

Abstract
The discovery of a new SARS-CoV-2 virus strain in South Africa presents a major public health threat, therefore contributing to increased infections and transmission rates during the second wave of the global pandemic. This study lays the groundwork for the development of a novel subunit vaccine candidate from the circulating strains of South African SARS-CoV-2 and provides an understanding of the molecular epidemiological trend of the circulating strains. A total of 475 whole-genome nucleotide sequences from South Africa submitted between December 1, 2020 and February 15, 2021 available at the GISAID database were retrieved based on its size, coverage level and hosts. To obtain the distribution of the clades and lineages of South African SARS-CoV-2 circulating strains, the metadata of the sequence retrieved were subjected to an epidemiological analysis. There was a prediction of the cytotoxic T lymphocytes (CTL), Helper T cells (HTL) and B-cell epitopes. Furthermore, there was allergenicity, antigenicity and toxicity predictions on the epitopes. The analysis of the physicochemical properties of the vaccine construct was performed; the secondary structure, tertiary structure and B-cell 3D conformational structure of the vaccine construct were predicted. Also, molecular binding simulations and dynamics simulations were adopted in the prediction of the vaccine construct's stability and binding affinity with TLRs. Result obtained from the metadata analysis indicated lineage B.1.351 to be in higher circulation among various circulating strains of SARS-CoV-2 in South Africa and GH has the highest number of circulating clades. The construct of the novel vaccine was antigenic, non-allergenic and non-toxic. The Instability index (II) score and aliphatic index were estimated as 41.74 and 78.72 respectively. The computed half-life in mammalian reticulocytes was 4.4 h in vitro, for yeast and in E. coli was >20 h and >10 h in vivo respectively. The grand average of hydropathicity (GRAVY) score is estimated to be −0.129, signifying the hydrophilic nature of the protein. The molecular docking indicates that the vaccine construct has a high binding affinity towards the TLRs with TLR 3 having the highest binding energy (−1203.2 kcal/mol) and TLR 9 with the lowest (−1559.5 kcal/mol). These results show that the vaccine construct is promising and should be evaluated using animal model.
Keywords: COVID-19, Computational construction, Control, South Africa, Bioinformatics, Vaccine candidate, SARS-CoV-2 virus
1. Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (also known as COVID -19) is currently a global pandemic affecting different continents of the world. Initially, in 2019, the virus emerged from Wuhan, China [1,2]. In December 2019, the virus was first detected in a pneumonia-infected patient in Wuhan, China. The diagnosis stipulated that it was a newly identified strain of coronavirus (β-coronavirus (nCoV)) [3,4]. Globally, the virus has been extensively transmitted to over 140 countries. United States of America, India, and Brazil, amongst others, are some of the badly affected countries. SARS-Cov-2 virus infection has resulted in many deaths globally. In South Africa, rates of infection of COVID -19 are the highest, leaving thousands of people dead [5]. Situation reports from the World Health Organization (WHO), as at 2:00 a.m., March 30, 2021, revealed that South Africa had more than 1.5 million COVID -19 cases, which represented 50.87% of all 3,039,220 cases within Africa [6,7]. Similarly, records from the South African department of health, as of March 30, 2021, revealed that the total number of COVID-19 cases stood at 1545,979 [7]. In year 2020, the 20H/501Y.V2 or B.1.351 strain of the virus (now called the beta β-variant), was discovered in South Africa. The variant became rapidly virulent, transmissible, and largely predominant [8]. It also contributed to rapid infection and transmission rates during the pandemic's second wave in South Africa [8]. A grave concern is that the Beta (β) strain is not effectively neutralized by convalescent plasma, and the sera of people who have been inoculated with different vaccines in development [[8], [9], [10], [11]]. This fear could create a tough situation in which the COVID-19 strains from South Africa may be too difficult to combat with existing vaccines. [12]. SARS-CoV-2 beta (β), delta (δ) and alpha (α) variants are some of the variants of concern. Our focus in this research article is on the Beta (β) variant because it was one of the pioneer variants that was virulent, very easy to transmit and more contagious, when it emerged. Thus, the continual emergence of other new SARS-CoV-2 virus strains in South Africa is an issue of great concern and risk to public health [12]. Consequently, we ask the following pertinent research questions: how will the new Beta (β) virulent strain of SARS-CoV-2 virus in South Africa be combated? What computational methods can be implemented to design and develop effective, efficient, and suitable vaccine candidates for South Africa, based on the old and the new Beta (β) virus strains? The aim here is to computationally construct a glycoprotein multi-epitope subunit vaccine candidate for existing and SARS-CoV-2 Beta (β) virus strains of South-Africa by focusing on genome data spanning the year 2020 to early year 2021. The motivation to develop this vaccine candidate stems from the fact that, most of the existing vaccines that have been developed are less effective and less potent in specifically addressing this new Beta (β) South African SARS-CoV-2 virus strain. This study provides a viable platform for discovering novel vaccine candidates for this new strain of SARS-CoV-2 virus. This will offer the opportunity to develop a country-specific vaccine for South Africa towards tackling the menace of the SARS-CoV-2 virus pandemic.
Many scientific literatures have adopted the knowledge of computational genomics and immunoinformatic to proffer solutions to different diseases [[85], [86], [87]]. Many pieces of scientific literature have been published on the computational design of glycoprotein multi-epitope subunit vaccine candidates [[13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]], but none has been specifically focused yet on the computational design of vaccine candidate for the old and new variants of the entire South African SARS-CoV-2 virus data strains from the year 2020–2021 [[13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]]. Our preference for adopting a spike-like glycoprotein as a potential choice for vaccine design is due to its capacity to produce vaccine candidates that are potent, efficacious, effective, and characterized by the absence of allergic reactions. Moreover, another justification for the choice of this vaccine candidate design is that in humans, coronavirus infections involve the interactions between spike glycoprotein(S-protein) of CoV and the host cell's receptor's angiotensin-converting enzyme 2 (ACE2). Existing literature shows that the coronavirus S-protein determines the entry of virus into the cells of hosts [[31], [32], [33]]. This research manuscript is structured as follows: Introduction is the subject of section 1. The focus of section 2 is the methodology. Results are the subject of section 3. Discussion is the focus of section 4. Conclusion is the subject of section 5.
2. Methodology
2.1. Sequence retrieval
To generate a data set that indicates various mutant strains of coronavirus circulating in South Africa, data of all the whole-genome nucleotide sequences submitted between December 1, 2020, and February 15, 2021, which can be found in the GISAID Database (https://www.gisaid.org/), were collected. The data were sorted based on the following criteria: (a) availability of complete genomes, (b) coverage level that is high, (c) Low coverage exclusion, and (d) human hosts only (there was no host such as cell and animal culture, or environmental samples present). Four hundred seventy-five whole-genome nucleotide sequences from South Africa were retrieved based on the criteria mentioned earlier, along with their metadata. A reference SARS-CoV-2 genome sequence of a Wuhan isolate, that has an accession number NC_045512.2, and was deposited on the January 17, 2020, was recovered from NCBI (https://www.ncbi.nlm.nih.gov), to utilize genome annotation to refine the alignment between the genomes later. See Supplementary material 1.
2.2. Annotation of retrieved sequences
The retrieved whole-genome nucleotide sequences were annotated using the Wuhan SARS-CoV-2 reference isolate (accession number: NC_045512.2) to select the spike glycoprotein coding region. EMBOSS tool (https://www.ebi.ac.uk/Tools/psa/emboss_needle/), was used to carry out an alignment process of the sequence. EMBOSS transeq (https://www.ebi.ac.uk/Tools/st/emboss_transeq/), was used to carry out the translation of the annotated coding regions to protein sequences. See Supplementary material 1.
2.3. Metadata analysis for clades and lineages
The metadata retrieved from GISAID was subjected to epidemiological analysis using R Studio to obtain the distribution of the clades and lineages for the downloaded sequences.
2.4. Antigenicity prediction, cytotoxic T lymphocytes (CTL), helper T lymphocytes (HTL) and B-cell epitopes prediction
