

Abstract
Background
The bioactivities of commensal duodenal microbiota greatly influence the biofunction of hosts. We investigated the role of Helicobacter pylori infection in extra-gastroduodenal diseases by determining the impact of H. pylori infection on the duodenal microbiota. We sequenced 16 S rRNA genes in samples aspirated from the descending duodenum of 47 (male, 20; female, 27) individuals who were screened for gastric cancer. Samples were analysed using 16 S rRNA gene amplicon sequencing, and the LEFSe and Kyoto Encyclopaedia of Genes and Genomes methods were used to determine whether the duodenal microflora and microbial biofunctions were affected using H. pylori infection.
Results
Thirteen and 34 participants tested positive and negative for H. pylori, respectively. We identified 1,404 bacterial operational taxonomic units from 23 phyla and 253 genera. H. pylori infection changed the relative mean abundance of three phyla (Proteobacteria, Actinobacteria, and TM7) and ten genera (Neisseria, Rothia, TM7-3, Leptotrichia, Lachnospiraceae, Megasphaera, F16, Moryella, Filifactor, and Paludibacter). Microbiota features were significantly influenced in H. pylori-positive participants by 12 taxa mostly classified as Gammaproteobacteria. Microbial functional annotation revealed that H. pylori significantly affected 12 microbial metabolic pathways.
Conclusions
H. pylori disrupted normal bacterial communities in the duodenum and changed the biofunctions of commensal microbiota primarily by upregulating specific metabolic pathways. Such upregulation may be involved in the onset of diseases associated with H. pylori infection.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12866-022-02437-w.
Keywords: Helicobacter pylori, Duodenal microbiota, LEfSe, KEGG, Microbial metabolic pathway
Background
Helicobacter pylori infection can cause chronic gastritis or peptic ulcers and is associated with the development of certain gastric cancers [1]. Recent epidemiological findings suggest that the prevalence of cardiovascular disease, haematological disease, neurodegenerative disease, liver disease, and metabolic syndrome is high in patients with H. pylori infection [2–6]. However, causal relationships between the pathogenesis factors in H. pylori infections and various other extra-gastroduodenal diseases remain obscure [4, 7, 8].
The theory that the bioactivities of commensal gut microbiota greatly influence host biofunction has attracted considerable attention in elucidating the pathophysiology of various diseases. In addition, the duodenum plays a key role in the crosstalk between the gut and the central nervous system, particularly as the release of brain-gut hormones and neurotransmitters in the small intestine, including the duodenum, are regulated by eating stimuli and information arising from the intraluminal environment. These hormones regulate widespread biofunctions, such as metabolism, biosynthesis, feeding behaviour, and gastrointestinal functions [9, 10].
Schulz et al. [11] found that H. pylori infection alters the duodenal microbiota based on evidence from reverse-transcribed 16 S rRNA and that the same results were derived from duodenal biopsies and aspirates. These findings suggest that alteration in the duodenal microbiota induced by H. pylori infection is related to the onset of various extra-gastroduodenal diseases. This is because some degradation products of digestion, attributed to duodenal microbial biofunction, act as chemical effectors on host biofunctions [12, 13].
Therefore, we aimed to elucidate the impact of H. pylori infection on the structure of commensal duodenal microbiota and their biofunctions using conventional analyses of microbial taxonomic diversity and the novel linear discriminant analysis (LDA) effect size (LEfSe) algorithm method to discover metagenomic biomarkers that could explain differences among microbial communities [14]. We also applied metagenomic functional prediction using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database to infer the microbial genetic features associated with biological functions and metabolic pathways [15–17], which facilitate the extraction of specific genetic information related to microbial biofunction from a chaotic metagenomic bin.
Results
Thirteen and 34 samples were H. pylori-positive and negative, respectively. Considering the Kimura–Takemoto classification of endoscopic atrophy, 30 of the 34 participants (88.2%) in the H. pylori-negative group had C0 (non-atrophy)–C2 atrophy, and most of the remaining four participants had a history of H. pylori eradication therapy. In contrast, C0–C2 atrophy was observed in five of the 13 participants (38.5%) in the H. pylori-positive group (Supplementary Information 1). These results indicate that gastric acid secretion decreased in the H. pylori-positive group.
MiSeq sequencing produced 3,312,084 reads with a mean of 70,470 ± 14,833 sequences per sample. These analyses were based on a rarefied table determined from 8,804 gene reads per sample. We identified 1,404 bacterial operational taxonomy units (OTU) from 23 phyla and 253 genera. The numbers of bacterial OTU per participant were 263.86 ± 78.46 and 284.53 ± 67.20 in the H. pylori-negative and -positive groups, respectively. These sequence data are available in the DDBJ Sequence Read Archive under the accession number DRA011815 (DRX275800 to DRX275846).
Association of H. pylori with microbial diversity
Neither α- nor β-diversity significantly differed between the H. pylori-positive and -negative groups (Supplementary Fig. 2).
