

Abstract
Sites of early neuropathologic change provide important clues regarding the initial clinical features of Alzheimer’s disease (AD). We have shown significant reductions in hippocampal synaptic density in participants with AD, consistent with the early degeneration of entorhinal cortical (ERC) cells that project to hippocampus via the perforant path. In this study, [11C]UCB-J binding to synaptic vesicle glycoprotein 2A (SV2A) and [18F]flortaucipir binding to tau were measured via PET in 10 participants with AD (5 mild cognitive impairment, 5 mild dementia) and 10 cognitively normal participants. In the overall sample, ERC tau was inversely associated with hippocampal synaptic density (r=−0.59, P=0.009). After correction for partial volume effects, the association of ERC tau with hippocampal synaptic density was stronger in the overall sample (r=−0.61, P=0.007) and in the AD group where the effect size was large, but not statistically significant (r=−0.58, P=0.06). This inverse association of ERC tau and hippocampal synaptic density may reflect synaptic failure due to tau pathology in ERC neurons projecting to the hippocampus.
Keywords: SV2A, tau, synaptic density, Alzheimer’s disease, [11C]UCB-J, PET, [18F]flortaucipir
1. Background
In Alzheimer’s disease (AD), the sites of early neuropathologic change provide important clues regarding the initial clinical features of the disease. Postmortem work has established that the earliest cortical tau pathology and neuronal loss in AD occurs in the entorhinal cortex (ERC), likely contributing to memory impairment (Braak, et al., 2011,Gomez-Isla, et al., 1996). Since these degenerating neurons project via the perforant path to all fields of the hippocampal formation, including the dentate gyrus, Cornu Ammonis (CA) fields, and the subiculum, the earliest and largest loss of synapses would be expected in the hippocampus. Indeed, those postmortem studies that have examined the prodromal or mild stages of AD have reported large reductions in the outer molecular layer of dentate gyrus (Masliah, et al., 1994,Scheff, et al., 2006), as well as the CA1 field (Scheff, et al., 2007).
With the recent advent of synaptic Positron Emission Tomography (PET) imaging, we have begun to evaluate synaptic alterations and their neuropathologic correlates in vivo. Synaptic vesicle glycoprotein 2A (SV2A) is expressed in virtually all synapses and is located in synaptic vesicles at presynaptic terminals (Bajjalieh, et al., 1994,Bajjalieh, et al., 1993) with a conserved expression pattern of about 5 copies of SV2A per vesicle (Mutch, et al., 2011). Binding of [11C]UCB-J to SV2A may thus serve as an indirect marker of synapses. In our initial study of [11C]UCB-J PET in early AD (Chen, et al., 2018a), we reported reduced hippocampal SV2A binding in AD compared with cognitively normal (CN) participants. In a more recent study, we analyzed a substantially larger sample of participants across a somewhat broader range of disease using [11C]UCB-J PET and observed widespread reductions of synaptic density in medial temporal and neocortical brain regions in early AD compared to CN participants (Mecca, et al., 2020). Our findings are consistent with those of two other studies in which SV2A binding was found to be reduced in AD participants, most prominently in the hippocampus (Bastin, et al., 2019,Vanhaute, et al., 2020).
PET imaging affords the unique opportunity to examine the relationship between regional tau deposition and regional synaptic loss in vivo in early AD. This approach may ultimately provide insights into the sequence of early pathologic events. Neurofibrillary tangle pathology has been theorized to precede and lead to synaptic loss (Iqbal and Grundke-Iqbal, 2002); however, this temporal relationship is generally viewed as uncertain (Serrano-Pozo, et al., 2010). Previous research has examined the relationship between tau deposition and synaptic loss [reviewed in Jadhav 2015 (Jadhav, et al., 2015)] in mouse models (de Calignon, et al., 2009,Kimura, et al., 2010,Spires-Jones, et al., 2008) and human postmortem studies (Bussiere, et al., 2003b,Falke, et al., 2003,Gomez-Isla, et al., 1997,Hof, et al., 2003,Ingelsson, et al., 2004,Iqbal and Grundke-Iqbal, 2002,Montero-Crespo, et al., 2021). PET imaging has allowed extension of this work to in vivo human studies. One recent study found an inverse association between tau deposition measured with [18F]MK-6240 PET and synaptic density measured with [11C]UCB-J PET within the medial temporal lobe (Vanhaute, et al., 2020). However, no information was provided about associations between [18F]MK-6240 and [11C]UCB-J uptake between individual medial temporal lobe regions.
In the present investigation, we used [18F]flortaucipir PET imaging to measure brain tau deposition and [11C]UCB-J PET to measure synaptic density in CN participants and those with AD. We analyzed the association of tau deposition in the ERC with synaptic density in the hippocampus. Since synaptic density may be confounded by volume loss, we also conducted an exploratory analysis of the association between ERC tau and hippocampal volume and performed partial volume correction (PVC). Finally, we explored the association between ERC tau and synaptic density across a broader range of brain regions.
2. Methods
Detailed methods are described in the Supplemental Material.
2.1. Study Participants and Design
Participants aged 55–85 years underwent a screening diagnostic evaluation to ensure eligibility. Individuals with AD dementia were required to meet diagnostic criteria for probable dementia due to AD (McKhann, et al., 2011), have a Clinical Dementia Rating (CDR) score of 0.5–1.0, and a Mini-Mental Status Examination (MMSE) score ≤26. Participants with mild cognitive impairment (MCI) were included if they met diagnostic criteria for amnestic MCI (Albert, et al., 2011), have a CDR score of 0.5, and a MMSE score of 24–30, inclusive. Participants with AD dementia and MCI were included if they had impaired episodic memory as evidenced by a Logical Memory II (LMII) score 1.5 standard deviations below an education-adjusted norm. CN participants were required to have a CDR score of 0, a MMSE score >26, and a normal education-adjusted LMII score. All participants received a PET scan with [11C]Pittsburgh Compound B ([11C]PiB) to determine the presence of brain amyloid-β (Aβ) accumulation as previously described (Chen, et al., 2018b). All participants provided written informed consent as approved by the Yale University Human Investigation Committee prior to participating in the study.
2.2. Brain Imaging
T1-weighted magnetic resonance imaging (MRI) was performed to define regions of interest (ROI), and to perform PVC using the Iterative Yang approach (Erlandsson, et al., 2012,Shidahara, et al., 2017). PET scans were performed on the HRRT (207 slices, resolution <3 mm FWHM) (de Jong, et al., 2007) with event-by-event motion correction (Jin, et al., 2013). Dynamic [11C]PiB scans were acquired for 90 min following administration of a bolus of up to 555 MBq of tracer (Mecca, et al., 2017). Dynamic [18F]flortaucipir ([18F]AV1451 / TAUVID™, Avid Radiopharmaceuticals/ Eli Lilly and Company) scans were acquired from 80 to 100 min after administration of a bolus of up to 370 MBq (Pontecorvo, et al., 2017). Dynamic [11C]UCB-J scans were acquired for 60 min after administration of a bolus of up to 740 MBq (Finnema, et al., 2018). Dynamic [11C]PiB and [11C]UCB-J PET images were motion corrected (FSL-FLIRT) and then registered to the participant’s MRI. [18F]Flortaucipir PET images did not undergo additional motion correction (beyond that of event-by-event) due to low brain uptake in healthy participants. Visual inspection indicated that interframe registration was accurate without additional software motion correction. MRI cortical reconstruction and volumetric segmentation was performed using FreeSurfer [version 6.0] (Fischl, 2012). See Supplemental Tables 1 and 2 for a list of ROIs.
