

Abstract
The disruption of epigenetic regulation is common in tumors; the abnormal expression of epigenetic factors leads to cancer occurrence and development. In this study, to investigate the potential function of histone methylation regulators in lung adenocarcinoma (LUAD), we performed differential expression analysis using RNA-seq data downloaded from The Cancer Genome Atlas (TCGA) database, and identified CBX2 and EZH2 as obviously upregulated histone methylation regulators. CBX2 knockdown significantly inhibited LUAD cell growth and metastasis in vitro and in vivo. The combined high expression of CBX2 and EZH2 was an indicator of poor prognosis in LUAD. The inhibition of both CBX2 and EZH2 exerted cooperative suppressive effects on the growth and metastasis of LUAD cells. Mechanistically, we revealed that CBX2 and EZH2 downregulated several PPAR signaling pathway genes and tumor suppressor genes through binding to their promoter cooperatively or separately. Furthermore, knockdown of CBX2 improved the therapeutic efficiency of EZH2 inhibitor on A549 cells. Our study reveals the cooperative oncogenic role of CBX2 and EZH2 in promoting LUAD progression, thereby providing potential targets for LUAD diagnosis and therapy.
Keywords: Lung adenocarcinoma, CBX2, EZH2, epigenetic regulation, growth, metastasis
Graphical abstract
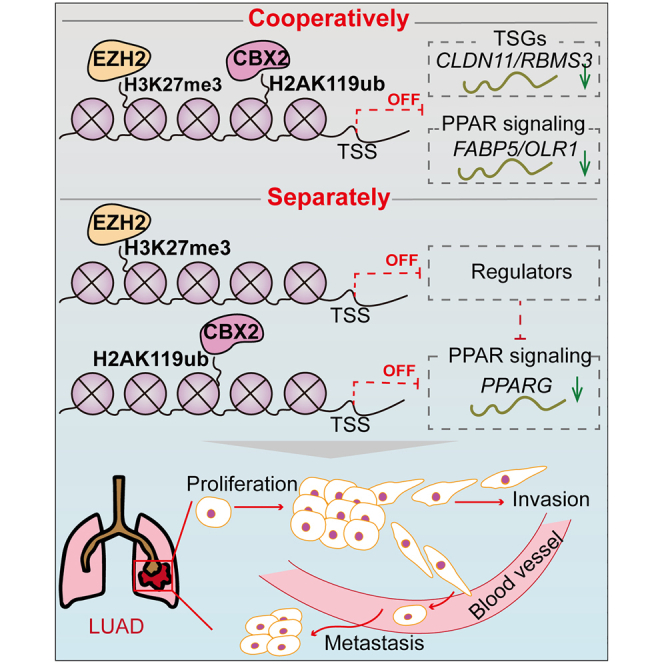
Two significantly upregulated histone methylation related genes, CBX2 and EZH2, suppress the transcription of several PPAR signaling pathway genes and tumor suppressor genes, showing a cooperative effect on suppressing lung adenocarcinomal development and revealing the potential clinical application of developing CBX2 inhibitors in combination with EZH2 inhibitors to treat LUAD.
Introduction
Epigenetic regulatory mechanisms, such as DNA methylation, histone modification, and RNA-mediated targeting, regulate gene expression and are fundamental to biological functions. Consequently, the abnormal expression of epigenetic factors can have a profound influence on cells and lead to tumorigenesis by affecting hallmarks of cancer, such as malignant proliferation and invasion.1 Moreover, the promising clinical and preclinical benefits obtained from drugs targeting epigenetic factors highlight the central role of epigenetics in cancer and the importance of exploring new epigenetic factors as therapeutic targets.2
Polycomb group (PcG) genes are a group of epigenetic regulators that play critical roles in the maintenance of cellular identity.3,4 During the past 70 years, a large number of PcG genes have been identified and intensively studied. PcG proteins function in several families of multiprotein complexes, such as polycomb repressive complex (PRC) 1 and PRC2. PRC2 catalyzes the methylation of K27 on histone H3. In mammals, PRC2 is mainly consisted of enhancer of zeste (EZH2 or EZH1), suppressor of zeste 12 (SUZ12), embryonic ectoderm development (EED), and retinoblastoma binding protein (RbAp46/48). EZH2 is the core component responsible for the catalytic activity of PRC2, as inhibition of EZH2 led to almost complete loss of H3K27me2/3. In recent years, PRC1 was classified into canonical (cPRC1) and noncanonical PRC1 (ncPRC1). The cPRC1 complex chiefly contains RING1 proteins (RING1A or RING1B), which catalyze the monoubiquitination of H2AK119 (H2AK119ub), one of the six PcG ring-finger domain proteins (PCGF1–PCGF6), polyhomeotic homologous proteins (PHC1–PHC3), and chromobox proteins (CBX2, 4, 6, 7, and 8).4,5 Polycomb-mediated gene silencing relies mostly on regulation of chromatin structure, in part through histone modifications, such as PRC2-catalyzed H3K27me2/3 and PRC1-catalyzed H2AK119ub. EZH2-catalyzed H3K27me3 functions as a recruitment signal for CBX proteins in the PRC1 complex.6 However, it has been reported that only a subset of PRC1 targets overlaps with H3K27me3-rich regions, and these regions are typically bound by cPRC1 complexes containing CBX2.7,8 Furthermore, there are genes targeted by PRC2 in the absence of H2AK119ub,9 and genes bound by PRC1 without PRC2.10,11 Notwithstanding, PRC1 and PRC2 are usually both required for the maintenance of gene repression. Although EZH2 has been reported to be upregulated in various tumor tissues,12 the cooperative effects of the PRC1 and PRC2 complexes in tumorigenesis have not been investigated. Therefore, exploring the combined tumorigenic function of CBX2 and EZH2, and integrating their downstream molecules could provide new therapeutic targets for cancer.
Lung cancer is a deadly malignancy and the leading cause of cancer-related death worldwide.13 Generally, lung cancer is classified into small cell lung cancer (SCLC, 20%) and non-small cell lung cancer (NSCLC, 80%).14 Lung adenocarcinoma (LUAD) is the most frequently diagnosed histological subtype of NSCLC.15 Great progress has been achieved in understanding the functions of epigenetic factors in LUAD progression. For example, high expression levels of HDAC1 and HDAC3 are associated with a poor prognosis in LUAD, and treatment with HDAC inhibitors has shown antiproliferative effects.16 However, identifying new epigenetic regulators as therapeutic targets in LUAD is still urgent.
