem.201603282. [DOI] [PubMed] [Google Scholar]
- Sheng W.; Zheng Y.-Q.; Wu Q.; Chen K.; Li M.; Jiao L.; Hao E.; Wang J.-Y.; Pei J. Synthesis, Characterization, and Tunable Semiconducting Properties of Aza-BODIPY Derived Polycyclic Aromatic Dyes. Sci. China: Chem. 2020, 63, 1240–1245. 10.1007/s11426-020-9807-7. [DOI] [Google Scholar]
- Tokimaru Y.; Ito S.; Nozaki K. Synthesis of Pyrrole-Fused Corannulenes: 1,3-Dipolar Cycloaddition of Azomethine Ylides to Corannulene. Angew. Chem., Int. Ed. 2017, 56, 15560–15564. 10.1002/anie.201707087. [DOI] [PubMed] [Google Scholar]
- Tokimaru Y.; Ito S.; Nozaki K. A Hybrid of Corannulene and Azacorannulene: Synthesis of a Highly Curved Nitrogen-Containing Buckybowl. Angew. Chem., Int. Ed. 2018, 57, 9818–9822. 10.1002/anie.201805678. [DOI] [PubMed] [Google Scholar]
- Kawahara K. P.; Matsuoka W.; Ito H.; Itami K. Synthesis of Nitrogen-Containing Polyaromatics by Aza-Annulative π-Extension of Unfunctionalized Aromatics. Angew. Chem., Int. Ed. 2020, 59, 6383–6388. 10.1002/anie.201913394. [DOI] [PubMed] [Google Scholar]
- Saha M.; Bao Y.-H.; Zhou C. A Diindole-Fused Corannulene Imide Derivative: Synthesis and Properties. Chem. Lett. 2018, 47, 1383–1386. 10.1246/cl.180680. [DOI] [Google Scholar]
- Rajeshkumar V.; Stuparu M. C. A Photochemical Approach to Aromatic Extension of the Corannulene Nucleus. Chem. Commun. 2016, 52, 9957–9960. 10.1039/C6CC04910A. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Csókás D.; Budanović M.; Webster R. D.; Pápai I.; Stuparu M. C. Synthesis of Azahelicenes through Mallory Reaction of Imine Precursors: Corannulene Substrates Provide an Exception to the Rule in Oxidative Photocyclizations of Diarylethenes. Chem. Sci. 2021, 12, 3977–3983. 10.1039/D0SC06730J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barát V.; Budanović M.; Halilovic D.; Huh J.; Webster R. D.; Mahadevegowda S. H.; Stuparu M. C. A General Approach to Non-Fullerene Electron Acceptors Based on the Corannulene Motif. Chem. Commun. 2019, 55, 3113–3116. 10.1039/C9CC00327D. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Allemann O.; Balaban T. S.; Vanthuyne N.; Linden A.; Baldridge K. K.; Siegel J. S. Chiral Atropisomeric Indenocorannulene Bowls: Critique of the Cahn-Ingold-Prelog Conception of Molecular Chirality. Angew. Chem., Int. Ed. 2018, 57, 6470–6474. 10.1002/anie.201801325. [DOI] [PubMed] [Google Scholar]
- Tian X.; Roch L. M.; Vanthuyne N.; Xu J.; Baldridge K. K.; Siegel J. S. Azaindenocorannulenes: Synthesis, Properties, and Chirality. Org. Lett. 2019, 21, 3510–3513. 10.1021/acs.orglett.9b00718. [DOI] [PubMed] [Google Scholar]
- Gu X.; Li H.; Shan B.; Liu Z.; Miao Q. Synthesis, Structure, and Properties of Tetrabenzo[7]Circulene. Org. Lett. 2017, 19, 2246–2249. 10.1021/acs.orglett.7b00714. [DOI] [PubMed] [Google Scholar]
- Tsefrikas V. M.; Greene A. K.; Scott L. T. 5-Azadibenzo[a,g]Corannulene. Org. Chem. Front. 2017, 4, 688–698. 10.1039/C6QO00831C. [DOI] [Google Scholar]
- Nakatsuka S.; Yasuda N.; Hatakeyama T. Four-Step Synthesis of B2N2-Embedded Corannulene. J. Am. Chem. Soc. 2018, 140, 13562–13565. 10.1021/jacs.8b08197. [DOI] [PubMed] [Google Scholar]
- Nagano T.; Nakamura K.; Tokimaru Y.; Ito S.; Miyajima D.; Aida T.; Nozaki K. Functionalization of Azapentabenzocorannulenes by Fivefold C-H Borylation and Cross-Coupling Arylation: Application to Columnar Liquid-Crystalline Materials. Chem. - Eur. J. 2018, 24, 14075–14078. 10.1002/chem.201803676. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Wei Z.; Tokimaru Y.; Ito S.; Nozaki K.; Petrukhina M. A. Stepwise Reduction of Azapentabenzocorannulene. Angew. Chem., Int. Ed. 2019, 58, 12107–12111. 10.1002/anie.201906748. [DOI] [PubMed] [Google Scholar]
- Yokoi H.; Hiroto S.; Sakamaki D.; Seki S.; Shinokubo H. Supramolecular Assemblies of a Nitrogen-Embedded Buckybowl Dimer with C60. Chem. Sci. 2018, 9, 819–824. 10.1039/C7SC04453D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M.; Hiroto S.; Yokoi H.; Lee S.; Kim D.; Shinokubo H. Azabuckybowl-Based Molecular Tweezers as C 60 and C 70 Receptors. J. Am. Chem. Soc. 2018, 140, 6336–6342. 10.1021/jacs.8b02327. [DOI] [PubMed] [Google Scholar]
- Yokoi H.; Hiroto S.; Shinokubo H. Reversible σ-Bond Formation in Bowl-Shaped π-Radical Cations: The Effects of Curved and Planar Structures. J. Am. Chem. Soc. 2018, 140, 4649–4655. 10.1021/jacs.8b00798. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhou D.; Zhang T.; Liu B.; Xu B. Iodine-Doped Sumanene and Its Application for the Synthesis of Chalcogenasumanenes and Silasumanenes. Chem. Commun. 2017, 53, 10279–10282. 10.1039/C7CC05885C. [DOI] [PubMed] [Google Scholar]
- Jiang M.; Guo J.; Liu B.; Tan Q.; Xu B. Synthesis of Tellurium-Containing π-Extended Aromatics with Room-Temperature Phosphorescence. Org. Lett. 2019, 21, 8328–8333. 10.1021/acs.orglett.9b03106. [DOI] [PubMed] [Google Scholar]
- Zhou D.; Gao Y.; Liu B.; Tan Q.; Xu B. Synthesis of Silicon and Germanium-Containing Heterosumanenes via Rhodium-Catalyzed Cyclodehydrogenation of Silicon/Germanium-Hydrogen and Carbon-Hydrogen Bonds. Org. Lett. 2017, 19, 4628–4631. 10.1021/acs.orglett.7b02254. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Deng G.; Li H.; Liu B.; Tan Q.; Xu B. Cyclization of 2-Biphenylthiols to Dibenzothiophenes under PdCl2/DMSO Catalysis. Org. Lett. 2018, 20, 5439–5443. 10.1021/acs.orglett.8b02347. [DOI] [PubMed] [Google Scholar]
- Liu Y.-M.; Xia D.; Li B.-W.; Zhang Q.-Y.; Sakurai T.; Tan Y.-Z.; Seki S.; Xie S.-Y.; Zheng L.-S. Functional Sulfur-Doped Buckybowls and Their Concave-Convex Supramolecular Assembly with Fullerenes. Angew. Chem., Int. Ed. 2016, 55, 13047–13051. 10.1002/anie.201606383. [DOI] [PubMed] [Google Scholar]
- Furukawa S.; Wu J.; Koyama M.; Hayashi K.; Hoshino N.; Takeda T.; Suzuki Y.; Kawamata J.; Saito M.; Akutagawa T. Ferroelectric Columnar Assemblies from the Bowl-to-Bowl Inversion of Aromatic Cores. Nat. Commun. 2021, 12, 768. 10.1038/s41467-021-21019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S.; Suda Y.; Kobayashi J.; Kawashima T.; Tada T.; Fujii S.; Kiguchi M.; Saito M. Triphosphasumanene Trisulfide: High Out-of-Plane Anisotropy and Janus-Type π-Surfaces. J. Am. Chem. Soc. 2017, 139, 5787–5792. 10.1021/jacs.6b12119. [DOI] [PubMed] [Google Scholar]
- Wang S.; Yan C.; Shang J.; Wang W.; Yuan C.; Zhang H.-L.; Shao X. Doping Sumanene with Both Chalcogens and Phosphorus(V): One-Step Synthesis, Coordination, and Selective Response Toward AgI. Angew. Chem., Int. Ed. 2019, 58, 3819–3823. 10.1002/anie.201813070. [DOI] [PubMed] [Google Scholar]
- Furukawa S.; Hayashi K.; Yamagishi K.; Saito M. Synthesis and Properties of Spiro-Type Heterasumanenes Containing Group 14 Elements as Bridging Atoms. Mater. Chem. Front. 2018, 2, 929–934. 10.1039/C7QM00590C. [DOI] [Google Scholar]
- Hou X.; Zhu Y.; Qin Y.; Chen L.; Li X.; Zhang H.-L.; Xu W.; Zhu D.; Shao X. Tris(S,S-Dioxide)-Trithiasumanene: Strong Fluorescence and Cocrystal with 1,2,6,7,10,11-Hexabutoxytriphenylene. Chem. Commun. 2017, 53, 1546–1549. 10.1039/C6CC09531C. [DOI] [PubMed] [Google Scholar]
- Wang S.; Li X.; Hou X.; Sun Y.; Shao X. Tritellurasumanene: Ultrasound Assisted One-Pot Synthesis and Extended Valence Adducts with Bromine. Chem. Commun. 2016, 52, 14486–14489. 10.1039/C6CC08170C. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Li X.; Sun C.; Shen H.; Hou X.; Lin D.; Zhang H.-L.; Di C.; Zhu D.; Shao X. Trichalcogenasumanene Ortho-Quinones: Synthesis, Properties, and Transformation into Various Heteropolycycles. Angew. Chem., Int. Ed. 2017, 56, 13470–13474. 10.1002/anie.201707397. [DOI] [PubMed] [Google Scholar]
- Wang S.; Shang J.; Yan C.; Wang W.; Yuan C.; Zhang H.-L.; Shao X. Trichalcogenasumanenes Containing Various Chalcogen Atoms: Synthesis, Structure, Properties, and Chemical Reactivity. Org. Chem. Front. 2019, 6, 263–272. 10.1039/C8QO01220B. [DOI] [Google Scholar]
- Wang Y.; Deng J.; Chen J.; Cao F.; Hou Y.; Yang Y.; Deng X.; Yang J.; Wu L.; Shao X.; et al. Dechalcogenization of Aryl Dichalcogenides to Synthesize Aryl Chalcogenides via Copper Catalysis. ACS Catal. 2020, 10, 2707–2712. 10.1021/acscatal.9b04931. [DOI] [Google Scholar]
- Geng R.; Hou X.; Sun Y.; Yan C.; Wu Y.; Zhang H.-L.; Shao X. Driving π-Plane to π-Bowl through Lateral Coordination at Room Temperature. Mater. Chem. Front. 2018, 2, 1456–1461. 10.1039/C8QM00168E. [DOI] [Google Scholar]
- Hou X.; Li X.; Sun C.; Chen L.; Sun Y.; Liu Z.; Zhang H.-L.; Shao X. Dissecting Trichalcogenasumanenes: π-Bowl to Planar, Invertible Curvature, and Chiral Polycycles. Chem. - Eur. J. 2017, 23, 14375–14383. 10.1002/chem.201703469. [DOI] [PubMed] [Google Scholar]
- Hou X.; Sun J.; Liu Z.; Yan C.; Song W.; Zhang H.-L.; Zhou S.; Shao X. Opening Two Benzene Rings on Trichalcogenasumanenes toward High Performance Organic Optical-Limiting Materials. Chem. Commun. 2018, 54, 10981–10984. 10.1039/C8CC05480K. [DOI] [PubMed] [Google Scholar]
- Sun J.; Sun Y.; Yan C.; Lin D.; Xie Z.; Zhou S.; Yuan C.; Zhang H.-L.; Shao X. Remarkable Nonlinear Optical Response of Pyrazine-Fused Trichalcogenasumanenes and Their Application for Optical Power Limiting. J. Mater. Chem. C 2018, 6, 13114–13119. 10.1039/C8TC04778B. [DOI] [Google Scholar]
- Fu B.; Wang C.; Sun Y.; Yao J.; Wang Y.; Ge F.; Yang F.; Liu Z.; Dang Y.; Zhang X.; et al. A “Phase Separation” Molecular Design Strategy Towards Large-Area 2D Molecular Crystals. Adv. Mater. 2019, 31, 1901437. 10.1002/adma.201901437. [DOI] [PubMed] [Google Scholar]
- Liu L.; Yan C.; Li Y.; Liu Z.; Yuan C.; Zhang H.-L.; Shao X. Tetrathiafulvalene-Fused Heterabuckybowl: Protonation-Induced Electron Transfer and Self-Sensitized Photooxidation. Chem. - Eur. J. 2020, 26, 7083–7091. 10.1002/chem.201905732. [DOI] [PubMed] [Google Scholar]
- Kaewmati P.; Tan Q.; Higashibayashi S.; Yakiyama Y.; Sakurai H. Synthesis of Triaryltriazasumanenes. Chem. Lett. 2017, 46, 146–148. 10.1246/cl.160978. [DOI] [Google Scholar]
- Kaewmati P.; Yakiyama Y.; Ohtsu H.; Kawano M.; Haesuwannakij S.; Higashibayashi S.; Sakurai H. Tris(2-Hydroxyphenyl)Triazasumanene: Bowl-Shaped Excited-State Intramolecular Proton Transfer (ESIPT) Fluorophore Coupled with Aggregation-Induced Enhanced Emission (AIEE). Mater. Chem. Front. 2018, 2, 514–519. 10.1039/C7QM00530J. [DOI] [Google Scholar]
- Tan Q.; Kaewmati P.; Higashibayashi S.; Kawano M.; Yakiyama Y.; Sakurai H. Triazasumanene: An Isoelectronic Heteroanalogue of Sumanene. Bull. Chem. Soc. Jpn. 2018, 91, 531–537. 10.1246/bcsj.20170384. [DOI] [Google Scholar]
- Chen F.; Tanaka T.; Osuka A. Exploring the “Fold-in” Strategy toward the Construction of a Highly-Strained Triazasumanene Skeleton. Chem. Commun. 2017, 53, 2705–2708. 10.1039/C7CC00329C. [DOI] [PubMed] [Google Scholar]
- Upadhyay G. M.; Talele H. R.; Bedekar A. V. Synthesis and Photophysical Properties of Aza[n]Helicenes. J. Org. Chem. 2016, 81, 7751–7759. 10.1021/acs.joc.6b01395. [DOI] [PubMed] [Google Scholar]
- Nagata Y.; Kato S.; Miyake Y.; Shinokubo H. Synthesis of Tetraaza[8]Circulenes from Tetrathia[8]Circulenes through an SNAr-Based Process. Org. Lett. 2017, 19, 2718–2721. 10.1021/acs.orglett.7b01074. [DOI] [PubMed] [Google Scholar]
- Murase H.; Nagata Y.; Akahori S.; Shinokubo H.; Miyake Y. Aggregation-Induced Emission in Tetrathia[8]Circulene Octaoxides via Restriction of the Dynamic Motion of Their Negatively Curved π-Frameworks. Chem. - Asian J. 2020, 15, 3873–3877. 10.1002/asia.202001129. [DOI] [PubMed] [Google Scholar]
- Matsuo Y.; Tanaka T.; Osuka A. Highly Stable Radical Cations of N,N’-Diarylated Tetrabenzotetraaza[8]Circulene. Chem. - Eur. J. 2020, 26, 8144–8152. 10.1002/chem.202001934. [DOI] [PubMed] [Google Scholar]
- Matsuo Y.; Tanaka T.; Osuka A. Diazadimethano[8]Circulene: Synthesis, Structure, Properties, and Isolation of Stable Radical Cation. Chem. Lett. 2020, 49, 959–962. 10.1246/cl.200336. [DOI] [Google Scholar]
- Morimoto Y.; Chen F.; Matsuo Y.; Kise K.; Tanaka T.; Osuka A. Improved Synthesis of Ortho-Phenylene-Bridged Cyclic Tetrapyrroles and Oxidative Fusion Reactions Toward Substituted Tetraaza[8]Circulenes. Chem. - Asian J. 2021, 16, 648–655. 10.1002/asia.202001459. [DOI] [PubMed] [Google Scholar]
- Chen F.; Hong Y. S.; Kim D.; Tanaka T.; Osuka A. Sequential N-Alkylations of Tetrabenzotetraaza[8]Circulene as a Tool To Tune Its Optical Properties. ChemPlusChem 2017, 82, 1048–1051. 10.1002/cplu.201600537. [DOI] [PubMed] [Google Scholar]
