

ABSTRACT
Querying massive functional genomic and annotation data collections, linking and summarizing the query results across data sources/data types are important steps in high-throughput genomic and genetic analytical workflows. However, these steps are made difficult by the heterogeneity and breadth of data sources, experimental assays, biological conditions/tissues/cell types and file formats. FILER (FunctIonaL gEnomics Repository) is a framework for querying large-scale genomics knowledge with a large, curated integrated catalog of harmonized functional genomic and annotation data coupled with a scalable genomic search and querying interface. FILER uniquely provides: (i) streamlined access to >50 000 harmonized, annotated genomic datasets across >20 integrated data sources, >1100 tissues/cell types and >20 experimental assays; (ii) a scalable genomic querying interface; and (iii) ability to analyze and annotate user’s experimental data. This rich resource spans >17 billion GRCh37/hg19 and GRCh38/hg38 genomic records. Our benchmark querying 7 × 109 hg19 FILER records shows FILER is highly scalable, with a sub-linear 32-fold increase in querying time when increasing the number of queries 1000-fold from 1000 to 1 000 000 intervals. Together, these features facilitate reproducible research and streamline integrating/querying large-scale genomic data within analyses/workflows. FILER can be deployed on cloud or local servers (https://bitbucket.org/wanglab-upenn/FILER) for integration with custom pipelines and is freely available (https://lisanwanglab.org/FILER).
INTRODUCTION
Functional genomic data and annotations are commonly used to provide the necessary functional evidence in various systems biology, genetic and genomic analyses, such as the analysis of the non-coding genome-wide association study (GWAS) signals or the analysis of the experimentally derived genomic regions (1–6). Such functional genomic annotation includes different types of data such as tissue-specific regulatory elements (enhancers) (7), transcription factor (TF) binding activity (8–10), chromatin states (11,12), genetic regulation (expression quantitative trait loci (eQTL), splicing QTLs (sQTL)) information (13,14) and chromatin conformation data (15). These data originate from a variety of sample sources including primary tissues, primary cells, immortalized cell lines, in vitro differentiated cells and others.
A primary source of such experimental data are the data collections from major functional genomics consortia. For example, ENCODE (8,9), GTEx (13,14), FANTOM5 (7) and NIH Roadmap Epigenomics (11) have generated datasets spanning over >50 000 experiments across >1000 tissues, cell types, biological conditions, with each dataset containing millions to billions of annotation or assay readout records across the genome. In order to pair these functional annotations with high-throughput analytical workflows, e.g. for processing current population-level studies such as UK Biobank (16) (500 000 individuals with >2500 phenotypes), we need a scalable, unified, high-throughput and robust access to these massive, heterogeneous genomic data collections.
To address these issues, we developed FILER (FunctIonaL gEnomics Repository), a large-scale, curated, integrated catalog of harmonized functional genomic and annotation data coupled with a scalable genomic search and querying interface to these data. The latest FILER release provides seamless integration of >58 000 harmonized genomic datasets (data tracks) across diverse (>20) primary data sources organized into >140 data collections, wide biological context (>1100 cell types) and various genomic and biological features (>30 experimental assays and data types). FILER provides unified access to this rich functional and annotation data resource spanning >17 billion records across genome with >2700× total genomic coverage for both GRCh37/hg19 and GRCh38/hg38. FILER can be used to perform flexible querying, staging and consolidation of these data for analyses. FILER is accessible via a web server (https://lisanwanglab.org/FILER) or can be deployed on users’ own cloud computing instances, local servers, high-performance computing clusters (https://bitbucket.org/wanglab-upenn/FILER) and integrated with genomic and genetic analysis workflows.
MATERIALS AND METHODS
FILER structure and data organization
FILER has been implemented using an easily updatable, extensible, and modular architecture. Figure 1 gives an overview of the FILER architecture and data organization (for more detailed information on the implementation, please refer to the ‘FILER implementation’ Section in the Supplementary). FILER contents are derived from the integration of many data and annotation resources as well as curation and processing of publicly available functional genomics datasets (Figure 1A). Supplementary Table S2 (‘Data sources’) and Supplementary Table S3 (‘Data collections’) show a detailed list of data sources and data collections available in FILER. First, primary annotation and functional genomics datasets (referred to as data tracks) from existing data sources are collected and compiled into a unified catalog, as shown in the diagram in Figure 1A. Second, individual genomic datasets are curated, processed, and imported into FILER (Figure 1A diagram) using the FILER data harmonization and annotation pipeline (see ‘FILER data harmonization and annotation pipeline’ for details). Data-source-specific metadata schemas were matched across data sources with the FILER schema (Supplementary Table S5 provides details of the schema matching) to generate standardized, consistent meta-data descriptions for each of the FILER data tracks (sets of genomic/annotation records).
Figure 1.
