

SUMMARY:
ATRX, a chromatin remodeler protein, is recurrently mutated in H3F3A-mutant pediatric glioblastoma (GBM) and IDH-mutant grade 2/3 adult glioma. Previous work has shown that ATRX-deficient GBM cells show enhanced sensitivity to irradiation, but the etiology remains unclear. We find that ATRX binds regulatory elements of cell cycle phase transition genes in GBM cells, and there is a marked reduction in Checkpoint Kinase 1 (CHEK1) expression with ATRX loss, leading to early release of G2/M entry after irradiation. ATRX-deficient cells exhibit enhanced activation of master cell cycle regulator ATM with irradiation. Addition of the ATM inhibitor AZD0156 doubles median survival in mice intra-cranially implanted with ATRX-deficient GBM cells, which is not seen in ATRX-wildtype controls. This study demonstrates that ATRX-deficient high-grade gliomas (HGGs) display Chk1-mediated dysregulation of cell cycle phase transitions, which opens a window for therapies targeting this phenotype.
Keywords: Glioma, Cell-cycle, Epigenetics, ATRX, ATM inhibitor, CHEK1
Graphical Abstract
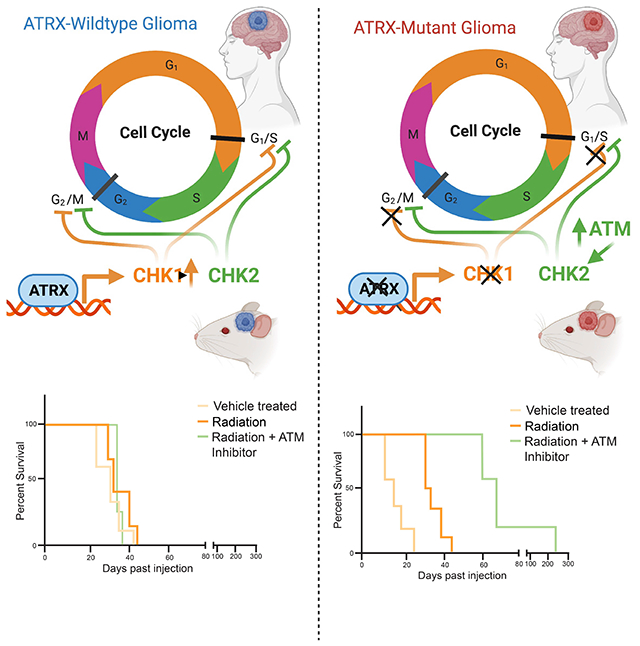
Qin et al. show that ATRX binds the gene regulatory elements and promotes expression of the cell cycle regulator of CHEK1 in glioma and glioma precursor cells. ATRX loss in high-grade glioma results in decreased Chk1, dysregulation of cell cycle checkpoints after radiation, and enhanced radio-sensitization with ATM inhibitors.
INTRODUCTION:
ATRX is a histone chaperone protein that loads histones onto telomeres and maintains heterochromatin environments (Berube et al., 2005; Heaphy et al., 2011; Lewis et al., 2010; Voon et al., 2015). ATRX is a member of the SWI/SNF superfamily of chromatin remodeler proteins, and contains two highly conserved domains (Valle-Garcia et al., 2016). An ADD (ATRX-DNMT3-DNMT3L) domain binds to and regulates H3K9me3-modified chromatin, and a SWI/SNF domain utilizes ATP to perform chromatin remodeling duties of ATRX (Valle-Garcia et al., 2016). ATRX also forms a complex with Death Domain Associated Protein (DAXX) to deposit the histone variant H3.3 at nucleosomes (Lewis et al., 2010). ATRX plays a key role in brain development. Germline mutations in ATRX cause α-thalassemia mental retardation X-linked (ATR-X) syndrome, a rare disorder that is marked by severe cognitive deficits (Gibbons and Higgs, 2000; Gibbons et al., 1995). In mice, ATRX deficiency in the forebrain leads to congenital reduction in cellular proliferation within the cerebral cortex and hippocampus (Berube et al., 2005). It was later demonstrated that this phenotype could be partially explained by p53-dependent neuronal apoptosis, which is rescued by the loss of p53 (Seah et al., 2008).
Recent work in human gliomas has documented recurrent loss-of-function mutations in ATRX (Koschmann et al., 2016a; Koschmann et al., 2016b; Koschmann et al., 2017; Schwartzentruber et al., 2012). In pediatric patients, ATRX mutation is reported in 31% of patients with primary glioblastoma (GBM) [World Health Organization (WHO) grade IV glioma], virtually always with concurrent mutation of TP53, and frequently with mutations in the histone variant gene H3F3A (Jiao et al., 2012; Miklja et al., 2019; Schwartzentruber et al., 2012). In adult patients, ATRX is mutated in WHO grade II/III astrocytic glioma and secondary GBM (Eckel-Passow et al., 2015; Jiao et al., 2012; Suzuki et al., 2015), where 75% of astrocytic gliomas with IDH1 and TP53 mutations also carry ATRX mutations (Eckel-Passow et al., 2015). ATRX-deficient tumors maintain telomere length in a telomerase-independent manner, labeled Alternative Lengthening of Telomeres (ALT) (Lovejoy et al., 2012). The ATRX protein has been shown to inhibit ALT, and when mutated, the affected tumors cells are able to maintain telomere extension through homologous recombination (HR) (Clynes and Gibbons, 2013; Clynes et al., 2015). Currently, treatments for ATRX-deficient glioma remain non-targeted and ineffective.
We previously established a genetically-engineered mouse model of ATRX-deficient GBM, and established that ATRX loss results in both glioma tumor growth and sensitivity to irradiation therapy (Koschmann et al., 2016a; Koschmann et al., 2016b; Koschmann et al., 2017). However, the mechanism driving these findings has not been fully established or replicated in human glioma models. In non-brain tumor settings such as embryonic stem cells (ESCs), ATRX primarily binds telomeres and repetitive (non-coding) heterochromatin (Valle-Garcia et al., 2016; Voon et al., 2015). Recent data have shown that in certain developmental contexts, such as in neuronal progenitor cells (NPCs), a potential cell of origin for glioma, ATRX plays a more prominent role in gene regulatory element (e.g. promoter, enhancer) binding and transcriptional regulation when compared to that in ESCs (Danussi et al., 2018).
The mechanism driving radiation sensitivity in ATRX-deficient glioma has not been established. We sought to clarify whether specific epigenetically driven transcriptional changes, wherein chromatin is modified without alteration to the gene itself, may explain the proliferative changes and responsivity to irradiation seen in ATRX-mutant human glioma. In this study, we demonstrate that in mNPCs and murine GBM (mGBM) neurospheres, ATRX binds to the regulatory elements of genes involved in cell cycle phase transition, including the cell cycle checkpoint regulatory gene Checkpoint Kinase 1 (CHEK1), and that ATRX loss results in down-regulation of Chk1 in multiple high-grade glioma (HGG) models. Additionally, we found that ATRX-deficient human and mouse GBM cells demonstrate reduced ability to maintain the G2/M cell cycle checkpoint in response to irradiation, and targeted sensitization with ATM inhibition. Our findings support a role for Chk1 reduction in the cell cycle dysregulation in ATRX-deficient glioma, and provide important clarity in the optimization of targeted therapies for ATRX-deficient glioma.
RESULTS:
ATRX loss in human glioma leads to increased sensitivity and proliferation following irradiation treatment in vitro
In order to clarify whether ATRX loss in human models of glioma contributes to changes in sensitivity to irradiation, we obtained three human GBM cell lines, U251, SF188 and UW479, with isogenic CRISPR ATRX knockout (U251-ATRXKO, SF188-ATRXKO, UW479-ATRXKO) or empty vector control (U251-ATRXEV, SF188-ATRXEV, UW479-ATRXEV) (Brosnan-Cashman et al., 2018). We also utilized two primary pHGG cell cultures, SJ-GBM2, an ATRX-mutant, IDH-wildtype cell line, and KNS42, an ATRX- and IDH-wildtype cell line (Figure 1A) (Project, 2016). All cells demonstrated loss of ATRX protein by western blot and positive c-circle (ALT) assay; in particular U251-ATRXKO cells (Figures 1B and S1A–B), confirming a critical phenotypic attribute seen in human ATRX-deficient gliomas (Heaphy et al., 2011). We found that ATRX loss did not intrinsically affect cellular proliferation in U251 cells as compared to isogenic controls (Figure S1C), consistent with published data of ATRXKO NPCs (Danussi et al., 2018).
Figure 1. ATRX-deficient human glioma cells demonstrate inappropriate cell cycling after treatment with irradiation.
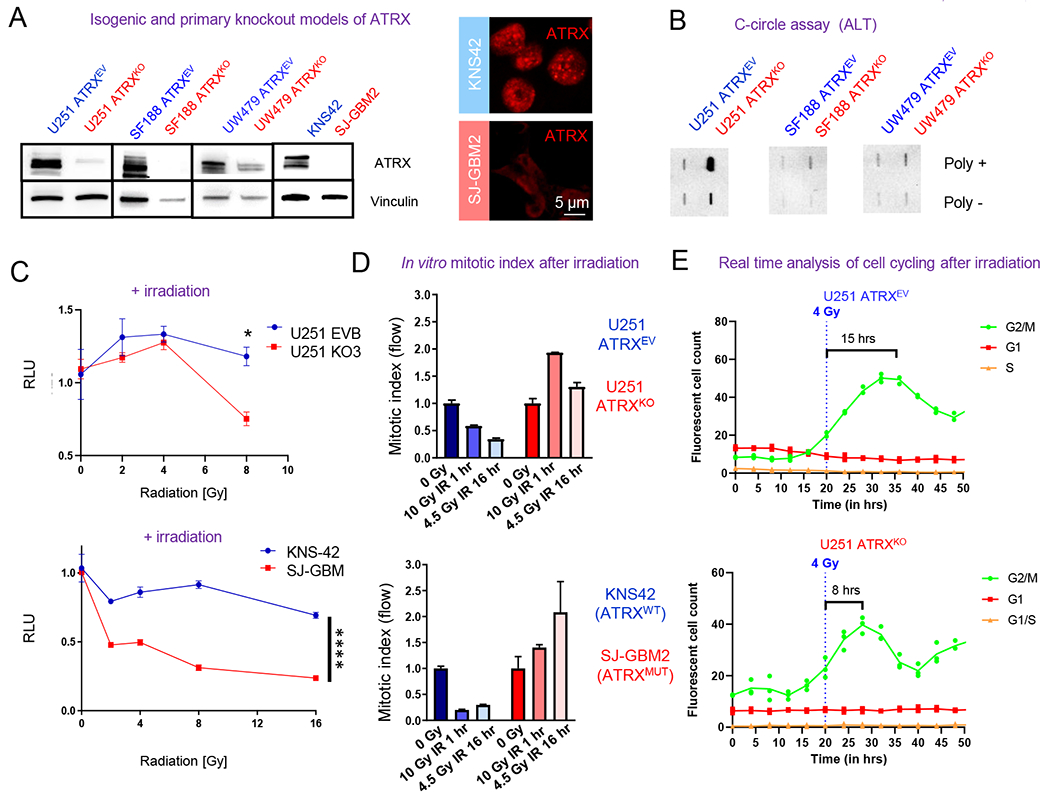
(A) Western blot of U251, SF188 and UW479 isogenic ARTXKO cells illustrating ATRX reduction. Immunoblots of KNS42 and SJ-GBM2 are also included (right two columns) illustrating ATRX reduction in ATRX-mutant SJ-GBM2 cells and immunocytochemistry images (right image) demonstrating their punctate nuclear staining of ATRX only in ATRX-wildtype KNS42 cells. (B) C-circle assay results demonstrating DNA from each sample in the presence (+) or absence (−) of Φ29 polymerase; positive staining demonstrates ALT in U251-ATRXKO cells only. (C) In vitro data showing proliferation of U251 ATRXKO cells and its isogenic control, as well as KNS42 (ATRX wildtype) and SJ-GBM2 (ATRX mutant), after exposure to increasing doses of irradiation (IR). (D) In vitro mitotic index assay at 1 hour after 10 Gy IR and 16 hours after 4.5 Gy IR demonstrates that U251 (ATRXKO) and SJ-GBM2 (ATRX mutant) cells display increased proliferation following treatment with IR. All values normalized to respective 0 Gy value. 0 Gy is normalized to 1.0. (E) Real-time analysis of cell cycle transitions using the Incucyte imaging system and the FastFUCCI reporter plasmid shows the U251 ATRXKO cells return to active cycling 2X faster after 4 Gy IR. [Mean ± SEM for triplicate experiments are shown. *P≤0.05, **P≤ 0.01, ***P≤0.001, and ****P≤0.0001 using 2-Way ANOVA.] For additional data, see also Figure S1.
