

Abstract
Background
Infiltrative astrocytic tumors with and without isocitrate dehydrogenase (IDH) mutation frequently contain mutations in the TP53 tumor suppressor gene. Disruption of normal p53 protein activity confers neoplastic cells with a number of oncogenic properties and is a common feature of aggressive malignancies. However, the high prevalence of TP53 mutation and its pathogenic role in IDH-mutant (IDHmut) astrocytoma is not well understood.
Methods
We performed a retrospective analysis of molecular and clinical data from patients with IDHmut astrocytoma at the University of Pittsburgh Medical Center between 2015 and 2019 as our initial cohort. We validated and expanded our findings using molecular and clinical data from The Cancer Genome Atlas.
Results
We show that the TP53 mutational spectrum in IDHmut astrocytomas is dominated by a single hotspot mutation that codes for the R273C amino acid change. This mutation is not enriched in IDH-wildtype astrocytomas. The high prevalence of TP53R273C mutation is not readily explained by known mutagenic mechanisms, and TP53R273C mutant tumors have lower transcriptional levels of proliferation-related genes compared to IDHmut astrocytomas harboring other forms of mutant p53. Despite lower proliferation, TP53R273C mutant tumors tend to progress more quickly and have a shorter overall survival than those with other TP53 mutations, particularly in male patients.
Conclusions
Our findings suggest that compared to other TP53 mutations, IDHmut astrocytomas may select for TP53R273C mutations during tumorigenesis. The genotype, sex, and mutation-specific findings are clinically relevant and should prompt further investigation of TP53R273C.
Keywords: astrocytoma, IDH, p53, TP53
Key Points.
TP53 R273C is a highly enriched hotspot mutation specifically in IDHmut astrocytoma.
IDHmut astrocytomas with TP53R273C have different gene expression profiles than those lacking this mutation.
Despite lower expression of proliferation-related genes, these tumors have worse prognosis.
Importance of the Study.
Despite decades of work, the role of TP53 alteration in tumorigenesis remains incompletely understood. Our study has identified a single TP53 gene mutation (R273C) that accounts for 20–30% of all TP53 gene mutations in IDH-mutant astrocytoma. Such a high prevalence of a single TP53 mutation has not been seen in other tumor types. Moreover, IDH-mutant astrocytoma with TP53R273C have sex dependent altered gene expression and overall poorer prognosis when compared to IDH-mutant astrocytomas with other TP53 alterations. These clinical findings are not accounted for by known mutagenic mechanisms and suggest that the hypothesized ability of certain TP53 mutations to impart gain-of-function properties in tumorigenesis may play a clinical role in IDH-mutant astrocytoma, laying the foundation for future functional studies.
Diffusely infiltrating gliomas are the most common primary malignant neoplasm of the central nervous system (CNS) and exhibit high morbidity and mortality. Curative therapeutic strategies for these tumors remain elusive.1,2 Despite the overall grim prognosis, a wide spectrum of biologic behavior exists among this category of tumors, ranging from the relatively indolent course of low-grade neoplasms to the much more aggressive glioblastoma (GBM). The distinctive patterns of molecular alterations that drive the initiation and progression of these neoplasms are increasingly recognized as a critical determinant of tumor behavior, and the World Health Organization (WHO) now formally incorporates molecular findings into the glioma classification scheme.3 Under this system, the isocitrate dehydrogenase (IDH)-mutant astrocytoma is recognized as a specific diagnostic entity, distinct from all other tumors of astrocytic lineage. Compared to their IDH-wild type (IDHwt) counterparts, astrocytomas with mutations in the IDH1 or IDH2 gene (IDHmut) tend to occur in younger patients, have a lower histologic grade at presentation, and have an improved prognosis.
The large majority of IDHmut astrocytomas of all grades harbor two additional molecular characteristics: TP53 gene mutations, identified in over 90% of tumors, and inactivating mutations of the ATRX gene, identified in over 70% of tumors.4–6 In contrast to events such as CDKN2A deletion that often occur later in the evolution of disease, the changes in TP53 and ATRX are early driver-type events, the ubiquity of which suggests they play an essential role in initiating and maintaining tumor growth. The high prevalence of TP53 mutation during early gliomagenesis in this relatively less aggressive neoplasm is especially curious, as other TP53-mutated tumors of the brain and other anatomic locations typically exhibit highly malignant behavior.
The markedly elevated rate of TP53 mutations found in IDHmut astrocytoma is not identified in most other glial tumors, nor in non-CNS neoplasms driven by IDH1/2 mutations. In the IDHwt diffuse astrocytic tumor category, a substantially smaller subset of tumors (<30%) also acquire TP53 mutations, and unlike IDHmut tumors, these tend to present with higher histologic grade, have a more aggressive clinical course, and may acquire the TP53 mutation later in tumor evolution.3,7–9 In oligodendroglioma, which shares both glial origins and IDH mutation, TP53 mutations are quite rare (<3%).4–6 Likewise, of the non-CNS tumors that harbor mutant IDH, including subsets of leukemia, cholangiocarcinoma, and chondrosarcoma, none seem to require TP53 mutation as an early driver event.10,11 In fact, TP53 mutation is rare in IDHmut cholangiocarcinoma, occurring at far lower frequency than in IDHwt cholangiocarcinoma.12 These comparisons suggest that mutant p53 plays a uniquely critical role in IDHmut astrocytoma, for reasons unrelated to either IDH mutation or glial lineage alone.
Previous studies have suggested that different subsets of TP53 mutations might have differential relevance in certain subtypes of glioma. Using largely outdated categories that separated GBMs into primary (de novo) and secondary (arising from lower-grade gliomas), it was observed that secondary GBMs harbored a higher rate of certain hotspot TP53 mutations than primary GBMs.13,14 With the introduction of the molecular-driven classification scheme in 2016, we now know that the vast majority of secondary GBMs represent IDHmut tumors. Given these findings, we hypothesized that the specific TP53 mutational spectra of IDHmut and IDHwt astrocytomas may differ considerably, and that the characterization of such differences could provide unique insight into the pathogenesis of both diseases.
