

Keywords: energy balance, gastrointestinal tract, motor control, obesity, vagus nerve
Abstract
During the past 30 yr, investigating the physiology of eating behaviors has generated a truly vast literature. This is fueled in part by a dramatic increase in obesity and its comorbidities that has coincided with an ever increasing sophistication of genetically based manipulations. These techniques have produced results with a remarkable degree of cell specificity, particularly at the cell signaling level, and have played a lead role in advancing the field. However, putting these findings into a brain-wide context that connects physiological signals and neurons to behavior and somatic physiology requires a thorough consideration of neuronal connections: a field that has also seen an extraordinary technological revolution. Our goal is to present a comprehensive and balanced assessment of how physiological signals associated with energy homeostasis interact at many brain levels to control eating behaviors. A major theme is that these signals engage sets of interacting neural networks throughout the brain that are defined by specific neural connections. We begin by discussing some fundamental concepts, including ones that still engender vigorous debate, that provide the necessary frameworks for understanding how the brain controls meal initiation and termination. These include key word definitions, ATP availability as the pivotal regulated variable in energy homeostasis, neuropeptide signaling, homeostatic and hedonic eating, and meal structure. Within this context, we discuss network models of how key regions in the endbrain (or telencephalon), hypothalamus, hindbrain, medulla, vagus nerve, and spinal cord work together with the gastrointestinal tract to enable the complex motor events that permit animals to eat in diverse situations.
CLINICAL HIGHLIGHTS
How eating behaviors are controlled by physiological systems is at the heart of understanding the etiologies of metabolic diseases. This review addresses the way physiological signals from the gastroinstestinal tract, adipose tissue, pancreas, etc. engage sets of interacting neural networks located throughout the brain to enable the complex motor events that lead animals to eat. A deeper understanding of how the brain is organized to control eating behaviors in a variety in diverse situations should help guide future investigations into conditions where aberrant eating leads to disease.
1. INTRODUCTION
1.1. Preface
Given the numerous reviews in the literature that deal with eating, food intake, and body weight, why do we need another? This review aims in a different direction from most others written in the past few years. Rather than focusing on the signal integration that occurs at the cellular and intracellular levels by way of genes, enzymes, receptors, etc., our goal is to use an integrative, systems-wide approach that considers the brain and periphery collectively. How are they structurally linked, and how do they work together? Rather than simply relating the effects of manipulations to food intake, we give primacy to understanding how physiological signals contribute to the different motor actions that constitute eating behavior. We identify these signals and then discuss how the brain uses them to modify the motor components of eating: foraging, approach behavior, and then direct interactions with food. This integration not only leads to behaviors that can fulfil immediate energy requirements but also ones that anticipate future energy demands. However, the complexity of the brain mechanisms that control eating can also generate behaviors that are uncoupled from energy requirements. Eating behaviors expressed in these circumstances can lead to obesity and other metabolic complications.
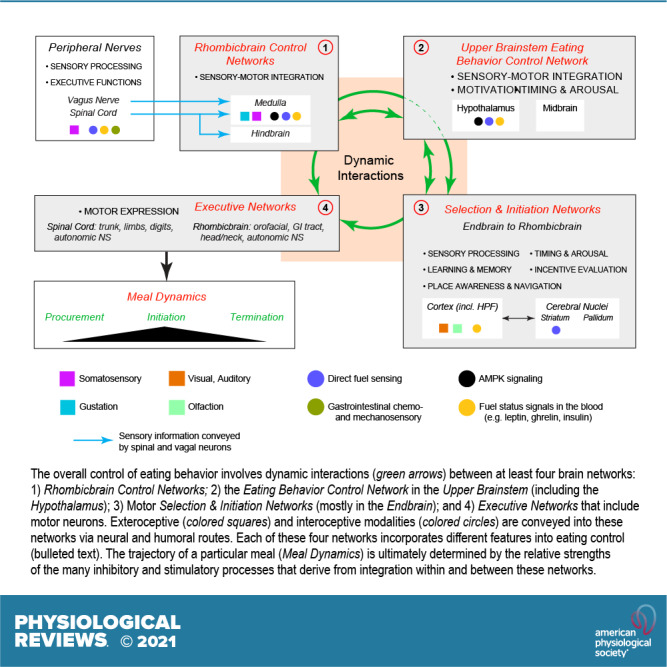
Technology has advanced to allow the targeted manipulation of rodent genes to determine which proteins and cellular control processes contribute to eating behaviors: the role of signals, receptors, transcription factors, enzymes, and their phosphorylation states, etc. However, because these types of results are limited to specific cell types or brain regions, they can only take us so far in understanding how physiological signals control eating behaviors. Ultimately, these signals determine how the various cell components (neurons, glia, tanycytes, etc.) in certain brain regions interact with their downstream targets in the networks that organize eating behaviors. Of particular importance are two eating control networks that we identify in the rhombicbrain and upper brainstem. Understanding the organization of these networks is therefore essential for assembling a bigger picture framework for the neurobiological control of eating behaviors.
To help define these brain networks, a revolution similar to the one that has enabled genetically targeted manipulations has occurred in neuroanatomy. This is being driven by two advances: 1) the use of increasingly sophisticated chemical and viral tract tracers (1–3); and 2) the application of network analyses to determine the organization of the vast numbers of connections already identified in the rodent brain (4–6). Consequently, we believe that understanding how the brain controls eating behaviors must include careful consideration of its connectional organization. Given the depth of our current knowledge about these connections, a central theme of this review is that connectional complexity cannot be ignored when interpreting functional results. We cannot understand the sophistication of eating behavior control without taking into account the structure-function relationships within the brain-wide networks that connect physiological signals to behavior. These considerations have yet to be fully incorporated into most functional models.
A central consideration is how the interactions between the body’s physiological signals and key brain networks, including their transmitters and neuromodulators, engage and coordinate eating-associated behavioral motor events. We take a network approach with the belief that considering the constituent neurons of hypothalamic, rhombicbrain, or indeed any other brain region as stand-alone control units or “centers,” to use an outdated and rather imprecise term, cannot fully account for the way the brain integrates different sensory modalities to produce appropriate motor actions (see also Refs. 7–9). This problem was usefully summarized many years ago (7, 10). In short, interpreting functional studies of neurons without considering their connectivity risks missing the wider context of understanding the central network controls of eating (11).
1.2. The Aim and Organization of This Review
Our aim is to present a thorough, balanced, and critical analysis of the vast literature from the past half century. We begin with some key concepts and definitions to establish our discussion parameters (sect. 2). These include control mechanisms, regulated variables, sensors, and homeostatic and hedonic eating. Because much of this review involves brain mechanisms, we also consider the increasing importance of consistent neuroanatomical nomenclature in an era of connectomics and neuroinformatics. The primacy of energetics and the position of ATP availability as the key homeostatically regulated variable are also central themes. We take this position because the sole purpose of eating in terms of energetics is to acquire the oxidizable fuels that cells use to maintain ATP availability. The dynamic relationships between energy acquisition, energy partitioning, energy storage, and energy expenditure are all directed toward this end. The next sections characterize what eating behaviors are (sect. 3) and how the brain is organized to control them (sect. 4). We then describe meal initiation (sect. 5) and meal termination (sect. 6) in terms of where and how signals interact with neurons, and the organization of the various neural networks to which they belong. We do not discuss some important topics because of space limitations (see sect. 8 for abbreviations). These include sex differences, eating pathologies, developmental aspects of eating behaviors, and the impact of the gut microbiota. The predominance of literature reviewed is from rodents, although some emphasis is also given to work using human participants and nonhuman primates.
1.3. Historical Background
We refer to the historical aspects of many topics throughout this review, and therefore, we only touch briefly upon some key milestones here.
1.3.1. The shift from clinical reports to experimental interventions.
The role that specific parts of the brain play in controlling motivated behaviors and arousal state first emerged from clinical reports ∼120 yr ago. These described patients who had brain lesions, tumors, or other pathologies, particularly in the hypothalamus, and also aberrant sleep/wake cycles, eating, drinking, and sexual behaviors as well as obesity, diabetes insipidus, and diabetes mellitus (12–14). This clinical foundation led to more experimentally based investigations of hypothalamic functions in mammals. One of the most important occurred in the 1930s when Steven Ranson and his colleagues at the Northwestern University in the US employed stereotaxic lesions for the first time to investigate the role of the hypothalamus in controlling ingestive behaviors in cats, rats, and monkeys (15–17). This work provided the foundation for many of the experimental interventions in the following decades, including the so-called dual center hypothesis, which assigned hunger and satiety to the lateral hypothalamic area (LHA) and ventromedial hypothalamic nucleus (VMH), respectively (18).
1.3.2. Discovery of physiological factors: brain transmitters and peripheral peptides.
For approximately the next 40 yr, experimentally investigating the control of eating was driven primarily by physiological psychologists, including Stellar, Miller, and others. A major breakthrough came in 1960 when Grossman (19) found that injections of either norepinephrine or carbachol into the perifornical LHA specifically stimulated eating or drinking, respectively. These results were the first to ascribe a neurochemical identity to the brain systems responsible for controlling ingestive behaviors. Early evidence of peptide influences in eating behaviors came from separate discoveries in 1973 and then in 1984. These showed that intraperitoneally applied cholecystokinin (CCK) and hypothalamically applied neuropeptide Y (NPY) had rapid but opposing effects on food intake (20, 21). In 1982, Langhans and colleagues (22) provided the first experimental evidence demonstrating that interfering with an endogenous peripheral peptide (glucagon), rather than its administration, can affect eating.
The molecular genetic characterization of leptin (23) by Jeffrey Friedman and colleagues, and its receptor (24, 25) in the mid-1990s, put a name to the factor whose existence Coleman had predicted almost 30 yr earlier from his famous parabiosis experiments (26, 27). The discovery of leptin in 1994 spurred an unprecedented explosion of experiments that have focused on its physiological relevance to energy balance and the mechanisms by which this is achieved (e.g., Refs. 28–30).
1.3.3. Motivation to action: reward, learning, memory, and navigation.
During the 1970s and 1980s work on the neurochemical bases of ingestive behaviors, reward, intracranial electrical and chemical self-stimulation, and the actions of drugs of abuse began to converge on a number of forebrain mediator sites (for reviews, see Refs. 31, 32). Foremost among these was the accumbens nucleus (ACB) in the ventral striatum, whose function is influenced by dopaminergic inputs from the midbrain ventral tegmental area (VTA), glutamatergic inputs from the prefrontal region of the cerebral cortex (PFC), and various other inputs.
A link between eating behaviors and dopaminergic systems, whose targets include the ACB, first emerged in the 1970s (33) as an explanation for the lateral hypothalamic syndrome. The apparent loss of motivation to eat that followed electrolytic lesions of the LHA turned out to be related more to destroying dopaminergic projections from VTA neurons to the ACB, rather than the loss of LHA neurons themselves (34). This was further supported by Mogenson and colleagues (35) who identified the ACB as a key player in the “limbic-motor interface” between “motivation and action.” These and later findings from the groups of Hoebel (36), Kelley (32), and many others highlighted the similarities (and differences) between the neurochemical mechanisms and neural connections that drive eating behaviors and drug abuse. These foundations helped pave the way for research on eating control to expand from physiological psychology to include learning and memory (e.g., Ref. 37), and now broader aspects of cognitive neuroscience, a trajectory that continues unabated.
1.3.4. Genetically driven cell manipulations: the 1980s to the present day.
Since the mid-1980s the application of genetic manipulations has been the biggest technical advance for how we investigate eating behaviors. It heralded the seemingly inexorable shift toward mice as the species of choice for studying the control of eating behaviors and other aspects of energy balance. Rather than choosing the mouse as a superior behavioral or physiological animal model, indeed it could be argued that rats provide higher resolution results of this type than mice, this move has been driven primarily by the ability to use gene manipulations to investigate a diverse range of molecular components. The initial method that enabled gene manipulation was the ability to incorporate foreign DNA into the germline of transgenic mice (38, 39), which quickly led to transgenic mice with altered growth hormone expression (40, 41). During the 1990s it became possible to manipulate the expression of enzymes, signals, and receptors, first at the whole animal level (42–46), and then in specific neuron populations (47–49). The ability to choose the optimal rodent model based on ethological and/or physiological considerations has been facilitated by the recent development of transgenic rats, as well as tools accessible in both mice and rats that allow for cell-specific and reversible activation or inhibition of eating control networks, for example, pharmacogenetics, optogenetics, and CRISPR-cas9 gene editing.
2. CONCEPTS AND DEFINITIONS
2.1. Word Usage
Four sets of terms contain words that are widely used, often interchangeably, in the ingestive behavior field. Despite their apparent equivalence, the words in each set are, like sex/gender, not synonymic; each provides nuanced but important distinctions that help explain how physiological and behavioral variables impact food intake and energy expenditure. We believe that applying clear meanings of these terms is more accurate and appropriate. These word sets are described in the following sections.
2.1.1. Eating/feeding.
We use the term eating to refer to the motor actions that animals, and humans, use to consume food. Although feeding is widely used in this manner, it more accurately refers to the actions executed by an individual to provide food to another.
2.1.2. Hunger/appetite.
Hunger is a sensation [Is there anything to eat? (50)]. The term hunger functions best as an intervening variable in the stimulus-response sequence between proactive signals and motor actions responsible for initiating eating (51, 52). Thus hunger drives us to eat; but there is no single specific mechanism of hunger. Rather, hunger connects the physiological state of negative energy balance, or any sign of a threatening energy deficit, to the various possible eating responses that are ultimately engaged by the brain’s motor control mechanisms. Negative energy balance or its anticipation generates the proactive physiological signals for eating, which are the independent variables in Miller’s schema that lead to hunger (52). Appetite, on the other hand, targets a specific food item. It may or may not be accompanied by hunger, but it is always associated with the expectation of reward. Analogous to the intervening variable concept for hunger, appetite functions as the intervening variable between a set of signals generating the desire to eat something specific and the resulting consumption of this particular food item [What do I want to eat? (50)].
2.1.3. Satiation/satiety.
Satiation develops during eating from the cumulative effects of inhibitory signals generated by the ingestion of food items. In other words, satiation signals ultimately bring eating to an end (I can no longer eat anything). They have many origins and are of diverse neural, endocrine, metabolic nature. After meal termination, a period of satiety begins, which lasts for some time before hunger and/or appetite return, i.e., satiety determines the length of the intermeal interval (I am still full). Again, different signals of varied origin determine the duration and intensity of satiety, as conceptualized in the notion of the satiety cascade (53). During satiety, sensory and cognitive processes interact with postingestive and postabsorptive peripheral and central mechanisms to inhibit further eating. Because satiation and satiety are concerned with the inhibition of eating, they can potentially affect total intake and facilitate body weight control.
2.1.4. Regulation/control.
These two terms are discussed in more detail in sect. 2.4.
2.2. Naming Brain Parts and Their Abbreviations
A significant part of this review focuses on the brain networks that control eating behaviors. Many brain regions, cell groups, nerves, and fiber tracts comprise these networks. During the past decade our understanding of how they are organized has taken a huge leap forward, in part because of genetically guided tracing techniques (2, 3) and sophisticated imaging (e.g., Refs. 54–57). Putting these results into larger conceptual frameworks involves generating maps that derive from meaningful comparisons between many neuroanatomical datasets, including ones generated by different research groups. To do this effectively requires using standardized brain atlases and established naming conventions (58, 59), without which there is significant risk of ambiguity, misinterpretation, and confusion between different experiments and investigators. For example, although the terms neocortex, neostriatum and neopallidum, basal ganglia, amygdala and extended amygdala, septum, and limbic system, etc. are all widely used, none have universally accepted definitions (60–62); they mean different things to different people. Throughout this review we therefore use a formalized nomenclature and abbreviations for the divisions, parts, and regions of the mammalian brain (FIGURE 1) (see Table A, Supplementary Item 7 from Ref. 59). Its foundation is a hierarchically organized set of brain and spinal cord parts (collectively the cerebrospinal trunk) whose parcellation derives from historical convention, and embryological and developmental principles (59, 63–65). It also favors English rather than Latin or Greek names, e.g., ‘endbrain’ rather than the Greek synonym “telencephalon.”
FIGURE 1.
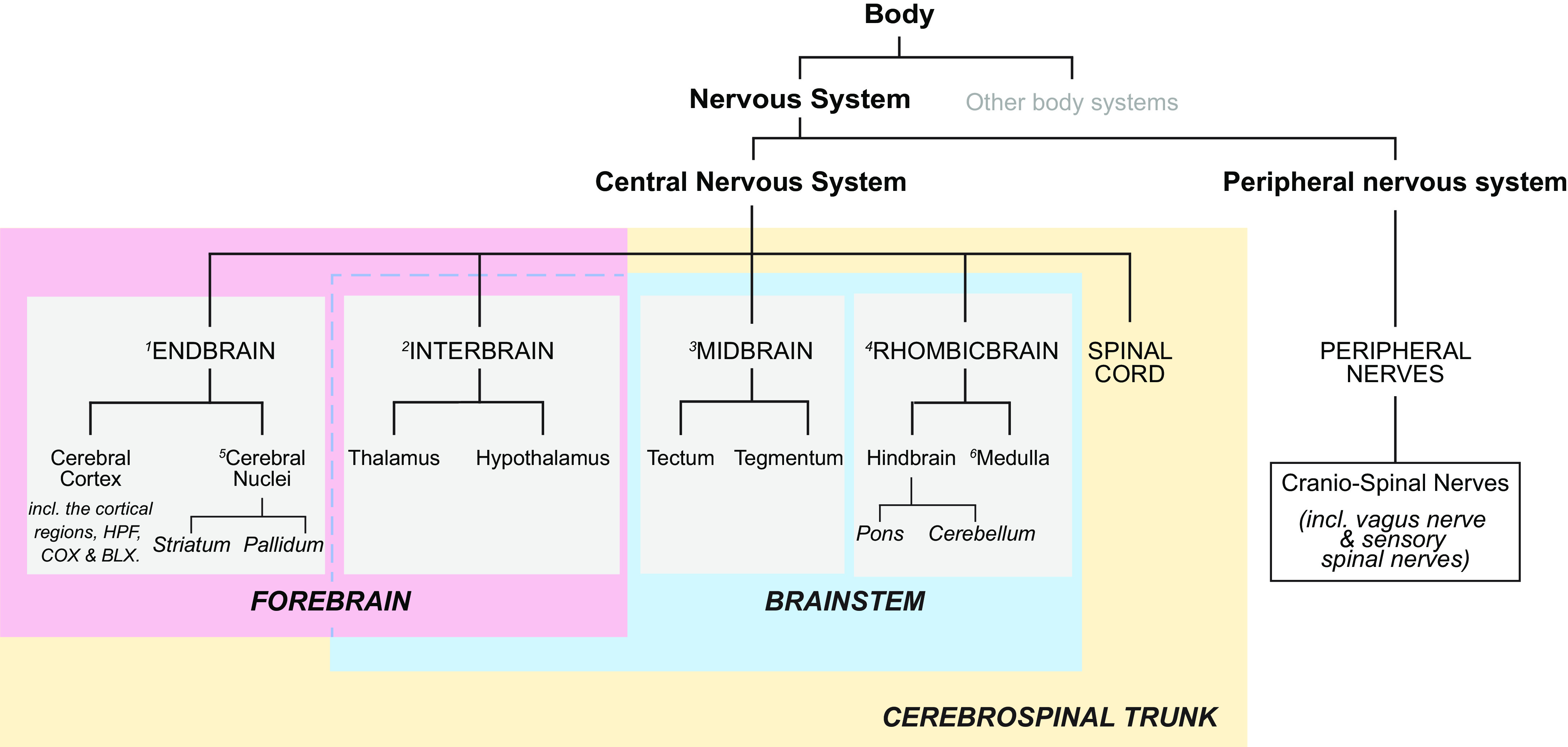
The hierarchy for the standardized nomenclature of brain parts used in this review is from previously published schemes for the rat and mouse brains. For more detailed descriptions and derivations, see Refs. 59, 63–66. Also known as 1: telencephalon; 2: diencephalon; 3: mesencephalon; 4: rhombencephalon; 5: basal ganglia; and 6: afterbrain. BLX, basolateral amygdalar complex; COX, cortical amygdalar complex; HPF, hippocampal formation.
2.3. The Importance of Homeostasis in the Context of Eating Behaviors
Homeostasis has been the defining concept since the first efforts to understand the contribution physiology makes to eating behaviors. Because so much has been written over the years about this keystone principle, we will only emphasize two aspects here. First, homeostasis does not imply that any constituent of the internal environment is held at a constant level; second, it does not define, as is sometimes inferred, a single process that somehow maintains overall stability of bodily functions. Instead, the idea that Cannon called “homeostasis” is that certain constituents of the body’s internal environments are each maintained within a controlled homeostatic range (67). Cannon defended his word choice as follows:
“Objection might be offered to the use of the term stasis, as implying something set and immobile, a stagnation. Stasis means however, not only that, but also a condition; it is in this sense that the term is employed. Homeo, the abbreviated form of homoio, is prefixed instead of homo, because the former indicates ‘like’ or ‘similar’ and admits some variation, whereas the latter, meaning the ‘same,’ indicates a fixed and rigid constancy. As in the branch of mechanics called ‘statics,’ the central concept is that of a steady state produced by the action of forces; homeostatics might therefore be regarded as preferable to homeostasis. The factors which operate in the body to maintain uniformity are often so peculiarly physiological that any hint of immediate explanation in terms of relatively simple mechanics seems misleading.” (67).
Our emphases (in italics) point out that Cannon explicitly provided for system flexibility. He also cautioned against looking to mechanics or engineering designs for explanations of physiology.
2.4. Regulated Variables and Control Mechanisms
The notion that the activity or amounts of the particular constituents of physiological systems fluctuate within relatively narrow ranges requires specific mechanisms that oppose their drift outside of this range. Correcting the drift above and below this range usually involves separate mechanisms. This brings us to the important distinction between regulation and control. Again, much has been written about using these two terms in physiology (e.g., Refs. 68–72). In part they grew out of efforts to apply control theory, as notably described by Wiener (73), to the interactions between homeostasis, physiological control processes, and motivated behaviors (see Ref. 74). However, some authors (e.g., Refs. 70, 75–77) have pointed out that applying control theory in this way continues to distract, particularly when eating behavior is viewed through the lens of molecular biology (77) or other reductionist approaches, by encouraging what may be futile searches for error signals, control centers, neurally embedded set points, etc.
The temporal organization of control mechanisms for eating is another important consideration. Many of these processes exhibit features that are anticipatory, proactive, or preemptive (69, 75, 78). These are properties that imply some degree of learning (69, 75, 79). The idea of anticipating a physiological challenge led Moore-Ede more than 30 yr ago to explicitly identify two temporally defined aspects to homeostasis (78). First, reactive homeostasis, as in the classic homeostatic mechanisms first described by Cannon (67, 80) and then many others. These are post hoc responses that occur at a time dictated by the challenge. Second, proactive homeostasis involving “corrective responses initiated in anticipation of a predictably timed challenge” (78). For eating, proactive mechanisms are perhaps best exemplified by the habitual eating seen in controlled environments that have unrestricted food supplies. These regular meals are anticipatory in that they allow animals to preempt entry into negative energy balance. The ability to time meal onset with the local environment is therefore critical, and so these meals are initiated by a combination of circadian timing signals and small excursions in proactive signals (see sect. 3.2).
To help provide the framework for eating behaviors into which we can place the role of physiological signals, we stress the following: regulation refers to the ability to maintain a variable within a narrow range, i.e., the performance of that variable rather than a specific mechanism directed to that end (72). Control mechanisms are those that maintain the narrow range of the regulated variable.
2.4.1. What is regulated in energy balance? What is controlled?
Carbohydrates, fats, and proteins are all acquired by eating. Digestion then releases their breakdown products (monomers) that are used in two ways: 1) for catabolism, where glucose and free fatty acids (together with their metabolites) are the principal oxidizable monomers for the ATP production required for both cell and whole organism function; and 2) for anabolism, which produces the macromolecules needed for storing energy, growth, reproduction, movement, etc., and therefore contribute to the function of the whole organism (FIGURE 2).
FIGURE 2.
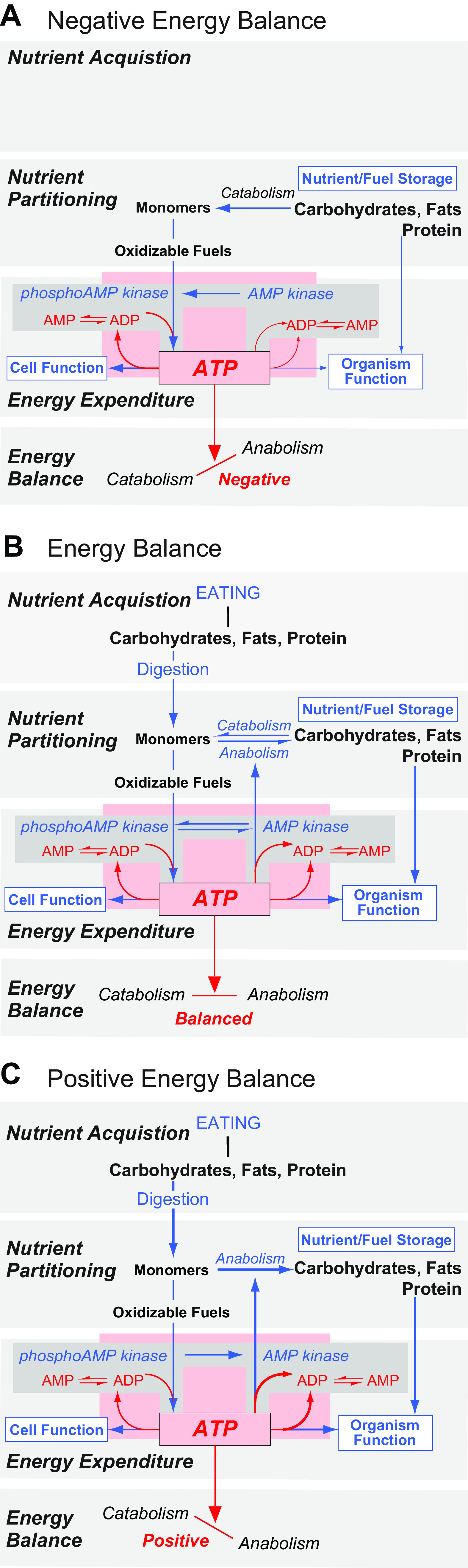
Energy Balance is determined by the collective state of 3 interactive sets of control processes: Nutrient Acquisition, Nutrient Partitioning, and Energy Expenditure. It fluctuates between three states: Negative (A), in Balance (B), and Positive (C). ATP availability (pink box) is the principal regulated variable in Energy Homeostasis and acts at the pivot point of Energy Balance (bottom box in A, B, and C). The various control processes that regulate ATP availability are shown with blue text and arrows, the size of which indicates their relative activity in a particular state. They are as follows: Eating/Digestion (Nutrient Acquisition); the synthesis (anabolism) and breakdown (catabolism) of energy storage polymers from/to their constituent oxidizable monomers (Nutrient Partitioning); and the rate of Energy Expenditure. In Nutrient Partitioning these monomers are appropriately distributed between Oxidizable Fuels and Nutrient/Fuel Storage depending on the current state of Energy Balance. ATP generated by oxidizing fuel molecules drives 2 general processes, the sum of which determines overall Energy Expenditure: internal Cell Function (active transport, secretion, endergonic reactions, etc.); and Organism Function (movement, growth, reproduction, etc.). In an extended absence of eating (A), ATP availability is maintained by catabolizing fuel stores and reducing energy expenditure. The phosphorylated state of AMP kinase (the sensor of ATP availability; gray boxes) is favored under these circumstances. When energy stores deplete, Cell Functions are maintained by reducing the anabolism of macromolecules to an extent that can compromise growth, reproduction, and other Organism Functions. When Eating provides oxidizable fuels that are equal to the requirements to maintain ATP availability (B) then the rate of anabolic and catabolic processes is equal, i.e., a state of energy balance. If Eating provides an excess of fuels required to maintain ATP availability within its narrow range (C), then energy balance favors anabolism and the dephosphorylated state of AMP kinase then predominates.
Whether anabolism or catabolism has overall precedence at a particular time determines the direction of an individual’s energy balance. Because ATP maintains life, ATP availability, as represented by cellular ATP:ADP ratios (81, 82), acts as the pivot point for energy balance (FIGURE 2). The ATP:ADP ratio is maintained close to 10:1 at all times for most cells (81–84). As such, a compelling case was made by Mark Friedman that ATP availability is the principal regulated variable in energy homeostasis (72).
To sustain life all living organisms require a minimum rate of ATP production. For animals, when food intake stops and fuel stores begin to deplete, ATP is increasingly directed inwards toward cell function at the expense of maintaining whole organism function (FIGURE 2A). This means that in the absence of eating, sets of control mechanisms increase catabolism to release monomers from fuel stores and thereby provide the oxidizable fuels to maintain ATP production (FIGURE 2A). Part of this process increases the phosphorylation of AMP kinase (AMPK), a fundamentally important enzyme for energy balance. In turn, phosphorylated (p)-AMPK increases AMP and ADP availability for ATP production (FIGURE 2A) (82). More broadly, when oxidizable fuel supplies are challenged, the collective outcome of many processes, including those that control blood glucose, adiposity, heat production, eating behaviors, etc., are directed toward maintaining ATP availability within a narrow range (FIGURE 2).
When animals acquire fuels and nutrients from food, their energy balance shifts toward neutrality (FIGURE 2B). As they continue to acquire food, energy balance begins to favor anabolism, which increases fuel stores and the biosynthesis of other macromolecules to help sustain those processes that have high energy demands such as growth and reproduction (FIGURE 2C). To enable certain life stages there are proactive signals that push energy balance toward the anabolic state that facilitates growth, reproduction, hibernation, and nurturing, etc. (85–87).
The control processes that maintain ATP availability (i.e., energy homeostasis) can be categorized as Nutrient Acquisition (eating and digestion), Nutrient Partitioning (which includes Nutrient Storage and Mobilization), and Energy Expenditure. Sets of control processes for each of these categories are shown in FIGURE 2. ATP availability in all cells is the apex regulated variable (FIGURE 2, pink box) that directly impacts Energy Balance. Unlike the narrow target range for ATP availability, the outcome ranges of these control processes vary widely. This key property permits mammals to adapt to perturbations and challenges from the external environment. For example, the rate, amount, type, and frequency of food consumption; adiposity levels; the rate of energy expenditure in response to ambient temperature fluctuations, etc. all have control processes that enable wide adaptive ranges. Failure to provide a high degree of adaptation would be catastrophic.
Unlike fuels, ATP cannot be stored. This is dramatically illustrated by the fact that an animal can survive for days or longer if it does not eat, whereas its survival time is measured in seconds if a toxin, for example, shuts down oxidative phosphorylation and ATP production. The lack of any ATP storage capacity also means that daily ATP turnover in humans is dramatic, and approximates total body weight (88). These characteristics mean that to maintain cell function, ATP availability is kept within a narrow range by dynamic interactions between nutrient acquisition (i.e., all types of eating behaviors), nutrient partitioning, and energy expenditure (89). In this way, none of these controlled processes assumes a primary role in maintaining ATP availability (energy homeostasis) all of the time; their respective contributions necessarily vary depending on circumstances.
To summarize this scheme: ATP availability, adiposity (and body weight), energy balance, and blood glucose are homeostatic variables that are regulated. Eating, nutrient partitioning, energy expenditure, etc. are not regulated. Instead they should be considered as highly adaptable control mechanisms for regulated variables. These processes are ultimately directed toward regulating ATP availability in cells, i.e., energy homeostasis (also see Refs. 72, 85, 90). Energy balance describes the state of the relationship at a particular time between the origins and destinations of the ATP (i.e., energy) derived from the monomers used as oxidizable fuels (FIGURE 2). Therefore, energy balance and energy homeostasis are not synonymous.
Although making a distinction between control and regulation may seem overly fastidious, it very usefully emphasizes the relationships between mechanisms and variables, and their contributions to the overall goal of enabling efficient and adaptable cell and organism function.
2.5. Sensors, Set Points, and Settling Points
2.5.1. Are certain fuel molecules preferred for maintaining energy balance?
Consistent with ATP availability being the principal regulated variable in energy balance, evidence supports the idea that the combined availability of all fuel sources is the primary controlling process for ATP production, rather than a preferred fuel, carbohydrate, fat, or protein (91). This has led to the idea of an energostatic (91) or ischymetric (92) control of eating, which is based on the continuous monitoring of either the energy derived from metabolism of absorbed nutrients, or some predictor of the energy yield (91). Nicolaidis and Rowland (92) emphasized time as an important factor of the sensing mechanism, i.e., the idea that the rate of energy turnover, or power, is metered rather than the total energy yield.
2.5.2. AMPK/pAMPK as the energy sensor.
If ATP availability is the principal regulated variable in energy balance, then its control processes require a sensor (e.g., Refs. 68, 71, 72). There is good evidence that changes in the phosphorylation state of AMPK (FIGURE 2) functions in this manner (82, 93). This enzyme is considered an evolutionarily conserved fuel gauge in all cells (82, 93). It can act in the brain as a sensor of energy availability and intracellular glucose (82, 93–97). To maintain adequate ATP availability the ATP:ADP ratio (FIGURE 2, dark gray box) favors ATP by ∼10:1 when ATP hydrolysis is at equilibrium. This ratio is maintained by way of allosteric control mechanisms involving ATP and AMP concentration that alter pAMPK activity, which can be further modulated by glucose and hormones including leptin and ghrelin (see Refs. 82, 98–100 for reviews). Hardie (82) makes the case that the ATP:AMP ratio is a more sensitive indicator of ATP availability than the ATP:ADP ratio. We return later to the idea of AMPK as a key energy sensor when we discuss how the brain organizes eating behaviors (sect. 4).
Although a peripheral, and particularly, a hepatic sensor of energy availability, has been implicated in the control of eating (72), most of the available evidence indicates that ATP:AMP or AMPK in hepatocytes may not serve this function (see Ref. 101). In fact, as far as food intake control is concerned, the enterocyte is the more likely peripheral candidate (101, 102). More work would therefore help clarify the relative contributions of these two sites. It is, however, worth mentioning that peripheral energy sensing does contribute to glycemic control. In this way, glucosensors in the hepatic portal vein wall associated with the splanchnic (spinal) and vagal nerve are an integral part of blood glucose control mechanisms (103, 104) and may also sense the catabolism of other energy-yielding substrates as well (103, 104). Similar mechanisms may be located in all sensory nerves that relay information from the gut, but this has yet to be thoroughly investigated.
Like AMPK, the mammalian target of rapamycin (mTOR; an evolutionally conserved serine–threonine kinase) also serves as a fuel sensor (105). It specifically monitors amino acid availability and stimulates protein synthesis and thus cell growth and proliferation (106). The reciprocal activity of AMPK and mTOR in peripheral cells suggests that mTOR promotes growth if energy and amino acids are available (107, 108).
2.5.3. Do “set points” and “stats” for physiological parameters exist?
2.5.3.1. thermostats, glucostats, and ponderostats.
Control theory introduced the idea of set points, error signals, and “… stats,” e.g., thermostats, lipostats, glucostats, ponderostats, etc. to physiology. Although a thorough discussion of the pros and cons of “set points” or “stats” in physiological regulation is outside the scope of this review, some comment is warranted because of the importance of eating behavior for the pertinent variables. Much evidence now favors actively controlled mechanisms that not only correct, but ideally prevent major deviations of these physiological parameters. These mechanisms are partly based on negative feedback but also, and perhaps mainly, on anticipatory, learned modulations of the target variable. These processes, however, do not require a reference set point or stat to be effective (e.g., Refs. 70, 109). The active part of this regulation comprises sensors that monitor the target variable. Any changes will trigger corrective responses, but this does not require comparison of the current level of the variable to a reference (set) point, and therefore, no error signal to activate the correction process. Rather, a regulated variable is the functional result of physiological control systems whose level reflects a balance or settling point, in which control of the system is determined by a combination of active (feedback and anticipatory) and passive mechanisms. Such systems describe the usual variability of most biological variables much better than the comparatively static concept of a set point or some form of “… stat.” Interestingly, although often implied, the idea of a fixed or static set point was never part of Cannon’s original idea of homeostasis; that is why he termed it “homeostasis” and not “homostasis” (see sect. 2.3).
2.5.3.2. a lipostat or set point for body weight.
The existence of a set point for body weight has been debated for over 60 yr (110). Kennedy’s lipostatic theory of food intake control posited that a certain level of body fat is defended by changes in food intake (111). Since then, most of the set-point models for body weight have focused on adipose tissue or adiposity as the regulated parameter. The argument usually is that changes in body weight of adult individuals mainly result from changes in adiposity, and that adipose tissue constitutes the body’s largest energy reservoir. While this concept led eventually to the discovery of leptin, it can hardly explain all the phenomena related to the relative constancy of body weight in adult individuals. Moreover, as for other physiological parameters, the notion of a set point for body weight is not consistent with most of the experimental findings. In a classic conceptual paper, Wirtshafter and Davis (74) pointed out that no specific externally controlled set point is required to explain the relative constancy of adult body weight by simple negative-feedback control. Referring to several previous papers arguing for a set-point regulation of body weight, they “describe a simple feedback control model which contains no set point, and yet is able to account in full for these and other data which have been cited in support of the existence of a body weight set point.” Wirtshafter and Davis (74) coined the term “settling point” (instead of set point) to reflect the fact that body weight, although actively regulated, can vary substantially depending on a myriad of open-loop variables that modulate the whole control system. For instance, this settling point concept can easily incorporate the sensory attractiveness of food and other environmental as well as socioeconomic influences. One problem with invoking the settling point concept as part of active feedback mechanisms is that it can be misinterpreted to mean that body weight is the result of a totally passive regulation, i.e., simply the result of a dynamic equilibrium of various influencing factors (e.g., Ref. 112). This assumption often compares the body weight settling point to the level in a water reservoir filled by rain, rivers, etc. and empties through various outflows. The water in the reservoir will simply settle passively at a level that is the net result of the inflow and outflow rates.
Body weight regulation does not work according to this principle. Clearly, some regulation exists. However, it is equally clear that there is no strict and efficiently defended set point for body weight; otherwise, obesity would not occur. In fact, many studies in different species (e.g., Refs. 113–115) including humans (e.g., Refs. 116–118) show that when environmental conditions are fixed, experimentally induced changes in body weight usually result in compensatory changes in energy intake and energy expenditure that tend to bring body weight back to normal. In humans, these responses are mainly observed after decreases in body weight and often appear to be absent after body weight increases. One of the more recent convincing demonstrations of such compensatory changes in humans is the finding that type-2-diabetic patients in a placebo-controlled trial with canaglifozin (an inhibitor of the sodium-glucose cotransporter 2) compensated for the weight loss by increasing food intake ∼100 kcal/day for every kg of body weight that was lost (119). The body weight loss with canaglifozin treatment is primarily driven by urinary glucose loss. The compensatory changes in food intake, which were calculated using a previously validated mathematical model, were much larger than the concomitant decreases in energy expenditure (119). The beauty of this study is that the patients were not aware of the body weight loss, thus in effect excluding any possible volitional change in food intake.
One important component of active feedback control is the feedback gain, i.e., how strong or powerful is the feedback signal that is triggered by deviations from the current/present level of the regulated parameter. A prominent feature of body weight regulation is that the feedback gain function is not linear, i.e., small deviations from the current level of body weight will trigger only weak compensatory responses. With increasing changes, these feedback signals become more powerful. Last, but not least, a decrease in body weight triggers more powerful feedback signals than an increase. As a result, the compensation works better after experimental decreases in body weight (mass) than after increases. This probably reflects the fact that, from an evolutionary point of view, the defense against a potentially dangerous decrease in body weight was more important, and more often required in human history, than a defense against an increase in body weight (mass). Unlike the decrease in body weight, which would eventually compromise reproductive success and survival during the inevitable periods of food scarcity, the increase might have had hardly any severe consequences except for the possibility of an increased danger of predation (see Refs. 112, 120). The nonlinear feedback gain function together with the lopsided efficiency has prompted speculations that there might be no compensation at all within a certain range of body weight, with compensatory mechanisms only operating at lower and upper intervention points (112). Whereas the mechanisms of the compensation, including the relevance of changes in energy intake and expenditure, may well differ between increases and decreases in body weight, the abundance of evidence argues against such a model. An excellent and thorough discussion of the body weight set point/settling point issue has recently been published (76).
A final point to note in this ongoing and important debate about the existence of set points is that there has been little evidence so far for a neural mechanism that can encode set point values. However, recent work from humans and rodents suggests that the ensemble activity of neurons in the insular (INS) and ventromedial PFC can represent the value of an animal’s replete state. This value can then be used to compute a behavioral trajectory that can reduce the magnitude of a deviated physiological state (121–123). This representation certainly bears more than a passing resemblance to the neural coding of a set point.
2.6. Homoeostatic and Hedonic Control of Eating
2.6.1. Preamble.
Much of the pioneering work that investigated the physiological control of eating in mammals used experimental designs that involved periods of food restriction. Because deficit-induced eating was correlated to what were considered negative feedback signals that reported reduced energy stores, this type of eating was, and still is, commonly referred to as homeostatic. This label was applied with the idea that in these circumstances eating reversed the decline of a homeostatically regulated variable that had resulted in negative energy balance. Earlier, we considered that the apex regulated variable in energy balance is ATP availability, which as FIGURE 2 shows is very tightly controlled by a variety of processes, including eating. In this regard, deficit-induced eating would quickly provide oxidizable fuels to maintain ATP availability as sensed by, for example, hypothalamic pAMPK. Although simple negative feedback is an attractive explanation, it does not provide a compelling framework for any type of eating behavior for animals in complex environments (69).
Conceptually, hedonic control is used to explain why some foods are highly preferred to others regardless of their caloric density. Seeking these desired foods occurs without a perceived or actual energy deficit and, moreover, can lead to calorie consumption beyond that needed to restore energy availability back into the regulated range (i.e., close to energy balance). The prevailing view is that some sort of hedonic forces override homeostatic mechanisms, thereby contributing to excessive calorie consumption and eventually body weight gain (124–126). However, again, as with homeostatic eating, the complex interactions between different and distributed control mechanisms belies simple pigeonholing.
2.6.2. Reward and eating behaviors.
Food reward or reward-based eating are commonly used but loosely defined psychological constructs. They refer to behavioral patterns associated with excessive food seeking and consumption that occur despite animals being in energy balance. Using terms such as reward-based eating in the context of caloric overconsumption is complicated by the fact that both food preferences and the capacity of certain foods or events to stimulate excessive eating behavior are specific to individuals and extremely dynamic. Food preferences are further modulated by other factors: physiological status (e.g., overall/general health, energy balance); recent consumption history (e.g., sensory specific satiety); previous experience [e.g., conditioned taste aversion or avoidance (CTA)]; together with various other exteroceptive and interoceptive factors.
We therefore argue that food reward and related constructs such as food addiction have no predictive or explanatory value for understanding eating and associated behaviors and thus function only as circular descriptive terms. Instead, we deconstruct the concepts of food reward by focusing on three conceptual domains that are at least partially distinguishable based on observable behavioral profiles, and/or can be characterized based on stimulus-reinforcement and response-reinforcement associations. These are as follows: Incentive Salience (effort-based food-directed behavior), Hedonic Evaluation (palatability, food preference), and the loss of Inhibitory Control (associative inhibition, impulsivity). We note that while these three constructs are not mutually exclusive with regards to their underlying psychological and neurobiological substrates, they do represent categorically distinct behavioral profiles that are each relevant to the type of foraging and consumption that occurs in the absence of an energy deficit and in spite of potential adverse biological consequences.
2.6.2.1. incentive salience.
Eating behavior inherently requires motivated behavioral responses to acquire the food. Incentive salience refers to the motivational value attributed to reinforcers and their predictive cues (also referred to as wanting by Berridge and colleagues (51). Incentive salience is frequently examined in rodents by measuring performance in effort-based instrumental/operant conditioning procedures that involve response-reinforcement (i.e., action-outcome) associations, typically using food reinforcers (e.g., sucrose) that are preferred to the maintenance chow provided in the home cage. Learning that a food-motivated action reliably results in a specific consequence, however, is not sufficient to determine whether that action should be performed or not. Rather, both the consequence and the value of the consequences of various alternative actions are critical components of how action-outcome associations influence behavior.
Food-directed motivated responses are not only determined by learned action-outcome associations, but also by the animal’s current evaluation of the affective properties of a specific food reinforcer, i.e., its incentive salience. Outcome devaluation studies (e.g., Refs. 127, 128), for example, demonstrate that hunger and satiety states can increase or reduce, respectively, the incentive salience of a food reinforcer and its associated operant response. However, the capacity of nutritive status to influence appetitive behavior is not absolute but rather involves what Balleine and colleagues (129–131) refer to as incentive learning, a process through which the incentive value of a specific food is modified through specific outcome-nutritive state learning (132). These findings collectively support the notion that incentive salience for specific foods is a dynamic property shaped by both nutritive state and by an animal’s previous experiences with specific food-nutritive state interactions.
Dopaminergic (mesolimbic) projections from the VTA to the ACB are a critical substrate in the neurobiological processes that govern incentive salience (see Refs. 133, 134 for further review). The bursts of action potentials from VTA dopamine cell bodies and the dopamine release associated with them are termed phasic dopamine signaling. Both sugar consumption (135) and postconditioning presentation of a Pavlovian conditioned stimulus associated with sucrose delivery (136) elicit the release of dopamine in the ACB from VTA neurons, analogous to responses evoked by amphetamine, cocaine, or alcohol (137, 138).
Functional evidence that dopamine signaling controls incentive salience for food is provided by work from Zhuang and colleagues (139–141) using the hyperdopaminergic dopamine transporter knockout mouse, which has elevated extracellular striatal dopamine levels. These mice show elevated levels of food-directed operant responding with minimal effects on total caloric consumption, Pavlovian (stimulus-stimulus) or instrumental (stimulus-response) learning, or hedonic orofacial responses to the reinforcer itself (139–141) (see below for further discussion of orosensory hedonic evaluation). These results are consistent with a framework where the VTA to ACB dopamine projections predominantly controls appetitive motivational components of eating behavior. This framework is further supported by results showing that rats with pharmacologically impaired dopamine signaling reallocate their effort-based operant behavior for more preferred foods toward less preferred foods that require less effortful food-seeking behaviors (reviewed in Ref. 142).
That incentive salience for specific food reinforcers is dynamically modified by nutritive state and postingestive factors is consistent with the pattern of phasic dopamine responses in the VTA to food and food-associated cues. For example, food restriction increases the magnitude of dopamine evoked by food consumption (143, 144). A recent study has also shown that VTA dopamine neuronal activity was higher in protein-deficient rats compared with controls when they consumed a preferred protein food rather than carbohydrate (145). Work from both rodents and human neuroimaging studies is consistent in identifying the dorsal striatum as a key region receiving postoral nutrient sensing (146–149). Furthermore, the dopamine 1 receptor is a likely target for the mesolimbic modulation of postoral nutrients, as pharmacological blockade of this receptor (but not the dopamine 2 receptor) blocks the acquisition of flavor-nutrient preference learning following intragastric carbohydrate infusions (150). More recently a potential upstream mediator of those VTA dopamine projections that can control eating-related incentive salience was identified. Thus activation of LHA GABAergic projections to VTA dopamine neurons stimulates food-motivated responding without influencing levels of food consumption (151). These findings collectively highlight a conceptual framework in which the psychological and neurobiological substrates that govern incentive motivation are distinct from those controlling caloric consumption once food is acquired and consumption has commenced.
2.6.2.2. hedonic evaluation.
Hedonic evaluation [also referred to as liking by Berridge and colleagues (51)] is another psychological construct that is applied to caloric overconsumption. It is distinct from incentive salience. Independent of their caloric density, some foods are more preferred to others based on orosensory properties, postingestive mechanisms, and learned interactions between these and other physiological processes. Such preferences are dynamic, are individual specific, and are directly influenced by the subjective hedonic properties of specific foods.
In rodents, two frequently used indirect readouts of the comparative hedonic evaluative properties of specific foods is to either compare the number of calories consumed in one or two bottle preference tests (with liquid nutrients) or to measure consumption of multiple solid foods presented simultaneously or separately. However, these tests require that animals first initiate and then maintain eating (or drinking), meaning that they cannot be applied under circumstances where animals are unable to eat or drink spontaneously or when ingestion is maintained at low and/or inconsistent levels. Moreover, such tests do not allow experimental separation of volitional motivated behaviors related to the incentive salience of the reinforcer (e.g., approach behaviors) and the consummatory behaviors that are directly influenced by hedonic evaluation. To circumvent these issues, Grill and Norgren (152, 153) developed the taste reactivity test in rodents, which assesses the response to gustatory stimuli by examining stereotyped responses based on various mimetic and body response components. Sapid sucrose and NaCl stimuli applied via an oral catheter elicit positive hedonic orofacial reactions that are qualitatively dissociable from the pattern of rejection/disgust reactions observed following aversive concentrations of quinine and other bitter tastants. Support that these taste reactivity measures are directly related to hedonic evaluation is based, in part, on data showing that orofacial reactions to concentrated NaCl change from negative disgust to positive liking following hormonally induced sodium depletion (154, 155).
Similar to incentive salience, hedonic evaluation of food reinforcement is also influenced by energy status. For example, satiation in rats reduces positive hedonic orofacial reactions to sweet tastes below control levels, whereas 48-h (but not 24-h) food restriction increases hedonic taste reactivity (156). Using computer reaction-based implicit tests of liking and wanting in human participants. Finlayson and colleagues (157) reported that hunger-satiety status differentially affected liking versus wanting for specific foods in a macronutrient-dependent manner.
Central opioid signaling represents a critical neurobiological substrate that mediates the hedonic evaluation associated with food consumption. Pharmacological work established a hyperphagic role for central opioid signaling, particularly through the mu-opioid receptor (158–161). The hyperphagic effects of central opioid signaling, however, appear to be specific, in that they preferentially promote consumption of foods with higher hedonic value that is independent of their macronutrient composition (162, 163). Kelley and colleagues (164–167) identified the rostrodorsal medial region of the ACB and the caudal ventral pallidum as critical sites where mu-opioid receptor signaling potently enhances food consumption and hedonic taste reactivity measures, particularly for high-fat or sucrose-enriched palatable foods.
2.6.2.3. inhibitory control.
As discussed above, incentive salience and hedonic evaluation are psychological constructs that can be measured separately, do not always correlate/covary, are each uniquely dynamic based on associative contingencies, and mediate different components of eating behavior (preprandial and prandial, respectively). However, it is often the case that foods with a high incentive value also have a high hedonic value. Thus one can easily imagine a perfect storm scenario for triggering excessive caloric consumption, i.e., consumption beyond immediate or long-term energetic need. Here, a food (or combination of foods, beverages) has 1) both a high incentive and hedonic value; 2) is easily accessible with minimal foraging effort; and 3) is available in a portion size that is sufficient to allow virtually unrestricted consumption. It could be argued that never before has humankind encountered the scenario commonly experienced in many modern cultures where there is an increasing prevalence of highly processed, highly palatable, yet easily prepared and affordable foods. Excessive consumption of palatable processed foods that have a high calorie content can contribute to maladaptive body weight gain that is associated with harmful metabolic and physiological outcomes. It is therefore important to consider the behavioral, psychological, and biological processes that permit an animal to inhibit impulses and prepotent responses to powerful food reinforcers and/or to stimuli associated with these reinforcers. This process is commonly referred to as inhibitory control or response inhibition.
Similar to food reward, reward-based eating, and food addiction, the phrase inhibitory control itself has minimal value for explaining or predicting complex eating behaviors, and like these other phrases, is merely a circular descriptor of an observed behavior. Here we discuss two domains that are conceptually related to the general idea of inhibitory control: 1) inhibitory associative learning, and 2) impulsivity. While they are not mutually exclusive with regards to underlying psychological and neurobiological substrates, they are distinguishable based on observable behavioral profiles and/or can be characterized based on stimulus-reinforcement and response-reinforcement associations.
2.6.2.3.1 Inhibitory associative learning.
In Pavlovian conditioning procedures, the delivery of the food reinforcer is not contingent on the animal’s response. Thus impaired associative inhibition is not manifest as an inappropriate response resulting in immediate negative consequences (e.g., punishment) but rather is typically measured as increased anticipatory responding to cues under conditions where the cues are either no longer reinforced, or the magnitude of associated reinforcement is greatly reduced. Davidson and colleagues (168–171) have proposed that normal mammalian eating behavior is largely under the control of conditional associative learning processes. Briefly, in their experimental design, food-associated stimuli function as the conditioned stimuli, whereas the postingestive nutritive reinforcement serves as the unconditioned stimulus. Interoceptive hunger and satiety states function as occasion setting stimuli that modulate the associative strength between external food-associated cues and the postingestive reinforcing effects of the food. Impaired associative inhibition is manifest as increased appetitive and/or consummatory behavior during satiety, a physiological state that under normal conditions signals the reduced magnitude of food reinforcement. The hippocampal formation (HPF) and the medial part of the PFC (PFCm) have been identified as critical substrates for this type of food reinforcement-based associative inhibitory learning process (see Refs. 170, 172, 173 for review).
2.6.2.3.2. Impulsivity.
An impulsive action or responding without apparent forethought for its consequences can often lead to immediate consequences that are undesired or unintended by the individual. In addition to being linked with various psychiatric disorders, including excessive gambling and drug addiction (174), impulsivity is related to excessive food intake (175, 176), binge eating disorder (177), weight gain (178), and obesity (179, 180).
Impulsivity can be subdivided into two distinct behavioral categories: 1) impulsive action, and 2) impulsive choice (181). Impulsive action refers to a failure to inhibit an inappropriate response to a stimulus that generally results in an immediate undesired outcome. Impulsive choice is characterized by decision-making that is driven by a distorted consideration of future behavioral consequences. Again this generally results in either an undesired outcome, or a less than optimal outcome when compared with outcomes associated with having made an alternative (less impulsive) choice. Reliable rodent models have been established to study both impulsive action and impulsive choice. The literature supports a strong neurobiological and behavioral homogeneity in these measures across species, with general commonalties observed between the neural systems underlying impulsive responding for food, inhibitory associative conditioning, and food-related incentive and hedonic processing (182–187). Neurobiological systems associated with food-directed impulse control based on rodent data include (but are not limited to) GABAergic neurons in the ACB core (188), opioid receptor signaling in the PFCm (189), ACB delta FosB-associated signaling pathways (190), and projections from the ventral part of the HPF (HPFv) to the PFCm (191, 192) and the ACB (193). Human neuroimaging studies also highlight the importance of the dorsolateral region of the PFC in impulse control (194), as well as striatal connections with the HPF, amygdala, and parahippocampal gyri (195).
2.6.3. Conclusion.
Results summarized above deconstruct the circular descriptive concept of food reward with a specific focus on deciphering the measurable behavioral profiles and neurobiological substrates of relevance to excessive caloric consumption beyond energetic need. The constructs of incentive salience, hedonic evaluation, and inhibition (associative and impulse control) were discussed separately based on distinct behavioral assessment parameters and, in some (but not all) cases, distinct biological substrates. Critical neural pathways associated with these constructs are the dopaminergic projections from the VTA to the ACB (incentive salience), mu-opioid receptor signaling in the ACB (hedonic evaluation), and HPFv glutamatergic projections to the PFCm and ACB (associative inhibition and food impulsivity).
2.7. Peptide Signaling
2.7.1. Preamble.
How we currently view the neural mechanisms that control eating behaviors has been driven to a large extent by investigating neuropeptide function. The impact on the field made by the discovery of eating-active peptides in the 1980s was perhaps only surpassed by the discovery of leptin a decade later. Even so, leptin’s actions in the brain are still mostly interpreted in the context of peptidergic neurons. Given the centrality of peptides in eating control networks, it remains rather surprising that, compared with the signaling mechanisms engaged by fast-acting neurotransmitters, the complex and diverse signaling mechanisms used by peptides are often poorly acknowledged. This seems to be a consequence of the widespread position of only viewing neural network function through the synapse, i.e., wired transmission (196, 197).
While fast-acting neurotransmission operates between neurons across the synaptic cleft, peptide release and action mostly does not. Fast-acting transmitters are released from small electro-lucent vesicles directly into the synaptic cleft. On the other hand, peptides are released from large dense core vesicles from sites that, while still being on the axon terminal, are mostly distal from the synaptic cleft (198). The physical distance between the release, ionotropic action, and termination of fast-acting neurotransmission is on the order of angstroms, which markedly limits its temporal and spatial range. The actions of fast-acting neurotransmitters is also limited by their rapid reuptake into the presynaptic terminal. These properties are different for peptides, which may diffuse some distance from their release site before being degraded by proteases. The collective actions of peptides and fast-acting transmitters therefore provide neurons with a remarkably diverse set of chemical signals (e.g., Ref. 199) that adds considerable flexibility to all the neural networks that control eating behaviors.
A further complication when considering peptide actions is that some neurons release peptides into the cerebrospinal fluid (CSF) by way of terminals in the ventricular ependyma (200). Others are released from dendrites (201). Some peptides therefore act on receptors that are located at considerable distances from their release sites. All these findings are incompatible with interpretations based only on synaptic signaling. Instead, these properties enable a second mode of chemical communication in neural networks: volume transmission.
We now consider how three underappreciated aspects of peptidergic signaling produce challenges to understanding eating behavior control networks.
2.7.2. Central and peripheral peptides: same molecules, different contexts.
The view that peptides function as sophisticated chemical signals began almost 100 yr ago with the discovery of substance P by von Euler and Gaddum. They found the highest concentrations of substance P in the brain and small intestine. The widespread distribution of a particular peptide quickly became a common feature as more were added to the signaling roster. In this way, some of the peptides we associate with the control of energy balance are synthesized by and have signaling properties within many tissues. CCK, glucagon-like peptide-1 (GLP-1), somatostatin, and NPY, for example, are synthesized in neurons as well as being important signals that originate in the pancreas, gastrointestinal (GI) tract, etc. Consequently, these peptides have equally varied functions that are determined by the nature of the signaling system in which they operate. We draw attention to these properties not only to highlight the signaling flexibility of peptides but also to caution against drawing functional inferences between the peripheral effects of a peptide and the actions of brain neurons that synthesize the same peptide; there may be little to no connection between these two independent functions. For example, recent results show that brain GLP-1 neurons have no role in encoding the actions of circulating GLP-1 in the brain (202).
2.7.3. Peptide receptor localization.
Most peptides signal using G protein-coupled receptors (GPCRs) that are located widely on target neurons, and mostly away from the synaptic cleft. They are broadly distributed presynaptically on axon terminals, postsynaptically on dendrites and neuronal somata (198) and on glial, epithelial, and ependymal cells. These diverse locations, coupled with the lack of specific reagents, particularly antibodies, or precisely targeted delivery systems mean that accurately locating, characterizing, and experimentally targeting GPCR function in the brain remains a significant challenge that currently limits how well we can interpret the contributions peptides make to network function (203). For example, two commonly used proxy markers for peptide receptor locations, in situ hybridization and the gene promoter-driven expression of fluorescent markers, are adequate for identifying target cell locations or their overall morphologies. However, neither indicates receptor location at the subcellular level because both report the expression of a receptor gene, not the receptor itself. This presents important interpretational caveats for many experimental approaches. For example, the effects of knocking down the expression of a GPCR mRNA in the neurons of a particular region does not limit knockdown effects on receptor function to that area because of possible presynaptic and postsynaptic locations of the translated receptor protein. The recent technical development of reporters for GPCR function (e.g., Refs. 204–206) offers the prospect of investigating neuropeptide signaling with much higher spatiotemporal resolution than is currently possible (203).
2.7.4. Volume transmission and dendritic release: thinking outside the synapse.
In contrast to wired synaptic signal transmission, where fast intercellular communication occurs between neurons separated by synapses or gap junctions, volume transmission is a slower form of intercellular communication where cell-to-cell transmission of signaling molecules in the brain occurs via the interstitial space and/or the CSF (196, 207). This type of signaling mechanism is supported by at least three sets of evidence. First, the fact that unidentified neuromodulators are present in human CSF in sufficient quantities to excite neurons (208). Second, the widespread and long-recognized mismatch between peptide innervation patterns in the brain and peptide receptor locations (209). Third, the release of peptides from nonsynaptic structures, particularly nerve terminals in the ventricular ependyma (200) and dendrites. In this regard, Leng and colleagues (210, 211) have shown that oxytocin (OXY) is somato-dendritically released from magnocellular neuroendocrine neurons in the supraoptic nucleus (SO), a topic that is discussed further in sect. 5.5.2.5.
Even though peptidergic signaling at the synapse is clearly a major way for peptides to alter neuronal function, much evidence now supports volume transmission as an additional signaling mechanism used by many peptides involved with eating control. These include melanin-concentrating hormone (MCH) and orexin (ORX) (200), GLP-1 (211), OXY (212–214), beta-endorphin (215, 216), as well as other chemical signals less closely involved, e.g., melatonin (217) and gonadotropin-releasing hormone (218). Volume transmission can therefore broaden the range of peptidergic actions in the brain compared with those that use synaptic mechanisms (212, 219).
3. WHAT ARE EATING BEHAVIORS?
To understand how the brain organizes eating behaviors and how physiological signals control these actions, it is important to first describe 1) the component events that constitute eating behaviors, 2) how these are temporally organized, and 3) the various circumstances during which they are expressed. It is also important to consider that the somatomotor actions that allow mammals to find and consume food items (i.e., eating behaviors) are accompanied by neuroendocrine and autonomic actions that appropriately coordinate digestion, fuel absorption, postabsorptive fuel partitioning, and energy expenditure.
3.1. The Organization of Eating Behaviors
3.1.1. The temporal organization of motivated behaviors.
The fundamental temporal organization of eating behaviors is, like all motivated behaviors, described by a scheme developed about 100 years ago by Craig (220, 221). This arrangement states that in the appetitive phase animals locate food items in their environments by employing species-specific, adaptive, and sometimes individually tailored procurement/foraging behaviors and approach strategies. Physiological meal initiation signals inform the brain about the status of fuel stores, circulating fuels, and gastrointestinal (GI) status. They influence decisions about activating or suppressing future eating episodes. Exteroceptive signals also make significant feed-forward contributions (222). Once food is located, more stereotypic movements (manipulating, licking, biting, chewing, swallowing) are used to interact with and ingest the food during the consummatory phase. Meal termination signals encode the internal and external consequences of ongoing eating behaviors. As feedback signals, they provide the brain with information about ingested fuels and satiation levels, as well as (real time) information about the oral, head, and body muscle movements involved with eating (e.g., Ref. 223). When eating episodes end, the animal proceeds to initiate another behavior (behavioral switching) based on motivational priorities. The next behavior may or may not be another eating episode.
The cyclic nature of what Craig (221) called “instinctive behavior” was an explicit part of his scheme. FIGURE 3 is consistent with this idea and shows that the individual behavioral phases can, if the behavior is repeated, be expressed cyclically while maintaining the same sequence.
FIGURE 3.

The phases of motivated behaviors progress in a series of cycles that change from one behavior to another at switching points (solid green circles). The timing and nature of the switches are determined by interactions between brain mechanisms and different exteroceptive and interoceptive signals. Eating is initiated from the previous behavior when these processes select the motor actions that enable eating. Eating then progresses until meals are terminated by combination of satiation signals and the predominance of other signals that favor a subsequent behavior, which may or may not be another meal.
Taking this one step further, eating behaviors are just one of many motivated behaviors that animals express over time. This means that behavioral expression patterns consist of a temporally ordered series of interconnected motivated behavior cycles. FIGURE 3 summarizes this arrangement. Note that an ongoing behavior could be the same as a previous one, as in the case of a series of eating episodes, or it could be different. FIGURE 3 shows the nodes where switching occurs from one behavior to the next as a consequence of changing behavioral prioritizations. With regard to composite meals, FIGURE 4 shows that these consist of a close relationship between eating and drinking episodes according to the above scheme (224), meaning that the previous and subsequent behaviors in FIGURE 3 could be drinking.
FIGURE 4.

The structure of 2 consecutive habitual meals eaten by a male rat about 5 h into the 12-h dark phase of the light cycle. As defined by Zorrilla et al. (224), a rat meal is a composite of clusters made up of short episodes (bouts) of food and water intake. The height and width of these bouts denote their amounts and duration. Composite meals tend to start and finish with a drinking cluster. Meals are defined by intermeal intervals of at least 5 min. Adapted from Ref. 225 with permission.
3.1.2. Structural organization of meals.
The meal is the main unit of eating for most experimental animals. Identifying a meal’s components and how they are temporally expressed provides the foundation for determining how the brain is organized to control eating behaviors. If the goal is to understand how physiological signals interact with the brain to control meal components, then we need to know how a meal is structured.
The simplest way to determine whether an animal has eaten or not is to measure the amount of food eaten per unit time. Although this allows gross levels of analyses, experimenter-imposed time bins have insufficient resolution if their duration is hours or more; they cannot identify individual meal components and how a particular manipulation or intervention changes them. More fine-grained analyses of rat meal structure (e.g., Refs. 224, 226, 227) have revealed individual bouts of food intake that are clustered together. These components are differentiated by their size, duration, and the intervals between them. Further details were added to meal structure by considering the drinking that is often closely associated with eating to make up a composite meal (224). FIGURE 4 shows two composite meals eaten by an unmanipulated ∼300 g rat ∼5.5 h into the dark period (from Ref. 225). Each meal is separated by an intermeal interval of ∼10 min. Meal latencies (time from a designated event to meal onset), size, frequency, and temporal distribution within a 24-h period are all variables that are affected by physiological signals and the brain regions and networks with which they interact.
The experimental design likely influences the tight structure shown in FIGURE 4. Meal structures in wild rats probably vary more because they face pressures and challenges not usually present in laboratory settings, particularly for singly house animals. Conspecific social interactions, learning, and other complexities (e.g., Ref. 228) may well impact eating and meal structure in more natural circumstances.
Meal patterns have been characterized in many different species (e.g., Refs. 229–234) including, of course, humans, in whom a broad array of external factors in addition to physiological signals influences the size and timing of meals (reviewed in Ref. 235).
3.2. Types of Eating Behavior
Given the sophistication with which we can now manipulate brain function to investigate eating behavior, beyond measuring gross intake little attention is often directed at determining what exactly a given manipulation is doing to the various components of eating behavior. For example, are manipulations influencing foraging behaviors, changing the latency to eat, meal size, meal frequency, and/or meal distributions within the light/dark period? Simply measuring intake at set intervals after a manipulation will miss some of these important variables, particularly in the short term. This means that ascribing the functional importance of a physiological signal or other manipulation to a particular aspect of behavior will be difficult.
With respect to the circumstances of meal initiation, we identify the three categories in the following sections.
3.2.1. Deficit-induced eating.
Deficit-induced eating is reactive and occurs in response to a clear negative energy balance that usually occurs with fasting or restricted food availability. Therefore, it is sometimes called homeostatic eating (sect. 2.6). The timing and frequency of meals are driven by the availability of food. Physiological signals (e.g., hypoglycemia, hypoleptinemia, and hyperghrelinemia) play a key role in initiating meals by interacting with brain networks to generate the feeling of hunger and stimulate eating. However, sufficient motor control networks are present in the rhombicbrain to sustain an adequate repertoire of deficit-dependent intake behavior (236). Deficit-induced eating is apparently much less important for species that hoard food (237) and possibly also for modern day humans, where available evidence suggests that significant compensatory eating rarely occurs following short periods of food abstinence (238).
3.2.2. Habitual eating
Habitual eating occurs at regular times of day if food is readily available (239). The timing and frequency of meals vary between species. Habitual eating is a proactive (anticipatory) event that generally occurs in the absence of significant energy deficits and their associated physiological signals, particularly hypoglycemia and hypoleptinemia. Habitual eating anticipates and prevents an energy deficit. Nevertheless, it may well be triggered at least in part by peripheral metabolic signals such as changes in nutrient absorption or a metabolic switch. One example of a proactive or anticipatory metabolic signal is the so called premeal decline in blood glucose originally described in rats (240, 241) but also observed in humans who are isolated from all time cues (242). There is also evidence that metabolic rate, if closely recorded, decreases before the onset of habitual meals in rats with unrestricted access to food (243). In addition to internal signals, circadian timing and altered arousal states make significant contributions to initiating meals in most mammals. However, a host of complex social and sensory factors can modify the timing and size of habitual meals in humans (244).
3.2.3. Opportunistic eating.
Opportunistic eating can occur in the absence of any metabolic or energy deficit signal. It can arise as a consequence of two situations: the recalled memory of a previous encounter with favored food items, which leads to increased foraging and eating behaviors, or a direct encounter with favored food items, which leads to increased consummatory behavior. Opportunistic eating involves processing information about previous eating encounters that are encoded within the neural networks responsible for learning, memory, and reward assignment. This processing leads to the activation of motor control systems that initiate appropriate foraging and/or consummatory events. The term hedonic eating is often associated with this type of eating (sect. 2.5). Rather than a primary influence from physiological signals (as occurs with deficit-induced eating), the timing and frequency of opportunistic meals depend on the availability of food or importantly, the presence of environmental cues that predict food (245). However, physiological signals certainly provide important modulatory influences on the expression of opportunistic eating (e.g., Refs. 246, 247). Opportunistic eating plays a significant role in human eating behaviors and may be a major contributor to the dramatic increase in obesity seen in many populations since 1980 (248).
3.3. Species Differences and Their Implications: Rats Versus Mice; Rodents Versus Humans
3.3.1. Preamble.
Human studies provide important information about the role of external and psychological factors in the control of eating, but in-depth studies of the underlying physiological and molecular mechanisms require animal models. As eating is an evolutionary conserved behavior, many physiological control mechanisms exhibit remarkable similarities among different mammals, and some basic principles can even be studied in nonmammals. Nevertheless, substantial species differences exist, and the selection of an animal model usually amounts to a tradeoff between relevance for human physiology and experimental throughput (249). The fact that rats, mice, and other rodents occupy different ecological niches, exhibit different social and ingestive behaviors (250), and are evolutionarily further apart than are humans and apes (251) means that these differences will affect our ability to explain eating behavior at a brain network-wide integrative level. The rat has traditionally been used for physiological and behavioral studies (252). Over the last 30 years however, the mouse has become the standard model used in biomedical research because of the availability of molecular genetic techniques. This has led to an increasingly important but sometimes poorly recognized disconnect between mice, rats, and other species, particularly humans (203).
3.3.2. Brain connectomes.
Although detailed descriptions of mouse brain connectivities are steadily increasing (e.g., Refs. 5, 55, 253–258), most investigators still have to rely to some extent on the rat literature for interpreting results for many brain regions. This approach makes the assumption that rat and mouse connectomes are largely interchangeable. While this may be true at the macroconnection level (region to region), more caution is warranted at the mesoconnection level (neuron to neuron) (see: Refs. 6, 63) for a more detailed explanation of these terms). For example, the rat (259) and mouse (260) paraventricular hypothalamic nucleus (PVH) have significant organizational differences. In addition, while the midbrain and rhombicbrain projections of the PVH have been carefully described in rats (261), there is currently no equivalent study in mice.
3.3.3. Physiology.
Body size is a key example of physiological differences between rodents and humans that have important implications for understanding eating behavior control. This is because the surface-to-volume ratio determines an individual’s thermoneutrality zone, which is the ambient temperature range where core temperature is maintained only by heat loss through the skin. At ambient temperatures below thermoneutrality, energy intake and expenditure must contribute toward maintaining core body temperatures. For mice and rats, the thermoneutral zone is 27–31°C (262, 263). However, even in thermoneutrality these animals must maintain a very high metabolic rate to compensate for the inevitable loss of energy caused by their relatively large surface area. Under common laboratory housing conditions, i.e., at an environmental temperature of 21–24°C, and in particular when they are singly housed, mice and rats are cold stressed (263) and must invest a substantial amount of energy to stay warm. Dressed humans on the other hand usually live in approximately thermoneutral conditions. This appears to translate into general differences between rodents and humans with respect to energy homeostasis. In other words, for mice it may be more efficient to regulate energy homeostasis via changes in energy expenditure, whereas for humans changes in food intake appear to be the better approach (264). One example of these different strategies may be the differences among mice, rats, and humans with respect to the mechanisms causing the body weight loss after bariatric surgery. Common surgical procedures such as Roux-en-Y gastric bypass or vertical sleeve gastrectomy cause a substantial body weight loss in all three species. In humans this body weight loss is mainly the result of a sustained decrease in food intake, whereas mice reduce food intake only temporarily, and the body weight loss is primarily the response to a substantial increase in energy expenditure (see Ref. 264). The rat seems to be somewhere between mouse and human in that there is a sustained decrease in food intake with some relative increase in energy expenditure (264). It is obvious that such differences limit translation of many mouse results to rats and humans.
3.3.4. Behavior.
Evolution dictates that an optimal forager has to maximize benefits (gain) in relation to costs/effort for obtaining food (265). Thus the procurement costs of the food are a major determinant of the species-specific eating behavior in a certain ecological space. As the effort to obtain food increases, meal size increases (e.g., Refs. 230, 266). Similar evolutionary principles apply to special adaptations in eating behavior such as hibernation and food hoarding, as well as strategies to cope with low temperatures and/or seasonally variable or unpredictable food supply (267, 268). Unlike rats and mice, humans and hamsters respond to food deprivation by increasing food hoarding more than intake (237, 269). Consequently, although investigating the nature of the neural mechanisms of these different strategies is inherently useful from a scientific perspective, the results from mice and rats are not so easily translated into understanding of how human ingestive behaviors are controlled by the physiological signals that encode an imminent or existing deficit.
3.3.5. Conclusion.
Using literature from one species to interpret results from another can be misleading without clarification. Species-specific differences are becoming increasingly important and without forethought will most likely be cumulative to the point where they will impair our ability to interpret results. These differences therefore need to be carefully described, and carefully and rigorously incorporated into experimental interpretations.
4. HOW IS THE BRAIN ORGANIZED TO CONTROL EATING BEHAVIORS?
4.1. Neurons and Behavior: Where Is the Connection?
During the past four decades much of what we know about how the brain controls eating behaviors comes from the increasingly sophisticated ways of manipulating cellular signaling and gene control processes. These studies, and particularly those using mouse genetics, have dominated the field for the past 15 yr. However, to some extent this has been at the expense of more in-depth behavioral analyses. That the focus of these genetically targeted manipulations is currently more on food intake rather than the broader aspects of eating behaviors is understandable, but it is anticipated that these approaches will at some point be directed to exploring the complexities of the appetitive actions, meal patterning, behavioral switching, etc. that comprise eating behavior.
Building on the foundation of what was once called physiological psychology, this more recent molecularly focused body of work has continued to draw attention to a relatively small number of brain regions. These include the LHA, PVH, VMH, dorsomedial (DMH), the arcuate (ARH) nuclei in the hypothalamus, and the parabrachial nucleus (PB) and nucleus of the solitary tract (NTS) in the rhombicbrain. The reason for this is clear: these regions are without doubt key contributors to the control of eating behaviors. However, while we know that particular sets of neurons within these regions help control eating behavior, often with an astonishing degree of molecular detail, our ability to define the specific roles of many neurons within a broader brain-wide context, remains elusive.
An important reason for this lack of clarity is that many studies do not report which specific aspects of eating behavior are affected when a particular set of neurons is manipulated; they simply report intake per unit of time rather than their effects on the structure of meals where food is freely available. As we have discussed earlier (sect. 3.1.2), although intake can be a useful parameter for some designs, it cannot be interpreted in terms of eating behaviors that involve learned associations and effort-based responses. Another reason is that the connectional organization of many brain regions remains poorly documented at the mesoscale (neuron type to neuron type, e.g., ARHAgRP neurons to PVHCRH neurons) and in some instances at the lower resolution macroscale (region to region, e.g., ARH to PVH). This is particularly true in mice, although our understanding is steadily and significantly improving (e.g., Refs. 5, 11, 55, 253). Another complication for defining networks is that communication between brain regions is not exclusively synaptic, as is shown by the ability of volume transmitted peptide signals (207, 212, 270) to alter eating behavior (200, 271) (see sect. 2.7.4).
Functional clarity is emerging for some regions because they can be manipulated with exquisite precision in mice (e.g., the PVH and ARH; Refs. 272–276). However, we still lack good functional models for more structurally complex regions like the LHA, the bed nuclei of the terminal stria (BST), and the PB. Their diverse neuronal populations, regional connectivity, and functional parcellation in mice remain incompletely characterized when compared with rats. This relative lack of detailed species-specific connectional information becomes important for contextualizing cell groups that have not previously been closely associated with eating behaviors, for example, the parasubthalamic nucleus (PSTN) and zona incerta (ZI) (277). Although this knowledge gap makes it difficult to move network analyses beyond the few already well-characterized regions in mice, it is an inevitable consequence of the advanced state of genetic manipulations in mice compared those in rats. However, current improvements in the ability to perform these types of manipulations in rats could soon help close this gap, which would be a benefit to the field.
As our ability to understand brain mechanisms becomes more fine-grained, appreciating species specificity is increasingly important because of the varied behavioral expression patterns seen in different species to the same challenges (see Ref. 278) for a wide-ranging discussion of species differences) (also see sect. 3.3.). Do connectional variations between these species contribute to these differences? Because we do not know, it may be better not to assume that the organization of the relevant neural connections are the same in each, particularly at meso- and microscale (synaptic connections between neurons) levels.
Despite these issues, it is still possible to put together a broad framework into which existing and new results can be placed to help understand how the brain controls eating behaviors. A framework of this kind is discussed in sect. 4.2. It provides the foundation for the more detailed mechanisms that are discussed later.
4.2. Integration and Action
4.2.1. Sensory-motor integration.
The fundamental principle of how the brain controls eating, and indeed all motivated behaviors, is that many types of sensory information must be integrated with motor control mechanisms in ways that enable the appropriate behavioral actions for eating. That the cerebrospinal trunk must link sensation with motor action was implicit in 19th century physiology. Ferrier (279) pointed out the mechanistic importance of “associations” between the brain’s motor and sensory regions for what he called “volitional behaviors,” an idea that was developed into the more explicit concept of sensory-motor integration by Sherrington ∼1900 (280, 281). Indeed, Sherrington (280) may have been the first to use the term integration in this context. Sensory-motor integration of this type raises a fundamental organizational question about how the brain uses the many and varied physiological signals to prioritize and organize the behavioral motor actions that collectively make up eating behavior. Put another way, how are physiological signals integrated with motivation and drive mechanisms so that the most appropriate motor actions are engaged for eating?
At the simplest level, there are two principal ways that physiological signals can influence motor control mechanisms (8, 32, 35, 282, 283). First, Reflex Control of motor outputs by sensory information occurs more or less directly with little to no intervening processing. Although motivated behaviors are, by definition, not reflexes, the sets of orofacial muscle movements associated with eating can be expressed with little or no processed control. For example, these are seen in decerebrate rats where orofacial movements are appropriately expressed to accept or reject food items placed into their mouths (284). Second, Processed Control of motor outputs by sensory information occurs after it has interacted with two substantial and highly complex brain domains: one responsible for Behavioral State and Circadian Timing; the other for the Neural Representation of Sensory Objects (29, 283, 285, 286). The network bases for these interactions is discussed in sect. 4.2.4.2.
4.2.2. Physiological state.
The expression of all behaviors takes place upon the backdrop of an animal’s physiological state. Physiological state varies depending on sex, age and development stage, and environmental conditions (temperature, photoperiod, etc.). It impacts all levels of motivated behavioral expression. Hormones play key roles in mediating changes in physiological state, which in turn modulates the functions of the brain domains for processed control of motor actions and how these respond to proactive and reactive signals.
4.2.3. Exteroceptive and interoceptive signal modalities.
Two modalities of sensory signals control eating behaviors: exteroceptive and interoceptive. Signals in both groups are each transduced by sets of specific receptors that are distributed throughout the body. Reflex and processed control mechanisms use both modalities.
Exteroceptive modalities are signals that originate from sources in the external environment. They encode the flavor (the result of integrating taste, smell, and touch), appearance, and sound of food. They enter the brain by way of defined or labeled line neural pathways. Exteroceptive signals are processed in cortical and subcortical brain regions and have a major impact on eating behavior control mechanisms often in combination with internally generated physiological signals.
Interoceptive modalities derive from within the body. They are humorally and neurally conveyed into the brain, and they make critical operational contributions to virtually all functional brain domains.
The nature of exteroceptive and interoceptive signals is not exclusive; some signals fit into both categories. For example, some molecules act as exteroceptive signals because they are present in foods or they are products of digestion (e.g., glucose, free fatty acids, some amino acids). They are detected by oral cavity taste receptors and other chemosensory receptors distributed throughout the gut. However, some of these same molecules are also internally generated by metabolism and so are interoceptive signals. These are sensed by sensory nerve mechanisms in the vasculature and by specific receptors in the brain. The important point about the different signal modalities is that the source of a particular signal determines the variety and locations of its transduction systems and therefore the route that transduced information takes into the brain (see sect. 4.3).
4.2.4. Integrating sensory information.
A thorough analysis of the available evidence supports a more or less hierarchical organization of how the brain controls eating. In this way, peripheral sensors represented by, or connected to, vagal (VSN) or spinal sensory (SSN) nerves register small physiological changes in those signals that can control eating (e.g., glucose, fatty acid oxidation, GLP-1). The brain itself usually does not detect such changes directly. Instead, peripheral signals are first transduced and integrated by VSNs and SSNs before being conveyed to and then processed by parts of the rhombicbrain. This processed information is then further integrated with those brain regions that control eating behavior. This whole process allows for proactive eating (i.e., habitual meals), thereby avoiding more dramatic swings in energy balance. Pharmacological manipulations of these physiological signals, however, often produce substantial and rapid changes in signal intensity that are more akin to an emergency threat (glucoprivation, lipoprivation, insulin-induced hypoglycemia). These manipulations are directly detected by the brain, and they trigger immediate reactive changes in eating (287). Such a hierarchical organization appears to be a general physiological principle because similar features can be observed in the mechanisms that control blood osmolality (288), body temperature (109, 289), blood pressure (290), and, of course, blood glucose (291, 292). Consistent with this hierarchical organization, substantial integration of signals that control eating occurs in the periphery. We now discuss these peripheral and brain integrative mechanism in more detail.
4.2.4.1. integration in the periphery.
Sensory nerves and in particular VSNs carry a major part of the sensory information from the gut to the brain (293). The cell bodies of VSNs and SSNs are both pseudounipolar cells located, respectively, in the bilateral nodose ganglia and the dorsal root ganglia (DRG). They are able to sense mechanical, a variety of chemical, thermal, and some noxious stimuli. Because of their intrinsic plasticity, VSNs are ideally suited to react to and integrate different peripheral stimuli that affect eating behavior and autonomic responses. Different mechanisms account for this plasticity, and some of them are particularly relevant for the issues discussed here: 1) the large number of polymodal nerve fibers that are able to respond to and integrate different stimuli, e.g., mechanical and chemical; 2) the large number of different receptors expressed on single nerve fibers, which allow for integration of the signals triggered by their cognate stimuli; and 3) the rapid changes in conductance and changes or switches in the expression of neuropeptides and neuropeptide receptors in response to various stimuli. Taken together, these points emphasize that neurally conveyed sensory information from the gut has already undergone substantial integration before it reaches the brain. This means that VSNs and SSNs represent important integrative units that are distinct from those in the rhombicbrain and more rostral brain regions (FIGURE 5).
FIGURE 5.