- Serizawa Y.; Akahori S.; Kato S.; Sakai H.; Hasobe T.; Miyake Y.; Shinokubo H. Synthesis of Tetrasilatetrathia[8]Circulenes by a Fourfold Intramolecular Dehydrogenative Silylation of C-H Bonds. Chem. - Eur. J. 2017, 23, 6948–6952. 10.1002/chem.201700729. [DOI] [PubMed] [Google Scholar]
- Kato S.; Akahori S.; Serizawa Y.; Lin X.; Yamauchi M.; Yagai S.; Sakurai T.; Matsuda W.; Seki S.; Shinokubo H.; et al. Systematic Synthesis of Tetrathia[8]Circulenes: The Influence of Peripheral Substituents on the Structures and Properties in Solution and Solid States. J. Org. Chem. 2020, 85, 62–69. 10.1021/acs.joc.9b01655. [DOI] [PubMed] [Google Scholar]
- Akahori S.; Sasamori T.; Shinokubo H.; Miyake Y. Enthalpically and Entropically Favorable Self-Assembly: Synthesis of C4h-Symmetric Tetraazatetrathia[8]Circulenes by Regioselective Introduction of Pyridine Rings. Chem. - Eur. J. 2021, 27, 5675–5682. 10.1002/chem.202005077. [DOI] [PubMed] [Google Scholar]
- Akahori S.; Sakai H.; Hasobe T.; Shinokubo H.; Miyake Y. Synthesis and Photodynamics of Tetragermatetrathia[8]Circulene. Org. Lett. 2018, 20, 304–307. 10.1021/acs.orglett.7b03764. [DOI] [PubMed] [Google Scholar]
- Xiong X.; Deng C.-L.; Li Z.; Peng X.-S.; Wong H. N. C. Quasi-Planar Diazadithio and Diazodiseleno[8]Circulenes: Synthesis, Structures and Properties. Org. Chem. Front. 2017, 4, 682–687. 10.1039/C6QO00662K. [DOI] [Google Scholar]
- Lousen B.; Pedersen S. K.; Bols P.; Hansen K. H.; Pedersen M. R.; Hammerich O.; Bondarchuk S.; Minaev B.; Baryshnikov G. V.; Ågren H.; et al. Compressing a Non-Planar Aromatic Heterocyclic [7]Helicene to a Planar Hetero[8]Circulene. Chem. - Eur. J. 2020, 26, 4935–4940. 10.1002/chem.201905339. [DOI] [PubMed] [Google Scholar]
- Chen F.; Tanaka T.; Mori T.; Osuka A. Synthesis, Structures, and Optical Properties of Azahelicene Derivatives and Unexpected Formation of Azahepta[8]Circulenes. Chem. - Eur. J. 2018, 24, 7489–7497. 10.1002/chem.201800617. [DOI] [PubMed] [Google Scholar]
- Matsuo Y.; Chen F.; Kise K.; Tanaka T.; Osuka A. Facile Synthesis of Fluorescent Hetero[8]Circulene Analogues with Tunable Solubilities and Optical Properties. Chem. Sci. 2019, 10, 11006–11012. 10.1039/C9SC05087F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Liu X.; Li C.; Li L.; Song J.; Shi J.; Morton M.; Rajca S.; Rajca A.; Wang H. Thiophene-Based Double Helices: Syntheses, X-Ray Structures, and Chiroptical Properties. J. Am. Chem. Soc. 2016, 138, 10002–10010. 10.1021/jacs.6b05709. [DOI] [PubMed] [Google Scholar]
- Li B.; Zhang S.; Li L.; Ma Z.; Li C.; Xu L.; Wang H. All-Thiophene-Based Double Helix: Synthesis, Crystal Structure, Chiroptical Property and Arylation. ACS Omega 2018, 3, 16014–16020. 10.1021/acsomega.8b02492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Zhao S.; Li B.; Xu L.; Li C.; Shi J.; Wang H. From Saddle-Shaped to Planar Cyclic Oligothienoacenes: Stepped-Cyclization and Their Applications in OFETs. Org. Lett. 2018, 20, 2181–2185. 10.1021/acs.orglett.8b00482. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Li Q.-Q.; Krejčí O.; Foster A. S.; Sun K.; Kawai S.; Ito S. On-Surface Synthesis of a π-Extended Diaza[8]Circulene. J. Am. Chem. Soc. 2020, 142, 11363–11369. 10.1021/jacs.0c02534. [DOI] [PubMed] [Google Scholar]
- Pedersen S. K.; Eriksen K.; Karaush-Karmazin N. N.; Minaev B.; Ågren H.; Baryshnikov G. V.; Pittelkow M. Anti-Aromatic versus Induced Paratropicity: Synthesis and Interrogation of a Dihydro-Diazatrioxa[9]Circulene with a Proton Placed Directly above the Central Ring. Angew. Chem., Int. Ed. 2020, 59, 5144–5150. 10.1002/anie.201913552. [DOI] [PubMed] [Google Scholar]
- Pedersen S. K.; Eriksen K.; Ågren H.; Minaev B. F.; Karaush-Karmazin N. N.; Hammerich O.; Baryshnikov G. V.; Pittelkow M. A Fully Conjugated Planar Heterocyclic [9]Circulene. J. Am. Chem. Soc. 2020, 142, 14058–14063. 10.1021/jacs.0c05898. [DOI] [PubMed] [Google Scholar]
- Das S.; Herng T. S.; Zafra J. L.; Burrezo P. M.; Kitano M.; Ishida M.; Gopalakrishna T. Y.; Hu P.; Osuka A.; Casado J.; et al. Fully Fused Quinoidal/Aromatic Carbazole Macrocycles with Poly-Radical Characters. J. Am. Chem. Soc. 2016, 138, 7782–7790. 10.1021/jacs.6b04539. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Chu M.; Miao Q. From Phenanthrylene Butadiynylene Macrocycles to S-Heterocycloarenes. Org. Lett. 2018, 20, 4259–4262. 10.1021/acs.orglett.8b01668. [DOI] [PubMed] [Google Scholar]
- Yang L.; Zhang N.; Han Y.; Zou Y.; Qiao Y.; Chang D.; Zhao Y.; Lu X.; Wu J.; Liu Y. A Sulfur-Containing Hetero-Octulene: Synthesis, Host-Guest Properties, and Transistor Applications. Chem. Commun. 2020, 56, 9990–9993. 10.1039/D0CC04289G. [DOI] [PubMed] [Google Scholar]
- Lu X.; An D.; Han Y.; Zou Y.; Qiao Y.; Zhang N.; Chang D.; Wu J.; Liu Y. A Cyclopenta-Fused Dibenzo[b, d]Thiophene-Co-Phenanthrene Macrocyclic Tetraradicaloid. Chem. Sci. 2021, 12, 3952–3957. 10.1039/D0SC06185A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A. B.; Cortizo-Lacalle D.; Perez-Miqueo I.; Valenti G.; Boni A.; Plas J.; Strutyński K.; De Feyter S.; Paolucci F.; Montes M.; et al. Twisted Aromatic Frameworks: Readily Exfoliable and Solution-Processable Two-Dimensional Conjugated Microporous Polymers. Angew. Chem., Int. Ed. 2017, 56, 6946–6951. 10.1002/anie.201700271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood J.; Lee E. K.; Noh H.-J.; Ahmad I.; Seo J.-M.; Im Y.-K.; Jeon J.-P.; Kim S.-J.; Oh J. H.; Baek J.-B. Fused Aromatic Network with Exceptionally High Carrier Mobility. Adv. Mater. 2021, 33, 2004707. 10.1002/adma.202004707. [DOI] [PubMed] [Google Scholar]
- An C.; Zhou S.; Baumgarten M. Condensed Derivatives of Thiadiazoloquinoxaline as Strong Acceptors. Cryst. Growth Des. 2015, 15, 1934–1938. 10.1021/acs.cgd.5b00105. [DOI] [Google Scholar]
- Biegger P.; Schaffroth M.; Brödner K.; Tverskoy O.; Rominger F.; Bunz U. H. F. Bisalkynylated 3,6-Diiminocyclohexa-1,4-Diene-1,4-Diamine. Chem. Commun. 2015, 51, 14844–14847. 10.1039/C5CC05427C. [DOI] [PubMed] [Google Scholar]
- Ganschow M.; Koser S.; Hodecker M.; Rominger F.; Freudenberg J.; Dreuw A.; Bunz U. H. F. Azaacenes Bearing Five-Membered Rings. Chem. - Eur. J. 2018, 24, 13667–13675. 10.1002/chem.201802900. [DOI] [PubMed] [Google Scholar]
- Kothavale S.; Sekar N. Novel Pyrazino-Phenanthroline Based Rigid Donor-π-Acceptor Compounds: A Detail Study of Optical Properties, Acidochromism, Solvatochromism and Structure-Property Relationship. Dyes Pigm. 2017, 136, 31–45. 10.1016/j.dyepig.2016.08.032. [DOI] [Google Scholar]
- Zhang C.; Zeng C.-C.; Lai S.-H.; Xing D.-G.; Li W.; Han B.-J.; Liu Y.-J. Synthesis, Cytotoxicity in Vitro, Apoptosis, Cell Cycle Arrest and Comet Assay of Asymmetry Ruthenium(II) Complexes. Polyhedron 2016, 106, 115–124. 10.1016/j.poly.2015.12.058. [DOI] [Google Scholar]
- Shuler W. G.; Parvathaneni S. P.; Rodriguez J. B.; Lewis T. N.; Berges A. J.; Bardeen C. J.; Krische M. J. Synthesis and Photophysical Properties of Soluble N-Doped Rubicenes via Ruthenium-Catalyzed Transfer Hydrogenative Benzannulation. Chem. - Eur. J. 2021, 27, 4898–4902. 10.1002/chem.202100134. [DOI] [PubMed] [Google Scholar]
- Tkachenko Yu. N.; Popov L. D.; Pozharskii A. F.; Borodkin S. A.; Levchenkov S. I. Nitro Derivatives of Pyrrolo[3,2-d]Pyrimidine-2,4-Diones: Synthesis of Amines and New Polynuclear Heterocycles Based Thereon. Russ. J. Org. Chem. 2017, 53, 1564–1572. 10.1134/S1070428017100128. [DOI] [Google Scholar]
- Rodríguez-Sanz A.; Sánchez-Alonso P.; Bellón T.; Alajarín R.; Martínez-Cabeza V.; Selgas R.; Vaquero J. J.; Álvarez-Builla J. Synthesis and Biological Evaluation of Pyridazino[1’,6’:1,2]Pyrido[3,4-b]Indolinium and Pyridazino[1,6-a]Benzimidazolium Salts as Anti-Inflammatory Agents. Eur. J. Med. Chem. 2015, 93, 83–92. 10.1016/j.ejmech.2015.01.060. [DOI] [PubMed] [Google Scholar]
- Gang M.-Y.; Liu J.-Q.; Wang X.-S. CuI-Catalyzed Sonogashira Reaction for the Efficient Synthesis of 1H-Imidazo[2,1-a]Isoquinoline Derivatives. Tetrahedron 2017, 73, 4698–4705. 10.1016/j.tet.2017.06.043. [DOI] [Google Scholar]
- Filatova E. A.; Gulevskaya A. V.; Pozharskii A. F.; Suslonov V. V. The Sonogashira Coupling of 2- and 4-Ethynyl Derivatives of Proton Sponge with 1,8-Diiodonaphthalene: Novel Cascade Transformations into Naphtho[1,2-k]Fluoranthenes and Acenaphtho[1,2-b]Benzo[g]Indoles. Tetrahedron 2018, 74, 165–173. 10.1016/j.tet.2017.11.058. [DOI] [Google Scholar]
- Wu H.; Fang R.; Tao J.; Wang D.; Qiao X.; Yang X.; Hartl F.; Li H. Diacenaphthylene-Fused Benzo[1,2-b:4,5-B’]Dithiophenes: Polycyclic Heteroacenes Containing Full-Carbon Five-Membered Aromatic Rings. Chem. Commun. 2017, 53, 751–754. 10.1039/C6CC09184A. [DOI] [PubMed] [Google Scholar]
- Murata M.; Togo M.; Mishima D.; Harada A.; Muraoka M. Benzo- and Thieno-Annulated Tetracenes: A One-Pot Synthesis via Cross-Dehydrogenative Annulation. Org. Lett. 2020, 22, 4160–4163. 10.1021/acs.orglett.0c01244. [DOI] [PubMed] [Google Scholar]
- Chaolumen Murata M.; Wakamiya A.; Murata Y. Dithieno-Fused Polycyclic Aromatic Hydrocarbon with a Pyracylene Moiety: Strong Antiaromatic Contribution to the Electronic Structure. Org. Lett. 2017, 19, 826–829. 10.1021/acs.orglett.6b03819. [DOI] [PubMed] [Google Scholar]
- Smet M.; Van Dijk J.; Dehaen W. An Improved Synthesis of Substituted Rubicenes Providing Access to Heterocyclic Rubicene Analogues. Synlett 1999, 1999, 495–497. 10.1055/s-1999-2624. [DOI] [Google Scholar]
- Mohebbi A. R.; Wudl F. Electron-Accepting Dithiarubicene (Emeraldicene) and Derivatives Prepared by Unprecedented Nucleophilic Hydrogen Substitution by Alkyllithium Reagents. Chem. - Eur. J. 2011, 17, 2642–2646. 10.1002/chem.201002608. [DOI] [PubMed] [Google Scholar]
- Wang M.; Mohebbi A. R.; Sun Y.; Wudl F. Ribbons, Vesicles, and Baskets: Supramolecular Assembly of a Coil-Plate-Coil Emeraldicene Derivative. Angew. Chem., Int. Ed. 2012, 51, 6920–6924. 10.1002/anie.201201796. [DOI] [PubMed] [Google Scholar]
- Chao Y.-C.; Yeh S.-C.; Hsu H.-L.; Jiang B.-H.; Sun K.-H.; Chen C.-T.; Chen C.-P.; Jeng R.-J. Visibly Transparent Conjugated Polymers Based on Non-Alternant Cyclopenta-Fused Emeraldicene for Polymer Solar Cells. Org. Electron. 2017, 49, 114–122. 10.1016/j.orgel.2017.06.049. [DOI] [Google Scholar]
- Bheemireddy S. R.; Hautzinger M. P.; Li T.; Lee B.; Plunkett K. N. Conjugated Ladder Polymers by a Cyclopentannulation Polymerization. J. Am. Chem. Soc. 2017, 139, 5801–5807. 10.1021/jacs.6b12916. [DOI] [PubMed] [Google Scholar]
- Ogawa N.; Yamaoka Y.; Yamada K.; Takasu K. Synthesis of π-Extended Fluoranthenes via a KHMDS-Promoted Anionic-Radical Reaction Cascade. Org. Lett. 2017, 19, 3327–3330. 10.1021/acs.orglett.7b01538. [DOI] [PubMed] [Google Scholar]
- Martínez Á. M.; Alonso I.; Rodríguez N.; Gómez Arrayás R.; Carretero J. C. Rhodium-Catalyzed Copper-Assisted Intermolecular Domino C-H Annulation of 1,3-Diynes with Picolinamides: Access to Pentacyclic π-Extended Systems. Chem. - Eur. J. 2019, 25, 5733–5742. 10.1002/chem.201900162. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Shi X.; Feng C.; Wu Y.; Zheng N.; Liu J.; Wu X.; Chen H.; Peng A.; Li J.; et al. Synthetic Routes for Heteroatom-Containing Alkylated/Arylated Polycyclic Aromatic Hydrocarbons. Angew. Chem., Int. Ed. 2021, 60, 2924–2928. 10.1002/anie.202014108. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Rominger F.; Mastalerz M. Fused π-Extended Truxenes via a Threefold Borylation as the Key Step. Chem. - Eur. J. 2016, 22, 3084–3093. 10.1002/chem.201504621. [DOI] [PubMed] [Google Scholar]
- Bheemireddy S. R.; Hussain W. A.; Uddin A.; Du Y.; Hautzinger M. P.; Kevorkian P. V.; Petrie F. A.; Plunkett K. N. Cyclopentannulation and Cyclodehydrogenation of Isomerically Pure 5,11-Dibromo-Anthradithiophenes Leading to Contorted Aromatics. Chem. Commun. 2018, 54, 14140–14143. 10.1039/C8CC07327A. [DOI] [PubMed] [Google Scholar]
- Stadlbauer W.; Dang V. H.; Guttenberger N. 5-Unsubstituted Pyrido[3,2,1-Jk]Carbazol-6-Ones: Syntheses, Substitution, and Cyclization Reactions. J. Heterocycl. Chem. 2015, 52, 114–123. 10.1002/jhet.1994. [DOI] [Google Scholar]
- Sen’ko O. A.; Dybenko A. G.; Garazd M. M.; Kartsev V. G. Synthesis of Benzoannelated Canthin-6-One Analogs. Chem. Nat. Compd. 2017, 53, 523–528. 10.1007/s10600-017-2037-9. [DOI] [Google Scholar]
