

Abstract
Abstract
The unavailability of a proper drug against SARS-CoV-2 infections and the emergence of various variants created a global crisis. In the present work, we have studied the antiviral behavior of feverfew plant in treating COVID-19. We have reported a systematic in silico study with the antiviral effects of various phytoconstituents Borneol (C10H18O), Camphene (C10H16), Camphor (C10H16O), Alpha-thujene (C10H16), Eugenol (C10H14O), Carvacrol (C10H14O) and Parthenolide (C15H20O3) of feverfew on the viral protein of SARS-CoV-2. Parthenolide shows the best binding affinity with both main protease (Mpro) and papain-like protease (PLpro). The molecular electrostatic potential and Mulliken atomic charges of the Parthenolide molecule shows the high chemical reactivity of the molecule. The docking of Parthenolide with PLpro give score of −8.0 kcal/mol that validates the good binding of Parthenolide molecule with PLpro. This complex was further considered for molecular dynamics simulations. The binding energy of the complex seems to range in between −3.85 to −11.07 kcal/mol that is high enough to validate the stability of the complex. Free energy decomposition analysis have been also performed to understand the contribution of residues that reside into the binding site. Good binding affinity and reactivity response suggested that Parthenolide can be used as a promising drug against the COVID-19.
Graphical Abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s11696-022-02067-6.
Keywords: COVID-19, DFT, MD simulations, Molecular docking, Parthenolide
Introduction
COVID-19 has emerged as a global pandemic disaster in 2019, and approximately forty-four Lakh people in the world had lost their lives. It is a contagious disease spread like the common cold (Asadi 2020; Mittal 2020). Its spike-like protein structures get latched to human angiotensin-converting enzyme 2 (ACE2) and dysfunction the organ (Hachke 2013; Hoffman 2020; Khelfaouis 2020). The ACE2 is considered as the main functional receptor of SARS-CoV-2. The envelope surface of SARS-CoV-2 comprises of spike glycoproteins, membrane proteins and envelope proteins. The virus enters inside the human body through ACE2 and its spike-like glycoprotein get bind to specific receptors on the cell membrane of the host cells (Ni 2020). When the virus enters the host cell, it got split by airway trypsin-like protease (HAT), cathepsins and transmembrane protease serine 2 (TMPRSS2) and make the penetration (Yadav 2021). This makes the selection of ACE2 receptor as a target protein against SARS-CoV-2 a fruitful option. Scientists have observed constant mutation of COVID-19 virus and emergence of various variants. The severity of COVID-19 is associated with these genetic variants. NKG2C is an integral membrane protein that leads to the extension of SARS-CoV-2 infection by binding to HLA-E in humans (Vietzen 2021). Alpha-B.1.1.7 (United Kingdom), Beta-B.1.351 (South Africa), Gamma-P.1 (Japan and Brazil) and Delta-B.1.617.2 (India) are some variants of COVID-19 (Bernal 2021). It has been observed that among the different variants, delta variant is highly contagious and likely more severe. Food and Drug Administration (FDA) has authorized many vaccines that are proven remedies against Delta and other known variants, but it is not sure whether these vaccines will cure against new upcoming variants. By mid-2021, National Regulatory Agencies (NRA) have approved twelve vaccines Pfizer-BioNTech (Oliver 2020), Moderna (Mahase 2020), Sputnik V (Tulleken 2021) Oxford-AstraZeneca, J&J (Earle 2021), Convidecia (Funk 2021), Sinopharm (BBIBP) (Wang 2020), CoronaVac (Wu 2021), Covaxin (Sapkal 2021) and RBD-Dimer (Mitra 2021) for the public. As per World health organization (WHO)’s, June 2021 report, vaccines like AstraZeneca/Oxford vaccine, J&J, Moderna, Pfizer/BioNTech and Sinopharm have met the necessary criteria for safety and efficacy (WHO 2021). Most of these vaccines are currently in use in different countries. Recently Drug Controller General of India (DCGI) has approved trials of 2-Deoxy-d-glucose (2-DG) medicine introduced by Defence Research and Development Organization (DRDO) (Balakrishna 2020). 2-DG is given to many COVID-19 patients to prevent the use of supplemental oxygen (Verma 2020). Chemists worldwide are trying hard to synthesize an ideal medicine that will work as complete treatment for COVID-19. Some commonly repurposed drugs and their combination used to treat COVID-19 infection are: ivermectin, doxycycline, nitazoxanide/azithromycin, remdesivir, oseltamivir, hydroxychloroquine, favipiravir, lopinavir/ritonavir, boceprevir, telaprevir, etc. (Yadav 2021). However, these drugs and their combination have possible side effects. The outbreak of COVID-19 has brought a positive impact on the herbal medicine market globally as herbal extracts are the best ways to boost up immunity (Panyod 2020). People are turning toward herbal medicines and herbal medicine therapy, which can also be a complementary option in treatment (Sharma 2020; Manzano 2020; Enioutina 2017). Many medicines comprising herbal extracts like ashwagandha, neem, tulsi, Triphala, turmeric, Tinospora cordifolia and ginger available in markets are taken in the medicinal form to boost up immunity (Verma 2011; Tripathi 2020; Akriti 2021; Gilani 2005; Chowdhury 2020). These studies reveal that plants and herbal extracts play a vital role in drug synthesis and medicinal sciences. We have chosen perennial flowering herb feverfew, which is biologically called Tanacetum parthenium L and belongs to the family of Asteraceae (Pablos 2017; Pareek 2011). Feverfew is a widely known medicinal plant having many uses. It has a wide range of secondary metabolites like lactones, flavonoids, eudesmanolide, germacranolides, guaianolides and many volatile oils having medicinal applications (Pourianezhad 2016). Along with antiviral capability, it possesses many other qualities also as anti-inflammatory, anticancer, effects in migraines, headaches and platelets (Wider 2015). European herbalists used feverfew as a folk medicine in treating fevers, cancers, migraine headaches, allergies, asthma, stomach and toothaches, nausea, vomiting and even in menstruating problems and labor pain during childbirth (Ferrero 2018; Behbahani 2018). Numerous works had been recorded based on it and few of them done so far are for the treatment of cancer, migraine, diabetes, leukemia and many more (Sur 2009; Curry 2004; Maizels 2004; Galeotti 2014; Guzman 2005; Özbilgin 2018; Halberstein 2005). For this work, we have reported molecular docking for the selected phytochemicals of feverfew with both the main protease (Mpro) and papain-like protease (PLpro) of COVID-19. The ligand structure was optimized using Density functional theory (DFT). The optimization was carried out as the optimized structure give better results in the docking (Rana 2021). High chemical reactivity of the ligand is established by UV–Vis spectrum and calculating the global reactivity parameters. Molecular dynamics (MD) simulations are also performed for the selected phytochemical with the best binding score to study the molecular stability and complexity of the drug-like component.
Materials and methods
Potential target protein structure for SARS-CoV-2 and protein receptor preparation
Coronavirus is a polyprotease receptor having two types of proteases Mpro and PLpro (Fig. 1) (Yang 2020; Mouffouk 2021). These proteases are responsible for the replication of the virus in the human body and enclosing ubiquitin and ISG15 into the host cell preventing regulation of protein circulation in human cells, respectively (Mengist 2020; Barretto 2005). It causes swift death of the healthy cells and rapid fall in the immunity of the patient (Shree 2022; Lakhera 2021, Altay 2020). In this study, we have taken 3D coordinates of Mpro (PDB ID: 6LU7) and PLpro (PDB ID: 4OVZ) in their native states derived by X-ray crystallography from protein data bank with resolution 2.16 Å and 2.50 Å, respectively, as drug target macromolecule. Structures of target protein 6LU7 with an inhibitor N3 and 4OVZ with lead inhibitor (Fig. 1) are downloaded from the “Protein data bank” (https://www.rcsb.org/), and the binding cavity residues are determined from the surrounding of co-crystallized ligands. Process of protein synthesis is done with the help of software “Biovia discovery studio visualizer” (https://discover.3ds.com/discovery-studio-visualizer-download), and the prepared protein is used for molecular docking.
Fig. 1.

Crystal structure of Human coronavirus captured from Biovia discovery studio visualizer a Mpro:6LU7; b PLpro:4OVZ
Potential inhibitor: feverfew
For the present study, we have taken seven different phytochemicals Borneol, Camphene, Camphor, Alpha-thujene, Eugenol, Carvacrol and Parthenolide having antiviral properties (Table 1) (Anastasiya 2015; Stevanović 2009; Pareek 2011). These phytochemicals have antiviral applications in treating influenza (Sokolova 2017; Sokolova 2017), diarrhea (Madeddu 2021), herpesvirus (Benensia 2000), human papillomavirus (Jeyamohan 2016), human norovirus (Gilling 2014), etc. 3D structures of all considered phytoconstituents are downloaded with the help of the online database “PubChem” (SD 1) in SDF format and converted into PDB format by “Open babel GUI” (http://openbabel.org/wiki/Main_Page). Morphology of feverfew plant is shown in the Fig. 2.
