

Abstract
Cancer transcriptomes frequently exhibit RNA dysregulation. As the resulting aberrant transcripts may be translated into cancer-specific proteins, there is growing interest in exploiting RNA dysregulation as a source of tumor antigens and thus novel immunotherapy targets. Recent advances in high-throughput technologies and rapid accumulations of multi-omic cancer profiling data in public repositories have provided opportunities to systematically characterize RNA dysregulation in cancer and identify antigen targets for immunotherapy. However, given the complexity of cancer transcriptomes and proteomes, important conceptual and technological challenges exist. Here, we highlight the expanding repertoire of tumor antigens arising from RNA dysregulation and introduce multi-omic and big-data strategies for identifying optimal immunotherapy targets. We discuss extant barriers for translating these targets into effective therapies, as well as the implications for future research.
Keywords: Cancer immunotherapy, CAR-T, TCR, tumor antigen, transcriptome, proteome, RNA processing, RNA-seq, proteogenomics
RNA Dysregulation as a Source of Cancer Immunotherapy Targets
Transcriptomic and proteomic outputs of human cells are controlled by multiple RNA-level regulatory processes. Mechanisms such as pre-mRNA alternative splicing (AS, see Glossary) and RNA editing can generate multiple protein isoforms from a single gene, greatly expanding the coding capacity and protein repertoire of human cells. Cancer cells exhibit widespread abnormalities in RNA processing, including driver alterations that functionally contribute to cancer development and progression [1, 2]. Through prevalent RNA dysregulation, cancer cells can express a distinct set of transcripts and proteins, some of which may represent therapeutic targets.
Targeted cancer immunotherapy has had tremendous successes in treating aggressive malignancies, but important challenges and unmet needs remain [3, 4]. One critical aspect in developing immunotherapies with safe and effective antitumor immunity is to discover targetable tumor antigens (TAs). Current TA discovery efforts focus primarily on somatic mutations in protein-coding regions (i.e. neoantigens) [5], along with several well-known alternative sources such as lineage-specific antigens and cancer/testis antigens [3, 6]. These strategies search only a small fraction of proteomic variations in cancer cells and are ineffective for many cancer types [7]. Compelling evidence now suggests that TAs arising from RNA dysregulation may represent an extensive but largely unexplored repertoire of novel immunotherapy targets [1, 2, 7]. Through this review, we endeavor to raise awareness of this emerging class of TAs, with the goal of motivating conceptual and technological advances that translate these novel targets into new cancer immunotherapies.
Targeting Tumor Antigens for Adoptive Cell Therapies
By augmenting and engineering patients’ antitumor immunity, cancer immunotherapies with immune checkpoint blockade (ICB) and adoptive cell therapies (ACTs) (Figure 1A, Box 1) have created a paradigm shift in cancer treatment. Immunotherapies have achieved improved patient survival, durable response, and other benefits for aggressive and previously untreatable cancers [3]. TAs play critical roles in inducing strong antitumor immunity [6]. The clinical efficacy of ICB is thought to originate from its ability to reactivate T cell responses to TAs [6, 8]. In ACTs, T cells are engineered to recognize TAs through distinct mechanisms, either by targeting extracellular peptides of cell surface proteins for chimeric antigen receptor (CAR) T cell (CAR-T) therapy, or by targeting peptide epitopes presented on the cell surface via major histocompatibility complex (MHC) molecules for T-cell receptor (TCR) therapy (Figure 1A) [9].
Figure 1. Major Types of Cancer Immunotherapies and Conventional Workflow for Neoantigen Discovery.

(A) Illustration of two major types of cancer immunotherapies. Immune checkpoint blockade (ICB) (e.g., immune checkpoint inhibitors against PD-1) augments the suppressed immune response against tumor cells. Adoptive cell therapies (ACTs) direct or engineer immune cells to improve the antitumor immunity. Engineered ACTs include chimeric antigen receptor T-cell (CAR-T) and T-cell receptor (TCR) therapies, in which the patient’s T cells are engineered ex-vivo to target known tumor antigens (TAs).
(B) Conventional workflow for neoantigen discovery. In a conventional workflow, tumor and tumor-adjacent normal tissues from a patient are sampled. Next, whole-genome or whole-exome sequencing is performed to identify somatic mutations in tumor DNA. The tumor tissue is also optionally analyzed by RNA-seq or MS-based proteomics. Downstream computational analyses are used to prioritize candidate neoantigens. Utility of candidate neoantigens as immunotherapy targets is experimentally validated by testing their ability to bind or functionally activate T cells through the TCR. Abbreviations: MHC, major histocompatibility complex; PD-1, programmed cell death 1; PD-L1, programmed cell death 1 ligand 1.
Box 1. Major Types of Cancer Immunotherapy.
There are two major types of cancer immunotherapy [3] (see Figure 1A in main text). ICB works by augmenting the suppressed immune response against tumor cells. The ICB strategy uses immune checkpoint inhibitors (e.g., neutralizing antibodies against PD-1 or CTLA-4) to reactivate tumor-specific T cells. Therapeutic antibodies and ACTs work by directing or engineering immune cells to improve the antitumor immunity. Therapeutic antibodies (e.g., anti-CD20 rituximab) bind TAs directly to direct the immune response. For ACTs, the patient’s own immune cells are harvested and modified ex-vivo to enhance their antitumor ability [9]. CAR-T and TCR therapies are examples of engineered ACTs, in which the patient’s T cells are engineered ex-vivo to target known TAs. Successful cases of ACTs include using tumor-infiltrating lymphocyte (TIL) therapy to treat metastatic melanoma, and CD19 CAR-T therapy to treat B-cell acute lymphoblastic leukemia.
TAs can be generated by DNA, RNA, or protein alterations in cancer cells. Certain features of TAs are critical for ensuring immunotherapy success [6]. Ideally, the expression of TAs should be exclusive to cancer cells or restricted to non-vital cell lineages, a feature related to their expression selectivity and cancer specificity (Box 2). Another important consideration is the TA foreignness – the extent to which the antigen is recognized by the patient’s immune system as ‘non-self’ [6, 8, 10].
Box 2. Expression Selectivity/Cancer Specificity of TAs.
The expression selectivity and cancer specificity of TAs can be grouped into three major classes, based on previous definitions in the literature [6]. The most specific TAs are the tumor-specific antigens (TSAs), or ‘neoantigens’, which are expressed exclusively by tumor cells. TSAs offer the highest potential for effective targeting with low off-tumor toxicity but may be difficult to find for certain cancer types. Tumor-associated antigens (TAAs) are overexpressed in tumor cells but expressed (to some extent) in normal cells. TAAs are the least tumor-selective TA type. Although immunogenicity and toxicity are concerns, TAAs can be useful targets because they are easy to identify and may be generically expressed across patients and tumor types. ERBB2 (HER2/neu) is a prominent TAA in breast cancer. Cancer/testis antigens (CTAs) are present at elevated levels in tumors and reproductive tissues, while showing limited expression in normal adult tissues. CTAs can be therapeutically targeted by taking advantage of the fact that normal reproductive cells do not express MHC class I molecules. New York esophageal squamous cell carcinoma 1 (NY-ESO-1) is a well-known CTA expressed in several cancer types.