Our previous work had shown enhanced irradiation sensitivity in ATRX-deficient GBM cells (Koschmann et al., 2016a). Consistent with this, ATRX-knockout cells show a higher sensitivity to irradiation as compared to their isogenic controls (Figures 1C and S1D). Primary pHGG cells carrying TP53 and ATRX mutations (SJ-GBM2 cells) were found to be more sensitive to irradiation than pHGG cells carrying TP53 mutation alone (KNS42 cells) (Figure 1C). Lovejoy et al. previously demonstrated that non-brain tumor ATRX-deficient (ALT+) cells show impaired G2/M checkpoint maintenance following irradiation (Lovejoy et al., 2012). We assessed this in our isogenic human GBM cells. We focused on the G2/M checkpoint as our ATRX-deficient models all carry concurrent TP53 mutation, and therefore expectant reduced G1/S function (Lovejoy et al., 2012). ATRX-wildtype KNS42 and U251 ATRXEV cells show appropriate G2/M cell cycle checkpoints (Figure 1D), with reduction in proliferation when compared to 0 Gray values after irradiation at previously described “initiation” (1 hour) and “maintenance” (16 hour) time points (Lovejoy et al., 2012). However, ATRX-deficient cells (SJ-GBM and U251-ATRXKO) demonstrate impaired reduction or increased mitosis at both time points when compared to 0 Gy, consistent with cell cycle checkpoint dysfunction in response to irradiation (Figure 1D). This experiment was repeated with isogenic UW479 and SF188 cells with results that support the trend seen in both the isogenic U251 cells and the primary pHGG cells. In SF188 isogenic cells, the ATRXEV cells showed some return to cycling at 16 hours after radiation, which was slightly increased above background mitotic index in ATRXKO cells. UW479 cells showed reductions in mitotic index after radiation in ATRXEV and ATRXKO cells, but late time point (16 hours) index was higher in ATRXKO cells (Figure S1E–F). In order to assess cell cycling kinetics of glioma cells, by ATRX status, we utilized the FastFUCCI reporter plasmid system, which tags Geminin (G2/M phase, GFP) and CDT1 (G1 phase, RFP) (Koh et al., 2017). We observed that when irradiated, the ATRX-knockout cells returned to active cell cycling about two times faster than ATRX-wildtype control cells (Figure 1E), indicating that the loss of ATRX impairs the ability of these cells to maintain G2/M cell cycle checkpoint when treated with irradiation.
ATRX preferentially binds to genes involved in cell cycle processes in mNPCs and mGBMs
We next set out to determine the etiology of dysfunctional cell cycle checkpoints in ATRX-deficient GBM cells. Recent work has established that in mNPCs, ATRX binds promoter and enhancer elements, and that its loss results in widespread transcriptional changes (Danussi et al., 2018). This phenotype has not been assessed or confirmed in GBM cells. Employing our previous model (Koschmann et al., 2016a), we generated mouse GBM cells (mGBM cells) from tumors generated by injecting Sleeping Beauty (SB) plasmids encoding shp53 and NRAS [with (NPA) or without shATRX (NP)] into the ventricles of neonatal mice (Figure 2A) and confirmed ATRX loss (Figure 2B–C).
Figure 2. ATRX preferentially binds to genes involved in cell cycle processes in mNPCs and mGBMs.
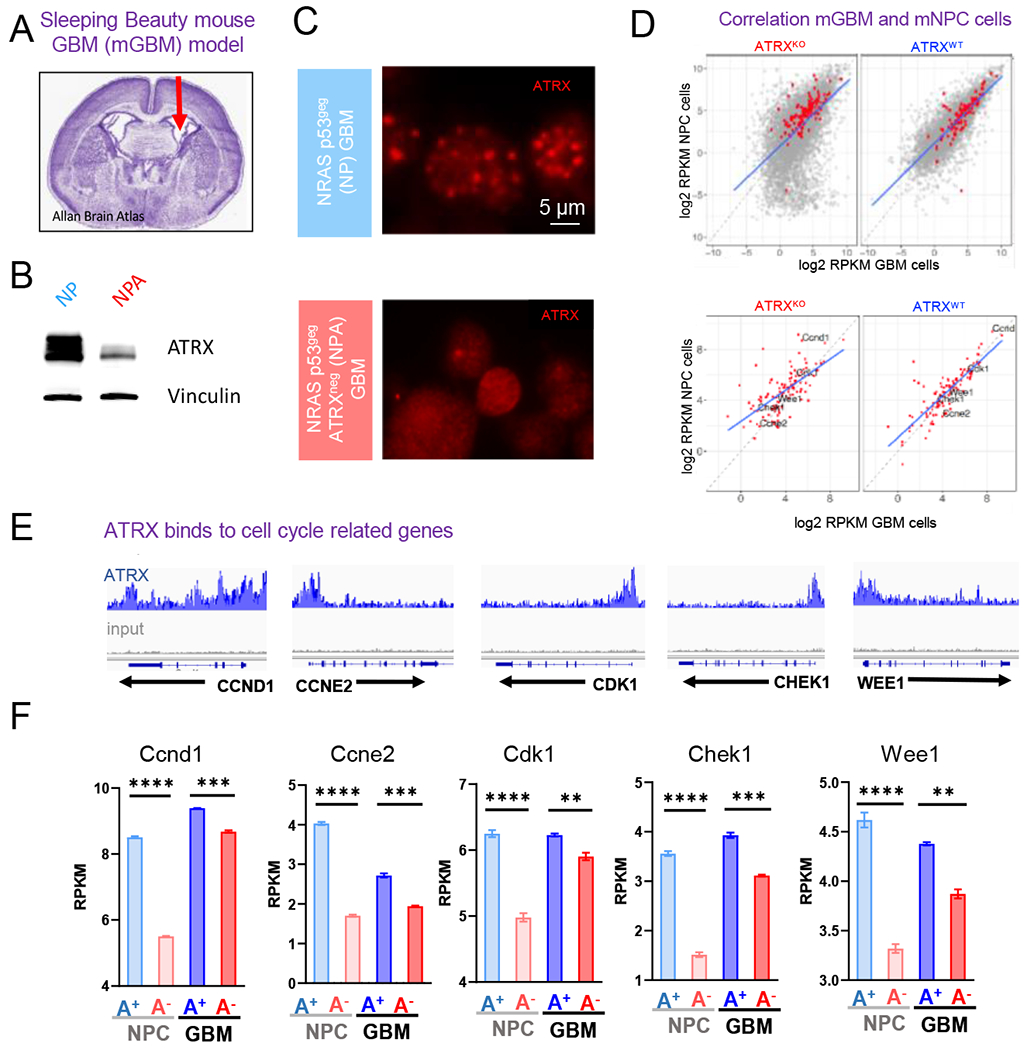
(A) Mouse GBM neurospheres (“NP” and “NPA”) are generated from tumors harvested from mice who underwent neonatal implantation of plasmids injected in the lateral ventricle, denoted by the arrow in the H&E stained section of a mouse brain. (B-C) Western blot (B) and Immunocytochemistry (C) analysis of NP and NPA mGBM neurospheres illustrating the loss of ATRX. (D) Correlation of average gene expression (log2 RPKM) between mGBM cells and mNPC cells (TP53−/− ATRXpos and TP53−/− ATRXneg) stratified by ATRX status (ATRXKO vs. ATRXWT). Top: the correlation of genome-wide genes with KEGG cell cycle genes denoted in red; bottom: the correlation of KEGG Cell cycle genes. (E) ATRX ChIP-seq tracks (n=3) in mNPC cells demonstrate ATRX promoter binding (gray track is input). (F) Expression levels (RPKM) of select cell cycle regulatory genes (Ccnd1, Ccne2, Cdk1, Chek1 and Wee1) with isogenic loss of ATRX in mNPC and mGBM cells. [Mean ± SEM for triplicate experiments are shown. *P≤0.05, ****p≤0 0001 using Welch’s t-test.] For additional data, see also Figure S2 and Tables S1–4, 7)
We then performed an integrated analysis of existing mNPC ATRX ChIP-seq/RNA-seq data (Danussi et al., 2018) and newly generated mGBM RNAseq data (+/− ATRX loss). We found a significant correlation between mNPC and mGBM neurosphere RNA expression profiles in both ATRX-deficient and ATRX-wildtype conditions (Figure 2D). In mNPC cells, ATRX significantly bound both DNA repair and cell cycle regulatory gene sets and showed differential expression with ATRX loss (Figure S2A–C and Table S1–S4). Of cell cycle regulatory gene sets identified in the mNPC dataset, we identified five genes that showed significant ATRX binding in mNPC cells (Figure 2E) and reduced expression with loss of ATRX in mNPC and mGBM cells, including Ccnd1, Ccne2, Cdk1, Chek1, and Wee1 (Figure 2F and Table S7).
ATRX-deficient human GBM cells show dysregulated expression of cell cycle related genes at single-cell level
ATRX mutation is found in two populations of human gliomas and corresponding bulk RNA-seq datasets have been previously generated on two large cohorts: i) pediatric HGG (pHGG, primarily concurrent with H3F3A mutation, PedcBioPortal, n=220) and ii) adult grade 2/3 glioma (“lower-grade” glioma, LGG, concurrent with IDH mutation, TCGA, n=414) (Mackay et al., 2017; Suzuki et al., 2015). We found that few cell cycle-related genes were differentially regulated by ATRX status using the two public bulk RNA-seq datasets (Figure S2D–E). We wondered if bulk RNA-seq of cancer cells may not be able to capture transcriptional alterations in cycling cell subgroups as only a minority (<5-10%) of tumor cells are dividing at any time in most human gliomas, and cell cycle genes are primarily not expressed in dormant cells (Tirosh et al., 2016b; Venteicher et al., 2017).
We performed single-cell RNA-seq (scRNA-seq) on isogenic human GBM U251-ATRXKO and U251-ATRXEV cells. By unsupervised clustering, we identified 13 clusters of cell populations (Figure 3A) and noted most prominent changes in two clusters: cluster 1 (U251-ATRXEV: 22% vs. U251-ATRXKO: 1%) and cluster 8 (U251-ATRXEV: 0.02% vs. U251-ATRXKO: 14%). By classifying cycling cells into S or G2/M phases based on the scores (see Materials and Methods), we recognized a distribution of cycling stages across the identified clusters, with a mixture of S and G2M phases in most clusters (Figures 3B).
Figure 3. ATRX-deficient human glioma cells display changes in cell-cycle associated gene expression and an increase in S and G2/M cycling phase.
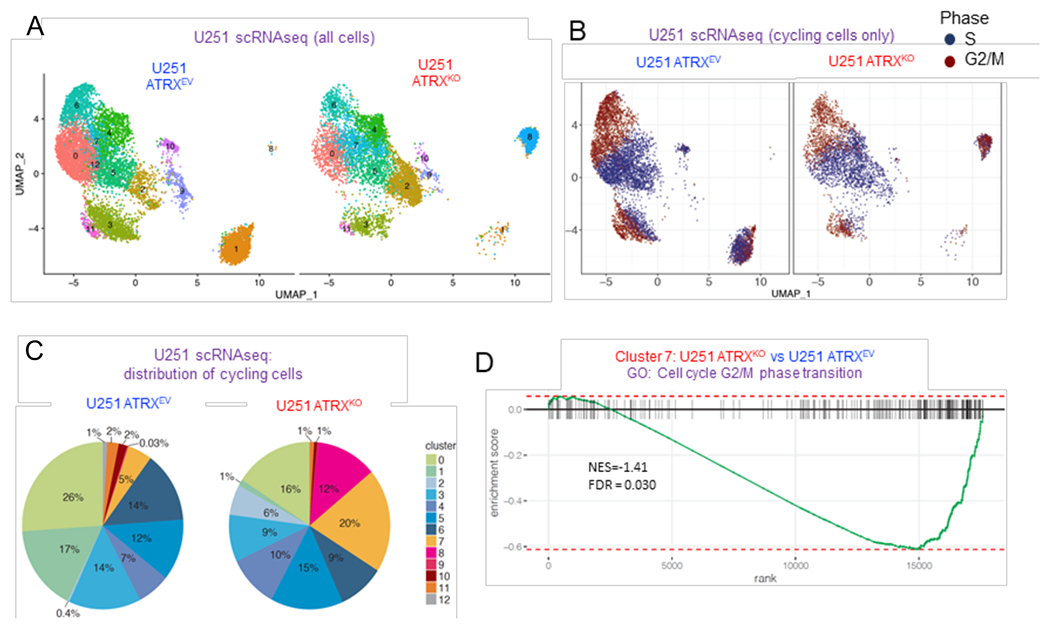
(A) UMAP visualization of cell clusters in U251-ARTXKO and U251-ARTXEV cells (number of PCs = 24). (B) UMAP visualization of cycling cells annotated by S (dark blue) or G2M (dark red) cycling phase. (C) Distribution of cycling cell proportion within each cluster in U251-ARTXKO and U251-ARTXEV cells respectively. (D) GSEA enrichment plot of GO term cell cycle G2/M phase transition that was significantly down-regulated in cluster 7 with the loss of ATRX. For additional data, see also Figure S3 and Tables S5–6.
Interestingly, Cluster 8 showed a significant number of cycling cells (12%), almost exclusively in ATRXKO cells (Figure 3C). ATRXKO cluster 7 cells showed a reduction in expression of the cell cycle G2/M phase transition gene set (Figure 3D and Table S5) and a four-fold increase in cycling cells (20% in ATRXKO vs. 5% in ATRXEV). CHEK1 was identified as one of the leading edge genes (i.e. the top or bottom ranked gene set) of several mitotic/cell cycle regulation processes in multiple clusters (Table S5–S6).
