

Abstract
Protein arginine methylation is an abundant post-translational modification involved in the modulation of essential cellular processes ranging from transcription, post-transcriptional RNA metabolism, and propagation of signaling cascades to the regulation of the DNA damage response.
Excitingly for the field, in the past few years there have been remarkable advances in the development of molecular tools and clinical compounds able to selectively and potently inhibit protein arginine methyltransferase (PRMT) functions.
In this review, we first discuss how the somatic mutations that confer advantages to cancer cells are often associated with vulnerabilities that can be exploited by PRMTs’ inhibition. In a second part, we discuss strategies to uncover synthetic lethal combinations between existing therapies and PRMT inhibitors.
1. Overview of protein arginine methylation
Positively charged amino acids (i.e. arginines, lysines and histidines) can be methylated (i.e. addition of -CH3 group(s)) by members of the protein methyltransferase family. Specifically, the guanidinium moiety of arginines, can be post-translationally modified to monomethylarginine (Rme1/MMA), asymmetrical dimethylarginine (Rme2a/ADMA) and/or symmetrical dimethylarginine (Rme2s/SDMA) by Protein Arginine MethylTransferases (PRMTs), using S-adenosyl methionine (SAM) as a cofactor 1 (Fig.1A). All PRMTs are able to catalyze the addition of a first -CH3 moiety (from Rme0 to Rme1/MMA). PRMT7, the only type III PRMT, is not able to add additional CH3 groups, while PRMT1/PRMT2/PRMT3/PRMT4/PRMT6 and PRMT8 (Type I PRMTs) are able to catalyze Rme2a/ADMA and PRMT5 and PRMT9 (Type II PRMTs) are able to catalyze Rme2s/SDMA (Fig.1B). The addition of one or two methyl moieties maintains the positive charge of the guanidine group, while adding hydrophobicity to the side chain and diminishing its ability to form hydrogen bonds.
Fig.1.
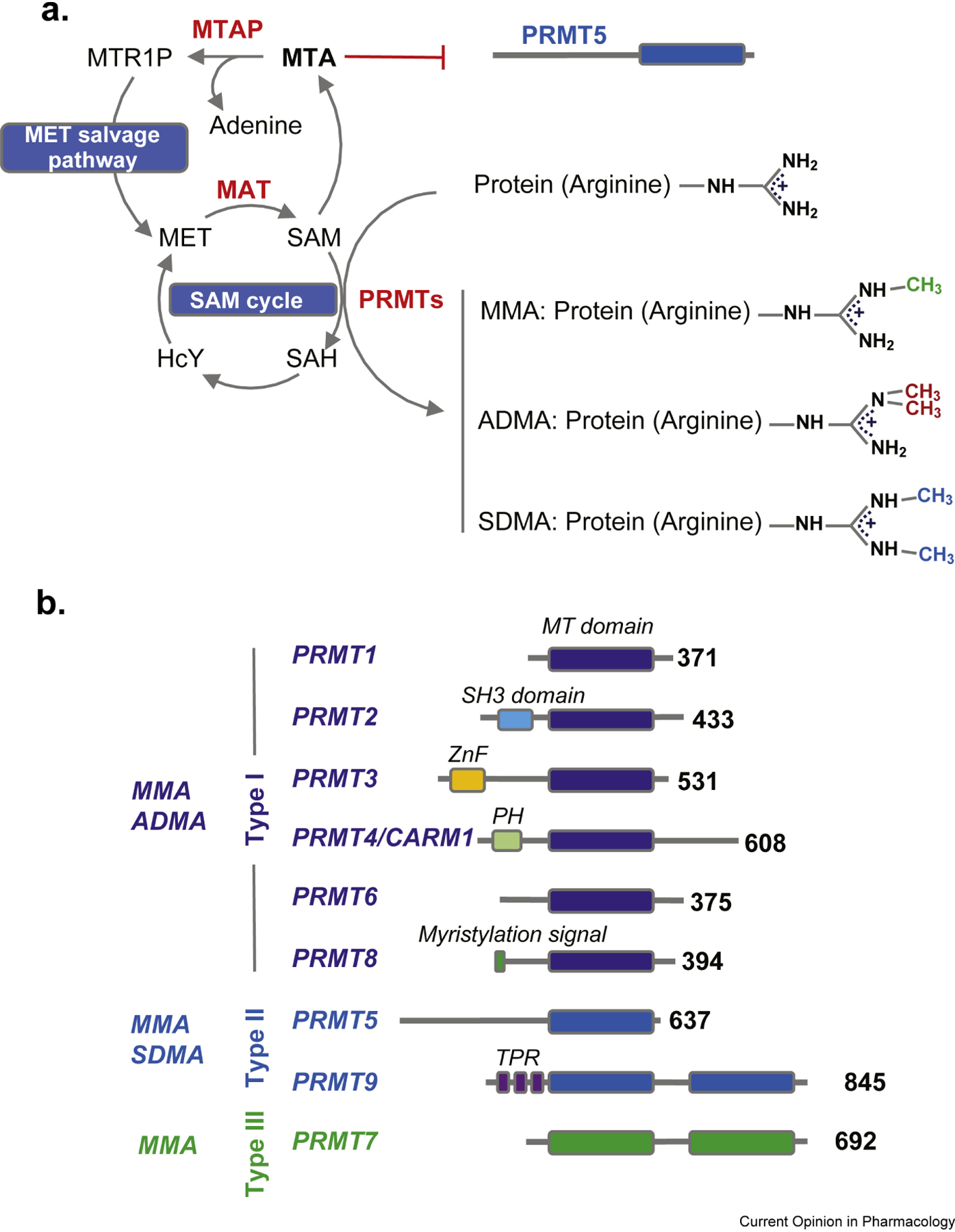
A. The SAM cycle and MET salvage pathway. All PRMTs utilize SAM to catalyze their reaction, leading to the synthesis of monomethylarginine (MMA), asymmetrical dimethylarginine (ADMA) and symmetrical dimethylarginine (SDMA). MTA accumulates in MTAP null tumors and inhibits PRMT5. Abbreviations: MET=Methionine, SAM=S-adenosylmethionine, SAH=S-adenosylhomocysteine, HcY=Homocysteine, MTR1P=Methythioribose-1-phosphate, MTA=Methylthioadenosine, MAT=Methionine adenosyltransferases, MTAP=Methylthioadenosine phosphorylase B. The PRMT family. All PRMTs share a MT domain. Type I PRMTs (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6 and 8 are Type I enzymes catalyzing MMA and ADMA. PRMT5 and PRMT9 are type II enzymes generating MMA and SDMA. PRMT7 is a type III enzyme generating only MMA. Abbreviations: MT=MethylTransferase domain; SH3=Src homology 3; ZnF=zinc finger; TPR=tetratricopeptide. Numbers at the C-terminus correspond to the aminoacid length of human PRMTs.
