

Abstract
Breast cancer ecosystems are composed of complex cell types, including tumor, stromal and immune cells, each of which can assume diverse phenotypes. Both the heterogeneous composition and spatially distinct tumor microenvironment impact breast cancer progression, treatment response and therapeutic resistance. Thus, a deeper understanding of breast cancer heterogeneity may help facilitate the development of novel therapies and improve outcomes for patients. The advent of paradigm shifting single-cell analysis and spatial pathologies allows for a comprehensive analysis of the tumor ecosystem as well as the interactions between its components at unprecedented resolution. In this review, we discuss the insights gained through single-cell analysis and spatial pathologies on breast cancer heterogeneity.
Keywords: Breast cancer, Tumor heterogeneity, Single-cell, Spatial pathology, Tumor microenvironment
1. Introduction
Breast cancer is the most frequently diagnosed cancer and the leading cause of cancer death in women worldwide (1). Breast cancer is composed of mosaic populations of tumor, immune and stromal cells with varying genetic, epigenetic, and phenotypic characteristics. Heterogeneous tumor cell subpopulations allow for selection and Darwinian evolution and enable beneficial cooperative interactions that may promote tumor progression and therapy resistance (2–4). The tumor immune and stroma context, i.e. the composition, spatial organization, and functional orientation of various immune and stromal cell subsets also strongly influences the disease course and patient outcome. The recent advancement of antibody-based immune-checkpoint blockage (ICB) therapies has paradigm shifted cancer treatment (5). However, only a minority of breast cancer patients respond to immunotherapy (6–8). A deeper analysis of the complexity and diversity of the tumor ecosystem holds promise for stratification of patients, prediction of therapeutic responsiveness, and potentially the identification of new druggable targets. Traditional bulk tumor profiling can reveal global tumor features, but cannot decode the cellular origin of gene expression, the spatial organization within tumors or the variability of individual cellular programs. Cutting-edge single-cell analysis and spatial pathologies are paving the way for comprehensively studying tumor heterogeneity. In this review, we summarize initially the conventional view of breast cancer heterogeneity. We then provide an overview of the recent technological developments of single-cell analysis and spatial pathologies. Key examples that leverage these technologies are provided to help elucidate the manifestations, drivers, and consequences of breast tumor heterogeneity. Finally, we discuss how this knowledge might be translated into novel therapeutic approaches.
2. Conventional breast cancer classification
Multiple efforts have been made to assess the intertumor and intratumor heterogeneity of breast cancer. Historically in the 1990s, breast cancer was defined as a disease with variations in prognosis and response to therapy. In the decade beginning in 2000, breast cancer was shown to be comprised of multiple subtypes. Beginning in 2014, based on a combination of histopathology, molecular profiles, and mutational repertoires, each tumor was shown to be genetically heterogeneous and composed of multiple clones (9).
Breast cancer can be categorized using different parameters. When classified by histological features, invasive ductal carcinoma not otherwise specified (IDC NOS) and invasive lobular carcinoma (ILC) are the two most common subtypes making up approximately 90% of all breast cancers (10). Immunopathological classification based on the presence of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal receptor 2 (HER2) defines subtypes with distinct prognosis and targeted treatment options. The more recently described intrinsic subtypes of breast cancer rely on mRNA expression that reflect tumor cell intrinsic properties from bulk tissue, and separate breast tumors into luminal A, luminal B, HER2-enriched, basal-like, and normal-like subtypes (11, 12). A variety of related classifiers also have been being proposed (13–19). Relationships among the above-mentioned classifications have been reviewed elsewhere (20, 21).
Besides tumor cell intrinsic features, the tumor microenvironment also allows for stratification and has prognostic significance (22, 23). Immunogenomic analysis of The Cancer Genome Atlas (TCGA) data which profiled bulk tumors identified six immune subtypes across cancer types: Wound healing, IFN-γ dominant, Inflammatory, Lymphocyte depleted, Immunologically quiet, and TGF-β dominant (24). Luminal A breast cancer is enriched in the wound healing subtype whereas highly mutated breast tumors are enriched in the IFN-γ dominant subtype (24). Tumor microenvironment (TME) features also have been associated with triple-negative breast cancer (TNBC) molecular subtypes and overall survival (25).
Intratumor heterogeneity poses a significant challenge in applying molecular prognostic markers and classifying patients that might benefit from specific therapies. The IHC threshold for ER/PR positivity is defined as greater than 1% of cells display expression (26, 27). Positive HER2 status is reported when tumors exhibit amplification of the ERBB2 (HER2) gene or the proportion of HER2+ tumor cells within the tumor exceeds a 10% threshold (28). This leaves the majority of cells uncharacterized. Besides tumor cells, a better characterization of tumor infiltrating immune and stromal cells may provide a better strategy for stratifying patients and overcoming immune suppression.