Influence of H. pylori on bacterial community structures
Figure 1 shows bacterial community structures at the phylum level. The relative mean abundance of Actinobacteria (negative 9.5%: positive 5.1%) and TM7 (negative 3.6%: positive 1.5%) was significantly higher in the H. pylori-negative group than in the H. pylori-positive group. In contrast, the abundance of Proteobacteria (negative 11.3%: positive 23%) was significantly higher in the H. pylori-positive group, and Acidobacteria and Planctomycetes were evident only in the H. pylori-negative group. The relative mean abundance of 14 phyla did not significantly differ between the H. pylori-negative and -positive groups. The results for the other four phyla were invalid for statistical analysis (Supplementary Information 2).
Fig. 1.
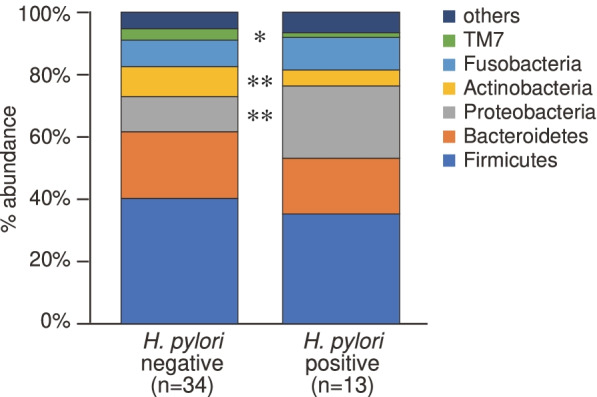
Differences in bacterial community structures at phylum level in Helicobacter pylori-positive and -negative groups. *P < 0.05 and **P < 0.01 (Welch’s t-tests)
The relative mean abundance of ten genera significantly differed between the H. pylori-negative and -positive groups (Table 1). Only the relative abundance of Neisseria was significantly higher in the H. pylori-positive group. In addition, the Mann–Whitney U test was used to compare the H. pylori-positive and H. pylori-negative groups of Neisseria. The median was 0.025 in the negative group, 0.140 in the positive group, and the P-value was 0.002. The relative abundance of the other nine genera (Rothia, [unknown order] TM7-3, Leptotrichia, [unknown genus] Lachnospiraceae, Megasphaera, [unknown genus] F16, Moryella, Filifactor, and Paludibacter) was significantly higher in the H. pylori-negative group. Furthermore, 188 and 143 genera were detected in the H. pylori-negative and -positive groups, respectively. These differences were attributed to the different microbial community structures of each group. Specifically, 60 and 15 genera were found only in the H. pylori-negative and -positive groups, respectively. Helicobacter was detected only in the H. pylori-positive group, with an abundance of 2.79 ± 6.82% (Supplementary Information 3).
Table 1.
Difference in the relative mean abundance of taxa at genus level in Helicobacter pylori-positive and -negative groups
| Genus | Relative mean abundance (%) ± SD | ||
|---|---|---|---|
| H. pylori | H. pylori | Pa | |
| negative (n = 34) | positive (n = 13) | ||
| Neisseria | 4.76 ± 5.93 | 11.74 ± 7.10 | <0.01 |
| Rothia | 6.83 ± 7.44 | 1.81 ± 1.36 | <0.001 |
| {Unknown Order} TM7-3 | 2.74 ± 3.54 | 0.80 ± 0.89 | <0.01 |
| Leptotrichia | 2.06 ± 1.84 | 1.18 ± 1.02 | <0.05 |
| {Unknown Genus} Lachnospiraceae | 0.90 ± 0.84 | 0.33 ± 0.30 | <0.01 |
| Megasphaera | 0.72 ± 0.76 | 0.38 ± 0.33 | <0.05 |
| {Unknown Genus} F16 | 0.49 ± 0.60 | 0.21 ± 0.23 | <0.05 |
| Moryella | 0.32 ± 0.39 | 0.11 ± 0.13 | <0.05 |
| Filifactor | 0.28 ± 0.45 | 0.09 ± 0.16 | <0.05 |
| Paludibacter | 0.04 ± 0.06 | 0.01 ± 0.02 | <0.05 |
Ten taxa with significant differences among 253 detected are shown. Some taxa that could not be identified at genus level were classified at higher levels. Notations in parentheses indicate classification level. aCalculated using Welch’s t-test
Influence of H. pylori on biologically relevant features
The LDA score derived from LEfSe analyses indicated that 12 taxa significantly influenced the biological features of the duodenal microbiota in the H. pylori-positive group (Fig. 2 A). These 12 taxa comprised three phyla (Streptophyta, Cyanobacteria, and TG5), one class (Gammaproteobacteria), one order (Pasteurellales), one family (Enterobacteriaceae), three genera (Pseudomonas, Moraxella, and Actinobacillus), and three species (Streptococcus porcinus, Haemophilus segnis, and Paenibacillus durum) of bacteria. Figure 2B shows the information on biological classification. Six of these taxa belonged to the class Gammaproteobacteria (phylum Proteobacteria): Pasteurellales, Pseudomonas, Moraxella, Actinobacillus, H. segnis, and Enterobacteriaceae. Moraxella and H. segnis are notable intraoral bacteria. Two taxa, S. porcinus and P. durum, belonged to the class Bacilli (belonging to Firmicutes). These results suggest Gammaproteobacteria can affect duodenal microbial features.