PRMTs are involved in the modulation of essential cellular processes such as transcription, post-transcriptional RNA metabolism (i.e. splicing), intracellular signaling, and DNA damage response (DDR).
Given such fundamental functions, it is no surprise that their homozygous deletion in mice results in embryonic or perinatal lethality and that human germline or somatic inactivating mutations in PRMT genes are rare. On the contrary, upregulation of PRMTs by a variety of mechanisms is a common feature of cancer (see 1 for review).
In this commentary, we discuss how cancer vulnerabilities, whether associated with somatic mutations or with existing therapies, can be exploited by targeting PRMT function. The plethora of available PRMT inhibitors, with different modes of action, used for research or in early clinical trials makes such a strategy appealing 1,2 (Table.1).
Table. 1.
PRMT, protein arginine methyltransferase; SAM, S-adenosyl methionine; MDS, myelodysplastic syndromes; MTAP, methylthioadenosine phosphorylase
| MANUFACTURER | COMPOUND | TARGET | MECHANISM OF ACTION | REFERENCE | CLINICAL INDICATION |
|---|---|---|---|---|---|
| AMI-1 | type I PRMT | Binding to substrates, prevents substrate recognition | 3 4 | ||
| JIN LAB-SGC | MS023 | type I PRMT | substrate-binding pocket | 5 | |
| GSK | GSK3368715 | type I PRMT | substrate-binding pocket | 6 | PhI: NCT03666988: Relapsed or refractory DLBCL and MTAP null solid tumors |
| JIN LAB-SGC | SGC707 | PRMT3 | allosteric | 7 | |
| TAKEDA | TP-064 | PRMT4 | substrate-binding pocket | 8 | |
| GSK/EPIZYME | EZM2302 or GSK3359088 | PRMT4 | substrate-binding pocket and making additional contact with the SAM-binding pocket | 9 | |
| JIN LAB-SGC | MS049 | PRMT4/6 | substrate-binding pocket | 10 | |
| EPIZYME | EPZ020411 | PRMT6 | substrate-binding pocket | 11 | |
| GSK/EPIZYME | EPZ015666 | PRMT5 | substrate-binding pocket and making additional contact with the SAM-binding pocket | 12 | |
| GSK/EPIZYME | GSK3326595 | substrate-binding pocket and making additional contact with the SAM-binding pocket | 13 | PhI NCT02783300: Solid tumours and NHL; PHI/II NCT03614728: MDS and AML PHII: NCT04676516 Early stage breast cancer |
|
| Eli LILLY | LLY-283 | PRMT5 | SAM-binding pocket | 14 | |
| JANSSEN | JNJ- 64619178 | PRMT5 | SAM-binding pocket and reaches the substrate- binding pocket | 15 | PhI NCT03573310: Relapsed or refractory B cell non-Hodgkin lymphoma (NHL) and advanced solid tumors |
| PFIZER | PF-06939999 | PRMT5 | SAM competitive | PhI NCT03854227: Advanced solid and metastatic tumors | |
| PFIZER | PF-06829927 | PRMT5 | SAM competitive | 16 | |
| PRELUDE | PRT543 | PRMT5 | SAM competitive | 17;18 | PhI NCT03886831 |
| PRELUDE | PRT811 | PRMT5 | SAM competitive | PhI NCT04089449 | |
| PRELUDE | C449 | PRMT5 | covalent inhibitor | 19 | |
| ARGONAUT | T1–44 | PRMT5 | SAM cooperative & peptide substrate competitive | 20 | |
| JIN LAB | MS4322 | PRMT5 | PROTAC degrader | 21 | |
| MERCK | Comp 1A | PRMT5 | Allosteric inhibitor | 22 | |
| AURIGENE | AU-14755 | PRMT5 | SAM cooperative & peptide substrate competitive | WO2019180628 WO2019180631 |
|
| CTXT | PRMT5 | WO2016034673 | |||
| ANGEX | PRMT5 | SAM competitive | WO2019112719 WO2020243178 |
||
| JUBILANT | JPRMT5i | PRMT5 | SAM cooperative & peptide substrate competitive | WO2019102494 | |
| SELLERS/IANARI LABs | BRD0639 | PRMT5 | PBM-competitive Disrupts PRMT5-RIOK1 complex | 23 | |
| SGC | SGC3027 | PRMT7 | cell permeable pro-drug converted to SGC8158 binding to the SAM-binding pocket | 24 |
2. Synthetic lethality with PRMT inhibitors
The classical concept of synthetic lethality states that simultaneous, but not individual, genetic loss of two genes impacts cell viability. This has been extended in cancer biology to encompass pharmacological inhibition or degradation of a particular target, leading to the selective killing of cancer cells bearing mutations or loss of the other gene, but not of normal cells retaining a wildtype copy 25(Fig.2).
Fig.2.