2.3. Tracer Kinetic Modeling
For [18F]flortaucipir, we computed SUVR images by summing the image frames from 80 to 100 min and dividing by the summed signal in an inferior cerebellum gray matter reference region (Baker, et al., 2017). For [11C]UCB-J, we generated parametric images of BPND using a simplified reference tissue model – 2 step (SRTM2) (Wu and Carson, 2002) and a small ROI (2 mL) in the core of the centrum semiovale (CS) as reference region (Mertens, et al., 2019,Rossano, et al., 2019). The use of SRTM2 requires a global value (clearance rate constant, k2, of the reference region), which was computed as a population average of k2 of the CS obtained using the 1TC model (; from a previous group of subjects with arterial blood sampling) (Mecca, et al., 2020). DVR values using a whole cerebellum reference region were computed for each voxel as (BPND +1)/(BPND[cerebellum]+1) as previously described (Mecca, et al., 2020) and further validated (O’Dell, et al., 2021). PVC images of [18F]flortaucipir SUVR and [11C]UCB-J DVR utilized a non-PVC reference region.
2.4. Statistical analyses
Statistical methods are detailed in the Supplemental Materials. Briefly, χ2 test were used for group comparisons of categorical variables and unpaired t-tests for continuous variables. For the primary hypotheses, univariate linear regression analysis and Pearson’s correlations were used to assess the association between ERC tau (SUVR) and hippocampal synaptic density (DVR). Since synaptic density may be confounded by volume loss, we also conducted an exploratory analysis of the association between ERC tau and hippocampal volume and performed PVC. The Benjamini-Hochberg procedure was used to control the false discovery rate (FDR) for multiple comparisons (3 comparisons for all participants, AD only, and CN only) diagnostic groups). Primary hypothesis testing was one-tailed. For sensitivity analyses, separate multiple linear regression models were fit that also included covariates of age, sex, and years of education. For secondary analyses, linear mixed models were used to compare [18F]flortaucipir SUVR or [11C]UCB-J DVR between AD and CN groups in multiple ROIs. Post-hoc comparisons utilized unpaired t-tests with FDR correction for multiple comparisons (16 comparisons for ROIs). Exploratory analyses assessed the relationship between ERC-tau and regional synaptic density using Pearson’s correlations. Correction for multiple comparisons were not performed for exploratory analyses and hypothesis testing was two-tailed. For display, Pearson’s r (effect size) maps were created by producing images with the voxels in each FreeSurfer region set uniformly to the calculated effect size for that region. In all analyses, P < 0.05 was used as a threshold for significance.
3. Results
3.1. Participant characteristics
The study sample consisted of 20 participants–10 who were CN and 10 with amnestic MCI due to AD or mild AD dementia. Diagnostic groups were well balanced for age, sex, and education (Table 1). AD participants had clinical characteristics typical of amnestic MCI and mild AD dementia with MMSE=23.1 ± 4.1 and CDR = 0.7 ± 0.3. The interscan interval for [11C]UCB-J and [18F]flortaucipir PET scans did not differ between the CN (5.8 ± 7.8 weeks) and AD (8.8 ± 8.8 weeks) groups (Table 1). APOE genotypes reflected expected patterns with higher copy numbers of the APOE ε4 allele in AD participants. Two of the CN participants were amyloid positive based on [11C]PiB PET criteria. Thus, the overall sample provided a spectrum of AD stages, including CN amyloid negative (n=8), CN amyloid positive (n=2), amnestic MCI due to AD (n=5), AD dementia (n=5).
Table 1.
Participant characteristics and clinical assessments
| Cognitively Normal | Alzheimer’s Disease | P | |
|---|---|---|---|
| Participants (n) | 10 | 10 (mild dementia: 5, MCI: 5) | - |
| Sex (F/M) | 6/4 | 6/4 | 1.0 |
| Age (years) | 72.1 (7.9) (61–81) | 68.8 (6.6) (58–79) | 0.32 |
| Education (years) | 17.7 (1.9) (14–20) | 16.6 (2.0) (13–19) | 0.22 |
| CDR-global | 0 (0) | 0.7 (0.3) (0.5–1.0) | < 0.00001 |
| CDR-SB | 0 (0) | 3.6 (1.4) (0.5–5.0) | < 0.00001 |
| MMSE | 29.3 (1.1) (27–30) | 23.1 (4.1) (14–28) | 0.0002 |
| LMII | 12.3 (4.4) (7–19) | 1.9 (2.5) (0–8) | < 0.00001 |
| RAVLT-delay | 8.8 (2.1) (6–12) | 2.4 (2.8) (0–9) | 0.00002 |
| Amyloid +/− | 2/8 | 10/0 | - |
| Interscan Interval (weeks) | 5.8 (7.8) (0.6–25.7) | 8.8 (8.8) (1.2 – 26.0) | 0.44 |
| APOE genotype (n) | 0.09 | ||
| ε3ε3 | 5 | 3 | - |
| ε2ε3 | 2 | 0 | |
| ε3ε4 | 3 | 5 | - |
| ε4ε4 | 0 | 2 | - |
Data are mean (SD) (range) for continuous variables. P values are for an unpaired t-test (continuous variables) or χ2 test (categorical variables). CDR-global, clinical dementia rating global score; CDR-SB, clinical dementia rating sum of boxes; MMSE, Mini-Mental State Examination; LMII, logical memory II; RAVLT, Rey Auditory Verbal Learning Test. Interscan Interval is the time between [11C]UCB-J and [18F]flortaucipir PET scans
3.2. Relationship between ERC tau and hippocampal synaptic density
For the primary analyses, we focused on the relationship between ERC tau deposition (SUVR) and hippocampal synaptic density (SV2A DVR). AD compared to CN participants demonstrated significantly higher ERC tau deposition (AD SUVR = 1.68 ± 0.16, CN SUVR = 1.11 ± 0.12, P < 0.00001) and significantly lower hippocampal SV2A binding (AD DVR = 0.88 ± 0.13, CN DVR = 1.01 ± 0.11, P = 0.02). In the overall sample that included CN and AD participants, consistent with our primary hypothesis, ERC tau demonstrated significant inverse associations with hippocampal synaptic density (r = −0.59, P = 0.009, Figure 1A, Table 2), but also with hippocampal volume (r = −0.55, P = 0.02, Figure 1B, Table 2). PVC strengthened the associations for both hippocampal synaptic density (r = −0.61, P = 0.007, Supplemental Figure 1A, Table 2) and volume (r = −0.62, P = 0.0006). Within the CN and AD groups individually, ERC tau was not associated with either hippocampal synaptic density or volume (Figure 1, Table 2). In the AD group, PVC also increased the strength of the associations of ERC tau with hippocampal synaptic density (r = −0.58, P = 0.06) and volume (r = −0.44, P = 0.15) (Supplemental Figure 1, Table 2). The strength of association was large for ERC tau with synaptic density and medium for ERC tau with hippocampal volume, but neither was significant in this small sample. Finally, additional multiple variable linear regression models that included covariates of age, sex, and education did not alter the results.
Figure 1. Association between ERC tau and hippocampal synaptic density or hippocampal volume.