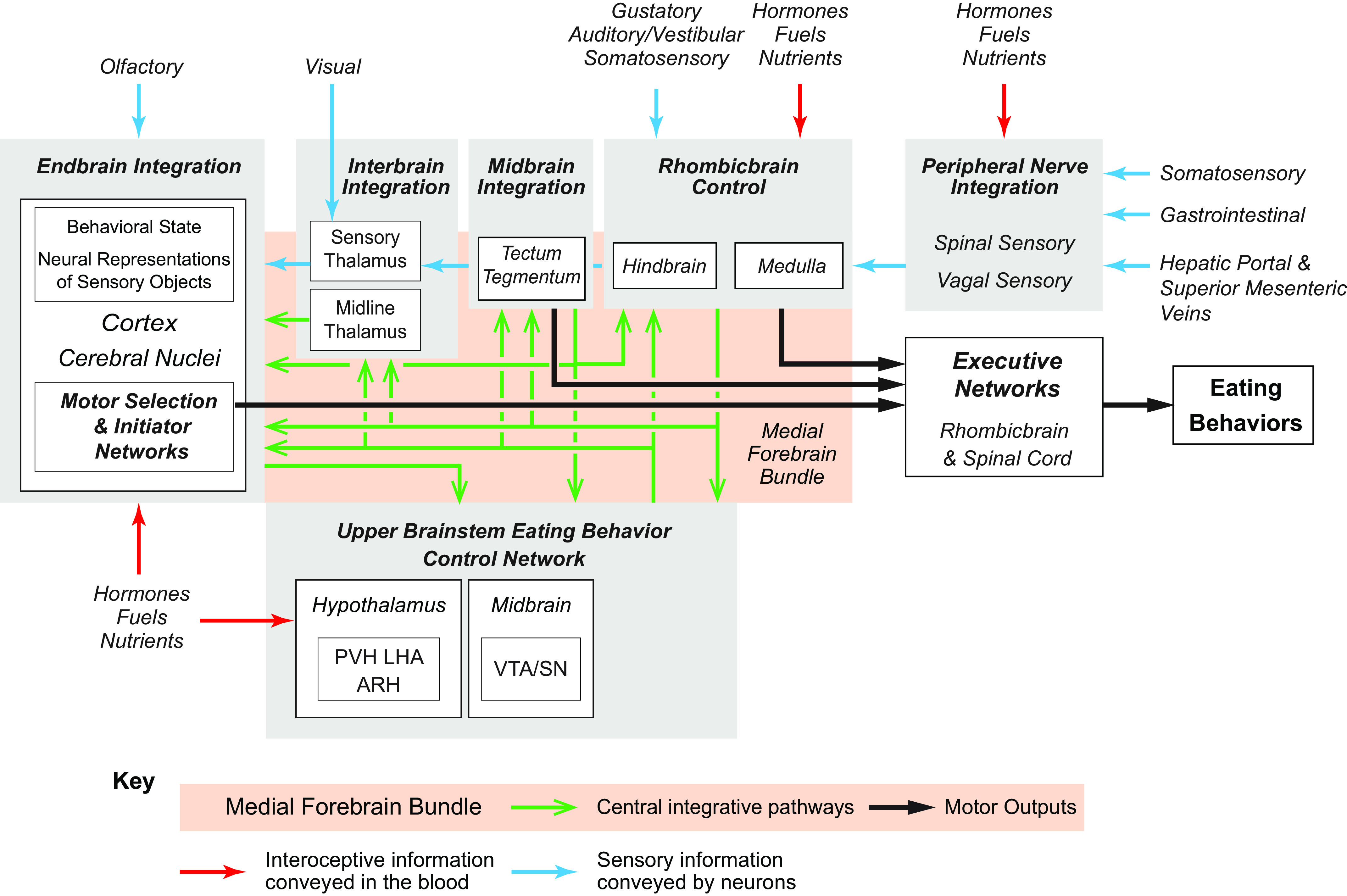
A schematic representation of the sensory-motor integration that occurs across the entire cerebrospinal trunk to control eating behaviors. The integration of interoceptive and exteroceptive information occurs in networks found at all brain levels (gray boxes). This ranges from the spinal and vagal sensory nerves, whose information is first received by the medulla, to cortical regions in the endbrain. Some of this sensory information is conveyed by neurons (blue arrows) and some in the blood (red arrows). The Upper Brainstem Eating Control Network located in the hypothalamus and the rostal parts of the midbrain plays a particularly important role in prioritizing and organizing motivated eating behaviors. It sends information to the endbrain, the midline thalamus (primarily to the paraventricular nucleus of the thalamus), the midbrain, and the rhombicbrain. Information flow within and between these brain networks is enabled by numerous and complex sets of connections (green arrows), many of which travel in the medial forebrain bundle (orange box), one of the largest and most complex fiber tracts in the mammalian brain (667). Motor control is enabled by projections (black arrows) that originate from integrative networks in the endbrain, midbrain, and rhombicbrain (also see FIGURE 6). See FIGURE 1 for the hierarchical organization and naming of the brain parts shown here. PVN, paraventricular nucleus; LHA, lateral hypothalamic area; ARH, arcuate nuclei in the hypothalamus; VTA, ventral tegmental area; SN, substantia nigra.
4.2.4.2. integration in the brain.
While many parts of the brain are capable of responding directly to blood-borne signals, only the medulla receives GI information conveyed by VSNs and SSNs. This region is therefore the brain’s gateway for this information (FIGURE 5). Furthermore, because some medullary neurons and glia have specific receptor systems for nutrients and hormones, this part of the brain can directly integrate humorally and neurally conveyed sensory signals (FIGURE 5). For example, some NTS neurons that receive information about gastric distention from VSNs also express leptin receptors (295).
Although receptors for nutrients, hormones, and ions are widely expressed by forebrain neurons and glia, these regions can only assimilate vagal and spinal sensory information by way of the complex and neurochemically diverse sets of connections they receive from the rhombicbrain (FIGURE 5). Without these connections, the forebrain does not perceive spinal and vagally conveyed interoceptive information from the periphery.
FIGURE 5 shows that much of the sensory information relevant to eating behaviors is integrated with two large and highly complex forebrain domains. One is responsible for Behavioral State (sleep/wake, attention and vigilance). The other is responsible for encoding Neural Representations of Sensory Objects (283, 285, 286). This hugely complex and widely distributed processing system is responsible for the following functions:
Reward/aversion assignment.
Learning. Memory consolidation and retrieval.
Processing egocentric (the relative position and orientation of the body and body parts) and allocentric (the location of the body in the environment) representations (see Refs. 283, 296) for further discussion).
Equally important from an integrative standpoint are the caudally directed connections from the forebrain to the rhombicbrain (FIGURE 5), which are also critical contributors to the way the brain controls eating. The organization and functional significance of all these connections are discussed later (sect. 5.4. and 6.2).
4.2.5. The motor control of eating: how do we connect eating control networks with the motor actions of eating?
4.2.5.1. general organization.
To provide the foundation and context for considering how various brain components interact to control eating behaviors (sects. 5 and 6), it is worth summarizing current ideas about how the brain’s motor control systems are organized.
Modeling these functions derives in part from two groups of studies whose origins go back almost a century. The first are the large-scale brain lesions and sections used to determine which aspects of eating behaviors are supported by regions distributed along the cerebrospinal trunk. These began with Ranson’s pioneering studies in the 1930s (297) that were later refined in the 1970s and 80s (298–300). They effectively showed which components of eating behaviors are maintained by isolated brain regions. The second group comprises neuroanatomical studies whose numbers expanded dramatically in the 1980s. These have been used by many groups to identify thousands of brain macroconnections in the rat and more recently, the mouse.
The collective outcome of these and many other functional and neuroanatomical studies is a minimal model of sensory-motor integration for motivated behaviors (FIGURE 6A) (8, 62). It involves a triple descending projection from the cerebral cortex and cerebral nuclei onto motor control systems located in the hypothalamus and further caudally (FIGURE 6Ai). These connections encode the output from the networks responsible for the Neural Representations of Sensory Objects in the endbrain (FIGURE 5). Section 5.4.5 presents more details about how these descending projections relate to eating behaviors. When combined with numerous experiments that have revealed many details of cerebrospinal motor control processes (reviewed by Refs. 301, 302), this minimal model proposes that the motor actions of eating are controlled by brain networks that operate at three functional levels (FIGURE 6Aii) (reviewed in Refs. 8, 62). These levels are described in the following sections.
FIGURE 6.
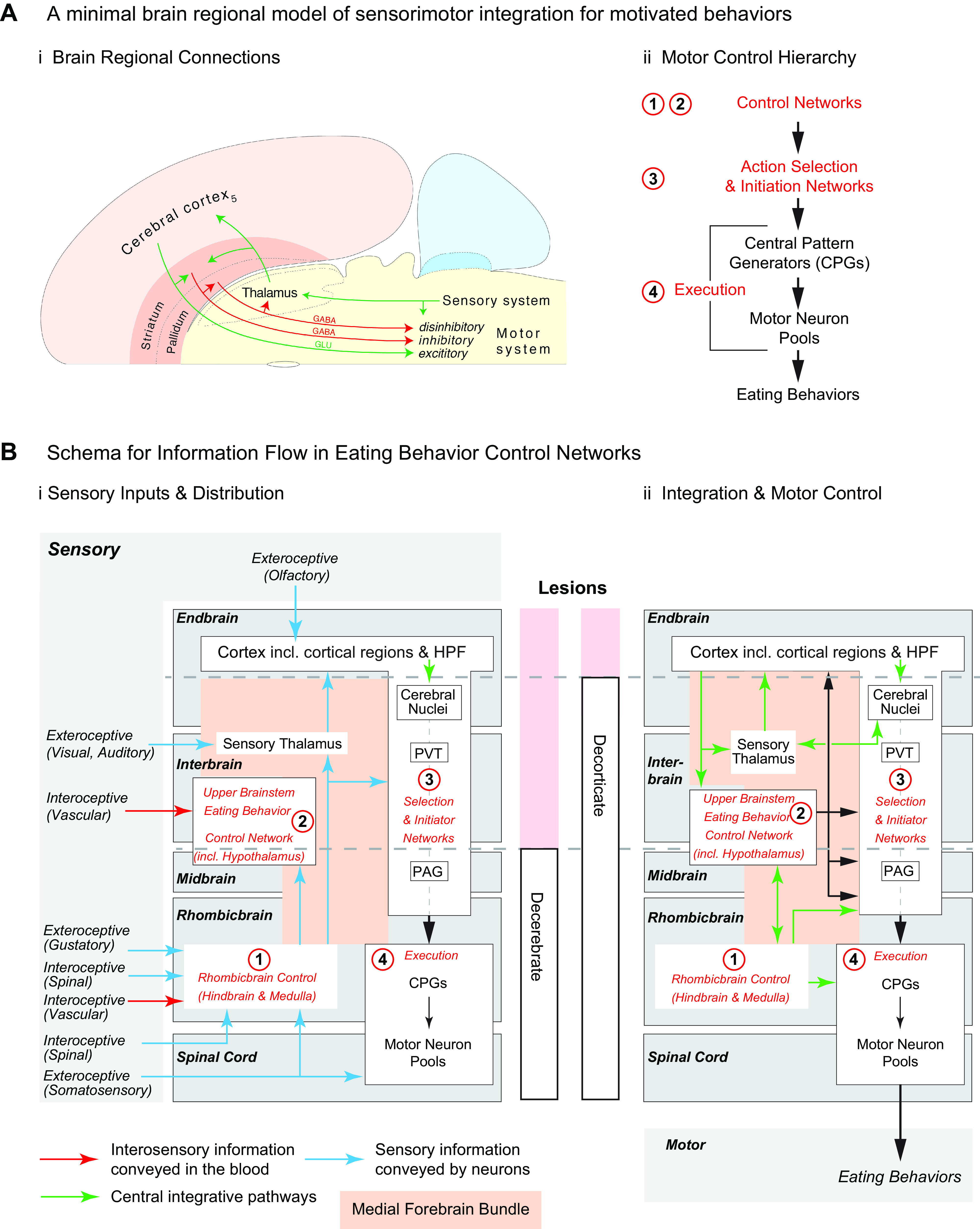
Ai: the connections from the cerebral cortex (light orange) and cortical nuclei (dark orange) into the sensory and motor systems of the brainstem (yellow) as described by Swanson (8, 62). The motor components are organized as a triple descending system of glutamatergic (green arrow) and GABAergic (red arrows) connections. Aii: the functional hierarchy of motor control for Eating Behaviors involves four networks that are organized at 3 levels (red): Control (1 and 2); Selection & Initiation (3); and Execution (4), which consists of central pattern generators and motor neuron pools. B: 2 schema to illustrate the organization of information flow related to sensory inputs (Bi) and motor outputs (Bii) of the 4 networks (red) identified in Aii. They are distributed across the endbrain, interbrain, midbrain, rhombicbrain, and spinal cord. The dashed horizontal lines represent the locations of the lesions used in rats to generate decerebrate and decorticate animals. See text for more details. CPG, central pattern generators; PAG, periaqueductal gray; PVT, paraventricular nucleus of the thalamus; HPF, hippocampal formation.
4.2.5.1.1. Control networks.
Control networks perform sensory-motor integration of varying degrees of sophistication to inform downstream mechanisms, which then initiate and execute appropriate motor events (FIGURE 6Aii). The control networks for eating behavior contain core components that exhibit all of the following properties:
They must consistently and robustly influence food intake when experimentally manipulated;
Because ATP availability is the pivotal regulated variable in energy balance (FIGURE 2), AMPK function in core network components should have direct effects on eating behaviors;
They should receive direct humoral and neural inputs that provide the means for physiological signals to influence their activity;
They should possess verified functional outputs to Behavior (Action) Selection and Initiation networks (see FIGURE 6).
We identify two control networks for eating behaviors with these properties: one in the rhombicbrain, and one in the upper brainstem. We note that these two networks may not be the only ones that fit these criteria, but they are currently the best characterized.
The rhombicbrain control network for eating involves the PB in the hindbrain and the NTS in the medulla. It is essential for integrating information from the GI tract and the spinal trigeminal complex (STC), including aversive stimuli, into motor control. As demonstrated by the primitive consummatory actions of decerebrate animals, this rhombicbrain network connects to Selection and Initiation, and Execution mechanisms located caudal to the brain cut to enable these movements (FIGURE 6Bii). However, in intact animals bidirectional, robust, and complex rhombicbrain connections with forebrain regions enable a far more sophisticated and motivated control of eating (FIGURE 6Bii).
Sets of behavior control networks in the upper brainstem, which includes the hypothalamus and part of the midbrain, instill appetitive and consummatory movements with a particular purpose, direction, or drive (62, 283, 303, 304). In this way these movements become part of a motivated behavior sequence (FIGURE 3). These control networks continuously integrate sets of interoceptive and preprocessed exteroceptive information from many sources to prioritize and organize the particular motivated behavior that is currently the most appropriate, or put another way, the one with the highest drive level.
The more rostral components of these control networks are located in the hypothalamus. They are organized in ways that enable specific motivated behaviors, including eating. The more caudally located components are in the caudal hypothalamus and midbrain (62). They consist of networks distributed between the mammillary bodies, the VTA, reticular substantia nigra (SN), and the parvicellular midbrain reticular nucleus, which is also known as the midbrain locomotor region. These components help organize body movements used for foraging (e.g., head and eye orienting movements, body locomotion, etc.) and so are less behavior-specific than the more rostral hypothalamic components. Collectively the hypothalamic and midbrain components form the upper brainstem behavior control networks.
The eating behavior control network in the upper brainstem, like the ones for other motivated behaviors, provides ascending projections to the endbrain, and descending projections to the midbrain, and rhombicbrain (FIGURE 6Bii) (8, 62). Notably however, these descending projections do not innervate any neurons that are directly responsible for motor execution (FIGURE 6Bii). Instead, the information encoded in descending projections is routed to executive premotor and motor neurons through Selection and Initiation networks (FIGURE 6Bii), primarily in the cerebral nuclei but possibly also in the midbrain periaqueductal gray (PAG) (305) and superior colliculus (258). The specific structure of the upper brainstem eating behavior control network is discussed in more detail in sect. 5.5.4.
4.2.5.1.2. Action selection and initiation.
This involves selecting and temporally sequencing the appropriate sets of motor programs that execute the highest priority behavior (e.g., Refs. 301, 306). For eating, these include approach, licking, biting, chewing, and swallowing. Arranging motor programs into a correctly sequenced and successfully executed behavior has been called behavioral syntax (307), which, in the case of chewing, breathing, and swallowing, for example, obviously have to be sequenced correctly for survival. While primitive motor action selection can be organized by rhombicbrain networks, the striatum and pallidum are crucial for motivated behavior sequences. Some striatal regions use dopaminergic mechanisms to assign incentive salience (reward/aversion) to objects in their internal and external environments. Importantly, these mechanisms are not dedicated to specific behaviors. Instead, they are targeted toward a particular behavior by inputs from cortical regions, parts of the thalamus, particularly the paraventricular thalamic nucleus (PVT), and the upper brainstem behavior control networks, including neurons in the LHA and VTA. Therefore, when these forebrain components work together they provide the context for selecting, sequencing, and instilling the required commitment and vigor (301, 308, 309) to the appropriate motor programs for appetitive and consummatory eating actions.
4.2.5.1.3. Execution.
Movements are executed by muscles that are controlled by motor neurons and their premotor control networks (FIGURE 6Aii). The motor neurons responsible for locomotion are located in the dorsal horn of the spinal cord, while the cranial nerve motor neurons responsible for the orofacial movements of eating are located in the hindbrain and medulla. All these motor neurons share a common premotor control mechanism: the generation of rhythmic movements by proximally located central pattern generators (CPGs). These directly control the skeletal muscles that enable the appetitive and consummatory actions of eating (301, 302, 310, 311). Movement during foraging requires obvious rhythmic and repetitive actions, running, walking, etc. However, as described by Tinbergen (303), the motor actions of the consummatory phase of motivated behaviors are also simple, rhythmic, and stereotypic. During eating this involves manipulating food items, salivation, licking, biting, chewing, swallowing, and the other proximate motor events required for food interactions. They must be expressed in a particular sequence to ingest the chosen food item successfully; chew before you swallow, for example (312). Also, orofacial movements during eating have to be coordinated with breathing to avoid choking (313).
Obviously, motor neurons and their proximal premotor control networks can be used for many behaviors. This is clear for whole body movements, but is also true for the orofacial movements of eating. For example, tongue (lingual) muscles are not only used for ingestive behaviors, but also for offspring grooming in maternal behavior, and for other motivated behaviors. Motor neurons in the hypoglossal nucleus initiate tongue movements, but their precise mode of engagement is controlled by sets of premotor neurons located in the rhombicbrain (314) that are responsible for the rhythmic movements characteristic of particular consummatory actions. In turn, selecting the premotor networks that are behaviorally most appropriate requires inputs from the Selection and Initiator networks we just described (FIGURE 6). Selection processes are specifically engaged for particular behaviors; for example, the program used by a female rat for rapid licking during drinking is different from the one she uses to groom her offspring. The upstream commands for selecting which particular licking pattern is engaged most likely originate in the upper brainstem eating control network which, by way of its projections to Selection and Initiator networks, ultimately bias the actions of downstream execution neurons and CPGs for hypoglossal neurons. Once licking and chewing begin, sensory feedback from these actions becomes a major controlling factor for continued eating (315).
4.2.5.2. decerebrate and decorticate animals.
FIGURE 6B illustrates which motor control components can still influence motor execution in decerebrate animals, where a cut is made just rostral to the superior colliculus (299) and in decorticate animals where the entire cerebral cortex, including the HPF, is removed (300, 316).
Decerebrate animals have all ascending and descending connections severed at the midbrain-interbrain boundary, meaning that signals from the upper brainstem eating control and initiation-selection networks rostral to the cut cannot reach executive networks in the rhombicbrain. However, these animals retain connections between rhombicbrain control networks that process incoming interoceptive and exteroceptive information, and those that execute orofacial motor patterns for licking, chewing, swallowing etc. Decorticate animals retain all bidirectional connections between the cerebral nuclei and all regions located further caudal, including the thalamus and hypothalamus (300). While these gross lesions clearly have limited ability to resolve fine-grained questions, they do provide useful pointers about which motor functions survive large-scale brain disconnections.
If decerebrate animals are challenged by negative energy balance (298), including insulin-induced hypoglycemia (317), they can execute appropriate orofacial movements to consume favored food items and reject aversive foods, providing food is placed directly into their mouths. However, they cannot compensate by overconsuming calories following acute energy restriction, or when maintained on a meal entrainment schedule (318, 319). They are also incapable of initiating any appetitive or consummatory actions in response to external stimuli, i.e., motivated eating (298). Interestingly, anencephalic newborn humans also show gustofacial responses of ingestion when presented with taste solutions (320). Decerebrate rats can, however, control meal size (298) and react to satiating signals (e.g., exogenous CCK, GLP-1 agonists, gastric distension) (321) in the same way as intact rats.
Although decorticate animals initially require some coaching to eat and drink because of impaired orofacial movement deficits, they eventually recover sufficiently to self-maintain their body weights at ∼90% of controls, including the ability to eat hard food pellets (300). Their deficits with regards to executing eating behavior fall into two groups: first, motor deficits, including orofacial movements, particularly licking, and the skilled movements associated with food manipulation; and second, the inability to execute tasks that require place learning and generating search strategies (322), which is likely a consequence of losing hippocampally mediated processes (323).
4.2.5.3. connecting hypothalamic eating control networks to the motor actions of eating.
The central role the hypothalamus plays in controlling motivated behavior is now universally accepted. This position has been accompanied by a dramatic increase in our knowledge of the functional organization of both the hypothalamus and the brain’s motor control systems. However, which neural connections provide the links between the hypothalamus and the forebrain motor control systems responsible for selecting and initiating eating behaviors? Despite this arguably being one of the most important questions for understanding the neural control of motivated behaviors, detailed answers are still sparse. At the most fundamental level, where this interaction occurs must be constrained by the destinations of output signals—both synaptic and nonsynaptic—from the various hypothalamic regions involved with controlling eating behaviors. Section 5.5.2 describes these outputs in greater detail, but those from the PVH, ARH, and particularly the LHA and VTA, seem best placed to provide motivational bias to the regions that organize the various motor actions of eating behaviors.
4.3. Peripheral Physiological Signals
4.3.1. How and where do physiological signals enter the brain?
The access points through which transduced sensory modalities enter the brain have long been considered as guides for identifying which downstream neural systems process this information. Two classic examples are the principal termination points of retinal sensory nerves in the optic nerve: the lateral geniculate nucleus (LGN) in the thalamus, and the suprachiasmatic nucleus (SCH) in the hypothalamus. Examining LGN function provided the first step toward a deeper understanding of how the brain processes visual information (324), while identifying the SCH pinpointed the location of the brain’s circadian oscillator (325–327). The access points used by physiological signals to enter the brain are therefore a useful starting point for thinking about how these signals ultimately control eating behaviors.
Physiological signals related to eating, energy balance, and energy homeostasis reach the brain by two principal pathways. First, information in the GI tract, hepatic portal vein, and adipose tissue is transduced by sensory mechanisms into neural signals that are conveyed to the brain by VSNs and SSNs. VSNs are well characterized, and although the overall scheme of SSNs is clear, the details of their trajectories from most organs associated with eating remain poorly understood. Second, the blood carries information that is related to metabolism to the brain in the form of hormones, nutrients, and ions. How these two modes of communication, neural and vascular, engage the brain is therefore quite different.
4.3.2. Neural pathways: vagal and spinal sensory nerves.
4.3.2.1. vagal sensory nerves.
Vagal sensory nerves (VSNs) carry a major part of the sensory information from the gut to the brain. The vagus nerve (the Xth cranial nerve) is a mixed nerve and an essential component of the physiological control system for eating. Up to 20% of its axons are parasympathetic (cholinergic) preganglionic motor fibers that originate in the dorsal motor nucleus of the vagus (DMX) and the ambiguous nucleus (AMB). These DMX preganglionic axons innervate parasympathetic ganglia close to the target organs, including those in the GI tract. Postganglionic cholinergic axons then provide the motor innervation of these organs. The remaining 75–80% are axons of the VSN (328). The sensory/motor fiber ratio in the vagus nerve is dramatically different to that in sympathetic nerves, where in the lumbar splanchnic nerve for example only ∼20% are sensory (329). VSNs are pseudounipolar cells whose cell bodies are located in the paired (left/right) nodose ganglia in the cervical region close to the internal carotid arteries. VSNs innervate all thoracic and abdominal organs including those in the hepatoportal region, the pancreas, and the GI tract. Their distribution within these target organs has been very well characterized using a variety of spatially and genetically directed neuroanatomical tracers (328, 330–336). As discussed in sect. 4.2.4.1, VSNs are sensitive to various chemical, thermal, and mechanical stimuli (330, 337). Because of this intrinsic plasticity, VSNs are ideally suited to react to and integrate different peripheral stimuli that affect eating behavior and autonomic responses. A striking example of the integrative capacity and plasticity of VSNs is the fact that activation of the same vagal fibers by different stimuli can either promote or inhibit eating, presumably depending on context and/or different dynamics of the neural response (see sect. 5.6.5). The important contribution of VSNs to the physiological control of eating behaviors is therefore unquestioned (e.g., Refs. 293, 334, 337–353).
Last, but not least, the vagus is certainly involved in mediating many effects of gut microbiota on central nervous system functions, including behavior. In fact, vagal nerve stimulation is a Food and Drug Administration-approved and well-established therapy for otherwise treatment-resistant epilepsy and other neuropsychiatric diseases (see Ref. 354). On the other hand, subdiaphragmatic vagal deafferentation, a surgical technique that eliminates all VSNs from below the diaphragm but does not heavily compromise GI functions because it leaves ∼50% of the vagal motor nerves intact (355), produces substantial effects on rat innate anxiety and learned fear, as well as on cognitive functions and affective behaviors (356–358). Gut microbiota can affect VSN signaling and, hence, modulate brain functions by stimulating the release of cytokines from mucosal immune cells or the release of gut hormones from enteroendocrine cells (359, 360). Short-chain fatty acids and other bioactive molecules produced by gut microbiota may trigger the release of these gut peptides.
VSNs project directly into the dorsolateral medulla. From there they join the solitary tract, which distributes to second-order neurons located along the length of the NTS (361). Some idea of their widespread rostrocaudal distribution in the NTS is shown by the extent of IB4 binding (FIGURE 7). IB4 is a lectin that binds to small unmyelinated primary sensory nerves (176, 325). The precise topographic distribution of vagal sensory information to second order NTS neurons is difficult to define precisely because the dendritic arbors of these NTS neurons can extend across several cytoarchitectonically defined NTS regions (361). This results in what has been called (361) a blurred rather than the more precise viscerotopic representation in the NTS that is often described. It emphasizes that it is not possible to assess how particular NTS neurons integrate vagal and spinal sensory information to impact eating behaviors without considering their dendritic organizations; a feature that is equally applicable to other brain regions associated with the control of energy balance (e.g., Refs. 72, 110, 272, 362–364).
FIGURE 7.

Dark field (A–E) and fluorescent (A’–E’) photomicrographs of 5 rostral to caudal coronal sections of the medulla from a single male rat injected intraperitoneally with fluorogold to retrogradely label neurons that project outside the blood-brain barrier (white neurons in A’–E’). Isolectin B4 (IB4) binding was used to label primary sensory inputs, including those in the vagus nerve (green). Parasympathetic pre-ganglionic neurons are identified using choline acetyl transferase immunohistochemistry (blue), some of which also colocalize fluorogold. The photomicrographs show the distribution of vagal sensory nerves as they enter the dorsal medulla (A’), the solitary tract (A’ and B’), and then the entire rostrocaudal extent of the nucleus of the solitary tract (C’ and D’). Labeling in the area postrema is a mixture of fluorogold accumulated from the circulation and IB4. We thank Myrtha Arnold (ETH Zürich) for preparing the rat brain used in this figure. Red scale bars = 500 µm. AMBv, ambiguous nucleus, ventral division; AP, area postrema; DMX, dorsal motor nucleus of the vagus; GR, gracile nucleus; Icp, internal cerebellar peduncle; NTS, nucleus of the solitary tract; co, NTS, commissural part; ge, NTS, gelatinous part; m, NTS, medial part; sptV, spinal tract of the trigeminal nerve; ts, solitary tract; Xa, afferent nerve of the vagus; XII, hypoglossal nucleus; Xe, efferent nerve of the vagus; XIIe, efferent nerve of the hypoglossal.
Neuroanatomical tracing with the herpes simplex-1 H129 (H129) virus reveals how vagal sensory information is conveyed from the NTS to other brain regions. H129 is an anterogradely transported polysynaptic neurotropic virus that sequentially infects neural networks connected to the primary infected neurons (365). When injected into peripheral targets, H129 anterogradely infects their associated sensory network. Two studies have injected H129 either into both nodose ganglia of mice (345), or into the left nodose of rats (366). In addition to the primary projections to the NTS seen in both species, downstream projections were seen to the area postrema (AP), PB, and parts of the hypothalamus, including the PVH, DMH, the PSTN and other parts of the LHA. Somewhat different projection patterns were seen from the right and left nodose ganglia in the mouse (345).
4.3.2.2. spinal sensory nerves.
The fact that spinal senory nerves (SSNs) convey sensory information from abdominal organs, including the stomach, intestine, and their blood vessels, has been known for over a century (345, 367) (for reviews see Refs. 368–370). Although they have been called sympathetic afferents because they track with sympathetic efferents in the splanchnic nerves, the value of the sympathetic label has been questioned (371–373). Nonetheless, these SSNs are well positioned to convey important information to the brain about GI/hepatoportal etc. state, particularly with regards to physiological threats, to help regulate energy balance. The majority of research about their organization has focused on their role in nociception and visceral pain (374, 375). However, recognition that some of these SSNs convey chemosensory information that relates to energy balance has gradually accumulated (103, 288, 336, 376–378).
The cell bodies of small diameter abdominal SSNs (A∂ and C fibers) are in the thoracic and lumbar DRG. Their distal processes run in the splanchnic nerves through the celiac and superior mesenteric ganglia. The majority of these DRG neurons terminate on lamina I neurons in the dorsal horn of the thoracic spinal cord (379). However, some C fibers also terminate in laminae II, V, and X, at least in the rat and guinea pig (369, 379, 380). The distribution of these small diameter SSNs can cover five or more spinal segments (380), while the dendritic arborization of the dorsal horn neurons upon which they terminate is similarly extensive. Interoceptive (viscerosensory) information from these nerves therefore distributes rather broadly within the spinal cord.
Information about the spinal sensory innervation of particular GI organs is steadily accumulating. To date, only three peripheral targets related to energy balance control have been investigated using H129 injections in rats, Siberian hamsters, or shrews: the stomach wall (381, 382), brown and white adipose tissue (383–386), and the wall of the hepatic portal vein (387). In all cases, infected neurons were found in the DRGs or dorsal horn of the spinal cord indicating that interoceptive input to the brain from these sites is conveyed at least in part by SSNs. A recent study using genetically targeted tracing shows SSN innervation in parts of the GI tract (336). However, more studies are required to improve our understanding of the role of SSNs that may innervate other peripheral organs related to energy balance.
Dorsal horn neurons project locally within the spinal cord to mediate spinal autonomic reflexes (373). Importantly, however, they also project into the brain using three routes: a spinothalamic projection (388, 389); a spinoparabrachial projection (390, 391); and projections to the NTS (391, 392). Although the majority of studies addressing these pathways has focused on pain, there is currently no evidence that spinal interoceptive and nociceptive pathways enter the brain by substantially different routes.
Craig (393) has proposed that pain (including visceral pain) is conveyed by SSNs to the dorsal horn as part of a multimodal “homeostatic afferent pathway.” Considered in this way it seems reasonable to include other interoceptive signals, particularly those that carry a physiological threat [e.g., hypoglycemia or hypernatremia (103, 376)], in this same category (394–397). Therefore, this peripheral sensory receptor-SSN-spinal cord-brain network conveys physiologically threatening interoceptive information to the brain. With regard to the control of eating, it is notable that rat spinal/trigeminal dorsal horn neurons appear to participate in suppressing food intake that results from pain by way of their projections to the PB (398) before being conveyed to the hypothalamus and other parts of the forebrain (also see sect. 6.3.1.1.3). Of interest here is the recent finding that ARHAGRP projections to the PB can modulate nociceptive processing during hunger (399). Finally, caution should be exercised in attempting cross-species comparisons because of the substantial species differences in spinothalamic-cortical projections (400).
4.3.3. Integration in the hindbrain, medulla, and peripheral nerves.
The medulla is prominently positioned to integrate peripheral sensory information because it is the first entry point into the brain for all neural signals from the GI tract, hepatic portal vein, and the liver (FIGURE 5). VSNs and SSNs convey eating-relevant information from the gut to the medial NTS, where the VSNs connect to NTS neurons mainly via glutamatergic synapses (401, 402). The NTS is also the brain’s entry point for taste information from the oral cavity. Although taste and GI sensory nerves do not directly interact in the NTS, they project from there to the same target neurons in the intermediate reticular formation (IRt) and in the PB.
In addition to multiple neural signals, the medulla has many receptors and sensors that can sample blood-borne humoral signals through the AP, a circumventricular organ that lacks a tight blood-brain barrier. The AP is located immediately dorsal to the medial portion of the NTS that receives all the sensory nerve information. Whether this part of the NTS has direct access to signals in the blood because of a leaky blood-barrier or diffusion from the AP has been controversial. Despite evidence that the microvasculature in this region appears to feature fenestrated endothelial cells (403), the presence of a tight glial-containing diffusion barrier between the AP and the NTS favors very limited access of NTS neurons to humoral signals (404). NTS pro-opiomelanocortin (POMC) neurons that are important for integrating all these signals (see below) are primarily located in this medial portion of the NTS. Of particular importance is that the NTS connects to the preoral motor neurons of the parvocellular and IRt, i.e., to neurons that ultimately govern the oral motor responses of ingestion (311).
4.3.4. Vascular pathways and the blood-brain barrier: physiological signal entry into cerebrospinal and interstitial fluids.
4.3.4.1. preamble.
The first reports showing that the ARH is a key access point for leptin’s actions in the hypothalamus (405–408) have spawned a huge literature about how leptin controls energy balance, including eating behaviors (e.g., reviewed by (222, 409). As was described in two seminal papers from the Elmquist and Hokfelt groups (410–412) for leptin more than 20 yr ago and then more generally by Sternson (413), considering these access points as network nodes has proved very useful for exploring how different physiological signals engage downstream projections to control eating behaviors. Since then, more detail has emerged about how circulating signals gain entry to the brain across the blood-brain barrier. To function as brain access points these regions must have the means to permit the movement of hormones, nutrients, and fuel molecules from the blood into the brain. Two processes contribute here. First, specific transport across the blood-brain barrier, which protects the brain from the potentially damaging ionic and nutrient fluxes that can occur in blood. The basal lamina of endothelial cells held together by tight-junctions forms the blood-brain barrier in the walls of brain capillaries. Astrocytic processes (end feet) and pericytes help maintain blood-brain barrier function. Many specific transport mechanisms enable the access of hormones, nutrients, and other metabolically important molecules across the blood-brain barrier and into the brain (414, 415). Second, circulating molecules have direct access to neurons in the brain’s circumventricular organs (CVOs) without the need to cross the blood-brain barrier. CVOs contain fenestrated capillaries that act as portals through which vascular signals have privileged access to proximal neurons and glia. These cells then transduce these signals in ways that can influence eating behaviors.
The forebrain has three CVOs: the subfornical organ (SFO), the vascular organ of the lamina terminalis, and the median eminence (ME). The AP is in the medulla. Two others are outgrowths of either the hypothalamus (the posterior pituitary) or the medulla (the pineal gland). Another, the subcommissural organ, is located at the caudal end of the third ventricle as it connects into the central canal.
The three CVOs that contribute most to controlling eating behavior and energy balance are the SFO, ME, and AP. The SFO and AP both contain the somata of neurons that have extensive projections into the hypothalamus and rhrombicbrain (416–419). These neurons, some of which have classic glucosensing properties, express an array of receptors that transduce information from circulating peptides and other hormones (415, 420–422).
Unlike the SFO and AP, the rodent and primate ME contains very few neuronal somata. Instead, the ME is the site where axon terminals of parvicellular neuroendocrine motor neurons release chemical signals that access hypophysial portal vasculature through fenestrated capillaries to control hormone release from the pars distalis of the pituitary gland. The permeability of these capillaries is controlled by MCH neurons, but not MCH itself, thereby providing a way to control the exposure of neurons in the adjacent part of the hypothalamus to circulating physiological signals (423).
The ME and the ventral walls of the third ventricle contain tanycyte somata, which are specialized glial cells that act as important brain entry portals for nutrients and hormones into the CSF and neuropil (reviewed by Ref. 424). Tanycyte processes extend ventrally toward the fenestrated capillaries of the hypophysial vasculature in the ME and the ventral surface of the brain, and laterally toward the ARH and VMH (363). Tanycytes can mediate the transport of leptin, ghrelin, and possibly GLP-1 into the CSF (363) the rate of which is influenced by energy status, at least for leptin (425), and is differentially controlled by ME- and ARH-associated tanycytes (426). However, some controversy still surrounds the leptin-associated mechanisms responsible for these observations (427). Tanycytes are also sensory elements for circulating glucose, amino acids, and possibly fatty acids (363, 428).
4.3.4.2. categories of physiological signals conveyed in the blood.
Circulating physiological signals that interact with the brain to impact eating behaviors fall into three broad categories. Metabolites, hormones, and a third category that is unrelated to oxidizable fuel metabolism or energy homeostasis but yet profoundly impacts eating behaviors. Blood osmolarity is an example of this third category.
All of these signals interact with a wide range of cellular transduction mechanisms located at various brain and peripheral sites. The physiology of many of these different signals with regard to eating behaviors are addressed elsewhere in this review, as well as in other publications (e.g., Refs. 340, 428–432) and will not be discussed here in more detail. Instead, we discuss how a key physiological signal from each of these three categories, glucose, leptin, and blood osmolality, engages the brain at the network level to control eating behaviors and meal structure.
4.3.4.2.1. Glucose.
Two sets of findings in the early 1980s suggested that falling levels of blood glucose and/or brain glucose utilization could trigger a meal. First, a small but significant drop in blood glucose precedes meal initiation in rats and humans (240–242, 433, 434); second, injecting 5-thioglucose, a nonmetabolized glucose analog, into the fourth but not the third ventricle strongly stimulated food intake (435). The notion that a reduction in brain cellular glucose utilization triggers meal initiation also has a long history (436, 437). How might these changes in blood and brain glucose levels, perhaps together with changes in cellular glucose utilization in the brain, translate into altered eating behaviors?
Substantial evidence shows that certain neurons can transduce ambient glucose concentration into meaningful changes in their firing rates. In turn these signaling events control the autonomic and behavioral actions that are required to regulate blood glucose and thereby its cellular availability (438). During the past 50 yr or so, the physiology of these glucosensing neurons has been extensively scrutinized (439, 440). This work began with the seminal work of Oomura, who first identified glucosensing neurons in the hypothalamus (441, 442) and then later developed the first schema for a brain glucose monitoring network (443).
The number of brain sites with identified glucosensing neurons continues to grow, and includes neurons in various hypothalamic, amygdalar, and medullary regions (287, 438, 444). No single cellular mechanism accounts for all glucosensing neurons (440). Although the ATP-sensitive potassium channel is the most studied, other cellular mechanisms have been identified (440) including those involving AMPK, whose phosphorylation state can be controlled directly by glucose (98).
While many glucosensing neurons are found inside the blood-brain barrier, others are located outside, including in the AP and SFO (420, 422, 445). These are important because glucose concentrations inside the blood-brain barrier are only ∼20% of those in the general circulation, and also because glucose concentration fluxes differ between these two compartments in a variety of situations (FIGURE 8) (433, 446, 447). Furthermore, neuronal glucosensing capability is not limited to the brain. Niijima (104) identified glucosensitive VSNs in the hepatic portal area as early as 1969, and Donovan and colleagues (103) have shown that glucosensing by SSNs in the rat hepatic portal vein wall mediates endocrine counterregulatory responses to slow-onset hypoglycemia. A similar capability is present in the dog hepatic portal vein wall (448). Sensory information conveyed from the hepatic portal vein wall by VSNs (104, 292) and SSNs converge onto neurons in the dorsal medulla, particularly those in the dorsal motor nucleus of the vagus (DMX) (387). Because the hepatic portal vein drains the small intestine into the liver, its blood glucose concentrations are significantly higher after a meal than in the posthepatic circulation (449, 450) (FIGURE 8). The dynamic range of these blood glucose fluxes encoded by sensory endings in the hepatic portal vein wall is therefore different from that encoded by brain glucosensory elements located outside the blood-brain barrier (i.e., in the posthepatic circulation), again emphasizing the complexity of the glucosensory information received by the brain at any particular time. The importance of hepatic portal vein wall glucosensing for reducing the activity of ARHAgRP neurons has recently been elegantly demonstrated in fasted mice (451). Interestingly, this study also showed that glucose and fat sensing by the gut epithelium and sensory nerve endings in the hepatic portal vein wall are conveyed to ARHAgRP neurons by different pathways into the brain: glucose via SSNs, and fat via VSNs (451).
FIGURE 8.
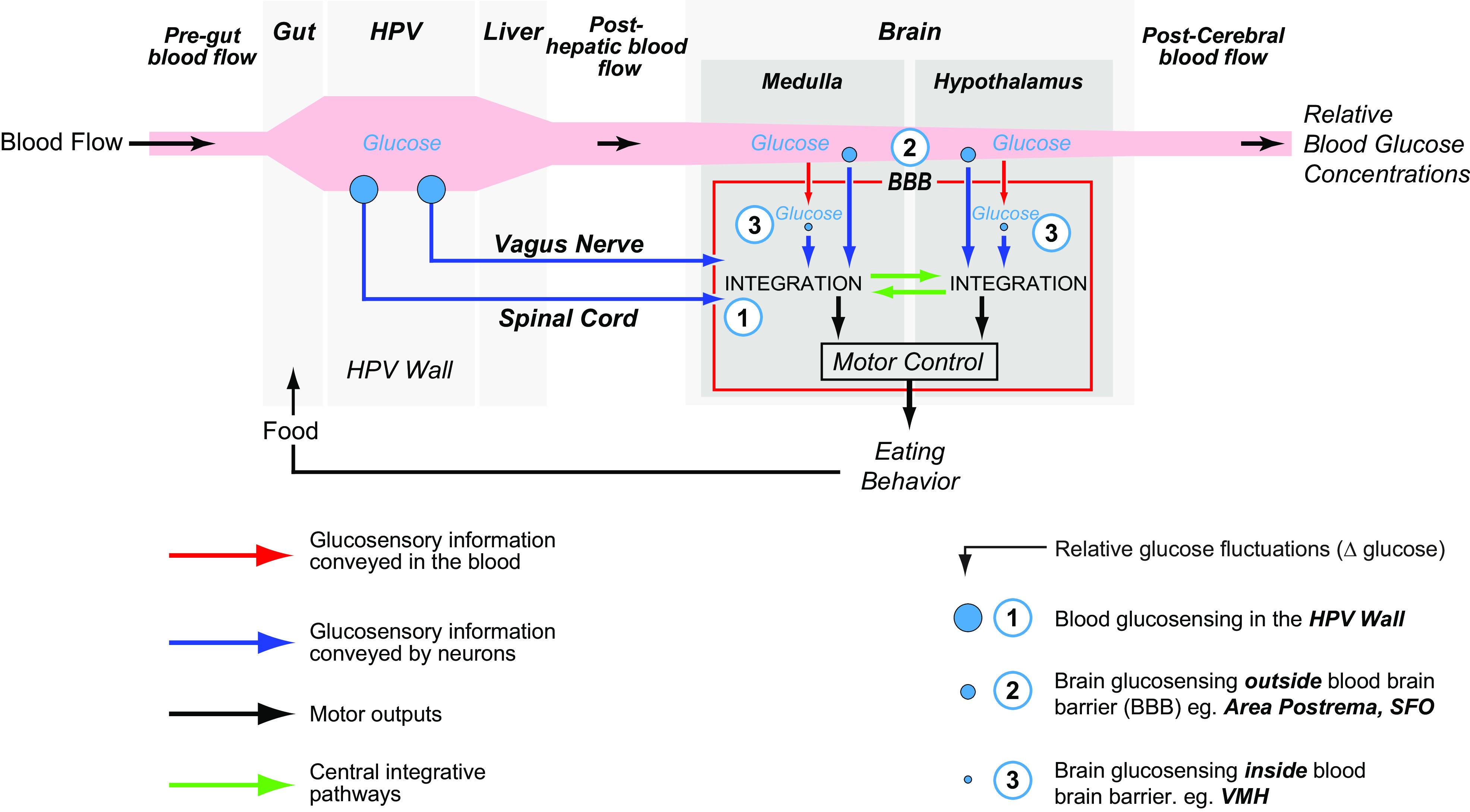
The brain receives sensory information about blood glucose from at least three distinct sites: the ambient glucose environment inside the blood-brain barrier; posthepatic blood glucose concentrations outside the blood-brain barrier (BBB), which are ∼5 times greater than those inside the blood-brain barrier; and postgut/pre-hepatic blood glucose concentrations from the wall of the hepatic portal vein. The glucose concentrations at each of these sites exhibit very different dynamic ranges, as indicated by the different sized blue circles, and the varying width of the pink blood flow “stripe.” This means that information from these sites is integrated by neurons in the medulla and hypothalamus to engage the full repertoire of appropriate autonomic and eating behavior responses to changing glucose. See text for more details. HPV, hepatic portal vein; SFO, subfornical organ; VMH, ventromedial hypothalamic nucleus.
FIGURE 8 illustrates the three different groups of glucosensing inputs that enable this integration. Those from 1) the hepatic portal vein wall; 2) those outside the blood-brain barrier; and 3) those inside the blood-brain barrier. A fourth group of possible vagal and spinal glucosensors has been reported in the small intestine (452, 453). However, it remains undetermined whether these are true glucosensors, or if their responses are dependent on an intervening signaling mechanism. We note that taste (gustatory) receptors in the oral cavity may provide another source of neurally conveyed glucosensory information to the rhombicbrain (454).
The distributed locations of these various glucosensing sites mean that comprehensive control by glucose of motor functions associated with eating and other aspects of energy balance most likely does not emerge from a single glucosensing center, but from glucosensory-motor integration within a brain-wide neural network. The principal components of this network are located in the hypothalamus and medulla (438), with each seeing different aspects of the glucose fluxes in the various physiological compartments (FIGURE 8).
The hypothalamus receives information about ambient extracellular glucose directly from its own glucosensory neurons that are mainly in the VMH, PVH, LHA, and indirectly from sites in the medulla. Two brain sources outside the blood-brain barrier also contribute (FIGURE 8): the SFO (445), whose neurons project to the PVH, LHA, and ARH (416, 418); and the movement of glucose from the blood into the adjacent ARH through fenestrated capillaries in the ME, the permeability of which is influenced dynamically by metabolic state (425).
Like the hypothalamus, the medulla has glucosensory neurons outside the blood-brain barrier, in this case the AP (422), as well as glucosensory neurons and glia inside the blood-brain barrier in the DMX, the NTS (455, 456), and possibly other locations. There is some evidence that glial cells may be part of the glucosensing mechanisms in these regions (456). Unlike the hypothalamus however, medullary neurons receive neurally conveyed glucosensory information via the VSN and SSN (FIGURE 8). This information arrives in the hypothalamus by way of a large contingent of ascending projections, most prominently from catecholaminergic neurons in the ventrolateral medulla and NTS (287, 457–462). These projections innervate forebrain cell groups that make up a core network for integrating medulla-to-hypothalamus conveyed information (see sect. 5.5.4) (457, 459, 463), including the long-term maintenance of glucose metabolism (464).
Which regions are responsible for initiating eating responses to altered glycemia or glucose utilization? To answer this question, it is important to emphasize that the dynamic properties of glycemia and glucose utilization during particular physiological challenges determine the relative importance of the glucosensing regions shown in FIGURE 8. The small changes that are associated with habitual meals are likely transduced by sensory endings in the hepatic portal vein wall (see sect. 5.6.3). However, a substantial body of evidence primarily obtained using pharmacological manipulations of glucose utilization supports the medulla rather than the hypothalamus as the primary integrative site for the eating responses to large and rapid changes in this variable (435, 465). Decerebrate rats (FIGURE 6) still show increased intake of a liquid diet following insulin-induced hypoglycemia or 2-deoxy-d-glucose (2-DG), consistent with the primacy of a medullary glucosensing locus (317, 466). Ritter and her colleagues (287, 460–462) elaborated this result by demonstrating the necessity of catecholaminergic projections to the hypothalamus for eating response in intact rats. The ARH (467) and LHA lateral to the fornix (462, 468, 469), rather than the PVH (470) appear to be the primary hypothalamic loci required for eating driven by cytoglucopenia (e.g., intracellular glucose deficiency caused by 2-DG injections).
Despite a great deal of work, the contributions of VMH glucosensing neurons to habitual meal initiation and to hypoglycemic- or cytoglucopenic-induced eating behaviors remain difficult to determine. Although blood glucose levels in rats show a small but consistent fall immediately before their habitual meals early in the dark phase (240–242, 433), local VMH glucose concentrations do not change (433), suggesting that the VMH is not involved with transducing the premeal decline in blood glucose into meal initiation (see sect. 5.5.2.6). Similarly, although VMH glucose concentrations fall after insulin-induced hypoglycemia (433, 447), direct injections of 2-DG into the VMH are not followed by eating (465). These results, taken together with the fact that catecholaminergic neurons do not innervate the main body of the VMH (457, 464), point to a coordinating rather than primary initiating role for the VMH in energy balance control. That is, a function that controls the autonomic motor responses (e.g., increased sympathoadrenal and glucagon secretion) required to maintain energy-demanding conspecific and antagonistic behavioral interactions, rather than one that is primarily engaged on a short-term basis to maintain daily energy balance (see sect. 5.5.2.6).
4.3.4.2.2. Leptin.
Leptin is a 167 amino acid cytokine-related peptide primarily synthesized by adipocytes. Leptin is not a short-term control signal for eating. Instead, because of the strong nonlinear correlation between its circulating levels and body adiposity (471), it acts primarily as a fuel gauge readout that updates the brain about levels of peripheral fat stores. Its brain actions decrease the attractiveness of food when energy stores are high (246, 472). It also acts as a tonic background signal that has a permissive effect for satiation signals from the gut, particularly CCK (473), gastric distension (295), and GLP-1 (474).
The mRNA encoding the long (signaling) form of the leptin receptor (LepRb) is found in neurons of many rat and mouse brain regions (475, 476) and astrocytes in the ARH (477). It is also found in VSNs (473) and SSNs (478), possibly including those supplying the hepatoportal region (479). Although LepRb function in the DMH, VMH, LHA, and the NTS has garnered much attention in how leptin regulates energy balance (e.g., Refs. 472, 480–485), the ARH has long been regarded as the prime brain target for investigating leptin’s actions on eating behaviors. This is due in no small part because two of its neuronal populations are fortuitously defined by peptides, agouti-related peptide (AgRP) and alpha-melanocyte-stimulating hormone (MSH), found nowhere else in the forebrain (see sect. 5.5.2.3.3). These have enabled precise genetically driven manipulations that have revealed leptin’s actions in the ARH in some detail. Leptin’s signaling properties in ARHLepRb neurons as well as the nature of the transport mechanisms that permit leptin to enter the brain in close proximity to the ARH (363, 426, 486) have been reviewed previously and the reader is referred to these for further information. Leptin also targets the HPF to modify memory functions in ways that can impact foraging strategies and food intake (323), as well as parts of the LHA to modify brain self-stimulation responses (487).
LepRb is expressed by some NTS neurons that are important controllers of eating behaviors, particularly with regard to meal size (236, 321, 480, 488). ARHLepRb and NTSLepRb neurons each mediate their effects by way of projections into distinct but partially overlapping leptin-sensitive networks that involve substantial bidirectional medulla-forebrain connections (see sects. 4.2.4.2 and 5.5.2.3.5). Although the functional arrangement of this leptin-sensitive medullary-hypothalamic network is not yet fully understood, the importance of these interactions for determining leptin’s actions on food intake is becoming apparent (489, 490). Thus Harris and colleagues (491) showed subthreshold doses of leptin had no effect on food intake when given separately into the third or fourth ventricles but were effective when given together. These cooperative effects appeared to be mediated via neural connections rather than the ventricular circulation (492). This group went on to show that the VMH was the most sensitive forebrain site in terms of pSTAT3, a marker of leptin signaling, responses (493, 494). The sensitizing effects of fourth ventricular leptin were abolished when VMHLepRb neurons were ablated (495). A separate study (496) had previously shown that intraperitoneal leptin dose-dependently increased the number of pSTAT3-containing neurons in the medial NTS (NTSm), dorsomedial (dm) VMH, and the ARH. However, the leptin sensitivity of these three regions was different. Although maximum pSTAT3 activation was apparent in all three regions, the ARH showed maximum responses at the lowest dose (50 µg/kg), whereas the VMHdm continued to respond in a dose-dependent manner up to 800 µg/kg of leptin. Taken together, these results suggest that NTSLepRb signaling is able to increase the sensitivity of VMHLepRb neurons to leptin via a set of forebrain-directed projections. This mechanism may increase the responsivity of eating-related output systems to a wider range of circulating leptin than would be possible without NTS-mediated sensitization (495, 497).
How the sensitizing effects of NTSLepRb neuronal signaling are conveyed to VMHLepRb neurons is unclear, but given the known organization of PB and PVT projections (see sects. 5.5.3.1 and 6.3.1.1), it may involve NTS to PB to VMH, and NTS to PVT to VMH projections. It is also uncertain where VMHLepRb neurons project to mediate these sensitized effects on food intake, but the PAG, the PVHap, and BST are possibilities (498–500). These results highlight the importance of considering leptin’s function in a broader network context rather than more narrowly at the level of individual forebrain or medullary regions.
4.3.4.2.3. Osmolality and dehydration anorexia.
Many species maintain a close interaction between water and food intake. In rats this is evident during the dark period as a tight linear relationship when food intake is greatest, but not during the light period when minimal food intake occurs (501). In a laboratory setting, nocturnal meals are structured to include prandial drinking bouts (224). However, during cellular dehydration (when water is lost from inside cells rather than the extracellular space), digestion is compromised because water is moved out of the gut to help maintain extracellular fluid composition. In turn, food intake is actively suppressed in proportion to increased blood osmolality, i.e., dehydration (DE) anorexia. In these circumstances energy balance shifts toward catabolism (502–504). Elevated osmolality is one of the most powerful physiological signals to suppress eating. This vital adaptive behavioral response therefore maintains fluid balance at the expense of energy stores.
DE-anorexia is evident not only as a decrease in habitual meal size (225) but also in the intake responses to LHA or PVH NPY injections (505), ACB muscimol injections (504), 2-DG-induced cytoglucopenia, and overnight food restriction (506). However, the latencies to initiate eating after all these manipulations is unchanged in DE-anorexic animals. Furthermore, as shown by the responses of DE-anorexic rats to 2-DG-induced cytoglucopenia, elevated osmolality primarily targets food intake mechanisms rather than associated neuroendocrine and autonomic responses (506). Therefore, the most important factor responsible for DE-anorexia is a failure to maintain eating behavior once a meal has started, rather than the ability to initiate a meal, which remains virtually intact.
Although details of how osmolality interacts with the upper brainstem eating control network to suppress eating are still unknown, attenuated Fos responses to 2-DG in the preautonomic part of the PVH and particularly in the LHA suggest key roles for these two regions (507). LHACART and LHAORX neurons may contribute to the progression and reversal of DE-anorexia (508, 509). However, of particular interest is that a subset of an extensive and heterogenous GABA/neurotensin (NT) population in the LHA (see sect. 5.5.2.2.5 for more details) is well-placed to contribute by virtue of their ability to suppress food intake when stimulated chemogenetically (510). Elevated osmolality activates neurons in the SFO, and parts of the BST and rostral hypothalamus, the projections of which (511) increase NT mRNA and corticotropin-releasing hormone (CRH) mRNA levels in some LHANT neurons (508, 512, 513). DE-sensitive LHANT neurons do not express LepRb (514) and project to the PB (515), whereas LHANT neurons that do express LepRb are DE-insensitive and target the VTA and SN (514). Collectively, these results show that increasing osmolality engages forebrain projections to LHA and PVH neurons to decrease meal size. These neurons are now less responsive to meal maintenance signals, including NPY-containing inputs from the ARH and medulla (505, 516).
Despite the strong suppression of food intake by elevated osmolality, DE-anorexic animals are primed to begin eating once water becomes available. Their hormonal profile together with the state of their ARHAgRP and ARHPOMC neurons is commensurate with a hungry animal that should, given the appropriate circumstances, begin eating (501). Indeed, once water is returned DE-anorexic animals begin eating vigorously within 8–9 min (501, 508). Given that blood osmolality is still significantly elevated at the time eating begins (502), it seems likely that water entering the gut generates the primary disinhibitory signal for eating, which is conveyed to the medulla by SSNs (376). This signal rapidly disinhibits the forebrain-generated inhibitory effects of blood osmolality on eating, which is then initiated (504). In summary, DE anorexia develops over many hours as slowly increasing blood osmolality enables forebrain projections to act on hypothalamic eating control neurons to reduce meal size. Once drinking water begins again, eating is stimulated within minutes as signals generated in the gut are conveyed to the medulla.
5. MEAL INITIATION
5.1. Preamble
In this section we use the conceptual frameworks we have developed earlier to discuss the processes responsible for initiating meals. Here we describe the various signals, the structural and functional components in the brain and periphery, together with how and where they interact so that appropriately expressed motor programs can direct animals toward food items and then provide them with the means to eat.
Understanding the physiological processes that initiate a meal requires remembering how motivated behaviors are temporally organized. FIGURE 3 shows that meals cannot begin until a previous behavior is terminated and a new appetitive phase is initiated. This usually involves some form of foraging. We therefore begin by describing our current understanding of how extero- and interoceptive signals interact with brain mechanisms, particularly those in the endbrain, to enable food procurement. We then describe the fundamentally important integrative mechanisms used by the hypothalamus, midbrain, rhombicbrain, and VSNs and SSNs to initiate meals.
5.2. Food Procurement: Foraging, Approach, and Interaction
5.2.1. Experimental strategies.
Section 4.2.5.1 described how meal initiation requires selecting and activating the appropriate motor programs for appetitive behaviors. This is achieved via interactions between exteroceptive and interoceptive signals, and those brain regions involved with motivation, memory, and movement control. However, until relatively recently how the brain was organized at the network level to do this was poorly understood. Despite the fact that defining the neuronal bases of food procurement that leads to food intake is central to understanding eating behaviors as a whole, the amount of experimental effort expended in this direction has been small compared with that for consummatory actions. A major reason is that there are no simple rodent-in-a-cage designs that capture the complexity of procurement behaviors. Instead, because there is neither physical nor cognitive distance between animals and food items in small home cages, this type of experiment primarily captures information about the consummatory phase. Important appetitive responses to experimental manipulations are much more challenging to acquire. A good example is that measuring intake responses to manipulating hypothalamic NPY signaling can only inform its possible role in the consummatory phases of eating. They miss its more prominent role in organizing procurement because this is bypassed in simple environments where food is immediately available (237, 517–519).
More complex experimental environments have enabled our ability to explore procurement actions. However, species and individual variations that engender the considerable adaptability in food procurement continue to make it difficult to determine the physiological signals and mechanisms used by the brain to organize these behaviors. Nonetheless, predator hunting and food hoarding provide insights about brain mechanisms. Predator hunting is a widely used strategy by omnivores and carnivores. Hoarding food for future rather than immediate consumption illustrates a completely different type of food interaction. Each has useful perspectives about how animals use similar brain mechanisms to engage very different motor programs to first locate and then interact with targeted food items. Because foraging for, hoarding, and then consuming food are temporally separated in hamsters, they have proved particularly insightful for investigating this dissociation (237, 520). However, the fact that some predator species also hoard food further illustrates the complexity of procurements strategies.
5.2.2. Predator hunting.
Rats and mice are omnivorous and will readily hunt and kill insects for food if they are available. During the past few years there has been an increasing interest on the neural bases of rodent predatory hunting as an exemplary foraging and food approach behavior. Although work on rodent hunting/prey catching behavior has a long history (e.g., Ref. 521), Canteras and his colleagues (522) were the first to investigate in more detail which brain regions organize rat predatory (insect) hunting and how they are functionally connected. By combining Fos mapping with neuroanatomical tract tracing and N-methyl-d-aspartic acid (NMDA) lesions, this group identified a set of amygdalar nuclei that showed greater activation during predator hunting than circadian timed eating (523). The medial part of the central nucleus of the amygdala (CEAm) is the main amygdalar output, with projections to the PSTN, lateral part of the intermediate gray of the superior colliculus (SCig) and retrorubral area, and from there the ventrolateral caudoputamen (VLPC). The VPLC is implicated in orofacial and forepaw movements during eating (524–526). NMDA lesions later confirmed the involvement of the SCig and the VLPC in organizing the motor actions of predatory hunting (305, 527).
The rostrolateral PAG is also a component in wider PAG functions that may contribute to appropriate bidirectional switching between hunting and other behaviors, including predator avoidance and maternal behavior (522, 528–531). The PAG also receives inputs from the VMH. Because of its many glucose- and leptin-sensing neurons (see sect. 5.5.2.6), the VMH may help integrate metabolic control information with the PAG as a means to rapidly switch behaviors in the face of changing environmental cues (532), with VMH to PAG connections coordinating energy balance with the energy demands of predation, conspecific interactions, or fight or flight actions.
More recently, significant and substantial refinements to our understanding of this network have been made using specific gene-driven techniques (345, 533–536). These studies identified additional components, including the medial preoptic area, parts of the LHA, the parvocellular reticular formation, pendunculopontine and cuneiform nuclei (components of the midbrain locomotor region), and the rostromedial ZI. These studies have also identified the functional significance of these various regions for predator hunting.
The collective outcome of this work is a complex interconnected brainwide (amygdalar-hypothalamic/thalamic-midbrain-rhombicbrain) network that can control the approach, attack, and direct interaction with chosen prey. Some of the components of this network appear to be nodes whose function can be modulated by physiological signals. For example, the PVT receives significant projections from ARH neurons, many of which are targets of circulating signals (see sect. 5.5.2.3). Furthermore, the CEA and PSTN, both of which are preferentially activated during predator hunting (523), are infected when HSV-1 H129 is injected into the nodose ganglia (345). These findings are consistent with the ability of vagally conveyed interoceptive information to modulate this predator eating network by way of PSTN and CEA projections.
5.2.3. Food hoarding.
When rats, mice, and Syrian and Siberian hamsters are denied access to food they all develop identical endocrine and hypothalamic neurochemical responses: increased levels of circulating ghrelin, ARH NPY, and AgRP mRNA and decreased levels of circulating leptin and ARH POMC mRNA (reviewed in Refs. 237, 537). These responses have been foundational for our understanding of eating control mechanisms. However, despite the similarity of these responses, the nature of the eating behaviors of these four rodent species following food deprivation is markedly different. Rats and mice show an immediate and robust compensatory hyperphagia when access to food is reinstated. Fasted hamsters do not overeat. Instead they decrease their energy expenditure (538) and increase the amount of food they hoard for future consumption (269). This was shown by Bartness and his colleagues (520, 539) using a housing environment where interacting with food required navigating between a burrow where nesting materials were located and a separate cage where food was provided. An intervening running wheel allowed the assessment of foraging effort (539), which was also increased by fasting (520). Furthermore, unlike rats and mice, male Siberian hamsters do not show increased food intake or hoarding in response to glucoprivic (cytoglucopenic) or lipoprivic metabolic blockers (237, 269). However, female Siberian hamsters do show increased food intake after cytoglucopenia, although this is far less than what occurs with rats (86). Siberian hamsters also show complex eating response interactions with the photoperiod (86, 237), which are most likely related to the fact that these animals hibernate.
As with compensatory eating responses in rats and mice, hypothalamic NPY and ghrelin signaling, together with ARHAgRP neuronal mechanisms, is strongly implicated in increased foraging and hoarding. In hamsters peripheral ghrelin injections increase foraging and food intake, but hoarding is elevated to a greater degree (540). These effects, as well as those activated by fasting, appear to involve NPY signaling (541). NPY injections targeting the PVH or perifornical LHA stimulate food hoarding to a greater extent than intake, but foraging is only increased after perifornical LHA injections (542). Third ventricular AgRP injections progressively increase the magnitude of intake, foraging, and hoarding in that order (543), results that are substantiated by the effects of actively reducing AgRP mRNA in ARH neurons (544). Collectively these results implicate the LHA, PVH, and ARH in hamster procurement behaviors, although ARH contributions may be limited to deficit-induced behaviors rather than those associated with habitual meals (545). The prominent role of NPY signaling in the perifornical LHA and the importance of this region for glucoprivic eating (34) (see sect. 5.5.2.2.6), suggests that NPY signaling from medullary catecholalaminergic inputs may contribute (237), but this remains untested.
Given the considerable commonality between physiological signals and their brain targets in rats, mice, and hamsters, how do these engender such different behavioral responses in these species? We will see later that the LHA, PVH, and ARH are the core components of the hypothalamic eating behavior control network (sect. 5.5.4), the output of which biases mechanisms in the VTA, SN, and the cerebral nuclei to select and initiate the appropriate procurement programs for the ongoing state of energy balance (cf. FIGURE 6B). The fact that AgRP stimulates food intake in rats and mice, but hoarding in hamsters (543) emphasizes this point. Recent advances in understanding how ARHAgRP neurons contribute to selecting context-dependent behavioral strategies may provide some pointers here (123, 274, 275, 546), particularly with regard to the relative roles of AgRP and NPY signaling (516). Section 5.5.2.3.5 discusses these aspects of ARH function in more detail.
Rats, mice, and hamsters each occupy different ecological niches, within which either hoarding or the immediate consumption of food provides the greatest adaptive value. Presumably nuanced species differences in the way that this eating control network interacts with downstream motor control mechanisms determines why each expresses distinct procurement behaviors to the same energetic challenges. However, a more substantial experimental focus on the appetitive phase of eating will be required before we can understand how this occurs.
5.3. Food Choices
5.3.1. Flavor.
In addition to food availability, food choice is largely determined by an interaction between flavor (gustation + olfaction + texture) perception, postingestive nutrient consequences, and previous learned experiences based on associations between these factors. Conditioned flavor avoidance (or aversion) learning is the classic example of associative learning processes affecting food choice by way of flavor-postingestive interactions. Here animals will avoid (or reject) flavors that have been previously associated with visceral malaise (547). Neutral or even aversive flavor cues can also become appetite-enhancing based on their learned associations with positive nutritive consequences. For example, nonnutritive flavors paired with gastric nutrient infusions are subsequently preferred compared with otherwise equally preferred flavors (548). In addition, taste stimuli that evoke aversive orofacial responses (e.g., quinine) evoke ingestive orofacial responses when associated with a nutritive consequence (549), further highlighting the plasticity of gustatory-hedonic interactions in guiding food choice.
5.3.2. Reactive macronutrient-specific mechanisms.
Experimental rodents and humans compensate for the calories consumed in a preload meal or snack by reducing caloric intake at a subsequent eating occasion. However, under controlled experimental conditions this compensation is generally independent of the macronutrient composition of the preload or the subsequent test meal, regardless of whether the preload was consumed normally or delivered intragastrically (550, 551). These findings suggest that the macronutrient composition of a meal has little influence over immediate subsequent eating bouts. However, in free-eating conditions, there is evidence from dietary records of a negative correlation between macronutrients consumed on 2 different days separated by 2 intervening days (552). This suggests that a possible negative feedback mechanism with a delay of 2 days may exist, although this could be based on cognitive control factors and not physiological negative feedback cues, per se.
While there is minimal evidence for macronutrient-specific negative feedback mechanisms in a state of energy sufficiency/balance, dietary protein restriction leads to physiological and behavioral changes, including changes in macronutrient preference toward protein, increased energy expenditure, and altered glucose metabolism (553). Furthermore, animals increase consumption of diets moderately restricted in amino acid composition (absent one or a few amino acids) and selectively find and consume missing amino acids when given multiple food options (554). The liver-derived metabolic hormone FGF21 appears to be a critical signal acting as a sensor to variations in amino acid consumption (555), as recent findings reveal that central FGF21 signaling is essential for protein-restricted mice to shift preference toward protein-containing foods (556).
Eating most commonly involves choosing between food options with mixed macronutrient composition. Thus food choice with regards to macronutrient selection may be less influenced by reactive processes (e.g., negative feedback systems for macronutrient deficiency) and more by proactive processes guided by hedonic factors and previous experiences. Indeed, recent work revealed that human participants are willing to pay more for snacks with fat + carbohydrate, versus equally familiar, preferred, and nutritive snacks comprised of fat or carbohydrate alone. This choice is associated with a blood-oxygen-level-dependent imaging response in the dorsal striatum and mediodorsal thalamus and is independent of the participants’ ability to estimate the energy density of the foods (557). In addition to macronutrient interactions, food choice is also determined largely by one’s expectation for a meal to deliver a far greater reduction in desire for food between meals, termed “expected satiety” (558). However, expected satiety influences on food choice are only critical determinants when small equicaloric portions are compared. Food choice with larger portions are motivated predominantly by palatability (559), which appears to be driven, at least in part, by the supra-additive effects of combining fat and carbohydrate (557).
5.3.3. Investigating food choice mechanisms with spatially targeted manipulations.
It is common for rodent behavioral pharmacological approaches to measure consumption of nutritionally adequate foods with a mixed macronutrient profile compared with offering a choice based on isolated macronutrient (or other) variations. However, rodent pharmacological studies that have used specific food choices provide evidence that some neurochemical systems can control eating differently based on macronutrient composition and/or hedonic properties. For example, results from several studies indicate that NPY selectively promotes carbohydrate intake when administered to the PVH (e.g., Refs. 560, 561; see also Ref. 562 for review), although various factors appear to modulate this effect. These include baseline macronutrient preferences, the choices offered, and the circadian time of the test (562, 563). On the other hand, NPY signaling in the ACB selectively promotes fat consumption (564). This suggests that neural systems-level modulation of eating behavior may involve common signals with divergent projection pathways that differentially guide food choice behavioral outcomes.
Activating opioid receptors in the ACB potently increases food intake (160). This hyperphagic response is directly related to the relative preference for a particular food, with larger effects correlated with higher preference. For example, with both high-fat and high-sugar foods simultaneously available (free choice procedure), ACB opioid receptor stimulation selectively increases carbohydrate intake in carbohydrate-preferring rat, and vice versa for fat-preferring rats (162), an effect also observed following PVH-directed mu-opioid agonism (163). Endogenous OXY signaling, on the other hand, acts as a carbohydrate-specific inhibitor of eating with minimal effect on lipid consumption and is independent of preference and hedonic properties (565).
5.4. Forebrain–Endbrain
5.4.1. Preamble.
This section will deal with where and how physiological signals interact with the endbrain regions that help organize eating, and particularly those involved with foraging strategies and food choices. Key processes include the following: learning/memory mechanisms that are responsible for identifying the locations of food sources and organizing strategies for navigating toward these locations, as well as memory and reward-based processes relevant for food selection and meal initiation. We then discuss the organization of endbrain projections that distribute eating-related information to more caudal parts of the brain, primarily the hypothalamus, to enable integration with the eating behavior control processes found in these locations.
5.4.2. Visuospatial navigation.
To achieve and maintain energy balance it is adaptive to remember the precise physical location of food sources, and then efficiently navigate back to a shelter/dwelling. Thus it follows that the neural substrates that mediate visuospatial navigation are important during the preprandial and foraging stages of eating behavior. These neural substrates are predominantly located in the endbrain. In particular, the HPF plays an essential role in processing relative information about an animal’s external environment and spatial orientation (566). The pyramidal neurons located within CA1-3 in Ammon’s horn of the HPF have spatially selective firing fields that reflect the animal’s location in an environment, and jointly form a continuously updated map-like representation of the external environment (567, 568). In rodents, the scale of representation increases almost linearly from <1 m at the dorsal (anterior) pole to ∼10 m at the ventral (posterior) pole (569) (note that rodent dorsal and ventral subregions are analogous to the primate posterior and anterior HPF, respectively). This place-cell mapping system allows for environments to be represented in the HPF dynamically and at a topographically graded continuum of scales.
A mental map of a particular environment by itself is not sufficient for locating a food source: egocentric position and orientation in the environment are also critical for effective navigation. These processes are encoded primarily by the medial entorhinal cortex (MEC) and the adjacent pre- and parasubiculum. The firing correlates of dedicated neuron populations within these regions have names that describe their function in navigation and positioning: grid cells, border cells, speed cells, and head direction cells (570). Of these, most attention has been directed to grid cells in the MEC. In rodents these neurons have relatively small hexagonally arranged firing fields that tile the two-dimensional surface available in an open environment. The MEC provides the main excitatory input to the dorsal HPF field CA1, and the physical distance between MEC firing fields, which is quite similar across different environments, helps encode dynamic information about self-motion and sensory information to contribute to the spatial map generated in HPF place cells (571).
The MEC (but not the HPF) has direct projections to the posterior parietal cortex (PPC) (572), a region that represents a critical downstream integrator of visuospatial and egocentric information relevant to navigation (573). Electrophysiological recordings in behaving primates (574, 575) and rats (576) indicate that PPC firing rates are determined by a complex coregistration of current location, target location, movement type, and position of various body parts. Fine-grained analyses of deficits in humans with cortical damage further support the framework that allocentric world-referenced and egocentric body-referenced signals come together in PPC neurons (577). Collective findings from multiple species support a larger framework that these integrated visuospatial and egocentric representations are translated into goal-directed movements via network interactions between PPC, retrosplenial cortex (see Ref. 578 for further review), HPF, and MEC.
Much of the rodent literature on spatial learning and memory has used either passive behavioral procedures (freely exploring a familiar environment) or procedures that require escaping aversive reinforcement (e.g., escaping water/swimming or bright lights, loud noises, predatory urine, etc.). Thus extending much of this literature to foraging behaviors rests on the assumption that the neural substrates involved in learning the spatial location of food are common with those involved in either passive or aversive reinforcement-based spatial learning. However, there is direct evidence that the HPF is required for foraging-related behaviors that necessitate learning and remembering the spatial location of food. For example, selective HPF lesions impair meal-related spatial memory used to relocate previously stored food sources in scrub jays (579). Similarly, intact rhesus monkeys preferentially return to a foraging site where food was previously obtained in a matching-to-location task, whereas monkeys with HPF lesions do not show this learned matching tendency (580). In rats HPF lesions produce errors in both reference memory (remembering where food was located on previous days) and working memory (remembering where food was very recently consumed within a session) in the appetitive eight-arm radial maze spatial memory procedure (581). However, in a variation of the task that relies on nonspatial, tactile cues, HPF lesions impair working memory but not reference memory (581), indicating that the HPF is critical for remembering where food was just consumed, even when solving this problem does not require visuospatial navigation.
5.4.3. Episodic memory.
In parallel to its role in visuospatial navigation discussed above, HPF neurons encode exteroceptive and interoceptive contextual information that forms the basis of episodic memory (the autobiographic memory of events, including who, what, when, where). The integrity of episodic memory for a recent eating occasions can profoundly influence foraging-related appetitive behaviors. For example, amnesic humans with bilateral nonselective damage to the HPF and surrounding medial temporal lobe structures (including the amygdala) will eat a full second meal that is offered immediately after consuming a meal (582). Similar findings have been observed in rats where reversible postprandial inactivation of either dorsal or ventral HPF using either muscimol infusions (583, 584) or optogenetic inhibition (585) decreases latency to initiate the subsequent meal, results that are hypothesized to be based on disrupted meal-related episodic memory consolidation. Consistent with these outcomes indicating reduced latency between eating episodes with impaired HPF function, rats with HPF lesions will consume more frequent habitual meals under free-eating conditions (586), an outcome that may be associated with increased overall energy intake and body weight (587).
5.4.4. Pavlovian conditioning: stimulus-induced eating.
Through learned associations, otherwise neutral environmental stimuli can acquire the capacity to promote eating that would not otherwise occur in the absence of exposure to the stimuli. Generally speaking, this phenomenon, termed cue-potentiated eating, is based on Pavlovian associations between external stimuli and highly palatable food or between external stimuli and food previously consumed during periods of chronic or acute energy restriction. Cue-potentiated eating occurs in both humans and rodents under experimental conditions (588, 589). Early work from Weingarten (590) characterized three key behavioral components of cue-potentiated eating in rodents: 1) the size of meals initiated following cue exposure resembles spontaneous meal size that occurs under free-eating conditions; 2) while external cues can override interoceptive satiety signals, the potency of cue-induced eating is nevertheless influenced by satiety signals from previous meals; and 3) the presentation of conditioned cues can affect meal patterns, but rats compensate by controlling the overall amount of calories consumed in a 24-h period (590). However, it should be noted that Weingarten’s work (591) involved the presentation of discrete conditioned cues (auditory visual compound stimuli), whereas elevations in longer-term (24 h) total caloric intake have been observed in rats exposed to contextual cues previously associated with palatable food consumption.
Using an elegant combination of lesion-based disconnection, behavior, tract tracing, and immediate-early gene mapping approaches, Petrovich and colleagues (592–594) identified neural connections between the basolateral amygdala (BLA), the LHA, and the PFCm as critical for the expression of cue-potentiated eating. These findings are complemented by human neuoroimaging studies revealing that the BLA response to food cues in food-sated subjects is associated with increased weight gain one year later (595), suggesting that the brain regions controlling cue-induced eating may be of relevance to obesity. More recent work identified a connection between the BLA and the INS as critical for mediating hunger-dependent enhancement of food cue responses. This supports a model whereby the interactions between the BLA and INS can gate appetitive action selection in response to food cues by weighing the predicted interoceptive outcome value with the context of current physiological needs (546, 596–599).
Circulating levels of the stomach-derived hormone ghrelin are largely determined by levels of energy restriction. However, ghrelin is also released from the stomach as an anticipatory eating signal in response to conditioned circadian and/or orosensory cues (reviewed in Ref. 600). While it is not yet known whether cephalic ghrelin responses occur in response to visual and other discrete food cues, both pharmacological blockade (601) and genetic (602) blockade of the ghrelin receptor (GHSR1a) inhibit the capacity of conditioned food cues to stimulate eating in sated animals. Results from human functional neuroimaging studies identify the amygdala, the orbitofrontal region of the cortex (ORB), the anterior insula, and the striatum as candidate brain regions for ghrelin’s effects on food cue reactivity, as intravenous ghrelin increased the functional (f)MRI response to food pictures in these regions (603). The HPFv (field CA1; vCA1) is another candidate brain region mediating these effects as GHSR1a activation in this region enhances meal initiation in response to discrete food cues previously associated with palatable food consumption (604). These findings identify yet another brain region of importance in the control of cue-induced eating. The vCA1 has monosynaptic connections with LHA, BLA, PFCm, and INS (reviewed in Ref. 247), and, of these, vCA1 projections to LHA (271) and PFCm (192) have been identified as being relevant to eating behavior. However, their function with regards to cue-potentiated eating remains to be explored.
Several findings identify ORX neurons that are mostly found in the LHA as a key downstream system for mediating ghrelin’s hyperphagic effects (144, 271, 605). ORX’s role in cue-induced eating is shown by the fact that a dose of an ORX receptor antagonist that has no effect on baseline eating in rats blocks cue-potentiated eating (606). Melanin-concentrating hormone (MCH) is another neuropeptide produced predominantly by LHA neurons, although these are different from those that synthesize ORX (607). Genetic deletion of MCH in mice significantly impairs the ability of a cue associated with palatable food consumption to evoke overeating in food-sated mice (608), suggesting that both of these LHA neuropeptide systems are involved in cue-potentiated eating.
Collectively the work reviewed herein reveals that cue-potentiated eating is a robust phenomenon observed in rats and mice that can trigger food procurement and consumption in the absence of metabolic deficit. Moreover, the control of cue-potentiated is driven by a complex interconnected endbrain network (PFCm, BLA, vCA1, and INS) that interfaces with the LHA (including ORX and MCH neurons), as well as peripheral signals (e.g., ghrelin).
5.4.5. Endbrain outputs to the hypothalamus and midbrain.
Many endbrain regions provide robust and complex projections to the hypothalamus that influence how it controls eating behaviors. However,, rather than trying to deal with the input/output relationships of these regions individually, a simpler approach is first to consider them at a systems level. Swanson et al. (61, 62, 609) first proposed a framework of this kind ∼20 yr ago. This combined neuroanatomical and functional model has two important organizational features. First, endbrain regions are designated as either cortical, striatal or pallidal (FIGURE 9A). Thus the entire HPF is cortical, while the various parts of the amygdala are either cortical or striatal (60). The lateral septal complex (LS) is entirely striatal; the BST is entirely pallidal. Endbrain outputs now fit into a relatively simple triple descending projection (FIGURE 9B) where the outputs from each of the three parts distributes topographically within the hypothalamus to control the expression of various motivated behaviors, including eating (see also FIGURE 9Ai).
FIGURE 9.
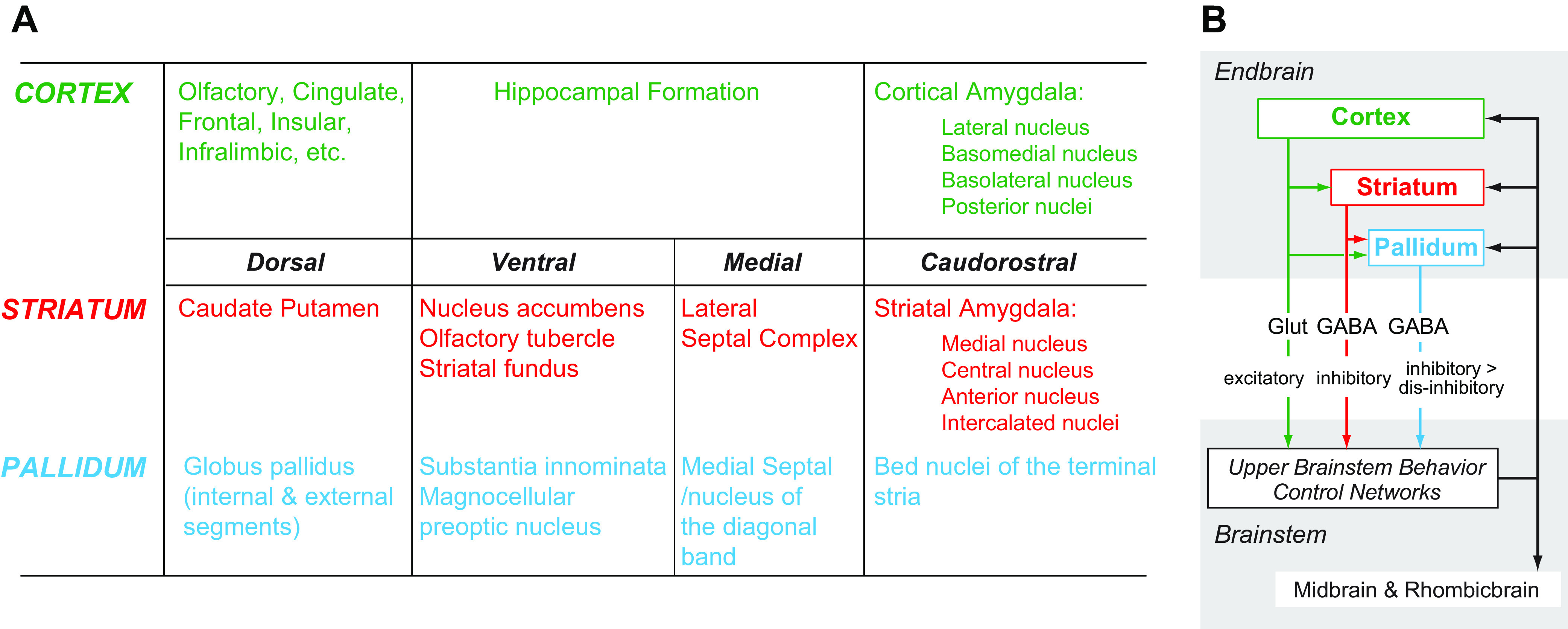
A: the brain regions that comprise the cortex, striatum, and pallidum. B: the connectional relations between the triple descending projections from the cortex (green), striatum (red), and pallidum (blue), and the components in the Upper Brainstem Behavior Control Networks. Note that the predominant neurotransmitters provide for excitatory, inhibitory, and disinhibitory influences on the behavior control networks in the upper brainstem. Projections from these control networks are shown in black. Also see FIGURES 5 and 6.