- Zhao J.; Li R.; Ai W.; Dong D.; Li J.; Chen L.; Xie L.; Yu T.; Huang W. Pi-Extended Diindole-Fused Azapentacenone: Synthesis, Characterization, and Photophysical and Lithium-Storage Properties. Chem. - Asian J. 2016, 11, 1382–1387. 10.1002/asia.201501366. [DOI] [PubMed] [Google Scholar]
- Pandit P.; Nakamura T.; Higashibayashi S. Synthesis and Acid-Responsive Electron-Transfer Disproportionation of Non- and Tetramesityl-Substituted 1,1’,9,9’-Bicarbazole. Chem. Lett. 2015, 44, 1336–1338. 10.1246/cl.150557. [DOI] [Google Scholar]
- Higashibayashi S.; Pandit P.; Haruki R.; Adachi S.; Kumai R. Redox-Dependent Transformation of a Hydrazinobuckybowl between Curved and Planar Geometries. Angew. Chem., Int. Ed. 2016, 55, 10830–10834. 10.1002/anie.201605340. [DOI] [PubMed] [Google Scholar]
- Petrova O. N.; Lipson V. V.; Zamigailo L. L.; Shirobokova M. G.; Musatov V. I.; Baumer V. N.; Sofronov D. S. Synthesis and Chemical Properties of 4-Aroyl-3-Methyl-4,10-Dihydroindeno[1,2-b]Pyrazolo-[4,3-e]Pyridin-5-Ones. Russ. J. Org. Chem. 2015, 51, 1597–1605. 10.1134/S1070428015110147. [DOI] [Google Scholar]
- Mule R. D.; Shaikh A. C.; Gade A. B.; Patil N. T. A New Class of N-Doped Ionic PAHs via Intramolecular [4 + 2]-Cycloaddition between Arylpyridines and Alkynes. Chem. Commun. 2018, 54, 11909–11912. 10.1039/C8CC05743E. [DOI] [PubMed] [Google Scholar]
- Shaikh A. C.; Banerjee S.; Mule R. D.; Bera S.; Patil N. T. External Oxidant-Dependent Reactivity Switch in Copper-Mediated Intramolecular Carboamination of Alkynes: Access to a Different Class of Fluorescent Ionic Nitrogen-Doped Polycyclic Aromatic Hydrocarbons. J. Org. Chem. 2019, 84, 4120–4130. 10.1021/acs.joc.9b00120. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Wu Z.; Liang L.; Sun Y.; Luo B.; Huang P.; Wen S. Heterocyclic Iodoniums as Versatile Synthons to Approach Diversified Polycyclic Heteroarenes. RSC Adv. 2019, 9, 33170–33179. 10.1039/C9RA07288H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W.-L.; Li S.-Q.; Wang J.; Zhang Z.-Y.; Yang Z.-W.; Xu D.; Xu C.; Lan H.-T.; Chen Z.-Z.; Xu Z.-G. An Efficient and Facile Method for the Synthesis of Benzimidazoisoquinoline Derivatives via a Multicomponent Reaction. ACS Comb. Sci. 2016, 18, 65–69. 10.1021/acscombsci.5b00145. [DOI] [PubMed] [Google Scholar]
- Chen Z.-Z.; Li S.-Q.; Zhang Y.-J.; Tang D.-Y.; Meng J.-P.; Lei J.; Li H.-Y.; Xu Z.-G. Synthesis of Pyridodiindoles with Anticancer Activity by a Three-Component Cascade Condensation. Org. Lett. 2018, 20, 7811–7815. 10.1021/acs.orglett.8b03245. [DOI] [PubMed] [Google Scholar]
- Zou S.; Liu Y.; Xi C. Concise and Efficient Synthesis of Indole-Indolone Scaffolds through MeOTf-Induced Annulation of N-(2-Cyanoaryl)Indoles. ACS Omega 2019, 4, 18734–18740. 10.1021/acsomega.9b02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volvoikar P. S.; Tilve S. G.; Zubkov F. I. A Concise Approach for the Synthesis of the ABCD Ring System of Alpkinidine. ChemistrySelect 2019, 4, 7187–7189. 10.1002/slct.201900357. [DOI] [Google Scholar]
- Benke B. P.; Hertwig L.; Yang X.; Rominger F.; Mastalerz M. Triptycene End-Capped Indigo Derivatives - Turning Insoluble Pigments to Soluble Dyes. Eur. J. Org. Chem. 2021, 2021, 72–76. 10.1002/ejoc.202001362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zissimou G. A.; Kourtellaris A.; Manoli M.; Koutentis P. A. Redox Active Quinoidal 1,2,4-Benzotriazines. J. Org. Chem. 2018, 83, 9391–9402. 10.1021/acs.joc.8b01311. [DOI] [PubMed] [Google Scholar]
- Gu P.-Y.; Liu G.; Zhao J.; Aratani N.; Ye X.; Liu Y.; Yamada H.; Nie L.; Zhang H.; Zhu J.; et al. Understanding the Structure-Determining Solid Fluorescence of an Azaacene Derivative. J. Mater. Chem. C 2017, 5, 8869–8874. 10.1039/C7TC03089D. [DOI] [Google Scholar]
- Kitamura K.; Asahina K.; Adachi K.; Hamura T. Intramolecular Benzoallene-Alkyne Cycloaddition Initiated by Site-Selective SN2’ Reaction of Epoxytetracene En Route to π-Extended Pyracylene. Chem. Commun. 2019, 55, 11021–11024. 10.1039/C9CC05500B. [DOI] [PubMed] [Google Scholar]
- Hu P.; He X.; Ng M.-F.; Ye J.; Zhao C.; Wang S.; Tan K.; Chaturvedi A.; Jiang H.; Kloc C.; et al. Trisulfide-Bond Acenes for Organic Batteries. Angew. Chem., Int. Ed. 2019, 58, 13513–13521. 10.1002/anie.201906301. [DOI] [PubMed] [Google Scholar]
- Delaunay W.; Szucs R.; Pascal S.; Mocanu A.; Bouit P.-A.; Nyulászi L.; Hissler M. Synthesis and Electronic Properties of Polycyclic Aromatic Hydrocarbons Doped with Phosphorus and Sulfur. Dalton Trans. 2016, 45, 1896–1903. 10.1039/C5DT04154F. [DOI] [PubMed] [Google Scholar]
- Ooyama Y.; Enoki T.; Aoyama S.; Ohshita J. Synthesis and Optical and Electrochemical Properties of a Phenanthrodithiophene (Fused-Bibenzo[c]Thiophene) Derivative. Org. Biomol. Chem. 2017, 15, 7302–7307. 10.1039/C7OB01695F. [DOI] [PubMed] [Google Scholar]
- Liu S.; Huang C.; Zhang J.; Tian S.; Li C.; Fu N.; Wang L.; Zhao B.; Huang W. Synthesis of Sulfur-Hybridized Pyracylene and the Unexpected Phenyl Shift Mediated Rearrangement of Scholl Reaction. Eur. J. Org. Chem. 2019, 2019, 3061–3070. 10.1002/ejoc.201900344. [DOI] [Google Scholar]
- Wu X.; Yang Y.; Han J.; Wang L. Palladium Catalyzed C-I and Vicinal C-H Dual Activation of Diaryliodonium Salts for Diarylation: Synthesis of 4,5-Benzocoumarins. Org. Lett. 2015, 17, 5654–5657. 10.1021/acs.orglett.5b02938. [DOI] [PubMed] [Google Scholar]
- Patil V. V.; Lee K. H.; Lee J. Y. Dibenzo[c, g]Indolo[3,2,1-Jk]Carbazole as a New Chromophore for Blue Organic Light-Emitting Diodes. J. Mater. Chem. C 2019, 7, 14301–14305. 10.1039/C9TC04783B. [DOI] [Google Scholar]
- Lee H. L.; Chung W. J.; Lee J. Y. Narrowband and Pure Violet Organic Emitter with a Full Width at Half Maximum of 14 Nm and y Color Coordinate of Below 0.02. Small 2020, 16, 1907569. 10.1002/smll.201907569. [DOI] [PubMed] [Google Scholar]
- Venunath Patil V.; Hyung Lee K.; Yeob Lee J. A Novel Fluorene-Indolocarbazole Hybrid Chromophore to Assemble High Efficiency Deep-Blue Fluorescent Emitters with Extended Device Lifetime. J. Mater. Chem. C 2020, 8, 3051–3057. 10.1039/C9TC06434F. [DOI] [Google Scholar]
- Kader T.; Stöger B.; Fröhlich J.; Kautny P. Azaindolo[3,2,1-Jk]Carbazoles: New Building Blocks for Functional Organic Materials. Chem. - Eur. J. 2019, 25, 4412–4425. 10.1002/chem.201805578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H. H.; Tran H. Q.; Ohlendorf L.; Ngo T. N.; Dang T. T.; Ehlers P.; Villinger A.; Langer P. One-Pot Palladium-Catalyzed Synthesis of Benzo[b]Carbazolediones. Synlett 2015, 26, 2429–2433. 10.1055/s-0035-1560212. [DOI] [Google Scholar]
- Deng N.; Zhang G. Nitrogen-Centered Concave Molecules with Double Fused Pentagons. Org. Lett. 2019, 21, 5248–5251. 10.1021/acs.orglett.9b01861. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Zhang G. A Nanoboat with Fused Concave N-Heterotriangulene. Angew. Chem., Int. Ed. 2020, 59, 8963–8968. 10.1002/anie.202002869. [DOI] [PubMed] [Google Scholar]
- Song Y.; Zhang G. Effect of Fusion Manner of Concave Molecules on the Properties of Resulting Nanoboats. Org. Lett. 2021, 23, 491–496. 10.1021/acs.orglett.0c04008. [DOI] [PubMed] [Google Scholar]
- Jones A. W.; Louillat-Habermeyer M.-L.; Patureau F. W. Strained Dehydrogenative Ring Closure of Phenylcarbazoles. Adv. Synth. Catal. 2015, 357, 945–949. 10.1002/adsc.201401136. [DOI] [Google Scholar]
- Rank C. K.; Jones A. W.; Wall T.; Martino-Fumo P. D.; Schröck S.; Gerhards M.; Patureau F. W. An Intermolecular C-H Oxidizing Strategy to Access Highly Fused Carbazole Skeletons from Simple Naphthylamines. Chem. Commun. 2019, 55, 13749–13752. 10.1039/C9CC05240B. [DOI] [PubMed] [Google Scholar]
- Taniguchi T.; Itai Y.; Nishii Y.; Tohnai N.; Miura M. Construction of Nitrogen-Containing Polycyclic Aromatic Compounds by Intramolecular Oxidative C-H/C-H Coupling of Bis(9H-Carbazol-9-Yl)Benzenes and Their Properties. Chem. Lett. 2019, 48, 1160–1163. 10.1246/cl.190494. [DOI] [Google Scholar]
- Puntscher H.; Kautny P.; Stöger B.; Tissot A.; Hametner C.; Hagemann H. R.; Fröhlich J.; Baumgartner T.; Lumpi D. Structure-Property Studies of P-Triarylamine-Substituted Dithieno[3,2-b:2’,3′-d]Phospholes. RSC Adv. 2015, 5, 93797–93807. 10.1039/C5RA13651B. [DOI] [Google Scholar]
- Kautny P.; Wu Z.; Eichelter J.; Horkel E.; Stöger B.; Chen J.; Ma D.; Fröhlich J.; Lumpi D. Indolo[3,2,1-Jk]Carbazole Based Planarized CBP Derivatives as Host Materials for PhOLEDs with Low Efficiency Roll-Off. Org. Electron. 2016, 34, 237–245. 10.1016/j.orgel.2016.04.036. [DOI] [Google Scholar]
- Seo J.-A.; Im Y.; Han S. H.; Lee C. W.; Lee J. Y. Unconventional Molecular Design Approach of High-Efficiency Deep Blue Thermally Activated Delayed Fluorescent Emitters Using Indolocarbazole as an Acceptor. ACS Appl. Mater. Interfaces 2017, 9, 37864–37872. 10.1021/acsami.7b09351. [DOI] [PubMed] [Google Scholar]
- Patil V. V.; Lee K. H.; Lee J. Y. Universal Blue Emitters for High Efficiency Thermally Activated Delayed Fluorescence and Fluorescent Organic Light-Emitting Diodes. Dyes Pigm. 2020, 174, 108070. 10.1016/j.dyepig.2019.108070. [DOI] [Google Scholar]
- Im Y.; Han S. H.; Lee J. Y. CN Substituted Indolocarbazole as a Core Structure of Exciton Harvesting and Lifetime Extending Host for Green Thermally Activated Delayed Fluorescent Emitter. Dyes Pigm. 2019, 164, 233–236. 10.1016/j.dyepig.2019.01.024. [DOI] [Google Scholar]
- Im Y.; Han S. H.; Lee J. Y. Deep Blue Thermally Activated Delayed Fluorescent Emitters Using CN-Modified Indolocarbazole as an Acceptor and Carbazole-Derived Donors. J. Mater. Chem. C 2018, 6, 5012–5017. 10.1039/C8TC00546J. [DOI] [Google Scholar]
- Konidena R. K.; Lee K. H.; Lee J. Y.; Hong W. P. 15H-Diindolo[2,3-b:1’,2’,3′-Lm]Carbazole: A Novel Rigid Donor for Highly Efficient Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C 2019, 7, 8037–8044. 10.1039/C9TC02195G. [DOI] [Google Scholar]
- Im Y.; Han S. H.; Lee J. Y. Dibenzothiophene and Indolocarbazole Cored Bipolar Hosts for Blue Phosphorescent Organic Light-Emitting Diodes. Org. Electron. 2018, 62, 560–565. 10.1016/j.orgel.2018.06.031. [DOI] [Google Scholar]
- Ma X.-J.; Zhu X.-D.; Wang K.-L.; Igbari F.; Yuan Y.; Zhang Y.; Gao C.-H.; Jiang Z.-Q.; Wang Z.-K.; Liao L.-S. Planar Starburst Hole-Transporting Materials for Highly Efficient Perovskite Solar Cells. Nano Energy 2019, 63, 103865. 10.1016/j.nanoen.2019.103865. [DOI] [Google Scholar]
- Holzer B.; Bintinger J.; Lumpi D.; Choi C.; Kim Y.; Stöger B.; Hametner C.; Marchetti-Deschmann M.; Plasser F.; Horkel E.; et al. Color Fine-Tuning of Optical Materials Through Rational Design. ChemPhysChem 2017, 18, 549–563. 10.1002/cphc.201601204. [DOI] [PubMed] [Google Scholar]
- Im Y.; Han S. H.; Lee J. Y. Bipolar Type Indolocarbazole Host for Green Phosphorescent Organic Light-Emitting Diodes. J. Ind. Eng. Chem. 2018, 66, 381–386. 10.1016/j.jiec.2018.06.004. [DOI] [Google Scholar]
- Lévesque É.; Bechara W. S.; Constantineau-Forget L.; Pelletier G.; Rachel N. M.; Pelletier J. N.; Charette A. B. General C-H Arylation Strategy for the Synthesis of Tunable Visible Light-Emitting Benzo[a]Imidazo[2,1,5-c, d]Indolizine Fluorophores. J. Org. Chem. 2017, 82, 5046–5067. 10.1021/acs.joc.6b02928. [DOI] [PubMed] [Google Scholar]
- Wu D.; Chen L.; Ma S.; Luo H.; Cao J.; Chen R.; Duan Z.; Mathey F. Synthesis of 1,3-Azaphospholes with Pyrrolo[1,2-a]Quinoline Skeleton and Their Optical Applications. Org. Lett. 2018, 20, 4103–4106. 10.1021/acs.orglett.8b01663. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Zhao Y.; Zhuang W.-H.; Wu J.-J.; Wang F.-Z.; Li C.-H.; Zuo J.-L. Phthalorubines: Fused-Ring Compounds Synthesized from Phthalonitrile. Angew. Chem., Int. Ed. 2018, 57, 15384–15389. 10.1002/anie.201807281. [DOI] [PubMed] [Google Scholar]
- Fu W. C.; Wang Z.; Chan W. T. K.; Lin Z.; Kwong F. Y. Regioselective Synthesis of Polycyclic and Heptagon-Embedded Aromatic Compounds through a Versatile π-Extension of Aryl Halides. Angew. Chem., Int. Ed. 2017, 56, 7166–7170. 10.1002/anie.201703551. [DOI] [PubMed] [Google Scholar]
- Samineni R.; Srihari P.; Mehta G. Versatile Route to Benzoannulated Medium-Ring Carbocycles via Aryne Insertion into Cyclic 1,3-Diketones: Application to a Synthesis of Radermachol. Org. Lett. 2016, 18, 2832–2835. 10.1021/acs.orglett.6b01078. [DOI] [PubMed] [Google Scholar]
