

ABSTRACT
Single-nucleotide polymorphism (SNPs) may cause the diverse functional impact on RNA or protein changing genotype and phenotype, which may lead to common or complex diseases like cancers. Accurate prediction of the functional impact of SNPs is crucial to discover the ‘influential’ (deleterious, pathogenic, disease-causing, and predisposing) variants from massive background polymorphisms in the human genome. Increasing computational methods have been developed to predict the functional impact of variants. However, predictive performances of these computational methods on massive genomic variants are still unclear. In this regard, we systematically evaluated 14 important computational methods including specific methods for one type of variant and general methods for multiple types of variants from several aspects; none of these methods achieved excellent (AUC ≥ 0.9) performance in both data sets. CADD and REVEL achieved excellent performance on multiple types of variants and missense variants, respectively. This comparison aims to assist researchers and clinicians to select appropriate methods or develop better predictive methods.
INTRODUCTION
With the rapid development of next-generation sequencing technologies, massive genomic variants in the human genome have been detected (1–3). Among them, a small subset of variants may be involved in common and complex diseases such as cancers and Mendelian diseases (4). How to distinguish which variants are ‘influential’ to the normal activities of life from the massive genomic variants, is meaningful and challenging research work. The functional impact (deleterious, pathogenic, disease-causing, and predisposing) of variants is that a genetic alteration may increase an individual’s susceptibility or predisposition to a certain disease or disorder (5). For example, a variant that occurred at the coding region of the DNA sequence may lead to the different amino acid translation or protein truncation, which may result in protein function weakening, association instability, or loss of protein function. The identification of the functional impact of massive variants is insufficiently efficient and usually time-consuming using experimental validation and manual curation. To the identification process, a growing number of computational methods and platforms have been developed to prioritize massive variants based on sequence homology/conservation (6–8), GC content (9), transcription factor binding sites (10,11), histone modification (12,13) and so on. According to the types of variants, all the methods can be classified into two types: (i) general methods applicable to all types of SNPs and (ii) specific methods applicable to a kind of variants.
Several surveys or comparisons (14–16) have been made to evaluate and analyze these prediction methods for different types of variants such as non-synonymous or synonymous variants. However, no studies have focused on the difference in prediction ability of computational methods of different types of SNPs within our knowledge. In this article, we provide a comprehensive comparison of general and specific methods in large-scale computational studies on predicting the functional impact of variants. In addition, one of the keys to a consistent and accurate comparison lies on unbiased test datasets. Thus, we constructed two independent test datasets based on the ClinVar and VariBench databases, which are widely used (14,16–18), reliable in quality and easily accessible. On these two datasets, we performed a comprehensive comparison of 14 functional impact prediction methods including CADD (19,20), DANN (21), FATHMM-MKL (22), FunSeq2 (23), PredictSNP2 (24), SIFT (25), PROVEAN (26), MetaLR (14), MetaSVM (14), MutationAssessor (27), PrimateAI (28), M-CAP (29), REVEL (30) and MISTIC (17). Based on the performance evaluation of these two datasets for 14 prediction methods, CADD and REVEL, obtained the best performance, respectively.
MATERIALS AND METHODS
Dataset resource
ClinVar
The ClinVar (31–35) database is a freely accessible, comprehensive, and public archive of human variations, phenotypes, and annotations of the functional impact of variants. ClinVar divides all human variants into 14 categories of clinical significance. Among them, five terms (benign, likely benign, uncertain significance, likely pathogenic and pathogenic) are used to indicate whether a variant is harmless or harmful. ClinVar provides continuous mutation information update to support researchers’ continuous research work.
VariBench
The VariBench (36) database provides multiple benchmark datasets from different resources such as ClinVar and Swiss-Prot. VariBench contains annotation information for experimentally verified effects and datasets that have been used to evaluate the performance of prediction methods. All variants are divided into two categories: pathogenic and neutral.