Vaxijen online server software was used to predict the spike glycoprotein's antigenicity. This can be accessed through http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html [34]. The default threshold for viral antigens (0.4) was used as the criterion for the screening. Likewise, the CTL epitopes of the sequences were predicted using NetCTL 1.2 server (https://services.healthtech.dtu.dk/service.php?NetCTL-1.2), [35]. The server predicted the CTL epitopes utilizing the proteasomal C-terminal cleavage, Transport associated with antigen processing (TAP) efficiency, and Major Histocompatibility Class I (MHC-I) affinity parameters. The value for weights on C-terminal cleavage was 0.15, TAP transport efficiency was 0.05, and the threshold value for epitopes was 0.75. Furthermore, Immune Epitope Database (IEDB) was used to predict HTL epitopes. IEDB can be found in the weblink: http://tools.iedb.org/mhcii. These mouse alleles, H2-IAb, H2-IEd, H2-IAd, and the IEDB recommended prediction method that utilizes the best approach for a given MHC molecule, were selected for the epitope prediction. [36]. Epitopes with two lowest percentile ranks for each allele were chosen for further processing.
Also, the server with acronym BCPREDS, is for B-cell Epitope Prediction, with weblink: (http://ailab-projects1.ist.psu.edu:8080/bcpred/index.html)was used for predicting the B-Cell epitopes [37]. The length and specificity of each epitope were set at 20 and 75%, respectively. The first four non-overlapping epitopes with the highest score were selected.
2.5. Selection of overlapping epitopes for CTL, HTL, and B cells
All the predicted epitopes in the previous steps were inspected to determine the epitopes that overlapped in each HTL, CTL, and B cells pool. Epitopes with a frequency of overlap occurring 60 times and above in each pool were selected for further processing.
2.6. Antigenicity and allergenicity prediction of overlapped epitopes
The selected epitopes that occurred 60 times or more in each pool of epitopes were subjected to antigenicity and allergenicity tests for the CTL and HTL epitopes and allergenicity tests for the B-cell epitopes. Antigenicity and allergenicity predictions were performed using Vaxijen (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) and Allertop (https://www.ddg-pharmfac.net/AllerTOP) servers, respectively. The model used by AllerTOP is dependent on the auto cross-covariance (ACC) modification of sequences of proteins into equal-length vectors. The model uses a classification by a k-nearest neighbor algorithm (kNN, k = 1), with a training set consisting of 2427 known allergens and 2427 non-allergens from different species.
2.7. Multi-epitope sequence construction
GPGPG and AAY linkers were used to link antigenic CTL and HTL epitopes and non-allergenic B-cell, CTL, and HTL epitopes.
AAY was used to link the CTL epitopes, while the GPGPG linker was used to link HTL and B cell epitopes.
2.8. Antigenicity, allergenicity and toxicity testing of multi-epitope construct
Epitopes were linked Vaxijen, AllerTOP, and ToxinPred (http://crdd.osdd.net/raghava/toxinpred/) servers were used to predict the toxicity, allergenicity and antigenicity of the multi-epitope vaccine construct. ToxinPred make use of dataset comprising of 1805 toxic peptides from 35 or less residues to test/predict the toxicity of query peptides [38].
2.9. Physicochemical properties of vaccine construct
There are many Physicochemical properties associated with vaccine construct. These include: the composition of the amino acid, the theoretical protrusion index (PI), aliphatic index, atomic composition, coefficient of extinction, weight of the molecule, grand average of hydropathicity, estimated half-life and the instability index. ProtParam (https://web.expasy.org/protparam/), was used to determine these properties (in vitro and in vivo). ProtParam is an Expasy suite that consists of bioinformatics tools [39].
2.10. Prediction of the secondary structure of multi-epitope construct
The secondary structure of the vaccine construct was predicted using the Self-Optimized Prediction Method (SOPMA) (https://nspa-prabi.ibcp.fr/cgi-bin/nspaautomat.pl?page=nspa sopma.html). The conformational states assessed by SOPMA were the helices, sheet, turn and coil of the construct [40].
2.11. Tertiary structure prediction of multi-epitope construct
RaptorX (http://raptorx.uchicago.edu/) was employed for the prediction of the tertiary structure of the vaccine construct. The server predicts functional annotation, disordered regions, solvent accessibility, protein tertiary structures, contact and distance map, and binding sites. These parameters are checked for quality using the global distance test, p-value, un-normalized global distance test, and the modeling error that occurs at each residue.
2.12. Prediction of B-Cell 3D conformational structure
The B-cell 3D conformational structure utilizing the already projected tertiary structure of the vaccine construct was determined by using Ellipro (http://tools.iedb.org/ellipro/). Each predicted epitope was designated with a PI (Protrusion index) value that was averaged over the epitope residue. The discontinuous epitopes on the protein structure were visualized using an open-source molecular viewer Jmol [41].
2.13. Tertiary structure refinement and validation
GalaxyRefine tool of the GalaxyWeb server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) was employed for refinement of the tertiary structure of the vaccine construct obtained from the preceding step. GalaxyRefine performs structure perturbation multiple times, and subsequently, the overall structural relaxation by molecular dynamics simulation [42]. The proSA server was used for the validation of the tertiary structure. The proSA server (https://prosa.services.came.sbg.ac.at/prosa.php) uses the atomic coordinates of the inputted structure to compute an overall quality score and validate the structure of the protein input [43].
2.14. Disulfide engineering
A design web server known as Disulfide (http://cptweb.cpt.wayne.edu/DbD2/), was used to the disulfide engineering of the vaccine construct to confer stability and strength on the protein. The server works by rapidly assessing residue pairs in the input for their proximity and geometry consistence for disulfide formation, on the assumption that all residues were mutated to cysteine. The procedure not only proves useful for increasing the stability of proteins, but also for the investigation of protein dynamics and interactions [44].
2.15. Protein-Protein docking using ClusPro web server
Protein-Protein docking was performed to assist with the predicting the interaction between the Toll-like Receptors (TLRs) of the innate immune system and the vaccine construct. The protein data bank (PDB) files for TLRs 2, 3, 4 and 9 were retrieved from PDB (https://www.wwpdb.org/) and fed into the ClusPro server (https://cluspro.org/login.php?redir=/qeueue.php) together with the vaccine construct, one at a time for each TLR. The procedure enables prediction of both the binding energy between the ligand and receptor, as well as the site of interaction in the formation of receptor-ligand complex [45].
2.16. Stability prediction of receptor-ligand complex
The iMOD server (http://imods.chaconlab.org/) was utilized for the prediction of the stability of interaction between the toll-like receptors and construct. The stability of the complex was interpreted from output parameters from the server, such as deformability, eigenvalues, and covariance [46].
2.17. Immune response simulation
C-ImmSim (http://150.146.2.1/C-IMMSIM/index.php) was used for simulation of humoral and cell-mediated immune response to the vaccine construct. Many of the existing vaccines require a minimum time interval between taking dose 1 and dose 2 to be four (4) weeks (over 28 days) apart [102]. However, some existing vaccines such as Janssen Pharmaceuticals vaccine (Johnson and Johnson) require a single dose [[103], [104], [105], [106], [107]]. Our simulation for this procedure took 1000 time-steps (with a time step of about 8 h), a random seed was set to 12,345, simulation volume and steps to 1000, and all other parameters [47]. The immune simulations we carried out was for a single dose of the vaccine [102], which has produced a good performance.
2.18. Computational cloning of vaccine construct
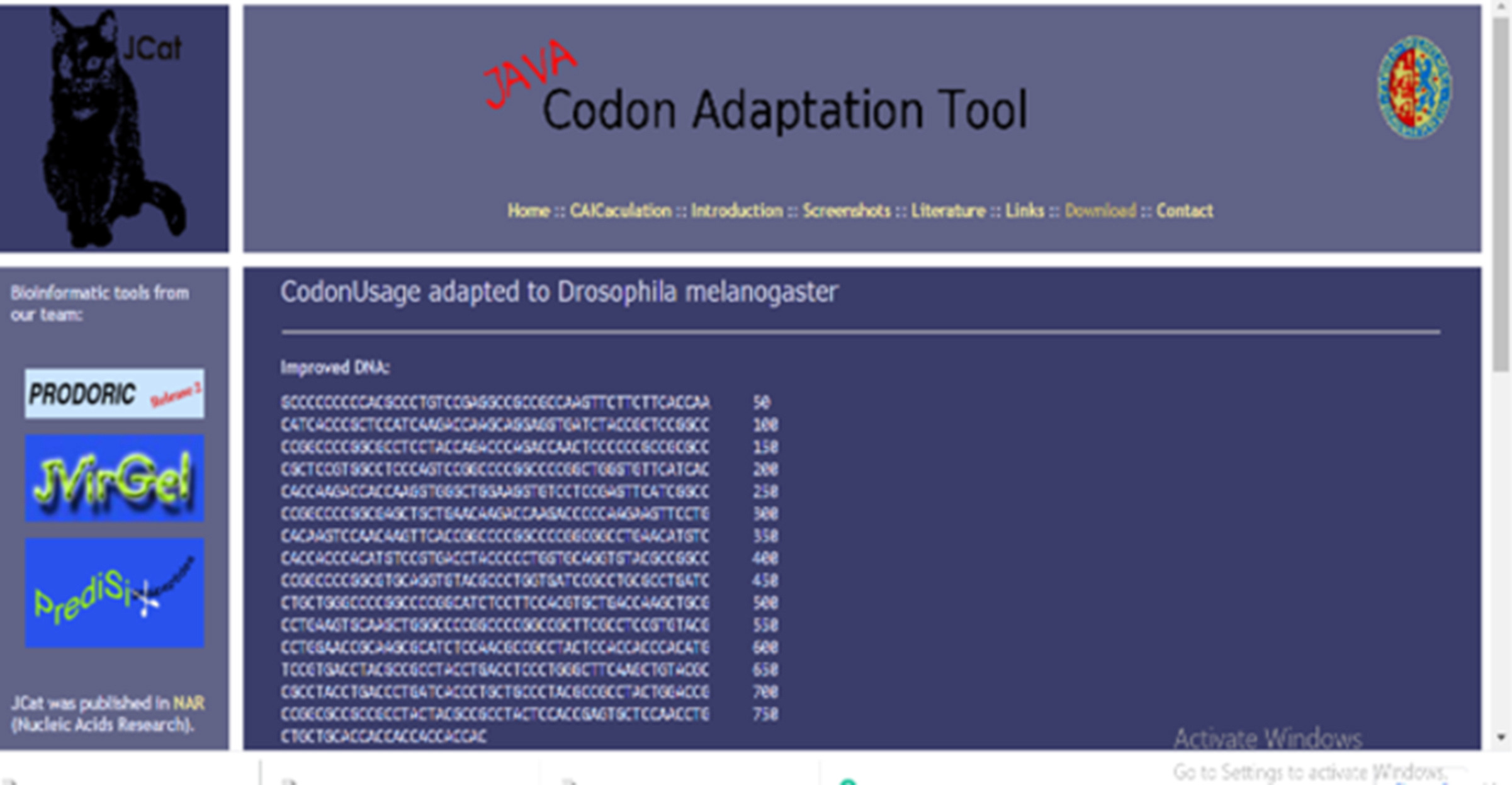
Cloning of the vaccine construct was simulated using SnapGene (https://www.snapgene.com/) after Java Codon Adaptation Tool (JCat, http://www.jcat.de/) was used for adaptation of the vaccine construct to the E.coli and Drosophila melanogaster expression system [48].
3. Results
3.1. Lineage of South African isolates
From Fig. 1 a that there were 28 different circulating lineages of SARS-CoV-2 under which the analyzed nucleotide sequences obtained could be classified. This is to provide us with an understanding of the lineages circulating in South Africa. The lineages are as follows; (Beta) B.1.351 (175), B.1 (33), B.1.381 (31), B.1.237 (30), B.1.1.54 (24), B.1.1.34 (16), B.1.1.53 (11), B.1.1.57 (10), B.1.1.74 (9), B.1.1.75 (8), C.1 (6), H.1 (6). B.1.1.4, B.1.1.40, and B.1.1.62 have four lineages each, and B.1.1.117 has 3. B.1.1.254, B.1.1.317, B.1.106, and B.1.417 all have two lineages while A, B.1.1.1, B.1.1.220, B.1.1.269 and B.1.1.273 has one lineage each.
Fig. 1a.

