

Abstract
Coenzyme Q (CoQ) analogs with a variable number of isoprenoid units have exhibited as anti-inflammatory as well as antioxidant molecules. Using novel quinone derivative CoQ0 (2,3-dimethoxy-5-methyl-1,4-benzoquinone, zero side chain isoprenoid), we studied its molecular activities against LPS/ATP-induced inflammation and redox imbalance in murine RAW264.7 macrophages. CoQ0's non- or subcytotoxic concentration suppressed the NLRP3 inflammasome and procaspase-1 activation, followed by downregulation of IL1β expression in LPS/ATP-stimulated RAW264.7 macrophages. Similarly, treatment of CoQ0 led to LC3-I/II accumulation and p62/SQSTM1 activation. An increase in the Beclin-1/Bcl-2 ratio and a decrease in the expression of phosphorylated PI3K/AKT, p70 S6 kinase, and mTOR showed that autophagy was activated. Besides, CoQ0 increased Parkin protein to recruit damaged mitochondria and induced mitophagy in LPS/ATP-stimulated RAW264.7 macrophages. CoQ0 inhibited LPS/ATP-stimulated ROS generation in RAW264.7 macrophages. Notably, when LPS/ATP-stimulated RAW264.7 macrophages were treated with CoQ0, Mito-TEMPO (a mitochondrial ROS inhibitor), or N-acetylcysteine (NAC, a ROS inhibitor), there was a significant reduction of LPS/ATP-stimulated NLRP3 inflammasome activation and IL1β expression. Interestingly, treatment with CoQ0 or Mito-TEMPO, but not NAC, significantly increased LPS/ATP-induced LC3-II accumulation indicating that mitophagy plays a key role in the regulation of CoQ0-inhibited NLRP3 inflammasome activation. Nrf2 knockdown significantly decreased IL1β expression in LPS/ATP-stimulated RAW264.7 macrophages suggesting that CoQ0 inhibited ROS-mediated NLRP3 inflammasome activation and IL1β expression was suppressed due to the Nrf2 activation. Hence, this study showed that CoQ0 might be a promising candidate for the therapeutics of inflammatory disorders due to its effective anti-inflammatory as well as antioxidant properties.
1. Introduction
NLRP3 is a commonly studied inflammasome complex that is named after the NLRP3 protein of the Nod-like receptor (NLR) family [1]. NLRP3 is a cytosolic protein of 115 kDa which is expressed in monocytes, neutrophils, dendritic cells, epithelial cells, and lymphocytes [2]. The NLRP3 inflammasome activation is a tightly regulated process that needs priming as well as activation signals [3]. The NLRP3 inflammasome activation is associated with age-related diseases [4, 5] as well as different types of cancer [6]. A variety of targets can be applied for its repression by taking benefits of the complex NLRP3 inflammasome signaling cascades, such as inhibition of NLRP3 inflammasome activation, inhibition of caspase-1 activation, suppression of upstream signals, and neutralization of inflammatory cytokines secreted by NLRP3 inflammasome [7].
Autophagy is an evolutionarily conserved catabolic mechanism that involves the generation of vesicles known as autophagosomes which engulf macromolecules and organelles of the cell and fuses with lysosomes for their breakdown [8]. Autophagic cell death is often referred to as programmed cell death of Type II which differs from Type I cell death including apoptosis and necrosis-like cell death mechanism [9]. One of the cases of autophagic cell death is due to the production of an increased amount of reactive oxygen species (ROS) that results from autophagic degradation of catalase [10]. Defects in autophagy play a vital role in the pathogenesis of several diseases including aging [11] and cancer [12]. Recent evidence revealed that autophagy has an important function in the development as well as the pathogenesis of inflammation and immunity response [13]. In inflammation, autophagy plays a critical role by affecting the development, survival, and homeostasis of inflammatory cells such as neutrophils, macrophages, and lymphocytes [14]. Removal of NLRP3 inflammasome and cytokine components by autophagy can suppress the activation of the inflammasome and inflammatory response. Similarly, pathways related to inflammasome can control autophagy for the balance between host defense inflammatory response and prevention of excessive and harmful inflammation [15]. The NF-E2-related factor 2 (Nrf2) transcription factor is a key mediator of the expression of cytoprotective genes which are activated during stress conditions due to the generation of ROS [16]. The increasing number of evidence supports that there is a crosstalk between the Nrf2 and inflammasome pathways at various levels. Inflammasomes and thus inflammation are inhibited by Nrf2-activating compounds [17].
Mitophagy is the process of removing damaged mitochondria through autophagy [18]. The injured mitochondria are engulfed in the autophagosomal membrane to turn into the autophagosome and then fuse with lysosomes, which are later degraded by hydroxylases [19]. Parkin and PTEN-induced kinase 1 (PINK1) play an important role in mitochondrial homeostasis and mitophagy [20]. Parkin is an E3 ligase that ubiquitinates outer mitochondrial membrane proteins, allowing the autophagic removal of the damaged organelles [21]. Parkin can regulate autophagy of damaged mitochondria, and its overexpression leads to induction of complete removal of mitochondria from cells by the process of mitophagy where membrane potential of mitochondria is lost [22]. Since Parkin selectively binds only to damaged mitochondria, there are speculations that it can mediate the mitophagy quality control pathway [23]. Disruption in Parkin-PINK1 signaling leads to impaired mitophagy [24]. Hence, gaining insight into the Parkin/PINK1/mitophagy pathway might help to understand the pathogenic signaling pathways.
Coenzyme (CoQ) is a ubiquinone analog available in all cells and membranes, and being a member of the mitochondrial respiratory chain, it functions in cellular metabolism [25]. CoQ0 consists of a benzoquinone ring conjugated to an isoprenoid chain. Depending upon the number of isoprenoid side chains, CoQ varies from CoQ0 to CoQ10 [26]. The antioxidant property of any compound is represented when it prevents oxidative stress-induced cell death [27]. Various analogs of CoQ0 have shown antioxidant or proantioxidant properties [28, 29]. CoQ0 is a coenzyme and redox-active compound without an isoprenoid side chain occurring mostly within mitochondria that suppresses the activity of complex 1 of the mitochondrial respiratory chain and prevents the opening of the mitochondrial permeability transition pore [30]. Recently, many studies have reported that CoQ0 has therapeutic effects on inflammation, metabolic disorders, and cancer [31–34]. CoQ0 improved atopic dermatitis-like wounds by reducing IL1β, IL4, IL6, IL10, and interferon (IFN) γ and by infiltrating neutrophils in the lesional skin [35]. ROS production by external stimuli along with lipopolysaccharide (LPS) promotes inflammatory response in cultured macrophages [36]. However, the main pharmacological efficacy against inflammation and redox imbalance of CoQ0 molecule has not been thoroughly examined, and the signaling pathways regulated by it remain largely unknown. Therefore, we examined if CoQ0 treatment could reduce the LPS-induced inflammatory response as well as redox imbalance in LPS- and ATP-induced RAW264.7 macrophages.
Lipopolysaccharide (LPS) is a powerful monocyte and macrophage activator and induces the secretion of numerous proinflammatory molecules, cytokines, nitric oxide (NO), tumor necrosis factor-alpha, and interleukins including IL1 or IL6 [37], which are responsible for the development and progression of inflammatory disease and cancer. One of the most characterized pathogen-associated molecular patterns (PAMPs) is LPS, which is the major constituent of the external membrane of Gram-negative bacteria [38]. Macrophage activation by LPS has been broadly studied to explore the inflammatory phenomenon in both cell culture and animal models [39]. During inflammation and infection, LPS can activate various signals within macrophages [40]. LPS activation of macrophages can cause an increase in oxygen absorption, leading to a range of reactive oxygen species (ROS), which are the key factors that drive oxidative stress-stimulated inflammation in immune cells [41]. In the extracellular environment, ATP is actively released in response to tissue harm and cellular stress [42]. ATP, generally secreted from dying and stressed cells, is used as a damage-associated molecular pattern to activate NLRP3 inflammation [43]. Nrf2 belongs to the base-leucine zipper (bZIP) which is the family of transcriptional activator proteins and triggered by endogenous oxidative stress [44]. It mediates cellular antioxidant responses by controlling the expression of genes that encode detoxifying enzymes and antioxidants [45]. Several pieces of evidence suggest that Nrf2 plays a crucial role in guarding macrophages against LPS-stimulated inflammation [46].
Previously, our in vitro and in vivo studies showed that CoQ0 (2,3-dimethoxy-5-methyl-1,4-benzoquinone), a novel quinone derivative, regulated NFκB/AP-1 activation and enhanced Nrf2 stabilization in attenuation of LPS-induced inflammation and redox imbalance [29]. The noncytotoxic concentrations of CoQ0 (2.5-10 μM) inhibited iNOS/COX-2 protein expression in LPS-stimulated macrophages, resulting in lower NO, PGE2, INF, and IL1 secretions. Moreover, in LPS-stimulated macrophages, CoQ0 induced the expression of HO-1 and NQO-1 genes by increasing Nrf2 nuclear translocation and Nrf2/ARE signaling [29]. However, studies regarding the effect of CoQ0 against LPS/ATP-induced inflammation and redox imbalance using murine macrophages have still not been carried out. In this study, we explored CoQ0 molecular activities against LPS/ATP-induced inflammation as well as redox imbalance in murine RAW264.7 macrophages. These results proposed that CoQ0 negatively regulates activation of macrophage by inducing autophagy and activation of Nrf2 and hindering a positive feedback loop of NLRP3 inflammasome pathways and may be a potential therapeutic target for inflammatory diseases because of its potent anti-inflammatory and antioxidant properties.