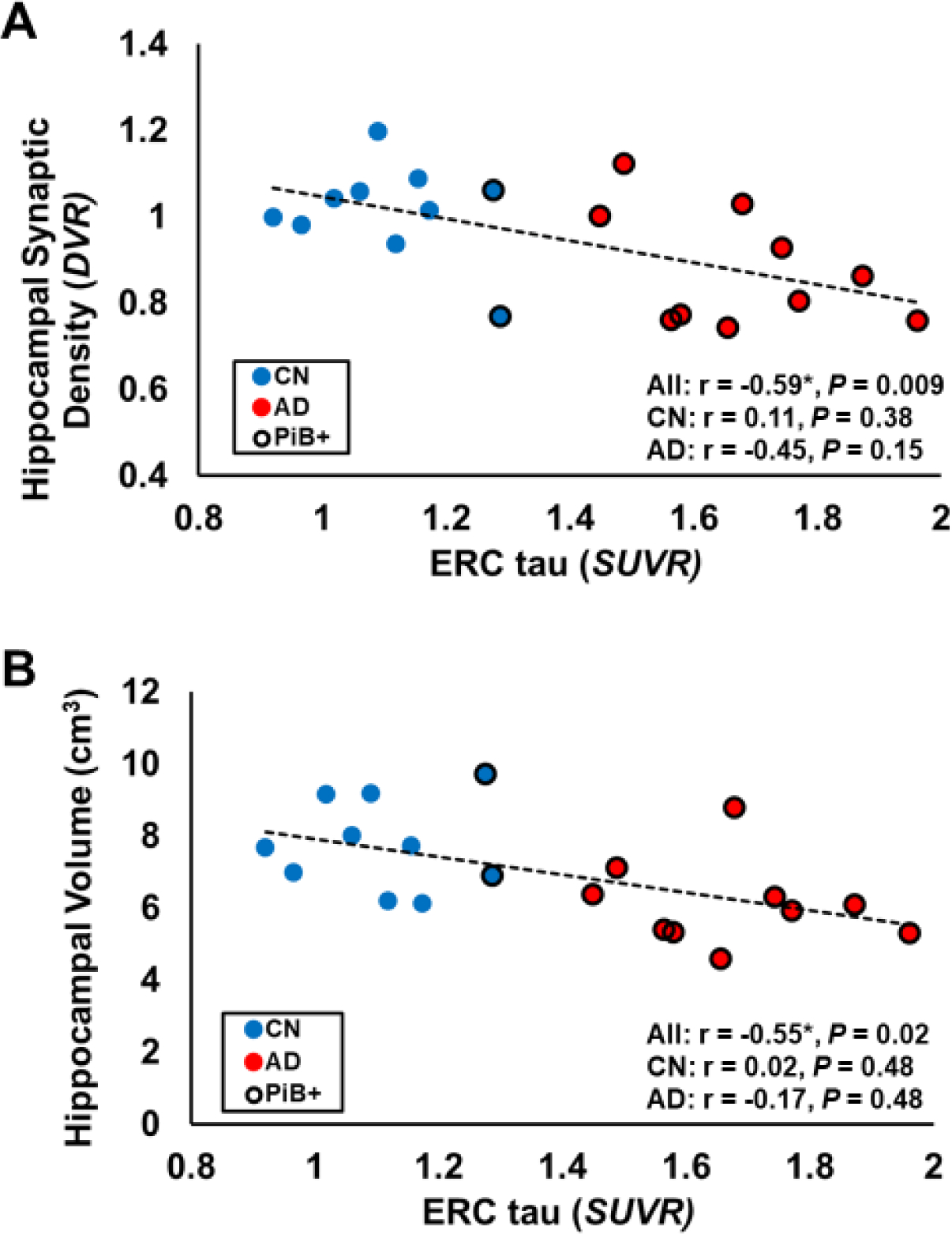
Higher ERC tau [18F]flortaucipir (SUVR) was significantly associated with (A) lower hippocampal synaptic density ([11C]UCB-J DVR) and (B) hippocampal volume in the overall sample that included both cognitively normal (n = 10) and Alzheimer’s disease (n = 10) participants. The figure displays a linear regression line. Values of r and P are for Pearson’s correlations with false discovery rate correction for 3 comparisons and one-tailed hypothesis testing. Amyloid positive individuals (PiB+) are indicated by a black circle border. * P<0.05 for Pearson’s correlation. Abbreviations: ERC, entorhinal cortical; SUVR, standard uptake value ratio of [18F]flortaucipir calculated with an inferior cerebellum reference region; DVR, distribution volume ratio of [11C]UCB-J calculated with a whole cerebellum reference region; All, all participants; CN, cognitively normal; AD, Alzheimer’s disease; PiB, [11C]Pittsburg h Compound B.
Table 2.
Correlation of ERC-tau (SUVR) and Hippocampal Synaptic Density (DVR) or Volume (cm3)
| All (n = 20) | AD (n = 10) | CN (n = 10) | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| PVC | Pearson’s r | P | Pearson’s r | P | Pearson’s r | P | |
| ERC tau with Hippocampal Synaptic Density | No | −0.59 | 0.009 | −0.45 | 0.15 | 0.11 | 0.38 |
| Yes | −0.61 | 0.007 | −0.58 | 0.06 | 0.16 | 0.33 | |
| ERC tau with Hippocampal Volume | No | −0.55 | 0.02 | −0.17 | 0.48 | 0.02 | 0.48 |
| Yes | −0.62 | 0.006 | −0.44 | 0.15 | −0.07 | 0.42 | |
P-values for Pearson’s r are false discovery rate corrected for 3 comparisons (All, AD, CN). All P-values are one-tailed since there were directional hypotheses. Abbreviations: ERC, entorhinal cortical; SUVR, standard uptake value ratio of [18F]flortaucipir calculated with an inferior cerebellum reference region; DVR, distribution volume ratio of [11C]UCB-J calculated with a whole cerebellum reference region; All, all participants; CN, cognitively normal; AD, Alzheimer’s disease; PVC, partial volume correction.
3.3. Pattern of tau and synaptic density differences in AD compared to CN groups
Next, we characterized group differences more broadly by comparing tau deposition (SUVR) and synaptic density reductions (DVR) between the AD and CN groups. A linear mixed model analysis of tau, including multiple regions spanning the Braak pathological stages of AD (Braak and Braak, 1991), demonstrated a significant effect of group (F(1,18) = 20.9, P < 0.0002) and group*region (F(15,270) = 5.2, P = < 0.0001) as predictors of tau deposition (SUVR). Post-hoc comparisons with correction for multiple comparisons revealed significantly greater tau deposition in AD compared to CN participants in all tested regions except for the medial occipital cortex (Figure 2A, Supplemental Table 3). These group differences were apparent when visualizing the average [18F]flortaucipir SUVR images registered and overlaid on the MNI template (Figure 3A). A second linear mixed model analysis for synaptic density demonstrated no significant effect of group (F(1,18) = 1.1, P = 0.30), but a significant effect of group*region (F(15,270) = 2.6, P = 0.001) as predictors of synaptic density (DVR). Although the primary analysis indicated that hippocampal synaptic density was significantly lower in AD (as compared to CN), post-hoc comparisons with correction for multiple comparisons were not significant for any tested regions (Figure 2B, Supplemental Table 3). Nonetheless, non-significant group differences in synaptic density (DVR) can be appreciated by visualizing the average [11C]UCB-J DVR images registered and overlaid on the MNI template (Figure 3B).
Figure 2. Comparison of tau deposition and synaptic density in AD and CN groups.