3. Enabling technologies for assessing tumor heterogeneity
Traditional bulk genomic, transcriptomic, and epigenetic analyses have provided valuable information, but this is integrated from multiple tumor clones and both tumor and nontumor cells derived from the microenvironment. This information, therefore, is difficult to completely deconvolve and critical differences between individual cells may be obscured. Recent advancements in single-cell analysis, spatial pathologies, and accompanying computational methodologies have enabled a comprehensive analysis of cellular heterogeneity, spatial organization, and cell-cell interactions (Table 1). These transformative technologies have prompted several large-scale initiatives to generate single cell and multiparametric atlases for understanding the tissue ecosystem in health and disease (29–34). For example, the Human Cell Atlas, the Human Tumor Atlas Network and the Tumor Profiler studies adopted a two-pronged strategy that pairs single-cell sequencing from dissociated specimens with spatially resolved multiplexed imaging assays in situ (29, 33, 34). Since single-cell profiling approaches often do not preserve the spatial organization and spatially resolved imaging approaches currently multiplex considerably fewer measurements, the combination of these two complementary methods can interrogate both the composition and architecture of tumor ecosystems (Fig. 1). A comprehensive summary of the current single-cell and spatial omics methodologies has been provided elsewhere (35–37).
Table 1.
Summary of enabling technologies for assessing tumor heterogeneity
| Technology | scRNA-seq | Suspension mass cytometry | Imaging mass cytometry | Spatial transcriptomics |
|---|---|---|---|---|
|
| ||||
| Molecule detected | RNA | Protein | Protein | RNA |
| Resolution | Single cell | Single cell | Single cell | 1 to 10 cells |
| Multiplexicity | Whole transcriptome | Around 40 markers | Around 40 markers | From 100 RNA species to whole transcriptome |
| Spatial | No, unless combined with laser capture microdissection | No | Yes | Yes |
| Limitations | Dropout of low abundant transcripts; Doublets contamination | Depends on the availability and specificity of antibodies; Relies on a prior knowledge of cell markers | Depends on the availability and specificity of antibodies; Relies on a prior knowledge of cell markers; Multiple fields need to be analyzed to minimize regional bias | Not single cell resolution in some platforms |
| cost-effective (calculated per cell) | *** | ***** | ** | * |
Fig. 1.
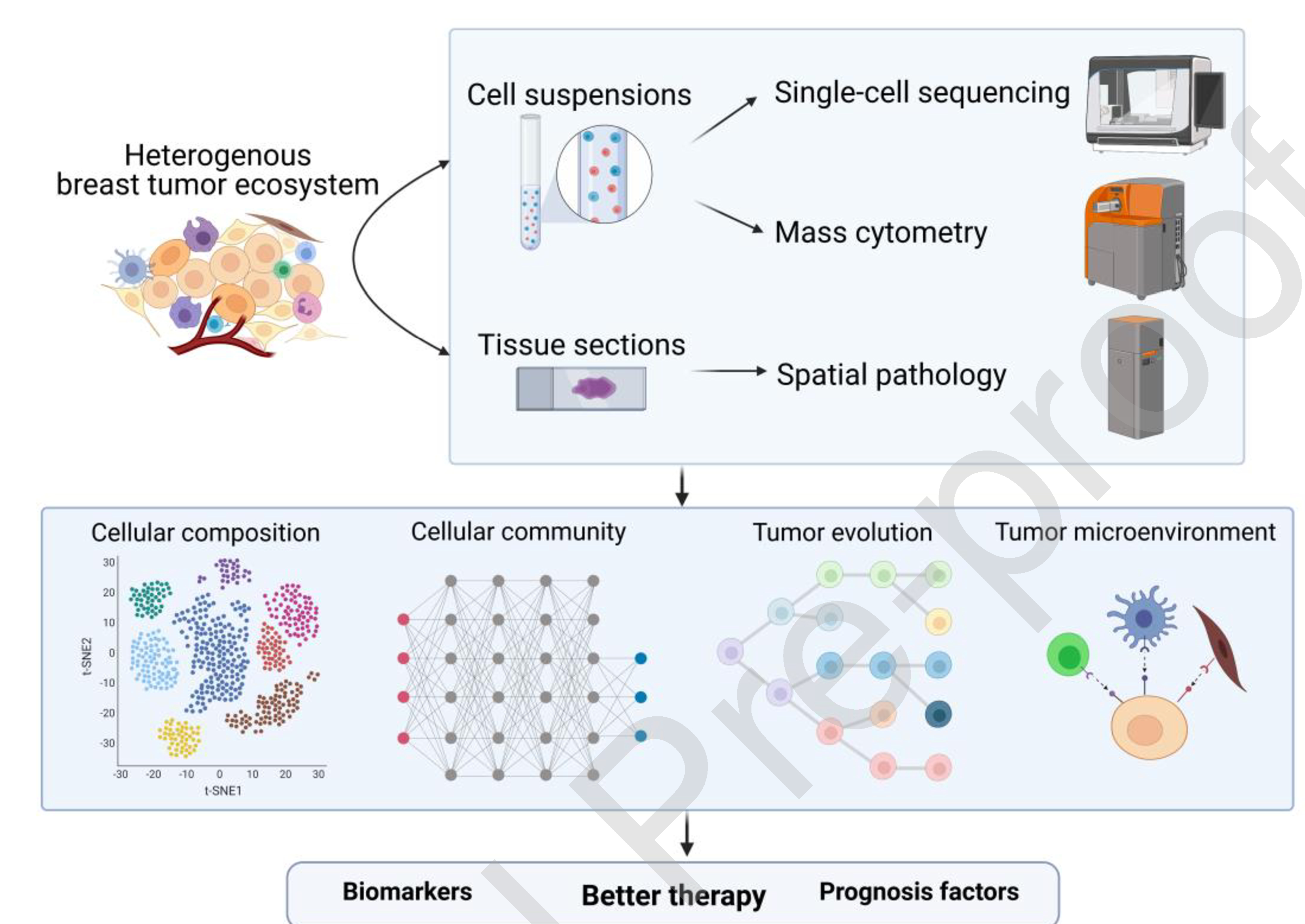
The applications of single-cell analysis and spatial pathologies in studies of breast cancer heterogeneity.