2. Materials and Methods
2.1. Chemicals and Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), glutamine, penicillin-streptomycin, ATP, and Mito-TEMPO were bought from the Invitrogen/GIBCO BRL (Grand Island, NY, USA). LPS (from Escherichia coli 055: B5), Coenzyme Q0 (2,3-dimethoxy-5-methyl-1,4-benzoquinone), 2′,7′-dihydrofluorescein-diacetate (DCFH2-DA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), N-acetylcysteine (NAC), and cycloheximide were bought from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against NLRP3 were obtained from Biorbyt (Cambridge, UK). Antibodies against IL1β and Parkin were acquired from Abcam (Cambridge, UK). Antibodies against Nrf2, p70 S6 kinase, p-p70 S6 kinase, procaspase-1, GAPDH, and β-actin were procured from Santa Cruz (Heidelberg, Germany). Antibodies against p62, Beclin-1, Bax, mTOR, AKT, PI3K, p-mTOR, p-AKT, p-PI3K, and histone H3 were brought from Cell Signaling Technology Inc. (Danvers, MA). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was purchased from Calbiochem (La Jolla, CA, USA). Antibodies against PINK1 were purchased from Genetic Technology Inc. (Miami, FL, USA). The rest of the chemicals were of the highest commercially available grade and supplied by either Sigma (St. Louis, MO, USA) or Merck (Darmstadt, Germany).
2.2. Cell Culture and Sample Treatment
RAW264.7 cells which were derived from murine macrophage were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were then cultured in DMEM that contained 2 mM glutamine, 1% penicillin-streptomycin, and 10% FBS at 37°C in a humidified atmosphere of 5% CO2. After incubating with CoQ0 for 1 h, the supernatant was removed and the cells were washed with phosphate buffer saline (PBS). After washing with PBS, the culture medium was displaced with a new medium with or without LPS (1 μg/mL) dissolved in PBS having pH 7.2 and ATP (5 mM) for the designated time.
2.3. MTT Assay
1 × 105 RAW264.7 cells/well were cultured on a 24-well plate till confluence and then incubated with 2.5-20 μM CoQ0 for 24 h. To monitor cell viability, after treatment with CoQ0, the cells were further incubated with 400 μL of 0.5 mg/mL of MTT along with medium for 1 h. Post incubation, the supernatant was discarded, and thus, formed formazan crystals were dissolved in 400 μL of dimethyl sulfoxide (DMSO). The absorbance was estimated using an enzyme-linked immunosorbent assay (ELISA) microplate reader (BioTek Instruments, Winooski, VT, USA) at 570 nm. To know the effects of CoQ0 on cells, viability was evaluated as the percentage of viable cells by comparing with vehicle-treated control whose arbitrary value was taken as 100%.
2.4. Determination of NO Levels in Cultured Media
The concentration of NO in the culture medium was estimated by using Griess reagents (Sigma-Aldrich, St. Louis, MO), NO being a major stable product which was based on the accumulation of nitrite. 4 × 105 RAW264.7 cells were grown in a 12-well plate in DMEM which contained 5 mM arginine. The cultured cells were pretreated with 2.5-12.5 μM of CoQ0 for 1 h and later stimulated for 18 h by LPS (1 μg/mL) or 17 h by LPS (1 μg/mL) followed by ATP (5 mM) for 1 h (total 18 h). Posttreatment, 100 μL supernatant was collected and dissolved in an equal amount of Griess reagents (0.75% sulfanilamide in 0.5 N HCl and 0.075% naphthylethylenediamine dihydrochloride in water = 1 : 1 mixture). Then, by using an ELISA microplate reader, the absorbance was recorded at 540 nm.
2.5. Cell Extract Preparation and Western Blot Analysis
1 × 106 RAW264.7 cells/dish were cultured in a 6 cm dish and treated with CoQ0 (2.5-10 μM) with or without LPS (1 μg/mL) and ATP (5 mM) for the designated period. Posttreatment, all the cells were detached from the culture dish and washed one time in cold PBS. Then, cytoplasmic, nuclear, and total extracts were prepared following the protocols as given by extraction reagents (Pierce Biotechnology, Rockford, IL, USA). Taking bovine serum albumin as standard, the amount of protein in every sample was calculated using the Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA). Using 8-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), an equal volume (50 μg) of denatured protein samples was first electrophoresed and later transferred to polyvinylidene fluoride (PVDF) and left overnight. On a subsequent day, blocking of the membranes was done for 30 min at room temperature by using 5% nonfat dry milk. Following blocking at first, using primary antibodies, the membranes were incubated for 2 h and later with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibody for 2 h (Pierce Biotechnology, Rockford, IL, USA). For measuring band intensities, a densitometric graph was developed by using commercial software (AlphaEase, Genetic Technology Inc., Miami, FL, USA) representing control as 100%.
2.6. Caspase-1 Activity Assay
The caspase-1 activity assay was performed at first by scraping RAW264.7 cells in cell lysis buffer and then later adding reaction buffer and YVAD-AFC substrate by following the instructions provided by a commercially available caspase-1 activity assay kit (Abcam, Cambridge, UK).
2.7. Immunofluorescence Staining
Prior to culture, 1 × 104 RAW264.7 cells/well were treated with CoQ0 (10 μM) for 1 h and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h and then seeded in an eight-well glass Tek chamber. Post culture using 2% paraformaldehyde, the cells were fixed for 15 min and then permeabilized with 0.1% Triton X-100 for 10 min and then washed and blocked with 10% FBS in PBS. Primary antibodies of anti-NLRP3 and anti-LC3B were incubated with 1.5% FBS for 2 h and then with fluorescein (FITC) (488 nm)-conjugated secondary antibody for 1 h in 6% bovine serum albumin (BSA). Cells were later stained with 1 μg/mL of DAPI for 5 min and washed with PBS and observed using a confocal microscope (630x magnification) (Leica TCS SP2, Heidelberg, Germany).
2.8. Intracellular ROS Production Measurement
Using DCFH2-DA, ROS that was generated intracellularly was estimated by a fluorescence spectrophotometer as described previously [47]. In brief, 1 × 105 RAW264.7 cells per well were pretreated in a 24-well plate with CoQ0 (10 μM) for 1 h with or without LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h. Later, 10 μM DCFH2-DA was provided to the growth medium and incubated at 37°C for 30 min more. Following incubation, warm PBS washing was done for the cells and using fluorescence microscopy (Olympus, Center Valley, PA, USA); thus, generated ROS was measured by observing the alterations in fluorescence which was caused by DCF production by the oxidation of DCFH2 [48]. The ROS generated was estimated in fold increase compared with the vehicle-treated cells, which were arbitrarily considered as 1-fold.
2.9. Transient siRNA Transfection for LC3B or Nrf2 Silencing
siRNA for silencing LC3B or Nrf2 was transfected in RAW264.7 cells using Lipofectamine RNAiMAX (Invitrogen, Grand Island, NY, USA). In order to carry out transfection, RAW264.7 cells were cultured using a 6-well plate with DMEM having 10% FBS. Before carrying out transfection, the cells were grown to 60% confluence. The culture medium was then replaced with 500 μL of Opti-MEM on the next day, and the cells were transfected with the RNAiMAX transfection reagent. 5 μL RNAiMAX and 250 μL of Opti-MEM were mixed and incubated together at room temperature for 5 min. In another tube, siRNA (100 pM) was prepared and added to the tube containing 250 μL of Opti-MEM. Thus, the obtained mixture was added to the diluted RNAiMAX. The siRNA/RNAiMAX mixture (500 μL) was allowed to incubate 25 min extra at room temperature to form a transfection complex. Thus, the obtained complex was subsequently added to the 6-well plate—making 1 mL as the final transfection volume. 6 h posttransfection, the medium was substituted by 2 mL standard culture medium and grown at 37°C. Ultimately, the cells were incubated along with CoQ0 (10 μM) for 1 h with or without LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h. Expression of LC3-I/II and pro-IL1β was quantified.
2.10. Statistical Analysis
All the results were expressed as the mean ± standard deviation (SD). Analysis of variance (ANOVA) followed by Dunnett's pair-wise comparisons was used for the analysis of all data. ∗p < 0.05, ∗∗p < 0.01, and∗∗∗p < 0.001 when compared with untreated control cells, and #p < 0.05, ##p < 0.01, and ###p < 0.001 when compared with 3-methyladenine (3-MA), or LPS/ATP-stimulated cells were considered to be statistically significant.