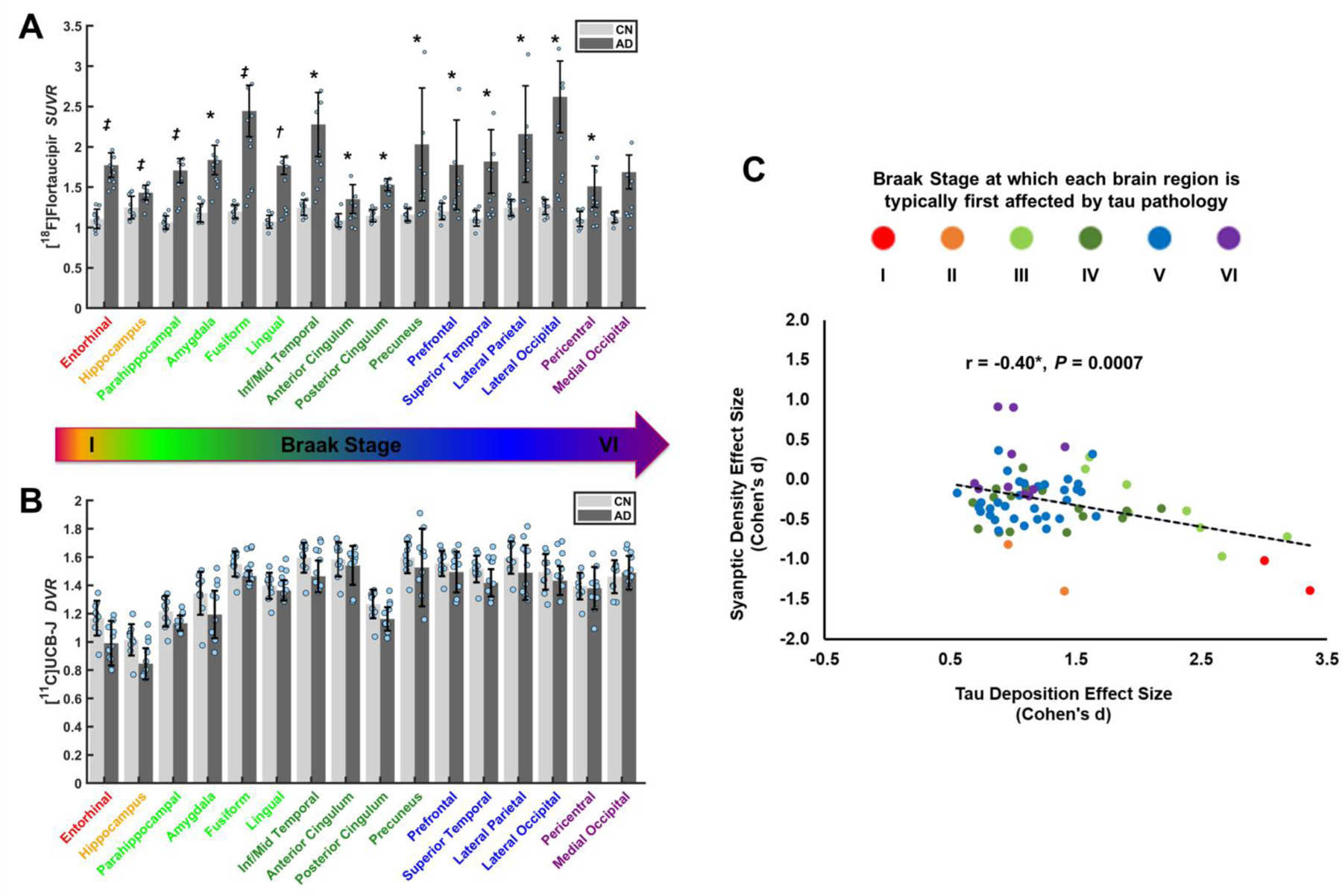
(A) Tau deposition was measured using [18F]flortaucipir SUVR and (B) synaptic density with [11C]UCB-J DVR. * P<0.05, † P<0.001, ‡ P<0.0001 for post hoc t-tests comparing AD (n=10) and CN (n=10) groups after false discovery rate correction for multiple comparisons. Dots represent the SUVR or DVR for each participant. Error bars represent standard deviations. (C) The effect sizes (Cohen’s d) to detect a group difference (AD < CN) in each brain region were plotted for tau deposition and synaptic density. * P<0.05 for Pearson’s correlation. Dots represent 70 brain regions that comprise the Braak stage regions form A and B. Colors represent Braak stages at which each brain region is typically first affected by tau pathology. Abbreviations: SUVR, standard uptake value ratio of [18F]flortaucipir calculated with an inferior cerebellum reference region; DVR, distribution volume ratio of [11C]UCB-J calculated with a whole cerebellum reference region; CN, cognitively normal, AD, Alzheimer’s disease; Inf/Mid Temporal, inferior and middle temporal gyri.
Figure 3. Average tau and synaptic density in AD and CN groups.
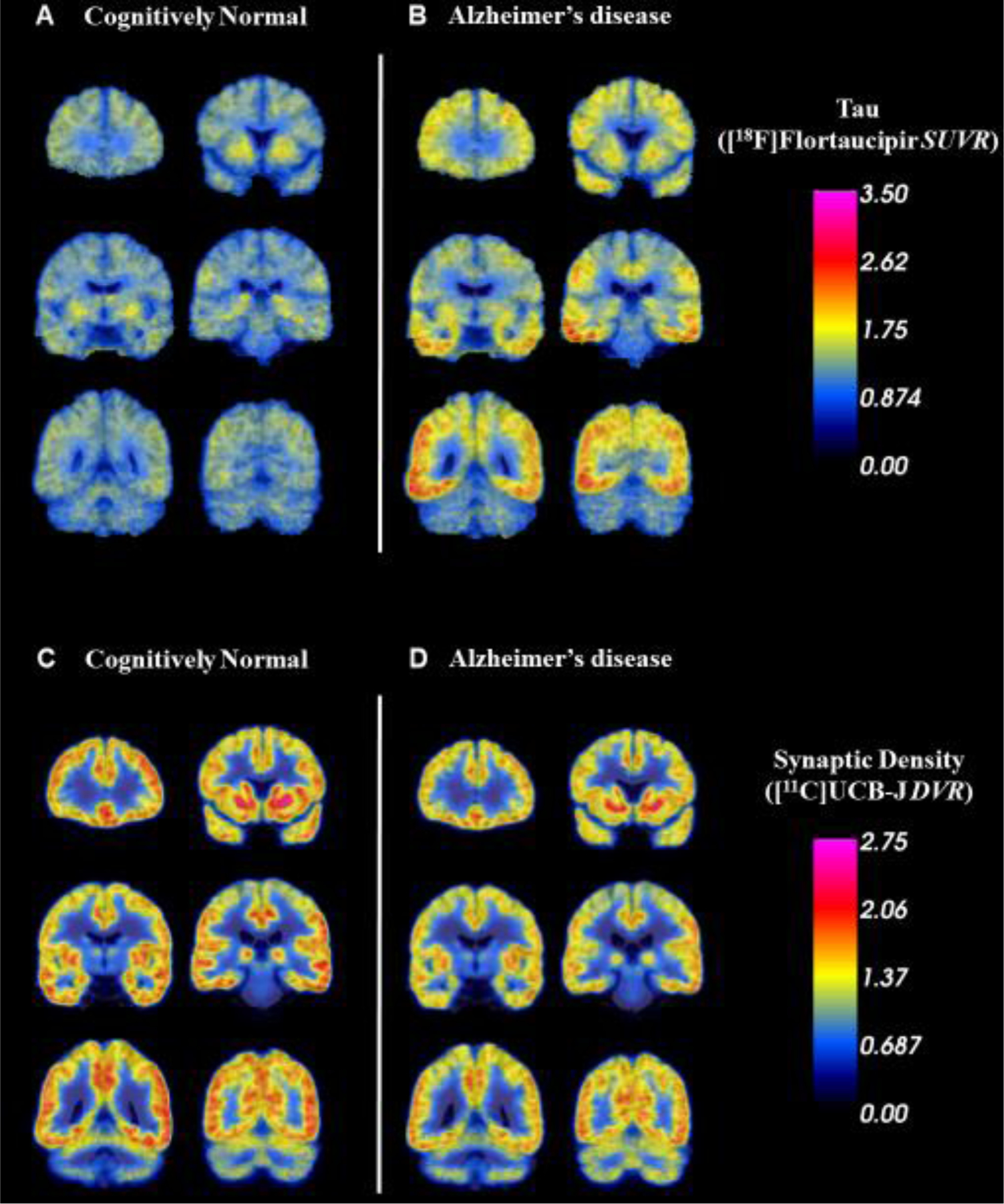
Coronal sections of average parametric images of tau deposition ([18F]flortaucipir SUVR) in 10 CN (A) and 10 AD (B) participants and of synaptic density ([11C]UCB-J DVR) in 10 CN (C) and 10 AD (D) participants. Average images were created after co-registration to a common MNI template. The average parametric PET scans are displayed in pseudocolor and overlaid on the MNI template T1 MRI. Higher tau deposition and lower synaptic density are apparent in AD compared to CN participants. Abbreviations: SUVR, standard uptake value ratio of [18F]flortaucipir calculated with an inferior cerebellum reference region; DVR, distribution volume ratio of [11C]UCB-J calculated with a whole cerebellum reference region.