Result showing the Lineages of South African Isolates. This result shows the lineage distribution of the SARs-CoV-2 South African isolates. This is Fig.1a legend.
It could be deduced that GH has the highest number of circulating clades, with a total of 174 (44.6%), followed by GR with a sum of 114 (29.2%). G has 97 (24.9%), while both GV and S have 4 (1.0%) and 1 (0.3%), respectively, as shown in Fig. 1 b below.
Fig. 1b.

Results showing the Clades of South African Isolates. This result shows the clades of the SARs-CoV-2 South African isolates. This is Fig.1b legend.
3.2. Prediction of cytotoxic T lymphocytes (CTL) epitopes, helper T lymphocytes (HTL, MHC-II) epitopes, and B-cells epitopes
The predicted spike glycoprotein was antigenic, by subjecting to different servers B-cell, HTL and CTL epitopes were predicted. The result of CTL which showed 9mer was displayed with numerous epitopes and which was within the threshold of 0.75. HTL epitopes were predicted, and low percentile was the basis of selection. Furthermore, there was a selection of the B-cell epitopes, and all the epitopes were used for vaccine construction.
3.3. Overlapping epitopes selection, antigenicity and allergenicity prediction for CTL, HTL and B cells epitopes
The result of the epitopes gotten for CTL, HTL and B cells was numerous so overlapping occurred, therefore, epitopes with a frequency of overlap occurring 60 times and above was the criteria used to screen down the epitopes, and these were the epitopes that was selected for further analysis. Also, the antigenicity and allergenicity test done for the selected epitopes of CTL, HTL and B cell reduced the epitopes to 5, 3 and 5 respectively, and these was subjected to further analysis.
3.4. Multi-epitope subunit vaccine construction
Five CTL epitopes, three HTL epitopes, and five B-cell epitopes were chosen to construct the vaccine, and these met the criteria of binding affinity. Linkers were utilized in the epitopes linking process, and to aid the efficacy of the vaccine, an adjuvant was linked to the construct. GPGPG linkers were utilized to join HTL and B-cell epitopes, AAY was used to connect CTL epitopes, and EAAAK was utilized for the attachment of the adjuvant to the whole design. This design is the ultimate vaccine construct with its schematic presentation depicted in Fig. 2 .
Fig. 2.

Vaccine construct. This figure shows the multi-epitope vaccine construction.
3.5. Antigenicity, allergenicity, and toxicity prediction of the vaccine construct
The antigenicity, allergenicity, and toxicity of the vaccine construct were screened, and the construct as reported on VaxiJen 2.0 and ANTIGENpro server was antigenic (which means the vaccine can stimulate an antigenic reaction). AllerTOP and Algpred server also revealed that the construct was non-allergenic. ToxinPred revealed its non-toxicity. Furthermore, it was revealed that there was insignificant similarity between the vaccine and the proteome when BLASTp server analysis was conducted.
3.6. Physicochemical properties results
The computed value for the molecular weight of the vaccine construct was 27998.35Da the theoretical isoelectric point (pI) value of 10.18 was predicted, the molecular formula for the construct was C1287H1986N348O344S5. It also estimated the Instability index (II) score and aliphatic index as 41.74 and 78.72, respectively. The moderate aliphatic index obtained here reveals that the protein has stability in a broad temperature spectrum [83].
The computed half-life in mammalian reticulocytes was 4.4 h in vitro, for yeast and in E. coli was >20 h and >10 h in vivo. The grand average of hydropathicity (GRAVY) score is estimated to be −0.129, signifying that protein is hydrophilic in nature. It is very important for proteins to be hydrophilic to allow them to be easily dispersed and moved around. The negative value of the GRAVY value also indicates that better interaction of the protein is promoted with surrounding molecules of water [82]. This also reveals that the protein has stable and soluble characteristics which are very significant properties for epitope-based vaccine design. All these characteristics makes this vaccine construct a potential success against the old and new South African SARS-CoV-2 virus. See Table 2 for the physicochemical properties results.
Table 2.
Antigenic, allergenic, and physicochemical characteristics of the vaccine construct.
| S/N | Properties | Discovery | Remarks and reference |
|---|---|---|---|
| 1 | Molecular weight | 27,998.35 kDa | Average |
| 2 | Theoretical pI | 10.18 | |
| 3 | Chemical formula | C1287H1986N348O344S5 | |
| 4 | Estimated half-life (mammalian reticulocytes, in vitro) | 4.4 h in vitro | |
| 5 | Estimated half-life (yeast-cells, in vivo) | >20 h in vivo | |
| 6 | Estimated half-life (E. coli, in vivo) | >10 h in vivo | |
| 7 | Instability index of vaccine | 41.74 | |
| 8 | Aliphatic index of vaccine | 78.72 | that the protein has stability in a broad temperature spectrum (Shey et al., 2019); |
| 9 | Grand average of hydropathicity (GRAVY) | −0.129 | This signifies that protein is hydrophilic in nature. It is very important for proteins to be hydrophilic to allow them to be easily dispersed and moved around. The negative value of the GRAVY value also indicates that better interaction of the protein is promoted with surrounding molecules of water (Droppa-Almeida et al., 2018). |
Other previous studies with similar results such as the research conducted by Chukwudozie and colleagues [80] revealed that the molecular weight of their vaccine was 60728.51 Da, while the theoretical pI was 9.30 with an estimated half-life of 30 h. The instability index of their results was 27.84 which showed that the vaccine is stable in a solvent environment, while the aliphatic index is 88.21, with a negative value GRAVY score of −0.056.
In another research [42], insilico analysis results revealed that all the proteins of the SARS-CoV-2, apart from nsp-16 encoded by orf1ab, were predicted as antigen. S protein was chosen as better vaccine candidate protein. The results from the research [42] revealed a negative GRAVY value. The S protein is hydrophilic, with an aliphatic index that is of moderate value, and thus a better potential vaccine candidate.
Some researchers [81], conducted an immunoinformatics and genome-wide analysis on the Indian SARS-CoV-2 genome sequence to extract conserved regions towards designing an epitope-based synthetic vaccine. The results of their research produced MHC-I and MHC-II restricted T-cell and B-cell epitopes which were used in designing the epitope-based synthetic vaccines.
Other researcherss [79], conducted research to identify T and B-cell epitopes and to determine potential vaccine candidate against SARS-CoV-2. The functional amino acids of S protein were also analyzed in their research. The Physicochemical constituents from the analysis of the S protein from their research results revealed that: there were 1273 amino acids(aa); the molecular weight was 141178.47 kDa. Their results produced a 6.24 value for the theoretical isolelectric point (pI) of subject protein. Their result produced a negatively charged protein because the isoelectric point is under 7. Other research works are those of Safavi and colleagues [84,88].
Further works include that of Wang and colleagues [94], that focused on the immunoinformatic analysis of the vaccine design of SARS-CoV-2. Their focus on was on the T-cell and B-cell epitopes. Rahman and colleagues [25], conducted immunoinformatic analysis on vaccine design by focusing on the surface glycoprotein epitopes. Feng and colleagues [95], conducted immunoinformatics analysis towards producing a vaccine design of the SARS-CoV-2 isolated from Wuhan-Hu-1. Mukherjee and colleagues [96], conducted immunoinformatic analysis to identify immunodominant epitopes as potential vaccine targets from the whole genome sequence and proteome of SARS-CoV-2 from different geographical locations via the NCBI database. Tahir Ul Qamar and colleagues [30], adopted insilico approaches to design multi-epitope vaccine against SARS-CoV-2. Oliveira and colleagues [97] conducted insilico analysis on the Nucleocapsid Protein of SARS-CoV-2 to design vaccine targets. The research conducted by Sarkar and colleagues [98], also engaged in designing epitope-based vaccines against SARS-CoV-2. Chakraborty and colleagues [99], adopted immunoinformatic approach to develop peptide-based vaccines against SARS-CoV-2. Fathollahi and colleagues also engaged in immunoinformatics approaches for vaccine design [100].
3.7. The prediction of the secondary structure
The SOPMA server predicted the constructed vaccine secondary structure. Other information provided by this server includes 25.19% alpha (α) helix, 45.74% Random coil, 1.55% Beta (β) turn, and 27.52% Extended strand. All these are represented in Fig. 3 (a and b) below:
Fig. 3.

Secondary structure Prediction of the vaccine construct; (a) indicating the score of alpha-helix, extended strand, random coil, and beta structure and (b) OMPL predictions of the secondary structure depicted by different colors. Blue color represents alpha-helix, and green color represents Beta strands, red color represents extended strand, and yellow color represents random coil.
3.8. Prediction of 3D configuration and conformational B-Cell epitopes]
The prediction of the 3D structure modeling of the vaccine construct was achieved by using RaptorX server (http://raptorx.uchicago.edu/StructPredV2/predict/).
The predicted 3D model showed an estimated RMSD (Å) 8.5787. The predicted 3-class secondary structure revealed that the alpha helix was more predominant, and for the individual residue at position 241, it displayed 87.8% helix, 9.2% coiled, and 3 β as shown in Fig. 4 a. Also, the Ellipro server was used in identifying the conformational B-Cell epitopes for the vaccine construct. Fig. 4 b.
Fig. 4a.

Showing predicted distribution for each residue as arrow goes towards each residue.
Fig. 4b.

The conformational B-cell epitopes of the final vaccine construct, grey sticks represent the bulk of the polyprotein, and a yellow surface represents the conformational B cell epitopes.
3.9. Tertiary structure refinement
For the consistency enhancement and configuration refinement of the vaccine, GalaxyRefine web-server was employed. On the basis of structure quality and several significant factors, which are GDT-HA (0.9593), RMSD (0.384), MolProbity (2.137), Clash score of 13.6, Poor rotamers of 1.4 and Rama favored (94.5), model 3 as shown in Fig. 5 was chosen out of five models displayed.
Fig. 5.