Test datasets
We constructed two independent datasets (reference gnome version is GRCh37/Hg19) in this study to conduct the performance comparison among the prediction methods listed in Table 1: (i) multiple types of variants such as missense variants, splice variants and 5′ UTR variants from the ClinVar database. Single-nucleotide variants with the clinically significant terms (‘pathogenic’, ‘likely pathogenic’, ‘benign’, ‘likely benign’) were collected as our tested benchmark data; (ii) missense variants from ClinVar and VariBench (the filtered VariBench datasets consists of HumVar (37), ExoVar (38), VariBench (36), predictSNP (39) and SwissVar (40)). The overlapping variants between the training set of compared methods and these two databases were removed to avoid the biased performance evaluation.
Table 1.
Summary of functional impact prediction methods analyzed in our study
| Order | Prediction Method | The Employed Model | Feature Set | Variant Type | Update (Y/N) | Published Journal | Web Site |
|---|---|---|---|---|---|---|---|
| 1 | CADD | Support Vector Machine | 63 distinct variant annotation retrieved from Ensembl Variant Effect Predictor (VEP), ENCODE project and UCSC genome browser tracks | All types of SNPs | Y | Nature Genetics (2014) | https://cadd.gs.washington.edu |
| 2 | DANN | Deep Learning | 63 distinct variant annotation retrieved from Ensembl Variant Effect Predictor (VEP), ENCODE project and UCSC genome browser tracks | All types of SNPs | N | Bioinformatics (2015) | https://cbcl.ics.uci.edu/public_data/DANN/ |
| 3 | FATHMM-MKL | Support Vector Machine | 10 feature groups including 46-way sequence conservation, histone modification (ChIP-Seq), transcription factor binding sites (TFBS PeakSeq), open chromatin (DNase-Seq), 100-way sequence conservation, GC content, Open Chromatin (FAIRE), transcription factor binding sites (TFBS SPP), genome segmentation and footprint from ENCODE project | All types of SNPs | Y | Bioinformatics (2015) | http://fathmm.biocompute.org.uk/ |
| 4 | FunSeq2 | Scoring System | 4 feature groups including variants in potential regulatory elements, nucleotide-level impact of regulatory variants, variants in conserved regions and network analysis of variants associated with genes | All types of SNPs (It is designed for non-coding variants) | Y | Genome Biology (2014) | http://funseq2.gersteinlab.org/ |
| 5 | PredictSNP2 | Ensemble Method | 5 functional prediction scores of variants | All types of SNPs | Y | PLOS Computational Biology (2016) | https://loschmidt.chemi.muni.cz/predictsnp2 |
| 6 | SIFT | Probability Estimation | Protein sequence conservation among homologs | Non-synonymous | Y | Nature Protocol (2009) | http://sift.jcvi.org |
| 7 | PROVEAN | Scoring System | Protein sequence conservation among homologs | Non-synonymous | Y | PLOS ONE (2012) | http://provean.jcvi.org/index.php |
| 8 | MetaLR | Logistic Regression | 9 functional prediction scores of variants | Non-synonymous | N | Human Molecular Genetics (2015) | No website |
| 9 | MetaSVM | Support Vector Machine | 9 functional prediction scores of variants | Non-synonymous | N | Human Molecular Genetics (2015) | No website |
| 10 | MutationAssessor | Scoring System | Sequence homology of protein | Non-synonymous | Y | Nucleic Acids Research (2011) | http://mutationassessor.org/r3/ |
| 11 | PrimateAI | Deep Learning | The protein structure and 51-length amino acid sequence centered at the variant of interest | Non-synonymous | Y | Nature Genetics (2018) | https://basespace.illumina.com/s/cPgCSmecvhb4 |
| 12 | M-CAP | Gradient Boosting Tree | Some pre-existing pathogenicity scores such as SIFT, CADD. Some pre-existing conservation scores. Four custom amino acid level features. | Missense | Y | Nature Genetics (2016) | http://bejerano.stanford.edu/MCAP/ |
| 13 | REVEL | Random Forest | Multiple functional prediction scores of variants and sequence conservation scores | Missense | N | The American Journal of Human Genetics (2016) | https://sites.google.com/site/revelgenomics/ |
| 14 | MISTIC | Ensemble Method | 4 feature groups including 690 functional measures, 8 multi-ethnic MAF, 8 conservation measures and 18 functional prediction scores | Missense | N | PLOS ONE (2020) | http://lbgi.fr/mistic/ |
Performance evaluation
The performance of the functional impact prediction methods was evaluated using the following measures:

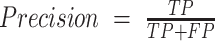

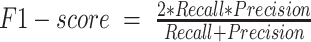
AUC = Area Under the ROC Curve
In the equations above, the following evaluation criteria are defined as follows: TP: True Positive; TN: True Negative; FP: False Positive; FN: False Negative. The Accuracy is the rate at which the prediction method correctly classifies the positive and negative cases. The Precision and Recall represent the ratio of real positive cases to predicted positive cases and correctly predicted positive to correctly predicted cases, respectively. F1-score is a compromise between precision and recall. The Area Under the Receiver Operating Characteristic curve (AUC) is a numerical representation of the ROC curve to indicate the performance of the prediction method more conveniently. Here, we employed ‘excellent’ (AUC ≥ 0.9), ‘very good’ (0.9 > AUC ≥ 0.8), ‘good’ (0.8 > AUC ≥ 0.7), ‘sufficient’ (0.7 > AUC ≥ 0.6) and ‘bad’ (0.6 > AUC ≥ 0.5) to evaluate the performance of computational prediction methods (41). The AUC, accuracy, precision, recall, and F1-score were obtained using the pROC (42) package implemented by the R language and the evaluation used the best accuracy to determine the thresholds.