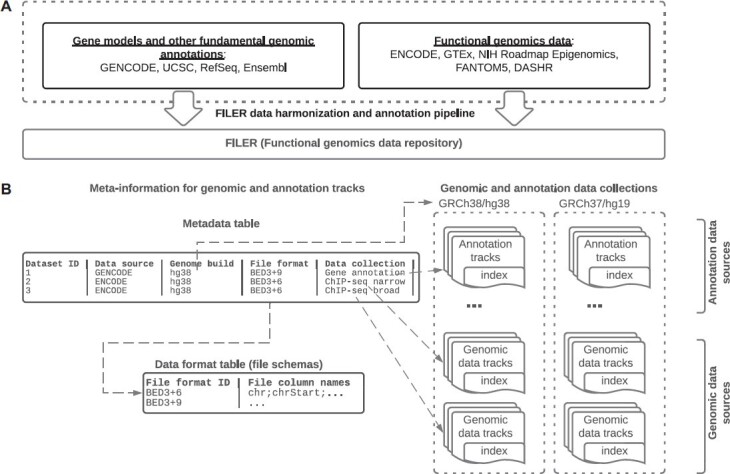
(A) Overview of the FILER architecture. Annotation resources and genomic data sources are collected and compiled into a unified data catalog. All datasets are curated, systematically processed, and imported into FILER using the data harmonization and annotation pipeline (see ‘FILER data harmonization and annotation pipeline’, Figure 2 for details). (B) Organization of FILER. Metadata and file schemas tables (shown on the left-hand side) contain detailed, standardized meta-information for individual datasets and descriptions of the data file schemas.
All datasets in FILER are organized into data collections (shown on the right-hand side) by data source, experimental assay, data type, file format, and genome build. For each FILER dataset, the metadata table contains a reference to the data collection containing the dataset (references are shown with dashed arrow lines in the figure). Data collections are indexed to enable efficient search and retrieval.
Each data source available in FILER (e.g. ENCODE, Roadmap and GTEx) is organized into one or more specific data collections corresponding to a particular experimental assay, data type/file format and genome build. This ensures that each data collection only contains tracks sharing the same file format, the same genome assembly, and the same experimental protocol, thus allowing all such tracks to be indexed together and, importantly, allowing query (e.g., overlap) results to be combined across tracks. Some data collection examples in FILER include ENCODE ChIP-seq called peaks in the narrow peak format, ENCODE DNase-seq peaks, or called small RNA loci from the DASHR database (17–19) of small non-coding RNAs across tissues/cell types. ‘FILER data harmonization and annotation pipeline’ describes FILER data collections and organization in more details. Supplementary Tables S2 and S3 provide details on data sources and data collections available in FILER.
Internally, FILER stores information in several tables:
Meta-information table (Figure 1B; Supplementary Table S4) containing the standardized information for each data track
File schema table (Figure 1B)
External, indexed data collections holding the actual genomic data (right-hand side of Figure 1B) referenced in the meta-information table
We describe each of these tables in the next sections below.
FILER meta-information table
The FILER metadata table stores detailed information for each data track including the data source (e.g. ENCODE, Roadmap and FANTOM5), experimental assay (e.g. ChIP-seq, DNase-seq and ATAC-seq), type of genomic records (e.g. called peaks, transcription start sites and gene models), biological source (e.g. cell type, tissue and cell line), data provenance (e.g. the data source version [if applicable], the date data were downloaded from the source and download URL). Both the original (as obtained from the data source) data files and the processed files are stored in FILER in separate folder locations. The metadata table store the references to the original file including the download URL and the local file location. Each data track is uniquely identified by a FILER identifier assigned when the data is added to FILER (see, e.g., Figure 1B and Supplementary Table S8).
Additionally, the metadata table stores a number of derived and computed properties used for data integration and organization purposes, including tissue category and data category. Tissue category is used to define terms corresponding to individual cell types/tissues into a broader, standardized tissue/cell type category (see Supplementary Methods, section on tissue categorization). The data category is designed to provide a biologically meaningful description of each track. The data category for each track in each of the data sources is systematically generated based on a combination of the data attributes, including the experimental assay (e.g., ChIP-seq, DNase-seq), experimental target (antibody, if applicable; e.g., CTCF protein, or H3K27ac histone mark) and the type of genomic records (e.g., narrow peaks, transcription start sites) (see Supplementary Methods; ‘FILER data harmonization and annotation pipeline’) on data track classification/categorization for details).
FILER file schema information table
Data tracks in FILER are stored in formats specific to particular data sources and/or experimental assays. The file schema table stores the schemas (see Figure 1B, bottom) for each type of data tracks. These file schemas can be used to extract additional information from the genomic records contained in the track files. Each record in the file schema table defines the number of standard BED fields, the number of extra fields, as well as the names of all the fields in the genomic record (see Figure 1B [bottom left] for an example; for more detailed information on the FILER database schema table please refer to Supplementary Table S6).
FILER indexed data collections
The external indexed data collections (see Figure 1B, right panel) are referenced in the FILER metadata table using pointers to the file directories holding the genomic/annotation data for each of the FILER data collections. Data indexes are created for each data collection and are used to accelerate access/search within data collections. The data indexes for each of the data collections are located within the data collection file directories in the standardized location (the ‘giggle_index’ sub-folders). Supplementary Table S3 provides details on the indexed data collections available in FILER. Data collections in FILER thus are stored in separate folders with each folder holding both the actual genomic datasets the corresponding genomic data index.
FILER data harmonization and annotation pipeline
The main steps in the FILER data harmonization and annotation pipeline (Figure 2) include (i) data annotation (metadata extraction and generation), (ii) data pre-processing and normalization, (iii) data classification and organization into data collections and (iv) genomic interval-based data collection indexing (20).