Advances in cancer genomic sequencing have enabled a clinically successful strategy for TA discovery focusing on somatic mutations (Figure 1B) [5, 6, 11]. Specifically, cancer genomic alterations – including single-nucleotide variations (SNVs), insertion-deletion variations, and gene fusions – can be detected by sequencing and comparing the tumor versus normal DNA of a given patient [11]. Protein sequence analyses can then identify somatic mutations in protein-coding regions that generate antigenic peptides.
Although this strategy is well-established, it has important limitations. Somatic mutations are usually specific to a small subset of patients, necessitating therapy development on an individualized (‘personalized’) basis [6]. Furthermore, only a small proportion of somatic mutations alter protein sequences and have strong immunogenicity, thus limiting the utility of this strategy for many cancer types with moderate or low mutation loads [5]. Given these inherent limitations, new types of TAs need to be systematically explored.
RNA Dysregulation-derived TAs as Targets for Immunotherapy
Post-transcriptional RNA processing plays key roles in regulating gene expression and generating the diverse transcript and protein isoforms required for essential biological functions [12]. RNA processing is governed by cis regulatory elements and trans regulatory factors, whose functions can be disrupted by somatic mutations and oncogenic signaling pathways in cancer [1, 2]. Dysregulation of RNA-related processes can reshape the cancer transcriptomic and proteomic landscape. Consequently, this dysregulation can modulate cellular phenotypes, by changing the abundance and diversity of transcripts and proteins (Figure 2). Some of these proteins could harbor cancer-specific antigenic peptides. Below, we summarize the main mechanisms of RNA dysregulation and recent progress in exploring RNA dysregulation as a source of cancer immunotherapy targets.
Figure 2. Mechanisms of RNA Dysregulation in Cancer.
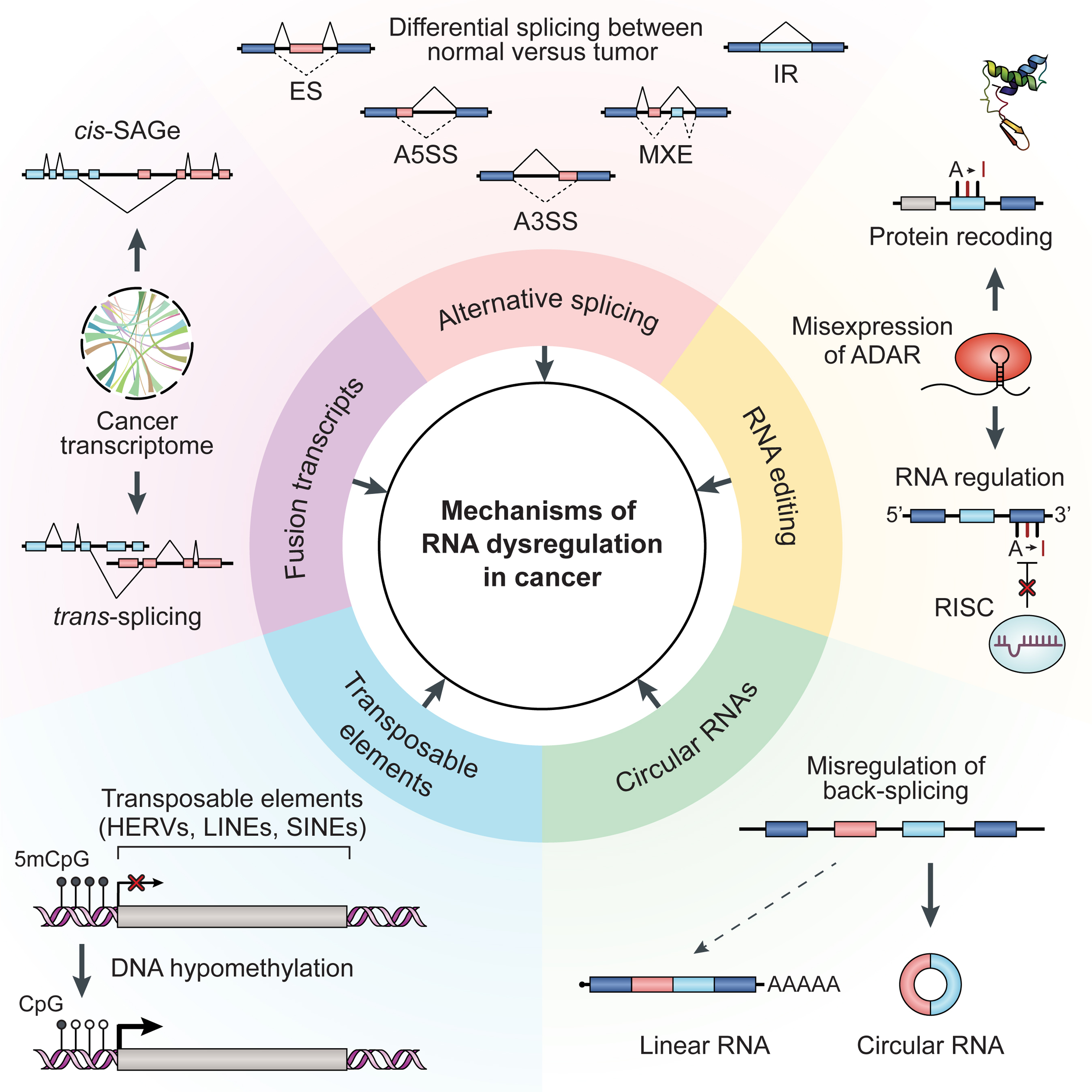
Dysregulation of RNA processing plays important roles in cancer development and progression. Aberrant RNA processing events that are important in cancer include alternative splicing, RNA editing, noncanonical splicing events producing circular RNAs and fusion transcripts, and expression of transposable elements. Abbreviations: 5mCpG, 5-methylcytosine-pG (methylated CpG); ADAR, adenosine deaminase acting on RNA; A3SS, alternative 3’ splice sites; A5SS, alternative 5’ splice sites; cis-SAGe, cis-splicing between adjacent genes; CpG, unmethylated CpG; ES, exon skipping; HERVs, human endogenous retroviruses; IR, intron retention; LINEs, long interspersed nuclear elements; MXE, mutually exclusive exons; RISC, RNA-induced silencing complex; SINEs, short interspersed nuclear elements.
Altered RNA Expression
Altered RNA expression is a major source of cancer transcriptome variation, sometimes playing essential oncogenic roles [13]. RNA overexpression in cancer cells can be caused by transcriptional or post-transcriptional processes that enhance gene transcription or RNA stability. If the corresponding protein product is consistently overexpressed across patients with a cancer type of interest, or even across multiple cancer types, and if the protein expression is low and sparse in normal tissues, then this protein may serve as a candidate TA [6]. Several clinically targeted TAs follow this pattern, with consistent overexpression in tumors and low or lineage-restricted expression in normal cells (e.g., CD19 [14], GPC2 [15]). However, issues of limited immunogenicity and off-tumor toxicity remain [3, 4].
Alternative Splicing
Pre-mRNA AS is a prevalent mechanism for diversifying gene products in human cells (Figure 2) [16]. AS is widespread in cancer transcriptomes and contributes to each of the ‘hallmarks of cancer’, the key phenotypic characteristics of cancer cells compared to normal cells [17, 18]. Cancer-associated AS can functionally modulate cancer development and progression through multiple mechanisms, such as by producing protein isoforms that promote cell proliferation, inhibit cell death, avoid antitumor immunity, or enable invasion and metastasis [17]. Additionally, AS events may predict patient prognosis and therapy response [2].