- Li X.; Pan J.; Song S.; Jiao N. Pd-Catalyzed Dehydrogenative Annulation Approach for the Efficient Synthesis of Phenanthridinones. Chem. Sci. 2016, 7, 5384–5389. 10.1039/C6SC01148A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Roisnel T.; Soulé J.-F.; Doucet H. Convenient Access to C10- and C11-(Di)Arylated Dibenzo[b, f]Azepines via Palladium-Catalyzed C-H Bonds Cleavages. Adv. Synth. Catal. 2019, 361, 791–802. 10.1002/adsc.201801366. [DOI] [Google Scholar]
- Ito M.; Kawasaki R.; Kanyiva K. S.; Shibata T. Construction of a Polycyclic Conjugated System Containing a Dibenzazepine Moiety by Cationic Gold(I)-Catalyzed Cycloisomerization. Eur. J. Org. Chem. 2016, 2016, 5234–5237. 10.1002/ejoc.201601147. [DOI] [Google Scholar]
- Skonieczny K.; Gryko D. T. Photochemical Conversion of Phenanthro[9,10-d]Imidazoles into π-Expanded Heterocycles. J. Org. Chem. 2015, 80, 5753–5763. 10.1021/acs.joc.5b00714. [DOI] [PubMed] [Google Scholar]
- Hayakawa S.; Kawasaki A.; Hong Y.; Uraguchi D.; Ooi T.; Kim D.; Akutagawa T.; Fukui N.; Shinokubo H. Inserting Nitrogen: An Effective Concept To Create Nonplanar and Stimuli-Responsive Perylene Bisimide Analogues. J. Am. Chem. Soc. 2019, 141, 19807–19816. 10.1021/jacs.9b09556. [DOI] [PubMed] [Google Scholar]
- Gadekar S. C.; Reddy B. K.; Panchal S. P.; Anand V. G. Metal Assisted Cyclomerization of Benzodipyrrins into Expanded Norroles, Aza-Heptalene and Acyclic Dimers. Chem. Commun. 2016, 52, 4565–4568. 10.1039/C5CC10356H. [DOI] [PubMed] [Google Scholar]
- Petrovskii P. P.; Tomashenko O. A.; Novikov M. S.; Khlebnikov A. F.; Stoeckli-Evans H. Synthesis, Crystal Structure, and Photophysical Properties of Dimethyl 7-Oxa-2a1-Azabenzo[b]Cyclopenta[Pq]Pleiadene-1,2-Dicarboxylate - Novel Fused Oxazapolycyclic Skeleton. Chem. Heterocycl. Compd. 2017, 53, 909–912. 10.1007/s10593-017-2144-3. [DOI] [Google Scholar]
- Hayakawa S.; Matsuo K.; Yamada H.; Fukui N.; Shinokubo H. Dinaphthothiepine Bisimide and Its Sulfoxide: Soluble Precursors for Perylene Bisimide. J. Am. Chem. Soc. 2020, 142, 11663–11668. 10.1021/jacs.0c04096. [DOI] [PubMed] [Google Scholar]
- Kirschbaum T.; Rominger F.; Mastalerz M. An Isosteric Triaza Analogue of a Polycyclic Aromatic Hydrocarbon Monkey Saddle. Chem. - Eur. J. 2020, 26, 14560–14564. 10.1002/chem.202002826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A. Y.; Sarg M. T.; El Deeb M. A.; Bayoumi A. H.; El Rabeb S. I. Facile Synthesis and Anticancer Activity Study of Novel Series of Substituted and Fused Coumarin Derivatives. J. Heterocycl. Chem. 2018, 55, 1426–1443. 10.1002/jhet.3179. [DOI] [Google Scholar]
- Igarashi T.; Tobisu M.; Chatani N. Catalytic Double Carbon-Boron Bond Formation for the Synthesis of Cyclic Diarylborinic Acids as Versatile Building Blocks for π-Extended Heteroarenes. Angew. Chem., Int. Ed. 2017, 56, 2069–2073. 10.1002/anie.201612535. [DOI] [PubMed] [Google Scholar]
- Mehra M. K.; Sharma S.; Rangan K.; Kumar D. Substrate or Solvent-Controlled PdII-Catalyzed Regioselective Arylation of Quinolin-4(1H)-Ones Using Diaryliodonium Salts: Facile Access to Benzoxocine and Aaptamine Analogues. Eur. J. Org. Chem. 2020, 2020, 2409–2413. 10.1002/ejoc.202000013. [DOI] [Google Scholar]
- Tanaka Y.; Tajima K.; Fukui N.; Shinokubo H. Dinaphtho[1,8-Bc:1’,8’-Fg][1,5]Dithiocine Bisimide. Asian J. Org. Chem. 2021, 10, 541–544. 10.1002/ajoc.202000722. [DOI] [Google Scholar]
- Bromby A. D.; Hogan D. T.; Sutherland T. C. Core Expanded, 21,23-Dithiadiacenaphtho[1,2-c]Porphyrin Interactions with [60]Fullerene. New J. Chem. 2017, 41, 4802–4805. 10.1039/C6NJ03353A. [DOI] [Google Scholar]
- Rauen P. J.; Lash T. D. Dihydropyracyloporphyrins. Tetrahedron Lett. 2020, 61, 152662. 10.1016/j.tetlet.2020.152662. [DOI] [Google Scholar]
- Okujima T.; Mack J.; Nakamura J.; Kubheka G.; Nyokong T.; Zhu H.; Komobuchi N.; Ono N.; Yamada H.; Uno H.; et al. Synthesis, Characterization, and Electronic Structures of Porphyrins Fused with Polycyclic Aromatic Ring Systems. Chem. - Eur. J. 2016, 22, 14730–14738. 10.1002/chem.201602213. [DOI] [PubMed] [Google Scholar]
- Gao R.; AbuSalim D. I.; Lash T. D. Pyreniporphyrins: Porphyrin Analogues That Incorporate a Polycyclic Aromatic Hydrocarbon Subunit within the Macrocyclic Framework. J. Org. Chem. 2017, 82, 6680–6688. 10.1021/acs.joc.7b00829. [DOI] [PubMed] [Google Scholar]
- Galstyan A.; Maurya Y. K.; Zhylitskaya H.; Bae Y. J.; Wu Y.-L.; Wasielewski M. R.; Lis T.; Dobrindt U.; Stępień M. π-Extended Donor-Acceptor Porphyrins and Metalloporphyrins for Antimicrobial Photodynamic Inactivation. Chem. - Eur. J. 2020, 26, 8262–8266. 10.1002/chem.201905372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiga E.; Kim G.; Chmielewski P. J.; Lis T.; Kim D.; Stępień M. Porphyrin-Ryleneimide Hybrids: Low-Bandgap Acceptors in Energy-Transfer Cassettes. Chem. - Asian J. 2020, 15, 2854–2858. 10.1002/asia.202000762. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Maurya Y. K.; Kang S.; Chmielewski P.; Lis T.; Cybińska J.; Kim D.; Stępień M. Porphyrin-Ryleneimide Hybrids: Tuning of Visible and Near-Infrared Absorption by Chromophore Desymmetrization. Org. Lett. 2020, 22, 7202–7207. 10.1021/acs.orglett.0c02544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Fu L.; Huang T.; Geng H.; Jiang W.; Wang Z. Rylene Annulated Phthalocyanine: A Fully Conjugated Block for the Construction of a Supramolecular Two-Dimensional Framework. Chem. Commun. 2018, 54, 7294–7297. 10.1039/C8CC01216D. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Jiang K.; Li C.; Liu Y.; Xu C.; Zheng W.; Guan D.; Li Y.; Zheng H.; Liu C.; et al. Precise Control of π-Electron Magnetism in Metal-Free Porphyrins. J. Am. Chem. Soc. 2020, 142, 18532–18540. 10.1021/jacs.0c07791. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Mateo L. M.; Robles R.; Ruffieux P.; Lorente N.; Bottari G.; Torres T.; Fasel R. Inducing Open-Shell Character in Porphyrins through Surface-Assisted Phenalenyl π-Extension. J. Am. Chem. Soc. 2020, 142, 18109–18117. 10.1021/jacs.0c07781. [DOI] [PubMed] [Google Scholar]
- Xiang F.; Gemeinhardt A.; Schneider M. A. Competition between Dehydrogenative Organometallic Bonding and Covalent Coupling of an Unfunctionalized Porphyrin on Cu(111). ACS Nano 2018, 12, 1203–1210. 10.1021/acsnano.7b06998. [DOI] [PubMed] [Google Scholar]
- Shu C.-H.; Xie Y.-L.; Wang A.; Shi K.-J.; Zhang W.-F.; Li D.-Y.; Liu P.-N. On-Surface Reactions of Aryl Chloride and Porphyrin Macrocycles via Merging Two Reactive Sites into a Single Precursor. Chem. Commun. 2018, 54, 12626–12629. 10.1039/C8CC07652A. [DOI] [PubMed] [Google Scholar]
- Seufert K.; McBride F.; Jaekel S.; Wit B.; Haq S.; Steiner A.; Poli P.; Persson M.; Raval R.; Grill L. Porphine Homocoupling on Au(111). J. Phys. Chem. C 2019, 123, 16690–16698. 10.1021/acs.jpcc.9b02770. [DOI] [Google Scholar]
- Xiang F.; Lu Y.; Wang Z.; Ju H.; Di Filippo G.; Li C.; Liu X.; Leng X.; Zhu J.; Wang L.; et al. On-Surface Synthesis of Chiral π-Conjugate Porphyrin Tapes by Substrate-Regulated Dehydrogenative Coupling. J. Phys. Chem. C 2019, 123, 23007–23013. 10.1021/acs.jpcc.9b06025. [DOI] [Google Scholar]
- Yin C.; Peng Z.; Liu D.; Song H.; Zhu H.; Chen Q.; Wu K. Selective Intramolecular Dehydrocyclization of Co-Porphyrin on Au(111). Molecules 2020, 25, 3766. 10.3390/molecules25173766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugavel M.; Adinarayana B.; Das M.; Peruncheralathan S.; Palepu N. R.; Srinivasan A. PtCl2Mediated Peripheral Transformation of Carbatriphyrin(3.1.1) into a Meso-Fused β-B’ Dimer and Its Monomer Analogue. Chem. Commun. 2020, 56, 12809–12812. 10.1039/D0CC05309K. [DOI] [PubMed] [Google Scholar]
- Lungerich D.; Hitzenberger J. F.; Ruppel M.; Döpper T.; Witt M.; Ivanović-Burmazović I.; Görling A.; Jux N.; Drewello T. Gas-Phase Transformation of Fluorinated Benzoporphyrins to Porphyrin-Embedded Conical Nanocarbons. Chem. - Eur. J. 2020, 26, 12180–12187. 10.1002/chem.202002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H.; Jing H.; Krayer M.; Allu S.; Veeraraghavaiah G.; Wu Z.; Jiang J.; Diers J. R.; Magdaong N. C. M.; Mandal A. K.; et al. Annulated Bacteriochlorins for Near-Infrared Photophysical Studies. New J. Chem. 2019, 43, 7209–7232. 10.1039/C9NJ01113G. [DOI] [Google Scholar]
- Hooper R. W.; Zhang A.; Koszelewski D.; Lewtak J. P.; Koszarna B.; Levy C. J.; Gryko D. T.; Stillman M. J. Differential Quenching of the Angular Momentum of the B and Q Bands of a Porphyrin as a Result of Extended Ring π-Conjugation. J. Porphyrins Phthalocyanines 2018, 22, 1111–1128. 10.1142/S1088424618501110. [DOI] [Google Scholar]
- Lewtak J. P.; Koszarna B.; Charyton M. K.; Gryko D. T. Extending a Porphyrin Chromophore via Fusion with Naphthalene. J. Porphyrins Phthalocyanines 2020, 24, 448–455. 10.1142/S1088424619501530. [DOI] [Google Scholar]
- Achary B. S.; Gokulnath S.; Ghosh S.; Mrinalini M.; Prasanthkumar S.; Giribabu L. Unprecedented Charge-Transfer Complex of Fused Diporphyrin as Near-Infrared Absorption-Induced High-Aspect-Ratio Nanorods. Chem. - Asian J. 2016, 11, 3498–3502. 10.1002/asia.201601363. [DOI] [PubMed] [Google Scholar]
- Osterloh W. R.; Kumar S.; Chaudhri N.; Fang Y.; Sankar M.; Kadish K. M. Facile Heterogeneous and Homogeneous Anion Induced Electrosynthesis: An Efficient Method for Obtaining π-Extended Porphyrins. Inorg. Chem. 2020, 59, 16737–16746. 10.1021/acs.inorgchem.0c02770. [DOI] [PubMed] [Google Scholar]
- Hurej K.; Stawski W.; Latos-Grażyński L.; Pawlicki M. Meso-N-Pyrrole as a Versatile Substituent Influencing the Optical Properties of Porphyrin. Chem. - Asian J. 2016, 11, 3329–3333. 10.1002/asia.201601210. [DOI] [PubMed] [Google Scholar]
- Fukui N.; Cha W.; Shimizu D.; Oh J.; Furukawa K.; Yorimitsu H.; Kim D.; Osuka A. Highly Planar Diarylamine-Fused Porphyrins and Their Remarkably Stable Radical Cations. Chem. Sci. 2017, 8, 189–199. 10.1039/C6SC02721K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kise K.; Hong Y.; Fukui N.; Shimizu D.; Kim D.; Osuka A. Diarylamine-Fused Subporphyrins: Proof of Twisted Intramolecular Charge Transfer (TICT) Mechanism. Chem. - Eur. J. 2018, 24, 8306–8310. 10.1002/chem.201801576. [DOI] [PubMed] [Google Scholar]
- Fukui N.; Yorimitsu H.; Osuka A. Meso-Meso-Linked Diarylamine-Fused Porphyrin Dimers. Chem. - Eur. J. 2016, 22, 18476–18483. 10.1002/chem.201604301. [DOI] [PubMed] [Google Scholar]
- Kato K.; Osuka A. Meta- and Para-Phenylenediamine-Fused Porphyrin Dimers: Synthesis and Magnetic Interactions of Their Dication Diradicals. Angew. Chem., Int. Ed. 2019, 58, 8546–8550. 10.1002/anie.201901939. [DOI] [PubMed] [Google Scholar]
- Kato K.; Osuka A. Helically Twisted Benzene-1,3,5-Triamine-Fused Porphyrin Dimers. Chem. Lett. 2020, 49, 517–520. 10.1246/cl.200115. [DOI] [Google Scholar]
- Fujimoto K.; Osuka A. A 1,5-Naphthyridine-Fused Porphyrin Dimer: Intense NIR Absorption and Facile Redox Interconversion with Its Reduced Congener. Chem. - Eur. J. 2018, 24, 6530–6533. 10.1002/chem.201800854. [DOI] [PubMed] [Google Scholar]
- Thuita D.; Guberman-Pfeffer M. J.; Brückner C. SNAr Reaction Toward the Synthesis of Fluorinated Quinolino[2,3,4-at]Porphyrins. Eur. J. Org. Chem. 2021, 2021, 318–323. 10.1002/ejoc.202001347. [DOI] [Google Scholar]
- Batalha P. N.; Gomes A. T. P. C.; Forezi L. S. M.; Costa L.; de Souza M. C. B. V.; Boechat F. d. C. S.; Ferreira V. F.; Almeida A.; Faustino M. A. F.; Neves M. G. P. M. S.; Cavaleiro J. A. S.; et al. Synthesis of New Porphyrin/4-Quinolone Conjugates and Evaluation of Their Efficiency in the Photoinactivation of Staphylococcus Aureus. RSC Adv. 2015, 5, 71228–71239. 10.1039/C5RA11070J. [DOI] [Google Scholar]
- Ahmad Dar T.; Mandeep Sankar M. Synthesis, Spectral and Electrochemical Redox Properties of N-Methyl Fused Nickel(II) Porphyrin. J. Porphyrins Phthalocyanines 2018, 22, 1106–1110. 10.1142/S1088424618501109. [DOI] [Google Scholar]
- Chevrier M.; Richeter S.; Coulembier O.; Surin M.; Mehdi A.; Lazzaroni R.; Evans R. C.; Dubois P.; Clément S. Expanding the Light Absorption of Poly(3-Hexylthiophene) by End-Functionalization with π-Extended Porphyrins. Chem. Commun. 2016, 52, 171–174. 10.1039/C5CC06290J. [DOI] [PubMed] [Google Scholar]
- Pereira A. M. V. M.; Lacerda P. S. S.; Silva B. N. M.; Neves M. G. P. M. S.; Silva A. M. S.; Silva B. V.; da Silva F. C.; Ferreira V. F.; Pinto A. C.; Cavaleiro J. A. S. One-Pot Synthesis of New Isatin-Porphyrin Conjugates by the Palladium Buchwald-Hartwig Methodology Involving β-Aminoporphyrinatonickel(II) and 3-Ketal Isatin Derivatives. Dyes Pigm. 2017, 139, 247–254. 10.1016/j.dyepig.2016.12.010. [DOI] [Google Scholar]