Fig. 2.

LEfSe analysis of differences in biologically relevant features between Helicobacter pylori-positive and -negative groups. Green and red: H. pylori-positive and -negative, respectively. (A) Rank of effect size of each taxa. (B) Taxonomic cladogram considering hierarchy and systematic closeness porcinus: Streptococcus porcinus, segnis: Haemophilus segnis, durum: Paenibacillus durum
Influence of H. pylori on duodenal microbial biofunctions
Among 327 investigated KEGG pathways (Supplementary Information 4), 163 were metabolic. The ko-abundance of 12 of these metabolic pathways significantly differed in the presence or absence of H. pylori infection (Table 2), and the ko-abundance of nine of these 12 was significantly greater in the H. pylori-positive group (synthesis and degradation of ketone bodies, tryptophan metabolism, ether lipid metabolism, linoleic acid metabolism, alpha-linolenic acid metabolism, biotin metabolism, carotenoid biosynthesis, phenylpropanoid biosynthesis, biosynthesis of terpenoids and steroids). This suggests that H. pylori generally upregulates metabolic functional activities in duodenal microbes. The remaining pathways comprised those associated with genetic information processing (n = 16), environmental information processing (n = 21), cellular processes (n = 19), organismal systems (n = 48), and human diseases (n = 60). Among these 164 pathways, 18 significantly differed between the individuals with and without H. pylori. However, it was impossible to infer whether these pathways were functioning for microbial biofunctions and affecting biofunctions of the host.
Table 2.
Differences in KEGG metabolic pathways between Helicobacter pylori-positive and -negative groups
| Class | KEGG pathway | Median ko-abundance | |||
|---|---|---|---|---|---|
| (ko number) | Pathway | H. pylori | H. pylori | ||
| negative (n = 34) | positive (n = 13) | Pb | |||
| M | ko00072† | Synthesis and degradation of ketone bodies | 1335885.8 | 1823878.4 | <0.05 |
| M | ko00380† | Tryptophan metabolism | 2254680.9 | 2759405.6 | <0.05 |
| M | ko00510 | N-glycan biosynthesis | 907554.4 | 700748.0 | <0.05 |
| M | ko00565a | Ether lipid metabolism | 150966.1 | 394815.2 | <0.001 |
| M | ko00571 | Lipoarabinomannan (LAM) biosynthesis | 397200.0 | 191311.5 | <0.05 |
| M | ko00591a | Linoleic acid metabolism | 85148.3 | 389478.0 | <0.001 |
| M | ko00592a | alpha-Linolenic acid metabolism | 132544.9 | 405656.4 | <0.001 |
| M | ko00780a | Biotin metabolism | 15103400 | 19564468 | <0.05 |
| M | ko00906a | Carotenoid biosynthesis | 129310.3 | 510048.5 | <0.01 |
| M | ko00940† | Phenylpropanoid biosynthesis | 348411.2 | 598460.1 | <0.01 |
| M | ko01053 | Biosynthesis of siderophore group nonribosomal peptides | 2025414.2 | 1127312.8 | <0.01 |
| M | ko01062a | Biosynthesis of terpenoids and steroids | 92469.4 | 428251.7 | <0.01 |
Twelve metabolic pathways that significantly differed among 327 detected pathways. M, metabolism. aMost abundant pathways in H. pylori-positive group. bCalculated using Mann–Whitney U tests
Discussion
Although a causal relationship has long been suspected between H. pylori infection and cardiovascular disease, haematologic disease, and metabolic syndrome, the roles of commensal microbiota in these diseases have remained obscure [3]. This study found that H. pylori infection significantly influenced the relative abundance of three phyla and ten genera in the duodenal microbiota and that the altered duodenal microbiota was characterised by increased Neisseria abundance and an enhanced impact of Gammaproteobacteria. The abundance of multiple commensal microbial metabolic pathways was also significantly altered, suggesting that H. pylori altered aspects of microbial metabolites that may affect host biofunctions. Many studies have investigated the gut microbiota before and after therapy for various diseases [6, 18]. Although many comparative studies on the gut microbiota have associated differences in the gut microbiota with certain diseases, the results remain inadequate, especially for factors originating in different microbiota that are substantial etiological effectors [5].
Although a study comparing biopsied gastric tissue with and without H. pylori reported differences in diversity [11], the present α- and β-diversity analyses in this limited sample size revealed no significant differences between H. pylori-positive and -negative groups (Supplementary Fig. 2). Although the results of the diversity analyses indicate no differences, this does not necessarily indicate that an identical abundance or representation of bacterial species exists in each group. In fact, the duodenal bacterial community structures differed at the phylum level between the two groups, with a greater abundance of Proteobacteria and a lower abundance of Actinobacteria and TM7 (Saccharibacteria) in the H. pylori-positive group. However, since the phylum to which H. pylori belongs has recently been changed from Proteobacteria to the newly established Epsilonbacteraeota [19], the results for the relative abundance of the bacterial community structure at the phylum level may change. The bacterial community structure at the genus level and LEfSe results suggested that H. pylori infection altered the microbial features by increasing the abundance of Neisseria and enhancing the impact of Gammaproteobacteria in the duodenum (Table 1; Fig. 2). These results appear to be inconsistent, as the abundance of Neisseria was significantly higher in the positive group in the genus-level community structure analysis, but not significantly different in the phylum and genus-level LEfSe analysis. This inconsistency likely originates from the different methods used and the purpose of each analysis. The results for the bacterial community structure were derived by relating the relative abundance of the number of reads for each taxon (or OTUs) to the number of reads for the entire sequence. These results do not indicate the actual number of bacteria because the method aligns the DNA concentrations between samples to equal concentrations. LEfSe is designed to increase the detection power compared to community structure analysis, which compares simple ratios [14]. Therefore, the two methods yield different results.