Figure 2.
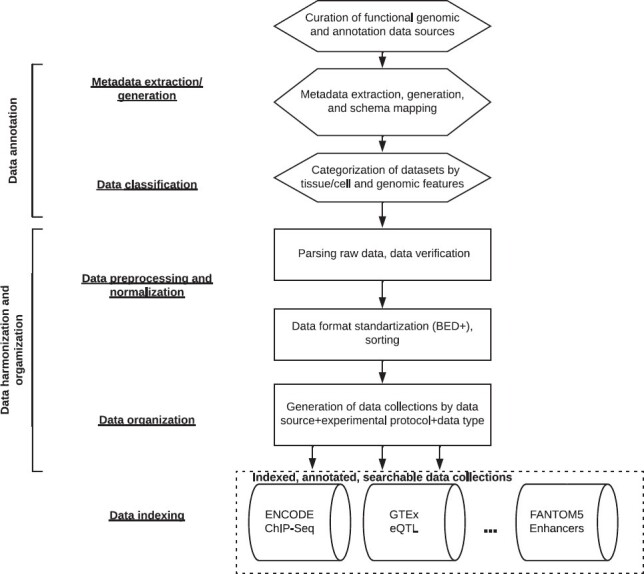
FILER data annotation and harmonization pipeline. All functional genomic datasets from the curated data sources are annotated with a standardized, consistent meta-information to allow for integration across diverse data sources (see ‘FILER structure and data organization’ and ‘FILER data harmonization and annotation pipeline’). All individual genomic datasets are systematically pre-processed into standard, uniform BED-like file formats, and grouped into data collections based on the data source, experimental data type and data format (see ‘FILER structure and data organization’). Each of the resulting data collections is indexed to allow efficient access using genomic-interval based indexing and query engine (see ‘FILER structure and data organization’ for details).
Data annotation
All genomic tracks were annotated with the standard set of attributes (meta-information) including data type (ChIP-seq peaks, transcription factor binding sites [TFBS], gene models etc.), assay type (ChIP-seq, DNAse-seq, ATAC-seq etc.), cell/tissue type, data source, data source version and other relevant meta-information (see Supplementary Table S4 describing FILER metadata schema). To extract/infer the necessary meta-information for each data track, the information provided in the original data source was parsed and matched to the standardized schema employed by FILER (see Supplementary Table S5 for details).
Data pre-processing
Uniform pre-processing (format standardization, file schema inference and normalization, genomic coordinated-based sorting, compression and indexing) is performed to convert each original data track (e.g, in GFF (21), BigBed (22) formats) into a standard, BED-like format. Supplementary Table S6 describes the file schemas for all data file formats integrated into FILER. Additionally, conversion of the genomic coordinates (liftover (23)) from GRCh37/hg37 to GRCh38/hg38 was performed for datasets available exclusively in the earlier GRCh37/hg19 genomic build. Currently, FILER includes genomic tracks for GRCh37/hg19 (hg19) and GRC38hg38 (hg38, hg38-lifted) genomic builds.
Data classification and organization into data collections
To further organize data and enable efficient search and retrieval of the functional genomics and annotation tracks, datasets from the same data source, same assay, same type of genomic records, same data format were physically grouped into data collections by data source, assay type, data type and genome build (see Supplementary Table S3 for details of data collections available in FILER). Each data collection is stored in a separate directory and contains files in the same format (e.g. BED3, BED6 etc.) and is indexed using genomic interval-based indexing (20) (see ‘Genome interval-based data indexing’ for details on data indexing in FILER). This genomic interval-based indexing schema created for each genomic collection facilitates cross-data collection search (e.g. genomic interval-based query) in FILER.
Additionally, all genomic and annotation data tracks included in FILER were categorized based on the biological source, tissue/cell type into broader tissue/cell type categories to further enable cross-data source integration of the data tracks (see Supplementary Methods, Tissue categorization section for details of tissue/cell type-based categorization employed by FILER). All FILER data tracks were annotated with a major tissue and organ systems category they belong to. FILER tracks were also systematically classified into biologically meaningful data categories (‘FILER meta-information table’) to allow the users to more easily access datasets of interest.
Genomic-interval based data indexing
Each data collection in FILER (Supplementary Table S3; ‘Data classification and organization into data collections’) is indexed using a customized genomic-interval based indexing (20). Indexing creates B + tree-like structures over genomic intervals contained in the data collection. The leaf nodes contain pointers to the data files and file offsets for individual genomic records (intervals) overlapping with the coordinate range covered by each leaf. The generated index for each data collection allows for efficient access to the genomic records overlapping a query interval across all the tracks stored in the data collection. Using these data indexes, the data collection tracks can then be efficiently accessed using the FILER Giggle-based query engine (see Supplementary Methods).
Data access and availability
Data is added to the FILER periodically, with public versioned releases (data freezes) every 6 months. Sustainability is ensured by the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) as maintainer of FILER. All data are stored in Amazon Cloud (24). A local copy of FILER can be deployed on user-own servers or cloud computing instances (https://bitbucket.org/wanglab-upenn/FILER) to enable integration with existing or new analysis pipelines using the provided installation and data scripts. Additionally, access to the data is provided using FILER web server (https://lisanwanglab.org/FILER).