- Jimenez A. J.; Mesa N. S.; Pereira A. M. V. M.; Jean M.; Vincent B.; Jeandon C.; Gisselbrecht J.-P.; Ruppert R. Oxidative Coupling of an Enaminoporphyrin: C-C, N-N Linkages or Both?. Asian J. Org. Chem. 2015, 4, 1294–1300. 10.1002/ajoc.201500279. [DOI] [Google Scholar]
- Pawlicki M.; Hurej K.; Kwiecińska K.; Szterenberg L.; Latos-Grażyński L. A Fused Meso-Aminoporphyrin: A Switchable near-IR Chromophore. Chem. Commun. 2015, 51, 11362–11365. 10.1039/C5CC01231G. [DOI] [PubMed] [Google Scholar]
- Abuteen A.; Zanganeh S.; Akhigbe J.; Samankumara L. P.; Aguirre A.; Biswal N.; Braune M.; Vollertsen A.; Röder B.; Brückner C.; et al. The Evaluation of NIR-Absorbing Porphyrin Derivatives as Contrast Agents in Photoacoustic Imaging. Phys. Chem. Chem. Phys. 2013, 15, 18502–18509. 10.1039/c3cp52193a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano M.; Erfanzadeh M.; Zhou F.; Zhu H.; Bornhütter T.; Röder B.; Zhu Q.; Brückner C. In Vivo Photoacoustic Tumor Tomography Using a Quinoline-Annulated Porphyrin as NIR Molecular Contrast Agent. Org. Biomol. Chem. 2017, 15, 972–983. 10.1039/C6OB02640K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhigbe J.; Yang M.; Luciano M.; Brückner C. Quinoline-Annulated Chlorins and Chlorin-Analogs. J. Porphyrins Phthalocyanines 2016, 20, 265–273. 10.1142/S1088424616500036. [DOI] [Google Scholar]
- Berthelot M.; Hoffmann G.; Bousfiha A.; Echaubard J.; Roger J.; Cattey H.; Romieu A.; Lucas D.; Fleurat-Lessard P.; Devillers C. H. Oxidative C-N Fusion of Pyridinyl-Substituted Porphyrins. Chem. Commun. 2018, 54, 5414–5417. 10.1039/C8CC01375F. [DOI] [PubMed] [Google Scholar]
- Li C.; Zhu L.; Liang W.; Su R.; Yin J.; Hu Y.; Lan Y.; Wu D.; You J. An Unusual [4 + 2] Fusion Strategy to Forge Meso-N/O -Heteroarene-Fused (Quinoidal) Porphyrins with Intense near-Infrared Q-Bands. Chem. Sci. 2019, 10, 7274–7280. 10.1039/C9SC01596E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi S.; Tanaka T.; Park K. H.; Kim D.; Osuka A. Triply Linked Corrole Dimers. Angew. Chem., Int. Ed. 2016, 55, 6535–6539. 10.1002/anie.201601864. [DOI] [PubMed] [Google Scholar]
- Okuda Y.; Fukui N.; Kim J.; Kim T.; Jiang H.-W.; Copley G.; Kitano M.; Kim D.; Osuka A. A Meso-Meso β-β β-β Triply Linked Subporphyrin Dimer. Angew. Chem., Int. Ed. 2017, 56, 12317–12321. 10.1002/anie.201707123. [DOI] [PubMed] [Google Scholar]
- Kato K.; Furukawa K.; Osuka A. A Stable Trimethylenemethane Triplet Diradical Based on a Trimeric Porphyrin Fused π-System. Angew. Chem., Int. Ed. 2018, 57, 9491–9494. 10.1002/anie.201804644. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Kim J.; Phan H.; Herng T. S.; Gopalakrishna T. Y.; Zeng W.; Ding J.; Kim D.; Wu J. 2,6-/1,5-Naphthoquinodimethane Bridged Porphyrin Dimer Diradicaloids. J. Porphyrins Phthalocyanines 2020, 24, 220–229. 10.1142/S1088424619500998. [DOI] [Google Scholar]
- Wang K.; Liu P.; Zhang F.; Xu L.; Zhou M.; Nakai A.; Kato K.; Furukawa K.; Tanaka T.; Osuka A.; et al. A Robust Porphyrin-Stabilized Triplet Carbon Diradical. Angew. Chem., Int. Ed. 2021, 60, 7002–7006. 10.1002/anie.202015356. [DOI] [PubMed] [Google Scholar]
- Kozyrev A. N.; Alderfer J. L.; Srikrishnan T.; Pandey R. K. Synthesis of Novel Laterally-Bridged Pyropheophorbide a Dimers. J. Chem. Soc., Perkin Trans. 1 1998, 837–838. 10.1039/a800609a. [DOI] [Google Scholar]
- Kozyrev A. N.; Suresh V.; Das S.; Senge M. O.; Shibata M.; Dougherty T. J.; Pandey R. K. Syntheses and Spectroscopic Studies of Novel Chlorins with Fused Quinoxaline or Benzimidazole Ring Systems and the Related Dimers with Extended Conjugation1. Tetrahedron 2000, 56, 3353–3364. 10.1016/S0040-4020(00)00256-8. [DOI] [Google Scholar]
- Kozyrev A.; Ethirajan M.; Chen P.; Ohkubo K.; Robinson B. C.; Barkigia K. M.; Fukuzumi S.; Kadish K. M.; Pandey R. K. Synthesis, Photophysical and Electrochemistry of Near-IR Absorbing Bacteriochlorins Related to Bacteriochlorophyll a. J. Org. Chem. 2012, 77, 10260–10271. 10.1021/jo301895p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko A. J.; Dukh M.; Patel N. J.; Missert J. R.; Ohulchanskyy T.; Tabaczynski W. A.; Ohkubo K.; Fukuzumi S.; Yao R.; Sajjad M.; et al. A Pyropheophorbide Analogue Containing a Fused Methoxy Cyclohexenone Ring System Shows Promising Cancer-Imaging Ability. ChemMedChem 2019, 14, 1503–1513. 10.1002/cmdc.201900352. [DOI] [PubMed] [Google Scholar]
- Dekkiche H.; Buisson A.; Langlois A.; Karsenti P.-L.; Ruhlmann L.; Ruppert R.; Harvey P. Metal Linkage Effects on Ultrafast Energy Transfer. Chem. - Eur. J. 2016, 22, 10484–10493. 10.1002/chem.201601322. [DOI] [PubMed] [Google Scholar]
- Dekkiche H.; Buisson A.; Langlois A.; Karsenti P.-L.; Ruhlmann L.; Harvey P. D.; Ruppert R. Ultrafast Singlet Energy Transfer in Porphyrin Dyads. Inorg. Chem. 2016, 55, 10329–10336. 10.1021/acs.inorgchem.6b01594. [DOI] [PubMed] [Google Scholar]
- Carvalho M.-A.; Dekkiche H.; Karmazin L.; Sanchez F.; Vincent B.; Kanesato M.; Kikkawa Y.; Ruppert R. Synthesis and Study at a Solid/Liquid Interface of Porphyrin Dimers Linked by Metal Ions. Inorg. Chem. 2017, 56, 15081–15090. 10.1021/acs.inorgchem.7b02422. [DOI] [PubMed] [Google Scholar]
- Carvalho M.-A.; Dekkiche H.; Nagasaki M.; Kikkawa Y.; Ruppert R. Coordination-Driven Construction of Porphyrin Nanoribbons at a Highly Oriented Pyrolytic Graphite (HOPG)/Liquid Interface. J. Am. Chem. Soc. 2019, 141, 10137–10141. 10.1021/jacs.9b02145. [DOI] [PubMed] [Google Scholar]
- Chan J. Y. M.; Kawata T.; Kobayashi N.; Ng D. K. P. Boron(III) Carbazosubphthalocyanines: Core-Expanded Antiaromatic Boron(III) Subphthalocyanine Analogues. Angew. Chem., Int. Ed. 2019, 58, 2272–2277. 10.1002/anie.201811420. [DOI] [PubMed] [Google Scholar]
- Maeda C.; Tanaka Y.; Shirakawa T.; Ema T. Synthesis and Electronic Properties of π-Expanded Carbazole-Based Porphyrins. Chem. Commun. 2019, 55, 10162–10165. 10.1039/C9CC05079E. [DOI] [PubMed] [Google Scholar]
- Maeda C.; Kurihara K.; Masuda M.; Yoshioka N. Effects of Cyano, Ethynyl and Ethylenedioxy Groups on the Photophysical Properties of Carbazole-Based Porphyrins. Org. Biomol. Chem. 2015, 13, 11286–11291. 10.1039/C5OB01824B. [DOI] [PubMed] [Google Scholar]
- Maeda C.; Ema T. Chiral Carbazole-Based Porphyrins Showing Absorption and Circular Dichroism in the near-Infrared Region. J. Porphyrins Phthalocyanines 2020, 24, 247–251. 10.1142/S1088424619500937. [DOI] [Google Scholar]
- Maeda C.; Takaishi K.; Ema T. Palladium Complexes of Carbazole-Based Chalcogenaisophlorins: Synthesis, Structure, and Solid-State NIR Absorption Spectra. ChemPlusChem 2017, 82, 1368–1371. 10.1002/cplu.201700430. [DOI] [PubMed] [Google Scholar]
- Wu T.; Kim T.; Yin B.; Wang K.; Xu L.; Zhou M.; Kim D.; Song J. Carbazole-Containing Porphyrinoid and Its Oligomers. Chem. Commun. 2019, 55, 11454–11457. 10.1039/C9CC06114B. [DOI] [PubMed] [Google Scholar]
- Kalaiselvan A.; Vamsi Krishna I. S.; Nambiar A. P.; Edwin A.; Reddy V. S.; Gokulnath S. Carbazole-Based Porphyrins: Synthesis, Structure-Photophysical Property Correlations, and Mercury Ion Sensing. Org. Lett. 2020, 22, 4494–4499. 10.1021/acs.orglett.0c01500. [DOI] [PubMed] [Google Scholar]
- Maeda C.; Takata M.; Honsho A.; Ema T. Intramolecular Electronic Coupling in the Thiophene-Bridged Carbazole-Based Diporphyrin. Org. Lett. 2016, 18, 6070–6073. 10.1021/acs.orglett.6b03054. [DOI] [PubMed] [Google Scholar]
- Maeda C.; Shirakawa T.; Ema T. Synthesis and Electronic Properties of Carbazole-Based Core-Modified Diporphyrins Showing near Infrared Absorption. Chem. Commun. 2020, 56, 15048–15051. 10.1039/D0CC06289H. [DOI] [PubMed] [Google Scholar]
- Maulbetsch T.; Kunz D. Carbenaporphyrins: No Longer Missing Ligands in N-Heterocyclic Carbene Chemistry. Angew. Chem., Int. Ed. 2021, 60, 2007–2012. 10.1002/anie.202013434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Yu C.; Yin Y.; Droste J.; Klabunde S.; Hansen M. R.; Mai Y. Bis-Anthracene Fused Porphyrin as an Efficient Photocatalyst: Facile Synthesis and Visible-Light-Driven Oxidative Coupling of Amines. Chem. - Eur. J. 2020, 26, 16497–16503. 10.1002/chem.202003398. [DOI] [PubMed] [Google Scholar]
- Kato K.; Cha W.; Oh J.; Furukawa K.; Yorimitsu H.; Kim D.; Osuka A. Spontaneous Formation of an Air-Stable Radical upon the Direct Fusion of Diphenylmethane to a Triarylporphyrin. Angew. Chem., Int. Ed. 2016, 55, 8711–8714. 10.1002/anie.201602683. [DOI] [PubMed] [Google Scholar]
- Kato K.; Furukawa K.; Mori T.; Osuka A. Porphyrin-Based Air-Stable Helical Radicals. Chem. - Eur. J. 2018, 24, 572–575. 10.1002/chem.201705291. [DOI] [PubMed] [Google Scholar]
- Umetani M.; Naoda K.; Tanaka T.; Lee S.-K.; Oh J.; Kim D.; Osuka A. Synthesis of Di-Peri-Dinaphthoporphyrins by PtCl2-Mediated Cyclization of Quinodimethane-Type Porphyrins. Angew. Chem., Int. Ed. 2016, 55, 6305–6309. 10.1002/anie.201601303. [DOI] [PubMed] [Google Scholar]
- Umetani M.; Naoda K.; Tanaka T.; Osuka A. Synthesis and Antiaromatic Character of Alkyl-Substituted Di-Peri-Dinaphthoporphyrin Ni(II) Complex. J. Porphyrins Phthalocyanines 2017, 21, 850–856. 10.1142/S1088424617500808. [DOI] [Google Scholar]
- Fujimoto K.; Oh J.; Yorimitsu H.; Kim D.; Osuka A. Directly Diphenylborane-Fused Porphyrins. Angew. Chem., Int. Ed. 2016, 55, 3196–3199. 10.1002/anie.201511981. [DOI] [PubMed] [Google Scholar]
- Kato K.; Kim J. O.; Yorimitsu H.; Kim D.; Osuka A. Triphenylsilane-Fused Porphyrins. Chem. - Asian J. 2016, 11, 1738–1746. 10.1002/asia.201600424. [DOI] [PubMed] [Google Scholar]
- Fujimoto K.; Kasuga Y.; Fukui N.; Osuka A. Diphenylphosphine-Oxide-Fused and Diphenylphosphine-Fused Porphyrins: Synthesis, Tunable Electronic Properties, and Formation of Cofacial Dimers. Chem. - Eur. J. 2017, 23, 6741–6745. 10.1002/chem.201700909. [DOI] [PubMed] [Google Scholar]
- Fujimoto K.; Osuka A. Effective Stabilization of a Planar Phosphorus(III) Center Embedded in a Porphyrin-Based Fused Aromatic Skeleton. Chem. Sci. 2017, 8, 8231–8239. 10.1039/C7SC03882H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Brambilla L.; Daukiya L.; Mali K. S.; De Feyter S.; Tommasini M.; Müllen K.; Narita A. Synthesis of Triply Fused Porphyrin-Nanographene Conjugates. Angew. Chem., Int. Ed. 2018, 57, 11233–11237. 10.1002/anie.201805063. [DOI] [PubMed] [Google Scholar]
- Fukui N.; Lee S.-K.; Kato K.; Shimizu D.; Tanaka T.; Lee S.; Yorimitsu H.; Kim D.; Osuka A. Regioselective Phenylene-Fusion Reactions of Ni(II)-Porphyrins Controlled by an Electron-Withdrawing Meso-Substituent. Chem. Sci. 2016, 7, 4059–4066. 10.1039/C5SC04748J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. M.; Oleszak C.; Hampel F.; Jux N. Oxidative Cyclodehydrogenation Reactions with Tetraarylporphyrins. Eur. J. Org. Chem. 2020, 2020, 6758–6762. 10.1002/ejoc.202001174. [DOI] [Google Scholar]
- Kopp S. M.; Gotfredsen H.; Deng J.-R.; Claridge T. D. W.; Anderson H. L. Global Aromaticity in a Partially Fused 8-Porphyrin Nanoring. J. Am. Chem. Soc. 2020, 142, 19393–19401. 10.1021/jacs.0c09973. [DOI] [PubMed] [Google Scholar]
- Ooi S.; Adinarayana B.; Shimizu D.; Tanaka T.; Osuka A. Stable Meso-Meso-Linked 2NH-Corrole Radical Dimers as a Key Intermediate to Corrole Tape. Angew. Chem., Int. Ed. 2020, 59, 9423–9427. 10.1002/anie.202002976. [DOI] [PubMed] [Google Scholar]
- Ooi S.; Tanaka T.; Ikeue T.; Yamasumi K.; Ueta K.; Shimizu D.; Ishida M.; Furuta H.; Osuka A. Bis-Copper(II) Complex of Triply-Linked Corrole Dimer and Its Dication. Chem. - Asian J. 2019, 14, 1771–1776. 10.1002/asia.201801467. [DOI] [PubMed] [Google Scholar]
- Ooi S.; Shimizu D.; Furukawa K.; Tanaka T.; Osuka A. Stable Face-to-Face Singlet Diradicaloids: Triply Linked Corrole Dimer Gallium(III) Complexes with Two μ-Hydroxo-Bridges. Angew. Chem., Int. Ed. 2018, 57, 14916–14920. 10.1002/anie.201810200. [DOI] [PubMed] [Google Scholar]
- Lee S.; Yamashita K.; Sakata N.; Hirao Y.; Ogawa K.; Ogawa T. Stable Singlet Biradicals of Rare-Earth-Fused Diporphyrin-Triple-Decker Complexes with Low Energy Gaps and Multi-Redox States. Chem. - Eur. J. 2019, 10.1002/chem.201805927. [DOI] [PubMed] [Google Scholar]
- Mori H.; Kim T.; Kim D.; Osuka A. Trimeric and Tetrameric Electron-Deficient Porphyrin Tapes. Chem. - Asian J. 2016, 11, 1454–1463. 10.1002/asia.201600241. [DOI] [PubMed] [Google Scholar]
- Yamashita K.; Hirano D.; Fujimaki K.; Sugiura K. Synthesis of Porphyrinquinone and Doubly-Fused Diporphyrin Quinone Through Oxidation of Diarylporphyrins Using a Hypervalent Iodine Compound. Chem. - Asian J. 2020, 15, 3037–3043. 10.1002/asia.202000781. [DOI] [PubMed] [Google Scholar]