Showing the refined tertiary structure showing predicted distribution for each residue as arrow goes towards each residue.
3.10. Tertiary structure validation
A Ramachandran plot was used to analyze the refined structure, by using the PROCHECK web-server. It was revealed that 88.6% was in favored regions, 10.9% in the allowed regions, and 0.5% in the disallowed regions (See Fig. 6 b in supplementary material). For the vaccine model inputted, a Z-score of −3.79 was predicted by proSA web (as shown in Fig. 6a and b), and 83.871% score was revealed by ERRAT online server. The following previously published scientific research papers adopted this method for validation [23,28,30,93].
Fig. 6.

Showing (a) Z-score graph and (b) Ramachandran plot.
See supplementary file for Fig. 6b Ramachandran plot.
3.11. Disulfide engineering
To improve and aid how stable the structure of the refined vaccine will be, disulfide engineering was conducted by using the Design v2.0 server. Out of 22 pair of residue suitable for disulfide engineering, 4 pair of residues which are Gln 43-Arg 49, Val 64-Gly83, Phe 65-Glu 80, and Asn249-His255, were chosen based on their energy, Chi3 value, and high B-factor as indicated in Fig. 7 .
Fig. 7.

Disulfide engineering showing disulfide adherence by disulfide by design 2.
3.12. Molecular docking
Molecular docking was performed by using ClusPro 2 [92], for this vaccine. ClusPro2 is based on the PIPER docking algorithm. PIPER depicts energy interactions between proteins using an expression or equation of the form: E = w1Erep + w2Eattr + w3Eelec + w4EDARS, where Erep and Eattr, to represent the repulsive and attractive contributions to the van der Waals energy interactions. Where Eelec is the electrostatics energy, and EDARS is a pairwise structure-based potential constructed by the Decoys as the Reference State (DARS) approach. The significance of the equation of this PIPER docking algorithm within ClusPro 2 cannot be underestimated. The coefficients within the equation define the weights of the terms, depicted as w1, w2, w3, and w4. The weights within the equation can be optimally applied for different types of docking problems. Molecular docking was conducted to assess and estimate the interactions between the refined model and the toll-like receptor (TLR). TLR2 (2z82), TLR3 (3CIG), TLR4 (2z64) and TLR9 (3WPG) individually as the receptor for the vaccine, generated different models from which the selected model for each receptor met desired criteria. This process was based on their binding energy weight, as shown in Fig. 8 a, b, 8c and 8d.
Fig. 8.

Showing Molecular docking of the construct with TLR: (a) Docked complexes for vaccine-TLR2 complex with vaccine colored blue and TLR2 colored red.(b) Docked complexes for vaccine-TLR3 complex with vaccine colored blue and TLR3 colored red. (c) Docked complexes for vaccine-TLR4 complex with vaccine colored blue and TLR4 colored red. (d) Docked complexes for vaccine-TLR9 complex with vaccine colored blue and TLR9 colored red.
See Table S1; The interaction and the selected docked model's binding energy weights.
3.13. Molecular dynamic simulation
The simulation study was conducted to determine the stability, mobility as well as atoms and molecules movements in the vaccine construct. The results of normal mode analysis (NMA), the molecular dynamics simulation of the vaccine construct, and TLR2 (2z82), TLR3 (3CIG), TLR4 (2z64) and TLR9 (3WPG) docked complexes are illustrated as obtained from the iMODS server. For the TLR2, TLR3, TLR4 and TLR9, Eigenvalue for the vaccine-receptor complexes were 2.27e-05, 2.06e-06 and 1.53e-05 for TLR2, TLR3, TLR4 and TLR9 respectively Fig. 9 a, b, 9c, and 9d. Our results are similar to the results obtained in other existing studies [28,110], and [99],where an eigenvalue of 2.129046e-05 was predicted for the vaccine-TLRs complex and it implied the stability of the vaccine-TLRs complex (See Fig. 11, Fig. 12, Fig. 13, Fig. 10, Fig. 14, Fig. 15 ).
Fig. 9.

Spin prediction result of the Molecular energy simulation: (a) Vaccine–TLR2 interaction spin prediction. (b) Vaccine–TLR3 interaction spin prediction. (c) Vaccine–TLR4 interaction spin prediction (d) Vaccine– TLR9 interaction spin prediction.
Fig. 11.

Molecular energy simulation deformability B-factor result: (a) deformability B-factor region of the vaccine–TLR2 interaction. (b)Deformability B-factor region of the vaccine–TLR3 interaction. (c) Deformability B-factor region of the vaccine–TLR4 interaction. (d) Deformability B-factor region of the vaccine–TLR9 interaction.
Fig. 12.

Molecular energy simulation variance result: (a) the vaccine–TLR2 interaction's variance. (b) the vaccine–TLR3 interaction's variance. (c) the vaccine–TLR4 interaction's variance. (d) the vaccine–TLR9 interaction's variance.
Fig. 13.

Molecular dynamics simulation elastic network result: (a) the Vaccine–TLR2 interaction's elastic network. (b) the vaccine–TLR3 interaction's elastic network (c) the vaccine–TLR4 interaction's elastic network. (d) the vaccine– TLR9 interaction's elastic network.
Fig. 10.

Molecular energy simulation Eigenvalue result: (a) The Vaccine–TLR2 interaction's Eigenvalue. (b) The Vaccine – TLR3 interaction's Eigenvalue. (c) The Vaccine–TLR4 interaction's Eigenvalue. (d) TheVaccine–TLR9 interaction's Eigenvalue.
Fig. 14.

Molecular dynamics simulation mobility B-factor result: (a) the vaccine–TLR2 interaction's mobility B-factor's result. (b) the vaccine–TLR3 interaction's mobility B-factor's result. (c) the vaccine–TLR4 interaction's mobility B-factor's result. (d) the vaccine–TLR9 interaction's mobility B-factor's result.
Fig. 15.

Molecular energy simulation residue index result: (a) the Vaccine–TLR2 interaction's residue index. (b) the vaccine–TLR3 interaction's residue index (c) the vaccine–TLR4 interaction's residue index (d) the vaccine– TLR9 interaction's residue index.
3.14. Immune simulation
The C-ImmSim server (http://150.146.2.1/C-IMMSIM/) [47] was used to reveal the successful system of immune response and how candidate vaccine half-life increased. Many of the existing vaccines require a sminimum time interval between taking dose 1 and dose 2 to be four (4) weeks (over 28 days) apart [102]. Fig. 16a – 16n shows the results obtained from the presentation of computational immune simulation of the projected vaccine peptide using C-ImmSim server.
Fig. 16.