The second aspect of this model is that those parts of the hypothalamus that control various motivated behaviors are broadly organized into control networks in the upper brainstem that include particular hypothalamic nuclei, each of which contributes to controlling a different motivated behavior, including eating (61, 62). Each of the three groups of descending endbrain projections then distributes topographically onto the appropriate regions of the control networks. Importantly, cortical outputs also provide projections to striatal and pallidal regions, and striatal outputs also project to the pallidum. The predominant neurotransmitter signature in each component provides a range of control from excitatory to inhibitory and disinhibitory, at least at the macroconnectional level, that is determined by the relative activity of a particular projection (FIGURE 9B). As a recent report shows, combinations of high resolution tracing techniques hold the promise of determining whether these transmitter signatures are retained at meso- and microconnectional levels (610).
These triple descending projections (FIGURE 10A) are organized for sets of projections from the amygdala, PFCm and HPF to the hypothalamus. As an example FIGURE 10B shows how amygdalar information reaches the hypothalamus via five sets of projections, either directly or indirectly via various cortical, striatal, and pallidal components (612–614). FIGURE 10C shows the various cortical, striatal, and pallidal routes through which eating relevant information from the PFCm or HPF can reach the hypothalamus. The anatomical details of these regional projections are found in the following papers: (364, 609–611, 615–622).
FIGURE 10.

A: information from the cortex and cortical nuclei (striatum and pallldum) is conveyed to the hypothalamus by way of a triple descending projection pathway (see FIGURES 6 and 9). Cortical regions are in green, striatal in red, and pallidal in blue. B: a summary of 5 major routes used to convey amygdalar (AMY) information to the hypothalamus (611). These can be either direct, or they involve the triple descending projectional scheme via the hippocampal formation (HPF), the medial pre-frontal region of cortex (PFCm), the lateral septal nuclei (LS), or the bed nuclei of the terminal stria (BST). C: 2 examples of how cortical (endbrain) projections to the hypothalamus involve the triple descending connectional scheme shown in FIGURE 9. They show how the medial part of the prefrontal cortex (PFCm) and the hippocampal formation (HPF) can communicate with the hypothalamus via striatal regions (red), or via striatal and pallidal (blue) regions. See text for more details and the references to the original studies. CEA, central nucleus of the amygdala; MEA, medial nucleus of the amygdala; LHA, lateral hypothalamic area; ACB, medial nucleus of the amygdala.
5.5. Forebrain the Hypothalamus as an Integrator for Meal Initiation
As we have described earlier (FIGURE 5), the hypothalamus integrates signals from three sources to enable the motivation to eat and to initiate meals.
Interoceptive humoral signals that circulate in the blood.
Signals from the GI tract that are transmitted to the rhombicbrain by the spinal cord and vagus nerve, which can then be conveyed to the hypothalamus by a series of prominent ascending connections.
A wealth of information that is related to exteroceptive modalities is conveyed by descending connections from the endbrain to the hypothalamus. This information is extensively processed in cortical, pallidal, and striatal regions, and can be modulated by sources 1 and 2.
We will see that how and where this integration occurs in the hypothalamus are central to understanding how it operates as the brain’s main integrator or controller for motivated eating.
5.5.1. Four experimental approaches have identified which hypothalamic components control eating behaviors.
5.5.1.1. spatially targeted chemical manipulations.
We have known for ∼60 yr that site-specific chemical manipulations of hypothalamic regions will initiate eating in animals that have unrestricted access to food, i.e., in the absence of signals encoding negative energy balance (19, 623). In this way, numerous studies have shown that manipulating a variety of peptidergic, monoaminergic, and amino acid-derived fast-acting neurotransmitter systems in the hypothalamus will initiate eating in a site-specific manner (for review see Ref. 624).
5.5.1.2. identifying regions where neural activity is altered by physiological signals: fos mapping.
The introduction of Fos as a marker of altered neuronal activity in 1988 (625) rapidly inspired many studies that revealed which neurons altered their activity following a particular stimulus. The primacy of Fos mapping has continued to the present time. Numerous studies that have located Fos activated neurons have helped identify which hypothalamic regions are impacted by circulating physiological signals. This approach has also revealed regions that are engaged after various types of eating behaviors are initiated (e.g., Refs. 505, 508, 523, 626, 627).
5.5.1.3. functional interrogation of network components and their connections.
Site-specific injections and Fos mapping have collectively identified those hypothalamic regions from which eating behavior, or at least increased food intake, is consistently stimulated. Importantly however, this work cannot address the functional organization of the associated neural networks. Tackling this problem began ∼10 yr ago with two sets of techniques whose specificity is gene-guided rather than location-guided (100, 413, 628). Since then pharmacogenetically or optogenetically controlling the activity of particular hypothalamic neurons has helped clarify their role in eating behaviors (54, 629, 630). Although these methods have largely confirmed the fundamentals of earlier findings, these gene-guided tools have added far greater temporal, functional, and often spatial resolution to neurochemically defined neural systems in the hypothalamus than has been previously possible.
5.5.1.4. neural connectivity and connectomics.
Understanding how eating behaviors are controlled at the network level requires a detailed knowledge of the connectional organization of network components. Our current understanding derives from four approaches. The first is traditional pathway tracing using spatially targeted chemical agents (reviewed by Refs. 6, 624, 631). The advent of the more sophisticated of these techniques began ∼50 yr ago. The remaining three, pathway tracing with transneuronal neurotropic viruses, specific neuronal targeting using genetically driven agents, and neuroinformatics, have all reached maturity more recently (3, 6, 624, 632).
To reveal complex network organization, pathway tracing with spatially targeted chemical tracing agents has provided by far the largest numbers of defined macroconnections (brain region to brain region), with the rat being the predominant species (633). These agents identify one-step connections rather than the multistep networks (or circuits) that are revealed by neurotropic viruses (3, 634). However, it is possible to establish relationships between one-step connections of many brain regions using combined anterograde and retrograde injections (e.g., Ref. 622), and network analyses tools (4, 635). This type of analysis has identified macroconnectional relationships in the hypothalamus and other forebrain regions on a scale not previously possible (e.g., Refs. 6, 55, 258, 632, 636). More detailed pathway tracing is possible with neurotropic viruses and specific neuronal targeting using genetically driven agents (for review see Ref. 413), which are adding ever increasing detail to meso (neuron type to neuron type)- and microconnections (specific synaptic connections between neurons).
5.5.2. Hypothalamic regions and meal initiation.
5.5.2.1. general considerations.
Many hypothalamic regions are implicated in eating behavior control by virtue of the increased or decreased food intake seen after targeted manipulations. However, unless the latency to begin eating after a manipulation is recorded it is very difficult to assign an initiating role to a particular region. All that can be said is that it plays some part in the eating process. Meal onset times after a manipulation can vary widely. For example, eating begins in less than 2 min when TRPV-1 channels in ARHAgRP neurons are activated by capsaicin (637), which is similar to the time taken to begin eating after blocking non-NMDA receptors in the ACB shell (638). Meal onset times after glutamate agonist injections into the LHA are <10 min (639), which is comparable to those seen after GABAergic antagonist injections into the ACB (640). NPY injections into the PVH or LHA are in the same range, with a mean latency of 8–10 min (21, 505). By comparison, optogenetically stimulating ZI neurons and their projections to the PVT (277) leads to eating within 2–3 s, which is astonishingly fast. Although these various delivery methods make precise comparisons difficult, the different meal onsets after these various manipulations give some indication about a region’s relative position within the networks that initiate meals.
Another consideration is whether manipulations activate eating alone, whether eating occurs in series with other behaviors (e.g., drinking), or whether it is a secondary consequence of another behavior. These complications make it difficult to tag a specific hypothalamic region as being a meal initiator as opposed to one that, for example, increases general arousal, within which eating is a component. Unfortunately, these constructs can be challenging to address in experimental designs. Cumulative food intake within defined time bins is often the only recorded variable, not the latency to eat or meal duration. Moreover, interactions between eating and other behaviors are difficult to address, particularly in simple environments, meaning that it is not always possible to place the functional significance of a particular region within the control hierarchy for meal initiation. Nevertheless what has emerged about individual regions is that there is no such thing as a dedicated hypothalamic eating center (see also Ref. 640).
As many studies have shown, all hypothalamic regions that are recognized as being intimately involved with controlling eating also control other motivated behaviors and, to a greater or lesser degree, autonomic function (for reviews, see Refs. 641–645, as well as the references in the following sections describing individual hypothalamic regions). Furthermore, even if the function of specific neurons within a particular region is closely associated with eating behaviors, their mesoconnections can allow interactions with other systems. For example, ARHPOMC and ARHAgRP neurons are tightly allied with eating behaviors and energy balance. Yet, they receive synaptic input from ARHkisspeptin neurons (646, 647) that form part of the GnRH pulse generator for luteinizing hormone secretion (642). This type of relationship not only illustrates the interactive complexity of these behavior control networks (648) but also the limitations of assigning restrictive functional labels (e.g., orexigenic or anorexigenic) to a region or its constituent cell types.
Numerous studies have shown that meal initiation in motivated eating is an output property of a defined network whose principal components are in the upper brainstem. The core components of this Upper Brainstem Eating Control Network each possesses the four properties we described earlier (sect. 4.2.5.1.1). We now describe the three hypothalamic regions most closely associated with meal initiation: specific parts of the LHA, the ARH, and the PVH. These contain the core hypothalamic components of this particular control network. We then discuss other hypothalamic regions that can, to a greater or lesser degree, influence eating behaviors: the SO, VMH, and DMH. We continue by discussing which parts of the hypothalamus are involved with integrating timing signals into meal initiation, and finish by describing how the LHA, ARH, and PVH bias the selection and initiation of the motor components of eating behavior.
5.5.2.2. lateral hypothalamic area.
The LHA makes up a sizable portion of the hypothalamus. It extends rostrocaudally for ∼75% of the rat hypothalamus and 55% of the mouse hypothalamus (59, 66). It is by far the most structurally and functionally complex hypothalamic region. It has extensive intrahypothalamic connections (632), and its extrahypothalamic connections extend throughout the entire cerebrospinal trunk. No other hypothalamic region has these properties. These regional connections enable LHA neurons to integrate three principal sets of information required for controlling eating behaviors: a wide-range of humorally and neurally conveyed interoceptive information; information processed by the cortex and cortical nuclei that encodes the memory, location, and incentive value of food items; and circadian timing and arousal state information. In short, the LHA may be the primary hypothalamic region for conveying the appropriate motivational drive to those brain regions responsible for selecting and initiating the motor components of eating behaviors (FIGURE 6Bii) (reviewed in Refs. 594, 649–652). While it is not the only decisive control region for initiating habitual and opportunistic meals, it certainly occupies a key position for this purpose.
All parts of the hypothalamus have difficulties when trying to determine their structure/function relationships with eating behaviors, but the LHA is by far the most challenging. Although our understanding of the LHA’s contributions to eating behaviors has dramatically improved during the past decade, its structural complexity still presents considerable technical and interpretative challenges. As a consequence, many facets of its overall role remain poorly understood. To provide a context for these challenges, we begin this section by looking at the LHA’s anatomy and the substantial impact this feature has on how we interpret the LHA’s controlling role in eating behaviors.
5.5.2.2.1. Anatomical considerations.
The LHA has over 16 identified gray matter regions in the rat (59), many of which have yet to be functionally defined. Many studies that manipulate LHA neuronal function use general or vague terms to describe location, and to complicate matters further, those that do use a parcellation scheme for the LHA often do so inconsistently, meaning that comparing results from different studies is very challenging. An in-depth review discusses the problems and implications for eating behaviors of accurately locating neuronal manipulations to brain reference spaces (624).
In an attempt to standardize LHA structure, the most systematic parcellation scheme is the one developed by Swanson and his colleagues based on its cytoarchitecture in the rat (59). FIGURE 11 shows that the LHA is divided into three rostrocaudally arranged groups, each of which contains up to eight regions. The most rostral of these is the anterior region of the LHA (LHAa), with the PSTN and subthalamic nucleus the most caudal. The connections of these LHA regions have been described to varying levels of detail in the rat, with comprehensive descriptions for regions in the LHA middle group (FIGURE 11, entries in red). Whether the LHA’s architecture is the same in rats, mice, and other mammals is undetermined.
FIGURE 11.

The hierarchical structure of the rat lateral hypothalamic area (LHA) as described in the Swanson Rat Brain Atlas (59). Those regions whose connections have already been described in detail are shown in red, with the corresponding references in square brackets.
5.5.2.2.2. Interpretive challenges imposed by the architecture of the lateral hypothalamic area.
Unlike the ARH, PVH, or VMH the heterogenous cell types of the LHA show a characteristically loose distribution with respect to the cytoarchitectonic boundaries of its gray matter regions. This is particularly true for four LHA neuronal cell types that are closely associated with eating behaviors: those that synthesize ORX, MCH, NT, and cocaine-amphetamine regulated transcript (CART) (508, 607, 653–655). LHA neurons do not appear to synthesize peptides, transcription factors, receptors, or other cellular components in a way that coincides with recognized cytoarchitectonic boundaries. In addition, the neurochemical makeup of LHA neurons is highly complex. For example, depending on their location some LHAMCH neurons also synthesize CART (655), while LHANT neurons are plastic and varied with regards to their peptide configuration (509, 656). These factors greatly complicate our understanding of how the LHA controls eating behaviors.
To simplify interpretations, we need to consider how we can cluster groups of LHA neurons. As an example, neurons that synthesize ORX are the only ones that are mostly confined to the LHA. They form a functionally heterogeneous population that distributes across multiple LHA regions (607, 654, 657–659). How can we unpick this population with regards to its projections and thereby its function? At least two nonexclusive schemes are apparent: one is based on the LHA’s cytoarchitecturally defined regions and their connectivity (FIGURE 11); the other is based on the connectivity of neurochemically defined neurons. To illustrate these two schemes, FIGURE 12A shows one LHA level where LHAORX neurons are distributed across multiple regions, each of which has distinct output projections. Two adjacent LHA regions, the suprafornical (LHAs; FIGURE 12A, yellow) and the juxtadorsomedial (LHAjd; FIGURE 12A, pink), together contain over 40% of the LHAORX population (654). Both regions locate to a part of the LHA closely associated with eating behaviors that is often loosely referred to as the perifornical area, a widely used label that is rarely defined empirically (however, see Ref. 661). Yet, the projection patterns of the LHAs and LHAjd are quite different (662), and both are different again from the nearby subfornical region (LHAsf, FIGURE 12A, blue) (663). Untangling the functional organization of LHAORX neurons is further complicated by the fact that they send projections to at least four different targets (FIGURE 12B), subsets of which are collateralized (660). These four groups are intermingled rather than being distributed in discrete LHA regions (FIGURE 12B) meaning that their connectional organization does not adhere to regional borders. Therefore, using genetically guided tools to manipulate neurons based solely on ORX expression or indeed any of the markers that are widely distributed in the LHA, including those for GABAergic and glutamatergic neurons, will generate effects across the multiple functional systems to which these neurons contribute.
FIGURE 12.

A: the locations of lateral hypothalamic area orexin (LHAORX) neurons (blue dots) at level 29 of the Swanson Rat Brain Atlas (59). Adapted from Ref. 508 with permission. B: the projections and collateralization of LHAORX neurons. Adapted from Ref. 660 with permission. See text for more details. ZI, zona incerta; RE, reuniens nucleus; PH, posterior hypothalamus; TUI, lateral part of the tuberal nucleus; VTA, ventral tegmental area; ARH, arcuate nucleus; TMN, tuberomammillary nucleus; ACBc, accumbens nucleus, core; opt, optic tract; jvv, juxtaventromedial region; jvd, juxtaventromedial region, dorsal zone; ventral zone; fx, fornix; ma, magnocellular nucleus; d, dorsal region; LC, locus ceruleus; jd, juxtadorsomedial; s, suprafornical region, sfp, subfornical region, posterior zone; vm, ventral region, medial zone; p, posterior; VMH, ventromedial hypothalamic nucleus.
The outcome of the LHA’s intricate anatomical structure is that it is obviously of no value to regard it as a single entity when considering its controlling role in eating behaviors, or indeed any of its functions. However, in defense of this broad view is the fact that manipulating specific LHA regions, although possible (e.g., Refs. 664, 665), remains a major challenge. Therefore, how do we deal with these problems? A reasonable starting point is to examine what spatially targeted manipulations have already told us about differences in the ability of LHA regions to initiate meals. While spatially targeting hypothalamic regions has lost its allure to genetic targeting in the past decade, mismatches between the chemical phenotypes of LHA neurons and the complex underlying organization of the LHA means that spatial targeting retains some value as a starting point for assessing the function of the LHA’s various parts.
5.5.2.2.3. The lateral hypothalamic area and meal initiation: what we already know from spatially targeted manipulations.
5.5.2.2.3.1. lesions.
Anand and Brobeck (18, 666) used large electrolytic lesions to identify a “satiety center” in the ventromedial hypothalamus, lesions of which produced hyperphagia. These actions were counterbalanced by a “feeding center” in the LHA where lesions led to an apparent loss of the motivation to eat. This dual center model included the lateral hypothalamic syndrome characterized by adipsia and aphagia. However, later studies using axon-sparing excitotoxins produced more nuanced results, demonstrating that the lateral hypothalamic syndrome was not in fact the result of damage to lateral hypothalamic neurons but was a consequence of destroying dopamine projections from the VTA and the compact part of the SN that traversed the LHA in the medial forebrain bundle (667). In particular, Winn (34) used NMDA, an axon-sparing glutamatergic excitotoxin, to lesion large areas of the LHA lateral to the fornix from about the level of the rostral PVH to immediately caudal of the DMH (FIGURE 13). Remarkably, after an initial recovery period NMDA lesioned rats maintain their food intake and rate of body weight increase at the same levels as unlesioned controls (469, 668). Therefore, unlike the original electrolytic and less specific kianate/ibotenate excitoxic lesions that extended beyond the boundaries of the LHA, NMDA LHA-lesioned rats do not have profound eating deficits. Importantly however, these NMDA lesions did eliminate eating responses to 2-DG-induced cytoglucopenia (468), perhaps in part, because they remove LHAORX neurons that are driven by catecholaminergic projections that encode glucosensing information from the medulla (462).
FIGURE 13.
Coronal sections of a rat brain from the work of Winn and colleagues (Ref. 469) with permission) showing the part of the lateral hypothalamic area with a bilateral neuron-specific lesion produced by N-methyl-d-aspartic acid (NMDA). See text for a description of the deficits produced by these lesions.
Considered together, these lesion studies show that animals with a substantial loss of LHA neurons lateral to the fornix (FIGURE 13) retain their ability to organize habitual meals but cannot develop eating responses after a challenge to internal energy balance. On the other hand, lateral hypothalamic syndrome animals have lost ascending dopaminergic projections that traverse the LHA and so are impaired in their ability to convert proactive signals processed by the hypothalamus, particularly in the ARH and PVH, and elsewhere in the brain into organized eating behaviors; a point to which we return later (sect. 5.5.4).
5.5.2.2.3.2. injections.
The first neurochemical injections targeting the LHA fostered the idea that the control of different motivated behaviors might be chemically coded, a notion that now lacks support (669). During the time when acetylcholine and norepinephrine were the only verified neurotransmitters, Grossman (19, 623) showed that adrenergic and cholinergic mechanisms in the LHA were associated with eating and drinking, respectively. Grossman’s experiments were the first to identify the perifornical LHA as a potential controller for eating and drinking. Using microinjections to assign function to hypothalamic locations was developed further by others (reviewed in Ref. 624). In particular, Stanley’s systematic work (624, 639, 670–674) in the LHA on the ability of NPY, glutamate, and GABA to alter food intake produced about as high a level of spatial resolution as is possible in this region with this technique. This work identified the perifornical LHA, rather than the PVH, as the most sensitive hypothalamic site for NPY-stimulated eating, and an area of the LHA lateral to the fornix between the caudal PVH and caudal DMH as a sensitive site that mediates glutamate-stimulated- or GABAA-receptor-suppression of eating. These two regions remain active research targets for understanding the LHA’s role in controlling eating behaviors.
5.5.2.2.4. Understanding the lateral hypothalamic area and meal initiation: where do we go from here?
The reason spatially targeted manipulations have a limited ability to reveal how the various parts of the LHA contribute to eating behaviors is obvious when seen from our current viewpoint: these approaches lack the required neuronal specificity and spatial resolution to account for what we now know about the LHA’s organization and its relationship to eating behaviors. Two developments during the past 25 yr have shifted the baseline in this regard. First, we know substantially more about the detailed outputs (FIGURE 11) and inputs of many LHA regions than we did when MCH, NT, CART, and ORX neurons were first identified in the LHA. Second, for the past 15 yr or so it has been possible to manipulate specific components of LHA neurons with genetically guided precision. This specificity, and the relative ease with which these manipulations are now accomplished, means that the principal technical focus for investigating what the LHA controls is the chemical phenotype of its neurons, rather than its regional and connectional architectures.
The complexity of LHA neurons has recently been raised to a new level by two single cell RNA sequence analyses. In the first (675), ∼3,500 mouse LHA cells were sampled from a 450-µm LHA slice (∼21% of the total length of the mouse LHA) at the level of the DMH that is dorsal, lateral, and ventrolateral to the fornix. This study found 30 different populations of GABAergic and glutamatergic neurons that were organized into 20 separable clusters based on the relative expression of many hundreds of genes. The second study (676) found that the transcriptional profile a series of genes associated with an array of intracellular signaling pathways in LHAGlut neurons was altered by high-fat-diet feeding. These changes were accompanied by reduced neuronal activation when a sucrose solution was consumed. The authors speculate that the reward encoding properties of LHAGlut neurons is altered by high-fat-diet feeding in a way that reduces their ability to restrain food intake, but both are key features that need to be elaborated. How the neuron clusters in both these studies map onto the LHA’s structural architecture is not yet known.
Given the ever-increasing depth and sophistication of how we comprehend the LHA’s two architectures, connectional and neurochemical, we now need to find ways to link these two frameworks more closely. A complementary approach that takes advantage of each framework’s strengths will undoubtedly provide more compelling insights about the LHA’s role in eating and other behaviors than independent analyses. To encourage this goal, our focus in the next three sections is to emphasize, where possible, the network context of the many functional studies that have appeared in the past decade. However, the preponderance of results from neurochemically targeted manipulations means that we use chemical phenotype as the basis for discussing the role of the LHA.
5.5.2.2.5. LHA neurons and meal initiation.
To approach this topic it is first worth describing in more detail the effects of eliminating LHA neurons, rather than fibers-of-passage in the medial forebrain bundle, on the ability of animals to initiate habitual meals. As mentioned earlier, Winn’s group (34) removed neurons in the LHA lateral to the fornix in rats that were individually housed with unrestricted access to food and water (FIGURE 13). They found 1) no motor deficits; 2) that food intake and the rate of weight gain in lesioned rats was indistinguishable from intact controls; and 3) they retained a normal compensatory eating response to deprivation (34, 468). These results make two points: first, that in a simple nonchallenging environment these LHA neurons, unlike ARHAgRP neurons, are clearly not required for habitual or deficit-induced meal initiation; and second, that lesioned rats took about a week to recover normal habitual food intake, meaning that the loss of LHA neuronal function is not immediately compensated by other regions, and requires complex network interactions. However, these experiments did not address how these lesions affected meal structure or 24-h intake distribution, or how well these animals performed in more challenging settings where complex foraging strategies are required. Given our current understanding of LHA structure and function, it seems reasonable to speculate the presence of defects to both variables.
5.5.2.2.5.1. gaba and glutamatergic neurons.
Which of these NMDA lesioned neurons mediate effects on meal initiation? Genetically guided manipulations of mouse LHAGABA (677) or LHAGlut (678) neurons lateral to the fornix at the level of the DMH suggest a complex role for these two populations, some of which synthesize ORX or MCH (675, 679). Jennings and coworkers (677) showed that either activating LHAGABA neurons or inhibiting LHAGlut neurons (678) led to both the appetitive and consummatory phases of eating behavior. Switching around the activation state of these two sets of neurons decreased these actions. Impressively, this group also produced evidence suggesting that the control functions for appetitive and consummatory actions were distributed between separable populations of LHAGABA neurons (677).
The ability of neurons in the LHA and the nearby innominate substance (SI) to rapidly respond to the sight and/or the flavor of food in a satiety-specific manner was first identified by Rolls and his colleagues (680–683) in a comprehensive set of monkey experiments in the mid-1970s. These neurons altered their responses as monkeys learned to associate a visual cue with the presentation of a food item (684) and were not related to feedback from orofacial motor actions (681). More recently, rat LHAGABA neurons have been shown to play an active role in a learning process that associates the sensory information of food cues with the rewarding effects of consumption (685). Although the precise location of these manipulated LHAGABA neurons is not clear, as is also true for the monkey LHA neurons recorded by Rolls and his colleagues (685), their optogenetic stimulation implicates LHAGABA projections to the VTA for signaling learned reward predictions, as opposed to controlling motor execution. One function of these LHAGABA projections maybe to exert disinhibitory control of VTA dopaminergic projections to the ACB (686). Whether these LHAGABA neurons also synthesize NT is not known (see sect. 5.5.2.2.5.4.).
If we now consider the collective outcomes from NMDA lesions and genetically guided manipulations, we see that this part of the LHA contains populations of neurons that counterbalance each other with regard to meal initiation (687). Activating or inhibiting LHAGlut or LHAGABA neurons will appropriately increase or decrease meal initiation. The work of Stanley and colleagues highlights a role for glutamatergic and GABAergic inputs to this region to help mediate these effects (624, 639, 670–674). Eliminating both populations (34, 468, 668) appears to cancel out their opposing influences, and animals retain the ability to initiate meals in a nonchallenging environment. However, as we have speculated above, their ability to obtain food in more complex situations may well be compromised. This is as yet untested.
5.5.2.2.5.2. mch and cart neurons.
LHAMCH neurons are found in multiple LHA regions and the ZI. Smaller numbers occur in the posterior hypothalamus and the DMH (607, 654). Importantly, they are distinct from LHAORX and LHANT neurons (200, 654, 688). Like LHAORX neurons, LHAMCH neurons project throughout the cerebrospinal trunk (689–692). They are also glutamatergic (693).
MCH both increases food intake and reduces energy expenditure through its seven-transmembrane domain G protein-coupled receptor, MCH 1 R, thereby promoting overall elevated weight gain in rodents (694–698). Rodents lacking MCH (699) or MCH 1 R (700) are lean, whereas transgenic MCH overexpression produces hyperphagia and obesity (701). Similar to the ORX system, the central distribution of MCH 1 R is extensive and spans throughout the cerebrospinal trunk (689, 697, 702, 703). Whereas LHAORX neurons promote eating, at least in part, based on promoting appetitive preprandial processes, LHAMCH neurons appear to predominantly promote consumption based on postoral sensory mechanisms that enhance the reward value of nutrient absorption, a phenomenon termed “appetition” by Sclafani and colleagues (704). For example, pairing ingestion of the nonnutritive sweetener sucralose with optogenetic activation of LHAMCH neurons in a two-bottle preference test of sucralose versus sucrose reversed the normal preference for sucrose over the nonnutritive sweetener sucralose in two-bottle tests (705). This study also showed that the postingestive reinforcing effects of sucrose in sweet-blind Trpm5-/- mice require LHAMCH neurons. Consistent with a role for these neurons in promoting appetition, the orexigenic action of LHAMCH neuron activation is enhanced when optogenetic stimulation is paired with food consumption (706).
The downstream neural targets through which LHAMCH neurons stimulate eating are incompletely understood but appear to involve communication within hypothalamus as well as those forebrain structures associated with incentive motivation and food reward. Hyperphagic responses to spatially targeted MCH are restricted to regions containing the PVH and DMH but not the LHA, VMH, or SO among others (707). The ACB has also been identified as an eating-relevant target of LHAMCH neurons via putative interactions with both dopaminergic (708) and opioid signaling pathways (709). A recent study showed that LHAMCH neurons can stimulate eating through both synaptic mechanisms and the direct release of MCH into the CSF (200). MCH levels in the CSF are elevated before the onset of nocturnal eating and after selective chemogenetic activation of LHAMCH neurons. Activation of these CSF-projecting neurons increases food intake (200). Approximately one-third of all MCH- and ORX-producing neurons in the LHA and ZI communicate directly with the CSF meaning that volume transmission is likely a significant communication route (200). Therefore, MCH effects on food intake not only occur at multiple sites of action but also through multiple neuropeptidergic signaling mechanisms (see sect. 2.7.4).
5.5.2.2.5.3. orexin neurons.
LHAORX neurons distribute widely throughout the entire cerebrospinal trunk and, together with LHAMCH neurons, may well have the most extensive projections of any hypothalamic neurons. Most regions from the frontal cortexes to the caudal spinal cord (657, 710) are moderately to heavily innervated, while a few, including the HPF and the cerebellum, have only sparse ORX inputs.
Although it has been clear for decades that the LHA is a key structure for eating behaviors, its size and complexity were barriers to clarifying its role when using the techniques of the time. The discovery of the ORX (hypocretin) peptide system in 1998 (711, 712) was a big step forward because it identified a peptide component in a large group of neurons whose brain location was largely confined to an LHA area that was already known to control eating behaviors (410).
The LHAORX system has three seemingly discrete functions. First, many of the initial studies focused on its ability to stimulate food intake. However, a report soon followed noting that on an equimolar basis, centrally administered ORX was approximately six times less potent than NPY and so, despite its name, it did not appear to be a significant orexigen (713). It is also notable that increased food intake has yet to be a reported outcome of pharmaco- or optogenetically stimulating ORX neurons (714, 715). Second, the discovery of a dysfunctional LHAORX system in human and canine narcolepsy (reviewed in Ref. 716) gave rise to many studies showing strong correlations between the activity of LHAORX neurons and arousal and sleep states (reviewed in Refs. 717, 718). Third, LHAORX neuronal projections to the ACB and the VTA were involved with associating environmental cues with the consumption of items of high incentive value, including food and drugs (reviewed in Ref. 658). The weight of this evidence therefore supports LHAORX neurons as being significant contributors to arousal, sleep state transitions, and the increased drive for behaviors of high motivational relevance.
We have already included parts of the LHA in the upper brainstem controller for eating behaviors. However, do LHAORX neurons themselves contribute to meal initiation? Direct evidence for their involvement in meal initiation comes from mice with LHAORX neuron ablation, who lack food anticipatory activity under daily restricted eating conditions (719). Furthermore, LHAORX neuron activity (recorded in vivo using fluorescent calcium indicators) increases before anticipated eating occurs and then diminishes within milliseconds after eating onset (720). However, these results are still not consistent with LHAORX neurons being involved solely with initiating eating. Instead these neurons appear to help orchestrate the behavior that has the highest priority for an animal’s current environmental location, its arousal state, and the particular time of its day, properties that are mediated by a wide range of forebrain inputs (721).
Therefore at the simplest level, if the signals received by LHAORX neurons collectively favor eating, then they will promote the appropriate appetitive actions to direct the animal toward a food source; if the signals favor another behavior, then that one will be initiated. This means that at least some LHAORX neurons should be able to integrate information about interoceptive and exteroceptive information from the endbrain and rhombicbrain with arousal state information to bias the most relevant motivated behavior (650, 722–724). Outputs from LHAORX neurons to the VTA, ACB, PVT PFCm, and other forebrain targets (282, 657, 725–728), together with LHAORX neuronal interactions with LHAMCH, LHANT (485, 729), and other neurons in the PVH and ARH (657) are all likely key players to enable this capability.
LHAORX neurons are not only involved in initiating eating in the appropriate circumstances but also play a role in increasing meal size via descending midbrain and rhombicbrain projections. ORX delivery into the fourth ventricle (730, 731) or direct ORX administration to the VTA (732) enhances meal size, whereas ORX receptor antagonist delivery has the opposite effect. Orexin’s meal size enhancing effects appear to involve an interaction with circulating endocrine factors. A recent neural pathway was identified through which ghrelin acts in HPFv neurons to selectively enhance meal size (without affecting meal frequency) through downstream communications to LHAORX neurons that project to the hindbrain laterodorsal tegmental nucleus (733). It remains to be determined whether distinct populations of LHAORX neurons mediate appetitive goal-directed behaviors versus satiation processing, as well as the extent to which these putative distinct populations of LHAORX neurons are differentiated based on anatomical LHA subregion location, neurotransmitter phenotype, and/or downstream projection targets.
5.5.2.2.5.4. neurotensin neurons.
LHANT neurons form a large GABAergic population that distributes across its medial and perifornical tiers and extends rostrally into the lateral preoptic area (514, 734, 735). They are separate from, but tightly intermingled with and outnumber LHAMCH and LHAORX neurons (508, 736). Rat LHANT neurons were first characterized as part of a broader set of forebrain projections to the VTA (734), and by their response to cellular dehydration, thereby implicating them in the control of eating and drinking behaviors during DE-anorexia (see sect. 4.3.4.2.3) (508, 511–514, 653). A major advance occurred when it was found that ∼15% of LHANT neurons express LepRb and project to the VTA and SN in a way that supports reduced food intake (514, 688, 736, 737). LHANT-LepRb neurons and those LHANT neurons that are activated by dehydration form separate populations (514). During the past decade, Leinninger and colleagues (294, 510, 688, 738–740) have systematically investigated these neurons to show their key role in transducing leptin signaling into eating behavior by way of altered VTA/SN function. There is also evidence that LHANT neurons exert local control on LHAORX neurons (741). Collectively, these results demonstrate the central position that LHANT neurons occupy in the hypothalamic control of meal initiation.
5.5.2.2.6. The actions of physiological signals on lateral hypothalamic neurons.
5.5.2.2.6.1. glucose.
Brain glucosensing neurons were first found in the LHA in 1969 (441, 442). Some of these neurons have since been identified as LHAORX and LHAMCH neurons (742, 743). Work by Burdakov and others (742, 744, 746) have characterized LHAORX glucosensing neurons as adaptive functional links between metabolic and arousal states. They can be gated by other metabolic fuels (1272) and provide predictive value for metabolic control (746, 747). These transduction capabilities of LHAORX neurons may directly rely on changes in their ATP concentrations and therefore their energy status (745). Burdakov and his colleagues (749) also showed that LHAORX neurons have amino acid-sensing capabilities.
5.5.2.2.6.2. leptin and ghrelin.
We have already seen how leptin can directly alter the function of LHANT neurons. Although LHAORX neuronal activity is influenced by leptin (742), this control is exerted in a complex indirect manner because neither these nor LHAMCH neurons express LepRb (481, 485, 748, 750). Importantly, however, leptin can influence presynaptic inputs to those LHAORX but not LHAMCH neurons that project to the VTA (751). Inputs from the ARH and LHANT neurons (736, 750, 752) figure prominently in these functions. Evidence also supports the idea that ghrelin can alter VTA dopaminergic transmission by acting directly on LHA neurons, and particularly LHAORX neurons (748), that project to the VTA (144, 605, 752, 753, 754).
5.5.2.2.6.3. vagal sensory information.
Recent viral tracing results show that the PSTN, one of the most caudal regions in the LHA, is a prominent forebrain target of vagal sensory information, an attribute it shares with the BST, PVH, DMH, other parts of the LHA, and the CEA (345, 366). The PSTN shows conspicuous Fos expression after deficit-activated eating (626) and is part of the projection network that rapidly leads to eating following optogenetic stimulation of ZIGABA neurons (277). By virtue of its outputs to the BST, PVT, CEA, PB, and the dorsal medulla, the PSTN may be a key LHA component through which vagal sensory information is incorporated into the upper brainstem eating control network and is then distributed throughout the brain (254, 277, 755, 756).
5.5.2.2.7. Which regions project to LHA neurons to control eating behaviors?
A region as large, and as structurally and functionally diverse as the LHA, has an equivalently large and diverse set of inputs, many of which are implicated in controlling eating behaviors (651). Those originating from within the hypothalamus and the rhombicbrain are discussed later. Here we consider those from the endbrain.
The LHA as a whole is the recipient of a major set of inputs from the endbrain, a detailed description of which is beyond the scope of this review. Therefore, before highlighting two examples from the work of Kelley and Petrovich that emphasize endbrain inputs, we note studies that either review or show evidence of inputs to the LHA from endbrain regions relevant for controlling eating behaviors: ACB (622) and amygdala and PFCm (621), BST (609, 757, 758), and parts of the lateral septal complex (511, 613).
A seminal group of findings from Kelley and her colleagues (638, 759, 760) first linked the outputs from ventral striatal regions to the LHA with meal initiation. They showed that manipulating fast-acting neurotransmission in the ACB shell rapidly increased food intake (638, 759, 760). This occurred in less than a minute after dose dependently blocking non-NMDA receptors in the medial part of the ACB shell (638). A major effector pathway for these findings is to the LHA (627, 638, 761), but this output may also involve projections to the SI (ventral pallidum) and PVT (762–764), by way of the LHA (622).
We have already discussed that the basis of cue potentiated eating is associative learning that requires a network involving the PFCm, BLA, and the LHA (sect. 5.4.4), and further work from Petrovich and her colleagues (592, 594, 621, 727) has elegantly shown the structure-function relationships within this network, including sets of inputs to the LHA (also see sect. 5.4.4).
Recent work has revealed the importance of projections from the INS to the PSTN. The PSTN shows conspicuous Fos expression after deficit-activated eating (626), during predator hunting (523), neophobic eating responses, and after lipopolysaccharide or cisplatin administration (765), which are agents that illicit strong sickness responses. The PSTN is part of a network that receives direct and indirect (via the CEA) inputs from the INS (765, 766), and then projects to the INS, CEA, PVT, PB and NTS (755, 765, 766). The PSTN appears well placed to act as a gate controller (go/no go) for eating behaviors (765). Taking these results together with the fact that the PTSN also receives interoceptive information via VSN (345) and PBlCGRP neurons (767), this key LHA nucleus may help determine whether preferred food items are consumed in response to conflicting interoceptive and exteroceptive information (765).
What is evident from all these reports is that of the three core hypothalamic components of the upper brainstem eating control network, the LHA is the prime recipient of inputs from the endbrain. Consequently, it is able to perform the widest range of integrative functions on this information.
5.5.2.3. arcuate nucleus.
The ARH is a long compact bilateral periventricular nucleus located between the most ventral part of the third ventricular wall and the ventral surface of the brain adjacent to the ME. It occupies ∼28% of the total length of the hypothalamus of both rats and mice. Since the discovery of leptin the ARH is a strong contender for the most popular brain target for investigating food intake. However, this has not always been the case. Because of its proximity to the ME and the hypophysial portal vasculature, the major focus of earlier studies of the ARH was as a mediator of reproductive function and anterior pituitary secretion, particularly prolactin and growth hormone secretion (e.g., Refs. 768–770). Indeed, the fact that ARHkisspeptin neurons are part of the GnRH pulse generator (642) usefully reminds us that this nucleus is involved with functions other than energy balance.
A role for the ARH in energy balance and food intake emerged ∼50 yr ago with reports of the obesogenic effects of ARH lesions generated by neonatal exposure to monosodium glutamate, an excitotoxin that has little to no blood-brain barrier penetrance in adult animals (771, 772). Interest increased dramatically with the discovery of leptin in 1994 (23) and the identification of the ARH as a major brain target for leptin signaling (405–408). Investigations rapidly proceeded in novel directions when significant numbers of ARH neurons were found to express one of two peptides: AgRP, which is only found in the ARH; and POMC, which, in the forebrain is only found in the ARH (773, 774). The other brain location of POMC neurons is the NTS. POMC can be further processed into seven different peptides, the nature of which depends on the cell type (775); ARHPOMC neurons predominantly produce aMSH and beta-endorphin (775, 776).
Many rat and mouse ARH neurons are GABAergic (777–779) particularly in the more rostral ARH; whereas in mice glutamatergic neurons are found in its more caudal regions (780) where they are involved with suppressing food intake (780, 781). In addition to being GABAergic or glutamatergic, ARH neurons as a group coexpress a rather impressive array of neuroactive molecules, including more than a dozen peptides and one catecholamine, dopamine (655, 782). Recent signal cell transcriptomic analyses of the arcuate-ME region (783, 784) reveal an extremely complex array of cell types and mixed chemical phenotypes. These results emphasize that the region of the hypothalamus around the base of the third ventricle is one of the most structurally complex regions of the entire brain (see also Refs. 363).
ARHAgRP neurons coexpress NPY, ARHPOMC neurons coexpress CART, and both can signal postsynaptically using GABA. As with virtually any neuron, coexpressed neuromodulatory signals, particularly those using GPCRs, increase the signaling flexibility of ARH neurons above and beyond that possible with fast-acting ionotropic neurotransmitters (785, 786). ARHAgRP and ARHPOMC neurons are often labeled orexigenic and anorexigenic, respectively. However, it is important to note that while these labels might be convenient, they are somewhat of an oversimplification that masks the underlying diversity and complexity of the processes these neurons use to control energy balance.
Three other non-AgRP, non-POMC ARH neuron populations have been implicated in controlling food intake: ARH tyrosine hydroxylase (TH) neurons; and two sets of ARH neurons that express cre under the control of pancreas-specific promoters, the rat insulin-2 (Ins2) promoter (the so-called RIP-cre neurons), and the pancreas-duodenum homeobox promoter PDX-1-cre (787–790).
5.5.2.3.1. The arcuate nucleus as an entry point for circulating physiological signals.
During the past 25 yr, there has been a vast amount of work on the cellular and intracellular mechanisms used by physiological signals to engage ARH neurons, the outcome of which then affects many aspects of energy balance. Because our goal is to discuss the wider context of the way ARH neurons interact with other regions of the brain to enable physiological signals to control eating behaviors, we refer readers to some of the many outstanding research reports and reviews on these cellular and intracellular mechanisms (e.g., Refs. 222, 304, 409, 426, 791–805).
5.5.2.3.2. AMP kinase and the arcuate nucleus.
As we discussed earlier (sect. 2.5.2), the enzyme AMPK is a ubiquitous cellular fuel gauge that helps control ATP availability and energy balance (FIGURE 2) (82, 99, 806–808). Changing AMPK’s phosphorylation state in ARH neurons, and thereby its activity, leads to corresponding increases or decreases in food intake (809). Fasting and ghrelin administration both increase AMPK activity (100, 809), whereas leptin decreases its activity (810, 811). Glucose availability also controls AMPK activity in ARH neurons (812–814), which may contribute to their glucosensing capability (813).
Switching AMPK’s phosphorylation state helps control ATP availability in all cells (FIGURE 2). When AMPK activity changes in those ARH neurons that directly influence eating behaviors, AMPK’s phosphorylation state is effectively transducing information related to ATP availability into altered neuronal activity, thereby affecting how downstream neural networks operate to control eating. Although all neurons, and very likely all cells, use AMPK to monitor and control their ATP availability, ARH neurons can couple changes in AMPK’s phosphorylation state to altered eating behaviors. This property may be limited to these ARH neurons along with other key hypothalamic regions. In principle, this property appears similar to brain glucosensing mechanisms in the VMH and elsewhere. Although all cells react to changes in glucose availability by altering their energy balance, only certain neurons detect glucose in a way that alters their firing rates to allow them to signal altered local glucose availability and thereby control counterregulatory responses (103, 438–440) (also see sect. 4.3.4.2.1).
The underlying cellular mechanisms in ARH neurons that change AMPK phosphorylation in response to ghrelin and fasting and how these alter downstream synaptic plasticity are not completely understood (100, 809). One possibility is that they involve glutamate signaling from PVHTRH neurons to the ARH (815). In turn this alters AMPK phosphorylation in target neurons by way of ghrelin signaling and intracellular calcium concentrations, which then alter downstream synaptic plasticity in ARHAgRP neurons (100, 811). AMPK-related mechanisms would therefore involve interactions between the PVH and ARH, emphasizing the precedence of a core hypothalamic eating network over individual regions to control the multiple aspects of eating behaviors (see sect. 5.5.4).
5.5.2.3.3. Arcuate neurons and meal initiation.
About 15 yr ago a series of landmark studies used genetically guided techniques to investigate the roles of ARH neurons in controlling food intake and the arrangement of their projections through which this control is exerted. The first of these studies ablated at least 85% of ARHAgRP neurons with gene-guided diphtheria toxin (816, 817). This approach demonstrated the necessity of these neurons in adult but not neonatal mice for both the appetitive and consummatory aspects of 24-h intake of a liquid diet (817, 818). Reduced food intake quickly became apparent after ablation, and starvation developed within 7 days (816, 817). Further work showed that starvation was mediated by GABAergic signaling in ARHAgRP projections to the PB (819), but it did not require melanocortin signaling (818).
Two other aspects have been revealed by ablating ARHAgRP neurons. First, their priority for controlling eating behaviors is influenced by the palatability of the available food (820). This emerged by examining the requirement of ARHAgRP neurons for eating in response to either a 24-h fast, ghrelin, or serotonin (5HT) receptor agonists. ARHAgRP neurons were required for eating responses to all three challenges when regular chow was presented but not for foods with high novelty or palatability. A role for dopaminergic mechanisms in these effects, possibly involving the VTA, was also strongly implicated (820), which emphasizes the involvement of ARH mechanisms in the networks associated with conveying motivation-related information to motor control networks (see sect. 5.5.4). Second, a recent report showing that catecholaminergic projections from the NTS to ARHAgRP neurons are required for glucoprivic eating in mice (467) means that the mechanisms responsible for this particular eating behavior are most likely different from those driving deficit-induced eating and daily habitual eating, neither of which in rats requires catecholaminergic inputs to the ARH (460, 464, 821).
While a role for the PB in meal control has been recognized for many years, particularly in response to aversive tastants and other noxious stimuli (822–824), attention has centered mostly around the PB’s extensive projections to the forebrain (825–829). However, the work of Wu and colleagues (819), together with other more recent findings in mice (272, 830–833), emphasize that projections from the ARH and PVH to the PB also have important actions for controlling food intake. We discuss these later (see sect. 6.3.1.1.3).
Genetically guided methods have demonstrated that optogenetically stimulating mouse ARHAgRP neurons quickly increases food intake (628, 834), showing that these neurons are part of a primary control network to initiate eating behavior. This particular action does not require inhibiting ARHPOMC neurons as an intervening step (628) meaning that ARHAgRP neurons themselves directly engage those downstream targets that initiate meals.
In contrast to ARHAgRP neurons, ablating ARHPOMC neurons with gene-guided diphtheria toxin increases food intake and body weight (816). However, short-term optical stimulation of ARHPOMC neurons does not suppress the habitual meals seen at the light/dark transition (628). Instead, reduced food intake only occurs when POMC neurons are stimulated for 24 h, an effect that is dependent on melanocortin signaling (628). Chemogenetic stimulation of ARHPOMC neurons over a 2-day period also suppressed food intake (835). On the other hand, chemogenetically stimulating NTSPOMC neurons did suppress those meals taken within 2 h of the light/dark transition (835), meaning that activating these two POMC cell groups reduces food intake, but it happens over very different timescales and in response to different physiological signals.
As we noted earlier, the ARH contains groups of neurons that do not express AgRP or POMC, and the ability of these neurons to control eating behaviors and adiposity has recently been characterized. One study (836) showed that chronically stimulating ARHGABA neurons leads to hyperphagia and obesity for up to 11 wk whose magnitude was similar to leptin deficiency. Interestingly, these effects were achieved independently of ARHAgRP neurons. Opposing actions on food intake and adiposity occurred when ARHGABA neurons were inhibited.
In another study, optically stimulating ARHTH neurons in mice increases food intake (837), but this may not be as rapid as when ARHAgRP neurons are stimulated. Suppressing ARHTH neurons for 5 mo reduces body weight (837), although it is not clear whether this effect is mediated exclusively by reduced food intake. ARHTH neurons are activated by food restriction, and their firing rates are increased directly by ghrelin (837). In terms of signaling mechanisms, ARHTH neurons, like other catecholaminergic neurons (838) release a fast-acting transmitter as well as dopamine, in this case GABA. They appear to exert their effects at least in part, by acting directly at three locations: local ARHPOMC (but not ARHAgRP neurons), on the PVH, and possibly by controlling ME function (837). Both of these studies (836, 837) emphasize the importance of considering neurons in the ARH that are not defined by AgRP or POMC expression as important components of food intake control (839).
The now classic view of the ARH’s role in eating behavior is that it acts as an entry point into the brain for circulating physiological signals to influence the way hypothalamic networks control eating (see sect. 5.5.4). While this is clearly true, a number of groundbreaking studies published since 2015 have added surprising and important new twists that have greatly expanded our appreciation of ARH function (652, 840). Although some of these used genetically guided techniques to explore the effects of manipulating ARH neuronal function, others took advantage of the ability to directly measure the real time activity of ARH neurons during challenges to energy balance or during the presentation of sensory cues related to meal initiation.
In one set of experiments by Chen and colleagues (805) optical recordings were obtained from populations of ARHAgRP or ARHPOMC neurons using a genetically guided calcium-sensitive dye to detect real-time calcium fluctuations in freely behaving mice. This technique showed that food presentation to food-deprived mice rapidly decreased ARHAgRP and increased ARHPOMC neuronal activities during subsequent ingestion. Remarkably, the onset of these neuronal activity changes was tightly correlated to food presentation rather than ingestion. Altered activity dynamics were completed in <45 s after food presentation, often before ingestion began, and did not require that mice were trained in the procedure. Similar real-time results were obtained from ARHAgRP neurons by Mandelblat-Cerf and colleagues (841) using a multiple electrode extracellular recording technique. This group also showed a significant firing rate increase in ARHAgRP neurons between the early and the later light phase when meal time approached. These high-resolution recordings correlated with both neuronal firing rate changes and the temporal organization of liquid meals. The magnitude of these neuronal activity dynamics was dependent on food quality, with nonfood items having no effect (805). Highly palatable foods continued to alter activity rates in recently fed mice, which was not the case for a less preferred food.
Measuring how removing food at different intervals into a meal altered neuronal activity revealed that ARH neurons reverse their activity dynamics with a relationship that was inversely proportional to meal duration: the longer the meal, the less effect removing food had on reversing the activity decline (805). This attribute may allow ARH neurons to quickly evaluate the amounts of food already consumed thereby providing a means for ARH neurons to determine the temporal status of a meal. These neurons can then rapidly communicate ongoing meal status to other brain regions that mediate foraging strategies (805), either inhibiting foraging in the event of a successful meal or reinitiating it in the event of a failed meal. This organization is also consistent with the fact that ARH neurons have projections that extend beyond those that might be expected if their only function is to control the simple meal initiation. We discuss ARH neuronal connections that may mediate these properties in sect. 5.5.2.3.5.
Consistent with a more expansive meal control function, three studies have shown that ARH neurons are apparently able to influence the expression of foraging behaviors in a variety of circumstances (637, 842, 843). In the absence of food ARHAgRP neurons use a NPY-dependent mechanism to initiate stereotypic behaviors similar to foraging (digging and walking) (637, 842). However, the food intake that is stimulated by ARHAgRP neurons is NPY independent. The NPY component of these stereotypic behaviors is interesting in that NPY signaling appears to have a more complex role in eating behavior, including its appetitive aspects, than one that simply stimulates the oral components of eating (517, 519, 844, 845). Padilla and colleagues (842) used a combination of tools to show that ARHAgRP neurons use projections to the medial nucleus of the amygdala (MEA) to mediate behavioral adaptations to negative energy balance in a way that encourages foraging behavior. Yet another aspect of ARHAgRP neuronal function was revealed when fed or food-restricted mice were challenged with sensory cues that predicted food or a threat (843). The subsequent food seeking behaviors were then measured after activating or silencing ARHAgRP neurons either before or during the threat challenge. Photostimulating ARHAgRP neurons before, but not after, entering the threatening environment was able to overcome the reluctance of fed mice to avoid the aversive threat. This supports the idea that when a mouse is hungry, or when ARHAgRP neurons are active, the mouse adapts its response to a threat to enable continued eating.
Another important study showed that optically stimulating ARHAgRP neurons carries a negative valence for the animal, while not being aversive (846). In this way mice actively avoid the location where ARHAgRP neuron stimulation occurs, but they do not show a CTA using the same stimulation parameters. However, ARHAgRP neurons were able to condition a flavor preference. This study also showed that ARHAgRP neuronal activity decreased before eating occurred when mice were presented with food-related cues. False-food related cues were ineffective. The authors interpreted these results in terms of ARHAgRP neurons generating a teaching signal that may be important for associating energy deficits with the negative feelings evident during hunger (847).
A more recent study showed that NPY signaling from ARHAgRP neurons rather than AgRP or GABA is responsible for the delayed and long-lasting effects of brief optogenetic stimulation of these neurons (516). This phenomenon is proposed as a way for these neurons to vary their signaling dynamics in a way that maintains eating behavior in the face of exteroceptive food-derived sensory signals that quickly inhibit ARHAgRP neurons (516, 805, 841).
Collectively, these highly innovative studies show that ARH neurons do not act merely as transducers of circulating physiological signals to initiate meals (652). Instead they can influence motivational aspects of eating behaviors in ways that have usually been ascribed to regions more closely associated with hedonic eating. In this way they helpfully blur the boundaries between homeostatic and hedonic eating (see sect. 2.6) to reveal the more complex integrated network arrangement for how eating behaviors are controlled.
5.5.2.3.4. Which regions project to arcuate neurons to control eating behaviors?
The fact that the ARH is located in close proximity to the base of the third ventricle has made it very difficult to use spatially guided tracers in rats and mice to identify its projections (848). The first comprehensive report of ARH inputs used fluorogold injections in sheep (849). Many of these inputs have since been confirmed specifically for ARHPOMC and ARHAgRP neurons in mice. These include significant intrahypothalamic projections from the DMH, PVH, and SO (11, 815, 847, 850, 851), as well as cholinergic inputs to ARHPOMC neurons from the diagonal band nucleus (852). Together, these reports identify ARH inputs ranging from the infralimbic and orbital cortexes rostrally, to the NTS caudally. Those extrahypothalamic regions reported by Wang and colleagues (11) to have significant connections to the ARH include the LS, ventral subiculum, and parts of the BST. However, it should be noted that some of these projections (e.g., from the ventral subiculum) have not been confirmed by anterograde tracing techniques, meaning that a detailed comprehensive map of all ARH inputs is not yet apparent. Nonetheless, it is clear that the widespread nature of these ARH inputs is consistent with the highly complex functions of ARH neurons.
5.5.2.3.5. Where do arcuate neurons project to control eating behaviors?
The fact that AgRP and forebrain POMC neurons are only found in the ARH facilitated the first immunohistochemical investigations of the ARH’s output projections. Using this approach, rat, monkey, and mouse ARHAgRP neurons project quite widely in the forebrain, including to the medial parts of the LS, CEA, and MEA, those parts of the anterior BST located medial and ventral to the anterior commissure, the preoptic area, PVH (particularly its parvicellular parts; see FIGURE 14), DMH (but only its anterior and ventral parts), parts of the LHA (including to MCH and ORX neurons), PVT, and the VTA (273, 411, 854–857). For the most part, the forebrain projections of ARHPOMC neurons match those of ARHAgRP neurons (409). Further caudally, strong ARHAgRP projections were detected by immunohistochemistry to the dorsal raphe, PAG, and PB but not the medulla or spinal cord (854). However, ARHPOMC projections are present in parts of the medulla, including the NTS (11, 490). Of these, ARHAgRP neuronal interactions with dorsal raphe have recently emerged as contributors to body weight and energy expenditure control without effects on food intake (858). ARHAgRP neuronal projections to the PB are discussed in sect. 6.3.1.1.3.3.
FIGURE 14.

Photomicrographs showing where some output projections originate (A) and some inputs distribute (B) in the paraventricular hypothalamic nucleus (PVH) as defined by immunohistochemistry and in situ hybridization. A: Nissl panel identifies the PVH subdivisions shown in each of the other panels. Fluorogold (FG; white) and Fast Blue (FB; blue), respectively, label PVH neurons that project to the thoracic spinal cord and outside the blood-brain barrier. Virtually all PVH neurons express vesicular glutamate transporter 2 (VGlut2), but not glutamate decarboxylase 65 (GAD65), thereby identifying a glutamatergic phenotype (853). B: agouti-related peptide (AgRP) and alpha-melanocyte-stimulating hormone (aMSH) originate from the arcuate nucleus and nucleus of the solitary tract (459); DBH defines catecholaminergic inputs mostly from the nucleus of the solitary tract (NTS) and ventrolateral medulla (366); the left nodose ganglion (LNG) as defined by HSV1-H129 injections (366); glucagon-like peptide 1 receptor (GLP)-1R mRNA identifies PVH neurons that express the GLP-1R protein, which itself is identified by GLP-1R immunohistochemistry. Bracketed panels show the same section with different markers. Uncredited panels are unpublished results from the authors’ laboratory (A.G.W. and G.S.W.). See text for more details. AHN, anterior hypothalamic nucleus; SBPV, subparaventricular zone; LNG, left nodose ganglion; DBH, dopamine-beta-hydroxylase; ap, anterior parvicellular part; dp, dorsal parvicellular part; mp, medial parvicellular part; pm, posterior magnocellular part; pv, periventricular part; vp, ventral parvicellular part.
While genetically targeted tracing techniques have confirmed output projections, they have added significantly more organizational detail, including intra-ARH connections, to these known forebrain and rhombicbrain projections (222, 546, 664, 788, 819, 842, 859, 860). In particular, Betley and colleagues (664) showed that groups of ARHAgRP neurons that project to the anterior BST, PVT, CEA, PVH, and parts of the PAG, are topographically distributed along the length of the ARH. The number of neurons in these groups is unequal, with the anterior BST and PVH receiving inputs from the most ARHAgRP neurons, while the PVT, CEA, PAG, and PB receive far fewer. The numbers for other ARHAgRP targets were not reported. Although functional results from this study seem to rule out a one-to-all organization, the degree of collateralization exhibited by these neurons is not known meaning that it is currently not possible to distinguish between a one-to-one or a one-to-many arrangement. How the outputs from other ARH neuron populations are organized is not yet known. Of great interest is that these various projection groups appear to control not only different aspects of meal initiation but also the relationships between meal initiation, foraging, and other noneating motivated behaviors (546, 664, 842, 859). This overall organization highlights the complex functional capabilities of ARH neurons, and the fact that these are conveyed to other parts of the brain by way of differentially organized sets of outputs.
When considering ARH outputs, it is worth remembering that unlike adjacent parts of the ventral hypothalamus, a significant number of ARH neurons project outside the blood brain barrier, most likely to the ME (861). This means that their axon terminals, but not their soma, are outside the blood-brain barrier, and so dispels the notion that entire ARH is outside the blood-brain barrier. In the rat and guinea pig, some of these ARH neurons synthesize POMC and its cleavage products (782, 862), thereby contributing to the β-endorphin released into the hypophysial portal vasculature (863, 864). It is not known whether these neuroendocrine ARHPOMC neurons have centrally projecting collaterals that contribute to the ARH’s more well-known effects on energy balance or if they are a separate population. Few if any ARHAgRP neurons project to the ME (854).
5.5.2.4. paraventricular hypothalamic nucleus.
The rodent paraventricular hypothalamic nucleus (PVH) is a large, complex, and well defined cell group located adjacent to the wall of the dorsal part of the third ventricle (259, 260, 865). Although it extends laterally into the medial hypothalamus, particularly the more caudal parts of the PVH, it is, like the ARH and DMH, considered part of the periventricular zone of the hypothalamus (59). In addition to being involved with eating behaviors, the PVH is implicated in controlling remarkably broad sets of autonomic, behavioral, and neuroendocrine motor functions (866–876).
The first hints that the PVH can control eating came in the early 1970s when experiments began to question whether the VMH was a component of the dual center hypothesis for eating control (18, 877). These studies shifted focus away from the VMH to the nearby ventral noradrenergic bundle and the PVH (878–880). Combining these findings with those implicating adrenergic mechanisms for eating (623, 881) revealed the PVH as the most sensitive hypothalamic site for adrenergic-stimulated eating (882).
5.5.2.4.1. The effects of manipulating paraventricular function.
For many years, the PVH has been targeted with a battery of spatially guided manipulations ranging from lesions of varying spatial accuracy to the effects of numerous chemical and pharmacological agents. At the same time, its inputs from the ARH and other forebrain regions, as well as its downstream projections to the midbrain, rhombicbrain, and spinal cord, were being characterized (260, 261, 607, 865, 883–885). Although the collective outcome of these studies has firmly established the PVH as a major hypothalamic node in eating control networks (62), more fine-grained levels of analyses are technically beyond most of these spatially guided techniques.
The development of genetically guided manipulations has dramatically improved our understanding of the PVH’s role in controlling eating. Many of these more recent studies have taken advantage of a basic helix-loop-helix-PAS nuclear transcription factor, single-minded 1 (SIM1), whose forebrain expression is strong in the PVH, the adjacent periventricular nucleus (PV), the SO, and the basomedial nucleus of the amygdala (BMA). More scattered SIM1 expression is also seen in parts of the LHA and other hypothalamic regions (47, 886).
The first investigations of SIM1 established that it may itself play a role in controlling eating behaviors (886–889). However, taking advantage of SIM1 expression in the PVH has enabled cre-driven manipulations in specific PVH cell types. These have not only helped clarify their role in food intake, they have also provided new high-resolution insights into the output pathways used by the PVH to control eating behaviors (272, 815, 890, 891). The nature of these pathways and their contributions to PVH function are discussed further in sect. 5.5.2.4.4.
The diversity of PVH functions, including eating behavior control, is reflected in its structural and neurochemical compartmentalization (259, 260). How might these properties be organized to enable functional adaption during changing behavioral states? Xu and colleagues (276) have recently provided a striking example of how neuronal ensembles are organized following a variety of challenges associated with eating, drinking, or fear. They used calcium imaging to show that molecularly defined sets of PVH neurons (e.g., the expression of Crh, vGLuT2, Npy1r, etc.) are arranged into ensemble groups whose constituency is dependent on behavioral state.
5.5.2.4.2. Direct modulation of paraventricular function by leptin, ghrelin, and glucose.
Whether leptin acts directly on PVH neurons has been controversial. Relatively low levels of leptin receptor mRNAs are found within the PVH (475, 476), and some PVH neurons show increased Fos but not pSTAT3 expression following intravenous leptin (494, 892). These results are consistent with indirect effects on PVH neurons (893), which we now know from many studies are mediated by leptin-sensitive projections, particularly from the ARH and from the DMH (894–896). Other evidence suggests, however, that leptin can directly affect PVH neuronal function. Bath applied leptin will dose dependently depolarize some PVH neurons in rat hypothalamic slices (897). The firing rate of some melanocortin 4 receptor (MC4R)-expressing neurons in PVH slices is inhibited by direct leptin application (898), whereas the firing rates of PVHTRH neurons in the PVHap is increased by leptin (899).
Virtually all PVHTRH neurons in the rat PVHap are nonneuroendocrine (259) and therefore contribute to PVH projections both within the hypothalamus and more caudally in the brain. PVHTRH neurons, together with PVHPACAP neurons, form part of a PVH projection to ARHAgRP neurons that decreases food intake when inhibited (815).
Some PVH neurons express brain-derived neurotrophic factor (BDNF), a peptide that has effects on energy balance and food intake (900–903). However, how they influence eating depends on their location (901). PVHBDNF neurons in more rostral parts of the PVH are involved with controlling food intake, whereas those located more caudally are involved with energy expenditure. Taken together with the differential actions of leptin within the PVH, these results demonstrate a functional parcellation of the PVH with regard to the control of food intake and other aspects of energy balance that illustrates the complexity of the hypothalamic networks in which the PVH is situated.
When considering the contribution that the PVH makes to leptin’s effects on food intake, it is worth noting a recent study showing that leptin’s ability to reduce the hyperphagic effects of starvation requires an inverse relationship with corticosterone secretion (904). Starvation-induced falls in leptin increase corticosterone secretion, which in turn increases the drive to eat by way of its actions on ARHAgRP neurons (904). Although the mechanisms behind this effect are unclear, a number of findings point to key roles for neuroendocrine PVHCRH neurons and their medullary catecholaminergic inputs. These include the ability of leptin to decrease CRH mRNA levels in the PVH (905), potential leptin/corticosterone interactions in controlling ARH AgRP mRNA levels (905), and the requirement of medullary catecholaminergic inputs to the PVH to maintain PVHCRH neuronal sensitivity to corticosterone in different states of energy balance (464, 906).
Although ghrelin can affect the firing rate of all three functional types of ex vivo PVH neurons (907), how these effects are mediated in vivo is unclear. Transport into the CSF and subsequent action in the PVH is one possibility (908), but it remains uncertain whether ghrelin can directly influence the PVH’s actions on eating behavior.
Electrophysiologically characterized glucosensing neurons are found in the rat PVH (909). While most are nonneuroendocrine, any functional role they may have in controlling energy balance and eating has not been found.
5.5.2.4.3. Which regions project to PVH neurons to control eating behaviors?
Projections to the PVH that influence eating behaviors originate from the endbrain, hypothalamus, midbrain, and rhombicbrain. Each source provides distinct sets of inputs to each functional compartment of the PVH.
5.5.2.4.3.1 endbrain.
The number of endbrain regions that provide eating-related input information directly into the PVH is small. Their main source is the BST in the rostral pallidum (59). Of the more than a dozen identified regions of the BST, those that project directly into the PVH are found in the parts of its anterior division that are located dorsally, ventrally, and laterally adjacent to the decussation of the anterior commissure (609, 758, 910, 911). Notably, these are some of the same nuclei that receive projections from ARHAgRP neurons (see sect. 5.5.2.3.5).
There are no reports using spatially guided tracers of significant PVH inputs in rats from any amygdalar (611, 756, 884) or cortical areas, including the HPF (498, 617, 884, 912). In this regard, the BST provides the main link between amygdalar and cortical areas and the PVH (e.g., Refs. 364, 618, 758, 913) (FIGURE 10). A somewhat contrary picture has emerged more recently from PVHBDNF neurons in mice using genetically guided tracing techniques that identified inputs from regions previously associated with energy balance control (913). However, this study also identified inputs from the infralimbic area and the lateral septal complex. Although these two regions innervate areas very close to the rat PVH, they clearly avoid it when spatially guided tracers are used (613, 914). Notably this same study (915) also identified many rostrally directed outputs from PVHBDNF neurons that have not been identified in rats using spatially guided tracers. Whether these discrepant sets of results derive from species differences or technical issues means they will need to be resolved before we can fully appreciate how connections between the PVH and endbrain regions control food intake and energy balance.
Collectively the pattern of PVH inputs means that it is not the main hypothalamic region that integrates endbrain eating-related information into hypothalamic control mechanisms. As described in sect. 5.5.2.2.7, LHA regions appear to be the principal recipients of these inputs.
5.5.2.4.3.2. hypothalamus.
Strong inputs to the PVH originate from other parts of the hypothalamus that are implicated in controlling energy balance and eating behavior, of which ARHAgRP and ARHPOMC are the best characterized (FIGURE 14). Other hypothalamic inputs originate from the following: 1) a set of preoptic nuclei that appear to form part of a visceromotor pattern generator (916); 2) the DMH) including its neurons that express LepRb (484, 895, 916, 917); 3) some but not all parts of the LHA (653, 662, 663, 918, 919); and 4) the VMH, which mostly targets the PVHap (498, 920), although a role, if any, for these VMH projections in eating behavior has yet to be fully characterized.
5.5.2.4.3.3. midbrain and rhombicbrain.
A key feature of the PVH is that it receives sets of inputs that are distinct from its immediately adjacent regions (FIGURE 14). These include a massive set of catecholamine-containing projections from the NTS and ventrolateral medulla that are heavily implicated in the eating and endocrine responses to cytoglucopenia and hypoglycemia (287, 458, 460, 461). Many of these neurons contain the adrenaline-synthesizing enzyme phenylethanolamine N-methyltransferase and some appear to be glutamatergic (921). Some catecholaminergic inputs that originate in the more caudal parts of the NTS contain prolactin-releasing peptide (PrRP) (922). PrRP suppresses food intake when delivered to forebrain and medullary sites (923, 924). A second significant medullary to PVH projection contains GLP-1 (925–927). Although distinct from catecholaminergic neurons originating in the NTS (927), GLP-1-containing inputs are also glutamatergic (928). Limited projections from the lateral PB target the nonneuroendocrine parts of the PVH (827) (see sect. 6.3.1.1.4).
Injecting the nodose ganglia with the anterogradely transported neurotropic virus HSV-1 H129 infects a restricted population of PVH neurons showing that the PVH receives information from VSNs (FIGURE 14) (345, 366). Following HSV-1 H129 injections into the rat left nodose ganglia, infected neurons appear to innervate a ventral population of parvicellular preautonomic neurons (FIGURE 14) (366). Although the signaling makeup of these projections is not known, they are clearly very well positioned to inform the PVH about GI status.
5.5.2.4.4. Where do PVH neurons project to control eating behaviors?
PVH outputs originate from two groups of neurons, the vast majority of which express vesicular glutamate transporter 2 (vGluT2), a marker for glutamatergic neurotransmission, rather than glutamate decarboxylase 65 (GAD65), which identifies GABAergic neurons (FIGURE 14) (853, 929,930). First, neuroendocrine control is enabled by two sets of projections to the ME and posterior pituitary (FIGURE 14) (865). Second, extensive PVH projections to the midbrain, rhombicbrain, and spinal cord originate from parvicellular neurons that are found mostly in its mid- and caudal levels (259, 260, 865). This output pattern differs from the ARH and LHA, both of which have prominent rostrally directed outputs. All rat PVH outputs identified so far and corroborated by more than one technique are either directed intrahypothalamically or caudally (e.g., Refs. 261, 613, 931, 932) and are heavily implicated in autonomic and behavior control (260, 317, 865, 933, 934). These neurons have received the most attention with regards to how the PVH controls eating behaviors. Collectively, these attributes make the PVH one of the three principal hypothalamic output portals, the LHA and ARH are the others, that enable it to coordinate neuroendocrine and autonomic function with motivated behaviors.
5.5.2.4.4.1. hypothalamus.
Intrahypothalamic PVH outputs target at least two key regions that control eating behaviors: the LHA and ARH. Rat PVH outputs target some middle group LHA regions (662, 918, 919), some of which may include those PVHCRH outputs that can control complex stress-associated behaviors in mice (935). PVH outputs to the ARH (11, 815) have been discussed previously in sect. 5.5.2.3.4.
5.5.2.4.4.2. the ventral tegmental area and substantia nigra.
Unlike those from the ARH and certainly the LHA, PVH outputs to the VTA and SN are not well documented. The most comprehensive study of descending PVH outputs in the rat used a spatially targeted anterograde tracer. It identified a relatively sparse projection to the VTA but not the SN (261). The rat VTA projection has been corroborated with retrograde tracers (936, 937) and perhaps most convincingly, in the mouse using a rabies virus whose monosynaptic retrograde transport was driven by Cre in dopaminergic neurons in the VTA and SNc. This study show labeled neurons in what appear to be those PVH regions with descending connections (938).
5.5.2.4.4.3. parabrachial nucleus.
The parabrachial nucleus (PB) is a major hindbrain region involved with controlling eating behaviors (see sect. 6.3.1.1.1.4). It receives large sets of sensory information from the spinal cord and NTS that encode visceroceptive, nociceptive, somatosensory, and gustatory modalities. Although it has long been associated with CTA, it has more recently assumed a wider role in eating behavior control largely as a result of genetically driven manipulations of inputs from the ARH and PVH (272, 819, 939).
Geerling and colleagues (261) have provided the most detailed description of PVH inputs to the rat PB. They show that it provides a major input to the lateral (viscerosensory) part of the PB, but a far sparser input to the medial (gustatory) PB (FIGURE 15, A and B). Of these projections, Li and colleagues have recently identified distinct galanin- and MC4R-expressing PVHSIM1 projections to the PB, each of which is responsive to energy state. These neurons target separate regions in the PB and the prelocus ceruleus (pre-LC). This area is situated between locus ceruleus and the medial PB (255, 261, 272) and has recently been shown to link food and water intake (940). Both of the PB regional targets of these particular PVHSIM1 neurons also receive projections from ARHAgRP neurons. When stimulated, each of these two sets of PVHSIM1neurons acts independently but in an additive manner to restrain eating (272). Not only that, they appear to account for the entire satiety- and body-weight regulating capacity of the PVH for habitual meals eaten during the first 3 h of the dark phase (272). In many respects these results are quite surprising given that the majority of PVH effects on food intake have long been assumed to target the dorsomedial medulla rather than the PB. However, it should be remembered that habitual meals constitute only one, but an obviously important, form of eating behavior; PVH projections to more caudal parts of the rhombicbrain are implicated in other types of eating behaviors.
FIGURE 15.

Inputs from the rat paraventricular hypothalamic nucleus labeled with Phaseolus vulgaris leucoagglutinin (PHAL; red) to the parabrachial region (A and B) and dorsal medulla (C–F). Adapted from Ref. 261 with permission. PB, parabrachial nucleus; PBm, PB medial; lc (or cl), central part of the lateral division; ld (or dl), dorsal part of the lateral division; le, external part of the lateral division; lv, ventral part of the lateral division; KF, Kölliker-Fuse subnucleus; mm, medial part of the medial division; NTS, nucleus of the solitary tract; com, commissural part; CU, cuneate nucleus; MeV, midbrain nucleus of trigeminal nerve; GR, gracile nucleus; IRt, intermediate reticular nucleus; DMX, dorsal motor nucleus of the vagus; AP, area postrem; SpV, spinal tract of the trigeminal nerve; sct, spinocerebellar tract; LC, locus ceruleus; scp, superior cerebellar penduncle; sct, spinocerebellar tract.
5.5.2.4.4.4. the nucleus of the solitary tract and the dorsal motor nucleus of the vagus.
FIGURE 15, C–F, shows that the nucleus of solitary tract (NTS) and the dorsal motor nucleus (DMX) are both heavily innervated by the PVH. Indeed parts of the NTS have the densest set of PVH inputs in the rhombicbrain (261). A primary role for these projections in autonomic control is strongly implicated. However, it remains difficult to distinguish whether these PVH projections have a primary effect on meal initiation rather than being a consequence of their ability to influence autonomic function, for example, by acting as a gain control on vagal function (941, 942).
One of the best characterized PVH neuronal populations with regard to food intake are those that synthesize OXY (see Refs. 222, 859, 943 for reviews). PVHoxy neurons project into the dorsomedial medulla, and further caudally into the spinal cord (883, 933, 944). OXY was identified ∼30 yr ago as a peptide that could suppress food intake, possibly by way of its interactions with gut-derived satiation signals (945, 946). Although subsequent studies have further characterized OXY’s actions on food intake, particularly meal size (947–949), the mechanisms it engages have turned out to be rather complex. The ability of parvicellular PVHOXY neurons to control food intake has been clearly demonstrated by Atasoy and colleagues (950) using genetically guided methods. This study used axonal photostimulation to show that projections from ARHAGRP neurons inhibit PVH neurons in a way that potently stimulates food intake. Nonneuroendocrine PVHOXY neurons are part of this process. However, there are two other ways that PVHOXY neurons may function to control eating, both of which involve magnocellular neuroendocrine PVHOXY neurons together with SOOXY neurons. These are discussed in sect. 5.5.2.5.
5.5.2.4.5. The paraventricular nucleus as part of the hypothalamic eating behavior control network.
As we described in sect. 4.2.5.1, the core structures that constitute the hypothalamic part of the upper brainstem eating control network must interact with selection and initiation networks to enable meal onset (FIGURE 6B). The ARH and LHA have direct outputs to the VTA and SN, as well as to targets in the pallidum and striatum that are well positioned for this purpose. However, PVH outputs are organized somewhat differently. It has no corroborated projections to cortical nuclei but does have a projection to the VTA, although this is currently uncharacterized. The PVH must therefore control eating behaviors in a different way to the ARH and LHA. Because its outputs are strongly directed into the rhombicbrain, particularly the dorsal medulla and PB (FIGURE 15), the PVH is well positioned to exert strong control on how information from the GI tract is processed to influence meal termination (272). The PVH may also exert some control on consummatory aspects of eating by way of projections to those parts of the reticular formation, the AMB, and paratrigeminal nucleus (261) involved with orofacial, laryngeal, and esophageal function.
5.5.2.5. supraoptic nucleus.
The SO is a morphologically simple nucleus that contains neuroendocrine magnocellular OXY and vasopressin neurons. Although not usually perceived as an important hypothalamic contributor to eating behavior control, SO neurons possess some hallmarks of metabolic sensors, including the expression of leptin, insulin, and possibly ghrelin receptors (951, 952). They also receive projections from the ARH, and are activated by aMSH (953), directly and indirectly by leptin (954, 955), by eating (956), and by gastric distension (957).
Neuroendocrine magnocellular axons terminate in the posterior pituitary to release OXY and vasopressin into the general vasculature. Two observations made ∼30 yr ago suggested that these neurons participate in control of food intake. The first was the very tight correlation between circulating OXY and CCK-driven suppression of food intake (958), suggesting a functional role for OXY released from magnocellular neurons into the general vasculature. One explanation for this correlation is that specific uptake mechanisms would permit the transport of circulating OXY across the blood-brain barrier thereby allowing it to access its cognate receptor in regions that are some distance from PVHOXY and SOOXY neurons. However, despite the presence of specific transport system in the blood-brain barrier (959), brain penetration by OXY appears to be poor (960, 961), which would favor additional mechanisms to explain the strong correlation observed by Verbalis and colleagues (958). These may involve OXY receptor mechanisms in parts of the rhombicbrain accessible to the circulation (962). However, a recent study (202) shows that the ability of OXY to suppress food intake requires OXY receptors expressed by those VSNs (963) that engage NTSGLP-1 neurons. These findings (202) help close a functional loop between OXY release from the posterior pituitary into the general circulation, and VSNs and NTSGLP-1 neurons, which then project rostrally into the forebrain.
The second observation was that magnocellular neuroendocrine neurons can release OXY or vasopressin from their dendrites into the local environment (212), thereby providing a structural basis for these neurons to use nonsynaptic peptidergic signaling. Large amounts of peptide are released in a manner that is independent of axonal transmission and can act at some distance from release sites (212, 213, 964). These features highlight the need to consider that the way neuropeptides control eating behaviors is not restricted to point-to-point synapse-based signaling mechanisms (965). Therefore, the SO should be considered a significant source of neurally released OXY that can have a functional impact on regions that do not receive a clear synaptic input from PVHOXY and SOOXY neurons (213, 270, 964). These results underscore the complex signaling mechanisms that can be used by OXY neurons to control eating behaviors. They caution against interpreting neural peptidergic mechanisms strictly in terms of synaptic transmission (see sect. 2.7.4).
5.5.2.6. ventromedial hypothalamic nucleus.
The VMH is well placed to respond to humoral physiological signals that control energy balance. Its neurons express receptors for ghrelin (966–968) and leptin, particularly in the VMHdm (475, 476). Amylin receptors are found in low abundance in its ventrolateral part, unlike the much higher levels seen in the DMH and ARH (969, 970). The VMH’s glucosensing capability is probably the best characterized of any brain region (433, 438, 439, 971, 972). Manipulating AMPK activity in the VMH affects energy expenditure and glucose metabolism (808, 973–975).
These attributes, together with many functional studies, are consistent with an influence of VMH manipulations on glycemic control and energy balance. However, functional and structural evidence only supports a secondary role for the VMH in meal initiation. Activating steroidogenic factor-1-expressing VMH neurons opto- or chemogenetically has no immediate effect on food intake but instead leads to gradually reduced intake over a period of hours (532, 976). The organization of VMH outputs also makes it difficult to resolve how, and particularly in what context, the VMH influences eating and energy balance. Some VMH outputs innervate the PVHap, periventricular PVH, the ventrolateral rather than the medial ARH, and the perifornical and medial tiers of the LHA (498, 499, 662, 918, 977). However, they only weakly target medullary or spinal cord autonomic control regions (498, 499, 865). Unlike the ARH and LHA, VMH outputs avoid the VTA and SN (498, 499). Instead VMH outputs primarily target hypothalamic medial zone nuclei, and particularly the anterior hypothalamic nucleus (AHN). VMH outputs also target the PAG, parts of the pallidum (BST) and striatum (CEA, MEA), the cortical amygdala (498–500), and the superior colliculus (258).
The pattern of VMH inputs therefore differs markedly from adjacent regions in the ventromedial hypothalamus. Unlike the PVH, DMH, ARH, and parts of the LHA, the main body of the VMH receives little to no catecholaminergic, AgRP, aMSH, or GLP-1 innervation (464, 854, 926, 927, 978). Instead, it receives substantial inputs from the PVT, PB, the BMA and MEA, and the ventral CA1 of the HPF (498, 617, 620, 827, 979, 980).
Two other complications are evident when analyzing how inputs influence VMH neuronal function. First, its dendrites extend well outside its Nissl-defined boundary (362, 981) meaning that peri-VMH inputs can form axo-dendritic synapses with VMH neurons. These may mediate effects from regions such as the ventral subiculum and medullary catecholaminergic inputs that strongly innervate the peri-VMH but not the nucleus itself (457, 616). Second, some peptides released from neurons whose axonal projections do not innervate the VMH, OXY from the SO, for example, may reach the VMH by volume transmission (213) to suppress food intake and energy expenditure (982).
One way to interpret these VMH properties is to consider that the metabolic information detected by its neurons impacts actions that are rather different from those controlled by neurons in the hypothalamic part of the eating control network. In this way the VMH’s role may not be as a primary controller of energy balance. Instead, it principally controls conspecific and antagonistic behaviors (530, 983–987). In this context it is notable that stimulating the ventromedial hypothalamus in humans elicits feelings of panic and anxiety, not hunger or satiety (988). This finding is consistent with the idea that the prominent metabosensory functions of the VMH help coordinate those behavioral and autonomic actions required to fulfill the energy demands of conspecific and antagonistic behaviors (532), perhaps in part by way of its outputs to the PAG, the PVHap, BST, and superior colliculus (258, 498–500). The VMH can certainly monitor energy status, including adiposity via leptin, and ambient glucose. However, rather than use this information primarily to coordinate eating behaviors with energy balance, as do the ARH, PVH, and parts of the LHA, its neurons ensure that the energy demands of conspecific and antagonistic behaviors are satisfied. The impact that the VMH has on eating behavior is perhaps best considered as a means to this end (532).
5.5.2.7. dorsomedial hypothalamic nucleus.
The DMH is located caudal to the PVH in the dorsal periventricular hypothalamic zone. Cytoarchitectonically it is more poorly defined than the PVH, particularly at its lateral border. It extends laterally toward the LHA and ventrally toward the VMH. It has three subdivisions with apparently similar outputs (989) but different chemoarchitectures. These include GABAergic, glutamatergic, cholinergic, and NPY-synthesizing neurons, with smaller populations of MCH and ORX-synthesizing neurons (484, 607, 654, 990).