Alternatively spliced exons or splice junctions can be translated into antigenic peptides that are enriched in cancer cells [19]. Some of the cancer-associated exons or splice junctions may arise from somatic mutations creating novel splice sites that are not present in normal cells, making them highly cancer-specific [20]. Additionally, AS introduces larger sequence changes to the final protein products, potentially making AS-derived TAs more ‘foreign’ and immunogenic than single amino acid changes introduced by somatic mutations.
RNA sequencing (RNA-seq) studies of many cancer types have revealed transcriptome-wide changes in AS [13, 20, 21]. Comparing tumor tissues to normal tissues, Kahles et al. found elevated levels of AS in tumor tissues, including numerous previously unannotated AS events with the majority being exon skipping and alternative 3’ splice site events [21]. Considering only splice junction peptides as candidate TAs, they validated the expression of some peptides using mass spectrometry (MS) proteomics data and predicted candidate antigens based on expected binding to MHC class I molecules. However, immunogenicity of the candidate TAs was not tested experimentally. A recent pan-cancer study showed that aberrant cancer-specific exons may be introduced by splice site-creating somatic mutations within intronic sequences, through a process called exonization [13]. Dysregulation of intron retention, a specific type of AS, is also common in cancer and provides a mechanism for inactivating tumor suppressors [22]. Smart et al. experimentally validated MHC class I presentation of intron retention-derived peptides, suggesting their potential immunogenicity [23]. Collectively, these studies provide compelling evidence for AS as a promising source of novel immunotherapy targets. The repertoire of AS-derived TAs may be particularly large for certain cancer types and patients carrying specific cancer driver mutations, considering that genes encoding splicing regulatory factors are frequently mutated in cancer [2].
Not all AS isoforms are capable of generating protein products. The nonsense-mediated mRNA decay (NMD) pathway is an important mechanism of post-transcriptional regulation, degrading mRNA transcripts containing premature termination codons (PTCs) [24–26]. Novel AS isoforms in tumors may harbor PTCs, resulting in transcript degradation via NMD [7]. Nonetheless, evidence suggests that peptides generated during the pioneer round of mRNA translation prior to NMD may still be presented by the MHC class I pathway [27]. Additionally, the NMD machinery is known to be dysregulated in cancers [28], potentially allowing the translation of PTC-containing transcripts into protein products and TAs.
Noncanonical Splicing
Noncanonical splicing events can generate novel transcripts and proteins [29]. Chimeric RNAs (fusion transcripts) can be generated from trans-splicing between distant gene loci or cis-splicing between adjacent genes (cis-SAGe), in the absence of chromosomal rearrangements (Figure 2) [30]. Chimeric RNAs resulting from trans-splicing and cis-SAGe have been found in embryonic stem cells [31] and other non-cancer tissues [32]. Many chimeric RNAs have been identified as biomarkers or potential therapeutic targets for cancer [30]. If a cancer-specific chimeric RNA is translated into a protein, then the cancer-specific peptide derived from the chimeric RNA may be a candidate TA [33]. Multiple tools exist to detect chimeric RNAs [34]. However, the authenticity of chimeric RNAs detected from cancer RNA-seq data remains a concern, as many such RNAs could represent sequencing or detection artifacts [34]. The cancer specificity and immunogenicity of chimeric RNA-derived TAs must be evaluated, considering that chimeric RNAs are also expressed in normal tissues [32].
Circular RNAs (circRNAs) are a unique class of RNA molecules with a covalently closed loop, formed by back-splicing events during pre-mRNA splicing (Figure 2) [35]. CircRNAs perform biological functions in normal cells but can be dysregulated in cancer cells, resulting in distinct circRNA expression profiles in tumor tissues [36, 37]. Although circRNAs lack the 5’ cap, they can use mechanisms of cap-independent translation [35, 38, 39]. This raises the intriguing possibility that some cancer-specific circRNAs may be translated to generate antigenic peptides. Much is still unknown about the regulation and functions of circRNA in cancer [40]. However, researchers have created databases of cancer-associated circRNAs (e.g., MiOncoCirc [36]), with the hope that circRNAs can be used as biomarkers or therapeutic targets [40].
RNA Editing
RNA editing is a prevalent post-transcriptional regulatory process in human cells. The most abundant type of RNA editing is adenosine-to-inosine (A-to-I) RNA editing [41]. A-to-I RNA editing plays important roles in RNA regulation and protein recoding in normal biological processes, and its dysregulation underlies many human diseases including cancer (Figure 2) [42–44]. Recent large-scale cancer transcriptome studies investigated global RNA editing profiles across diverse cancer types [45, 46]. A-to-I RNA-editing is associated with clinically relevant features, including patient survival [45, 46], drug sensitivity [45], and cancer progression [47].
RNA editing contributes to proteomic diversity of cancer cells [48]. Using MS immunopeptidomic data and a T-cell-mediated cell-killing assay, Zhang et al. provided experimental evidence that RNA editing-derived peptides can be presented by MHC molecules and elicit immune responses [49]. RNA editing-derived TAs were recognized by tumor-infiltrating T cells physiologically present in tumor tissues. Nonetheless, a careful evaluation of cancer specificity and off-tumor toxicity is warranted, considering the somewhat promiscuous nature of RNA editing regulation by RNA editing enzymes [41].
Expressed Transposable Elements
Transposable elements (TEs) occupy approximately half of the human genome and have many roles in human biology and disease [50]. Dysregulation of TEs due to global loss of DNA methylation is common in human cancers (Figure 2) [51]. Hypomethylation of TE promoters allows epigenetically silenced TEs to be re-expressed in cancer. LINE-1 promoter hypomethylation has been associated with poor clinical features (e.g., poor prognosis, drug resistance, tumor aggressiveness) for several cancer types [51]. To systematically investigate the activation of TE promoters and its impact on gene expression, Jang et al. recently analyzed RNA-seq data from The Cancer Genome Atlas (TCGA) project for 7,769 tumor samples across 15 cancer types [52]. They showed that epigenetic reactivation of cryptic regulatory elements within TEs drives oncogene activation [52]. TE-associated transcripts may be derived entirely from expressed TEs as independent transcriptional units, or partially from TE-derived alternative promoters and first exons as part of host genes [53].
Peptides derived from aberrantly expressed TEs may be foreign to the immune system and immunogenic. Integrating RNA-seq data with MS immunopeptidomic data, Laumont et al. found that aberrantly expressed but nonmutated transcripts derived from endogenous TEs can be translated into targetable TAs [54]. Similarly, Kong et al. assessed TE expression in cancer transcriptomes and demonstrated that peptides derived from TEs such as HERV, LINE, SINE, and SVA elements are presented on MHC class I molecules [55]. One difficulty in studying expressed TEs is the accurate discovery and quantitation of TE-derived transcripts, as the repetitive nature of their sequences across the human genome poses a challenge for standard short-read RNA-seq [53]. Future studies using long-read RNA-seq may provide a better picture of expressed TEs and their resulting TA repertoire in cancer [56].