To assess if similar features were found in human glioma tumors, we analyzed two previously published scRNA-seq datasets of IDH-mutant gliomas with ATRX mutation (n=10) (Venteicher et al., 2017) and ATRX wildtype (n=6) (Tirosh et al., 2016b). Using age and grade matched tumors, we adapted the previous approach (Tirosh et al., 2016b) to classify individual cells into five cell cycle phases (Figure S3). ATRX-mutant tumors showed an increased proportion of cycling cells (Beta regression p = 1.21×10−6) as well as a four-fold increase in aberrant “double positive” cells with expression of genes associated with both S and G2/M phase, consistent with dysregulated expression of cell cycle-associated genes. Altogether, these findings point to dysregulation of cell cycle regulatory genes in sub-populations of ATRX-deficient glioma cells.
ATRX loss in mNPC and mGBM cells results in down-regulation of Chk1
The above studies consistently identified Chk1 loss as a potential mediator of irradiation response in ATRX –deficient glioma cells. Chk1 is activated by DNA damage in order to halt DNA replication in the S phase and G1/S and G2/M transitions (Otto and Sicinski, 2017; Patil et al., 2013). ATRX has previously been shown to either reduce (Leung et al., 2013) or increase (Wang et al., 2019) activation of Chk1 (pChk1) following DNA damage in non-glioma models; but the epigenetic impact of ATRX loss on CHEK1 expression has not yet been addressed. We found that mGBM cells with ATRX loss (NPA) demonstrate reduced ATRX and H3.3 binding at Chek1 promoter/gene loci (Figures 4A–B). In addition to reduced expression of Chek1 at the RNA levels in NPC and mGBM cells (Figure 2F, above), mGBM cells show reduced Chk1 and pChk1 at the protein level (Figure 4C). Human GBM cells demonstrate a decrease in pChk1 in cells lacking ATRX (Figure 4D). Additional human cell lines UW479 and SF188 recapitulate what was seen in NPC and mGBM cells at the RNA level, as well, where ATRX KO cells demonstrate overall lower Chk1 RNA levels (Figure S4A). We next overexpressed Chek1 in ATRX KO cells (U251 ATRXKOChk1OE) (Figure 4E) and assessed cell cycle kinetics (FastFUCCI) following irradiation. Overexpression of Chek1 in ATRX KO cells (U251 ATRXKOChk1OE) showed slower return to cycling, similar to ATRX-wildtype cells (Figures 4F–G). This phenotype was further confirmed using flow cytometric analysis of BrdU staining G1-S transition following irradiation in isogenic UW479 ATRXKO cells with or without Chek1 overexpression (Figure S4B–C). These data support that Chk1 plays a role in ATRX-mediated cell cycle dysregulation following irradiation.
Figure 4. Loss of ATRX in murine neuronal precursor cells (mNPC) and murine GBM cells (mGBM) results in down-regulation of Chk1.
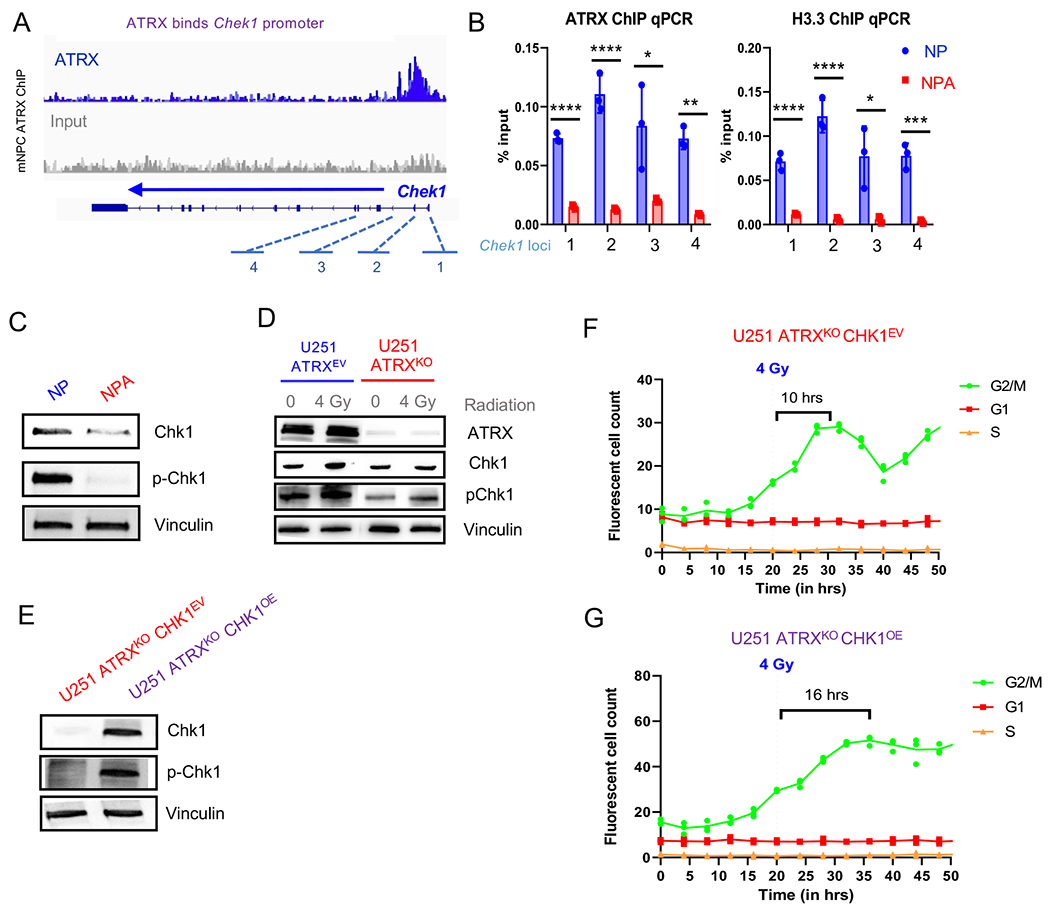
(A) ATRX promotor binding at Chek1 loci in mNPC cells (ChIP-seq, n=3 from Danussi et al., 2018). (B) mGBM cells with ATRXKO (“NPA”) show reduced ATRX and H3.3 binding at Chek1 gene loci (“1” through “4” from A) compared to mGBM cell controls without ATRXKO (“NP”), n=3. (C) Chek1 expression is downregulated in mNPC cells with ATRX loss. (D) Western blot of U251 ATRXEV and U251 ATRXKO cells with and without 4 Gy IR. ATR pathway proteins are marked in purple. (E) Western blot of U251 ATRXKO cells with isogenic Chk1 overexpression or empty vector (n=3 replicates for C and D). (F-G) Incucyte live cell imaging analysis of U251 ATRXKOChk1OE cells incorporated with the FastFUCCI reporter plasmid show a gradual return (more than 1.5X slower) to cycling after 4 Gy IR. (Mean ± SEM for triplicate experiments are shown. *P≤ 0.05, **P≤ 0.01, and ***P≤0.001 using Welch’s t-test). For additional data, see also Figure S4.
ATM inhibitors improve the radiation sensitivity in ATRX-mutated glioma
The involvement of ATRX in the expression of genes involved in cell cycle regulation such as Chek1 suggested that ATRX-deficient gliomas may be particularly dependent on upstream regulators of cell cycle checkpoint activation to keep cells from aberrant mitosis and cell death after irradiation. To explore this, we investigated two master cell cycle regulators that promote cell cycle checkpoint activation, ATR and ATM (Figure 5A). Chk1 is activated by phosphorylation via ATR, which is recruited to regulate CDK1 activity and can subsequently block the G2/M transition (Patil et al., 2013). ATM is recruited to sites of DNA double-stranded breaks (DSBs) and is responsible for phosphorylating (and activating) Chk2, subsequently halting progression through S phase in preparation for DNA damage repair (Durant et al., 2018). However, the activity of ATM and ATR is not mutually exclusive and Chk1 and Chk2 can be activated by either (Patil et al., 2013).
Figure 5. ATRX-deficient human glioma cells demonstrate selective radio-sensitization with ATM inhibition.
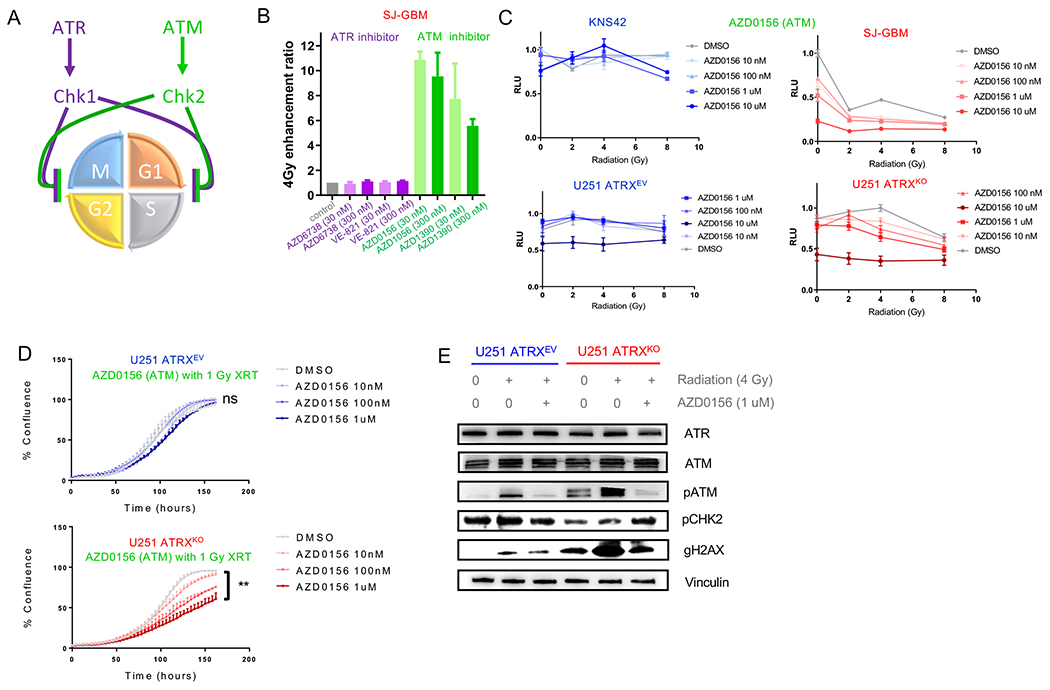
(A) Illustration of mechanism by which ATR and ATM proteins enforce cell cycle checkpoints. (B) Enhancement ratio after 4 Gy IR (clonogenic assay) performed on SJGBM2 cells against ATR (purple) and ATM (green) inhibitors. (C) In vitro proliferation assay plot of various radiation doses of ATM inhibitor AZD0156 for KNS42 and SJGBM2 (above) and ATRX isogenic U251 cells (below). (D) Plot of tumor cell confluence over time for U251 isogenic ATRXKO cell cultures treated with increasing concentrations of AZD0156 at 1 Gy XRT. (E) Immunoblots for ATR (purple) and ATM pathway (green) at baseline, by IR with DMSO and IR with 1 uM AZD0156 in U251 isogenic ATRXKO cells. [Mean ± SEM for triplicate experiments are shown. **p≤ 0.01 using Welch’s t-test] For additional data, see also Figures S5 and S6.
The ATM inhibitors, AZD0156 and AZD1390, exhibited significant radiation sensitivity, even at 30 nM (Figure 5B). Only ATRX-deficient cells showed a dose-dependent and synergistic pattern between ATM inhibitors and radiation in both human and mouse cells (Figures 5C–D and S5A–D). We treated isogenic cells (U251 and UW479) with two additional ATM inhibitors (KU-60019 and KU-55933) and found minimal radio-sensitization in nM-uM range, regardless of ATRX status, consistent with previous data showing significant increase in potency (300-2,000x) in the newer AZ compounds over these previous generation inhibitors (Riches et al., 2020) (Figure S5E–H).
Western blots of treated isogenic human cells demonstrated overall similar levels of total ATR and total ATM protein, but an increase in phosphorylated (activated)-ATM and its substrate gH2AX (Burma et al., 2001) after irradiation in ATRXKO cells compared to controls (Figure 5E and Figure S6A). Interestingly, we did find that ATRX binds the promoter of both ATM and ATR and both genes were down-regulated in mNPC cells when compared by ATRX status. However, we did not see notable differences in expression of ATM or ATR in mGBM cells (Figure S6B–C) or in human glial tumors by ATRX status (data not shown). Overall, these data support minimal difference in expression of ATM and ATR but differential activation of ATM in ATRX-deificent GBM cells. Importantly, both pATM and gH2AX were reduced by treatment with AZD0156 in knockout cells, consistent with inhibition of ATM kinase activity by AZD0156.
ATM is a master regulator of the DNA-damage response (DDR), which includes both cell cycle and DNA-damage repair processes (Marechal and Zou, 2013). Previous work by our group has shown that ATRX loss can affect DNA-damage repair in glioma cells (Koschmann et al., 2016a), and that glioma cells with co-occurring IDH and ATRX mutations are sensitized to radiation damage through impaired DNA-damage repair (Nunez et al., 2019). In order to clarify whether ATM inhibition was impacting cell cycle control, we performed cell cycle analysis by flow cytometry and found that only ATRX–deficient cells (SJ-GBM and U251-ATRXKO) showed an increase in cycling cells with ATM inhibitor (AZD-0156) treatment in both irradiated and non-irradiated settings (Figure 6A). This provides evidence of a cell-cycle specific impact of ATM inhibition in ATRX-deficient cells.
Figure 6. ATM inhibition results in selective increase in cell cycling in ATRX-deficient glioma cells and ATM-specific inhibition in vitro and in vivo.