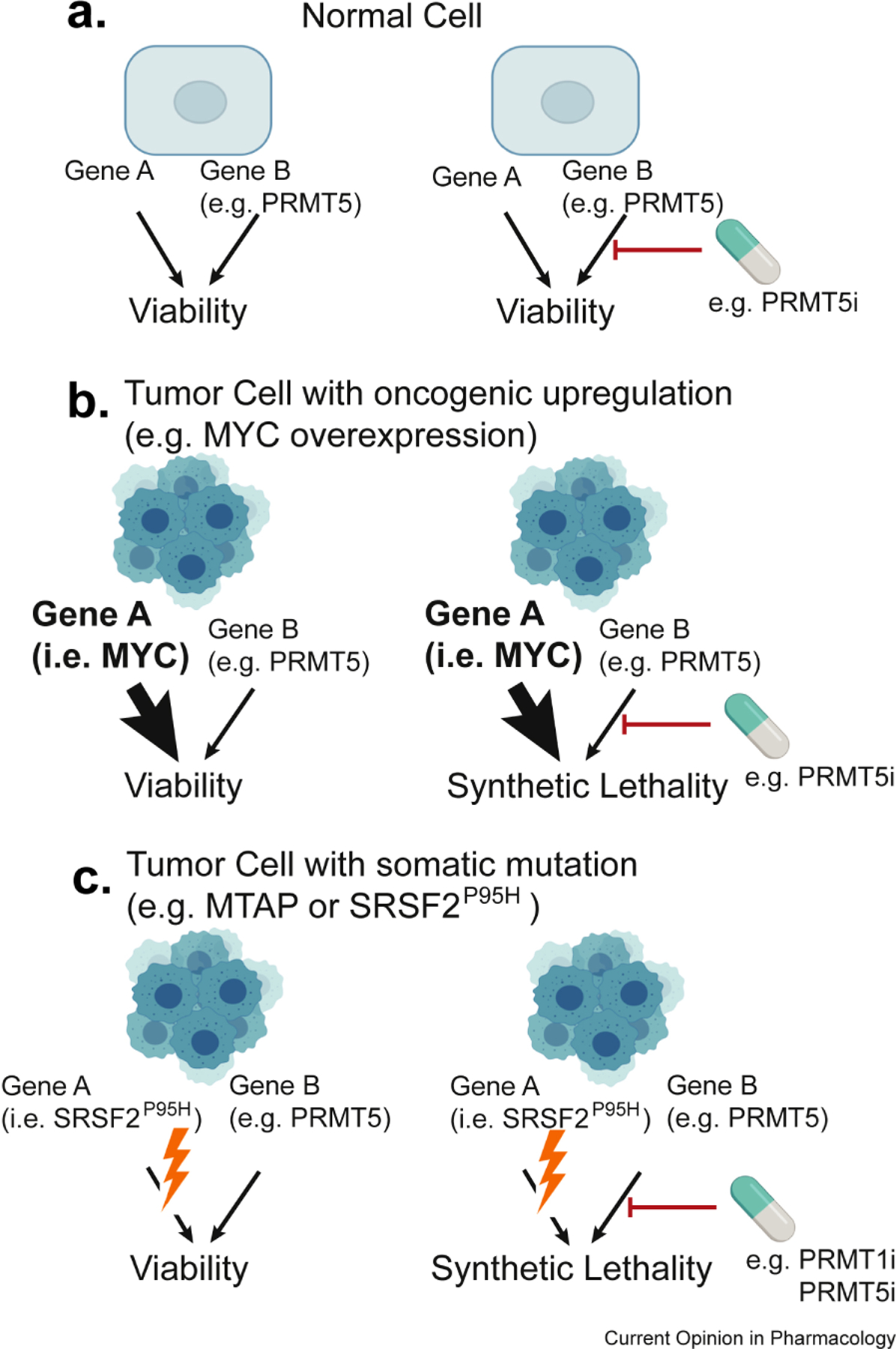
Synthetic vulnerabilities to PRMT inhibitors in cancer. A. In normal cells, the inhibition of gene B (in this example PRMT5), is compatible with cellular viability, granted that gene A is active. B. Synthetic dose lethality. In cells with oncogenic upregulation of a driver (in this example MYC), there is cellular dependence on gene B (i.e. PRMT5). C. Synthetic or Collateral vulnerability. In cells with mutation in a driver (e.g. SRSF2P95H; U2AF1S34F; SF3B1K700E) there is cellular dependence on gene B (i.e. PRMT5). A specific sub-example of synthetic lethality, is when the deleted gene A is a passenger deletion (i.e. MTAP, co-deleted with the CDKN2A/B locus).
Synthetic lethality was first described in Drosophila and yeast, where large scale genetic manipulations have historically been easier to achieve. With the advent of siRNA, shRNA, and, most recently, CRISPR/Cas9 technologies, multiple studies have extended the concept to mammalian cells 26.
Here we summarize three examples of this concept that are relevant to PRMT inhibition.
2a. Tumors overexpressing MYC
MYC (or c-MYC) is part of the proto-oncogenic family of basic-helix–loop–helix leucine-zipper (bHLH-LZ) transcription factors together with NMYC and LMYC. They all recognize an E-box DNA element (i.e. CACGTG or non-canonical variants differing by one base pair) to promote or, less commonly, prevent transcription.
MYC levels are highly regulated at multiple stages (i.e. transcriptional, post-transcriptional, translational and post-translational), to allow rapid induction and degradation in response to signaling cascades influencing fundamental cellular processes (i.e. growth, proliferation, death/apoptosis, anabolism, nucleotide and protein biosynthetic pathways)27.
MYC upregulation contributes to tumorigenesis and is a hallmark of multiple human malignancies 27. Mechanistically, MYC regulates the differential expression of a discrete set of genes, mainly involved in metabolic processes such as RNA processing, ribosome biogenesis, and de novo purine synthesis 28,29. In turn, this MYC-induced transcriptional program results in cellular rewiring, which ultimately affects global transcription levels as a secondary effect 30.
An important set of genes directly activated by MYC is components of the splicing machinery, among which PRMT5 is a critical enzyme sustaining the kinetics of spliceosomal assembly 31. PRMT5 depletion results in cell cycle arrest followed by apoptosis, due to substantial deregulation of cellular splicing. Specifically, in the absence of PRMT5, Sm proteins are not symmetrically dimethylated and fail to be correctly assembled into snRNPs by the SMN complex. As a consequence, cells with reduced U1 levels fail to recognize weak 5′ splice sites, resulting in aberrant accumulation of intron retention and selective skipping of weak alternative exons31.