Table 1.
Active phytochemicals of feverfew plant with their PubChem IDs, chemical formulae and 3D structures

Fig. 2.

Morphology of feverfew plant
Drug-likeness properties and ADMET properties
To verify the pharmacokinetic ability of the drug-like molecule, drug-likeness properties of phytochemicals are screened. Some main important drug-likeness rules are Lipinski’s rule, Veber’s rule, Ghose filter, Egan rule, Muegge rule, lipophilicity (iLOGP, WLOGP, XLOGP3, MLOGP, log Po/w), water solubility (Log S (SILICOS-IT)), etc. (Lipinski 2004). Screening for PAINS is also necessary as they give false positive results while predicting the binding site and the presence of any such PAINS can alter the results (Baell JB 2016). Compounds like hydroxyphenyl hydrazones, quinones, etc., called pan-assay interference compounds (PAINS), should be necessarily removed from the target protein (Baell JB 2017). All these properties are studied with the help of the online database SWISS-ADME (http://www.swissadme.ch/). Analyzing ADMET properties is a vital step for drug discovery (Cheng 2012; Mary 2021). Gastrointestinal absorption (GI) and blood brain barrier (BBB) penetration are the important metabolic properties that predicts the oral bioavailability of molecule. Along with the RO5 and other pharmacokinetic rules, the considered inhibitor must follow the ADMET properties also. The ADMET features are listed using ADMETsar (http://lmmd.ecust.edu.cn/admetsar1) open-source tool.
Molecular docking and visualization
We have preferred Auto Dock Vina (http://vina.scripps.edu/) for our study. Docking with Auto Dock Vina sets a specific algorithm like setting up energy difference = 4 kcal/mol and exhaustiveness = 8. The grid box which specifies the location for ligands to get attached to binding sites is centered with coordinates x = -26.283, y = 12.599 and z = 58.965 for 6LU7 and x = -9.600, y = 41.005 and z = -29.793 for 4OVZ. The protein thus synthesized is saved in pdbqt format. Ligands are also converted into pdbqt format. Furthermore, the most stable receptor–ligand complex is chosen among the nine poses obtained after docking by analyzing binding affinity (kcal/mol), the dipole moment of ligand (Debye), number of hydrogen bonds, inhibition constant (µM) and dreiding energy. The Dreiding energy is the energy showing the stability of the 3D geometry of the molecule. For more authenticity in our docking results, we have performed docking for both the proteins with selected ligand using Pyrx (https://sourceforge.net/projects/pyrx/) software.
Computational method for structural analysis
All the theoretical calculations, including the optimization of ground-state geometry, are carried out using the Gaussian 09 program. We have downloaded the structure of the Parthenolide molecule from online database “PubChem (https://pubchem.ncbi.nlm.nih.gov/)” in PDB format. The graphic user interface “OpenBabel (http://openbabel.org/wiki/Main_Page)” is used to convert the structure into gaussian input file. Optimization is done for the selected phytochemical with DFT using Becke3-Lee–Yang–Parr (B3) exchange functional combined with the (LYP) correlation functional with the standard 6-311G (d, p) basis set (Parr 1980). In the present paper, we have performed the DFT calculations for the Parthenolide molecule to investigate its interaction with target protein. The more the ligand will be reactive, more it will participate in chemical reactions and bond formation with protein. We have performed geometry optimization and computed energy of the probe system at room temperature as a relaxed model where geometry coordinates are not rigid. We have not determined the potential barriers in the present case. Excited states calculation of molecule was performed using time-dependent DFT (TD-DFT). The frontier molecular orbital (FMOs) is simulated for the molecule using Koopman’s theorem (Koopman 1933).
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
The electron affinity (EA) is computed as -ELUMO, while the ionization potential (IP) is computed as -EHOMO (Sahoo 2020). IP and EA are useful in analyzing the electronegativity (χ), hardness (η), electrophilicity (ω) and other FMO parameters, which are important in assessing the reactivity of a molecule (Deng 2020). The probe system's constant electrostatic potential surface map is known as molecular electrostatic potential (MEP) (Priya 2021). MEP surface mapped with electrostatic potential surface and atomic charges (Mulliken) are derived by using an optimized structure.
Molecular dynamics
MD simulation is also performed for best protein–ligand complex monitoring physical movements of atoms in the compound. For this job, we used Linux-based platform GROMACS 5.1 package with GROMOS43A2 force fields. We have used MD simulations for calculating thermodynamical parameters, root mean square deviation (RMSD), root mean square fluctuation (RMSF), intermolecular hydrogen bonds, potential energy (Epot) and binding energy of the receptor–inhibitor complex. Parameters obtained by the MD approach explain the potentiality and stability of the considered drug-like molecule. To maintain the neutrality of ligand TIP3P water model has been used with a buffer distance of 10 Å and volume as 893,000 Å3and 4Na + ions are added to maintain the neutrality of receptor–inhibitor complex structure in a cubic box. Time-varying (1 ps-100000 ps) steepest descent algorithm with 50,000 steps was used to do energy minimization of the complex. Molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method adaptive Poisson–Boltzmann solver (APBS) and GROMACS packages are used for calculating interaction free energies (ΔGbind). MD simulation of the complex was accomplished after collecting snapshots at every 100 ps between 0 and 100 ns and ΔGbind was calculated by the below-given equations.
| 6 |
| 7 |
| 8 |
| 9 |
where TΔS, ΔGbind,solv, ΔEcovalent, ΔEelectrostatic and ΔEvanderwaals are conformational energy change due to binding, solvation free energy change, molecular mechanical energy changes in gas phase, the covalent energy, electrostatic energy and van der Waals energy changes, respectively, and ΔEMM is the sum of ΔEcovalent, ΔEelectrostatic and ΔEvanderwaals changes. ΔGbind,solv is the sum of polar and nonpolar contributions. Intermolecular hydrogen bonds give us an account of the metabolism and adsorption of the complex. The compactness of the structure of apoprotein and protein–ligand complex is expressed by the radius of gyration (Rg). Binding energy explains the intensity of the bond formation between protein and ligand, and thus, the stability of the complex formed is accounted by RMSD. RMSF shows the heterogeneity of our compound in a complex state. All these parameters explain how our biomolecule is going to behave when bounded with the protein. Hence, MD simulation explains the potentiality of biomolecule as a drug-like compound.
Results and discussions
Virtual screening result analysis
Other than Camphene and Alpha-thujene, Borneol, Myrtenal, Camphor, Eugenol, Carvacrol and Parthenolide fully follows the Lipinski rule, Ghose and Mugge’s rule (SD 2). For all phytochemicals other than Parthenolide, Mugge’s rule is violated with molecular weight less than 200 g/mol and heteroatoms less than two, but Parthenolide follows Mugge’s rule with molecular weight 248.32 g/mol. Ghose’s rule is also violated by Camphene, Alpha-thujene, Borneol, Myrtenal, Camphor and Carvacrol, but followed by Eugenol and Parthenolide. Zero value of TPSA and low GI absorption of Camphene and Alpha-thujene shows unavailability of polar atoms and inappropriate 3D molecular geometry of these compounds can be predicted. Lipophilicity parameters for all phytochemicals falls between -0.4 to 5.6. BBB permeability of all the phytochemicals is high, but GI absorption for Camphene and Alpha-thujene is low. Inhibitor Parthenolide follows most of the drug-likeness rules. Its molar refractivity is 69.34 falling between 40 and 130, is weighted 248.32 g/mol which is less than 500 g/mol and has a total number of atoms 21. (Table 2). There is no presence of any PAINS compounds traced in Parthenolide which shows accuracy in the binding sites. Along with these, it shows the solubility of 4.02 × 10–1 mg/ml which is quite good. Moreover, it does not show any carcinogenetic nature, so we can conclude that it does not support the formation of cancerous elements in the body after consumption. The logP values of the compound shows the permeability of the drug to reach the target tissue inside the body. The logS value shows the extent of the solubility of the drug-like molecule. All these values are termed as the lipophilicity properties of the molecule. These are a kind of filter that improves the quality of the docking and probably increases the docking score, when followed. Thus, the logP values should be around 2 (Nurisso 2012). For Parthenolide, the lipophilicity values are around 2. Thus, Parthenolide molecule has all its drug-like properties validated as a drug-like molecule.
Table 2.