In this study, to investigate the potential function of histone methylation regulators in LUAD progression, we performed differential expression analysis of 58 paired tumor/normal LUAD samples from The Cancer Genome Atlas (TCGA) and identified CBX2 and EZH2 as obviously upregulated genes in the tumor samples. We showed that depletion of CBX2 significantly suppressed the growth and metastasis of LUAD in vitro and in vivo. The Kaplan-Meier univariate survival analysis showed that the high expression of both CBX2 and EZH2 was more significantly associated with a poor prognosis than high expression of CBX2 or EZH2 alone; in addition, the expression of CBX2 and EZH2 in LUAD was positively correlated. The double inhibition of CBX2 and EZH2 led to more significant cellular effects in LUAD cells than inhibition of either alone. Regarding the downstream targets, we showed that CBX2 and EZH2 downregulated several peroxisome proliferator-activated receptors (PPAR) signaling pathway genes and tumor suppressor genes (TSGs) by binding to their promoters together or separately. Knockdown of PPARG, a key regulator in the PPAR signaling pathway, partially rescued the decrease in LUAD cell proliferation and invasion caused by the CBX2 and EZH2 depletion. Furthermore, knockdown of CBX2 improved the therapeutic efficiency of EZH2 inhibitor on A549 cells. Together, our study results reveal the cooperative role of CBX2 and EZH2 in promoting LUAD progression, providing potential new targets for LUAD diagnosis and therapy.
Results
The PcG protein CBX2 is significantly upregulated in LUAD, and its depletion suppresses the growth and invasion of LUAD cells
Histone methylation is a very important type of histone modifications that regulates gene expression. To investigate the potential function of histone methylation in LUAD, we downloaded RNA sequencing (RNA-seq) data of LUAD from TCGA database and analyzed the expression level of genes encoding writers (methyltransferases), readers, and erasers (demethylases) of histone methylation (Table S1) in LUAD. The differential expression analysis of 58 paired tumor/normal samples revealed that 7 of 129 histone methylation-related genes were dysregulated (six genes were upregulated, and one gene was downregulated) in tumor samples (Figure 1A). Among these differentially expressed genes (DEGs), the most obviously upregulated genes include CBX2 and EZH2 (Figures 1A and 1B). EZH2 was previously reported to be overexpressed in LUAD and play an oncogenic role in the progression of LUAD.17,18 Similar to the observed expression pattern of EZH2, the mRNA expression level of CBX2 was increased 5-fold in tumor tissues compared with that in normal tissues (Figure 1B). Moreover, we found that the CBX2 expression continuously increased from normal samples to distant metastasis-free (DMF) samples and from DMF samples to distant metastasis (DM) samples (Figure 1C). However, the expression of EZH2 was not significantly different between DMF and DM patients (Figure 1C). These results indicated that the overexpression of CBX2 is potentially associated with LUAD progression.
Figure 1.

The PcG protein CBX2 is significantly upregulated in LUAD, and its depletion suppresses the growth and invasion of LUAD cells
(A) Heatmap showing the expression of 7 differentially expressed (FDR <0.01, fold change ≥2 and CPM >30) histone methylation-related genes in LUAD tumor samples and normal samples. (B) The differences of CBX2 and EZH2 mRNA expression between the paired tumor (n = 58) and normal (n = 58) samples (pairs are connected by gray dash lines). The p values were estimated using the R package NOISeq. ∗∗∗p ≤ 0.001. (C) The differences of CBX2 and EZH2 mRNA expression between normal (n = 58, tumor adjacent), DMF (n = 353), and DM (n = 25) samples. (D-G) A549 cells transfected with control, CBX2, or EZH2 siRNAs were applied. (D) CCK-8 assays were used to examine the cell viability of the transfected A549 cells. (E) EdU incorporation assays were used to examine the proliferation of the transfected A549 cells. (F) FITC-labeled annexin V and propidium iodide were used to stain the transfected A549 cells, and flow cytometry was used to detect apoptosis. (G) The cell invasion was examined by counting the transmigrated cells under a microscope. In (D-G), each bar represents the mean ±SD of three independent biological replicates. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (Student's t test). (H) Protein expression of EMT markers was determined by western blotting in A549 cells transfected with control, CBX2, or EZH2 siRNAs. The molecular weights are indicated on the left side of the plot.
To validate the potential carcinogenic role of CBX2 in LUAD, we investigated its function in cell viability, proliferation, apoptosis, and invasion in A549 cells. Cell Counting Kit-8 (CCK-8) assays were first used to detect the viability of A549 cells with CBX2 knockdown. The results showed that CBX2 depletion led to a marked decrease in the viability of A549 cells, similar to the effects of the EZH2 knockdown (Figure 1D). Then, 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays were performed to observe the effects of the CBX2 knockdown on the proliferation of A549 cells (Figure 1E). The results demonstrated that the CBX2 silencing inhibited cell proliferation. The apoptosis analysis using annexin V-FITC (fluoresceine isothiocyanate) and propidium iodide staining (Figure 1F) showed that the knockdown of CBX2 promoted the apoptosis of A549 cells. Furthermore, we investigated the role of CBX2 in the invasion of LUAD cells (Figure 1G). The results of transwell assays showed that CBX2 deficiency led to a significant decrease in A549 cell invasion, which is similar to the effect of EZH2 depletion. Moreover, the expression of epithelial-to-mesenchymal transition (EMT) markers in A549 cells was analyzed by western blot after transfection of small interfering RNAs (siRNAs) against CBX2 or EZH2. Similar to the EZH2 knockdown, depletion of CBX2 led to increased protein levels of epithelial markers, including E-cadherin, α-catenin, β-catenin, as well as γ-catenin (Figure 1H), and decreased protein level of a mesenchymal marker, N-cadherin, demonstrating the role of CBX2 in promoting EMT. Overall, the in vitro experiments demonstrated that CBX2 plays an important role in promoting the growth and invasion of LUAD cells.
CBX2 knockdown significantly inhibits LUAD growth and metastasis in vivo
After confirming the carcinogenic role of CBX2 at the cellular level, we explored its functions in vivo. We transplanted lung tumors derived from normal A549 cells or A549 cells expressing CBX2 single guide RNAs (sgRNAs), which greatly inhibited CBX2 protein expression compared with that in control cells (Figure 2A), into nude mice (BALB/c; Charles River, Beijing, China; n = 6 per group) (Figure 2B). Then we monitored the growth of the implanted tumors over a period of four weeks. We found that the tumor growth, including tumor volume (Figure 2C) and weight (Figure 2D), were significantly decreased in athymic mice that received cells expressing CBX2 sgRNAs. To investigate the role of CBX2 in LUAD metastasis in vivo, normal A549-luc cells or A549-luc cells expressing CBX2 sgRNAs were injected into female severe combined immunodeficiency (SCID) (six-week-old) mice via the lateral tail vein, and metastatic tumor cells were monitored weekly using a bioluminescence imaging system (Xenogen) (Figure 2E). The results showed that tumor metastasis was significantly decreased in mice that received A549-luc cells expressing CBX2 sgRNAs compared with in mice that received normal A549-luc cells (Figure 2E). Overall, these results showed that CBX2 depletion suppresses LUAD tumor growth and metastasis in vivo.