New technologies such as single-cell sequencing, mass cytometry and spatial pathologies have helped to dissect the cellular composition and community, evolution and microenvironment of breast tumors. The elucidation of the heterogenous ecosystem of breast cancer should facilitate the development of novel predictive biomarkers, prognostic features, and therapeutic targets.
3.1. Single-cell sequencing
Single-cell DNA-sequencing (scDNA-seq) detects copy-number aberrations or mutations in individual cells circumventing the problem of low sensitivity in detecting rare mutations in bulk DNA-seq (38). Applications of scDNA-seq include clonal substructure dissection, clonal lineage reconstruction, tumor evolution inference, and mutation co-occurrence or mutual exclusivity interrogation (36). However, scDNA-seq cannot reveal cell type or state and may be limited in its coverage.
Single-cell RNA-sequencing (scRNA-seq) provides transcriptional profiles of individual cells at a snapshot in time (39). The development of droplet-based systems greatly increases reaction throughput, reduces the cost, and improves the utility of scRNA-seq (40). Potential limitations of scRNA-seq include doublet contamination and dropout of low abundance transcripts. The applications of this technology include identifying cell populations, dissecting cell states, elucidating gene signatures, categorizing expression subtypes, inferring tumor lineages, and analyzing cell type specific differential expression (36). Multiple studies have utilized scRNA-seq to characterize mammary cell subpopulations and differentiation trajectories across different stages of mammary gland development (41).
Single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) measures chromatin accessibility across the genome of single cells and has an application range similar to scRNA-seq (42). A major limitation of this method is the low coverage with an estimated 1–10% extraction of the total accessible peaks (43). However, compared with scRNA-seq, scATAC-seq may provide deeper insights into gene regulation, and more faithfully distinguishes cell lineages and identities (44).
As standard single-cell sequencing approaches use disaggregated cells as the input, the spatial organization of single-cell phenotypes is not preserved. To reflect spatial context, laser capture microdissection has been used together with scDNA-seq (45) or scRNA-seq (46), which is often referred to as topographic single-cell sequencing. scRNA-seq alone may not always differentiate between cell populations with subtle transcriptomic differences, and direct transcriptional profiles may not directly correlate with protein expression due to post-transcriptional regulation. To overcome these limitations, several single-cell multi-omics methods have been developed to improve cell type and state characterization. For example, scRNA-seq has been combined with highly multiplexed protein marker detection in Cellular Indexing of Transcriptomes and Epitopes by sequencing (CITE-seq) and RNA Expression and Protein sequencing assay (REAP-seq). In both methods, oligonucleotide-barcoded antibodies are used to provide a quantitative readout of cellular proteins in combination with existing scRNA-seq (47,48).
3.2. Suspension mass cytometry
Another typical single-cell analysis is flow cytometry, which employs fluorophore-conjugated antibodies to detect and quantify protein abundance in individual cells. However, due to the overlap of the fluorescent spectra of the labelling dyes, the number of proteins that can be analyzed simultaneously by flow cytometry is restricted to around 10 in conventional flow and up to 30 in spectral flow. Mass cytometry, or cytometry by time-of-flight (CyTOF), is a high-dimensional single-cell proteomic analysis method which uses rare earth metal ion tags rather than fluorochromes for antibody labeling. Analysis of metal abundance using the mass cytometer allows evaluation of marker levels with minimal spillover between channels (49, 50). In mass cytometry assays, over 40 protein parameters can be simultaneously quantified at single-cell resolution enabling the evaluation of cell subsets in the TME (51). Mass cytometry allows for the detection of low abundance proteins which otherwise may not be detectable in scRNA-seq. However, it is dependent on the availability and specificity of antibodies and relies on a prior knowledge of cell markers which limits its utility for biomarker discoveries. Similar to single-cell sequencing, suspension mass cytometry uses dissociated tissues and therefore does not capture spatial information.