Importantly, by utilizing both genetic (heterozygous or homozygous PRMT5 deletion) and pharmacological (PRMT5 inhibitors) approaches, the community has reached a consensus on a “threshold model” to explain the role of PRMT5 in cancer (Fig.3A). In short, cancer cells typically require 2–5 fold more PRMT5 activity than normal cells. Critically, normal cells can still function with up to 15–20% residual PRMT5 activity, while cancer cells require >50%. Consistent with this model, heterozygous deletion of Prmt5 is well tolerated in mice while extending the latency of B cell lymphoma significantly. In the MYC-driven Eμ‐myc lymphoma model, the increased dependency on highly active core splicing machinery represents a specific vulnerability associated with MYC overexpression. A possible explanation for this increased dependency on PRMT5 and the splicing machinery in general 31,32 is that in order to sustain the high metabolic rates, MYC-overexpressing cells have higher levels of transcribed mRNAs, which need to be correctly spliced to ensure cell survival. The vulnerability of MYC overexpressing cells to subtle perturbations of the splicing machinery (i.e. PRMT5 inhibition, or depletion of essential components such as SmB or PHF5A) has also been observed in patient-derived glioblastoma multiforme stem cells (GSCs)33. Interestingly, this vulnerability can be recapitulated in Neural Stem Cells (NSC) ectopically overexpressing MYC, but not other oncogenic drivers (i.e. hTERT and dominant-negative p53, among others)33.
Fig.3.
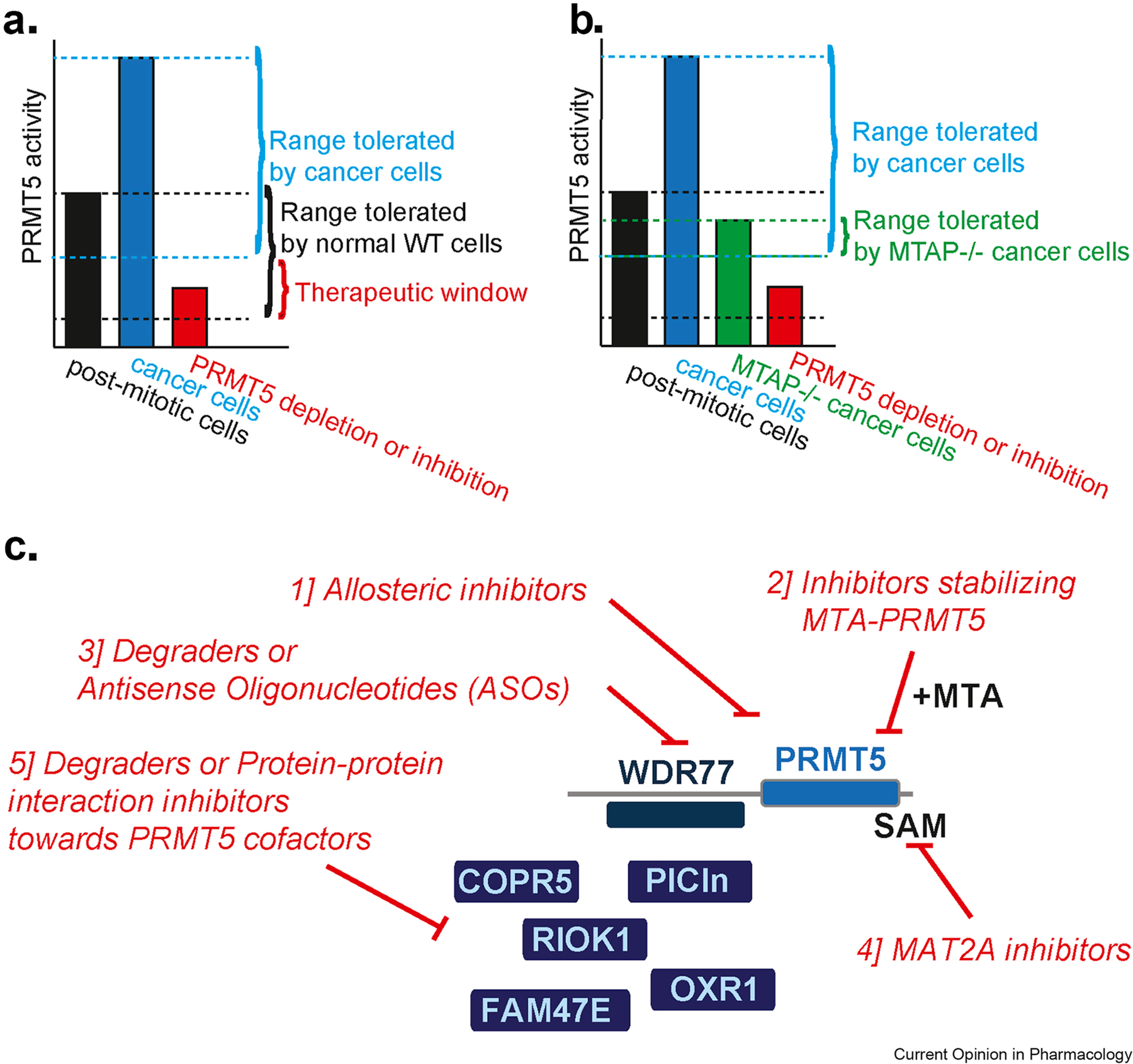
A. Threshold model of PRMT5 activity. Normal post-mitotic cells (black bar) require a certain PRMT5 activity, cancer cells typically require 2–5 fold more (blue bar). In addition, normal cells can tolerate low PRMT5 activity (indicated by the bottom black line), while cancer cells require more. The therapeutic window in red is indicated. B. Threshold model of PRMT5 activity in MTAP null cells. As in A. with the addition of the basal lower activity of PRMT5 in MTAP null cells. C. Modalities of PRMT5 inhibition.
An alternative explanation of the synthetic lethality between MYC overexpression and PRMT5 depletion is that MYC leads to transcriptional upregulation of several components of the translation machinery, including ribosomal RNAs/proteins and elongation factors (e.g. eIF4E). Consistent with this dependency, MYC oncogenic activity is suppressed by haploinsufficiency of ribosomal protein rpl24 34, and PRMT5 activity is required for cap‐dependent translation by eIF4E1.