- Fukui N.; Kim T.; Kim D.; Osuka A. Porphyrin Arch-Tapes: Synthesis, Contorted Structures, and Full Conjugation. J. Am. Chem. Soc. 2017, 139, 9075–9088. 10.1021/jacs.7b05332. [DOI] [PubMed] [Google Scholar]
- Fukui N.; Osuka A. Singly and Doubly 1,2-Phenylene-Inserted Porphyrin Arch-Tape Dimers: Synthesis and Highly Contorted Structures. Angew. Chem., Int. Ed. 2018, 57, 6304–6308. 10.1002/anie.201802494. [DOI] [PubMed] [Google Scholar]
- Fukui N.; Osuka A. Singly and Doubly Sulfone-Inserted Porphyrin Arch-Tape Dimers. Bull. Chem. Soc. Jpn. 2018, 91, 1131–1137. 10.1246/bcsj.20180103. [DOI] [PubMed] [Google Scholar]
- Dimé A. K. D.; Devillers C. H.; Cattey H.; Lucas D. Versatile Redox Reactivity of Triaryl-Meso-Substituted Ni(II) Porphyrin. Dalton Trans. 2014, 43, 14554–14564. 10.1039/C4DT00221K. [DOI] [PubMed] [Google Scholar]
- Konev D. V.; Devillers C. H.; Lizgina K. V.; Baulin V. E.; Vorotyntsev M. A. Electropolymerization of Non-Substituted Mg(II) Porphine: Effects of Proton Acceptor Addition. J. Electroanal. Chem. 2015, 737, 235–242. 10.1016/j.jelechem.2014.09.018. [DOI] [Google Scholar]
- Rolle S. D.; Konev D. V.; Devillers C. H.; Lizgina K. V.; Lucas D.; Stern C.; Herbst F.; Heintz O.; Vorotyntsev M. A. Efficient Synthesis of a New Electroactive Polymer of Co(II) Porphine by in-Situ Replacement of Mg(II) inside Mg(II) Polyporphine Film. Electrochim. Acta 2016, 204, 276–286. 10.1016/j.electacta.2016.03.039. [DOI] [Google Scholar]
- Rolle S. D.; Devillers C. H.; Fournier S.; Heintz O.; Gibault H.; Lucas D. A Glassy Carbon Electrode Modified by a Triply-Fused-like Co(II) Polyporphine and Its Ability for Sulphite Oxidation and Detection. New J. Chem. 2018, 42, 8180–8189. 10.1039/C7NJ04370H. [DOI] [Google Scholar]
- Bengasi G.; Baba K.; Frache G.; Desport J.; Gratia P.; Heinze K.; Boscher N. D. Conductive Fused Porphyrin Tapes on Sensitive Substrates by a Chemical Vapor Deposition Approach. Angew. Chem., Int. Ed. 2019, 58, 2103–2108. 10.1002/anie.201814034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba K.; Bengasi G.; El Assad D.; Grysan P.; Lentzen E.; Heinze K.; Frache G.; Boscher N. D. Conductive Directly Fused Poly(Porphyrin) Coatings by Oxidative Chemical Vapour Deposition - From Single- to Triple-Fused. Eur. J. Org. Chem. 2019, 2019, 2368–2375. 10.1002/ejoc.201900045. [DOI] [Google Scholar]
- Bengasi G.; Baba K.; Back O.; Frache G.; Heinze K.; Boscher N. D. Reactivity of Nickel(II) Porphyrins in OCVD Processes—Polymerisation, Intramolecular Cyclisation and Chlorination. Chem. - Eur. J. 2019, 25, 8313–8320. 10.1002/chem.201900793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengasi G.; Desport J. S.; Baba K.; Cosas Fernandes J. P.; De Castro O.; Heinze K.; Boscher N. D. Molecular Flattening Effect to Enhance the Conductivity of Fused Porphyrin Tape Thin Films. RSC Adv. 2020, 10, 7048–7057. 10.1039/C9RA09711B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengasi G.; Quétu L.; Baba K.; Ost A.; Fernandes J. P. C.; Grysan P.; Heinze K.; Boscher N. D. Constitution and Conductivity of Metalloporphyrin Tapes. Eur. J. Inorg. Chem. 2020, 2020, 1938–1945. 10.1002/ejic.202000243. [DOI] [Google Scholar]
- Baba K.; Bengasi G.; Loyer F.; Fernandes J. P. C.; El Assad D.; De Castro O.; Boscher N. D. Fused Metalloporphyrin Thin Film with Tunable Porosity via Chemical Vapor Deposition. ACS Appl. Mater. Interfaces 2020, 12, 37732–37740. 10.1021/acsami.0c09630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Flores A. M.; Bengasi G.; Baba K.; Boscher N. D. Fused Porphyrin Thin Films as Heterogeneous Visible-Light Active Photocatalysts with Well-Defined Active Metal Sites for Hydrogen Generation. ACS Appl. Energy Mater. 2020, 3, 9848–9855. 10.1021/acsaem.0c01545. [DOI] [Google Scholar]
- Bengasi G.; Meunier-Prest R.; Baba K.; Kumar A.; Pellegrino A. L.; Boscher N. D.; Bouvet M. Molecular Engineering of Porphyrin-Tapes/Phthalocyanine Heterojunctions for a Highly Sensitive Ammonia Sensor. Adv. Electron. Mater. 2020, 6, 2000812. 10.1002/aelm.202000812. [DOI] [Google Scholar]
- Mateo L. M.; Sun Q.; Liu S.-X.; Bergkamp J. J.; Eimre K.; Pignedoli C. A.; Ruffieux P.; Decurtins S.; Bottari G.; Fasel R.; et al. On-Surface Synthesis and Characterization of Triply Fused Porphyrin-Graphene Nanoribbon Hybrids. Angew. Chem., Int. Ed. 2020, 59, 1334–1339. 10.1002/anie.201913024. [DOI] [PubMed] [Google Scholar]
- Mateo L. M.; Sun Q.; Eimre K.; Pignedoli C. A.; Torres T.; Fasel R.; Bottari G. On-Surface Synthesis of Singly and Doubly Porphyrin-Capped Graphene Nanoribbon Segments. Chem. Sci. 2021, 12, 247–252. 10.1039/D0SC04316H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forezi L. d. S. M.; Gomes A. T. P. C.; Neves M. G. P. M. S.; Ferreira V. F.; Boechat F. d. C. S.; de Souza M. C. B. V.; Cavaleiro J. A. S. Synthesis of β-Substituted Meso-Tetraaryl-21,23-Dithiaporphyrins by Heck Reaction. Eur. J. Org. Chem. 2015, 2015, 5909–5913. 10.1002/ejoc.201500812. [DOI] [Google Scholar]
- Tebi S.; Paszkiewicz M.; Aldahhak H.; Allegretti F.; Gonglach S.; Haas M.; Waser M.; Deimel P. S.; Aguilar P. C.; Zhang Y.-Q.; et al. On-Surface Site-Selective Cyclization of Corrole Radicals. ACS Nano 2017, 11, 3383–3391. 10.1021/acsnano.7b00766. [DOI] [PubMed] [Google Scholar]
- Saegusa Y.; Ishizuka T.; Shiota Y.; Yoshizawa K.; Kojima T. Acid-Base Properties of a Freebase Form of a Quadruply Ring-Fused Porphyrin—Stepwise Protonation Induced by Rigid Ring-Fused Structure. J. Org. Chem. 2017, 82, 322–330. 10.1021/acs.joc.6b02419. [DOI] [PubMed] [Google Scholar]
- Saegusa Y.; Ishizuka T.; Kojima T. Substituent Effects at the β-Positions of the Nonfused Pyrroles in a Quadruply Fused Porphyrin on the Structure and Optical and Electrochemical Properties. Inorg. Chem. 2018, 57, 1106–1115. 10.1021/acs.inorgchem.7b02551. [DOI] [PubMed] [Google Scholar]
- Saegusa Y.; Ishizuka T.; Shiota Y.; Yoshizawa K.; Kojima T. NH Tautomerism of a Quadruply Fused Porphyrin: Rigid Fused Structure Delays the Proton Transfer. J. Phys. Chem. B 2018, 122, 316–327. 10.1021/acs.jpcb.7b10945. [DOI] [PubMed] [Google Scholar]
- Li J.; He N.; Liu Y.; Zhang Z.; Zhang X.; Han X.; Gai Y.; Liu Y.; Yin J.; Wang J. Synthesis and Photophysical Properties of Novel Pyridine Fused Chlorophyll a Derivatives. Dyes Pigm. 2017, 146, 189–198. 10.1016/j.dyepig.2017.07.005. [DOI] [Google Scholar]
- Higashino T.; Nishimura I.; Imahori H. Phosphole-Fused Dehydropurpurins via Titanium-Mediated [2 + 2+1] Cyclization Strategy. Chem. - Eur. J. 2019, 25, 13816–13823. 10.1002/chem.201903269. [DOI] [PubMed] [Google Scholar]
- Higashino T.; Nishimura I.; Imahori H. Unique Role of Heterole-Fused Structures in Aromaticity and Physicochemical Properties of 7,8-Dehydropurpurins. Chem. - Eur. J. 2020, 26, 12043–12049. 10.1002/chem.202001361. [DOI] [PubMed] [Google Scholar]
- Nagai T.; Takiguchi A.; Ueda M.; Oda K.; Hiroto S.; Shinokubo H. X-Shaped Cyclobutane-Linked Tetraporphyrins through a Thermal [2 + 2] Cycloaddition of Etheno-Fused Diporphyrins. J. Am. Chem. Soc. 2018, 140, 8392–8395. 10.1021/jacs.8b04673. [DOI] [PubMed] [Google Scholar]
- Miyagawa K.; Hisaki I.; Fukui N.; Shinokubo H. Redox-Induced Reversible [2 + 2] Cycloaddition of an Etheno-Fused Diporphyrin. Chem. Sci. 2021, 12, 5224–5229. 10.1039/D1SC00438G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Wei P.; Ishida M.; Li X.; Savage M.; Guo R.; Ou Z.; Yang S.; Furuta H.; Xie Y. Macrocyclic Transformations from Norrole to Isonorrole and an N-Confused Corrole with a Fused Hexacyclic Ring System Triggered by a Pyrrole Substituent. Angew. Chem., Int. Ed. 2016, 55, 3063–3067. 10.1002/anie.201510879. [DOI] [PubMed] [Google Scholar]
- Kumar Maurya Y.; Wei P.; Shimada T.; Yamasumi K.; Mori S.; Furukawa K.; Kusaba H.; Ishihara T.; Xie Y.; Ishida M.; Furuta H.; et al. Chiral Interlocked Corrole Dimers Directly Linked at Inner Carbon Atoms of Confused Pyrrole Rings. Chem. - Asian J. 2021, 16, 743–747. 10.1002/asia.202100083. [DOI] [PubMed] [Google Scholar]
- Fujimoto K.; Shimizu D.; Osuka A. Porphyrin-Stabilized Nitrenium Dication. Chem. - Eur. J. 2018, 25, 521–525. 10.1002/chem.201805491. [DOI] [PubMed] [Google Scholar]
- Fujimoto K.; Shimizu D.; Mori T.; Li Y.; Zhou M.; Song J.; Osuka A. Selective Formation of Helical Tetrapyrrin-Fused Porphyrins by Oxidation of β-to-β Linked Meso-Aminoporphyrin Dimers. Chem. - Eur. J. 2018, 25, 1711–1715. 10.1002/chem.201805659. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhou M.; Xu L.; Zhou B.; Rao Y.; Nie H.; Gu T.; Zhou J.; Liang X.; Yin B.; et al. Simultaneous Implementation of N-Heterocycle-Fused Bridge and Modified Pyrrole Unit on Ni(II) Porphyrin Dimers. Org. Lett. 2020, 22, 6001–6005. 10.1021/acs.orglett.0c02084. [DOI] [PubMed] [Google Scholar]
- Isar P.; Ravikanth M. Dibenzoylbenzodipyrroles: Key Precursors for the Synthesis of Fused Meso -Aryl Sapphyrins. J. Org. Chem. 2020, 85, 7287–7296. 10.1021/acs.joc.0c00663. [DOI] [PubMed] [Google Scholar]
- Firmansyah D.; Hong S.-J.; Dutta R.; He Q.; Bae J.; Jo H.; Kim H.; Ok K. M.; Lynch V. M.; Byon H. R.; et al. Trapping of Stable [4n+1] π-Electron Species from Peripherally Substituted, Conformationally Rigid, Antiaromatic Hexaphyrins. Chem. - Eur. J. 2019, 25, 3525–3531. 10.1002/chem.201900022. [DOI] [PubMed] [Google Scholar]
- Ishida M.; Furuyama T.; Lim J. M.; Lee S.; Zhang Z.; Ghosh S. K.; Lynch V. M.; Lee C.-H.; Kobayashi N.; Kim D.; et al. Structural, Photophysical, and Magnetic Circular Dichroism Studies of Three Rigidified Meso-Pentafluorophenyl-Substituted Hexaphyrin Analogues. Chem. - Eur. J. 2017, 23, 6682–6692. 10.1002/chem.201700759. [DOI] [PubMed] [Google Scholar]
- Kim G.; Dutta R.; Cha W.-Y.; Hong S.-J.; Oh J.; Firmansyah D.; Jo H.; Ok K. M.; Lee C.-H.; Kim D. Noncovalent Intermolecular Interaction in Cofacially Stacked 24π Antiaromatic Hexaphyrin Dimer. Chem. - Eur. J. 2020, 26, 16434–16440. 10.1002/chem.202002884. [DOI] [PubMed] [Google Scholar]
- Dutta R.; Chandra B.; Hong S.-J.; Park Y.; Jung Y. M.; Lee C.-H. Post Synthetic Modification of Planar Antiaromatic Hexaphyrin (1.0.1.0.1.0) by Regio-Selective, Sequential SNAr. Molecules 2021, 26, 1025. 10.3390/molecules26041025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samala S.; Dutta R.; He Q.; Park Y.; Chandra B.; Lynch V. M.; Jung Y. M.; Sessler J. L.; Lee C.-H. A Robust Bis-Rhodium(I) Complex of π-Extended Planar, Anti-Aromatic Hexaphyrin[1.0.1.0.1.0]. Chem. Commun. 2020, 56, 758–761. 10.1039/C9CC09221H. [DOI] [PubMed] [Google Scholar]
- Sarma T.; Kim G.; Sen S.; Cha W.-Y.; Duan Z.; Moore M. D.; Lynch V. M.; Zhang Z.; Kim D.; Sessler J. L. Proton-Coupled Redox Switching in an Annulated π-Extended Core-Modified Octaphyrin. J. Am. Chem. Soc. 2018, 140, 12111–12119. 10.1021/jacs.8b06938. [DOI] [PubMed] [Google Scholar]
- Bhowmik S.; Kosa M.; Mizrahi A.; Fridman N.; Saphier M.; Stanger A.; Gross Z. The Planar Cyclooctatetraene Bridge in Bis-Metallic Macrocycles: Isolating or Conjugating?. Inorg. Chem. 2017, 56, 2287–2296. 10.1021/acs.inorgchem.6b02944. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Chaudhary A.; Srinivasan A.; Suresh C. H.; Chandrashekar T. K. [32]π Fused Core-Modified Heptaphyrin with Möbius Aromaticity. Chem. - Eur. J. 2016, 22, 3942–3946. 10.1002/chem.201505116. [DOI] [PubMed] [Google Scholar]
- Dash S.; Ghosh A.; Chandrashekar T. K. Dithienothiophene Fused 30π Heptaphyrin and 34π Octaphyrins: Syntheses, Characterization and Spectral Properties. J. Porphyrins Phthalocyanines 2020, 24, 98–104. 10.1142/S1088424619500974. [DOI] [Google Scholar]
- Ghosh A.; Srinivasan A.; Suresh C. H.; Chandrashekar T. K. [40]π Fused and Nonfused Core-Modified Nonaphyrins: Syntheses and Structural Diversity. Chem. - Eur. J. 2016, 22, 11152–11155. 10.1002/chem.201602245. [DOI] [PubMed] [Google Scholar]
- Cha W.-Y.; Kim T.; Ghosh A.; Zhang Z.; Ke X.-S.; Ali R.; Lynch V. M.; Jung J.; Kim W.; Lee S.; et al. Bicyclic Baird-Type Aromaticity. Nat. Chem. 2017, 9, 1243–1248. 10.1038/nchem.2834. [DOI] [PubMed] [Google Scholar]
- Cha W.-Y.; Ahn A.; Kim T.; Oh J.; Ali R.; Park J. S.; Kim D. Changes in Macrocyclic Aromaticity and Formation of a Charge-Separated State by Complexation of Expanded Porphyrin and C60. Chem. Commun. 2019, 55, 8301–8304. 10.1039/C9CC03928G. [DOI] [PubMed] [Google Scholar]