Molecular configuration, drug-likeness properties and ADMET properties of preferred phytochemical Parthenolide (Data collected by ADMETsar)
| Physiochemical properties | Parthenolide | Lipophilicity | Parthenolide | Drug-likeness | Parthenolide |
|---|---|---|---|---|---|
| Molecular weight | 248.32 g/mol | iLOGP | 2.49 | Lipinski | Yes, 0 violation |
| Heavy atoms | 18 | XLOGP3 | 2.34 | Ghose | Yes |
| Rotatable bonds | 0 | WLOGP | 2.76 | Veber | Yes |
| H-Acceptor bonds | 3 | MLOGP | 2.47 | Egan | Yes |
| H-donor bonds | 0 | SILICOS-IT | 3.12 | Mugge’s | Yes |
| Molar refractivity | 69.34 | Consensus | 2.64 | Bioavailability Score | 0.55 |
| TPSA | 38.83 Å2 | Solubility | 4.02 × 10–1 mg/ml | PAINS | 0 alert |
Molecular docking result analysis
Multiple times docking is performed for each one of the ligands with both proteases of COVID-19 to get variation in binding affinity. Docking results for all phytochemicals are mentioned in SD 3. The ligands with large structure are considered to produce many hypothetical interactions with the protein through the binding sites. So, being the largest in the size among the other considered phytochemicals, Parthenolide molecule has the tendency to generate better binding scores. Thus, it can be said that larger the structure, larger will be the interactions between the ligand and the binding sites, and higher will be the binding score of that ligand (Kitchen 2004). Binding affinity of Borneol, Camphene, Myrtenal, Camphor, Alpha-thujene, Eugenol, Carvacrol and Parthenolide for 4OVZ is -5.7 kcal/mol, -5.7 kcal/mol, -5.8 kcal/mol, -6.0 kcal/mol, -6.1 kcal/mol, -6.6 kcal/mol, -6.8 kcal/mol and -8.0 kcal/mol and for 6LU7, binding affinity score is -4.6 kcal/mol, -4.7 kcal/mol, -5.1 kcal/mol, -4.8 kcal/mol, -4.5 kcal/mol, -5.1 kcal/mol, -5.3 kcal/mol and -6.1 kcal/mol, respectively. Among all the considered phytochemicals the docking score for Parthenolide is found the best. The most stable inhibitor–receptor structure for both Mpro and PLpro is chosen based on minimum drieding energy, maximum dipole moment and maximum hydrogen bonds. 6th pose for both Mpro (Residues: LYS102, ASP153) and PLpro (Residues: HIS172, GLN175) is chosen as the most stable complex and the interactions details are mentioned in Table 3. For 6LU7, the best pose shows binding affinity of -6.1 kcal/mol, dreiding energy 186.21, dipole moment 3.190 Debye (SD 4). It has two hydrogen bonds comprising one conventional and one carbon bind at LYS102 and ASP153 amino acid binding site (SD 7). The best pose of receptor 4OVZ shows dipole moment and dreiding energy of 3.189 and 186.43 (SD 5), respectively, same as in case of Mpro. With the binding affinity score of -8.0 kcal/mol, it has four conventional hydrogen bonds. The 2D structure of PLpro in complex form has four hydrogen bonds and is shown in Fig. 3. When docked with Pyrx, docking score does not seem to vary much. For Mpro docking score is -6.3 kcal/mol and that for PLpro is -8.2 kcal/mol, which is very near to the docking scores of respective proteins, in the case of Auto Dock Vina (SD 6(a) and 6(b)). Donor–acceptor interactions obtained by docking of Parthenolide with receptor PLpro (Fig. 4) and with receptor Mpro (SD 8) show the bound state of ligand at the binding site.
Table 3.
Interaction details of the best pose for Parthenolide (as P) targeted with receptor protein 6LU7 and 4OVZ
| Protein | Binding affinity (kcal/mol) |
Hydrogen-bonded interaction (donor: acceptor, distance in A) [Type of bond] |
Total number of Hydrogen bonds | Dipole moment (ligand) (Debye) |
Dreiding energy (protein + ligand) |
Inhibition constant (M) (× 10–8) |
|---|---|---|---|---|---|---|
| Parthenolide | ||||||
| 6LU7 | -6.1 |
(A:LYS102:HZ2-: P:O, 2.04) [Conventional Hydrogen Bond] (P:C-: A:ASP153:OD2, 3.25) [Carbon Hydrogen Bond] |
2 | 3.190 | 186.21 | 3390.5 |
| 4OVZ | -8.0 |
(A:HIS172:HD1-:P:O, 2.37) [Conventional Hydrogen Bond] (A:GLN175:HE21-:P:O,2.50) [Conventional Hydrogen Bond] (A:GLN175:HE22-:P:O, 2.64) [Conventional Hydrogen Bond] (B:GLN175:HE22-:P:O, 2.43 [Conventional Hydrogen Bond] |
4 | 3.189 | 186.43 | 135.83 |
Fig. 3.

2-D structures showing hydrogen bond interactions between Parthenolide and PLpro. Conventional hydrogen bonds are shown by dark green color
Fig. 4.

Donor–acceptor interactions obtained by docking of Parthenolide and receptor 4OVZ of PLpro protease of COVID-19
Among both the receptors, PLpro shows the best binding score, hence receptor PLpro is further taken for MD simulation. Some common medicines like chloroquine, favipiravir, remdesivir, nelfinavir, paritaprevir, raltegravir, praziquantel, interferon, etc. are used for COVID-19 treatment (Kandeel 2020; Patel 2020). But most of these drugs show a binding affinity score less than that of Parthenolide (Table 4).
Table 4.
FDA-approved drugs used for COVID-19 treatment with low binding affinity score than Parthenolide and are used as medicine
Optimized structure
The probe structure was optimized in the ground state using the B3LYP/6-311G (d, p) level of theory. The optimized structure of Parthenolide is shown in Fig. 5. SD8 and SD9 display the different bond lengths (Å) and bond angles (°) of Parthenolide. The single-point energy can be given by using following equation:
| 10 |
where Eelec is electronic energy, ZPE is zero-point energy, Evib is vibrational energy, Erot is rotational energy and Etransl is translational energy. Basically, ZPE is the correction to the electronic energy of the molecule for the effects of molecular vibrations. In the present work, the rigid model was not taken into account, so translational and rotational energy were included in the internal molecule energy. The single-point energy of the optimized structure of Parthenolide is observed as -507,645.464 kcal/mol. The optimized structure of Parthenolide shows a non-planar geometry with C1 point group symmetry. Non-planar geometry of the probe molecule creates a high value of dipole moment of 4.814 Debye. The probe system's bioactivity could be boosted by the high dipole moment value, which is a property of a drug that facilitates the formation of a bond or complex between the drug and the target protein (Sagaama 2020; Rad 2021).
Fig. 5.
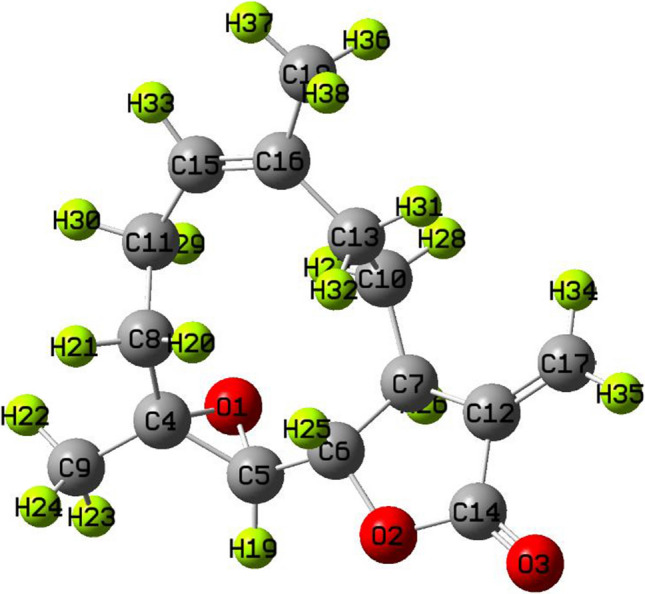
Optimized structure of Parthenolide with atomic symbols
Frontier molecular orbitals analysis
The highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbital (LUMO) of Parthenolide (Fig. 6) helps in calculating other frontier molecular parameters. With the help of the following formulae in the framework of Koopmans' theorem, we can calculate different FMO related molecular parameters (Jasmine 2016). The value of Eg for Parthenolide is 4.88 eV (Table 5). We have found a moderate value of Eg, indicating that Parthenolide is chemically reactive and optically polarizable. Our probe system has a high IP (6.42 eV) and χ (3.98 eV) value, confirming Parthenolide's increased reactivity. The reactivity of organic molecules in a system may be classified based on the parameter (Jasmine 2016). It is a low electrophile if its value is less than 0.8 eV; a moderate electrophile if it is between 0.8 and 1.5 eV; and a heavy electrophile if it is higher than 1.5 eV. The probe system in this case has a value of 3.24 eV, suggesting a significant electrophile presence. Parthenolide is also an electron-withdrawing agent, as evidenced by its chemical potential of -3.721 eV (Dormingo 2002). Chemical hardness, which is associated with reactivity, is a measure of a system's tolerance to changes in electron distribution. In our probe system, the hardness was found to be rather high. All the mentioned FMO parameters suggest the chemical reactiveness of the Parthenolide and its high probability of interacting with the target protein.