3.3. High-dimensional imaging
Although single-cell sequencing or suspension mass cytometry enables enumeration of cell subsets found in the TME, the properties of tumors are determined not only by the presence of heterogeneous cell populations, but also by complex cellular interactions (52). Highly multiplexed imaging fills this gap through quantitative measurement of dozens of proteins simultaneously in intact tissue. This technique enables the generation of a detailed spatial map of single-cell phenotypes and cellular communities. This is instrumental for understanding the spatial and phenotypic features affecting tumor progression and therapeutic response. Early generations of multiplexed imaging technologies such as cyclic immunofluorescence (CycIF) were based on traditional immunofluorescence (IF) and rely on cycles of epitope staining followed by chemical quenching of fluorophores and restaining to progressively build a multichannel image (53, 54). Such methods only allow assessment of a dozen antigens at a given time. The advent of mass spectroscopy immunohistochemistry (IHC) has enabled robust measurement of around 40 markers simultaneously. In this method, primary antibodies for protein detection are conjugated to metal isotopes and then incubated with tissue sections similarly with IHC. Next, either laser ablation (in imaging mass cytometry (IMC)) (55) or ion beam (in multiplexed ion beam imaging by time-of-flight (MIBI-TOF)) (52, 56) are employed to liberate metals for detection by the mass spectrometer. Of note, both lineage markers and signaling molecules, especially phosphorylated proteins, can be interrogated in high-dimensional imaging, which enables the analysis of cell signaling networks in a cell-by-cell spatially resolved manner (35). Similar to suspension mass cytometry and all other antibody-based detection methods, IMC is limited by antibody specificity. Furthermore, because of tumor heterogeneity, multiple regions need to be analyzed to minimize bias.
3.4. Spatial transcriptomics
Emerging technologies for spatial transcriptomics characterize gene expression profiles while retaining spatial information that overcome limitations associated with scRNA-seq. Such methodologies provide detailed molecular maps, enabling a further understanding of the relationship between cellular gene expression and the interactions with the local environment. Some of these technologies reach single-cell or even subcellular resolution. For example, fluorescent in situ RNA sequencing (FISSEQ) and its adapted version expansion sequencing (ExSeq) can provide high dimensional gene expression in a single cell in situ by sequencing cross-linked complementary DNA amplicons (57, 58). Multiplexed error-robust fluorescence in situ hybridization (MERFISH) can simultaneously image 100 to 1000 RNA species in an individual cell by using combinatorial FISH labeling with error-robust encoding schemes (59). Other spatial transcriptomic approaches that do not have single-cell resolution such as spatial transcriptomic microarrays (60) and Slide-Seq (61) offer high throughput and genome-wide spatial transcriptomics profile at a 10–100 cell resolution. The recently released 10x Genomics Visium platform expands the spatial resolution fivefold beyond the first-generation spatial transcriptomic microarrays, reaching a 1–10 cell resolution depending on the tissue type (62, 63). Immunofluorescence can also be combined with this method for protein co-detection. Further development of these spatial technologies will be instrumental in constructing high-resolution cellular atlases from heterogeneous tumor ecosystems and for defining the molecular pathways involved in tumorigenesis. In the following sections, we will discuss new insights obtained from these new technologies that improved our understanding of breast cancer heterogeneity.
4. The spectrum of breast cancer heterogeneity
The breast tumor ecosystem is extremely complex. Cell-to-cell variations of breast cancer cells manifest in genetic backgrounds, epigenetic profiles, cellular phenotypes, spatial distributions, and interactions with the TME. Immune and stromal cells in the breast TME also exhibit substantial molecular and functional diversity. To highlight the applications to breast cancer of the technologies mentioned in Section 3, a few representative examples are provided below. Readers are directed to a recent review on this subject for additional information (64).