RESULTS
FILER contents
FILER contains functional genomics and annotation data characterizing various biological features of the human genome. Figure 3 shows the distribution of functional genomic datasets integrated into FILER by data source, experimental data types, tissue/cell type categories, and biological sample types. FILER integrates functional genomic and annotation data across many primary data sources (Figure 3A) with the genomic/annotation records spanning a wide range of experimental assays/data types (Figure 3B). The data tracks in FILER are generated from a variety of biological sources (Figure 3C) and span a variety of tissue/cell type categories (Figure 3D) corresponding to >1000 tissues/cell types. Supplementary Figure S1 shows the distribution FILER genomic/annotation records across tissue/cell types categories, individual types of cells/tissues and data sources. Currently, FILER includes datasets across 14 human tissue and organ systems including cardiovascular (19.5%), nervous (15.9%), reproductive (8.9%), digestive (13.1%), stem cell (9.1%) and other categories.
Figure 3.
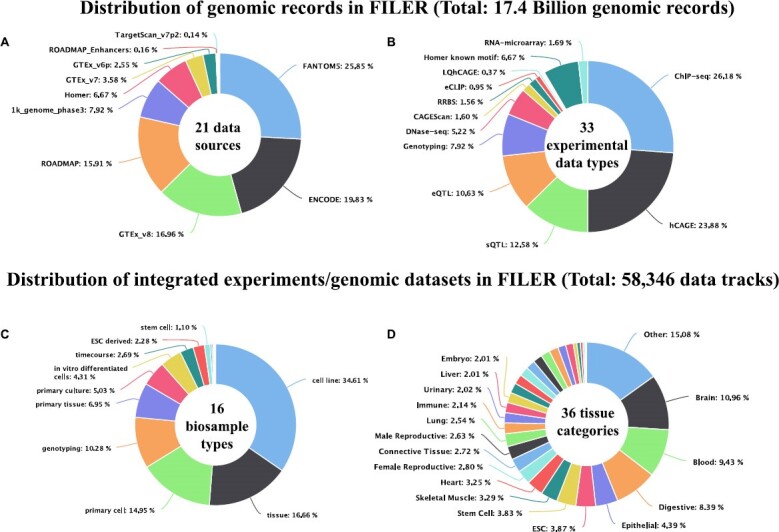
FILER data composition. (A) Composition of functional genomic records by data source/consortium. (B) Distribution of genomic records across experimental assays and data types. C) Distribution of datasets by biological source. D) Distribution of datasets across FILER tissue/cell type categories.
Supplementary Table S7 provides a summary of the FILER datasets/records by the type of biological annotations and genomic features. FILER rich genomic and annotation data collection includes functional data capturing regulatory elements such as enhancers, transcription factor activity (transcription factor binding sites [TFBS] profiled by ChIP-seq (8,9,25) and computational analyses (10,26)), quantitative trait loci [QTL] including expression QTL [eQTL] and splicing QTL [sQTL], chromatin state [ChIP-seq (8,9,25), DNase-seq (12), ATAC-seq (27)], gene transcription activity (transcription start sites [TSS] profiled by CAGE (7)), long-noncoding RNAs, small RNA loci (17,18), miRNA-mRNA interactions (28), histone marks (ChIP-seq) (8,9,11,25) and others (Figure 3). In the case of replicated experiments, e.g. for histone and transcription factor ChIP-seq ENCODE experiments, in addition to peaks obtained for each replicate, more stringent/reproducible peak sets may be available including sets of replicated peaks, optimal or conservative IDR (irreproducible discovery rate) peaks sets (29).
FILER data aggregation and integration
FILER data are derived by harmonizing and integrating functional genomics and annotation datasets (Methods; ‘FILER structure and data organization’, ‘FILER data harmonization and annotation pipeline’) across various primary data sources/consortiums (21 in FILER v1.0). Supplementary Table S2 provides details of the primary data sources and experimental data types integrated into FILER.
FILER performs data integration at three levels: (i) individual dataset description (consistent meta information describing specifics of each of individual datasets), (ii) grouping of datasets into broader categories across data sources (see Materials and Methods) and (iii) data file formats (consistent and uniform file formats) (see Materials and Methods).
In particular, each individual FILER dataset is described by a consistent, standardized set of information attributes (metadata, >30 features; Supplementary Table S4), including biological sample, tissue/cell type, experimental protocol, library characteristics, along with file meta-information. This standard set of FILER attributes allows for consistent organization, presentation and retrieval of various datasets across data sources, biological conditions, and experimental data types (see ‘FILER features’; Supplementary Figure S4).
Further integration across data sources included in FILER is achieved by categorization of individual datasets into broader tissue/cell type, systems, and biological data categories (Materials and Methods; Supplementary Methods). Currently, FILER tracks span 36 tissue/cell type categories (e.g. see Supplementary Figure S1 for a summary of data tracks by tissue/cell type category) and include data across 162 fine-level biological data categories (see Supplementary Table S7 for a summary of biological data classification in FILER) and 14 system-level categories. This integrative categorization allows FILER to be used for viewing, filtering and aggregating data and query results across various tissue/cell types contexts and data sources (see, e.g., Supplementary Figure S4). Furthermore, all datasets integrated into FILER use consistent, uniform data file formats (BED-based) (Materials and Methods; see Supplementary Table S6 for details of BED formats available in FILER) allowing for seamless integration, retrieval, and comparison of datasets across data sources, experimental data types (see ‘FILER features’ and ‘FILER example use cases’ for examples).