- Higashino T.; Kumagai A.; Imahori H. Thiophene-Fused Dithiaoctaphyrins: π-System Switching between Cross-Conjugated and Macrocyclic π-Networks. Chem. Commun. 2017, 53, 5091–5094. 10.1039/C7CC01273J. [DOI] [PubMed] [Google Scholar]
- Higashino T.; Kumagai A.; Sakaki S.; Imahori H. Reversible π-System Switching of Thiophene-Fused Thiahexaphyrins by Solvent and Oxidation/Reduction. Chem. Sci. 2018, 9, 7528–7539. 10.1039/C8SC02448K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B. K.; Rawson J.; Gadekar S. C.; Kögerler P.; Anand V. G. A Naphthalene-Fused Dimer of an Anti-Aromatic Expanded Isophlorin. Chem. Commun. 2017, 53, 8211–8214. 10.1039/C7CC04050D. [DOI] [PubMed] [Google Scholar]
- Ke X.-S.; Hong Y.; Tu P.; He Q.; Lynch V. M.; Kim D.; Sessler J. L. Hetero Cu(III)-Pd(II) Complex of a Dibenzo[g, p]Chrysene-Fused Bis-Dicarbacorrole with Stable Organic Radical Character. J. Am. Chem. Soc. 2017, 139, 15232–15238. 10.1021/jacs.7b09167. [DOI] [PubMed] [Google Scholar]
- Ke X.-S.; Hong Y.; Lynch V. M.; Kim D.; Sessler J. L. Metal-Stabilized Quinoidal Dibenzo[g, p]Chrysene-Fused Bis-Dicarbacorrole System. J. Am. Chem. Soc. 2018, 140, 7579–7586. 10.1021/jacs.8b02718. [DOI] [PubMed] [Google Scholar]
- Ke X.-S.; Kim T.; He Q.; Lynch V. M.; Kim D.; Sessler J. L. Three-Dimensional Fully Conjugated Carbaporphyrin Cage. J. Am. Chem. Soc. 2018, 140, 16455–16459. 10.1021/jacs.8b11158. [DOI] [PubMed] [Google Scholar]
- Szyszko B.; Przewoznik M.; Białek M. J.; Białońska A.; Chmielewski P. J.; Cichos J.; Latos-Grazyński L. Helicenophyrins: Expanded Carbaporphyrins Incorporating Aza[5]Helicene and Heptacyclic S-Shaped Aza[5]Helicene Motifs. Angew. Chem., Int. Ed. 2018, 57, 4030–4034. 10.1002/anie.201800879. [DOI] [PubMed] [Google Scholar]
- Kupietz K.; Białek M. J.; Białońska A.; Szyszko B.; Latos-Grazyński L. Aromaticity Control via Modifications of a Macrocyclic Frame: 5,6-Dimethoxyphenanthriporphyrin and 5,6-Dioxophenanthriporphyrin. Org. Chem. Front. 2018, 5, 3068–3076. 10.1039/C8QO00751A. [DOI] [Google Scholar]
- Kupietz K.; Białek M. J.; Białońska A.; Szyszko B.; Latos-Grazyński L. Organocopper(III) Phenanthriporphyrin—Exocyclic Transformations. Inorg. Chem. 2019, 58, 1451–1461. 10.1021/acs.inorgchem.8b02997. [DOI] [PubMed] [Google Scholar]
- Szyszko B.; Drózdz D.; Sarwa A.; Mucha S. G.; Białońska A.; Białek M. J.; Matczyszyn K.; Latos-Grazyński L. An Exocyclic π-System Extension of the Phenanthriporphyrin Framework: Towards Azaaceneporphyrinoids. Org. Chem. Front. 2020, 7, 1430–1436. 10.1039/D0QO00436G. [DOI] [Google Scholar]
- Szyszko B.; Małecki M.; Berlicka A.; Białek M. J.; Białońska A.; Kupietz K.; Pacholska-Dudziak E.; Latos-Grazyński L. Incorporation of a Phenanthrene Subunit into a Sapphyrin Framework: Synthesis of Expanded Aceneporphyrinoids. Chem. - Eur. J. 2016, 22, 7602–7608. 10.1002/chem.201600606. [DOI] [PubMed] [Google Scholar]
- Szyszko B.; Chmielewski P. J.; Przewoznik M.; Białek M. J.; Kupietz K.; Białońska A.; Latos-Grazyński L. Diphenanthrioctaphyrin(1.1.1.0.1.1.1.0): Conformational Switching Controls the Stereochemical Dynamics of the Topologically Chiral System. J. Am. Chem. Soc. 2019, 141, 6060–6072. 10.1021/jacs.9b01357. [DOI] [PubMed] [Google Scholar]
- Szyszko B.; Przewoznik M.; Białek M. J.; Białońska A.; Chmielewski P. J.; Latos-Grazyński L. Conformation-Dependent Response to the Protonation of Diphenanthrioctaphyrin(1.1.1.0.1.1.1.0): A Route to Pseudorotaxane-Like Structures. Chem. - Eur. J. 2020, 26, 8555–8566. 10.1002/chem.202000940. [DOI] [PubMed] [Google Scholar]
- Szyszko B.; Rymut P.; Matviyishyn M.; Białońska A.; Latos-Grazyński L. Kinetic versus Thermodynamic Control Over Multiple Conformations of Di-2,7-Naphthihexaphyrin(1.1.1.1.1.1). Angew. Chem., Int. Ed. 2020, 59, 20137–20146. 10.1002/anie.202008518. [DOI] [PubMed] [Google Scholar]
- Aslam A. S.; Hong J.-H.; Shin J.-H.; Cho D.-G. Synthesis of a Phlorin from a Meso-Fused Anthriporphyrin by a Diels-Alder Strategy. Angew. Chem., Int. Ed. 2017, 56, 16247–16251. 10.1002/anie.201709026. [DOI] [PubMed] [Google Scholar]
- Hong J.-H.; Aslam A. S.; Ishida M.; Mori S.; Furuta H.; Cho D.-G. 2-(Naphthalen-1-yl)Thiophene as a New Motif for Porphyrinoids: Meso-Fused Carbaporphyrin. J. Am. Chem. Soc. 2016, 138, 4992–4995. 10.1021/jacs.6b01063. [DOI] [PubMed] [Google Scholar]
- Hong J.-H.; Aslam A. S.; Cho B.; Ko M.-S.; Kim Y.; Heo J.; Cho D.-G. Carbaporphyrin Dimers That Bear a Rigid Naphthalene Motif as an Internal Strap. Org. Lett. 2021, 23, 1846–1850. 10.1021/acs.orglett.1c00252. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Thorat K. G.; Sinha A.; Butcher R. J.; Ravikanth M. Meso-Fused Carbatriphyrins(2.1.1) and Its Organo Phosphorus(V) Complex. J. Org. Chem. 2019, 84, 9067–9074. 10.1021/acs.joc.9b01015. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Laxman K.; Ravikanth M. Antiaromatic Carbaporphyrinoids: Fluorene as a Fused Motif toward the Synthesis of Meso-Fused Heterobenziporphyrins. Org. Lett. 2019, 21, 8726–8730. 10.1021/acs.orglett.9b03329. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Thorat K. G.; Ravikanth M. Synthesis and Studies of Strained Fluorenophyrins. J. Org. Chem. 2019, 84, 10321–10327. 10.1021/acs.joc.9b01486. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Rajeswara Rao M.; Lee W.-Z.; Ravikanth M. Hybrid Macrocycles of Subporphyrins and Triphyrins. Org. Lett. 2017, 19, 5924–5927. 10.1021/acs.orglett.7b02919. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Thorat K. G.; Ravikanth M. Benzofuran-/Benzothiophene-Incorporated NIR-Absorbing Triphyrins(2.1.1). Org. Lett. 2018, 20, 4871–4874. 10.1021/acs.orglett.8b02012. [DOI] [PubMed] [Google Scholar]
- Szyszko B.; Matviyishyn M.; Hirka S.; Pacholska-Dudziak E.; Białońska A.; Latos-Grazyński L. 28-Hetero-2,7-Naphthiporphyrins: Horizontal Expansion of the m-Benziporphyrin Macrocycle. Org. Lett. 2019, 21, 7009–7014. 10.1021/acs.orglett.9b02587. [DOI] [PubMed] [Google Scholar]
- Rao Y.; Kim J. O.; Kim W.; Zhong G.; Yin B.; Zhou M.; Shinokubo H.; Aratani N.; Tanaka T.; Liu S.; et al. β-to-β 2,5-Pyrrolylene-Linked Cyclic Porphyrin Oligomers. Chem. - Eur. J. 2016, 22, 8801–8804. 10.1002/chem.201601306. [DOI] [PubMed] [Google Scholar]
- Yin B.; Liang X.; Zhu W.; Xu L.; Zhou M.; Song J. β to β Terpyridylene-Bridged Porphyrin Nanorings. Chin. Chem. Lett. 2018, 29, 99–101. 10.1016/j.cclet.2017.05.003. [DOI] [Google Scholar]
- Xu Y.; Gsänger S.; Minameyer M. B.; Imaz I.; Maspoch D.; Shyshov O.; Schwer F.; Ribas X.; Drewello T.; Meyer B.; et al. Highly Strained, Radially π-Conjugated Porphyrinylene Nanohoops. J. Am. Chem. Soc. 2019, 141, 18500–18507. 10.1021/jacs.9b08584. [DOI] [PubMed] [Google Scholar]
- Ren L.; Gopalakrishna T. Y.; Park I.-H.; Han Y.; Wu J. Porphyrin/Quinoidal-Bithiophene-Based Macrocycles and Their Dications: Template-Free Synthesis and Global Aromaticity. Angew. Chem., Int. Ed. 2020, 59, 2230–2234. 10.1002/anie.201911269. [DOI] [PubMed] [Google Scholar]
- Sung Y. M.; Oh J.; Kim W.; Mori H.; Osuka A.; Kim D. Switching between Aromatic and Antiaromatic 1,3-Phenylene-Strapped [26]- and [28]Hexaphyrins upon Passage to the Singlet Excited State. J. Am. Chem. Soc. 2015, 137, 11856–11859. 10.1021/jacs.5b04047. [DOI] [PubMed] [Google Scholar]
- Oh J.; Sung Y. M.; Mori H.; Park S.; Jorner K.; Ottosson H.; Lim M.; Osuka A.; Kim D. Unraveling Excited-Singlet-State Aromaticity via Vibrational Analysis. Chem. 2017, 3, 870–880. 10.1016/j.chempr.2017.09.005. [DOI] [Google Scholar]
- Soya T.; Mori H.; Osuka A. Quadruply Twisted Hückel-Aromatic Dodecaphyrin. Angew. Chem., Int. Ed. 2018, 57, 15882–15886. 10.1002/anie.201811433. [DOI] [PubMed] [Google Scholar]
- Soya T.; Mori H.; Hong Y.; Koo Y. H.; Kim D.; Osuka A. Internally 2,5-Thienylene-Bridged [46]Decaphyrin: (Annuleno)Annulene Network Consisting of Möbius Aromatic Thia[28]Hexaphyrins and Strong Hückel Aromaticity of Its Protonated Form. Angew. Chem., Int. Ed. 2017, 56, 3232–3236. 10.1002/anie.201700607. [DOI] [PubMed] [Google Scholar]
- Soya T.; Osuka A. Internally Bridged Hückel Aromatic [46]Decaphyrins: (Doubly-Twisted-Annuleno)Doubly-Twisted-Annulene Variants. Chem. - Eur. J. 2019, 25, 5173–5176. 10.1002/chem.201900819. [DOI] [PubMed] [Google Scholar]
- Ni Y.; Gopalakrishna T. Y.; Phan H.; Kim T.; Herng T. S.; Han Y.; Tao T.; Ding J.; Kim D.; Wu J. 3D Global Aromaticity in a Fully Conjugated Diradicaloid Cage at Different Oxidation States. Nat. Chem. 2020, 12, 242–248. 10.1038/s41557-019-0399-2. [DOI] [PubMed] [Google Scholar]
- Białek M.ł J.; Latos-Grazynski L.ła. Aromaticity Switching via Azulene Transformations in Azulene-Bridged A,D-Dithiahexaphyrin. Chem. Commun. 2018, 54, 1837–1840. 10.1039/C7CC08754C. [DOI] [PubMed] [Google Scholar]
- Rao Y.; Kim T.; Park K. H.; Peng F.; Liu L.; Liu Y.; Wen B.; Liu S.; Kirk S. R.; Wu L.; et al. π-Extended “Earring” Porphyrins with Multiple Cavities and Near-Infrared Absorption. Angew. Chem., Int. Ed. 2016, 55, 6438–6442. 10.1002/anie.201600955. [DOI] [PubMed] [Google Scholar]
- Guan H.; Zhou M.; Yin B.; Xu L.; Song J. Synthesis and Characterization of π-Extended “Earring” Subporphyrins. Beilstein J. Org. Chem. 2018, 14, 1956–1960. 10.3762/bjoc.14.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Li F.; Rao Y.; Wen B.; Xu L.; Zhou M.; Tanaka T.; Osuka A.; Song J. Synthesis, Structures, and Near-IR Absorption of Heterole-Fused Earring Porphyrins. Angew. Chem., Int. Ed. 2019, 58, 8124–8128. 10.1002/anie.201903446. [DOI] [PubMed] [Google Scholar]
- Stawski W.; Hurej K.; Skonieczny J.; Pawlicki M. Organoboron Complexes in Edge-Sharing Macrocycles: The Triphyrin(2.1.1)-Tetraphyrin(1.1.1.1) Hybrid. Angew. Chem., Int. Ed. 2019, 58, 10946–10950. 10.1002/anie.201904819. [DOI] [PubMed] [Google Scholar]
- Basumatary B.; Ramana Reddy R. V.; Rahul; Sankar J. The Curious Case of a Parasitic Twin of the Corroles. Angew. Chem., Int. Ed. 2018, 57, 5052–5056. 10.1002/anie.201801555. [DOI] [PubMed] [Google Scholar]
- Xue S.; Kuzuhara D.; Aratani N.; Yamada H. Synthesis of a Porphyrin(2.1.2.1) Nanobelt and Its Ability To Bind Fullerene. Org. Lett. 2019, 21, 2069–2072. 10.1021/acs.orglett.9b00329. [DOI] [PubMed] [Google Scholar]
- Meng Z.; Stolz R. M.; Mirica K. A. Two-Dimensional Chemiresistive Covalent Organic Framework with High Intrinsic Conductivity. J. Am. Chem. Soc. 2019, 141, 11929–11937. 10.1021/jacs.9b03441. [DOI] [PubMed] [Google Scholar]
- Wang M.; Ballabio M.; Wang M.; Lin H.-H.; Biswal B. P.; Han X.; Paasch S.; Brunner E.; Liu P.; Chen M.; et al. Unveiling Electronic Properties in Metal-Phthalocyanine-Based Pyrazine-Linked Conjugated Two-Dimensional Covalent Organic Frameworks. J. Am. Chem. Soc. 2019, 141, 16810–16816. 10.1021/jacs.9b07644. [DOI] [PubMed] [Google Scholar]
- Wang M.; Wang M.; Lin H.-H.; Ballabio M.; Zhong H.; Bonn M.; Zhou S.; Heine T.; Cánovas E.; Dong R.; et al. High-Mobility Semiconducting Two-Dimensional Conjugated Covalent Organic Frameworks with p-Type Doping. J. Am. Chem. Soc. 2020, 142, 21622–21627. 10.1021/jacs.0c10482. [DOI] [PubMed] [Google Scholar]
- Uno H.; Muramatsu K.; Hiraoka S.; Tahara H.; Hirose M.; Tamura E.; Shiraishi T.; Mack J.; Kobayashi N.; Mori S.; et al. Synthesis and Aromaticity of Benzene-Fused Doubly N-Confused Porphyrins. Chem. - Eur. J. 2020, 26, 5701–5708. 10.1002/chem.202000339. [DOI] [PubMed] [Google Scholar]
- Yoneda T.; Hoshino T.; Neya S. Stable Non-Fused [22]Pentaphyrins and A Fused [24]Pentaphyrin Displaying Crystal Polymorphism between Hückel and Möbius Structures. Chem. - Asian J. 2017, 12, 405–409. 10.1002/asia.201601689. [DOI] [PubMed] [Google Scholar]
- Ishida S.; Kim J. O.; Kim D.; Osuka A. Doubly N-Fused [24]Pentaphyrin Silicon Complex and Its Fluorosilicate: Enhanced Möbius Aromaticity in the Fluorosilicate. Chem. - Eur. J. 2016, 22, 16554–16561. 10.1002/chem.201603598. [DOI] [PubMed] [Google Scholar]
- Nakai A.; Yoneda T.; Tanaka T.; Osuka A. PdII Insertion-Triggered Meso-Carbon Extrusion of N-Fused Pentaphyrin to Form N-Fused Sapphyrin PdII Complexes. Chem. Commun. 2021, 57, 3034–3037. 10.1039/D1CC00518A. [DOI] [PubMed] [Google Scholar]
- Li Q.; Ishida M.; Kai H.; Gu T.; Li C.; Li X.; Baryshnikov G.; Liang X.; Zhu W.; Ågren H.; et al. Skeletal Rearrangement of Twisted Thia-Norhexaphyrin: Multiply Annulated Polypyrrolic Aromatic Macrocycles. Angew. Chem., Int. Ed. 2019, 58, 5925–5929. 10.1002/anie.201900010. [DOI] [PubMed] [Google Scholar]