The increase in Neisseria in the duodenum is probably related to the gastric acid output owing to atrophic gastritis induced by H. pylori. Intraoral indigenous bacteria in the genus Neisseria are not generally highly pathogenic, except for Neisseria gonorrhoeae and Neisseria meningitidis, which cause gonorrhoea and meningitis, respectively [20]. However, excessive Neisseria proliferation in the duodenum may be pathogenic through changes in microbial community structure [21, 22]. The subclass Gammaproteobacteria comprises several medically important bacteria, such as
Enterobacteriaceae, Vibrionaceae, and Pseudomonadaceae
Many studies on the relationships between H. pylori infections and extra-gastric diseases have identified increased short-chain fatty acid (SCFA) production induced by the proliferation of Bacteroidetes [23]. These SCFAs induce the release of gut hormones such as peptide YY and glucose like peptide-1, activation of host metabolic pathways, mucosal immune response, and inflammation [18, 23, 24]. This study revealed that H. pylori did not significantly change the abundance of Bacteroidetes in the duodenum (Fig. 1). The LDA scores also indicated that taxa belonging to Bacteroidetes did not significantly affect duodenal microbial features (Fig. 2). These findings were consistent with previous analyses of duodenal aspirates [10]. Although the hypothesis that increased SCFA production causes various diseases is attractive, SCFAs are generated primarily through the fermentation of nonhost-digestible dietary fibres by the colonic microbiota. Therefore, other factors associated with upper gastrointestinal microbial functions should be considered.
The KEGG pathway analysis showed that 12 bacterial metabolic pathways were affected by the presence or absence of H. pylori infection. Two pathways that were upregulated in the H. pylori-positive group, synthesis and degradation of ketone bodies (ko00072) and ether lipid metabolism (ko00565), are important for degradation of fatty acids, butyrate and acetic acid synthesis, and the production of phosphocholine or seminolipid, which functions in the maintenance of mucosal integrity and immune homeostasis [25, 26].
Notably, the ko-abundance of the tryptophan metabolic pathway (ko00380) was significantly greater in the H. pylori-positive group, suggesting that an abnormal tryptophan supply from the intestine impaired serotonin production. Serotonin is a paracrine messenger expressed primarily in enterochromaffin cells and enteric neurons. This information would help to clarify the causal relationship between H. pylori infection, the duodenal microbiota, and the pathophysiology of functional dyspepsia [27–29]. In addition, serotonin production issues may alter local serotonin concentrations in portal blood, which can also affect the gut-liver axis [30].
The pathways of linoleic (ko00591) and α-linolenic (ko00592) acid metabolism were also upregulated in the H. pylori-positive group (Table 2). Such upregulation may cause an imbalance between ω-3 and ω-6 fatty acids and affect the arachidonic acid cascade associated with inflammation [31]. The biotin metabolic pathway (ko00780) was also upregulated in the H. pylori-positive group. Bacteria synthesise biotin, which is an indispensable essential cofactor for fatty acid biosynthesis. Vitamin A production may also be affected by H. pylori because the biosynthetic pathway of the vitamin A precursor, carotenoid (ko00906), was upregulated in the H. pylori-positive group [32, 33]. The phenylpropanoid biosynthesis pathway (ko00940) was also upregulated significantly in this group. However, the physiological significance of this upregulation in humans is difficult to determine because the roles of metabolites (such as chavicol, eugenol, lignin) originating in this pathway have not been fully elucidated. The terpenoid and steroid biosynthesis pathway (ko01062) was upregulated in the H. pylori-positive group. This could extensively affect host functions because terpenoids are steroid precursors and closely related to cytochrome P450 that functions as an oxidase in terpenoid biosynthesis [34, 35].
Yap et al. [36] found 45 upregulated and 551 downregulated serum metabolites 18 months after H. pylori eradication. The affected metabolites were mapped to various biochemical pathways, including tryptophan metabolism, biosynthesis of unsaturated fatty acids, and linoleic acid metabolism. Although whether these alterations affect host biofunctions remains obscure, our findings confirmed that the metabolomic findings reported by Yap et al. originated from microbial metabolic pathways affected by H. pylori infection. Notably, KEGG analysis is only a prediction due to the presence of DNA. Transcriptome analysis will be needed to determine if these metabolic pathways are indeed upregulated.