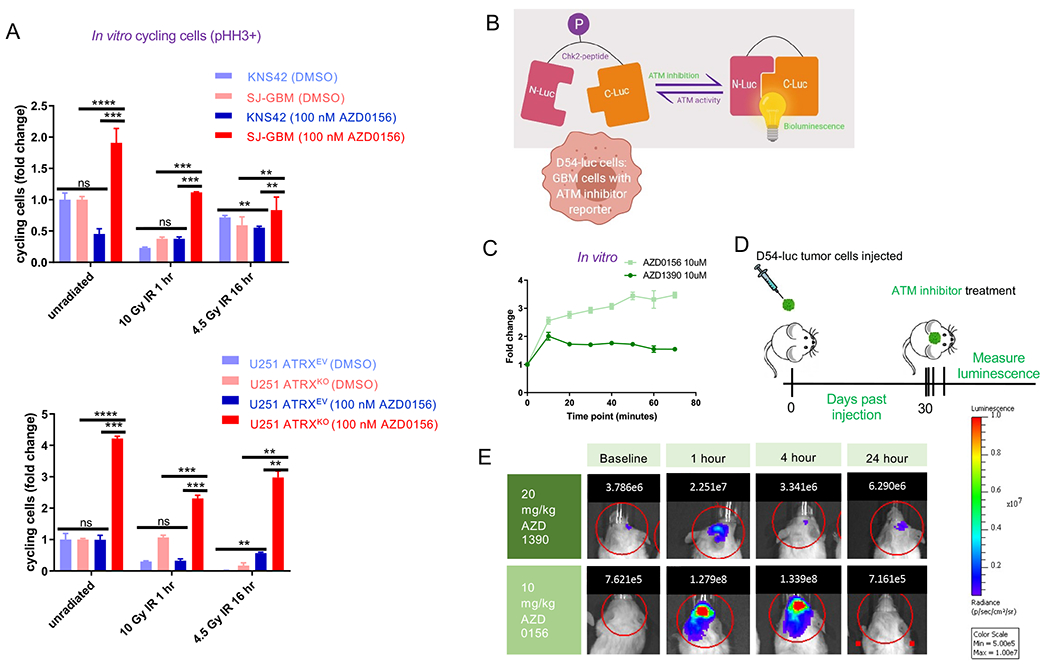
(A) Flow cytometric cell cycle analysis [y-axis represents fold change in cycling cells, or percentage phospho-histone H3 (“pHH3”) positive] demonstrating selective increase in cycling cells with ATM inhibitor treatment in ATRX-deficient cells only. (B) Schematic of ATM inhibition reporter assay. (C) Graph depicting bioluminescence change compared to baseline after treatment with ATM inhibitor in D54 cells expressing ATM-Luciferase inhibition reporter in vitro (higher value represents more inhibition). Line represents mean ± SEM for triplicate experiments. (D) Schematic of in vivo D54-ATMR assay to assess in vivo intra-cranial on target changes with ATM inhibition. (E) Representative mice showing bioluminescence (photons) at various time points before and after treatment with ATM inhibitors. [Mean ± SEM for triplicate experiments are shown. *P≤0.05, **P≤ 0.01, ***P≤0.001, ****P≤0.0001 using Welch’s t-test.] For additional data, see also Figure S7.
ATM inhibition target effects is confirmed in vivo
To clarify the translational relevance of this finding, we sought to determine whether these inhibitors could cross the blood-brain barrier at concentrations that are sufficient to inhibit ATM in brain tumor cells. We worked with a human GBM cell line, D54-ATMR (Nyati et al., 2017), that was modified to endogenously express an ATM-Luciferase inhibition reporter (Figure 6B). In vitro, both AZD0156 and AZD1390 showed a sustained increase in ATM inhibition signal over time compared to baseline (Figure 6C). We intra-cranially implanted D54-ATMR cells into the cortex of mice (Figure 6D), then ~30 days later measured bioluminescence of the tumors at baseline and treated them with both ATM inhibitors separately. Treatment of mice intra-cranially implanted with D54-ATMR cells (Figure 6D) with both ATM inhibitors increased bioluminescence compared to baseline, which then reverted to baseline by 24 hours for AZD0156 and 4 hours for AZD1390 (Figure 6E), consistent with an in vivo on-target effect. A brain pharmacokinetic study of AZD0156 in non-tumor bearing mice demonstrated a peak level of ~300 nM at one-hour post intraperitoneal (IP) dosing, and a half-life of approximately 4 hours (Figure S7A).
ATM inhibitors significantly improve survival in the ATRX-deficient orthotopic murine brain tumor model
We next assessed whether treatment with ATM inhibitors could extend survival when added to radiation in an aggressive ATRX-deficient orthotopic murine brain tumor model. Mice with implanted ATRX sufficient (“NP”) and ATRX-deficient mouse GBM neurospheres (“NPA”) tumors underwent treatment with irradiation with or without ATM inhibitor over two weeks (Figure 7A–B). Mice with NP tumors showed a similar average median survival irrespective of treatment condition, demonstrating insensitivity to irradiation and/or ATM inhibitor treatment (Figure 7C).
Figure 7. Treatment of implanted ATRX-deficient mouse GBM cells with ATM inhibitors.
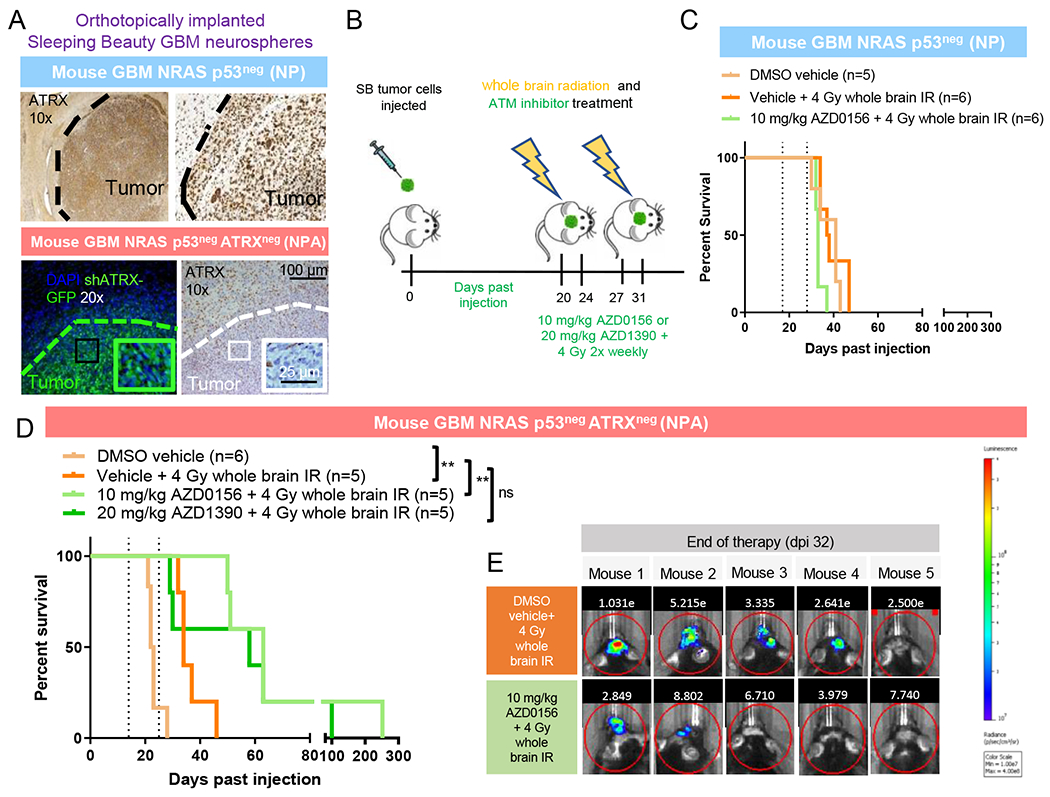
(A) Immunofluorescence and IHC staining of tumors of mice implanted with mouse GBM neurospheres with (NPA) and without (NP) ATRX knockdown (GFP stains for shATRX plasmid). Tumor shows robust ATRX loss which is not seen in surrounding normal cortex. Boundary between tumor and non-tumor tissue is denoted by dotted line. (B) Schematic of treatment with whole brain IR and ATM inhibitor for mice implanted with tumor cells. (C) Kaplan-Meier survival curves of C57BL/6 mice bearing SB implanted NP tumors with no response to irradiation +/− ATM inhibition. (D) Kaplan-Meier survival curves of C57BL/6 mice bearing SB implanted NPA tumors show significant radio-sensitization with AZD0156. **P≤ 0.01 using Log-rank (Mantel-Cox) test. (E) Tumor bioluminescence for tumor-bearing NPA mice treated with 4 Gy whole brain irradiation with and without AZD0156. For additional data, see also Figure S7.
Vehicle-treated NPA-implanted mice had a slightly reduced median survival (24 days) compared to vehicle-treated NP controls (median survival 41 days), consistent with our previous data showing that ATRX loss resulted in increased tumor growth (Koschmann et al., 2016a) (Figure 7D). NPA mice treated with 4 Gy irradiation showed a significant increase in survival compared to untreated mice (Figure 7D). Treatment with AZD0156 or AZD1390 with 4 Gy irradiation in NPA mice resulted in a near tripling of median survival (Figure 7D) and a decrease in average luminescent tumor size, although this did not reach significance (Figure 7E). Both groups (AZD0156 and AZD1390) had long term survivors (>100 days). Treatment with radiation and AZD0156 of mice with flank-implanted primary human pHGG cells with (SJ-GBM) and without (KNS42) ATRX mutation also demonstrated reduction in tumor growth only in SJ-GBM tumors (Figure S7B–C).
DISCUSSION:
Our study extends recent work demonstrating the broad impact of ATRX loss on tumor cell biology. We demonstrate that ATRX binds to the gene regulatory elements of cell cycle regulatory genes, notably Chek1, in both GBM and glioma precursor cells, resulting in changes in transcription with ATRX loss, including reduced expression of Chk1 at both the RNA and protein levels. In line with this, ATRX-deficient human and mouse glioma models demonstrate impaired cell cycle phase control which was exacerbated by treatment with ATM inhibitors. This association between ATRX loss, transcriptional changes, and cell cycle dysregulation provides additional mechanistic insight into the selective pressure for ATRX loss/mutation in human glioma and also its irradiation sensitivity.
Previous work in mouse embryonic stem cells (mESCs) demonstrated that ATRX binding occurs primarily within tandem repeat sequences of telomeric regions and within euchromatin (Law et al., 2010). Our analysis of mNPCs confirms recent data that ATRX binding occurs frequently at gene promoters, enhancers, and CpG islands/shores (Danussi et al., 2018). However, the enrichment of ATRX at genes involved in cell cycle regulation has not been reported. ATRX has been shown to impact the activation of cell cycle regulatory genes in various ways, depending on the tumor type and model. Graham et al. demonstrated that ATRX-mutant prostate cancer cells show reduced expression of cell cycle gene sets, but did not identify a specific gene or pathway that mediates cell cycle control (Graham et al., 2019).
ATRX binding leads to the deposition of the transcriptionally active histone variant H3.3 affecting chromatin compaction (Lewis et al., 2010). We see changes in H3.3 depositions at some of these loci. Through ChIP qPCR, we discovered ATRX binding at promotors/enhancers of Chek1, and a decrease in this binding and H3.3 deposition with ATRX loss. We therefore propose a model in which ATRX binds the promoters and participates in the coordination of the expression of cell cycle genes through deposition of H3.3, which is subsequently post-translationally modified. Additionally, the mutation/loss of ATRX associated with dysregulated expression of cell-cycle associated genes in ATRX-deficient glioma cells, with Chek1 being the most significantly affected of these genes. ChIP-seq experiments to confirm these findings could not be performed due to the transient genomic binding of ATRX and the inaccessibility of an optimal antibody, though we are working to optimize this for future studies.
Our data show that bulk RNA-seq analysis has a reduced ability to detect changes in cell cycle-associated gene expression in sub-populations of ATRX-deficient glioma cells, likely because GBM cell populations are more heterogenous and therefore cycle less consistently. In contrast, the scRNA-seq performed on human glioma cells and tumors successfully captured the heterogeneous cell cycling within sub-population of cells, and changes associated with the loss of ATRX. These discoveries confirm the cell cycle dysregulation by ATRXKO in human glioma cells. Our data support that ATRX/Chk1-mediated G2/M dysfunction allows ATRX deficient cells to maintain normal proliferation through up-regulation of ATM, which prevents cells from cycling inappropriately. Liang et al. recently showed that knockdown of cell cycle genes, including WEE1, result in lethality in ATRX knockout hepatocellular cancer cells (Liang et al., 2020). While our studies support the implication of ATRX loss on cell cycle regulation, we found reduced WEE1 expression in mNPCs and mGBMs with ATRX loss pointing to this gene not being as essential for ATRX-deficient GBM.
Importantly, our study shows that ATRX-mutant cells are uniquely sensitive to ATM inhibition. We hypothesize that these cells are inherently sensitive to irradiation because of their ATRX deficiency, but are able to partially divert from mitotic catastrophe by increasing ATM activity. Active ATM is capable of activating both Chk1 and Chk2, driving appropriate cell cycle checkpoints. By inhibiting ATM, the cells are no longer able to avoid dysfunctional cell cycling and apoptotic cell death. Other studies have supported ATRX-mutant cells’ sensitivity to irradiation after ATR inhibition (Flynn et al., 2015). We observed that ATRX deficient cells from both mouse and human tumor models exhibited a more robust sensitization to ATM inhibition, but our data does not preclude the efficacy of ATR inhibitors, as both are involved in cell cycle control.