There are also species differences. Although NPY mRNA is found in the posterior part (p) of the DMH of lean rats (484, 991) and primates (991), it is only seen in the DMHp of mice with diet-induced obesity (991, 992), and these DMH NPY neurons are leptin-sensitive during diet-induced obesity (992). In rats NPY mRNA is increased by food deprivation and lactation but not with diet-induced obesity (464, 991). However, no differences between rat and mouse DMH output patterns have been reported (895, 989).
Although lesion (993) and more recent optogenetic studies (896, 994) implicate some involvement of the DMH in food intake, the circumstances during which this occurs are complex and most likely are not a primary drive to eat (991). Instead, the weight of evidence supports a more prominent controlling role for the DMH in a range of other functions including brown adipose tissue (BAT) and shivering thermogenesis (995, 996); timing functions associated with energy balance, including circadian timed and food anticipatory activity (see sect. 5.5.3), and CCK-mediated satiation (997, 998). These functions are likely enabled by substantial inputs from the SCH and subparaventricular zone (SBPZ) (999–1001), ARH (855, 989, 1002), the median preoptic nucleus and the preoptic area (107, 989, 1003), and the hindbrain and medulla, including the PB and those medullary neurons containing GLP-1 or catecholamines (464, 926, 989). Some ascending inputs also convey vagally mediated signals from the gut to the DMH (366). Notably, catecholaminergic inputs to the DMH originate from the same medullary sources as those innervating the PVH, ARH, LHA, BST, and PVT that may indicate extensive collateralization (464).
The DMH is a major target of leptin signaling (475, 482, 894). However, unlike the ARH and LHA, the DMH may not be a significant contributor to the network engaged by leptin to control habitual food intake (483). Instead, leptin’s action in the DMH appears more closely allied to its wider role in thermogenesis and energy expenditure (482, 991, 992, 996, 1004, 1005), although a recent report implicates GABAergic DMH projections to ARHAgRP neurons in leptin’s ability to restrain deficit-induced food intake should be noted (851).
The major outputs of the DMH are mostly restricted to the forebrain, with strong projections to the PVH, parts of the LHA, the ARH, and a series of smaller nuclei in the rostral hypothalamus (847, 850, 851, 895, 916, 989). Part of this output pattern has been associated with a DMH contribution to a visceromotor pattern generator that helps coordinate autonomic function during disturbances to energy balance and other challenges (916). Few descending projections have been identified, although the PAG and the rostral raphe pallidus appear important targets (107, 1006). DMH outputs to the medulla are more difficult to reconcile. Although NPY-containing DMH projections to the dorsal medulla have been reported using virally mediated methods (998, 1007), these have not yet been corroborated with other anterograde or retrograde methods (895, 989, 1008) and so their significance currently remains uncertain.
Finally, it is worth noting the difficulty that the structure of the DMH impose on investigating its function in eating and energy balance control. Because of the loose application of nomenclature in this part of the hypothalamus, there remains some confusion in the literature about exactly where some manipulations are made. It is unclear whether some target the dorsomedial hypothalamus, which is a loosely defined region that includes parts of the LHA, or the dorsomedial nucleus itself. A recent study on DMH projections to the PAG emphasizes these problems (1006).
5.5.3. The coordinating role of timing signals in meal initiation.
Timing signals control and coordinate an animal’s arousal state, body temperature, metabolism etc. with its environmental conditions. Meal timing is part of this process, particularly for those meals that are habitual and anticipatory in nature. While our understanding of the molecular basis of cellular clocks has improved dramatically during the past decade (e.g., Refs. 1009–1014), how the various timing signals are coordinated at a brain network level to control meal initiation remains less clear. We address this question by considering how two types of timing signals contribute to meal initiation: photoperiodically entrained circadian timing, and those mechanisms responsible for food anticipatory activity.
5.5.3.1. photoperiodically entrained circadian timing: the suprachiasmatic nucleus.
Linking meal initiation to the prevailing photoperiod is vital for most mammals. This property is easily seen in nocturnal laboratory rodents. With unlimited food availability, the initiation of their main set of meals is coordinated with the time lights are turned off. In the real world, this coordination maximizes opportunities to find food items, but for many prey species it also limits their exposure risks to predators. Photoperiodically entrained meals also contribute to proactive energy homoeostasis, which allows animals to anticipate and then avoid energy deficits (78).
Information about the timing and duration of the photoperiod is processed by the SCH in the ventral periventricular hypothalamus. Interactions between its direct retinal input and the cellular clock mechanisms in SCH neurons (1009, 1015–1017) enable this small cell group to transmit a photoperiodically entrained circadian timing signal to a relatively restricted but highly interconnected set of targets in the interbrain that make up a Circadian Timing Network for eating behaviors (FIGURE 16): the adjacent SBPZ, PVT, and DMH (989, 999, 1000, 1018–1020).
FIGURE 16.
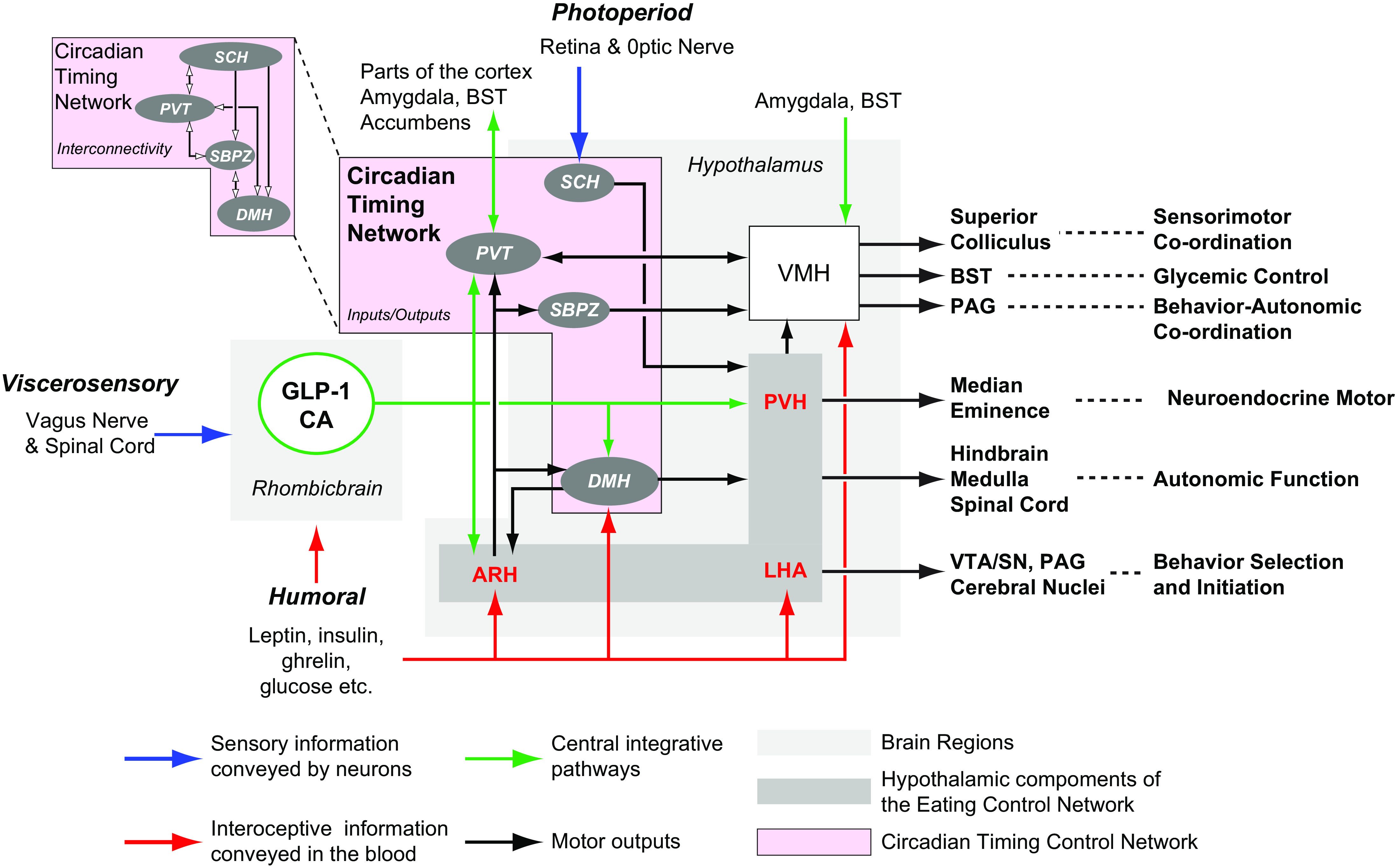
A schematic summarizing the organization of a circadian timing network (pink box) whose main components are located in the hypothalamus (SCH, SBPZ, DMH) and dorsal midline thalamus (PVT). These nuclei are highly interconnected (pink box, Interconnectivity). The principal input for photoperiodic entrainment is from the retina to the SCH, which contains the main circadian clock neurons. Outputs from the circadian timing network tend to avoid motor control regions. Instead, they project collectively to the ARH, LHA, and PVH, which are the core hypothalamic components (gray box) of the Upper Brainstem Eating Control Network (see FIGURE 17). Humoral signals target the DMH directly, while robust catecholaminergic (CAs) and glucagon-like peptide 1 (GLPR-1) containing inputs from the medulla target the PVT and DMH, and are well positioned to bring other humoral information from the medulla, including that conveyed by the spinal cord and the vagus nerve. See text for more details. SCH, suprachiasmatic nucleus; SBPZ, subparaventricular zone; DMH, dorsomedial hypothalamic nucleus; ARH, arcuate nucleus; LHA, lateral hypothalamic area; PVH, paraventricular hypothalamic nucleus; SN, substantia nigra; PAG, periaqueductal gray; BST, bed nuclei of the terminal stria; VMH, ventromedial hypothalamic nucleus.
The SBPZ is the principal distributor of circadian-related information (1001, 1020). FIGURE 16 shows that it projects to the DMH and PVT, and to other regions not innervated by the SCH (1001, 1003, 1020). The topography of the SBPZ is functionally organized (1001) thereby providing a means to control different physiological functions (1021). Notably, the dorsolateral SBPZ has bidirectional connections with the DMH and VMH (984, 1001). The DMH then projects to the ARH, PVH (as does the SCH), and LHA. It is also worth noting that the SBPZ to VMH projection is also implicated in modulating attack behaviors (984).
By virtue of their connections, the DMH and PVT are nodal points for directing circadian and arousal state influences to the hypothalamic components of the upper brainstem eating control network (see FIGURE 17). The PVT is one of the few brain regions with bidirectional connections with the SCH (979, 999, 1000, 1018, 1022). It is linked to arousal, motivation, and reward functions through its bidirectional projections with parts of the LHA, cortex, and cortical nuclei, including the amygdala and ventral striatum (282, 726, 979, 1023–1031).
FIGURE 17.

A schematic of the Upper Brainstem Eating Behavior Control Network showing the interconnectivity (red) and outputs (black) of its 3 core components, the ARH, PVH, and LHA. See text for more details. ARH, arcuate nucleus; LHA, lateral hypothalamic area; PVH, paraventricular hypothalamic nucleus; SN, substantia nigra; VTA, ventral tegmental area.
The DMH receives strong inputs from the SCH, SBPZ, and the PVT (989, 999–1001, 1018). In addition, the way intrahypothalamic DMH outputs are distributed suggests this region is a component of two functional networks important for controlling energy balance. The first is a visceromotor controller that appropriately organizes and coordinates autonomic and neuroendocrine motor output with eating and other behaviors (632, 916). A place for the DMH in a circadian timing network is consistent with this function (916). The second is a network involved with distributing circadian information that controls arousal state (1032, 1033).
The DMH and PVT each receive two additional inputs that provide information related to energy balance (FIGURE 16). First, catecholamine-containing and GLP-1-containing projections from the medulla (464, 926, 989) that can relay the integrated output of medullary processing of humoral and neural (spinal and vagal) information to the DMH and PVT (1005, 1028). Catecholamine-containing projections to the PVT and DMH encode information related to glycemia and glucocorticoid feedback that can control both short- and long-term aspects of energy balance, including eating behavior and energy expenditure (287, 458, 459, 462, 464, 821, 906, 1034). CA- and GLP-1-containing inputs to the SBPZ and VMH are meagre or absent (FIGURE 16) (457, 464, 926, 927). Second, the ARH projects strongly to the DMH and PVT. ARH neurons directly encode humoral information, while some DMH neurons express leptin receptors. The ARH also receives the same catecholamine and GLP-1-containing inputs from the medulla that innervate the DMH and PVH (459, 464, 926). Collectively, these inputs provide the PVT and DMH with the capability to integrate a wide range of information into the circadian control of eating behaviors (cf. FIGURE 16).
This circadian timing network in the interbrain is therefore well placed to distribute timing-influenced information to those parts of the forebrain that control eating behaviors, including the PVH and LHA, as well as to parts of the cortex, BST, and amygdala (FIGURE 16), which are all key endbrain regions for initiating eating behavior.
5.5.3.2. food-entrainable oscillators and meal initiation.
The ability of animals to anticipate the timed presentation of food with increased activity in the presence or absence of photoperiodic cues has long been recognized (1035, 1036). This behavior not only includes increased arousal and approach to the vicinity of food presentation (1037) but also shifts daily endocrine secretory patterns. Glucocorticoids were first identified in this regard (1038), but later work also identified leptin, insulin, glucagon, GLP-1, and ghrelin (1039). Further work has shown that food presentation will not only act as an entraining signal for SCH timing mechanisms, but can also entrain SCH-independent oscillators, the so-called food-entrainable oscillators (1040, 1041). These systems involve the effects of investigator-timed meals on activity rather than endogenously timed mechanisms for meal initiation, which puts their detailed examination outside the scope of this review. However, we note that although searching for the neural bases of food-entrainable oscillators has proved difficult and quite controversial (1040), available evidence points to the involvement of some of the same brain regions already prominent in photoperiodically entrained circadian timing, particularly the ARH and DMH (1012, 1041–1045). Interactions between metabolic hormones and mechanisms in the hypothalamus and dorsal striatum have also been implicated in metabolic timing (1011, 1039, 1046, 1047).
5.5.4. An upper brainstem network that controls eating behaviors.
Building on the seminal work of Ranson (15, 1048) and Stellar (877), Swanson (8, 62, 283) proposed that the hypothalamus provides motivational drive to ingestive and other behaviors by way of ascending and descending outputs from a behavior control column comprised of nuclei in the hypothalamic medial zone and adjacent midbrain regions in the upper brainstem, particularly the VTA and parts of the SN. During the past 20 yr what we know about the functional, structural, and connectional organization of the hypothalamus has developed to such an extent that we can now add considerably more detail to this upper brainstem eating control network (FIGURE 17).
The weight of evidence favors three regions as its core hypothalamic constituents (FIGURE 17): the ARH, PVH, and some LHA regions, particularly the PSTN and those in its middle tier around the fornix (i.e., perifornical). Defining these three regions as core components is determined by the fact that they possess all four of the properties we introduced in sect. 4.2.5.1.1.
Core components must consistently and robustly influence food intake when experimentally manipulated. Designs that assess appetitive actions are particularly persuasive in this regard. There is no doubt that the ARH, PVH, and parts of the LHA all possess this property.
Given our emphasis on ATP availability as the pivotal regulated variable in energy homeostasis (FIGURE 2), AMPK function in core network components should have direct effects on eating behaviors. We have already discussed AMPK’s role in the ARH (sect. 5.5.2.3.2). Studies show similar attributes in PVH neurons (95, 810, 811). However, the evidence is less convincing for LHA neurons, perhaps because the LHA’s much greater structural complexity makes them less experimentally accessible with techniques with low-spatial resolution (e.g. analyses of protein, mRNA, etc. in grossly dissected tissue).
Core components must receive direct humoral and neural inputs that provide the means for physiological signals to influence their activity. We have seen that the ARH, PVH, and key parts of the LHA each receive significant inputs from the rhombicbrain that can shape eating behaviors and other functions that control energy balance. These include inputs containing catecholamines and GLP-1, as well as those that convey sensory information from the vagus nerve (345, 366). The ARH, PVH, and the LHA also have cellular mechanisms that transduce leptin, glucose, ghrelin, and other signals that circulate in the blood into modified neuronal function.
Core components must possess verified functional outputs to motor Selection and Initiation networks (FIGURE 6Bii). These projections are instrumental for conveying information from the hypothalamus to brain regions that organize the appetitive actions that direct animals toward food sources, and then enable approach and consummatory actions (FIGURE 3). Each hypothalamic component of the core network projects to the VTA and/or the SN, which are the midbrain parts of this network (FIGURE 17) (8, 62, 283). In turn, the VTA and SN have substantial connections to those parts of the striatum and pallidum (i.e. the cerebral nuclei) implicated in motor control (FIGURE 17).
Importantly, each of these four properties is not exclusive to the ARH, PVH, or LHA. In this way quite a few hypothalamic regions can alter food intake when manipulated, but many of these have either sparse or no outputs that can easily facilitate direct control of eating. The VMH, and particularly the DMH, fit into this category. These two regions, along with others (e.g., the SCH, anteroventral periventricular nucleus, AHN, and ventral premammilary nucleus), are best considered as hypothalamic components that provide additional information to eating behavior control (e.g., circadian timing as discussed in sect. 5.5.3.1), as well as enabling coordination between eating and other motivated behaviors such as conspecific, maternal, or aggressive/defensive behaviors (283). Similarly, not all of these secondary regions receive neurally conveyed signals or have access to circulating humoral signals by virtue of their position inside the blood-brain barrier.
The distinct connectional and functional attributes of the ARH, PVH, and LHA mean that each contributes different qualities to the core network, a feature that adds considerable functional flexibility. Their striking degree of interconnectivity (FIGURE 17) provides for information sharing that again adds flexibility for controlling eating behaviors. This point is well illustrated by the very different behavioral endpoints that occur in rats, mice, and hamsters after food deprivation (see sect. 5.2.3), despite the apparently similar key roles of the ARH, PVH, and LHA have to control eating in each species.
Three other organizational features are worth noting. First, the ARH and LHA, but not the PVH (which has no corroborated ascending projections), have major and direct outputs to the striatum and pallidum, as well as to the PVT, which itself has pronounced striatal projections (1023). This arrangement means that the ARH and LHA can directly access the striatum and pallidum without engaging the VTA or SN (FIGURE 17), adding yet more network flexibility. Second, the PVH has robust descending outputs that are strongly implicated in autonomic control, a role it shares with ARH and parts of the LHA. The descending connections from the PVH may also allow it to directly control meal termination mechanisms in the dorsal medulla and PB, characteristics that it again shares with the ARH and LHA. PVH outputs to the LHA may be another way that it influences eating behaviors. Third, unlike the LHA, the PVH and ARH both have diverse groups of neuroendocrine motor neurons. Collectively, these arrangements emphasize the wider integrative functions of this control network, and that full control of eating behavior requires the interplay between the hypothalamus, endbrain, and rhombicbrain that is mediated by robust bidirectional pathways (FIGURE 5).
5.6. Hindbrain, Medulla, and Sensory Nerves
5.6.1. Preamble.
The medulla contains important sensory and motor cell groups for meal initiation. The NTS, is the main recipient of vagally and spinally transmitted viscerosensory information (see sect. 4.3.2), gustatory information from the oral cavity, and humoral information from the AP. Groups of neurons in the hindbrain and medulla constitute the orofacial and laryngeal central pattern generators for biting, chewing, and swallowing (311). Here we focus on the role played by the medulla in processing viscerosensory information for meal initiation.
5.6.2. Glucosensing and the medulla in meal initiation.
Glucose acts in at least two ways to initiate meals: when cellular levels of glucose fall below a threshold (cytoglucopenia), and when glycemia falls before habitual meals begin. Each constitutes a different state and uses different mechanisms to initiate eating.
Cytoglucopenia in certain medullary neurons leads to a set of counterregulatory responses, including eating (435, 461). These are mediated by catecholaminergic neurons in the ventrolateral medulla and NTS that project into the hypothalamic part of the upper brainstem eating control network (FIGURE 17) (461). However, cytoglucopenia triggers an emergency reaction, meaning that the ensuing deficit-induced eating does not recapitulate habitual meal initiation. Nevertheless, glucose seems to be involved in habitual meal initiation because the often observed premeal decline in glycemia (240–242, 433, 434) is causally related to habitual meal initiation. Unlike cytoglucopenia, premeal declines in glycemia do not reflect an energy or glucose deficit; rather, they are a proactive signal or pattern. As Campfield and Smith stated, they are ‘… signals that “interrogate” the peripheral metabolic state and signal meal initiation if additional energy intake will be needed to maintain glucose homeostasis for the coming time interval’ (1049). In other words, the transient declines in glycemia that are involved in habitual meal initiation are not the reaction to an energy deficit; instead, metabolic patterns that trigger meal initiation do so to avoid an energy deficit. In this way well-timed glucose infusions, but not infusions of other metabolic fuels during the premeal decline in glycemia, delay the following meal (1049). This not only indicates a causal relationship between the premeal decline in glycemia and meal initiation but also its specificity for glucose.
5.6.3. Sensing of glucose availability by vagal sensory nerves.
Those catecholaminergic neurons in the ventrolateral medulla that initiate eating in response to cytoglucopenia by way of their hypothalamic projections also connect to NTS catecholaminergic neurons (NTSCA) where they modulate the satiating effect to CCK that is mediated by these neurons (461). Many of these NTSCA receive monosynaptic inputs from VSNs (1050). Cutting abdominal and, particularly, hepatic portal vein VSNs compromised the otherwise reliable coupling between the premeal decline in glycemia and meal initiation (1049). However, it did not completely eliminate the coupling, i.e., some premeal declines still predicted habitual meals. It is worth noting that cytoglucopenia in hepatic portal vein VSNs can apparently also trigger eating. In one study, 2-DG stimulated eating in rabbits faster and more potently after infusion into the hepatic portal vein than after infusion into other blood vessels in intact animals or into the hepatic portal vein after vagotomy (1051). In another study, a low dose of 2-DG injected intraperitoneally into rats with hepatic branch vagomtomy failed to significantly stimulate eating after dark onset and after the rats had ingested a meal (1052).
Together these findings suggest that peripheral and medullary glucose sensors are involved in translating the premeal decline in glycemia into meal initiation, with the peripheral, presumably hepatic portal sensors, featuring a lower threshold than the medullary glucose sensors connected to catecholaminergic neurons. As mentioned above, these medullary glucoreceptors are crucial for initiating deficit-induced eating and endocrine counterregulatory responses to cerebral cytoglucopenia (435), which is a clear emergency for the animal. Together with ascending catecholaminergic neurons, they are also the major mediators of the appetitive and consummatory responses to systemic cytoglucopenia (460, 1053). Although catecholaminergic forebrain projections are not required for habitual meal initiation (821), they become important for restraining eating and other aspects of energy balance when animals eat a high-calorie diet (464). These results emphasize that the medulla not only contributes to short term emergency responses but also to the long-term control of food intake and energy balance.
5.6.4. Sensing of other stimuli that initiate eating by vagal sensory nerves.
There is evidence for VSNs detecting and hence, the medulla being involved with processing other metabolic signals that lead to meal initiation. The fatty acid oxidation inhibitors 2-mercaptoacetate (MA; an acyl-CoA dehydrogenase inhibitor), methyl palmoxirate, and etomoxir (both inhibitors of carnitine palmitoyl transferase-1) have long been known to increase food intake (1054, 1055) and do so mainly by initiating meals (1056). The eating that is stimulated by these compounds depends at least in part on intact VSNs (1057–1059). Peripheral administrations of the fructose analogue 2,5-anhydromannitol, which blocks oxidative phosphorylation and thus decreases ATP production, and the sodium pump inhibitor ouabain produced similar findings (1058, 1060, 1061). Again, the eating responses to all these compounds constitute emergency reactions to a dramatic reduction in oxidizable fuel availability and so are examples of deficit-induced eating that do not necessarily recapitulate the onset of habitual meals. However, unlike the emergency response to systemic cytoglucopenia, they do depend on intact abdominal VSNs (1058, 1059, 1061). This may reflect the fact that neurons usually depend on glucose to cover their energy needs and do not metabolize fatty acids. The evidence for a causal relationship between the inhibition of fatty acid oxidation and stimulation of eating appears to be solid for the CPT-1 inhibitors. The acyl-CoA dehydrogenase inhibitor MA has meanwhile been shown to stimulate eating mainly by acting on the GPR40 and not by inhibiting peripheral fatty acid oxidation (1062). However, MA, like the other fatty acid inhibitors, clearly requires intact abdominal VSNs to initiate a meal.
A widely distributed network of metabolic sensors in the periphery (intestine, hepatic portal vein) and brain (medulla and hypothalamus) therefore monitors the fuel status of the organism to initiate proactive (habitual) or reactive (deficit-induced) eating responses when necessary. One example is habitual eating seen in response to the premeal decline in blood glucose. Others include the physiological fluctuations in the metabolic state of enterocytes (101, 102) and deficit-induced eating in response to pharmacologically driven energy deprivation (e.g., Refs. 461, 1055, 1060). As shown in FIGURES 5 and 8, the medulla is clearly a key part of the integrative mechanism for these signals and their translation into meal initiation.
5.6.5. The plasticity of vagal sensory nerves.
The fact that substances can stimulate eating via signaling in VSNs is remarkable because it has primarily been implicated in meal termination/inhibition of eating (see below). That the same neural substrate can mediate signals triggering exactly opposite behavioral responses is yet another example for the enormous plasticity and versatility of these data highways between gut and medulla. Interestingly, single unit recordings from intestinal VSNs showed that 90% of the fibers increased their firing rate in response to MA, a meal initiator. However, these same VSNs also increased their firing rate in response to CCK or 5HT (1063), which inhibit eating, and do so, at least as far as CCK is concerned, primarily via VSNs. This indicates that context and/or differences in the firing dynamics of the VSNs determine whether a meal is initiated or terminated in response to the VSN signal. Another possibility is that the solution of this puzzle resides in the enormous plasticity of the medullary synapses that VSNs make with NTS neurons (see below). This plasticity enables opposite behavioral outcomes depending on the dynamics of the incoming signal. For instance, CCK increased single unit VSN activity within seconds, whereas MA required a few minutes to produce an increase (1063). In addition, while the CCK effect was only present for approximately 2 min, the MA effect continued for ∼30 min.
5.6.6. Eating in response to ghrelin.
The stomach hormone ghrelin has often been termed a hunger hormone because it increases food intake reliably after administration in animals and humans (1064, 1065). Ghrelin does so by initiating meals (1066) and by increasing meal size via increased gastric emptying rate and attenuation of vagally mediated satiation signals (733, 1067, 1068). Acylated ghrelin (which is the form of ghrelin that stimulates eating) activates the growth hormone secretagogue receptor (GHS‐R) (1064). GHS-R is expressed in the brain and on VSNs (1065). The plasma concentration of ghrelin has often been reported to increase with energy restriction (see Ref. 1069). However, in humans and mice, this increase is actually in des-acylated ghrelin (1070, 1071), which is a hormone in its own right that has metabolic effects but does not stimulate eating. Plasma levels of acylated ghrelin peak before meals and decrease after eating (1072, 1073). In rodents and humans, plasma levels of acylated ghrelin are highest before anticipated meals (1072, 1074), indicating that the release of acylated ghrelin before meals is mainly conditioned.
Although exogenous ghrelin reliably stimulates eating, acylated ghrelin does not appear to act as a physiological hunger signal (1075). For example, mimicking the physiological premeal rise in circulating ghrelin is not sufficient to increase subjective hunger or trigger eating in humans (1076), and ghrelin-deficient rodents are not anorectic and do not lose body weight (1069, 1071). Furthermore, the activity of the membrane‐bound enzyme ghrelin‐O‐acyl‐transferase (GOAT), which produces the acylated form of ghrelin, is diminished with prolonged fasting (1071), scarcely something to expect if acylated ghrelin were a hunger signal. Last, but not least, GOAT relies heavily on dietary medium-chain fatty acids to produce acylated ghrelin (1071); its formation therefore depends more on the presence than the absence of food, which does not fit a hunger function either. Thus the GOAT-ghrelin system may sense dietary fat and make food more attractive thereby promoting ingestion of food that is available. Peripheral, intracerebroventricular, or direct hippocampal administration of ghrelin each produces interoceptive cues that generalize to a state of 24-h food restriction in otherwise nonrestricted rats. Moreover, pharmacological blockade of GHS-R directed to specific brain regions (e.g., VTA, HPF, LHA, amygdala) reduces food intake and food-motivated responses (271, 1077, 1078), thereby supporting a role for ghrelin as an appetite-promoting signal.
The eating-stimulatory effect of ghrelin was originally proposed to require intact VSNs (1079). It is unclear, however, whether the rats in these studies were able to eat large solid food meals only 7 days after total subdiaphragmatic vagotomy. A different study (1080) used vagal nerve ablation to examine very thoroughly exogenous ghrelin’s eating stimulatory effect during the light phase. It found that neither total subdiaphragmatic vagotomy nor subdiaphragmatic vagal deafferentation had any effect on the eating stimulatory effect of intraperitoneally administered ghrelin. Importantly, these animals had fully recovered from surgeries, their ablations were functionally and histologically verified, and the body weights and baseline food intakes of the lesioned groups in the critical time period were similar to their surgical controls. These findings indicate that exogenous ghrelin does not require intact VSNs to stimulate eating during the light phase. However, a recent paper showed that the food intake stimulated by peripheral ghrelin during the dark phase was attenuated in animals with total subdiaphragmatic vagotomy (1081). This same study also showed that knockdown of GHS-R in VSNs influenced meal patterns during the dark but not light phase. Thus the light-dark cycle may be a critical variable in determining whether ghrelin-stimulated eating involves VSN signaling. Additional results reveal that ghrelin administration antagonizes the stimulatory effect of CCK on VSNs and on the expression of CART in VSNs, as well as on the nuclear translocation of the early growth response factor-1 (EGR1). Ghrelin therefore has the potential to influence eating by modulating the effects of other, satiating gut peptides acting through VSN signaling (see Refs. 340, 1069, 1082).
To summarize this section, excepting some emergency signals like rapid-onset hypoglycemia, it appears that those eating-stimulatory signals that act in the medulla mainly modulate peripheral input and, thus, increase the probability that eating occurs in case of an adequate opportunity.
6. MEAL TERMINATION
6.1. Preamble
At the simplest level, meals are temporally defined by the ongoing relationship between those physiological signals and brain processes that stimulate eating, those that suppress eating, and the competing processes involved with expressing other motivated behaviors. These interactions include what is going on in the animal’s environment by way of exteroceptive processing in the endbrain, gustatory processing in the rhombicbrain, competing motivational drives (hypothalamus), and gut-derived satiety signals (rhombicbrain). The outcome of these brain-wide interactions over time determines the size and temporal structure of a meal, i.e., its volume, duration, and caloric value. This interaction also determines a meal’s relationship to the motivational states and their associated behaviors that precede and follow a meal (FIGURE 3), that is, when a meal starts and finishes, and what event follows a meal. This could be another meal or a completely different behavior. Increasing satiation signals most likely terminate meals in a controlled environment, but in the real world any number of local challenges may terminate meals before satiety mechanisms predominate.
The first outcome of meal termination is the cessation of any direct interactions with food items that are not resumed within the limits of a species-defined meal. This means that biting, chewing, and swallowing, together with any reaching, grasping, and related movements must stop to be replaced with the motor actions associated with another behavior. What controls this switching? Primarily it is the outcome of how various interoceptive and exteroceptive signals are processed dynamically by the brain-wide networks we now describe.
6.2. Forebrain and Midbrain
The hindbrain and medulla are important brain parts for integrating GI signals, blood-borne humoral signals, and descending hypothalamic input to control meal size (1083). However, the amount of food consumed during an eating episode also has two major determinants that involve the forebrain and midbrain. First, physiological signals of negative energy balance that influence deficit-induced eating; and second influences from the external environment that engage cognitive processes in the endbrain.
Deficit-induced eating is widely used to explore behavior and physiological mechanisms. As discussed earlier, the modified levels of circulating leptin and other signals seen in rodents after a fast alter neuron activation in, for example, the ARH as part of the mechanisms that drive eating. Eating is then terminated by reactive signals acting in the medulla and elsewhere. A useful way to investigate which brain regions and networks are involved with terminating these types of meals is to characterize those neurons whose Fos expression changes after food is returned and eating is complete. This approach has identified a set of interconnected forebrain regions that can curtail deficit-induced meals (626, 756). These regions include the CEA, ARH, PVT, ZI, PB, and parts of the LHA (including the PSTN), BST, and agranular and prelimbic cortices. Notably, projections from the CEAm but not the CEAl are capable of decreasing meal size and prolonging intermeal intervals when pharmacogenetically stimulated (626, 756). However, additional complexity to this arrangement is evident because CEA neurons that express 5-HT2a receptors and apparently distribute throughout the CEA will promote food intake by inhibiting target neurons in the PBl (766). This effect is evident during food intake rather than procurement (766).
Meal size can be manipulated in humans under controlled experimental conditions through the portion size effect, social facilitation of eating, and food variety versus food monotony (1084). However, the neural substrates mediating these effects are not fully understood. In addition to exteroceptive and cognitive factors, humoral, and central neuropeptidergic signals act directly in midbrain and endbrain regions to control meal size. Both amylin (1085) and GLP-1 (1086), for example, act on receptors expressed in the VTA to reduce food intake specifically through a reduction in meal size. Similarly, GLP-1 receptor activation in the LS (1087) and HPFv (1088) reduces intake via a specific reduction in meal size. LSNT neurons that contain GLP-1 receptor mRNA are well positioned to mediate these effects particularly in stressful situations by way of projections to the LHA (1089). GLP-1 released from neurons may access the HPFv by way of volume transmission (1088). Within-meal satiation signals also interface with brain networks involved in reward and cognition through a recently identified multisynaptic neural pathway connecting GI-innervating VSNs to dopamine-producing neurons in the SN (345).
In addition to engaging midbrain dopaminergic populations, within-meal satiation signals also communicate to rodent HPF neurons because gastric distension or intestinal nutrient infusion robustly increases cerebral blood flow in the HPF (1090, 1091). Furthermore, HPF blood flow in humans also increases following gastric vagal nerve stimulation (1092). The medial septal nucleus was recently identified as connecting gut VSN signaling to glutamatergic neurons in the dorsal CA3 and dentate gyrus subfields of the dorsal HPF, and moreover, this pathway is required for episodic memory function (346). While food-specific learning was not directly examined in this study, the purpose of this vagal-HPF communication may be to promote the formation of meal-related episodic memories, which, in turn, have a potent influence on meal size across subsequent eating bouts. For example, Higgs and colleagues (1093–1095) show in humans that priming explicit episodic memory recall of a recent meal decreases the amount of food that is consumed at the subsequent meal. Similarly, reversible postprandial inactivation of either dorsal HPF or HPFv in rats increases meal size at the subsequent meal, an outcome likely based on disrupted meal-related episodic memory consolidation (583, 584).
Gustatory-hedonic interactions during a meal can also potently affect meal size through striatal and cortical processing. For example, electrophysiological recordings from neurons in the primate ORB reveal that flavor-responsive single neurons terminate activity when the animal is fed to satiety with a specific food (e.g., fatty cream), and further, that the same neuron will reinvigorate responding when a new flavor/food is introduced (e.g., glucose) (1096). This modulation of flavor-evoked signals by motivational state, termed sensory specific satiety, is not a property commonly observed in early stages of the gustatory system (1097). Similarly, a response in the human ORB detected by fMRI is correlated with subjective pleasantness and is reduced when liquid food is eaten to satiety. Importantly, this decrease in ORB response is specific to the particular liquid food consumed within the meal (1098). These neurophysiological and functional neuroimaging findings may have relevance to the food variety (1099) and food monotony (1100) effects in humans. For the variety effect, meal size is increased or reduced, respectively, when different foods are presented either simultaneously or sequentially, whereas the amount of food consumed generally decreases over time with limited variety or choice (e.g., military, prison, etc.). The ORB is not uniquely positioned, however, to process gustatory-hedonic interactions during a meal. For example, within-meal pleasantness ratings in humans are also correlated with PET response in the dorsal putamen and caudate nucleus (148), as well as the INS (1101). Moreover, electrophysiological neural activities in the rat LHA, ORB, INS, and amygdala are sensitive to changes in satiety state through different phases of a complete eating cycle (1102). Of great interest is that INS neurons not only appear to respond differentially based on varying hunger and satiety states but also shift neural responses from a hunger- to a satiety-related pattern in response to food-predictive cues, suggesting a pattern of dynamic activity in this region that is based on the prediction of a forthcoming energetic state (123).
6.3. Hindbrain, Medulla, and Sensory Nerves
6.3.1. Brain regions.
Two parts of the rhombicbrain play key roles in terminating meals: the NTS and the PB. We have already described the NTS as a major recipient and integrator of humorally and neurally conveyed viscerosensory information (sect. 4.3.3). We therefore begin this section by describing how the PB makes significant contributions to meal termination by acting as an integrative link between various sensory modalities that are processed in the NTS and networks in the forebrain.
6.3.1.1. the parabrachial nucleus.
6.3.1.1.1. Preamble.
The parabrachial nucleus (PB) is a large cell group located in a complex region of the dorsal pons that also contains the locus ceruleus (LC), Barrington’s nucleus, the lateral dorsal tegmental area, and the pontine central gray (FIGURE 18A). The importance of another of these other dorsal pontine cell groups has recently been highlighted by the identification of a group of glutamatergic neurons adjacent to the PB and LC (the peri-LC) that uses palatability information to modulate food and water intake depending on deficit-driven need (940).
FIGURE 18.
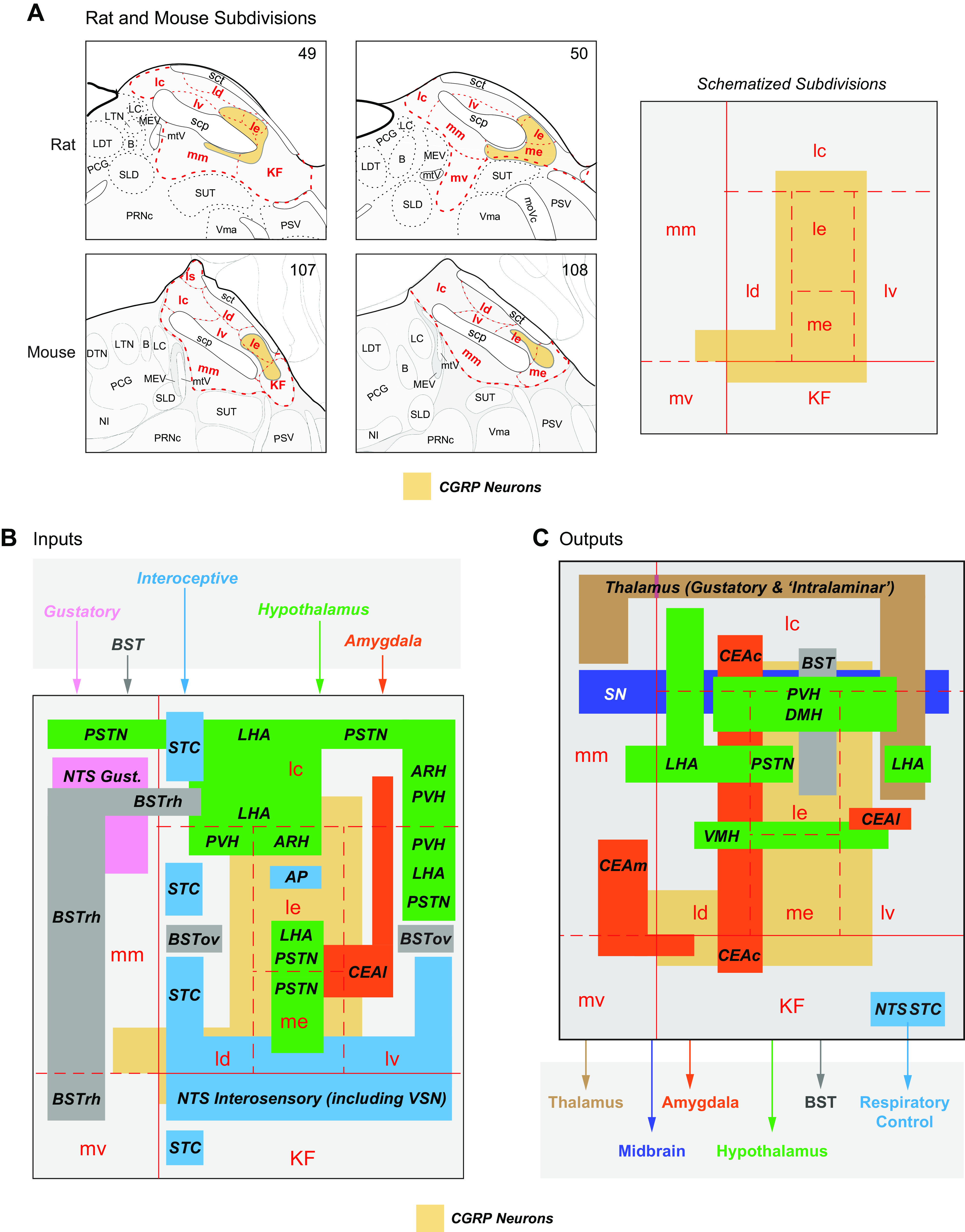
A: subdivisions of the parabrachial nucleus (PB) at levels 49 and 50 of Swanson Rat Brain Atlas (59) and levels 107 and 108 of the Allen Mouse Brain Atlas (66). The single panel on the right shows a schematized version of the rat and mouse PB showing the relative positions of the subdivisions based on the Atlas maps. Note that the size of the subdivisions is not to scale. The PB is part of a rhombicbrain network that makes significant contributions to eating behaviors control (see FIGURE 5). This control is mediated by its numerous wide-ranging connections. Their targets and origins in the various parts of the PB are summarized in B and C, respectively. Note that only those PB connections implicated in eating behavior control are shown in A and B. Source and target regions are color coded as follows: amygdala, orange; BST, bed nuclei of the terminal stria, gray; gustatory inputs, pink; hypothalamus, green; interoceptive inputs and autonomic outputs, blue; midbrain, dark blue; thalamus, brown. The regional locations of each set of connections derive from many papers that are cited in the text. calcitonin gene-related peptide (CGRP) neurons are an important source of PB outputs, and their locations are shown in yellow in A–C. Their locations in the rat PB have been transcribed from Refs. 1117, 1143 and in the mouse PB from Refs. 767, 830, 1118. LTN, lateral tegmental nucleus; LC, locus ceruleus; KF, Kölliker-Fuse subnucleus; PSV, principal sensory nucleus of trigeminal nerve; PRNc, pontine reticular nucleus, caudal part; MEV, midbrain nucleus of trigeminal nerve; PCG, pontine central gray; BST, bed nuclei of the terminal stria; PSTN, parasubthalamic nucleus; CEAm, central nucleus of the amygdala, middle part; NTS, nucleus of the solitary tract; LHA, lateral hypothalamic area; lc, central part of the lateral division; ld, dorsal part of the lateral division; le, external part of the lateral division; lv, ventral part of the lateral division; KF, Kölliker-Fuse subnucleus; mm, medial part of the medial division; CEAL, central nucleus of the amygdala, lateral; SN, substantia nigra; STC, spinal trigeminal comple; LHA, lateral hypothalamic area; ARH, arcuate nucleus; mm, medial part of the medial division; AP, area postrema; SUT, supratrigeminal nucleus; SLD, sublaterodorsal nucleus; scp, superior cerebellar penduncle; me, median eminence; LDT, laterodorsal tegmental nucleus; PSV, principal sensory nucleus of trigeminal nerve; Vma, motor nucleus of trigeminal nerve, magnocellular part; B, Barrington’s nucleus; mtV, midbrain tract of trigeminal nerve.
The PB comprises the Kölliker-Fuse subnucleus (KF) and medial (PBm) and lateral (PBl) divisions that are separated by the superior cerebellar peduncle (scp). Collectively the PBm, PBl, and KF contain ∼10 subdivisions in the rat (59, 829), and 8 subdivisions in the mouse (66) (FIGURE 18A). As with many regions of the brain, we have a greater understanding of the connectional organization of the rat PB subdivisions than those in any other species.
Connectional analyses in the 1970s and 1980s had already identified the PB as a target for interoceptive and gustatory information from the NTS and for nociceptive information from the dorsal horn and STC. However, a clear appreciation of how the PB functions in eating behaviors did not appear until about a decade later with the advent of higher resolution analytical techniques (for reviews see Refs. 390, 1103–1105). These identified three controlling roles for the PB, all of which manifest as inhibitory influences on food intake: 1) terminating deficit-induced and habitual meals; 2) a role in appetite suppression following aversive environmental and physiological stimuli, for example, pain, general malaise, infection (824); and 3) establishing a CTA, an important taste-driven learning mechanism for food avoidance (see Ref. 823) for a comprehensive review) that, in some ways is related to role 2.
6.3.1.1.2. Physiological signals and neuronal activation in the parabrachial nucleus.
A significant number of eating-related physiological signals and stimuli activate PB neurons as evidenced by increased Fos expression. Virtually all of these neurons are located in its lateral external (le) and lateral central (lc) subdivisions (see FIGURE 18A for the locations of the PB subdivisions in the rat and mouse). These stimuli include the increased blood osmolality that is associated with DE-anorexia (507), and those that establish a CTA (1106). Increased numbers of Fos-expressing neurons also implicate a role for the PB in autonomic responses to cold exposure (1107) including its interaction with des-acyl ghrelin (1108). Fos expression increases after peripherally administered eating-active antimetabolites, hormones, and pharmaceuticals. They include MA and 2-DG (507, 1109), amylin (1110), exendin-4 (1111), CCK (1112, 1113), lipopolysaccharide (LPS) (1114), and leptin (892, 1115, 1116). Many of these Fos-expressing neurons colocalize with CGRP (830, 1117, 1118). Leptin can also act directly on PB neurons. Some CCK neurons in the rostral part of the mouse PBlc express LepRb (476) and show pSTAT3 responses to peripherally injected leptin (476, 1119). Leptin injections into the PBl dose dependently reduce cumulative food intake and habitual meal size but not meal number (1120).
6.3.1.1.3. Parabrachial inputs: how do physiological signals access parabrachial neurons?
The PB is innervated by sets of inputs from the dorsal medulla, spinal cord (see Ref. 823 for review), and forebrain. Each set encodes mostly different physiological signals and stimuli, and targets different PB subdivisions (FIGURE 18B). Collectively they are responsible for much of the neuronal activation we have just described thereby enabling the PB to influence many aspects of eating behaviors.
6.3.1.1.3.1. inputs from the dorsal medulla.
Inputs from mid to caudal levels of the NTS encode interoceptive information from neurons that receive vagal and spinal sensory inputs. These inputs distribute widely within the PBl and KF (345, 1121, 1122) (FIGURE 18B) and include CCK-, galanin-, CRH-, dopamine beta-hydroxylase (DBH)-, and GLP-1-containing projections (1120, 1123–1125). Pharmacologically activating or inhibiting the GLP-1 component of this projection respectively decreases or increases cumulative food intake in habitual meals in ways that may involve altered motivational mechanisms (1120, 1124). Moreover, a recent study has shown that dynorphin (DYN) neurons in the laterodorsal subdivision (ld) (1126) are responsive to oro-gastric signals (349). These PBldDYN neurons suppress intake by way of projections to the PVH (349).
A second medullary projection most likely carries information to the PBl from a wide variety of humorally conveyed signals that are transduced by neurons in the AP (1121, 1127, 1128), including the cytokine GDF-15 (1129). A third set of medullary inputs originates in the rostral NTS and conveys gustatory information to the PBm (FIGURE 18B) (1129).
6.3.1.1.3.2. inputs from the spinal trigeminal complex.
The spinoparabrachial tract conveys topographically organized mostly nociceptive information from the spinal trigeminal nuclei and dorsal horn of the spinal cord to the PBld, lc, and le parts of the PB (FIGURE 18B) (1130–1133). These projections are implicated in mediating the effects of visceral, cutaneous, and thermal pain on food intake (398, 824, 1134).
6.3.1.1.3.3. inputs from the forebrain.
The PB receives sets of inputs from the CEA, BST, ARH, parts of the LHA (including the PSTN), the PVH (255, 261, 515, 609, 662, 755–757, 766, 918, 919, 1135, 1136), and the lateral PFC, and INS infralimbic cortex (1135, 1137) (FIGURE 18B). Forebrain inputs are found in virtually all PB subdivisions. Like inputs from the STC, NTS, and AP, the PBle, lc, and lv are the predominant targets from forebrain neurons (FIGURE 18B). Inputs from the CEAm to the PB are implicated as part of a brain-wide network that terminates deficit-induced eating (756).
6.3.1.1.4. Where do parabrachial neurons project to control eating behaviors?
The extensive outputs of the rat PB to the BST, amygdala, PAG, hypothalamus, and thalamus have been carefully described by the groups of Saper and Bernard (Refs. 825–829, 1121, 1131, 1132, 1138 but also see Ref. 1139). The projections of mouse PBCGRP neurons have recently been described in some detail (767) and conform to those of the corresponding rat PB subdivisions. The origins of some of these outputs are shown in FIGURE 18C, which highlights the complex overlap they have with PB inputs in the various PB subdivisions. PB connections therefore form a complex input/output mosaic across the entire nucleus (FIGURE 18, B and C). A more detailed description of these projections in relation to eating behaviors is beyond the scope of this review, but they have been reviewed recently with respect to gustation (823).
Two aspects of PB outputs are worth noting. First, the PB projects massively to the VMH and to a lesser extent, the DMH (827), as well as to some components associated with the postulated hypothalamic visceromotor pattern generator (916). Significant projections from the PBl are also present in three of the primary components of the upper brainstem eating control network (FIGURE 17). The SN (345, 1140), PVH, and parts of the LHA each receive robust inputs (FIGURE 18C), but any inputs to the ARH are much more sparse (827). Second, primates differ significantly from rats and mice in that gustatory information from the primate NTS bypasses the PB and is conveyed directly to the gustatory thalamus (1141). This organization means that unlike rodents, there is little to no opportunity in primates to convey gustatory information from the rostral NTS to the hypothalamus and some cerebral nuclei. One possible basis for this major evolutionary divergence in the gustatory pathway is that rodents, unlike primates, lack the physiology necessary for emesis. Thus an additional processing node in the gustatory pathway upstream of thalamic and cortical processing (the PB), while not essential for mammals capable of emesis, may be critical for the associative learning mechanisms required for rodents to effectively avoid ingesting toxic compounds. Consistent with this framework, CTA learning in rodents is quite robust, quickly established, and difficult to extinguish, and so the rodent PB is critical for processing visceral distress and CTA learning (1142).
6.3.1.1.5. The effects of genetically targeted manipulations on parabrachial function.
CGRP neurons are located in the PBle, lc, lv, and parts of the KF (FIGURE 18) (767, 830, 1117, 1118, 1143), thereby providing an inviting target for experimental manipulations. A comprehensive series of studies from Palmiter and his colleagues (824, 830, 1118, 1134, 1144, 1145) has used this arrangement to reveal a key integrative role for PBlCGRP neurons in controlling various aspects of food intake. One of the most important of these findings from a physiological perspective is that pharmacogenetic activation of PBlCGRP neurons just before the beginning of the dark period reduces habitual meal size but not number in normally eating animals (1118). Moreover, inhibiting PBlCGRP neurons also reduces the ability of peripherally delivered CCK, amylin, leptin, LiCl, GDF-15, or LPS, some of which encode aversive stimuli, to terminate food intake (830, 1118, 1128). These effects are most likely mediated in part by inputs from APGFRAL neurons (1128), NTSCCK, NTSDBH, and NTSGLP-1 neurons (1120, 1124, 1125, 1146), and PBlCGRP outputs to the CEAl (767, 830).
A second NTS to PBl projection involves NTS neurons that express calcitonin receptors (CalcR) (1147). The location of these neurons overlaps with NTSTH neurons. Although their destinations in the various PBl subdivisions is unclear, they do not appear to target PBCGRP neurons. Importantly, NTSCalcR pathways promote meal termination without engendering aversion (1147). Thus NTSCalcR neurons are distinct from those NTSCCK neurons that are important for mediating eating suppression by aversive stimuli (1146, 1147).
Part of the PBl’s integrative role also involves counterbalanced interactions between PBlCGRP and ARHAGRP neurons. As we saw earlier (sect. 5.5.2.3.3), adult mice starve after ARHAGRP neuronal ablation (817). This response is prevented by pharmacogenetic inhibition of PBlCGRP neurons (830). Palmiter’s group (1118) then went on to show that ARHAGRP projections to PBlCGRP neurons delay satiation thereby increasing food intake. This mechanism is separate from the one engaged by ARHAGRP to initiate food intake through their projections to PVHMC4R neurons (832). Thus in adult mice the loss of ARHAGRP neurons removes their interactions with both the PVH and the PBl thereby massively compromising the animal’s ability to initiate meals (824).
6.3.1.1.6. The role of parabrachial nucleus in meal termination.
Results from the functional studies we have just described show that the PB (together with the NTS) occupies a pivotal position in the rhombicbrain network that helps control eating behavior (box 1 in FIGURE 19), particularly with regards to the timing of meal termination. Parts of both nuclei possess all four of the core component criteria we described earlier (sect. 4.2.5.1.1), including a key role for AMPK (97, 321, 1083). As part of this network, the PBl has key inputs from those parts of the NTS and STC that process interoceptive information from the GI tract, hepatic portal vein, etc. FIGURE 19 also shows that the PB has strong connections with two other eating control networks located elsewhere in the brain. These are: the ARH, PVH, LHA (including the PSTN), and SN in the upper brainstem eating control network (box 2 in FIGURE 19); and the CEA and parts of the BST in the cerebral nuclei associated with behavioral selection and initiation (box 3 in FIGURE 19). These bidirectional network connections enable the PBl to have major influences on meal duration.
FIGURE 19.

1: schematic showing how the lateral parabrachial nucleus (PBl) is part of the Rhombicbrain Control Network (1) within which it has connections with medullary regions including the nucleus of the solitary tract (NTS), the area postrema (AP), and the spinal trigeminal complex (STC). The PBl has robust bidirectional connections with two other brain networks that contribute to the timing of meal termination. 2: Upper Brainstem Eating Behavior Control network that includes the hypothalamus (see FIGURE 17 for more details of this network). 3: the motor Selection and Initiator Networks in the cortex and cerebral nuclei. Additional signals are provided by gustatory information that is conveyed to the motor selection and initiation networks by way of connections involving the medial part of the PB (PBm) and the thalamus. 4: appropriate behavioral actions are then executed using selected motor neurons in the Executive Networks. It seems reasonable to assume that during meals ongoing changes in the relative activity states of the connections between these control networks and the PB help determine when meals are terminated in a wide variety of situations. CEA, central nucleus of the amygdala; BST, bed nuclei of the terminal stria; PVH, paraventricular hypothalamic nucleus; ARH, arcuate nucleus; SN, sunstantia nigra; VTN, ventral tegmental area; LHA, lateral hypothalamic area.
Three sets of recent studies contribute to the organization shown in FIGURE 19. First, two NTS to PBl projections promote meal termination with (NTSCCK) or without (NTSCalcR) engendering aversion (1125, 1147). Considering these projections together with the NTS to PB projections that convey gustatory information (at least in rodents), and projections in the spinoparabrachial tract that convey nociceptive influences on food intake, there at least four distinct pathways from the medulla that can affect PB function. Second, projections to the PBl from ARHAgRP neurons (817, 824, 830, 1118), from galanin- and MC4R-expressing PVHSIM1 neurons (272), and possibly some LHNT/CRH neurons (508, 515) strongly highlight the role of hypothalamic to PBl connections on determining when meals are terminated. Third, the importance of CEA outputs to the BST, PSTN, and the PBl for terminating deficit-induced meals has been shown after chemogenetic stimulation of the CEAm but not the CEAl (756). Taken together with the neuroanatomical studies that describe the complexities of PB connections (summarized in FIGURE 18), these and other functional studies suggest that after a meal is initiated ongoing changes in the relative activity states of the PBl’s bidirectional connections (FIGURE 19) help determine when meals are terminated in a wide variety of situations.
6.3.2. Which signals?
Many peripheral nerve and humoral signals can terminate meals. The medulla is ideally positioned to receive and integrate all eating-related information arising from ingesting a meal, and then to translate it in a way that terminates the meal. To discuss all these signals in detail would be beyond the scope of this review. However, important principles are the multiple interactions among these signals, their interactions with adiposity or metabolic signals, (which, notably, are features targeted by promising pharmacologic obesity treatments), and their effects on brain network function. To highlight some of these principles, we focus on the following: 1) gastrointestinal fill or distension; 2) glucose; 3) CCK, a gut hormone that has an established physiological satiating function; 4) GLP-1, a gut hormone with strong satiating function; 5) oxytocin, a hormone synthesized in the PVH and SO, and released from the posterior pituitary; and 6) amylin, a pancreatic hormone that acts in the medulla as a satiation factor.
6.3.2.1. gastrointestinal distension.
The stomach functions as a reservoir of ingested food, performs further (after chewing) mechanical breakdown of the food, and begins chemical degradation (primarily of protein) to deliver the semiliquid chyme to the duodenum at a controlled rate. A vago-vagal accommodation reflex that is activated during eating, triggered mainly by mechanosensors in the stomach wall, but also by intestinal nutrient receptors and CCK, allows gastric volume to increase without a significant increase in intragastric pressure or stomach wall distension (e.g., Ref. 1148). Given these functions, its dense innervation, and prime location, the stomach is ideally positioned to monitor and control food intake. It therefore seems obvious that gastric distension should play a major role in meal termination. Experiments in rats with pyloric cuffs, which prevent ingested food from leaving the stomach, demonstrate that gastric distension per se will inhibit eating (1149). Within meal extension of a gastric balloon also inhibits eating (e.g., Ref. 295). In both situations, however, the gastric volumes needed to inhibit eating are usually larger than the volumes reached during a normal meal. Likewise, neither gastric fundus nor gastric antrum distension reduced subsequent caloric intake in humans (1150). Fundus distension beyond what would be expected during a normal meal still did not affect intake; instead, it caused reduced feelings of hunger and increased fullness as assessed by visual analog scale (1150). The physiological relevance of gastric distension alone as a satiation signal is therefore questionable.
Recent evidence indicates that distension of the small intestine can also inhibit eating (49), but as with gastric distension, supraphysiological volumes are apparently needed for this effect (347). Thus activation of small intestinal VSN mechanosensors rather than gastric distension alone was sufficient to inhibit eating potently. Also recently, Kim and colleagues (349) identified the neural pathway that conveys graded, eating-inhibitory signals from distension of the upper digestive tract via VSNs and the medulla to the PB. The authors speculate that “because distension of the digestive tract does not fundamentally resolve hunger or thirst …,” these “mechanosensory feedback signals would work in concert with other feedback mechanisms that are based on diverse features of the ingesta, …” (349). In fact, gastric volumetric and postgastric mechanical and chemical (primarily nutritional) signals are activated concurrently during a meal. Substantial evidence indicates that normal gastric and upper small intestinal distension during a meal synergizes with postgastric signals to produce satiation. In rats, up to 40% of a liquid meal empties into the small intestine before meal termination (1151). Measuring gastric emptying in humans is not trivial, but the available literature indicates that there are substantial differences between the emptying of liquid and solid meals, with the former emptying much faster than the latter (1152). However, many of the pertinent studies used unrealistically different meals. In a study that used magnetic resonance imaging (MRI) with scintigraphic validation to measure gastric emptying, realistic liquid and solid meals with identical nutrient composition and nutrient density (1153) produced little difference. In both conditions ∼20% of the ingested meal had left the stomach ∼30 min after meal onset. Moreover, gastric emptying was approximately linear throughout the experiment. This indicates that the nutrients from solid meals empty into the duodenum more rapidly than commonly assumed. Under normal conditions, i.e., when liquids are consumed together with a solid meal, the entry of soluble nutrients (e.g., sugar) into the duodenum may even be faster than measured in this experiment. In any case, these findings support the concept that also during normal eating in humans, and not only during ingestion of a liquid experimental meal in laboratory animals, mechanical and chemical signals from the stomach and small intestine are activated concurrently.
6.3.2.2. glucose.
Russek was the first to propose a role for hepatic glucosensors in the control of eating (1154). The virtual absence of VSN terminals in liver parenchyma (330) argues against the existence of hepatic parenchymal glucosensors as conceptualized by Russek. Clearly, however, VSNs terminating in the wall of the hepatic portal vein function as sensors for absorbed glucose generating signals that can influence insulin release (103, 292, 1155, 1156) and glucose metabolism (1157). VSNs that innervate the hepatic portal vein wall directly innervate the DMX (387) and so are well placed to mediate these effects. Activating these glucosensors may also inhibit eating. Glucose reduced short-term food intake in rats when infused into the hepatic portal vein, but not when infused into the jugular vein (1158, 1159). Later, intrameal hepatic portal vein infusions of lower glucose doses (1 mM vs. 3 mM) specifically reduced meal size (1160). Together, these findings indicate that the prandial increase in hepatic portal vein glucose concentrations stimulates hepatic portal vein glucosensors in ways that contribute to meal termination. Whether or not this is related to the same mechanism as the recently reported silencing of ARHAgRP neurons in fasted mice by hepatic portal vein glucose via SSNs (451) remains to be determined.
6.3.2.3. cholecystokinin.
Fatty acids and aromatic amino acids from digested food stimulate cholecystokinin (CCK) secretion from I cells that are mainly located in the duodenum. CCK has several physiological effects, such as inhibiting gastric emptying, and stimulating exocrine pancreatic secretion and gall bladder emptying. A landmark paper by Gibbs and colleagues in 1973 (20) showed that intraperitoneal injections of the synthetic octapeptide form of CCK (CCK‐8) inhibits eating in rats. Interestingly, chronic intraperitoneal administration of CCK-8 before each single spontaneous meal in rats persistently reduced meal size, but over time meal frequency increased such that the decrease in meal size was compensated and 24-h food intake was no longer affected (1157). This emphasizes that CCK-8 specifically reduces meal size and that meal initiation and meal termination are independently controlled behavioral phenomena.
CCK research still focuses almost exclusively on the effects of CCK-8, which is somewhat surprising because CCK-8 is the form of CCK mainly released by nerve terminals and not I cells. Several longer forms of bioactive CCK featuring the crucial 7 amino acid carboxyl terminus, i.e., CCK‐22, CCK‐33, CCK‐39, and CCK‐58, have been identified in vivo (1161). The predominant peripheral forms, and hence, the major circulating (i.e., endocrine) forms in most species appear to be CCK-33 and CCK-58. Immediately after their release, they may act on VSNs that terminate in the gut wall, the effect presumably recapitulated by intraperitoneal injections of CCK-8. However, after entering the blood, they may also have an endocrine effect by targeting CCK receptors elsewhere, perhaps in the hepatic portal vein (1162), in the medulla (1163), or in the hypothalamus (1164). The fourth ventricular injections of the CCK-1 receptor antagonist lorglumide that Lo et al. employed (1163) might also antagonize a neural CCK link in the satiating actions of peripheral CCK, for instance in the form of NTSCCK neurons projecting to the PVH (1165). However, this appears unlikely given the rostral to caudal flow of the cerebrospinal fluid. Therefore, these and other (1161, 1164, 1166) findings suggest that hindbrain CCK receptors are involved in the eating-inhibitory effect of endogenous peripheral CCK. Consistent with this possibility, CCK-33 and CCK-58 have a longer half-life in the circulation than the shorter CCK forms. Furthermore, CCK-58 is not degraded in the liver (1161) and is more lipophilic than CCK-8, favoring both an endocrine effect and a lymphatic route of distribution bypassing the liver. CCK-58 also leads to longer activation of VSNs than CCK-8. All these features may be relevant for the fact that CCK-58 also prolongs the intermeal interval, i.e., that it affects satiety in addition to satiation (1161), whereas exogenous CCK-8 selectively reduces meal size (1167).
Thus a general picture emerges indicating that endogenous peripheral CCK inhibits eating via VSN activation and via an endocrine effect, presumably in the medulla (1163). Recent evidence indicates that intestinal CCK1 receptors are located primarily on mechanosensitive VSNs (347, 348), suggesting that these mechanosensitive VSNs mediate the satiating effect of CCK. Although this seems somewhat counterintuitive as the chemosensitive mucosal vagal endings are closer to the CCK secreting enteroendocrine cells (347, 348), it fits the observation that CCK and GI distension synergize to inhibit eating (see below). In sum, evidence from hundreds of papers since 1973 shows that CCK is part of the physiological mechanism of satiation, and that it fulfills the criteria for a physiological satiating signal in humans (1167).
6.3.2.4. glucagon-like peptide-1.
As with CCK, we focus here on circulating glucagon-like peptide-1 (GLP-1). During a meal, different luminal nutrients stimulate the release of the 30 amino acid peptide GLP‐1 from L‐cells located in the small intestine (1168). The density of L cells in the intestine increases from proximal to distal (1169), but because the mucosal area is larger in the proximal than in the distal small intestine, the number of L cells is also high in the proximal part, and likely contribute to GLP-1 secretion (1170). Whether there is also an early release of GLP-1 from distal small intestinal L cells triggered by a neuroendocrine reflex from the proximal intestine (1171) is still unresolved. GLP‐1 inhibits GI motility, particularly gastric emptying (the ileal brake mechanism), and it enhances glucose-stimulated insulin release (see Ref. 1168). Exogenously administered GLP-1 inhibits eating in animals and humans after acute and chronic administration (for reviews, see Refs. 1166, 1168, 1172), as well as in obese individuals without (1173) or with type II diabetes (1174). In one study intraperitoneally, but not intracerebroventricularly, administered exendin-9 (Ex-9; a GLP-1 receptor antagonist) reduced the satiating effect of intraperitoneally administered GLP‐1 (1175). These and other findings indicate that native GLP-1 administered exogenously (intraperitoneally) does not target the brain to reduce food intake.
There is strong evidence that endogenously secreted GLP-1 contributes to physiological meal termination (satiation). It most likely acts via abdominal VSNs because the bilateral knockdown of GLP-1 receptor (GLP-1R) mRNA in nodose ganglia that harbor VSN cell bodies caused a persistent increase in meal size (338) (FIGURE 20). As with intestinal CCK1 receptors (see above), the GLP-1Rs that help control meal size are located primarily on mechanosensitive VSNs (347, 348, 353). GLP-1R located on mucosal, i.e., chemosensitive VSNs (347), may be involved in the vagally mediated component of the GLP-1’s incretin effect (338). In this regard, a recent study has shown that GLP-1Rs on the VSN that innervate the GI tract are capable of influencing glycemia and glucose tolerance independent of effects on food intake (336). As with the persistent meal size reduction evident after chronic, meal contingent CCK-8 administration (1157), the meal size increasing effect of the nodose ganglia GLP-1R mRNA knockdown was compensated by a change in meal number, such that 24-h food intake was unaffected (338).
FIGURE 20.
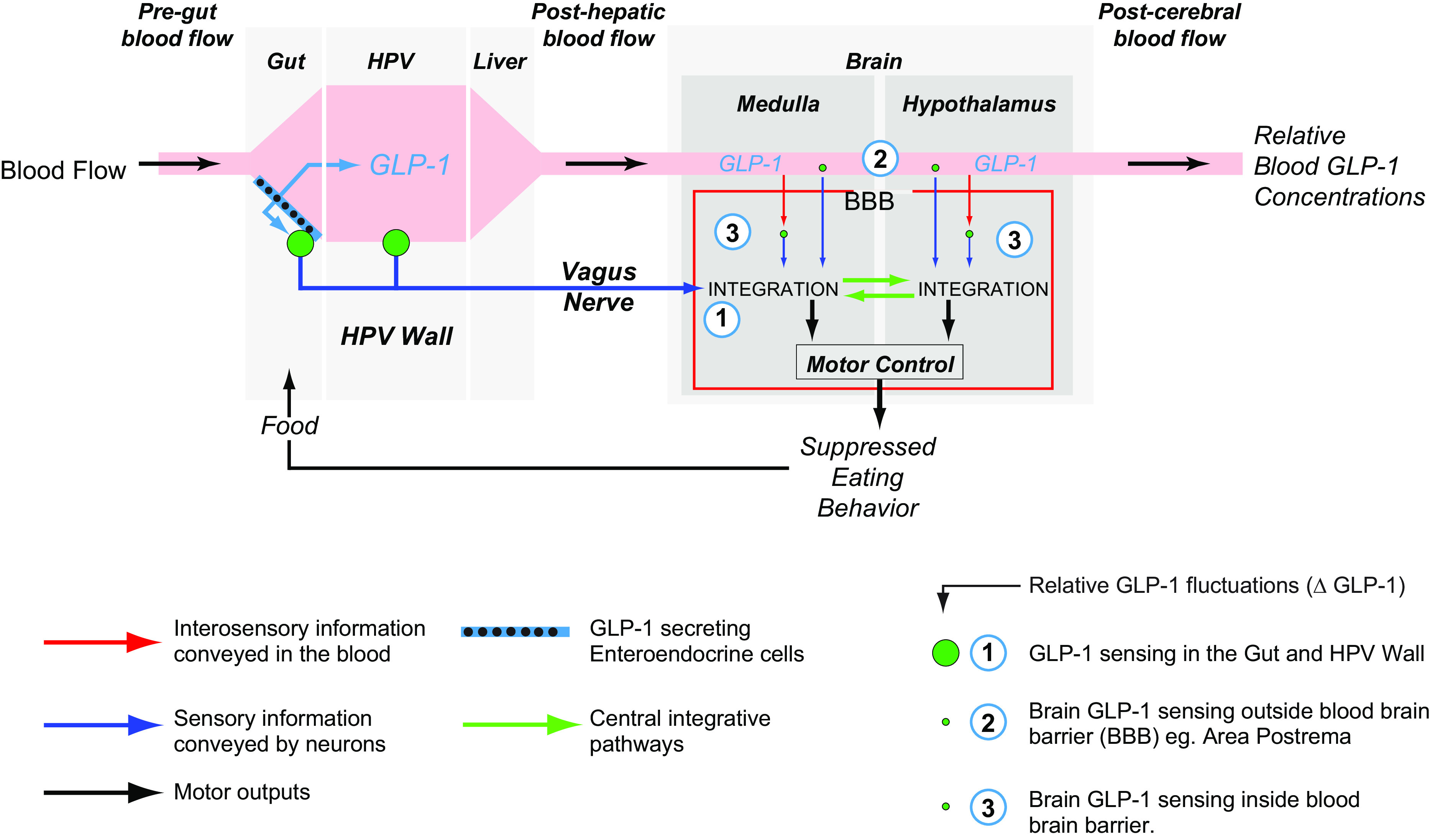
Glucagon-like peptide 1 (GLP-1), as a peripheral signal that controls eating behavior, is released from enteroendocrine cells in the gut, from where it enters the hepatic portal vein (HPV). Vagal sensory endings are the principal means by which this GLP-1 information is conveyed to the brain. These are found in the gut and stomach walls mainly on mechanosensitive vagal sensory neurons (VSNs) (347, 353) and in the HPV wall (1176). The majority of vascular GLP-1 is degraded by DPP-IV in the liver and blood, meaning that the vascular half-life of GLP-1 and its concentrations in the posthepatic vasculature are usually very low, as indicated by the varying width of the pink blood flow “stripe.” This means that opportunities are limited for any direct interactions of vascular GLP-1 with the brain (indicated by the different sized green circles). These interactions may only occur at its highest circulating concentrations. Note that GLP-1 is also synthesized by some medullary glutamatergic neurons that have widespread projections within the brain. Although these neurons have major influences on eating behaviors, they have little to no role in conveying information about vascular GLP-1 within the brain (202). See text for more details.
In most experiments exogenous native GLP-1 reduced meal size (see Ref. 340). Moreover, intact abdominal VSNs are necessary for the satiating effect of intraperitoneally administered exogenous GLP-1 (340), indicating that such GLP-1 administrations to a certain degree mimic the effects of endogenous intestinal GLP-1. However, if the same dose of GLP-1 in the same animals was administered into the hepatic portal vein, abdominal VSNs were not necessary for the effect of GLP-1 on meal size (341), indicating that, depending on the route of administration, exogenous GLP-1 can activate at least two separate pathways to limit meal size. Intravenously infused GLP-1 most likely acts primarily in the medulla to inhibit eating because lesions of the AP and intrafourth ventricular infusions of EX-9 blocked this effect (1176). However, the physiological relevance of this direct effect of circulating exogenous GLP-1 on the medulla to inhibit eating is questionable because once GLP-1 enters the blood stream, it is rapidly degraded by DPP-IV (1177). Efficient degradation also occurs in the liver. As a result, only ∼10–15% of the released endogenous GLP-1 reaches the general circulation (1168) (FIGURE 20). Together with rapid renal excretion, the degradation accounts for a biological half-life of GLP-1 in the circulation of just 1–2 min. In relation to relatively spontaneous chow meals in the rat the active GLP-1 concentration increased in the hepatic portal vein but not in the general circulation (1176) (FIGURE 20), indicating that a direct central effect of circulating GLP-1 on eating may only be relevant when GLP-1 levels are extremely high.
It is important to emphasize in this context that the eating-inhibitory effects of the therapeutically employed GLP-1R agonists are primarily due to a direct effect of these substances on the hypothalamus and the medulla (1172). For instance, while abdominal VSNs are necessary for the satiating effect of intraperitoneally administered native GLP-1 (341), they mediate only part of the acute eating-inhibitory effect of GLP-1R agonists (1178, 1179). On the other hand, subcutaneously administered liraglutide failed to reduce 24-h food intake in mice with a CNS-specific deletion of the GLP-1R mRNA (1180), and intracerebroventricularly administration of the GLP-1R antagonist EX-9 antagonized the eating-inhibitory effect of intraperitoneally injected GLP-1R agonists in rats (1178). Other findings indicate that intravenously infused liraglutide inhibits eating in mice by acting on the ARHPOMC/CART neurons (1181). Thus systemically administered GLP-1R agonists do not simply recapitulate the mechanisms of the physiological satiating effect of endogenous peripheral GLP-1.
6.3.2.5. oxytocin.
While the ability of oxytocin to suppress food intake has long been recognized, most investigations into this action have been focused within the brain (see sect. 5.5.2.5). However, OXY released from neuroendocrine terminals in the posterior pituitary has also been postulated as an agent that can reduce food intake, perhaps by way of VSNs (963). A recent report provides evidence that OXY-sensitive VSNs directly engage NTSGLP-1 neurons to suppress food intake (202). Some VSNs that express OXY receptors (OXYR) but not GLP-1R, and project to prepro-glucagon expressing neurons in the NTS that synthesize GLP-1 (FIGURE 21). Ablation of NTSGLP-1 neurons abolishes the ability of peripherally delivered OXY but not GLP-1 agonists to reduce food intake. These findings show that VSNOXYR and NTSGLP-1 neurons form an important link between circulating OXY and medullary mechanisms that can terminate meals (202). Importantly, this study also shows that NTSGLP-1 neurons are not part of the mechanisms used by circulating GLP-1 to suppress food intake.
FIGURE 21.

Axon terminals of vagal sensory neurons (VSNs; blue) form synapses with nucleus of the solitary tract (NTS) neurons, including those that synthesize pre-proglucagon [PPG; the precursor of glucagon-like protein-1 (GLP-1)], catecholamines (CA), and pro-opiomelanocortin (POMC) (the precursor of aMSH). These NTS neurons, many of which are themselves glutamatergic, have projections (green) to eating behavior related networks located more rostrally in the brain (see also FIGURES 5 and 16). For POMC neurons, these targets also include the dorsal motor nucleus of the vagus (DMX). VSNs are glutamatergic (blue) and also synthesize melanin-concentrating hormone (MCH; black) and cocaine-amphetamine regulated transcript [cocaine-amphetamine regulated transcript (CART); brown], which can be independently modulated by various stimuli. Various physiological signals (red) act on VSN terminals in the NTS and on their endings in the gasterointestinal (GI) tract and hepatic portal vein wall to alter VSN signaling properties. All of these factors provide VSN signaling with a high degree of integrative plasticity. See text for more details. OXY, oxytocin.; MC4R, melanocortin 4 receptor; GI, gastrointestinal.
6.3.2.6. amylin.
Amylin (islet amyloid polypeptide) is released together with insulin in response to eating from the pancreatic beta cells (1182, 1183). Several lines of evidence from behavioral, electrophysiological and imaging experiments indicate that circulating amylin acts primarily on the AP to inhibit eating (see Refs. 430, 1127, 1184 for review and Refs. 1185, 1186). In addition, amylin acts also on ARHPOMC neurons, where it enhances the effect of leptin. Amylin also reduces blood glucose by a combined effect on food intake, gastric emptying, digestive secretions, and nutrient absorption. The amylin analog pramlintide is an approved antidiabetic and is also used to treat obesity (430, 1184). The amylin receptor is unusual in that it combines the calcitonin receptor (CTR) with one of three receptor activity-modifying proteins (RAMP1-3). These RAMPs substantially increase the affinity of the CTR for amylin and, in effect, turn the CTR into an amylin receptor (1184). The binding of amylin to its receptors in the AP then activates an ascending neural pathway that comprises the NTS, the PBl, the CEA and, presumably, the BST (1127). Another possible site of action for circulating amylin and its interaction with leptin is the VTA (1085, 1127). Together these findings may provide the basis for a recently proposed action of amylin on food reward (430). Finally, amylin appears to act as a leptin sensitizer in the VMH by way of its ability to increase IL-6 production from microglia (970) (see below).
6.3.3. Interactions among satiation signals.
6.3.3.1. gastrointestinal distension and cck.
From a physiological point of view, it is important to emphasize that eating never activates satiation signals in isolation. For instance, eating activates GI volumetric and intestinal nutritional signals concurrently. In fact, the integration of multiple signals is crucial for the appropriate control of meal size, with different types of foods and in varying situations. This integrative function of the medulla continues the integration that has already begun in the periphery, where sensory nerves, particularly VSNs, detect many different signals that affect meal size. Polymodal VSNs that react to mechanical as well as to chemical stimuli provide a classic example for peripheral vagal sensory integration of such signals. In the early 1990s, Schwartz and Moran (350, 351) documented a whole range of such interactions between CCK-8 and gastric or duodenal distension as well as between the presence of nutrients in the small intestine and gastric distension. For instance, they showed that the combination of subthreshold distension of the stomach in a rat and near celiac artery infusion of CCK-8 at a subthreshold dose caused a dramatic increase in the firing rate of gastric VSN single units. They also demonstrated priming of load-sensitive VSNs by CCK-8, i.e., the prior exposure of such fibers to CCK-8 substantially enhanced their response to a subsequent gastric load (351). These findings nicely demonstrate that mechanosensitive VSNs that have receptive fields in the stomach or duodenum also react to CCK-8. These combined mechanical and peptidergic actions then generate a synergistic electrophysiological response in VSNs, followed by a synergistic activation of NTS neurons (1187). Ultimately this translates into the synergistic inhibition of eating by CCK-8 and stomach distension observed in animals (e.g., Refs. 1188, 1189) and humans (1190, 1191).
More recently, molecular profiling and single cell RNA sequencing of VSNs showed that CCK receptors are in fact primarily expressed on mechanosensitive VSNs from the upper GI tract (347, 348), and that activation of these fibers is critical and sufficient for an inhibition of eating (347) providing a molecular basis for the electrophysiological and functional phenomena described above. It is worth mentioning in this context that the mechanosensitive intestinal sensory nerves are also very sensitive to chemical stimuli (1192). Moreover, the expression patterns of peptide receptors and small molecule transmitter receptors on sensory nerves from the GI tract appear to be generally region specific, depending on the functional features of the various parts of the gut.
6.3.3.2. other gut peptides.
Vagal sensory receptor expression is sensitive to eating or metabolic state in the sense that fasting increases the expression of receptors for eating stimulatory peptides, whereas eating increases the expression of receptors for satiating peptides. Some of this effect appears to reflect an action of gut peptides on the expression of other gut peptide receptors, thereby enhancing interactions among different peptidergic ligands. Thus CCK and peptide tyrosine tyrosine (PYY) both inhibit food intake, and CCK, like eating, enhances the expression of the Y2 receptor in gastric VSNs (1193). CCK also restores the expression of other satiating gut peptide receptors after fasting. It is questionable whether the satiating effect of endogenous PYY requires intact VSNs. Nevertheless, the effect of CCK on the Y2 receptors was only shown for Y2 receptors on gastric VSNs, suggesting that there is an interaction with respect to the influence of CCK and PYY on the effects of gastric fill and/or on gastric emptying, i.e., on nutrient delivery into the small intestine.
6.3.3.3. gut peptides and serotonin.
Two examples of interactions between gut peptides and a small molecule transmitter are those between CCK or GLP-1, and serotonin (5HT). Already 30 yr ago, Cooper and Dourish (1194) postulated an interaction of CCK and 5HT in the control of eating. The original idea of this interaction was that peripheral CCK would modulate central 5HT circuits involved in eating control (1194, 1195). In fact, peripherally administered CCK facilitated hypothalamic 5HT release (1196), the pharmacological inhibition of hypothalamic 5HT release antagonized peripheral CCK-induced satiation (1197), and the eating-inhibitory effect of intraperitoneally injected 5HT was attenuated in 5HT2C receptor knockout mice (1198). Nevertheless, several lines of evidence indicate that there is also an interaction between CCK and 5HT or serotoninergic drugs, presumably via CCK1 and 5HT3 receptors at the level of the medulla and VSNs. The majority of endogenous 5HT is located in the GI tract (1199, 1200), and VSNs heavily express 5HT3 receptors (347, 1201). Chemical and mechanical stimuli release 5HT from intestinal enterochromaffin cells (EC) (1202). The EC-derived 5HT is sufficient, but not necessary for triggering the peristaltic reflex (1200), and it can activate mechanosensitive VSNs in two ways: 1) by initiating muscle contraction via an activation of intrinsic sensory nerves, the mechanism involved in peristaltic reflexes, and 2) by acting directly on 5HT3 receptors on mechanosensitive sensory nerves. Neither peripherally administered 5HT (1273, 1274) nor the selective 5HT3 receptor antagonist ondansetron (1275) readily cross the blood brain barrier. Nevertheless, peripherally administered 5HT inhibits eating (e.g., Refs. 1276, 1277) and ondansetron has been shown to antagonize peripheral CCK-induced satiation. Results from a series of ondansetron studies indicate that CCK and 5HT inhibit eating synergistically by activating CCK1 and 5HT3 receptors (1203–1205) and that this interaction is also instrumental for the satiating effect of intraintestinally administered lipids (1206).
A 5HT-CCK interaction could also occur in intestinal VSN terminals and the medulla (1207). Systemic 5HT levels increase substantially after a meal (1208), and 5HT3 receptors are also located on the somata of VSNs in the nodose ganglia (1209) as well as on the central terminals of these neurons in the medulla (1209, 1210), where their activation enhances glutamatergic transmission. Circulating 5HT appears to have access to these NTS 5HT3 receptors (1211), raising the possibility that circulating 5HT in response to a meal may modulate the CCK-based satiation signal at the level of the NTS (1207) in addition to any effect it may have at the intestinal terminals of VSNs.
Enteroendocrine L cells release GLP-1 in response to luminal nutrient stimulation and some evidence indicates that a significant part of this GLP-1 acts on GLP-1R that are highly expressed on neighboring enteroendocrine cells (1212). In turn, enteroendocrine cells release 5HT, which then acts on VSNs. As mentioned above, mucosal and mechanosensitive GI VSNs heavily express the 5HT3 receptor (347), which seems ideally placed to enable serotonergic interactions between enteroendocrine cells and VSNs and thereby between chemical and mechanical signals in the control of eating. 5HT may also enhance any direct effect of GLP-1 on VSNs. As mentioned earlier, VSNs also express the GLP-1R and VSN GLP-1R that are involved in the physiological control of meal size (338), but it is conceivable or even likely that there is also an interaction between 5HT derived from enteroendocrine cells and GLP-1 at the level of the sensory nerves. Considering the many different factors that stimulate 5HT release from enteroendocrine cells (e.g., Ref. 1202), the mediation and/or modulation of GLP-1 signaling by 5HT raises the possibility of multiple interactions or integration of several diverse stimuli by this mechanism.