This study has several limitations and issues. First, we could not exclude the possibility that some subjects in the H. pylori-negative group might have already experienced significant changes in the structure and biofunctions of the commensal duodenal microbiota due to previous H. pylori infections. In fact, nine patients with a history of infection and eradication were included in the H. pylori-negative group (Supplementary Information 1). To eliminate this concern, it is necessary to analyse changes in microbial features before and after H. pylori eradication therapy in the same subject.
In addition, we could not comprehensively evaluate the effect of gastric acid on the duodenal microbiota because we did not quantify gastric acid secretion. The extent of gastric mucosal atrophy caused by H. pylori infection depends on various factors, such as age, duration of infection, differences between individual immune responses, and the number of bacteria. The gastric acid output depends on the extent of gastric mucosal atrophy, and the extent of atrophic gastritis is closely related to a history of H. pylori infection [37]. In fact, the endoscopic findings in this study indicated that an extended atrophic change was likely to be observed in the H. pylori-positive group. Moreover, the duodenal microbiota might be affected by a decrease in gastric acid output.
Another limitation is that contamination of the gastric microbiota could not be completely ruled out because of the sampling method and due to the lack of bacterial culture. A concern has been raised that aspirate samples include only floating microbiota, which may have little to do with host biofunctions, and that the microbiota originating from biopsy samples (mucosa-associated microbial community structure) inhabit the mucosa [38, 39]. Our findings suggest that microbial metabolite production may fluctuate depending on changes in commensal duodenal microbiota and this phenomenon may affect host biofunctions. These action mechanisms may not depend on areas inhabited by microbes, such as duodenal juice or mucosa, because microbial metabolites act as chemical effectors. This study focused on changes in duodenal bacterial flora caused by the presence of H. pylori rather than direct changes in the duodenal bacterial flora.
Finally, differential abundance (DA) analysis methods for microbiome data are controversial in terms of consistency and reliability. For example, some have pointed out that the false discovery rate could sometimes not be controlled in LEfSe analysis, which was used in this study [40]. The problem originates in the biases due to differences in sampling fractions among collected samples, and it would be difficult to correct the biases adequately. Recently, the Analysis of Compositions of Microbiomes with Bias Correction (ANCOM-BC) has been proposed as a solution to overcome this shortcoming and is considered having a potential as a more reliable DA analysis method for microbiome data [41].
In conclusion, H. pylori infection changed the aspects of the microbiota in the descending part of the duodenum. This dysbiosis, characterised primarily by the upregulation of microbial metabolic pathways, altered commensal microbial biofunctions, which may affect host biofunctions. The gut microbiota can be regarded as an independent organ within the gut lumen, and an investigation of biofunctions originating from this “commensal bacterial organ” would help elucidating the aetiology of various diseases.
Methods
Participants
This study included 20 males and 27 female patients living in Ishigaki Island, Okinawa, Japan (mean age: 58.8 ± 11.3 years), who were screened for gastric cancer. We obtained information from all participants about treatment with gastric acid inhibitors, antibiotics, and medical history of H. pylori eradication (Supplementary Information 1). Patients treated with antibiotics within 4 weeks before sampling were excluded. To establish a valid standard deviation that could provide a 95% confidence interval of the mean value, the sample size of H. pylori-negative participants was over 30. The obtained data were used as normal control data for statistical analysis. We collected duodenal aspiration in order of examinee, and the sample size of H. pylori-positive participants was settled when the sample size of participants in the negative group reached 30 and beyond. The study protocol (Supplementary Fig. 1) was implemented under the approval of the Ethics Committee of the Toho University School of Medicine (authorisation number: A16080), in accordance with current good clinical practice and the Declaration of Helsinki (2013). All participants provided written informed consent to participate before enrolment.
Patient and public involvement
The design of this study proceeded without public involvement. Patient involvement was restricted to sample collection at the time of enrolment. Patients were neither consulted to interpret the results nor invited to contribute to the writing or editing of this article.
Collection of duodenal fluids and esophagogastroduodenoscopy
Duodenal fluids were collected from the descending part of the duodenum using a PW-2 L-1 fluororesin tube (Olympus, Tokyo, Japan) under standard video endoscopy with the Olympus GIF-XQ260 or GIF-XP260N video gastroscope (Olympus, Tokyo, Japan). The tube was sterilised and changed for each patient. Duodenal fluid was aspirated immediately after injecting 5 mL of saline into the descending part of the duodenum, and the aspirate was immediately cryopreserved at −80 °C. One certified endoscopist (HZ) conducted all endoscopic procedures and sampling to avoid bias. The endoscopic findings were recorded simultaneously, and the extent of atrophic gastritis was evaluated in accordance with Kimura–Takemoto classification for endoscopic atrophy [42].
Extraction of genomic DNA
Genomic DNA (gDNA) was extracted from duodenal fluid using PowerFecal DNA Isolation Kit (Mo Bio Laboratories, Inc., Carlsbad, CA, USA) as described in the protocol provided by the manufacturer.
Identification of H. pylori
We applied conventional nested polymerase chain reactions (PCR) of extracted gDNA [43] and then sequenced amplicons to verify the presence of H. pylori. The participants were then assigned to groups based on the presence or absence of H. pylori.