3. Results
3.1. The Effects of Coenzyme Q0 (CoQ0) on Cell Viability of RAW264.7 Macrophages
In order to investigate the anti-inflammatory, as well as antioxidant, properties of CoQ0, first of all, the cytotoxic effects of different doses of CoQ0 (Figure 1(a)) ranging from 2.5 to 20 μM on RAW264.7 macrophages were examined. The results of MTT analysis revealed that when treating with CoQ0 for 24 h, there was no obvious effect on the viability of macrophage up to 10 μM concentration, but a significant downregulation was observed with 20 μM treatment (Figure 1(b)). So, depending on this result, the non- or subcytotoxic concentrations of CoQ0, i.e., ≤15 μM, were taken for further carrying out in vitro studies and examining its response on LPS/ATP-induced inflammation and redox imbalance.
Figure 1.
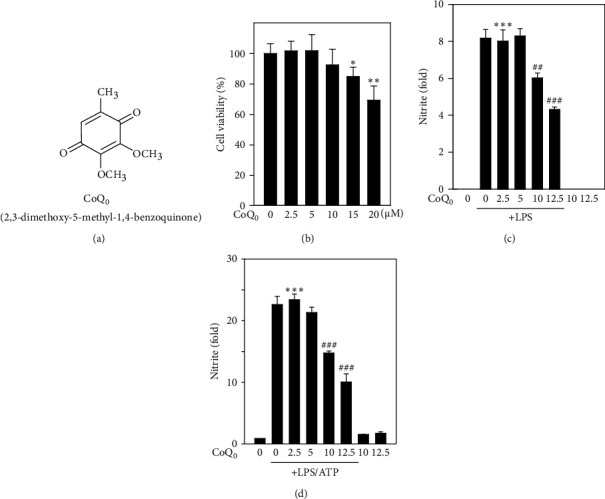
Coenzyme Q0 (CoQ0) suppresses NO production in LPS- or LPS/ATP-stimulated RAW264.7 macrophages. (a) Structure of Coenzyme Q0 (CoQ0, 2,3-dimethoxy-5-methyl-1,4-benzoquinone). (b) MTT assay carried out by treating RAW264.7 cells with CoQ0 (2.5-20 μM) for 24 h. (c, d) Production of NO was estimated by measuring the nitrite formation and stable end-metabolic of NO in the culture medium. Prior to NO estimation, cells were treated with different doses of CoQ0 ranging from 2.5 to 12.5 μM for 1 h, and then, LPS (1 μg/mL) was stimulated for 18 h or LPS (1 μg/mL) for 17 h followed by 5 mM ATP treatment for 1 h. The results were calculated as the mean ± SD of three independent experiments. ∗∗p < 0.01; ∗∗∗p < 0.001, compared with untreated control cells, and ##p < 0.01; ###p < 0.001 compared with LPS or LPS/ATP-stimulated cells assigned as statistically significant.
3.2. CoQ0 Suppresses Production of NO in LPS- or LPS/ATP-Stimulated RAW264.7 Macrophages
To investigate the anti-inflammatory properties of CoQ0, macrophages were first treated with CoQ0 (2.5-12.5 μM) for 1 h and later stimulated either with LPS (1 μg/mL) for 18 h or LPS (1 μg/mL) for (17 h) and then by ATP (5 mM) for 1 h. The results revealed that LPS stimulation alone significantly increased nitric oxide (NO) production in culture medium, but treatment with CoQ0 significantly decreased NO production in a dose-dependent fashion (Figure 1(c)), but this effect was reversed when macrophages were LPS/ATP-stimulated (Figure 1(d)). However, there were no significant changes observed with CoQ0 alone-treated cells (Figures 1(c) and 1(d)).
3.3. CoQ0 Inhibits LPS/ATP-Stimulated IL1β Expression through NLRP3 Inflammasome and Procaspase-1 Activation in RAW264.7 Macrophages
At first, we analyzed whether CoQ0 inhibits activation of NLRP3 inflammasome in RAW264.7 macrophages. For this, we used a well-known in vitro model of NLRP3 inflammasome activation, where ATP propels cleavage of caspase-1 in macrophages that are LPS primed [49]. RAW264.7 cells were pretreated with LPS and later stimulated with ATP along with different concentrations of CoQ0. Here, we investigated whether CoQ0 could repress the LPS/ATP-induced NLRP3 and procaspase-1 activation in macrophages. As anticipated and proven by the Western blot assay, the procaspase-1 was noticeably increased in macrophages stimulated with LPS/ATP corroborating that NLRP3 inflammasome was activated (Figure 2(a)). CoQ0 treatment significantly suppressed NLRP3 and procaspase-1 in a dose-dependent manner (Figure 2(a)), and the reduced activity of caspase-1 was observed (Figure 2(b)). To further prove the anti-inflammatory effects of CoQ0, using the Western blot assay, we examined the expression of pro-IL1β in lysis fractions and mature and pro-IL1β in supernatant fractions of macrophages. Upon LPS/ATP stimulation, there was a significant upregulation in the expression of pro-IL1β in lysis and both mature and pro-IL1β in supernatant fractions which were substantially inhibited by the treatment of CoQ0 (2.5-10 μM, 1 h) in a dose-dependent manner (Figure 2(c)).
Figure 2.
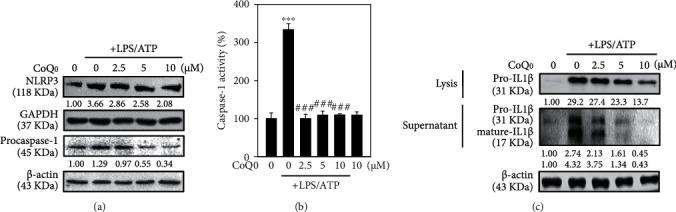
CoQ0 inhibits NLRP3 inflammasome activation in LPS/ATP-stimulated RAW264.7 macrophages. Cells were first treated with CoQ0 (2.5-10 μM) and later stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) treatment for 1 h. (a) The expression of NLRP3 and procaspase-1 protein was determined by Western blotting. (b) Caspase-1 activity was measured using a caspase-1 activity assay kit. (c) Pro-IL1β expressions in both lysis and supernatant and mature-IL1β in the supernatant were determined by Western blot using β-actin as internal control as well as band intensities were calculated by AlphaEaseFCTM (Genetic Technologies, Inc., Florida, USA) software. The data were calculated from the mean ± standard deviation (SD) of three independent experiments and ∗∗∗p < 0.001, compared with untreated control cells, and ###p < 0.001 compared with LPS/ATP-stimulated cells which was significant.
3.4. CoQ0 Increases the Accumulation of LC3-II and Activates Autophagy in LPS/ATP-Stimulated RAW264.7 Macrophages
LC3-II is a well-known marker for autophagy that reveals the lysosomal turnover and autophagy inside the cells, so monitoring of LC3-II is essential to know about autophagy. Methods such as Western blot and immunofluorescence are widely used for detecting autophagy in cells [50]. Thus, we investigated if the inhibition of NLRP3 inflammasome by CoQ0 is regulated by the induction of autophagy. Recently, it has been disclosed that p62/SQSTM1 remains at the interface that links autophagy along with oxidative stress signaling [51]. The expression of p62, which is also known as sequestosome 1 (SQSTM1), is used to measure the autophagy flux [52]. It is responsible for the proteasomal degradation of ubiquitinated proteins and is found all over the cell as well as in various signaling pathways. During autophagy, this protein binds with LC3 through a specific motif and degrades itself [53]. CoQ0 (2.5-10 μg/mL) along with LPS/ATP stimulation in RAW264.7 macrophages and its effect on the expression of LC3-I/II and p62 were explored in this study. Western blot data revealed that with increasing concentrations of CoQ0, LC3-I/II and p62 expression went on increasing (Figure 3(a)). Employing the fluorescence method, accumulation of LC3 in RAW264.7 cells was further investigated. CoQ0 (10 μg/mL) upregulated LC3 without or with LPS/ATP-stimulated RAW264.7 cells which was consistent with Western blot results (Figure 3(b)). This effect was found to be statistically significant and measured approximately 3- or 5-fold without or with LPS/ATP stimulation-treated cells in comparison to control cells (Figure 3(c)). Additionally, to examine the role of CoQ0 (10 μg/mL) mediating autophagy in LPS/ATP-stimulated RAW264.7 cells, 3-MA was used which being a pharmacological inhibitor of autophagy interrupts the lysosomal function throughout autophagy. The fluorescence data revealed that CoQ0 alone treatment exhibited that 3-MA (2.5 mM) inhibited the LC3-II accumulation in the early autophagy (Figures 3(b) and 3(c)). These results suggested that CoQ0 activated autophagy through LC3-II signaling cascades in LPS/ATP-stimulated RAW264.7 macrophages.
Figure 3.