6.3.4. Interactions between satiation signals and leptin.
Leptin acts mainly in the hypothalamus and in the medulla (see sect. 4.3.4.2.2). It affects meal size by modulating the actions of peripheral satiation signals at both sites. VSNs also express the LepRb, providing the basis for interactions among leptin and gut peptides already at the level of VSNs. The current concept is that leptin provides the tonic (background) signal that modulates the sensitivity of the fibers for short-acting peptides such as CCK, gastric distension, and GLP-1. One example for a molecular mechanism of this interaction may be the cooperative effects of leptin and CCK on the immediate early gene EGR1. Whereas leptin stimulates EGR1 expression, CCK stimulates its nuclear translocation, an effect that is enhanced by leptin and inhibited by ghrelin (344). EGR1, on the other hand, mediates the stimulatory effect of CCK on the expression of CART in VSNs, which is again enhanced by leptin (344). The CART found within VSNs, finally, is considered a major modulator at the level of the synapse between VSN and NTS neurons (see sect. 6.3). In line with these molecular aspects, electrophysiological, neuroanatomical and behavioral data show that leptin already interacts with CCK at the level of the VSN (1213) and that this interaction continues in the medulla (see below) and the hypothalamus. This indicates that the integration of short-term meal-related and long-term energy balance-related signals commences in the periphery, i.e., in sensory nerves, and continues at many locations throughout the central nervous system.
A potent interaction has also been shown for amylin and leptin. Thus amylin coadministration restored leptin responsiveness in dietary-induced obese rats (1214). These and other findings indicate that amylin and leptin synergistically activate neuronal signaling pathways to enhance metabolism and to inhibit eating (1215). The AP is the primary site where circulating amylin acts to inhibit eating (see above and Refs. 430, 1184), and some AP neurons express all components of the amylin receptor and LepRb (1278). Moreover, evidence from electrophysiological experiments indicates that amylin and leptin synergistically activate these AP neurons (1217), which may provide one of the mechanisms for the synergistic effect of amylin and leptin on eating and metabolism. Leptin and amylin also act synergistically in the VTA (1218). VTA neurons express receptors for amylin (1216) and leptin (1219), and coadministration of both into the VTA exerts a synergistic inhibitory effect on eating (1220), which is presumably mediated by their effects on VTA dopaminergic neurons. Additional synergism has been shown for amylin and leptin acting in the VMH (1218). Coadministration of amylin with leptin enhanced leptin-induced phosphorylation of STAT3 in the ARH and LepRb binding in the ARH, VMH, and DMH (1215). Interestingly, several lines of evidence indicate that astrocyte-derived interleukin-6 mediates the synergy of amylin and leptin in the VMH (see Ref. 1218).
6.3.5. Interactions with glucose.
A complex bidirectional interaction exists between systemic blood glucose levels and gastric emptying (1221). Thus variations in gastric emptying account for ∼35% of the variations in postprandial blood glucose concentrations in healthy humans. The prandial and postprandial release of incretins, insulin, and other factors are responsible for this relationship (1221). In turn, hyperglycemia and hypoglycemia substantially inhibit and accelerate gastric emptying, respectively. The effects of circulating glucose on gastric emptying are also complex. Pancreatic polypeptide, ghrelin, and nitric oxide may be involved (1221). In general, these reciprocal influences between gastric emptying and blood glucose presumably represent an additional component of the regulation of blood glucose by which the systemic blood glucose level exerts a feedback control over the delivery of glucose into the small intestine. Not surprisingly, all components involved in this feedback relationship can also affect eating. The described mechanism is therefore yet another example for the complex and delicate functional circuitry that balances eating, the GI transit of food, the processing of the digested nutrients, and the metabolic needs of the organism.
Part of this glucose effect may be a consequence of its interactions with the 5HT that is derived from enteroendocrine cells. 5HT acts as a paracrine signal to mediate the effects of luminal stimuli, including glucose, on VSNs (1222). In addition to this peripheral interaction, circulating glucose can also interact with VSN signaling at the level of the NTS by modulating the excitability of GI nodose ganglion neurons via KATP channels (1223) and by increasing the number of 5HT3 receptors on central terminals of VSNs, which enhances glutamate release (1210).
Circulating glucose also interacts with gut peptides. Burcelin and colleagues (1224) demonstrated that the hepatic portal vein glucosensor that is involved in the control of postprandial glycemia and presumably, the control of eating (1159) requires GLP-1 for proper functioning. They showed that hepatic portal vein glucose infusions increased the whole body clearance of glucose compared with saline infusion in mice. Coinfusion of the GLP-1 receptor antagonist EX-9 blocked the effect of hepatic portal vein glucose on glucose clearance. Likewise, the effect was absent in GLP-1 receptor knockout mice (1224). The authors concluded that “the GLP-1 receptor is part of the hepatoportal glucose sensor and that basal fasting levels of GLP-1 sufficiently activate the receptor to confer maximum glucose competence to the sensor.” This is also interesting because it suggests that different activation mechanisms form the basis of this interaction, i.e., one compound, in this case GLP-1, may activate sensory nerves by binding to cell membrane GPCR, whereas the other compound, in this case glucose, may have to be taken up and metabolized to influence sensory nerve activity. Glucose transporters, glucokinase (GK), and KATP channels, in other words, all elements of glucosensing known from pancreatic beta-cells or from hypothalamic glucosensing neurons have been detected in VSNs (1225), and glucose transporter-2 in VSNs appears to be required for hepatic portal glucosensing (1226). Fifty years ago, Niijima (104) had already proposed that hepatic portal glucosensing mechanisms monitor intracellular glucose utilization, which requires previous uptake via glucose transporters. In later studies, Niijima (1227) showed that the firing frequency of the hepatic portal sensory units was inversely related to the glucose concentration in the hepatic portal vein and potently stimulated by 2-DG, pointing toward glucose utilization as the critical mechanism.
6.4. Integration of Satiation Signals in the Medulla
Any environmental factor or endogenous mechanism that influences energy intake can only do so by modulating the initiation or termination of individual meals. Many factors therefore influence the meal taking control networks in the rhombicbrain. We have already alluded to some of these interactions among endogenous factors. To discuss all of them in depth would be beyond the scope of this review. To exemplify the enormous integrative capacity of the rhombicbrain in integrating meal-related information, we focus here on the multiple factors that influence the monosynaptic glutamatergic transmission between VSN and NTS neurons in the medulla (FIGURE 21).
Glutamate is the primary excitatory fast neurotransmitter in VSN and SSN signaling in the NTS (1228–1231). Acting through ionotropic and metabotropic glutamate receptors on NTSGLP-1, NTSPOMC, and NTSCA neurons (1050, 1232, 1233), it accounts for a large part of medullary neurotransmission (see Ref. 1230). It is clear that medullary glutamate is involved in the control of eating (1234–1236), and in particular in the mediation of VSN satiating signals from the GI tract (1230). Thus CCK exerts its eating-inhibitory action by modulating glutamate release from the central terminals of VSNs (1234), which appears to influence eating via downstream catecholaminergic and POMC neurons (1050, 1233). Ritter and his colleagues (e.g., Refs. 1237, 1238) showed that NMDA receptors are involved in mediating CCK satiation. Interestingly, pre and postsynaptic NMDA receptors appear to be involved in this function (1239). Several lines of evidence indicate, however, that glutamate cannot be the only neurotransmitter involved in transmission of the multiple and diverse VSN signals that are processed in the medulla (see Ref. 342). De Lartigue (342) emphasizes three points in the context of this plasticity: 1) magnesium ions block NMDA receptors at resting potential (1240), which requires interaction with another ion or peptide to remove the block for signal transmission; in fact, an interaction with a neuropeptide is crucial for the proper functioning of glutamatergic synapses (1241); 2) glutamate is involved in many other functions, not just eating; as there is no morphological separation of NTS neurons with different functions in the NTS, any leakage of glutamate out of the synapse could cause inappropriate functional responses to peripheral signals; and 3) ingesting similar foods and nutrients results in varying meal sizes depending on time of day and many other factors. Glutamate alone is poorly placed to encode this plasticity. Instead, modulatory interactions between glutamate and neuropeptides make key contributions to the integration of information conveyed by VSN to the NTS.
In addition to glutamate VSNs express several peptides that are involved in medullary synaptic transmission (see Ref. 342). CART (1242), MCH (689, 1243), and CGRP (1244) are probably the most prominent with respect to the control of eating. CART acts primarily in the rhombicbrain to inhibit eating. Blocking the cerebral aqueduct prevented the eating-inhibitory effect of CART injected into the third ventricle, whereas intrafourth ventricular CART still reduced food intake (1245). Recently, Lee et al. (1246) reported that viral-mediated nodose ganglia (i.e., presumably VSNs) knockdown of CART in rats caused a sustained increase in food intake that was due to larger meals and resulted in increased body weight. Furthermore, injection of CART directly into the NTS decreased food intake in ad libitum-fed rats (1246). These findings establish endogenous medullary CART as a necessary mediator of normal meal termination in rats. CART colocalizes with CCK-1 receptors in VSNs (1242). Eating and satiating hormones such as CCK enhance CART synthesis and release from VSNs in fasted animals (343). Behavioral studies indicate that CART is involved in the transmission of CCK-related satiation signals from VSN to NTS neurons in rats (1279). CART not only mediates CCK-induced satiation, it also appears to be involved with mediating the satiating effects of GLP-1. Thus everything mentioned about CCK and CART may also apply to GLP-1 and CART. Silencing CART in the nodose ganglia not only increases meal size, it also antagonizes exogenous CCK- and GLP-1-induced satiation (1246). On the other hand, ghrelin antagonizes the effect of CCK on CART (343), and it antagonizes its satiating effect. It is important to note that in response to a switch from fasting to eating, CART modulation of neuronal transmission may only be evident 2 h after the switch because of the delay produced by axonal transport (342).
MCH is mainly expressed in LHA neurons, which project to many brain areas (see sect. 5.5.2.2.5) (690). However, it is also another key peptide for modulating the glutamatergic transmission of satiating signals in the medulla (FIGURE 21). Interestingly, the MCH1 receptor is expressed in the NTS (690), and by VSN neurons that also express MCH (1243). The same VSNs coexpress MCH and CART (343). Eating status also modulates VSN MCH expression, i.e., it is high in fasting animals and it decreases after a meal (1243). Moreover, many MCH expressing VSNs also express the CCK1 receptor (343). CCK shuts down MCH and increases CART expression (342), effects that leptin enhances and ghrelin antagonizes. MCH-deficient mice (1247) as well as rats treated with a MCH1 receptor antagonist (694) both show reduced meal size. Some evidence indicates that MCH inhibits glutamate release from VSNs by acting at presynaptic receptors (342), which could be one component of its eating stimulatory effect.
As mentioned above, VSNs activate NTSPOMC and NTSCA neurons neurons using glutamatergic synapses (1050, 1233), and several findings argue for the involvement of NTSPOMC and NTSCA neurons in the eating-inhibitory effect of GI satiation signals (FIGURE 21). NTS neurons also heavily express MC4R (1248), and the activation or inhibition of medullary MC4R decreases or increase food intake, respectively, by producing effects on meal size, not frequency (1249–1251). Interestingly, MC4R are found on VSNs in the medulla and therefore perfectly positioned for presynaptic enhancement of glutamate synaptic transmission (FIGURE 21). This mechanism appears to be the major contributor to the MC4R activation-induced inhibition of eating that results from the enhancement of GI satiation signals (1251, 1252). There is a functionally important difference between NTSCA and NTSPOMC neurons. NTSCA neurons project heavily to more rostral areas of the brain including the hypothalamus (401, 1253, 1254) and are therefore perfectly placed to relay meal-related GI signals to the brain areas involved with energy homeostasis and modifying reward. On the other hand, NTSPOMC neurons mainly project to the medulla (11), hence forming local reciprocal circuits. In fact, many outputs from NTSPOMC neurons go to rhombicbrain structures that control consummatory aspects of ingestion (11, 1255–1257), thus providing a direct link between sensory input and motor output with an enormous integrative capacity. In essence, all these data indicate that the glutamatergic synapses between VSN and NTSPOMC and NTSCA neurons are a major integrative site for diverse food-related oral and GI signals. Furthermore, these NTSPOMC and NTSCA neurons relay information into the forebrain but also directly influence local medullary networks that control meal taking.
LepRb and leptin signaling are active in the NTSm. In fact, the LepRb expressing NTS POMC neurons (1258) described above are the major targets of leptin in the medulla (FIGURE 21). It is relevant in this context that exogenous leptin when administered peripherally or centrally specifically affects meal size (1259, 1260). Likewise, deficits in the leptin system that lead to a loss of leptin action always manifest themselves in changes in meal size and not frequency (1261–1263). Specific medullary LepRb knockdown increased body weight (temporarily on chow and permanently on a high-fat diet) by increasing meal size (1264). This effect was presumably related to a reduction of the satiating effect of GI satiation signals such as gut peptides because rats with medullary LepRb knockdown were also less sensitive to the satiating effect of exogenous CCK (1264). This same study provided evidence that AMPK contributes to the effects of manipulating leptin signaling in the NTS (1264), again showing the importance of this key energy sensor in the rhombicbrain eating control network. By acting in the medulla, leptin also enhances the eating-inhibitory effect of gastric distension (295). Gastric distension activated ∼40% of the neurons that are also activated by fourth ventricular administration of leptin and there was a synergistic effect of gastric distension and leptin with respect to inhibition of eating. Again, these results are consistent with the hypothesis that leptin acts on the NTSPOMC neurons to enhance the effects of GI satiation signals.
Leptin can affect medullary neurons directly (see above) or via descending projections from the hypothalamus (FIGURE 21). A significant contingent of POMC fibers in the medulla is from the ARH with the remainder originating locally in the NTS (11, 490). One major distinction between leptin effects on the NTS and the ARH seems to be that the medullary effects of leptin on eating and energy balance are primarily due to the activation of NTSPOMC neurons (see above), whereas the ARH effects of leptin on energy balance appear to be primarily mediated by the inhibition of ARHAgRP neurons. Thus Xu and colleagues (850) showed that selective deletion of the leptin receptor in ARHAgRP neurons reproduced the effects of global leptin deficiency and eliminated the effect of chronic leptin administration on food intake and body weight. This is consistent with findings indicating that the LepRb in the ARH are already saturated at normal leptin concentrations (496), and that a decrease in circulating leptin, which indicates reduced fat stores, is the physiologically relevant signal. Decreased leptin is detected by the hypothalamus to mount a systemic emergency response, which manifests as prompt eating and a shutdown of all nonessential energy-consuming activities. In contrast, the direct effect of leptin on medullary neurons, and the descending projections from the hypothalamus to the rhombicbrain that are downstream of ARHPOMC neurons, provide mainly a tonic background signal, hence increasing the dynamic range of responses to leptin and adjusting meal size by modulating the action of meal-related satiation signals.
Fasting and ghrelin enhance while leptin inhibits AMPK activity in the ARH, thereby reducing AgRP signaling and antagonizing the fasting- or ghrelin-induced stimulation of eating. Similarly, leptin reduces food intake by acting in the rhombicbrain at least in part by reducing AMPK activity (97). Pharmacological manipulation of medullary AMPK activity affected food intake and energy expenditure, and stimulation of medullary AMPK by fourth ventricular injection of 5-aminoimidazole-4-carboxamide-riboside (AICAR), attenuated the eating-inhibitory effect of 4th ventricular administration of leptin (97). These results indicate that the interaction between cellular energy sensing (AMPK) and endocrine signals reflecting the body’s energy stores are not limited to the hypothalamus, but extend to regions in the rhombicbrain control network.
Glucose modulates glutamate transmission from VSN to catecholaminergic neurons in the medulla in two ways: 1) directly, when extracellular glucose is sensed through a presynaptic mechanism that depends on VSN GK and 5HT3 receptors (1265); and 2) indirectly, via inputs from catecholaminergic neurons in the ventrolateral medulla involved with sensing cytoglucopenia (see above, Ref. 461). In other words, a decrease in glucose availability directly and indirectly silences the NTSCA neurons and thus weakens their response to VSN signals triggered by CCK and presumably other satiating agents. This feature may link meal termination to glucose availability. In addition, glucose also modifies the response of NTS neurons to aMSH (1266). Thus glucose appears to modulate several steps in the medullary integration of meal-related information (FIGURE 21). Finally, 5HT, in addition to its role in modulating glutamate release via 5HT3 receptors on the NTS terminals of VSNs (see above; FIGURE 21), can also modulate the postsynaptic NTSPOMC neurons via 5HT2C receptors (1267).
7. SUMMARY, CONCLUSIONS, AND FUTURE DIRECTIONS
In this section we return to our central theme of signals, neurons, and networks, and reiterate key concepts in each category. In so doing, we also provide some pointers for future investigations.
As we mention throughout, our goal has been to consider how various physiological signals and neurons from multiple brain regions operate together at a network level to control eating behaviors. We hope we have substantiated the idea that the brain does not exert this control by way of centers, which imply dedicated components that are in close proximity to one another. Instead, control signals are mediated by way of the integrated output from various networks that not only distribute across parts of the brain, the hypothalamus and rhombicbrain, for example, but also involve integration within the spinal cord and vagus nerve (FIGURE 22). Some of these networks distribute information to others that are primarily located in the endbrain, which then select and initiate the motor programs for eating.
FIGURE 22.
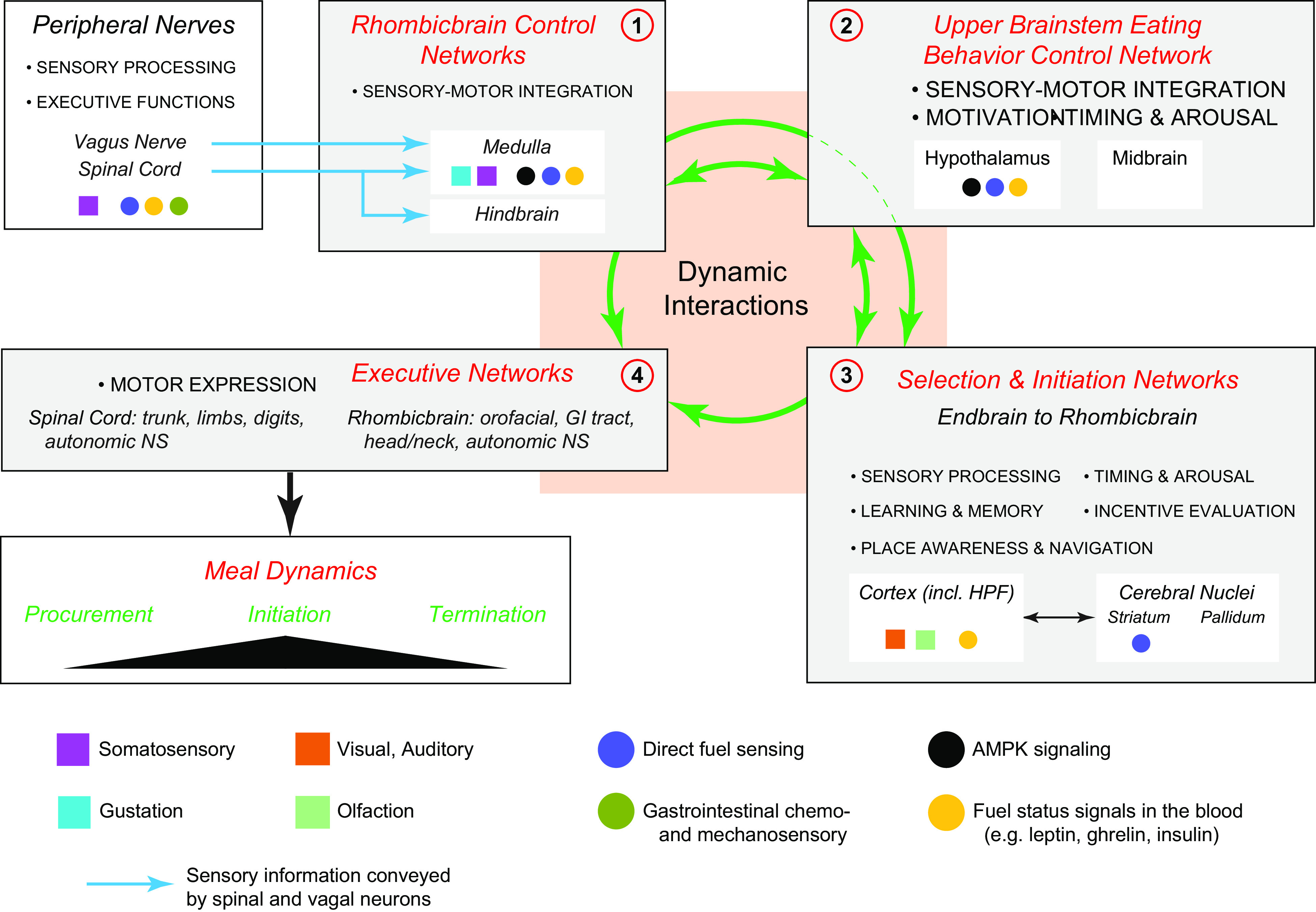
The control of eating from its beginning in the procurement (appetitive) phase to meal initiation (consummatory phase) and then meal termination involves dynamic interactions (green arrows) between at least 4 brain networks: 1) Rhombicbrain Control Networks; 2) the Eating Behavior Control Network in the Upper Brainstem (primarily in the hypothalamus); 3) Selection & Initiation Networks (primarily in the Endbrain but also as far caudal as the rhombicbrain) that organize the appropriate motor programs that allow animals to forage for and then interact with food items; and 4) Executive Networks that include the cranial and spinal motor neurons that enable these programs. Exteroceptive (colored squares) and interoceptive modalities (colored circles) are conveyed into, within, and between these networks via different routes, including brain connections and the vasculature, together with the spinal cord and the vagus nerve for a variety of sensory modalities. Each of these 4 networks incorporates different features into eating control (bulleted text). Many of the interactions between networks occur using connections (green arrows) that travel within the medial forebrain bundle (orange box). The trajectory of a particular meal (Meal Dynamics) is ultimately determined by the relative strengths of the many inhibitory and stimulatory processes that derive from integration within and between these networks. HPF, hippocampal formation; GI, gastrointestinal; NS, nervous system.
The ARH, PVH, and LHA are the core components in a crucial upper brainstem eating control network (FIGURE 17). Significant inputs to these components come from endbrain regions that are responsible for learning and memory, as well as assigning incentive value to food items. Other inputs come from circulating hormones and nutrients and from the ascending connections of the rhombicbrain control network. The collective output from the ARH, PVH, and LHA biases motor control systems located elsewhere in the brain toward eating behaviors. This process then involves selecting and initiating appropriate motor programs to enable animals to engage in foraging strategies that guide them to food sources. Parts of the endbrain, particularly some of the cortical nuclei, are intimately involved with this function (301). Our understanding of the way physiological signals influence these selection mechanisms, as well as the nature of the connections that enable information transfer from the cortex and cortical nuclei to the hypothalamus and further caudally, has improved dramatically in recent years (e.g., Refs. 323, 594). Furthermore, although some midbrain and hindbrain regions are implicated in the control of eating behaviors, for example, the periaqueductal gray (1268) and dorsal raphe (1269), their significance within the various brain control networks remains unclear. This means that exactly where the links are between the eating control and the motor selection and initiation networks and how they operate remains unresolved, thereby posing important questions for future investigations.
The ARH, PVH, and LHA also provide substantial descending projections to those parts of the hindbrain and medulla in the rhombicbrain control network that receive information from the GI tract via VSNs and SSNs, and are particularly important for terminating meals. These hindbrain and medullary regions also provide robust projections back to the forebrain, including the ARH, PVH, and LHA. Therefore, it seems reasonable to conclude that these ascending and descending connections determine the relative dynamics in the hypothalamic and rhombicbrain networks that help bias meals toward either initiation or termination (FIGURE 22). In certain circumstances, disrupting these interactions can have a major impact on long-term control of energy balance (e.g., Ref. 464). Further work into the nature of this biasing process and how it operates is warranted.
It is worth remembering that eating behaviors involve autonomic and neuroendocrine outputs that also require control networks (e.g., Refs. 875, 916). The activity of these networks needs to be coordinated with overt eating behavior. Similarly, to maximize their anticipatory advantage, habitual meals are best expressed in coordination with the prevailing photoperiod, a property that involves a circadian timing network (FIGURE 16).
How is this coordination achieved? One way to enable rapid flexibility and information flow between these multiple interactive networks is by way of shared components or nodes. One example is the DMH, which contributes to a proposed forebrain visceromotor pattern generator (916), and contributes to body temperature control, and therefore energy expenditure (996). However, the DMH also has a prominent place in the forebrain circadian timing network (FIGURE 16). The DMH has strong connections to the PVH, which as we have seen, is a major hypothalamic component of the upper brainstem eating control network (FIGURE 17). The nodal properties of various brain regions like the DMH are poorly understood, but identifying other common nodes between connectional networks may help reveal their functional significance. Neuroinformatics methods now provide one way to do this (e.g., Ref. 632).
In terms of signals, virtually all neurons coexpress neuropeptides together with fast-acting ionotropic transmitters. Physiological signals confer considerable flexibility to peptide expression within individual neurons (e.g., Ref. 875). Therefore, it is not surprising that neuropeptide signaling is at the heart of how the brain controls eating behaviors. We have described how manipulating GPCR function can have a profound impact on the way eating behaviors are expressed. Sophisticated manipulations can now reduce or enhance the signaling activity of selected peptides within neurons (e.g., Ref. 516). However, as we pointed out at the beginning of this review (sect. 2.7), large gaps remain in our knowledge about how peptide signaling between neurons is achieved, and what the cellular consequences of peptide actions on target neurons mean for downstream network function. Recently developed indicators of GPCR function may help with this goal (204, 206, 254). In terms of how neuropeptides operate at axon terminals located away from the synaptic cleft, and particularly with regard to nonsynaptic volume transmission mechanisms, more detailed investigations of mechanisms and implications would seem warranted. Indeed going forward it may not be possible to make major advances in our understanding of the functional organization of eating control systems without revealing more details about these highly complex and flexible peptidergic signaling mechanisms.
Physiology and behavior are at the heart of this review. Therefore, an appropriate place to finish is to reiterate two points. First, understanding complex physiological mechanisms at the highest level often requires experiments that have been described as whole animal physiology. These techniques may involve sophisticated surgeries, measurements, and analyses. They are to be encouraged to help us understand how the brain and GI tract, etc. work together to control eating behaviors. Second, we cannot explain how eating behavior is controlled without knowing exactly what that behavior looks like. That is, in terms of what its constituent motor actions are and how they are temporally arranged; simply measuring food intake by itself does not describe eating behavior. The critical information gained by observing behaviors in detail was emphasized by ethologists almost 70 yr ago (e.g., Ref. 303), and its validity still remains important today (e.g., Ref. 983). Ethologists like Tinbergen and others (303, 1270) understood what was needed to fully explain how motivated behaviors are controlled by the brain, but they did not have the tools to work out the details. Now we do (203, 1271). It would therefore be worth our while to look at the complexities of eating behavior in more detail so that they provide a more expansive context for considering established and emerging knowledge about how brain components work together to enable animals to eat.
8. GLOSSARY
8.1. Anatomical Abbreviations
- ACB
accumbens nucleus
- ACBc
accumbens nucleus, core
- AHN
anterior hypothalamic nucleus
- AMB
ambiguous nucleus
- AMBv
ambiguous nucleus, ventral division
- AP
area postrema
- ARH
arcuate nucleus
- B
Barrington’s nucleus
- BLA
basolateral amygdala
- BMA
basomedial nucleus of the amygdala
- BST
-
bed nuclei of the terminal stria
Parts of the BST:- ov
- oval nucleus
- rh
- rhomboid nucleus
- CEA
central nucleus of the amygdala
- CEAl
CEA, lateral part
- CEAm
CEA, medial part
- CSF
cerebrospinal fluid
- CU
cuneate nucleus
- CVO
circumventricular organ
- dm
dorsomedial
- DMH
dorsomedial hypothalamic nucleus
- DMHa
anterior part of the DMH
- DMHp
posterior part of the DMH
- DMX
dorsal motor nucleus of the vagus
- DRG
dorsal root ganglia
- EC
enterochromaffin cells
- fx
fornix
- GI
gastrointestinal
- GR
gracile nucleus
- HPF
hippocampal formation
- HPFv
HPF, ventral part
- HPV
hepatic portal vein
- icp
internal cerebellar peduncle
- IML
intermediolateral column
- INS
insular region of the cerebral cortex
- IRt
intermediate reticular nucleus
- KF
Kölliker-Fuse subnucleus
- LC
locus ceruleus
- LDCV
large dense core vesicles
- LDT
laterodorsal tegmental nucleus
- LGN
lateral geniculate nucleus
- LHA
-
lateral hypothalamic area
Parts of the LHA:- d
- dorsal region
- jd
- juxtadorsomedial
- jvd
- juxtaventromedial region, dorsal zone
- jvv
- juxtaventromedial region, ventral zone
- ma
- magnocellular nucleus
- s
- suprafornical region
- sfp
- subfornical region, posterior zone
- vm
- ventral region, medial zone
- LNG
left nodose ganglion
- LS
lateral septal nucleus
- LTN
lateral tegmental nucleus
- m
medial
- ME
median eminence
- MEA
medial nucleus of the amygdala
- MEC
medial entorhinal area of the cerebral cortex
- MEV
midbrain nucleus of trigeminal nerve
- MoV
motor nucleus of the trigeminal nerve
- mtV
midbrain tract of trigeminal nerve
- NI
nucleus incertus
- NS
nervous system
- NTS
nucleus of the solitary tract
- NTSco
NTS, commissural part
- NTSge
NTS, gelatinous part
- NTSm
NTS, medial part
- opt
optic tract
- ORB
orbitofrontal region of the cortex
- PAG
periaqueductal gray
- PB
-
parabrachial nucleus
Parts of the PB:- lc (or cl)
- central part of the lateral division
- ld (or dl)
- dorsal part of the lateral division
- le (or el)
- external part of the lateral division
- lv (or vl)
- ventral part of the lateral division
- KF
- Kölliker-Fuse subnucleus
- mm (or m)
- medial part of the medial division
- wa
- waist area
- PCG
pontine central gray
- PFC
prefrontal region of the cerebral cortex
- PFCm
medial prefrontal region of the cerebral cortex
- PH
posterior hypothalamus
- PMRN
parvicellular midbrain reticular nucleus
- PPC
posterior parietal area of the cerebral cortex
- PRNc
pontine reticular nucleus, caudal part
- PSTN
parasubthalamic nucleus
- PSV
principal sensory nucleus of trigeminal nerve
- PVH
-
paraventricular hypothalamic nucleus
Parts of the PVH:- ap
- anterior parvicellular part
- dp
- dorsal parvicellular part
- mp
- medial parvicellular part
- pm
- posterior magnocellular part
- pv
- periventricular part
- vp
- ventral parvicellular part.
- PVT
paraventricular thalamic nucleus
- RE
reuniens nucleus
- SBPZ
subparaventricular zone
- SCH
suprachiasmatic nucleus
- SCig
lateral part of the intermediate gray of the superior colliculus
- scp
superior cerebellar penduncle
- sct
spinocerebellar tract
- SELV
small electro-lucent vesicles
- SFO
subfornical organ
- SI
innominate substance
- SLD
sublaterodorsal nucleus
- SN
substantia nigra
- SNc
compact part of the substantia nigra
- SNr
reticular part of the substantia nigra
- SO
supraoptic nucleus
- sptV
spinal tract of the trigeminal nerve
- SSN
spinal sensory nerves or neurons
- STC
spinal trigeminal complex
- SUT
supratrigeminal nucleus
- TMN
tuberomammillary nucleus
- ts (or t)
solitary tract
- TUI
lateral part of the tuberal nucleus
- VLPC
ventrolateral caudoputamen
- Vma
motor nucleus of trigeminal nerve, magnocellular part
- VMH
ventromedial hypothalamic nucleus.
- VMHa
anterior part of the VMH
- VMHc
central part of the VMH.
- VSN
vagal sensory nerves or neurons
- VTA
ventral tegmental area
- Xa
sensory nerve of the vagus
- Xe
motor nerve of the vagus
- XII
hypoglossal nucleus
- XIIe
motor nerve of the hypoglossal.
- ZI
zona incerta
8.2. Chemical Abbreviations
- 2DG
2-deoxy-d-glucose
- 5HT
serotonin
- AgRP
agouti-related peptide
- AMPK
adenosine monophosphate kinase
- aMSH
alpha-melanocyte-stimulating hormone
- BDNF
brain-derived neurotrophic factor
- CART
cocaine-amphetamine regulated transcript
- CCK
cholecystokinin
- CGRP
calcitonin gene-related peptideCRH(F)corticotropin-releasing hormone (factor)
- CTR
calcitonin receptor
- DBH
dopamine-beta-hydroxylase
- DYN
dynorphin
- EGR1
early growth response factor-1
- FB
fast blue
- FG
fluorogold
- GAD67
glutamate decarboxylase 67
- GHS‐R
growth hormone secretagogue receptor
- GLP-1
glucagon-like peptide-1
- GLP-1R
GLP-1 receptor
- Glut
glutamate
- GOAT
ghrelin‐O‐acyl‐transferase
- GPCR
G-protein coupled receptor
- H/O
hypocretin/orexin (ORX)
- LepRb
long form of the leptin receptor
- LPS
lipopolysaccharide
- MA
mercaptoacetate
- MC4R
melanocortin 4 receptor
- MCH
melanin-concentrating hormone
- MCH 1R
MCH receptor 1
- mTOR
mammalian target of rapamycin
- NMDA
N-methyl-d-aspartic acid
- NPY
neuropeptide Y
- NT
neurotensin
- ORX
orexin
- OXY
oxytocin
- p
phosphorylated
- PACAP
pituitary adenylate cyclase-activating peptide
- POMC
pro-opiomelanocortin
- PrRP
prolactin-releasing peptide
- PYY
peptide tyrosine tyrosine
- RAMP
receptor activity modifying protein
- SIM1
single-minded 1
- STAT3
signal transducer and activator of transcription 3
- TH
tyrosine hydroxylase
- TRH
thyrotropin-releasing hormone
- VGlut2
vesicular glutamate transporter 2
8.3. Physiological Abbreviations
- CPG
central pattern generator
- CTA
conditioned taste aversion/avoidance
- DE
dehydration
- LHS
lateral hypothalamic syndrome
GRANTS
The work from the authors’ laboratories discussed in this review was supported by the following HHS National Institutes of Health Grants: NS-029728, MH-066168, DK-118910, and DK-121531 (to A.G.W.); DK-104897, DK-118402, and DK-123423 (to S.E.K.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.G.W. prepared figures; A.G.W., S.E.K., G.S. and W.L. drafted manuscript; A.G.W., S.E.K., G.S. and W.L. edited and revised manuscript; A.G.W., S.E.K., G.S. and W.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the past and present members of their laboratories for outstanding contributions to the ideas, concepts, and results that we discuss here. We also acknowledge the much appreciated input, opinions, and guidance from mentors, colleagues, and friends in science.
REFERENCES
- 1.Zingg B, Chou XL, Zhang ZG, Mesik L, Liang F, Tao HW, Zhang LI. AAV-mediated anterograde transsynaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93: 33–47, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits: a decade of progress. Neuron 98: 256–281, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nassi JJ, Cepko CL, Born RT, Beier KT. Neuroanatomy goes viral! Front Neuroanat 9: 1–24, 2015. doi: 10.3389/fnana.2015.0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bota M, Sporns O, Swanson LW. Architecture of the cerebral cortical association connectome underlying cognition. Proc Natl Acad Sci U S A 112: E2093–E2101, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, Yang XW, Shih JC, Toga AW, Dong HW. The mouse cortico-striatal projectome. Nat Neurosci 19: 1100–1114, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swanson LW, Lichtman JW. From Cajal to connectome and beyond. Annu Rev Neurosci 39: 197–216, 2016. [DOI] [PubMed] [Google Scholar]
- 7.Swanson LW, Mogenson GJ. Neural mechanisms for the functional coupling of autonomic, endocrine and somatomotor responses in adaptive behavior. Brain Res 228: 1–34, 1981. [DOI] [PubMed] [Google Scholar]
- 8.Swanson LW. Cerebral hemisphere regulation of motivated behavior. Brain Res 886: 113–164, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Morgane PJ. Historical and modern concepts of hypothalamic organization and function. In: Handbook of the Hypothalamus Volume 1, Anatomy of the Hypothalamus, edited by Panksepp J, Morgane PJ.. New York: Marcel Dekker, Inc., 1979, p. 1–64. [Google Scholar]
- 10.Davis JD, Levine MW. A model for the control of ingestion. Psychol Rev 84: 379–412, 1977. [PubMed] [Google Scholar]
- 11.Wang D, He X, Zhao Z, Feng Q, Lin R, Sun Y, Ding T, Xu F, Luo M, Zhan C. Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat 9: 40, 2015. doi: 10.3389/fnana.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cushing H. Neurohypophysial mechanisms from a clinical standpoint. In: Papers Relating to the Pituitary Body, Hypothalamus, and Parasympathetic Nervous System. Springfield, IL: Charles C. Thomas, 1932. [Google Scholar]
- 13.Riddoch G. Clinical aspects of hypothalamic derangement. In: The Hypothalamus Morphological, Functional, Clinical and Surgical Aspects. Edinburgh: Oliver and Boyd, 1939, p. 101–130. [Google Scholar]
- 14.von Economo C. Sleep as a problem of localization. J Ner Ment Dis 71: 249–259, 1930. [Google Scholar]
- 15.Ranson SW, Magoun HW. The hypothalamus. Ergebnisse Physiologie Exper Pharmakologie 41: 56–163, 1939. [Google Scholar]
- 16.Fisher C, Ingram WR, Ranson SW. Diabetes Insipidus and the Neuro-Hormonal Control of Water Balance: a Contribution to the Structure and Function of the Hypothalamico-Hypophyseal System. Ann Arbor, MI: Edward Brothers, Inc., 1938. [Google Scholar]
- 17.Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec 78: 149–172, 1940. [Google Scholar]
- 18.Anaand BK, Brobeck JR. Hypothalamic control of food intake in rats and cats. Yale J Biol Med 24: 123–140, 1951. [PMC free article] [PubMed] [Google Scholar]
- 19.Grossman SP. Eating or drinking elicited by direct adrenergic or cholinergic stimulation of hypothalamus. Science 132: 301–302, 1960. [DOI] [PubMed] [Google Scholar]
- 20.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol 84: 488–495, 1973. [DOI] [PubMed] [Google Scholar]
- 21.Stanley BG, Leibowitz SF. Neuropeptide-y injected in the paraventricular hypothalamus–a powerful stimulant of feeding-behavior. Proc Natl Acad Sci U S A 82: 3940–3943, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langhans W, Zeiger U, Scharrer E, Geary N. Stimulation of feeding in rats by intraperitoneal injection of antibodies to glucagon. Science 218: 894–896, 1982. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 372: 425–432, 1994. [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell 83: 1263–1271, 1995. [DOI] [PubMed] [Google Scholar]
- 25.Chua SC Jr, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271: 994–996, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Coleman DL, Hummel KP. Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol 217: 1298–1304, 1969. [DOI] [PubMed] [Google Scholar]
- 27.Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 9: 294–298, 1973. [DOI] [PubMed] [Google Scholar]
- 28.Flier JS. Clinical review 94: what’s in a name? In search of leptin’s physiologic role. J Clin Endocrinol Metab 83: 1407–1413, 1998. doi: 10.1210/jcem.83.5.4779,10.1210/jc.83.5.1407. [DOI] [PubMed] [Google Scholar]
- 29.Flier JS. Starvation in the midst of plenty: reflections on the history and biology of insulin and leptin. Endocr Rev 40: 1–16, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenbaum M, Leibel RL. 20 years of leptin: role of leptin in energy homeostasis in humans. J Endocrinol 223: T83–96, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wise RA. Forebrain substrates of reward and motivation. J Comp Neurol 493: 115–121, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baldo BA, Kelley AE. Discrete neurochemical coding of distinguishable motivational processes: insights from nucleus accumbens control of feeding. Psychopharmacology (Berl) 191: 439–459, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Stricker EM, Zigmond MJ. Recovery of function after damage to central catecholamine- containing neurons: a neurochemical model for the lateral hypothalamic syndrome. In: Progress Psychobiology Physioliogy Psychology. New York: Academic Press, 1976, p. 121–188. [Google Scholar]
- 34.Winn P. The lateral hypothalamus and motivated behavior: an old syndrome reassessed and a new perspective gained. Curr Dir Psychol Sci 4: 182–187, 1995. doi: 10.1111/1467-8721.ep10772629. [DOI] [Google Scholar]
- 35.Mogenson GJ, Jones DL, Yim CY. From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14: 69–97, 1980. [DOI] [PubMed] [Google Scholar]
- 36.Hoebel BG. Brain neurotransmitters in food and drug reward. Am J Clin Nutr 42: 1133–1150, 1985. [DOI] [PubMed] [Google Scholar]
- 37.Davidson TL, Jarrard LE. A role for hippocampus in the utilization of hunger signals. Behav Neur Biol 59: 167–171, 1993. [DOI] [PubMed] [Google Scholar]
- 38.Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci U S A 77: 7380–7384, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinster RL, Chen HY, Trumbauer M, Senear AW, Warren R, Palmiter RD. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell 27: 223–231, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmiter RD, Brinster RL, Hammer RE, Trumbauer ME, Rosenfeld MG, Birnberg NC, Evans RM. Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature 300: 611–615, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans RM, Swanson L, Rosenfeld MG. Creation of transgenic animals to study development and as models for human disease. Recent Prog Horm Res 41: 317–337, 1985. doi: 10.1016/b978-0-12-571141-8.50011-1. [DOI] [PubMed] [Google Scholar]
- 42.Susulic VS, Frederich RC, Lawitts J, Tozzo E, Kahn BB, Harper ME, Himms-Hagen J, Flier JS, Lowell BB. Targeted disruption of the beta 3-adrenergic receptor gene. J Biol Chem 270: 29483–29492, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83: 1197–1209, 1995. [DOI] [PubMed] [Google Scholar]
- 44.Erickson JC, Hollopeter G, Palmiter RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274: 1704–1707, 1996. [DOI] [PubMed] [Google Scholar]
- 45.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88: 131–141, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Mantzoros CS, Qu D, Frederich RC, Susulic VS, Lowell BB, Maratos-Flier E, Flier JS. Activation of beta(3) adrenergic receptors suppresses leptin expression and mediates a leptin-independent inhibition of food intake in mice. Diabetes 45: 909–914, 1996. [DOI] [PubMed] [Google Scholar]
- 47.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang Cy, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123: 493–505, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 108: 1113–1121, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest 115: 3484–3493, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castonguay TW, Applegate EA, Upton DE, Stern JS. Hunger and appetite: old concepts/new distinctions. Nutr Rev 41: 101–110, 1983. doi: 10.1111/j.1753-4887.1983.tb07163.x. [DOI] [PubMed] [Google Scholar]
- 51.Berridge KC. Motivation concepts in behavioral neuroscience. Physiol Behav 81: 179–209, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Miller NE. Liberalization of basic S-R concepts: extensions to conflict behavior, motivation, and social learning. In: Psychology: A Study of a Science, edited by Koch S. New York: McGraw-Hill, 1959, p. 196–292. [Google Scholar]
- 53.Blundell JE, Hill AJ. Evaluating the satiating power of foods: implications for acceptance and consumption. In: Food Acceptance and Nutrition, edited by Solms JB, Pangbourne RM, Raunhardt O.. London: Academic Press, 1987, p. 205–219. [Google Scholar]
- 54.Kim CK, Adhikari A, Deisseroth K. Integration of optogenetics with complementary methodologies in systems neuroscience. Nat Rev Neurosci 18: 222–235, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bienkowski MS, Bowman I, Song MY, Gou L, Ard T, Cotter K, Zhu M, Benavidez NL, Yamashita S, Abu-Jaber J, Azam S, Lo D, Foster NN, Hintiryan H, Dong HW. Integration of gene expression and brain-wide connectivity reveals the multiscale organization of mouse hippocampal networks. Nat Neurosci 21: 1628–1623, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cazemier JL, Clascá F, Tiesinga PH. Connectomic analysis of brain networks: novel techniques and future directions. Front Neuroanat 10: 110, 2016. doi: 10.3389/fnana.2016.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winnubst J, Bas E, Ferreira TA, Wu Z, Economo MN, Edson P, Arthur BJ, Bruns C, Rokicki K, Schauder D, Olbris DJ, Murphy SD, Ackerman DG, Arshadi C, Baldwin P, Blake R, Elsayed A, Hasan M, Ramirez D, Dos Santos B, Weldon M, Zafar A, Dudman JT, Gerfen CR, Hantman AW, Korff W, Sternson SM, Spruston N, Svoboda K, Chandrashekar J. Reconstruction of 1,000 projection neurons reveals new cell types and organization of long-range connectivity in the mouse brain. Cell 179: 268–281, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ten Donkelaar HJ, Puelles L. Editorial: recent developments in neuroanatomical terminology. Front Neuroanat 13: 80, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swanson LW. Brain maps 4.0-structure of the rat brain: an open access atlas with global nervous system nomenclature ontology and flatmaps. J Comp Neurol 526: 935–943, 2018. doi: 10.1002/cne.24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swanson LW, Petrovich GD. What is the amygdala? Trends Neurosci 21: 323–331, 1998. [DOI] [PubMed] [Google Scholar]
- 61.Swanson LW. What is the brain? Trends Neurosci 23: 519–527, 2000. [DOI] [PubMed] [Google Scholar]
- 62.Swanson LW. Anatomy of the soul as reflected in the cerebral hemispheres: neural circuits underlying voluntary control of basic motivated behaviors. J Comp Neurol 493: 122–131, 2005. [DOI] [PubMed] [Google Scholar]
- 63.Swanson LW, Bota M. Foundational model of structural connectivity in the nervous system with a schema for wiring diagrams, connectome, and basic plan architecture. Proc Natl Acad Sci U S A 107: 20610–20617, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swanson LW. Neuroanatomical Terminology: a Lexicon of Classical Origins and Historical Foundations. Oxford: Oxford University Press, 2015. [Google Scholar]
- 65.Hahn JD, Swanson LW, Bowman I, Foster NN, Zingg B, Bienkowski MS, Hintiryan H, Dong HW. An open access mouse brain flatmap and upgraded rat and human brain flatmaps based on current reference atlases. J Comp Neurol 529: 576–594, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen Institute for Brain Science. Allen Mouse Brain Atlas. Seattle, WA: Allen Institute for Brain Science, 2008. [Google Scholar]
- 67.Cannon WB. Organization for physiological homeostasis. Physiol Rev 9: 399–431, 1929. [Google Scholar]
- 68.Cabanac M. Adjustable set point: to honor Harold T. Hammel. J Appl Physiol 100: 1338–1346, 2006. doi: 10.1152/japplphysiol.01021.2005. [DOI] [PubMed] [Google Scholar]
- 69.Woods SC, Ramsay DS. Homeostasis: beyond Curt Richter. Appetite 49: 388–398, 2007. doi: 10.1016/j.appet.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramsay DS, Woods SC. Clarifying the roles of homeostasis and allostasis in physiological regulation. Psychol Rev 121: 225–247, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brobeck JR. Exchange, control, and regulation. In: Physiological Controls and Regulations, edited by Yamamoto WS, Brobeck JR.. Philadelphia, PA: Saunders; 1965. p. 1–14. [Google Scholar]
- 72.Friedman MI. Food intake: control, regulation, and the illusion of dysregulation. In: Appetite and Food Intake-Behavioral and Physiological Considerations, edited by Harris RB, Mattes RD.. Boca Raton, FL: CRC Press, 2008, p. 1–19. [Google Scholar]
- 73.Wiener N. Cybernetics. Sci Am 179: 14–18, 1948. [DOI] [PubMed] [Google Scholar]
- 74.Wirtshafter D, Davis JD. Set points, settling points, and the control of body weight. Physiol Behav 19: 75–78, 1977. [DOI] [PubMed] [Google Scholar]
- 75.Ramsay DS, Woods SC. Physiological regulation: how it really works. Cell Metab 24: 361–364, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Geary N. Control-theory models of body-weight regulation and body-weight-regulatory appetite. Appetite 144: 104440, 2020. [DOI] [PubMed] [Google Scholar]
- 77.Geary N. A new way of looking at eating. Am J Physiol Regul Integr Comp Physiol 288: R1444–R1446, 2005. [DOI] [PubMed] [Google Scholar]
- 78.Moore-Ede MC. Physiology of the circadian timing system: predictive versus reactive homeostasis. Am J Physiol Regul Integr Comp Physiol 250: R737–R752, 1986. [DOI] [PubMed] [Google Scholar]
- 79.Dworkin BR. Learning and Physiological Regulation. Chicago, IL: University of Chicago Press, 1993. [Google Scholar]
- 80.Cannon WB. The Wisdom of the Body. New York: W. W. Norton & Company, 1932. [Google Scholar]
- 81.Krebs H. The Croonian Lecture, 1963. Gluconeogenesis. Proc R Soc Lond B Biol Sci 159: 545–564, 1964. [DOI] [PubMed] [Google Scholar]
- 82.Hardie DG. Keeping the home fires burning: AMP-activated protein kinase. J R Soc Interface 15: 20170774, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moore F, Weekes J, Hardie DG. Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur J Biochem 199: 691–697, 1991. [DOI] [PubMed] [Google Scholar]
- 84.Noda LA. Kinase. In: The Enzymes, edited by Boyer PD. New York: Academic Press, 1971, vol VIII, p. 279–305. [Google Scholar]
- 85.Schneider JE, Wise JD, Benton NA, Brozek JM, Keen-Rhinehart E. When do we eat? Ingestive behavior, survival, and reproductive success. Horm Behav 64: 702–728, 2013. [DOI] [PubMed] [Google Scholar]
- 86.Bartness TJ, Morley JE, Levine AS. Effects of food-deprivation and metabolic fuel utilization on the photoperiodic control of food-intake in Siberian hamsters. Physiol Behav 57: 61–68, 1995. [DOI] [PubMed] [Google Scholar]
- 87.Hill JW, Elias CF. Neuroanatomical framework of the metabolic control of reproduction. Physiol Rev 98: 2349–2380, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tornroth-Horsefield S, Neutze R. Opening and closing the metabolite gate. Proc Natl Acad Sci U S A 105: 19565–19566, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Flatt JP. Misconceptions in body weight regulation: implications for the obesity pandemic. Crit Rev Clin Lab Sci 49: 150–165, 2012. [DOI] [PubMed] [Google Scholar]
- 90.Schneider JE, Watts AG. Energy partitioning, ingestive behavior and reproductive success. In: Hormones, Brain, and Behavior, edited by Pfaff DW. New York: Academic Press, 2009, p. 205–258. [Google Scholar]
- 91.Booth DA. Postabsorptively induced suppression of appetite and the energostatic control of feeding. Physiol Behav 9: 199–202, 1972. [DOI] [PubMed] [Google Scholar]
- 92.Nicolaidis S, Rowland N. Metering of intravenous versus oral nutrients and regulation of energy balance. Am J Physiol 231: 661–668, 1976. [DOI] [PubMed] [Google Scholar]
- 93.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hardie DG. Organismal carbohydrate and lipid homeostasis. Cold Spring Harb Perspect Biol 4: a006031, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Okamoto S, Sato T, Tateyama M, Kageyama H, Maejima Y, Nakata M, Hirako S, Matsuo T, Kyaw S, Shiuchi T, Toda C, Sedbazar U, Saito K, Asgar NF, Zhang B, Yokota S, Kobayashi K, Foufelle F, Ferré P, Nakazato M, Masuzaki H, Shioda S, Yada T, Kahn BB, Minokoshi Y. Activation of AMPK-regulated CRH neurons in the PVH is sufficient and necessary to induce dietary preference for carbohydrate over fat. Cell Rep 22: 706–721, 2018. [DOI] [PubMed] [Google Scholar]
- 96.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25, 2005. [DOI] [PubMed] [Google Scholar]
- 97.Hayes MR, Skibicka KP, Bence KK, Grill HJ. Dorsal hindbrain 5′-adenosine monophosphate-activated protein kinase as an intracellular mediator of energy balance. Endocrinology 150: 2175–2182, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab 27: 299–313, 2018. [DOI] [PubMed] [Google Scholar]
- 99.Stark R, Ashley SE, Andrews ZB. AMPK and the neuroendocrine regulation of appetite and energy expenditure. Mol Cell Endocrinol 366: 215–223, 2013. [DOI] [PubMed] [Google Scholar]
- 100.Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146: 992–1003, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Langhans W, Leitner C, Arnold M. Dietary fat sensing via fatty acid oxidation in enterocytes: possible role in the control of eating. Am J Physiol Regul Integr Comp Physiol 300: R554–R565, 2011. [DOI] [PubMed] [Google Scholar]
- 102.Langhans W. The enterocyte as an energy flow sensor in the control of eating. Forum Nutr 63: 75–84, 2010. [DOI] [PubMed] [Google Scholar]
- 103.Donovan CM, Watts AG. Peripheral and central glucose sensing in hypoglycemic detection. Physiology (Bethesda) 29: 314–324, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niijima A. Afferent impulse discharges from glucoreceptors in the liver of the guinea pig. Ann N Y Acad Sci 157: 690–700, 1969. [DOI] [PubMed] [Google Scholar]
- 105.Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science 294: 1102–1105, 2001. [DOI] [PubMed] [Google Scholar]
- 106.Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol 4: 117–126, 2003. [DOI] [PubMed] [Google Scholar]
- 107.Morrison SF, Madden CJ, Tupone D. Central control of brown adipose tissue thermogenesis. Front Endocrinol (Lausanne) 3: 1–19, 2012. doi: 10.3389/fendo.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fromentin G, Darcel N, Chaumontet C, Marsset-Baglieri A, Nadkarni N, Tomé D. Peripheral and central mechanisms involved in the control of food intake by dietary amino acids and proteins. Nutr Res Rev 25: 29–39, 2012. doi: 10.1017/S0954422411000175. [DOI] [PubMed] [Google Scholar]
- 109.Romanovsky AA. Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. Am J Physiol Regul Integr Comp Physiol 292: R37–R46, 2007. doi: 10.1152/ajpregu.00668.2006. [DOI] [PubMed] [Google Scholar]
- 110.Keesey RE, Boyle PC, Kemnltz J, Mitchell JS. The role of the lateral hypothalamus in determining the body weight set point. In: Hunger: Basic Mechanisms and Clinical Implications, edited by Novin D, Wyrwicka W, Bray G.. New York: Raven Press, 1970. [Google Scholar]
- 111.Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proc R Soc Lond B Biol Sci 140: 578–596, 1953. [DOI] [PubMed] [Google Scholar]
- 112.Speakman JR, Levitsky DA, Allison DB, Bray MS, de Castro JM, Clegg DJ, Clapham JC, Dulloo AG, Gruer L, Haw S, Hebebrand J, Hetherington MM, Higgs S, Jebb SA, Loos RJ, Luckman S, Luke A, Mohammed-Ali V, O’Rahilly S, Pereira M, Perusse L, Robinson TN, Rolls B, Symonds ME, Westerterp-Plantenga MS. Westerterp-Plantenga MS. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech 4: 733–745, 2011. doi: 10.1242/dmm.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Harris RB, Kasser TR, Martin RJ. Dynamics of recovery of body composition after overfeeding, food restriction or starvation of mature female rats. J Nutr 116: 2536–2546, 1986. doi: 10.1093/jn/116.12.2536. [DOI] [PubMed] [Google Scholar]
- 114.Keesey RE, Hirvonen MD. Body weight set-points: determination and adjustment. J Nutr 127: 1875S–1883S, 1997. doi: 10.1093/jn/127.9.1875S. [DOI] [PubMed] [Google Scholar]
- 115.Egli G, Langhans W, Scharrer E. Selective hepatic vagotomy does not prevent compensatory feeding in response to body weight changes. J Auton Nerv Syst 15: 45–53, 1986. doi: 10.1016/0165-1838(86)90078-0. [DOI] [PubMed] [Google Scholar]
- 116.Heyman MB, Young VR, Fuss P, Tsay R, Joseph L, Roberts SB. Underfeeding and body weight regulation in normal-weight young men. Am J Physiol Regul Integr Comp Physiol 263: R250–R257, 1992. doi: 10.1152/ajpregu.1992.263.2.R250. [DOI] [PubMed] [Google Scholar]
- 117.Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med 332: 621–628, 1995. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- 118.Dulloo AG, Jacquet J, Girardier L. Poststarvation hyperphagia and body fat overshooting in humans: a role for feedback signals from lean and fat tissues. Am J Clin Nutr 65: 717–723, 1997. doi: 10.1093/ajcn/65.3.717. [DOI] [PubMed] [Google Scholar]
- 119.Polidori D, Sanghvi A, Seeley RJ, Hall KD. How strongly does appetite counter weight loss? quantification of the feedback control of human energy intake. Obesity (Silver Spring) 24: 2289–2295, 2016. doi: 10.1002/oby.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Speakman JR. Why lipostatic set point systems are unlikely to evolve. Mol Metab 7: 147–154, 2018. doi: 10.1016/j.molmet.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Juechems K, Balaguer J, Castanon Ruz HS, O’Reilly M, Summerfield JX. A network for computing value equilibrium in the human medial prefrontal cortx. Neuron 101: 977–987, 2019. doi: 10.1016/j.neuron.2018.12.029. [DOI] [PubMed] [Google Scholar]
- 122.Juechems K, Summerfield C. Where does value come from? Trends Cogn Sci 23: 836–850, 2019. doi: 10.1016/j.tics.2019.07.012. [DOI] [PubMed] [Google Scholar]
- 123.Livneh Y, Sugden AU, Madara JC, Essner RA, Flores VI, Sugden LA, Resch JM, Lowell BB, Andermann ML. Estimation of current and future physiological states in insular cortex. Neuron 105: 1094–1111, 2020. doi: 10.1016/j.neuron.2019.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.de Araujo IE, Schatzker M, Small DM. Rethinking food reward. Annu Rev Psychol 71: 139–164, 2020. doi: 10.1146/annurev-psych-122216-011643. [DOI] [PubMed] [Google Scholar]
- 125.Kenny PJ. Reward mechanisms in obesity: new insights and future directions. Neuron 69: 664–679, 2011. doi: 10.1016/j.neuron.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu CM, Kanoski SE. Homeostatic and non-homeostatic controls of feeding behavior: distinct vs. common neural systems. Physiol Behav 193: 223–231, 2018. doi: 10.1016/j.physbeh.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rescorla RA. The role of information about the response-outcome relation in instrumental discrimination learning. J Exp Psychol Anim Behav Process 16: 262–270, 1990. doi: 10.1037/0097-7403.16.3.262. [DOI] [PubMed] [Google Scholar]
- 128.Colwill RM, Rescorla RA. Effect of reinforcer devaluation on discriminative control of instrumental behavior. J Exp Psychol Anim Behav Process 16: 40–47, 1990. doi: 10.1037/0097-7403.16.1.40. [DOI] [PubMed] [Google Scholar]
- 129.Balleine BW, Incentive processes in instrumental conditioning. In: Handbook of Contemporary Learning Theories, edited by Mowrer RR, Klein SB. Mahwah, NJ: Lawrence Erlbaum Associates, 2001, p. 307–366. [Google Scholar]
- 130.Balleine BW, Dickinson A. The role of incentive learning in instrumental outcome revaluation by sensory-specific satiety. Anim Learn Behav 26: 46–59, 1998. doi: 10.3758/BF03199161. [DOI] [Google Scholar]
- 131.Balleine BW, Dickinson A. Goal-directed instrumental action: contingency and incentive learning and their cortical substrates. Neuropharmacology 37: 407–419, 1998. doi: 10.1016/S0028-3908(98)00033-1. [DOI] [PubMed] [Google Scholar]
- 132.Balleine B. Instrumental performance following a shift in primary motivation depends on incentive learning. J Exp Psychol Anim Behav Process 18: 236–250, 1992. doi: 10.1037/0097-7403.18.3.236. [DOI] [PubMed] [Google Scholar]
- 133.Berridge KC. The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacol (Berl) 191: 391–431, 2007. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- 134.Hsu TM, McCutcheon JE, Roitman MF. Parallels and overlap: the integration of homeostatic signals by mesolimbic dopamine neurons. Front Psychiatry 9: 410, 2018. doi: 10.3389/fpsyt.2018.00410,10.3389/fpsyg.2018.00410,10.3389/fphys.2018.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hernandez L, Stanley BG, Hoebel BG. A small, removable microdialysis probe. Life Sci 39: 2629–2637, 1986. doi: 10.1016/0024-3205(86)90119-0. [DOI] [PubMed] [Google Scholar]
- 136.Roitman MF, Stuber GD, Phillips PE, Wightman RM, Carelli RM. Dopamine operates as a subsecond modulator of food seeking. J Neurosci 24: 1265–1271, 2004. doi: 10.1523/JNEUROSCI.3823-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hernandez L, Hoebel BG. Food reward and cocaine increase extracellular dopamine in the nucleus accumbens as measured by microdialysis. Life Sci 42: 1705–1712, 1988. doi: 10.1016/0024-3205(88)90036-7. [DOI] [PubMed] [Google Scholar]
- 138.Hernandez L, Hoebel BG. Feeding and hypothalamic stimulation increase dopamine turnover in the accumbens. Physiol Behav 44: 599–606, 1988. doi: 10.1016/0031-9384(88)90324-1. [DOI] [PubMed] [Google Scholar]
- 139.Cagniard B, Balsam PD, Brunner D, Zhuang X. Mice with chronically elevated dopamine exhibit enhanced motivation, but not learning, for a food reward. Neuropsychopharmacology 31: 1362–1370, 2006. [DOI] [PubMed] [Google Scholar]
- 140.Pecina S, Cagniard B, Berridge KC, Aldridge JW, Zhuang X. Hyperdopaminergic mutant mice have higher “wanting” but not “liking” for sweet rewards. J Neurosci 23: 9395–9402, 2003. doi: 10.1523/JNEUROSCI.23-28-09395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yin HH, Zhuang X, Balleine BW. Instrumental learning in hyperdopaminergic mice. Neurobiol Learn Mem 85: 283–288, 2006. doi: 10.1016/j.nlm.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 142.Salamone JD, Correa M, Ferrigno S, Yang JH, Rotolo RA, Presby RE. The psychopharmacology of effort-related decision making: dopamine, adenosine, and insights into the neurochemistry of motivation. Pharmacol Rev 70: 747–762, 2018. doi: 10.1124/pr.117.015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wilson C, Nomikos GG, Collu M, Fibiger HC. Dopaminergic correlates of motivated behavior: importance of drive. J Neurosci 15: 5169–5178, 1995. doi: 10.1523/JNEUROSCI.15-07-05169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cone JJ, McCutcheon JE, Roitman MF. Ghrelin acts as an interface between physiological state and phasic dopamine signaling. J Neurosci 34: 4905–4913, 2014. doi: 10.1523/JNEUROSCI.4404-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chiacchierini G, Naneix F, Peters KZ, Apergis-Schoute J, Snoeren EM, McCutcheon JE. Protein appetite drives macronutrient-related differences in ventral tegmental area neural activity. J Neurosci 41: 5080–5092, 2021. doi: 10.1523/JNEUROSCI.3082-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tellez LA, Han W, Zhang X, Ferreira TL, Perez IO, Shammah-Lagnado SJ, van den Pol AN, de Araujo IE. Separate circuitries encode the hedonic and nutritional values of sugar. Nat Neurosci 19: 465–470, 2016. doi: 10.1038/nn.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tellez LA, Ren X, Han W, Medina S, Ferreira JG, Yeckel CW, de Araujo IE. Glucose utilization rates regulate intake levels of artificial sweeteners. J Physiol 591: 5727–5744, 2013. doi: 10.1113/jphysiol.2013.263103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Small DM, Jones-Gotman M, Dagher A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage 19: 1709–1715, 2003. doi: 10.1016/S1053-8119(03)00253-2. [DOI] [PubMed] [Google Scholar]
- 149.Thanarajah SE, Backes H, DiFeliceantonio AG, Albus K, Cremer AL, Hanssen R, Lippert RN, Cornely OA, Small DM, Bruning JC, Tittgemeyer M. Food intake recruits orosensory and post-ingestive dopaminergic circuits to affect eating desire in humans. Cell Metab 29: 695–706, 2019. doi: 10.1016/j.cmet.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 150.Azzara AV, Bodnar RJ, Delamater AR, Sclafani A. D1 but not D2 dopamine receptor antagonism blocks the acquisition of a flavor preference conditioned by intragastric carbohydrate infusions. Pharmacol Biochem Behav 68: 709–720, 2001. doi: 10.1016/S0091-3057(01)00484-1. [DOI] [PubMed] [Google Scholar]
- 151.Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, Neve R, Wildes CP, Tye KM. Decoding neural circuits that control compulsive sucrose seeking. Cell 160: 528–541, 2015. doi: 10.1016/j.cell.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Grill HJ, Norgren R. The taste reactivity test. I. Mimetic responses to gustatory stimuli in neurologically normal rats. Brain Res 143: 263–279, 1978. doi: 10.1016/0006-8993(78)90568-1. [DOI] [PubMed] [Google Scholar]
- 153.Grill HJ, Norgren R. The taste reactivity test. II. Mimetic responses to gustatory stimuli in chronic thalamic and chronic decerebrate rats. Brain Res 143: 281–297, 1978. doi: 10.1016/0006-8993(78)90569-3. [DOI] [PubMed] [Google Scholar]
- 154.Berridge KC, Flynn FW, Schulkin J, Grill HJ. Sodium depletion enhances salt palatability in rats. Behav Neurosci 98: 652–660, 1984. doi: 10.1037/0735-7044.98.4.652. [DOI] [PubMed] [Google Scholar]
- 155.Berridge KC, Schulkin J. Palatability shift of a salt-associated incentive during sodium depletion. Q J Exp Psychol B 41: 121–138, 1989. [PubMed] [Google Scholar]
- 156.Berridge KC. Modulation of taste affect by hunger, caloric satiety, and sensory-specific satiety in the rat. Appetite 16: 103–120, 1991. doi: 10.1016/0195-6663(91)90036-R. [DOI] [PubMed] [Google Scholar]
- 157.Finlayson G, King N, Blundell JE. Is it possible to dissociate ‘liking’ and ‘wanting’ for foods in humans? A novel experimental procedure. Physiol Behav 90: 36–42, 2007. doi: 10.1016/j.physbeh.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 158.Levine AS, Billington CJ. Opioids. Are they regulators of feeding? Ann N Y Acad Sci 575: 209–219, 1989. doi: 10.1111/j.1749-6632.1989.tb53244.x. [DOI] [PubMed] [Google Scholar]
- 159.Hoebel BG. Neurotransmitters in the control of feeding and its rewards: monoamines, opiates, and brain-gut peptides. Res Publ Assoc Res Nerv Ment Dis 62: 15–38, 1984. [PubMed] [Google Scholar]
- 160.Bakshi VP, Kelley AE. Feeding induced by opioid stimulation of the ventral striatum: role of opiate receptor subtypes. J Pharmacol Exp Ther 265: 1253–1260, 1993. [PubMed] [Google Scholar]
- 161.Leibowitz SF. Brain neurotransmitters and appetite regulation. Psychopharmacol Bull 21: 412–418, 1985. [PubMed] [Google Scholar]
- 162.Gosnell BA, Krahn DD, Majchrzak MJ. The effects of morphine on diet selection are dependent upon baseline diet preferences. Pharmacol Biochem Behav 37: 207–212, 1990. doi: 10.1016/0091-3057(90)90322-9. [DOI] [PubMed] [Google Scholar]
- 163.Naleid AM, Grace MK, Chimukangara M, Billington CJ, Levine AS. Paraventricular opioids alter intake of high-fat but not high-sucrose diet depending on diet preference in a binge model of feeding. Am J Physiol Regul Integr Comp Physiol 293: R99–R105, 2007. doi: 10.1152/ajpregu.00675.2006. [DOI] [PubMed] [Google Scholar]
- 164.Zhang M, Gosnell BA, Kelley AE. Intake of high-fat food is selectively enhanced by mu opioid receptor stimulation within the nucleus accumbens. J Pharmacol Exp Ther 285: 908–914, 1998. [PubMed] [Google Scholar]
- 165.Pecina S, Berridge KC. Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J Neurosci 25: 11777–11786, 2005. doi: 10.1523/JNEUROSCI.2329-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Castro DC, Berridge KC. Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting”. J Neurosci 34: 4239–4250, 2014. doi: 10.1523/JNEUROSCI.4458-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Smith KS, Berridge KC. The ventral pallidum and hedonic reward: neurochemical maps of sucrose “liking” and food intake. J Neurosci 25: 8637–8649, 2005. doi: 10.1523/JNEUROSCI.1902-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Benoit SC, Davis JF, Davidson TL. Learned and cognitive controls of food intake. Brain Res 1350: 71–76, 2010. doi: 10.1016/j.brainres.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Davidson TL. Hunger cues as modulatory stimuli. In: Occasion Setting: Associative Learning and Cognition in Animals, edited by Schmajuk NA, Holland PC.. Washington, DC: American Psychological Association, p. 223–248, 1998. [Google Scholar]
- 170.Davidson TL, Kanoski SE, Schier LA, Clegg DJ, Benoit SC. A potential role for the hippocampus in energy intake and body weight regulation. Curr Opin Pharmacol 7: 613–616, 2007. doi: 10.1016/j.coph.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kanoski SE. Cognitive and neuronal systems underlying obesity. Physiol Behav 106: 337–344, 2012. doi: 10.1016/j.physbeh.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Giustino TF, Maren S. The role of the medial prefrontal cortex in the conditioning and extinction of fear. Front Behav Neurosci 9: 298, 2015. doi: 10.3389/fnbeh.2015.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yizhar O, Klavir O. Reciprocal amygdala-prefrontal interactions in learning. Curr Opin Neurobiol 52: 149–155, 2018. doi: 10.1016/j.conb.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 174.Dalley JW, Robbins TW. Fractionating impulsivity: neuropsychiatric implications. Nat Rev Neurosci 18: 158–171, 2017. doi: 10.1038/nrn.2017.8,10.1038/nrn.2016.158. [DOI] [PubMed] [Google Scholar]
- 175.Guerrieri R, Nederkoorn C, Jansen A. How impulsiveness and variety influence food intake in a sample of healthy women. Appetite 48: 119–122, 2007. doi: 10.1016/j.appet.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 176.Guerrieri R, Nederkoorn C, Jansen A. The interaction between impulsivity and a varied food environment: its influence on food intake and overweight. Int J Obes 32: 708–714, 2008. doi: 10.1038/sj.ijo.0803770. [DOI] [PubMed] [Google Scholar]
- 177.Giel KE, Teufel M, Junne F, Zipfel S, Schag K. Food-related impulsivity in obesity and binge eating disorder-a systematic update of the evidence. Nutrients 9: 1170, 2017. doi: 10.3390/nu9111170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nederkoorn C, Houben K, Hofmann W, Roefs A, Jansen A. Control yourself or just eat what you like? Weight gain over a year is predicted by an interactive effect of response inhibition and implicit preference for snack foods. Health Psychol 29: 389–393, 2010. doi: 10.1037/a0019921. [DOI] [PubMed] [Google Scholar]
- 179.Mole TB, Irvine MA, Worbe Y, Collins P, Mitchell SP, Bolton S, Harrison NA, Robbins TW, Voon V. Impulsivity in disorders of food and drug misuse. Psychol Med 45: 771–782, 2015. doi: 10.1017/S0033291714001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Amlung M, Petker T, Jackson J, Balodis I, MacKillop J. Steep discounting of delayed monetary and food rewards in obesity: a meta-analysis. Psychol Med 46: 2423–2434, 2016. doi: 10.1017/S0033291716000866. [DOI] [PubMed] [Google Scholar]
- 181.MacKillop J, Weafer J, C Gray J, Oshri A, Palmer A, de Wit H. The latent structure of impulsivity: impulsive choice, impulsive action, and impulsive personality traits. Psychopharmacology (Berl) 233: 3361–3370, 2016. doi: 10.1007/s00213-016-4372-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Winstanley CA. The utility of rat models of impulsivity in developing pharmacotherapies for impulse control disorders. Brit J Pharmacol 164: 1301–1321, 2011. doi: 10.1111/j.1476-5381.2011.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Broos N, Schmaal L, Wiskerke J, Kostelijk L, Lam T, Stoop N, Weierink L, Ham J, de Geus EJ, Schoffelmeer AN, van den Brink W, Veltman DJ, de Vries TJ, Pattij T, Goudriaan AE. The relationship between impulsive choice and impulsive action: a cross-species translational study. PLoS One 7: e36781, 2012. doi: 10.1371/journal.pone.0036781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dalley JW, Everitt BJ, Robbins TW. Impulsivity, compulsivity, and top-down cognitive control. Neuron 69: 680–694, 2011. doi: 10.1016/j.neuron.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 185.Baldo BA. Prefrontal cortical opioids and dysregulated motivation: a network hypothesis. Trends Neurosci 39: 366–377, 2016. doi: 10.1016/j.tins.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Selleck RA, Baldo BA. Feeding-modulatory effects of mu-opioids in the medial prefrontal cortex: a review of recent findings and comparison to opioid actions in the nucleus accumbens. Psychopharmacology (Berl) 234: 1439–1449, 2017. doi: 10.1007/s00213-016-4522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Torregrossa MM, Quinn JJ, Taylor JR. Impulsivity, compulsivity, and habit: the role of orbitofrontal cortex revisited. Biol Psychiatry 63: 253–255, 2008. doi: 10.1016/j.biopsych.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Caprioli D, Sawiak SJ, Merlo E, Theobald DE, Spoelder M, Jupp B, Voon V, Carpenter TA, Everitt BJ, Robbins TW, Dalley JW. Gamma aminobutyric acidergic and neuronal structural markers in the nucleus accumbens core underlie trait-like impulsive behavior. Biol Psych 75: 115–123, 2014. doi: 10.1016/j.biopsych.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Selleck RA, Lake C, Estrada V, Riederer J, Andrzejewski M, Sadeghian K, Baldo BA. Endogenous opioid signaling in the medial prefrontal cortex is required for the expression of hunger-induced impulsive action. Neuropsychopharmacology 40: 2464–2474, 2015. doi: 10.1038/npp.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Velazquez-Sanchez C, Ferragud A, Moore CF, Everitt BJ, Sabino V, Cottone P. High trait impulsivity predicts food addiction-like behavior in the rat. Neuropsychopharmacology 39: 2463–2472, 2014. doi: 10.1038/npp.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chudasama Y, Doobay VM, Liu Y. Hippocampal-prefrontal cortical circuit mediates inhibitory response control in the rat. J Neurosci 32: 10915–10924, 2012. doi: 10.1523/JNEUROSCI.1463-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hsu TM, Noble EE, Liu CM, Cortella AM, Konanur VR, Suarez AN, Reiner DJ, Hahn JD, Hayes MR, Kanoski SE. A hippocampus to prefrontal cortex neural pathway inhibits food motivation through glucagon-like peptide-1 signaling. Mol Psychiatry 23: 1555–1565, 2018. doi: 10.1038/mp.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Noble EE, Wang Z, Liu CM, Davis EA, Suarez AN, Stein LM, Tsan L, Terrill SJ, Hsu TM, Jung AH, Raycraft LM, Hahn JD, Darvas M, Cortella AM, Schier LA, Johnson AW, Hayes MR, Holschneider DP, Kanoski SE. Hypothalamus-hippocampus circuitry regulates impulsivity via melanin-concentrating hormone. Nature Commun 10: 4923, 2019. doi: 10.1038/s41467-019-12895-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kekic M, McClelland J, Bartholdy S, Boysen E, Musiat P, Dalton B, Tiza M, David AS, Campbell IC, Schmidt U. Single-session transcranial direct current stimulation temporarily improves symptoms, mood, and self-regulatory control in bulimia nervosa: a randomised controlled trial. PLoS One 12: e0167606, 2017. doi: 10.1371/journal.pone.0167606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sharkey RJ, Bourque J, Larcher K, Misic B, Zhang Y, Altinkaya A, Sadikot A, Conrod P, Evans AC, Garavan H, Leyton M, Seguin JR, Pihl R, Dagher A. Mesolimbic connectivity signatures of impulsivity and BMI in early adolescence. Appetite 132: 25–36, 2019. doi: 10.1016/j.appet.2018.09.019. [DOI] [PubMed] [Google Scholar]
- 196.Agnati LF, Leo G, Zanardi A, Genedani S, Rivera A, Fuxe K, Guidolin D. Volume transmission and wiring transmission from cellular to molecular networks: history and perspectives. Acta Physiol (Oxf) 187: 329–344, 2006. doi: 10.1111/j.1748-1716.2006.01579.x. [DOI] [PubMed] [Google Scholar]
- 197.Agnati LF, Marcoli M, Maura G, Woods A, Guidolin D. The brain as a ‘hyper-network’: the key role of neural networks as main producers of the integrated brain actions especially via the ‘broadcasted’ neuroconnectomics. J Neural Transm (Vienna) 125: 883–897, 2018. doi: 10.1007/s00702-018-1855-7. [DOI] [PubMed] [Google Scholar]
- 198.Hökfelt T, Barde S, Xu ZQ, Kuteeva E, Rüegg J, Le Maitre E, Risling M, Kehr J, Ihnatko R, Theodorsson E, Palkovits M, Deakin W, Bagdy G, Juhasz G, Prud’homme HJ, Mechawar N, Diaz-Heijtz R, Ögren SO. Neuropeptide and small transmitter coexistence: fundamental studies and relevance to mental illness. Front Neural Circuits 12: 106, 2018. doi: 10.3389/fncir.2018.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Smith SJ, Sümbül U, Graybuck LT, Collman F, Seshamani S, Gala R, Gliko O, Elabbady L, Miller JA, Bakken TE, Rossier J, Yao Z, Lein E, Zeng H, Tasic B, Hawrylycz M. Single-cell transcriptomic evidence for dense intracortical neuropeptide networks. Elife 8: e47889, 2019. doi: 10.7554/elife.47889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Noble EE, Hahn JD, Konanur VR, Hsu TM, Page SJ, Cortella AM, Liu CM, Song MY, Suarez AN, Szujewski CC, Rider D, Clarke JE, Darvas M, Appleyard SM, Kanoski SE. Control of feeding behavior by cerebral ventricular volume transmission of melanin-concentrating hormone. Cell Metab 28: 55–68, 2018. doi: 10.1016/j.cmet.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ludwig M, Apps D, Menzies J, Patel JC, Rice ME. Dendritic release of neurotransmitters. Compr Physiol 7: 235–252, 2016. doi: 10.1002/cphy.c160007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Brierley DI, Holt MK, Singh A, de Araujo A, McDougle M, Vergara M, Afaghani MH, Lee SJ, Scott K, Maske C, Langhans W, Krause E, de Kloet A, Gribble FM, Reimann F, Rinaman L, de Lartigue G, Trapp S. Central and peripheral GLP-1 systems independently suppress eating. Nat Metab 3: 258–273, 2021. doi: 10.1038/s42255-021-00344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Langhans WA, Arnold M, Banks WA, Card JP, Dailey MJ, Daniels D, de Kloet AD, et al. New horizons for future research-critical issues to consider for maximizing research excellence and impact. Mol Metab 14: 53–59, 2018. doi: 10.1016/j.molmet.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ding K, Han Y, Seid TW, Buser C, Karigo T, Zhang S, Dickman DK, Anderson DJ. Imaging neuropeptide release at synapses with a genetically engineered reporter. Elife 8: e46421, 2019. doi: 10.7554/elife.46421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Patriarchi T, Cho JR, Merten K, Marley A, Broussard GJ, Liang R, Williams J, Nimmerjahn A, von Zastrow M, Gradinaru V, Tian L. Imaging neuromodulators with high spatiotemporal resolution using genetically encoded indicators. Nat Protoc 14: 3471–3505, 2019. doi: 10.1038/s41596-019-0239-2. [DOI] [PubMed] [Google Scholar]
- 206.Sun F, Zeng J, Jing M, Zhou J, Feng J, Owen SF, Luo Y, Li F, Wang H, Yamaguchi T, Yong Z, Gao Y, Peng W, Wang L, Zhang S, Du J, Lin D, Xu M, Kreitzer AC, Cui G, Li Y. A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell 174: 481–496, 2018. doi: 10.1016/j.cell.2018.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Agnati LF, Fuxe K, Zoli M, Ozini I, Toffano G, Ferraguti F. A correlation analysis of the regional distribution of central enkephalin and beta-endorphin immunoreactive terminals and of opiate receptors in adult and old male rats. Evidence for the existence of two main types of communication in the central nervous system: the volume transmission and the wiring transmission. Acta Physiol Scand 128: 201–207, 1986. doi: 10.1111/j.1748-1716.1986.tb07967.x. [DOI] [PubMed] [Google Scholar]
- 208.Bjorefeldt A, Andreasson U, Daborg J, Riebe I, Wasling P, Zetterberg H, Hanse E. Human cerebrospinal fluid increases the excitability of pyramidal neurons in the in vitro brain slice. J Physiol 593: 231–243, 2015. doi: 10.1113/jphysiol.2014.284711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Palacios JM, Dietl MM. Regulatory peptide receptors: visualization by autoradiography. Experientia 43: 750–761, 1987. doi: 10.1007/BF01945352. [DOI] [PubMed] [Google Scholar]
- 210.Tobin V, Leng G, Ludwig M. The involvement of actin, calcium channels and exocytosis proteins in somato-dendritic oxytocin and vasopressin release. Front Physiol 3: 261, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tobin VA, Douglas AJ, Leng G, Ludwig M. The involvement of voltage-operated calcium channels in somato-dendritic oxytocin release. PLoS One 6: e25366, 2011. doi: 10.1371/journal.pone.0025366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Leng G, Ludwig M, Jacques Benoit Lecture. Information processing in the hypothalamus: peptides and analogue computation. J Neuroendocrinol 18: 379–392, 2006. doi: 10.1111/j.1365-2826.2006.01428.x. [DOI] [PubMed] [Google Scholar]