Methods and Materials
Cohort Descriptions
Diffusely infiltrating glioma specimens from the University of Pittsburgh Medical Center (UPMC) archives were identified (UPMC cohort). Data were collected from specimens that underwent molecular analysis by the GlioSeq™ next-generation sequencing panel between 2015 and 2019. Details on the GlioSeq panel can be found in the Supplementary Methods. 668 infiltrating astrocytic tumors were identified, of which 108 were IDHmut. Demographic data, diagnosis, and grade were collected from the surgical pathology reports. Blinded re-review of histologic slides was performed by two neuropathologists (DFM and TMP), confirming diagnosis and WHO grade in all cases. Ki67 was estimated to the nearest percent in the area of highest proliferation. These estimates were largely concordant with the originally reported value in the pathology report, with the exception of an apparent typographical error in one original report. One case (tumor 19) did not have a Ki67-stained slide available for review, and the originally reported value was used. All data were collected with the approval of the University of Pittsburgh Institutional Review Board (study numbers PRO07010097 and STUDY20040135). Detailed data for the UPMC cohort including patient demographic information, tumor characteristics, details of molecular alterations, and survival status and intervals, are available in Supplementary Table 1.
Additional cases were identified from The Cancer Genome Atlas (TCGA) lower-grade glioma (LGG) and GBM datasets (TCGA cohort). Clinical outcome and molecular data were downloaded from cBioPortal (https://cbioportal.org, last accessed 10/13/2020) and the NIH Genomic Data Commons (https://portal.gdc.cancer.gov/, last accessed 10/13/2020). All tumors were reclassified based on IDH and 1p/19q status to reflect current diagnostic categories.
Exclusion Criteria
For analyses comparing different TP53 mutations, samples classified as astrocytomas by absence of 1p/19q codeletion but lacking pathogenic TP53 mutation were excluded (UPMC cohort: 12 of 108, 11%; TCGA cohort: 11 of 160, 7%).
Data Processing and Statistical Analysis
The Python 3 programming language was used for data processing and statistical analyses. The specific packages are provided in the Supplementary Methods.
TP53 Mutation Spectrum Analysis
The frequency of various TP53 mutations was compared by codon position using jsProteinMapper.15 In-frame insertions and deletions and frame-shift mutations are plotted at the position of the first altered codon.
Mutational Signature Analysis
COSMIC mutational signatures (v3.1, GRCh37) for single base substitutions (SBS) were downloaded from https://cancer.sanger.ac.uk/signatures/downloads/ (accessed 2/5/2021). Signatures were derived as previously described.16
Methylation Analysis
Methylation data for the TCGA cohort were obtained as previously described.17 Processed methylation data were downloaded from the NIH genomic data commons portal. Methylation sites on sex chromosomes were excluded. Dimensionality reduction via t-distributed Stochastic Neighbor Embedding (t-SNE) was performed using the implementation included in the scikit-learn Python package.
Gene Expression Profiling
Level 3 gene expression data were downloaded from TCGA data coordination center. This dataset shows the gene-level transcription estimates as log2(x+1) transformed RSEM normalized count. Genes are mapped onto the human genome coordinates using UCSC Xena HUGO probeMap. Differential expression analysis was performed using the LIMMA (Linear Methods for Microarray Analysis18) module of the WebMeV analysis platform (http://mev.tm4.org, last accessed 6/17/2020). The resulting rank-ordered gene lists were subjected to Gene Set Enrichment Analysis (GSEA) to identify significantly up- and down-regulated hallmark gene sets.19
Comutation Analysis
Molecular alterations identified by next-generation sequencing of the UPMC cohort were extracted from the pathology report. To allow a direct comparison between the TCGA and UPMC cohorts, mutation and copy number alteration data for the TCGA cohort was limited to those genes included in the GlioSeq20 panel plus CDK4, a frequently altered gene in IDHmut gliomas that is not included in GlioSeq. Of note, the version of GlioSeq used during this time period did not distinguish between homozygous and hemizygous/complex CDKN2A deletion (for additional explanation, see Supplementary Methods). Data exploration and visualization was performed with the interactive browser-based widget jsComut.15 The TCGA cohort contained only a small number (15) of IDHmut, TP53-mutant glioblastomas, of which only 1 harbored a TP53R273C mutation, making it unsuitable for comparative analyses with the UPMC cohort.
Immunohistochemistry
Immunohistochemical staining of formalin fixed, paraffin embedded (FFPE) tissue sections was performed at the UPMC clinical laboratory as part of routine diagnostic evaluation of all cases in the UPMC cohort. Additional details can be found in the Supplementary Methods.
Survival Analysis
Survival status and date of death/last known alive for patients in the UPMC cohort was determined from the electronic medical record and publicly searchable online sources. Survival intervals were measured from the date of initial pathologic diagnosis. Subsequent specimens demonstrating tumor progression to a higher WHO grade were used to calculate progression-free intervals. Survival status and interval for the TCGA cohort were downloaded directly from the TCGA. Kaplan–Meier survival curves, log-rank statistical testing, and multivariate Cox-Proportional Hazards (Cox-PH) analysis were performed using the lifelines Python module.