Overview of prediction methods for functional impact of variants
Several computational methods have been developed to predict the functional impact of variants. In this article, we evaluated 14 important state-of-the-art prediction methods (Table 1) including CADD (19,20), DANN (21), FATHMM-MKL (22), FunSeq2 (23), PredictSNP2 (24), SIFT (25), PROVEAN (26), MetaLR (14), MetaSVM (14), MutationAssessor (27), PrimateAI (28), M-CAP (29), REVEL (30) and MISTIC (17) (we obtained prediction scores for each genomic variant for 14 methods by running their stand-alone programs, publicly available web servers, ANNOVAR (43) or the dbNSFP (44) database). According to the employed features and model, all of the prediction methods are divided into two types: (i) general methods applicable to all types of SNPs and (ii) specific methods applicable to a kind of variants.
General methods
CADD
The Combined Annotation–Dependent Depletion (CADD) is a general framework, which integrates diverse genome annotations and scores of any possible human single-nucleotide variant or small insertion-deletion event. CADD employs 63 distinct variant annotations retrieved from Ensembl (45), Variant Effect Predictor (VEP), ENCODE project, and UCSC genome browser tracks (46–48) and implemented a support vector machine (SVM) as the predictive model, which was trained to differentiate 14.7 million high-frequency human-derived alleles from 14.7 million simulated variants.
DANN
Deleterious annotation of genetic variants using neural networks (DANN) is a deep neural network model. Instead of SVM model of CADD, DANN performed an artificial neural network with several hidden layers of units using the training data of CADD (16627775 ‘observed’ variants and 49407057 ‘simulated’ variants).
FATHMM-MKL
FATHMM-MKL is an integrative approach that predicts the functional impacts of coding and non-coding variants. Ten coding and non-coding feature sets (such as sequence conservation, histone modification, and transcription factor binding sites) are employed to train the SVM model to prioritize the coding and non-coding variants.
FunSeq2
FunSeq2 implements a scoring system that consists of coding scoring scheme and noncoding scoring scheme to prioritize variants in cancer. Four feature groups including variants in potential regulatory elements, the nucleotide-level impact of regulatory variants, variants in conserved regions, and network analysis of variants associated with genes were used as the input of the scoring system.
PredictSNP2
PredictSNP2 integrates five prediction methods (CADD, DANN, FATHMM, FunSeq2 and GWAWA) to predict the functional impact of variants. PredictSNP2 employs a consensus classifier to build the prediction model. The consensus was determined based on a majority vote, with the composition of classifier being weighted by their confidences.
Specific methods
SIFT
Sorting tolerant from intolerant (SIFT) is designed to prioritize non-synonymous single nucleotide polymorphism (nsSNP) occurring in the coding region of genome may cause an amino acid substitution (AAS) of the corresponding gene product, and this change may affect the function of host gene product and the phenotype of host organism. SIFT calculates a prediction score derived from the distribution of amino acid residues observed at the given position in the sequence alignment and the estimated unobserved frequencies of amino acid distribution calculated from a Dirichlet mixture.