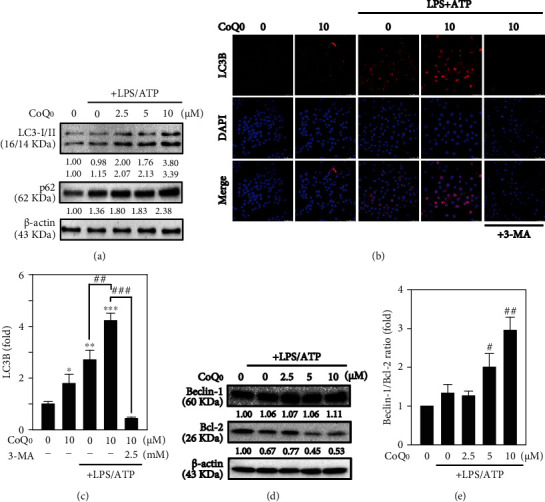
CoQ0 induces autophagy in LPS/ATP-stimulated RAW264.7 macrophages. Cells were first treated with 2.5-10 μM of CoQ0 and/or 2.5 mM 3-MA (autophagy inhibitor) for 1 h and then followed by stimulation of LPS (1 μg/mL) for 5 h and ATP (5 mM) for 1 h. (a) The expression of LC3-I/LC3-II and p62/SQSTM1 protein was estimated by Western blot. (b) The modifications in LC3B expression were observed by immunofluorescence staining. Cells were incubated with anti-LC3B antibody followed by secondary antibody labelled with FITC. 630x magnification of a confocal microscope was used to visualize the subcellular localization of LC3B. (c) Fold changes in LC3B were determined. (d) CoQ0 affects Beclin-1 and Bcl-2 expression in a dose-dependent manner shown by Western blot analysis. (e) Beclin-1/Bcl-2 ratio relative changes were measured by commercial software representing control as 1-fold. The results were calculated as the mean ± SD of three independent experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, compared with untreated control cells, and #p < 0.05; ##p < 0.01; ###p < 0.001 compared with 3-MA- or LPS/ATP-stimulated cells assigned statistically significant.
3.5. CoQ0 Dysregulates Beclin-1 and Bcl-2 Ratio Leading to Autophagy in LPS/ATP-Stimulated RAW264.7 Macrophages
Beclin-1 is one of the important proteins to initiate autophagy which recruits crucial autophagy proteins to a preautophagosomal structure. In addition, Bcl-2 combines with Beclin-1 and decreases its proautophagy profile, but the apoptotic role cannot be neutralized [54]. The effects of various doses of CoQ0 in Beclin-1 and Bcl-2 protein expression were examined through Western blot analysis which demonstrated that CoQ0 (2.5-10 μM for 6 h) dose-dependently suppressed Bcl-2 expression in LPS/ATP-stimulated RAW264.7 macrophages (Figures 3(d) and 3(e)). In contrast, Beclin-1 expression did not show significant changes. Remarkably, due to low Bcl-2 expression, CoQ0 dysregulated Beclin-1 and Bcl-2 protein ratio thus guiding to autophagy (Figures 3(d) and 3(e)).
3.6. CoQ0 Reduces the Phosphorylation of PI3K/AKT, p70 S6 Kinase, and mTOR Expressions Leading to Autophagy in LPS/ATP-Stimulated RAW264.7 Macrophages
The PI3K/AKT/p70 S6 kinase/mTOR signaling pathway is a key regulator and affects the regulation of autophagy [55]. In this study, the effects of CoQ0 on PI3K/AKT/P70 S6 kinase/mTOR expression in LPS/ATP-stimulated RAW264.7 macrophages were investigated. In comparison to untreated control, for CoQ0 (10 μg/mL) treated for 0-60 min, the expression of phosphorylated PI3K (Tyr467/Tyr199), AKT (Ser473), p70 S6 kinase (Thr389), and mTOR (Ser2448) was downregulated in a time-dependent manner (Figure 4(a)). Additionally, for CoQ0 (2.5-10 μg/mL) treated for 60 min, the expression of p70 S6 kinase (Thr389) and AKT (Ser473) was downregulated in a dose-dependent manner (Figure 4(b)). So, the results revealed that the inhibition of PI3K/AKT/p70 S6 kinase/mTOR was due to autophagy activation by CoQ0 in LPS/ATP-induced RAW264.7 macrophages.
Figure 4.
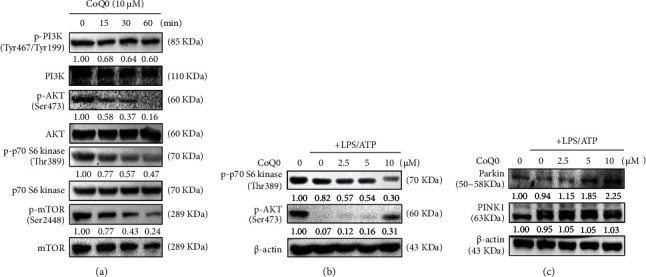
CoQ0 induces mitophagy in LPS/ATP-stimulated RAW264.7 macrophages. (a) Time-dependent expression of p-PI3K, PI3K, p-AKT, AKT, p-p70 S6 kinase, p70 S6 kinase, p-mTOR, and mTOR was determined by Western blot. Cells were first treated with CoQ0 (10 μM) for 0-60 min and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h. (b) Dose-dependent expression of p-p70 S6 kinase and p-AKT was determined by Western blot. Cells were pretreated with CoQ0 (2.5-10 μM) for 60 min and then LPS (1 μg/mL) stimulation for 5 h followed by ATP (5 mM) for 1 h, and the changes in the intensities of protein bands were measured by commercial quantitative software. (c) The cells were first treated with CoQ0 (2.5-10 μM) for 60 min and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h, and lastly, expression of Parkin and PINK1 was determined by Western blot.
3.7. CoQ0 Induces Mitophagy in LPS/ATP-Stimulated RAW264.7 Macrophages
Cells remove defective mitochondria through a special type of autophagy known as mitophagy [56]. Evidence showed that Parkin is recruited from the cytosol to damaged mitochondria to regulate the removal of the damaged organelles [21]. In order to know whether Parkin was recruited into the mitochondria, Western blot analysis was performed. CoQ0 treatment reduced the Parkin expression hence suggesting that it impaired the mitochondria (Figure 4(c)). But, in cells pretreated with CoQ0, there were no significant changes observed in PINK1 expression (Figure 4(c)), thus suggesting that CoQ0 pretreatment upregulated the expression of Parkin protein to recruit the damaged mitochondria.
3.8. CoQ0 Inhibits LPS/ATP-Stimulated NLRP3 Inflammasome Activation through Autophagy Induction in RAW264.7 Macrophages
In order to know if there was a role of autophagy in NLRP3 inflammasome suppression, an autophagy inhibitor, 3-MA, which blocks the formation of autophagosome was supplied to the culture medium. Remarkably, 3-MA reversed the effects of CoQ0 in reducing the expressions of NLRP3, procaspase-1, and pro-IL1β which was significantly upregulated in LPS/ATP-stimulated macrophages due to activation of NLRP3 inflammasome (Figure 5(a)). Furthermore, siLC3B reversed the results of CoQ0 in failing to suppress pro-IL1β expression when autophagy was suppressed by LC3 silencing or knockdown (Figure 5(b)). Hence, the data indicated that autophagy acts as a cell-intrinsic phenomenon to restrict the activation of NLRP3 inflammasome, and by inducing autophagy, CoQ0 potentiates this regulatory mechanism.
Figure 5.
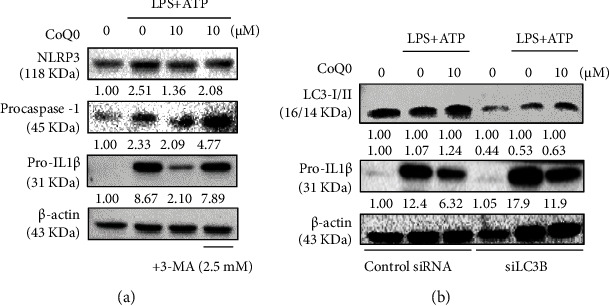
CoQ0 inhibits NLRP3 inflammasome activation through autophagy induction in LPS/ATP-stimulated RAW264.7 macrophages. (a) Cells were pretreated with CoQ0 (10 μM) and/or autophagy inhibitor 3-MA (2.5 mM) for 1 h and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h. NLRP3, procaspase-1, pro-IL1β, and LC3-I/II were determined by Western blot. (b) LC3 knockdown attenuated the protective effects of CoQ0. Cells were first transfected with siRNA that is specific to either LC3B or a nonsilencing control then pretreated with CoQ0 (10 μM) for 1 h and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h, and the expression of LC3-I/II or pro-IL1β proteins in both control and siLC3B was determined using Western blot analysis.
3.9. CoQ0 Attenuates LPS/ATP-Stimulated ROS Generation in RAW264.7 Macrophages
ROS plays a crucial role in the regulation of different inflammatory mediators. The accumulation of LPS-stimulated ROS in macrophages can increase the inflammatory responses [57]. When LPS/ATP was stimulated for 6 h to the RAW264.7 macrophages, it triggered the intracellular ROS generation as shown in Figures 6(a) and 6(b). But treatment with CoQ0 (10 μM) for 1 h earlier to LPS/ATP stimulation significantly reduced the ROS production despite the cells incubated with CoQ0 alone, i.e., without stimulation, revealing that there were no changes in ROS levels when compared to control (Figures 6(a) and 6(b)).
Figure 6.