Multi-omic and Big-data Strategies to Discover RNA Dysregulation-Derived TAs
Due to the complexity of the human transcriptome and proteome, reliable discovery of RNA dysregulation-derived TAs is technically difficult. At minimum, an effective strategy must: 1) precisely characterize transcript sequences and abundances in cancer, 2) reliably identify translated protein products, 3) robustly determine the expression selectivity and cancer specificity of putative targets, and 4) systematically evaluate the likelihood that identified cancer-specific peptides can be targeted by immunotherapies. However, several challenges exist. For cancer-specific transcripts that are unannotated or derived from complex RNA processing events, it is not trivial to identify the exact full-length RNA sequences and their corresponding protein products. Furthermore, it is difficult to ascertain the expression selectivity and cancer specificity of candidate TAs (see Box 2), an important feature for the efficacy and off-tumor toxicity of targeted immunotherapies. For TCR targets, epitope prediction is still an actively researched topic, and existing tools have limited accuracy and reproducibility [57]. For CAR-T targets, cell surface localization and topology of proteins need to be predicted and confirmed.
Given these challenges, an integrated multi-omic and big-data strategy incorporating state-of-the-art experimental and computational tools, as well as large-scale reference data resources for tumor and normal tissues, is needed for comprehensive discovery and prioritization of RNA dysregulation-derived TAs. Below, we describe tools that can be used, improved, and synthesized to create an integrated TA discovery workflow (Figure 3, Key Figure).
Figure 3, Key Figure. Multi-omic and Big-data Strategies to Discover RNA Dysregulation-Derived Immunotherapy Targets.
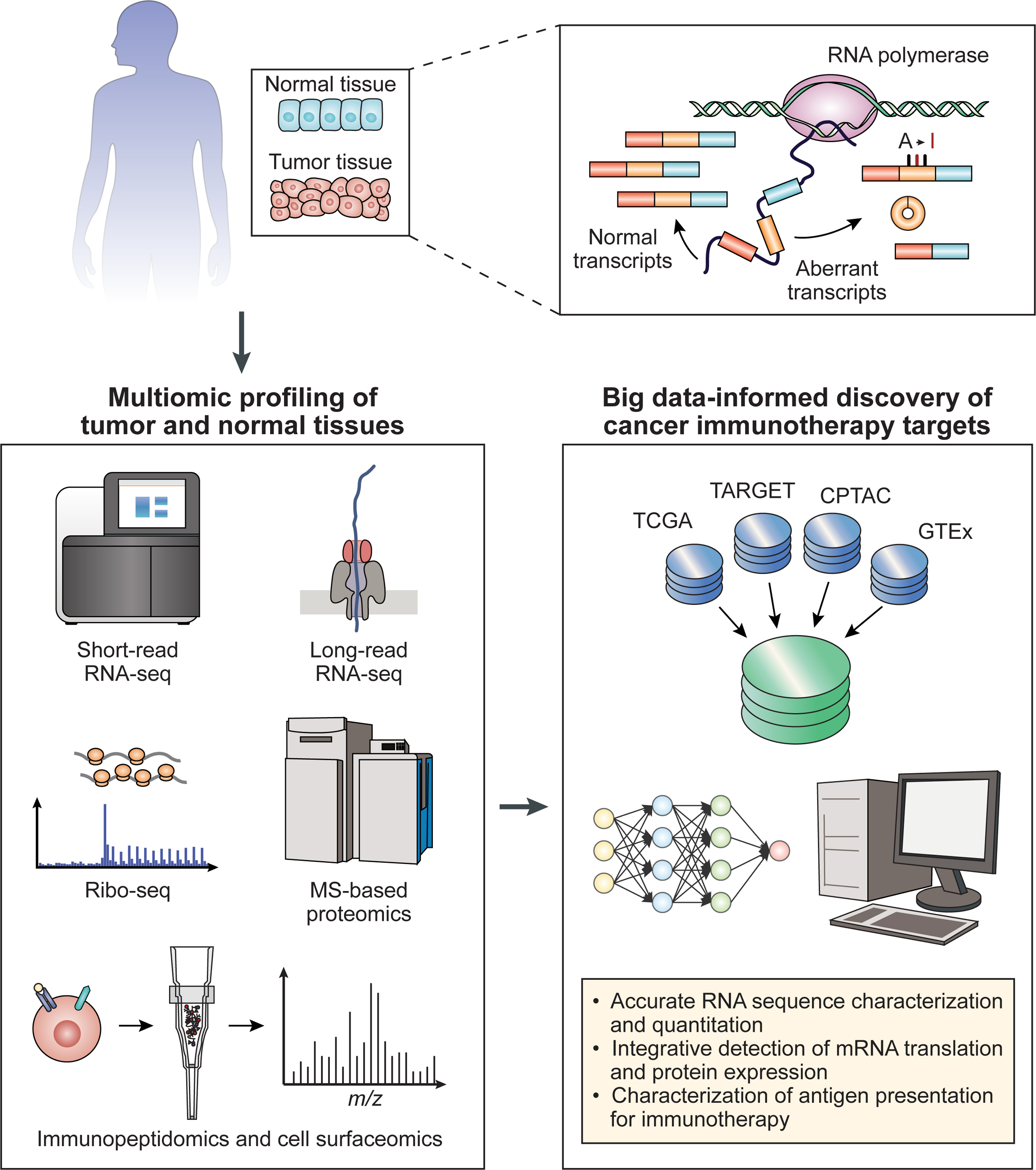
In an integrated workflow, tumor and tumor-adjacent normal tissues from a patient are subject to multi-omic profiling, followed by a big data-informed computational analysis to discover RNA dysregulation-derived immunotherapy targets. In addition to standard short-read RNA-seq, patient tissues may be subject to long-read RNA-seq, for accurate RNA sequence characterization and quantitation combining short-read and long-read RNA-seq data. Ribo-seq and MS-based proteomics enable integrative detection of mRNA translation and protein expression. Immunopeptidomics and cell surfaceomics characterize antigen presentation for immunotherapy. A big data-informed computational analysis should efficiently integrate these multi-omic data on patient tissues with large-scale reference data resources for tumor and normal tissues (Table 1), to guide the selection of candidate TAs with low off-tumor toxicity and broad clinical applicability. Abbreviations: CPTAC, Clinical Proteomic Tumor Analysis Consortium; GTEx, Genotype-Tissue Expression; MS, mass spectrometry; Ribo-seq, ribosome profiling; TARGET, Therapeutically Applicable Research to Generate Effective Treatments; TCGA, The Cancer Genome Atlas.
Accurate RNA Sequence Characterization and Quantitation
Short-read RNA-seq is the most widely used tool to profile the human transcriptome, with extensive applications in large-scale molecular profiling of cancer and normal tissues [33, 58, 59]. In addition to quantifying overall gene expression levels, short-read RNA-seq detects many types of transcript alterations, such as AS and RNA editing. For this reason, short-read RNA-seq is the primary approach used today to study dysregulated RNA processing in cancer transcriptomes [33].
Advanced computational and statistical algorithms have been developed to accurately analyze short-read RNA-seq data [33, 58]. These include general algorithms to align RNA-seq reads and quantify gene expression levels, as well as specialized tools to characterize specific types of transcript alterations (e.g., AS, RNA editing, chimeric RNAs, circRNAs, etc.). Combining RNA-seq analysis with downstream analytical tools for protein inference and antigen prediction, researchers can detect candidate TAs derived from multiple types of RNA dysregulation [21, 23, 49, 54, 55].