PROVEAN
Protein Variation Effect Analyzer (PROVEAN) is a prediction method based on the delta alignment score of pairwise sequence alignment. The delta alignment score represents that it can interpret a change in the alignment score caused by an amino acid variation as the functional impact of host protein of the variant. PROVEAN also can be used to predict the functional impact of all classes of protein sequence variations such as insertions, deletions and multiple substitutions.
MetaLR and MetaSVM
MetaLR and MetaSVM are two ensemble methods, logistic regression and support vector machine, which integrate multiple scores of prediction methods. These two methods only focused on the non-synonymous variants.
MutationAssessor
MutationAssessor predicts the functional impact of variants based on the assumption that protein family sequences reflect the continuity of functional constraints and can be treated as a statistical ensemble, that is, the observed distributions of residues in aligned positions of homologous sequences reflect the functional constraints on these residues. Thus, evolutionarily unfavorable variants/residues are not observed or observed less frequently than neutral variants/residues, while critically important residues are conserved in diverse evolutionary settings. As a result, the functional impact score consists of the conservation score and the specificity score is used as the prediction score.
PrimateAI
PrimateAI was proposed by the researchers of Illumina Artificial Intelligence Laboratory. PrimateAI predicts the functional impact of variants based on the architecture of deep learning network and takes the 54-length amino acid sequence centered at the variants of interest as input of deep learning model. In addition to using linear sequence data, the secondary structure of proteins is also used as input data for deep learning models.
M-CAP
Mendelian clinically applicable pathogenicity (M-CAP) produces likelihood scores that aim to misclassify no >5% of pathogenic variants while aggressively reducing the list of variants of uncertain significance. The feature set of M-CAP consists of 9 functional prediction scores such as SIFT and CADD, 7 pre-existing conservation and variant intolerance scores and 4 custom amino acid scores. The prediction model of M-CAP is gradient boosting tree classifier.
REVEL
REVEL is an ensemble method, which integrates multiple functional prediction scores and sequence conservation scores such as SIFT, PROVEAN, FATHMM, MutationAssessor, GERP++ and phyloP. The employed model of REVEL is random forest classifier.
MISTIC
MISTIC also is an ensemble method, which integrates four feature groups: (i) 8 multi-ethnic MAF; (ii) 8 conservation scores; (iii) 690 functional measures; (iv) 7 functional prediction scores such as SIFT and CADD. MISTIC employs random forest classifier and logistic regression classifier as the prediction model.
RESULTS
Experiments based on multiple types of variants in ClinVar and VariBench
Experimental results on multiple types of variants (e.g. intron variants and missense variants) are shown in Figure 1 and Table 2. Figure 1 shows the ROC curves for five general methods. The CADD (AUC: 0.948) showed the best performance and the AUCs for the other four methods (PredictSNP2, DANN, FATHMM-MKL and FunSeq2) were >0.7 (AUC for PredictSNP2 is 0.787, AUC for DANN is 0.883, AUC for FATHMM-MKL is 0.857 and AUC for FunSeq2 is 0.822). Table 2 shows the accuracy, precision, recall and F1-score for five general methods. The CADD (accuracy: 0.8796, precision: 0.761 and F1-score: 0.8242) showed best performances on accuracy, precision and F1-score. The DANN (recall: 0.9223) showed the best performance on recall. Overall, the CADD method achieved the best performance based on the multiple types of variants.
Figure 1.

The AUCs of different prediction methods on the multiple types of variants.
Table 2.
The performance of prediction methods using multiple types of variants
| Order | Methods | Accuracy | Precision | Recall | F1-score |
|---|---|---|---|---|---|
| 1 | PredictSNP2 | 0.7142 | 0.5266 | 0.8940 | 0.6628 |
| 2 | DANN | 0.8258 | 0.6592 | 0.9223 | 0.7688 |
| 3 | FATHMM-MKL | 0.7855 | 0.6134 | 0.8576 | 0.7152 |
| 4 | FunSeq2 | 0.7235 | 0.5391 | 0.8264 | 0.6525 |
| 5 | CADD | 0.8796 | 0.7610 | 0.8988 | 0.8242 |
To visualize the distribution of scores of PredictSNP2, DANN, FATHMM-MKL, FunSeq2 and CADD, we plotted raw prediction scores of deleterious and neutral variants as shown in Figure 2. As demonstrated in Figure 2, CADD scores are distributed in low score areas for neutral variants and in high score areas for deleterious variants. Thus, CADD achieved the best performance in most evaluation criteria. PredictSNP2 scores are highly concentrated near −1 for neutral variants and 1 for deleterious variants. However, there are many deleterious and neutral variants densely clustered around 1 leading to the average performance of predictSNP2. The distribution of scores analysis facilitated the selection of threshold for clinicians and researchers.