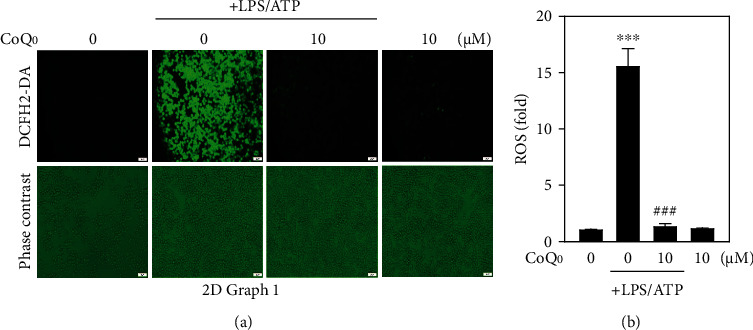
CoQ0 attenuates LPS/ATP-stimulated ROS generation in RAW264.7 macrophages. (a) Cells were pretreated with CoQ0 (10 μM) for 1 h and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h. The level of intracellular ROS was measured by DCF fluorescence using fluorescence microscopy (200x magnification). (b) Data are presented as fold change, and the results were calculated as the mean ± SD of three experiments, where ∗∗∗p < 0.001, compared with untreated control cells, and ###p < 0.001 compared with LPS/ATP-stimulated cells.
3.10. CoQ0 Inhibits ROS-Mediated NLRP3 Inflammasome Activation and IL1β Expression in LPS/ATP-Stimulated RAW264.7 Macrophages
Recent evidence suggests that mitochondria-derived reactive oxygen species (mtROS) are linked to IL1β expression through the Nod-like receptor pyrin domain-containing 3 (NLRP3) inflammasome, which is a redox sensor [58]. NLRP3 inflammasome activation in response to LPS and ATP needs mtROS produced from defective mitochondria, and mitochondrial DNA is released into the cytosol in an NLRP3- and mtROS-dependent manner [59]. The expression of NLRP3 was monitored using a fluorescence microscope. We found that CoQ0 (10 μM), Mito-TEMPO (0.5 mM), and NAC (2 mM) treatment suppressed NLRP3 expression in LPS/ATP-induced RAW264.7 macrophages (Figure 7(a)). Likewise, the expression of pro-IL1β was estimated using Western blot. CoQ0, Mito-TEMPO, and NAC treatment inhibited the expression of pro-IL1β in comparison to LPS/ATP alone treatment. The effects of CoQ0 and Mito-TEMPO were found to be significant in comparison to NAC treatment (Figure 7(b)). These results indicated that ROS including mtROS signaling cascades are involved in the regulation of CoQ0-inhibited NLRP3 inflammasome and pro-IL1β expression.
Figure 7.
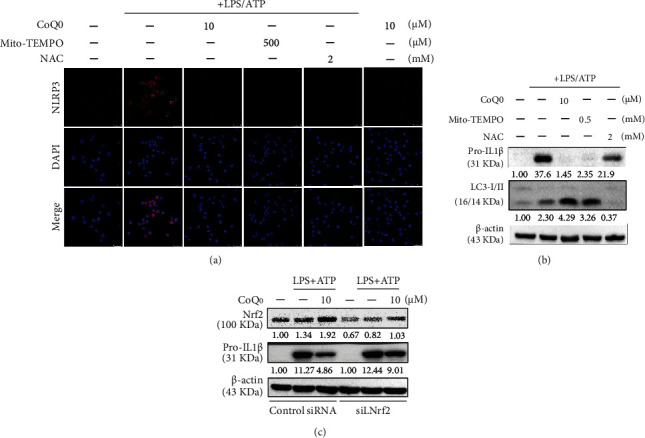
CoQ0 inhibits ROS-mediated NLRP3 inflammasome activation through autophagy induction and Nrf2 activation in LPS/ATP-stimulated RAW264.7 macrophages. Cells were pretreated with CoQ0 (2.5-10 μM), Mito-TEMPO (0.5 mM), or NAC (2 mM) for 1 h and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h. (a) Immunofluorescence staining of RAW264.7 cells and the nuclear localization of NLRP3 were visualized by the immunofluorescence method. Cells were stained with DAPI (1 μg/mL) for 5 min and examined by fluorescence microscopy. (b) The expression of pro-IL1β or LC3-I/II proteins was measured by Western blot analysis. (c) Nrf2 knockdown attenuated the protective effects of CoQ0. Cells were transfected with siRNA that is specific to either Nrf2 or a nonsilencing control. Transfected cells were pretreated with CoQ0 (2.5-10 μM) for 1 h and then stimulated with LPS (1 μg/mL) for 5 h followed by ATP (5 mM) for 1 h, and the expression of Nrf2 or pro-IL1β proteins in both control and siNrf2 was measured by Western blot analysis.
3.11. CoQ0 Inhibits mtROS-Mediated NLRP3 Inflammasome Activation through Mitophagy Induction in LPS/ATP-Stimulated RAW264.7 Macrophages
Mitophagy is a mitochondria-selective autophagic mechanism that exists within cells to remove damaged mitochondria and preserve mitochondrial homeostasis in the face of stress [60]. During mitophagy, mitochondria are engulfed into the vesicles which are coated with autophagosomal marker MAP1 light chain 3 (LC3) [61]. Blocking mitophagy triggers an accumulation of damaged, ROS-producing mitochondria, which activates the NLRP3 inflammasome [49]. In order to confirm the influence of mitophagy, cells were first treated with CoQ0, Mito-TEMPO, or NAC, and then, changes in expression of LC3-II were determined by Western blot. Our analysis depicted that LPS/ATP-induced LC3-II expression was significantly enhanced by CoQ0 and Mito-TEMPO, but this effect was not observed in NAC treatment (Figure 7(b)). These findings exhibited that mitophagy is involved in the regulation of CoQ0-inhibited NLRP3 inflammasome activation and pro-IL1β expression in LPS/ATP-stimulated RAW264.7 macrophages.
3.12. Nrf2 Knockdown Suppresses CoQ0 Mediated Anti-NLRP3 Inflammasome Activation
In our previous study, CoQ0 increased Nrf2 nuclear translocation and Nrf2/ARE-signaling in LPS-stimulated macrophages [29]. Recent studies have revealed that Nrf2 could negatively regulate the activity of NLRP3 inflammasome by suppressing activation of NLRP3 inflammasome induced by ROS [62]. Recently, it has been demonstrated that pro-IL1β is required for autophagic degradation [63]. We addressed this possibility, by quantifying pro-IL1β in LPS/ATP-stimulated lysates of macrophages treated with CoQ0 by Western blot. Cells were first transfected with Nrf2-specific siRNA or a nonsilencing control and posttransfection treated with 10 μM of CoQ0 for 1 h and then stimulated with LPS (1 μg/mL) for 5 h and ATP (5 mM) for 1 h, and the expression of Nrf2 or pro-IL1β proteins in control as well as siNrf2 was estimated via Western blot. When Nrf2 was knocked down using siRNA, it attenuated the protective effects of CoQ0. CoQ0 (10 μM) treatment decreased the level of pro-IL1β in macrophages when Nrf2 is silenced in comparison to control siRNA (Figure 7(c)) suggesting that CoQ0-mediated anti-NLRP3 inflammasome activation was suppressed because of Nrf2 knockdown.
4. Discussions
Earlier, we have shown that CoQ0 (2,3-dimethoxy-5-methyl-1,4-benzoquinone) regulated NFκB/AP-1 activation and enhanced Nrf2 stabilization in the attenuation of LPS-induced inflammation and redox imbalance [29]. The noncytotoxic concentrations of CoQ0 (2.5-10 μM) inhibited iNOS/COX-2 protein expression in LPS-stimulated macrophages, resulting in lower NO, PGE2, INF, and IL1 secretions. Moreover, in LPS-stimulated macrophages, CoQ0 induced HO-1 and NQO-1 gene expression by increasing Nrf2 nuclear translocation and Nrf2/ARE signaling [29]. To our knowledge, this is the first study that CoQ0 inhibited ROS-mediated NLRP3 inflammasome activation through mitophagy induction and Nrf2 activation in LPS/ATP-stimulated macrophages.
LPS is a potent activator of monocytes and macrophages that triggers the secretion of several proinflammatory cytokines [37]. We first tested if CoQ0 could exhibit an anti-inflammatory effect on LPS/ATP-stimulated macrophages. Data suggested that CoQ0 dose-dependently and significantly decreased NO production and inferred the protective effect of CoQ0 on macrophages. The inflammasome NLRP3 is a multiprotein complex that regulates caspase-1 activation and assists in releasing proinflammatory cytokine IL1β [64, 65], one of the most characterized cytokines known to play an important role in autoimmune diseases [64, 65]. Our data revealed that LPS/ATP alone-induced macrophage showed the increased expression of NLRP3 inflammasome and procaspase-1; however, CoQ0 treatment could repress the LPS/ATP-induced NLRP3 and procaspase-1 activation. Caspase-1 also called as an interleukin-1β converting enzyme (ICE) is a proteolytic enzyme that processes the mature form of the inactive precursor IL1β [66, 67]. The inflammasome assembly of NLRP3 and subsequent self-processing of proteolytics will activate caspase-1 itself as an inactive cytoplasm precursor [68]. The caspase-1 activity measurement assay revealed that LPS/ATP stimulation on macrophages increased caspase activation, which was then decreased by CoQ0 treatment. Likewise, increased expression of pro-IL1β and mature IL1β in lysis and supernatant and pro-IL1β in lysis was observed in LPS/ATP-stimulated macrophages. However, this effect was found to be downregulated with increasing dose treatment of CoQ0 thus suggesting that CoQ0 can inhibit LPS/ATP-stimulated NLRP3 inflammasome activation in RAW264.7 macrophages. Our data revealed that CoQ0 regulates the activation of macrophages by enhancing a negative regulatory loop amongst NLRP3 inflammasome.