C-ImmSim presentation of computational immune simulation of the projected vaccine peptide. (a) The production of Immunoglobulin in response to antigen injection; certain subclasses are depicted as colored peaks. (b) B-cell population evolution after the injection. (c) T-helper cells' population after injection. (d) T-helper cells evolution (e) Natural Killer cells' population after injection and depiction of TR (Regulatory) cell population per state (f) Depiction of TH cell population (g) Depiction of TH cell population per state (h) Depiction of TC cell population (cells per mm3) (i) depicts ng/nl versus days plot (j) depicts MA population cells per state (cells per mm3) (k) depicts NK cell population (cells per mm3) (l) depicts PLB cell population (cells per mm3) (m) depicts DC population per state (cells per mm3) (n) depicts EP population per state (cells per mm3).
Interpretation of these results are as follows: Fig. 16a shows the production of immunoglobulin in response to antigen injections; here, certain subclasses are depicted as colored peaks. Fig. 16b shows the B-cell population evolution after the injections. Fig. 16c shows the T-helper cells' population after injections. Fig. 16d shows the helper's cell evolution. Fig. 16e reveals the depiction of TR (Regulatory) cell population per state and the depiction of the Natural Killer cells' population after injections. Fig. 16f depicts of TH cell population. The diagram in Fig. 16g reveals the Depiction of TH cell population per state. Fig. 16h shows a depiction of TC cell population (cells per mm3). Fig. 16i depicts the plot of ng/nl versus days. Fig. 16j shows the representation of MA population cells per state (cells per mm3). The diagram in Fig. 16k shows NK cell population (cells per mm3). Fig. 16l reveals PLB cell population (cells per mm3). Fig. 16m depicts DC population per state (cells per mm3). Fig. 16n depicts EP population per state (cells per mm3). Results obtained in our study are similar in pattern to the results obtained in the following works [101,108,109] in which a similar approach was adopted.
3.15. Codon modification and computational cloning
E.coli was used to express the COVID-19 vaccine candidate by using the JCAT and SnapGene server. The GC content of the improved sequence 53.61% and the Codon Adaptation Index (CAI) of 1.0 were predicted after the adaptation of the vaccine into the Escherichia coli strain k12. Adapting the vaccine into E. coli strain predicted the GC content of 53.61%, Codon Adaptation Index (CAI) of 1.0; and back-translated the protein sequence to a E. coli codon compatible nucleotide. The back-translated nucleotide was adapted into the E.coli expression.
4. Discussion
The elongated duration and consequences of the SARS-CoV2 pandemic beyond the world's imagination have left humans and scientists alike in a state of confusion and despair. Ever since the declaration of a new strain of the coronavirus breed, scientists all over the globe have been tirelessly involved in various research to put an end to the menace. The goal first to curb its spread and to make it extinct. Vaccine has over the years proved to be a means of infectious disease prevention and considering the varying case mortality rate of (0.9%–15.44%) [49], infection rate (estimated 40–81%), and the degree of infection fatality (0.9% or 1%) [50] of the SARS-CoV 2, it was necessary to find a means to curtail its propagation. In South Africa, however, the proportion of cases is 50.87% [51], representing the highest burdened in Africa.
The advent of the computational approach in research has helped improve research turnaround time, which has aided many vaccine developments and the COVID 19 vaccine in circulation. We employed this computational approach to develop a glycoprotein multi-epitope vaccine for all the variants of SARS-CoV 2 in South Africa, particularly the B.1.351(which WHO renamed as Beta variant), which is a variant of concern noted for its virulence (8). As it stands, the world does not know when this pandemic will come to an end, for the earth to be in a state of rest. Although vaccines have been developed, yet some nations have been reported to be experiencing the third wave [52]; hence it is necessary to be prepared as many have predicted that the third wave may cripple African health care [53], with South Africa leading in the total number of the reported cases from the continent. The government is on the race to ensure vaccination among the population in a bid to return the world to normalcy because people have relented in the effort of not getting infected by neglecting the precaution guides.
More than 320 variants and lineages of SARS-CoV-2 have been described globally [54], and the most occurring strain of SARS-CoV 2 from this study is the lineage B.1.351 (beta variant) (Fig. 1a), which is within the GH clades (Fig. 1b) circulating in the South-African isolates recovered from GISAID database. The variation in a lineage occurs when there is a deletion or insertion along the stretch of the nucleotide sequences, which differs from the parent isolate. These changes in the order of the nucleotide allow grouping into class and family, which is also the basis of speciation. Positively stranded RNA viruses such as SARS-CoV-2 are characterized by the constant mutations occurring in their genome, which is attributed to the lack of proofreading capacity of its open reading frame. The resulting variants are referred to as quasi-species. The clade GH and GR are the most frequent clade in this study and are similar to [55], who reported that the GH and GR clade is the most common in the world. Howbeit, the B.1.351(beta) strain was first reported in South Africa and continues to be the greater part in circulation. This is suggestive that some of the SARS-CoV-2 variants may downplay the immune response and negatively contribute to the efficacy of the available vaccines against COVID-19 [56], reported that most population of viral clade belongs to the G, GR, or GH; this is true regarding the most occurring clade in our study. The lowest reported lineage and clade from our isolates were C2 and S, respectively.
The mechanism which informed the selection of the spike glycoprotein for vaccine development stems from its ability to initiate immune response at the point of SARS-CoV-2 internalization [28]. Spike glycoprotein is generally regarded as conserved; however, it is not without mutation. Dominant mutation has been reported at D614G position on the SARS-CoV-2 genome [57] but not without conservation. The epitopes were predicted, and the conserved epitopes were used for the construction of the vaccine. Epitopes are referred to as antigenic determinants encoding the protein region capable of stimulating cell-mediated immunity and are the hallmark of vaccine design [58]. Antigen definite T-cell is cell-mediated, Cell-mediated, and innate immunity is important in the clearance of invasive organisms such as SARS-CoV-2. The cell-mediated immune response (CMI) is most effective in destroying virus-infected cells. CMI activates the natural killer cells, macrophages, cytotoxic T-lymphocytes that secrete cytokines that can digest invading antigen. Also, B-cells produce antibodies against infection after receiving the signal from T-cells; they are effectors of humoral immunity defending against pathogens by the process of antibody production [59,60]. The determinants of adaptive and humoral responses are B-cells. The epitopes outlined in this study encompass the B-cell, HTL, and CTL, which are necessary to initiate defense against invading antigen. The epitopes have proved to be incapable of infection, easily generated, and chemically reliable [28]. All selected epitopes in the vaccine construct that met the scrutiny were used to build up the vaccine. A robust immune response necessitates the use of an adjuvant which hones the antigens visibility to identifiable immune response using a proven RS 09 adjuvant ([74], and attaching an adjuvant to a vaccine necessitates the use of a linker, resulting in the entire vaccine design, and between two epitopes, linkers such as EAAAK, AAY and GPGPG are inserted to achieve the effective separation required for the epitope's effectiveness; these linkers functions differently for rigidity and plasticity in effective vaccine construct [61, [75]. EAAAK linker is a rigid α-helix forming peptide, with intramolecular hydrogen bonding having a closed packed backbone. Rigid linkers are beneficial over flexible linkers by defining the functional sites between the epitopes to avoid meddling of their respective functional properties. The GPGPG linkers is crucial in decoding the junctional immunogenicity which confers expression of epitopes immunogenicity while AAY linker is the binding site for the proteasomes in mammalian cells thereby enhancing the immunogenicity of the multi-epitope vaccine [76]). A multi-epitope vaccine devoid of linkers may yield a new protein with unknown properties downstream [76]. The other vaccine component is the immunoadjuvant, which serves as supplement agents for the target immune response for a vaccine antigen [60,61]. All the elucidated characteristics of a good vaccine to be generally regarded as safe were tested in the epitopes; it was ascertained to be non-toxic, anti-antigen, anti-allergen, and capable of accelerating immunity. In another research, [62], it was asserted that all epitopes generated are not capable of uniform response despite the frequency of occurrence; nonetheless, all the epitopes considered for selection were immune stimulants, and the inclusion of an adjuvant resolves weak immunogens.
Molecule size is a factor to be considered when designing a vaccine, as it determines the route of administration. To direct antibodies to the lymph nodes, bigger molecules are encouraged, but for better diffusion, smaller molecule sizes are encouraged (≤3.5). The molecular weight observed in this study was 27.99 kDa and has proved to be soluble when expressed, which is similar to the submission of [63] the theoretical isoelectric point (pI) value of 10.18 was predicted, implying a basic protein and its similar to the work of Safavi and colleagues [84]. The grand average of hydropathicity (GRAVY) score is estimated to be −0.129, denoting a hydrophilic protein.
The analysis of the results obtained by secondary structure server SOPMA indicated that the protein consists of α-helix, Random coil, β-turn with extended strand which are 25.19%, 45.74%, 1.55%, and 27.52%, respectively, as shown in Fig. 7, this results especially for the random coil is like [64] revealing the copiousness of the random coil region determines the stability of the model. The conformational B-Cell epitopes for the vaccine construct were also identified to be appropriate. A better quality with enhanced structure of vaccine model was produced through refinement and validation. Validation of a candidate vaccine helps in screening for immunoreactivity over serological study; this is one of the principal significances of validation [65].
The refined structure revealed the Ramachandran plot analysis, which indicated that 88.6% of the residue was in favored regions, 10.9% in the allowed regions, and 0.5% in the disallowed regions, as shown in Fig. 6 (See Fig. 6 in the supplementary material). The proSA web predicted the Z-score of −3.79 and 83.871% score and ERRAT online server, respectively, to further establish the quality of the model, and based on the score given by ERRAT; the model was regarded to be suitable. Both proSA and ERRAT server corroborate the quality and the detection of potential errors that could arise in the polished model [66]. Furthermore, the model was used for several studies which include, disulfide prediction for stability when exposed to host system biochemical stress. Stability, mobility, atoms, and molecules movements were determined in the vaccine construct by using a molecular dynamics simulation study. Also, molecular docking was done on ClusPro 2 Fig. 7 to evaluate the binding efficiency and the interaction between the refined model and the toll-like receptor (TLR).
Toll-like receptor (TLRs) are a vital component of the innate immunity which include pattern recognition receptors (PPRs) especially for the identification of pathogen, they are specialized in distinguishing molecular structures that are conserved [67] and plays an essential role in the linkage of innate and adaptive immunity. Among different host cells, expression of TLRs varies, and the expression and location are controlled in response to a specific ligand; the ligands that bind to TLRs are efficient in activating specific intracellular signaling flows that will instigate host defense reactions [67]. In antiviral immunity, various TLRs showed significance. According to [68], out of ten (10), TLRs encoded by human innate immunity, four TLRs (TLR 3, 7, 8, and 9) recognizes RNA viruses. TLR 2 recognizes structural and non-structural viral protein, which stimulates inflammatory cytokine production. Animal model studies has revealed that TLR 3 activation enables a protective effect against SARS-CoV and MERS [65]. Specific modes of viral nucleic acids are responsible for antiviral signaling recognizable by TLR 2, 3, 4, and 9 to identify genetic materials of virus in the endolysosomal pathway.
Molecular docking [[89], [90], [91],111] was performed to evaluate the affinity between the vaccine and TLR2, TLR3, TLR4, and TLR9. The TLRs (Fig. 8) that were docked with the vaccine indicated a high vaccine-TLR binding affinity.
The mechanism for immune simulation presents a pathogen within the organisms' system, which then gives a glimpse of the body's defense against the invading antigen. It is necessary because it enables us to have an idea of the interactions within the immune cells. Non-arguable fact is the complexity of the hosts' reaction to a foreign body [69]. In Fig. 9 above, in a bid to counteract the introduced antigen, some immune components were secreted because of the innate and adaptive immune response [74]. Innate immunity acts as the first line of defense against encroaching organisms. It operates with the aid of pattern recognition receptors (PRR), which stimulates the biochemical flow that promotes cellular immune components' activation [70]. Adaptive immunity, however, is antigen-specific, and its optimum activation is by collaboration with natural immunity. B-cells differentiate into antigen-specific antibodies (immunoglobulins), which are usually activated by the T-helper cells. The memory cells of the B-lymphocytes mark the fingerprint of the antigen against future exposure [54]. The above Fig. 16 b and c show the perceived association result, the memory cells upsurge was steady with time, and it also revealed that the B-cell was active over the duration. This is indicative that a boost dose might not be required. This study proved and satisfied the theory of immune response, and it's similar to the finding of [28]. Several responses were generated in the insilico immune analysis; Fig. 15i revealed a striking response of IFN-g, an important cytokine in the natural and adaptive immunity which activates NK cells, neutrophils, and macrophages [71], also Interleukin-2 (IL-2), which modulates leucocytes activities, it enhances the components of the immune system such as the B-cells, T-cell, and NK-cells. Transforming growth factors and macrophages are convoluted to function properly by the stimuli of IL-2. All the immune secretions in this model are the characteristic feature of an established immunity.
The advent of molecular biology has led to advances in deciphering developmental regulations within the host. The expression of a gene within the cell is pivotal to understanding the effect of therapy. Hence, vectors, which are the conveyance of pathogens or genes between two or more hosts, are important. Biological vectors such as Escherichia coli have been the plasmid vector for most discoveries in genetics. The vaccine protein was expressed in Escherichia coli by aligning the codon sequences into the expression pattern E. coli (See Fig. 17 b) for expression by codon adaptation.
Fig. 17.