First, moving into the single-cell analysis era, Chung et al. performed scRNA-seq for 515 cells which contained both tumor cells and non-tumor immune cells from 11 breast cancer patients across the intrinsic molecular subtypes (65). They found that tumor cells variably expressed aggressive cancer signatures such as epithelial-mesenchymal transition (EMT), stemness, angiogenesis, proliferation and recurrence, whereas most T cells and macrophages displayed immunosuppressive characteristics (65). Although informational, this study is restricted by the low cell number analyzed. To elucidate the immune cell phenotypic diversity in breast cancer, Azizi et al. performed scRNA-seq on 45,000 sorted immune cells from 8 breast tumors as well as matched normal breast tissue, blood, and lymph nodes (66). All the major populations of immune cells are present in breast cancer. These include both lymphocytes such as T cells, B cells and natural killer cells, and myeloid cells such as macrophages, dendritic cells, granulocytes such as neutrophils and eosinophils, and mast cells. Analyses by Azizi et al. further revealed significantly increased heterogeneity of lymphoid and myeloid cells in tumor in comparison to normal breast tissue. This heterogeneity was characterized by combinatorial expression of genes reflecting responses to diverse environmental stimuli. They also observed a continuum of T cell states which suggests that the conventional notion that only a few discrete states of differentiation or activation shape the TME is oversimplified (66). To characterize stromal heterogeneity, Wu et al. performed scRNA-seq in 5 TNBCs and observed two cancer-associated fibroblast (CAF) and two perivascular-like (PVL) subpopulations. CAFs clustered into myofibroblast-like CAFs or inflammatory-like CAFs and PVL cells clustered into differentiated PVL or immature PVL cells. These stromal subpopulations differed in their morphology, surface marker expression, spatial localization, and functional properties in regulating the ECM. Moreover, the expression of gene signatures derived from inflammatory-like CAF and differentiated PVL cells is associated with cytotoxic T-cell dysfunction and exclusion in independent TNBC cohorts (67). Such findings suggest crosstalk of stromal-immune compartments and may provide potential strategies for overcoming immune therapy resistance.
Second, studies by Wagner et al. utilized suspension mass cytometry which has a much higher throughput to reveal the tumor and immune cell diversity in breast tumor ecosystems (68). These investigators analyzed the expression of 73 proteins using tumor and immune cell-centric antibody panels in 26 million cells from 144 human breast tumor and 50 non-tumor tissue samples. For the tumor cell compartment, they established three computational scores to represent heterogeneity: 1) Tumor individuality describes whether tumor cells of a sample were more similar to cells of the same sample; 2) Phenotypic abnormality measures the tumor cell phenotypic deviation from non-tumor epithelial cells; and 3) Tumor richness quantifies the number of different co-existing tumor cell phenotypes. The results of these studies indicated that tumors exhibit individuality in tumor cell composition, including phenotypic abnormalities and phenotype dominance. In addition, for the immune cell compartment, a high abundance of PD-L1+ tumor associated macrophages (TAMs) and exhausted T cells was observed in high-grade ER+ and ER- tumors. The hierarchical clustering of both tumor and immune components revealed distinct groups which were able to stratify patients (66).
Third, to map the spatial organization of single-cell phenotypes of breast cancer, Bodenmiller and colleagues utilized IMC to simultaneously quantify over 30 biomarkers spanning multiple cell lineages in hundreds of patients (69, 70). They reported that tumors in general were spatially segregated into separate tumor communities as opposed to interspersed heterogeneous tumor masses, and that patients with tumors with spatiophenotypic heterogeneity had a worse prognosis (69). The observation of the presence of stromal cells in every clinical subtype at similar densities prompted the authors to investigate whether the tumor-stromal microenvironment communities were more informative than the tumor or stromal phenotype content alone in predicting patient survival. Indeed, spatially defined cell communities were independently associated with patient outcome but not single-cell phenotypes or cellular metaclusters (69). In their IMC analyses of samples from METABRIC, a large breast-cancer cohort with an extensive genomic annotation, they found that distinct combinations of single-cell phenotypes and multi-cellular communities were associated with genomic subtypes and aberrations of breast cancer. For example, epithelial luminal cell phenotypes are divided into those driven by either mutations or copy-number alterations. A model that combines contributions of both cell phenotypes and their neighborhoods improved outcome prediction as compared to cell composition alone, highlighting the clinical relevance of spatial statistics (70).
Finally, with the goal of depicting the immune landscape of the TNBC microenvironment, Keren et al. used MIBI-TOF to simultaneously measure in situ expression of 36 proteins covering cell identity, function, and immune regulation in 41 patients at sub-cellular resolution (71). As expected, they found that the composition of tumor-immune populations varied across individuals. More interestingly, the spatial infiltration by immune cells varied from compartmentalized to mixed patterns, and a compartmentalized immune structure along the tumor-immune border was associated with better patient survival (71). Taking a different approach, Park and colleagues profiled the tumor immune compartment by laser capture microdissection-derived gene expression to stratify TNBC based on the tumor immune microenvironment (72). They found that stromal restriction of CD8+ T cells and stromal expression of PD-L1 defined a distinct poor-outcome immunomodulatory microenvironment, whereas infiltration of granzyme B+ CD8+ T cells and a type 1 IFN signature defined an immunoreactive microenvironment with good patient outcomes (72).