The majority of our correlational analyses were circuit-based—tau deposition in ERC compared to synaptic density in hippocampus (3.2) and other regions (3.4). However, to provide a broad characterization of the relationship between AD-related tau deposition and AD-related synaptic loss intra-regionally, we also compared the effect size (Cohen’s d) to detect a difference between CN and AD groups for both tau deposition and synaptic density for each of the brain regions spanning the range of Braak stages listed in Supplemental Table 2. We observed a significant negative association between the effect sizes for tau deposition and synaptic density (r = 0.40, P = 0.0007, Figure 2C), i.e., the brain regions with the greatest AD-related tau deposition also had the greatest AD-related synaptic loss. Moreover, we found a general tendency for larger effect sizes—and thus more robust associations—in regions already affected in early Braak stages (Figure 2C), although the hippocampus (Braak II region) appeared to be a relative exception to this relationship.
3.4. ERC tau associations with synaptic density in all brain regions
Additional exploratory analyses were performed to assess the association of ERC tau with synaptic density in all ipsilateral FreeSurfer subcortical and Desikan-Killiany atlas regions. Pearson’s r was calculated for the correlation between ERC tau and synaptic density in all regions for all participants (Figure 4A), CN participants (Figure 4B), and participants with AD (Figure 4C). For these analyses, left and right hemisphere regions were analyzed separately. Similar correlation maps were also produced after PVC of both [18F]flortaucipir and [11C]UCB-J PET scans (Figure 4D–F). Images were thresholded for regions that had a P < 0.05. These exploratory analyses were not corrected for multiple comparisons. Consistent with the primary regional analysis, in the entire sample, ERC tau was inversely associated with synaptic density in the hippocampi (Figure 4A, Supplemental Figure 2A–B). These inverse associations extended to several other regions (Supplemental Figure 2C–G): left entorhinal cortex, left amygdala, left banks of the superior temporal sulcus, left middle temporal gyrus, and left pars opercularis. ERC tau was not associated with regional synaptic density in the CN group (Figure 4B). Interestingly, in analyses restricted to the AD group, higher ERC tau was inversely associated with synaptic density in the left hippocampus, left amygdala, left banks of the superior temporal sulcus, left lingual cortex, and left lateral occipital cortex (Figure 4C, Supplemental Figure 2[A,D,F,H,I]). Some of these exploratory associations remained significant after PVC (Supplemental Figure 3) in the entire sample (hippocampi, Figure 4D) and in the AD group (left hippocampus, left temporal pole, left amygdala, left lingual cortex; Figure 4F).
Figure 4. Correlation maps of ERC tau deposition and synaptic density in all regions.
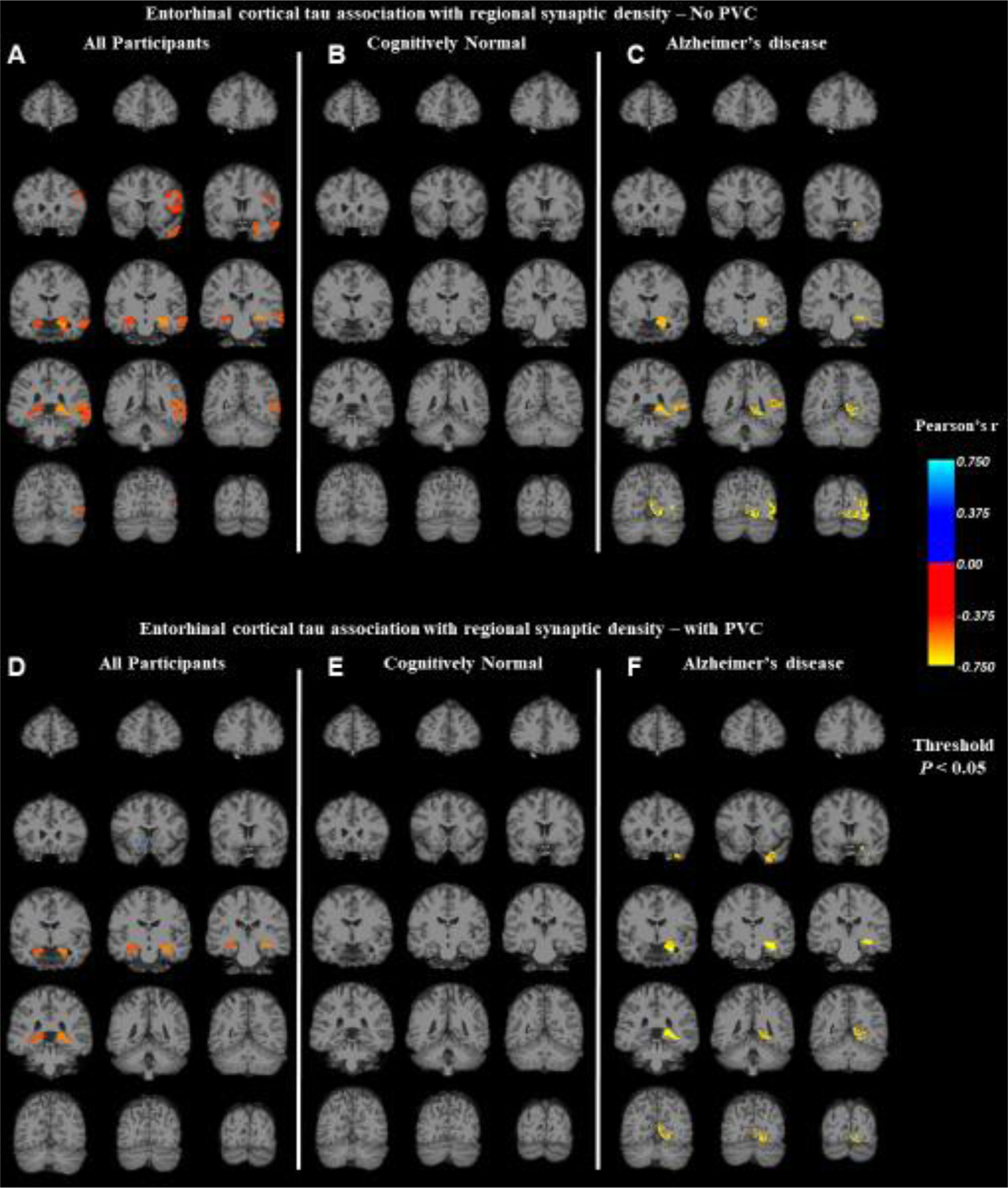
Pearson’s r was calculated for the correlation between ERC tau deposition ([18F]flortaucipir SUVR) and synaptic density ([11C]UCB-J DVR) in all FreeSurfer regions for (A) all participants (n = 20), (B) cognitively normal participants (n = 10), and (C) participants with Alzheimer’s disease (n = 10). Similar correlation maps were also produced with partial volume correction of both [18F]flortaucipir and [11C]UCB-J PET scans (D, E, and F). The color scale represents Pearson’s r, which is displayed only for regions that had an uncorrected P < 0.05. Abbreviations: PVC, partial volume correction.