Results
TP53 Mutational Profile Depends Critically on IDHmut Status
We first examined whether IDHmut and IDHwt astrocytomas had distinct TP53 mutational profiles, as was suggested by previous comparisons of primary versus secondary GBM13 and GBM versus lower-grade gliomas.14 Using the interactive browser-based visualization tool jsProteinMapper,15 we explored the distribution of mutations along the TP53 gene, with cases stratified by WHO grade and IDH status (Figure 1A). A striking finding emerged: IDHmut astrocytomas of all grades showed a single dominant hotspot at codon 273 which accounted for 38% of all TP53 mutations, whereas IDHwt tumors showed a broader, more conventional mix of mutations that more closely resembled the pan-cancer mutation profile, with 4.8% of mutations occurring at codon 273 (P < .001, chi-squared test). To corroborate this observation made on the UPMC cohort, we turned to the TCGA LGG and GBM datasets, and observed the same findings, with codon 273 accounting for a much higher proportion of all TP53 mutations than any other hotspot, in IDHmut but not IDHwt astrocytomas (21% vs 5.3%, P < .01, chi-squared test).
Figure 1.

R273C is the single dominant TP53 hotspot mutation in IDHmut but not IDHwt astrocytomas. (A) TP53 mutational profiles of IDHmut (left column) and IDHwt (right column) astrocytomas reveal a dominant hotspot at codon 273 which is specific to the IDHmut tumor subset. Note that y-axes are scaled per-plot to clearly show the distributions. (B) Mutations at TP53 codon 273 in IDHmut astrocytomas are dominated by the R273C amino acid change, which occurs at a far higher frequency than other pan-cancer hotspot mutations, including R273H. (C) In contrast, IDHwt astrocytomas are not enriched for this mutation.
R273C is the Only Dominant TP53 Hotspot Mutation in IDHmut Astrocytomas
Mutations at codon 273 can lead to multiple possible amino acid changes depending on the nucleotide-level alteration (Figure 2A). Two of these, R273H and R273C, are considered hotspot mutants across all cancers, each accounting for ~2.5–3% of all TP53 mutations across cancer subtypes.21,22 To determine whether the dominant codon 273 hotspot in IDHmut astrocytomas was due to one or both of these possible mutations, we next examined the specific frequencies of each possible hotspot amino acid change (Figure 1B and C). Notably, only the R273C amino acid change was highly enriched, while R273H showed a similar frequency to the other unenriched hotspot mutations. This is not a common phenomenon: across all TCGA studies, the only organ with highly imbalanced frequencies of these two mutations is the brain (Figure 3A).
Figure 2.

Enrichment of TP53 R273C is not accounted for by known mutational mechanism signatures. (A–B) DNA context of select point mutations at hotspot codons in TP53 (A) and IDH1 (B). (C) Consequences of C>T transitions at methylated CpG site at these codons, grouped by trinucleotide context. At right, the frequency at which each trinucleotide is found within the four COSMIC SBS mutational signatures that are enriched for the G(C>T)G or A(C>T)G transitions (corresponding to TP53R273C and IDH1R132H respectively). (D) The profile of TP53 mutations in IDH-mutant astrocytomas does not match these SBS signatures (UPMC cohort). To avoid confounding effects, mutations at the dominant hotspot codon 273 are excluded.
Figure 3.

TP53 R273C mutant tumors are overrepresented in brain tumors but are not different than other TP53 mutants in DNA methylation or heterozygosity. (A) Across all TCGA studies, TP53R273C is seen at much higher frequency than TP53R273H specifically in brain tumors. (B) Analysis of genome-wide methylation patterns of the TCGA cohort shows that IDHmut astrocytomas with the TP53R273C mutation do not form a distinct cluster, but are distributed through the methylation space shared by other IDHmut astrocytomas. (C) For tumors with a single TP53 mutation, TP53 variant allele frequency (VAF) is plotted against the corresponding IDH1/2 VAF.
Examination of the mutational spectra across patient demographics revealed that R273C mutations were more common in females versus males, to varying degrees. In the UPMC cohort the R273C mutation accounted for 34% of all TP53 mutations in females and 27% in males, though this was not statistically significant. In the TCGA data set, the difference was more marked, with R273C accounting for 26% of mutations in females versus 11% in males (P < .05, chi-squared test).
Enrichment of the TP53R273C Mutation is Not Explained by Known Mutational Signatures
We first considered whether the high prevalence of TP53R273C might be due to a particular mechanism of mutagenesis that favors this mutation over other TP53 mutations. A selective mechanism of mutation might also explain why IDHmut astrocytomas are also highly enriched for the IDH1R132H mutation compared other functionally equivalent IDH mutations.23,24 To examine this possibility, we turned to the emerging literature of mutational signatures in human cancers, in which mutagenic mechanisms are identified by the patterns of base substitutions that result from different causes of unrepaired DNA damage.16,25TP53R273C and IDH1R132H both occur due to cytosine (C) to thymidine (T) transitions (C>T) at methylated CpG sites, as do other oncogenic mutations at TP53 codons 273 and 175 and IDH1 codon 132, which are pan-cancer hotspots but not highly enriched in IDHmut astrocytoma (Figure 2A and B). Since C>T transitions are highly represented in certain COSMIC single base substitution (SBS) signatures (eg clock-like signature 1), we asked whether the trinucleotide context (TC) of the highly enriched versus nonenriched mutations might match any of these previously identified signatures. To plausibly explain the frequencies at which TP53R273C and IDH1R132H are found in IDHmut astrocytomas, the TCs of these mutations (ie GCG or ACG) would need to account for a reasonable proportion of all mutations within candidate signatures. We identified four signatures in which one of these TCs exceeded 5% of all mutations (SBS1, SBS6, SBS15, SBS87). Of these, SBS6 and SBS15 would favor transitions causing TP53R273C over TP53R273H and IDH1R132H over IDH1R132C, recapitulating the enrichments seen in clinical samples (Figure 2C). Two lines of evidence argue against these signatures being a sufficient explanation, however. First, the TP53R175H hotspot mutation has the same GCG TC as TP53R273C yet is not equivalently enriched. Second, the profile of base transitions of all other noncodon-273 TP53 mutations is not a good match (Figure 2D), and it is implausible that mutagenic mechanisms would preferentially act only at specific codons of a gene.