Despite the broad applications and successes of short-read RNA-seq in cancer research, this approach falls short in the inference of complex RNA processing events and their resulting transcript and protein products [60]. Reliable inference of protein products requires knowledge of full-length transcripts. Because short-read RNA-seq only examines fragments of full-length transcripts, it can be difficult to infer the protein products that correspond to the identified RNA processing events, especially for novel, unannotated events. Certain types of cancer-associated transcripts, such as chimeric RNAs and expressed TEs, are particularly challenging to characterize by short-read RNA-seq [34, 56].
In recent years, there has been a growing interest in long-read RNA-seq technologies that can overcome many of the inherent limitations of short-read RNA-seq [58]. Short-read RNA-seq typically generates reads of up to 600 bases. By contrast, long-read RNA-seq on third-generation sequencing platforms, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), can generate reads exceeding 10 kb [60]. Thus, long-read RNA-seq could be an ideal tool for resolving full-length transcript structures and highly repetitive transcript sequences [56, 58, 60]. Long-read RNA-seq is also useful for accurate inference of open reading frames (ORFs) and reliable identification of potential NMD targets. Despite its potential, long-read RNA-seq remains an under-used technology for human transcriptome analysis due to its high error rate and low throughput, along with the associated computational challenges [60]. Nonetheless, with continued technological advances, we expect that long-read RNA-seq will become a powerful and broadly used tool for cancer transcriptome analysis and TA discovery. For example, a recent study used long-read RNA-seq to identify aberrant AS isoforms and potential TAs in non-small cell lung cancer [61].
Integrative Detection of mRNA Translation and Protein Expression
Not all transcripts are translated into stable protein products [62]. For TA discovery, it is important to confirm the protein expression of cancer-specific transcripts discovered by transcriptome analysis. Several experimental and computational approaches provide evidence for mRNA translation and protein expression. These approaches can be incorporated into a TA discovery workflow.
Ribo-seq is an RNA-seq-based ribosome profiling strategy in which ribosome-protected mRNA fragments are captured and sequenced [63]. Ribo-seq has been widely used to profile ribosome association with different RNA species and to quantify mRNA translational efficiency, providing insights into the role of translational control in regulating protein expression [64, 65]. Ribo-seq is an especially powerful tool for identifying ORFs, as Ribo-seq signals within translated ORFs display unique characteristics [66–68]. Therefore, Ribo-seq can greatly constrain the search space and reduce the false positive rate during the in-silico search for candidate TAs. Indeed, a recent Ribo-seq study identified MHC class I-bound peptides from thousands of novel unannotated ORFs [69].
MS-based proteomics characterizes protein expression and modifications at a global scale [70]. Various sample preparation and labeling methods, combined with different MS instruments, have been developed for analyzing protein expression, modifications, protein-protein interactions, and other proteome features. MS-based proteomics can be applied to whole-cell proteomes, or specific subsets such as MHC-bound peptides (immunopeptidome [71]) and cell surface proteins (cell surfaceome [72]). Large-scale studies have generated MS-based proteomics data across cancer and normal tissues [73–76]. These data provide important resources for confirming protein expression of putative TAs identified from RNA-seq data.
Proteogenomics is an increasingly popular approach for peptide identification, in which MS-based proteomics data are augmented with corresponding genomic and/or transcriptomic data [77, 78]. In MS-based proteomics, peptide identification requires searching MS spectra against a library (database) of known proteins. Thus, unlike RNA-seq, which can discover novel events, MS-based proteomics relies on the completeness and accuracy of the MS search library, as proteins not included in the search library cannot be detected [79]. Customized sample-specific search libraries, generated based on transcriptomics data (e.g., RNA-seq and Ribo-seq data), can substantially increase detection rates and reduce false positives [77, 78]. Indeed, as a data-driven approach, proteogenomics can enable protein-level detection of unannotated events (e.g., novel AS or RNA editing events), which would be absent in a standard MS search library. Additionally, transcriptomics data can be used to remove protein products corresponding to non-expressed (RNA-seq) or non-translated (Ribo-seq) mRNAs from the MS search library, thereby improving peptide identification. A number of studies have applied proteogenomics to examine cancer transcriptomes and proteomes, resulting in the identification of novel cancer-specific peptides and epitopes [21, 23, 49, 54, 55, 80]. Proteogenomics could be particularly useful for identifying products of “atypical” translational events, such as proteins translated from circRNAs [39] or from PTC-containing transcripts evading NMD [81]. Additionally, this approach can be applied in a retrospective manner to existing MS-based proteomics datasets, to confirm expressed proteins derived from novel types of RNA dysregulation in cancer cells.
Characterization of Antigen Presentation for Immunotherapy
Not all peptides from expressed proteins are accessible to T cells via TCRs or CARs. To be recognized, a peptide must be presented by MHC molecules or located on the cell surface for recognition by TCRs or CARs, respectively [9]. To confirm antigen presentation, the immunopeptidome or cell surfaceome can be profiled using specialized MS-based proteomics approaches. Immunopeptidomics examines peptides bound to antigen-presenting molecules, providing experimental evidence for peptide presentation by MHC molecules on the cell surface [71]. This approach has gained popularity in immuno-oncology as a tool to discover and validate TAs [23, 54, 82]. Cell surfaceomics is used to profile expression of proteins on the cell surface. Using this technique, researchers have discovered cell surface proteins as TAs for CAR-T therapies of advanced prostate cancers [72]. To facilitate peptide identification, immunopeptidomics and cell surfaceomics data may be paired with RNA-seq data in a proteogenomic workflow [23, 54]. As MS-based approaches, both immunopeptidomics and cell surfaceomics are limited by modest sensitivity [83]. Nonetheless, they hold promise for many applications in antigen discovery and validation for targeted immunotherapies.
Big Data-informed Discovery and Prioritization of RNA Dysregulation-derived TAs
Massive datasets containing DNA sequencing, RNA-seq, and proteomics data from tumor and normal tissues along with clinical annotations have been accumulated in publically available repositories (Table 1). These datasets provide a rich resource for discovery and prioritization of TAs derived from RNA dysregulation.
Table 1.
Publically Available Genomic, Transcriptomic, and Proteomic Datasets for Cancer Research.a
| Project name | Sample source | Phenotype | Data typesb | Sample size |
|---|---|---|---|---|
| The Cancer Genome Atlas (TCGA)i | Tissue-derived | Adult tumor & adjacent normal | WES, RNA-seq, Other | >10,000 |
| Clinical Proteomic Tumor Analysis Consortium (CPTAC)ii | Tissue-derived | Adult tumor & adjacent normal | MS proteomics, WGS, WES, RNA-seq, Other | ~3,000 (Ongoing) |
| Cancer Cell Line Encyclopedia (CCLE)iii | Cell lines | Adult & pediatric tumor | WGS, WES, RNA-seq, Other | ~1,000 |
| International Cancer Genome Consortium (ICGC)iv | Tissue-derived | Adult tumor | WGS, WES, RNA-seq, Other | >24,000 donors (86 cancer projects) |
| Therapeutically Applicable Research to Generate Effective Treatments (TARGET)v | Tissue-derived | Pediatric tumor | WGS, WES, RNA-seq, Other | ~1,700 |
| St. Jude Pediatric Cancer Genome Project (PCGP)vi | Tissue-derived | Pediatric tumor | WGS, WES, RNA-seq | ~2,000 |
| Human Tumor Atlas Network (HTAN) vii | Tissue-derived | Adult & pediatric tumor | WES, bulk & single-cell RNAseq, Other | Ongoing |
| Genotype-Tissue Expression (GTEx)viii | Tissue-derived & cell lines | Adult normal | WGS, RNA-seq, Other | >17,000 (>900 individuals) |
Abbreviations: WES, whole-exome sequencing; WGS, whole-genome sequencing.