As LC3 is a promising autophagy marker [69], its intracellular distribution has been examined to find out whether CoQ0 can induce autophagy in LPS/ATP-stimulated RAW264.7 macrophages. Our results revealed that the accumulation of LC3-I and LC3-II was dose-dependently increased following CoQ0 treatment. The high dose (10 μM) of CoQ0 notably increased with the high accumulation of LC3-II which suggested that CoQ0 activated autophagy through LC3-II signaling cascades in LPS/ATP-stimulated RAW264.7 macrophages. Recently, p62/SQSTM1 is found to be at the interface that links autophagy as well as oxidative stress [51]. p62 or sequestosome 1 (SQSTM1) is an important protein that binds directly to LC3 and then undergoes self-degradation while autophagy occurs [70]. Being a multifunctional ubiquitin-binding protein, p62/SQSTM1 involves many important processes of autophagy [71]. p62/SQSTM1 plays a crucial part in the oxidative stress response pathway through its direct association with the ubiquitin ligase adaptor Kelch-like ECH-associated protein 1 (Keap-1), resulting in Nrf2 activation [51]. p62/SQSTM1 being an autophagy adaptor combines with protein aggregates which are ubiquitinated and convey them into the autophagosome thus enhancing selective autophagy [72]. Furthermore, p62/SQSTM1 in recent time appeared as a mediator of the Nrf2-Keap-1-ARE (antioxidant response element) axis by competing with the relationship between Nrf2 and Keap-1 as well as activating Nrf2, the target genes of which are antioxidant proteins and detoxification enzymes [73]. From our results, it was known that the expression level of p62/SQSTM1 remarkably increased with CoQ0 (2.5-10 μM) incubation after 24 h in a dose-dependent fashion suggesting that CoQ0 induced autophagy in LPS/ATP-stimulated RAW264.7 macrophages. The increase in p62/SQSTM1 levels was linked with the increasing accumulation of LC3 in RAW264.7 macrophages, and p62/SQSTM1 had an important role in regulating the CoQ0 action in suppression of NLRP3 inflammasome.
The proteins of the Bcl-2 family serve as key regulators of mitochondrial-mediated apoptosis and work as either activators or inhibitors [74]. The relationship between Beclin-1 and Bcl-2 is complex, and the Beclin-1 proautophagic property can be reduced by Bcl-2 [75]. Hence, the effect of CoQ0 on Bcl-2 protein and its function in Beclin-1 expression in LPS/ATP-stimulated RAW264.7 macrophages was studied. Western blotting data revealed that Beclin-1 proteins dramatically increased with CoQ0 in a dose-dependent manner. In contrast, Bcl-2 expression was suppressed with CoQ0. The increase in the ratio of Beclin-1/Bcl-2 with increasing concentration of CoQ0 suggested that there was an autophagy induction in LPS/ATP-stimulated macrophages. Pretreatment of cells with 3-MA (2.5 mM) successfully decreased LC3B indicating that autophagy was caused by CoQ0.
Later, we demonstrated the signaling pathways leading to the CoQ0-mediated effects on LPS/ATP-stimulated RAW264.7 macrophages. The PI3K/AKT, p70 S6 kinase, and mTOR pathways are critical for inflammation and different diseases [76]. mTOR collaborates with PI3K effectors to phosphorylate the p70 S6 kinase which is associated with protein translation of an mRNA transcript family that encodes the fundamental components of protein synthesis apparatus [77]. Our data indicated that CoQ0 (10 μM) time- and dose-dependently downregulated the phosphorylations of PI3K (Tyr467/Tyr199)/AKT (Ser473), p70 S6 kinase (Thr389), and/or mTOR (Ser 2448) proteins in LPS/ATP-stimulated cells inferring that autophagy plays a critical role in CoQ0-mediated effects on RAW264.7 macrophages (Figure 4).
Using a specialized type of autophagy also called mitophagy, cells eliminate defective mitochondria [56]. Evidence has demonstrated that Parkin is recruited from the cytosol to depolarized mitochondria to guide the elimination of the damaged organelles also known as selective autophagy or mitophagy [21]. The PINK1 and E3 ubiquitin ligase Parkin pathway mediate the removal of damaged mitochondria. Accumulation of Parkin and PINK1 takes place in impaired mitochondria to boost their segregation from the mitochondrial network and target these organelles for autophagic degradation in a process that involves ubiquitination of Parkin-dependent mitochondrial proteins [56]. Our analysis showed that Parkin expression was reduced by the treatment of CoQ0 indicating that it impaired the mitochondria. However, there were no significant changes observed in PINK1 expression when pretreated with CoQ0 thus suggesting that CoQ0 pretreatment upregulated the expression of Parkin protein to recruit the damaged mitochondria. We exhibited that CoQ0 has mitigated LPS/ATP-induced inflammation along with redox imbalance in RAW264.7 macrophages. This result was similar to our previous study which showed that induction of LPS in RAW264.7 cells increased levels of ROS [57]. To know whether there was any function of autophagy in NLRP3 inflammasome suppression, we treated the LPS/ATP-stimulated RAW264.7 macrophages with autophagy inhibitor (3-MA) which blocks autophagosome formation. Notably, 3-MA reversed the effects of CoQ0 on reducing the NLRP3 inflammasome, procaspase-1, and pro-IL1β expression. Additionally, when autophagy was suppressed by LC3 silencing, siLC3 reversed the results of CoQ0 in failing to suppress pro-IL1β expression. So, from this result, we came to know that mitophagy acts as a cell-intrinsic phenomenon to restrict NLRP3 inflammasome activation and CoQ0 potentiates this regulatory mechanism through mitophagy induction.
Immune cells such as monocytes and macrophages respond first to any injury in tissue by recognizing danger signals and triggering the inflammatory process [78]. Evidence showed that cell-autonomous regulatory feedback loops are employed in NLRP3 inflammasome regulation, of which autophagy is the most dominant. Former studies have manifested that NLRP3 inflammasome activation is limited by the induction of autophagy of inflammatory signals because of the removal of damaged mitochondria and prevention of mitochondrial ROS release [49]. Consistent with this existing evidence, our study revealed that a noncytotoxic concentration of CoQ0 is capable of suppressing intracellular ROS generation and NLRP3 inflammasome against LPS/ATP stimulation in RAW264.7 macrophages. Stress and inflammatory conditions are associated with both the Nrf2 transcription factor and NLRP3 inflammasome, while inflammatory activation of NLRP3 induces inflammation and eventually death of inflammation-activating cells. Nrf2 activation promotes cell survival and prevents inflammation [17]. Nrf2, a transcription factor, activators result in cytoprotective proteins and enzyme expression that, under stress conditions, facilitates the survival of Nrf2-activating cells [17]. In response to LPS and ATP, NLRP3 inflammasome activation requires mitochondrial ROS generated from dysfunctional mitochondria, and mitochondrial DNA is secreted into the cytosol in both NLRP3 and mitochondrial ROS-dependent ways [59]. Mito-TEMPO, which is a mitochondria-specific ROS scavenger, suppressed both IL1β secretion after nigericin or ATP exposure [79]. In our study, when LPS/ATP-stimulated RAW264.7 macrophages were treated with CoQ0, Mito-TEMPO, and NAC, a significant reduction of LPS/ATP-stimulated NLRP3 inflammasome activation and pro-IL1β expression was observed which indicates that ROS signaling cascades are involved in CoQ0-inhibited NLRP3 inflammasome activation and pro-IL1β expression. Surprisingly, when LPS/ATP-stimulated RAW264.7 macrophages were treated with CoQ0 or Mito-TEMPO, but not NAC, there is significantly increased LPS/ATP-induced LC3-II accumulation indicating the occurrence of mitophagy. Following LPS/ATP stimulation, Nrf2 knockdown significantly decreased pro-IL1β expression in RAW264.7 macrophages indicating that CoQ0 inhibited ROS-mediated NLRP3 inflammasome activation and pro-IL1β expression was suppressed due to the Nrf2 activation (Figure 7). From the above, we proposed that CoQ0 negatively regulates macrophage activation through mitophagy induction and Nrf2 pathways that hinder a positive feedback loop of NLRP3 inflammasome mechanisms.