Molecular Characteristics of TP53R273C-Mutant Astrocytomas Versus Those with Other TP53 Mutations
We next hypothesized that the high prevalence of TP53R273C mutations within IDHmut astrocytomas could arise from selective advantage, and asked whether TP53R273C mutant tumors have different molecular characteristics than those harboring other types of mutant p53 protein. Several possible mechanisms exist. Given the critical epigenetic remodeling effects of mutant IDH, we wondered whether TP53R273C mutation might systematically affect the tumor methylome. However, TP53R273C mutant tumors did not form a distinct methylation cluster, as visualized by t-SNE dimensionality reduction (Figure 3B). Excluding IDHwt and oligodendroglioma samples from the analysis did not substantially change this distribution (not shown). Additional methylation-based analyses also failed to segregate these tumors – the global methylation levels in TP53R273C mutant tumors versus others were not different, and TP53R273C tumors showed no significant predilection for either the glioma CpG island methylator (G-CIMP) -low or -high methylation clusters.17,26
We then investigated the possibility that TP53R273C preferentially produces dominant-negative mutant p53 protein (ie not necessitating loss of the wild-type allele), reasoning that this could provide a selective advantage to transformed precursor cells during early gliomagenesis. To answer this, we took advantage of the ubiquity of IDH mutations in all tumor cells and the allelic heterozygosity necessary for tumor cell survival and D-2HG oncometabolite production, which provides a reference measure of tumor cellularity.10,27–30 Leveraging this, we compared the variant allele frequency (VAF) of each TP53 mutation to the VAF of the IDH1/2 mutation in the same tumor (Figure 3C). Concordant with recent analyses that have demonstrated frequent (>90%) loss of the wild type p53 allele across all cancer subtypes,31 we find that nearly all tumors show loss of the wild type TP53 allele: the TP53:IDH1/2 VAF ratio is ~2:1 in most cases, with only a handful of tumors falling near the unity line that indicates heterozygous TP53 mutation. Importantly, none of the TP53R273C mutant tumors retained a wild-type allele, arguing against this possible selective mechanism.
Examining the landscape of molecular alterations seen in IDHmut astrocytomas stratified by sex and TP53R273C mutation status (Figure 4), we found a small but statistically significant interaction between the R273C mutation and sex, as was hinted at by the increased frequency of this mutation in female patients. First considering WHO grades 2 and 3 tumors (Figure 4A) in the combined UPMC and TCGA cohorts, we found that female tumors harboring TP53R273C showed fewer additional genomic alterations beyond the canonical IDH-TP53-ATRX triad than those with alternative TP53 mutations, while the relationship was opposite in males (Figure 3C; P < .05, chi-squared test). Turning to the smaller set of IDHmut GBMs, we found that by this stage in tumor evolution virtually all tumors had acquired additional genomic hits, regardless of TP53R273C status (Figure 4B). However, a notable difference was identified in the pattern of cooccurring alterations in GBMs, with the TP53R273C mutants having a lower rate of CDKN2A copy number loss compared to tumors lacking a TP53R273C mutation (Figure 3D; P < .01, chi-squared test), an important genomic event that correlates with more aggressive clinical behavior in IDHmut astrocytomas.32–35
Figure 4.

Comutation analysis of TP53 R273C mutants and tumors with other TP53 mutations. (A–B) The genomic landscape of IDHmut astrocytomas, grouped by sex and TP53R273C status, for WHO grades 2-3 (A) and WHO grade 4 (B) tumors. (C) Fewer female tumors with TP53R273C have additional genomic alterations than those with other TP53 mutations; this pattern is reversed in male tumors (P < .05, chi-squared test). (D) TP53R273C mutant WHO grade 4 tumors show lower incidence of CDKN2A copy number loss than those with other TP53 mutations (P < .01, chi-squared test); RB1 loss is more common (see (B)).