Data types marked as ‘Other’ refer to data other than DNA sequencing, RNA sequencing, or MS proteomics data, e.g., methylation profiling, miRNA profiling, kinome sequencing, ChIP-seq, etc.
Efficient integration of large-scale multi-omic data is critical for solving complex problems in cancer transcriptomics and immunotherapy. Trained on big data, advanced statistical and machine-learning models can enhance our ability to interpret data and make accurate predictions. For instance, the sensitivity of RNA-seq-based AS analysis can be enhanced by using deep-learning models trained on large-scale RNA-seq data covering many cellular states and perturbation conditions [84]. Similarly, the prediction accuracy for antigen binding to MHC class I and II molecules can be improved by deep-learning models trained on immunopeptidomics data combined with RNA-seq data [85, 86].
Just as reference genomes aid in the discovery of somatic mutations [87], large-scale reference RNA-seq and proteomics datasets of tumor and normal tissues can aid in the discovery and prioritization of RNA dysregulation-derived TAs. Each tumor sample may harbor thousands of aberrantly expressed RNAs with protein-coding potential. Thus, candidate TAs must be efficiently prioritized. By querying large-scale reference datasets of tumor and normal tissues, one can assess and rank TAs based on critical features, including their expression selectivity and cancer specificity. Similarly, by examining data from the same cancer type, one can discover ‘public’ TAs commonly shared by patients. These features would guide the selection of candidate TAs with low off-tumor toxicity and broad clinical applicability for therapeutic development.
Concluding Remarks and Future Perspectives
RNA dysregulation provides a potentially extensive source of TAs for targeted immunotherapy. Specific patterns of AS, chimeric transcripts, circRNAs, RNA editing, and expressed TEs are known to be altered in cancer transcriptomes, thereby generating a large repertoire of cancer-specific proteins. Nonetheless, substantial conceptual barriers and technological challenges need to be addressed before these candidate TAs can be reliably identified and translated into effective therapies. While neoantigens derived from somatic mutations have been extensively characterized [5], RNA dysregulation as a source of TAs represents an emerging concept that is still under active exploration. As a result, existing studies often use ad hoc criteria to define and report this class of TAs. Integrated computational platforms for identifying RNA dysregulation-derived TAs are lacking, and there is no well-established standard or go-to tool yet to identify this class of TAs. In this review, we outline a conceptual framework along with general principles for identifying RNA dysregulation-derived TAs. We anticipate that these discussions will help motivate the development of new experimental strategies and computational tools, which are highly demanded by the research community.
Several mechanistic and immunological features of RNA dysregulation-derived TAs remain obscure (see Outstanding Questions). Most importantly, the expression selectivity and cancer specificity of candidate TAs must be systematically examined to ensure high immunogenicity and low off-tumor toxicity. Current studies primarily choose datasets in an ad hoc manner to define normal transcriptome profiles for common RNA processing events (e.g., AS [19]). Future work should establish standardized transcriptome references for different types of RNA dysregulation and patient populations. For example, GTEx is a commonly used reference database of normal transcriptome profiles [21, 88], but the GTEx RNA-seq data are exclusively on adult tissues [59]. Transcriptome references for normal pediatric tissues would be instrumental for TA discovery in pediatric cancers. New single-cell RNA-seq (scRNA-seq) datasets [89, 90] may further increase the resolution of transcriptome references by adding cell type-specific and spatial information. Importantly, as these transcriptome references may be collected from disparate data sources, standardized and reproducible computational workflows and best practices are needed to control for biological and technical confounding factors, including batch effects, and to aggregate such heterogeneous data into a unified resource.
Outstanding Questions.
What strategies can be used to systematically determine the expression selectivity and cancer specificity of RNA dysregulation-derived tumor antigens (TAs)?
How do we establish standardized transcriptome references for different types of RNA dysregulation and distinct patient populations?
What computational infrastructure (i.e., data standards, best practices, and centralized data repositories) is needed to enable discovery of RNA dysregulation-derived cancer immunotherapy targets?
What are the mechanisms (cis versus trans) by which RNA dysregulation-derived TAs are generated?
Can TAs derived from RNA dysregulation be lost during treatment, leading to therapy resistance and relapse?
How does the clonality of RNA dysregulation-derived TAs affect the immune response and treatment outcome?
What technologies must be developed for preclinical validation of CAR-T and TCR targets arising from RNA dysregulation?
A critical question for developing immunotherapies against RNA dysregulation-derived TAs is the stability of their expression in response to treatment. AS has been reported as a mechanism for antigen escape and treatment resistance in response to CD19-targeted CAR-T therapy [91]. In a study of patients with relapsed B-ALL following CD19 CAR-T therapy, Sotillo et al. reported that CD19 mRNA can be alternatively spliced, causing loss of the epitope required for CAR-T recognition as well as loss of cell surface expression of the CD19 protein (Figure 4) [91]. Extrapolating from this finding, it is conceivable that a TA arising from AS or other types of RNA dysregulation may be lost in cancer as a resistance mechanism during the course of treatment. To begin to address this question, we must understand how a given TA is generated. Cancer-specific aberrant transcripts may result from somatic mutations disrupting cis regulatory elements, or oncogenic pathways and cellular signals altering the expression or activity of trans regulatory factors. It is possible that TAs derived from these different mechanisms (cis versus trans) may have different trajectories in response to treatment. One possible strategy to target ‘stable’ TAs is to identify and select aberrant transcripts that functionally drive cancer development. Alternatively, it is possible to target transcripts directly regulated by oncogenic drivers. For example, Phillips et al. recently identified approximately 500 AS events consistently regulated by the MYC oncogene across multiple cancer types [92]. As MYC plays a critical role in many aggressive cancers, these MYC-regulated AS events may represent attractive targets for immunotherapies. A related issue regarding treatment resistance is antigen clonality [8]. In the context of RNA dysregulation-derived TAs, it is important to know the portion of tumor cells expressing a given TA. The heterogeneity of RNA processing events has not been well characterized at the single-cell level [93]. Future development and application of isoform-resolved single-cell transcriptomics technologies (e.g., single-cell long-read RNA-seq [94]) may decipher the clonal structure of cancer transcriptomes at the isoform resolution and aid in TA discovery and prioritization.
Figure 4. Alternative splicing of CD19 as a mechanism for antigen escape and treatment resistance in response to CD19-targeted CAR-T therapy.