Last, cells with high MYC levels are subject to replicative stress 27. Reduction in PRMT5 activity could exacerbate accumulation of R-loops and DNA damage, ultimately leading to cell death (see section 3 for more detail)35,36
2b. Tumors with point mutations in splicing factors
More than 90% of transcriptional units express alternatively spliced isoforms and deregulated alternatively spliced (AS) oncogenes and tumor suppressors are linked to cancer initiation or maintenance37. Myelodysplastic syndromes (MDS) are particularly affected by this phenomenon. Specifically, SF3B1, U2AF1, SRSF2, and ZRSR2 constitute the four most commonly mutated spliceosomal components in MDS (i.e. SRSF2P95H; U2AF1S34F; SF3B1K700E and ZRSR2 loss of function mutations), as well as other liquid and solid malignancies 37. Splicing factor mutations occur as heterozygous point mutations at distinct residues and are generally mutually exclusive. These mutations alter the normal RNA binding and splicing preferences of splicing factor proteins in a sequence-specific manner, that is distinct from loss-of-function 37. Importantly, cells become dependent on expression of the wildtype (WT) allele, which explains the heterozygous nature of these mutations38. From a therapeutic standpoint, splicing factor mutant cells are preferentially sensitive to further perturbations of the splicing machinery, such as those obtained using SF3B1 inhibitors 39, which led to early phase clinical trials in patients with CMML and MDS (NCT02841540). The initial report of this study showed no major adverse effects, but disappointingly no objective complete or partial remission, highlighting the need to inhibit spliceosome function in alternative and more efficient ways 40.
Perturbation of asymmetric (ADMA) and symmetric arginine dimethylation (SDMA) of multiple RNA binding proteins and splicing factors, through inhibition of PRMT1 and PRMT5, respectively, provides such alternate means of therapeutic splicing inhibition 31,35,41. Indeed, reducing splicing fidelity by inhibiting ADMA/SDMA results in strong preferential killing of splicing factor mutant AMLs over their wildtype counterparts 42, consistent with what was previously observed with SF3B1 inhibitors. Mechanistically, both type I (PRMT1, PRMT4 and PRMT6) as well as type II PRMT enzymes (i.e. PRMT5), methylate distinct arginines on an overlapping set of core and alternative splicing factors. These RBPs converge to regulate the alternative splicing of key downstream targets (e.g. a poison exon on EZH2), leading to the observed synthetic lethality in SRSF2 or SF3B1 mutant cells, and the synergistic effects between PRMT1 and PRMT5 inhibitors42.
Besides the convergence on alternative splicing events, there are at least two other possible explanations for the synthetic lethality. First, both SRSF2 and SF3B1 perturbation lead to the mis-splicing of genes important in regulating the NF-KB pathway 38. Given that PRMT5 activity is necessary for proper NF-KB function, PRMT5 inhibition could lead to downregulation of an essential pro-survival pathway in AML and MDS (Fig.2).
Second, splicing factor mutant tumors are known to upregulate the formation of R-loops, leading to DNA damage 43. Both type I and II PRMTs have been linked to R-loop biology, and their inhibition leads to R-loop accumulation 36,44, potentially explaining the synthetic lethality observed in SRSF2 or SF3B1 mutant AMLs42. Specifically, PRMT5 mediated methylation of PolII is important to prevent R-loop formation, by mediating the recruitment of Senataxin to chromatin via methylation of RNAPolII R1810me2s and the SMN reader protein 36. Type I PRMTs (i.e. PRMT1 and PRMT4) have instead been implicated in TOP3b recruitment, via methylation of H4R3me2a and RNAPolII R1810me2a and the subsequent binding of the TDRD3 reader protein 45. As a consequence, PRMT inhibition would lead to excessive R-loop formation, tilting the balance towards apoptotic cell death (Fig.2).
2c. MTAP null tumors
Despite the high frequency of loss-of-function genetic mutations in cancer, very few therapeutic approaches target tumor suppressor genes (TSGs) directly; there is instead a major focus on gain-of–function oncogenic events. This is often due to practical reasons, such as the absence of a protein to target upon TSG deletion, or the challenge of identifying small molecules able to restore TSG function in the case of inactivating mutations. These difficulties have led scientists to look at the problem from alternative angles: in particular, deletions or mutations of either TSG or neighboring genes might rewire cellular metabolism or the cancer epigenomic landscape, making tumor cells selectively vulnerable to specific drug treatments, concepts that have been referred to as synthetic and collateral lethality, respectively.
Chromosome 9p21 (chr9p21) is known to contain important TSGs such as p16/INK4a and p19/ARF and is homozygously deleted in a broad variety of solid tumors. The deletion varies widely both in frequency (e.g. reaching over 50% in mesotheliomas, gliomas and melanomas) and in amplitude, often including neighboring genes such as the methylthio-adenosine phosphorylase (MTAP) gene 46.
MTAP is an enzyme that is part of the methionine salvage pathway and, as the name suggests, it generates methionine and adenine by phosphorylating 5′-methylthioadenosine (MTA)(Fig.1A). In an attempt to identify collateral vulnerabilities in MTAP null cells, three independent groups 47–49 have identified PRMT5 and its interacting partners (i.e. WDR77/MEP50, RIOK1, COPR5, pICln), as key targets that, if downregulated, will selectively kill MTAP deficient tumors.
Mechanistically, all three groups were able to demonstrate that MTA (MTAP substrate), which accumulates in MTAP null cells at a concentration of 100 μM and additionally diffuses out in the tumor microenvironment, is a selective PRMT5 inhibitor, which occupies the SAM binding pocket and leads to downregulation of PRMT5-mediated symmetrically dimethylated proteins in the cell.
Importantly, from a therapeutic standpoint, the first PRMT5 inhibitor used in preclinical studies (EPZ015666) did not show any preferential killing of MTAP null cells. While at first these findings were surprising, it can be explained by the mechanistic action of this compound, which requires SAM in order to bind to the PRMT5 pocket 12. Thus, EPZ015666 will not bind to a preformed PRMT5-MTA complex. For similar reasons, additional PRMT5 inhibitors that are currently in development and that target the SAM binding pockets are also predicted to show no or little selectivity toward MTAP null tumors.