Figure 2.

CBX2 knockdown significantly suppresses LUAD growth and metastasis in vivo
(A) Western blot analysis validating the inhibitory effect of CBX2 sgRNAs on the protein expression of CBX2 in A549 cells. (B) Control A549 cells or A549 cells expressing CBX2 sgRNAs (A549-sgCBX2) were transplanted into female athymic nude mice (BALB/c, Charles River; ages between five and six weeks; six mice per group). (C) Tumors were measured at the indicated times using a vernier caliper. According to the formula, V = π/6 × length × width2, the volume of tumor was calculated. Error bars represent the mean ± SD of six animal measurements. ∗p ≤ 0.05; ∗∗p ≤ 0.01 (Student's t test). (D) The tumors dissected from the mice were weighed. Each bar represents the mean ± SD of six animal measurements. ∗∗∗p ≤ 0.001 (Student's t test). (E) Representative in vivo bioluminescent images showing the difference in metastasis between the A549-Luc and A549-sgCBX2-Luc mice group. After six weeks of initial implantation, the metastasis of tumor cells was quantified using bioluminescence imaging. Each bar represents the mean ± SD of three animal measurements. ∗p ≤ 0.05 (Student's t test).
The potential cooperative function of CBX2 and EZH2 in promoting LUAD
Then we performed the univariate survival analysis and found that the overexpression of either CBX2 or EZH2 was slightly associated with a poor prognosis (Figure 3A), and the multivariate survival analysis showed that these associations were dependent of TNM stage, years of smoking, metastasis, and mutation of KRAS and EGFR (Figure S1A). However, the combined high expression of CBX2 and EZH2 was significantly associated with a poor prognosis (p = 0.0085; Figure 3B) and was more valuable in predicting prognosis than the high expression of CBX2 (p = 0.067) or EZH2 (p = 0.021) alone (Figure 3A). The multivariate survival analysis indicated that the combined high expression of CBX2 and EZH2 was a predictor of poor prognosis independent of TNM stage, years of smoking, metastasis, and mutation of KRAS and EGFR (Figure S1B).
Figure 3.

Potential functional relevance between CBX2 and EZH2
(A) Kaplan-Meier survival curve for patients with high (>median expression) or low (<median expression) expression of CBX2 or EZH2. The survival difference was determined by log rank test. According to a standard procedure, tumor samples were grouped into high and low groups by the median expression of specific gene. The median survival times were marked by a dashed line. (B) Kaplan-Meier survival curve of the combined CBX2 and EZH2 expression. (C) The proteins expression of CBX2 and EZH2 detected by immunohistochemistry of LUAD microarrays (scale bar, 50 μm) and (D) their quantification in human LUAD (n = 75) and adjacent normal tissues (n = 75). ∗∗∗p ≤ 0.001. (E) The Spearman correlation between CBX2 and EZH2 protein expression levels in LUAD tissue microarrays (n = 75). (F) Spearman correlation between CBX2 and EZH2 mRNA expression levels in TCGA tumor samples. In (E) and (F), r: correlation coefficient; n: number of samples.
Given the reported role of CBX2 as a reader of EZH2-catalyzed H3K27me3,4 we further investigated the functional relevance of EZH2 and CBX2 in LUAD. The immunohistochemical assays showed that the protein levels of both CBX2 and EZH2 were higher in LUAD tumor samples than in adjacent normal samples (Figures 3C and 3D). The correlation analysis revealed that the protein levels of EZH2 and CBX2 in LUAD samples were positively correlated (r = 0.41; p = 4.1 × 10−5; Figure 3E). Moreover, their mRNA levels were also positively correlated (r = 0.34; p = 2.5 × 10−15; Figure 3F). Together, these results revealed a potential cooperative carcinogenic role of CBX2 and EZH2 in promoting LUAD.
Inhibition of both CBX2 and EZH2 leads to more significant effects on cell behaviors in LUAD cells
To validate the cooperative carcinogenic effect of CBX2 and EZH2 in LUAD, we used a double knockdown strategy to investigate their cooperative roles in regulating LUAD cell functions. The effect of CBX2, EZH2, or combined CBX2/EZH2 knockdown by specific siRNAs was examined by western blot analysis (Figure S2A). MTT assays (Figure 4A), EdU incorporation assays (Figure 4B), apoptosis assays (Figure 4C), migration assays (Figure S2B), and transwell assays (Figure 4D) were carried out to assess the viability, proliferation, apoptosis, migration, and invasion, respectively, of A549 and H1299 cells. The results showed that the knockdown of both CBX2 and EZH2 led to a more marked decrease in the viability, proliferation, migration, and invasion of A549 and H1299 cells, and to a more significant increase in apoptosis, than knockdown of either CBX2 or EZH2 alone (Figures 4A–4D, and S2B), indicating the cooperative functions of these genes in promoting LUAD.
Figure 4.

Inhibition of both CBX2 and EZH2 led to more significant cellular effects in LUAD cells
A549 or H1299 cells were transfected with control, CBX2, EZH2, or double gene siRNAs. (A) Transfected A549 cells were applied to detect cell viability using MTT assays. (B) Transfected A549 cells were used to detect cell proliferation using the EdU incorporation assay. Scale bar, 100 μm. (C) FITC-labeled annexin V and propidium iodide were used to stain the transfected A549 cells. Flow cytometry was then performed to detect apoptosis. (D) Transfected A549 cells were used to detect invasion using transwell assays. The images showing one field under a microscope. The transmigrated cells were counted. Scale bar, 200 μm. In (A-D), each bar represents the mean ±SD of three independent biological replicates. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (Student's t test).
ChIP-seq reveals potential targets of CBX2 and EZH2
The cPRC1 complex is recruited to target genes partially by binding of the CBX protein to H3K27me3,4 which is catalyzed mainly by EZH2.10 Furthermore, we found that CBX2 and EZH2 cooperatively promoted LUAD (Figures 3 and 4). Therefore, we performed CBX2 chromatin immunoprecipitation sequencing (ChIP-seq) and downloaded H3K27me3 ChIP-seq data (GSE75903) of A549 cells to explore the transcriptional targets of CBX2 and EZH2 in LUAD. The analysis of the ChIP-seq data of CBX2 and H3K27me3 showed that 6.4% of the CBX2 peaks overlapped with H3K27me3 peaks and that 0.2% of the H3K27me3 peaks overlapped with CBX2 peaks (Figure 5A). This finding was consistent with published studies indicating that a small set of PRC1 targets overlaps with H3K27me3-rich regions.7,8,19 Regarding protein-coding genes, 3,166 and 5,577 genes were occupied by CBX2 and EZH2 (H3K27me3), respectively (Figures 5B and 5C). There were 822 genes occupied by both CBX2 and EZH2 (Figure 5D), among which, 145 genes were downregulated in LUAD samples (obtained through analyzing TCGA LUAD RNAseq data). The binding profile of CBX2 to these 822 genes showed that the binding signal of CBX2 was located exactly at the transcription start site (TSS) (Figure 5E).