Histologic Features of TP53R273C Mutant Astrocytomas
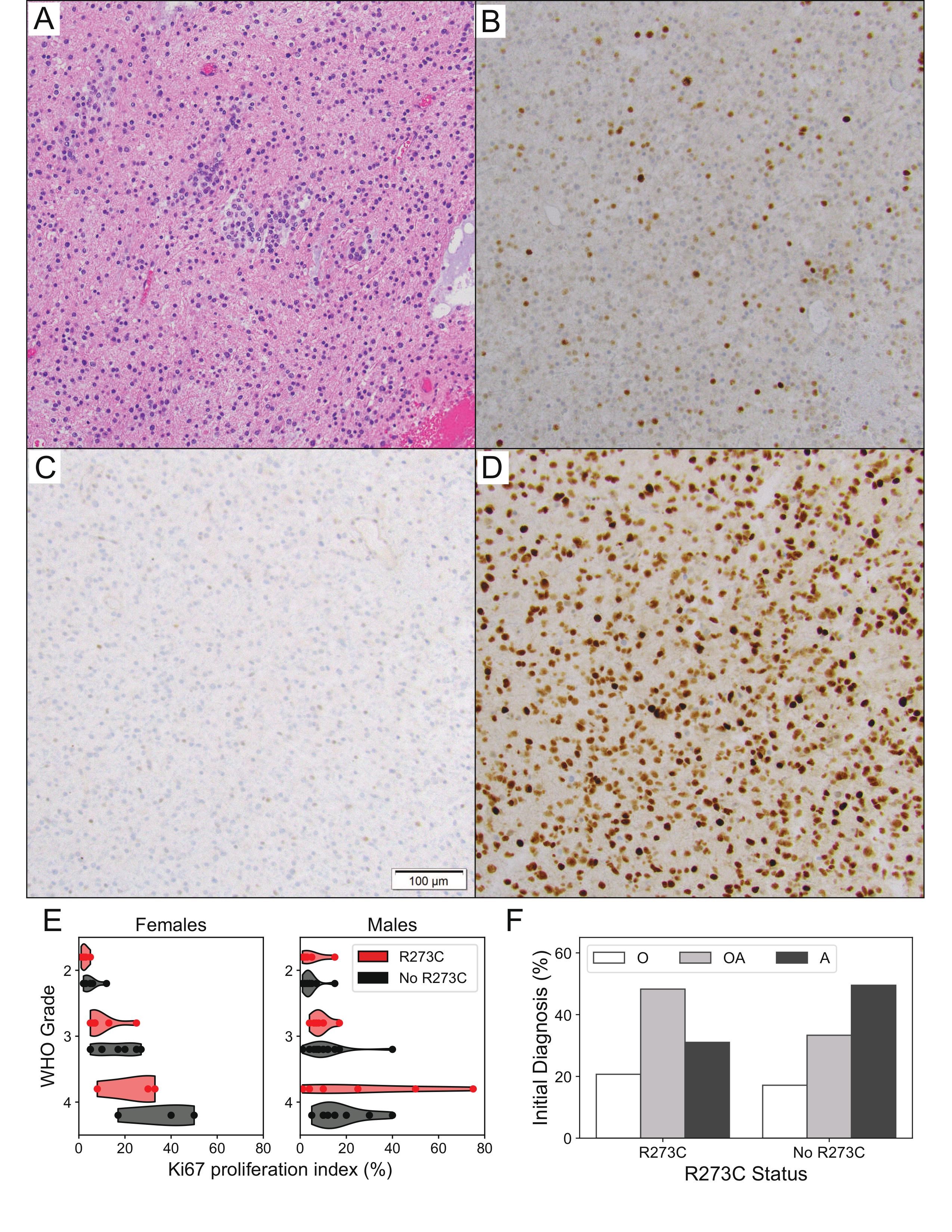
Supplementary Figure S1 reviews the histopathologic features of the TP53R273C-mutant astrocytomas. Low-grade TP53R273C mutant tumors frequently demonstrated an oligodendroglioma-like morphology, with monotonous round nuclei and variable cytoplasmic clearing in FFPE tissue sections stained with hematoxylin and eosin (H&E; Supplementary Figure S1A). p53 immunostaining was often indeterminate (Supplementary Figure S1B-D). In addition, the Ki67 proliferation index for p53R273C-mutant tumors in female patients tended to be lower compared to cases lacking this mutation, grade for grade (Supplementary Figure S1E), though this did not reach statistical significance (P > .05, Mann–Whitney U-test). Taking advantage of the fact that the clinical data for the TCGA LGG cohort includes the initial histologic (premolecular) diagnosis of each tumor at the time of case submission, we also found that IDHmut astrocytomas with TP53R273C mutation were diagnosed most frequently as the now-obsolete “oligoastrocytoma,” while tumors harboring other forms of mutant p53 were most commonly diagnosed as “astrocytoma” (Supplementary Figure S1F).
Sex-Dependent Gene Expression Differences in TP53R273C Mutants Versus Other TP53 Mutants
We next examined gene expression profiles for potential differences in these tumors compared to those with other TP53 mutations. Because of the sex difference in mutational frequency, our initial differential expression analyses were dominated by genes encoded on sex chromosomes, leading us to stratify patients by sex for further analyses. Differential expression and gene set enrichment analysis revealed significant associations between the TP53R273C mutation and gene expression, and an interaction with sex. Most strikingly, the Hallmark gene sets associated with proliferation and cell cycle, including the G2M checkpoint, E2F targets, and mitotic spindle gene sets, are down-regulated in TP53R273C mutants tumors compared to TP53 mutations at other codons (Figure 5A and B), supporting the observation of lower Ki67 immunohistochemical labeling (Supplementary Figure S1E). This effect was larger and more statistically significant in females, but was also present in males. Since TP53R273C is known to have a minor amount of residual binding capacity at the p53 response element,36 we next asked whether direct p53 target genes,37 or genes in the Hallmark p53 pathway, were differentially expressed in these tumors. Only a modest effect was seen, arguing against residual wild type function playing a significant role (Figure 5C). Notably, expression of CDKN1A (p21) was increased in TP53R273C tumors specifically in females, but not males. The single most significantly up-regulated Hallmark gene set in TP53R273C tumors is TNF-⍺ signaling via NF-κB, which was similarly seen only seen in female tumors (Figure 5D).
Figure 5.

TP53 R273C -mutant tumors show differential gene expression relative to other TP53 mutations, with sex-specific differences. LIMMA analysis of RNA expression data from the TCGA LGG cohort was used to compare gene expression in TP53R273C astrocytomas to those lacking a codon 273 mutation. (A–B) Analysis of female tumors (left column) and male tumors (right column) demonstrates a cloud of down-regulated genes, more prominent in females, which gene set enrichment analysis showed to be composed of proliferation-related pathways. (C) Direct p53 target genes and the Hallmark p53 pathway gene set showed modest effects, mainly in females, including significant upregulation of p21. (D) The largest up-regulated Hallmark gene set is TNF-α signaling via NF-κB.
Given the possibility that R273H, the other hotspot mutation at codon 273, might show similar effects to R273C, we performed differential expression analysis in two stages. Data shown in Figure 5 are a comparison of R273C mutants versus TP53 mutations at codons other than 273. We then directly compared R273C to R273H, and found the same pattern of differential expression (albeit noisy and not statistically significant due to the very small number of R273H-mutant tumors in the data set; not shown), arguing that the effect is due to the specific amino acid change, not just the codon position.