In order to take advantage of the collateral vulnerability in MTAP null cells, we thus predict that several approaches could be successful to reduce PRMT5 activity in the context of MTAP null tumors (Fig.3B–C): 1] Develop allosteric PRMT5 inhibitors 22, not targeting the SAM pocket or not requiring a PRMT5/SAM complex to bind; 2] Develop PRMT5 inhibitors that selectively recognize and stabilize the PRMT5/MTA complex; 3] Develop therapeutics that will reduce PRMT5 levels in cells. These can be small molecule degraders 21, or alternative modalities such as antisense oligonucleotides (ASOs)50. Both would mimic the effect reported in the case of PRMT5 shRNA-mediated downregulation 47–49; 4] Develop inhibitors that interfere with PRMT5 activity by impacting the availability of SAM or other cofactors. One such example is the recently reported MAT2A inhibitor, able to impair growth of MTAP null tumor cells by perturbing PRMT5-mediated splicing and DNA damage control51; 5] Develop degraders or protein-protein interaction inhibitors directed against PRMT5 cofactors. These include: Methylosome Protein 50- MEP50/WDR77 (PRMT5 obligatory and stoichiometric partner, affecting PRMT5 enzymatic activity), Methylosome subunit pICln (directing PRMT5 methylation activity towards Sm proteins), RIO Kinase 1-RIOK1 (directing PRMT5 methylation activity towards Nucleolin), cooperator of PRMT5- COPR5 (directing PRMT5 methylation activity towards histone substrates), oxidation resistance gene 1 (OXR1) (directing PRMT5 methylation activity towards H3R2 methylation)52, Family With Sequence Similarity 47 Member E-FAM47E (enhancing PRMT5 chromatin association and histone methylation activity) 53, and possibly others yet to be identified1. The first in class of such class of inhibitors has recently been reported 23.
In addition to testing PRMT5 inhibitors, several groups have now highlighted the synergistic effect of combining PRMT5 selective inhibitors with type I inhibitors 42,54. Indeed, type I PRMT inhibitors preferentially kill MTAP null tumors, which have an impaired PRMT5 activity, highlighting an additional vulnerability of such tumors6,54. It will be interesting to test in the future whether MTAP null tumors will be preferentially sensitive to other inhibitors of the spliceosome or to inhibitors converging on additional PRMT5-regulated pathways (i.e. DDR).
3. Synergistic drug combinations with PRMT inhibitors
In addition to identifying synthetic lethality between PRMT inhibition and genetic alternations in cancer (as described above), several groups have uncovered potential drug combinations that would synergize with PRMT inhibitors.
3a. PRMTi and the DNA double-strand break response (DDR) pathway
Different PRMT family members are able to modulate the DDR pathway, and as such, multiple groups have first characterized the mechanism of action and then tried to combine PRMT inhibition with DNA damage-inducing chemotherapy agents, ionizing radiation (IR), or PARP inhibitors, to mention a few.
It is well established that PRMT5 inhibition leads to DNA damage accumulation and induction of a p53 response 35. This can be due to several proposed mechanisms, which are likely happening simultaneously to coordinate a robust DDR. First, PRMT5 can directly methylate RUVBL1 at R205; this SDMA event is essential to modulate 53BP1 release at sites of homologous recombination (HR)-mediated double-strand break (DSB) repair 55. Additionally, PRMT5 inhibition leads to aberrant splicing of multiple genes involved in HR, including Tip60 56. An important corollary of this latter study is that, similarly to other documented HR deficiencies (i.e. BRCA1 loss of function), PARP inhibition can be used in combination with PRMT5 inhibitors to synergistically kill tumor cells. Third, PRMT5 directly methylates 53BP1, affecting its stability, and coordinating non-homologous end joining (NHEJ) repair following etoposide treatment 57. Last, as pointed out earlier, both type I PRMTs and PRMT5 are important to prevent R-loop accumulation and subsequent DNA damage36,45.
In breast cancer, PRMT5 regulates cancer cell stemness 58, a function that has recently been linked to doxorubicin resistance59, while in pancreatic cancer, PRMT5 was identified by a CRISPR screen as a druggable target whose inhibition synergizes with gemcitabine (Gem) to induce DNA damage. PRMT5 can also directly methylate p53, Rad9 (cell cycle checkpoint control protein), Fen1 (Flap endonuclease-1), and TDP (tyrosyl-DNA phosphodiesterase)(summarized in1), further corroborating the relevance of PRMT5 in DDR.
It is noteworthy that PRMT5-mediated suppression of p53 activity is also relevant to the PRMT5/DDR axis. This was first documented in the developing brain, where PRMT5 depletion led to mis-splicing of a key exon in Mdm4, leading to depletion of MDM4 and activation of a P53 transcriptional program 35. This was later confirmed by multiple studies in the context of solid and hematopoietic tumors, highlighting the importance of an intact MDM4/p53 axis for the activity of PRMT5 inhibitors 13,31,60,61.
PRMT1 function has also been clearly linked to the DDR. Specifically, the MRE11 and 53BP1 GAR motif, methylated by PRMT1, is important for proper DNA repair, making cells and organisms deficient in such ADMAs more sensitive to IR 62–66.
In summary, clinically promising strategies include combination of PRMT inhibitors with DNA damage-inducing agents or drugs blocking an effective DDR response.
3b. PRMTi and combination with other precision therapies
Both PRMT1 and PRMT5 activity is important for pro-survival and proliferative signaling cascades, such as BMP, TGFbeta, EGFR, and PDGFR (recently reviewed in 1), making the case for combining PRMT inhibitors with available kinase inhibitors or biologics (i.e. cetuximab) targeting these signaling cascades. As an example, PRMT5 methylation of proteins in the PI3K/AKT pathways led to testing a combination of PRMT5 and mTOR inhibitors as a potential therapeutic approach in glioblastoma (GBM) 67.