Figure 5.

ChIP-seq profile of CBX2 and EZH2
(A) Venn plots showing the CBX2 peaks overlapping with the H3K27me3 peaks (left) and the H3K27me3 peaks overlapping with the CBX2 peaks (right). (B) CBX2 target genes and (C) EZH2 target genes annotated by the Ensembl and Gencode databases. (D) Venn diagram showing the overlap between the CBX2 target genes and the EZH2 target genes. For the overlapping genes, red, blue, and gray represents genes upregulated, downregulated, and unchanged genes in LUAD, respectively. (E) Normalized signal of CBX2 and H3K27me3 and their input peaks on 822 overlapping target genes identified by ChIP-seq and their relative position to the TSS. (F) Pathway enrichment analysis (p < 0.05) of 145 downregulated genes (FDR <0.01, fold change ≥1.5, and CPM >30). (G-H) Genome browser showing the binding peaks of CBX2 and EZH2 on (G) genes in the PPAR signaling pathway and (H) TSGs. The green bars indicate the TSS regions (TSS-8 kb ∼ TSS+2 kb) of different transcripts. The narrowPeak row presents the peak of the signals obtained by MACS2. Black frames highlight the peak regions among the TSS regions.
Given the inhibitory roles of CBX2 and EZH2 in regulating gene expression, we investigated the pathways enriched in the 145 LUAD-downregulated genes targeted by both CBX2 and EZH2. These downregulated genes overlapped with several pathways, including the PPAR signaling pathway (Figure 5F), which was also the most downregulated pathway in LUAD (Figure S3A). In addition, CBX2 and EZH2 could also bind and regulate target genes independently. For example, we found that CBX2 bound the TSS regions of PPARG (Figure 5G) and CD36 (Figure S3B) and that EZH2 bound the TSS regions of SORBS1, SLC27A6, RXRG, LPL, FABP4,and ACADL (Figure S3B) in the PPAR signaling pathway. Therefore, among the 16 downregulated PPAR signaling genes in LUAD, 11 genes might be bound and regulated by CBX2 and/or EZH2 (Figures 5G and S3B). In addition to these genes in the PPAR signaling pathway, CBX2 and EZH2 co-bound 14 other TSGs, namely, RBMS3, CLDN11, AGTR1, PCDH10, PPP2CB, NRG1, SMARCA2, ITIH5, CADM1, NR4A1, DCN, NDRG4, SOCS3, and FHL1; the ChIP-seq peaks of RBMS3 and CLDN11 are shown in Figure 5H.
Transcriptional regulation of target genes by CBX2 and EZH2
To confirm the ChIP-seq results, we performed ChIP assays using specific antibodies against CBX2, H2AK119ub, EZH2, and H3K27me3, with non-specific IgG as the negative control. The results indicated the occupancy of CBX2, H2AK119ub, EZH2, and H3K27me3 on the promoters of their common targets: RBMS3 and CLDN11 (Figure 6A). The results also verified the enrichment of H2AK119ub in the PPARG promoter (Figures 6C and 6E), whereas the enrichment of H3K27me3 in the promoter of PPARG was very weak (Figures 6D and 6F), which is consistent with the H3K27me3 ChIP-seq data (Figure 5G). To investigate the mutual influence of H2AK119ub and H3K27me3 on their common target genes, we knocked down the expression of CBX2 or EZH2 by their specific siRNA or shRNA, respectively (Figure 6B) and then performed ChIP assays using primers targeting CLDN11. The results showed that knockdown of CBX2 not only significantly decreased PRC1-catalyzed H2A119ub (Figure 6E) but also markedly decreased EZH2-catalyzed H3K27me3 on the promoter of CLDN11 (Figure 6F), indicating that the CBX2-mediated H2AK119ub assists the occupation of H3K27me3 in the promoter of CLDN11. Similarly, knockdown of EZH2 significantly decreased the level of H3K27me3 (Figure 6D), as well as the level of H2AK119ub on the promoter of CLDN11 (Figure 6C), suggesting that the EZH2-catalyzed H3K27me3 also assists the occupation of H2AK119ub in the promoter of CLDN11. However, knockdown of EZH2 did not change the H2AK119ub level in PPARG (Figure 6C), whose promoter was only modified by H2AK119ub but not EZH2-catalzyed H3K27me3. Our results validated the binding of CBX2 and EZH2 to their target genes and indicated that the enrichments of H2AK119ub and H3K27me3 are mutually required for the occupancy of CBX2 and EZH2 on their co-targeted gene, i.e., CLDN11.
Figure 6.

Transcriptional regulation of their targets by CBX2 and EZH2
(A) ChIP assays were performed using antibodies against EZH2, CBX2, H3K27me3, H2AK119ub, or non-specific IgG, and primers targeting the RBMS3 and CLDN11 promoters. (B) The mRNA expression of CBX2 or EZH2 after the transfection of CBX2 siRNAs or EZH2 shRNAs (n = 3). (C-F) ChIP assays were performed, n = 4. (C) H2AK119ub antibodies and non-specific IgG were used in EZH2 depleted cells. (D) H3K27me3 antibodies and non-specific IgG were used in EZH2 depleted cells. (E) H2AK119ub antibodies and non-specific IgG were used in CBX2 depleted cells. (F) H3K27me3 antibodies and normal IgG were used in CBX2 depleted cells. (G) Quantitative real-time RT-PCR assays were performed to examine the transcription of indicated genes in the A549 cells transfected with specific siRNAs (n = 3). In (A-D) , ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (Student's t test).