- Jana A.; Ishida M.; Furuta H. Benzo-Tetrathiafulvalene- (BTTF-) Annulated Expanded Porphyrins: Potential Next-Generation Multielectron Reservoirs. Chem. - Eur. J. 2021, 27, 4466–4472. 10.1002/chem.202005021. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Liu X.; Li C.; Tong Z.; Baryshnikov G.; Ågren H.; Li Q.; Xie Y. Rational Synthesis of 5,5,5-Tricyclic Fused Thia-Heptaphyrin (1.1.1.1.1.1.0) From a Helical Oligopyrrin Hybrid. Chem. - Asian J. 2020, 15, 1285–1289. 10.1002/asia.202000100. [DOI] [PubMed] [Google Scholar]
- Saltsman I.; Goldberg I.; Gross Z. One-Step Conversions of a Simple Corrole into Chiral and Amphiphilic Derivatives. Tetrahedron Lett. 2003, 44, 5669–5673. 10.1016/S0040-4039(03)01356-X. [DOI] [Google Scholar]
- Saltsman I.; Simkhovich L.; Balazs Y.; Goldberg I.; Gross Z. Synthesis, Spectroscopy, and Structures of New Rhodium(I) and Rhodium(III) Corroles and Catalysis Thereby. Inorg. Chim. Acta 2004, 357, 3038–3046. 10.1016/j.ica.2004.05.002. [DOI] [Google Scholar]
- Patra S. K.; Sahu K.; Patra B.; Sahoo D. K.; Mondal S.; Mukherjee P.; Biswal H. S.; Kar S. Synthesis of Urea Derivatives via Reductive Carbon Dioxide Fixation into Contracted Porphyrin Analogues. Green Chem. 2017, 19, 5772–5776. 10.1039/C7GC02798B. [DOI] [Google Scholar]
- Idec A.; Pawlicki M.; Latos-Grazyński L. Three-Stage Aromaticity Switching in Boron(III) and Phosphorus(V) N-Fused p-Benziporphyrin. Chem. - Eur. J. 2019, 25, 200–204. 10.1002/chem.201804983. [DOI] [PubMed] [Google Scholar]
- Marshall-Roth T.; Libretto N. J.; Wrobel A. T.; Anderton K. J.; Pegis M. L.; Ricke N. D.; Voorhis T. V.; Miller J. T.; Surendranath Y. A Pyridinic Fe-N4Macrocycle Models the Active Sites in Fe/N-Doped Carbon Electrocatalysts. Nat. Commun. 2020, 11, 5283. 10.1038/s41467-020-18969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya M.; Takahama R.; Kamoi K.; Ohyama J.; Kawashima S.; Kojima R.; Okada M.; Hayakawa T.; Nabae Y. Fourteen-Membered Macrocyclic Fe Complexes Inspired by FeN 4 -Center-Embedded Graphene for Oxygen Reduction Catalysis. J. Phys. Chem. C 2020, 124, 20730–20735. 10.1021/acs.jpcc.0c05536. [DOI] [Google Scholar]
- Adachi S.; Shibasaki M.; Kumagai N. TriQuinoline. Nat. Commun. 2019, 10, 3820. 10.1038/s41467-019-11818-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimasaki T.; Kuroda R.; Akao M.; Akimoto T.; Ishikawa T.; Iwanaga T.; Teramoto N.; Shibata M. Synthesis and Properties of a Conjugated Macrocyclic Molecule Incorporating Two Quinoline Moieties. Chem. Lett. 2019, 48, 133–136. 10.1246/cl.180888. [DOI] [Google Scholar]
- Ikemoto K.; Yang S.; Naito H.; Kotani M.; Sato S.; Isobe H. A Nitrogen-Doped Nanotube Molecule with Atom Vacancy Defects. Nat. Commun. 2020, 11, 1807. 10.1038/s41467-020-15662-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Tanaka T.; Hong Y. S.; Mori T.; Kim D.; Osuka A. Closed Pentaaza[9]Helicene and Hexathia[9]/[5]Helicene: Oxidative Fusion Reactions of Ortho-Phenylene-Bridged Cyclic Hexapyrroles and Hexathiophenes. Angew. Chem., Int. Ed. 2017, 56, 14688–14693. 10.1002/anie.201708429. [DOI] [PubMed] [Google Scholar]
- Yamamoto K.; Pandit P.; Higashibayashi S. Non-Planar [n]Cyclo-1,8-Carbazolylenes (N = 3,4,6) and [3]Cyclo-1,8-Carbazolylenyl B, P, PO, SiPh Complexes. Chem. - Eur. J. 2017, 23, 14011–14016. 10.1002/chem.201702853. [DOI] [PubMed] [Google Scholar]
- Yasui M.; Ohtsu H.; Kawano M.; Hanaya K.; Sugai T.; Higashibayashi S. Dearomative Oxidative Rearrangement of [3]Cyclo-1,8-Carbazolylene. Chem. Lett. 2018, 47, 1357–1359. 10.1246/cl.180637. [DOI] [Google Scholar]
- Kuroda Y.; Sakamoto Y.; Suzuki T.; Kayahara E.; Yamago S. Tetracyclo(2,7-Carbazole)s: Diatropicity and Paratropicity of Inner Regions of Nanohoops. J. Org. Chem. 2016, 81, 3356–3363. 10.1021/acs.joc.6b00425. [DOI] [PubMed] [Google Scholar]
- Lucas F.; Sicard L.; Jeannin O.; Rault-Berthelot J.; Jacques E.; Quinton C.; Poriel C. [4]Cyclo-N-Ethyl-2,7-Carbazole: Synthesis, Structural, Electronic and Charge Transport Properties. Chem. - Eur. J. 2019, 25, 7740–7748. 10.1002/chem.201901066. [DOI] [PubMed] [Google Scholar]
- Kayahara E.; Zhai X.; Yamago S. Synthesis and Physical Properties of [4]Cyclo-3,7-Dibenzo[b, d]Thiophene and Its S,S-Dioxide. Can. J. Chem. 2017, 95, 351–356. 10.1139/cjc-2016-0474. [DOI] [Google Scholar]
- Shimasaki T.; Okajima S.; Ishikawa R.; Kawaguchi S.; Akimoto T.; Asano N.; Iwanaga T.; Watanabe M.; Teramoto N.; Shibata M. Synthesis and Properties of Fully Conjugated Macrocycles Composed of M-Diethynylene-Phenylene-Bridged Two Dibenzofuran, Dibenzothiophene and Carbazole Units. Tetrahedron 2018, 74, 2454–2465. 10.1016/j.tet.2018.03.072. [DOI] [Google Scholar]
- Klajn J.; Stawski W.; Chmielewski P. J.; Cybińska J.; Pawlicki M. A Route to a Cyclobutane-Linked Double-Looped System via a Helical Macrocycle. Chem. Commun. 2019, 55, 4558–4561. 10.1039/C9CC01201J. [DOI] [PubMed] [Google Scholar]
- Kawano S.; Ishida Y.; Tanaka K. Columnar Liquid-Crystalline Metallomacrocycles. J. Am. Chem. Soc. 2015, 137, 2295–2302. 10.1021/ja510585u. [DOI] [PubMed] [Google Scholar]
- Kawano S.; Kato M.; Soumiya S.; Nakaya M.; Onoe J.; Tanaka K. Columnar Liquid Crystals from a Giant Macrocycle Mesogen. Angew. Chem., Int. Ed. 2018, 57, 167–171. 10.1002/anie.201709542. [DOI] [PubMed] [Google Scholar]
- Dobscha J. R.; Debnath S.; Fadler R. E.; Fatila E. M.; Pink M.; Raghavachari K.; Flood A. H. Host-Host Interactions Control Self-Assembly and Switching of Triple and Double Decker Stacks of Tricarbazole Macrocycles Co-Assembled with Anti-Electrostatic Bisulfate Dimers. Chem. - Eur. J. 2018, 24, 9841–9852. 10.1002/chem.201800827. [DOI] [PubMed] [Google Scholar]
- Dobscha J. R.; Castillo H. D.; Li Y.; Fadler R. E.; Taylor R. D.; Brown A. A.; Trainor C. Q.; Tait S. L.; Flood A. H. Sequence-Defined Macrocycles for Understanding and Controlling the Build-up of Hierarchical Order in Self-Assembled 2D Arrays. J. Am. Chem. Soc. 2019, 141, 17588–17600. 10.1021/jacs.9b06410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath S.; Yang J.; Ortoleva P.; Raghavachari K. Understanding the Origin of 2D Self-Assembly of Tricarbazole Macrocycles: An Integrated Quantum Mechanical/Molecular Dynamics Study. J. Phys. Chem. C 2019, 123, 17616–17623. 10.1021/acs.jpcc.9b04290. [DOI] [Google Scholar]
- Castillo H. D.; Yang J.; Debnath S.; Dobscha J. R.; Trainor C. Q.; Mortensen R. D.; Raghavachari K.; Flood A. H.; Ortoleva P. J.; Tait S. L. Solution-Mediated Annealing Pathways Are Critical for Supramolecular Ordering of Complex Macrocycles at Surfaces. J. Phys. Chem. C 2020, 124, 6689–6699. 10.1021/acs.jpcc.9b11867. [DOI] [Google Scholar]
- Jin S.; Kato S.; Nakamura Y. Synthesis, Self-Association, and Anion Recognition of Conjugated Macrocycles Composed of Carbazole and Triazolium Moieties. Chem. Lett. 2016, 45, 869–871. 10.1246/cl.160400. [DOI] [Google Scholar]
- Ball M. L.; Zhang B.; Xu Q.; Paley D. W.; Ritter V. C.; Ng F.; Steigerwald M. L.; Nuckolls C. Influence of Molecular Conformation on Electron Transport in Giant, Conjugated Macrocycles. J. Am. Chem. Soc. 2018, 140, 10135–10139. 10.1021/jacs.8b06565. [DOI] [PubMed] [Google Scholar]
- Phulwale B. V.; Mishra S. K.; Nečas M.; Mazal C. Phenanthrylene-Butadiynylene and Phenanthrylene-Thienylene Macrocycles: Synthesis, Structure, and Properties. J. Org. Chem. 2016, 81, 6244–6252. 10.1021/acs.joc.6b00814. [DOI] [PubMed] [Google Scholar]
- Phulwale B. V.; Mishra S. K.; Mazal C. Synthesis and Properties of π-Conjugated Donor-Acceptor Macrocycles Derived from Phenanthrylene Building Blocks. Tetrahedron 2018, 74, 3616–3623. 10.1016/j.tet.2018.05.025. [DOI] [Google Scholar]
- Kanagalatha R.; Rajakumar P.; Senthamil Selvi C.S.; Mohan N. Synthesis and Antimicrobicidal Activity of Phenothiazinophanes: A New Class of Permanent Fluorescence Sensing Stilbenophanes. Asian J. Chem. 2015, 27, 4373–4378. 10.14233/ajchem.2015.19128. [DOI] [Google Scholar]
- Xie J.; Li X.; Wang S.; Li A.; Jiang L.; Zhu K. Heteroatom-Bridged Molecular Belts as Containers. Nat. Commun. 2020, 11, 3348. 10.1038/s41467-020-17134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Gopalakrishna T. Y.; Phan H.; Herng T. S.; Ding J.; Wu J. From Open-Shell Singlet Diradicaloid to Closed-Shell Global Antiaromatic Macrocycles. Angew. Chem., Int. Ed. 2018, 57, 7166–7170. 10.1002/anie.201803949. [DOI] [PubMed] [Google Scholar]
- Rana A.; Hong Y.; Gopalakrishna T. Y.; Phan H.; Herng T. S.; Yadav P.; Ding J.; Kim D.; Wu J. Stable Expanded Porphycene-Based Diradicaloid and Tetraradicaloid. Angew. Chem., Int. Ed. 2018, 57, 12534–12537. 10.1002/anie.201807411. [DOI] [PubMed] [Google Scholar]
- Shouda T.; Nakanishi K.; Sasamori T.; Tokitoh N.; Kuramochi K.; Tsubaki K. Synthesis and Structures of Zigzag Shaped [12]Cyclo-p-Phenylene Composed of Dinaphthofuran Units and Biphenyl Units. J. Org. Chem. 2017, 82, 7850–7855. 10.1021/acs.joc.7b01053. [DOI] [PubMed] [Google Scholar]
- Higashibayashi S. Synthesis of Flake-Shaped [3]Cyclo-4,6-Dibenzofuranylene. Chem. Lett. 2018, 47, 95–96. 10.1246/cl.170925. [DOI] [Google Scholar]
- Kurata R.; Sakamaki D.; Ito A. Tetraaza[1.1.1.1]m, p, m, p-Cyclophane Diradical Dications Revisited: Tuning Spin States by Confronted Arenes. Org. Lett. 2017, 19, 3115–3118. 10.1021/acs.orglett.7b01229. [DOI] [PubMed] [Google Scholar]
- Kurata R.; Sakamaki D.; Uebe M.; Kinoshita M.; Iwanaga T.; Matsumoto T.; Ito A. Isolable Triradical Trication of Hexaaza[1 6 ]Paracyclophane with Embedded 9,10-Anthrylenes: A Frustrated Three-Spin System. Org. Lett. 2017, 19, 4371–4374. 10.1021/acs.orglett.7b02088. [DOI] [PubMed] [Google Scholar]
- Wang W.; Chen C.; Shu C.; Rajca S.; Wang X.; Rajca A. S = 1 Tetraazacyclophane Diradical Dication with Robust Stability: A Case of Low-Temperature One-Dimensional Antiferromagnetic Chain. J. Am. Chem. Soc. 2018, 140, 7820–7826. 10.1021/jacs.8b02415. [DOI] [PubMed] [Google Scholar]
- Dong S.; Gopalakrishna T. Y.; Han Y.; Chi C. Cyclobis(7,8-(Para -quinodimethane)-4,4’-triphenylamine) and Its Cationic Species Showing Annulene-Like Global (Anti)Aromaticity. Angew. Chem., Int. Ed. 2019, 58, 11742–11746. 10.1002/anie.201905763. [DOI] [PubMed] [Google Scholar]
- Li G.; Matsuno T.; Han Y.; Phan H.; Wu S.; Jiang Q.; Zou Y.; Isobe H.; Wu J. Benzidine/Quinoidal-Benzidine-Linked, Superbenzene-Based Π-Conjugated Chiral Macrocycles and Cyclophanes. Angew. Chem., Int. Ed. 2020, 59, 9727–9735. 10.1002/anie.202002447. [DOI] [PubMed] [Google Scholar]
- Li Y.; Yagi A.; Itami K. Synthesis of Highly Twisted, Nonplanar Aromatic Macrocycles Enabled by an Axially Chiral 4,5-Diphenylphenanthrene Building Block. J. Am. Chem. Soc. 2020, 142, 3246–3253. 10.1021/jacs.9b13549. [DOI] [PubMed] [Google Scholar]
- Kurosaki R.; Hayashi H.; Suzuki M.; Jiang J.; Hatanaka M.; Aratani N.; Yamada H. A Remarkably Strained Cyclopyrenylene Trimer That Undergoes Metal-Free Direct Oxygen Insertion into the Biaryl C-C σ-Bond. Chem. Sci. 2019, 10, 6785–6790. 10.1039/C9SC01777A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R.; Hasegawa M.; Nishinaga T.; Yoza K.; Mazaki Y. Efficient Synthesis, Structure, and Complexation Studies of Electron-Donating Thiacalix[ n ]Dithienothiophene. Angew. Chem., Int. Ed. 2015, 54, 2734–2738. 10.1002/anie.201410970. [DOI] [PubMed] [Google Scholar]
- Hasegawa M.; Takahashi K.; Inoue R.; Haga S.; Mazaki Y. Selenacalix[4]Dithienothiophene: Synthesis, Structure, and Complexation of a Cyclic Tetramer of Selenide-Bridging Dithienothiophene. Chem. - Asian J. 2019, 14, 1647–1650. 10.1002/asia.201801105. [DOI] [PubMed] [Google Scholar]
- Landovský T.; Dvořáková H.; Eigner V.; Babor M.; Krupička M.; Kohout M.; Lhoták P. Chemoselective Oxidation of Phenoxathiin-Based Thiacalix[4]Arene and the Stereoselective Alkylation of Products. New J. Chem. 2018, 42, 20074–20086. 10.1039/C8NJ04690E. [DOI] [Google Scholar]
- Vrzal L.; Kratochvílová-Šimánová M.; Landovský T.; Polívková K.; Budka J.; Dvořáková H.; Lhoták P. Application of RDC Enhanced NMR Spectroscopy in Structural Analysis of Thiacalix[4]Arene Derivatives. Org. Biomol. Chem. 2015, 13, 9610–9618. 10.1039/C5OB01424G. [DOI] [PubMed] [Google Scholar]
- Landovský T.; Tichotová M.; Vrzal L.; Budka J.; Eigner V.; Dvořáková H.; Lhoták P. Structure Elucidation of Phenoxathiin-Based Thiacalix[4]Arene Conformations Using NOE and RDC Data. Tetrahedron 2018, 74, 902–907. 10.1016/j.tet.2018.01.020. [DOI] [Google Scholar]