PRMT1-mediated methylation of EGFR promotes EGFR signaling. To exploit this in a therapeutic setting, several groups have combined PRMT1 knockdown (KD) or inhibitors with EGFR inhibitors (i.e. erlotinib or cetuximab). In one study, the authors show that PRMT1 inhibition increased non-small cell lung cancer (NSCLC) sensitivity to erlotinib and reversed EMT to overcome resistance to the kinase inhibitor68. In other studies, PRMT1 depletion sensitized cells to cetuximab treatment 69,70. While the authors used PRMT1 KD, it will be important in the future to test if similar effects are observed with PRMT1 inhibitors (i.e. GSK3368715 or MS023). Overall, these studies have demonstrated encouraging efficacy in vitro and in mouse models. However, future investigations are necessary to assess the degree of cumulative toxicity, or potential mechanisms of tumor resistance.
PRMT1-mediated FLT3 methylation sustains AML maintenance. As such, combination of a potent FLT3 inhibitor, Quizartinib (AC220) with MS023 (a pan-type I PRMT inhibitor, primarily inhibiting PRMT1) efficiently eliminated FLT3-ITD(+) AML cells and would be an interesting approach to test in the clinic 71.
Additionally, a recent publication assessed the efficacy of a novel PRMT5 inhibitor (C220) in JAKV617F-mutant myeloproliferative neoplasm (MPN). C220 was very effective in inducing DNA damage and apoptosis, both as a monotherapy, and in combination with JAK inhibition (ruxolitinib) 18.
4. Future outlook
Overall, it is still early days for PRMT inhibitors in the clinic and these coming years should be focused on identifying additional synthetic lethality strategies to widen the therapeutic windows for these compounds. An important aspect to take into consideration for the effectiveness of synthetic lethality strategies will be tumor heterogeneity. Ideally, the majority of tumor cells should carry the given somatic alteration, such that the vulnerability being exploited is clonal, rather than subclonal. In the examples discussed above, splicing factor mutations tend to occur early, typically in the founding clone of MDS and other myeloid malignancies 72; similarly, MTAP deletion occurs early on in tumor progression, concurrently with that of the CDKN2A/B locus 73. Last, MYC amplification and/or its increased expression is a hallmark of cancer, and is also present clonally in multiple cancer types 27, making all of these alterations viable options to target with PRMT inhibitors to exploit their synthetic lethality.
In the near future, in addition to cell intrinsic effects of PRMT inhibitors, it will be paramount to test PRMT inhibitors in combination with immunotherapy. The rationale for such combination stems from the known link between splicing deregulation and creation of neo-antigens 74–76 and the fact that PRMT1 and PRMT5 inhibition leads to widespread inclusion of novel exons as well as intron retention. A recent study has indeed tested the combination of PRMT5 inhibition with anti-PD-1 therapies, demonstrating high efficacy in a murine melanoma model (B16cells) due to the negative correlation between PRMT5 activity and both MHCI antigen presentation and PD-L1 expression 77.
At the same time, the potential effect of PRMT inhibitors on T-cell fitness and activation of an interferon response will have to be carefully evaluated 16,17,78,79 to further pave the way for more extensive testing in the clinic.
ACKNOWLEDGEMENTS
Research reported in this publication was supported in part by National Cancer Institute of the NIH (R01 CA249204 and R01CA248984) and ISMMS seed fund to EG. MS was supported by an NCI training grant (T32CA078207).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT(S)
EG is co-founders and scientific advisors of IMMUNOA Pte Ltd. EG has served as scientific advisor for Lion TCR Pte Ltd and Prelude Therapeutics. The rest of the authors declare no competing financial interests.
References
- 1.Guccione E & Richard S The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 20, 642–657 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Kaniskan HU, Martini ML & Jin J Inhibitors of Protein Methyltransferases and Demethylases. Chem Rev 118, 989–1068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng D, et al. Small molecule regulators of protein arginine methyltransferases. J Biol Chem 279, 23892–23899 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Castellano S, et al. Design, synthesis and biological evaluation of carboxy analogues of arginine methyltransferase inhibitor 1 (AMI-1). ChemMedChem 5, 398–414 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Eram MS, et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem Biol 11, 772–781 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fedoriw A, et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 36, 100–114 e125 (2019).31257072 * Together with Fong et al and Gao et al, characterizes the syntheteic lethality between PRMT1 and PRMT5 inhibition.
- 7.Kaniskan HU, et al. A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew Chem Int Ed Engl 54, 5166–5170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakayama K, et al. TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 9, 18480–18493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drew AE, et al. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci Rep 7, 17993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen Y, et al. Discovery of a Potent, Selective, and Cell-Active Dual Inhibitor of Protein Arginine Methyltransferase 4 and Protein Arginine Methyltransferase 6. J Med Chem 59, 9124–9139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell LH, et al. Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med Chem Lett 6, 655–659 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chan-Penebre E, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 432–437 (2015).25915199 * First in class PRMT5 inhibitor.
- 13.Gerhart SV, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep 8, 9711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonday ZQ, et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med Chem Lett 9, 612–617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.al TW e. Abstract 4859: JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivo activity, demonstrated in several lung cancer models. Cancer Res (2018).
- 16.Metz PJ, et al. Symmetric Arginine Dimethylation Is Selectively Required for mRNA Splicing and the Initiation of Type I and Type III Interferon Signaling. Cell Rep 30, 1935–1950 e1938 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Snyder KJ, et al. PRMT5 regulates T cell interferon response and is a target for acute graft-versus-host disease. JCI Insight 5(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pastore F, et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene-Regulatory Networks Leading to Therapeutic Efficacy in JAK2(V617F)-Mutant MPN. Cancer Discov 10, 1742–1757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, et al. Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med Chem Lett 10, 1033–1038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barczak W, et al. PRMT5 promotes cancer cell migration and invasion through the E2F pathway. Cell Death Dis 11, 572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen Y, et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J Med Chem 63, 9977–9989 (2020).32787082 * First in class PRMT5 degrader.
- 22. Palte RL, et al. Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5). ACS Med Chem Lett 11, 1688–1693 (2020).32944135 * First in class PRMT5 allosteric inhibitor.