Based on the results showing that CBX2 and/or EZH2 occupy the promoters of PPAR signaling genes (PPARG, FABP5, and OLR1) and TSGs (RBMS3 and CLDN11), we further investigated whether CBX2 and EZH2 regulate the transcription of these genes in A549 cells. Therefore, total mRNA was extracted from A549 cells transfected with CBX2 siRNAs, EZH2 siRNAs, combined siRNAs, or control siRNAs. Then, quantitative real-time RT-PCR assays were performed. The results showed that depletion of CBX2, EZH2, or both led to the increased transcription of PPARG, OLR1, FABP5, RBMS3, and CLDN11 (Figure 6G). For OLR1, PPARG, FABP5, and CLDN11, knockdown of both CBX2 and EZH2 had a more significant inhibitory effect than knockdown of CBX2 or EZH2 alone (Figure 6G). Interestingly, although the enrichment of H3K27me3 in the promoter of PPARG was very weak (Figures 6D and 6F), indicating that PPARG was not a direct target of EZH2, the knockdown of EZH2 also led to an obvious upregulation of PPARG. This upregulation could be an indirect effect caused by EZH2 depletion-mediated transcriptional changes of PPARG upstream genes. Together, our data showed that CBX2 and EZH2 downregulated some PPAR signaling pathway genes and several TSGs by binding to their promoters cooperatively or separately.
A downstream target of CBX2 and EZH2 suppresses the growth and invasion of LUAD cells
As stated above, 11 genes in the PPAR signaling pathway (downregulated genes in LUAD) were co- or separately bound by CBX2 and EZH2 in A549 cells, among which PPARG, OLR1, and FABP5 were confirmed to be downregulated by CBX2 and EZH2 in our experiments, implying a tumor-suppressive function of the PPAR signaling pathway. However, PPARG, a key gene of the PPAR signaling pathway, was reported to play an ambiguous functional role in different cancers.20 To determine the role of PPARG in LUAD, we knocked down the mRNA expression of PPARG by its specific siRNAs (Figure 7A). In vitro experiments indicated that PPARG deficiency led to a significant increase in the viability (Figure 7B) and invasion (Figure 7C) of LUAD cells. Furthermore, PPARG knockdown partially rescued the decrease in the viability (Figure 7D) and invasive activity (Figure 7E) of LUAD cells caused by the CBX2 and EZH2 knockdown, demonstrating that PPARG, which plays a role in inhibiting LUAD cell viability and invasion, is one of the functional downstream targets of CBX2 and EZH2.
Figure 7.

The PPAR pathway suppresses the viability and invasion of LUAD cells
(A) Real-time quantitative RT-PCR was performed to detect the knockdown effect of the PPARG siRNAs transfection. (B) MTT assays were used to detect the viability of cells transfected with control or PPARG siRNAs. (C) Transwell assays were used to examine invasion of A549 cells transfected with the control or the PPARG siRNAs. The images show one field under a microscope. The invaded cells were counted. Scale bar, 200 μm. (D) MTT assays were performed to detect the viability of cells transfected with the indicated siRNAs. (E) Transwell assays were used to examine the invasion of A549 cells transfected with the indicated siRNAs. Scale bar, 200 μm. In (A-D), each bar in the bar plots represents the mean ±SD of three independent biological replicates; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (Student's t test). (F) Kaplan-Meier survival curve of OLR1 based on TCGA data. Tumor samples were grouped into high and low groups by the median expression of OLR1. (G) Kaplan-Meier survival curve of the combination of CBX2, EZH2, and OLR1. Tumor samples were grouped into high and low groups by median expression of CBX2, EZH2, and OLR1.
Furthermore, to further confirm the role of PPAR signaling in LUAD suppression, we also performed survival analysis of patients stratified by the expression levels of these 11 genes to reveal the clinical relevance of the PPAR signaling pathway. The results showed that overexpression of OLR1, CD36, and RXRG was significantly (p < 0.05) associated with good prognosis in patients with LUAD (Figures 7F and S3C). Interestingly, the combination of higher expression of these three genes with lower expression of CBX2 and/or EZH2 was more significantly (p < 0.05) associated with good prognosis (Figures 7G and S3D). These results indicated the potential tumor suppressor role of these PPAR signaling genes in LUAD.
Application potential in clinic: CBX2 knockdown enhances the therapeutic efficiency of tazemetostat
To investigate the importance of the cooperative effects of CBX2 and EZH2 inhibition in the clinical treatment of LUAD, we examined the effects of combined CBX2 knockdown and tazemetostat (TAZ, an oral EZH2 inhibitor in clinical phase 2 trial21) treatment on LUAD cell growth and invasion. We found that the inhibitory effect induced by CBX2 knockdown was approximately equal to that of TAZ in inhibiting the viability (Figure 8A) and invasion (Figure 8B) of A549 cells. In addition, when CBX2 siRNA was used in combination with TAZ treatment, cell viability (Figure 8A) and invasion (Figure 8B) decreased more significantly than those in the cells treated with TAZ alone. In summary, CBX2 inhibition improved the therapeutic efficiency of TAZ.
Figure 8.

CBX2 knockdown enhances the therapeutic efficiency of tazemetostat
A549 cells transfected with control or CBX2 siRNAs were treated with DMSO or TAZ (200 nmol/L). (A) MTT assays were applied to examine the viability of the indicated cells (n = 4). (B) Transwell assays were applied to detect the invasion of A549 cells (n = 3). The images represent one field under a microscope. Scale bar, 200 μm. The invaded cells were counted.
Discussion
Abnormal epigenetic regulation profoundly influences cellular processes, leading to tumorigenesis. Drugs targeting epigenetic factors have been used in clinical trials to treat tumors. For example, EZH2 performs oncogenic functions, and its inhibitor has been used to treat a variety of cancers, such as breast, prostate, and lung cancer.22, 23, 24 In our study, to investigate the potential function of histone methylation regulators in LUAD, we performed differential expression analysis of 58 paired tumor/normal LUAD samples from TCGA and found marked upregulation of CBX2 and EZH2 in LUAD samples (Figure 1). Then, we validated the oncogenic role of CBX2 in vitro (Figure 1) and in vivo (Figure 2). The combined high expression of CBX2 and EZH2 served as an indicator of poor survival in LUAD patients (Figure 3B, p = 0.008), and their expressions were positively correlated (Figures 3E and 3F). These results suggested that CBX2 and EZH2 play a potential oncogenic role in LUAD through cooperation. We also validated the cooperative role of CBX2 and EZH2 in promoting LUAD cell growth and invasion (Figure 4). Moreover, CBX2 inhibition enhanced the effect of TAZ, an EZH2 inhibitor in clinical phase 2 trial, on inhibiting survival and invasion of LUAD cells. This study elucidated the oncogenic role of CBX2 in LUAD and the cooperative oncogenic role of CBX2 and EZH2, which reveals a potential combined target for therapy.