Fig. 6.
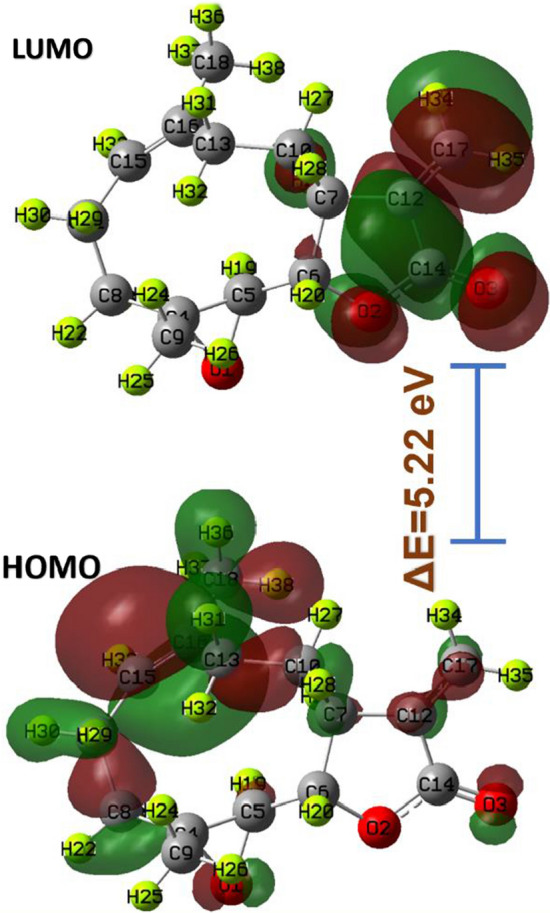
HOMO and LUMO of Parthenolide
Table 5.
Calculated electronic properties of the Parthenolide. All values are in eV and S is in eV−1
| S. No | Molecular Properties | Values (eV) |
|---|---|---|
| 1 | HOMO | -6.42 |
| 2 | LUMO | -1.54 |
| 3 | Energy gap (Eg) | 4.88 |
| 4 | Ionization potential (IP) | 6.42 |
| 5 | Electron affinity (EA) | 1.54 |
| 6 | Electrophilicity Index (ω) | 3.24 |
| 7 | Chemical Potential (μ) | -3.98 |
| 8 | Electronegativity (χ) | 3.98 |
| 9 | Softness (S) | 0.40 |
| 10 | Hardness (η) | 2.44 |
The gross population of HOMO–LUMO for the Parthenolide molecule was also examined (Fig. 6). We find localization in HOMO although their LUMO has a significant degree of delocalization. This may presume that any charge transfer interaction is a result of the localization and delocalization in HOMO and LUMO. According to both FMO related metrics and HOMO–LUMO, it is seen that the Parthenolide probe has a stronger reactivity.
Charge analysis
Mulliken charges of a system are important factors in forecasting a molecule's nucleophilic and electrophilic reactive regions (Sagaama 2020; Sheikhi 2018). Parthenolide has electron donor and acceptor groups, indicating that it can form a complex with the target protein. Figure 7 depicts different Mulliken charges computed for the Parthenolide (SD 10). According to the observed charge analysis, all the hydrogen atoms have positive charges. The H26, H27 and H29 atoms have the most positive charge (0.18034 e, 0.18338 e and 0.18251 e) than other hydrogen atoms (Fig. 7). Carbon atoms in the Parthenolide possess both positive and negative charges. C14 atom has the maximum positive Mulliken charge of 0.49564 e, while the C18 atom have the maximum negative Mulliken charge of -0.47408 e (Fig. 7). Oxygen atoms consist high value of the negative charge. O1, O2 and O3 atoms possess charge accordingly. The charge fluctuation of the probe molecule represents the molecule's capacity to accept/donate electrons, and hence, the system's increased reactiveness. Parthenolide has electron donor and acceptor groups, indicating that it can bind to the target protein and form a complex.
Fig. 7.

Mulliken charges of the Parthenolide molecule
MEP analysis
The MEP surface marks the molecule's electrophilic and nucleophilic reactive sites (Koopman 1933). Various colors in the MEP represent different electrostatic potential locations, such as red color for electron-rich regions, orange color for the partially negative charge, yellow for the moderate electron-rich region and blue for positive charges. The reactive site of the system is shown by the red, orange, yellow and blue colors in MEP. While the MEP surface's green color shows neutral sites, this signifies that a portion of the system will not participate in the chemical reaction. Figure 8 shows the MEP surface of the Parthenolide molecule. The red color on the MEP surface of Parthenolide suggests electron-rich areas above the O3 oxygen atom, indicating the presence of lone pair electrons. A yellow color around the O1 oxygen atom indicates a moderately electron-rich region (Fig. 8). The light blue color of hydrogen atoms such as H20, H31, H26 and H36 indicates electrophilic zones, also known as electron-poor zones. In the other parts of the Parthenolide, the zero potential or neutral zones are depicted (Fig. 8). Electrophile and nucleophile part of Parthenolide indicates that the molecule and the target protein system might have favorable chemical interactions.
Fig. 8.
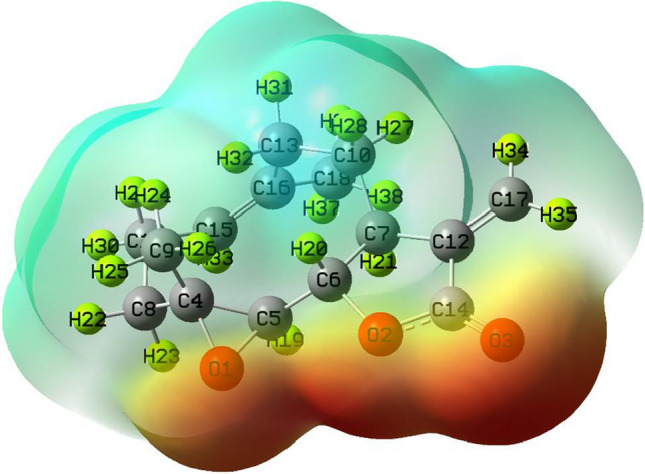
Molecular electrostatic potential map of the molecule Parthenolide
Absorption analysis
The electronic structure and reactivity of materials determine their optical properties. The computed UV–Vis spectrum of the Parthenolide molecule is shown in Fig. 9, and computed absorption excitation energies (E) and oscillator strength (f) are represented in Table 5. The maximum intensity peak for the Parthenolide molecule is observed at 285.60 nm, and f is 0.0047 for the S0 → S1 transition. This wavelength is responsible for the majority of the absorption band's formation. S0 → S2 transition is observed at 280.64 nm, and f is 0.0035 and S0 → S3 transition is found at 232.41 nm wavelength with 0.0024 value of f (Table 6). The π → π* and n → π* correspond to these electronic transitions. As a result of these electronic transformations, the probe mechanism becomes increasingly unstable, confirming its binding ability to the target protein.
Fig. 9.
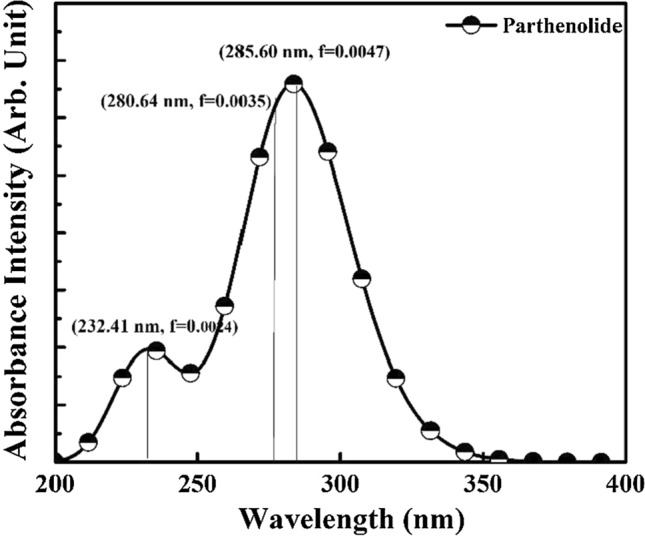
UV–Vis spectra of Parthenolide molecule
Table 6.