The current version of FILER includes 152 indexed data collections (Materials and Methods). Supplementary Table S3 provides details on the data collections available in FILER.
Indexing of the individual data collections allows FILER to provide a scalable interface for querying (Materials and Methods; Supplementary Methods) genomic datasets by genomic coordinates or analysis and annotation of users’ own data (‘FILER features’ and ‘FILER example use cases’).
FILER provides query/search functions (‘FILER features’), allowing to easily integrate and query datasets using custom genetic and genomic analysis workflows such as INFERNO (1,2) (see ‘FILER example use cases’ for an example use cases). FILER data can be accessed using the website or deployed (‘FILER deployment’) at user’s own site and accessed through a command-line interface (‘FILER data access and querying API’). The scalable interface of FILER allows to find overlaps of user-generated experimental data (e.g. for a particular biological condition) with reference FILER datasets across various tissues/cell types and data sources (‘FILER features’), facilitating research studies across different human diseases.
FILER features
FILER aims to provide a unified functional genomics and annotation resource to the scientific community. Currently, FILER allows the users to
quickly stage (identify and retrieve) relevant experimental datasets in specific tissue contexts for downstream analyses (Supplementary Figure S4; ‘FILER deployment’)
efficiently search and retrieve all genomic records (annotations and experimental features) across various data sources within a genomic region of interest or a set of genomic regions (Supplementary Figure S2; ‘FILER data access and querying API’)
analyze and annotate user-provided experimental data using the reference datasets in FILER (Supplementary Figure S5; ‘FILER data access and querying API’)
deploy locally or on the cloud the entire FILER or a custom/selected subset of the FILER data (‘FILER deployment’)
Supplementary Table S1 shows a comparison of the FILER features with currently existing tools and web servers.
Search
FILER allows users to search for relevant genomic datasets based on their properties (meta-information). The genomic records (genomic features and annotations) across datasets can also be searched using FILER based on (i) genomic coordinates of the region of interest or (ii) a set of coordinates for the genomic loci, e.g., experimentally derived regions. Next, we describe the search functionality provided by FILER in more detail.
Search by genomic interval: Search by genomic interval (Supplementary Figure 3a) in FILER allows users to locate genomics data tracks and features overlapping with the given genomic coordinates. All the overlapping genomic features, data tracks, and their associated meta-data will be returned (see Supplementary Table S9 for an example of annotated overlap BED file; descriptions of the file columns are provided in Supplementary Table S4).
Analyze and annotate user’s own data: As shown in Supplementary Figure S2, the user’s genomic interval data are provided in BED format. The user BED file can either be uploaded to the FILER web server, or provided through a web-accessible URL (‘Analyze your own data’ tab).
Browse and search genomic datasets by data source, tissue/cell type, assay, data type and other properties: FILER allows users to locate relevant data tracks by a given metadata attribute or a combination of attributes (see Supplementary Figure S4; ‘FILER example use cases’, Use Case 1 for an example). The selected data tracks matching the search parameters can be then downloaded including the processed, as well as the original (raw) datasets along with their associated meta-data.
Analysis and annotation of user-provided data
FILER allows users to locate all data tracks and genomic features overlapping user-provided experimental/genomic features/intervals. The genomic/experimental intervals in the user-provided BED file are compared with all FILER datasets and all overlapping genomic features, data tracks and meta-data are reported (Supplementary Figure S3). The full set of results are available for download in the compressed (zip) format. The main annotated BED file containing all overlaps and FILER metadata is also available for download. FILER outputs a set of results tables and figures generated by matching the set of genomic loci in the user/input experiment against sets of genomic loci in each tissue and cell type in the reference FILER datasets (Supplementary Figure S3). As FILER performs this head-to-head comparison of the genomic loci in the input BED file with all functional genomics datasets in FILER, this functionality allows users to annotate and compare easily any experimental data of their own interest against all the FILER reference datasets.
FILER example use cases
Broad tissue/cell type coverage and a wide range of experimental data types available in FILER enable systematic analyses of genome-wide studies such as experimentally-derived ChIP-seq or RNA-seq genomic intervals or association signals and loci observed in GWASs.
We next describe example use cases for FILER, including (i) using a FILER web server for identification and retrieval of relevant experimental datasets (Use case 1), (ii) using custom/user experimental/genomic interval data with FILER webserver (Use case 2) and (iii) integration into high-throughput analysis workflows (Use case 3) using FILER command-line data access scripts.
Use case 1: relevant experimental data retrieval/data staging.
FILER can be used to identify and download relevant datasets using various criteria, including a particular human tissue context (tissue category), a particular experimental data type (assay) or a specific data source. As shown in Supplementary Figure S4, via the web-interface, data selectors allow users to choose specific data of interest. All the data tracks matching the selection criteria can be download in bulk or individually, including the associated track meta-information.
Alternatively, the command line interface provided by FILER (see Supplementary Methods, ‘Deploying FILER data’ section) allows users to deploy the relevant data subset of interest locally or on a cloud using the FILER meta-information table. The metadata will serve as a guide to download individual data tracks, re-create FILER data structure, and generate data indexes (see also Use case 3 for an example of integrating FILER data with a high-throughput workflow; Supplementary Methods, ‘Deploying FILER data’).