Sequencing
Sequencing libraries were prepared for the Illumina MiSeq platform (Illumina, San Diego, CA, USA). The V3 and V4 regions of the 16 S rRNA gene were targeted using the primer pair 341 F (5′-CCTACGGGNGGCWGCAG-3′) and 806R (5′-GACTACHVGGGTATCTAATCC-3′) [44], and a 16 S rRNA gene library was prepared for sequencing as described in the protocol provided by the manufacturer (https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf). The PCR amplicons were purified using Wizard SV Gel and PCR Clean-Up System (Promega, St. Louis, MO, USA), then sequenced and quantified using the MiSeq system [45].
Bioinformatic and statistical analyses
Sequencing data were processed using CLC Genomic Workbench 10.0.1 and CLC Microbial Genomics Module 2.5 (Qiagen, Hilden, Germany). Overlapping paired-end reads were merged and trimmed, and chimeric reads were filtered using default parameters. The remaining reads were clustered into OTU with 97% identity using the Greengenes database (version 13.5) as the reference [46]. We evaluated bacterial diversity by calculating α- and β-diversity from rarefied OTU tables. The α-diversity was evaluated using richness based on the number of OTU and evenness appraised using the Shannon diversity index [47, 48]. Statistical analyses were performed using EZR, a graphical user interface for R [49]. β-diversity was evaluated based on the OTU Table [50] as the unweighted UniFrac distance can distinguish dissimilarities between microbial profiles of two samples. The β-diversity results were analysed via permutational multivariate analyses of variance (PERMANOVA) using the CLC Genomics Workbench 10.0.1. The relative abundances of phyla and genera in the H. pylori-positive and -negative groups were compared based on unrarefied OTU tables using Welch’s t-tests in Microsoft Excel for Windows 10.
The LEfSe algorithm can identify genomic taxa with a relative abundance that differs between groups. We computed LEfSe using the Galaxy web application and workflow framework (https://huttenhower.sph.harvard.edu/galaxy/) to support high-dimensional class comparisons with a focus on metagenomic analysis.
The biofunctions of the duodenal microbiota were inferred via metagenomic functional annotation. The OTU abundance table was uploaded to the Piphillin server (https://piphillin.secondgenome.com/) with the KEGG Orthology (KO) database as the reference genomic database [51]. Thereafter, KEGG pathways were identified based on the gene information in the OTU, enabling the interpretations of high-level biofunctions of the microbiota.
The KEGG pathway analysis results identified the ko-abundances that corresponded to the abundance of specific KEGG pathways and quantitatively represented microbial biofunction characteristics. Then, pathways classified under metabolism were investigated in detail because the gut microbiota could be regarded as an independent organ that produces various metabolites with metabolic functions in the gut lumen [13]. Each ko-abundance was compared between the H. pylori-positive and -negative groups using Mann–Whitney U tests in Microsoft Excel for Windows 10. All values with P < 0.05 were considered statistically significant.
Supplementary Information
Acknowledgements
The authors are grateful to the staff of the Department of Microbiology and Infectious Diseases, Toho University School of Medicine, and all participants. Thanks to Professor Hidekazu Suzuki, Department of Gastroenterology, Tokai University School of Medicine, for useful advice. This study was supported by JSPS KAKENHI Grant Number JP20K17003 (Grant-in-Aid for Early-Career Scientists), Japan Hospital General Medical Society, and Toho University School of Medicine Research Project Grant Numbers 18–21. We thank Editage (www.editage.com) for English language editing.
Abbreviations
- DA
DA, Differential abundance
- KEGG
Kyoto Encyclopaedia of Genes and Genomes
- KO
KEGG Orthology
- LDA
Linear discriminant analysis
- OTU
Operational taxonomy units
- PCR
Polymerase chain reactions
- PERMANOVA
Permutational multivariate analyses of variance
- SCFA
Short-chain fatty acid
Authors' contributions
TM, HZ, and YF contributed to the study design, analysis, and interpretation of the data. TM, HZ, YF, and EK drafted the manuscript. TM, HZ, YK, TW, and HN collected samples and identified suitable subjects. HZ, NF, HN, KA, YI, KT, and YU supervised the study procedures. TM, HZ, and YF conducted laboratory experiments and analyses. TM, HZ, and YF conducted bioinformatic and statistical analyses. All authors read and approved the final version of the manuscript. TM and HZ contributed equally to this study.
Funding
Japan Society for the Promotion of Science Grant-in-Aid for Early-Career Scientists (KAKENHI), Japan Hospital General Medical Society, and Toho University School of Medicine Research Project.
Availability of data and materials
Data are available in a public, open access repository. Sequence data are available from DDBJ (https://www.ddbj.nig.ac.jp/) under the accession number DRA011815 (DRX275800 to DRX275846).
Declarations
Ethics approval and consent to participate
The study protocol was implemented under the approval of the Ethics Committee of the Toho University School of Medicine (authorisation number: A16080), in accordance with current good clinical practice and the Declaration of Helsinki (2013). All participants provided written informed consent to participate before enrolment.