UV–Vis absorption data of Parthenolide molecule
| Absorption | |||
|---|---|---|---|
| Theoretical | |||
| Transition | λ (nm) | E (eV) | (f) |
| S0 → S1 | 285.60 | 4.3412 | 0.0047 |
| S0 → S2 | 280.64 | 4.4178 | 0.0035 |
| S0 → S3 | 232.41 | 5.3347 | 0.0024 |
Molecular dynamics result analysis
Following the optimization process, simulation is performed for apoprotein and 4OVZ: Parthenolide complex for a time trajectory of 100 ns to study the stability of the complex. Different parameters like hydrogen bonds, a radius of gyration, RMSD, RSMF and binding free energy of apoprotein and 4OVZ: Parthenolide complex are traced and compared based on average values and range of fluctuations.
Hydrogen bond analysis
Hydrogen bonds are known to contribute to protein–ligand complex formation and explains the stability of the complex. For ligand, in this case, the average value of hydrogen bond interaction is 2. 4OVZ: Parthenolide complex has hydrogen bond interaction ranging between 0 and 3 and the simulation trajectory is shown in Fig. 10 (Table 7). A high number of hydrogen bond interactions show the stability of our complex.
Fig. 10.
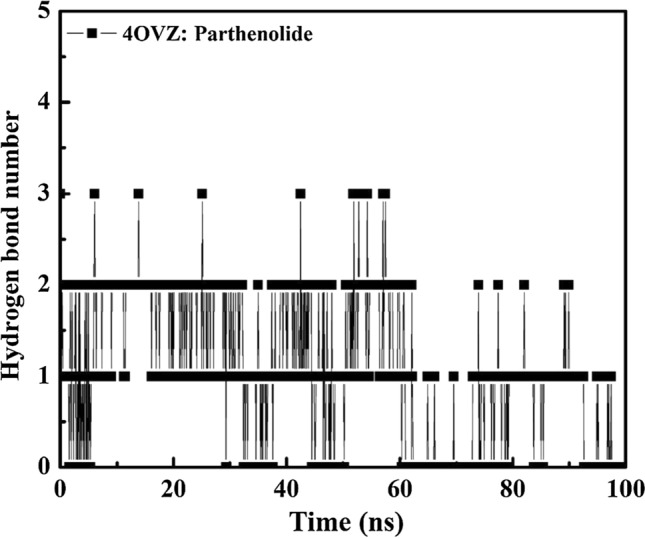
Intermolecular hydrogen bond numbers between apoprotein and protein: ligand complex for time trajectory from 0 to 100 ns
Table 7.
MD simulation output of time resolved trajectory of 4OVZ in its apostate and in complex state with Parthenolide for time trajectory from 0 to 100 ns
| S No | Parameter | Apo-4OVZ | 4OVZ + Parthenolide complex | ||
|---|---|---|---|---|---|
| Mean | Range | Mean | Range | ||
| 1 | Radius of gyration (nm) | 23.81 | 23.58–24.05 | 24.34 | 23.71–24.85 |
| 2 | RMSD (nm) | 3.33 | 1.82–4.29 | 2.89 | 2.5–3.37 |
| 3 | RMSF (nm) | 1.906 | 0.59–3.85 | 2.29 | 0.69–9.44 |
| 4 | Binding energy (Kcal/mol) | NA | NA | –8.23 | -3.85 to -11.07 |
| 5 | Hydrogen bond | NA | NA | 2 | 0–5 |
Radius of gyration (Rg)
Average Rg values for apo-4OVZ and complex for 100 ns of simulation time are 23.8 nm and 24.34 nm (Fig. 11), respectively, which are very near and do not show any observable difference. The lesser the gap between average values of apoprotein and protein–ligand complex, the more it will be compact throughout the trajectory. Rg value of apoprotein ranges from 23.58 to 24.05 nm, complex shows variation between 23.71 and 24.85 nm. Rg values for apo-4OVZ and complex do not show any dissimilarity showing that the complex remains quite compressed and stable during the full-time trajectory of simulation.
Fig. 11.
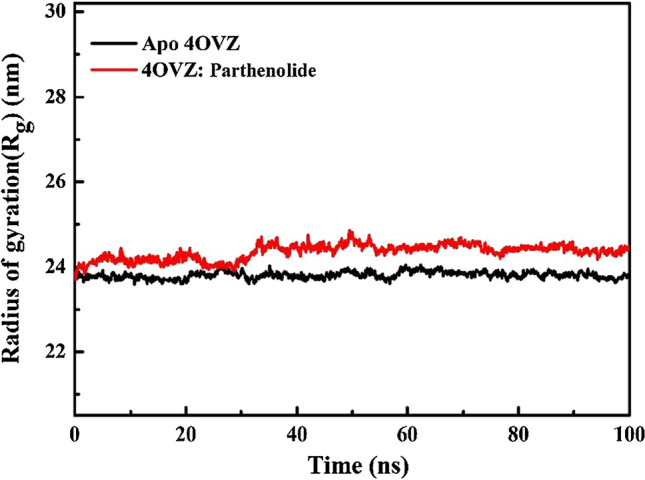
Total radius of gyration for apo-4OVZ and 4OVZ-Parthenolide complex for time trajectory from 0 to 100 ns
Root mean square deviation
RMSD of apoprotein and PLpro complexed with Parthenolide is shown in Fig. 12. Where the apoprotein shows rise in between 40 and 80 ns of the simulation process, the complex on the other hand, shows a linear graph and less rise. The complex shows very low variation in the range of 2.5–3.37 nm. But in the case of complex, the range of fluctuations falls between 1.82 nm and 4.29 nm. Hence, the RMSD value of the complex is less than the average value of RMSD of apoprotein (3.33 nm) is bit higher than that of the complex (2.89 nm). The difference in average RMSD values of protein and complex of 0.44 nm is seen. This imperfection observed is due to position restrains in the structure.
Fig. 12.
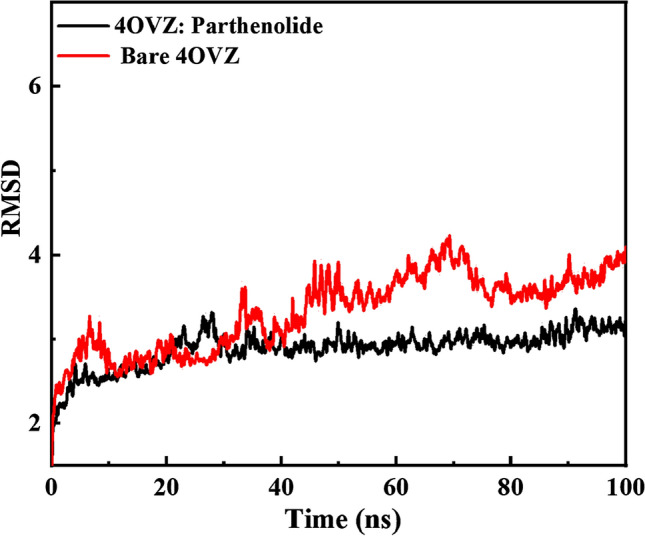
Root mean square deviation graphs of 4OVZ in apostate and its complex with Parthenolide for time trajectory from 0 to 100 ns
Root mean square fluctuations
RMSF justifies the steady state of a molecule. With an average value of 2.29 nm for the entire trajectory, the RMSF shows fluctuations in the range between 0.69 nm and 9.44 nm. 1.90 nm is the average value of RMSF of apoprotein that is less than the protein–ligand complex average value. The trajectory of apoprotein found to be ranging between 0.591 and 3.85 nm. Between 175 and 250 residue numbers, the RMSF value of complex shows major peaks with high stability of the complex (Fig. 13). From all the data obtained by RMSD and RMSF analysis, we can say that the complex formed by 4OVZ and Parthenolide is stable and attains its stability without getting disturbed and dislocated throughout the simulation.
Fig. 13.
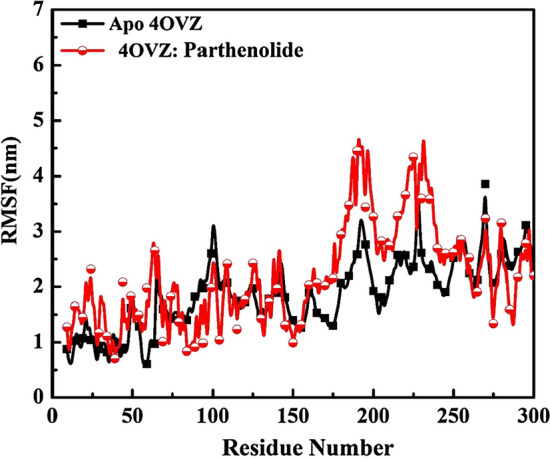
Root mean square fluctuations graphs of 4OVZ in apostate and its complex with Parthenolide for time trajectory from 0 to 100 ns
Binding energy
For 4OVZ: Parthenolide complex, the average binding energy from 0 to 100 ns is calculated to -8.23 kcal/mol. Figure 14 shows the binding energy of the complex and its range between -3.85 and -11.07 kcal/mol. A high value of binding energy for complex validates the strength of bond formation in between protein and ligand. It also helps in interpreting whether the compound has boundness to be used as a potential drug or not.