Use case 2: analyzing and annotating user’s own data.
FILER can be used for analysis of users’ own genomic interval data (e.g. top regions from GWAS analysis). In particular, FILER can be used to find overlaps with custom genomic data (in the form of genomic intervals) from other types of user experiments, such as ChIP-seq peaks, RNA-seq small RNA peaks, or ATAC-seq open chromatin regions. Figure 4 shows running time of FILER for various input data types (ChIP-seq peaks and Factorbook TFBSs) with input sizes ranging from 1000 to 1 000 000 query intervals. As can be seen from the figure, FILER scales well with input size: for example, increasing input size 90× from 1000 to 90 000 ChIP-seq peaks only results in 7× increase in the running time. Similarly, increasing number of input intervals 1000× from 1000 to 1 000 000 for Factorbook TFBSs only results in a sublinear 32× increase in the running time.
Figure 4.
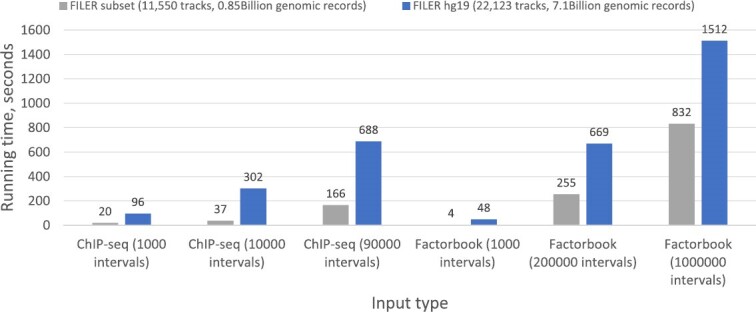
FILER highly efficient genomic search and retrieval. Shown is the running time (in seconds) as a function of the input size and type (from 1000 to 1 000 000 query intervals, ChIP-seq and transcription factor binding site [TFBS] data) and the number of reference data tracks/files (all hg19 FILER tracks with 7.1 Billion records and a ∼10% subsample of the FILER data with 0.8 Billion records). FILER demonstrates great efficiency as the observed running time for querying 1000× more of TFBS (Factorbook) genomic intervals (106 versus 103) against all 7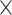 109 hg19 FILER records (blue series) increased sub-linearly by only a factor of 32×. Running time is measured on an AWS m5.4xlarge instance.
109 hg19 FILER records (blue series) increased sub-linearly by only a factor of 32×. Running time is measured on an AWS m5.4xlarge instance.
Using the provided BED file with select genomic intervals as input, FILER will generate an annotated BED file containing all genomic records overlapping with the user’s own list of variants or genomic regions of interest (available for download under ‘Annotated overlaps’ subsection). The annotated overlap file will contain overlap records consisting of the three sections: (i) all the data fields in the user input, (ii) overlapping FILER data track information and (iii) the associated FILER track meta-information. Along with the main annotated overlap file in BED format, FILER will generate a report detailing the distribution of overlapping genomic records across tissue categories, data sources and experimental data types. Additionally, overlapping genomic records corresponding to a particular tissue category/data source, or tissue category/experimental assay are available for individual download and further analysis.
In Supplementary Figure S5, we showed the FILER results via the web-portal when analyzed using a sample of ChIP-seq ENCODE data (ENCODE accession ENCFF000AIA; 92 008 ChIP-seq peaks) as an example of genomic interval data. This interactive heatmap allows users to download results in BED format for any specific tissue category and data source.
Use case 3: integration with a genetic and genomic analyses workflow.
Figure 5 shows an example of integrating FILER with a custom aggregation and analysis pipeline. In this example, FILER data are deployed on Amazon AWS cloud (see Supplementary Methods, ‘Deploying FILER data’ for details) and the SparkINFERNO (1) pipeline for non-coding variant analysis is using FILER datasets and FILER genomic query engine to characterize the input GWAS genetic variants. The annotated overlap data output spans across several biological data types, including enhancer, transcription factor (TF) binding and open chromatin genomic features.
Figure 5.
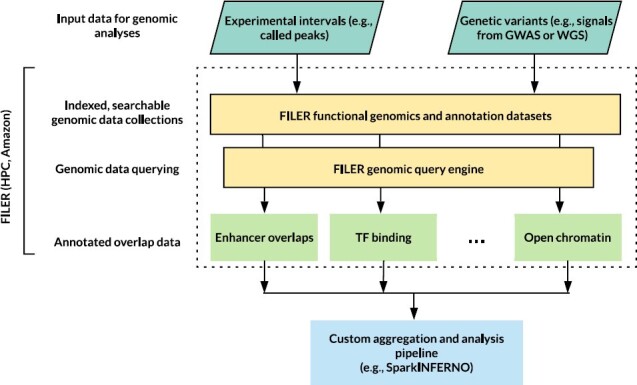
Example of integrating FILER (middle part of the figure) with a custom aggregation and analysis workflow (bottom of the figure), SparkINFERNO (1) for high-throughput non-coding variant analysis (inputs at the top).