- 213.Sabatier N, Leng G, Menzies J. Oxytocin, feeding, and satiety. Front Endocrinol (Lausanne) 4: 35, 2013. doi: 10.3389/fendo.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Veening JG, de JT, Barendregt HP. Oxytocin-messages via the cerebrospinal fluid: behavioral effects; a review. Physiol Behav 101: 193–210, 2010. [DOI] [PubMed] [Google Scholar]
- 215.Hoistad M, Samskog J, Jacobsen KX, Olsson A, Hansson HA, Brodin E, Fuxe K. Detection of beta-endorphin in the cerebrospinal fluid after intrastriatal microinjection into the rat brain. Brain Res 1041: 167–180, 2005. doi: 10.1016/j.brainres.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 216.MacMillan SJ, Mark MA, Duggan AW. The release of beta-endorphin and the neuropeptide-receptor mismatch in the brain. Brain Res 794: 127–136, 1998. [DOI] [PubMed] [Google Scholar]
- 217.Skinner DC, Malpaux B. High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood recirculating through the choroid plexus. Endocrinology 140: 4399–4405, 1999. doi: 10.1210/endo.140.10.7074. [DOI] [PubMed] [Google Scholar]
- 218.Caraty A, Skinner DC. Gonadotropin-releasing hormone in third ventricular cerebrospinal fluid: endogenous distribution and exogenous uptake. Endocrinology 149: 5227–5234, 2008. doi: 10.1210/en.2007-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Veening JG, Barendregt HP. The regulation of brain states by neuroactive substances distributed via the cerebrospinal fluid; a review. Cerebrospinal Fluid Res 7: 1, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Burghardt GM, Burkhardt RW. Wallace Craig’s appetites and aversions as constituents of instincts: a centennial appreciation. J Comp Psychol 132: 361–372, 2018. doi: 10.1037/com0000155. [DOI] [PubMed] [Google Scholar]
- 221.Craig W. Appetites and aversions as constituents of instincts. Proc Natl Acad Sci U S A 3: 685–688, 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Andermann ML, Lowell BB. Toward a wiring diagram understanding of appetite control. Neuron 95: 757–778, 2017. doi: 10.1016/j.neuron.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Carpenter RH. Homeostasis: a plea for a unified approach. Adv Physiol Educ 28: 180–187, 2004. doi: 10.1152/advan.00012.2004. [DOI] [PubMed] [Google Scholar]
- 224.Zorrilla EP, Inoue K, Fekete EM, Tabarin A, Valdez GR, Koob GF. Measuring meals: structure of prandial food and water intake of rats. Am J Physiol Regul Integr Comp Physiol 288: R1450–R1467, 2005. [DOI] [PubMed] [Google Scholar]
- 225.Boyle CN, Lorenzen SM, Compton D, Watts AG. Dehydration-anorexia derives from a reduction in meal size, but not meal number. Physiol Behav 105: 305–314, 2012. doi: 10.1016/j.physbeh.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Smith JC. Microstructure of the rat’s intake of food, sucrose and saccharin in 24-hour tests. Neurosci Biobehav Rev 24: 199–212, 2000. doi: 10.1016/S0149-7634(99)00073-1. [DOI] [PubMed] [Google Scholar]
- 227.Clifton PG. Meal patterning in rodents: psychopharmacological and neuroanatomical studies. Neurosci Biobehav Rev 24: 213–222, 2000. doi: 10.1016/S0149-7634(99)00074-3. [DOI] [PubMed] [Google Scholar]
- 228.Hsu TM, Noble EE, Reiner DJ, Liu CM, Suarez AN, Konanur VR, Hayes MR, Kanoski SE. Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology 131: 487–496, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ralston SL, Van den Broek G, Baile CA. Feed intake patterns and associated blood glucose, free fatty acid and insulin changes in ponies. J Anim Sci 49: 838–845, 1979. [DOI] [PubMed] [Google Scholar]
- 230.Collier G, Johnson DF, Morgan C. Meal patterns of cats encountering variable food procurement cost. J Exp Anal Behav 67: 303–310, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Geiselman PJ, Martin JR, VanderWeele DA, Novin D. Multivariate analysis of meal patterning in intact and vagotomized rabbits. J Comp Physiol Psychol 94: 388–399, 1980. [DOI] [PubMed] [Google Scholar]
- 232.Morgan CA, Emmans GC, Tolkamp BJ, Kyriazakis I. Analysis of the feeding behavior of pigs using different models. Physiol Behav 68: 395–403, 2000. [DOI] [PubMed] [Google Scholar]
- 233.Durst B, Senn M, Langhans W. Eating patterns of lactating dairy cows of three different breeds fed grass ad lib. Physiol Behav 54: 625–631, 1993. doi: 10.1016/0031-9384(93)90069-R. [DOI] [PubMed] [Google Scholar]
- 234.Senn M, Langhans W, Scharrer E. Meal patterns of pygmy goats fed hay and concentrate ad lib. Physiol Behav 48: 49–53, 1990. [DOI] [PubMed] [Google Scholar]
- 235.Leech RM, Worsley A, Timperio A, McNaughton SA. Understanding meal patterns: definitions, methodology and impact on nutrient intake and diet quality. Nutr Res Rev 28: 1–21, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Grill HJ. Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity (Silver Spring) 14: 216S–221S, 2006. doi: 10.1038/oby.2006.312. [DOI] [PubMed] [Google Scholar]
- 237.Bartness TJ, Keen-Rhinehart E, Dailey MJ, Teubner BJ. Neural and hormonal control of food hoarding. Am J Physiol Regul Integr Comp Physiol 301: R641–R655, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Levitsky DA, DeRosimo L. One day of food restriction does not result in an increase in subsequent daily food intake in humans. Physiol Behav 99: 495–499, 2010. doi: 10.1016/j.physbeh.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 239.Strubbe JH, Woods SC. The timing of meals. Psychol Rev 111: 128–141, 2004. [DOI] [PubMed] [Google Scholar]
- 240.Louis-Sylvestre J, Le Magnen J. Fall in blood glucose level precedes meal onset in free-feeding rats. Neurosci Biobehav Rev 4: 13–15, 1980. doi: 10.1016/0149-7634(80)90041-X. [DOI] [PubMed] [Google Scholar]
- 241.Louis-Sylvestre J, Le Magnen J. A fall in blood glucose level precedes meal onset in free-feeding rats. Obesity Res 4: 497–500, 1996. doi: 10.1002/j.1550-8528.1996.tb00261.x. [DOI] [PubMed] [Google Scholar]
- 242.Melanson KJ, Westerterp-Plantenga MS, Campfield LA, Saris WH. Blood glucose and meal patterns in time-blinded males, after aspartame, carbohydrate, and fat consumption, in relation to sweetness perception. Br J Nutr 82: 437–446, 1999. doi: 10.1017/S0007114599001695. [DOI] [PubMed] [Google Scholar]
- 243.Nicolaidis S, Even P. Physiological determinant of hunger, satiation, and satiety. Am J Clin Nutr 42: 1083–1092, 1985. [DOI] [PubMed] [Google Scholar]
- 244.de Castro JM. The control of food intake of free-living humans: putting the pieces back together. Physiol Behav 100: 446–453, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Petrovich GD. Feeding behavior survival circuit: anticipation & competition. Curr Opin Behavi Sci 24: 137–142, 2018. doi: 10.1016/j.cobeha.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Fulton S. Appetite and reward. Front Neuroendoc 31: 85–103, 2010. doi: 10.1016/j.yfrne.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 247.Kanoski SE, Grill HJ. Hippocampus contributions to food intake control: mnemonic, neuroanatomical, and endocrine mechanisms. Biol Psychiatry 81: 748–756, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.DA L, Pacanowski CR. Free will and the obesity epidemic. Public Health Nutr 15: 126–141, 2012. doi: 10.1017/S1368980011002187. [DOI] [PubMed] [Google Scholar]
- 249.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schurmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Muller TD, Tschop MH. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 14: 140–162, 2018. [DOI] [PubMed] [Google Scholar]
- 250.Kondrakiewicz K, Kostecki M, Szadzińska W, Knapska E. Ecological validity of social interaction tests in rats and mice. Genes Brain Behav 18: e12525, 2019. [DOI] [PubMed] [Google Scholar]
- 251.Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521, 2004. [DOI] [PubMed] [Google Scholar]
- 252.Hok V, Poucet B, Duvelle E, Save E, Sargolini F. Spatial cognition in mice and rats: similarities and differences in brain and behavior. Wiley Interdiscip Rev Cogn Sci 7: 406–421, 2016. [DOI] [PubMed] [Google Scholar]
- 253.Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, et al. A mesoscale connectome of the mouse brain. Nature 508: 207–214, 2014. doi: 10.1038/nature13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Gasparini S, Howland JM, Thatcher AJ, Geerling JC. Central afferents to the nucleus of the solitary tract in rats and mice. J Comp Neurol 528: 2708–2728, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gasparini S, Resch JM, Gore AM, Peltekian L, Geerling JC. Pre-locus coeruleus neurons in rat and mouse. Am J Physiol Regul Integr Comp Physiol. 320: R342–R361, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Whitesell JD, Liska A, Coletta L, Hirokawa KE, Bohn P, Williford A, Groblewski PA, Graddis N, Kuan L, Knox JE, Ho A, Wakeman W, Nicovich PR, Nguyen TN, van Velthoven CT, Garren E, Fong O, Naeemi M, Henry AM, Dee N, Smith KA, Levi B, Feng D, Ng L, Tasic B, Zeng H, Mihalas S, Gozzi A, Harris JA. Regional, layer, and cell-type-specific connectivity of the mouse default mode network. Neuron 109: 545–559, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zingg B, Hintiryan H, Gou L, Song MY, Bay M, Bienkowski MS, Foster NN, Yamashita S, Bowman I, Toga AW, Dong HW. Neural networks of the mouse neocortex. Cell 156: 1096–1111, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Benavidez NL, Bienkowski MS, Zhu M, Garcia LH, Fayzullina M, Gao L, Bowman I, Gou L, Khanjani N, Cotter KR, Korobkova L, Becerra M, Cao C, Song MY, Zhang B, Yamashita S, Tugangui AJ, Zingg B, Rose K, Lo D, Foster NN, Boesen T, Mun HS, Aquino S, Wickersham IR, Ascoli GA, Hintiryan H, Dong HW. Organization of the inputs and outputs of the mouse superior colliculus. Nature Commun 12: 4004, 2021. doi: 10.1038/s41467-021-24241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Simmons DM, Swanson LW. Comparison of the spatial distribution of seven types of neuroendocrine neurons in the rat paraventricular nucleus: toward a global 3D model. J Comp Neurol 516: 423–441, 2009. doi: 10.1002/cne.22126. [DOI] [PubMed] [Google Scholar]
- 260.Biag J, Huang Y, Gou L, Hintiryan H, Askarinam A, Hahn JD, Toga AW, Dong HW. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J Comp Neurol 520: 6–33, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Geerling JC, Shin JW, Chimenti PC, Loewy AD. Paraventricular hypothalamic nucleus: axonal projections to the brainstem. J Comp Neurol 518: 1460–1499, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Overton JM. Phenotyping small animals as models for the human metabolic syndrome: thermoneutrality matters. Int J Obes 34: S53–58, 2010. doi: 10.1038/ijo.2010.240. [DOI] [PubMed] [Google Scholar]
- 263.Fischer AW, Cannon B, Nedergaard J. Optimal housing temperatures for mice to mimic the thermal environment of humans: an experimental study. Mol Metab 7: 161–170, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lutz TA. Considering our methods: methodological issues with rodent models of appetite and obesity research. Physiol Behav 192: 182–187, 2018. [DOI] [PubMed] [Google Scholar]
- 265.Schoener TW. Theory of feeding strategies. Annu Rev Ecol Syst 2: 369–404, 1971. [Google Scholar]
- 266.Collier G, Johnson DF, Mathis C. The currency of procurement cost. J Exp Anal Behav 78: 31–61, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Pravosudov VV, Clayton NS. A test of the adaptive specialization hypothesis: population differences in caching, memory, and the hippocampus in black-capped chickadees (Poecile atricapilla). Behav Neurosci 116: 515–522, 2002. [PubMed] [Google Scholar]
- 268.Geiser F. Metabolic rate and body temperature reduction during hibernation and daily torpor. Annu Rev Physiol 66: 239–274, 2004. [DOI] [PubMed] [Google Scholar]
- 269.Bartness TJ, Clein MR. Effects of food deprivation and restriction, and metabolic blockers on food hoarding in Siberian hamsters. Am J Physiol Regul Integr Physiol 266: R1111–R1117, 1994. [DOI] [PubMed] [Google Scholar]
- 270.Leng G, Ludwig M. Neurotransmitters and peptides: whispered secrets and public announcements. J Physiol 586: 5625–5632, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Hsu TM, Hahn JD, Konanur VR, Noble EE, Suarez AN, Thai J, Nakamoto EM, Kanoski SE. Hippocampus ghrelin signaling mediates appetite through lateral hypothalamic orexin pathways. Elife 4: e1190, 2015. doi: 10.7554/elife.11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Li MM, Madara JC, Steger JS, Krashes MJ, Balthasar N, Campbell JN, Resch JM, Conley NJ, Garfield AS, Lowell BB. The paraventricular hypothalamus regulates satiety and prevents obesity via two genetically distinct circuits. Neuron 102: 653–667.e6, 2019. doi: 10.1016/j.neuron.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Li XY, Han Y, Zhang W, Wang SR, Wei YC, Li SS, Lin JK, Yan JJ, Chen AX, Zhang X, Zhao ZD, Shen WL, Xu XH. AGRP neurons project to the medial preoptic area and modulate maternal nest-building. J Neurosci 39: 456–471, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Burnett CJ, Li C, Webber E, Tsaousidou E, Xue SY, Brüning JC, Krashes MJ. Hunger-driven motivational state competition. Neuron 92: 187–201, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Burnett CJ, Funderburk SC, Navarrete J, Sabol A, Liang-Guallpa J, Desrochers TM, Krashes MJ. Need-based prioritization of behavior. eLife 8: e44527, 2019. doi: 10.7554/elife.44527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Xu S, Yang H, Menon V, Lemire AL, Wang L, Henry FE, Turaga SC, Sternson SM. Behavioral state coding by molecularly defined paraventricular hypothalamic cell type ensembles. Science 370: eabb2494, 2020. doi: 10.1126/science.abb2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Zhang X, Van Den Pol AN. Rapid binge-like eating and body weight gain driven by zona incerta GABA neuron activation. Science 356: 853–859, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Bartness TJ. Studies of food intake: lessons from nontraditionally studied species. In: Handbook of Behavioral Neurobiology Neurobiology of Food and Fluid Intake, edited by Woods ES. New York: Kluwer Academic/Plenum Publishers, 2004, p. 423–467. [Google Scholar]
- 279.Ferrier D. The Functions of the Brain. London: Smith Elder & Co., 1886. [Google Scholar]
- 280.Sherrington CS. Lecture 1 Introductory–co-ordination of the simple reflex. In: The Integrative Action of the Nervous System. New York: Charles Scribner’s Sons, 1906, p. 1–35. [Google Scholar]
- 281.Swazey JP. Reflexes and Motor Integration: Sherrington’s Concept of Integrative Action. Cambridge, MA: Harvard University Press, 1969. [Google Scholar]
- 282.Kelley AE, Baldo BA, Pratt WE, Will MJ. Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiol Behav 86: 773–795, 2005. [DOI] [PubMed] [Google Scholar]
- 283.Watts AG, Swanson LW. Anatomy of motivation. In: Stevens’ Handbook of Experimental Psychology, Volume 3, Learning, Motivation, and Emotion (3rd ed.), edited by Pashler H, Gallistel R.. New York, John Wiley & Sons, p. 563–631, 2002. [Google Scholar]
- 284.Grill HJ, Kaplan JM. Caudal brainstem participates in the distributed neural control of feeding. In: Handbook of Behavioral Neurobiology Neurobiology of Food and Fluid Intake, edited by Stricker E. New York: Plenum Press, 1990, p. 125–149. [Google Scholar]
- 285.Watts AG. Neuropeptides and the integration of motor responses to dehydration. Annu Rev Neurosci 24: 357–384, 2001. [DOI] [PubMed] [Google Scholar]
- 286.Watts AG. Neuroendocrine regulation of food intake. In: The Handbook of Neuroendocrinology, edited by Fink G, Pfaff DW, Levine JE.. London, Academic Press, Elsevier, 2012, p. 331–354. [Google Scholar]
- 287.Ritter S, Li AJ, Wang Q, Dinh TT. Minireview: the value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology 152: 4019–4032, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci 9: 519–531, 2008. [DOI] [PubMed] [Google Scholar]
- 289.Benarroch EE. Thermoregulation: recent concepts and remaining questions. Neurology 69: 1293–1297, 2008. doi: 10.1212/01.wnl.0000315313.25316.0f. [DOI] [PubMed] [Google Scholar]
- 290.Hall JE, Brands MW, Henegar JR. Angiotensin II and long-term arterial pressure regulation: the overriding dominance of the kidney. J Am Soc Nephrol 10: S258–265, 1999. [PubMed] [Google Scholar]
- 291.Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) 22: 241–251, 2007. doi: 10.1152/physiol.00010.2007. [DOI] [PubMed] [Google Scholar]
- 292.Niijima A. Glucose-sensitive afferent nerve fibers in the liver and their role in food intake and blood glucose regulation. J Auton Nerv Syst 9: 207–220, 1983. [DOI] [PubMed] [Google Scholar]
- 293.Berthoud HR, Vl A, Neuhuber WL. Gut-brain communication and obesity: understanding functions of the vagus nerve. J Clin Invest 131: e143770, 2021. doi: 10.1172/JCI143770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Opland D, Sutton A, Woodworth H, Brown J, Bugescu R, Garcia A, Christensen L, Rhodes C, Myers M Jr, Leinninger G. Loss of neurotensin receptor-1 disrupts the control of the mesolimbic dopamine system by leptin and promotes hedonic feeding and obesity. Mol Metab 2: 423–434, 2013. doi: 10.1016/j.molmet.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Huo L, Maeng L, Bjorbaek C, Grill HJ. Leptin and the control of food intake: neurons in the nucleus of the solitary tract are activated by both gastric distension and leptin. Endocrinology 148: 2189–2197, 2007. doi: 10.1210/en.2006-1572. [DOI] [PubMed] [Google Scholar]
- 296.Wang C, Chen X, Knierim JJ. Egocentric and allocentric representations of space in the rodent brain. Curr Opin Neurobiol 60: 12–20, 2020. doi: 10.1016/j.conb.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Hinsey JC, Ranson SW, McNattin RF. The rôle of the hypothalamus and mesencephalon in locomotion. Arch NeurPsych 23: 1–43, 1930. doi: 10.1001/archneurpsyc.1930.02220070004001. [DOI] [Google Scholar]
- 298.Grill HJ, Norgren R. Chronically decerebrate rats demonstrate satiation but not bait shyness. Science 201: 267–269, 1978. doi: 10.1126/science.663655. [DOI] [PubMed] [Google Scholar]
- 299.Grill HJ, Norgren R. Neurological tests and behavioral deficits in chronic thalamic and chronic decerebrate rats. Brain Res 143: 299–312, 1978. doi: 10.1016/0006-8993(78)90570-X. [DOI] [PubMed] [Google Scholar]
- 300.Whishaw IQ. The decorticate rat. In: The Cerebral Cortex of the Rat, edited by Kolb B, Tees RC.. Cambridge, MA: MIT Press, 1990, p. 239–267. [Google Scholar]
- 301.Arber S, Costa RM. Connecting neuronal circuits for movement. Science 360: 1403–1404, 2018. doi: 10.1126/science.aat5994. [DOI] [PubMed] [Google Scholar]
- 302.Grillner S, Manira E. Current principles of motor control, with special reference to vertebrate locomotion. Physiol Rev 100: 271–320, 2020. doi: 10.1152/physrev.00015.2019. [DOI] [PubMed] [Google Scholar]
- 303.Tinbergen N. The Study of Instinct. Oxford: Oxford University Press, 1951. [Google Scholar]
- 304.Anderson DJ. Circuit modules linking internal states and social behaviour in flies and mice. Nature Rev Neurosci 17: 692–704, 2016. doi: 10.1038/nrn.2016.125. [DOI] [PubMed] [Google Scholar]
- 305.dos Santos LM, Boschen SL, Bortolanza M, de Oliveira WF, Furigo IC, Mota-Ortiz SR, Da Cunha C, Canteras NS. The role of the ventrolateral caudoputamen in predatory hunting. Physiol Behav 105: 893–898, 2012. doi: 10.1016/j.physbeh.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 306.Klaus A, Alves da Silva J, Costa RM. What, if, and when to move: basal ganglia circuits and self-paced action initiation. Annu Rev Neurosci 42: 459–483, 2019. doi: 10.1146/annurev-neuro-072116-031033. [DOI] [PubMed] [Google Scholar]
- 307.Aldridge JW, Berridge KC. Coding of serial order by neostriatal neurons: a ‘natural action’ approach to movement sequence. J Neurosci 18: 2777–2787, 1998. doi: 10.1523/JNEUROSCI.18-07-02777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Jin X, Tecuapetla F, Costa RM. Basal ganglia subcircuits distinctively encode the parsing and concatenation of action sequences. Nat Neurosci 17: 423–430, 2014. doi: 10.1038/nn.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Jin X, Costa RM. Shaping action sequences in basal ganglia circuits. Curr Opin Neurobiol 33: 188–196, 2015. doi: 10.1016/j.conb.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Westberg KG, Kolta A. The trigeminal circuits responsible for chewing. Int Rev Neurobiol 97: 77–98, 2011. doi: 10.1016/B978-0-12-385198-7.00004-7. [DOI] [PubMed] [Google Scholar]
- 311.Moore JD, Kleinfeld D, Wang F. How the brainstem controls orofacial behaviors comprised of rhythmic actions. Trends Neurosci 37: 370–380, 2014. doi: 10.1016/j.tins.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Lund JP. Chew before you swallow. Prog Brain Res 188: 219–228, 2011. doi: 10.1016/B978-0-444-53825-3.00020-6. [DOI] [PubMed] [Google Scholar]
- 313.Ruder L, Arber S. Brainstem circuits controlling action diversification. Annu Rev Neurosci 42: 485–504, 2019. doi: 10.1146/annurev-neuro-070918-050201. [DOI] [PubMed] [Google Scholar]
- 314.Travers JB, Rinaman L. Identification of lingual motor control circuits using two strains of pseudorabies virus. Neuroscience 115: 1139–1151, 2002. doi: 10.1016/S0306-4522(02)00489-X. [DOI] [PubMed] [Google Scholar]
- 315.Lund JP, Kolta A. Generation of the central masticatory pattern and its modification by sensory feedback. Dysphagia 21: 167–174, 2006. doi: 10.1007/s00455-006-9027-6. [DOI] [PubMed] [Google Scholar]
- 316.Whishaw IQ, Schallert T, Kolb B. An analysis of feeding and sensorimotor abilities of rats after decortication. J Comp Physiol Psych 95: 85–103, 1981. doi: 10.1037/h0077760. [DOI] [PubMed] [Google Scholar]
- 317.Flynn FW, Grill HJ. Insulin elicits ingestion in decerebrate rats. Science 221: 188–190, 1983. doi: 10.1126/science.6344221. [DOI] [PubMed] [Google Scholar]
- 318.Kaplan JM, Roitman M, Grill HJ. Food deprivation does not potentiate glucose taste reactivity responses of chronic decerebrate rats. Brain Res 870: 102–108, 2000. doi: 10.1016/S0006-8993(00)02406-9. [DOI] [PubMed] [Google Scholar]
- 319.Kaplan JM, Seeley RJ, Grill HJ. Daily caloric intake in intact and chronic decerebrate rats. Behav Neurosci 107: 876–881, 1993. doi: 10.1037/0735-7044.107.5.876. [DOI] [PubMed] [Google Scholar]
- 320.Steiner JE. The gustofacial response: observation on normal and anencephalic newborn infants. Symp Oral Sens Percept 4: 254–278, 1973. [PubMed] [Google Scholar]
- 321.Grill HJ, Hayes MR. The nucleus tractus solitarius: a portal for visceral afferent signal processing, energy status assessment and integration of their combined effects on food intake. Int J Obes 33: S11–15, 2009. doi: 10.1038/ijo.2009.10. [DOI] [PubMed] [Google Scholar]
- 322.Whishaw IQ, Kolb B. Decortication abolishes place but not cue learning in rats. Behav Brain Res 11: 123–134, 1984. doi: 10.1016/0166-4328(84)90135-9. [DOI] [PubMed] [Google Scholar]
- 323.Suarez AN, Noble EE, Kanoski SE. Regulation of memory function by feeding-relevant biological systems: following the breadcrumbs to the hippocampus. Front Mol Neurosci 12: 1–21, 2019. doi: 10.3389/fnmol.2019.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Hubel DH, Wiesel TN. Receptive fields, binocular interaction and functional architecture in the cat’s visual cortex. J Physiol 160: 106–154, 1962. doi: 10.1113/jphysiol.1962.sp006837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Hendrickson AE, Wagoner N, Cowan WM. An autoradiographic and electron microscopic study of retino-hypothalamic connections. Z Zellforsch Mikrosk Anat 135: 1–26, 1972. doi: 10.1007/BF00307084. [DOI] [PubMed] [Google Scholar]
- 326.Moore RY, Lenn NJ. A retinohypothalamic projection in the rat. J Comp Neurol 146: 1–14, 1972. doi: 10.1002/cne.901460102. [DOI] [PubMed] [Google Scholar]
- 327.Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci U S A 69: 1583–1586, 1972. doi: 10.1073/pnas.69.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci 85: 1–17, 2000. [DOI] [PubMed] [Google Scholar]
- 329.Janig W, Morrison JF. Functional properties of spinal visceral afferents supplying abdominal and pelvic organs, with special emphasis on visceral nociception. Prog Brain Res 67: 87–114, 1986. [DOI] [PubMed] [Google Scholar]
- 330.Berthoud HR. Anatomy and function of sensory hepatic nerves. Anat Rec A Discov Mol Cell Evol Biol 280: 827–835, 2004. [DOI] [PubMed] [Google Scholar]
- 331.Berthoud HR. Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol Motil 20, Suppl 1: 64–72, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Bohórquez DV, Liddle RA. The gut connectome: making sense of what you eat. J Clin Invest 125: 888–890, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Kaelberer MM, Buchanan KL, Klein ME, Barth BB, Montoya MM, Shen X, Bohórquez DV. A gut-brain neural circuit for nutrient sensory transduction. Science 361: eaat5236, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Powley TL, Jaffey DM, McAdams J, Baronowsky EA, Black D, Chesney L, Evans C, Phillips RJ. Vagal innervation of the stomach reassessed: brain-gut connectome uses smart terminals. Ann N Y Acad Sci 1454: 14–30, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Powley TL, Phillips RJ. Musings on the wanderer: what’s new in our understanding of vago-vagal reflexes? I. Morphology and topography of vagal afferents innervating the GI tract. Am J Physiol Gastrointest Liver Physiol 283: G1217–G1225, 2002. [DOI] [PubMed] [Google Scholar]
- 336.Borgmann D, Ciglieri E, Biglari N, Brandt C, Cremer AL, Backes H, Tittgemeyer M, Wunderlich FT, Brüning JC, Fenselau H. Gut-brain communication by distinct sensory neurons differently controls feeding and glucose metabolism. Cell Metab 33: 1466–1482, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Berthoud HR. The vagus nerve, food intake and obesity. Regul Pept 149: 15–25, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 65: 34–43, 2016. [DOI] [PubMed] [Google Scholar]
- 339.Langhans W, Geary N. Overview of the physiological control of eating. Forum Nutr 63: 9–53, 2010. [DOI] [PubMed] [Google Scholar]
- 340.Langhans WH, Holst JJ. Afferent endocrine control of eating. In: Neuroendocrinology of Appetite, edited by Dickson SL, Mercer JG.. Chichester, UK: John Wiley & Sons, Ltd., 2016, p. 24–54. [Google Scholar]
- 341.Ruttimann EB, Arnold M, Hillebrand JJ, Geary N, Langhans W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology 150: 1174–1181, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.de Lartigue G. Putative roles of neuropeptides in vagal afferent signaling. Physiol Behav 136: 155–169, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.de Lartigue G, Dimaline R, Varro A, Dockray GJ. Cocaine- and amphetamine-regulated transcript: stimulation of expression in rat vagal afferent neurons by cholecystokinin and suppression by ghrelin. J Neurosci 27: 2876–2882, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.de Lartigue G, Lur G, Dimaline R, Varro A, Raybould H, Dockray GJ. EGR1 Is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology 151: 3589–3599, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Han W, Tellez LA, Perkins MH, Perez IO, Qu T, Ferreira J, Ferreira TL, Quinn D, Liu ZW, Gao XB, Kaelberer MM, Bohórquez DV, Shammah-Lagnado SJ, de Lartigue G, de Araujo IE. A neural circuit for gut-induced reward. Cell 175: 665–678, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Suarez AN, Hsu TM, Liu CM, Noble EE, Cortella AM, Nakamoto EM, Hahn JD, de Lartigue G, Kanoski SE. Gut vagal sensory signaling regulates hippocampus function through multi-order pathways. Nat Commun 9: 2181, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Bai L, Mesgarzadeh S, Ramesh KS, Huey EL, Liu Y, Gray LA, Aitken TJ, Chen Y, Beutler LR, Ahn JS, Madisen L, Zeng H, Krasnow MA, Knight ZA. Genetic identification of vagal sensory neurons that control feeding. Cell 179: 1129–1143, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Egerod KL, Petersen N, Timshel PN, Rekling JC, Wang Y, Liu Q, Schwartz TW, Gautron L. Profiling of G protein-coupled receptors in vagal afferents reveals novel gut-to-brain sensing mechanisms. Mol Metab 12: 62–75, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Kim DY, Heo G, Kim M, Kim H, Jin JA, Kim HK, Jung S, An M, Ahn BH, Park JH, Park HE, Lee M, Lee JW, Schwartz GJ, Kim SY. A neural circuit mechanism for mechanosensory feedback control of ingestion. Nature 580: 376–380, 2020. [DOI] [PubMed] [Google Scholar]
- 350.Schwartz GJ, McHugh PR, Moran TH. Integration of vagal afferent responses to gastric loads and cholecystokinin in rats. Am J Physiol Regul Integr Comp Physiol 261: R64–R69, 1991. [DOI] [PubMed] [Google Scholar]
- 351.Schwartz GJ, Moran TH. Sub-diaphragmatic vagal afferent integration of meal-related gastrointestinal signals. Neurosci Biobehav Rev 20: 47–56, 1996. [DOI] [PubMed] [Google Scholar]
- 352.Page AJ, Kentish SJ. Plasticity of gastrointestinal vagal afferent satiety signals. Neurogastroenterol Motil 29: e12973, 2017. [DOI] [PubMed] [Google Scholar]
- 353.Williams EK, Chang RB, Strochlic DE, Umans BD, Lowell BB, Liberles SD. Sensory neurons that detect stretch and nutrients in the digestive system. Cell 166: 209–221, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Beekwilder JP, Beems T. Overview of the clinical applications of vagus nerve stimulation. J Clin Neurophysiol 27: 130–138, 2010. [DOI] [PubMed] [Google Scholar]
- 355.Norgren R, Smith GP. A method for selective section of vagal afferent or efferent axons in the rat. Am J Physiol Regul Integr Comp Physiol 267: R1136–R1141, 1994. [DOI] [PubMed] [Google Scholar]
- 356.Klarer M, Weber-Stadlbauer U, Arnold M, Langhans W, Meyer U. Abdominal vagal deafferentation alters affective behaviors in rats. J Affect Disord 252: 404–412, 2019. [DOI] [PubMed] [Google Scholar]
- 357.Klarer M, Weber-Stadlbauer U, Arnold M, Langhans W, Meyer U. Cognitive effects of subdiaphragmatic vagal deafferentation in rats. Neurobiol Learn Mem 142: 190–199, 2017. [DOI] [PubMed] [Google Scholar]
- 358.Klarer M, Arnold M, Gunther L, Winter C, Langhans W, Meyer U. Gut vagal afferents differentially modulate innate anxiety and learned fear. J Neurosci 34: 7067–7076, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 10: 735–742, 2012. [DOI] [PubMed] [Google Scholar]
- 360.Montiel-Castro AJ, Gonzalez-Cervantes RM, Bravo-Ruiseco G, Pacheco-Lopez G. The microbiota-gut-brain axis: neurobehavioral correlates, health and sociality. Front Integr Neurosci 7: 70, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Andresen MC, Paton JF, The nucleus of the solitary tract: processing information from viscerosensory afferents. In: Central Regulation of Autonomic Functions, edited by Llewellyn-Smith IJ, Verberne AJ. New York: Oxford University Press, 2011, p. 23–46. [Google Scholar]
- 362.Millhouse OE. A Golgi anatomy of the rodent hypothalamus. In: Handbook of the Hypothalamus Volume 1, Anatomy of the Hypothalamus, edited by Panksepp J, Morgane PJ.. New York: Marcel Dekker, Inc., 1979, p. 221–265. [Google Scholar]
- 363.Prevot V, Dehouck B, Sharif A, Ciofi P, Giacobini P, Clasadonte J. The versatile tanycyte: a hypothalamic integrator of reproduction and energy metabolism. Endoc Rev 39: 333–368, 2018. doi: 10.1210/er.2017-00235. [DOI] [PubMed] [Google Scholar]
- 364.Radley JJ, Sawchenko PE. A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J Neurosci 31: 9683–9695, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Wojaczynski GJ, Engel EA, Steren KE, Enquist LW, Patrick Card J. The neuroinvasive profiles of H129 (herpes simplex virus type 1) recombinants with putative anterograde-only transneuronal spread properties. Brain Struct Funct 220: 1395–1420, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Krieger JP, da Conceição EP, Sanchez-Watts G, Arnold M, Pettersen KG, Mohammed M, Modica S, Lossel P, Morrison SF, Madden CJ, Watts AG, Langhans W, Lee SJ. Glucagon-like peptide-1 regulates brown adipose tissue thermogenesis via the gut-brain axis in rats. Am J Physiol Regul Integr Comp Physiol 315: R708–R720, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Langley JN. The sympathetic and other related systems of nerves. In: Text-Book of Physiology, edited by Schafer EA. Edinburgh & London: Young J. Pentland, 1900, p. 616–696. [Google Scholar]
- 368.Bain WA, Irving JT, McSwiney BA. The afferent fibres from the abdomen in the splanchnic nerves. J Physiol 84: 323–333, 1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Cervero FF, Foreman RD. Sensory innervation of the viscera. In: Central Regulation of Autonomic Functions, edited by Spyer KM, Loewy AD.. New York: Oxford University Press, 1990, p. 104–125. [Google Scholar]
- 370.Ranson SW. Afferent paths for visceral reflexes. Physiol Rev 1: 477–522, 1921. [Google Scholar]
- 371.Langley JN. The autonomic nervous system. Brain 26: 1–26, 1903. [Google Scholar]
- 372.Blessing WW. The Lower Brainstem and Bodily Homeostasis. New York: Oxford University Press, 1997. [Google Scholar]
- 373.Jänig W. The Integrative Action of the Autonomic Nervous System. Neurobiology of Homeostasis. Cambridge, UK: Cambridge University Press, 2006. [Google Scholar]
- 374.Gauriau C, Bernard JF. Pain pathways and parabrachial circuits in the rat. Exp Physiol 87: 251–258, 2002. [DOI] [PubMed] [Google Scholar]
- 375.Craig AD. Pain mechanisms: labeled lines versus convergence in central processing. Annu Rev Neurosci 26: 1–30, 2003. [DOI] [PubMed] [Google Scholar]
- 376.Vallet PG, Baertschi AJ. Spinal afferents for peripheral osmoreceptors in the rat. Brain Res 239: 271–274, 1982. doi: 10.1016/0006-8993(82)90850-2. [DOI] [PubMed] [Google Scholar]
- 377.Fujita S, Donovan CM. Celiac-superior mesenteric ganglionectomy, but not vagotomy, suppresses the sympathoadrenal response to insulin-induced hypoglycemia. Diabetes 54: 3258–3264, 2005. doi: 10.2337/diabetes.54.11.3258. [DOI] [PubMed] [Google Scholar]
- 378.Fujita S, Bohland M, Sanchez-Watts G, Watts AG, Donovan CM. Hypoglycemic detection at the portal vein is mediated by capsaicin-sensitive primary sensory neurons. Am J Physiol Endocrinol Metab 293: E96–E101, 2007. doi: 10.1152/ajpendo.00415.2006. [DOI] [PubMed] [Google Scholar]
- 379.Sugiura Y, Terui N, Hosoya Y, Tonosaki Y, Nishiyama K, Honda T. Quantitative-analysis of central terminal projections of visceral and somatic unmyelinated (c) primary afferent-fibers in the guinea-pig. J Comp Neurol 332: 315–325, 1993. doi: 10.1002/cne.903320305. [DOI] [PubMed] [Google Scholar]
- 380.Sugiura Y, Terui N, Hosoya Y. Difference in distribution of central terminals between visceral and somatic unmyelinated(C) primary afferent-fibers. J Neurophysiol 62: 834–840, 1989. doi: 10.1152/jn.1989.62.4.834. [DOI] [PubMed] [Google Scholar]
- 381.Rinaman L, Schwartz G. Anterograde transneuronal viral tracing of central viscerosensory pathways in rats. J Neurosci 24: 2782–2786, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Horn CC, Meyers K, Lim A, Dye M, Pak D, Rinaman L, Yates BJ. Delineation of vagal emetic pathways: intragastric copper sulfate-induced emesis and viral tract tracing in musk shrews. Am J Physiol Regul Integr Comp Physiol 306: R341–R351, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Bartness TJ, Vaughan CH, Song CK. Sympathetic and sensory innervation of brown adipose tissue. Int J Obes 34: S36–42, 2010. doi: 10.1038/ijo.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Vaughan CH, Bartness TJ. Anterograde transneuronal viral tract tracing reveals central sensory circuits from brown fat and sensory denervation alters its thermogenic responses. Am J Physiol Regul Integr Comp Physiol 302: R1049–R1058, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Ryu V, Watts AG, Xue B, Bartness TJ. Bidirectional crosstalk between the sensory and sympathetic motor systems innervating brown and white adipose tissue in male Siberian hamsters. Am J Physiol Regul Integr Comp Physiol 312: R324–R337, 2017. doi: 10.1152/ajpregu.00456.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Song CK, Schwartz GJ, Bartness TJ. Anterograde transneuronal viral tract tracing reveals central sensory circuits from white adipose tissue. Am J Physiol Regul Integr Comp Physiol 296: R501–R511, 2009. doi: 10.1152/ajpregu.90786.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Garcia-Luna C, Sanchez-Watts G, Arnold M, de Lartigue G, DeWalt N, Langhans W, Watts AG. The medullary targets of neurally conveyed sensory information from the rat hepatic portal and superior mesenteric veins. eNeuro 8: ENEURO.0419-20.2021, 2021. doi: 10.1523/ENEURO.0419-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Cliffer KD, Burstein R, Giesler GJ. Distributions of spinothalamic, spinohypothalamic, and spinotelencephalic fibers revealed by anterograde transport of PHA-L in rats. J Neurosci 11: 852–868, 1991. doi: 10.1523/JNEUROSCI.11-03-00852.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Gauriau C, Bernard JF. A comparative reappraisal of projections from the superficial laminae of the dorsal horn in the rat: the forebrain. J Comp Neurol 468: 24–56, 2004. doi: 10.1002/cne.10873. [DOI] [PubMed] [Google Scholar]
- 390.Saper CB. The spinoparabrachial pathway–shedding new light on an old path. J Comp Neurol 353: 477–479, 1995. doi: 10.1002/cne.903530402. [DOI] [PubMed] [Google Scholar]
- 391.Craig AD. Distribution of brainstem projections from spinal lamina I neurons in the cat and the monkey. J Comp Neurol 361: 225–248, 1995. doi: 10.1002/cne.903610204. [DOI] [PubMed] [Google Scholar]
- 392.Gamboa-Esteves FO, Tavares I, Almeida A, Batten TF, McWilliam PN, Lima D. Projection sites of superficial and deep spinal dorsal horn cells in the nucleus tractus solitarii of the rat. Brain Res 921: 195–205, 2001. doi: 10.1016/S0006-8993(01)03118-3. [DOI] [PubMed] [Google Scholar]
- 393.Craig AD. A new view of pain as a homeostatic emotion. Trends Neurosci 26: 303–307, 2003. doi: 10.1016/S0166-2236(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 394.Strigo IA, Craig AD. Interoception, homeostatic emotions and sympathovagal balance. Philos Trans R Soc Lond B Biol Sci 371, 20160010, 2016. doi: 10.1098/rstb.2016.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Craig AD. How do you feel? Interoception: the sense of the physiological condition of the body. Nat Rev Neurosci 3: 655–666, 2002. doi: 10.1038/nrn894. [DOI] [PubMed] [Google Scholar]
- 396.Craig AD. Interoception: the sense of the physiological condition of the body. Curr Opin Neurobiol 13: 500–505, 2003. doi: 10.1016/S0959-4388(03)00090-4. [DOI] [PubMed] [Google Scholar]
- 397.Craig AD. The sentient self. Brain Struct Funct 214: 563–577, 2010. doi: 10.1007/s00429-010-0248-y. [DOI] [PubMed] [Google Scholar]
- 398.Malick A, Jakubowski M, Elmquist JK, Saper CB, Burstein R. A neurohistochemical blueprint for pain-induced loss of appetite. Proc Natl Acad Sci U S A 98: 9930–9935, 2001. doi: 10.1073/pnas.171616898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Alhadeff AL, Su Z, Hernandez E, Klima ML, Phillips SZ, Holland RA, Guo C, Hantman AW, De Jonghe BC, Betley JN. A Neural circuit for the suppression of pain by a competing need state. Cell 173: 140–152, 2018. doi: 10.1016/j.cell.2018.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Craig AD. A rat is not a monkey is not a human: comment on Mogil (Nature Rev. Neurosci. 10, 283-294 (2009)). Nat Rev Neurosci 10: 466–466, 2009. doi: 10.1038/nrn2606-c1. [DOI] [PubMed] [Google Scholar]
- 401.Travagli RA, Anselmi L. Vagal neurocircuitry and its influence on gastric motility. Nat Rev Gastroenterol Hepatol 13: 389–401, 2016. doi: 10.1038/nrgastro.2016.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Zheng H, Kelly L, Patterson LM, Berthoud HR. Effect of brain stem NMDA-receptor blockade by MK-801 on behavioral and fos responses to vagal satiety signals. Am J Physiol Regul Integr Comp Physiol 277: R1104–R1111, 1999. doi: 10.1152/ajpregu.1999.277.4.R1104. [DOI] [PubMed] [Google Scholar]
- 403.Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 259: R1131–R1138, 1990. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- 404.Wang QP, Guan JL, Pan W, Kastin AJ, Shioda S. A diffusion barrier between the area postrema and nucleus tractus solitarius. Neurochem Res 33: 2035–2043, 2008. doi: 10.1007/s11064-008-9676-y. [DOI] [PubMed] [Google Scholar]
- 405.Glaum SR, Hara M, Bindokas VP, Lee CC, Polonsky KS, Bell GI, Miller RJ. Leptin, the obese gene product, rapidly modulates synaptic transmission in the hypothalamus. Mol Pharmacol 50: 230–235, 1996. [PubMed] [Google Scholar]
- 406.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett 387: 113–116, 1996. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- 407.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest 98: 1101–1106, 1996. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Morgan PJ, Trayhurn P. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendoc 8: 733–735, 1996. doi: 10.1046/j.1365-2826.1996.05161.x. [DOI] [PubMed] [Google Scholar]
- 409.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci 8: 571–578, 2005. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 410.Sawchenko PE. Toward a new neurobiology of energy balance, appetite, and obesity: the anatomists weigh in. J Comp Neurol 402: 435–441, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 411.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol 402: 442–459, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 412.Broberger C, De Lecea L, Sutcliffe JG, Hokfelt T. Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol 402: 460–474, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 413.Sternson SM. Hypothalamic survival circuits: blueprints for purposive behaviors. Neuron 77: 810–824, 2013. doi: 10.1016/j.neuron.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Banks WA. Blood-brain barrier as a regulatory interface. Forum Nutr 63: 102– 110, 2010. doi: 10.1159/000264398. [DOI] [PubMed] [Google Scholar]
- 415.Miyata S. New aspects in fenestrated capillary and tissue dynamics in the sensory circumventricular organs of adult brains. Front Neurosci 9: 390, 2015. doi: 10.3389/fnins.2015.00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Kawano H, Masuko S. Region-specific projections from the subfornical organ to the paraventricular hypothalamic nucleus in the rat. Neuroscience 169: 1227–1234, 2010. doi: 10.1016/j.neuroscience.2010.05.065. [DOI] [PubMed] [Google Scholar]
- 417.Shapiro RE, Miselis RR. The central neural connections of the area postrema of the rat. J Comp Neurol 234: 344–364, 1985. doi: 10.1002/cne.902340306. [DOI] [PubMed] [Google Scholar]
- 418.Swanson LW, Lind RW. Neural projections subserving the initiation of a specific motivated behavior in the rat: new projections from the subfornical organ. Brain Res 379: 399–403, 1986. doi: 10.1016/0006-8993(86)90799-7. [DOI] [PubMed] [Google Scholar]
- 419.Tokita K, Inoue T, Boughter JD. Afferent connections of the parabrachial nucleus in C57BL/6J mice. Neuroscience 161: 475–488, 2009. doi: 10.1016/j.neuroscience.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Cancelliere NM, Ferguson AV. Subfornical organ neurons integrate cardiovascular and metabolic signals. Am J Physiol Regul Integr Comp Physiol 312: R253–R262, 2017. doi: 10.1152/ajpregu.00423.2016. [DOI] [PubMed] [Google Scholar]
- 421.McKinley MJ, Denton DA, Ryan PJ, Yao ST, Stefanidis A, Oldfield BJ. From sensory circumventricular organs to cerebral cortex: neural pathways controlling thirst and hunger. J Neuroendocrinol 31: e12689, 2019. doi: 10.1111/jne.12689. [DOI] [PubMed] [Google Scholar]
- 422.Riediger T, Schmid HA, Lutz TA, Simon E. Amylin and glucose co-activate area postrema neurons of the rat. Neurosci Lett 328: 121–124, 2002. doi: 10.1016/S0304-3940(02)00482-2. [DOI] [PubMed] [Google Scholar]
- 423.Jiang H, Gallet S, Klemm P, Scholl P, Folz-Donahue K, Altmüller J, Alber J, Heilinger C, Kukat C, Loyens A, Müller-Fielitz H, Sundaram S, Schwaninger M, Prevot V, Brüning JC. MCH neurons regulate permeability of the median eminence barrier. Neuron 107: 306–314, 2020. doi: 10.1016/j.neuron.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Morita-Takemura S, Wanaka A. Blood-to-brain communication in the hypothalamus for energy intake regulation. Neurochem Intl 128: 135–142, 2019. doi: 10.1016/j.neuint.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 425.Langlet F, Levin BE, Luquet S, Mazzone M, Messina A, Dunn-Meynell AA, Balland E, Lacombe A, Mazur D, Carmeliet P, Bouret SG, Prevot V, Dehouck B. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab 17: 607–617, 2013. doi: 10.1016/j.cmet.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, Rasika S, Falluel-Morel A, Anouar Y, Dehouck B, Trinquet E, Jockers R, Bouret SG, Prévot VV. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab 19: 293–301, 2014. doi: 10.1016/j.cmet.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Yoo S, Cha D, Kim DW, Hoang TV, Blackshaw S. Tanycyte-independent control of hypothalamic leptin signaling. Front Neurosci 13: 240, 2019. doi: 10.3389/fnhum.2019.00240,10.3389/fnins.2019.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Elizondo-Vega RJ, Oyarce RA. Nutrient sensing by hypothalamic tanycytes. Front Endoc 10: 491–498, 2019. doi: 10.3389/fendo.2019.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Woods SC, Lutz TA, Geary N, Langhans W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Phil Trans R Soc Lond B Biol Sci 361: 1219–1235, 2006. doi: 10.1098/rstb.2006.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Boyle CN, Lutz TA, Le Foll C. Amylin-Its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol Metab 8: 203–210, 2018. doi: 10.1016/j.molmet.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Heeley N, Blouet C. Central amino acid sensing in the control of feeding behavior. Front Endocrinol (Lausanne) 7: 148, 2016. doi: 10.3389/fendo.2016.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Milstein JL, Ferris HA. The brain as an insulin-sensitive metabolic organ. Mol Metab 52: 101234, 2021. doi: 10.1016/j.molmet.2021.101234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Dunn-Meynell AA, Sanders NM, Compton D, Becker TC, Eiki JI, Zhang BB, Levin BE. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J Neurosci 29: 7015–7022, 2009. doi: 10.1523/JNEUROSCI.0334-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Campfield LA, Brandon P, Smith FJ. On-line continuous measurement of blood glucose and meal pattern in free-feeding rats: the role of glucose in meal initiation. Brain Res Bull 14: 605–616, 1985. doi: 10.1016/0361-9230(85)90110-8. [DOI] [PubMed] [Google Scholar]
- 435.Ritter RC, Slusser PG, Stone S. Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213: 451–452, 1981. doi: 10.1126/science.6264602. [DOI] [PubMed] [Google Scholar]
- 436.Mayer J. Glucostatic mechanism of regulation of food intake. N Engl J Med 249: 13–16, 1953. doi: 10.1056/nejm195307022490104. [DOI] [PubMed] [Google Scholar]
- 437.Levin BE. Mayer and the Glucostatic Hypothesis. SSIB Ingestive Classics. https://www.ssib.org/web/classic1.php. [2013]. [Google Scholar]
- 438.Watts AG, Donovan CM. Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendoc 31: 32–43, 2010. doi: 10.1016/j.yfrne.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes 53: 2521–2528, 2004. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- 440.Routh VH, Hao L, Santiago AM, Sheng Z, Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci 8: 1–13, 2014. doi: 10.3389/fnsys.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Oomura Y, Ono T, Ooyama H, Wayner MJ. Glucose and osmosensitive neurones of the rat hypothalamus. Nature 222: 282–284, 1969. doi: 10.1038/222282a0. [DOI] [PubMed] [Google Scholar]
- 442.Oomura Y, Ooyama H, Sugimori M, Nakamura T, Yamada Y. Glucose inhibition of the glucose-sensitive neurone in the rat lateral hypothalamus. Nature 247: 284–286, 1974. doi: 10.1038/247284a0. [DOI] [PubMed] [Google Scholar]
- 443.Oomura Y, Yoshimatsu H. Neural network of glucose monitoring system. J Auton Nerv Syst 10: 359–372, 1984. doi: 10.1016/0165-1838(84)90033-X. [DOI] [PubMed] [Google Scholar]
- 444.Koekkoek LL, Mul JD, la Fleur SE. Glucose-sensing in the reward system. Front Neurosci 11: 341–311, 2017. doi: 10.3389/fnhum.2017.00341,10.3389/fnins.2017.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Medeiros N, Dai L, Ferguson AV. Glucose-responsive neurons in the subfornical organ of the rat–a novel site for direct CNS monitoring of circulating glucose. Neuroscience 201: 157–165, 2012. doi: 10.1016/j.neuroscience.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 446.Barnes MB, Lawson MA, Beverly JL. Rate of fall in blood glucose and recurrent hypoglycemia affect glucose dynamics and noradrenergic activation in the ventromedial hypothalamus. Am J Physiol Regul Integr Comp Physiol 301: R1815–R1820, 2011. doi: 10.1152/ajpregu.00171.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.de Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 52: 2767–2773, 2003. doi: 10.2337/diabetes.52.11.2767. [DOI] [PubMed] [Google Scholar]
- 448.Ionut V, Castro AV, Woolcott OO, Stefanovski D, Iyer MS, Broussard JL, Mkrtchyan H, Burch M, Elazary R, Kirkman E, Bergman RN. Essentiality of portal vein receptors in hypoglycemic counterregulation: direct proof via denervation in male canines. Endocrinology 155: 1247–1254, 2014. doi: 10.1210/en.2013-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Strubbe JH, Steffens AB. Blood glucose levels in portal and peripheral circulation and their relation to food intake in the rat. Physiol Behav 19: 303–307, 1977. doi: 10.1016/0031-9384(77)90342-0. [DOI] [PubMed] [Google Scholar]
- 450.Langhans W. Hepatic and intestinal handling of metabolites during feeding in rats. Physiol Behav 49: 1203–1209, 1991. doi: 10.1016/0031-9384(91)90352-O. [DOI] [PubMed] [Google Scholar]
- 451.Goldstein N, McKnight AD, Carty JR, Arnold M, Betley JN, Alhadeff AL. Hypothalamic detection of macronutrients via multiple gut-brain pathways. Cell Metab 33: 676–687, 2021. doi: 10.1016/j.cmet.2020.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Mei N. Vagal glucoreceptors in the small intestine of the cat. J Physiol 282: 485–506, 1978. doi: 10.1113/jphysiol.1978.sp012477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Perrin J, Crousillat J, Mei N. Assessment of true splanchnic glucoreceptors in the jejuno-ileum of the cat. Brain Res Bull 7: 625–628, 1981. doi: 10.1016/0361-9230(81)90108-8. [DOI] [PubMed] [Google Scholar]
- 454.Schier LA, Spector AC. Post-oral sugar detection rapidly and chemospecifically modulates taste-guided behavior. Am J Physiol Regul Integr Comp Physiol 311: R742–R755, 2016. doi: 10.1152/ajpregu.00155.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570: 469–484, 2006. doi: 10.1113/jphysiol.2005.098822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Rogers RC, Hermann GE. Hindbrain astrocytes and glucose counter-regulation. Physiol Behav 204: 140–150, 2019. doi: 10.1016/j.physbeh.2019.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Jokiaho AJ, Donovan CM, Watts AG. The rate of fall of blood glucose determines the necessity of forebrain-projecting catecholaminergic neurons for male rat sympathoadrenal responses. Diabetes 63: 2854–2865, 2014. doi: 10.2337/db13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Khan AM, Kaminski KL, Sanchez-Watts G, Ponzio TA, Kuzmiski JB, Bains JS, Watts AG. MAP kinases couple hindbrain-derived catecholamine signals to hypothalamic adrenocortical control mechanisms during glycemia-related challenges. J Neurosci 31: 18479–18491, 2011. doi: 10.1523/JNEUROSCI.4785-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Khan AM, Walker EM, Dominguez N, Watts AG. Neural input is critical for arcuate hypothalamic neurons to mount intracellular signaling responses to systemic insulin and deoxyglucose challenges in male rats: implications for communication within feeding and metabolic control networks. Endocrinology 155: 405–416, 2014. doi: 10.1210/en.2013-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol 432: 197–216, 2001. doi: 10.1002/cne.1097. [DOI] [PubMed] [Google Scholar]
- 461.Ritter S, Li AJ, Wang Q. Hindbrain glucoregulatory mechanisms: critical role of catecholamine neurons in the ventrolateral medulla. Physiol Behav 208: 112568, 2019. doi: 10.1016/j.physbeh.2019.112568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Li AJ, Wang Q, Elsarelli MM, Brown RL, Ritter S. Hindbrain catecholamine neurons activate orexin neurons during systemic glucoprivation in male rats. Endocrinology 156: 2807–2820, 2015. doi: 10.1210/en.2015-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Lee SJ, Diener K, Kaufman S, Krieger JP, Pettersen KG, Jejelava N, Arnold M, Watts AG, Langhans W. Limiting glucocorticoid secretion increases the anorexigenic property of Exendin-4. Mol Metab 5: 552–565, 2016. doi: 10.1016/j.molmet.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Lee SJ, Jokiaho AJ, Sanchez-Watts G, Watts AG. Catecholaminergic projections into an interconnected forebrain network control the sensitivity of male rats to diet-induced obesity. Am J Physiol Regul Integr Comp Physiol 314: R811–R823, 2018. doi: 10.1152/ajpregu.00423.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Ritter S, Dinh TT, Zhang YB. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856: 37–47, 2000. doi: 10.1016/S0006-8993(99)02327-6. [DOI] [PubMed] [Google Scholar]
- 466.Darling RA, Ritter S. 2-Deoxy-d-glucose, but not mercaptoacetate, increases food intake in decerebrate rats. Am J Physiol Regul Integr Comp Physiol 297: R382–R386, 2009. doi: 10.1152/ajpregu.90827.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Aklan I, Sayar Atasoy N, Yavuz Y, Ates T, Coban I, Koksalar F, Filiz G, Topcu IC, Oncul M, Dilsiz P, Cebecioglu U, Alp MI, Yilmaz B, Davis DR, Hajdukiewicz K, Saito K, Konopka W, Cui H, Atasoy D. NTS catecholamine neurons mediate hypoglycemic hunger via medial hypothalamic feeding pathways. Cell Metab 31: 313–326, 2020. doi: 10.1016/j.cmet.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Clark AJ, Clark JM, Winn P. NMDA lesions of rat lateral hypothalamus: effects of dietary and physiological challenges. Neuroreport 1: 263–266, 1990. doi: 10.1097/00001756-199011000-00024. [DOI] [PubMed] [Google Scholar]
- 469.Clark JM, Clark A, Warne D, Rugg EL, Lightman SL, Winn P. Neuroendocrine and behavioural responses to hyperosmolality in rats with lesions of the lateral hypothalamus made by N-methyl-d-aspartate. Neuroscience 45: 625–629, 1991. doi: 10.1016/0306-4522(91)90275-S. [DOI] [PubMed] [Google Scholar]
- 470.Calingasan NY, Ritter S. Hypothalamic paraventricular nucleus lesions do not abolish glucoprivic or lipoprivic feeding. Brain Res 595: 25–31, 1992. doi: 10.1016/0006-8993(92)91448-N. [DOI] [PubMed] [Google Scholar]
- 471.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334: 292–295, 1996. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 472.Davis JF, Choi DL, Schurdak JD, Fitzgerald MF, Clegg DJ, Lipton JW, Figlewicz DP, Benoit SC. Leptin regulates energy balance and motivation through action at distinct neural circuits. Biol Psychiatry 69: 668–674, 2011. doi: 10.1016/j.biopsych.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Ritter RC, Campos CA, Nasse J, Peters JH. Vagal afferent signaling and the integration of direct and indirect controls of food intake. In: Appetite and Food Intake-Central Control, edited by Harris RB. Boca Raton, FL: CRC Press, 2017, p. 229–257. [PubMed] [Google Scholar]
- 474.Williams DL, Baskin DG, Schwartz MW. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes 55: 3387–3393, 2006. doi: 10.2337/db06-0558. [DOI] [PubMed] [Google Scholar]
- 475.Elmquist JK, BjoRboK C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol 395: 535–547, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 476.Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol 514: 518–532, 2009. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, Liu ZW, Zimmer MR, Jeong JK, Szigeti-Buck K, Gao Y, Garcia-Caceres C, Yi CX, Salmaso N, Vaccarino FM, Chowen J, Diano S, Dietrich MO, Tschöp MH, Horvath TL. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17: 908–910, 2014. doi: 10.1038/nn.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Murphy KT, Schwartz GJ, Nguyen NL, Mendez JM, Ryu V, Bartness TJ. Leptin-sensitive sensory nerves innervate white fat. Am J Physiol Endocrinol Metab 304: E1338–E1347, 2013. doi: 10.1152/ajpendo.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Niijima A. Hepatoportal leptin sensors and their reflex effects on autonomic outflow in the rat. J Obesity 2011: 516842, 2011. doi: 10.1155/2011/516842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Grill HJ. Leptin and the systems neuroscience of meal size control. Front Neuroendoc 31: 61–78, 2010. doi: 10.1016/j.yfrne.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S, Diano S, Horvath TL, Seeley RJ, Becker JB, Münzberg H, Myers MG. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab 10: 89–98, 2009. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Zhang Y, Kerman IA, Laque A, Nguyen P, Faouzi M, Louis GW, Jones JC, Rhodes C, Münzberg H. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J Neurosci 31: 1873–1884, 2011. doi: 10.1523/JNEUROSCI.3223-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Rezai-Zadeh K, Yu S, Jiang Y, Laque A, Schwartzenburg C, Morrison CD, Derbenev AV, Zsombok A, Münzberg H. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol Metab 3: 681–693, 2014. doi: 10.1016/j.molmet.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Lee SJ, Kirigiti M, Lindsley SR, Loche A, Madden CJ, Morrison SF, Smith MS, Grove KL. Efferent projections of neuropeptide Y-expressing neurons of the dorsomedial hypothalamus in chronic hyperphagic models. J Comp Neurol 521: 1891–1914, 2013. doi: 10.1002/cne.23265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Goforth PB, Leinninger GM, Patterson CM, Satin LS, Myers MG. Leptin acts via lateral hypothalamic area neurotensin neurons to inhibit orexin neurons by multiple GABA-independent mechanisms. J Neurosci 34: 11405–11415, 2014. doi: 10.1523/JNEUROSCI.5167-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Villanueva EC, Myers MG. Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes 32: S8–12, 2008. doi: 10.1038/ijo.2008.82,10.1038/ijo.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Fulton S, Woodside B, Shizgal P. Modulation of brain reward circuitry by leptin. Science 287: 125–128, 2000. doi: 10.1126/science.287.5450.125. [DOI] [PubMed] [Google Scholar]
- 488.Scott MM, Williams KW, Rossi J, Lee CE, Elmquist JK. Leptin receptor expression in hindbrain Glp-1 neurons regulates food intake and energy balance in mice. J Clin Invest 121: 2413–2421, 2011. doi: 10.1172/JCI43703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Harris RB. Loss of leptin receptor-expressing cells in the hindbrain decreases forebrain leptin sensitivity. Am J Physiol Endocrinol Metab 318: E806–E816, 2020. doi: 10.1152/ajpendo.00020.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Zheng H, Patterson LM, Rhodes CJ, Louis GW, Skibicka KP, Grill HJ, Myers MG Jr, Berthoud HR. A potential role for hypothalamomedullary POMC projections in leptin-induced suppression of food intake. Am J Physiol Regul Integr Comp Physiol 298: R720–R728, 2010. doi: 10.1152/ajpregu.00619.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Desai BN, Harris RB. Integrated effects of leptin in the forebrain and hindbrain of male rats. Endocrinology 154: 2663–2675, 2013. doi: 10.1210/en.2013-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Vaill MI, Desai BN, Harris RB. Blockade of the cerebral aqueduct in rats provides evidence of antagonistic leptin responses in the forebrain and hindbrain. Am J Physiol Endocrinol Metab 306: E414–E423, 2014. doi: 10.1152/ajpendo.00661.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Harris RB, Desai BN. Fourth-ventricle leptin infusions dose-dependently activate hypothalamic signal transducer and activator of transcription 3. Am J Physiol Endocrinol Metab 311: E939–E948, 2016. doi: 10.1152/ajpendo.00343.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Desai BN, Harris RB. Leptin in the hindbrain facilitates phosphorylation of STAT3 in the hypothalamus. Am J Physiol Endocrinol Metab 308: E351–E361, 2015. doi: 10.1152/ajpendo.00501.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Seamon M, Ahn WM, Li AJ, Ritter S, Harris RB. Leptin receptor-expressing neurons in ventromedial nucleus of the hypothalamus contribute to weight loss caused by fourth ventricle leptin infusions. Am J Physiol Endocrinol Metab 317: E586–E596, 2019. doi: 10.1152/ajpendo.00205.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Maniscalco JW, Rinaman L. Systemic leptin dose-dependently increases STAT3 phosphorylation within hypothalamic and hindbrain nuclei. Am J Physiol Regul Integr Comp Physiol 306: R576–R585, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Harris RB. Low-dose leptin infusion in the fourth ventricle of rats enhances the response to third-ventricle leptin injection. Am J Physiol Endocrinol Metab 313: E134–E147, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris‐Leucoagglutinin study in the rat. J Comp Neurol 348: 41–79, 1994. doi: 10.1002/cne.903480103. [DOI] [PubMed] [Google Scholar]
- 499.Lindberg D, Chen P, Li C. Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J Comp Neurol 521: 3167–3190, 2013. [DOI] [PubMed] [Google Scholar]
- 500.Meek TH, Nelson JT, Matsen ME, Dorfman MD, Guyenet SJ, Damian V, Allison MB, Scarlett JM, Nguyen HT, Thaler JP, Olson DP, Myers MG, Schwartz MW, Morton GJ. Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A 113: E2073–E2082, 2016. doi: 10.1073/pnas.1521160113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Watts AG. Dehydration-associated anorexia: development and rapid reversal. Physiol Behav 65: 871–878, 1999. [DOI] [PubMed] [Google Scholar]
- 502.Watts AG. Understanding the neural control of ingestive behaviors: helping to separate cause from effect with dehydration-associated anorexia. Horm Behav 37: 261–283, 2000. [DOI] [PubMed] [Google Scholar]
- 503.Watts AG, Salter DS, Neuner CM. Neural network interactions and ingestive behavior control during anorexia. Physiol Behav 91: 389–396, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Watts AG, Boyle CN. The functional architecture of dehydration-anorexia. Physiol Behav 100: 472–477, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Salter-Venzon D, Watts AG. Site-specific attenuation of food intake but not the latency to eat after hypothalamic injections of neuropeptide Y in dehydrated-anorexic rats. Am J Physiol Regul Integr Comp Physiol 297: R1813–R1821, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Salter D, Watts AG. Differential suppression of hyperglycemic, feeding, and neuroendocrine responses in anorexia. Am J Physiol Regul Integr Comp Physiol 284: R174–R182, 2003. [DOI] [PubMed] [Google Scholar]
- 507.Salter-Venzon D, Watts AG. The role of hypothalamic ingestive behavior controllers in generating dehydration anorexia: a Fos mapping study. Am J Physiol Regul Integr Comp Physiol 295: R1009–R1019, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Watts AG, Sanchez-Watts G. Rapid and preferential activation of Fos protein in hypocretin/orexin neurons following the reversal of dehydration-anorexia. J Comp Neurol 502: 768–782, 2007. [DOI] [PubMed] [Google Scholar]
- 509.García-Luna C, Soberanes-Chávez P, de Gortari P. Impaired hypothalamic cocaine- and amphetamine-regulated transcript expression in lateral hypothalamic area and paraventricular nuclei of dehydration-induced anorexic rats. J Neuroendoc 29: e12541, 2017. doi: 10.1111/jne.12541. [DOI] [PubMed] [Google Scholar]
- 510.Woodworth HL, Brown JA, Batchelor HM, Bugescu R, Leinninger GM. Determination of neurotensin projections to the ventral tegmental area in mice. Neuropeptides 68: 57–74, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Kelly AB, Watts AG. Mediation of dehydration-induced peptidergic gene expression in the rat lateral hypothalamic area by forebrain afferent projections. J Comp Neurol 370: 231–246, 1996. [CrossR doi:. [DOI] [PubMed] [Google Scholar]
- 512.Watts AG. Osmotic stimulation differentially affects cellular levels of corticotropin-releasing hormone and neurotensin/neuromedin N mRNAs in the lateral hypothalamic area and central nucleus of the amygdala. Brain Res 581: 208–216, 1992. [DOI] [PubMed] [Google Scholar]
- 513.Watts AG, Kelly AB, Sanchez-Watts G. Neuropeptides and thirst: the temporal response of corticotropin-releasing hormone and neurotensin/neuromedin N gene expression in rat limbic forebrain neurons to drinking hypertonic saline. Behav Neurosci 109: 1146–1157, 1995. [DOI] [PubMed] [Google Scholar]
- 514.Brown JA, Wright A, Bugescu R, Christensen L, Olson DP, Leinninger GM. Distinct subsets of lateral hypothalamic neurotensin neurons are activated by leptin or dehydration. Sci Rep 9: 1873, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Kelly AB, Watts AG. The region of the pontine parabrachial nucleus is a major target of dehydration-sensitive CRH neurons in the rat lateral hypothalamic area. J Comp Neurol 394: 48–63, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 516.Chen Y, Essner RA, Kosar S, Miller OH, Lin YC, Mesgarzadeh S, Knight ZA. Sustained NPY signaling enables AgRP neurons to drive feeding. eLife 8: 1–15, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Seeley RJ, Payne CJ, Woods SC. Neuropeptide-Y fails to increase intraoral intake in rats. Am J Physiol Regul Integr Comp Physiol 268: R423–R427, 1995. [DOI] [PubMed] [Google Scholar]
- 518.Woods SC, Seeley RJ, Porte D Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science 280: 1378–1383, 1998. [DOI] [PubMed] [Google Scholar]
- 519.Ammar AA, Sederholm F, Saito TR, Scheurink A, Johnson AE, Södersten P. NPY-leptin: opposing effects on appetitive and consummatory ingestive behavior and sexual behavior. Am J Physiol Regul Integr Comp Physiol 278: R1627–R1633, 2000. [DOI] [PubMed] [Google Scholar]
- 520.Day DE, Bartness TJ. Fasting-induced increases in food hoarding are dependent on the foraging-effort level. Physiol Behav 78: 655–668, 2003. [DOI] [PubMed] [Google Scholar]
- 521.Butler K. Predatory behavior in laboratory mice: strain and sex comparisons. J Comp Physiol Psych 85: 243–249, 1973. doi: 10.1037/h0035008. [DOI] [PubMed] [Google Scholar]
- 522.Comoli E, Ribeiro-Barbosa ER, Canteras NS. Predatory hunting and exposure to a live predator induce opposite patterns of Fos immunoreactivity in the PAG. Behav Brain Res 138: 17–28, 2003. [DOI] [PubMed] [Google Scholar]
- 523.Comoli E, Ribeiro-Barbosa ER, Negrao N, Goto M, Canteras NS. Functional mapping of the prosencephalic systems involved in organizing predatory behavior in rats. Neuroscience 130: 1055–1067, 2005. [DOI] [PubMed] [Google Scholar]
- 524.Ikeda H, Adachi K, Fujita S, Tomiyama K, Saigusa T, Kobayashi M, Koshikawa N, Waddington JL. Investigating complex basal ganglia circuitry in the regulation of motor behaviour, with particular focus on orofacial movement. Behav Pharmacol 26: 18–32, 2015. [DOI] [PubMed] [Google Scholar]
- 525.Pisa M. Motor functions of the striatum in the rat-critical role of the lateral region in tongue and forelimb reaching. Neuroscience 24: 453–463, 1988. [DOI] [PubMed] [Google Scholar]
- 526.Whishaw IQ, Faraji J, Kuntz JR, Mirza Agha B, Metz GA, Mohajerani MH. The syntactic organization of pasta-eating and the structure of reach movements in the head-fixed mouse. Sci Rep 7: 10987, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Furigo IC, de Oliveira WF, de Oliveira AR, Comoli E, Baldo MV, Mota-Ortiz SR, Canteras NS. The role of the superior colliculus in predatory hunting. Neuroscience 165: 1–15, 2010. [DOI] [PubMed] [Google Scholar]
- 528.Canteras NS. Hypothalamic survival circuits related to social and predatory defenses and their interactions with metabolic control, reproductive behaviors and memory systems. Curr Opin Behavi Sci 24: 7–13, 2018. [Google Scholar]
- 529.Franklin TB. Recent advancements surrounding the role of the periaqueductal gray in predators and prey. Front Behav Neurosci 13: 60, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Gross CT, Canteras NS. The many paths to fear. Nat Rev Neurosci 13: 651–658, 2012. [DOI] [PubMed] [Google Scholar]
- 531.Mota-Ortiz SR, Sukikara MH, Felicio LF, Canteras NS. Afferent connections to the rostrolateral part of the periaqueductal gray: a critical region influencing the motivation drive to hunt and forage. Neural Plast 2009: 612698, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Viskaitis P, Irvine EE, Smith MA, Choudhury AI, Alvarez-Curto E, Glegola JA, Hardy DG, Pedroni SM, Paiva Pessoa MR, Fernando AB, Katsouri L, Sardini A, Ungless MA, Milligan G, Withers DJ. Modulation of SF1 neuron activity coordinately regulates both feeding behavior and associated emotional states. Cell Rep 21: 3559–3572, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Li Y, Zeng J, Zhang J, Yue C, Zhong W, Liu Z, Feng Q, Luo M. Hypothalamic circuits for predation and evasion. Neuron 97: 911–924, 2018. [DOI] [PubMed] [Google Scholar]
- 534.Park SG, Jeong YC, Kim DG, Lee MH, Shin A, Park G, Ryoo J, Hong J, Bae S, Kim CH, Lee PS, Kim D. Medial preoptic circuit induces hunting-like actions to target objects and prey. Nat Neurosci 21: 364–372, 2018. [DOI] [PubMed] [Google Scholar]
- 535.Shang C, Liu A, Li D, Xie Z, Chen Z, Huang M, Li Y, Wang Y, Shen WL, Cao P. A subcortical excitatory circuit for sensory-triggered predatory hunting in mice. Nat Neurosci 22: 909–920, 2019. [DOI] [PubMed] [Google Scholar]
- 536.Zhao ZD, Chen Z, Xiang X, Hu M, Xie H, Jia X, Cai F, Cui Y, Chen Z, Qian L, Liu J, Shang C, Yang Y, Ni X, Sun W, Hu J, Cao P, Li H, Shen WL. Zona incerta GABAergic neurons integrate prey-related sensory signals and induce an appetitive drive to promote hunting. Nat Neurosci 22: 921–932, 2019. [DOI] [PubMed] [Google Scholar]
- 537.Keen-Rhinehart E, Dailey MJ, Bartness T. Physiological mechanisms for food-hoarding motivation in animals. Philos Trans R Soc Lond B Biol Sci 365: 961–975, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Borer KT, Allen ER, Smalley RE, Lundell L, Stockton J. Recovery from energy deficit in golden hamsters. Am J Physiol Regul Comp Physiol 248: R439–R446, 1985. [DOI] [PubMed] [Google Scholar]
- 539.Day DE, Bartness TJ. Effects of foraging effort on body fat and food hoarding in Siberian hamsters. J Exp Zool 289: 162–171, 2001. doi:. [DOI] [PubMed] [Google Scholar]
- 540.Keen-Rhinehart E, Bartness TJ. Peripheral ghrelin injections stimulate food intake, foraging, and food hoarding in Siberian hamsters. Am J Physiol Regul Integr Comp Physiol 288: R716–R722, 2005. [DOI] [PubMed] [Google Scholar]
- 541.Keen-Rhinehart E, Bartness TJ. NPY Y1 receptor is involved in ghrelin- and fasting-induced increases in foraging, food hoarding, and food intake. Am J Physiol Regul Integr Comp Physiol 292: R1728–R1737, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Dailey MJ, Bartness TJ. Appetitive and consummatory ingestive behaviors stimulated by PVH and perifornical area NPY injections. Am J Physiol Regul Integr Comp Physiol 296: R877–R892, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Day DE, Bartness TJ. Agouti-related protein increases food hoarding more than food intake in Siberian hamsters. Am J Physiol Regul Integr Comp Physiol 286: R38–R45, 2004. doi: 10.1152/ajpregu.00284.2003. [DOI] [PubMed] [Google Scholar]
- 544.Thomas MA, Tran V, Ryu V, Xue B, Bartness TJ. AgRP knockdown blocks long-term appetitive, but not consummatory, feeding behaviors in Siberian hamsters. Physiol Behav 190: 61–70, 2018. doi: 10.1016/j.physbeh.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Dailey MJ, Bartness TJ. Arcuate nucleus destruction does not block food deprivation-induced increases in food foraging and hoarding. Brain Res 1323: 94–108, 2010. doi: 10.1016/j.brainres.2010.01.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Livneh Y, Ramesh RN, Burgess CR, Levandowski KM, Madara JC, Fenselau H, Goldey GJ, Diaz VE, Jikomes N, Resch JM, Lowell BB, Andermann ML. Homeostatic circuits selectively gate food cue responses in insular cortex. Nature 546: 611–616, 2017. doi: 10.1038/nature22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Garcia J, Kimeldorf DJ, Koelling RA. Conditioned aversion to saccharin resulting from exposure to gamma radiation. Science 122: 157–158, 1955. doi: 10.1126/science.122.3160.157. [DOI] [PubMed] [Google Scholar]
- 548.Sclafani A. Post-ingestive positive controls of ingestive behavior. Appetite 36: 79–83, 2001. doi: 10.1006/appe.2000.0370. [DOI] [PubMed] [Google Scholar]
- 549.Breslin PA, Davidson TL, Grill HJ. Conditioned reversal of reactions to normally avoided tastes. Physiol Behav 47: 535–538, 1990. doi: 10.1016/0031-9384(90)90122-K. [DOI] [PubMed] [Google Scholar]
- 550.Foltin RW, Rolls BJ, Moran TH, Kelly TH, McNelis AL, Fischman MW. Caloric, but not macronutrient, compensation by humans for required-eating occasions with meals and snack varying in fat and carbohydrate. Am J Clin Nutr 55: 331–342, 1992. doi: 10.1093/ajcn/55.2.331. [DOI] [PubMed] [Google Scholar]
- 551.Shide DJ, Rolls BJ. Information about the fat content of preloads influences energy intake in healthy women. J Am Diet Assoc 95: 993–998, 1995. doi: 10.1016/S0002-8223(95)00273-1. [DOI] [PubMed] [Google Scholar]
- 552.de Castro JM. Prior day’s intake has macronutrient-specific delayed negative feedback effects on the spontaneous food intake of free-living humans. J Nutr 128: 61–67, 1998. doi: 10.1093/jn/128.1.61. [DOI] [PubMed] [Google Scholar]
- 553.Maida A, Zota A, Sjoberg KA, Schumacher J, Sijmonsma TP, Pfenninger A, Christensen MM, Gantert T, Fuhrmeister J, Rothermel U, Schmoll D, Heikenwalder M, Iovanna JL, Stemmer K, Kiens B, Herzig S, Rose AJ. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J Clin Invest 126: 3263–3278, 2016. doi: 10.1172/JCI85946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Hrupka BJ, Lin YM, Gietzen DW, Rogers QR. Small changes in essential amino acid concentrations alter diet selection in amino acid-deficient rats. J Nutr 127: 777–784, 1997. doi: 10.1093/jn/127.5.777. [DOI] [PubMed] [Google Scholar]
- 555.Hill CM, Berthoud HR, Munzberg H, Morrison CD. Homeostatic sensing of dietary protein restriction: a case for FGF21. Front Neuroendoc 51: 125–131, 2018. doi: 10.1016/j.yfrne.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Hill CM, Laeger T, Dehner M, Albarado DC, Clarke B, Wanders D, Burke SJ, Collier JJ, Qualls-Creekmore E, Solon-Biet SM, Simpson SJ, Berthoud HR, Munzberg H, Morrison CD. FGF21 signals protein status to the brain and adaptively regulates food choice and metabolism. Cell Rep 27: 2934–2947, 2019. doi: 10.1016/j.celrep.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.DiFeliceantonio AG, Coppin G, Rigoux L, Thanarajah SE, Dagher A, Tittgemeyer M, Small DM. Supra-additive effects of combining fat and carbohydrate on food reward. Cell Metab 28: 33–44, 2018. doi: 10.1016/j.cmet.2018.05.018. [DOI] [PubMed] [Google Scholar]
- 558.Brunstrom JM, Shakeshaft NG, Scott-Samuel NE. Measuring ‘expected satiety’ in a range of common foods using a method of constant stimuli. Appetite 51: 604–614, 2008. doi: 10.1016/j.appet.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 559.Brunstrom JM, Jarvstad A, Griggs RL, Potter C, Evans NR, Martin AA, Brooks JC, Rogers PJ. Large portions encourage the selection of palatable rather than filling foods. J Nutr 146: 2117–2123, 2016. doi: 10.3945/jn.116.235184. [DOI] [PubMed] [Google Scholar]
- 560.Stanley BG, Daniel DR, Chin AS, Leibowitz SF. Paraventricular nucleus injections of peptide YY and neuropeptide Y preferentially enhance carbohydrate ingestion. Peptides 6: 1205–1211, 1985. doi: 10.1016/0196-9781(85)90452-8. [DOI] [PubMed] [Google Scholar]
- 561.Leibowitz SF, Alexander JT. Analysis of neuropeptide Y-induced feeding: dissociation of Y1 and Y2 receptor effects on natural meal patterns. Peptides 12: 1251–1260, 1991. doi: 10.1016/0196-9781(91)90203-2. [DOI] [PubMed] [Google Scholar]
- 562.Thibault L, Booth DA. Macronutrient-specific dietary selection in rodents and its neural bases. Neurosci Biobehav Rev 23: 457–528, 1999. doi: 10.1016/S0149-7634(98)00047-5. [DOI] [PubMed] [Google Scholar]
- 563.Smith BK, Berthoud HR, York DA, Bray GA. Differential effects of baseline macronutrient preferences on macronutrient selection after galanin, NPY, and an overnight fast. Peptides 18: 207–211, 1997. doi: 10.1016/S0196-9781(96)00318-X. [DOI] [PubMed] [Google Scholar]
- 564.van den Heuvel JK, Furman K, Gumbs MC, Eggels L, Opland DM, Land BB, Kolk SM, Narayanan NS, Fliers E, Kalsbeek A, DiLeone RJ, la Fleur SE. Neuropeptide Y activity in the nucleus accumbens modulates feeding behavior and neuronal activity. Biol Psychiatry 77: 633–641, 2015. doi: 10.1016/j.biopsych.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Olszewski PK, Klockars A, Olszewska AM, Fredriksson R, SchiöTh HB, Levine AS. Molecular, immunohistochemical, and pharmacological evidence of oxytocin’s role as inhibitor of carbohydrate but not fat intake. Endocrinology 151: 4736–4744, 2010. doi: 10.1210/en.2010-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Morris RG, Frey U. Hippocampal synaptic plasticity: role in spatial learning or the automatic recording of attended experience? In: The Hippocampal and Parietal Foundations of Spatial Cognition, edited by Burgess N, Jeffery KJ, O’Keefe J.. London: Oxford University Press, 1999, p. 220–246. [Google Scholar]
- 567.O’Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res 34: 171–175, 1971. doi: 10.1016/S0079-6123(08)63963-1,10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- 568.O’Keefe J, Nadel L. Precis of O’Keefe and Nadel’s the hippocampus as a cognitive map. Behav Brain Sci 2: 487–533, 1979. doi: 10.1017/S0140525X00063949. [DOI] [Google Scholar]
- 569.Kjelstrup KB, Solstad T, Brun VH, Hafting T, Leutgeb S, Witter MP, Moser EI, Moser MB. Finite scale of spatial representation in the hippocampus. Science 321: 140–143, 2008. doi: 10.1126/science.1157086. [DOI] [PubMed] [Google Scholar]
- 570.Rowland DC, Roudi Y, Moser MB, Moser EI. Ten years of grid cells. Annu Rev Neurosci 39: 19–40, 2016. doi: 10.1146/annurev-neuro-070815-013824. [DOI] [PubMed] [Google Scholar]
- 571.Moser EI, Moser MB, McNaughton BL. Spatial representation in the hippocampal formation: a history. Nat Neurosci 20: 1448–1464, 2017. doi: 10.1038/nn.4653. [DOI] [PubMed] [Google Scholar]
- 572.Burwell RD, Amaral DG. Cortical afferents of the perirhinal, postrhinal, and entorhinal cortices of the rat. J Comp Neurol 398: 179–205, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 573.Whitlock JR, Sutherland RJ, Witter MP, Moser MB, Moser EI. Navigating from hippocampus to parietal cortex. Proc Natl Acad Sci U S A 105: 14755–14762, 2008. doi: 10.1073/pnas.0804216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Sato N, Sakata H, Tanaka YL, Taira M. Navigation-associated medial parietal neurons in monkeys. Proc Natl Acad Sci U S A 103: 17001–17006, 2006. doi: 10.1073/pnas.0604277103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Taira M, Mine S, Georgopoulos AP, Murata A, Sakata H. Parietal cortex neurons of the monkey related to the visual guidance of hand movement. Exp Brain Res 83: 29–36, 1990. doi: 10.1007/bf00232190. [DOI] [PubMed] [Google Scholar]
- 576.McNaughton BL, Mizumori SJ, Barnes CA, Leonard BJ, Marquis M, Green EJ. Cortical representation of motion during unrestrained spatial navigation in the rat. Cereb Cortex 4: 27–39, 1994. doi: 10.1093/cercor/4.1.27. [DOI] [PubMed] [Google Scholar]
- 577.Goodale MA, Milner AD. Separate visual pathways for perception and action. Trends Neurosci 15: 20–25, 1992. doi: 10.1016/0166-2236(92)90344-8. [DOI] [PubMed] [Google Scholar]
- 578.Ekstrom AD, Huffman DJ, Starrett M. Interacting networks of brain regions underlie human spatial navigation: a review and novel synthesis of the literature. J Neurophysiol 118: 3328–3344, 2017. doi: 10.1152/jn.00531.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Ns C, Dickinson A. Episodic-like memory during cache recovery by scrub jays. Nature 395: 272–274, 1998. doi: 10.1038/26216. [DOI] [PubMed] [Google Scholar]