Fig. 14.
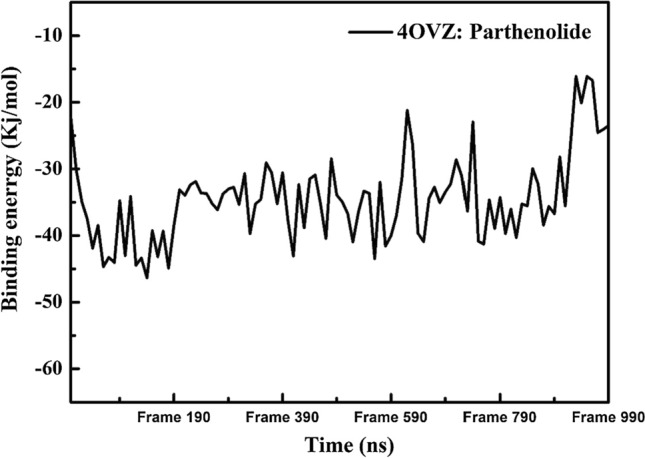
Graphical representation showing binding free energy for protein–ligand complex for time trajectory from 0 to 100 ns
Free energy decomposition
Free energy of individual residues in ligand binding has been determined using free energy decomposition studies. In this study, energy decomposition of individual residues at the binding pocket of PLpro substantiated the significant contribution from His172 and Gln175 -24.43 kcal/mol and -60.34 kcal/mol involved in conventional hydrogen bonding as confirmed in molecular docking and MD simulation (Fig. 15). Although, hydrogen bonding formed between PLpro and the ligand by significant energy contribution from amino acids, other weak interactions such as van der Waal’s and hydrophobic interactions contributed by His74, Thr75 with weak binding energies while Asp77, Gln98, Gln123, Glu135, Glu168 are involved in making salt bridges with strong binding with the ligand molecule.
Fig. 15.

Free energy decomposition of individual residue of PLpro at its binding cavity while interacting with the ligand
Conclusion
The present study we carried is in the search for a new natural drug that could not only inhibit SARS-CoV-2 infection but also boost up immunity of individuals. Docking results for Parthenolide verified that it could bind more efficiently with protease PLpro (with -8.0 kcal/mol binding affinity) as compared to Mpro (-6.1 kcal/mol binding affinity). Due to having antiviral properties and molecular stability, Parthenolide is considered as the most active phytoconstituent. Screening shows the better capability of Parthenolide for being an inhibitor drug against coronavirus as it is non-toxic and non-carcinogenic. Also, it showed anti-inflammatory and immuno-modulatory potential. Optimization of Parthenolide showed the value of dipole moment (7.93 Debye) and explained the bioactivity of the probe molecule. Being hydrogen-rich and carbon-rich, it showed a strong bonding possibility with protein. Parameters computed by optimized structure such as MEP showed the presence of electrophile and nucleophile region which justified the strong possibility of charge transfer in Parthenolide. Its absorption spectra validate its optical behavior and hence stability. MD simulation was performed, due to which we can justify the molecular stability of the protein–ligand complex. Parameters like hydrogen bond interactions and radius of gyration (Rg) showed the stability of the complex throughout simulation time. RMSD and RMSF values justify that simulation has not interrupted the structures of the protein–ligand complex and apo-4OVZ. All the above-given properties show that Parthenolide might get utilized for further innovation and development of a potential drug against coronavirus. With this in-silico study, we can expect to accelerate the creation of a drug candidate targeting PLpro of SARS-CoV-2.
Supplementary Information
Below is the link to the electronic supplementary material.
Authors' contributions
SL: Data curation, Writing- Original draft preparation, Visualization, Investigation, Software, Validation. KD: Conceptualization, Writing- Reviewing and Editing. AG: Data curation, Software, Validation. PC: Conceptualization, Methodology, Supervision. MR: Conceptualization, Methodology, Writing- Reviewing and Editing, Supervision.
Funding
Not applicable.
Declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Akriti, Chandurkar P, Gujar N, Murab T, Choudhary A, Tripathi N (2021) A survey on the effect of indian herbs and spices in boosting immunity against COVID 19 on Bhopal population. Int J Res Mark 2(1):267–275. https://www.ijrpr.com/uploads/V2ISSUE1/IJRPR0154.pdf
- Altay O, Mohammadi E, Lam S, Turkez H, Boren J, Nielsen J, Uhlen M, Mardinoglu A. Current status of COVID-19 therapies and drug repositioning applications. iScience. 2020;23(7):101303. doi: 10.1016/j.isci.2020.101303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiya SS, Olga IY, Andrey VS, Yuriy VG, Yuliya VR, Vladimir VZ, Tatiana ST, Andrey GP, Oleg IK, Nariman FS. Discovery of a new class of antiviral compounds: camphor imine derivatives. Eur J Med Chem. 2015;105(13):263–273. doi: 10.1016/j.ejmech.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Asadi S, Bouvier N, Wexler A, Ristenpart WD. The coronavirus pandemic and aerosols: does COVID-19 transmit via expiratory particles. Aerosol Sci Tech. 2020;54(6):635–638. doi: 10.1080/02786826.2020.1749229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB. Feeling nature’s PAINS: natural products, natural product drugs, and pan assay interference compounds (PAINS) J Nat Prod. 2016;79(3):616–628. doi: 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Baell JB, Nissink JWM. Seven year itch: pan-assay interference compounds (PAINS) in utility and limitations. ACS Chem Bio. 2017;13(1):36–44. doi: 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna A, Thakur P, Singh S, Dev SN, Jain V, Varshney A, Sharma RK (2020) Glucose antimetabolite 2-Deoxy-D-Glucose and its derivative as promising candidates for tackling COVID-19: insights derived from in silico docking and molecular simulations. 10.22541/au.158567174.40895611
- Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. Papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79(24):15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbahani AB, Fooladi AAI. Development of a novel edible coating made by Balangu seed mucilage and Feverfew essential oil and investigation of its effect on the shelf life of beef slices during refrigerated storage through intelligent modelling. J Food Saf. 2018;38(3):e12443. doi: 10.1111/jfs.12443. [DOI] [Google Scholar]
- Benencia F, Courrèges MC (2000) In vitro and in vivo activity of eugenol on human herpesvirus. Wiley online library 14(7):495–500. 10.1002/1099-1573(200011)14:7<495::AID-PTR650>3.0.CO;2-8 [DOI] [PubMed]
- Bernal JL, Andrews N, Gower C, Gallagher G, Simmons R, Thelwall S, Stowe J, Tessier E, Groves N, Dabrera G, Myers R, Campbell CNJ. Effectiveness of Covid-19 vaccines against the B.1.617.2 (Delta) variant. N Engl J Med. 2021;385:585–594. doi: 10.1056/NEJMoa2108891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y. A comprehensive source and free tool for assessment of chemical ADMET properties. ACS Publication Chem Inf Model. 2012;52:3099–3105. doi: 10.1016/j.molstruc.2017.09.108. [DOI] [PubMed] [Google Scholar]
- Chowdhury P. In silico investigation of phytoconstituents from Indian medicinal herb ‘Tinospora cordifolia (giloy)’ against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1803968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry EA, Murry DJ, Yoder C, Fife K, Armstrong V, Nakshatri H, Connell M, Sweeney CJ. Phase I dose escalation trial of feverfew with standardized doses of Parthenolide in patients with cancer. Invest New Drugs. 2004;22:299–305. doi: 10.1023/B:DRUG.0000026256.38560.be. [DOI] [PubMed] [Google Scholar]
- Deng Y, Wang L, Ge Y, Huang L, Gao D, Bi J, Zou G. (NH4)3[B(OH)3]2(COOH)3: a graphite-like UV nonlinear optical material with a large birefringence via structural optimization. Chem Commun. 2020;56:9982–9985. doi: 10.1039/D0CC01881C. [DOI] [PubMed] [Google Scholar]
- Domingo LR, Aurell MJ, Pacrez P, Renato Contreras R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron. 2002;58(22):4417–4423. doi: 10.1016/S0040-4020(02)00410-6. [DOI] [Google Scholar]
- Earle KA, Ambrosino DM, Gartland AF, Goldblatt D, Gilbert PB, Siber GR, Dull P, Plotkin SA. Evidence for antibody as a protective correlate for COVID-19 vaccines. Vaccine. 2021;39(32):4423–4428. doi: 10.1016/j.vaccine.2021.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enioutina EY, Salis ER, Job KM, Gubarev ML, Krepkova LV, Catherine MT. Herbal Medicines: challenges in the modern world. Part 5. status and current directions of complementary and alternative herbal medicine worldwide. Expert Rev Clin Pharmacol. 2017;10(3):327–338. doi: 10.1080/17512433.2017.1268917. [DOI] [PubMed] [Google Scholar]