4. Discussion
We used [18F]flortaucipir and [11C]UCB-J PET to investigate the relationship between tau deposition and synaptic density in individuals with normal cognition or AD, spanning the continuum of CN amyloid negative, CN amyloid positive, amnestic MCI due to AD, and AD dementia. We first examined the association between tau deposition in the ERC, a region of early neurofibrillary tangle formation and neuronal loss, and synaptic loss in the hippocampus, a region with dense projections from the ERC. Consistent with our hypothesis, in the full sample we observed a significant inverse association between ERC tau and hippocampal synaptic density that was slightly increased after correction for partial volume effects. While this association was not significant in the AD group alone, it was still of medium strength prior to PVC, and large afterward. Similar inverse associations were found between ERC tau and hippocampal volume, but of smaller magnitude. Our exploratory analyses suggested that ERC tau may be associated with synaptic density in other regions, which are discussed below.
4.1. ERC tau and the perforant path
This study supports our hypothesis of an association between ERC tau deposition and hippocampal SV2A binding with evidence of a significant correlation in the overall sample, and a trend for a correlation in the smaller AD group. We focused on the relationship between tau pathology in the ERC and synaptic loss in the hippocampus and other regions based on substantial postmortem and recent in vivo research. Postmortem work has established that the earliest cortical tau pathology and neuronal loss in AD occurs in the ERC (Braak, et al., 2011,Gomez-Isla, et al., 1996). Compared to controls, individuals with mild AD have demonstrated a 60% reduction in cell count in layer II and 40% reduction in layer IV of the ERC (Gomez-Isla, et al., 1996). Since these neurons project via the perforant path to all fields of the hippocampal formation, including the dentate gyrus, CA fields, and the subiculum, the earliest and largest reductions in synapses and presynaptic markers are predicted in the hippocampus. Indeed, neuropathological studies have reported a reduction in synapses in the outer molecular layer of dentate gyrus of 44% in mild AD and 13% - 20% in MCI (Masliah, et al., 1994,Scheff, et al., 2006), as well in the CA1 field of 55% in mild AD and 18% in MCI (Scheff, et al., 2007). Consistent with postmortem work, using SV2A PET, we (Chen, et al., 2018a,Mecca, et al., 2020) and others (Bastin, et al., 2019) have observed the most robust reductions in synaptic density in the hippocampus, a finding also true for the present sample (Figure 2). Notably, the only study—to our knowledge—to measure SV2A in postmortem AD brain also revealed reductions in the hippocampus (Robinson, et al., 2014).
4.2. ERC tau and other brain regions
We also examined the specificity of the ERC-tau and hippocampal SV2A association by exploring the relationship between ERC tau and regional SV2A more broadly (Figure 4). Consistent with the primary regional analysis, in the entire sample, ERC tau was inversely associated with synaptic density in the hippocampus (Figure 4A), but also in several other regions: left entorhinal cortex, left amygdala, left banks of the superior temporal sulcus, left middle temporal gyrus, left pars opercularis. Interestingly, in analyses restricted to the AD group, ERC-tau was inversely associated with synaptic density in the left hippocampus, left amygdala, left banks of the superior temporal sulcus, left lingual cortex, and left lateral occipital cortex (Figure 4C). Some of these exploratory associations remained significant after PVC in the entire sample (right hippocampus, Figure 4D) and in the AD group (left hippocampus, left temporal pole, left amygdala, left lingual cortex; Figure 4F). Associations with synaptic density in some of these regions are also consistent with the known projections of ERC, which has strong output directed to amygdala (Stefanacci and Amaral, 2000), to the rostral part of the polysensory area of the superior temporal sulcus and the caudal superior temporal gyrus (Munoz and Insausti, 2005), and to the lateral occipital cortex (Munoz and Insausti, 2005,Orban, et al., 2004). Thus, our overall results would appear to reflect the known anatomical connectivity of ERC.
4.3. Association between tau deposition and synaptic density in previous studies
Previous research has examined the relationship between tau deposition and synaptic loss [reviewed in Jadhav 2015 (Jadhav, et al., 2015)]. Neuronal and synaptic loss have been shown to parallel NFT pathology in both AD mouse models (de Calignon, et al., 2009,Kimura, et al., 2010,Spires-Jones, et al., 2008) and human postmortem studies (Bussiere, et al., 2003a,Falke, et al., 2003,Gomez-Isla, et al., 1997,Hof, et al., 2003,Ingelsson, et al., 2004,Iqbal and Grundke-Iqbal, 2002,Montero-Crespo, et al., 2021). NFT pathology has been theorized to precede and lead to synaptic loss (Iqbal and Grundke-Iqbal, 2002). However, this temporal relationship is generally viewed as uncertain (Serrano-Pozo, et al., 2010) and requires verification in longitudinal studies in vivo. Quantitative associations between NFT pathology and synaptic loss are generally absent from human postmortem studies. Nonetheless, Falke et al. reported that the average dendritic arborization index in subiculum demonstrated a large inverse association with NFT density in the pyramidal layer and fusiform layer and approached significance in the molecular layer in a combined sample of AD and control participants (Falke, et al., 2003); and Montero-Crespo, et al. using scanning electron microscopy found that synaptic density in the CA1 field was inversely correlated with the degree of tau pathology in the stratum pyramidale of AD participants (Montero-Crespo, et al., 2021).
A recent human PET imaging study has examined the association between tau deposition with [18F]MK-6240 and synaptic density with [11C]UCB-J (Vanhaute, et al., 2020). and reported that [18F]MK-6240 binding was inversely related to [11C]UCB-J binding within the medial temporal lobe in a group of participants that included individuals with normal cognition and aMCI. However, no information was provided about associations between [18F]MK-6240 and [11C]UCB-J uptake between individual medial temporal lobe regions. Our primary analysis within the medial temporal lobe focused on the ERC, an early area of tau deposition, and detected associated synaptic loss in the hippocampus. Another recent report of 7 amyloid positive participants with AD (Coomans, et al., 2021) examined the relationship between tau deposition with [18F]flortaucipir and synaptic density with [11C]UCB-J in individual neocortical brain regions for individual subjects and observed significant inverse correlations. I.e., subject-regions with higher tau deposition tended to have lower synaptic density, but ERC and hippocampus were omitted from the analysis. Although our primary analyses were circuit-based (tau deposition in ERC compared to synaptic density in hippocampus and other regions), we also performed a correlational analysis within individual brain regions. However, unlike Coomans et al., we compared the effect sizes for AD-related tau deposition and AD-related synaptic loss using the full AD and CN sample for 70 brain regions (Figure 2C). We observed a significant negative association between the effect sizes for tau deposition and synaptic density (Figure 2C). Interestingly, larger effect sizes—and thus more robust associations—were seen in regions already affected in early Braak stages, which may be due to the fact that longer-standing tau pathogenesis in these regions has had more time to produce synaptic loss by the clinical stage of symptomatic AD. A relative exception to this relationship was hippocampus (Braak II region). However, this result is not inconsistent with our hypothesis that it is primarily tau pathology in the ERC that drives synaptic loss in the hippocampus rather than hippocampal-generated tau pathology.
4.4. Limitations
Important limitations of this study include the small sample size—especially of AD participants—that limited our power to analyze the relationship between tau deposition and synaptic density within the AD population alone. However, our results can guide the design of future studies in symptomatic AD. A power analysis using the results from this work suggested that a sample size of 29 participants may be needed to achieve 80% power to detect an association between ERC tau and hippocampal synaptic density in the setting of symptomatic AD. Our ability to analyze the relationship between tau deposition and synaptic density across the full continuum of AD is further limited by the paucity of CN participants with AD pathologic change. Within our sample, the two CN participants who were positive for brain amyloid (Figure 1A) had intermediate ERC tau and intermediate synaptic loss, suggesting the value of including such individuals to study this association in preclinical AD. A similar important limitation is the absence of longitudinal data, which would enable us to define stage of disease within participants and to analyze the relationship between change in tau deposition in conjunction with synaptic loss from the early stages of disease.