We previously reported the efficacy of ATM inhibition in a murine model of IDH-mutant glioma due to IDH1-driven epigenetic up-regulation of the DNA damage response (Nunez et al., 2019) . In this manuscript, we utilized high-grade glioma cells with isogenic ATRX loss/ALT positivity on a wildtype IDH background. The results of these studies are therefore most relevant to ATRX-mutant pediatric (and young adult) high-grade glioma, which harbor IDH-wildtype status (Schwartzentruber et al., 2012). However, these mechanistic insights of ATRX impact on glioma biology may have implications for gliomas with with co-mutation of ATRX and IDH or other solid tumors (e.g. osteosarcoma or neuroblastoma) with ATRX mutation.
CNS penetration and pre-clinical data of AZD1390 make this a promising translatable therapy (Durant et al., 2018). Unfortunately, current phase 1 trials in adult GBM with AZD1390 (NCT03423628) may miss this patient population, as adult grade IV glioma rarely carries ATRX mutation which appears to be required for ATM inhibitor radiosensitization based on our data. Indeed, no previous human studies have selected for ATRX mutation/loss as a targetable eligibility criterion. Our data will help provide the rationale for improved precision radiation sensitizing therapies for ATRX-deficient glioma.
In summary, this study shows that ATRX-deficient glioma demonstrates sensitivity to ATM inhibition via epigenetic dysregulation of cell cycle checkpoints. Currently, treatments for ATRX-deficient glioma remain non-targeted and ineffective. Our epigenetic and phenotypic results open a window for therapies targeting the unique features of ATRX-deficient glioma.
LIMITATIONS OF THE STUDY
Our data are supportive of a model where ATRX loss indicates sensitivity to ATM inhibition in glioma cells, however, further studies would serve to strengthen these results. While we performed in vivo efficacy studies with orthotopic (brain) implantation of mouse (NPA) cells, our treatment of implanted human pHGG cells (SJ-GBM and KNS42) was performed with flank imlantations. Further, other newer generation ATM inhibitors would need to be tested both in vitro and in vivo to confirm that our results stem from ATM inhibition broadly and is not just a result of AZD0156 itself. Additionally, ChIP-sequencing experiments in our mouse and human models to confirm the results seen in NPCs from a prior study (Danussi et al., 2018) could not be performed due to the transient genomic binding of ATRX and the inaccessibility of an optimal antibody, though we are working to optimize this for future studies.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Carl Koschmann (ckoschma@med.umich.edu).
Materials Availability
This study generated human cell lines, but these are unavailable due to issues with cell viability after freezing.
Data and code availability
The RNA-seq of mouse GBM neurospheres was deposited in Gene Expression Omnibus (GEO) with the accession number of GSE178113. The single cell RNA-seq of human isogenic cell line U251 was deposited in Gene Expression Omnibus (GEO) with the accession number of GSE178114.
The code used for the downstream analysis in this manuscript was deposited in Zenodo: https://zenodo.org/badge/latestdoi/429519345
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Mouse GBM neurosphere generation
Mouse GBM neurospheres were generated from GBMs dissociated from immune-competent mice (C57BL/6 or FVB) injected with Sleeping Beauty plasmids encoding: (1) a short hairpin against p53 (shp53), (2) a plasmid over-expressing NRAS, and (3) with or without a plasmid expressing a short hairpin against ATRX (shATRX) into the ventricles of neonatal mice. Mouse GBM primary cell cultures were maintained in neural stem-cell medium [DMEM/F12 with L-glutamine (Gibco, 11320-033), B-27 supplement (Gibco 12587-010), N-2 supplement (Gibco 17502-048), penicillin-streptomycin (Cellgro 30-001-CI), and Normocin (Invivogen)]. FGF and EGF (Shenandoah Biotech 100-26, 100-146) supplementation was performed two times per week at 1:l (20 ng/uL each) per 1 ml medium. ATRX expression in primary cell cultures was assessed by (1) immunocytochemistry (1:250) and (2) Western blotting (1:200) with ATRX antibody (Santa Cruz, sc-15408).
Primary and isogenic cell culture
Established pediatric glioma cell culture KNS42 (Grade IV, Glioblastoma, Male) was obtained from Dr. Alan Meeker on November 1st, 2015 (Johns Hopkins University, Baltimore, MD) and cultured in DMEM:F12 (#11995065) from Invitrogen with 2 mM GlutaMAX-I Supplement (#35050061) from Invitrogen and 10% fetal bovine serum (FBS). Established GBM cell culture SJ-GBM2 (Female) was obtained from the Children’s Oncology Group cell line repository and is cultured in IMDM medium (#12440053) from Invitrogen with 20% FBS. U251 ATRXEVA, ATRXEVB, ATRXKO1 and ATRXKO3, SF188 ATRXEV1, ATRXEV2, ATRXKO1 and ATRXKO3, and UW479 ATRXEV1, ATRXEV2, ATRXKO1 and ATRXKO2 cell cultures were also obtained from Dr. Alan Meeker. (U251 ATRXEV denotes ATRXEVB, U251). (ATRXKO denotes ATRXKO3, SF188 ATRXEV denotes ATRXEV1, SF188 ATRXKO denotes ATRXKO2, UW479 ATRXEV denotes ATRXEV1 and UW479 ATRXKO denotes ATRXKO1). U251 cells (Male) were cultured in RPMI (#SH3025502, ThermoFisher) supplemented with 10% FBS. SF188 (Male) and UW479 (Female) cells were cultured in DMEM:F12 (+L glut + HEPES) (#11330-032) from Invitrogen with 2mM GlutaMax-I Supplement (#35050061).
Mouse studies and treatment grouping
Male and female C57BL/6, FVB, and NOD scid gamma (NSG) mice (weighing 20 to 25 g) were obtained from Charles River or Jackson Labs and were 6-10 weeks of age at the start of surgery. All animal studies were conducted according to the guidelines approved by the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan. Mice were randomized into groups before treatment.
Method Details
Mouse GBM neurosphere generation
Mouse GBM neurospheres were generated from GBMs dissociated from immune-competent mice (C57BL/6 or FVB) injected with Sleeping Beauty plasmids encoding: (1) a short hairpin against p53 (shp53), (2) a plasmid over-expressing NRAS, and (3) with or without a plasmid expressing a short hairpin against ATRX (shATRX) into the ventricles of neonatal mice. Mouse GBM primary cell cultures were maintained in neural stem-cell medium [DMEM/F12 with L-glutamine (Gibco, 11320-033), B-27 supplement (Gibco 12587-010), N-2 supplement (Gibco 17502-048), penicillin-streptomycin (Cellgro 30-001-CI), and Normocin (Invivogen)]. FGF and EGF (Shenandoah Biotech 100-26, 100-146) supplementation was performed two times per week at 1:l (20 ng/uL each) per 1 ml medium. ATRX expression in primary cell cultures was assessed by (1) immunocytochemistry (1:250) and (2) Western blotting (1:200) with ATRX antibody (Santa Cruz, sc-15408).
Primary and isogenic cell culture
Established pediatric glioma cell culture KNS42 (Grade IV, Glioblastoma) was obtained from Dr. Alan Meeker on November 1st, 2015 (Johns Hopkins University, Baltimore, MD) and cultured in DMEM:F12 (#11995065) from Invitrogen with 2 mM GlutaMAX-I Supplement (#35050061) from Invitrogen and 10% fetal bovine serum (FBS). Established GBM cell culture SJ-GBM2 was obtained from the Children’s Oncology Group cell line repository and is cultured in IMDM medium (#12440053) from Invitrogen with 20% FBS. U251 ATRXEVA, ATRXEVB, ATRXKO1 and ATRXKO3, SF188 ATRXEV1, ATRXEV2, ATRXKO1 and ATRXKO3, and UW479 ATRXEV1, ATRXEV2, ATRXKO1 and ATRXKO2 cell cultures were also obtained from Dr. Alan Meeker. (U251 ATRXEV denotes ATRXEVB, U251. (ATRXKO denotes ATRXKO3, SF188 ATRXEV denotes ATRXEV1, SF188 ATRXKO denotes ATRXKO2, UW479 ATRXEV denotes ATRXEV1 and UW479 ATRXKO denotes ATRXKO1). U251 cells were cultured in RPMI (#SH3025502, ThermoFisher) supplemented with 10% FBS. SF188 and UW479 cells were cultured in DMEM:F12 (+L glut + HEPES) (#11330-032) from Invitrogen with 2mM GlutaMax-I Supplement (#35050061).
Chk1 isogenic overexpression cell lines were generated using the ATRX isogenic knockout cells – U251 ATRXKO, SF188 ATRXKO and UW479 ATRXKO cells. The Chk1 overexpression plasmid was obtained from Addgene (#22894) and was cloned into lentiviral particles at the Vector core at the University of Michigan. The ATRX isogenic knockout cells were then transduced with either the Chk1-containing lentivirus or an empty vector overnight and expression confirmed with western blot before starting experiments.
Mouse neuronal precursor cell (mNPC) ATRX ChIP-seq and RNA-seq analysis
We downloaded the Atrx peak (mm9) bed files and raw RNA-seq data from the GEO GSE100462 dataset, which was derived from Atrx ChIP-seq on Tp53 (positive and negative); Atrx (wildtype and knockout) mouse glioma cells (Danussi et al., 2018).
For ChIP-Seq, the Atrx peaks were annotated to different genomic regions by R package annotatr (version 1.0.3) (Cavalcante and Sartor, 2017). Gene Set Enrichment (GSE) testing was conducted by the chipenrich R package (version 2.6.1) using the polyenrich method and “1kb” locus definition (i.e. assigning peaks to gene promoters) (Welch et al., 2014). Enhancers were defined by FANTOM5 permissive enhancers (Andersson et al., 2014). To filter out the closely related GO terms for reporting purposes, we used the R package GO.db (Carlson et al., 2007) to determine relationships among significant terms. A GO term was filtered if one or more of its parents, children or siblings had a higher rank in the list (Koneva et al., 2018). To visualize the peaks, we also downloaded the corresponding raw sequencing data from SRA SRP110502 project. Reads were aligned to mm9 using Bowtie2 (version 2.2.1) with default parameters. The read alignment bam files were converted to bedGraph files by deepTools v.3.1.3 using RPKM normalization (Ramirez et al., 2016). To simplify the graphical tracks, the average coverage of replicate samples was calculated from bedGraph files, which were then converted to bigWig files by UCSC bedGraphToBigWig tools and visualized using the IGV browser (Robinson et al., 2011).
For RNA-seq, we downloaded the raw sequencing data from the GEO GSE100462 Tp53−/− RNA-seq dataset including 6 samples (Danussi et al., 2018). FASTQC was employed to assess overall quality of each sample followed by TrimGalore processing to trim low-quality bases and adapter sequences (Andrews, 2010). The trimmed reads were aligned to mm9 genome by STAR (version 2.5.3a) (Dobin et al., 2013), and assembled to exons by HTSeq (version 0.11.2) (Anders et al., 2015) with the following parameters: “--mode=union --stranded=no”. The differential expression analysis between TP53−/−/ATRX+ and TP53−/−/ATRX− groups was performed using the R package edgeR (version 3.20.9) (Robinson et al., 2010) and the generalized linear model (GLM) method. Genes with an absolute fold change greater than 2 and FDR less than 0.05 were reported. Gene set enrichment testing was performed by the RNA-Enrich method (http://lrpath.ncibi.org). To filter out closely related GO terms for reporting purposes, we used the same approach as described above.
Native ChIP-qPCR and RNA-seq for mouse GBM neurospheres
Native ChIP-qPCR was performed on primary mouse ATRX positive “NP” (C39E) and ATRX knockdown “NPA” (C54B) GBM neurosphere cells, generated according to previous methods (Koschmann et al., 2016a). The protocol for native ChIP-qPCR was adapted from previously described methods (Donovan LL, 2014), and optimized for mouse glioma primary cells. Anti-Histone H3.3 Antibody (2 μl, cat# 09838, Millipore Sigma), ATRX antibody (5 μl, cat# H-300, SC-15408, Santa Cruz), and control IgG (2 μg Cat#12370, Millipore Sigma) were used for immunoprecipitation. Quantitative-PCR was performed using 1μl of eluted ChIP DNA. Primers for the enhancer, promoter, and exon regions for CDKN1A, CCND1, CHEK1, and CDK1 were predicted based on ATRX ChIP-seq data on mouse NPC cells (Danussi et al., 2018). For a complete list of primer sequences, see (SI Appendix, Table S7). Enrichment at target sites was quantified using the percent input method as has been previously described (Bender et al., 2013).
Total RNA was isolated from the cells using the Qiagen RNeasy Mini Kit (Qiagen, cat# 74104). For each sample, 4 ug of total RNA was then used in Illumina’s TruSeq Stranded mRNA Library kit (Cat# 20020594). Libraries were sequenced on Illumina NextSeq 500 as paired-end 42-nt reads. The RNA-seq data was analyzed using the same pipeline as the one used for mNPC RNA-seq data (see above).