- Ferrero S, Barra F, Vigliercio GM, Scala C (2018) The efficacy of oral administration of resveratrol in association with feverfew (RevifastDol) for the treatment of primary dysmenorrhea: a retrospective cohort study. Women’s Health Research 2(1):1–13. http://www.starlingscience.com/article/the-efficacy-of-oral-administration-of-resveratrol-in-association-with-feverfew-revifastdol-for-the-treatment-of-primary-dysmenorrhea-a-retrospective-cohort-study.pdf
- Funk CD, Laferrière C, Ardakani A. Target product profile analysis of COVID-19 vaccines in phase III clinical trials and beyond: an early 2021 perspective. Viruses. 2021;13(3):418. doi: 10.3390/v13030418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti N, Maidecchi A, Mattoli L, Burico M, Ghelardini C. St. John's Wort seed and feverfew flower extracts relieve painful diabetic neuropathy in a rat model of diabetes. Fitoterapia. 2014;92:23–33. doi: 10.1016/j.fitote.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Gilani AH, Shah AJ, Ghayur MN, Majeed K. Pharmacological basis for the use of turmeric in gastrointestinal and respiratory disorders. Life Sci. 2005;76(26):3089–3105. doi: 10.1016/j.lfs.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Gilling DH, Kitajima M, Torrey JR, Bright KR. Antiviral efficacy and mechanisms of action of oregano essential oil and its primary component carvacrol against murine norovirus. J Appl Microbiol. 2014;116(5):1149–1163. doi: 10.1111/jam.12453. [DOI] [PubMed] [Google Scholar]
- Guzman ML, Jordan CT. Feverfew: weeding out the root of leukaemia. Expert Opinion on Biological Therapy. 2005;5(9):1147–1152. doi: 10.1517/14712598.5.9.1147. [DOI] [PubMed] [Google Scholar]
- Halberstein RA. Medicinal plants: historical and cross cultural usage patterns. Ann Epidemiol. 2005;15(9):686–699. doi: 10.1016/j.annepidem.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, Penninger J, Krähenbühl S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52:783–792. doi: 10.1007/s40262-013-0072-7. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Weber HK, Schroeder S, Kruger N, Herrler T, Erichen S, Schiergens TS, Herrler WuNH, Nitsche A, Muller MA, Drosten C, Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and Is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Tang, X, Wu, H, Zhang J, Wang W, Wang Z, Song L, Zhai M, Zhao L, Yang H, Ma X, Zhou S, Cai J (2020) Virtual screening and molecular dynamics on blockage of key drug targets as treatment for COVID-19 Caused by SARS-CoV-2, Preprints. https://www.preprints.org/manuscript/202003.0239
- Jasmine GF, Amalanathan M, Roy SDD. Molecular structure and charge transfer contributions to nonlinear optical property of 2-Methyl-4-nitroaniline: A DFT study. J Mol Struct. 2016;1112:63–70. doi: 10.1016/j.molstruc.2016.02.013. [DOI] [Google Scholar]
- Jeyamohan S, Moorthy RK, Kannan MK. Parthenolide induces apoptosis and autophagy through the suppression of PI3K/Akt signaling pathway in cervical cancer. Biotechnol Lett. 2016;38:1251–1260. doi: 10.1007/s10529-016-2102-7. [DOI] [PubMed] [Google Scholar]
- Kandeel M, Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. doi: 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelfaoui H, Harkati D, Saleh BA. Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1803967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen D, Decornez H, Furr J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Koopmans T. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica. 1933;1:104–113. doi: 10.1016/S0031-8914(34)90011-2. [DOI] [Google Scholar]
- Lakhera S, Devlal K, Ghosh A, Rana M. In Silico Investigation of phytoconstituents of medicinal herb piper longum against SARS-CoV-2 by molecular docking and molecular dynamics analysis. Results Chem. 2021 doi: 10.1016/j.rechem.2021.100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Madeddu S, Marongiu A, Sanna G, Zannella C, Falconieri D, Porcedda S, Manzin A, Piras A. Bovine viral diarrhea virus (BVDV): a preliminary study on antiviral properties of some aromatic and medicinal plants. Pathogens. 2021 doi: 10.3390/pathogens10040403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahase E (2020) COVID-19: moderna vaccine is nearly 95% effective, trial involving high risk and elderly people shows. BMJ 371(m4471):71. 10.1136/bmj.m4471
- Maizels M, Blumenfeld A, Burchette R. A combination of riboflavin, magnesium, and feverfew for MiGraine prophylaxis: a randomized trial. J Head Face Pain. 2004;44(9):885–890. doi: 10.1111/j.1526-4610.2004.04170.x. [DOI] [PubMed] [Google Scholar]
- Manzano SE, Cardenas JAG, Manzano AF. Worldwide research trends on medicinal plants. Int J Environ Res Public Health. 2020;17(10):3376. doi: 10.3390/ijerph17103376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary YS, Mary S, Thomas R, Narayana B, Samshuddin S, Sarojini BK, Armaković S, Armaković SJ, Pillai GG. Theoretical studies on the structure and various physico-chemical and biological properties of a terphenyl derivative with immense anti-protozoan activity. Polycycl Aromat Compd. 2021;41:825–840. doi: 10.1080/10406638.2019.1624974. [DOI] [Google Scholar]
- Mengist HM, Fan X, Jin T. Designing of improved drugs for COVID-19: crystal structure of SARS-CoV-2 main protease Mpro. Signal Transduct Target Ther. 2020;5:67. doi: 10.1038/s41392-020-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S (2021) New Generation Vaccine Strategies against COVID-19- Academia Letters. https://www.academia.edu/download/66288433/letter20210417-15616-oazot7.pdf
- Mittal A, Manjunath K, Ranjan RK, Kaushik S, Kumar S, Verma V. COVID-19 pandemic: insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020;16(8):e1008762. doi: 10.1371/journal.ppat.1008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouffouk C, Mouffouk S, Mouffouk S, Hambaba L, Haba H. Flavonols as potential antiviral drugs targeting SARS-CoV-2 proteases (3CLpro and PLpro), spike protein, RNA-dependent RNA polymerase (RdRp) and angiotensin-converting enzyme II receptor (ACE2) Eur J Pharmacol. 2021;891:173759. doi: 10.1016/j.ejphar.2020.173759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, Hou C, Wang H, Liu J, Yang D, Xu Y, Cao Z, Gao Z. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24:422. doi: 10.1186/s13054-020-03120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurisso A, Bravo J, Carrupt PA, Daina A. Molecular docking using the molecular lipophilicity potential as hydrophobic descriptor: impact on GOLD docking performance. J Chem Inf Model. 2012;52(5):1319–1327. doi: 10.1021/ci200515g. [DOI] [PubMed] [Google Scholar]
- Oliver SE, Gargano JW, Marin M, Wallace M, Curran KG, Chamberland M, McClung N, Campos-Outcalt D, Morgan RL, Mbaeyi S, Romero JR, Talbot HK, Lee GM, Bell BP, Dooling K. The advisory committee on immunization practices' interim recommendation for use of Pfizer-BioNTech COVID-19 vaccine-United States MMWR. Morbidity Mortality Weekly Report. 2020;69(50):1922–1924. doi: 10.15585/mmwr.mm6950e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özbilgin S, Akkol EK, Ergene Öz B, Ilhan M, Saltan G, Acıkara ÖB, Tekin M, Keleş H, Süntar I. In vivo activity assessment of some Tanacetum species used as traditional wound healer along with identification of the phytochemical profile by a new validated HPLC method. Iran J Basic Med Sci. 2018;21(2):145–152. doi: 10.22038/IJBMS.2018.24258.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablos I, Eichhorn S, Briza P, Claudia A, Gartner U, Wolf M, Ebner C, Bohle B, Arora N, Vieths S, Ferreira F, Gadermaier G. Proteomic profiling of the weed feverfew, a neglected pollen allergen source. Sci Rep. 2017;7:6049. doi: 10.1038/s41598-017-06213-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyod S, Ho CT, Sheen LH. Dietary therapy and herbal medicine for COVID-19 prevention: a review and perspective. J Tradit Complement Med. 2020;10(4):420–427. doi: 10.1016/j.jtcme.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek A, Suthar M, Rathore GS, Bansal V. Feverfew (Tanacetum parthenium L.): a systematic review. Pharmacogn Rev. 2011;9:103–110. doi: 10.4103/0973-7847.79105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr RG (1980) Functional theory of atoms and molecules, Oxford University Press, New York. 47 (10). 10.1007/978-94-009-9027-2_2