4.5. Conclusions
We observed a significant inverse association between tau deposition in the ERC measured with [18F]flortaucipir and synaptic density measured in the hippocampus with [11C]UCB-J PET in individuals with normal cognition and early AD, spanning the continuum of CN amyloid negative, CN amyloid positive, amnestic MCI due to AD, and AD dementia. This relationship was strengthened by PVC and thus was not attributable solely to an association between tau accumulation and brain tissue loss. Exploratory analyses suggested that ERC tau may be associated with synaptic density in other regions, many of which reflect the known anatomical connectivity of ERC. Larger longitudinal studies beginning at the preclinical stages of AD are needed to examine more fully the relationship between tau accumulation and synaptic loss. An understanding of the temporal course of tau accumulation and synaptic alterations may expand our understanding of AD pathogenesis and guide development of biomarkers for diagnosis and therapeutic efficacy.
Supplementary Material
Highlights.
Entorhinal cortical tau was inversely associated with hippocampal synaptic density.
The association was stronger after correction for partial volume effects.
Tau pathology in entorhinal cortex may cause synaptic failure in the hippocampus.
Acknowledgements.
We wish to thank the research participants for their contributions, and the staff of the Yale Alzheimer’s Disease Research Unit, and the Yale PET Center for their excellent study coordination and technical assistance. We also thank UCB for providing the [11C]UCB-J radiolabeling precursor and the unlabeled reference standard. [18F]flortaucipir doses were provided free of charge by Avid Radiopharmaceuticals, Inc., a wholly owned subsidiary of Eli Lilly and Company.
Funding. This research was supported by the National Institute on Aging (R01AG052560, R01AG062276, K23AG057794, P50AG047270, P30AG066508), Eli Lily and Company [MKC], and The Dana Foundation [MKC]). BCV receives support from Claude D. Pepper Older Americans Independence Center from the NIH/NIA (P30AG021342). This publication was made possible by CTSA Grant Number UL1 TR000142 from the National Center for Advancing Translational Science (NCATS), a component of NIH. The funding bodies had no role in the design of the study, data collection, analysis, interpretation or writing of the manuscript.
Competing interests. APM, REC, and CHvD report grants from National Institutes of Health for the conduct of the study. APM reports grants for clinical trials from Genentech and Eisai outside the submitted work. MKC reports research support from the Dana Foundation and research support from Eli Lilly. MKC reports consulting fees from Eisai and Actinium outside the submitted work. YH reports research grants from the UCB and Eli Lilly outside the submitted work. YH, NBN, and REC have a patent for a newer version of the tracer. REC has received research funding from UCB. REC reports having received grants from BMS, Pfizer, Siemens, and UCB, outside the submitted work. CHvD reports consulting fees from Kyowa Kirin, Roche, Merck, Eli Lilly, and Janssen and grants for clinical trials from Biogen, Novartis, Eli Lilly, Merck, Eisai, Janssen, Roche, Genentech, Toyama, and Biohaven, outside the submitted work. No other disclosures are reported.
Abbreviations
- PET
positron emission tomography
- AD
Alzheimer’s disease
- ERC
entorhinal cortex
- SV2A
Synaptic Vesicle Glycoprotein 2A
- CN
cognitively normal
- DVR
distribution volume ratio
- SUVR
standardized uptake value ratio
- CDR
clinical dementia rating
- MMSE
Mini-Mental Status Examination
- MCI
mild cognitive impairment
- LMII
Logical Memory II
- [11C]PiB
[11C]Pittsburgh Compound B
- Aβ
Amyloid-β
- MRI
magnetic resonance imaging
- ROI
region of interest
- PVC
partial volume correction
- SRTM2
simplified reference tissue model – 2 step
- CS
centrum semiovale
- FDR
false discovery rate
Footnotes
Declarations
Availability of data and material. The datasets used and/or analyzed during the current study are not publicly available due to ongoing analysis and manuscript preparation but are available from the corresponding author on reasonable request.
Data contained in this manuscript have not been previously published and have not been submitted elsewhere while under consideration at Neurobiology of Aging. If accepted, this work will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.
All authors have reviewed the contents of the manuscript, approved of its contents and have validated the accuracy of the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH 2011. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7(3), 270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajjalieh SM, Frantz GD, Weimann JM, McConnell SK, Scheller RH 1994. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J Neurosci 14(9), 5223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajjalieh SM, Peterson K, Linial M, Scheller RH 1993. Brain contains two forms of synaptic vesicle protein 2. Proc Natl Acad Sci U S A 90(6), 2150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ 2017. Considerations and code for partial volume correcting [(18)F]-AV-1451 tau PET data. Data Brief 15, 648–57. doi: 10.1016/j.dib.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin C, Bahri MA, Meyer F, Manard M, Delhaye E, Plenevaux A, Becker G, Seret A, Mella C, Giacomelli F, Degueldre C, Balteau E, Luxen A, Salmon E 2019. In vivo imaging of synaptic loss in Alzheimer’s disease with [18F]UCB-H positron emission tomography. Eur J Nucl Med Mol Imaging. doi: 10.1007/s00259-019-04461-x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4), 239–59. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K 2011. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70(11), 960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR 2003a. Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: stereologic analysis of prefrontal cortex area 9. J Comp Neurol 463(3), 281–302. doi: 10.1002/cne.10760. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Gold G, Kovari E, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR 2003b. Stereologic analysis of neurofibrillary tangle formation in prefrontal cortex area 9 in aging and Alzheimer’s disease. Neuroscience 117(3), 577–92. doi: 10.1016/s0306-4522(02)00942-9. [DOI] [PubMed] [Google Scholar]
- Chen MK, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin SF, Najafzadeh S, Ropchan J, Lu Y, McDonald JW, Michalak HR, Nabulsi NB, Arnsten AFT, Huang Y, Carson RE, van Dyck CH 2018a. Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA neurology. doi: 10.1001/jamaneurol.2018.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MK, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin SF, Najafzadeh S, Ropchan J, Lu Y, McDonald JW, Michalak HR, Nabulsi NB, Arnsten AFT, Huang Y, Carson RE, van Dyck CH 2018b. Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA neurology 75(10), 1215–24. doi: 10.1001/jamaneurol.2018.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomans EM, Schoonhoven DN, Tuncel H, Verfaillie SCJ, Wolters EE, Boellaard R, Ossenkoppele R, den Braber A, Scheper W, Schober P, Sweeney SP, Ryan JM, Schuit RC, Windhorst AD, Barkhof F, Scheltens P, Golla SSV, Hillebrand A, Gouw AA, van Berckel BNM 2021. In vivo tau pathology is associated with synaptic loss and altered synaptic function. Alzheimers Res Ther 13(1), 35. doi: 10.1186/s13195-021-00772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Spires-Jones TL, Pitstick R, Carlson GA, Hyman BT 2009. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol 68(7), 757–61. doi: 10.1097/NEN.0b013e3181a9fc66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong HW, van Velden FH, Kloet RW, Buijs FL, Boellaard R, Lammertsma AA 2007. Performance evaluation of the ECAT HRRT: an LSO-LYSO double layer high resolution, high sensitivity scanner. Phys Med Biol 52(5), 1505–26. doi: 10.1088/0031-9155/52/5/019. [DOI] [PubMed] [Google Scholar]