To identify factors responsible for the upregulation of CBX2 and EZH2, and their positive correlation in LUAD, we constructed the regulatory network of the transcription factor (TF)-miRNA feedforward loop using CBX2, EZH2, 78 upregulated TFs, and 24 downregulated miRNAs in LUAD. We obtained 20 regulators (6 miRNAs and 14 TFs) targeting EZH2 and 19 regulators (9 miRNAs and 10 TFs) targeting CBX2. Among them, 5 TFs (E2F1, E2F3, SOX4, SOX9, and FOXP3) and 5 miRNAs (miR-101-3p, miR-195-5p, let-7a-5p, let-7c-5p, and miR-30d-5p) regulated both CBX2 and EZH2. This may be the possible reason for the positive correlation between the expression of CBX2 and EZH2 in LUAD. The experimental verification of this part will be carried out in the future.
Through high-throughput sequencing, we found that CBX2 and EZH2 bound to 11 PPAR signaling pathway genes and 14 TSGs to add histone modification in the TSS regions of these target genes. Among the CBX2 and H3K27me3 peaks identified by ChIP-seq, 6.4% of the CBX2 peaks overlapped with the H3K27me3 peaks. This finding is consistent with published studies, suggesting that PRC1 can be recruited to chromatin independently of PRC2 and H3K27me3.7,8,19 One reason for this phenomenon is that, in addition to CBX2, other CBX proteins also function as components of PRC1 to bind to H3K27me3.4 Furthermore, the chromodomain in CBX proteins can specifically recognize both H3K27me3 and H3K9me3. Moreover, CBX proteins may regulate transcription independent of the PRC1 complex. In addition, our data further indicated that the binding of CBX2 and EZH2 to their common target genes and the establishment and maintenance of H2AK119ub and H3K27me3 were mutually required. This finding is consistent with previous reports showing that, for the common targets, EZH2 is needed for the binding of some CBX proteins to chromatin and that loss of H2AK119ub1 led to rapid dysregulation of PRC2 activity and the loss of H3K27me3 deposition.25,26
The PPAR signaling pathway was the most significantly downregulated pathway in LUAD, and among the 16 downregulated PPAR signaling genes, 11 genes might be bound and regulated by CBX2 and/or EZH2. PPARG is the core regulator gene in PPAR signaling pathway. Interestingly, PPARG showed an ambiguous functional role in different cancers20 and appeared to be a driver in different pathways,27 determining its role in LUAD is crucial, and may provide more insight into the role of PPARG in cancer development. Therefore, we focused on PPARG, which is one of the downstream genes of CBX2 and EZH2, for functional experiments. In our study, we confirmed the tumor suppressor role of PPARG in reducing cell viability and inhibiting invasion. This finding is consistent with a previous study showing that the activation of PPARG inhibits the proliferation of cancer cells through changing the metabolic, ultimately resulting in the arrest of cell cycle.28 We also confirmed that PPARG is one of the functional downstream targets of CBX2/EZH2, because PPARG knockdown partially rescues the phenotype caused by CBX2 and EZH2 depletion.
In summary, through bioinformatic data mining and a series of in vitro and in vivo studies, we revealed the role of CBX2 in promoting growth and metastasis in LUAD, indicating that CBX2 is a new potential therapy target. In addition, we found that CBX2 and EZH2 cooperatively promote the growth and metastasis of LUAD by adding histone modifications on the promoter region of several PPAR signaling pathway genes and TSGs, suggesting the potential clinical application of developing CBX2 inhibitors in combination with EZH2 inhibitors to treat LUAD. We further presented numerous gene combinations as prognostic indicators of LUAD, which performed well in the prognostic stratification of LUAD patients. Together, the results of this study provide insights into the mechanism of epigenetic regulation in LUAD progression, and identify several predictors of prognosis as well as potential novel therapeutic targets of LUAD.
Materials and methods
Differential expression analysis
The mRNA (576 samples, Illumina HiSeq level 3) sequencing data of LUAD were collected from TCGA database. We quantified the gene expression data by RSEM. The tumor samples (n = 58) with paired adjacent normal samples (n = 58) were screened to perform a differential expression analysis. The NOISeq29 R package was used to calculate the DEGs from the RNA-seq data of 58 tumor-normal paired samples. The DEGs were identified with the thresholds false discovery rate (FDR) <0.01, fold change ≥2 and counts per million (CPM) >30. In addition, the gene expression profile was scaled by row, and the heatmap was computed by the ComplexHeatmap30 R package. In addition, we relaxed the threshold of fold change ≥1.5 to identify the DEGs targeted by both CBX2 and EZH2.
Survival analysis
TCGA survival data of LUAD were collected from the literature,31 which provides four major clinical outcome endpoints and a summary of the endpoint usage recommendations for each cancer type. The progression-free interval (PFI) survival data of LUAD passed all tests in this study,31 and LUAD is a very aggressive cancer type; thus, five-year PFI survival data were extracted to analyze LUAD patient survival in this study. The univariate Kaplan-Meier (log rank test) and multivariate Cox analyses were performed to determine the independent risk characteristics. In detail, the gene expression data and the clinical data were merged through the TCGA sample barcode. Then, for single gene survival analysis, according to the median expression of a specific gene, the samples were classified into two groups (high and low expression). In the survival analysis that combines multiple genes' expression, first, the samples were classified into two groups according to the median expression of each gene; second, the samples were filtered depending on the gene function hypothesis. For example, CBX2 and EZH2 were both upregulated in LUAD and may function together as oncogenes; therefore, samples with both EZH2 and CBX2 highly expressed were included in the EZH2High/CBX2High group, and the EZH2Low/CBX2Low group was defined similarly. In the univariate survival analysis, the log rank test compared the survival differences between two groups. In multivariate survival analysis, a Cox regression model, including objective variable (such as CB×2 expression) and other covariates (TNM stage, smoking years, metastasis, and mutation of KRAS and EGFR), was constructed. The hazard ratios and 95% confidence intervals of these variables were estimated to quantify the strength of these associations. A p <0.05 was considered indicative of a significant independent prognostic predictor. The R package survival was implemented to perform survival analysis, and package survminer was used to draw the survival curves.
Immunohistochemistry and analysis
As we previously described,32 microarrays containing LUAD and adjacent normal tissue (HLugA150CS02) were obtained from Shanghai Outdo Biotech (Shanghai, China). Tissue sections were observed and scored by an investigator blinded to the clinicopathologic data. Paraffin-embedded tissue microarrays were deparaffinized using xylene and rehydrated through a graded alcohol series. Antigen was retrieved in 5 mM citrate buffer (pH 6.0) at a sub-boiling temperature for 10 min. After inactivating endogenous peroxidase using 3% H2O2 solution in methanol for 10 min at room temperature, the tissue microarrays were blocked with goat serum, followed by incubation with anti-EZH2 or anti-CBX2 antibodies. Then, the tissue microarrays were incubated with a biotin-conjugated secondary antibody and streptavidin-biotin-peroxidase. Diaminobenzidine was used as a chromogenic substrate to visualize the proteins. Finally, the tissue microarrays were counterstained with hematoxylin and examined under a microscope.