Competing interests
The authors have no disclosure or other conflicts of interest to report.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Clyne M, Dolan B, Reeves EP. Bacterial factors that mediate colonization of the stomach and virulence of Helicobacter pylori. FEMS Microbiol Lett. 2007;268:135–143. doi: 10.1111/j.1574-6968.2007.00648.x. [DOI] [PubMed] [Google Scholar]
- 2.Gasbarrini A, Fox J, Gasbarrini G. Helicobacter pylori and other Helicobacter spp. chronic infections and extragastric diseases. Eur J Gastroenterol Hepatol. 2000;12:1057–1060. [PubMed] [Google Scholar]
- 3.Ražuka-Ebela D, Giupponi B, Franceschi F. Helicobacter pylori and extragastric diseases. Helicobacter. 2018;23(Suppl 1):e12520. doi: 10.1111/hel.12520. [DOI] [PubMed] [Google Scholar]
- 4.Zendehdel A, Roham M. Biological evidence of the relationship between Helicobacter pylori and associated extragastric diseases. J Cell Biochem. 2019;120:12128–12140. doi: 10.1002/jcb.28681. [DOI] [PubMed] [Google Scholar]
- 5.Peng W, Yi P, Yang J, et al. Association of gut microbiota composition and function with a senescence-accelerated mouse model of Alzheimer’s disease using 16S rRNA gene and metagenomic sequencing analysis. Aging (Albany NY) 2018;10:4054–4065. doi: 10.18632/aging.101693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim MS, Kim Y, Choi H, et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut. 2020;69:283–294. doi: 10.1136/gutjnl-2018-317431. [DOI] [PubMed] [Google Scholar]
- 7.Waskito LA, Salama NR, Yamaoka Y. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2018;23(Suppl 1):e12516. doi: 10.1111/hel.12516. [DOI] [PubMed] [Google Scholar]
- 8.Schulz C, Schütte K, Malfertheiner P. Helicobacter pylori and other gastric microbiota in gastroduodenal pathologies. Dig Dis. 2016;34:210–216. doi: 10.1159/000443353. [DOI] [PubMed] [Google Scholar]
- 9.Jung HK, Talley NJ. Role of the duodenum in the pathogenesis of functional dyspepsia: A paradigm shift. J Neurogastroenterol Motil. 2018;24:345–354. doi: 10.5056/jnm18060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holtmann G, Shah A, Morrison M. Pathophysiology of functional gastrointestinal disorders: A holistic overview. Dig Dis. 2017;35(Suppl 1):5–13. doi: 10.1159/000485409. [DOI] [PubMed] [Google Scholar]
- 11.Schulz C, Schütte K, Koch N, et al. The active bacterial assemblages of the upper GI tract in individuals with and without Helicobacter infection. Gut. 2018;67:216–225. doi: 10.1136/gutjnl-2016-312904. [DOI] [PubMed] [Google Scholar]
- 12.Obata Y, Pachnis V. The effect of microbiota and the immune system on the development and organization of the enteric nervous system. Gastroenterology. 2016;151:836–844. doi: 10.1053/j.gastro.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shanahan F, van Sinderen D, O’Toole PW, Stanton C. Feeding the microbiota: transducer of nutrient signals for the host. Gut. 2017;66:1709–1717. doi: 10.1136/gutjnl-2017-313872. [DOI] [PubMed] [Google Scholar]
- 14.Segata N, Lizard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353-D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):D590-D595. doi: 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tilg H, Moschen AR. Microbiota and diabetes: An evolving relationship. Gut. 2014;63:1513–1521. doi: 10.1136/gutjnl-2014-306928. [DOI] [PubMed] [Google Scholar]
- 19.David WW, Inka V, Christian R. Comparative Genomic Analysis of the Class Epsilonproteobacteria and Proposed Reclassification to Epsilonbacteraeota (phyl. nov.) Front Microbiol. 2017;00682. [DOI] [PMC free article] [PubMed]
- 20.Liu G, Tang CM, Exley RM. Non-pathogenic Neisseria: Members of an abundant, multi-habitat, diverse genus. Microbiology. 2015;161:1297–1312. doi: 10.1099/mic.0.000086. [DOI] [PubMed] [Google Scholar]
- 21.Clemence MEA, Maiden MCJ, Harrison OB. Characterization of capsule genes in non-pathogenic Neisseria species. Microb Genom. 2018;4:e000208. doi: 10.1099/mgen.0.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donati C, Zolfo M, Albanese D, et al. Uncovering oral Neisseria tropism and persistence using metagenomic sequencing. Nat Microbiol. 2016;1:16070. doi: 10.1038/nmicrobiol.2016.70. [DOI] [PubMed] [Google Scholar]
- 23.Castaño-Rodríguez N, Mitchell HM, Kaakoush NO. NAFLD, Helicobacter species and the intestinal microbiome. Best Pract Res Clin Gastroenterol. 2017;31:657–668. doi: 10.1016/j.bpg.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 24.van de Wouw M, Schellekens H, Dinan TG, Cryan JF. Microbiota-gut-brain axis: modulator of host metabolism and appetite. J Nutr. 2017;147:727–745. doi: 10.3945/jn.116.240481. [DOI] [PubMed] [Google Scholar]
- 25.Gonçalves P, Araújo JR, Di Santo JP. A cross-talk between microbiota-derived short-chain fatty acids and the most mucosal immune system regulates intestinal homeostasis and inflammatory bowel disease. Inflam Bowel Dis. 2018;24:558–572. doi: 10.1093/ibd/izx029. [DOI] [PubMed] [Google Scholar]