- Patel D, Athar M, Jha PC. Computational investigation of binding of chloroquinone and hydroxychloroquinone against PLPro of SARS-CoV-2. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1844804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourianezhad F, Tahmasebi S, Nikfar S, Mirhoseini M, Abdusi V (2016) Review on feverfew, a valuable medicinal plant. J HerbMed Pharmacol 5(2): 45–49. http://eprints.skums.ac.ir/id/eprint/4684
- Prajapat M, Sarma P, Shekhar N, Avti P, Sinha S, Kaur H, Kumar S, Bhattacharyya A, Kumar H, Bansal S, Medhi B. Drug targets for corona virus: a systematic review. Indian J Pharmacol. 2020;52(1):56–65. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya S, Umadevi T, Gowri S, Vinitha G. Computational molecular structure analysis, electronic properties (HOMO-LUMO, MEP), Hirshfeld surface analysis and third order nonlinear optical profiling of ninhydrin derivative with Z-scan studies. Comput Theor Chem. 2021;1202:113345. doi: 10.1016/j.comptc.2021.113345. [DOI] [Google Scholar]
- Rad AS, Ardjmand M, Esfahani MR, Khodashenas B. DFT calculations towards the geometry optimization, electronic structure, infrared spectroscopy and UV vis analyses of Favipiravir adsorption on the first-row transition metals doped fullerenes; a new strategy for COVID-19 therapy. Spectrochimica Acta A Mol Biomol Spectrosc. 2021;24:119082. doi: 10.1016/j.saa.2020.119082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Y, Liang C, Rong L, Rong S, Peng L. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int J Antimicrob Agents. 2020;56:106012. doi: 10.1016/j.ijantimicag.2020.106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana M, Yadav P, Chowdhury P. A computational study of ivermectin and doxycycline combination drug against SARS-CoV-2 infection. Res Square. 2021 doi: 10.21203/rs.3.rs-755838/v1. [DOI] [Google Scholar]
- Sagaama A, Noureddine O, Brandain SA, Jadryka AJ, Flakus HT, Ghalla H, Issaoui N. Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Comput Biol Chem. 2020;87:107311. doi: 10.1016/j.compbiolchem.2020.107311. [DOI] [PubMed] [Google Scholar]
- Sahoo D, Priyadarshini P, Dandela R, Alagarasan D, Ganesa R, Varadharajaperumal S, Naik R. Optimization of linear and nonlinear optical parameters in As40Se60 film by annealing at different temperature. Optik. 2020;219:165286. doi: 10.1016/j.ijleo.2020.165286. [DOI] [Google Scholar]
- Sapkal GN, Yadav PD, Ella R, Deshpande GR, Sahay RS, Gupta N, Vadrevu KM, Abraham P, Panda S, Bhargava B. Inactivated COVID-19 vaccine BBV152/COVAXIN effectively neutralizes recently emerged B.1.1.7 variant of SARS-CoV-2. J Travel Med. 2021;28(4):051. doi: 10.1093/jtm/taab051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma L. Dietary management to build adaptive immunity against COVID-19. J PeerScientist. 2020;2:e1000016. doi: 10.5281/zenodo.3774086. [DOI] [Google Scholar]
- Sheikhi M, Shahab S, Filippovich L, Yahyaei H, Dikusar E, Khaleghian M. New derivatives of (E, E)-azomethines: Design, quantum chemical modeling, spectroscopic (FT-IR, UV/Vis, polarization) studies, synthesis and their applications: experimental and theoretical investigations. J Mol Struct. 2018;1152:368–385. doi: 10.1016/j.molstruc.2017.09.108. [DOI] [Google Scholar]
- Shree P, Mishra P, Selvaraj C, Singh SK, Chaube R, Garg N, Tripathi YB. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants – Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi)–a molecular docking study. J Biomol Struct Dyn. 2022;40(1):190–203. doi: 10.1080/07391102.2020.1810778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova AS, Yarovaya DI, Baev DS, Shernyukov DS, Shtro AA, Zarubaev VV, Salakhutdinov NF. Aliphatic and alicyclic camphor imines as effective inhibitors of influenza virus H1N1. Eur J Med Chem. 2017;1(27):661–670. doi: 10.1016/j.ejmech.2016.10.035. [DOI] [PubMed] [Google Scholar]
- Sokolova AS, Yarovaya OA, Semenova MD, Shtro AA, Orshanskaya IR, Zarubaev VV, Salakhutdinov NF. Synthesis and in vitro study of novel borneol derivatives as potent inhibitors of the influenza A virus. Med Chem Commun. 2017;8:960–963. doi: 10.1039/C6MD00657D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanović ZPD, Nastovski TL, Ristić MS, Radanović DS. Variability of Essential Oil Composition of Cultivated Feverfew (Tanacetum parthenium (L.) Schultz Bip.) Populations. J Essent Oil Res. 2009;21(4):292–294. doi: 10.1080/10412905.2009.9700174. [DOI] [Google Scholar]
- Sur K, Martin K, Liebel L, Lyte P, Shapiro S, Southall M. Anti-inflammatory activity of parthenolide-depleted feverfew (Tanacetum parthenium) Inflammopharmacol. 2009;17:42–49. doi: 10.1007/s10787-008-8040-9. [DOI] [PubMed] [Google Scholar]
- Tripathi MK, Singh P, Sharma S, Tej PS, Ethayathulla AS, Kaur P. Identification of bioactive molecule from Withania somnifera (Ashwagandha) as SARS-CoV-2 main protease inhibitor. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1790425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukkelen CV. COVID-19: Sputnik vaccine rockets, thanks to Lancet boost. BMJ. 2021;373:n1108. doi: 10.1136/bmj.n1108. [DOI] [PubMed] [Google Scholar]
- Verma A, Adhikary A, Woloschak G, Dwarakanath BS, Rao VLP. A combinatorial approach of a polypharmacological adjuvant 2-deoxy-D-glucose with low dose radiation therapy to quell the cytokine storm in COVID-19 management. Int J Radiat Biol. 2020;96(11):1323–1328. doi: 10.1080/09553002.2020.1818865. [DOI] [PubMed] [Google Scholar]
- Verma SK, Kumar A (2011) Therapeutic uses of Withania somnifera (Ashwagandha) with a note on withanolides and its pharmacological actions. Asian J Pharm Clin Res 4(1). https://www.researchgate.net/profile/Sitansu_Verma3/publication/260419415_Therapeutic_uses_of_Withania_somnifera_Ashwagandha_with_a_note_on_withanolides_and_its_pharmacological_actions/links/0f31753115cd32e430000000.pdf
- Vietzen H, Marianna A, Traugott M, Aberle J, Aberle SW, Puchhammer-Stöckl E. Deletion of the NKG2C receptor encoding KLRC2 gene and HLA-E variants are risk factors for severe COVID-19. Genet Med. 2021;23:963–967. doi: 10.1038/s41436-020-01077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang Y, Huang B, Deng W, Quan Y, Wang W, Xu W, Zhao W, Li N, Zhang J, Liang H, Bao L, Xu Y, Ding L, Zhou W, Gao H, Liu J, Niu P, Zhao L, Zhen W, Fu H, Yu S, Zhang Z, Xu G, Li C, Lou Z, Xu M, Qin C, Wu G, Gao GF, Tan W, Yang X. Development of an inactivated vaccine candidate, BBIBP-CorV, with potent protection against SARS-CoV-2. Cell. 2020;182(3):713–721. doi: 10.1016/j.cell.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wider B, Pittler MH, Ernst E. Feverfew for preventing migraine. Cochrane Database Syst Rev. 2015 doi: 10.1002/14651858.CD002286.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organisation- Coronavirus disease 2019 (COVID-19) Situation Report - 94. https://apps.who.int/iris/handle/10665/331865
- World Health Organization (2021). Interim recommendations for use of the Pfizer–BioNTech COVID-19 vaccine, BNT162b2, under emergency use listing: interim guidance, 8 January 2021. World Health Organization. https://apps.who.int/iris/handle/10665/338484
- Wu Z, Hu Y, Xu M, Chen Z, Yang W, Jiang Z, Li M, Jin H, Cui G, Chen P, Wang L, Zhao Y, Ding Y, Zhao Y, Yin W. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine (CoronaVac) in healthy adults aged 60 years and older: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect Dis. 2021;21(6):803–812. doi: 10.1016/S1473-3099(20)30987-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav P, Rana M, Chowdhury P. DFT and MD simulation investigation of favipiravir as an emerging antiviral option against viral protease (3CLpro) of SARS-CoV-2. J Mol Struct. 2021 doi: 10.1016/j.molstruc.2021.131253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, Wu Y, Zhang L, Yu Z, Fang M, Yu T, Wang Y, Pan S, Zou X, Yuan S, Shang Y. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–481. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.