For the data obtained from immunohistochemistry, the percent positive rate was scored as follows: 1, <25%; 2, 25%–50%; and 3, ≥50%. The expression score was calculated by multiplying the positive rate by the staining intensity (the median was used when the staining intensity was a range value; for instance, we used 0.75 to represent a range of 0.5–1) to represent the protein expression levels.
Cell line authentication, culture, inhibitor treatments, and transfections
As we previously described,32 A549 and H1299 cells were obtained from the National Infrastructure of Cell Line Resource (Shanghai, China). The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) at 37°C in a humidified atmosphere with 5% CO2. The cell lines were authenticated by an examination of their morphology and growth characteristics. All the cell lines were confirmed to be mycoplasma-free. For siRNA transfections, according to the manufacturer's standard protocol, Lipofectamine 2000 was used to transfect 30 nM siRNAs into 106 cells. The detailed methods of the cell viability assay, apoptosis assay, EdU incorporation assay, and migration assay are presented in the supplemental information.
Animal experiments
Statement: all procedures involving animals were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University and were conducted in accordance with the NIH's Guide for the Care and Use of Laboratory Animals (eighth ed., National Academies Press, 2011).
CBX2 single guide RNA (sgRNA) or control sequences were cloned into pLentiCRISPRv2 vectors, and then lentiviruses expressing sgRNAs and Cas9 were packaged and used to infect A549 cells. As we previously described,32 lung tumors derived from control A549 cells or A549 cells expressing CBX2 sgRNAs were transplanted into female athymic nude mice (BALB/c, Charles River; between five and six weeks of age; six mice per group). Tumors were measured using a vernier caliper over a period of four weeks, and tumor volumes were calculated according to the following formula: V = π/6 × length × width2. Tumor volumes were examined by an investigator blinded to the experiment procedure. Mice in which tumors did not form were removed from this study. Tumors were observed in all mice injected with A549 cells. Mice were sacrificed at 23 days post injection. Tumors were then excised and imaged.
To investigate LUAD metastasis in vivo, normal A549 cells or A549 cells stably expressing CBX2 sgRNAs were transduced with lentiviral vectors carrying the luciferase expression cassette. These cells were separately injected into six-week-old female SCID mice via the lateral tail vein. The metastasis of tumor cells was monitored weekly by a bioluminescence imaging system (Xenogen). The bioluminescence analysis was performed by a technician blinded to the subgroup allocations.
Antibody sources
The following antibodies were used: anti-CBX2 (A302-524A) from Bethyl Laboratories. (Montgomery, TX, USA); anti-EZH2 (ab191250) and anti-H3K27me3 (ab6002) from Abcam (Cambridge, MA, USA); anti-E-cadherin (610,404), anti-N-cadherin (610,921), and anti-α-catenin (610,194) from BD Biosciences (Franklin Lakes, NJ, USA); anti-Vimentin (SC32322), anti-β-catenin (SC7963), and anti-γ-catenin (SC33634) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and anti-H2AK119ub (ABE569) and anti-β-actin (A1978) from Sigma-Aldrich (St. Louis, MO, USA). Horseradish peroxidase-conjugated secondary antibodies (sc-2030 and sc-2031) were obtained from Santa Cruz Biotechnology.
ChIP and ChIP-seq
The ChIP experiments were performed as previously described.33,34 Briefly, A549 cells were washed with PBS twice and crosslinked with 1% formaldehyde for 10 min. Then, cells were rinsed with ice-cold PBS twice and collected. Cells were collected and resuspended in lysis buffer (50 mM Tris-HCl [pH 8.1], 10 mM EDTA, 1% SDS, 1× protease inhibitor cocktail), and sonicated for 10 cycles at the maximum amplitude using a water bath sonicator (Diagenode) before centrifugation for 10 min. Then, immunoprecipitation was performed using antibodies against CBX2, EZH2, H2AK119ub, and H3K27me3 or non-specific IgG as the negative control. The eluted DNA fragments were purified using a DNA purification kit (QIAquick Spin Kit; Qiagen, Valencia, CA, USA) and subjected to PCR or sent to BGI-Shenzhen (Shenzhen, China) for deep sequencing.33
ChIP-seq data analysis
Using Bowtie (version 1.0.1), millions of reads generated by the ChIP-seq of CBX2 and H3K27me3 (GEO: GSE75903) were aligned with the human reference genome (version GRCh38), and nonuniquely mapped reads were excluded. Then, MACS2 (version 2.1.0)35 was subsequently used to detect the genomic regions (namely, peaks) enriched with multiple overlapping DNA fragments. The positive binding sites were extracted using the FDR (cutoff: 1 × 10−3) estimated by MACS2, which was defined to compare the peaks obtained from the treated and control samples. The peak intersection analysis was performed by the intersectBed function of BEDtools (version 2.25.0), which was applied to identify overlaps between the H3K27me3 and the CBX2 peaks and between the H3K27me3 or CBX2 peak summits and the TSS regions (TSS-8 kb ∼ TSS+2 kb). In all analyses, a 1 bp intersection was considered a peak overlap. A gene was considered a target of H3K27me3 or CBX2 when its peak summit overlapped with the TSS region (GENCODE version 21) of at least one transcript of the gene. Then, deepTools (version 3.0.2) was used to generate the ChIP-seq profiles around the TSS regions through calculating the average coverage at each position. Finally, the data were visualized with the UCSC Genome Browser.36 The ChIP-seq data of CBX2 in A549 cells were deposited in the NGDC (National Genomics Data Center) database under accession number CRA001102.
Ethics approval and consent to participate
All procedures involving animals were approved by the ethics committee of Tianjin Medical University and were conducted in accordance with the NIH's Guide for the Care and Use of Laboratory Animals (eighth ed., National Academies Press, 2011).
Availability of data and materials
The data supporting the findings of this study are available from TCGA and the GEO. The ChIP-seq data of CBX2 in A549 cells were deposited in NGDC (National Genomics Data Center) with accession number CRA001102 (https://bigd.big.ac.cn/gsa/browse/CRA001102).
Acknowledgments
We thank TCGA and the Gene Expression Omnibus (GEO). for providing the public data supporting our study and the program for the HUST Academic Frontier Youth Team. We thank the high-performance computing center of Wuhan University of Science and Technology for supporting the numerical calculation in this study.
Role of the funding source: This work was supported by the National Natural Science Foundation of China (nos. 31822030, and 31771458); and Natural Science Foundation of Tianjin (no. 18JCQNJC81400) and Science and Technology Development Fund of Tianjin Education Commission for Higher Education (no. 2018KJ068). The funding agencies played no role in this study.