5. Sources of breast cancer intratumor heterogeneity
Phenotypic and functional heterogeneity among breast tumor cells is a complicated phenomenon that entails both genetic clonal diversity and nongenetic sources of heterogeneity (73).
5.1. Genetic events
Cancer is an evolutionary process and genetic diversification is the primary mechanism for clonal evolution. This genetic diversity arises from genomic instability that operates at multiple levels, ranging from single point mutations to chromosomal rearrangements (74, 75). Genomic instability might be caused by exposure to exogenous mutagens and aberrations in endogenous processes such as errors in DNA replication and repair (2). Under the assumption that mutational complexity increases with time, phylogenetic analysis can be applied to infer genetic lineages and to order the chronology of mutations that have occurred over time (76). Therefore, the genetic heterogeneity of tumor populations can be used to reconstruct tumor evolution from a single time-point sample.
Different models of clonal evolution have been proposed, including linear, branching, neutral, and punctuated evolution (77, 78). Linear evolution is defined by sequential clonal succession where new driver mutations provide a strong selective advantage that can lead to clonal expansion and that clone can outcompete previous populations and become dominant. In branching evolution, divergent subclones emerge independently from a common ancestor. In this model, acquired new driver mutations lead to expansion of tumor populations which do not outcompete all other populations. The simultaneous expansion of multiple clones raises the possibility of clonal cooperation which has been experimentally supported in several studies (79–82). Neutral evolution is defined by the absence of selection, wherein random mutations accumulate over time leading to genetic drift. In the neutral evolution model, heterogeneity is proposed to be a byproduct of tumor progression that results from stochastic processes and has no functional significance in driving tumor growth. All these three models assume that mutations are acquired sequentially and gradually over time. In contrast in the punctuated model, chromosomal aberrations are acquired in short evolutionary bursts at the early stage of tumor progression, and this leads to multiple genotypes some of which are stabilized without much later evolution (78).
To explore how the breast cancer genome evolves to generate heterogeneity, Navin and colleagues first used an early generation scDNA-seq to profile 1,000 single cells from 12 TNBC samples. They identified that punctuated genome-wide aneuploidy is acquired at an early stage of tumor evolution in short bursts of genomic instability. This is followed by stable clonal expansions that eventually constitute the tumor mass (83). It is possible that dysfunctional p53 causes genomic instability early in TNBC evolution since TP53 is the most commonly mutated gene (17). This punctuated evolution pattern has been described for other cancer types as well (84–86). However, due to the small number of cells that were sequenced and the low resolution in these early generation scDNA-seq method in that study, the question of whether copy number profiles continue to evolve at small scale after the initial burst of genome rearrangements could not be answered. With more recent advancements in scDNA-seq technologies, the Navin group performed copy number analysis of 16,178 single cells from 8 TNBC samples. They found that copy number evolution is ongoing during clonal expansion after the initial catastrophic event, suggesting that tumor cells never stop improving their fitness during the growth of the primary tumors (87)
5.2. Non-genetic events
Heterogeneity Importantly does not only manifest itself as genetic alterations such as somatic mutations and copy number aberrations. Non-genetic sources of heterogeneity also lead to cell-to-cell phenotypic variability. These include epigenetic, transcriptomic, proteomic and metabolic differences in tumor cells which possess similar genetic alterations. Numerous epigenetic alterations including DNA methylation, histone modifications, and chromatin remodeling contribute to heterogeneity within tumors (88). A diversity of chromatin landscapes within both the stromal and tumor cell populations was reported by Grosselin and colleagues, who performed single-cell chromatin immunoprecipitation followed by sequencing (ChIP-seq) in breast cancer patient-derived xenograft (PDX) samples (89). In ER-positive breast cancer, the expression of the histone H3 lysine 4 (H3K4) demethylase KDM5B promotes transcriptomic heterogeneity within tumor cells which may lead to endocrine resistance (90).
Another factor that contributes to cellular variability is the plasticity of cell states. EMT is the best-known example of cellular plasticity in breast cancer. Instead of being entirely comprised of cancer cells with either epithelial or mesenchymal features, most breast cancers exhibit a spectrum of epithelial and mesenchymal phenotypes (91). Intermediate or partial EMT, which involves a combination of epithelial and mesenchymal gene expression, may confer cancer cells with increased plasticity and cancer stem cell properties as well as therapeutic resistance (92, 93). For more information on the role of genetic and non-genetic clonal diversity in tumor evolution and the insights gained through single-cell analyses, readers are referred to several comprehensive recent reviews (74, 94).