(a) Adaptation of codon and computational cloning: a graph depicting sequence adaptation (See Fig. 17a in the Supplementary material), (b) computational cloning for the adapted vaccine sequence into pET28A (+) vector to ensure the expression of the vaccine protein in an E. coli system.
The vaccine proteins were inputted into E. coli k12 strain (JCAT server). Predicted GC content was 53.61% which is an immune stimulator and similar to the output obtained from the research of Oladipo and colleagues [28]) codon Adaptation Index (CAI) 1.0. Restriction enzyme XhoI (158) and XbaI (335) was the selected cloning site. Expression plasmid pET28A are necessary to stimulate T7 promoter driven expression for the massive production of recombinant proteins containing 6X His-tag [77,78]). The GC content of the improved sequences was 53.61% and codon Adaptation Index (CAI) of 1.0 in E. coli expression system. The advantages of using pET28A is that it produces strong expression, and it also produces expressions that are tightly controlled. The efficiency of translation, characterization of gene behavior, and optimization of DNA vaccines are usually measured by its CAI, which is ≤ 1 [72]. The codon adaptation reflects the vaccine efficacy within the host organism, and enhanced GC content might be a good policy in vaccine innovations [73].
5. Conclusion
A glycoprotein multi-epitope subunit vaccine candidate was computationally constructed for the old and new variant of the South African SARs-CoV-2 virus strains from the South African SARs-CoV-2 whole genome sequence data obtained from December 1, 2020 and February 15, 2021, from the GISAID scientific database. Computational genomics and immunoinformatics methods were adopted to analyze and process the data to obtain novel vaccine candidates for the South African 20H/501Y.V2 or B.1.351(Beta) variant. The outcome of this research will assist the relevant agencies (Biologists, Government, pharmaceutical companies) to transform the vaccine candidates into real-life COVID-19 vaccines to effectively manage the new COVID-19 variants for South Africa. This will reduce the rate of transmission of the disease. Hence, COVID-19 transmissions will be drastically reduced, thus protecting the lives of the uninfected and promoting public health. Future work will focus on the collation and the analysis of newer strains of the SARs-CoV-2 virus for South Africa to develop future vaccine candidates.
Author contributions
Conceiving and Conceptualization of research: O.O.O.
Design of the study: O.O.O. and E.K.O.
Methodology: O.O.O., E.K.O., E.O.D., A.E.A., B.A.I., M.P.O., B.H.O., B.M.O., H.M.A., E.M.J., A.J.O.
Data Curation: O.O.O., E.K.O., E.O.D., A.E.A., B.A.I., M.P.O., B.H.O., B.M.O., H.M.A., E.M.J., A.J.O.
Data analysis: O.O.O., E.K.O., E.O.D., A.E.A., B.A.I., M.P.O., B.H.O., B.M.O., H.M.A., E.M.J., A.J.O.
Writing of original draft: O.O.O., and E.K.O.
Writing – critical review, revised version, and re-editing: O.O.O.
Funding acquisition and Principal Investigator: O.O.O.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
All authors read and approved the final manuscript.
Data availability
All relevant data on the accession numbers of sequences used for the experiments in this article are within the manuscript and its Supporting Information files. Sequence Data used for this experiment can be found in GISAID Database (https://www.gisaid.org/).
This project information can be found at: https://github.com/oluwagbemi/COVID-19-Vaccine-Candidate-for-South-Africa---Phase-1.
This project information can also be found on the research project page with weblink: https://olugbengaoluwagbemi.weebly.com/research-projects.html.
All figures (larger and clearer versions) have been included in the supplementary material accompanying this manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The research of this article was supported by DAAD within the framework of the climapAfrica programme with funds of the Federal Ministry of Education and Research. The publisher is fully responsible for the content. The German DAAD personal research grant (with Grant Award/Scholarship Personal Reference Number: ST32/91769426) was awarded to Olugbenga Oluseun Oluwagbemi. This research was also fully funded by the Oppenheimer Memorial Trust (OMT) personal research grant (with Grant Award/Scholarship Reference Number: OMT Ref. 21563/01), awarded to Olugbenga Oluseun Oluwagbemi. Helix Biogen Institute (known as Helix Biogen Consult) provided some technical support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.imu.2022.100845.
Appendix A. Supplementary data
The following is the supplementary data to this article:
{kind=link}
{kind=link}
References
- 1.Thienemann F., Pinto F., Grobbee D.E., Boehm M., Bazargani N., Ge J., et al. World heart federation briefing on prevention: coronavirus disease 2019 (COVID-19) in low-income countries. Glob Heart. 2020;15(1):31. doi: 10.5334/gh.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization Novel coronavirus – China. 2020. https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ [Internet]. Available from:
- 3.Guo Y.R., Cao Q.D., Hong Z.S., Tan Y.Y., Chen S.D., Jin H.J. The origin, transmission, and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak–an update on the status. Mil Med Res. 2020;7:1–10. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian X., Li C., Huang A., Xia S., Lu S., Shi Z. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microb Infect. 2020;9:382–385. doi: 10.1080/22221751.2020.1729069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salgotra R., Gandomi M., Gandomi A.H. Evolutionary modelling of the COVID-19 pandemic in fifteen most affected countries. Chaos, Solit Fractals. 2020;140:1–45. doi: 10.1016/j.chaos.2020.110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization WHO 2021. https://who.maps.arcgis.com/apps/opsdashboard/index.html#/0c9b3a8b68d0437a8cf28581e9c063a9
- 7.dashboard W.C.O. COVID-19 alert number: 0800 029 999; MoH website: 2021. http://www.health.gov.za/
- 8.Tegally H, Wilkinson E, Giovanetti M., et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. Preprint published online December 22, doi:10.1101/2020.12.21.20248640.
- 9.Wibmer CK, Ayres F, Hermanus T, et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. bioRxiv. Preprint published online January 19, doi:10.1101/2021.01.18.427166. [DOI] [PubMed]
- 10.Wu K, Werner AP, Moliva JI, et al. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. bioRxiv. Preprint published online January 25, doi:10.1101/2021.01.25.427948.
- 11.Ho D., Wang P., Liu L., et al. Increased resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 to antibody neutralization. Res Sq. 2021 doi: 10.21203/rs.3.rs-155394/v1. Preprint published online January 29. [DOI] [Google Scholar]
- 12.Mascola J.R., Graham B.S., Fauci A.S. SARS-CoV-2 viral variants—tackling a moving target. JAMA. 2021 doi: 10.1001/jama.2021.2088. Published online February 11. [DOI] [PubMed] [Google Scholar]
- 13.Kar T., Narsaria U., Basak S., et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep. 2020;10:10895. doi: 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhardwaj A. Indian Journal of Pharmaceutical Sciences; 2021. In silico multi subunit vaccine design referring spike glycoprotein of SARS-COV-2 (COVID-19): the world pandemic. [Google Scholar]
- 15.Abraham Peele K., Srihansa T., Krupanidhi S., Sai Ayyagari Vijaya, CVenkateswarulu T. Design of multi-epitope vaccine candidate against SARS-CoV-2: a in-silico study. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1770127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padilla-Sanchez V. SARS-CoV-2 structural analysis of receptor binding domain new variants from United Kingdom and South Africa. Res Ideas and Outcomes. 2021;7 doi: 10.3897/rio.7.e62936. [DOI] [Google Scholar]
- 17.Yazdani Z., Rafiei A., Yazdani M., Valadan R. Design an efficient multi-epitope peptide vaccine candidate against SARS-CoV-2: an in silico analysis. Infect Drug Resist. 2020;13:3007–3022. doi: 10.2147/IDR.S264573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar A., Kumar P., Saumya K.U., Kapuganti S.K., Bhardwaj T., Giri R. Exploring the SARS-CoV-2 structural proteins for multi-epitope vaccine development: an in-silico approach. Expet Rev Vaccine. 2020;19(9):887–898. doi: 10.1080/14760584.2020.1813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abhishek Singh A., Thakur M., Sharma L.K., Chandra K. Designing a multi-epitope peptide-based vaccine against SARS-CoV-2. Sci Rep. 2020;10:16219. doi: 10.1038/s41598-020-73371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin W.R., Cheng F. A rational design of a multi-epitope vaccine against SARS-CoV-2 which accounts for the glycan shield of the spike glycoprotein. J Biomol Struct Dyn. 2021 doi: 10.1080/07391102.2021.1894986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitra D., Pandey J., Jain A., Swaroop S. In silico design of multi-epitope-based peptide vaccine against SARS-CoV-2 using its spike protein. J Biomol Struct Dyn. 2021 doi: 10.1080/07391102.2020.1869092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalita P., Padhi A.K., Zhang K.Y.J., Tripathi T. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb Pathog. 2020;145 doi: 10.1016/j.micpath.2020.104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samad A., Ahammad F., Nain Z., Alam R., Imon R.R., Hasan M., Shahedur M.d., Rahman Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J Biomol Struct Dyn. 2020:1–17. doi: 10.1080/07391102.2020.1792347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharya M., Sharma A.R., Patra P., Ghosh P., Sharma G., Patra B.C., Saha R.P., Lee S.S., Chakraborty C. A SARS-CoV-2 vaccine candidate: in-silico cloning and validation. Inf. Med. Unlocked. 2020;20:1–6. doi: 10.1016/j.imu.2020.100394. 100394; ISSN 2352-9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahman N., Ali F., Basharat Z., Shehroz M., Khan M.K., Jeandet P., Nepovimova E., Kuca K., Khan H. Vaccine design from the ensemble of surface glycoprotein epitopes of SARS-CoV-2: an immunoinformatics approach. Vaccines. 2020;8(3):1–17. doi: 10.3390/vaccines8030423. 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ul Qamar M.T., Shahid F., Aslam S., Ashfaq U.A., Aslam S., Fatima I., Fareed M.M., Zohaib A., Chen L.L. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect Dis Poverty. 2020;9:132. doi: 10.1186/s40249-020-00752-w. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanami S., Zandi M., Pourhossein B., Mobini G.-R., Safaei M., Abed A., Arvejeh P.M., Chermahini F.A., Alizadeh M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int J Biol Macromol. 2020;164:871–883. doi: 10.1016/j.ijbiomac.2020.07.117. ISSN 0141-8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oladipo E.K., Ajayi A.F., Ariyo O.E., Onile S.O., Jimah E.M., Ezediuno L.O., Adebayo O.I., Adebayo E.T., Odeyemi A.N., Oyeleke M.O., Oyewole M.P., Oguntomi A.S., Akindiya O.E., Olamoyegun B.O., Aremu V.O., Arowosaye A.O., Aboderin D.O., Bello H.B., Senbadejo T.Y., Awoyelu E.H., Oladipo A.A., Oladipo B.B., Ajayi L.O., Majolagbe O.N., Oyawoye O.M., Oloke J.K. Exploration of surface glycoprotein to design multi-epitope vaccine for the prevention of Covid-19. Inf. Med. Unlocked. 2020;21 doi: 10.1016/j.imu.2020.100438. 2020, 100438,1-15; ISSN 2352-9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Behmard E., Soleymani B., Najafi A., et al. Immunoinformatic design of a COVID-19 subunit vaccine using entire structural immunogenic epitopes of SARS-CoV-2. Sci Rep. 2020;10:20864. doi: 10.1038/s41598-020-77547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahir ul Qamar M., Rehman A., Tusleem K., Ashfaq U.A., Qasim M., Zhu X., et al. Designing of a next generation multi-epitope based vaccine (MEV) against SARS-COV-2: immunoinformatics and in silico approaches. PLoS One. 2020;15(12) doi: 10.1371/journal.pone.0244176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann M., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong S.K., Li W., Moore M.J., Choe H., Farzan M.A. 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen M.V., Lundegaard C., Lamberth K., Buus S., Lund O., Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vita R., Overton J.A., Greenbaum J.A., Ponomarenko J., Clark J.D., Cantrell J.R., Peters B. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:405–412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Manzalawy Y., Dobbs D., Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recogn. 2008;21(4):243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta S., Kapoor P., Chaudhary K., Gautam A., Kumar R. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9) doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. In: The proteomics protocols handbook. Walker John M., editor. Humana Press; 2005. Protein identification and analysis tools on the ExPASy server; pp. 571–607. [Google Scholar]
- 40.Geourjon C., Deléage G. Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. 1995;11(6):681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- 41.Julia P., Huynh-Hoa B., Li Wei, Nicholas F., Philip E.B., Alessandro S., Bjoern P. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heo L., Park H., Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41(W1):W384–W388. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Craig D.B., Dombkowski A.A. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinf. 2013;14:346. doi: 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desta I.T., Porter K.A., Xia B., Kozakov D., Vajda S. Performance and its limits in rigid body protein-protein docking. Structure. 2020;28(9):1071–1081. doi: 10.1016/j.str.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.López-Blanco J.R., Garzón J.I., Chacón P. iMod. Multipurpose normal mode analysis in internal coordinates. Bioinformatics. 2011;27(20):2843–2850. doi: 10.1093/bioinformatics/btr497. [DOI] [PubMed] [Google Scholar]
- 47.Rapin N., Lund O., Bernaschi M., Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grote A., Hiller K., Scheer M., Munch R., Nortemann B., Hempel D.C., Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33:W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durmaz B., Abdulmajed O., Durmaz R. Mutations observed in the SARS-CoV-2 spike glycoprotein and their effects in the interaction of virus with ACE-2 receptor. Medeniyet med j. 2020;35(3):253–260. doi: 10.5222/MMJ.2020.98048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ioannidis John P.A. World Health Organization. 2021. 99, 19-33F. [DOI]
- 51.Moutal A., Martin L.F., Boinon L., Gomez K., Ran D., Zhou Y., Stratton H.J., Cai S., Luo S., Gonzalez K.B., Perez-Miller S., Patwardhan A., Ibrahim M.M., Khanna R. SARS-CoV-2 spike protein co-opts VEGF-A/neuropilin-1 receptor signaling to induce analgesia. Pain. 2021;162(1):243–252. doi: 10.1097/j.pain.0000000000002097. Jan, PMID: 33009246; PMCID: PMC7737878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taboada M., González M., Alvarez A., Eiras M., Costa J., Álvarez J., Seoane-Pillado T. First, second and third wave of COVID-19. What have we changed in the ICU management of these patients? J Infect. 2021;S0163–4453(21):160–162. doi: 10.1016/j.jinf.2021.03.027. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aanuoluwapo, Adeyimika, Afolabi, Olayinka, Stephen, Ilesanmi. Dealing with vaccine hesitancy in Africa: the prospective COVID-19 vaccine context DO-10.11604/pamj..38.3.27401,2021-20201-05. [DOI] [PMC free article] [PubMed]
- 54.Happi A.N., Ugwu C.A., Happi C.T. Tracking the emergence of new SARS-CoV-2 variants in South Africa. Nat Med. 2021;27:372–373. doi: 10.1038/s41591-021-01265-1. 2021. [DOI] [PubMed] [Google Scholar]
- 55.Mercatelli, Giorgi . 2020. Geographic and genomic distribution of SARS-CoV-2 mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urhan A., Abeel T. Emergence of novel SARS-CoV-2 variants in The Netherlands. Sci Rep. 2021;11:6625. doi: 10.1038/s41598-021-85363-7. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Zyoud Walid, Haddad Hazem. Mutational sensitivity of D614G in spike protein of SARS-CoV-2 in Jordan. Biochem Bioph Rep. 2021;25:100896. doi: 10.1016/j.bbrep.2020.100896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kogay R., Schönbach C. In: Ranganathan S., Gribskov M., Nakai K., Schönbach C., editors. Elsevier; 2019. Epitope predictions. (Encyclopedia of bioinformatics and computational biology: ABC of bioinformatics). 1;2,952. [DOI] [Google Scholar]
- 59.Tobón G.J., Izquierdo J.H., Cañas C.A. B lymphocytes: development, tolerance, and their role in autoimmunity-focus on systemic lupus erythematosus. Autoimmune Dis. 2013:827254. doi: 10.1155/2013/827254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zupančič Eva, Curato Caterina, Paisana Maria, Rodrigues Catarina, Porat Ziv, Viana Ana S., Afonso Carlos A.M. João pinto, rogério gaspar, joão N. Moreira, ronit satchi-fainaro, steffen jung, helena F. Florindo. Rational design of nanoparticles towards targeting antigen-presenting cells and improved T cell priming. J Contr Release. 2017;258:182–195. doi: 10.1016/j.jconrel.2017.05.014. ISSN 0168-3659. [DOI] [PubMed] [Google Scholar]
- 61.Soria-Guerra R.E., Nieto-Gomez R., Govea-Alonso D.O., Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inf. 2015;53:405–414. doi: 10.1016/j.jbi.2014.11.003. ISSN 1532-0464. [DOI] [PubMed] [Google Scholar]
- 62.Birra D., Benucci M., Landolfi L., Merchionda A., Loi G., Amato P., Licata G., Quartuccio L., Triggiani M., Moscato P. COVID 19: a clue from innate immunity. Immunol Res. 2020;68:161–168. doi: 10.1007/s12026-020-09137-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raza M.T., Mizan S., Yasmin F., Akash A.S., Shahik S.M. Epitope-based universal vaccine for Human T-lymphotropic virus-1 (HTLV-1) PLoS One. 2021;16(4) doi: 10.1371/journal.pone.0248001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bibi S., Ullah I., Zhu B., Adnan M., Liaqat R., Kong W.B., Niu S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci Rep. 2021;11(1):1249. doi: 10.1038/s41598-020-80899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Majid M., Andleeb S. Designing a multi-epitopic vaccine against the enterotoxigenic Bacteroides fragilis based on immunoinformatics approach. Sci Rep. 2019;9:19780. doi: 10.1038/s41598-019-55613-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Zayat S.R., Sibaii H., Mannaa F.A. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Cent. 2019;43:187. doi: 10.1186/s42269-019-0227-2. [DOI] [Google Scholar]
- 67.Yu H., Wang Z., Jiang Q., Zeng T., Luo M., Zeng F., Liu J., Tian J., Long T., Yang X. Detection of serum IgM and IgG for COVID-19 diagnosis. Sci China Life Sci. 2020;63:1678. doi: 10.1007/s11427-020-1688-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tegally H., Wilkinson E., Lessells R.J., et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat Med. 2021;27:440–446. doi: 10.1038/s41591-021-01255-3. [DOI] [PubMed] [Google Scholar]
- 69.Biron M., Boon C. Temporal issues in person–organization fit, person–job fit and turnover: the role of leader–member exchange. Hum Relat. 2016;69(12):2177–2200. doi: 10.1177/0018726716636945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Payne S. Publ. Academic Press; 2017. Viruses: from understanding to investigation; pp. 1–352. 1. 9780128031094. [Google Scholar]
- 71.Janeway C.A., Jr., Travers P., Walport M., et al. Garland Science; New York: 2001. [Google Scholar]