5. Conclusions
In conclusion, our findings demonstrated that non- or subcytotoxic doses of CoQ0 exhibited anti-inflammatory and antioxidant properties (Figure 8). CoQ0 suppressed the NLRP3 inflammasome and procaspase-1 activation which in turn downregulated pro-IL1β expression levels. Accumulation of LC3-II, p62/SQSTM1, and AVO as well as dysregulation of Beclin1/Bcl-2 showed that CoQ0 treatment in LPS/ATP-stimulated macrophages induced autophagy. Additionally, CoQ0 reduced the expression of phosphorylated PI3K/AKT, p70 S6 kinase, and mTOR thus leading to autophagy. Besides, CoQ0 increased Parkin protein to recruit damaged mitochondria and induced mitophagy in LPS/ATP-stimulated RAW264.7 macrophages. Similarly, CoQ0 inhibited ROS-mediated NLRP3 inflammasome activation through mitophagy induction and Nrf2 activation in LPS/ATP-stimulated macrophages. Hence, this study showed that CoQ0 might be a promising candidate for the therapeutics of inflammatory disorders due to its effective anti-inflammatory as well as antioxidant properties.
Figure 8.
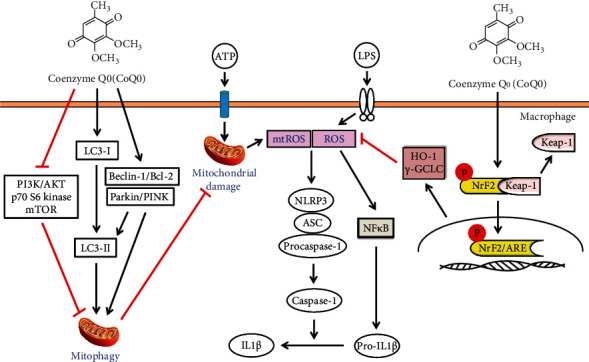
Graphical summary. Collectively, our results showed that subcytotoxic treatments of macrophages with CoQ0 displayed antioxidant and anti-inflammatory properties. CoQ0 inhibited the NLRP3 inflammasome and procaspase-1 activation which in turn suppressed pro-IL1β expression levels. On the other hand, CoQ0 exposure in LPS/ATP-stimulated macrophages incited autophagy which was evident by the accumulation of LC3-II and p62/SQSTM1 as well as dysregulation of Beclin1/Bcl-2. This was accompanied by reduced phosphorylation of PI3K/AKT, p70 S6 kinase, and mTOR. Besides, CoQ0 inhibited ROS-mediated NLRP3 inflammasome activation through mitophagy induction and Nrf2 activation in LPS/ATP-stimulated macrophages. Altogether, we propose that CoQ0 might be a promising candidate for the therapeutics of inflammatory abnormalities due to its effective anti-inflammatory as well as antioxidant properties.
Acknowledgments
This study was supported by the Ministry of Science and Technology, Asia University, and China Medical University, Taiwan (grants MOST-109-2320-B-039-057-MY3, MOST-107-2320-B-039-013-MY3, and CMU109-ASIA-10). This work was financially aided by the “Chinese Medicine Research Center, China Medical University,” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan (CMRC-CHM-1).
Data Availability
All the data used to support the findings of this study are included within the article and the supplementary information file(s), and they are available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
Y.C.H. worked on conceptualization, investigation, data curation, validation, writing of original draft, funding acquisition, and final manuscript checking. Y.F.T. worked on investigation and data curation. S.P. worked on data curation and review and editing of the manuscript. S.S. worked on investigation, data curation, and writing of the original draft. K.Y.L. worked on investigation and data curation. C.W.L. worked on investigation and data curation. C.C.L. worked on investigation and data curation. S.T.H. worked on investigation and data curation. H.L.Y. worked on conceptualization, experimental design, investigation, validation, writing of original draft, review and editing of the manuscript, and funding acquisition.
Supplementary Materials
The file contains original raw images of Western blot figures in the manuscript.
References
- 1.Inoue M., Shinohara M. L. The role of interferon-β in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis–in the perspective of inflammasomes. Immunology . 2013;139(1):11–18. doi: 10.1111/imm.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhong Y., Kinio A., Saleh M. Functions of NOD-like receptors in human diseases. Frontiers in Immunology . 2013;4:p. 333. doi: 10.3389/fimmu.2013.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKee C. M., Coll R. C. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. Journal of Leukocyte Biology . 2020;108(3):937–952. doi: 10.1002/JLB.3MR0720-513R. [DOI] [PubMed] [Google Scholar]
- 4.Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature . 2006;440(7081):237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 5.Duewell P., Kono H., Rayner K. J., et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature . 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolb R., Liu G. H., Janowski A. M., Sutterwala F. S., Zhang W. Inflammasomes in cancer: a double-edged sword. Protein & Cell . 2014;5(1):12–20. doi: 10.1007/s13238-013-0001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zahid A., Li B., Kombe A. J. K., Jin T., Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Frontiers in Immunology . 2019;10:p. 2538. doi: 10.3389/fimmu.2019.02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gump J. M., Thorburn A. Autophagy and apoptosis: what is the connection? Trends in Cell Biology . 2011;21(7):387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine B., Yuan J. Autophagy in cell death: an innocent convict? The Journal of Clinical Investigation . 2005;115(10):2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu L., Wan F., Dutta S., et al. Autophagic programmed cell death by selective catalase degradation. Proceedings of the National Academy of Sciences of the United States of America . 2006;103(13):4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yen W. L., Klionsky D. J. How to live long and prosper: autophagy, mitochondria, and aging. Physiology . 2008;23(5):248–262. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]
- 12.Mathew R., Karantza-Wadsworth V., White E. Role of autophagy in cancer. Nature Reviews Cancer . 2007;7(12):961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong Z., Sanchez-Lopez E., Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell . 2016;166(2):288–298. doi: 10.1016/j.cell.2016.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian M., Fang X., Wang X. Autophagy and inflammation. Clinical and Translational Medicine . 2017;6(1):1–11. doi: 10.1186/s40169-017-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biasizzo M., Kopitar-Jerala N. Interplay between NLRP3 Inflammasome and autophagy. Frontiers in Immunology . 2020;11:p. 2470. doi: 10.3389/fimmu.2020.591803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tonelli C., Chio I. I. C., Tuveson D. A. Transcriptional regulation by Nrf 2. Antioxidants & Redox Signaling . 2018;29(17):1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hennig P., Garstkiewicz M., Grossi S., di Filippo M., French L., Beer H. D. The crosstalk between Nrf2 and inflammasomes. International Journal of Molecular Sciences . 2018;19(2):p. 562. doi: 10.3390/ijms19020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding W. X., Yin X. M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biological Chemistry . 2012;393(7):547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubli D. A., Gustafsson Å. B. Mitochondria and mitophagy: the yin and yang of cell death control. Circulation Research . 2012;111(9):1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarraf S. A., Raman M., Guarani-Pereira V., et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature . 2013;496(7445):372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narendra D., Walker J. E., Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harbor Perspectives in Biology . 2012;4(11, article a011338) doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narendra D., Tanaka A., Suen D. F., Youle R. J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of Cell Biology . 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin S. M., Youle R. J. PINK1-and Parkin-mediated mitophagy at a glance. Journal of Cell Science . 2012;125(4):795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorn G. W., Kitsis R. N. The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circulation Research . 2015;116(1):167–182. doi: 10.1161/CIRCRESAHA.116.303554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochimica et Biophysica Acta (BBA) - Biomembranes . 2004;1660(1-2):171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 26.Quinzii C. M., Hirano M. Coenzyme Q and mitochondrial disease. Developmental Disabilities Research Reviews . 2010;16(2):183–188. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan B. L., Norhaizan M. E., Liew W. P. P., Sulaiman Rahman H. Antioxidant and oxidative stress: a mutual interplay in age-related diseases. Frontiers in Pharmacology . 2018;9:p. 1162. doi: 10.3389/fphar.2018.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentinger M., Brismar K., Dallner G. The antioxidant role of coenzyme Q. Mitochondrion . 2007;7:S41–S50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Yang H. L., Lin M. W., Korivi M., et al. Coenzyme Q0 regulates NFκB/AP-1 activation and enhances Nrf2 stabilization in attenuation of LPS-induced inflammation and redox imbalance: Evidence from in vitro and in vivo studies. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms . 2016;1859(2):246–261. doi: 10.1016/j.bbagrm.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Devun F., Walter L., Belliere J., Cottet-Rousselle C., Leverve X., Fontaine E. Ubiquinone analogs: a mitochondrial permeability transition pore-dependent pathway to selective cell death. PLoS One . 2010;5(7, article e11792) doi: 10.1371/journal.pone.0011792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Somers-Edgar T. J., Rosengren R. J. Coenzyme Q0 induces apoptosis and modulates the cell cycle in estrogen receptor negative breast cancer cells. Anti-Cancer Drugs . 2009;20(1):33–40. doi: 10.1097/CAD.0b013e328314b5c5. [DOI] [PubMed] [Google Scholar]