6. Consequences of intratumor heterogeneity
Tumor cell clonal heterogeneity and cooperative plasticity equip tumors with substantial functional adaptability which may potentiate tumor growth and metastasis. In a mouse mammary tumor model, Cleary et al. showed that aberrant expression of Wnt1 can generate tumors comprising both basal Hras-mutant Wnt1-low and luminal Hras wild-type Wnt1-high subclones. These subclones cooperate to maintain tumor growth as the basal cells depended on Wnt1 secreted by luminal cells for growth. When Wnt1 production is blocked, basal cells recruit heterologous Wnt-producing cells to restore tumor growth (80). In a basal-like Trp53-null mouse model, Zhang et al. reported that the presence of a mesenchymal-like subpopulation supported tumor growth by providing paracrine niche ligands to tumor-initiating cells. Knockdown of these ligands in the mesenchymal cells or their corresponding receptors in the tumor-initiating cells led to reduced tumorigenicity and prolonged tumor latency (81). Similarly, the Polyak group discovered that minor cancer cell subclones can drive tumor growth at both the primary and metastatic sites in a non-cell-autonomous manner (79, 82).
Besides the cooperation between tumor cell subpopulations, the complex interactions between tumor cells and stromal cells also promote tumor progression and can be exploited for therapy development. In a C3(1)/SV40 Tag-derived mouse mammary tumor model, Cazet et al. used scRNA-seq to find that CAFs are the primary population in the TME that respond to Hedgehog ligand secreted by tumor cells. The Hedgehog-activated CAFs in turn express Ffg5 and remodel the ECM to promote cancer stemness and chemoresistance. Interruption of this tumor-CAF interplay by treatment with inhibitors of Smoothened involved in Hedgehog signaling sensitized tumors to docetaxel chemotherapy in both PDX mouse models and in patients from a Phase I clinical trial (95). Similarly, crosstalk between other cell populations in the TME can be predicted computationally in scRNA-seq (67).
Another major consequence of intratumor heterogeneity is therapeutic resistance. The existence of varying genetic and phenotypic subpopulations within a tumor provides a substrate for Darwinian evolution (96). Janiszewska et al. developed a specific-to-allele PCR-FISH (STAR-FISH) method that allows for the combined detection of PIK3CA mutation encoding His1047Arg and HER2 copy number alterations in single cells in intact tissues. In untreated HER2-positive breast cancer, they identified a major population of cells with HER2 amplification and wild-type PIK3CA and a minor population of cells with mutant PIK3CA. However, neoadjuvant chemotherapy dramatically increased the frequency of PIK3CA mutant cells and reduced the dominant HER2-amplified population, which may confer resistance to the HER2-targeting antibody trastuzamab (97). Grosselin et al. performed scChIP-seq and discovered that a common chromatin signature was shared between a subclone of cells from treatment-naive tumors and cells from tamoxifen-resistant tumors derived from the same ER-positive PDX model (89). This implies that instead of epigenetic reprogramming of cancer cells, the selective expansion of rare cells with distinct chromatin features may underly therapy resistance.
Because of the paucity of well-defined specific therapeutic targets in TNBC, the standard of care for these patients in the neoadjuvant setting is chemotherapy (NAC), which usually includes a combination of anthracyclines and taxanes. Although 40–50% of patients with stage II to III TNBC may achieve a pathologic complete response (pCR), many patients develop chemoresistance (98). Two alternative hypotheses have been developed to explain the genomic basis of chemotherapy resistance: (1) adaptive resistance which results from the selection and expansion of rare pre-existing clones; or (2) acquired resistance wherein new genomic alterations are generated to confer a chemoresistant phenotype. Due to the lack of techniques to resolve intratumor heterogeneity and the difficulty to detect rare mutations in the bulk sequencing era, the question of which mechanism caused chemoresistance in TNBC was unresolved. With the advent of single-cell technology, Kim et al. tackled this question by combined scDNA-seq and single-nucleus RNA-seq (snRNA-seq) of longitudinal samples collected from TNBC patients before, during, or after neoadjuvant NAC treatment. In half of the patients, NAC led to clonal extinction wherein clones were completely eliminated and no residual tumor cells were detected; in the other half of patients, clones persisted after treatment with shifted frequencies. In the clonal persistence patients, copy-number aberrations were adaptively selected rather than de novo acquired in response to chemotherapy. Consistently, they did not observe an increase in mutation burden in response to NAC in the samples. In contrast to the adaptive evolution at genomic level, the chemoresistant transcriptomic profiles did not pre-exist and were acquired via transcriptional reprogramming after treatment. This implies that adaptive genomic evolution and acquired transcriptional evolution collectively lead to phenotypic adaptation. In their gene set variation analysis of the snRNA-seq data, gene signatures including AKT1/mTOR, hypoxia, EMT, extracellular matrix degradation, and angiogenesis, were upregulated in the chemoresistant tumor cells post NAC treatment (99). This suggests that it might be possible to overcome chemoresistance by targeting the Akt/mTOR pathway, reprogramming EMT or perhaps by inhibiting HIF-1α.