Transcriptome and somatic mutation data analysis on human glioma tumors
The processed gene expression and somatic mutation data were downloaded for two human glioma cohorts: i) the pediatric HGG cohort data accessed from the Pediatric HGG ICR London (220 samples) (Mackay et al., 2017), and ii) the adult TCGA-HGG cohort data (414 samples) (Koschmann et al., 2016a, Koschmann et al., 2016b, Schwartzentruber et al., 2012, Seah et al., 2008, Suzuki et al., 2015) accessed from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov). The ATRX-mut and ATRX-WT patients were identified using their somatic mutation data, and the normalized expression levels (RPKM) of cell cycle genes were compared between the two groups. Wilcoxon rank-sum tests were applied to identify genes with significant differential expression by ATRX status.
Single cell RNA-seq analysis on human glioma tumors
The processed gene expression data of two single cell RNA-seq (scRNA-seq) datasets were downloaded from the GEO database: GSE7063 oligodendroglioma samples (Tirosh et al., 2016b) and GSE89567 astrocytomas (Venteicher et al., 2017). All samples contain IDH1 mutation, the oligodendroglioma samples are all ATRX-wt (n=6), and the astrocytomas are all ATRX-mut (n=10). By following the methods applied by Tirosh et al (Tirosh et al., 2016b), we calculated the G1S and G2M phase scores of individual cells for each dataset separately. We assigned each cell to one of 6 cell cycle phases. To examine the difference in the percentage of cycling cells between ATRX-mut and ATRX-wt tumors, we first filtered out two tumors with the total number of cells ≤ 300 (MGH103 and MGH107 in ATRX-mut group), and then calculated the percentage of cycling cells in each tumor and performed Beta regression by R package betareg (Grün et al., 2011) to investigate the association between ATRX status and the percentage of cycling cells including cycling phases as a covariate. Since the number of tumors in each group is small (ATRX-wt: n = 6; ATRX-mut: n = 8), which limited the power to test the interactions between ATRX status and each cycling phase, we applied Wilcoxon sum-rank test to evaluate the difference in percentage of cycling cells by ATRX status at each phase.
Single cell RNA-seq analysis for human isogenic cell line U251
Single cell RNA-seq (scRNA-seq) was performed on isogenic U251 cells at the University of Michigan Advanced Genomics Core on the 10X Chromium system. Cells were prepared as a single cell suspension of 1000 cells/ ul in 100 ul volume. The target was specified at 10,000 cells at a 50,000 read depth. Libraries were sequenced on the Illumina NovaSeq 6000 sequencer to generate 28x98 bp paired end reads for the sequencing parameters. Data processing including quality control, read alignment using hg38 genome and gene quantification were conducted using the 10X Cell Ranger software. R package Seurat (Stuart et al., 2019) was used for normalization, data integration, and clustering analysis . Cells with mitochondrial expression > 25% or the number of expressed gene outside the 1.5×IQR (i.e. outliers) were filtered out. The optimal number of PCs were selected when its decrease in standard deviation < 0.5%, and used for cell visualization by UMAP. MAST approach (Finak et al., 2015) implemented in R Seurat package was applied to identify cluster markers and differential genes for each cluster between U251-ATRXKO and U251-ATRXEVcells. The genes that expressed in > 25% cells at both comparative conditions and showed FDR < 0.05 and fold change (FC) > 1.5 were defined as cluster markers or differentially expressed genes by ATRX status. The pre-ranked GSEA approach implemented in the R package rgsea (Korotkevich et al., 2019) was used to identify the up/down-regulated Gene Ontology Biological Process (GOBP) terms or KEGG pathways among the genes ranked by expression difference between ATRX-KO and ATRX-WT. To identify the cycling cells, we calculated S and G2M scores for each cell using the module scoring method implemented in the R Seurat package (Stuart et al., 2019) and the S or G2M cell markers from Tirosh et al (Tirosh et al., 2016a). The cells with S or G2M score > 0 were considered to be cycling cells, and those with S score > G2M score were at S phase, and vice versa. The cycling cells were annotated by S or G2M phase and then visualized in the same UMAP. To investigate the change in cycling cells by ATRX status, we calculate the percentage of cycling cells in each cluster (number of cycling cells in each cluster/total number of cycling cells) for U251-ATRXKO and U251-ATRXEVcells separately. Beta regression, implemented in the R package betareg (Grün et al., 2011), was performed to examine how the percentage of cycling cells was associated with cluster and ATRX status.
Western blot
Western blots were performed based on standard protocols on whole-cell protein lysates. Membranes were washed three times and proteins were imaged with Clarity™ Western ECL substrate (#170-5060) from Bio-Rad on FluorChem M system. Antibodies used were anti-ATRX (sc-15408) and anti-ATR (sc-51573) from Santa Cruz biotechnology; anti-ATRX (A301-045A) from Bethyl laboratories LLC; anti-Vinculin (#700062) from Invitrogen; anti-CHK1 (#2360), anti-pCHK1 Ser345 (#2348), anti-CHK2 pT68 (#2661), anti-Phospho-Histone H2A.X Ser139 (#2577), and anti-mouse IgG, HRP-linked Antibody (#7076) from Cell Signaling Technology; anti-ATM pS1981 (ab81292) from Abcam; and HRP Goat Anti-Rabbit IgG Antibody (P1-1000) from Vector Laboratories.
C-Circle assay for ALT
DNA from KNS42, SJ-GBM2, U251 ATRXEV, U251 ATRXKO, SF188 ATRXEV, SF188 ATRXKO UW479 ATRXEV, UW479 ATRXKO cells was isolated using the DNeasy Blood and Tissue Kit (#69504) from Qiagen. C-circle assay for ALT was performed on tumor DNA and cell culture DNA as previously described (Brosnan-Cashman et al., 2018). Briefly, 400 ng of digested DNA from each sample was added to a rolling circle amplification reaction in the presence or absence of Φ29 polymerase. Products were blotted onto a nylon membrane and detected with a DIG-labeled telomere probe. Samples were scored as positive for ALT when found to have qualitative change above background with addition of Φ29 DNA polymerase, as previously described (Brosnan-Cashman et al., 2018).
Immunocytochemistry
Cells were seeded in 12-well plates containing sterile coverslips and incubated at 37°C for 24 hours. Cells were washed with phosphate buffered saline (PBS) containing Ca2+ and Mg2+ and fixed with 4% PFA in PBS for 15 minutes at room temperature (RT) then washed with PBS until use. Cells were mounted on coverslips with Prolong™ Gold antifade reagent with DAPI (P36931) from Invitrogen. Antibodies used were anti ATRX (sc-15408) from Santa Cruz Biotechnology and Alexa-Fluor 488 (A-11034) from Thermo Fisher.
In vitro irradiation studies
For irradiation (IR) conditions, cells were plated in T-75 flasks to a density of 80% and incubated at 37°C for 24 hours. For radio-sensitization studies, cells were pretreated with ATM inhibitor or an equivalent volume of DMSO, as a control, for one hour then subsequently irradiated. For IR-treated protein characterization, whole-cell lysates were collected four hours after irradiation. Radiation was delivered using a Philips RT250 (Kimtron Medical) within the Experimental Irradiation Shared Resource of the University of Michigan Rogel Cancer Center.
Cell proliferation assay
Cells were seeded in 96 well tissue culture plates at 2,000 cells per well (adherent human cell cultures) and 4,000 cells per well (mouse GBM neurospheres) and incubated at 37°C for 24 hours. Proliferation assays were performed on primary mouse ATRX positive “NP” (C39E) and ATRX knockdown “NPA” (C54B) GBM neurosphere cells. Cells were then treated with increasing concentrations of drug or irradiation in triplicate and incubated at 37°C and 5% CO2, for 72 hours. For treatment conditions of ATM/ATR inhibitors, compounds were obtained from Selleck. For treatment with IR, cells were treated 1 hour prior to radiation. Cell viability was assessed with XTT Cell Proliferation Assay Kit (#10010200) from Cayman Chemical and was normalized against untreated (DMSO) values.
Incucyte proliferation assay
U251-ATRXEV, U251-ATRXKO SF188 ATRXEV, SF188 ATRXKO, UW479 ATRXEV and UW479 ATRXKO cells were plated into 96 well tissue culture plates (CellPro) with a seeding density of 2,000 cells per well. The plates were incubated for 24 hours at 37°C and 5% CO2 before being irradiated according to their irradiation condition (0 Gy, 1, Gy, or 2 Gy). Cells were also treated with various concentrations of ATM/ATR inhibitors (10nM, 100nM, or 1uM). For these experiments, the drug was added to the cell media one hour before irradiation. After irradiation, the plates were placed in an IncuCyte Zoom device for data collection (brightfield confluence) and imaged at 3-hour intervals.
Clonogenic assay
Cells treated with drugs and/or XRT were processed for clonogenic survival. Drugs were given for 24 hours, beginning 1 hour prior to XRT. Radiation survival was normalized for drug toxicity and the radiation enhancement ratio (RER) was calculated as the ratio of the normalized surviving fraction at 4Gy under control conditions divided by the normalized surviving fraction at 4Gy under drug treatment conditions. A value greater than 1 indicates radio-sensitization. Cytotoxicity in the absence of radiation treatment was calculated by normalizing the plating efficiencies of drug treated cells to non-drug treated cells.
Mitotic index: phospho-histone H3 staining
A mitotic index assay was performed utilizing phospho-histone H3 staining. This assay was conducted on the following human cells lines: SJ-GBM2, KNS42, U251 ATRXKO, and U251 ATRXEV. Each cell line was plated into three vented TC-treated cell culture flasks (CellPro) and allowed to incubate for 24 hours at 37°C and 5% CO2. Following the incubation period, the cells were irradiated and then fixed with 70% ethanol according to their assigned condition. In drug treated experiments, 100nM AZD0156 was added to the cell media one hour prior to irradiation. Each cell line had three different conditions: 0-hour fixation post 0 Gy irradiation, 1-hour fixation post 10 Gy irradiation, and 16-hour fixation post 4.5 Gy irradiation. The cells that underwent fixation were stored at 4°C until the final 16-hour fixation point occurred. The ethanol-fixed cells then underwent permeabilization with the addition of 0.25% Triton/PBS and were kept on ice for 10 minutes. After permeabilization, the cells were stained with a rabbit Phospho-Histone H3 (Ser10) primary antibody 3377T (Cell Signaling) followed by a goat anti-rabbit IgG (H+L) FITC-conjugated secondary antibody F2765 (ThermoFisher Scientific). Next, the cells were stained with propidium iodide (PI) P4864 (Sigma) (50ug/mL PI + 100mg/mL RNase A), or DAPI (1 ug/mL) and each cell culture flask was divided into three 5mL polypropylene round-bottom Falcon tubes (Corning). Data were acquired via flow cytometry (Bio-Rad Ze5 #2 Analyzer) and normalized to 0 Gy values within each cell line. We also measured G1 cell cycle transition using BrdU Flow Kit (BD Pharmingen cat. #559619).
Cell cycle analysis using FastFUCCI reporter gene
Isogenic cell lines containing ATRX KO and Chk1 OE as well as ATRX KO only were used in this study alongside their controls. These cells were transfected with the FastFUCCI reporter which tags CDT1 expressed in the G1 phase with RFP (red) and Geminin expressed in the G2/M phase with GFP (green) respectively (Koh et al., 2017). The IncuCyte live cell imaging system was used to monitor cell cycling in real time. Quantification of the data generated was analyzed using the IncuCyte Zoom 2018A software after specifying the desired threshold values. The values generated were plotted using GraphPad Prism.
Implantation of mouse cells and treatment
Male and female C57BL/6, FVB, and NOD scid gamma (NSG) mice (weighing 20 to 25 g) were obtained from Charles River or Jackson Labs and were 6-10 weeks of age at the start of surgery. All animal studies were conducted according to the guidelines approved by the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan. Mice were anesthetized with injection of 120 mg/kg ketamine and 0.5 mg/kg dexmedetomidine. Hair above scalp was shaven, disinfected with iodine, and a 1 cm incision was made above scalp to expose cranium. The periosteum was removed with scalpel. Next, a 0.6mm burr hole was drilled 2 mm right of midline and 0.2 mm anterior to the bregma with the Ideal Micro Drill (MD-1200 120V) from Braintree Scientific Inc. Implantation studies were performed on primary ATRX knockdown “NPA” (C54B) GBM neurosphere cells. Mice were placed in a Mouse/Neonatal Rat Adaptor stereotactic frame (#51615) from Stoelting. A 10 ul syringe (#7635-01) fitted with 33-gauge needle (#7762-06) from Hamilton, was filled with cell suspension (15,000 cells per uL) and penetrated 3 mm into brain tissue. After waiting two minutes, one microliter of cell suspension was injected over one minute and the needle was slowly removed after waiting 3 minutes after injection. Incision was closed with 4-0 nylon suture and mouse was given 1 mg/kg atipamezole for reversal and monitored for recovery. For survival studies, mice were monitored daily for symptoms of morbidity, including impaired mobility, scruffed fur, hunched posture, ataxia, and seizures. Mice displaying symptoms of morbidity were anesthetized and brains were removed for histological processing with Tyrode’s solution and 4% PFA as previously described.