TP53R273C Mutation-Specific Effects on Survival
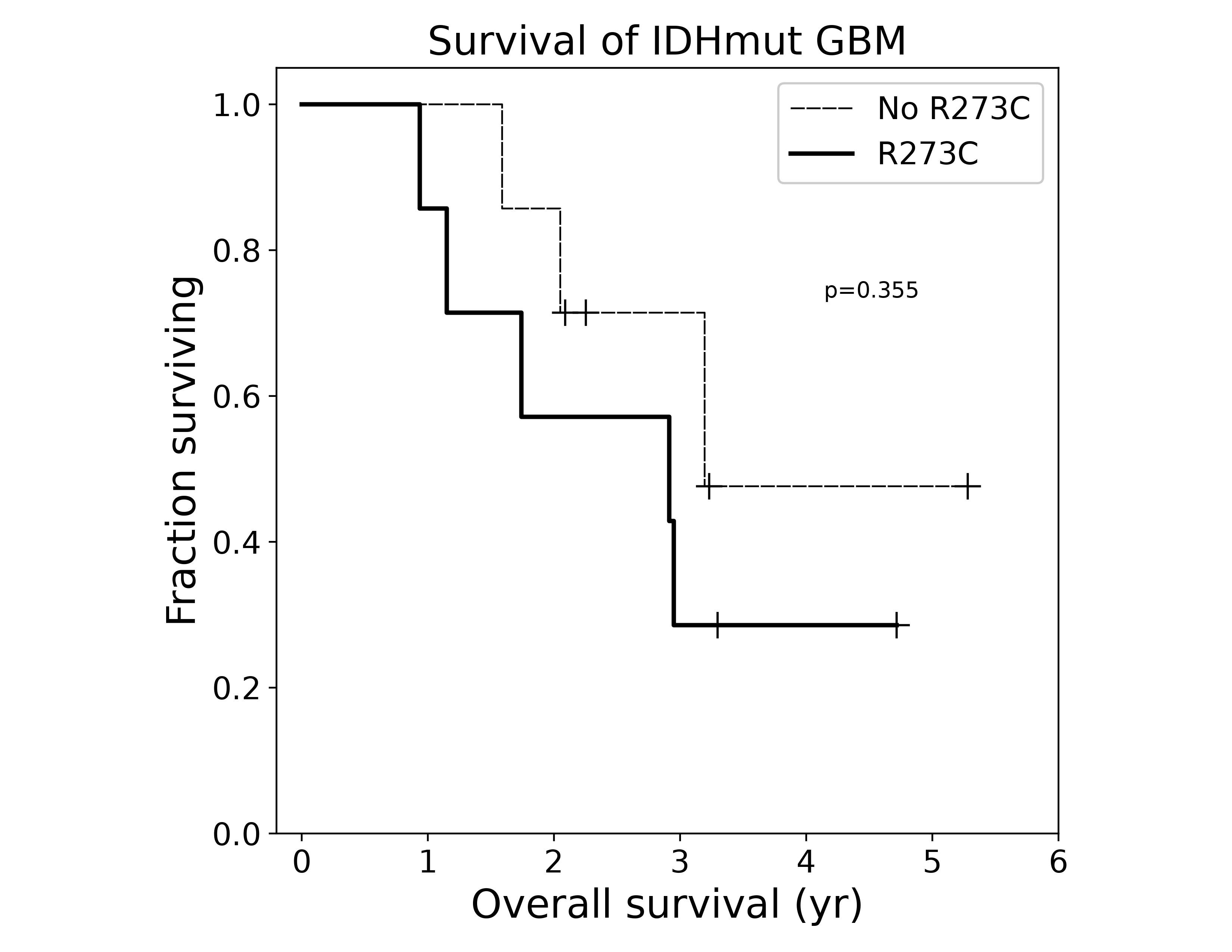
In light of the bland morphology and decreased proliferative signature of the TP53R273C mutant tumor subset, we wondered this molecular alteration might confer a better prognosis than other TP53 mutations. We instead found the opposite (Figure 6 and Supplementary Figure S2). Stratifying tumors by TP53R273C status, we find that lower-grade (grades 2-3) tumors with this mutation have shorter progression-free survival (PFS) and overall survival (OS), driven by significantly worse outcomes in male patients (Figure 6, bottom row). In the subset of tumors presenting as GBM, OS was shorter for TP53R273C mutant tumors, though numbers are small (N = 14) and this did not reach significance (Supplementary Figure S2). Additionally, TP53R273C approached significance in multivariate Cox-PH analyses for both PFS (P = .06–.08) and OS (P = .13–.23) with a hazard ratio of 1.63-2.59 (UPMC cohort; Supplementary Table 2).
Figure 6.

TP53 R273C confers poor prognosis compared to other TP53 mutations. Kaplan–Meier curves for WHO grades 2–3 tumors were constructed for progression-free (UPMC) and overall survival (UPMC and TCGA). Progression includes transformation to higher WHO grade and death. P-values were calculated using the log-rank test.
Discussion
Recognition of the importance of mutations in the IDH1 or IDH2 genes on the natural history of diffuse gliomas has been a defining advance in our understanding of glioma biology over the past decade. Pathogenic IDH mutations exclusively occur at the active site of the isocitrate dehydrogenase enzyme and lend the mutant protein the neomorphic ability to catalyze NADPH-dependent reduction of α-ketoglutarate to D-2-hydroxyglutarate (D-2HG).10 The resulting accumulation of D-2HG promotes a state of DNA and histone hypermethylation, leading to the CpG island methylator phenotype, a block of differentiation, and a host of oncogenic properties which have been extensively reviewed in the literature.10,27,28 How this abnormal cellular state interacts with the other characteristic molecular alterations in the two subcategories of IDHmut glioma are less clear. In particular, the high frequency of missense TP53 mutations in IDHmut astrocytoma is well described but lacking in explanation.