Author contributions
C.X. and A.-Y.G. designed and supervised the study. H.C., Y.D., B.L., X.D., Y.-M.C., and M.L. performed the experiments. F.-F.H., Y.D., and H.D. were responsible for experimental data analysis. F.-F.H., C.-J.L., H.H., and Q.Z. performed TCGA data analysis and interpretation. F.-F.H., A.-Y.G., and C.X. wrote, and/or revised the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.032.
Contributor Information
An-Yuan Guo, Email: guoay@hust.edu.cn.
Chenghao Xuan, Email: chenghaoxuan@tmu.edu.cn.
Supplemental information
References
- 1.Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Sparmann A., van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 4.Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simon J.A., Kingston R.E. Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 6.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 7.Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., Reinberg D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loubière V., Delest A., Thomas A., Bonev B., Schuettengruber B., Sati S., Martinez A.-M., Cavalli G. Coordinate redeployment of PRC1 proteins suppresses tumor formation during Drosophila development. Nat. Genet. 2016;48:1436–1442. doi: 10.1038/ng.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ku M., Koche R.P., Rheinbay E., Mendenhall E.M., Endoh M., Mikkelsen T.S., Presser A., Nusbaum C., Xie X., Chi A.S., et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sing A., Pannell D., Karaiskakis A., Sturgeon K., Djabali M., Ellis J., Lipshitz H.D., Cordes S.P. A vertebrate polycomb response element governs segmentation of the posterior hindbrain. Cell. 2009;138:885–897. doi: 10.1016/j.cell.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 11.Schoeftner S., Sengupta A.K., Kubicek S., Mechtler K., Spahn L., Koseki H., Jenuwein T., Wutz A. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 2006;25:3110–3122. doi: 10.1038/sj.emboj.7601187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamagishi M., Uchimaru K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 2017;29:375–381. doi: 10.1097/CCO.0000000000000390. [DOI] [PubMed] [Google Scholar]
- 13.Sangodkar J., Katz S., Melville H., Narla G. Lung adenocarcinoma: lessons in translation from bench to bedside. Mt. Sinai J. Med. 2010;77:597–605. doi: 10.1002/msj.20226. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 15.Rezaei M.K., Nolan N.J., Schwartz A.M. Surgical pathology of lung cancer. Semin. Respir. Crit. Care Med. 2013;34:770–786. doi: 10.1055/s-0033-1358558. [DOI] [PubMed] [Google Scholar]
- 16.Mamdani H., Jalal S.I. Histone deacetylase inhibition in non-small cell lung cancer: hype or hope? Front. Cell Dev. Biol. 2020;8:1126. doi: 10.3389/fcell.2020.582370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H., Qi J., Reyes J.M., Li L., Rao P.K., Li F., Lin C.Y., Perry J.A., Lawlor M.A., Federation A., et al. Oncogenic deregulation of EZH2 as an opportunity for targeted therapy in lung cancer. Cancer Discov. 2016;6:1006–1021. doi: 10.1158/2159-8290.CD-16-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poirier J.T., Gardner E.E., Connis N., Moreira A.L., de Stanchina E., Hann C.L., Rudin C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015;34:5869–5878. doi: 10.1038/onc.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz Y.B., Pirrotta V. Ruled by ubiquitylation: a new order for polycomb recruitment. Cell Rep. 2014;8:321–325. doi: 10.1016/j.celrep.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Lehrke M., Lazar M.A. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 21.Morschhauser F., Tilly H., Chaidos A., McKay P., Phillips T., Assouline S., Batlevi C.L., Campbell P., Ribrag V., Damaj G.L., et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21:1433–1442. doi: 10.1016/S1470-2045(20)30441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frankel A.E., Liu X., Minna J.D. Developing EZH2-targeted therapy for lung cancer. Cancer Discov. 2016;6:949–952. doi: 10.1158/2159-8290.CD-16-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bachmann I.M., Halvorsen O.J., Collett K., Stefansson I.M., Straume O., Haukaas S.A., Salvesen H.B., Otte A.P., Akslen L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 24.Hussain M., Rao M., Humphries A.E., Hong J.A., Liu F., Yang M., Caragacianu D., Schrump D.S. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009;69:3570–3578. doi: 10.1158/0008-5472.CAN-08-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhen C.Y., Tatavosian R., Huynh T.N., Duc H.N., Das R., Kokotovic M., Grimm J.B., Lavis L.D., Lee J., Mejia F.J., et al. Live-cell single-molecule tracking reveals co-recognition of H3K27me3 and DNA targets polycomb Cbx7-PRC1 to chromatin. Elife. 2016;5:e17667. doi: 10.7554/eLife.17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamburri S., Lavarone E., Fernández-Pérez D., Conway E., Zanotti M., Manganaro D., Pasini D. Histone H2AK119 mono-ubiquitination is essential for polycomb-mediated transcriptional repression. Mol. Cell. 2020;77:840–856.e5. doi: 10.1016/j.molcel.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fanale D., Amodeo V., Caruso S. The interplay between metabolism, PPAR signaling pathway, and cancer. PPAR Res. 2017;2017:1–2. doi: 10.1155/2017/1830626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava N., Kollipara R.K., Singh D.K., Sudderth J., Hu Z., Nguyen H., Wang S., Humphries C.G., Carstens R., Huffman K.E., et al. Inhibition of cancer cell proliferation by PPARγ is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. 2014;20:650–661. doi: 10.1016/j.cmet.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarazona S., Furió-Tarí P., Turrà D., Pietro A.D., Nueda M.J., Ferrer A., Conesa A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43:e140. doi: 10.1093/nar/gkv711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 31.Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V., et al. An integrated TCGA pan-cancer clinical data Resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan Y., Huo D., Gao J., Wu H., Ye Z., Liu Z., Zhang K., Shan L., Zhou X., Wang Y., et al. Ubiquitin ligase RNF20/40 facilitates spindle assembly and promotes breast carcinogenesis through stabilizing motor protein Eg5. Nat. Commun. 2016;7:12648. doi: 10.1038/ncomms12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shang Y., Hu X., DiRenzo J., Lazar M.A., Brown M. Cofactor dynamics and sufficiency in estrogen receptor–regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 34.Xuan C., Wang Q., Han X., Duan Y., Li L., Shi L., Wang Y., Shan L., Yao Z., Shang Y. RBB, a novel transcription repressor, represses the transcription of HDM2 oncogene. Oncogene. 2013;32:3711–3721. doi: 10.1038/onc.2012.386. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available from TCGA and the GEO. The ChIP-seq data of CBX2 in A549 cells were deposited in NGDC (National Genomics Data Center) with accession number CRA001102 (https://bigd.big.ac.cn/gsa/browse/CRA001102).