In a study of patients with relapsed B-ALL following CD19 CAR-T therapy, Sotillo et al. (2015) identified an increased level of alternatively spliced CD19 mRNA transcripts lacking exon 2 (CD19 Δex2) in relapsed tumors [91]. Production of the exon 2 skipping isoform can be caused by cis-acting somatic mutations in the exon and/or altered expression of a trans-acting splicing factor. This CD19 Δex2 mRNA isoform can generate a truncated CD19 protein isoform lacking the epitope required for CAR-T recognition as well as lacking cell surface expression of the CD19 protein, thus resulting in treatment resistance and relapse. Purple box represents CD19 exon 2. Yellow star represents somatic mutations in the exon.
Important technological and computational problems for TA discovery remain to be addressed. Antigen prediction algorithms require further upgrades. Algorithms trained on in vivo data from state-of-the-art epitope detection methods (e.g., immunopeptidomics) outperform older-generation prediction tools for antigen binding to MHC molecules [85, 86], but there is still substantial room for improvement. Similarly, transcript and protein identification remain a challenge in genomics and computational biology. Long-read RNA-seq coupled with multi-omic data integration (e.g., proteogenomics) may enable a more comprehensive and accurate characterization of cancer transcriptomes and proteomes, thus enhancing TA discovery.
This review focuses on strategies to discover and prioritize candidate TAs derived from RNA dysregulation. However, such candidate TAs must be rigorously tested at the preclinical stage. To examine the immunogenicity and efficacy of TCR targets, MHC multimer-based assays and other T-cell functional assays are needed. For CAR targets, it is important to confirm stable expression and topology of candidate TAs on the cell surface. The efficacy and potential toxicity of TCR or CAR-T therapies need to be assessed in model systems. If such studies are successful, subsequent clinical development could see new targeted cancer immunotherapies reach the clinic.
Highlights.
Discovery of optimal tumor antigens (TAs) is key for developing targeted immunotherapies with safe and effective antitumor immunity. Novel sources of TAs (beyond somatic mutations) are needed to expand the scope of current therapies.
Dysregulation of RNA processing reshapes the cancer transcriptome and proteome by changing the abundance and diversity of gene products. Antigens arising from RNA dysregulation are potential cancer immunotherapy targets.
Recent advances in high-throughput technologies and rapid accumulations of multi-omic cancer profiling data in public repositories have provided opportunities to systematically characterize RNA dysregulation in cancer.
To comprehensively discover and prioritize TAs derived from RNA dysregulation, we propose an integrated multi-omic and big data strategy incorporating state-of-the-art experimental and computational tools as well as large-scale reference data resources for tumor and normal tissues.
Acknowledgments
This research was supported by funds from NIH/National Cancer Institute under Awards R01CA220238, U01CA233074, U24CA232979, and P50CA092131; and the Parker Institute for Cancer Immunotherapy, Grant 20163828.
GLOSSARY
- Alternative mRNA splicing (AS)
A regulated biological process during gene expression, in which alternative choices of exons or splice sites generate multiple transcript isoforms from a single gene. AS is a major source of transcript and protein diversity in human cells. There are five basic types (exon skipping, alternative 5′ splice sites, alternative 3′ splice sites, mutually exclusive exons, and intron retention), as well as more complex types
- Chimeric RNAs (or fusion transcripts)
Transcripts generated from trans-splicing between distant genes or cis-splicing between adjacent genes. These RNA products are called ‘chimeric’ or ‘fusion’ transcripts because they comprise exons from different genes
- Circular RNAs (circRNAs)
Closed-loop RNAs resulting from the back-splicing of exons within pre-mRNA
- Foreignness
Ability of an antigen to be recognized as ‘non-self’ or ‘foreign’ by the host immune system. Foreignness is one important feature in determining the immunogenicity of an antigen
- Immunogenicity
Ability of an antigen to induce an immune response
- Immunopeptidome (or MHC/HLA peptidome/ligandome)
Collection of short peptides presented on the cell surface by MHC/HLA molecules
- Nonsense-mediated mRNA decay (NMD)
An mRNA surveillance mechanism to degrade transcripts containing premature termination codons (PTCs)
- RNA editing
RNA processing mechanism whereby RNA undergoes site-specific nucleotide modifications. The most common type of RNA editing is adenosine (A)-to-inosine (I) editing, in which adenosine deaminase acting on RNA (ADAR) enzymes catalyze conversion of A to I
- Surfaceome
Collection of proteins found on the cell surface
- Transposable elements (TEs)
DNA sequences that can move from one position to another in the genome. Approximately half of the human genome is occupied by TEs
- Tumor antigen (TA)
Molecular entity (usually a peptide) on the tumor capable of eliciting an immune response by the host
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: Disclaimer Statement
Y.X. is a scientific cofounder of Panorama Medicine and consulted for PACT Pharma. O.N.W. currently has consulting, equity, and/or board relationships with Trethera Corporation, Kronos Biosciences, Sofie Biosciences, Breakthrough Properties, Vida Ventures, Nammi Therapeutics, Two River, Iconovir, and Allogene Therapeutics. A.R. has received honoraria from consulting with Amgen, Bristol-Myers Squibb, Chugai, Genentech, Merck, Novartis, and Roche, and is or has been a member of the scientific advisory board and holds stock in Advaxis, Arcus Biosciences, Bioncotech Therapeutics, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Lutris Pharma, Merus, PACT Pharma, Rgenix, and Tango Therapeutics. None of these companies contributed to or directed the writing of this review article.