For flank implantations, the cells are prepared in a 1:1 ratio with 50 uL Matrigel (Corning #356234) and 50 uL of the single cell suspension (1x106 cells) in a 1 mL syringe fitted with a 26-gauge needle. The mice were anesthetized with isoflurane in a plastic desiccator places in an externally vented fume hood. The cell suspension is then injected into the flank of the mouse on the posterior/lateral aspect of the lower rib cage. The same procedure is repeated on the other flank to obtain n=2 tumors per mouse. The mice were observed for tumor growth and development and the tumor area was measured.
Treatment of intracranially-implanted mice with radiation and/or ATM inhibitors
Mice with implanted tumors were allowed to grow until the tumor bioluminescence score reached 10^7 radiance (p/s/cm3/sr). Mice were randomized into groups before treatment. Mice underwent two weeks of treatment, every 3-4 days for a total of 4 treatments. Treatment groups included vehicle (DMSO) alone, 4Gy whole brain XRT with vehicle, 4Gy whole brain XRT with 10 mg/kg AZD0156, and 4Gy whole-brain XRT with 20 mg/kg AZD1390. For in vivo preparation, AZD0156 was dissolved in DMSO at 7.2 mM with PBS at a ratio of 9:1 for dosing 10 mg/kg(Pike et al., 2018). For in vivo preparation, AZD1390 was dissolved in 5 mg/ml in vehicle solution (0.5%w/v HPMC/0.1%w/v Tween 80) and diluted to 2 mg/ml in vehicle for dosing 20 mg/kg (Durant et al., 2018). ATM inhibitors were given to mice- AZD1560 through IP and AZD1390 through oral-gavage- one hour prior to whole-brain XRT. Tumor growth and treatment response was monitored by IVIS bioluminescence once per week starting at 10 days post implantation.
Treatment of flank-implanted mice with radiation and/or ATM inhibitors
Mice with implanted flank tumors were allowed to grow until the tumor size reached 3x3 mm area. Mice were randomized into groups before treatment. Mice underwent one week of treatment, 3 days apart for a total of 2 treatments. Treatment groups included vehicle (DMSO) alone, 4Gy whole brain XRT with vehicle, 4Gy whole brain XRT with 5 mg/kg AZD0156, and 4Gy whole-brain XRT with 10 mg/kg AZD0156. For in vivo preparation, AZD0156 was dissolved in DMSO at 7.2 mM with PBS at a ratio of 9:1 for dosing (Pike et al., 2018). ATM inhibitors were given to mice- AZD1560 through IP one hour prior to flank tumor irradiation. Tumor growth and treatment response was monitored by measuring the tumor area twice per week starting at 15 days post implantation.
Bioluminescence imaging
Tumor bearing mice were imaged using the IVIS Spectrum machine at the University of Michigan Center for Molecular Imaging. Mice were injected with 160 mg/kg D-luciferin (#115144-35-9) from Gold Biotechnology and anesthetized with 2% isoflurane. Ten minutes after luciferin injection, mice were placed into the machine in a prone position and bioluminescence was measured. Mice were imaged until peak signal was obtained for each mouse. Tumor bioluminescent signal is measured in radiance (photons) (p/s/cm3/sr) in a circular region of interest (ROI) over the cranium of each mouse with Living Image Software (PerkinElmer Inc).
ATM inhibition reporter (in vitro D54 cells)
Human GBM D54 ATM-Luciferase inhibition reporter cells (D54-ATMR) were obtained from Dr. Alnawaz Rehemtulla (University of Michigan). D54-ATMR cells were plated into a 96 well, white bottom cell culture plate (Alkali Scientific Inc.) with a seeding density of 2,000 cells per well. The cells were then incubated for 24 hours at 37°C and 5% CO2 before being treated with different concentrations of AZD0156 and AZD1390 (10uM and 10nM). Next, 100uL of luciferin (150ug/mL) was added to each well, and the plate was briefly rocked on a plate shaker for one minute. The luminesce signal from the wells was then read by the Synergy HTX Multi-Mode microplate reader (BioTek). The plate was analyzed every ten minutes for 70 minutes.
ATM inhibition reporter (in vivo D54 cells)
D54-ATMR cells were implanted into the cortex of NSG mice as described in the above methods at 240,000 cells per mouse, and tumor monitoring was done as described in the above methods. After allowing tumors to grow large enough to detect baseline ATM-Luciferase inhibition reporter without treatment with ATM inhibitor, mice were imaged for baseline bioluminescence signal and randomized into two groups of three mice each. Mice were treated with either 20 mg/kg AZD1390 or 10 mg/kg AZD0156 intraperitoneally (IP) three hours after baseline imaging was conducted and then subsequently imaged for ATM-Luciferase inhibition reporter activity one hour after treatment, four hours after treatment, and 24 hours after treatment.
Pharmacokinetic (PK) analysis of AZD0156 for CNS retention
AZD0156 (Selleck) at 1 mg/mL in PBS containing 20% DMSO and 20% PEG-400 was given by IP injection (10 mg/kg). At the given time points (1h, 2h, 4h, and 7h), mice were sacrificed (n=3 each) and the brains were taken out and frozen at −80°C immediately for later preparation and analysis. At the given time points (1h, 2h, 4h, and 7h), blood samples were collected using heparinized calibrated pipettes. Samples were centrifuged at 15000 rpm for 10 min. Subsequently, blood plasma was collected from the upper layer. The plasma was frozen at −80°C for later analysis. Brains were taken out and frozen at −80°C immediately for later preparation and analysis.
Liquid chromatography–mass spectrometry (LC-MS) grade acetonitrile was purchased from Sigma-Aldrich. Formic acid (98%; LC-MS grade) was obtained from Fluka. A Milli-Q water system from Millipore was used to obtain ultrapure deionized water.
Stock solutions, working solutions, and quality control samples:
AZD0156 and the internal standard were individually weighed and dissolved in acetonitrile to stock solutions and then stored at −20°C. The AZD0156 stock solution was then diluted with acetonitrile to a series of working solutions from 2.5 to 5,000 ng/mL The quality control working solutions at low, medium, and high concentrations were prepared using a separately prepared stock solution. For sample preparation, the AZD0156 stock solution was diluted to 1,000 ng/mL with acetonitrile. Quality control samples were evenly distributed among samples from each batch.
The concentrations of AZD0156 were determined with a Sciex AB-5500 Qtrap mass spectrometer with electrospray ionization source, interfaced with a Shimadzu high-performance LC system. The LC-MS/MS system was controlled with Analyst Software version 1.6 from Applied Biosystems; this was also used for acquisition and processing of data. Separation was performed on a Waters Xbridge C18 column (50 × 2.1 mm ID, 3.5 μm); the flow rate was 0.4 mL/min. A (100% H2O with 0.1% formic acid) and B (100% acetonitrile with 0.1% formic acid) comprised the mobile phase. The gradient began with 5% B for 30 seconds and then linearly increased to 99% B at 2 minute and then reduced to 5% B at 4.1 minutes to 5.5 minutes with a runtime of 6 minutes in total. The mass spectrometer was operated in positive mode; multiple reaction monitoring was used for analysis. The Q1 m/z and Q3 m/z was 487.9 and 401.1, respectively.
Quantification and Statistical Analysis
All quantitative data are presented as mean ± SEM unless otherwise specified. Survival curves were plotted using the Kaplan–Meier method and compared with a log-rank test. Statistical significance for triplicate experiments were calculated using the Welch’s t-test and 2-Way ANOVA. Differences were statistically significant at P < 0.05. Statistical details are located in the figure legends. Statistical analysis was done utilizing GraphPad Prism.
Supplementary Material
Table S1: RNA-seq Analysis of Differentially Expressed Genes between ATRX-negative and ATRX-positive Glioma Samples. Related to Figure 2.
Table S2: ChIP-seq Analysis of Gene Set Enrichment Associated with ATRX Peak Binding. Related to Figure 2.
Table S3: RNA-seq Analysis of Gene Set Enrichment between ATRX-negative and ATRX-positive Glioma Samples. Related to Figure 2.
Table S4: Comparing Upregulated Genes with Peak ATRX Binding at Promoter Regions between ATRX-negative and ATRX-positive Glioma Samples. Related to Figure 2.
Table S5: Gene Cluster Markers. Related to Figure 3.
Table S6: Clustered Cell Cycle Gene Set Enrichment Analysis between ATRX wild-type and ATRX-knockout Glioma Samples. Related to Figure 3.
Table S7: Additional qPCR Primers. Related to Figure 2.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-ATRX | Santa Cruz Biotechnology | sc-15408 |
| anti-ATR | Santa Cruz Biotechnology | sc-51573 |
| anti-ATRX | Bethyl Laboratories, LLC | A301-045A |
| anti-Vinculin | Invitrogen | #700062 |
| anti-CHK1 | Cell Signaling Technology | #2360 |
| anti-pCHK1 Ser345 | Cell Signaling Technology | #2348 |
| anti-CHK2 pT68 | Cell Signaling Technology | #2661 |
| anti-Phospho-Histone H2A.X Ser139 | Cell Signaling Technology | #2577 |
| anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | #7076 |
| anti-ATM pS1981 | Abcam | ab81292 |
| HRP Goat Anti-Rabbit IgG Antibody | Vector Laboratories | P1-1000 |
| Anti-Histone H3.3 Antibody | Millipore Sigma | #09838 |
| Control IgG | Millipore Sigma | #12370 |
| Alexa-Fluor 488 | ThermoFisher | A-11034 |
| Rabbit Phospho-Histone H3 (Ser10) primary antibody | Cell Signaling | 3377T |
| Goat Anti-Rabbit IgG (H+L) FITC-conjugated secondary antibody | ThermoFisher | F2765 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| B-27 supplement | Gibco | 12587-010 |
| N-2 supplement | Gibco | 17502-048 |
| Penicillin-Streptomycin | Cellgro | 30-001-CI |
| Normocin | Invivogen | ant-nr-1 |
| FGF | Shenandoah Biotech | 100-26 |
| EGF | Shenandoah Biotech | 100-146 |
| GlutaMAX-I Supplement | Invitrogen | #35050061 |
| Fetal Bovine Serum (FBS) | Sigma | F4135-500ML |
| Clarity™ Western ECL substrate | Bio-rad | #170-5060 |
| DMSO | Sigma | D8418-250ML |
| Paraformaldehyde (PFA) | Sigma | 158127-500G |
| Prolong™ Gold antifade reagent with DAPI | Invitrogen | P36931 |
| AZD0156 | Selleck | S8375 |
| AZD1390 | Selleck | S8680 |
| KU-55933 | Selleck | S1092 |
| KU-60019 | Selleck | S1570 |
| Nocodazole | Sigma | M1404-10MG |
| Triton X-100 | Sigma | T8787 |
| Propidium Iodide | Sigma | P4864 |
| RNase A | Qiagen | 19101 |
| Formic acid | Fisher Scientific | 60-006-16 |
| D-luciferin | Gold Biotechnology | #115144-35-9 |
| Critical Commercial Assays | ||
| Qiagen RNeasy Mini Kit | Qiagen | #74104 |
| TruSeq Stranded mRNA Library Kit | Illumina | #20020594 |
| DNeasy Blood and Tissue Kit | Qiagen | #69504 |
| Deposited Data | ||
| Raw data | this paper | GSE178116 |
| Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling | Danussi et al., 2018 | GSE100462, GSE100464 |
| Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma | Mackay et al., 2017 | EGA - EGAS00001000226, EGAS0000100192, EGAS00001000575, EGAS00001000720, EGAS00001001139; the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/)-GSE19578, GSE26576, GSE21420, GSE34824, GSE36245, GSE36278 GSE50022, GSE50021, GSE50024, GSE55712; ArrayExpress - E-TABM-857, E-TABM-1107. |
| Genomic Data Commons (GDC) Data Portal | The Cancer Genome Atlas Program | https://portal.gdc.cancer.gov |
| single cell RNA-seq data of oligodendroglioma samples | Tirosh et al., 2016b | GSE7063 |
| single cell RNA-seq data of astrocytomas samples | Venteicher et al., 2017 | GSE89567 |
| Experimental Models: Cell Lines | ||
| KNS42 | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| SJ-GBM2 | Children’s Oncology Group cell line repository | #12440053 |
| U251 ATRXEV | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| U251 ATRXKO | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| SF188 ATRXEV | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| SF188 ATRXKO | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| UW479 ATRXEV | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| UW479 ATRXKO | Dr. Alan Meeker, Johns Hopkins University, Baltimore, MD | N/A |
| Human GBM D54 ATM-Luciferase inhibition reporter cells | Dr. Alnawaz Rehemtulla University of Michigan, Ann Arbor, MI | N/A |
| U251 ATRXKO Chk1EV | This paper | N/A |
| U251 ATRXKO Chk1OE | This paper | N/A |
| U251 ATRXKO FastFUCCI | This paper | N/A |
| U251 ATRXEV FastFUCCI | This paper | N/A |
| U251 ATRXKO Chk1EV FastFUCCI | This paper | N/A |
| U251 ATRXKO Chk1OE FastFUCCI | This paper | N/A |
| UW479 ATRXKO Chk1EV | This paper | N/A |
| UW479 ATRXKO Chk1KO | This paper | N/A |
| SF188 ATRXKO Chk1EV | This paper | N/A |
| SF188 ATRXKO Chk1KO | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River or Jackson Labs | N/A |
| Mouse: NOD scid gamma (NSG) | Charles River or Jackson Labs | N/A |
| Mouse: FVB | Charles River or Jackson Labs | N/A |
| Oligonucleotides | ||
| Human Chk1 Forward Primer 5′GGTGCCTATGGAGAAGTTCAA | IDT | N/A |
| Human Chk1 Reverse Primer 5′TCTACGGCACGCTTCATATC | IDT | N/A |
| Human GAPDH Forward Primer 5′GGATTTGGTCGTATTGGG | IDT | N/A |
| Human GAPDH Reverse Primer 5′GGAAGATGGTGATGGGATT | IDT | N/A |
| Primers (see Table S7) | This paper | N/A |
| Recombinant DNA | ||
| shp53 lentiviral plasmid | Dr. John Ohlfest, University of Minnesota, Minneapolis, Minnesota | N/A |
| shATRX lentiviral plasmid | generated for Koschmann et al., 2016 | N/A |
| NRAS overexpression plasmid | Dr. John Ohlfest, University of Minnesota, Minneapolis, Minnesota | N/A |
| pcDNA4-Chk1-Flag Overexpression Plasmid | Addgene | #22894 |
| pBOB-EF1-FastFUCCI-Puro Plasmid | Addgene | #86849 |
| Software and Algorithms | ||
| chipenrich (v2.6.1) | Welch et al., 2014 | https://bioconductor.org/packages/release/bioc/html/chipenrich.html |
| annotatr (v1.0.3) | Cavalcante and Sartor, 2017 | https://www.bioconductor.org/packages/release/bioc/html/annotatr.html |