- 72.Stachyra A., Redkiewicz P., Kosson P., Protasiuk A., Góra-Sochacka A., Kudla G., Sirko A. Codon optimization of antigen coding sequences improves the immune potential of DNA vaccines against avian influenza virus H5N1 in mice and chickens. Virol J. 2016;13(1):143. doi: 10.1186/s12985-016-0599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Folcik V.A., An G.C., Orosz C.G. The Basic Immune Simulator: an agent-based model to study the interactions between innate and adaptive immunity. Theor Biol Med Model. 2007;4:39. doi: 10.1186/1742-4682-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shanmugam A., Rajoria S., George A.L., Mittelman A., Suriano R., Tiwari R.K. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0030839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khan M.T., Islam R., Jerin T.J., Mahmud A., Khatun S., Kobir A., et al. Immunoinformatics and molecular dynamics approaches: next generation vaccine design against West Nile virus. PLoS One. 2021;16(6) doi: 10.1371/journal.pone.0253393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ayyagari V.S., T C V., K A.P., Srirama K. Design of a multi-epitope-based vaccine targeting M-protein of SARS-CoV2: an immunoinformatics approach. J Biomol Struct Dyn. 2020;1–15 doi: 10.1080/07391102.2020.1850357. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shilling P.J., Mirzadeh K., Cumming A.J., Widesheim M., Köck Z., Daley D.O. Improved designs for pET expression plasmids increase protein production yield in Escherichia coli. Commun biol. 2020;3(1):214. doi: 10.1038/s42003-020-0939-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shilling P.J., Daley D.O. Implementing novel designs in pET expression plasmids that increase protein production. Bio-protocol. 2021;11(16):e4133. doi: 10.21769/BioProtoc.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tohidinia M., Sefid F. Identifcation B and T-Cell epitopes and functional exposed amino acids of S protein as a potential vaccine candidate against SARS-CoV-2/COVID-19. Microb Pathog. 2020;148(2020):104459. doi: 10.1016/j.micpath.2020.104459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chukwudozie O.S., Gray C.M., Fagbayi T.A., Chukwuanukwu R.C., Oyebanji V.O., Bankole T.T., Adewole R.A., Daniel E.M. Immuno-informatics design of a multimeric epitope peptide based vaccine targeting SARS-CoV-2 spike glycoprotein. PLoS One. 2021;16(3) doi: 10.1371/journal.pone.0248061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghosh N., Sharma N., Saha I., Saha S. Genome-wide analysis of Indian SARS-CoV-2 genomes to identify T-cell and B-cell epitopes from conserved regions based on immunogenicity and antigenicity. Int Immunopharm. 2021;91(2021):107276. doi: 10.1016/j.intimp.2020.107276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Droppa-Almeida D., Franceschi E., Padilha F. Immune-informatic analysis and design of peptide vaccine from multi-epitopes against corynebacterium pseudotuberculosis. Bioinf Biol Insights. 2018;12 doi: 10.1177/1177932218755337. 1177932218755337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shey R.A., Ghogomu S.M., Esoh K.K., Nebangwa N.D., Shintouo C.M., Nongley N.F., Asa B.F., Ngale F.N., Vanhamme L., Souopgui J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. h Sci Rep. 2019;9(1):4409. doi: 10.1038/s41598-019-40833-x. 2019 Mar 13. PMID: 30867498; PMCID: PMC6416346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Safavi A., Kefayat A., Mahdevar E., Abiri A., Ghahremani F. Exploring the out of sight antigens of SARS-CoV-2 to design a candidate multi-epitope vaccine by utilizing immunoinformatics approaches. Vaccine. 2020;38(48):7612–7628. doi: 10.1016/j.vaccine.2020.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oluwagbemi O., Olaitan A. A comparative computational genomics of ebola virus disease strains: in-silico insight for ebola control. Inf. Med. Unlocked. 2018;12:106–119. doi: 10.1016/j.imu.2018.07.004. Published by Elsevier (Science Direct), 2018. [DOI] [Google Scholar]
- 86.Oluwagbemi O., Adewumi A., Misra S., Leon M. MAFODKM: mobile application framework for the management of omics data and knowledge mining. J Phys Conf. 2020;1566:1–7. doi: 10.1088/1742-6596/1566/1/012132. https://iopscience.iop.org/article/10.1088/1742-6596/1566/1/012132/meta 012132, 2020. [DOI] [Google Scholar]
- 87.Olaitan A., Oluwagbemi O., Fatumo S., Makolo A. A comparative analysis of the genetic relationships between the pathogens of ebola hemorrhagic fever, marburg virus, HIV, hepatitis A, hepatitis B, hepatitis C, hepatitis D, and hepatitis E. ISCB Afr ASBCB Conf Bioinf. 2015 https://zenodo.org/record/21406#.WCOIxtyko-Y March 09-11, 2015, Dar es Salaam, Tanzania. [Google Scholar]
- 88.Safavi A., Kefayat A., Abiri A., Mahdevar E., Behnia A.H., Ghahremani F. In silico analysis of transmembrane protein 31 (TMEM31) antigen to design novel multiepitope peptide and DNA cancer vaccines against melanoma. Mol Immunol. 2019;112:93–102. doi: 10.1016/j.molimm.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 89.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–1662. [Google Scholar]
- 90.Rarey M., Kramer B., Lengauer T., Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261(3):470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 91.Muegge I., Martin Y.C. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J Med Chem. 1999;42(5):791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 92.Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D., Yueh C., Beglov D., Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jahangirian E., Jamal G.A., Nouroozi M., Mohammadpour A. A reverse vaccinology and immunoinformatics approach for designing a multiepitope vaccine against SARS-CoV-2. Immunogenetics. 2021 doi: 10.1007/s00251-021-01228-3. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang D., Mai J., Zhou W., Yu W., Zhan Y., Wang N., Epstein N.D., Yang Y. Immunoinformatic analysis of T- and B-cell epitopes for SARS-CoV-2 vaccine design. Vaccines. 2020;8(3):355. doi: 10.3390/vaccines8030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feng Y., Jiang H., Qiu M., Liu L., Zou S., Li Y., Guo Q., Han N., Sun Y., Wang K., Lu L., Zhuang X., Zhang S., Chen S., Mo F. Multi-epitope vaccine design using an immunoinformatic approach for SARS-CoV-2. Pathogens. 2021;10(6):737. doi: 10.3390/pathogens10060737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mukherjee S., Tworowski D., Detroja R., Mukherjee S.B., Frenkel-Morgenstern M. Immunoinformatics and structural analysis for identification of immunodominant epitopes in SARS-CoV-2 as potential vaccine targets. Vaccines. 2020;8(2):290. doi: 10.3390/vaccines8020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oliveira S.C., de Magalhães M., Homan E.J. Immunoinformatic analysis of SARS-CoV-2 Nucleocapsid protein and identification of COVID-19 vaccine targets. Front Immunol. 2020;11:587615. doi: 10.3389/fimmu.2020.587615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sarkar B., Ullah M.A., Johora F.T., Taniya M.A., Araf Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2) Immunobiology. 2020;225(3) doi: 10.1016/j.imbio.2020.151955. 151955, ISSN 0171-2985, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chakraborty C., Sharma A.R., Bhattacharya M., Sharma G., Lee S.S. Immunoinformatics approach for the identification and characterization of T cell and B cell epitopes towards the peptide-based vaccine against SARS-CoV-2. Arch Med Res. 2021;52(4):362–370. doi: 10.1016/j.arcmed.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fathollahi M., Fathollahi A., Motamedi H., Moradi J., Alvandi A., Abiri R. In silico vaccine design and epitope mapping of New Delhi metallo-beta-lactamase (NDM): an immunoinformatics approach. BMC Bioinf. 2021;22:458. doi: 10.1186/s12859-021-04378-z. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Al Zamane S., Alam Nobel Fahim, Jebin Ruksana Akter, Amin Mohammed Badrul, Somadder Pratul Dipta, Antora Nusrat Jahan, Hossain Md Imam, Islam Mohammod Johirul, Ahmed Kawsar, Moni Mohammad Ali. Development of an in silico multi-epitope vaccine against SARS-COV-2 by précised immune-informatics approaches. Inf. Med. Unlocked. 2021;27 doi: 10.1016/j.imu.2021.100781. 100781,ISSN 2352-9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Castiglione F., Mantile P., De Berardinis, Prisco A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput Math Meth Med. 2013;2012:9. doi: 10.1155/2012/842329. Article ID 842329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mascellino M.T., Di Timoteo F., De Angelis M., Oliva A. Overview of the main anti-SARS-CoV-2 vaccines: mechanism of action, efficacy and safety. Infect Drug Resist. 2021;14:3459–3476. doi: 10.2147/IDR.S315727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mallajosyula J.K., Hiatt Ernie, Hume Steve, Johnson Ashley, Jeevan Trushar, Chikwamba Rachel, Pogue Gregory P., Bratcher Barry, Haydon Hugh, Webby Richard J., McCormick Alison A. Single-dose monomeric HA subunit vaccine generates full protection from influenza challenge. Hum Vaccines Immunother. 2014;10(3):586–595. doi: 10.4161/hv.27567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Burgos R.M., Badowski M.E., Drwiega E., Ghassemi S., Griffith N., Herald F., Johnson M., Smith R.O., Michienzi S.M. The race to a COVID-19 vaccine: opportunities and challenges in development and distribution. Drugs Context. 2021;10 doi: 10.7573/dic.2020-12-2. 2020-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagy A., Alhatlani B. An overview of current COVID-19 vaccine platforms. Comput Struct Biotechnol J. 2021;19(2021):2508–2517. doi: 10.1016/j.csbj.2021.04.061. ISSN 2001-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Excler J.L., Saville M., Berkley S., et al. Vaccine development for emerging infectious diseases. Nat Med. 2021;27:591–600. doi: 10.1038/s41591-021-01301-0. [DOI] [PubMed] [Google Scholar]
- 108.Saha R., Ghosh P., Burra V.L.S.P. Designing a next generation multi-epitope-based peptide vaccine candidate against SARS-CoV-2 using computational approaches. 3 Biotech. 2021;11:47. doi: 10.1007/s13205-020-02574-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rahmani A., Baee Masoud, Saleki Kiarash, Moradi Saead, Reza Nouri Hamid. Applying high throughput and comprehensive immunoinformatics approaches to design a trivalent subunit vaccine for induction of immune response against emerging human coronaviruses SARS-CoV, MERS-CoV and SARS-CoV-2. J Biomol Struct Dyn. 2021 doi: 10.1080/07391102.2021.1876774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ezediuno L.O., Onile Olugbenga S., Oladipo Elijah K., Majolagbe Olusola N., Jimah Esther M., Senbadejo Tosin Y. Designing multi-epitope subunit vaccine for ocular trachoma infection using Chlamydia trachomatis polymorphic membrane proteins G. Inf. Med. Unlocked. 2021;26(2021) 100764. [Google Scholar]
- 111.Meng X.Y., Zhang H.X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data on the accession numbers of sequences used for the experiments in this article are within the manuscript and its Supporting Information files. Sequence Data used for this experiment can be found in GISAID Database (https://www.gisaid.org/).
This project information can be found at: https://github.com/oluwagbemi/COVID-19-Vaccine-Candidate-for-South-Africa---Phase-1.
This project information can also be found on the research project page with weblink: https://olugbengaoluwagbemi.weebly.com/research-projects.html.
All figures (larger and clearer versions) have been included in the supplementary material accompanying this manuscript.