- 580.Hampton RR, Hampstead BM, Murray EA. Selective hippocampal damage in rhesus monkeys impairs spatial memory in an open-field test. Hippocampus 14: 808–818, 2004. doi: 10.1002/hipo.10217. [DOI] [PubMed] [Google Scholar]
- 581.Jarrard LE, Davidson TL, Bowring B. Functional differentiation within the medial temporal lobe in the rat. Hippocampus 14: 434–449, 2004. doi: 10.1002/hipo.10194. [DOI] [PubMed] [Google Scholar]
- 582.Hebben N, Corkin S, Eichenbaum H, Shedlack K. Diminished ability to interpret and report internal states after bilateral medial temporal resection: case H.M. Behav Neurosci 99: 1031–1039, 1985. doi: 10.1037/0735-7044.99.6.1031. [DOI] [PubMed] [Google Scholar]
- 583.Henderson YO, Smith GP, Parent MB. Hippocampal neurons inhibit meal onset. Hippocampus 23: 100–107, 2013. doi: 10.1002/hipo.22062. [DOI] [PubMed] [Google Scholar]
- 584.Hannapel RC, Henderson YH, Nalloor R, Vazdarjanova A, Parent MB. Ventral hippocampal neurons inhibit postprandial energy intake. Hippocampus 27: 274–284, 2017. doi: 10.1002/hipo.22692. [DOI] [PubMed] [Google Scholar]
- 585.Hannapel R, Ramesh J, Ross A, LaLumiere RT, Roseberry AG, Parent MB. Postmeal optogenetic inhibition of dorsal or ventral hippocampal pyramidal neurons increases future intake. eNeuro 6: ENEURO.0457-18.2018, 2019. doi: 10.1523/ENEURO.0457-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Clifton PG, Vickers SP, Somerville EM. Little and often: ingestive behavior patterns following hippocampal lesions in rats. Behav Neurosci 112: 502–511, 1998. doi: 10.1037/0735-7044.112.3.502. [DOI] [PubMed] [Google Scholar]
- 587.Davidson TL, Kanoski SE, Chan K, Clegg DJ, Benoit SC, Jarrard LE. Hippocampal lesions impair retention of discriminative responding based on energy state cues. Behav Neurosci 124: 97–105, 2010. doi: 10.1037/a0018402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Johnson AW. Eating beyond metabolic need: how environmental cues influence feeding behavior. Trends Neurosci 36: 101–109, 2013. doi: 10.1016/j.tins.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 589.Cornell CE, Rodin J, Weingarten H. Stimulus-induced eating when satiated. Physiol Behav 45: 695–704, 1989. doi: 10.1016/0031-9384(89)90281-3. [DOI] [PubMed] [Google Scholar]
- 590.Weingarten HP. Meal initiation controlled by learned cues: basic behavioral properties. Appetite 5: 147–158, 1984. doi: 10.1016/S0195-6663(84)80035-5. [DOI] [PubMed] [Google Scholar]
- 591.Boggiano MM, Dorsey JR, Thomas JM, Murdaugh DL. The Pavlovian power of palatable food: lessons for weight-loss adherence from a new rodent model of cue-induced overeating. Int J Obes 33: 693–701, 2009. doi: 10.1038/ijo.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Petrovich GD, Holland PC, Gallagher M. Amygdalar and prefrontal pathways to the lateral hypothalamus are activated by a learned cue that stimulates eating. J Neurosci 25: 8295–8302, 2005. doi: 10.1523/JNEUROSCI.2480-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Petrovich GD. Forebrain networks and the control of feeding by environmental learned cues. Physiol Behav 121: 10–18, 2013. doi: 10.1016/j.physbeh.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Petrovich GD. Lateral hypothalamus as a motivation-cognition interface in the control of feeding behavior. Front Syst Neurosci 12: 14, 2018. doi: 10.3389/fnsys.2018.00014,10.3389/fnhum.2018.00014,10.3389/fnins.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Sun X, Kroemer NB, Veldhuizen MG, Babbs AE, de Araujo IE, Gitelman DR, Sherwin RS, Sinha R, Small DM. Basolateral amygdala response to food cues in the absence of hunger is associated with weight gain susceptibility. J Neurosci 35: 7964–7976, 2015. doi: 10.1523/JNEUROSCI.3884-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Burgess CR, Livneh Y, Ramesh RN, Andermann ML. Gating of visual processing by physiological need. Curr Opin Neurobiol 49: 16–23, 2018. doi: 10.1016/j.conb.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Parkes SL, Balleine BW. Incentive memory: evidence the basolateral amygdala encodes and the insular cortex retrieves outcome values to guide choice between goal-directed actions. J Neurosci 33: 8753–8763, 2013. doi: 10.1523/JNEUROSCI.5071-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Azevedo EP, Pomeranz L, Cheng J, Schneeberger M, Vaughan R, Stern SA, Tan B, Doerig K, Greengard P, Friedman JM. A role of Drd2 hippocampal neurons in context-dependent food intake. Neuron 102: 873–886, 2019. doi: 10.1016/j.neuron.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 599.Stern SA, Doerig KR, Azevedo EP, Stoffel E, Friedman JM. Control of non-homeostatic feeding in sated mice using associative learning of contextual food cues. Mol Psychiatry 25: 666–679, 2020. doi: 10.1038/s41380-018-0072-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Hsu TM, Suarez AN, Kanoski SE. Ghrelin: a link between memory and ingestive behavior. Physiol Behav 162: 10–17, 2016. doi: 10.1016/j.physbeh.2016.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Dailey MJ, Moran TH, Holland PC, Johnson AW. The antagonism of ghrelin alters the appetitive response to learned cues associated with food. Behav Brain Res 303: 191–200, 2016. doi: 10.1016/j.bbr.2016.01.040. [DOI] [PubMed] [Google Scholar]
- 602.Walker AK, Ibia IE, Zigman JM. Disruption of cue-potentiated feeding in mice with blocked ghrelin signaling. Physiol Behav 108: 34–43, 2012. doi: 10.1016/j.physbeh.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab 7: 400–409, 2008. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 604.Kanoski SE, Fortin SM, Ricks KM, Grill HJ. Ghrelin signaling in the ventral hippocampus stimulates learned and motivational aspects of feeding via PI3K-Akt signaling. Biol Psychiatry 73: 915–923, 2013. doi: 10.1016/j.biopsych.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Woloszyn J, Yanagisawa M, Lutter M, Zigman JM. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry 67: 880–886, 2010. doi: 10.1016/j.biopsych.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Cole S, Mayer HS, Petrovich GD. Orexin/hypocretin-1 receptor antagonism selectively reduces cue-induced feeding in sated rats. Sci Rep 5: 16143, 2015. doi: 10.1038/srep16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Swanson LW, Sanchez-Watts G, Watts AG. Comparison of melanin-concentrating hormone and hypocretin/orexin mRNA expression patterns in a new parceling scheme of the lateral hypothalamic zone. Neurosci Lett 387: 80–84, 2005. doi: 10.1016/j.neulet.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 608.Sherwood A, Holland PC, Adamantidis A, Johnson AW. Deletion of melanin concentrating hormone receptor-1 disrupts overeating in the presence of food cues. Physiol Behav 152: 402–407, 2015. doi: 10.1016/j.physbeh.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 609.Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol 436: 430–455, 2001. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- 610.Wood M, Adil O, Wallace T, Fourman S, Wilson SP, Herman JP, Myers B. Infralimbic prefrontal cortex structural and functional connectivity with the limbic forebrain: a combined viral genetic and optogenetic analysis. Brain Struct Funct 224: 73–97, 2019. doi: 10.1007/s00429-018-1762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Petrovich GD, Canteras NS, Swanson LW. Combinatorial amygdalar inputs to hippocampal domains and hypothalamic behavior systems. Brain Res Brain Res Rev 38: 247–289, 2001. doi: 10.1016/S0165-0173(01)00080-7. [DOI] [PubMed] [Google Scholar]
- 612.Risold PY, Swanson LW. Structural evidence for functional domains in the rat hippocampus. Science 272: 1484–1486, 1996. doi: 10.1126/science.272.5267.1484. [DOI] [PubMed] [Google Scholar]
- 613.Risold PY, Swanson LW. Connections of the rat lateral septal complex. Brain Res Brain Res Rev 24: 115–195, 1997. doi: 10.1016/S0165-0173(97)00009-X. [DOI] [PubMed] [Google Scholar]
- 614.Risold PY, Thompson RH, Swanson LW. The structural organization of connections between hypothalamus and cerebral cortex. Brain Res Brain Res Rev 24: 197–254, 1997. doi: 10.1016/S0165-0173(97)00007-6. [DOI] [PubMed] [Google Scholar]
- 615.Barbier M, Fellmann D, Risold PY. Morphofunctional organization of the connections from the medial and intermediate parts of the central nucleus of the amygdala into distinct divisions of the lateral hypothalamic area in the rat. Front Neurol 9: 199–112, 2018. doi: 10.3389/fneur.2018.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Canteras NS, Swanson LW. Projections of the ventral subiculum to the amygdala, septum, and hypothalamus-a phal anterograde tract-tracing study in the rat. J Comp Neurol 324: 180–194, 1992. doi: 10.1002/cne.903240204. [DOI] [PubMed] [Google Scholar]
- 617.Cenquizca LA, Swanson LW. Analysis of direct hippocampal cortical field CA1 axonal projections to diencephalon in the rat. J Comp Neurol 497: 101–114, 2006. doi: 10.1002/cne.20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Cullinan WE, Herman JP, Watson SJ. Ventral subicular interaction with the hypothalamic paraventricular nucleus-evidence for a relay in the bed nucleus of the stria terminalis. J Comp Neurol 332: 1–20, 1993. doi: 10.1002/cne.903320102. [DOI] [PubMed] [Google Scholar]
- 619.Dong HW, Petrovich GD, Swanson LW. Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Brain Res Rev 38: 192–246, 2001. doi: 10.1016/S0165-0173(01)00079-0. [DOI] [PubMed] [Google Scholar]
- 620.Petrovich GD, Risold PY, Swanson LW. Organization of projections from the basomedial nucleus of the amygdala: a PHAL study in the rat. J Comp Neurol 374: 387–420, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 621.Reppucci CJ, Petrovich GD. Organization of connections between the amygdala, medial prefrontal cortex, and lateral hypothalamus: a single and double retrograde tracing study in rats. Brain Struct Funct 221: 2937–2962, 2016. doi: 10.1007/s00429-015-1081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Thompson RH, Swanson LW. Hypothesis-driven structural connectivity analysis supports network over hierarchical model of brain architecture. Proc Natl Acad Sci U S A 107: 15235–15239, 2010. doi: 10.1073/pnas.1009112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Grossman SP. Direct adrenergic and cholinergic stimulation of hypothalamic mechanisms. Am J Physiol 202: 872–882, 1962. doi: 10.1152/ajplegacy.1962.202.5.872. [DOI] [PubMed] [Google Scholar]
- 624.Khan AM. Controlling feeding behavior by chemical or gene-directed targeting in the brain: what’s so spatial about our methods? Front Neurosci 7: 182, 2013. doi: 10.3389/fnins.2013.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Sagar SM, Sharp FR, Curran T. Expression of c-fos protein in brain: metabolic mapping at the cellular level. Science 240: 1328–1331, 1988. doi: 10.1126/science.3131879. [DOI] [PubMed] [Google Scholar]
- 626.Zséli G, Vida B, Martinez A, Lechan RM, Khan AM, Fekete C. Elucidation of the anatomy of a satiety network: focus on connectivity of the parabrachial nucleus in the adult rat. J Comp Neurol 524: 2803–2827, 2016. doi: 10.1002/cne.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Stratford TR, Kelley AE. Evidence of a functional relationship between the nucleus accumbens shell and lateral hypothalamus subserving the control of feeding behavior. J Neurosci 19: 11040–11048, 1999. doi: 10.1523/JNEUROSCI.19-24-11040.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14: 351–355, 2011. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Sternson SM, Atasoy D, Betley JN, Henry FE, Xu S. An emerging technology framework for the neurobiology of appetite. Cell Metab 23: 234–253, 2016. doi: 10.1016/j.cmet.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 630.Atasoy D, Sternson SM. Chemogenetic tools for causal cellular and neuronal biology. Physiol Rev 98: 391–418, 2018. doi: 10.1152/physrev.00009.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Bota M, Hong-Wei D, Swanson LW. From gene networks to brain networks. Nat Neurosci 6: 795–799, 2003. doi: 10.1038/nn1096. [DOI] [PubMed] [Google Scholar]
- 632.Hahn JD, Sporns O, Watts AG, Swanson LW. Macroscale intrinsic network architecture of the hypothalamus. Proc Natl Acad Sci U S A 116: 8018–8027, 2019. doi: 10.1073/pnas.1819448116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Bota M, Dong HW, Swanson LW. Combining collation and annotation efforts toward completion of the rat and mouse connectomes in BAMS. Front Neuroinform 6: 2, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Card JP, Enquist LW. Transneuronal circuit analysis with pseudorabies viruses. Curr Protoc Neurosci 68: 1.5.1–1.5.39, 2014. doi: 10.1002/0471142301.ns0105s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Sales-Pardo M, Guimerà R, Moreira AA, Amaral LA. Extracting the hierarchical organization of complex systems. Proc Natl Acad Sci U S A 104: 15224–15229, 2007. doi: 10.1073/pnas.0703740104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Swanson LW, Sporns O, Hahn JD. The network organization of rat intrathalamic macroconnections and a comparison with other forebrain divisions. Proc Natl Acad Sci U S A 116: 13661–13669, 2019. doi: 10.1073/pnas.1905961116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Dietrich MO, Zimmer MR, Bober J, Horvath TL. Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell 160: 1222–1232, 2015. doi: 10.1016/j.cell.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Maldonado-Irizarry CS, Swanson CJ, Kelley AE. Glutamate receptors in the nucleus accumbens shell control feeding behavior via the lateral hypothalamus. J Neurosci 15: 6779–6788, 1995. doi: 10.1523/JNEUROSCI.15-10-06779.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Stanley BG, Ha LH, Spears LC, Dee MG. Lateral hypothalamic injections of glutamate, kainic acid,d,l-α-amino-3-hydroxy-5-methyl-isoxazole propionic acid or N-methyl-d-aspartic acid rapidly elicit intense transient eating in rats. Brain Res 613: 88–95, 1993.doi: 10.1016/0006-8993(93)90458-Y. [DOI] [PubMed] [Google Scholar]
- 640.Woods SC, Begg DP. Regulation of the motivation to eat. Curr Top Behav Neurosci 27: 15–34, 2016. doi: 10.1007/7854_2015_381. [DOI] [PubMed] [Google Scholar]
- 641.Micevych PE, Meisel RL. Integrating neural circuits controlling female sexual behavior. Front Syst Neurosci 11: 42, 2017. doi: 10.3389/fnsys.2017.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Herbison AE. The gonadotropin-releasing hormone pulse generator. Endocrinology 159: 3723–3736, 2018. [DOI] [PubMed] [Google Scholar]
- 643.Hashikawa Y, Hashikawa K, Falkner AL, Lin D. Ventromedial hypothalamus and the generation of aggression. Front Syst Neurosci 11: 94, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Canteras NS, Graeff FG. Executive and modulatory neural circuits of defensive reactions: implications for panic disorder. Neurosci Biobehav Rev 46: 352–364, 2014. doi: 10.1016/j.neubiorev.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 645.Lyons DJ, Broberger C. TIDAL WAVES: network mechanisms in the neuroendocrine control of prolactin release. Front Neuroendocrinol 35: 420–438, 2014. doi: 10.1016/j.yfrne.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 646.Fu LY, Van Den Pol AN. Kisspeptin directly excites anorexigenic proopiomelanocortin neurons but inhibits orexigenic neuropeptide Y cells by an indirect synaptic mechanism. J Neurosci 30: 10205–10219, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Nestor CC, Qiu J, Padilla SL, Zhang C, Bosch MA, Fan W, Aicher SA, Palmiter RD, Rønnekleiv OK, Kelly MJ. Optogenetic stimulation of arcuate nucleus Kiss1 neurons reveals a steroid-dependent glutamatergic input to POMC and AgRP neurons in male mice. Mol Endocrinol 30: 630–644, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Talbi R, Navarro VM. Novel insights into the metabolic action of Kiss1 neurons. Endocrine Connections 9: R124–R133, 2020. doi: 10.1530/EC-20-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Qualls-Creekmore E, Münzberg H. Modulation of feeding and associated behaviors by lateral hypothalamic circuits. Endocrinology 159: 3631–3642, 2018. doi: 10.1210/en.2018-00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Burdakov D. How orexin signals bias action: hypothalamic and accumbal circuits. Brain Res 1731: 145943, 2020. doi: 10.1016/j.brainres.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 651.Clarke RE, Verdejo-García A, Andrews ZB. The role of corticostriatal-hypothalamic neural circuits in feeding behaviour: implications for obesity. J Neurochem 147: 715–729, 2018. doi: 10.1111/jnc.14455. [DOI] [PubMed] [Google Scholar]
- 652.Sternson SM, Eiselt AK. Three pillars for the neural control of appetite. Annu Rev Physiol 79: 401–423, 2017. doi: 10.1146/annurev-physiol-021115-104948. [DOI] [PubMed] [Google Scholar]
- 653.Watts AG, Sanchez-Watts G, Kelly AB. Distinct patterns of neuropeptide gene expression in the lateral hypothalamic area and arcuate nucleus are associated with dehydration-induced anorexia. J. Neurosci 19: 6111–6121, 1999. doi: 10.1523/JNEUROSCI.19-14-06111.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Hahn JD. Comparison of melanin-concentrating hormone and hypocretin/orexin peptide expression patterns in a current parceling scheme of the lateral hypothalamic zone. Neurosci Lett 468: 12–17, 2010. doi: 10.1016/j.neulet.2009.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Vrang N, Larsen PJ, Clausen JT, Kristensen P. Neurochemical characterization of hypothalamic cocaine-amphetamine-regulated transcript neurons. J. Neurosci 19: RC5–RC5, 1999. doi: 10.1523/JNEUROSCI.19-10-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Laque A, Zhang Y, Gettys S, Nguyen TA, Bui K, Morrison CD, Münzberg H. Leptin receptor neurons in the mouse hypothalamus are colocalized with the neuropeptide galanin and mediate anorexigenic leptin action. Am J Physiol Endoc Metab 304: E999–E1011, 2013. doi: 10.1152/ajpendo.00643.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18: 9996–10015, 1998. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Harris GC, Jones A. G. Arousal and reward: a dichotomy in orexin function. Trends Neurosci 29: 571–577, 2006. doi: 10.1016/j.tins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 659.Harris GC, Wimmer M, Randall-Thompson JF, Jones A. G. Lateral hypothalamic orexin neurons are critically involved in learning to associate an environment with morphine reward. Behav Brain Res 183: 43–51, 2007. doi: 10.1016/j.bbr.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Iyer M, Essner RA, Klingenberg B, Carter ME. Identification of discrete, intermingled hypocretin neuronal populations. J Comp Neurol 526: 2937–2954, 2018. doi: 10.1002/cne.24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Papp RS, Palkovits M. Brainstem projections of neurons located in various subdivisions of the dorsolateral hypothalamic area-an anterograde tract-tracing study. Front Neuroanat 8: 34, 2014. doi: 10.3389/fnana.2014.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Hahn JD, Swanson LW. Distinct patterns of neuronal inputs and outputs of the juxtaparaventricular and suprafornical regions of the lateral hypothalamic area in the male rat. Brain Res Rev 64: 14–103, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Goto M, Canteras NS, Burns G, Swanson LW. Projections from the subfornical region of the lateral hypothalamic area. J Comp Neurol 493: 412–438, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Betley JN, Cao ZF, Ritola KD, Sternson SM. Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell 155: 1337–1350, 2013. doi: 10.1016/j.cell.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Rangel MJ, Baldo MV, Canteras NS, Hahn JD. Evidence of a role for the lateral hypothalamic area juxtadorsomedial region (LHAjd) in defensive behaviors associated with social defeat. Front Syst Neurosci 10: 92, 2016. doi: 10.3389/fnsys.2016.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Anand BK. Nervous regulation of food intake. Physiol Rev 41: 677–708, 1961. doi: 10.1152/physrev.1961.41.4.677. [DOI] [PubMed] [Google Scholar]
- 667.Nieuwenhuys R, Geeraedts LM, Veening JG. The medial forebrain bundle of the rat. I. General introduction. J Comp Neurol 206: 49–81, 1982. doi: 10.1002/cne.902060106. [DOI] [PubMed] [Google Scholar]
- 668.Winn P, Clark A, Hastings M, Clark J, Latimer M, Rugg E, Brownlee B. Excitotoxic lesions of the lateral hypothalamus made by N-methyl-d-aspartate in the rat: behavioural, histological and biochemical analyses. Exp Brain Res 82: 628–636, 1990. doi: 10.1007/bf00228804. [DOI] [PubMed] [Google Scholar]
- 669.Booth DA. The behavioral and neural sciences of ingestion. In: Handbook of Behavioral Neurobiology Neurobiology of Food and Fluid Intake, edited by Stricker E. New York: Plenum Press, 1990, p. 465–488. [Google Scholar]
- 670.Stanley BG, Urstadt KR, Charles JR, Kee T. Glutamate and GABA in lateral hypothalamic mechanisms controlling food intake. Physiol Behav 104: 40–46, 2011. doi: 10.1016/j.physbeh.2011.04.046. [DOI] [PubMed] [Google Scholar]
- 671.Duva MA, Tomkins EM, Moranda LM, Kaplan R, Sukhaseum A, Jimenez A, Stanley BG. Reverse microdialysis of N-methyl-D-aspartic acid into the lateral hypothalamus of rats: effects on feeding and other behaviors. Brain Res 921: 122–132, 2001. doi: 10.1016/S0006-8993(01)03108-0. [DOI] [PubMed] [Google Scholar]
- 672.Duva MA, Tomkins EM, Moranda LM, Kaplan R, Sukhaseum A, Bernardo JP, Stanley BG. Regional differences in feeding and other behaviors elicited by N-methyl-D-aspartic acid in the rodent hypothalamus: a reverse microdialysis mapping study. Brain Res 925: 141–147, 2002. doi: 10.1016/S0006-8993(01)03269-3. [DOI] [PubMed] [Google Scholar]
- 673.Duva MA, Tomkins EM, Moranda LM, Kaplan R, Sukhaseum A, Stanley BG. Origins of lateral hypothalamic afferents associated with N-methyl-d-aspartic acid-elicited eating studied using reverse microdialysis of NMDA and Fluorogold. Neurosci Res 52: 95–106, 2005. doi: 10.1016/j.neures.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 674.Turenius CI, Charles JR, Tsai DH, Ebersole PL, Htut MH, Ngo PT, Lara RN, Stanley BG. The tuberal lateral hypothalamus is a major target for GABAA–but not GABAB-mediated control of food intake. Brain Res 1283: 65–72, 2009. doi: 10.1016/j.brainres.2009.05.064. [DOI] [PubMed] [Google Scholar]
- 675.Mickelsen LE, Bolisetty M, Chimileski BR, Fujita A, Beltrami EJ, Costanzo JT, Naparstek JR, Robson P, Jackson AC. Single-cell transcriptomic analysis of the lateral hypothalamic area reveals molecularly distinct populations of inhibitory and excitatory neurons. Nat Neurosci 22: 642–656, 2019. doi: 10.1038/s41593-019-0349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Rossi MA, Basiri ML, McHenry JA, Kosyk O, Otis JM, van den Munkhof HE, Bryois J, Hübel C, Breen G, Guo W, Bulik CM, Sullivan PF, Stuber GD. Obesity remodels activity and transcriptional state of a lateral hypothalamic brake on feeding. Science 364: 1271–1274, 2019. doi: 10.1126/science.aax1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, Veleta K, Kantak PA, Aita M, Shilling-Scrivo K, Ramakrishnan C, Deisseroth K, Otte S, Stuber GD. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell 160: 516–527, 2015. doi: 10.1016/j.cell.2014.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341: 1517–1521, 2013. doi: 10.1126/science.1241812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Beekly BG, Frankel WC, Berg T, Allen SJ, Garcia-Galiano D, Vanini G, Elias CF. Dissociated Pmch and Cre expression in lactating Pmch-Cre BAC transgenic mice. Front Neuroanat 14: 60, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Rolls ET, Burton MJ, Mora F. Hypothalamic neuronal responses associated with the sight of food. Brain Res 111: 53–66, 1976. doi: 10.1016/0006-8993(76)91048-9. [DOI] [PubMed] [Google Scholar]
- 681.Rolls ET, Sanghera MK, Roper-Hall A. The latency of activation of neurones in the lateral hypothalamus and substantia innominata during feeding in the monkey. Brain Res 164: 121–135, 1979. doi: 10.1016/0006-8993(79)90010-6. [DOI] [PubMed] [Google Scholar]
- 682.Burton MJ, Rolls ET, Mora F. Effects of hunger on the responses of neurons in the lateral hypothalamus to the sight and taste of food. Exp Neurol 51: 668–677, 1976. doi: 10.1016/0014-4886(76)90189-8. [DOI] [PubMed] [Google Scholar]
- 683.Rolls ET. Reward systems in the brain and nutrition. Annu Rev Nutr 36: 435–470, 2016. doi: 10.1146/annurev-nutr-071715-050725. [DOI] [PubMed] [Google Scholar]
- 684.Mora F, Rolls ET, Burton MJ. Modulation during learning of the responses of neurons in the lateral hypothalamus to the sight of food. Exp Neurol 53: 508–519, 1976. doi: 10.1016/0014-4886(76)90089-3. [DOI] [PubMed] [Google Scholar]
- 685.Sharpe MJ, Marchant NJ, Whitaker LR, Richie CT, Zhang YJ, Campbell EJ, Koivula PP, Necarsulmer JC, Mejias-Aponte C, Morales M, Pickel J, Smith JC, Niv Y, Shaham Y, Harvey BK, Schoenbaum G. Lateral hypothalamic GABAergic neurons encode reward predictions that are relayed to the ventral tegmental area to regulate learning. Curr Biol 27: 2089–2100, 2017. doi: 10.1016/j.cub.2017.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 686.Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, Izadmehr EM, Tye KM. Inhibitory input from the lateral hypothalamus to the ventral tegmental area disinhibits dopamine neurons and promotes behavioral activation. Neuron 90: 1286–1298, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Stuber GD, Wise RA. Lateral hypothalamic circuits for feeding and reward. Nat Neurosci 19: 198–205, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Leinninger GM, Opland DM, Jo YH, Faouzi M, Christensen L, Cappellucci LA, Rhodes CJ, Gnegy ME, Becker JB, Pothos EN, Seasholtz AF, Thompson RC, Myers MG. Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab 14: 313–323, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Skofitsch G, Jacobowitz DM, Zamir N. Immunohistochemical localization of a melanin concentrating hormone-like peptide in the rat brain. Brain Res Bull 15: 635–649, 1985. [DOI] [PubMed] [Google Scholar]
- 690.Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. The melanin‐concentrating hormone system of the rat brain: an immuno‐ and hybridization histochemical characterization. J Comp Neurol 319: 218–245, 1992. [DOI] [PubMed] [Google Scholar]
- 691.Bittencourt JC, Elias CF. Melanin-concentrating hormone and neuropeptide EI projections from the lateral hypothalamic area and zona incerta to the medial septal nucleus and spinal cord: a study using multiple neuronal tracers. Brain Res 805: 1–19, 1998. [DOI] [PubMed] [Google Scholar]
- 692.Diniz GB, Bittencourt JC. The melanin-concentrating hormone as an integrative peptide driving motivated behaviors. Front Syst Neurosci 11: 32, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Schneeberger M, Tan K, Nectow AR, Parolari L, Caglar C, Azevedo E, Li Z, Domingos A, Friedman JM. Functional analysis reveals differential effects of glutamate and MCH neuropeptide in MCH neurons. Mol Metab 13: 83–89, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Kowalski TJ, Farley C, Cohen-Williams ME, Varty G, Spar BD. Melanin-concentrating hormone-1 receptor antagonism decreases feeding by reducing meal size. Eur J Pharmacol 497: 41–47, 2004. [DOI] [PubMed] [Google Scholar]
- 695.Shearman LP, Camacho RE, Sloan Stribling D, Zhou D, Bednarek MA, Hreniuk DL, Feighner SD, Tan CP, Howard AD, Van der Ploeg LH, MacIntyre DE, Hickey GJ, Strack AM. Chronic MCH-1 receptor modulation alters appetite, body weight and adiposity in rats. Eur J Pharmacol 475: 37–47, 2003. [DOI] [PubMed] [Google Scholar]
- 696.Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM. Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature 400: 261–265, 1999. [DOI] [PubMed] [Google Scholar]
- 697.Lembo PM, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, St-Onge S, Pou C, Labrecque J, Groblewski T, O’Donnell D, Payza K, Ahmad S, Walker P. The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nat Cell Biol 1: 267–271, 1999. [DOI] [PubMed] [Google Scholar]
- 698.Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 380: 243–247, 1996. [DOI] [PubMed] [Google Scholar]
- 699.Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396: 670–674, 1998. [DOI] [PubMed] [Google Scholar]
- 700.Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, Van der Ploeg LH, Qian S. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A 99: 3240–3245, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, Maratos-Flier E. Maratos-Flier E. Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 107: 379–386, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702.Chee MJ, Pissios P, Maratos-Flier E. Neurochemical characterization of neurons expressing melanin-concentrating hormone receptor 1 in the mouse hypothalamus. J Comp Neurol 521: 2208–2234, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Bittencourt JC. Anatomical organization of the melanin-concentrating hormone peptide family in the mammalian brain. Gen Comp Endocrinol 172: 185–197, 2011. [DOI] [PubMed] [Google Scholar]
- 704.Sclafani A. Gut-brain nutrient signaling. Appetition vs. satiation. Appetite 71: 454–458, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Domingos AI, Sordillo A, Dietrich MO, Liu ZW, Tellez LA, Vaynshteyn J, Ferreira JG, Ekstrand MI, Horvath TL, de Araujo IE, Friedman JM. Hypothalamic melanin concentrating hormone neurons communicate the nutrient value of sugar. Elife 2: e01462, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Dilsiz P, Aklan I, Sayar Atasoy N, Yavuz Y, Filiz G, Koksalar F, Ates T, Oncul M, Coban I, Ates OE, Cebecioglu U, Alp M, Yilmaz B, Atasoy D. MCH neuron activity is sufficient for reward and reinforces feeding. Neuroendocrinology 110: 258–270, 2020. doi: 10.1159/000501234. [DOI] [PubMed] [Google Scholar]
- 707.Abbott CR, Kennedy AR, Wren AM, Rossi M, Murphy KG, Seal LJ, Todd JF, Ghatei MA, Small CJ, Bloom SR. Identification of hypothalamic nuclei involved in the orexigenic effect of melanin-concentrating hormone. Endocrinology 144: 3943–3949, 2003. [DOI] [PubMed] [Google Scholar]
- 708.Georgescu D, Sears RM, Hommel JD, Barrot M, Bolanos CA, Marsh DJ, Bednarek MA, Bibb JA, Maratos-Flier E, Nestler EJ, DiLeone RJ. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci 25: 2933–2940, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Lopez CA, Guesdon B, Baraboi ED, Roffarello BM, Hetu M, Richard D. Involvement of the opioid system in the orexigenic and hedonic effects of melanin-concentrating hormone. Am J Physiol Regul Integr Comp Physiol 301: R1105–R1111, 2011. [DOI] [PubMed] [Google Scholar]
- 710.van den Pol AN. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. J. Neurosci 19: 3171–3182, 1999. doi: 10.1523/JNEUROSCI.19-08-03171.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.de Lecea L, Kilduff TS, Peyron C, Gao XB, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A 95: 322–327, 1998. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch J, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92: 573–585, 1998. doi: 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 713.Edwards CM, Abusnana S, Sunter D, Murphy KG, Ghatei MA, Bloom SR. The effect of the orexins on food intake: comparison with neuropeptide Y, melanin-concentrating hormone and galanin. J Endocrinol 160: R7–R12, 1999. doi: 10.1677/joe.0.160r007. [DOI] [PubMed] [Google Scholar]
- 714.Zink AN, Bunney PE, Holm AA, Billington CJ, Kotz CM. Neuromodulation of orexin neurons reduces diet-induced adiposity. Int J Obes (Lond) 42: 737–745, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715.Sargin D. The role of the orexin system in stress response. Neuropharmacology 154: 68–78, 2019. [DOI] [PubMed] [Google Scholar]
- 716.Liblau RS, Vassalli A, Seifinejad A, Tafti M. Hypocretin (orexin) biology and the pathophysiology of narcolepsy with cataplexy. Lancet Neurol 14: 318–328, 2015. [DOI] [PubMed] [Google Scholar]
- 717.Alexandre C, Andermann ML, Scammell TE. Control of arousal by the orexin neurons. Curr Opin Neurobiol 23: 752–759, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.Nevárez N, de Lecea L. Recent advances in understanding the roles of hypocretin/orexin in arousal, affect, and motivation. F1000Res 7: 1421, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.Akiyama M, Yuasa T, Hayasaka N, Horikawa K, Sakurai T, Shibata S. Reduced food anticipatory activity in genetically orexin (hypocretin) neuron-ablated mice. Eur J Neurosci 20: 3054–3062, 2004. [DOI] [PubMed] [Google Scholar]
- 720.Gonzalez JA, Jensen LT, Iordanidou P, Strom M, Fugger L, Burdakov D. Inhibitory interplay between orexin neurons and eating. Curr Biol 26: 2486–2491, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, Kageyama H, Kunita S, Takahashi S, Goto K, Koyama Y, Shioda S, Yanagisawa M. Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron 46: 297–308, 2005. doi: 10.1016/j.neuron.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 722.Graebner AK, Iyer M, Carter ME. Understanding how discrete populations of hypothalamic neurons orchestrate complicated behavioral states. Front Syst Neurosci 9: 1–16, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Mahler SV, Moorman DE, Smith RJ, James MH, Aston Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat Neurosci 17: 1298–1303, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Herrera CG, Ponomarenko A, Korotkova T, Burdakov D, Adamantidis A. Sleep & metabolism: The multitasking ability of lateral hypothalamic inhibitory circuitries. Front Neuroendoc 44: 27–34, 2017. doi: 10.1016/j.yfrne.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 725.Lee EY, Lee HS. Dual projections of single orexin- or CART-immunoreactive, lateral hypothalamic neurons to the paraventricular thalamic nucleus and nucleus accumbens shell in the rat: light microscopic study. Brain Res 1634: 104–118, 2016. [DOI] [PubMed] [Google Scholar]
- 726.Ren S, Wang Y, Yue F, Cheng X, Dang R, Qiao Q, Sun X, Li X, Jiang Q, Yao J, Qin H, Wang G, Liao X, Gao D, Xia J, Zhang J, Hu B, Yan J, Wang Y, Xu M, Han Y, Tang X, Chen X, He C, Hu Z. The paraventricular thalamus is a critical thalamic area for wakefulness. Science 362: 429–434, 2018. [DOI] [PubMed] [Google Scholar]
- 727.Cole S, Keefer SE, Anderson LC, Petrovich GD. Medial prefrontal cortex neural plasticity, orexin receptor 1 signaling, and connectivity with the lateral hypothalamus are necessary in cue-potentiated feeding. J Neurosci 40: 1744–1755, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Tyree SM, de Lecea L. Lateral hypothalamic control of the ventral tegmental area: reward evaluation and the driving of motivated behavior. Front Syst Neurosci 11: 50, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729.Rao Y, Lu M, Ge F, Marsh DJ, Qian S, Wang AH, Picciotto MR, Gao XB. Regulation of synaptic efficacy in hypocretin/orexin-containing neurons by melanin concentrating hormone in the lateral hypothalamus. J Neurosci 28: 9101–9110, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 730.Parise EM, Lilly N, Kay K, Dossat AM, Seth R, Overton JM, Williams DL. Evidence for the role of hindbrain orexin-1 receptors in the control of meal size. Am J Physiol Regul Integr Comp Physiol 301: R1692–R1699, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Baird JP, Choe A, Loveland JL, Beck J, Mahoney CE, Lord JS, Grigg LA. Orexin-A hyperphagia: hindbrain participation in consummatory feeding responses. Endocrinology 150: 1202–1216, 2009. doi: 10.1210/en.2008-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732.Terrill SJ, Hyde KM, Kay KE, Greene HE, Maske CB, Knierim AE, Davis JF, Williams DL. Ventral tegmental area orexin 1 receptors promote palatable food intake and oppose postingestive negative feedback. Am J Physiol Regul Integr Comp Physiol 311: R592–R599, 2016. doi: 10.1152/ajpregu.00097.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Suarez AN, Liu CM, Cortella AM, Noble EE, Kanoski SE. Ghrelin and orexin interact to increase meal size through a descending hippocampus to hindbrain signaling pathway. Biol Psychiatry 87: 1001–1011, 2020. doi: 10.1016/j.biopsych.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Zahm DS, Grosu S, Williams EA, Qin S, Bérod A. Neurons of origin of the neurotensinergic plexus enmeshing the ventral tegmental area in rat: retrograde labeling and in situ hybridization combined. Neuroscience 104: 841–851, 2001. doi: 10.1016/S0306-4522(01)00118-X. [DOI] [PubMed] [Google Scholar]
- 735.Schroeder LE, Furdock R, Quiles CR, Kurt G, Perez-Bonilla P, Garcia A, Colon-Ortiz C, Brown J, Bugescu R, Leinninger GM. Mapping the populations of neurotensin neurons in the male mouse brain. Neuropeptides 76: 101930, 2019. doi: 10.1016/j.npep.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Brown JA, Bugescu R, Mayer TA, Gata-Garcia A, Kurt G, Woodworth HL, Leinninger GM. Loss of action via neurotensin-leptin receptor neurons disrupts leptin and ghrelin-mediated control of energy balance. Endocrinology 158: 1271–1288, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Woodworth HL, Batchelor HM, Beekly BG, Bugescu R, Brown JA, Kurt G, Fuller PM, Leinninger GM. Neurotensin receptor-1 identifies a subset of ventral tegmental dopamine neurons that coordinates energy balance. Cell Rep 20: 1881–1892, 2017. doi: 10.1016/j.celrep.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738.Kempadoo KA, Tourino C, Cho SL, Magnani F, Leinninger GM, Stuber GD, Zhang F, Myers MG, Deisseroth K, de Lecea L, Bonci A. Hypothalamic neurotensin projections promote reward by enhancing glutamate transmission in the VTA. J Neurosci 33: 7618–7626, 2013. doi: 10.1523/JNEUROSCI.2588-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 739.Patterson CM, Wong JM, Leinninger GM, Allison MB, Mabrouk OS, Kasper CL, Gonzalez IE, Mackenzie A, Jones JC, Kennedy RT, Myers MG. Ventral tegmental area neurotensin signaling links the lateral hypothalamus to locomotor activity and striatal dopamine efflux in male mice. Endocrinology 156: 1692–1700, 2015. doi: 10.1210/en.2014-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740.Schroeder LE, Leinninger GM. Role of central neurotensin in regulating feeding: Implications for the development and treatment of body weight disorders. Biochim Biophys Acta Mol Basis Dis 1864: 900–916, 2018. doi: 10.1016/j.bbadis.2017.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Brown J, Sagante A, Mayer T, Wright A, Bugescu R, Fuller PM, Leinninger G. Lateral hypothalamic area neurotensin neurons are required for control of orexin neurons and energy balance. Endocrinology 159: 3158–3176, 2018. doi: 10.1210/en.2018-00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Ki Y, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38: 701–713, 2003. doi: 10.1016/S0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 743.Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci 25: 2429–2433, 2005. doi: 10.1523/JNEUROSCI.4925-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Williams KW, Scott MM, Elmquist JK. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. Eur J Pharmacol 660: 2–12, 2011. doi: 10.1016/j.ejphar.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Liu ZW, Gan G, Suyama S, Gao XB. Intracellular energy status regulates activity in hypocretin/orexin neurones: a link between energy and behavioural states. J Physiol 589: 4157–4166, 2011. doi: 10.1113/jphysiol.2011.212514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Kosse C, Gonzalez A, Burdakov D. Predictive models of glucose control: roles for glucose-sensing neurones. Acta Physiol (Oxf) 213: 7–18, 2015. doi: 10.1111/apha.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Burdakov D. Reactive and predictive homeostasis: roles of orexin/hypocretin neurons. Neuropharmacology 154: 61–67, 2019. [ doi: 10.1016/j.neuropharm.2018.10.024. [DOI] [PubMed] [Google Scholar]
- 748.Sheng Z, Santiago AM, Thomas MP, Routh VH. Metabolic regulation of lateral hypothalamic glucose-inhibited orexin neurons may influence midbrain reward neurocircuitry. Mol Cell Neurosci 62: 30–41, 2014. doi: 10.1016/j.mcn.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Karnani MM, Apergis-Schoute J, Adamantidis A, Jensen LT, de Lecea L, Fugger L, Burdakov D. Activation of central orexin/hypocretin neurons by dietary amino acids. Neuron 72: 616–629, 2011. [DOI] [PubMed] [Google Scholar]
- 750.Cui H, Sohn JW, Gautron L, Funahashi H, Williams KW, Elmquist JK, Lutter M. Neuroanatomy of melanocortin-4 receptor pathway in the lateral hypothalamic area. J Comp Neurol 520: 4168–4183, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Liu JJ, Bello NT, Pang ZP. Pre-synaptic regulation of leptin in a defined lateral hypothalamus-ventral tegmental area neurocircuitry depends on energy state. J. Neurosci 37: 11854–11866, 2017. doi: 10.1523/JNEUROSCI.1942-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Brown JA, Woodworth HL, Leinninger GM. To ingest or rest? Specialized roles of lateral hypothalamic area neurons in coordinating energy balance. Front Syst Neurosci 9: 9, 2015. doi: 10.3389/fnsys.2015.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Olszewski PK, Li D, Grace MK, Billington CJ, Kotz CM, Levine AS. Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides 24: 597–602, 2003. doi: 10.1016/S0196-9781(03)00105-0. [DOI] [PubMed] [Google Scholar]
- 754.Toshinai K, Date Y, Murakami N, Shimada M, Mondal MS, Shimbara T, Guan JL, Wang QP, Funahashi H, Sakurai T, Shioda S, Matsukura S, Kangawa K, Nakazato M. Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology 144: 1506–1512, 2003. doi: 10.1210/en.2002-220788. [DOI] [PubMed] [Google Scholar]
- 755.Goto M, Swanson LW. Axonal projections from the parasubthalamic nucleus. J Comp Neurol 469: 581–607, 2004. doi: 10.1002/cne.11036. [DOI] [PubMed] [Google Scholar]
- 756.Zséli G, Vida B, Szilvásy-Szabó A, Tóth M, Lechan RM, Fekete C. Neuronal connections of the central amygdalar nucleus with refeeding-activated brain areas in rats. Brain Struct Funct 223: 391–414, 2018. doi: 10.1007/s00429-017-1501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Dong HW, Swanson LW. Projections from the rhomboid nucleus of the bed nuclei of the stria terminalis: implications for cerebral hemisphere regulation of ingestive behaviors. J Comp Neurol 463: 434–472, 2003. doi: 10.1002/cne.10758. [DOI] [PubMed] [Google Scholar]
- 758.Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, anteromedial area: Cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J Comp Neurol 494: 142–178, 2006. doi: 10.1002/cne.20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.Stratford TR, Kelley AE. GABA in the nucleus accumbens shell participates in the central regulation of feeding behavior. J Neurosci 17: 4434–4440, 1997. doi: 10.1523/JNEUROSCI.17-11-04434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Kelley AE. Ventral striatal control of appetitive motivation: Role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev 27: 765–776, 2004. doi: 10.1016/j.neubiorev.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 761.Stratford TR, Wirtshafter D. Evidence that the nucleus accumbens shell, ventral pallidum, and lateral hypothalamus are components of a lateralized feeding circuit. Behav Brain Res 226: 548–554, 2012. doi: 10.1016/j.bbr.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Stratford TR, Wirtshafter D. Lateral hypothalamic involvement in feeding elicited from the ventral pallidum. Eur J Neurosci 37: 648–653, 2013. doi: 10.1111/ejn.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.Stratford TR, Wirtshafter D. Injections of muscimol into the paraventricular thalamic nucleus, but not mediodorsal thalamic nuclei, induce feeding in rats. Brain Res 1490: 128–133, 2013. doi: 10.1016/j.brainres.2012.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Choi DL, Davis JF, Magrisso IJ, Fitzgerald ME, Lipton JW, Benoit SC. Orexin signaling in the paraventricular thalamic nucleus modulates mesolimbic dopamine and hedonic feeding in the rat. Neuroscience 210: 243–248, 2012. doi: 10.1016/j.neuroscience.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Barbier M, Chometton S, Pautrat A, Miguet-Alfonsi C, Datiche F, Gascuel J, Fellmann D, Peterschmitt Y, Coizet V, Risold PY. A basal ganglia-like cortical-amygdalar-hypothalamic network mediates feeding behavior. Proc Natl Acad Sci U S A 117: 15967–15976, 2020. doi: 10.1073/pnas.2004914117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Douglass AM, Kucukdereli H, Ponserre M, Markovic M, Gründemann J, Strobel C, Alcala Morales PL, Conzelmann KK, Lüthi A, Klein R. Central amygdala circuits modulate food consumption through a positive-valence mechanism. Nat Neurosci 20: 1384–1394, 2017. doi: 10.1038/nn.4623. [DOI] [PubMed] [Google Scholar]
- 767.Huang D, Grady FS, Peltekian L, Laing JJ, Geerling JC. Efferent projections of CGRP/Calca-expressing parabrachial neurons in mice. J Comp Neurol 529: 2911–2957, 2021. doi: 10.1002/cne.25136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768.Bicknell RJ. Endogenous opioid peptides and hypothalamic neuroendocrine neurones. J Endoc 107: 437–446, 1985. doi: 10.1677/joe.0.1070437. [DOI] [PubMed] [Google Scholar]
- 769.Moore KE, Demarest KT, Lookingland KJ. Stress, prolactin and hypothalamic dopaminergic neurons. Neuropharmacology 26: 801–808, 1987. doi: 10.1016/0028-3908(87)90055-4. [DOI] [PubMed] [Google Scholar]
- 770.Epelbaum J. Intrahypothalamic neurohormonal interactions in the control of growth-hormone secretion. Ciba Found Symp 168: 54–68, 1992. doi: 10.1002/9780470514283.ch5. [DOI] [PubMed] [Google Scholar]
- 771.Olney JW, Sharpe LG. Brain lesions in an infant rhesus monkey treated with monosodium glutamate. Science 166: 386–388, 1969. doi: 10.1126/science.166.3903.386. [DOI] [PubMed] [Google Scholar]
- 772.Dawson R, Lorden JF. Behavioral and neurochemical effects of neonatal administration of monosodium L-glutamate in mice. J Comp Physiol Psychol 95: 71–84, 1981. doi: 10.1037/h0077761. [DOI] [PubMed] [Google Scholar]
- 773.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278: 135–138, 1997. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 774.Shutter JR, Graham M, Kinsey AC, Scully S, Lüthy R, Stark KL. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes Dev 11: 593–602, 1997. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- 775.Herbert E, Uhler M. Biosynthesis of polyprotein precursors to regulatory peptides. Cell 30: 1–2, 1982. doi: 10.1016/0092-8674(82)90002-2. [DOI] [PubMed] [Google Scholar]
- 776.Bloch B, Bugnon C, Fellmann D, Lenys D, Gouget A. Neurons of the rat hypothalamus reactive with antisera against endorphins, ACTH, MSH and beta-LPH. Cell Tissue Res 204: 1–15, 1979. doi: 10.1007/bf00235160. [DOI] [PubMed] [Google Scholar]
- 777.Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ. GABA release from proopiomelanocortin neurons. J Neurosci 24: 1578–1583, 2004. doi: 10.1523/JNEUROSCI.3952-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778.Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci 29: 13684–13690, 2009. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 779.Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11: 998–1000, 2008. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Van Den Pol AN, Acuna C, Davis JN, Huang H, Zhang X. Defining the caudal hypothalamic arcuate nucleus with a focus on anorexic excitatory neurons. J Physiol 597: 1605–1625, 2019. doi: 10.1113/JP277152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Fenselau H, Campbell JN, Verstegen AM, Madara JC, Xu J, Shah BP, Resch JM, Yang Z, Mandelblat-Cerf Y, Livneh Y, Lowell BB. A rapidly acting glutamatergic ARC→PVH satiety circuit postsynaptically regulated by α-MSH. Nat Neurosci 20: 42–51, 2017. doi: 10.1038/nn.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 782.Everitt BJ, Meister B, Hökfelt T, Melander T, Terenius L, Rökaeus A, Theodorsson-Norheim E, Dockray G, Edwardson J, Cuello C. The hypothalamic arcuate nucleus-median eminence complex: immunohistochemistry of transmitters, peptides and DARPP-32 with special reference to coexistence in dopamine neurons. Brain Res 396: 97–155, 1986. [DOI] [PubMed] [Google Scholar]
- 783.Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, Goldman M, Verstegen AM, Resch JM, McCarroll SA, Rosen ED, Lowell BB, Tsai LT. A molecular census of arcuate hypothalamus and median eminence cell types. Nat Neurosci 20: 484–496, 2017. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Romanov RA, Alpár A, Hökfelt T, Harkany T. Molecular diversity of corticotropin-releasing hormone mRNA-containing neurons in the hypothalamus. J Endocrinol 232: R161–R172, 2017. doi: 10.1530/JOE-16-0256. [DOI] [PubMed] [Google Scholar]
- 785.Dulcis D, Jamshidi P, Leutgeb S, Spitzer NC. Neurotransmitter switching in the adult brain regulates behavior. Science 340: 449–453, 2013. doi: 10.1126/science.1234152. [DOI] [PubMed] [Google Scholar]
- 786.Svensson E, Apergis-Schoute J, Burnstock G, Nusbaum MP, Parker D, Schiöth HB. General principles of neuronal co-transmission: insights from multiple model systems. Front Neural Circuits 12: 19–28, 2019. doi: 10.3389/fncir.2018.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 787.Song J, Xu Y, Hu X, Choi B, Tong Q. Brain expression of Cre recombinase driven by pancreas-specific promoters. Genesis 48: 628–634, 2010. doi: 10.1002/dvg.20672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Kong D, Tong Q, Ye C, Koda S, Fuller PM, Krashes MJ, Vong L, Ray RS, Olson DP, Lowell BB. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell 151: 645–657, 2012. doi: 10.1016/j.cell.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Kim ER, Wu Z, Sun H, Xu Y, Mangieri LR, Xu Y, Xu Y, Tong Q. Hypothalamic non-AgRP, non-POMC GABAergic neurons are required for postweaning feeding and NPY hyperphagia. J Neurosci 35: 10440–10450, 2015. doi: 10.1523/JNEUROSCI.1110-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, Bernal-Mizrachi E, Elghazi L, Roe MW, Labosky PA, Myers MG, Gannon M, Powers AC, Dempsey PJ. Conditional gene targeting in mouse pancreatic beta-cells analysis of ectopic cre transgene expression in the brain. Diabetes 59: 3090–3098, 2010. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Cabral A, Valdivia S, Fernandez G, Reynaldo M, Perelló M. Divergent neuronal circuitries underlying acute orexigenic effects of peripheral or central ghrelin: critical role of brain accessibility. J Neuroendocrinol 26: 542–554, 2014. doi: 10.1111/jne.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Wang Q, Liu C, Uchida A, Chuang JC, Walker A, Liu T, Osborne-Lawrence S, Mason BL, Mosher C, Berglund ED, Elmquist JK, Zigman JM. Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol Metab 3: 64–72, 2014. doi: 10.1016/j.molmet.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793.Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 70: 537–556, 2008. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 794.Myers MG. Outstanding Scientific Achievement Award Lecture 2010: Deconstructing leptin: from signals to circuits. Diabetes 59: 2708–2714, 2010. doi: 10.2337/db10-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795.Mercer AJ, Hentges ST, Meshul CK, Low MJ. Unraveling the central proopiomelanocortin neural circuits. Front Neurosci 7: 19, 2013. doi: 10.3389/fnins.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Jn F, Myers MG. Minireview: CNS mechanisms of leptin action. Mol Endocrinol 30: 3–12, 2016. doi: 10.1210/me.2015-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 797.Toda C, Santoro A, Kim JD, Diano S. POMC neurons: from birth to death. Annu Rev Physiol 79: 209–236, 2017. doi: 10.1146/annurev-physiol-022516-034110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798.Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol 10: 650–658, 2014. doi: 10.1038/nrendo.2014.160. [DOI] [PubMed] [Google Scholar]
- 799.Zhang L, Hernandez-Sanchez D, Herzog H. Regulation of feeding-related behaviors by arcuate neuropeptide Y neurons. Endocrinology 160: 1411–1420, 2019. doi: 10.1210/en.2019-00056. [DOI] [PubMed] [Google Scholar]
- 800.Joly-Amado A, Cansell C, Denis RG, Delbès AS, Castel J, Martinez S, Luquet S. The hypothalamic arcuate nucleus and the control of peripheral substrates. Best Prac Res Clinl Endoc Metab 28: 725–737, 2014. doi: 10.1016/j.beem.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 801.Krashes MJ, Lowell BB, Garfield AS. Melanocortin-4 receptor-regulated energy homeostasis. Nat Neurosci 19: 206–219, 2016. doi: 10.1038/nn.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802.Friedman J. 20 years of leptin: leptin at 20: an overview. J Endoc 223: T1–8, 2014. doi: 10.1530/JOE-14-0405. [DOI] [PubMed] [Google Scholar]
- 803.Schaeffer M, Langlet F, Lafont C, Molino F, Hodson DJ, Roux T, Lamarque L, Verdié P, Bourrier E, Dehouck B, Banères JL, Martinez J, Méry PF, Marie J, Trinquet E, Fehrentz JA, Prévot V, Mollard P. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci U S A 110: 1512–1517, 2013. doi: 10.1073/pnas.1212137110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804.Mason BL, Wang Q, Zigman JM. The central nervous system sites mediating the orexigenic actions of ghrelin. Annu Rev Physiol 76: 519–533, 2014. doi: 10.1146/annurev-physiol-021113-170310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Chen Y, Lin YC, Kuo TW, Knight ZA. Sensory detection of food rapidly modulates arcuate feeding circuits. Cell 160: 829–841, 2015. doi: 10.1016/j.cell.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Schneeberger M, Claret M. Recent insights into the role of hypothalamic AMPK signaling cascade upon metabolic control. Front Neurosci 6: 185, 2012. doi: 10.3389/fnins.2012.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66: 789–800, 2017. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808.López M. Hypothalamic AMPK and energy balance. Eur J Clin Invest 48: e12996, 2018. doi: 10.1111/eci.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Kong D, Dagon Y, Campbell JN, Guo Y, Yang Z, Yi X, Aryal P, Wellenstein K, Kahn BB, Sabatini BL, Lowell BB. A postsynaptic AMPK→p21-activated kinase pathway drives fasting-induced synaptic plasticity in AgRP neurons. Neuron 91: 25–33, 2016. doi: 10.1016/j.neuron.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 810.Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279: 12005–12008, 2004. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- 811.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428: 569–574, 2004. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 812.Alquier T, Kawashima J, Tsuji Y, Kahn BB. Role of hypothalamic adenosine 5’-monophosphate-activated protein kinase in the impaired counterregulatory response induced by repetitive neuroglucopenia. Endocrinology 148: 1367–1375, 2007. doi: 10.1210/en.2006-1039. [DOI] [PubMed] [Google Scholar]
- 813.Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117: 2325–2336, 2007. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Lockie SH, Stark R, Mequinion M, Ch’ng S, Kong D, Spanswick DC, Lawrence AJ, Andrews ZB. Glucose availability predicts the feeding response to ghrelin in male mice, an effect dependent on AMPK in AgRP neurons. Endocrinology 159: 3605–3614, 2018. doi: 10.1210/en.2018-00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, Vong L, Pei H, Watabe-Uchida M, Uchida N, Liberles SD, Lowell BB. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507: 238–242, 2014. doi: 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816.Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Brüning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci 8: 1289–1291, 2005. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 817.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310: 683–685, 2005. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 818.Wu Q, Howell MP, Cowley MA, Palmiter RD. Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc Natl Acad Sci U S A 105: 2687–2692, 2008. doi: 10.1073/pnas.0712062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 819.Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 137: 1225–1234, 2009. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 820.Denis RG, Joly-Amado A, Webber E, Langlet F, Schaeffer M, Padilla SL, Cansell C, Dehouck B, Castel J, Delbès AS, Martinez S, Lacombe A, Rouch C, Kassis N, Fehrentz JA, Martinez J, Verdié P, Hnasko TS, Palmiter RD, Krashes MJ, Güler AD, Magnan C, Luquet S. Palatability can drive feeding independent of AgRP neurons. Cell Metab 22: 646–657, 2015. doi: 10.1016/j.cmet.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 821.Li AJ, Wang Q, Dinh TT, Wiater MF, Eskelsen AK, Ritter S. Hindbrain catecholamine neurons control rapid switching of metabolic substrate use during glucoprivation in male rats. Endocrinology 154: 4570–4579, 2013. doi: 10.1210/en.2013-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 822.Saper CB. The house alarm. Cell Metab 23: 754–755, 2016. doi: 10.1016/j.cmet.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 823.Schier LA, Spector AC. The functional and neurobiological properties of bad taste. Physiol Rev 99: 605–663, 2019. doi: 10.1152/physrev.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 824.Palmiter RD. The parabrachial nucleus: CGRP neurons function as a general alarm. Trends Neurosci 41: 280–293, 2018. doi: 10.1016/j.tins.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Alden M, Besson JM, Bernard JF. Organization of the efferent projections from the pontine parabrachial area to the bed nucleus of the stria terminalis and neighboring regions: a PHA-L study in the rat. J Comp Neurol 341: 289–314, 1994. doi: 10.1002/cne.903410302. [DOI] [PubMed] [Google Scholar]
- 826.Bernard JF, Alden M, Besson JM. The organization of the efferent projections from the pontine parabrachial area to the amygdaloid complex: a Phaseolus vulgaris leucoagglutinin (PHA-L) study in the rat. J Comp Neurol 329: 201–229, 1993. doi: 10.1002/cne.903290205. [DOI] [PubMed] [Google Scholar]
- 827.Bester H, Besson JM, Bernard JF. Organization of efferent projections from the parabrachial area to the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol 383: 245–281, 1997. doi:. [DOI] [PubMed] [Google Scholar]
- 828.Bester H, Bourgeais L, Villanueva L, Besson JM, Bernard JF. Differential projections to the intralaminar and gustatory thalamus from the parabrachial area: a PHA-L study in the rat. J Comp Neurol 405: 421–449, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 829.Fulwiler CE, Saper CB. Subnuclear organization of the efferent connections of the parabrachial nucleus in the rat. Brain Res 319: 229–259, 1984. doi: 10.1016/0165-0173(84)90012-2. [DOI] [PubMed] [Google Scholar]
- 830.Carter ME, Soden ME, Zweifel LS, Palmiter RD. Genetic identification of a neural circuit that suppresses appetite. Nature 503: 111–114, 2013. doi: 10.1038/nature12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Shah BP, Vong L, Olson DP, Koda S, Krashes MJ, Ye C, Yang Z, Fuller PM, Elmquist JK, Lowell BB. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proc Natl Acad Sci U S A 111: 13193–13198, 2014. doi: 10.1073/pnas.1407843111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 832.Garfield AS, Li C, Madara JC, Shah BP, Webber E, Steger JS, Campbell JN, Gavrilova O, Lee CE, Olson DP, Elmquist JK, Tannous BA, Krashes MJ, Lowell BB. Neural basis for melanocortin-4 receptor–regulated appetite. Nat Neurosci 18: 863–871, 2015. doi: 10.1038/nn.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 833.Essner RA, Smith AG, Jamnik AA, Ryba AR, Trutner ZD, Carter ME. AgRP neurons can increase food intake during conditions of appetite suppression and inhibit anorexigenic parabrachial neurons. J Neurosci 37: 8678–8687, 2017. doi: 10.1523/JNEUROSCI.0798-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 834.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121: 1424–1428, 2011. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 835.Zhan C, Zhou J, Feng Q, Zhang JE, Lin S, Bao J, Wu P, Luo M. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J Neurosci 33: 3624–3632, 2013. doi: 10.1523/JNEUROSCI.2742-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836.Zhu C, Jiang Z, Xu Y, Cai ZL, Jiang Q, Xu Y, Xue M, Arenkiel BR, Wu Q, Shu G, Tong Q. Profound and redundant functions of arcuate neurons in obesity development. Nat Metab 2: 763–744, 2020. doi: 10.1038/s42255-020-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 837.Zhang X, Van Den Pol AN. Hypothalamic arcuate nucleus tyrosine hydroxylase neurons play orexigenic role in energy homeostasis. Nat Neurosci 19: 1341–1347, 2016. doi: 10.1038/nn.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 838.DePuy SD, Stornetta RL, Bochorishvili G, Deisseroth K, Witten I, Coates M, Guyenet PG. Glutamatergic neurotransmission between the C1 neurons and the parasympathetic preganglionic neurons of the dorsal motor nucleus of the vagus. J Neurosci 33: 1486–1497, 2013. doi: 10.1523/JNEUROSCI.4269-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 839.Sternson SM. Seeing the forest for the trees in obesity. Nat Metab 2: 1–2, 2020. doi: 10.1038/s42255-020-0168-y. [DOI] [PubMed] [Google Scholar]
- 840.Seeley RJ, Berridge KC. The hunger games. Cell 160: 805–806, 2015. doi: 10.1016/j.cell.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 841.Mandelblat-Cerf Y, Ramesh RN, Burgess CR, Patella P, Yang Z, Lowell BB, Andermann ML. Arcuate hypothalamic AgRP and putative POMC neurons show opposite changes in spiking across multiple timescales. Elife 4: e07122, 2015. doi: 10.7554/eLife.07122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Padilla SL, Qiu J, Soden ME, Sanz E, Nestor CC, Barker FD, Quintana A, Zweifel LS, Rønnekleiv OK, Kelly MJ, Palmiter RD. Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nat Neurosci 19: 734–741, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 843.Jikomes N, Ramesh RN, Mandelblat-Cerf Y, Andermann ML. Preemptive stimulation of AgRP neurons in fed mice enables conditioned food seeking under threat. Curr Biol 26: 2500–2507, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 844.Day DE, Keen-Rhinehart E, Bartness TJ. Role of NPY and its receptor subtypes in foraging, food hoarding, and food intake by Siberian hamsters. Am J Physiol Regul Integr Comp Physiol 289: R29–R36, 2005. [DOI] [PubMed] [Google Scholar]
- 845.Ammar AA, Nergårdh R, Fredholm BB, Brodin U, Södersten P. Intake inhibition by NPY and CCK-8: A challenge of the notion of NPY as an ‘Orexigen’. Behav Brain Res 161: 82–87, 2005. [DOI] [PubMed] [Google Scholar]
- 846.Betley JN, Xu S, Cao ZF, Gong R, Magnus CJ, Yu Y, Sternson SM. Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature 521: 180–185, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Yeo SH, Herbison AE. Projections of arcuate nucleus and rostral periventricular kisspeptin neurons in the adult female mouse brain. Endocrinology 152: 2387–2399, 2011. [DOI] [PubMed] [Google Scholar]
- 849.Qi Y, Iqbal J, Oldfield BJ, Clarke IJ. Neural connectivity in the mediobasal hypothalamus of the sheep brain. Neuroendocrinology 87: 91–112, 2008. [DOI] [PubMed] [Google Scholar]
- 847.Garfield AS, Shah BP, Burgess CR, Li MM, Li C, Steger JS, Madara JC, Campbell JN, Kroeger D, Scammell TE, Tannous BA, Myers MG, Andermann ML, Krashes MJ, Lowell BB. Dynamic GABAergic afferent modulation of AgRP neurons. Nat Neurosci 19: 1628–1635, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 850.Xu J, Bartolome CL, Low CS, Yi X, Chien CH, Wang P, Kong D. Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature 556: 505–509, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 851.Rau AR, Hentges ST. GABAergic inputs to POMC neurons originating from the dorsomedial hypothalamus are regulated by energy state. J Neurosci 39: 6449–6459, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 852.Herman AM, Ortiz-Guzman J, Kochukov M, Herman I, Quast KB, Patel JM, Tepe B, Carlson JC, Ung K, Selever J, Tong Q, Arenkiel BR. A cholinergic basal forebrain feeding circuit modulates appetite suppression. Nature 538: 253–256, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 854.Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T. The Neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A 95: 15043–15048, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 855.Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat. Endocrinology 140: 1408–1415, 1999. [DOI] [PubMed] [Google Scholar]
- 856.Dietrich MO, Bober J, Ferreira JG, Tellez LA, Mineur YS, Souza DO, Gao XB, Picciotto MR, Araújo I, Liu ZW, Horvath TL. AgRP neurons regulate development of dopamine neuronal plasticity and nonfood-associated behaviors. Nat Neurosci 15: 1108–1110, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Gumbs MC, Vuuregge AH, Eggels L, Unmehopa UA, Lamuadni K, Mul JD, la Fleur SE. Afferent neuropeptide Y projections to the ventral tegmental area in normal-weight male Wistar rats. J Comp Neurol 527: 2659–2674, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 858.Han Y, Xia G, Srisai D, Meng F, He Y, Ran Y, He Y, Farias M, Hoang G, Tóth I, Dietrich MO, Chen MH, Xu Y, Wu Q. Deciphering an AgRP-serotoninergic neural circuit in distinct control of energy metabolism from feeding. Nat Commun 12: 3525, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 859.Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature 488: 172–177, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 860.Sternson SM, Atasoy D. Agouti-related protein neuron circuits that regulate appetite. Neuroendocrinology 100: 95–102, 2014. [DOI] [PubMed] [Google Scholar]
- 861.Merchenthaler I. Neurons with access to the general circulation in the central nervous system of the rat: a retrograde tracing study with fluoro-gold. Neuroscience 44: 655–662, 1991. [DOI] [PubMed] [Google Scholar]
- 862.Loose MD, Niu JC, Nguyen TT, Thornton JE. Estrogen modulation of two subpopulations of ß-endorphin neurons in ovariectomized guinea pigs distinguished by peripherally injected fluorogold. Endocrine 3: 827–831, 1995. [DOI] [PubMed] [Google Scholar]
- 863.Sarkar DK, Yen SS. Changes in beta-endorphin-like immunoreactivity in pituitary portal blood during the estrous cycle and after ovariectomy in rats. Endocrinology 116: 2075–2079, 1985. [DOI] [PubMed] [Google Scholar]
- 864.Sheward WJ, Lim A, Alder B, Copolov D, Dow RC, Fink G. Hypothalamic release of atrial natriuretic factor and beta-endorphin into rat hypophysial portal plasma: relationship to oestrous cycle and effects of hypophysectomy. J Endoc 131: 113–125, 1991. doi: 10.1677/joe.0.1310113. [DOI] [PubMed] [Google Scholar]
- 865.Swanson LW, Kuypers HG. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double‐labeling methods. J Comp Neurol 194: 555–570, 1980. doi: 10.1002/cne.901940306. [DOI] [PubMed] [Google Scholar]
- 866.Coote JH. Cardiovascular function of the paraventricular nucleus of the hypothalamus. Biol Signals 4: 142–149, 1995. [DOI] [PubMed] [Google Scholar]
- 867.Coote JH. A role for the paraventricular nucleus of the hypothalamus in the autonomic control of heart and kidney. Exp Physiol 90: 169–173, 2005. [DOI] [PubMed] [Google Scholar]
- 868.Nunn N, Womack M, Dart C, Barrett-Jolley R. Function and pharmacology of spinally-projecting sympathetic pre-autonomic neurones in the paraventricular nucleus of the hypothalamus. Curr Neuropharmacol 9: 262–277, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Kc P, Dick TE. Modulation of cardiorespiratory function mediated by the paraventricular nucleus. Respir Physiol Neurobiol 174: 55–64, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 870.Madden CJ, Morrison SF. Neurons in the paraventricular nucleus of the hypothalamus inhibit sympathetic outflow to brown adipose tissue. Am J Physiol Regul Integr Comp Physiol 296: R831–R843, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 871.Zhang Z, Boelen A, Kalsbeek A, Fliers E. TRH neurons and thyroid hormone coordinate the hypothalamic response to cold. Eur Thyroid J 7: 279–288, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 872.Argiolas A, Melis MR. Central control of penile erection: role of the paraventricular nucleus of the hypothalamus. Prog Neurobiol 76: 1–21, 2005. [DOI] [PubMed] [Google Scholar]
- 873.Bisschop PH, Fliers E, Kalsbeek A. Autonomic regulation of hepatic glucose production. Compr Physiol 5: 147–165, 2015. doi: 10.1002/cphy.c140009. [DOI] [PubMed] [Google Scholar]
- 874.O’Hare JD, Zsombok A. Brain-liver connections: role of the preautonomic PVN neurons. Am J Physiol Endocrinol Metab 310: E183–E189, 2016. doi: 10.1152/ajpendo.00302.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 875.Watts AG. Glucocorticoid regulation of peptide genes in neuroendocrine CRH neurons: a complexity beyond negative feedback. Front Neuroendoc 26: 109–130, 2005. doi: 10.1016/j.yfrne.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 876.Badoer E. Role of the hypothalamic PVN in the regulation of renal sympathetic nerve activity and blood flow during hyperthermia and in heart failure. Am J Physiol Renal Physiol 298: F839–F846, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 877.Stellar E. The physiology of motivation. Psychol Rev 61: 5–22, 1954. [DOI] [PubMed] [Google Scholar]
- 878.Atrens DM, Vietinghoff-Riesch F. The motivational properties of electrical stimulation of the medial and paraventricular hypothalamic nuclei. Physiol Behav 9: 229–235, 1972. [DOI] [PubMed] [Google Scholar]
- 879.Gold RM. Hypothalamic obesity: the myth of the ventromedial nucleus. Science 182: 488–490, 1973. [DOI] [PubMed] [Google Scholar]
- 880.Gold RM, Jones AP, Sawchenko PE, Kapatos G. Paraventricular area-critical focus of a longitudinal neurocircuitry mediating food-intake. Physiol Behav 18: 1111–1119, 1977. [DOI] [PubMed] [Google Scholar]
- 881.Hoebel BG. Feeding–neural control of intake. Annu Rev Physiol 33: 533–568, 1971. [DOI] [PubMed] [Google Scholar]
- 882.Leibowitz SF. Paraventricular nucleus: A primary site mediating adrenergic stimulation of feeding and drinking. Pharm Biochem Behav 8: 163–175, 1978. doi: 10.1016/0091-3057(78)90333-7. [DOI] [PubMed] [Google Scholar]
- 883.Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal-cord in the rat. J Comp Neurol 205: 260–272, 1982. [DOI] [PubMed] [Google Scholar]
- 884.Sawchenko PE, Swanson LW. The organization of forebrain afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol 218: 121–144, 1983. [DOI] [PubMed] [Google Scholar]
- 885.Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci 6: 269–324, 1983. [DOI] [PubMed] [Google Scholar]
- 886.Holder JL, Zhang L, Kublaoui BM, DiLeone RJ, Oz OK, Bair CH, Lee YH, Zinn AR. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab 287: E105–E113, 2004. [DOI] [PubMed] [Google Scholar]
- 887.Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Levy E, Mitchell GA, Himms-Hagen J, Fan CM. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet 10: 1465–1473, 2001. [DOI] [PubMed] [Google Scholar]
- 888.Yang C, Gagnon D, Vachon P, Tremblay A, Levy E, Massie B, Michaud JL. Adenoviral-mediated modulation of Sim1 expression in the paraventricular nucleus affects food intake. J Neurosci 26: 7116–7120, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 889.Xi D, Gandhi N, Lai M, Kublaoui BM. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PLoS One 7: e36453, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890.Stachniak TJ, Ghosh A, Sternson SM. Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus→midbrain pathway for feeding behavior. Neuron 82: 797–808, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 891.Gonzalez IE, Ramirez-Matias J, Lu C, Pan W, Zhu A, Myers MG, Olson DP. Paraventricular calcitonin receptor-expressing neurons modulate energy homeostasis in male mice. Endocrinology 162: 1–15, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 892.Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol 423: 261–281, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 893.Sutton AK, Myers MG, Olson DP. The role of PVH circuits in leptin action and energy balance. Annu Rev Physiol 78: 207–221, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A 95: 741–746, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.Gautron L, Lazarus M, Scott MM, Saper CB, Elmquist JK. Identifying the efferent projections of leptin-responsive neurons in the dorsomedial hypothalamus using a novel conditional tracing approach. J Comp Neurol 518: 2090–2108, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 896.Otgon-Uul Z, Suyama S, Onodera H, Yada T. Optogenetic activation of leptin- and glucose-regulated GABAergic neurons in dorsomedial hypothalamus promotes food intake via inhibitory synaptic transmission to paraventricular nucleus of hypothalamus. Mol Metab 5: 709–715, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Powis JE, Bains JS, Ferguson AV. Leptin depolarizes rat hypothalamic paraventricular nucleus neurons. Am J Physiol Regul Integr Comp Physiol 274: R1468–R1472, 1998. [DOI] [PubMed] [Google Scholar]
- 898.Ghamari-Langroudi M, Srisai D, Cone RD. Multinodal regulation of the arcuate/paraventricular nucleus circuit by leptin. Proc Natl Acad Sci U S A 108: 355–360, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899.Ghamari-Langroudi M, Vella KR, Srisai D, Sugrue ML, Hollenberg AN, Cone RD. Regulation of thyrotropin-releasing hormone-expressing neurons in paraventricular nucleus of the hypothalamus by signals of adiposity. Mol Endocrinol 24: 2366–2381, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 900.Pelleymounter MA, Cullen MJ, Wellman CL. Characteristics of BDNF-induced weight loss. Exp Neurol 131: 229–238, 1995. [DOI] [PubMed] [Google Scholar]
- 901.An JJ, Liao GY, Kinney CE, Sahibzada N, Xu B. Discrete BDNF neurons in the paraventricular hypothalamus control feeding and energy expenditure. Cell Metab 22: 175–188, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 902.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci 6: 736–742, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 903.Wang C, Bomberg E, Billington C, Levine A, Kotz CM. Brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus reduces energy intake. Am J Physiol Regul Integr Comp Physiol 293: R1003–1012, 2007. [DOI] [PubMed] [Google Scholar]
- 904.Perry RJ, Resch JM, Douglass AM, Madara JC, Rabin-Court A, Kucukdereli H, Wu C, Song JD, Lowell BB, Shulman GI. Leptin’s hunger-suppressing effects are mediated by the hypothalamic-pituitary-adrenocortical axis in rodents. Proc Natl Acad Sci U S A 116: 13670–13679, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 905.Arvaniti K, Huang Q, Richard D. Effects of leptin and corticosterone on the expression of corticotropin-releasing hormone, agouti-related protein, and proopiomelanocortin in the brain of ob/ob mouse. Neuroendocrinology 73: 227–236, 2001. doi: 10.1159/000054639. [DOI] [PubMed] [Google Scholar]
- 906.Kaminski KL, Watts AG. Intact catecholamine inputs to the forebrain are required for appropriate regulation of corticotrophin-releasing hormone and vasopressin gene expression by corticosterone in the rat paraventricular nucleus. J Neuroendoc 24: 1517–1526, 2012. doi: 10.1111/j.1365-2826.2012.02363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 907.Dos Santos RC, Grover HM, Reis LC, Ferguson AV, Mecawi AS. Electrophysiological effects of ghrelin in the hypothalamic paraventricular nucleus neurons. Front Cell Neurosci 12: 275, 2018. doi: 10.3389/fnhum.2018.00275,10.3389/fnins.2018.00275,10.3389/fncel.2018.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 908.Uriarte M, De Francesco PN, Fernandez G, Cabral A, Castrogiovanni D, Lalonde T, Luyt LG, Trejo S, Perello M. Evidence supporting a role for the blood-cerebrospinal fluid barrier transporting circulating ghrelin into the brain. Mol Neurobiol 56: 4120–4134, 2019. doi: 10.1007/s12035-018-1362-8. [DOI] [PubMed] [Google Scholar]
- 909.Melnick IV, Price CJ, Colmers WF. Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci 34: 272–282, 2011. doi: 10.1111/j.1460-9568.2011.07742.x. [DOI] [PubMed] [Google Scholar]
- 910.Dong HW, Swanson LW. Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J Comp Neurol 468: 277–298, 2004. doi: 10.1002/cne.10949. [DOI] [PubMed] [Google Scholar]
- 911.Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, posterior division: Implications for cerebral hemisphere regulation of defensive and reproductive behaviors. J Comp Neurol 471: 396–433, 2004. doi: 10.1002/cne.20002. [DOI] [PubMed] [Google Scholar]
- 912.Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51: 32–58, 2003. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- 913.Radley JJ, Sawchenko PE. Evidence for involvement of a limbic paraventricular hypothalamic inhibitory network in hypothalamic-pituitary-adrenal axis adaptations to repeated stress. J Comp Neurol 523: 2769–2787, 2015. doi: 10.1002/cne.23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 915.Luo F, Mu Y, Gao C, Xiao Y, Zhou Q, Yang Y, Ni X, Shen WL, Yang J. Whole-brain patterns of the presynaptic inputs and axonal projections of BDNF neurons in the paraventricular nucleus. J Genet Genomics 46: 31–40, 2019. [DOI] [PubMed] [Google Scholar]
- 914.Hoover WB, Vertes RP. Projections of the medial orbital and ventral orbital cortex in the rat. J Comp Neurol 519: 3766–3801, 2011. doi: 10.1002/cne.22733. [DOI] [PubMed] [Google Scholar]
- 916.Thompson RH, Swanson LW. Structural characterization of a hypothalamic visceromotor pattern generator network. Brain Res Brain Res Rev 41: 153–202, 2003. doi: 10.1016/S0165-0173(02)00232-1. [DOI] [PubMed] [Google Scholar]
- 917.Thompson RH, Canteras NS, Swanson LW. Organization of projections from the dorsomedial nucleus of the hypothalamus: a PHA-L study in the rat. J Comp Neurol 376: 143–173, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 918.Hahn JD, Swanson LW. Connections of the lateral hypothalamic area juxtadorsomedial region in the male rat. J Comp Neurol 520: 1831–1890, 2012. doi: 10.1002/cne.23064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 919.Hahn JD, Swanson LW. Connections of the juxtaventromedial region of the lateral hypothalamic area in the male rat. Front Syst Neurosci 9: 1–53, 2015. doi: 10.3389/fnsys.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 920.van-Hover C, Li C. Stress-activated afferent inputs into the anterior parvicellular part of the paraventricular nucleus of the hypothalamus: Insights into urocortin 3 neuron activation. Brain Res 1611: 29–43, 2015. doi: 10.1016/j.brainres.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 921.Johnson CS, Bains JS, Watts AG. Neurotransmitter diversity in pre-synaptic terminals located in the parvicellular neuroendocrine paraventricular nucleus of the rat and mouse hypothalamus. J Comp Neurol 526: 1287–1306, 2018. doi: 10.1002/cne.24407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 922.Morales T, Hinuma S, Sawchenko PE. Prolactin-releasing peptide is expressed in afferents to the endocrine hypothalamus, but not in neurosecretory neurones. J Neuroendocrinol 12: 1 31–140, 2000. doi: 10.1046/j.1365-2826.2000.00428.x. [DOI] [PubMed] [Google Scholar]
- 923.Lawrence CB, Celsi F, Brennand J, Luckman SM. Alternative role for prolactin-releasing peptide in the regulation of food intake. Nat Rev Neurosci 3: 645–646, 2000. doi: 10.1038/76597. doi: 10.1038/76597. [DOI] [PubMed] [Google Scholar]
- 924.Davis XS, Grill HJ. The hindbrain is a site of energy balance action for prolactin-releasing peptide: feeding and thermic effects from GPR10 stimulation of the nucleus tractus solitarius/area postrema. Psychopharmacology (Berl) 235: 2287–2301, 2018. doi: 10.1007/s00213-018-4925-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 925.Vrang N, Hansen M, Larsen PJ, Tang-Christensen M. Characterization of brainstem preproglucagon projections to the paraventricular and dorsomedial hypothalamic nuclei. Brain Res 1149: 118–126, 2007. doi: 10.1016/j.brainres.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 926.Gu G, Roland B, Tomaselli K, Dolman CS, Lowe C, Heilig JS. Glucagon-like peptide-1 in the rat brain: distribution of expression and functional implication. J Comp Neurol 521: 2235–2261, 2013. doi: 10.1002/cne.23282. [DOI] [PubMed] [Google Scholar]