Figure 2.

The distribution histogram of scores of PredictSNP2, DANN, FATHMM-MKL, FunSeq2 and CADD for deleterious and neutral variants.
To evaluate the correlation of predictive results between any two computational methods, we computed the Spearman’s Rank Correlation Coefficientρ(rho) based on prediction scores of five general methods. As shown in Figure 3, the highest correlation was found between CADD and DANN (rho: 0.85). The prediction scores of CADD and DANN were also highly positively correlated, which may be related to the fact that both used the same training set and feature set. The prediction scores of FunSeq2 have relatively low correlation with other methods, which also coincided with the score distribution in Figure 2.
Figure 3.

Correlation coefficients between the 5 methods (PredictSNP2, DANN, FATHMM-MKL, FunSeq2 and CADD) based on multiple types of variants.
Experiments based on missense variants in ClinVar and VariBench
Missense variants produce the Amino Acid Substitutions (AASs) in the sequences of gene products. Usually, the AASs may affect the biological function of a gene product in many ways (49). However, compared with nonsense variants, the effect of AASs on sequences and structures of gene products is not intuitive enough. Thus, prediction of the possible functional impact of missense variants is an important and challenging problem. Therefore, we choose missense variants to evaluate these computational prediction methods. Experimental results on missense variants are shown in Figure 4 and Table 3. Figure 4 shows the ROC curves for 14 methods. The REVEL (AUC: 0.905) showed the best performance and FunSeq2 (AUC: 0.603) achieved the worst performance. The AUC for the other individual prediction methods ranged from 0.696 to 0.89. In the experiments on multiple types of variants, DANN, FATHMM-MKL and FunSeq2 have achieved 'very good' performance and CADD has achieved 'excellent' performance. In the experiments on missense variants, CADD and FATHMM-MKL just achieved 'good' performance, DANN and FunSeq2 even only achieved 'sufficient, performance. We attribute the different performances to insufficient/unoptimized feature set for missense variants in these general prediction methods.
Figure 4.

The AUCs of different prediction methods on the missense variants.
Table 3.
The performance of prediction methods using the missense variants
| Order | Methods | Accuracy | Precision | Recall | F1-score |
|---|---|---|---|---|---|
| 1 | PredictSNP2 | 0.6983 | 0.7042 | 0.7609 | 0.7315 |
| 2 | DANN | 0.6716 | 0.6642 | 0.7928 | 0.7228 |
| 3 | FATHMM-MKL | 0.6793 | 0.6884 | 0.7422 | 0.7143 |
| 4 | FunSeq2 | 0.5947 | 0.6046 | 0.7211 | 0.6577 |
| 5 | CADD | 0.7198 | 0.7453 | 0.7312 | 0.7382 |
| 6 | SIFT | 0.7146 | 0.7771 | 0.6612 | 0.7145 |
| 7 | PROVEAN | 0.7267 | 0.7476 | 0.7457 | 0.7467 |
| 8 | MetaLR | 0.7964 | 0.8360 | 0.7751 | 0.8044 |
| 9 | MetaSVM | 0.8015 | 0.8409 | 0.7802 | 0.8094 |
| 10 | MutationAssessor | 0.7163 | 0.7797 | 0.6617 | 0.7159 |
| 11 | PrimateAI | 0.6877 | 0.6939 | 0.7546 | 0.7230 |
| 12 | M-CAP | 0.8040 | 0.8494 | 0.7744 | 0.8101 |
| 13 | REVEL | 0.8305 | 0.8578 | 0.8226 | 0.8398 |
| 14 | MISTIC | 0.8216 | 0.8604 | 0.7994 | 0.8288 |
Then, we also plotted the distribution histogram of raw scores of 14 prediction methods for deleterious and neutral variants as shown in Supplementary Figure S1. The prediction scores of some methods, including MetaLR, MetaSVM, REVEL and MISTIC, can produce peaks in two relatively separate regions. These methods also showed good performances in the prediction of the functional impact of missense variants (Figure 4 and Table 3). Although the prediction scores of M-CAP did not call two separate peaks for deleterious and neutral variants, the prediction scores of neutral variants concentrated on the low-score area and the prediction scores of deleterious variants were evenly distributed in the whole prediction score range. Thus, the performance of M-CAP also was relatively good. However, the score distribution of this method may cause confusion in the selection of the threshold in practical applications. This analysis provided another way to illustrate the ability of different methods and another perspective to help researchers and users determine the classification threshold reasonably.