- 26.Hiippala K, Jouhten H, Ronkainen A, et al. The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients. 2018;10:988. doi: 10.3390/nu10080988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Vadder F, Grasset E, Holm LM, et al. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc Natl Acad Sci U S A. 2018;115:6458–6463. doi: 10.1073/pnas.1720017115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Israelyan N, Del Colle A, Li Z, et al. Effects of serotonin and slow-release 5-hydroxytryptophan on gastrointestinal motility in a mouse model of depression. Gastroenterology. 2019;157:507-521.e4. [DOI] [PMC free article] [PubMed]
- 29.Wauters L, Talley NJ, Walker MM, Tack J, Vanuytsel T. Novel concepts in the pathophysiology and treatment of functional dyspepsia. Gut. 2020;69:591–600. doi: 10.1136/gutjnl-2019-318536. [DOI] [PubMed] [Google Scholar]
- 30.Choi W, Namkung J, Hwang I, et al. Serotonin signals through a gut-liver axis to regulate hepatic steatosis. Nat Commun. 2018;9:4824. Erratum in: 2019;10:158. [DOI] [PMC free article] [PubMed]
- 31.Cunnane SC. Problems with essential fatty acids: time for a new paradigm? Prog Lipid Res. 2003;42:544–568. doi: 10.1016/s0163-7827(03)00038-9. [DOI] [PubMed] [Google Scholar]
- 32.Moran NE, Mohn ES, Hason N, Erdman JW, Jr, Johnson EJ. Intrinsic and extrinsic factors impacting absorption, metabolism, and health effects of dietary carotenoids. Adv Nutr. 2018;9:465–492. doi: 10.1093/advances/nmy025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Said HM. Biotin: biochemical, physiological, and clinical aspects. In: Stanger O, editor. Water Soluble Vitamins. Subcellular Biochemistry. Dordrecht: Springer; 2012. pp. 1–19. [DOI] [PubMed] [Google Scholar]
- 34.Bian G, Deng Z, Liu T. Strategies for terpenoid overproduction and new terpenoid discovery. Curr Opin Biotechnol. 2017;48:234–241. doi: 10.1016/j.copbio.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Loke MF, Chua EG, Gan HM, et al. Metabolomics and 16S rRNA sequencing of human colorectal cancers and adjacent mucosa. PLOS ONE. 2018;13:e0208584. doi: 10.1371/journal.pone.0208584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yap TW, Leow AHR, Azmi AN, et al. Global fecal and plasma metabolic dynamics related to Helicobacter pylori eradication. Front Microbiol. 2017;8:536. doi: 10.3389/fmicb.2017.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mihara M, Haruma K, Kamada T, et al. The role of endoscopic findings for the diagnosis of Helicobacter pylori infection: Evaluation in a country with high prevalence of atrophic gastritis. Helicobacter. 1999;4:40–48. doi: 10.1046/j.1523-5378.1999.09016.x. [DOI] [PubMed] [Google Scholar]
- 38.Nishino K, Nishia A, Inoue R, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol. 2018;53:95–106. doi: 10.1007/s00535-017-1384-4. [DOI] [PubMed] [Google Scholar]
- 39.Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: Causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14:573–584. doi: 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nearing JT, Douglas GM, Hayes M, et al. Microbiome differential abundance methods produce disturbingly different results across 38 datasets. bioRxiv Preprint posted online May 10, 2021. doi: 10.1101/2021.05.10.443486
- 41.Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11:3514. doi: 10.1038/s41467-020-17041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimura K, Takemoto T. An endoscopic recognition of the atrophic border and its significance in chronic gastritis. Endoscopy. 1969;1:87–97. [Google Scholar]
- 43.Rimbara E, Sasatsu M, Graham DY. PCR detection of Helicobacter pylori in clinical samples. In: Wilks M, editor. PCR Detection of Microbial Pathogens. Methods in Molecular Biology (Methods and Protocols) Totowa, NJ: Humana Press; 2013. pp. 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klindworth A, Pruesse E, Schweer T, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang H, Barker SC, Shao R. Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol Evol. 2013;5:1298–1308. doi: 10.1093/gbe/evt094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim BR, Shin J, Guevarra R, et al. Deciphering diversity indices for a better understanding of microbial communities. J Microbiol Biotechnol. 2017;27:2089–2093. doi: 10.4014/jmb.1709.09027. [DOI] [PubMed] [Google Scholar]
- 48.Ringel Y, Maharshak N, Ringel-Kulka T, et al. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes. 2015;6:173–181. doi: 10.1080/19490976.2015.1044711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwai S, Weinmaier T, Schmidt BL, et al. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLOS ONE. 2016;11:e0166104. doi: 10.1371/journal.pone.0166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in a public, open access repository. Sequence data are available from DDBJ (https://www.ddbj.nig.ac.jp/) under the accession number DRA011815 (DRX275800 to DRX275846).