References
- 1.Obeng EA et al. (2019) Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov 9 (11), 1493–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cherry S and Lynch KW (2020) Alternative splicing and cancer: insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev 34 (15–16), 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldman AD et al. (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol [DOI] [PMC free article] [PubMed]
- 4.Bonifant CL et al. (2016) Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 3, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schumacher TN and Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348 (6230), 69–74. [DOI] [PubMed] [Google Scholar]
- 6.Yarchoan M et al. (2017) Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 17 (4), 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith CC et al. (2019) Alternative tumour-specific antigens. Nat Rev Cancer 19 (8), 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keenan TE et al. (2019) Genomic correlates of response to immune checkpoint blockade. Nat Med 25 (3), 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg SA and Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348 (6230), 62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richman LP et al. (2019) Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade. Cell Syst 9 (4), 375–382 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richters MM et al. (2019) Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med 11 (1), 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Licatalosi DD and Darnell RB (2010) RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 11 (1), 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.PCAWG Transcriptome Core Group et al. (2020) Genomic basis for RNA alterations in cancer. Nature 578 (7793), 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grupp SA et al. (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368 (16), 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosse KR et al. (2017) Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell 32 (3), 295–309 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park E et al. (2018) The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am J Hum Genet 102 (1), 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oltean S and Bates DO (2014) Hallmarks of alternative splicing in cancer. Oncogene 33 (46), 5311–8. [DOI] [PubMed] [Google Scholar]
- 18.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 19.Frankiw L et al. (2019) Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol 19 (11), 675–687. [DOI] [PubMed] [Google Scholar]
- 20.Jayasinghe RG et al. (2018) Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep 23 (1), 270–281 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahles A et al. (2018) Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34 (2), 211–224 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung H et al. (2015) Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 47 (11), 1242–8. [DOI] [PubMed] [Google Scholar]
- 23.Smart AC et al. (2018) Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 36 (11), 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendell JT et al. (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36 (10), 1073–8. [DOI] [PubMed] [Google Scholar]
- 25.Lindeboom RG et al. (2016) The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 48 (10), 1112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindeboom RGH et al. (2019) The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat Genet 51 (11), 1645–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Apcher S et al. (2011) Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc Natl Acad Sci U S A 108 (28), 11572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popp MW and Maquat LE (2018) Nonsense-mediated mRNA Decay and Cancer. Curr Opin Genet Dev 48, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sibley CR et al. (2016) Lessons from non-canonical splicing. Nat Rev Genet 17 (7), 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia Y et al. (2016) Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2 (9), 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu CS et al. (2014) Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res 24 (1), 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babiceanu M et al. (2016) Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res 44 (6), 2859–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cieslik M and Chinnaiyan AM (2018) Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet 19 (2), 93–109. [DOI] [PubMed] [Google Scholar]
- 34.Haas BJ et al. (2019) Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol 20 (1), 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kristensen LS et al. (2019) The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20 (11), 675–691. [DOI] [PubMed] [Google Scholar]
- 36.Vo JN et al. (2019) The Landscape of Circular RNA in Cancer. Cell 176 (4), 869–881 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S et al. (2019) Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 176 (4), 831–843 e22. [DOI] [PubMed] [Google Scholar]
- 38.Pamudurti NR et al. (2017) Translation of CircRNAs. Mol Cell 66 (1), 9–21 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y et al. (2017) Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res 27 (5), 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J et al. (2020) Circular RNAs in Cancer: Biogenesis, Function, and Clinical Significance. Trends Cancer 6 (4), 319–336. [DOI] [PubMed] [Google Scholar]
- 41.Nishikura K (2016) A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17 (2), 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walkley CR and Li JB (2017) Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18 (1), 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eisenberg E and Levanon EY (2018) A-to-I RNA editing - immune protector and transcriptome diversifier. Nat Rev Genet 19 (8), 473–490. [DOI] [PubMed] [Google Scholar]
- 44.Xu X et al. (2018) The role of A-to-I RNA editing in cancer development. Curr Opin Genet Dev 48, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han L et al. (2015) The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 28 (4), 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paz-Yaacov N et al. (2015) Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep 13 (2), 267–76. [DOI] [PubMed] [Google Scholar]
- 47.Chen L et al. (2013) Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med 19 (2), 209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng X et al. (2018) A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 33 (5), 817–828 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang M et al. (2018) RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat Commun 9 (1), 3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chuong EB et al. (2017) Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18 (2), 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burns KH (2017) Transposable elements in cancer. Nat Rev Cancer 17 (7), 415–424. [DOI] [PubMed] [Google Scholar]
- 52.Jang HS et al. (2019) Transposable elements drive widespread expression of oncogenes in human cancers. Nat Genet 51 (4), 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lanciano S and Cristofari G (2020) Measuring and interpreting transposable element expression. Nat Rev Genet [DOI] [PubMed]
- 54.Laumont CM et al. (2018) Noncoding regions are the main source of targetable tumor-specific antigens. Science Translational Medicine 10, eaau5516. [DOI] [PubMed] [Google Scholar]
- 55.Kong Y et al. (2019) Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat Commun 10 (1), 5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shahid S and Slotkin RK (2020) The current revolution in transposable element biology enabled by long reads. Curr Opin Plant Biol 54, 49–56. [DOI] [PubMed] [Google Scholar]
- 57.(2017) The problem with neoantigen prediction. Nat Biotechnol 35 (2), 97. [DOI] [PubMed] [Google Scholar]
- 58.Stark R et al. (2019) RNA sequencing: the teenage years. Nat Rev Genet 20 (11), 631–656. [DOI] [PubMed] [Google Scholar]
- 59.GTEx Consortium et al. (2017) Genetic effects on gene expression across human tissues. Nature 550 (7675), 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amarasinghe SL et al. (2020) Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21 (1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oka M et al. (2021) Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol 22 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schuller AP and Green R (2018) Roadblocks and resolutions in eukaryotic translation. Nat Rev Mol Cell Biol 19 (8), 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ingolia NT et al. (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7 (8), 1534–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ingolia NT (2014) Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet 15 (3), 205–13. [DOI] [PubMed] [Google Scholar]
- 65.Brar GA and Weissman JS (2015) Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 16 (11), 651–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reixachs-Sole M et al. (2020) Ribosome profiling at isoform level reveals evolutionary conserved impacts of differential splicing on the proteome. Nat Commun 11 (1), 1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calviello L et al. (2020) Quantification of translation uncovers the functions of the alternative transcriptome. Nat Struct Mol Biol 27 (8), 717–725. [DOI] [PubMed] [Google Scholar]
- 68.Zhang P et al. (2017) Genome-wide identification and differential analysis of translational initiation. Nat Commun 8 (1), 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ouspenskaia T et al. (2020) Thousands of novel unannotated proteins expand the MHC I immunopeptidome in cancer. bioRxiv, 2020.02.12.945840
- 70.Aebersold R and Mann M (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537 (7620), 347–55. [DOI] [PubMed] [Google Scholar]
- 71.Caron E et al. (2015) Analysis of Major Histocompatibility Complex (MHC) Immunopeptidomes Using Mass Spectrometry. Mol Cell Proteomics 14 (12), 3105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JK et al. (2018) Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc Natl Acad Sci U S A 115 (19), E4473–E4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gillette MA et al. (2020) Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182 (1), 200–225 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dou Y et al. (2020) Proteogenomic Characterization of Endometrial Carcinoma. Cell 180 (4), 729–748 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim MS et al. (2014) A draft map of the human proteome. Nature 509 (7502), 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilhelm M et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509 (7502), 582–7. [DOI] [PubMed] [Google Scholar]
- 77.Nesvizhskii AI (2014) Proteogenomics: concepts, applications and computational strategies. Nat Methods 11 (11), 1114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruggles KV et al. (2017) Methods, Tools and Current Perspectives in Proteogenomics. Mol Cell Proteomics 16 (6), 959–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nesvizhskii AI and Aebersold R (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics 4 (10), 1419–40. [DOI] [PubMed] [Google Scholar]
- 80.Wen B et al. (2020) Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat Commun 11 (1), 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Laumont CM et al. (2016) Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun 7, 10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shao W et al. (2018) The SysteMHC Atlas project. Nucleic Acids Res 46 (D1), D1237–D1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Timp W and Timp G (2020) Beyond mass spectrometry, the next step in proteomics. Sci Adv 6 (2), eaax8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z et al. (2019) Deep-learning augmented RNA-seq analysis of transcript splicing. Nat Methods 16 (4), 307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sarkizova S et al. (2020) A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol 38 (2), 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen B et al. (2019) Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol 37 (11), 1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ellrott K et al. (2018) Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6 (3), 271–281 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan Y et al. (2019) IRIS: Big data-informed discovery of cancer immunotherapy targets arising from pre-mRNA alternative splicing. bioRxiv, 843268. [DOI] [PMC free article] [PubMed]
- 89.Rozenblatt-Rosen O et al. (2020) The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181 (2), 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Regev A et al. (2017) The Human Cell Atlas. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sotillo E et al. (2015) Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5 (12), 1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Phillips JW et al. (2020) Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc Natl Acad Sci U S A 117 (10), 5269–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arzalluz-Luque A and Conesa A (2018) Single-cell RNAseq for the study of isoforms-how is that possible? Genome Biol 19 (1), 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gupta I et al. (2018) Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat Biotechnol. </References> [DOI] [PubMed]