The existence of hotspot point mutations in TP53 has been recognized for decades, with codons 175, 245, 248, 249, 273, and 282 accounting for a much higher frequency of all point mutations compared to all other codons.22 Across cancers, each of these hotspot codons accounts for approximately 3–7% of the total point mutations; although 3–7% is far lower than the prevalence of single hotspot mutations in certain known oncogenes (eg BRAFV600E), this degree of enrichment is nonetheless highly significant, and mutations at these codons are frequently hypothesized to confer beneficial gain-of-function (GOF) properties.22,38,39 Given this baseline pan-cancer level of hotspot mutational frequencies, it is remarkable that in IDHmut astrocytomas the R273C amino acid change accounts for 20–30%+ of all TP53 mutations.
Somatic mutations in cancer can become enriched by two primary mechanisms: selective mutagenesis or selective advantage. We find both possibilities intriguing. Currently established signatures of mutagenic mechanisms do not appear to adequately explain the pattern of highly enriched TP53 and IDH1 hotspot mutations in IDHmut astrocytomas (Figure 2), but it remains possible that an as-yet unidentified mutational mechanism might be active in IDHmut gliomagenesis. However, we believe the selective advantage hypothesis is more likely, as multiple lines of evidence support the idea that mutant p53R273C protein may have unique effects on the tumor cell properties. At the level of gene expression, TP53R372C mutant tumors show differential downregulation of proliferation-related gene sets compared to tumors harboring other TP53 mutations, and upregulation of other gene sets, in a sex-dependent manner (Figure 5). The downregulation of proliferation-related gene sets is supported by lower Ki67 proliferation rates in these tumors, particularly in female patients (Supplementary Figure S1E). The frequency of additional molecular alterations also appears to subtly depend on TP53R273C status (Figure 4), though further analysis of larger cohorts is needed. Finally, there were notable clinical differences in patients with TP53R273C mutated astrocytomas, including increased prevalence in female patients (Figure 1B) and shorter survival, particularly in male patients (Figure 6). In light of the latter, we note that the apparent increased frequency of TP53R273C in female patients may in part be related to the combination of longer survival of females with this mutation and the cross-sectional nature of the cohorts, which may bias the observed incidence of the mutation.
Despite tens of thousands of publications spanning decades of research on TP53, this “guardian of the genome” continues to generate controversy and challenge understanding.40–42 While the high frequency of point mutations compared to truncating and deletion mutations is unquestioned, the relative contributions of GOF and dominant negative effects towards the oncogenic properties of TP53 mutation remains contentious.22,43–45 A recent study of TP53 mutations in myeloid malignancies performed a detailed multi-modality investigation of the role of mutant TP53 in these cancers.43 The authors showed that the mutational spectrum in myeloid lesions was similar to the pan-cancer spectrum, and that the most common mutations showed no evidence of GOF activity, but rather were seemingly selected for by dominant negative mechanisms. Notably, TP53R273C was not among the mutations studied in detail, as it was not highly prevalent in myeloid cancers. In contrast, this mutation is extremely prevalent in IDHmut astrocytomas, and tumors harboring this form of mutant p53 protein versus others appear to have a worse prognosis.
Although the precise mechanisms by which TP53R273C differs from other TP53 mutations in IDHmut astrocytoma are not clear from the clinical data, some possibilities appear more likely than others. The downregulation of proliferative pathways (Figure 5A and B) could indicate preservation of some degree of wild-type activity, but this is argued against by the lack of significant differential expression of other p53 target gene pathways (Figure 5C) as well as the loss of the wild-type allele in all TP53R273C tumors (Figure 3B and C). The biology of IDHmut astrocytomas is fundamentally shaped by the G-CIMP related epigenomic remodeling caused by IDH mutation. Although TP53R273C mutant tumors are not distinctive in DNA methylation signature (Figure 3B), an interaction with epigenetic mechanisms may occur at the level of the histone. In particular, differential interactions of p53R273C with sex chromosome encoded histone demethylases could potentially explain the differing tumor behavior in male and female patients with astrocytomas containing this mutation. Another intriguing possibility is that TP53R273C may differentially activate NF-κB related mechanisms (Figure 5D), as this signaling pathway has been implicated in a variety of aggressive glioma phenotypes including invasiveness and therapy resistance.46 We note, however, that significant activation of this pathway was only seen in female patients and therefore does not address the particularly poor prognosis in male patients. Experimental investigations of these hypotheses, and other possible mechanisms, would be of great interest.
In this study, we have shown that the TP53 mutation spectrum in IDHmut but not IDHwt astrocytoma is dominated by a single highly enriched hotspot mutation, TP53R273C. Survival analysis demonstrates mutation-specific poor outcome, particularly in male patients, despite histologic and transcriptomic evidence of lower proliferation. Intriguingly, sex-related differences identified in the UPMC and TCGA cohorts raise the possibility that TP53R273C may have different oncogenic mechanisms and/or therapeutic responses depending on the patient’s sex; it will be of great interest whether additional cohorts will further support these findings. Unfortunately, cell line and animal models of TP53 GOF properties have focused on other hotspot mutations and largely ignored TP53R273C. Based on our results, it seems likely that experimental work focused on TP53R273C in IDHmut astrocytoma, in both male and female model systems, has high potential for elucidating mechanisms of p53 GOF, astrocytoma oncogenesis, and sex differences in glioma biology. Incorporation of molecular findings into brain tumor classification systems has already enabled greater diagnostic and prognostic precision. Our findings here suggest that it may be wise, at least in some cancers, to consider that specific TP53 mutations may have distinctive properties, as we work to provide ever-more personalized medical care to patients.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Preliminary findings were presented at the 2019 American Association for Neuropathologists annual meeting.