| Bowtie2 (v2.2.1) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| deepTools (v.3.1.3) | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| IGV (v2.11.3) | Robinson et al., 2011 | https://igv.org |
| FastQC (v0.11.5) | Andrews, S., 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| TrimGalore (v0.6.0) | Github | https://github.com/FelixKrueger/TrimGalore |
| STAR (v2.5.3a) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq (v0.11.2) | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| edgeR (v3.20.9) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| RNA-Enrich | N/A | http://lrpath.ncibi.org |
| betareg | Grün et al., 2011 | https://cran.r-project.org/web/packages/betareg/betareg.pdf |
| Seurat (v4.0.5) | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| fgsea | Korotkevich et al., 2019 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
ATRX binds regulatory elements of CHEK1 in glioma and glioma precursor cells
ATRX loss is associated with loss of the cell cycle regulator Chk1
Chk1 loss increases reliance on ATM, an alternate cell cycle checkpoint modulator
ATM inhibition may sensitize ATRX-deficient gliomas to radiation therapy
Acknowledgements:
CK is supported by NIH/NINDS Grants K08-NS099427-01 and R01-NS119231, DOD Grant CA201129P1, The University of Michigan Chad Carr Pediatric Brain Tumor Center, the Evans Family, the Chad Tough Defeat DIPG Foundation, U CAN-CER VIVE, Catching Up with Jack, and the Morgan Behen Golf Classic. MGC is supported by NINDS R37NS094804, RO1NS105556; R21NS107894. PRL is supported by NINDS RO1NS082311 and RO1NS096756. TQ and MAS are partially supported by the University of Michigan Rogel Cancer Center, which is funded by the National Cancer Institute grant # P30 CA046592-29. SV is supported by NINDS R01NS110572. Experimental irradiation experiments were partially supported by Cancer Center Support Grant P30CA46592.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
REFERENCES:
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Andrews S (2010). FastQC: a quality control tool for high throughput sequence data. Version 0.11. 2 Website: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. [Google Scholar]
- Schmidl C, Suzuki T, et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461. 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Berube NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, Higgs DR, Slack RS, and Picketts DJ (2005). The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest 115, 258–267. 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, and Berman SH (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnan-Cashman JA, Yuan M, Graham MK, Rizzo AJ, Myers KM, Davis C, Zhang R, Esopi DM, Raabe EH, Eberhart CG, et al. (2018). ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS One 13, e0204159. 10.1371/journal.pone.0204159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, and Chen DJ (2001). ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276, 42462–42467. 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Carlson M, Falcon S, Pages H, Li N, biocViews AnnotationData F, Me adSplit I, ChIPpeakAnno E, GOstats P, and Agi4x44PreProcess, S.M. (2007). A set of annotation maps describing the entire Gene Ontology. R package version 3. [Google Scholar]
- Cavalcante RG, and Sartor MA (2017). annotatr: genomic regions in context. Bioinformatics 33, 2381–2383. 10.1093/bioinformatics/btx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes D, and Gibbons RJ (2013). ATRX and the replication of structured DNA. Curr Opin Genet Dev 23, 289–294. 10.1016/j.gde.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, and Gibbons RJ (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6, 7538. 10.1038/ncomms8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danussi C, Bose P, Parthasarathy PT, Silberman PC, Van Arnam JS, Vitucci M, Tang OY, Heguy A, Wang Y, Chan TA, et al. (2018). Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat Commun 9, 1057. 10.1038/s41467-018-03476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan LL LJ (2014). Native Chromatin Immunoprecipitation from Brain Tissue Using Magnetic Beads. Medicinal Chemistry 4, 3. 10.4172/2161-0444.1000223. [DOI] [Google Scholar]
- Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, Yang Z, Riches L, Trinidad AG, and Fok JH (2018). The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Science advances 4, eaat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. (2015). Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 372, 2499–2508. 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278. 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suva ML, Benes CH, et al. (2015). Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277. 10.1126/science.1257216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, and Higgs DR (2000). Molecular-clinical spectrum of the ATR-X syndrome. Am J Med Genet 97, 204–212. 10.1002/1096-8628(200023)97:3<204::AID-AJMG1038>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, Picketts DJ, Villard L, and Higgs DR (1995). Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 80, 837–845. 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Graham MK, Kim J, Da J, Brosnan-Cashman JA, Rizzo A, Baena Del Valle JA, Chia L, Rubenstein M, Davis C, Zheng Q, et al. (2019). Functional Loss of ATRX and TERC Activates Alternative Lengthening of Telomeres (ALT) in LAPC4 Prostate Cancer Cells. Mol Cancer Res 17, 2480–2491. 10.1158/1541-7786.MCR-19-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün B, Kosmidis I, and Zeileis A (2011). Extended beta regression in R: shaken, stirred, mixed, and partitioned. Working Papers in Economics and Statistics. [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, et al. (2012). Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3, 709–722. 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SB, Mascalchi P, Rodriguez E, Lin Y, Jodrell DI, Richards FM, and Lyons SK (2017). A quantitative FastFUCCI assay defines cell cycle dynamics at a single-cell level. J Cell Sci 130, 512–520. 10.1242/jcs.195164. [DOI] [PubMed] [Google Scholar]
- Koneva LA, Zhang Y, Virani S, Hall PB, McHugh JB, Chepeha DB, Wolf GT, Carey TE, Rozek LS, and Sartor MA (2018). HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol Cancer Res 16, 90–102. 10.1158/1541-7786.MCR-17-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, and Sergushichev A (2019). Fast gene set enrichment analysis. BioRxiv, 060012. [Google Scholar]
- Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, Mendez F, Kamran N, Dzaman M, Mulpuri L, et al. (2016a). ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Science translational medicine 8, 328ra328. 10.1126/scitranslmed.aac8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmann C, Lowenstein PR, and Castro MG (2016b). ATRX mutations and glioblastoma: Impaired DNA damage repair, alternative lengthening of telomeres, and genetic instability. Molecular & cellular oncology 3, e1167158. 10.1080/23723556.2016.1167158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmann C, Nunez FJ, Mendez F, Brosnan-Cashman JA, Meeker AK, Lowenstein PR, and Castro MG (2017). Mutated Chromatin Regulatory Factors as Tumor Drivers in Cancer. Cancer Res 77, 227–233. 10.1158/0008-5472.CAN-16-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A, et al. (2010). ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143, 367–378. 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Lee C, Patil S, and Sartor MA (2016). RNA-Enrich: a cut-off free functional enrichment testing method for RNA-seq with improved detection power. Bioinformatics 32, 1100–1102. 10.1093/bioinformatics/btv694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JW, Ghosal G, Wang W, Shen X, Wang J, Li L, and Chen J (2013). Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J Biol Chem 288, 6342–6350. 10.1074/jbc.M112.411603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, and Allis CD (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A 107, 14075–14080. 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zhao H, Diplas BH, Liu S, Liu J, Wang D, Lu Y, Zhu Q, Wu J, and Wang W (2020). Genome-wide CRISPR-Cas9 screen reveals selective vulnerability of ATRX-mutant cancers to WEE1 inhibition. Cancer research 80, 510–523. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M, et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8, e1002772. 10.1371/journal.pgen.1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. (2017). Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32, 520–537 e525. 10.1016/j.ccell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklja Z, Pasternak A, Stallard S, Nicolaides T, Kline-Nunnally C, Cole B, Beroukhim R, Bandopadhayay P, Chi S, Ramkissoon SH, et al. (2019). Molecular profiling and targeted therapy in pediatric gliomas: review and consensus recommendations. Neuro Oncol. 10.1093/neuonc/noz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez FJ, Mendez FM, Kadiyala P, Alghamri MS, Savelieff MG, Garcia-Fabiani MB, Haase S, Koschmann C, Calinescu AA, Kamran N, et al. (2019). IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Science translational medicine 11. 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyati S, Young G, Ross BD, and Rehemtulla A (2017). Quantitative and Dynamic Imaging of ATM Kinase Activity. Methods Mol Biol 1596, 131–145. 10.1007/978-1-4939-6940-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 17, 93–115. 10.1038/nrc.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil M, Pabla N, and Dong Z (2013). Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cellular and molecular life sciences 70, 4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike KG, Barlaam B, Cadogan E, Campbell A, Chen Y, Colclough N, Davies NL, de-Almeida C, Degorce SL, and Didelot M (2018). The identification of potent, selective, and orally available inhibitors of ataxia telangiectasia mutated (ATM) kinase: the discovery of AZD0156 (8-{6-[3-(Dimethylamino) propoxy] pyridin-3-yl}-3-methyl-1-(tetrahydro-2 H-pyran-4-yl)-1, 3-dihydro-2 H-imidazo [4, 5-c] quinolin-2-one). ACS Publications. [DOI] [PubMed] [Google Scholar]
- Project, I.C.G.C.P.T. (2016). Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riches LC, Trinidad AG, Hughes G, Jones GN, Hughes AM, Thomason AG, Gavine P, Cui A, Ling S, and Stott J (2020). Pharmacology of the ATM inhibitor AZD0156: potentiation of irradiation and olaparib responses preclinically. Molecular cancer therapeutics 19, 13–25. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Seah C, Levy MA, Jiang Y, Mokhtarzada S, Higgs DR, Gibbons RJ, and Berube NG (2008). Neuronal death resulting from targeted disruption of the Snf2 protein ATRX is mediated by p53. J Neurosci 28, 12570–12580. 10.1523/JNEUROSCI.4048-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, et al. (2015). Mutational landscape and clonal architecture in grade II and III gliomas. Nature genetics 47, 458–468. 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016a). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016b). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, and Kaufmann WK (2007). The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 27, 3131–3142. 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-Garcia D, Qadeer ZA, McHugh DS, Ghiraldini FG, Chowdhury AH, Hasson D, Dyer MA, Recillas-Targa F, and Bernstein E (2016). ATRX binds to atypical chromatin domains at the 3′ exons of zinc finger genes to preserve H3K9me3 enrichment. Epigenetics 11, 398–414. 10.1080/15592294.2016.1169351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355. 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon HP, Hughes JR, Rode C, De La Rosa-Velazquez IA, Jenuwein T, Feil R, Higgs DR, and Gibbons RJ (2015). ATRX Plays a Key Role in Maintaining Silencing at Interstitial Heterochromatic Loci and Imprinted Genes. Cell Rep 11, 405–418. 10.1016/j.celrep.2015.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang J, Wild AT, Wu WH, Shah R, Danussi C, Riggins GJ, Kannan K, Sulman EP, Chan TA, and Huse JT (2019). G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat Commun 10, 943. 10.1038/s41467-019-08905-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RP, Lee C, Imbriano PM, Patil S, Weymouth TE, Smith RA, Scott LJ, and Sartor MA (2014). ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res 42, e105. 10.1093/nar/gku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: RNA-seq Analysis of Differentially Expressed Genes between ATRX-negative and ATRX-positive Glioma Samples. Related to Figure 2.
Table S2: ChIP-seq Analysis of Gene Set Enrichment Associated with ATRX Peak Binding. Related to Figure 2.
Table S3: RNA-seq Analysis of Gene Set Enrichment between ATRX-negative and ATRX-positive Glioma Samples. Related to Figure 2.
Table S4: Comparing Upregulated Genes with Peak ATRX Binding at Promoter Regions between ATRX-negative and ATRX-positive Glioma Samples. Related to Figure 2.
Table S5: Gene Cluster Markers. Related to Figure 3.
Table S6: Clustered Cell Cycle Gene Set Enrichment Analysis between ATRX wild-type and ATRX-knockout Glioma Samples. Related to Figure 3.
Table S7: Additional qPCR Primers. Related to Figure 2.
Data Availability Statement
The RNA-seq of mouse GBM neurospheres was deposited in Gene Expression Omnibus (GEO) with the accession number of GSE178113. The single cell RNA-seq of human isogenic cell line U251 was deposited in Gene Expression Omnibus (GEO) with the accession number of GSE178114.
The code used for the downstream analysis in this manuscript was deposited in Zenodo: https://zenodo.org/badge/latestdoi/429519345
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.