- 32.Chung C. H., Yeh S. C., Chen C. J., Lee K. T. Coenzyme Q0 from Antrodia cinnamomea in submerged cultures induces reactive oxygen species-mediated apoptosis in A549 human lung cancer cells. Evidence-based Complementary and Alternative Medicine . 2014;2014:10. doi: 10.1155/2014/246748.246748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H. M., Yang H. L., Thiyagarajan V., et al. Coenzyme Q0 Enhances Ultraviolet B–Induced Apoptosis in Human Estrogen Receptor–Positive Breast (MCF-7) Cancer Cells. Integrative Cancer Therapies . 2017;16(3):385–396. doi: 10.1177/1534735416673907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H. L., Korivi M., Lin M. W., Chen S. C., Chou C. W., Hseu Y. C. Anti-angiogenic properties of coenzyme Q0 through downregulation of MMP-9/NF-κB and upregulation of HO-1 signaling in TNF-α- activated human endothelial cells. Biochemical Pharmacology . 2015;98(1):144–156. doi: 10.1016/j.bcp.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Chou W. L., Lee T. H., Huang T. H., et al. Coenzyme Q0 from Antrodia cinnamomea exhibits drug-resistant bacteria eradication and keratinocyte inflammation mitigation to ameliorate infected atopic dermatitis in mouse. Frontiers in Pharmacology . 2019;10:p. 1445. doi: 10.3389/fphar.2019.01445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanlioglu S., Williams C. M., Samavati L., et al. Lipopolysaccharide Induces Rac1-dependent Reactive Oxygen Species Formation and Coordinates Tumor Necrosis Factor-α Secretion through IKK Regulation of NF-κB. Journal of Biological Chemistry . 2001;276(32):30188–30198. doi: 10.1074/jbc.M102061200. [DOI] [PubMed] [Google Scholar]
- 37.Meng F., Lowell C. A. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. Journal of Experimental Medicine . 1997;185(9):1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu H. Y., Wen M. H. Lipopolysaccharide-mediated Reactive Oxygen Species and Signal Transduction in the Regulation of Interleukin-1 Gene Expression. Journal of Biological Chemistry . 2002;277(25):22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- 39.Hambleton J., Weinstein S. L., Lem L., DeFranco A. L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proceedings of the National Academy of Sciences of the United States of America . 1996;93(7):2774–2778. doi: 10.1073/pnas.93.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin T. R. Recognition of bacterial endotoxin in the lungs. American Journal of Respiratory Cell and Molecular Biology . 2000;23(2):128–132. doi: 10.1165/ajrcmb.23.2.f189. [DOI] [PubMed] [Google Scholar]
- 41.Valko M., Rhodes C. J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions . 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 42.Stagg J., Smyth M. J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene . 2010;29(39):5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 43.Mariathasan S., Weiss D. S., Newton K., et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature . 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi M., Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Advances in Enzyme Regulation . 2006;46(1):113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 45.Ma Q. Role of nrf 2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology . 2013;53(1):401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alcaraz M. J., Vicente A. M., Araico A., Dominguez J. N., Terencio M. C., Ferrándiz M. L. Role of nuclear factor-κB and heme oxygenase-1 in the mechanism of action of an anti-inflammatory chalcone derivative in RAW 264.7 cells. British Journal of Pharmacology . 2004;142(7):1191–1199. doi: 10.1038/sj.bjp.0705821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hseu Y. C., Lee M. S., Wu C. R., et al. The chalcone flavokawain B induces G2/M cell-cycle arrest and apoptosis in human oral carcinoma HSC-3 cells through the intracellular ROS generation and downregulation of the Akt/p 38 MAPK signaling pathway. Journal of Agricultural and Food Chemistry . 2012;60(9):2385–2397. doi: 10.1021/jf205053r. [DOI] [PubMed] [Google Scholar]
- 48.Wang H., Joseph J. A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical Biology and Medicine . 1999;27(5-6):612–616. doi: 10.1016/S0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- 49.Zhou R., Yazdi A. S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature . 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Z., Singh R., Aschner M. Methods for the detection of autophagy in mammalian cells. Current Protocols in Toxicology . 2016;69(1) doi: 10.1002/cptx.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nezis I. P., Stenmark H. P 62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxidants & Redox Signaling . 2012;17(5):786–793. doi: 10.1089/ars.2011.4394. [DOI] [PubMed] [Google Scholar]
- 52.Puissant A., Fenouille N., Auberger P. When autophagy meets cancer through p62/SQSTM1. American Journal of Cancer Research . 2012;2(4):397–413. [PMC free article] [PubMed] [Google Scholar]
- 53.Tanida I., Waguri S. Measurement of autophagy in cells and tissues. In: Bross P., Gregersen N., editors. Protein Misfolding and Cellular Stress in Disease and Aging . Vol. 648. Totowa, NJ: Humana Press; 2010. pp. 193–214. (Methods in Molecular Biology (Methods and Protocols)). [DOI] [PubMed] [Google Scholar]
- 54.Menon M. B., Dhamija S. Beclin 1 phosphorylation–at the center of autophagy regulation. Frontiers in Cell and Developmental Biology . 2018;6:p. 137. doi: 10.3389/fcell.2018.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang H., Liu Y., Wang D., et al. The upstream pathway of mTOR-mediated autophagy in liver diseases. Cell . 2019;8(12):p. 1597. doi: 10.3390/cells8121597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashrafi G., Schwarz T. L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death & Differentiation . 2013;20(1):31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang H. L., Lin S. W., Lee C. C., et al. Induction of Nrf2-mediated genes by Antrodia salmonea inhibits ROS generation and inflammatory effects in lipopolysaccharide-stimulated RAW264. 7 macrophages. Food & Function . 2015;6(1):229–240. doi: 10.1039/C4FO00869C. [DOI] [PubMed] [Google Scholar]
- 58.Wei P., Yang F., Zheng Q., Tang W., Li J. The potential role of the NLRP3 inflammasome activation as a link between mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Frontiers in Cellular Neuroscience . 2019;13:p. 73. doi: 10.3389/fncel.2019.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakahira K., Haspel J. A., Rathinam V. A., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology . 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim M. J., Yoon J. H., Ryu J. H. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Reports . 2016;49(10):529–535. doi: 10.5483/BMBRep.2016.49.10.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim I., Rodriguez-Enriquez S., Lemasters J. J. Selective degradation of mitochondria by mitophagy. Archives of Biochemistry and Biophysics . 2007;462(2):245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu X., Zhang X., Ding Y., et al. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxidants & Redox Signaling . 2017;26(1):28–43. doi: 10.1089/ars.2015.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harris J., Hartman M., Roche C., et al. Autophagy Controls IL-1β Secretion by Targeting Pro-IL-1β for Degradation. Journal of Biological Chemistry . 2011;286(11):9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van de Veerdonk F. L., Netea M. G. New insights in the immunobiology of IL-1 family members. Frontiers in Immunology . 2013;4:p. 167. doi: 10.3389/fimmu.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qi J., Ye X., Ren G., et al. Pharmacological efficacy of anti-IL-1β scFv, Fab and full-length antibodies in treatment of rheumatoid arthritis. Molecular Immunology . 2014;57(2):59–65. doi: 10.1016/j.molimm.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 66.Franchi L., Eigenbrod T., Muñoz-Planillo R., Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nature Immunology . 2009;10(3):241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bauernfeind F. G., Horvath G., Stutz A., et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. The Journal of Immunology . 2009;183(2):787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Latz E. The inflammasomes: mechanisms of activation and function. Current Opinion in Immunology . 2010;22(1):28–33. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kabeya Y., Mizushima N., Ueno T., et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO Journal . 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ichimura Y., Kumanomidou T., Sou Y. S., et al. Structural Basis for Sorting Mechanism of p62 in Selective Autophagy. Journal of Biological Chemistry . 2008;283(33):22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y., Li Q., Li Q., et al. P62/SQSTM1, a central but unexploited target: advances in its physiological/pathogenic functions and small molecular modulators. Journal of Medicinal Chemistry . 2020;63(18):10135–10157. doi: 10.1021/acs.jmedchem.9b02038. [DOI] [PubMed] [Google Scholar]
- 72.Liu W. J., Ye L., Huang W. F., et al. P62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cellular & Molecular Biology Letters . 2016;21(1):1–14. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jain A., Lamark T., Sjøttem E., et al. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. Journal of Biological Chemistry . 2010;285(29):22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shamas-Din A., Kale J., Leber B., Andrews D. W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harbor Perspectives in Biology . 2013;5(4, article a008714) doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marquez R. T., Xu L. Bcl-2: Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. American Journal of Cancer Research . 2012;2(2):214–221. [PMC free article] [PubMed] [Google Scholar]
- 76.Babchia N., Calipel A., Mouriaux F., Faussat A. M., Mascarelli F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: interaction with B-Raf/ERK. Investigative Ophthalmology & Visual Science . 2010;51(1):421–429. doi: 10.1167/iovs.09-3974. [DOI] [PubMed] [Google Scholar]
- 77.Pullen N., Dennis P. B., Andjelkovic M., et al. Phosphorylation and activation of p70s6kby PDK1. Science . 1998;279(5351):707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 78.Chiu S., Bharat A. Role of monocytes and macrophages in regulating immune response following lung transplantation. Current Opinion in Organ Transplantation . 2016;21(3):239–245. doi: 10.1097/MOT.0000000000000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heid M. E., Keyel P. A., Kamga C., Shiva S., Watkins S. C., Salter R. D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. The Journal of Immunology . 2013;191(10):5230–5238. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The file contains original raw images of Western blot figures in the manuscript.
Data Availability Statement
All the data used to support the findings of this study are included within the article and the supplementary information file(s), and they are available from the corresponding author upon request.