The rho coefficients between 14 prediction methods on missense variants are as shown in Figure 5. MetaLR and MetaSVM showed the highest positive correlation (rho: 0.95) because these two methods employed the same ensemble learning components. Besides, the scores of MetaLR, MetaSVM, REVEL and MISTIC also showed a high positive correlation, which was also consistent with the performance of evaluation criteria as shown in Figure 4 and Table 3. Compared with the other prediction methods, the lower scores for SIFT and PROVEAN indicate deleterious variants and higher scores indicate neutral variants. Therefore, the coefficients of SIFT /PROVEAN and other prediction methods are negative, that is, negative correlation. MetaLR and MetaSVM employ the same feature set but different machine learning methods (linear regression and support vector machine) to predict the functional impact of variants. However, both the experimental results and the correlation coefficients of the predicted scores of these two methods are similar. MetaSVM, CADD and FATHMM-MKL employ support vector machine as the predictive model, but different feature sets to predict the functional impact of variants. CADD and FATHMM-MKL have achieved ‘good’ performance, and MetaSVM has achieved ‘very good’ performance. SIFT, PROVEAN and MutationAssessor employ scoring system or probability estimation based on the protein sequence to predict the functional impact of variants. Although the three methods use different scoring systems or probability estimations, they achieve ‘good’ performance.
Figure 5.

Correlation coefficients between the 14 methods based on the missense variants.
DISCUSSION
Annotation and analysis of genomic variants are critical and interesting studies in the post-genome era. Among them, the study of functional impact of SNPs is an important research field. For example, Daboub et al. (50) claimed the discovery that two deleterious variants in RASA1 were associated with Parkes Weber syndrome. Timms et al. (51) discovered that some BACA1/2 deleterious variants occurred in all breast cancer subtypes. Based on the biological experiment methods, the identification of the functional impact of massive variants is insufficiently efficient and usually time-consuming. Thus, many computational prediction methods have been widely developed to investigate the functional impact of genomic variants. However, the predictive performance of these computational methods on massive genomic variants is still unclear. Here, we evaluated 14 state-of-the-art computational methods including general methods applicable to all types of SNPs and specific methods applicable to a kind of variants. For general methods, CADD achieved ‘excellent’ (AUC ≥ 0.9) performance on multiple types of variants but ‘good’ (0.8 > AUC ≥ 0.7) performance on missense variants. FATHMM-MKL and PredictSNP2 achieved ‘very good’ (0.9 > AUC ≥ 0.8) performance on multiple types of variants but also ‘good’ performance on missense variants. For specific methods, REVEL achieved ‘excellent’ performance and some ensemble learning methods (e.g. MISTIC, MetaSVM and MetaLR) achieved ‘very good’ performances on missense variants. Some methods that employed single type of feature (e.g. SIFT and PROVEAN) also achieved ‘good’ performance on missense variants.