- Erlandsson K, Buvat I, Pretorius PH, Thomas BA, Hutton BF 2012. A review of partial volume correction techniques for emission tomography and their applications in neurology, cardiology and oncology. Phys Med Biol 57(21), R119–59. doi: 10.1088/0031-9155/57/21/R119. [DOI] [PubMed] [Google Scholar]
- Falke E, Nissanov J, Mitchell TW, Bennett DA, Trojanowski JQ, Arnold SE 2003. Subicular dendritic arborization in Alzheimer’s disease correlates with neurofibrillary tangle density. Am J Pathol 163(4), 1615–21. doi: 10.1016/S0002-9440(10)63518-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnema SJ, Nabulsi NB, Mercier J, Lin SF, Chen MK, Matuskey D, Gallezot JD, Henry S, Hannestad J, Huang Y, Carson RE 2018. Kinetic evaluation and test-retest reproducibility of [(11)C]UCB-J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. J Cereb Blood Flow Metab 38(11), 2041–52. doi: 10.1177/0271678X17724947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B 2012. FreeSurfer. Neuroimage 62(2), 774–81. doi: 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT 1997. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41(1), 17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW Jr., Morris JC, Growdon JH, Hyman BT 1996. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 16(14), 4491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Bussiere T, Gold G, Kovari E, Giannakopoulos P, Bouras C, Perl DP, Morrison JH 2003. Stereologic evidence for persistence of viable neurons in layer II of the entorhinal cortex and the CA1 field in Alzheimer disease. J Neuropathol Exp Neurol 62(1), 55–67. doi: 10.1093/jnen/62.1.55. [DOI] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC 2004. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62(6), 925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I 2002. Neurofibrillary pathology leads to synaptic loss and not the other way around in Alzheimer disease. J Alzheimers Dis 4(3), 235–8. doi: 10.3233/jad-2002-4313. [DOI] [PubMed] [Google Scholar]
- Jadhav S, Cubinkova V, Zimova I, Brezovakova V, Madari A, Cigankova V, Zilka N 2015. Tau-mediated synaptic damage in Alzheimer’s disease. Transl Neurosci 6(1), 214–26. doi: 10.1515/tnsci-2015-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Mulnix T, Gallezot JD, Carson RE 2013. Evaluation of motion correction methods in human brain PET imaging--a simulation study based on human motion data. Medical physics 40(10), 102503. doi: 10.1118/1.4819820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A 2010. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem 285(49), 38692–9. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R 1994. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 174(1), 67–72. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7(3), 263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca AP, Barcelos NM, Wang S, Bruck A, Nabulsi N, Planeta-Wilson B, Nadelmann J, Benincasa AL, Ropchan J, Huang Y, Gelernter J, Van Ness PH, Carson RE, van Dyck CH 2017. Cortical beta-amyloid burden, gray matter, and memory in adults at varying APOE epsilon4 risk for Alzheimer’s disease. Neurobiol Aging 61, 207–14. doi: 10.1016/j.neurobiolaging.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca AP, Chen MK, O’Dell RS, Naganawa M, Toyonaga T, Godek TA, Harris JE, Bartlett HH, Zhao W, Nabulsi NB, Wyk BCV, Varma P, Arnsten AFT, Huang Y, Carson RE, van Dyck CH 2020. In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimers Dement. doi: 10.1002/alz.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens N, Maguire RP, Serdons K, Lacroix B, Mercier J, Sciberras D, Van Laere K, Koole M 2019. Validation of Parametric Methods for [(11)C]UCB-J PET Imaging Using Subcortical White Matter as Reference Tissue. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. doi: 10.1007/s11307-019-01387-6. [DOI] [PubMed] [Google Scholar]
- Montero-Crespo M, Dominguez-Alvaro M, Alonso-Nanclares L, DeFelipe J, Blazquez-Llorca L 2021. Three-dimensional analysis of synaptic organization in the hippocampal CA1 field in Alzheimer’s disease. Brain 144(2), 553–73. doi: 10.1093/brain/awaa406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz M, Insausti R 2005. Cortical efferents of the entorhinal cortex and the adjacent parahippocampal region in the monkey (Macaca fascicularis). Eur J Neurosci 22(6), 1368–88. doi: 10.1111/j.1460-9568.2005.04299.x. [DOI] [PubMed] [Google Scholar]
- Mutch SA, Kensel-Hammes P, Gadd JC, Fujimoto BS, Allen RW, Schiro PG, Lorenz RM, Kuyper CL, Kuo JS, Bajjalieh SM, Chiu DT 2011. Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J Neurosci 31(4), 1461–70. doi:31/4/1461 [pii] 10.1523/JNEUROSCI.3805-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell RS, Mecca AP, Chen MK, Naganawa M, Toyonaga T, Lu Y, Godek TA, Harris JE, Bartlett HH, Banks ER, Kominek VL, Zhao W, Nabulsi NB, Ropchan J, Ye Y, Vander Wyk BC, Huang Y, Arnsten AFT, Carson RE, van Dyck CH 2021. Association of Abeta deposition and regional synaptic density in early Alzheimer’s disease: a PET imaging study with [(11)C]UCB-J. Alzheimers Res Ther 13(1), 11. doi: 10.1186/s13195-020-00742-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orban GA, Van Essen D, Vanduffel W 2004. Comparative mapping of higher visual areas in monkeys and humans. Trends in cognitive sciences 8(7), 315–24. doi: 10.1016/j.tics.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Pontecorvo MJ, Devous MD Sr., Navitsky M, Lu M, Salloway S, Schaerf FW, Jennings D, Arora AK, McGeehan A, Lim NC, Xiong H, Joshi AD, Siderowf A, Mintun MA, investigators FA-A 2017. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140(3), 748–63. doi: 10.1093/brain/aww334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Molina-Porcel L, Corrada MM, Raible K, Lee EB, Lee VM, Kawas CH, Trojanowski JQ 2014. Perforant path synaptic loss correlates with cognitive impairment and Alzheimer’s disease in the oldest-old. Brain 137(Pt 9), 2578–87. doi:awu190 [pii] 10.1093/brain/awu190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossano S, Toyonaga T, Finnema SJ, Naganawa M, Lu Y, Nabulsi N, Ropchan J, De Bruyn S, Otoul C, Stockis A, Nicolas JM, Martin P, Mercier J, Huang Y, Maguire RP, Carson RE 2019. Assessment of a white matter reference region for (11)C-UCB-J PET quantification. J Cereb Blood Flow Metab, 271678X19879230. doi: 10.1177/0271678X19879230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ 2007. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68(18), 1501–8. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ 2006. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27(10), 1372–84. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT 2010. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain 133(Pt 5), 1312–27. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shidahara M, Thomas BA, Okamura N, Ibaraki M, Matsubara K, Oyama S, Ishikawa Y, Watanuki S, Iwata R, Furumoto S, Tashiro M, Yanai K, Gonda K, Watabe H 2017. A comparison of five partial volume correction methods for Tau and Amyloid PET imaging with [18F]THK5351 and [11C]PIB. Ann Nucl Med 31(7), 563–9. doi: 10.1007/s12149-017-1185-0. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT 2008. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci 28(4), 862–7. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanacci L, Amaral DG 2000. Topographic organization of cortical inputs to the lateral nucleus of the macaque monkey amygdala: a retrograde tracing study. J Comp Neurol 421(1), 52–79. doi:. [DOI] [PubMed] [Google Scholar]
- Vanhaute H, Ceccarini J, Michiels L, Koole M, Sunaert S, Lemmens R, Triau E, Emsell L, Vandenbulcke M, Van Laere K 2020. In vivo synaptic density loss is related to tau deposition in amnestic mild cognitive impairment. Neurology. doi: 10.1212/WNL.0000000000009818. [DOI] [PubMed] [Google Scholar]
- Wu Y, Carson RE 2002. Noise reduction in the simplified reference tissue model for neuroreceptor functional imaging. J Cereb Blood Flow Metab 22, 1440–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.