To demonstrate the utility of FILER in analyzing and characterizing genomic regions or features of interest, we used FILER together with SparkINFERNO pipeline (1) for functional characterization of non-coding variants in IBD GWAS (30). As part of the SparkINFERNO pipeline, all genome-wide significant GWAS variants (P-value < 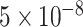 ) were used to obtain a set of candidate, potentially causal IBD variants by linkage disequilibrium (LD)-based pruning and expansion of the genome-wide significant GWAS SNPs. All the resulting 16 694 candidate variants were overlapped against GRCh37/hg19 FILER tracks (22 123 tracks, 7.1 Billion genomic records). In total, over 40 million overlaps with FILER genomic records were found. The overlap of these variants across functional genomic categories in different tissue types is shown in Supplementary Figure S6. As can be seen from the figure, overlap of IBD variants with the FILER data highlighted tissues that are likely important for IBD disease etiology, including blood, immune and digestive categories (2,30).
) were used to obtain a set of candidate, potentially causal IBD variants by linkage disequilibrium (LD)-based pruning and expansion of the genome-wide significant GWAS SNPs. All the resulting 16 694 candidate variants were overlapped against GRCh37/hg19 FILER tracks (22 123 tracks, 7.1 Billion genomic records). In total, over 40 million overlaps with FILER genomic records were found. The overlap of these variants across functional genomic categories in different tissue types is shown in Supplementary Figure S6. As can be seen from the figure, overlap of IBD variants with the FILER data highlighted tissues that are likely important for IBD disease etiology, including blood, immune and digestive categories (2,30).
FILER deployment
FILER is available in two different ways:
FILER can be deployed locally on a server or cluster (https://bitbucket.org/wanglab-upenn/FILER). This not only allows users to access harmonized functional genomics and annotation data stored in FILER but also integrates FILER into custom analysis pipelines. The scalable, genomic indexing-based interface allows to efficiently access FILER large-scale functional genomics data collection and use it in custom analyses.
FILER data are accessible through the web server (https://lisanwanglab.org/FILER). It allows users to search and retrieve relevant data, retrieve genomic features and annotation for genomic regions of interest, and analyze and annotate user’s experimental data with the reference FILER datasets.
FILER code repository (https://bitbucket.org/wanglab-upenn/FILER) provides installation, data querying and preprocessing scripts to download, prepare metadata and index FILER data. Supplementary Methods (‘Deploying FILER data’) provides details on how to install and use FILER locally.
FILER data access and querying API
All FILER data and metadata can be accessed programmatically using command-line interface. Command-line scripts for accessing/querying FILER data are available in the FILER code repository (https://bitbucket.org/wanglab-upenn/FILER). Individual track data and track metadata can be accessed, e.g. using the provided get_region_data.sh and get_metadata.sh scripts. Tracks in FILER can also be queried by a genomic interval of interest (e.g. get_overlapping_tracks_by_coord.sh script). For example, to find hg38 FILER tracks with genomic records overlapping a given genomic region (chr1:1103243–1203243), the following command can be used:
bash get_overlapping_tracks_by_coord.sh --region chr1:1103243–1203243 --outputDir query_output --genomeBuild hg38 --configFile filer.ini
For further details on the syntax, usage and example commands please refer to the README (https://bitbucket.org/wanglab-upenn/FILER) and the help (--help) for individual scripts.
DISCUSSION
Analyses of the results from GWASs and biological experiments require using external functional data as evidence for further interpretation, characterization and discovery. However, there is currently no single resource that provides unified access to a harmonized collection of such functional genomics and annotation data. This complicates the search and use of relevant functional genomic data, as well as the comparison and aggregation of these heterogeneous datasets for the analyses. More importantly, without a unified, harmonized data resource it is difficult to use these valuable resources in various genomic and genetic pipelines. We envision that the large-scale, harmonized FILER genomic and annotation data collection will facilitate the downstream genetic and genomic analyses, including but not limited to studying GWAS signals, via system biology, causal gene and other analyses. This allows researchers to focus on the creative analysis tasks, genomic analyses and discoveries rather than data collection and cleaning.
FILER uniquely provides an integrated and extensible repository of harmonized functional genomics and annotation data allowing for efficient and seamless retrieval, analysis and comparison across data sources, biological conditions, tissues/cell types and experimental data types. Using provided deployment and data interface, FILER allows integration with the existing or new analytical workflows. In addition to the web-based access, FILER can be installed on a local server, high-performance computing (HPC) cluster or cloud computing instances (see ‘FILER deployment’; Supplementary Methods).
Broad tissue/cell and experimental data type coverage in FILER enables systematic analyses of genome-wide experiments such as ChIP-seq or ATAC-seq, or genome-wide analyses of association signals observed in GWASs. Moreover, the modular, data collection-based FILER data architecture allows additional analysis-specific/user data or new data sources to be easily added without affecting other datasets/data collections.
We expect FILER to have a broad use in functional genomic research and genetic analyses for several reasons. First, FILER provides a new and flexible framework for integrating, harmonizing and efficiently querying large-scale genomics data from various sources. Second, FILER already integrates a broad range of genomic data types and biological conditions/tissues/cell types (>58 000 datasets) readily usable for genomic/genetic analyses and can be further expanded with additional data. Third, the framework, accompanying website, and the provided efficient data access and querying interfaces are all easy to use and will serve as convenient tools in broad range of applications and genomics-related research in general.