Advantages and disadvantages of 14 prediction methods
Different prediction methods have certain advantages and disadvantages. The methods such as REVEL, MISTIC, M-CAP, MetaLR and MetaSVM integrate the prediction scores of other computational methods as features. Their advantage is that their prediction performance is very good, but the types of variants that can be predicted are relatively limited. SIFT, PROVEAN and MutationAssessor mainly focus on the impact of changes in protein sequence sites. Their performance is relatively good, but the biological meaning of their prediction scores is clear and easy to understand. CADD, PredictSNP2 and FATHMM-MKL are able to predict the functional impact of multiple types of variants. However, the predictive ability of these methods is not good enough for missense variants. DANN and PrimateAI employ deep learning technology, but their performance does not significantly outperform other methods. Here, we recommend some specific methods, such as M-CAP and REVEL, when the variants concerned by the user are non-synonymous or missense. However, CADD and FATHMM-MKL may be the better choice when users need to predict the functional impact of large-scale uncertain types of variants.
Future work
Deep learning techniques have achieved an overwhelming advantage in some research fields of computer science and bioinformatics, such as computer vision and natural language process. In the field of prioritizing variants, some methods also employ deep learning techniques such as deep neural network (DNN) to predict the functional impact of variants. For example, PrimateAI employs a DNN model to facilitate the effect prediction of variants. However, PrimateAI did not significantly outperform other prediction methods that employed traditional machine learning techniques or scoring systems. We think that DNN model requires a larger number of variant data with deleterious and neutral labels, while variant data with deleterious and neutral labels are not enough now. With the increase of labeled variant samples, the performance of methods based on deep learning techniques should be better. In addition to increasing the number of labeled variants, variant feature mining and the development of deep learning technology are potential ways to improve the performance of computational prediction methods.
DATA AVAILABILITY
The datasets for this study can be found in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and VariBench (http://structure.bmc.lu.se/VariBench/GrimmDatasets.php).
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge the contributions of colleagues in the group.
Author Contributions: D.W. and J.L. designed and implemented the algorithm. D.W. and J.L. analyzed the results and wrote the manuscript. Y.W. and E.W. made suggestions. All authors read and approved the final manuscript.
Contributor Information
Dong Wang, School of Computer Science and Technology, Harbin Institute of Technology Harbin, Harbin, Heilongjiang, 150001,China.
Jie Li, School of Computer Science and Technology, Harbin Institute of Technology Harbin, Harbin, Heilongjiang, 150001,China.
Yadong Wang, School of Computer Science and Technology, Harbin Institute of Technology Harbin, Harbin, Heilongjiang, 150001,China.
Edwin Wang, Department of Medical Genetics, University of Calgary, Calgary, HSC 1185, Canada.
FUNDING
National Key Research and Development Program of China [2016YFC0901905].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1. Rabbani B., Tekin M., Mahdieh N.. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014; 59:5–15. [DOI] [PubMed] [Google Scholar]
- 2. Goodwin S., McPherson J.D., McCombie W.R.. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016; 17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lek M., Karczewski K.J., Minikel E.V, Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A.et al.. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014; 508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. National Cancer Institute Pathogenic Variant Difinition. 2021; https://www.cancer.gov/publications/dictionaries/genetics-dictionary/def/pathogenic-variant [Google Scholar]
- 6. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S.et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davydov E.V, Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S.. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010; 6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A.. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010; 20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D.et al.. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004; 14:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rozowsky J., Euskirchen G., Auerbach R.K., Zhang Z.D., Gibson T., Bjornson R., Carriero N., Snyder M., Gerstein M.B.. PeakSeq enables systematic scoring of chip-seq experiments relative to controls. Nat. Biotechnol. 2009; 27:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kharchenko P.V, Tolstorukov M.Y., Park P.J.. Design and analysis of chip-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008; 26:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kazachenka A., Bertozzi T.M., Sjoberg-Herrera M.K., Walker N., Gardner J., Gunning R., Pahita E., Adams S., Adams D., Ferguson-Smith A.C.. Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell. 2018; 175:1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inoue F., Kircher M., Martin B., Cooper G.M., Witten D.M., McManus M.T., Ahituv N., Shendure J.. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017; 27:38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X.. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015; 24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng N., Li M., Zhao L., Zhang B., Yang Y., Zheng C.-H., Xia J.. Comparison and integration of computational methods for deleterious synonymous mutation prediction. Brief. Bioinform. 2020; 21:970–981. [DOI] [PubMed] [Google Scholar]