- 927.Larsen PJ, TangChristensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 77: 257–270, 1997. doi: 10.1016/S0306-4522(96)00434-4. [DOI] [PubMed] [Google Scholar]
- 928.Zheng H, Stornetta RL, Agassandian K, Rinaman L. Glutamatergic phenotype of glucagon-like peptide 1 neurons in the caudal nucleus of the solitary tract in rats. Brain Struct Funct 220: 3011–3022, 2015. doi: 10.1007/s00429-014-0841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 929.Lin W, McKinney K, Liu L, Lakhlani S, Jennes L. Distribution of vesicular glutamate transporter-2 messenger ribonucleic acid and protein in the septum-hypothalamus of the rat. Endocrinology 144: 662–670, 2003. doi: 10.1210/en.2002-220908. [DOI] [PubMed] [Google Scholar]
- 930.Hrabovszky E, Deli L, Turi GF, Kalló I, Liposits Z. Glutamatergic innervation of the hypothalamic median eminence and posterior pituitary of the rat. Neuroscience 144: 1383–1392, 2007. doi: 10.1016/j.neuroscience.2006.10.053. [DOI] [PubMed] [Google Scholar]
- 853.Watts AG, Khan AM. Identifying links in the chain: the dynamic coupling of catecholamines, peptide synthesis, and peptide release in hypothalamic neuroendocrine neurons. Adv Pharmacol 68: 421–444, 2013. doi: 10.1016/B978-0-12-411512-5.00020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 931.Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct 212: 149–179, 2007. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- 932.Kampe J, Tschop MH, Hollis JH, Oldfield BJ. An anatomic basis for the communication of hypothalamic, cortical and mesolimbic circuitry in the regulation of energy balance. Eur J Neurosci 30: 415–430, 2009. doi: 10.1111/j.1460-9568.2009.06818.x. [DOI] [PubMed] [Google Scholar]
- 933.Swanson LW, McKellar S. The distribution of oxytocin- and neurophysin-stained fibers in the spinal cord of the rat and monkey. J Comp Neurol 188: 87–106, 1979. doi: 10.1002/cne.901880108. [DOI] [PubMed] [Google Scholar]
- 934.Cechetto DF, Saper CB. Neurochemical organization of the hypothalamic projection to the spinal cord in the rat. J Comp Neurol 272: 579–604, 1988. doi: 10.1002/cne.902720410. [DOI] [PubMed] [Google Scholar]
- 935.Füzesi T, Daviu N, Wamsteeker Cusulin JI, Bonin RP, Bains JS. Hypothalamic CRH neurons orchestrate complex behaviours after stress. Nat Commun 7: 11937, 2016. doi: 10.1038/ncomms11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 936.Geisler S, Zahm DS. Afferents of the ventral tegmental area in the rat-anatomical substratum for integrative functions. J Comp Neurol 490: 270–294, 2005. doi: 10.1002/cne.20668. [DOI] [PubMed] [Google Scholar]
- 937.Rodaros D, Caruana DA, Amir S, Stewart J. Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience 150: 8–13, 2007. doi: 10.1016/j.neuroscience.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 938.Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74: 858–873, 2012. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 939.Wu Q, Palmiter RD. GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. Eur J Pharmacol 660: 21–27, 2011. doi: 10.1016/j.ejphar.2010.10.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 940.Gong R, Xu S, Hermundstad A, Yu Y, Sternson SM. Hindbrain double-negative feedback mediates palatability-guided food and water consumption. Cell 182: 1589–1605, 2020. doi: 10.1016/j.cell.2020.07.031. [DOI] [PubMed] [Google Scholar]
- 941.Rogers RC, Hermann GE. Gastric-vagal solitary neurons excited by paraventricular nucleus microstimulation. J Auto Nerv Syst 14: 351–362, 1985. doi: 10.1016/0165-1838(85)90081-5. [DOI] [PubMed] [Google Scholar]
- 942.Banks D, Harris MC. Activation within dorsal medullary nuclei following stimulation in the hypothalamic paraventricular nucleus in rats. Pflugers Archiv 408: 619–627, 1987. doi: 10.1007/BF00581165. [DOI] [PubMed] [Google Scholar]
- 943.Lawson EA, Olszewski PK, Weller A, Blevins JE. The role of oxytocin in regulation of appetitive behaviour, body weight and glucose homeostasis. J Neuroendoc 24: 79–19, 2019. doi: 10.1111/jne.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 944.Buijs RM, van der Beek EM, Renaud LP, Day TA, Jhamandas JH. Oxytocin localization and function in the A1, noradrenergic cell group: ultrastructural and electrophysiological studies. Neuroscience 39: 717–725, 1990. doi: 10.1016/0306-4522(90)90255-3. [DOI] [PubMed] [Google Scholar]
- 945.Arletti R, Benelli A, Bertolini A. Influence of oxytocin on feeding-behavior in the rat. Peptides 10: 89–93, 1989. doi: 10.1016/0196-9781(89)90082-X. [DOI] [PubMed] [Google Scholar]
- 946.Olson BR, Drutarosky MD, Chow MS, Hruby VJ, Stricker EM, Verbalis JG. Oxytocin and an oxytocin agonist administered centrally decrease food-intake in rats. Peptides 12: 113–118, 1991. doi: 10.1016/0196-9781(91)90176-P. [DOI] [PubMed] [Google Scholar]
- 947.Blevins JE, Ho JM. Role of oxytocin signaling in the regulation of body weight. Rev Endoc Metab Disord 14: 311–329, 2013. doi: 10.1007/s11154-013-9260-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 948.Blevins JE, Schwartz MW, Baskin DG. Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol 287: R87–R96, 2004. doi: 10.1152/ajpregu.00604.2003. [DOI] [PubMed] [Google Scholar]
- 949.Leslie M, Silva P, Paloyelis Y, Blevins J, Treasure J. A systematic review and quantitative meta-analysis of the effects of oxytocin on feeding. J Neuroendocrinol 30: e12584, 2018. doi: 10.1111/jne.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 950.Atasoy D, Betley JN, Li WP, Su HH, Sertel SM, Scheffer LK, Simpson JH, Fetter RD, Sternson SM. A genetically specified connectomics approach applied to long-range feeding regulatory circuits. Nat Neurosci 17: 1830–1839, 2014. doi: 10.1038/nn.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 951.Yokoyama T, Saito T, Ohbuchi T, Suzuki H, Otsubo H, Okamoto T, Fujihara H, Nagatomo T, Ueta Y. Ghrelin potentiates miniature excitatory postsynaptic currents in supraoptic magnocellular neurones. J Neuroendoc 21: 910–920, 2009. doi: 10.1111/j.1365-2826.2009.01911.x. [DOI] [PubMed] [Google Scholar]
- 952.Song Z, Levin BE, Stevens W, Sladek CD. Supraoptic oxytocin and vasopressin neurons function as glucose and metabolic sensors. Am J Physiol Regul Integr Comp Physiol 306: R447–R456, 2014. doi: 10.1152/ajpregu.00520.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 953.Sabatier N, Caquineau C, Dayanithi G, Bull P, Douglas AJ, Guan X, Jiang M, Van der Ploeg L, Leng G. Alpha-melanocyte-stimulating hormone stimulates oxytocin release from the dendrites of hypothalamic neurons while inhibiting oxytocin release from their terminals in the neurohypophysis. J Neurosci 23: 10351–10358, 2003. doi: 10.1523/JNEUROSCI.23-32-10351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 954.Velmurugan S, Russell JA, Leng G. Systemic leptin increases the electrical activity of supraoptic nucleus oxytocin neurones in virgin and late pregnant rats. J Neuroendoc 25: 383–390, 2013. doi: 10.1111/jne.12016. [DOI] [PubMed] [Google Scholar]
- 955.Honda K, Narita K, Murata T, Higuchi T. Leptin affects the electrical activity of neurones in the hypothalamic supraoptic nucleus. Brain Res Bull 57: 721–725, 2002. doi: 10.1016/S0361-9230(01)00788-2. [DOI] [PubMed] [Google Scholar]
- 956.Johnstone LE, Fong TM, Leng G. Neuronal activation in the hypothalamus and brainstem during feeding in rats. Cell Metab 4: 313–321, 2006. doi: 10.1016/j.cmet.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 957.Renaud LP, Tang M, McCann MJ, Stricker EM, Verbalis JG. Cholecystokinin and gastric distension activate oxytocinergic cells in rat hypothalamus. Am J Physiol Regul Integr Comp Physiol 253: R661–R665, 1987. doi: 10.1152/ajpregu.1987.253.4.R661. [DOI] [PubMed] [Google Scholar]
- 958.Verbalis JG, McCann MJ, McHale CM, Stricker EM. Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science 232: 1417–1419, 1986. doi: 10.1126/science.3715453. [DOI] [PubMed] [Google Scholar]
- 959.Durham DA, Banks WA, Kastin AJ. Carrier-mediated transport of labeled oxytocin from brain to blood. Neuroendocrinology 53: 447–452, 1991. doi: 10.1159/000125756. [DOI] [PubMed] [Google Scholar]
- 960.Mens WB, Witter A, Van Wimersma Greidanus TB. Penetration of neurohypophyseal hormones from plasma into cerebrospinal fluid (CSF): half-times of disappearance of these neuropeptides from CSF. Brain Res 262: 143–149, 1983. doi: 10.1016/0006-8993(83)90478-X. [DOI] [PubMed] [Google Scholar]
- 961.Lee MR, Scheidweiler KB, Diao XX, Akhlaghi F, Cummins A, Huestis MA, Leggio L, Averbeck BB. Oxytocin by intranasal and intravenous routes reaches the cerebrospinal fluid in rhesus macaques: determination using a novel oxytocin assay. Mol Physchiatry 23: 115–122, 2018. doi: 10.1038/mp.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 962.Ho JM, Anekonda VT, Thompson BW, Zhu M, Curry RW, Hwang BH, Morton GJ, Schwartz MW, Baskin DG, Appleyard SM, Blevins JE. Hindbrain oxytocin receptors contribute to the effects of circulating oxytocin on food intake in male rats. Endocrinology 155: 2845–2857, 2014. doi: 10.1210/en.2014-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 963.Iwasaki Y, Maejima Y, Suyama S, Yoshida M, Arai T, Katsurada K, Kumari P, Nakabayashi H, Kakei M, Yada T. Peripheral oxytocin activates vagal afferent neurons to suppress feeding in normal and leptin-resistant mice: a route for ameliorating hyperphagia and obesity. Am J Physiol Regul Integr Comp Physiol 308: R360–R369, 2015. [DOI] [PubMed] [Google Scholar]
- 964.Sabatier N, Leng RI. Central release of oxytocin and the ventromedial hypothalamus. Biochem Soc Trans 35: 1247–1251, 2007. doi: 10.1042/BST0351247. [DOI] [PubMed] [Google Scholar]
- 965.Landgraf R, Neumann ID. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol 25: 150–176, 2004. [ doi: 10.1016/j.yfrne.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 966.Tannenbaum GS, Lapointe M, Beaudet A, Howard AD. Expression of growth hormone secretagogue-receptors by growth hormone-releasing hormone neurons in the mediobasal hypothalamus. Endocrinology 139: 4420–4423, 1998. doi: 10.1210/endo.139.10.6330. [DOI] [PubMed] [Google Scholar]
- 967.Mitchell V, Bouret S, Beauvillain JC, Schilling A, Perret M, Kordon C, Epelbaum J. Comparative distribution of mRNA encoding the growth hormone secretagogue-receptor (GHS-R) in Microcebus murinus (Primate, lemurian) and rat forebrain and pituitary. J Comp Neurol 429: 469–489, 2001. doi:. [DOI] [PubMed] [Google Scholar]
- 968.Harrold JA, Dovey T, Cai XJ, Halford JC, Pinkney J. Autoradiographic analysis of ghrelin receptors in the rat hypothalamus. Brain Res 1196: 59–64, 2008. doi: 10.1016/j.brainres.2007.12.055. [DOI] [PubMed] [Google Scholar]
- 969.Sexton PM, Paxinos G, Kenney MA, Wookey PJ, Beaumont K. In vitro autoradiographic localization of amylin binding sites in rat brain. Neuroscience 62: 553–567, 1994. doi: 10.1016/0306-4522(94)90388-3. [DOI] [PubMed] [Google Scholar]
- 970.Le Foll C, Johnson MD, Dunn-Meynell AA, Boyle CN, Lutz TA, Levin BE. Amylin-induced central IL-6 production enhances ventromedial hypothalamic leptin signaling. Diabetes 64: 1621–1631, 2015. doi: 10.2337/db14-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 971.Routh VH. Glucose sensing neurons in the ventromedial hypothalamus. Sensors (Basel) 10: 9002–9025, 2010. doi: 10.3390/s101009002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 972.Zhou C, Teegala SB, Khan BA, Gonzalez C, Routh VH. Hypoglycemia: role of hypothalamic glucose-inhibited (GI) neurons in detection and correction. Front Physiol 9: 901–908, 2018. doi: 10.3389/fpls.2018.00901,10.3389/fphys.2018.00901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 973.Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, Chun S, Kim SW, Park JY, Lee KU, Kim MS. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 48: 2170–2178, 2005. doi: 10.1007/s00125-005-1913-1. [DOI] [PubMed] [Google Scholar]
- 974.McCrimmon RJ, Shaw M, Fan X, Cheng H, Ding Y, Vella MC, Zhou L, McNay EC, Sherwin RS. Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes 57: 444–450, 2008. [DOI] [PubMed] [Google Scholar]
- 975.Alhamami HN, Uddin MM, Mahmood AS, Briski KP. Lateral but not medial hypothalamic ampk activation occurs at the hypoglycemic nadir in insulin-injected male rats: impact of caudal dorsomedial hindbrain catecholamine signaling. Neuroscience 379: 103–114, 2018. [DOI] [PubMed] [Google Scholar]
- 976.Coutinho EA, Okamoto S, Ishikawa AW, Yokota S, Wada N, Hirabayashi T, Saito K, Sato T, Takagi K, Wang CC, Kobayashi K, Ogawa Y, Shioda S, Yoshimura Y, Minokoshi Y. Activation of SF1 neurons in the ventromedial hypothalamus by DREADD technology increases insulin sensitivity in peripheral tissues. Diabetes 66: 2372–2386, 2017. [DOI] [PubMed] [Google Scholar]
- 977.Sternson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH > arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci 8: 1356–1363, 2005. [DOI] [PubMed] [Google Scholar]
- 978.Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine‐B‐hydroxylase as a marker. J Comp Neurol 163: 467–505, 1975. [DOI] [PubMed] [Google Scholar]
- 979.Moga MM, Weis RP, Moore RY. Efferent projections of the paraventricular thalamic nucleus in the rat. J Comp Neurol 359: 221–238, 1995. [DOI] [PubMed] [Google Scholar]
- 980.Shimogawa Y, Sakuma Y, Yamanouchi K. Efferent and afferent connections of the ventromedial hypothalamic nucleus determined by neural tracer analysis: implications for lordosis regulation in female rats. Neurosci Res 91: 19–33, 2015. doi: 10.1016/j.neures.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 981.Millhouse OE. The organization of the ventromedial hypothalamic nucleus. Brain Res 55: 71–87, 1973. doi: 10.1016/0006-8993(73)90489-7. [DOI] [PubMed] [Google Scholar]
- 982.Noble EE, Billington CJ, Kotz CM, Wang C. Oxytocin in the ventromedial hypothalamic nucleus reduces feeding and acutely increases energy expenditure. Am J Physiol Regul Integr Comp Physiol 307: R737–R745, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 983.Anderson DJ. Optogenetics, sex, and violence in the brain: implications for psychiatry. Biol Psychiatry 71: 1081–1089, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 984.Todd WD, Fenselau H, Wang JL, Zhang R, Machado NL, Venner A, Broadhurst RY, Kaur S, Lynagh T, Olson DP, Lowell BB, Fuller PM, Saper CB. A hypothalamic circuit for the circadian control of aggression. Nat Neurosci 21: 717–724, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985.Lin D, Boyle MP, Dollar P, Lee H, Lein ES, Perona P, Anderson DJ. Functional identification of an aggression locus in the mouse hypothalamus. Nature 470: 221–226, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 986.Kunwar PS, Zelikowsky M, Remedios R, Cai H. Ventromedial hypothalamic neurons control a defensive emotion state. Elife 4: e06633, 2015. doi: 10.7554/eLife.06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 987.Lo L, Yao S, Kim DW, Cetin A, Harris J, Zeng H, Anderson DJ, Weissbourd B. Connectional architecture of a mouse hypothalamic circuit node controlling social behavior. Proc Natl Acad Sci U S A 116: 7503–7512, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 988.Wilent WB, Oh MY, Buetefisch CM, Bailes JE, Cantella D, Angle C, Whiting DM. Induction of panic attack by stimulation of the ventromedial hypothalamus. J Neurosurg 112: 1295–1298, 2010. [DOI] [PubMed] [Google Scholar]
- 989.Thompson RH, Swanson LW. Organization of inputs to the dorsomedial nucleus of the hypothalamus: a reexamination with Fluorogold and PHAL in the rat. Brain Res Brain Res Rev 27: 89–118, 1998. [DOI] [PubMed] [Google Scholar]
- 990.Rao ZR, Yamano M, Wanaka A, Tatehata T, Shiosaka S, Tohyama M. Distribution of cholinergic neurons and fibers in the hypothalamus of the rat using choline acetyltransferase as a marker. Neuroscience 20: 923–934, 1987. [DOI] [PubMed] [Google Scholar]
- 991.Bi S, Kim YJ, Zheng F. Dorsomedial hypothalamic NPY and energy balance control. Neuropeptides 46: 309–314, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992.Lee SJ, Verma S, Simonds SE, Kirigiti MA, Kievit P, Lindsley SR, Loche A, Smith MS, Cowley MA, Grove KL. Leptin stimulates neuropeptide Y and cocaine amphetamine-regulated transcript coexpressing neuronal activity in the dorsomedial hypothalamus in diet-induced obese mice. J Neurosci 33: 15306–15317, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 993.Bellinger LL, Bernardis LL. The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol Behav 76: 431–442, 2002. [DOI] [PubMed] [Google Scholar]
- 994.Jeong JH, Lee DK, Jo YH. Cholinergic neurons in the dorsomedial hypothalamus regulate food intake. Mol Metab 6: 306–312, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 995.Pandit R, Beerens S, Adan RA. Role of leptin in energy expenditure: the hypothalamic perspective. Am J Physiol Regul Integr Comp Physiol 312: R938–R947, 2017. [DOI] [PubMed] [Google Scholar]
- 996.Morrison SF, Nakamura K. Central mechanisms for thermoregulation. Annu Rev Physiol 81: 285–308, 2019. doi: 10.1146/annurev-physiol-020518-114546. [DOI] [PubMed] [Google Scholar]
- 997.Yang L, Scott KA, Hyun J, Tamashiro KL, Tray N, Moran TH, Bi S. Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J Neurosci 29: 179–190, 2009. doi: 10.1523/JNEUROSCI.4379-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 998.de La Serre CB, Kim YJ, Moran TH, Bi S. Dorsomedial hypothalamic NPY affects cholecystokinin-induced satiety via modulation of brain stem catecholamine neuronal signaling. Am J Physiol Regul Integr Comp Physiol 311: R930–R939, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 999.Watts AG, Swanson LW, Sanchez-Watts G. Efferent projections of the suprachiasmatic nucleus: I. Studies using anterograde transport of Phaseolus vulgaris leucoagglutinin in the rat. J Comp Neurol 258: 204–229, 1987. doi: 10.1002/cne.902580204. [DOI] [PubMed] [Google Scholar]
- 1000.Leak RK, Moore RY. Topographic organization of suprachiasmatic nucleus projection neurons. J Comp Neurol 433: 312–334, 2001. [DOI] [PubMed] [Google Scholar]
- 1001.Vujovic N, Gooley JJ, Jhou TC, Saper CB. Projections from the subparaventricular zone define four channels of output from the circadian timing system. J Comp Neurol 523: 2714–2737, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1002.Shi Z, Madden CJ, Brooks VL. Arcuate neuropeptide Y inhibits sympathetic nerve activity via multiple neuropathways. J Clin Invest 127: 2868–2880, 2017. doi: 10.1172/JCI92008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1003.Morrison SF. Central control of body temperature. F1000Res 5: 880–2016, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1004.Chao PT, Yang L, Aja S, Moran TH, Bi S. Knockdown of NPY expression in the dorsomedial hypothalamus promotes development of brown adipocytes and prevents diet-induced obesity. Cell Metab 13: 573–583, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1005.Lee SJ, Sanchez-Watts G, Krieger JP, Pignalosa A, Norell PN, Cortella A, Pettersen KG, Vrdoljak D, Hayes MR, Kanoski SE, Langhans W, Watts AG. Loss of dorsomedial hypothalamic GLP-1 signaling reduces BAT thermogenesis and increases adiposity. Mol Metab 11: 33–46, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1006.de Git KC, van Tuijl DC, Luijendijk MC, Wolterink-Donselaar IG, Ghanem A, Conzelmann KK, Adan RA. Anatomical projections of the dorsomedial hypothalamus to the periaqueductal grey and their role in thermoregulation: a cautionary note. Physiol Rep 6: e13807, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1007.Li L, de La Serre CB, Zhang N, Yang L, Li H, Bi S. Knockdown of neuropeptide Y in the dorsomedial hypothalamus promotes hepatic insulin sensitivity in male rats. Endocrinology 157: 4842–4852, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1008.Schwanzel-Fukuda M, Morrell JI, Pfaff DW. Localization of forebrain neurons which project directly to the medulla and spinal cord of the rat by retrograde tracing with wheat germ agglutinin. J Comp Neurol 226: 1–20, 1984. [DOI] [PubMed] [Google Scholar]
- 1009.Bass J. Circadian topology of metabolism. Nature 491: 348–356, 2012. [DOI] [PubMed] [Google Scholar]
- 1010.Marcheva B, Ramsey KM, Peek CB, Affinati A, Bass ME. J. Circadian clocks and metabolism. Handb Exp Pharmacol 217: 127–155, 2013. doi: 10.1007/978-3-642-25950-0_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1011.Challet E. Keeping circadian time with hormones. Diabetes Obes Metab 17: 76–83, 2015. [DOI] [PubMed] [Google Scholar]
- 1012.Cedernaes J, Huang W, Ramsey KM, Waldeck N, Cheng L, Marcheva B, Omura C, Kobayashi Y, Peek CB, Levine DC, Dhir R, Awatramani R, Bradfield CA, Wang XA, Takahashi JS, Mokadem M, Ahima RS, Bass J. Transcriptional basis for rhythmic control of hunger and metabolism within the AgRP neuron. Cell Metab 29: 1078–1091, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1013.Panda S. Circadian physiology of metabolism. Science 354: 1008–1015, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1014.Kim P, Oster H, Lehnert H, Schmid SM, Salamat N, Barclay JL, Maronde E, Inder W, Rawashdeh O. Coupling the circadian clock to homeostasis: the role of period in timing. Physiology. Endoc Rev 40: 66–95, 2019. doi: 10.1210/er.2018-00049. [DOI] [PubMed] [Google Scholar]
- 1015.Asher G, Sassone-Corsi P. Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell 161: 84–92, 2015. [DOI] [PubMed] [Google Scholar]
- 1016.Hastings MH, Maywood ES, Brancaccio M. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat Rev Neurosci 19: 453–469, 2018. [DOI] [PubMed] [Google Scholar]
- 1017.Canteras NS, Ribeiro-Barbosa ÉR, Goto M, Cipolla-Neto J, Swanson LW. The retinohypothalamic tract: comparison of axonal projection patterns from four major targets. Brain Res Rev 65: 150–183, 2011. [DOI] [PubMed] [Google Scholar]
- 1018.Watts AG, Swanson LW. Efferent projections of the suprachiasmatic nucleus: II. Studies using retrograde transport of fluorescent dyes and simultaneous peptide immunohistochemistry in the rat. J Comp Neurol 258: 230–252, 1987. doi: 10.1002/cne.902580205. [DOI] [PubMed] [Google Scholar]
- 1019.Morin LP. Neuroanatomy of the extended circadian rhythm system. Exp Neurol 243: 4–20, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1020.Watts AG. The efferent projections of the suprachiasmatic nucleus: anatomical insights into the control of circadian rhythms. In: The Suprachiasmatic Nucleus: The Mind’s Clock edited by Klein D, Moore RY, Reppert SM.. Oxford University Press; New York, 1991, p. 77–104. [Google Scholar]
- 1021.Lu J, Zhang YH, Chou TC, Gaus SE, Elmquist JK, Shiromani P, Saper CB. Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep-wake cycle and temperature regulation. J. Neurosci 21: 4864–4874, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1022.Li S, Kirouac GJ. Projections from the paraventricular nucleus of the thalamus to the forebrain, with special emphasis on the extended amygdala. J Comp Neurol 506: 263–287, 2008. [DOI] [PubMed] [Google Scholar]
- 1023.Kirouac GJ. Placing the paraventricular nucleus of the thalamus within the brain circuits that control behavior. Neurosci Biobehav Rev 56: 315–329, 2015. [DOI] [PubMed] [Google Scholar]
- 1024.Millan EZ, Ong Z, McNally GP. Paraventricular thalamus: gateway to feeding, appetitive motivation, and drug addiction. Prog Brain Res 235: 113–137, 2017. [DOI] [PubMed] [Google Scholar]
- 1025.Otis JM, Zhu M, Namboodiri VM, Cook CA, Kosyk O, Matan AM, Ying R, Hashikawa Y, Hashikawa K, Trujillo-Pisanty I, Guo J, Ung RL, Rodriguez-Romaguera J, Anton ES, Stuber GD. Paraventricular thalamus projection neurons integrate cortical and hypothalamic signals for cue-reward processing. Neuron 103: 423–431, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1026.Labouèbe G, Boutrel B, Tarussio D, Thorens B. Glucose-responsive neurons of the paraventricular thalamus control sucrose-seeking behavior. Nat Neurosci 19: 999–1002, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1027.Do-Monte FH, Minier-Toribio A, Quiñones-Laracuente K, Medina-Colón EM, Quirk GJ. Thalamic regulation of sucrose seeking during unexpected reward omission. Neuron 94: 388–400, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1028.Ong ZY, Liu JJ, Pang ZP, Grill HJ. Paraventricular thalamic control of food intake and reward: Role of glucagon-like peptide-1 receptor signaling. Neuropsychopharmacology 42: 2387–2397, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1029.Cheng J, Wang J, Ma X, Ullah R, Shen Y, Zhou YD. Anterior paraventricular thalamus to nucleus accumbens projection is involved in feeding behavior in a novel environment. Front Mol Neurosci 11: 1–12, 2018. doi: 10.3389/fnmol.2018.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1030.Choi EA, Jean-Richard-dit-Bressel P, Clifford CW, McNally GP. Paraventricular thalamus controls behavior during motivational conflict. J Neurosci 39: 4945–4958, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1031.Dong X, Li S, Kirouac GJ. Collateralization of projections from the paraventricular nucleus of the thalamus to the nucleus accumbens, bed nucleus of the stria terminalis, and central nucleus of the amygdala. Brain Struct Funct 222: 3927–3943, 2017. [DOI] [PubMed] [Google Scholar]
- 1032.Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat Neurosci 4: 732–738, 2001. [DOI] [PubMed] [Google Scholar]
- 1033.Mahoney CE, Brewer J, Bittman EL. Central control of circadian phase in arousal-promoting neurons. PLoS ONE 8: e67173, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1034.Ritter S, Watts AG, Dinh TT, Sanchez-Watts G, Pedrow C. Immunotoxin lesion of hypothalamically projecting norepinephrine and epinephrine neurons differentially affects circadian and stressor-stimulated corticosterone secretion. Endocrinology 144: 1357–1367, 2003. [DOI] [PubMed] [Google Scholar]
- 1035.Bolles RC, De Lorge J. The rat’s adjustment to a-diurnal feeding cycles. J Comp Physiol Psychol 55: 760–762, 1962. [DOI] [PubMed] [Google Scholar]
- 1036.Richter CP. A behavioristic study of the activity of the rat. Comp Psych Monographs 1: 1–55, 1922. [Google Scholar]
- 1037.Boulos Z, Rosenwasser AM, Terman M. Feeding schedules and the circadian organization of behavior in the rat. Behav Brain Res 1: 39–65, 1980. [DOI] [PubMed] [Google Scholar]
- 1038.Krieger DT. Food and water restriction shifts corticosterone, temperature, activity and brain amine periodicity. Endocrinology 95: 1195–1201, 1974. [DOI] [PubMed] [Google Scholar]
- 1039.Patton DF, Mistlberger RE. Circadian adaptations to meal timing: neuroendocrine mechanisms. Front Neurosci 7: 1–14, 2013. doi: 10.3389/fnins.2013.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1040.Mistlberger RE. Food-anticipatory circadian rhythms: concepts and methods. Eur J Neurosci 30: 1718–1729, 2009. [DOI] [PubMed] [Google Scholar]
- 1041.Mistlberger RE. Neurobiology of food anticipatory circadian rhythms. Physiol Behav 104: 535–545, 2011. [DOI] [PubMed] [Google Scholar]
- 1042.Gooley JJ, Schomer A, Saper CB. The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nat Neurosci 9: 398–407, 2006. [DOI] [PubMed] [Google Scholar]
- 1043.Mieda M, Williams SC, Richardson JA, Tanaka K, Yanagisawa M. The dorsomedial hypothalamic nucleus as a putative food-entrainable circadian pacemaker. Proc Natl Acad Sci U S A 103: 12150–12155, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1044.Acosta-Galvan G, Yi CX, van der Vliet J, Jhamandas JH, Panula P, Angeles-Castellanos M, Del Carmen Basualdo M, Escobar C, Buijs RM. Interaction between hypothalamic dorsomedial nucleus and the suprachiasmatic nucleus determines intensity of food anticipatory behavior. Proc Natl Acad Sci U S A 108: 5813–5818, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1045.Rastogi A, Mintz EM. Neural correlates of food anticipatory activity in mice subjected to once- or twice-daily feeding periods. Eur J Neurosci 46: 2265–2275, 2017. [DOI] [PubMed] [Google Scholar]
- 1046.de Lartigue G, McDougle M. Dorsal striatum dopamine oscillations: setting the pace of food anticipatory activity. Acta Physiol (Oxford, England) 225: e13152, 2019. doi: 10.1111/apha.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1047.Mistlberger RE. Food as circadian time cue for appetitive behavior. F1000Res 9: F1000 Faculty Rev-61, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1048.Ranson SW. Somnolence caused by hypothalamic lesions in the monkey. Arch Neur Psych 71: 1–23, 1939. [Google Scholar]
- 1049.Campfield LA, Smith FJ. Blood glucose dynamics and control of meal initiation: a pattern detection and recognition theory. Physiol Rev 83: 25–58, 2003. doi: 10.1152/physrev.00019.2002. [DOI] [PubMed] [Google Scholar]
- 1050.Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. J Neurosci 27: 13292–13302, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1051.Novin D, VanderWeele DA, Rezek M. Infusion of 2-deoxy-D-glucose into the hepatic-portal system causes eating: evidence for peripheral glucoreceptors. Science 181: 858–860, 1973. [DOI] [PubMed] [Google Scholar]
- 1052.Del Prete E, Scharrer E. Circadian effects of hepatic branch vagotomy on the feeding response to 2-deoxy-D-glucose in rats. J Auton Nerv Syst 46: 27–36, 1994. [DOI] [PubMed] [Google Scholar]
- 1053.Hudson B, Ritter S. Hindbrain catecholamine neurons mediate consummatory responses to glucoprivation. Physiol Behav 82: 241–250, 2004. [DOI] [PubMed] [Google Scholar]
- 1054.Scharrer E, Langhans W. Control of food intake by fatty acid oxidation. Am J Physiol Regul Integr Comp Physiol 250: R1003–R1006, 1986. [DOI] [PubMed] [Google Scholar]
- 1055.Friedman MI, Tordoff MG. Fatty acid oxidation and glucose utilization interact to control food intake in rats. Am J Physiol Regul Integr Comp Physiol 251: R840–R845, 1986. [DOI] [PubMed] [Google Scholar]
- 1056.Langhans W, Scharrer E. Role of fatty acid oxidation in control of meal pattern. Behav Neural Biol 47: 7–16, 1987. [DOI] [PubMed] [Google Scholar]
- 1057.Langhans W, Scharrer E. Evidence for a vagally mediated satiety signal derived from hepatic fatty acid oxidation. J Auton Nerv Syst 18: 13–18, 1987. [DOI] [PubMed] [Google Scholar]
- 1058.Horn CC, Tordoff MG, Friedman MI. Role of vagal afferent innervation in feeding and brain Fos expression produced by metabolic inhibitors. Brain Res 919: 198–206, 2001. [DOI] [PubMed] [Google Scholar]
- 1059.Brandt K, Arnold M, Geary N, Langhans W, Leonhardt M. Vagal afferents mediate the feeding response to mercaptoacetate but not to the beta (3) adrenergic receptor agonist CL 316,243. Neurosci Lett 411: 104–107, 2007. [DOI] [PubMed] [Google Scholar]
- 1060.Tordoff MG, Rafka R, DiNovi MJ, Friedman MI. 2,5-Anhydro-d-mannitol: a fructose analogue that increases food intake in rats. Am J Physiol Regul Integr Physiol 254: R150–R153, 1988. [DOI] [PubMed] [Google Scholar]
- 1061.Langhans W, Scharrer E. Evidence for a role of the sodium pump of hepatocytes in the control of food intake. J Auton Nerv Syst 20: 199–205, 1987. [DOI] [PubMed] [Google Scholar]
- 1062.Darling RA, Zhao H, Kinch D, Li AJ, Simasko SM, Ritter S. Mercaptoacetate and fatty acids exert direct and antagonistic effects on nodose neurons via GPR40 fatty acid receptors. Am J Physiol Regul Integr Comp Physiol 307: R35–R43, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1063.Arnold M, Langhans W. Mercaptoacetate (MA) increases intestinal vagal afferent activity. Appetite 52: 545–870, 2009. doi: 10.1016/j.appet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1066.Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature 407: 908–913, 2000. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 1072.Tschop M, Wawarta R, Riepl RL, Friedrich S, Bidlingmaier M, Landgraf R, Folwaczny C. Post-prandial decrease of circulating human ghrelin levels. J Endocrinol Invest 24: RC19–21, 2001. doi: 10.1007/BF03351037. [DOI] [PubMed] [Google Scholar]
- 1067.Cabral A, Cornejo MP, Fernandez G, De Francesco PN, Garcia-Romero G, Uriarte M, Zigman JM, Portiansky E, Reynaldo M, Perello M. Circulating ghrelin acts on GABA neurons of the area postrema and mediates gastric emptying in male mice. Endocrinology 158: 1436–1449, 2017. doi: 10.1210/en.2016-1815. [DOI] [PubMed] [Google Scholar]
- 1068.Levin F, Edholm T, Schmidt PT, GrybäCk P, Jacobsson H, Degerblad M, HöYbye C, Holst JJ, Rehfeld JF, HellströM PM, NäSlund E, Ghrelin stimulates gastric emptying and hunger in normal-weight humans. J Clin Endocrinol Metab 91: 3296–3302, 2006. doi: 10.1210/jc.2005-2638. [DOI] [PubMed] [Google Scholar]
- 1064.Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273: 974–977, 1996. [PMC doi: 10.1126/science.273.5277.974. [DOI] [PubMed] [Google Scholar]
- 1065.Sakata I, Yamazaki M, Inoue K, Hayashi Y, Kangawa K, Sakai T. Growth hormone secretagogue receptor expression in the cells of the stomach-projected afferent nerve in the rat nodose ganglion. Neurosci Lett 342: 183–186, 2003. [DOI] [PubMed] [Google Scholar]
- 1069.Muller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, et al. Ghrelin. Mol Metab 4: 437–460, 2015. doi: 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1070.Liu J, Prudom CE, Nass R, Pezzoli SS, Oliveri MC, Johnson ML, Veldhuis P, Gordon DA, Howard AD, Witcher DR, Geysen HM, Gaylinn BD, Thorner MO. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J Clin Endocrinol Metab 93: 1980–1987, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1071.Kirchner H, Gutierrez JA, Solenberg PJ, Pfluger PT, Czyzyk TA, Willency JA, Schurmann A, Joost HG, Jandacek RJ, Hale JE, Heiman ML, Tschop MH. GOAT links dietary lipids with the endocrine control of energy balance. Nat Med 15: 741–745, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1073.Cummings DE, Weigle DS, Frayo RS, Breen PA, Ma MK, Dellinger EP, Purnell JQ. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 346: 1623–1630, 2002. doi: 10.1056/NEJMoa012908. [DOI] [PubMed] [Google Scholar]
- 1074.Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50: 1714–1719, 2001. [DOI] [PubMed] [Google Scholar]
- 1075.Kirchner H, Heppner KM, Tschop MH. The role of ghrelin in the control of energy balance. Handb Exp Pharmacol 209: 161–184, 2012. doi: 10.1007/978-3-642-24716-3_7. [DOI] [PubMed] [Google Scholar]
- 1076.Lippl F, Erdmann J, Steiger A, Lichter N, Czogalla-Peter C, Bidlingmaier M, Tholl S, Schusdziarra V. Low-dose ghrelin infusion–evidence against a hormonal role in food intake. Regul Pept 174: 26–31, 2012. [DOI] [PubMed] [Google Scholar]
- 1077.Lopez-Ferreras L, Richard JE, Anderberg RH, Nilsson FH, Olandersson K, Kanoski SE, Skibicka KP. Ghrelin’s control of food reward and body weight in the lateral hypothalamic area is sexually dimorphic. Physiol Behav 176: 40–49, 2017. doi: 10.1016/j.physbeh.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1078.Skibicka KP, Hansson C, Alvarez-Crespo M, Friberg PA, Dickson SL. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience 180: 129–137, 2011. [DOI] [PubMed] [Google Scholar]
- 1079.Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123: 1120–1128, 2002. [DOI] [PubMed] [Google Scholar]
- 1080.Arnold M, Mura A, Langhans W, Geary N. Gut vagal afferents are not necessary for the eating-stimulatory effect of intraperitoneally injected ghrelin in the rat. J Neurosci 26: 11052–11060, 2006. doi: 10.1523/JNEUROSCI.2606-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1081.Davis EA, Wald HS, Suarez AN, Zubcevic J, Liu CM, Cortella AM, Kamitakahara AK, Polson JW, Arnold M, Grill HJ, de Lartigue G, Kanoski SE. Ghrelin signaling affects feeding behavior, metabolism, and memory through the vagus nerve. Curr Biol 30: 4510–4518, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1082.Date Y, Toshinai K, Koda S, Miyazato M, Shimbara T, Tsuruta T, Niijima A, Kangawa K, Nakazato M. Peripheral interaction of ghrelin with cholecystokinin on feeding regulation. Endocrinology 146: 3518–3525, 2005. [DOI] [PubMed] [Google Scholar]
- 1083.Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 16: 296–309, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1084.Levitsky DA. The non-regulation of food intake in humans: hope for reversing the epidemic of obesity. Physiol Behav 86: 623–632, 2005. [DOI] [PubMed] [Google Scholar]
- 1085.Mietlicki-Baase EG, Reiner DJ, Cone JJ, Olivos DR, McGrath LE, Zimmer DJ, Roitman MF, Hayes MR. Amylin modulates the mesolimbic dopamine system to control energy balance. Neuropsychopharmacology 40: 372–385, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1086.Mietlicki-Baase EG, Ortinski PI, Rupprecht LE, Olivos DR, Alhadeff AL, Pierce RC, Hayes MR. The food intake-suppressive effects of glucagon-like peptide-1 receptor signaling in the ventral tegmental area are mediated by AMPA/kainate receptors. Am J Physiol Endocrinol Metab 305: E1367–E1374, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1087.Terrill SJ, Maske CB, Williams DL. Endogenous GLP-1 in lateral septum contributes to stress-induced hypophagia. Physiol Behav 192: 17–22, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1088.Hsu TM, Hahn JD, Konanur VR, Lam A, Kanoski SE. Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmission. Neuropsychopharmacol 40: 327–337, 2015. doi: 10.1038/npp.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1089.Azevedo EP, Tan B, Pomeranz LE, Ivan V, Fetcho R, Schneeberger M, Doerig KR, Liston C, Friedman JM, Stern SA. A limbic circuit selectively links active escape to food suppression. Elife 9: e58894, 2020. doi: 10.7554/elife.58894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1090.Min DK, Tuor UI, Chelikani PK. Gastric distention induced functional magnetic resonance signal changes in the rodent brain. Neuroscience 179: 151–158, 2011. [DOI] [PubMed] [Google Scholar]
- 1091.Min DK, Tuor UI, Koopmans HS, Chelikani PK. Changes in differential functional magnetic resonance signals in the rodent brain elicited by mixed-nutrient or protein-enriched meals. Gastroenterology 141: 1832–1841, 2011. [DOI] [PubMed] [Google Scholar]
- 1092.Wang GJ, Yang J, Volkow ND, Telang F, Ma Y, Zhu W, Wong CT, Tomasi D, Thanos PK, Fowler JS. Gastric stimulation in obese subjects activates the hippocampus and other regions involved in brain reward circuitry. PNAS Proc Natl Acad Sci U S A 103: 15641–15645, 2006. doi: 10.1073/pnas.0601977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1093.Higgs S. Memory for recent eating and its influence on subsequent food intake. Appetite 39: 159–166, 2002. [DOI] [PubMed] [Google Scholar]
- 1094.Higgs S. Cognitive influences on food intake: the effects of manipulating memory for recent eating. Physiol Behav 94: 734–739, 2008. [DOI] [PubMed] [Google Scholar]
- 1095.Brunstrom JM, Burn JF, Sell NR, Collingwood JM, Rogers PJ, Wilkinson LL, Hinton EC, Maynard OM, Ferriday D. Episodic memory and appetite regulation in humans. PLoS One 7: e50707, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096.Rolls ET, Sienkiewicz ZJ, Yaxley S. Hunger modulates the responses to gustatory stimuli of single neurons in the caudolateral orbitofrontal cortex of the macaque monkey. Eur J Neurosci 1: 53–60, 1989. [DOI] [PubMed] [Google Scholar]
- 1097.Yaxley S, Rolls ET, Sienkiewicz ZJ. The responsiveness of neurons in the insular gustatory cortex of the macaque monkey is independent of hunger. Physiol Behav 42: 223–229, 1988. [DOI] [PubMed] [Google Scholar]
- 1098.Kringelbach ML, O’Doherty J, Rolls ET, Andrews C. Activation of the human orbitofrontal cortex to a liquid food stimulus is correlated with its subjective pleasantness. Cereb Cortex 13: 1064–1071, 2003. doi: 10.1093/cercor/13.10.1064. [DOI] [PubMed] [Google Scholar]
- 1099.Norton GN, Anderson AS, Hetherington MM. Volume and variety: relative effects on food intake. Physiol Behav 87: 714–722, 2006. [DOI] [PubMed] [Google Scholar]
- 1100.Siegel PS, Pilgrim FJ. The effect of monotony on the acceptance of food. Am J Psychol 71: 756–759, 1958. [PubMed] [Google Scholar]
- 1101.Small DM, Zatorre RJ, Dagher A, Evans AC, Jones-Gotman M. Changes in brain activity related to eating chocolate: from pleasure to aversion. Brain 124: 1720–1733, 2001. [DOI] [PubMed] [Google Scholar]
- 1102.de Araujo IE, Gutierrez R, Oliveira-Maia AJ, Pereira A Jr, Nicolelis MA, Simon SA. Neural ensemble coding of satiety states. Neuron 51: 483–494, 2006. [DOI] [PubMed] [Google Scholar]
- 1103.Spector AC. Gustatory function in the parabrachial nuclei: implications from lesion studies in rats. Rev Neurosci 6: 143–175, 1995. doi: 10.1515/revneuro.1995.6.2.143. [DOI] [PubMed] [Google Scholar]
- 1104.Bernard JF, Bandler R. Parallel circuits for emotional coping behaviour: new pieces in the puzzle. J Comp Neurol 401: 429–436, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 1105.Reilly S. The parabrachial nucleus and conditioned taste aversion. Brain Res Bull 48: 239–254, 1999. [DOI] [PubMed] [Google Scholar]
- 1106.Sakai N, Yamamoto T. Conditioned taste aversion and c-fos expression in the rat brainstem after administration of various USs. Neuroreport 8: 2215–2220, 1997. [DOI] [PubMed] [Google Scholar]
- 1107.Cano G, Passerin AM, Schiltz JC, Card JP, Morrison SF, Sved AF. Anatomical substrates for the central control of sympathetic outflow to interscapular adipose tissue during cold exposure. J Comp Neurol 460: 303–326, 2003. [DOI] [PubMed] [Google Scholar]
- 1108.Uchida Y, Nagashima K, Yuri K. Fasting or systemic des-acyl ghrelin administration to rats facilitates thermoregulatory behavior in a cold environment. Brain Res 1696: 10–21, 2018. doi: 10.1016/j.brainres.2018.05.038. [DOI] [PubMed] [Google Scholar]
- 1109.Ritter S, Dinh TT. 2-Mercaptoacetate and 2-deoxy-D-glucose induce Fos-like immunoreactivity in rat-brain. Brain Res 641: 111–120, 1994. [DOI] [PubMed] [Google Scholar]
- 1110.Potes CS, Lutz TA, Riediger T. Identification of central projections from amylin-activated neurons to the lateral hypothalamus. Brain Res 1334: 31–44, 2010. [DOI] [PubMed] [Google Scholar]
- 1111.Baraboi ED, St-Pierre DH, Shooner J, Timofeeva E, Richard D. Brain activation following peripheral administration of the GLP-1 receptor agonist exendin-4. Am J Physiol Regul Integr Comp Physiol 301: R1011–R1024, 2011. [DOI] [PubMed] [Google Scholar]
- 1112.Li BH, Rowland NE. Cholecystokinin- and dexfenfluramine-induced anorexia compared using devazepide and c-fos expression in the rat brain. Reg Peptides 50: 223–233, 1994. doi: 10.1016/0167-0115(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 1113.Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci 23: 10084–10092, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1114.Elmquist JK, Scammell TE, Jacobson CD, Saper CB. Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. J Comp Neurol 371: 85–103, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 1115.Hosoi T, Kawagishi T, Okuma Y, Tanaka J, Nomura Y. Brain stem is a direct target for leptin’s action in the central nervous system. Endocrinology 143: 3498–3504, 2002. [DOI] [PubMed] [Google Scholar]
- 1116.Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, Saper CB. Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology 138: 839–842, 1997. [DOI] [PubMed] [Google Scholar]
- 1117.Paues J, Engblom D, Mackerlova L, Ericsson-Dahlstrand A, Blomqvist A. Feeding-related immune responsive brain stem neurons: association with CGRP. Neuroreport 12: 2399–2403, 2001. [DOI] [PubMed] [Google Scholar]
- 1118.Campos CA, Bowen AJ, Schwartz MW, Palmiter RD. Parabrachial CGRP neurons control meal termination. Cell Metab 23: 811–820, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1119.Flak JN, Patterson CM, Garfield AS, D’Agostino G, Goforth PB, Sutton AK, Malec PA, Wong JM, Germani M, Jones JC, Rajala M, Satin L, Rhodes CJ, Olson DP, Kennedy RT, Heisler LK, Myers MG. Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balance. Nat Neurosci 17: 1744–1750, 2014. doi: 10.1038/nn.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1120.Alhadeff AL, Hayes MR, Grill HJ. Leptin receptor signaling in the lateral parabrachial nucleus contributes to the control of food intake. Am J Physiol Regul Integr Comp Physiol 307: R1338–R1344, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1121.Herbert H, Moga MM, Saper CB. Connections of the parabrachial nucleus with the nucleus of the solitary tract and the medullary reticular formation in the rat. J Comp Neurol 293: 540–580, 1990. [DOI] [PubMed] [Google Scholar]
- 1122.Rinaman L. Ascending projections from the caudal visceral nucleus of the solitary tract to brain regions involved in food intake and energy expenditure. Brain Res 1350: 18–34, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1123.Herbert H, Saper CB. Cholecystokinin‐, galanin‐, and corticotropin‐releasing factor‐like immunoreactive projections from the nucleus of the solitary tract to the parabrachial nucleus in the rat. J Comp Neurol 293: 581–598, 1990. [DOI] [PubMed] [Google Scholar]
- 1124.Richard JE, Farkas I, Anesten F, Anderberg RH, Dickson SL, Gribble FM, Reimann F, Jansson JO, Liposits Z, Skibicka KP. GLP-1 receptor stimulation of the lateral parabrachial nucleus reduces food intake: neuroanatomical, electrophysiological, and behavioral evidence. Endocrinology 155: 4356–4367, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1125.Roman CW, Va D, Palmiter RD. Genetically and functionally defined NTS to PBN brain circuits mediating anorexia. Nature Commun 7: 11905, 2016. doi: 10.1038/ncomms11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1126.Hermanson O, Telkov M, Geijer T, Hallbeck M, Blomqvist A. Preprodynorphin mRNA-expressing neurones in the rat parabrachial nucleus: subnuclear localization, hypothalamic projections and colocalization with noxious-evoked fos-like immunoreactivity. Eur J Neurosci 10: 358–367, 1998. [DOI] [PubMed] [Google Scholar]
- 1127.Boccia L, Gamakharia S, Coester B, Whiting L, Lutz TA, Foll L. C. Amylin brain circuitry. Peptides 132: 170366, 2020. doi: 10.1016/j.peptides.2020.170366. [DOI] [PubMed] [Google Scholar]
- 1128.Sabatini PV, Frikke-Schmidt H, Arthurs J, Gordian DP, Atel A, Rupp AC, Adams JM, Wang J, Beck Jørgensen S, Olson DP, Palmiter RD, Myers MG, Seeley RJ. GFRAL-expressing neurons suppress food intake via aversive pathways. Proc Natl Acad Sci U S A 118: e2021357118, 2021. doi: 10.1073/pnas.2021357118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1129.Karimnamazi H, Travers SP, Travers JB. Oral and gastric input to the parabrachial nucleus of the rat. Brain Res 957: 193–206, 2002. [DOI] [PubMed] [Google Scholar]
- 1130.Cechetto DF, Standaert DG, Saper CB. Spinal and trigeminal dorsal horn projections to the parabrachial nucleus in the rat. J Comp Neurol 240: 153–160, 1985. [DOI] [PubMed] [Google Scholar]
- 1131.Standaert DG, Watson SJ, Houghten RA, Saper CB. Opioid peptide immunoreactivity in spinal and trigeminal dorsal horn neurons projecting to the parabrachial nucleus in the rat. J Neurosci 6: 1220–1226, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1132.Bernard JF, Dallel R, Raboisson P, Villanueva L, Bars DL. D. Organization of the efferent projections from the spinal cervical enlargement to the parabrachial area and periaqueductal gray: a PHA-L study in the rat. J Comp Neurol 353: 480–505, 1995. doi: 10.1002/cne.903530403. [DOI] [PubMed] [Google Scholar]
- 1133.Feil K, Herbert H. Topographic organization of spinal and trigeminal somatosensory pathways to the rat parabrachial and Kölliker-Fuse nuclei. J Comp Neurol 353: 506–528, 1995. [DOI] [PubMed] [Google Scholar]
- 1134.Campos CA, Bowen AJ, Roman CW, Palmiter RD. Encoding of danger by parabrachial CGRP neurons. Nature 555: 617–622, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1135.Moga MM, Herbert H, Hurley KM, Yasui Y, Gray TS, Saper CB. Organization of cortical, basal forebrain, and hypothalamic afferents to the parabrachial nucleus in the rat. J Comp Neurol 295: 624–661, 1990. [DOI] [PubMed] [Google Scholar]
- 1136.Petrovich GD, Swanson LW. Projections from the lateral part of the central amygdalar nucleus to the postulated fear conditioning circuit. Brain Res 763: 247–254, 1997. [DOI] [PubMed] [Google Scholar]
- 1137.Hurley KM, Herbert H, Moga MM, Saper CB. Efferent projections of the infralimbic cortex of the rat. J Comp Neurol 308: 249–276, 1991. [DOI] [PubMed] [Google Scholar]
- 1138.Cechetto DF, Saper CB. Evidence for a viscerotopic sensory representation in the cortex and thalamus in the rat. J Comp Neurol 262: 27–45, 1987. [DOI] [PubMed] [Google Scholar]
- 1139.Karimnamazi H, Travers JB. Differential projections from gustatory responsive regions of the parabrachial nucleus to the medulla and forebrain. Brain Res 813: 283–302, 1998. [DOI] [PubMed] [Google Scholar]
- 1140.Coizet V, Dommett EJ, Klop EM, Redgrave P, Overton PG. The parabrachial nucleus is a critical link in the transmission of short latency nociceptive information to midbrain dopaminergic neurons. Neuroscience 168: 263–272, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1141.Pritchard TC, Hamilton RB, Norgren R. Projections of the parabrachial nucleus in the old world monkey. Exp Neurol 165: 101–117, 2000. [DOI] [PubMed] [Google Scholar]
- 1142.Chiang MC, Bowen A, Schier LA, Tupone D, Uddin O, Heinricher MM. Parabrachial complex: a hub for pain and aversion. J Neurosci 39: 8225–8230, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1143.Yasui Y, Saper CB, Cechetto DF. Calcitonin gene-related peptide immunoreactivity in the visceral sensory cortex, thalamus, and related pathways in the rat. J Comp Neurol 290: 487–501, 1989. [DOI] [PubMed] [Google Scholar]
- 1144.Carter ME, Han S, Palmiter RD. Parabrachial calcitonin gene-related peptide neurons mediate conditioned taste aversion. J Neurosci 35: 4582–4586, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1145.Chen N, Callaway CW, Guyette FX, Rittenberger JC, Doshi AA, Dezfulian C, Elmer J, Pittsburgh Post-Cardiac Arrest Service. Arrest etiology among patients resuscitated from cardiac arrest. Resuscitation 130: 33–40, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1146.Roman CW, Sloat SR, Palmiter RD. A tale of two circuits: CCKNTS neuron stimulation controls appetite and induces opposing motivational states by projections to distinct brain regions. Neuroscience 358: 316–324, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1147.Cheng W, Gonzalez I, Pan W, Tsang AH, Adams J, Ndoka E, Gordian D, Khoury B, Roelofs K, Evers SS, MacKinnon A, Wu S, Frikke-Schmidt H, Flak JN, Trevaskis JL, Rhodes CJ, Fukada SI, Seeley RJ, Sandoval DA, Olson DP, Blouet C, Myers MG. Calcitonin receptor neurons in the mouse nucleus tractus solitarius control energy balance via the non-aversive suppression of feeding. Cell Metab 31: 301–312, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1148.Azpiroz F, Feinle-Bisset C, Grundy D, Tack J. Gastric sensitivity and reflexes: basic mechanisms underlying clinical problems. J Gastroenterol 49: 206–218, 2014. [DOI] [PubMed] [Google Scholar]
- 1149.Phillips RJ, Powley TL. Gastric volume rather than nutrient content inhibits food intake. Am J Physiol 271: R766–R769, 1996. [DOI] [PubMed] [Google Scholar]
- 1150.Oesch S, Ruegg C, Fischer B, Degen L, Beglinger C. Effect of gastric distension prior to eating on food intake and feelings of satiety in humans. Physiol Behav 87: 903–910, 2006. [DOI] [PubMed] [Google Scholar]
- 1151.Kaplan JM, Spector AC, Grill HJ. Dynamics of gastric emptying during and after stomach fill. Am J Physiol 263: R813–819, 1992. [DOI] [PubMed] [Google Scholar]
- 1152.Hellstrom PM, Gryback P, Jacobsson H. The physiology of gastric emptying. Best Pract Res Clin Anaesthesiol 20: 397–407, 2006. [DOI] [PubMed] [Google Scholar]
- 1153.Feinle C, Kunz P, Boesiger P, Fried M, Schwizer W. Scintigraphic validation of a magnetic resonance imaging method to study gastric emptying of a solid meal in humans. Gut 44: 106–111, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1154.Russek M. Participation of hepatic glucoreceptors in the control of intake of food. Nature 197: 79–80, 1963. [DOI] [PubMed] [Google Scholar]
- 1155.Balkan B, Li X. Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am J Physiol Regul Integr Comp Physiol 279: R1449–R1454, 2000. [DOI] [PubMed] [Google Scholar]
- 1156.Burcelin R, Dolci W, Thorens B. Portal glucose infusion in the mouse induces hypoglycemia: evidence that the hepatoportal glucose sensor stimulates glucose utilization. Diabetes 49: 1635–1642, 2000. [DOI] [PubMed] [Google Scholar]
- 1158.Tordoff MG, Friedman MI. Hepatic control of feeding: effect of glucose, fructose, and mannitol infusion. Am J Physiol Integr Comp Physiol 254: R969–R976, 1988. [DOI] [PubMed] [Google Scholar]
- 1159.Tordoff MG, Tluczek JP, Friedman MI. Effect of hepatic portal glucose concentration on food intake and metabolism. Am J Physiol Regul Integr Comp Physiol 257: R1474–R1480, 1989. [DOI] [PubMed] [Google Scholar]
- 1160.Langhans W, Grossmann F, Geary N. Intrameal hepatic-portal infusion of glucose reduces spontaneous meal size in rats. Physiol Behav 73: 499–507, 2001. [DOI] [PubMed] [Google Scholar]
- 1157.West DB, Fey D, Woods SC. Cholecystokinin persistently suppresses meal size but not food intake in free-feeding rats. Am J Physiol Integr Comp Physiol 246: R776–R787, 1984. doi: 10.1152/ajpregu.1984.246.5.R776. [DOI] [PubMed] [Google Scholar]
- 1161.Overduin J, Gibbs J, Cummings DE, Reeve JR Jr.. CCK-58 elicits both satiety and satiation in rats while CCK-8 elicits only satiation. Peptides 54: 71–80, 2014. doi: 10.1016/j.peptides.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1162.Eisen S, Phillips RJ, Geary N, Baronowsky EA, Powley TL, Smith GP. Inhibitory effects on intake of cholecystokinin-8 and cholecystokinin-33 in rats with hepatic proper or common hepatic branch vagal innervation. Am J Physiol Regul Integr Comp Physiol 289: R456–R462, 2005. [DOI] [PubMed] [Google Scholar]
- 1163.Lo CC, Davidson WS, Hibbard SK, Georgievsky M, Lee A, Tso P, Woods SC. Intraperitoneal CCK and fourth-intraventricular Apo AIV require both peripheral and NTS CCK1R to reduce food intake in male rats. Endocrinology 155: 1700–1707, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Blevins JE, Stanley BG, Reidelberger RD. Brain regions where cholecystokinin suppresses feeding in rats. Brain Res 860: 1–10, 2000. [DOI] [PubMed] [Google Scholar]
- 1165.D’Agostino G, Lyons DJ, Cristiano C, Burke LK, Madara JC, Campbell JN, Garcia AP, Land BB, Lowell BB, Dileone RJ, Heisler LK. Appetite controlled by a cholecystokinin nucleus of the solitary tract to hypothalamus neurocircuit. Elife 5: e12225, 2016. doi: 10.7554/elife.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1166.Steinert RE, Feinle-Bisset C, Asarian L, Horowitz M, Beglinger C, Geary N, Ghrelin CC. GLP-1, and PYY(3-36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol Rev 97: 411–463, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1167.Geary N. Endocrine controls of eating: CCK, leptin, and ghrelin. Physiol Behav 81: 719–733, 2004. [DOI] [PubMed] [Google Scholar]
- 1168.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 87: 1409–1439, 2007. [DOI] [PubMed] [Google Scholar]
- 1169.Svendsen B, Pedersen J, Albrechtsen NJ, Hartmann B, Torang S, Rehfeld JF, Poulsen SS, Holst JJ. An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 156: 847–857, 2015. [DOI] [PubMed] [Google Scholar]
- 1170.Panaro BL, Yusta B, Matthews D, Koehler JA, Song Y, Sandoval DA, Drucker DJ. Intestine-selective reduction of Gcg expression reveals the importance of the distal gut for GLP-1 secretion. Mol Metab 37: 100990, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1171.Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology 140: 1687–1694, 1999. doi: 10.1210/endo.140.4.6643. [DOI] [PubMed] [Google Scholar]
- 1172.Muller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, Fritsche A, Gribble F, Grill HJ, Habener JF, Holst JJ, Langhans W, Meier JJ, Nauck MA, Perez-Tilve D, Pocai A, Reimann F, Sandoval DA, Schwartz TW, Seeley RJ, Stemmer K, Tang-Christensen M, Woods SC, DiMarchi RD, Tschop MH. Glucagon-like peptide 1 (GLP-1). Mol Metab 30: 72–130, 2019. doi: 10.1016/j.molmet.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1173.Naslund E, Barkeling B, King N, Gutniak M, Blundell JE, Holst JJ, Rossner S, Hellstrom PM. Energy intake and appetite are suppressed by glucagon-like peptide-1 (GLP-1) in obese men. Int J Obes 23: 304–311, 1999. doi: 10.1038/sj.ijo.0800818. [DOI] [PubMed] [Google Scholar]
- 1174.Gutzwiller JP, Drewe J, Goke B, Schmidt H, Rohrer B, Lareida J, Beglinger C. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol Regul Integr Comp Physiol 276: R1541–R1544, 1999. [DOI] [PubMed] [Google Scholar]
- 1175.Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 150: 1680–1687, 2009. doi: 10.1210/en.2008-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1176.Punjabi M, Arnold M, Ruttimann E, Graber M, Geary N, Pacheco-Lopez G, Langhans W. Circulating glucagon-like peptide-1 (GLP-1) inhibits eating in male rats by acting in the hindbrain and without inducing avoidance. Endocrinology 155: 1690–1699, 2014. doi: 10.1210/en.2013-1447. [DOI] [PubMed] [Google Scholar]
- 1177.Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 214: 829–835, 1993. [DOI] [PubMed] [Google Scholar]
- 1178.Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 152: 3103–3112, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1179.Labouesse MA, Stadlbauer U, Weber E, Arnold M, Langhans W, Pacheco-Lopez G. Vagal afferents mediate early satiation and prevent flavour avoidance learning in response to intraperitoneally infused exendin-4. J Neuroendocrinol 24: 1505–1516, 2012. doi: 10.1111/j.1365-2826.2012.02364.x. [DOI] [PubMed] [Google Scholar]
- 1180.Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J Clin Invest 124: 2456–2463, 2014. doi: 10.1172/JCI72434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1181.Secher A, Jelsing J, Baquero AF, Hecksher-Sorensen J, Cowley MA, Dalboge LS, Hansen G, Grove KL, Pyke C, Raun K, Schaffer L, Tang-Christensen M, Verma S, Witgen BM, Vrang NB, Knudsen L. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest 124: 4473–4488, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1182.Butler PC, Chou J, Carter WB, Wang YN, Bu BH, Chang D, Chang JK, Rizza RA. Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes 39: 752–756, 1990. [DOI] [PubMed] [Google Scholar]
- 1183.Cooper GJ. Amylin compared with calcitonin gene-related peptide: structure, biology, and relevance to metabolic disease. Endocr Rev 15: 163–201, 1994. [DOI] [PubMed] [Google Scholar]
- 1184.Hay DL, Chen S, Lutz TA, Parkes DG, Roth JD. Amylin: pharmacology, physiology, and clinical potential. Pharmacol Rev 67: 564–600, 2015. [DOI] [PubMed] [Google Scholar]
- 1185.Lutz TA, Senn M, Althaus J, Prete Ehrensperger DE, Scharrer F. E. Lesion of the area postrema/nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides 19: 309–317, 1998. doi: 10.1016/S0196-9781(97)00292-1. [DOI] [PubMed] [Google Scholar]
- 1186.Mollet A, Gilg S, Riediger T, Lutz TA. Infusion of the amylin antagonist AC 187 into the area postrema increases food intake in rats. Physiol Behav 81: 149–155, 2004. doi: 10.1016/j.physbeh.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 1187.van de Wall EH, Duffy P, Ritter RC. CCK enhances response to gastric distension by acting on capsaicin-insensitive vagal afferents. Am J Physiol Regul Integr Comp Physiol 289: R695–R703, 2005. [DOI] [PubMed] [Google Scholar]
- 1188.McHugh PR, Moran TH. The stomach, cholecystokinin, and satiety. Fed Proc 45: 1384–1390, 1986. [PubMed] [Google Scholar]
- 1189.Schwartz GJ, Netterville LA, McHugh PR, Moran TH. Gastric loads potentiate inhibition of food intake produced by a cholecystokinin analogue. Am J Physiol Regul Integr Comp Physiol 261: R1141–R1146, 1991. [DOI] [PubMed] [Google Scholar]
- 1190.Kissileff HR, Carretta JC, Geliebter A, Pi-Sunyer FX. Cholecystokinin and stomach distension combine to reduce food intake in humans. Am J Physiol Regul Integr Comp Physiol 285: R992–R998, 2003. [DOI] [PubMed] [Google Scholar]
- 1191.Lal S, McLaughlin J, Barlow J, D’Amato M, Giacovelli G, Varro A, Dockray GJ, Thompson DG. Cholecystokinin pathways modulate sensations induced by gastric distension in humans. Am J Physiol Gastrointest Liver Physiol 287: G72–G79, 2004. [DOI] [PubMed] [Google Scholar]
- 1192.Brookes SJ, Spencer NJ, Costa M, Zagorodnyuk VP. Extrinsic primary afferent signalling in the gut. Nat Rev Gastroenterol Hepatol 10: 286–296, 2013. [DOI] [PubMed] [Google Scholar]
- 1193.Burdyga G, de Lartigue G, Raybould HE, Morris R, Dimaline R, Varro A, Thompson DG, Dockray GJ. Cholecystokinin regulates expression of Y2 receptors in vagal afferent neurons serving the stomach. J Neurosci 28: 11583–11592, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1194.Cooper SJ, Dourish CT. Multiple cholecystokinin (CCK) receptors and CCK-monoamine interactions are instrumental in the control of feeding. Physiol Behav 48: 849–857, 1990. [DOI] [PubMed] [Google Scholar]
- 1195.Cooper SJ. Cholecystokinin modulation of serotonergic control of feeding behavior. Ann N Y Acad Sci 780: 213–222, 1996. [DOI] [PubMed] [Google Scholar]
- 1196.Voigt JP, Sohr R, Fink H. CCK-8S facilitates 5-HT release in the rat hypothalamus. Pharmacol Biochem Behav 59: 179–182, 1998. [DOI] [PubMed] [Google Scholar]
- 1197.Poeschla B, Gibbs J, Simansky KJ, Greenberg D, Smith GP. Cholecystokinin-induced satiety depends on activation of 5-HT1C receptors. Am J Physiol Regul Integr Comp Physiol 264: R62–R64, 1993. [DOI] [PubMed] [Google Scholar]
- 1198.Asarian L. Loss of cholecystokinin and glucagon-like peptide-1-induced satiation in mice lacking serotonin 2C receptors. Am J Physiol Regul Integr Comp Physiol 296: R51–R56, 2009. [DOI] [PubMed] [Google Scholar]
- 1199.Mawe GM, Coates MD, Moses PL. Review article: intestinal serotonin signalling in irritable bowel syndrome. Aliment Pharmacol Ther 23: 1067–1076, 2006. [DOI] [PubMed] [Google Scholar]
- 1200.Mawe GM, Hoffman JM. Serotonin signalling in the gut–functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol 10: 473–486, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1201.Champaneria S, Costall B, Naylor RJ, Robertson DW. Identification and distribution of 5-HT3 recognition sites in the rat gastrointestinal tract. Br J Pharmacol 106: 693–696, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1202.Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O’Donnell TA, Brierley SM, Ingraham HA, Julius D. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 170: 185–198, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1203.Hayes MR, Covasa M. CCK and 5-HT act synergistically to suppress food intake through simultaneous activation of CCK-1 and 5-HT3 receptors. Peptides 26: 2322–2330, 2005. [DOI] [PubMed] [Google Scholar]
- 1204.Hayes MR, Moore RL, Shah SM, Covasa M. 5-HT3 receptors participate in CCK-induced suppression of food intake by delaying gastric emptying. Am J Physiol Regul Integr Comp Physiol 287: R817–R823, 2004. [DOI] [PubMed] [Google Scholar]
- 1205.Hayes MR, Savastano DM, Covasa M. Cholecystokinin-induced satiety is mediated through interdependent cooperation of CCK-A and 5-HT3 receptors. Physiol Behav 82: 663–669, 2004. [DOI] [PubMed] [Google Scholar]
- 1206.Savastano DM, Hayes MR, Covasa M. Serotonin-type 3 receptors mediate intestinal lipid-induced satiation and Fos-like immunoreactivity in the dorsal hindbrain. Am J Physiol Regul Integr Comp Physiol 292: R1063–R1070, 2007. [DOI] [PubMed] [Google Scholar]
- 1207.Browning KN. Role of central vagal 5-HT3 receptors in gastrointestinal physiology and pathophysiology. Front Neurosci 9: 413, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1208.Houghton LA, Atkinson W, Whitaker RP, Whorwell PJ, Rimmer MJ. Increased platelet depleted plasma 5-hydroxytryptamine concentration following meal ingestion in symptomatic female subjects with diarrhoea predominant irritable bowel syndrome. Gut 52: 663–670, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1209.Babic T, Troy AE, Fortna SR, Browning KN. Glucose-dependent trafficking of 5-HT3 receptors in rat gastrointestinal vagal afferent neurons. Neurogastroenterol Motil 24: e476-488–e488, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1210.Wan S, Browning KN. D-glucose modulates synaptic transmission from the central terminals of vagal afferent fibers. Am J Physiol Gastrointest Liver Physiol 294: G757–G763, 2008. [DOI] [PubMed] [Google Scholar]
- 1211.Lacolley P, Owen JR, Sandock K, Lewis TH, Bates JN, Robertson TP, Lewis SJ. Occipital artery injections of 5-HT may directly activate the cell bodies of vagal and glossopharyngeal afferent cell bodies in the rat. Neuroscience 143: 289–308, 2006. [DOI] [PubMed] [Google Scholar]
- 1212.Lund ML, Egerod KL, Engelstoft MS, Dmytriyeva O, Theodorsson E, Patel BA, Schwartz TW. Enterochromaffin 5-HT cells–a major target for GLP-1 and gut microbial metabolites. Mol Metab 11: 70–83, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1213.Wang L, Barachina MD, Martinez V, Wei JY, Tache Y. Synergistic interaction between CCK and leptin to regulate food intake. Regul Pept 92: 79–85, 2000. [DOI] [PubMed] [Google Scholar]
- 1214.Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, Baron AD. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A 105: 7257–7262, 2008. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1215.Turek VF, Trevaskis JL, Levin BE, Dunn-Meynell AA, Irani B, Gu G, Wittmer C, Griffin PS, Vu C, Parkes DG, Roth JD. Mechanisms of amylin/leptin synergy in rodent models. Endocrinology 151: 143–152, 2010. [DOI] [PubMed] [Google Scholar]
- 1217.Smith PM, Brzezinska P, Hubert F, Mimee A, Maurice DH, Ferguson AV. Leptin influences the excitability of area postrema neurons. Am J Physiol Regul Integr Comp Physiol 310: R440–R448, 2016. [DOI] [PubMed] [Google Scholar]
- 1218.Levin BE, Lutz TA. Amylin and leptin: co-regulators of energy homeostasis and neuronal development. Trends Endocrinol Metab 28: 153–164, 2017. [DOI] [PubMed] [Google Scholar]
- 1216.Beaumont K, Kenney MA, Young AA, Rink TJ. High affinity amylin binding sites in rat brain. Mol Pharmacol 44: 493–497, 1993. [PubMed] [Google Scholar]
- 1219.Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res 964: 107–115, 2003. [DOI] [PubMed] [Google Scholar]
- 1220.Mietlicki-Baase EG, Olivos DR, Jeffrey BA, Hayes MR. Cooperative interaction between leptin and amylin signaling in the ventral tegmental area for the control of food intake. Am J Physiol Endocrinol Metab 308: E1116–E1122, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1221.Phillips LK, Deane AM, Jones KL, Rayner CK, Horowitz M. Gastric emptying and glycaemia in health and diabetes mellitus. Nat Rev Endocrinol 11: 112–128, 2015. [DOI] [PubMed] [Google Scholar]
- 1222.Zhu JX, Zhu XY, Owyang C, Li Y. Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J Physiol 530: 431–442, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 1223.Grabauskas G, Song I, Zhou S, Owyang C. Electrophysiological identification of glucose-sensing neurons in rat nodose ganglia. J Physiol 588: 617–632, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1224.Burcelin R, Da Costa A, Drucker D, Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 50: 1720–1728, 2001. [DOI] [PubMed] [Google Scholar]
- 1225.Grabauskas G, Zhou SY, Lu Y, Song I, Owyang C. Essential elements for glucosensing by gastric vagal afferents: immunocytochemistry and electrophysiology studies in the rat. Endocrinology 154: 296–307, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1226.Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes 49: 1643–1648, 2000. [DOI] [PubMed] [Google Scholar]
- 1227.Niijima A. The effect of D-glucose on the firing rate of glucose-sensitive vagal afferents in the liver in comparison with the effect of 2-deoxy-D-glucose. J Auton Nerv Syst 10: 255–260, 1984. [DOI] [PubMed] [Google Scholar]
- 1228.Reis DJ, Granata AR, Perrone MH, Talman WT. Evidence that glutamic acid is the neurotransmitter of baroreceptor afferent terminating in the nucleus tractus solitarius (NTS). J Auton Nerv Syst 3: 321–334, 1981. [DOI] [PubMed] [Google Scholar]
- 1229.Allchin RE, Batten TF, McWilliam PN, Vaughan PF. Electrical stimulation of the vagus increases extracellular glutamate recovered from the nucleus tractus solitarii of the cat by in vivo microdialysis. Exp Physiol 79: 265–268, 1994. [DOI] [PubMed] [Google Scholar]
- 1230.Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol 68: 279–305, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1231.Lawrence AJ. Neurotransmitter mechanisms of rat vagal afferent neurons. Clin Exp Pharmacol Physiol 22: 869–873, 1995. [DOI] [PubMed] [Google Scholar]
- 1232.Berthoud HR, Earle T, Zheng H, Patterson LM, Phifer C. Food-related gastrointestinal signals activate caudal brainstem neurons expressing both NMDA and AMPA receptors. Brain Res 915: 143–154, 2001. [DOI] [PubMed] [Google Scholar]
- 1233.Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci 25: 3578–3585, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1234.Bednar I, Qian M, Qureshi GA, Kallstrom L, Johnson AE, Carrer H, Sodersten P. Glutamate inhibits ingestive behaviour. J Neuroendocrinol 6: 403–408, 1994. [DOI] [PubMed] [Google Scholar]
- 1235.Treece BR, Covasa M, Ritter RC, Burns GA. Delay in meal termination follows blockade of N-methyl-D-aspartate receptors in the dorsal hindbrain. Brain Res 810: 34–40, 1998. [DOI] [PubMed] [Google Scholar]
- 1236.Hung CY, Covasa M, Ritter RC, Burns GA. Hindbrain administration of NMDA receptor antagonist AP-5 increases food intake in the rat. Am J Physiol Regul Integr Comp Physiol 290: R642–R651, 2006. [DOI] [PubMed] [Google Scholar]
- 1237.Wright J, Campos C, Herzog T, Covasa M, Czaja K, Ritter RC. Reduction of food intake by cholecystokinin requires activation of hindbrain NMDA-type glutamate receptors. Am J Physiol Regul Integr Comp Physiol 301: R448–R455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1238.Campos CA, Wright JS, Czaja K, Ritter RC. CCK-induced reduction of food intake and hindbrain MAPK signaling are mediated by NMDA receptor activation. Endocrinology 153: 2633–2646, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1239.Ritter RC. A tale of two endings: modulation of satiation by NMDA receptors on or near central and peripheral vagal afferent terminals. Physiol Behav 105: 94–99, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1240.Ruppersberg JP, Kitzing E, Schoepfer R. The mechanism of magnesium block of NMDA receptors. Semin Neurosci 6: 87–96, 1994. [Google Scholar]
- 1241.Bains JS, Wamsteeker Cusulin JI, Inoue W. Stress-related synaptic plasticity in the hypothalamus. Nat Rev Neurosci 16: 377–388, 2015. [DOI] [PubMed] [Google Scholar]
- 1242.Broberger C, Holmberg K, Kuhar MJ, Hokfelt T. Cocaine- and amphetamine-regulated transcript in the rat vagus nerve: A putative mediator of cholecystokinin-induced satiety. Proc Natl Acad Sci U S A 96: 13506–13511, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1243.Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Feeding-dependent depression of melanin-concentrating hormone and melanin-concentrating hormone receptor-1 expression in vagal afferent neurones. Neuroscience 137: 1405–1415, 2006. [DOI] [PubMed] [Google Scholar]
- 1244.Franco-Cereceda A, Henke H, Lundberg JM, Petermann JB, Hokfelt T, Fischer JA. Calcitonin gene-related peptide (CGRP) in capsaicin-sensitive substance P-immunoreactive sensory neurons in animals and man: distribution and release by capsaicin. Peptides 8: 399–410, 1987. [DOI] [PubMed] [Google Scholar]
- 1245.Aja S, Sahandy S, Ladenheim EE, Schwartz GJ, Moran TH. Intracerebroventricular CART peptide reduces food intake and alters motor behavior at a hindbrain site. Am J Physiol Regul Integr Comp Physiol 281: R1862–R1867, 2001. [DOI] [PubMed] [Google Scholar]
- 1246.Lee SJ, Krieger JP, Vergara M, Quinn D, McDougle M, de Araujo A, Darling R, Zollinger B, Anderson S, Pan A, Simonnet EJ, Pignalosa A, Arnold M, Singh A, Langhans W, Raybould HE, de Lartigue G. Blunted vagal cocaine- and amphetamine-regulated transcript promotes hyperphagia and weight gain. Cell Rep 30: 2028–2039, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1247.Mul JD, la Fleur SE, Toonen PW, Afrasiab-Middelman A, Binnekade R, Schetters D, Verheij MM, Sears RM, Homberg JR, Schoffelmeer AN, Adan RA, Rj D, Tj DV, Cuppen E. Chronic loss of melanin-concentrating hormone affects motivational aspects of feeding in the rat. PLoS One 6: e19600, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1248.Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol 457: 213–235, 2003. [DOI] [PubMed] [Google Scholar]
- 1249.Grill HJ, Ginsberg AB, Seeley RJ, Kaplan JM. Brainstem application of melanocortin receptor ligands produces long-lasting effects on feeding and body weight. J Neurosci 18: 10128–10135, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1250.Zheng H, Patterson LM, Phifer CB, Berthoud HR. Brain stem melanocortinergic modulation of meal size and identification of hypothalamic POMC projections. Am J Physiol Regul Integr Comp Physiol 289: R247–R258, 2005. [DOI] [PubMed] [Google Scholar]
- 1251.Campos CA, Shiina H, Ritter RC. Central vagal afferent endings mediate reduction of food intake by melanocortin-3/4 receptor agonist. J Neurosci 34: 12636–12645, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1252.Wan S, Browning KN, Coleman FH, Sutton G, Zheng H, Butler A, Berthoud HR, Travagli RA. Presynaptic melanocortin-4 receptors on vagal afferent fibers modulate the excitability of rat nucleus tractus solitarius neurons. J Neurosci 28: 4957–4966, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1253.Riche D, De Pommery J, Menetrey D. D. Neuropeptides and catecholamines in efferent projections of the nuclei of the solitary tract in the rat. J Comp Neurol 293: 399–424, 1990. doi: 10.1002/cne.902930306. [DOI] [PubMed] [Google Scholar]
- 1254.Petrov T, Krukoff TL, Jhamandas JH. Branching projections of catecholaminergic brainstem neurons to the paraventricular hypothalamic nucleus and the central nucleus of the amygdala in the rat. Brain Res 609: 81–92, 1993. [DOI] [PubMed] [Google Scholar]
- 1255.Chen Z, Travers SP, Travers JB. Muscimol infusions in the brain stem reticular formation reversibly block ingestion in the awake rat. Am J Physiol Regul Integr Comp Physiol 280: R1085–1094, 2001. [DOI] [PubMed] [Google Scholar]
- 1256.Et S, Cheng S, Takatoh J, Han BX, Wang F. Monosynaptic premotor circuit tracing reveals neural substrates for oro-motor coordination. Elife 3: e02511, 2014. doi: 10.7554/elife.02511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1257.Travers JB, Herman K, Travers SP. Suppression of third ventricular NPY-elicited feeding following medullary reticular formation infusions of muscimol. Behav Neurosci 124: 225–233, 2010. doi: 10.1037/a0018928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1258.Garfield AS, Patterson C, Skora S, Gribble FM, Reimann F, Evans ML, Myers MG Jr, Heisler LK. Neurochemical characterization of body weight-regulating leptin receptor neurons in the nucleus of the solitary tract. Endocrinology 153: 4600–4607, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1259.Kahler A, Geary N, Eckel LA, Campfield LA, Smith FJ, Langhans W. Chronic administration of OB protein decreases food intake by selectively reducing meal size in male rats. Am J Physiol Regul Integr Comp Physiol 275: R180–R185, 1998. doi: 10.1152/ajpregu.1998.275.1.R180. [DOI] [PubMed] [Google Scholar]
- 1260.Flynn MC, Scott TR, Pritchard TC, Plata-Salaman CR. Mode of action of OB protein (leptin) on feeding. Am J Physiol Regul Integr Comp Physiol 275: R174–R179, 1998. doi: 10.1152/ajpregu.1998.275.1.R174. [DOI] [PubMed] [Google Scholar]
- 1261.Ho A, Chin A. Circadian feeding and drinking patterns of genetically obese mice fed solid chow diet. Physiol Behav 43: 651–656, 1988. [DOI] [PubMed] [Google Scholar]
- 1262.Alingh Prins A, de Jong-Nagelsmit A, Keijser J, Strubbe JH. Daily rhythms of feeding in the genetically obese and lean Zucker rats. Physiol Behav 38: 423–426, 1986. doi: 10.1016/0031-9384(86)90115-0. [DOI] [PubMed] [Google Scholar]
- 1263.Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, Baskin DG, Schwartz MW. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest 115: 703–710, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1264.Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, Grill HJ. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 11: 77–83, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1265.Roberts BL, Zhu M, Zhao H, Dillon C, Appleyard SM. High glucose increases action potential firing of catecholamine neurons in the nucleus of the solitary tract by increasing spontaneous glutamate inputs. Am J Physiol Regul Integr Comp Physiol 313: R229–R239, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1266.Mimee A, Ferguson AV. Glycemic state regulates melanocortin, but not nesfatin-1, responsiveness of glucose-sensing neurons in the nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol 308: R690–R699, 2015. doi: 10.1152/ajpregu.00477.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1267.D’Agostino G, Lyons D, Cristiano C, Lettieri M, Olarte-Sanchez C, Burke LK, Greenwald-Yarnell M, Cansell C, Doslikova B, Georgescu T, Martinez de Morentin PB, Myers MG Jr, Rochford JJ, Heisler LK. Nucleus of the solitary tract serotonin 5-HT2C receptors modulate food intake. Cell Metab 28: 619–630, 2018. doi: 10.1016/j.cmet.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1268.Hao S, Yang H, Wang X, He Y, Xu H, Wu X, Pan L, Liu Y, Lou H, Xu H, Ma H, Xi W, Zhou Y, Duan S, Wang H. The lateral hypothalamic and BNST GABAergic projections to the anterior ventrolateral periaqueductal gray regulate feeding. Cell Rep 28: 616–624, 2019. [DOI] [PubMed] [Google Scholar]
- 1269.Nectow AR, Schneeberger M, Zhang H, Field BC, Renier N, Azevedo E, Patel B, Liang Y, Mitra S, Tessier-Lavigne M, Han MH, Friedman JM. Identification of a brainstem circuit controlling feeding. Cell 170: 429–435, 2017. [DOI] [PubMed] [Google Scholar]
- 1270.Hinde RA. Animal Behavior. A Synthesis of Ethology and Comparative Psychology. New York: McGraw-Hill, 1970. [Google Scholar]
- 1271.Lewis AS, Calipari ES, Siciliano CA. Toward standardized guidelines for investigating neural circuit control of behavior in animal research. eNeuro 8: 10.1523/ENEURO.0498-20.2021, 2021. doi: 10.1523/eneuro.0498-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1272.Venner A, Karnani MM, Gonzalez JA, Jensen LT, Fugger L, Burdakov D. Orexin neurons as conditional glucosensors: paradoxical regulation of sugar sensing by intracellular fuels. J Physiol 589: 5701–5708, 2011. doi: 10.1113/jphysiol.2011.217000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1273.Axelrod J, Inscoe JK. The uptake and binding of circulating serotonin and the effect of drugs. J Pharmacol Exp Ther 141: 161–165, 1963. [PubMed] [Google Scholar]
- 1274.Oldendorf WH. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol 221: 1629–1639, 1971. doi: 10.1152/ajplegacy.1971.221.6.1629. [DOI] [PubMed] [Google Scholar]
- 1275.Simpson KH, Murphy P, Colthup PV, Whelan P. Concentration of ondansetron in cerebrospinal fluid following oral dosing in volunteers. Psychopharmacology (Berl) ) 109: 497–498, 1992. doi: 10.1007/BF02247730. [DOI] [PubMed] [Google Scholar]
- 1276.Pollock JD, Rowland N. Peripherally administered serotonin decreases food intake in rats. Pharmacol Biochem Behav 15: 179–183, 1981. doi: 10.1016/0091-3057(81)90174-x. [DOI] [PubMed] [Google Scholar]
- 1277.Bray GA, York DA. Studies on food intake of genetically obese rats. Am J Physiol 223: 176–179, 1972. doi: 10.1152/ajplegacy.1972.223.1.176. [DOI] [PubMed] [Google Scholar]
- 1278.Liberini CG, Boyle CN, Cifani C, Venniro M, Hope BT, Lutz TA. Amylin receptor components and the leptin receptor are co-expressed in single rat area postrema neurons. Eur J Neurosci 43: 653–661, 2016. doi: 10.1111/ejn.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1279.Heldsinger A, Lu Y, Zhou SY, Wu X, Grabauskas G, Song I, Owyang C. Cocaine- and amphetamine-regulated transcript is the neurotransmitter regulating the action of cholecystokinin and leptin on short-term satiety in rats. Am J Physiol Gastrointest Liver Physiol 303: G1042–G1051, 2012. doi: 10.1152/ajpgi.00231.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]