- 23. McKinney David C, B.J.M., Ranaghan Matthew, Moroco Jamie A, Brousseau Merissa, Mullin-Bernstein Zachary, O’Keefe Meghan, McCarren Patrick, Mesleh Michael F., Mulvaney Kathleen M., Singh Ritu, Bajrami Besnik, Skepner Adam, Timm David E., Porter Dale, Kaushik Virendar K., Sellers William R., Ianari Alessandra. Discovery of a first-in-class inhibitor of the PRMT5-substrate adaptor interaction. bioRxiv (2021). * First in class PRMT5-substrate adaptor interaction inhibitor.
- 24.Szewczyk MM, et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat Commun 11, 2396 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang A, Garraway LA, Ashworth A & Weber B Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 19, 23–38 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Behan FM, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Gabay M, Li Y & Felsher DW MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 4(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muhar M, et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabo A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kress TR, Sabo A & Amati B MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15, 593–607 (2015). [DOI] [PubMed] [Google Scholar]
- 31. Koh CM, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100 (2015).25970242 ** First in vivo characterization of the effects of PRMT5 deletion on tumor initiation and maintenance.
- 32.Hsu TY, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hubert CG, et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes & development 27, 1032–1045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barna M, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bezzi M, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes & development 27, 1903–1916 (2013).24013503 ** First in vivo characterization of the effects of PRMT5 deletion on splicing regulation.
- 36.Zhao DY, et al. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature 529, 48–53 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Obeng EA, Stewart C & Abdel-Wahab O Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov 9, 1493–1510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SC, et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 34, 225–241 e228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seiler M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24, 497–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steensma., et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 134 (Supplement_1): 673(2019). [Google Scholar]
- 41.Radzisheuskaya A, et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol 26, 999–1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fong JY, et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 36, 194–209 e199 (2019).31408619 ** Together with Fedoriw et al and Gao et al, characterizes the syntheteic lethality between PRMT1 and PRMT5 inhibition. Additionally, defines the synthetic lethality between splicing factor mutant AMLs and PRMT inhibition.
- 43.Chen L, et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell 69, 412–425 e416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villarreal OD, Mersaoui SY, Yu Z, Masson JY & Richard S Genome-wide R-loop analysis defines unique roles for DDX5, XRN2, and PRMT5 in DNA/RNA hybrid resolution. Life Sci Alliance 3(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, et al. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell 53, 484–497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kryukov GV, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mavrakis KJ, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213 (2016). [DOI] [PubMed] [Google Scholar]
- 49. Marjon K, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep 15, 574–587 (2016).27068473 ** Milestone papers on the synthetic vulnerability of MTAP null tumors to further inhibition of the MAT2A/PRMT5/RIOK1 axis.
- 50.Dhuri K, et al. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med 9(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kalev P, et al. MAT2A inhibition blocks the growth of MTAP-deleted cancer cells by reducing PRMT5-dependent mRNA splicing and inducing DNA damage. Cancer Cell (2021). * First in class MAT2A inhibitor, preferentially killing MTAP-deleted tumors.
- 52.Yang M, et al. OXR1A, a Coactivator of PRMT5 Regulating Histone Arginine Methylation. Cell Rep 30, 4165–4178 e4167 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Chakrapani B, et al. The uncharacterized protein FAM47E interacts with PRMT5 and regulates its functions. Life Sci Alliance 4(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao G, et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res 47, 5038–5048 (2019).30916320 * Together with Fedoriw et al and Fong et al, characterizes the syntheteic lethality between PRMT1 and PRMT5 inhibition.
- 55.Clarke TL, et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol Cell 65, 900–916 e907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hamard PJ, et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep 24, 2643–2657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hwang JW, et al. PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun Biol 3, 428 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiang K, et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep 21, 3498–3513 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, et al. PRMT5 determines the sensitivity to chemotherapeutics by governing stemness in breast cancer. Breast Cancer Res Treat 168, 531–542 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Dewaele M, et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest 126, 68–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.AbuHammad S, et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proceedings of the National Academy of Sciences of the United States of America 116, 17990–18000 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boisvert FM, Cote J, Boulanger MC & Richard S A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics 2, 1319–1330 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Yu Z, et al. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res 22, 305–320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan Q, Tian R, Zhao H, Li L & Bi X Multiple Arginine Residues Are Methylated in Drosophila Mre11 and Required for Survival Following Ionizing Radiation. G3 (Bethesda) 8, 2099–2106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boisvert FM, Rhie A, Richard S & Doherty AJ The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle 4, 1834–1841 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Vadnais C, et al. GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nat Commun 9, 1418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holmes B, et al. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J Neurooncol 145, 11–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iderzorig T, et al. Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem Biophys Res Commun 496, 770–777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liao HW, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest 125, 4529–4543 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakai K, et al. The role of PRMT1 in EGFR methylation and signaling in MDA-MB-468 triple-negative breast cancer cells. Breast Cancer 25, 74–80 (2018). [DOI] [PubMed] [Google Scholar]
- 71.He X, et al. PRMT1-mediated FLT3 arginine methylation promotes maintenance of FLT3-ITD(+) acute myeloid leukemia. Blood 134, 548–560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saez B, Walter MJ & Graubert TA Splicing factor gene mutations in hematologic malignancies. Blood 129, 1260–1269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krasinskas AM, Bartlett DL, Cieply K & Dacic S CDKN2A and MTAP deletions in peritoneal mesotheliomas are correlated with loss of p16 protein expression and poor survival. Mod Pathol 23, 531–538 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Zhang Z, et al. ASNEO: Identification of personalized alternative splicing based neoantigens with RNA-seq. Aging (Albany NY) 12, 14633–14648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oka M, et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol 22, 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frankiw L, Baltimore D & Li G Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol 19, 675–687 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Kim H, et al. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci Transl Med 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Webb LM, et al. Protein arginine methyltransferase 5 promotes cholesterol biosynthesis-mediated Th17 responses and autoimmunity. J Clin Invest 130, 1683–1698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanaka Y, Nagai Y, Okumura M, Greene MI & Kambayashi T PRMT5 Is Required for T Cell Survival and Proliferation by Maintaining Cytokine Signaling. Front Immunol 11, 621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]