- 16. Hassan M.S., Shaalan A.A., Dessouky M.I., Abdelnaiem A.E., ElHefnawi M.. A review study: computational techniques for expecting the impact of non-synonymous single nucleotide variants in human diseases. Gene. 2019; 680:20–33. [DOI] [PubMed] [Google Scholar]
- 17. Chennen K., Weber T., Lornage X., Kress A., Böhm J., Thompson J., Laporte J., Poch O.. MISTIC: a prediction tool to reveal disease-relevant deleterious missense variants. PLoS One. 2020; 15:e0236962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J., Zhao T., Zhang Y., Zhang K., Shi L., Chen Y., Wang X., Sun Z.. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018; 46:7793–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M.. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quang D., Chen Y., Xie X.. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015; 31:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shihab H.A., Rogers M.F., Gough J., Mort M., Cooper D.N., Day I.N.M., Gaunt T.R., Campbell C.. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015; 31:1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fu Y., Liu Z., Lou S., Bedford J., Mu X.J., Yip K.Y., Khurana E., Gerstein M.. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014; 15:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bendl J., Musil M., Štourač J., Zendulka J., Damborský J., Brezovský J.. PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput. Biol. 2016; 12:e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar P., Henikoff S., Ng P.C.. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009; 4:1073–1082. [DOI] [PubMed] [Google Scholar]
- 26. Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P.. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012; 7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reva B., Antipin Y., Sander C.. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sundaram L., Gao H., Padigepati S.R., McRae J.F., Li Y., Kosmicki J.A., Fritzilas N., Hakenberg J., Dutta A., Shon J.et al.. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018; 50:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jagadeesh K.A., Wenger A.M., Berger M.J., Guturu H., Stenson P.D., Cooper D.N., Bernstein J.A., Bejerano G.. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016; 48:1581. [DOI] [PubMed] [Google Scholar]
- 30. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D.et al.. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016; 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C.et al.. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020; 48:D835–D844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Landrum M.J., Kattman B.L.. ClinVar at five years: delivering on the promise. Hum. Mutat. 2018; 39:1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W.et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J.et al.. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nair P.S., Vihinen M.. VariBench: a benchmark database for variations. Hum. Mutat. 2013; 34:42–49. [DOI] [PubMed] [Google Scholar]
- 37. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li M.-X., Kwan J.S.H., Bao S.-Y., Yang W., Ho S.-L., Song Y.-Q., Sham P.C.. Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013; 9:e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bendl J., Stourac J., Salanda O., Pavelka A., Wieben E.D., Zendulka J., Brezovsky J., Damborsky J.. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014; 10:e1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mottaz A., David F.P.A., Veuthey A.-L., Yip Y.L.. Easy retrieval of single amino-acid polymorphisms and phenotype information using swissvar. Bioinformatics. 2010; 26:851–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Šimundić A.-M. Measures of diagnostic accuracy: basic definitions. Ejifcc. 2009; 19:203. [PMC free article] [PubMed] [Google Scholar]
- 42. Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M.. pROC: an open-source package for r and S+ to analyze and compare ROC curves. BMC Bioinform. 2011; 12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu X., Li C., Mou C., Dong Y., Tu Y.. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020; 12:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R.et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haeussler M., Zweig A.S., Tyner C., Speir M.L., Rosenbloom K.R., Raney B.J., Lee C.M., Lee B.T., Hinrichs A.S., Gonzalez J.N.et al.. The UCSC genome browser database: 2019 update. Nucleic Acids Res. 2019; 47:D853–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raney B.J., Dreszer T.R., Barber G.P., Clawson H., Fujita P.A., Wang T., Nguyen N., Paten B., Zweig A.S., Karolchik D.et al.. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC genome browser. Bioinformatics. 2014; 30:1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thusberg J., Vihinen M.. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum. Mutat. 2009; 30:703–714. [DOI] [PubMed] [Google Scholar]
- 50. Daboub J.A.F., Grimmer J.F., Frigerio A., Wooderchak-Donahue W., Arnold R., Szymanski J., Longo N., Bayrak-Toydemir P.. Parkes weber syndrome associated with two somatic pathogenic variants in RASA1. Mol. Case Stud. 2020; 6:a005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Timms K.M., Abkevich V., Hughes E., Neff C., Reid J., Morris B., Kalva S., Potter J., Tran T.V, Chen J.et al.. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014; 16:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets for this study can be found in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and VariBench (http://structure.bmc.lu.se/VariBench/GrimmDatasets.php).