Funding
No funding source supported this study.
Conflict of interest statement. The authors declare no conflict of interest.
Authorship statement: Study design: TMP and GHM. Analysis and interpretation: TMP, GHM, SA, NA, and DFM. Writing and revision: TMP, GHM, SA, NA, and DFM.
References
- 1. Shergalis A, Bankhead A 3rd, Luesakul U, Muangsin N, Neamati N. Current challenges and opportunities in treating Glioblastoma. Pharmacol Rev. 2018; 70(3):412–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajesh Y, Pal I, Banik P, et al. Insights into molecular therapy of glioma: current challenges and next generation blueprint. Acta Pharmacol Sin. 2017; 38(5):591–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016; 131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 4. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015; 47(5):458–468. [DOI] [PubMed] [Google Scholar]
- 7. Brennan CW, Verhaak RG, McKenna A, et al. ; TCGA Research Network . The somatic genomic landscape of glioblastoma. Cell. 2013; 155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Draaisma K, Chatzipli A, Taphoorn M, et al. Molecular evolution of IDH wild-type glioblastomas treated with standard of care affects survival and design of precision medicine trials: a report from the EORTC 1542 study. J Clin Oncol. 2020; 38(1):81–99. [DOI] [PubMed] [Google Scholar]
- 9. Berzero G, Di Stefano AL, Ronchi S, et al. IDH-wildtype lower grade diffuse gliomas: the importance of histological grade and molecular assessment for prognostic stratification. Neuro Oncol. 2021; 23(6):955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013; 3(7):730–741. [DOI] [PubMed] [Google Scholar]
- 11. Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. 2010; 16(9):387–397. [DOI] [PubMed] [Google Scholar]
- 12. Su L, Zhang X, Zheng L, Wang M, Zhu Z, Li P. Mutation of isocitrate dehydrogenase 1 in cholangiocarcinoma impairs tumor progression by inhibiting isocitrate metabolism. Front Endocrinol (Lausanne). 2020; 11:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004; 64(19):6892–6899. [DOI] [PubMed] [Google Scholar]
- 14. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017; 170(6):1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pearce TM, Nikiforova MN, Roy S. Interactive browser-based genomics data visualization tools for translational and clinical laboratory applications. J Mol Diagn. 2019; 21(6):985–993. [DOI] [PubMed] [Google Scholar]
- 16. Alexandrov LB, Kim J, Haradhvala NJ, et al. ; PCAWG Mutational Signatures Working Group; PCAWG Consortium . The repertoire of mutational signatures in human cancer. Nature. 2020; 578(7793):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ceccarelli M, Barthel FP, Malta TM, et al. ; TCGA Research Network . Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016; 164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1(6):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016; 18(3):379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018; 25(1):154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012; 26(12):1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010; 207(2):339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ward PS, Cross JR, Lu C, et al. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012; 31(19):2491–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. ; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain . Signatures of mutational processes in human cancer. Nature. 2013; 500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noushmehr H, Weisenberger DJ, Diefes K, et al. ; Cancer Genome Atlas Research Network . Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010; 17(5):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo C, Pirozzi CJ, Lopez GY, Yan H. Isocitrate dehydrogenase mutations in gliomas: mechanisms, biomarkers and therapeutic target. Curr Opin Neurol. 2011; 24(6):648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han S, Liu Y, Cai SJ, et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. 2020; 122(11):1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh A, Gurav M, Dhanavade S, Shetty O, Epari S. Diffuse glioma - rare homozygous IDH point mutation, is it an oncogenetic mechanism? Neuropathology. 2017; 37(6):582–585. [DOI] [PubMed] [Google Scholar]
- 30. Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012; 18(20):5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donehower LA, Soussi T, Korkut A, et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 2019; 28(5):1370–1384.e1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Appay R, Dehais C, Maurage CA, et al. ; POLA Network . CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019; 21(12):1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu VM, O’Connor KP, Shah AH, et al. The prognostic significance of CDKN2A homozygous deletion in IDH-mutant lower-grade glioma and glioblastoma: a systematic review of the contemporary literature. J Neurooncol. 2020; 148(2):221–229. [DOI] [PubMed] [Google Scholar]
- 34. Marker DF, Pearce TM. Homozygous deletion of CDKN2A by fluorescence in situ hybridization is prognostic in grade 4, but not grade 2 or 3, IDH-mutant astrocytomas. Acta Neuropathol Commun. 2020; 8(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shirahata M, Ono T, Stichel D, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018; 136(1):153–166. [DOI] [PubMed] [Google Scholar]
- 36. Fischer NW, Prodeus A, Gariépy J. Survival in males with glioma and gastric adenocarcinoma correlates with mutant p53 residual transcriptional activity. Jci Insight. 2018; 3(15):e121364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017; 36(28):3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bargonetti J, Prives C. Gain-of-function mutant p53: history and speculation. J Mol Cell Biol. 2019; 11(7):605–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Oijen MG, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000; 6(6):2138–2145. [PubMed] [Google Scholar]
- 40. Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010; 2(12):a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358(6381):15–16. [DOI] [PubMed] [Google Scholar]
- 42. Sammons MA, Nguyen TT, McDade SS, Fischer M. Tumor suppressor p53: from engaging DNA to target gene regulation. Nucleic Acids Res. 2020; 48(16):8848–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boettcher S, Miller PG, Sharma R, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019; 365(6453):599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene. 2004; 23(13):2330–2338. [DOI] [PubMed] [Google Scholar]
- 45. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010; 2(2):a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Soubannier V, Stifani S. NF-κB signalling in glioblastoma. Biomedicines. 2017; 5(2):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.