Future developments for FILER will include (i) broadening of tissue/cell type coverage, (ii) adding and expanding experimental data types including chromatin interaction/3D genome organization data, gene and RNA expression, and single-cell experiments, (iii) adding disease-specific datasets, (iv) variant-level annotations, (v) adding model organism (drosophila and mouse) data (e.g. from ADSP Functional Genomics Consortium https://www.nia.nih.gov/research/ad-genetics) and (vi) visualization, interactive display and exploration of functional genomic and annotation datasets.
DATA AVAILABILITY
All harmonized genomic datasets and the corresponding annotation meta-data are freely available from the FILER website https://lisanwanglab.org/FILER. An entire FILER database or a selected data subset can be deployed on local server, cloud or high-performance computing (HPC) clusters using installation and distribution scripts provided at https://bitbucket.org/wanglab-upenn/FILER.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mitchell Tang for setting up, collecting and organizing the initial collection of genomic and annotation tracks. We also thank the members of Wang Lab for their comments and insightful discussions.
FUNDING
National Institute on Aging [U24-AG041689, U54-AG052427, U01-AG032984]; Biomarkers Across Neurodegenerative Diseases (BAND 3) (award number 18062), co-funded by Michael J Fox Foundation, Alzheimer’s Association; Alzheimer’s Research UK; Weston Brain Institute. Funding for open access charge: National Institute on Aging [U24-AG041689, U54-AG052427].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kuksa P.P., Lee C.-Y., Amlie-Wolf A., Gangadharan P., Mlynarski E.E., Chou Y.-F., Lin H.-J., Issen H., Greenfest-Allen E., Valladares O. et al. SparkINFERNO: a scalable high-throughput pipeline for inferring molecular mechanisms of non-coding genetic variants. Bioinformatics. 2020; 36:3879–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amlie-Wolf A., Tang M., Mlynarski E.E., Kuksa P.P., Valladares O., Katanic Z., Tsuang D., Brown C.D., Schellenberg G.D., Wang L.-S. INFERNO: inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res. 2018; 46:8740–8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watanabe K., Taskesen E., van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017; 8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagraj V.P., Magee N.E., Sheffield N.C. LOLAweb: a containerized web server for interactive genomic locus overlap enrichment analysis. Nucleic Acids Res. 2018; 46:W194–W199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rouillard A.D., Gundersen G.W., Fernandez N.F., Wang Z., Monteiro C.D., McDermott M.G., Ma’ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford). 2016; 2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dozmorov M.G., Cara L.R., Giles C.B., Wren J.D. GenomeRunner web server: regulatory similarity and differences define the functional impact of SNP sets. Bioinformatics. 2016; 32:2256–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R. et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K. et al. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-Regulatory elements required for macrophage and b cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J. et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song L., Crawford G.E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010; 2010:pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aguet F., Brown A.A., Castel S.E., Davis J.R., He Y., Jo B., Mohammadi P., Park Y., Parsana P., Segrè A.V et al. Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aguet F., Barbeira A.N., Bonazzola R., Brown A., Castel S.E., Jo B., Kasela S., Kim-Hellmuth S., Liang Y., Oliva M. et al. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science (80-). 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-). 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J. et al. The UK biobank resource with deep phenotyping and genomic data. Nature. 2018; 562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuksa P.P., Amlie-Wolf A., Katanic Z., Valladares O., Wang L.S., Leung Y.Y. DASHR 2.0: integrated database of human small non-coding RNA genes and mature products. Bioinformatics. 2019; 35:1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leung Y.Y., Kuksa P.P., Amlie-Wolf A., Valladares O., Ungar L.H., Kannan S., Gregory B.D., Wang L.-S. DASHR: database of small human noncoding RNAs. Nucleic Acids Res. 2016; 44:D216–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuksa P.P., Amlie-Wolf A., Katanić Ž., Valladares O., Wang L.-S., Leung Y.Y. SPAR: small RNA-seq portal for analysis of sequencing experiments. Nucleic Acids Res. 2018; 46:W36–W42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Layer R.M., Pedersen B.S., Disera T., Marth G.T., Gertz J., Quinlan A.R. GIGGLE: a search engine for large-scale integrated genome analysis. Nat. Methods. 2018; 15:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. GFF3 General Feature Format 2021; (30 November 2021, date last accessed)http://gmod.org/wiki/GFF3
- 22. Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D. BigWig and bigbed: enabling browsing of large distributed datasets. Bioinformatics. 2010; 26:2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuhn R.M., Haussler D., James Kent W. The UCSC genome browser and associated tools. Brief. Bioinform. 2013; 14:144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amazon Web Services 2021; (30 November 2021, date last accessed)https://aws.amazon.com/
- 25. Park P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009; 10:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J., Zhuang J., Iyer S., Lin X.Y., Greven M.C., Kim B.H., Moore J., Pierce B.G., Dong X., Virgil D. et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013; 41:D171–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015; 2015:21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Agarwal V., Bell G.W., Nam J.-W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012; 22:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015; 47:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All harmonized genomic datasets and the corresponding annotation meta-data are freely available from the FILER website https://lisanwanglab.org/FILER. An entire FILER database or a selected data subset can be deployed on local server, cloud or high-performance computing (HPC) clusters using installation and distribution scripts provided at https://bitbucket.org/wanglab-upenn/FILER.