7. Concluding remarks
Intratumor heterogeneity represents a major challenge to effective cancer therapy and personalized medicine. However, with the aid of new technologies such as single-cell analysis and spatial pathologies, investigators are now beginning to elucidate the complex composition and dynamics of tumor ecosystem during disease progression and treatment. This should help reveal novel predictive biomarkers, prognostic features, and therapeutic targets, although the translation of this knowledge to the clinic still has yet to be fully realized. While targeted therapies will remain an important approach, in order to minimize resistance to a single drug, different combinations will need to be tailored based upon tumor heterogeneity and evolution. However, it is likely that no combination will suffice to target all tumor cells, and that the bystander effects elicited by the immune system will be required to eliminate residual disease caused by heterogeneity. Importantly, spatially resolved immunophenotypes may help inform the response and resistance to immunotherapies. Mapping which cells express immune checkpoint ligands and receptors and where these interactions occur within tumors will be key for treatment design. Combination therapy directed at both tumor cells and the TME will no doubt be required for improved treatment regimens for breast cancer.
Acknowledgement
This work was supported by grants CA148761 from the National Cancer Institute and RP170172 from the Cancer Prevention and Research Institute of Texas (J.M.R.). The figure in this review was created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021. [DOI] [PubMed] [Google Scholar]
- 2.Dagogo-Jack I, and Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018; 15(2):81 –94. [DOI] [PubMed] [Google Scholar]
- 3.McGranahan N, and Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27(1 ):15–26. [DOI] [PubMed] [Google Scholar]
- 4.Beca F, and Polyak K. Intratumor Heterogeneity in Breast Cancer. Adv Exp Med Biol. 2016;882:169–89. [DOI] [PubMed] [Google Scholar]
- 5.Waldman AD, Fritz JM, and Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wein L, Luen SJ, Savas P, Salgado R, and Loi S. Checkpoint blockade in the treatment of breast cancer: current status and future directions. Br J Cancer. 2018;119(1 ):4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379(22):2108–21. [DOI] [PubMed] [Google Scholar]
- 8.Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med. 2020;382(9):810–21. [DOI] [PubMed] [Google Scholar]
- 9.Martelotto LG, Ng CKY, Piscuoglio S, Weigelt B, and Reis-Filho JS. Breast cancer intratumor heterogeneity. Breast Cancer Research. 2014;16(3):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Cl, Uribe DJ, and Daling JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer. 2005;93(9):1046–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. [DOI] [PubMed] [Google Scholar]
- 12.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69(10):4116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107(15):6994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61 –70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali HR, Rueda OM, Chin SF, Curtis C, Dunning MJ, Aparicio SA, et al. Genome-driven integrated classification of breast cancer validated in over 7,500 samples. Genome Biol. 2014;15(8):431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell. 2018;33(4):690–705..e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertos NR, and Park M. Breast cancer - one term, many entities? The Journal of clinical investigation. 2011;121(10):3789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russnes HG, Navin N, Hicks J, and Borresen-Dale AL. Insight into the heterogeneity of breast cancer through next-generation sequencing. The Journal of clinical investigation. 2011;121(10):3810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–27. [DOI] [PubMed] [Google Scholar]
- 23.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity. 2018;48(4):812–30.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bareche Y, Buisseret L, Gruosso T, Girard E, Venet D, Dupont F, et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J Natl Cancer Inst. 2020;112(7):708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coates AS, Winer EP, Goldhirsch A, Gelber RD, Gnant M, Piccart-Gebhart M, et al. Tailoring therapies-improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol. 2015;26(8):1533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allison KH, Hammond MEH, Dowsett M, McKernin SE, Carey LA, Fitzgibbons PL, et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J Clin Oncol 2020;38(12):1346–66. [DOI] [PubMed] [Google Scholar]
- 28.Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36(20):2105–22. [DOI] [PubMed] [Google Scholar]
- 29.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The Human Cell Atlas. Elife. 2017;6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rozenblatt-Rosen O, Stubbington MJT, Regev A, and Teichmann SA. The Human Cell Atlas: from vision to reality. Nature. 2017;550(7677):451 –3. [DOI] [PubMed] [Google Scholar]
- 31.Hu BC. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature. 2019;574(7777): 187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajewsky N, Almouzni G, Gorski SA, Aerts S, Amit I, Bertero MG, et al. LifeTime and improving European healthcare through cell-basedinterceptive medicine. Nature. 2020;587(7834):377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell. 2020;181(2):236–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irmisch A, Bonilla X, Chevrier S, Lehmann KV, Singer F, Toussaint NC, et al. The Tumor Profiler Study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell. 2021;39(3):288–93. [DOI] [PubMed] [Google Scholar]
- 35.Lun XK, and Bodenmiller B. Profiling Cell Signaling Networks at Single-cell Resolution. Mol Cell Proteomics. 2020;19(5):744–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim B, Lin Y, and Navin N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell. 2020;37(4):456–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Longo SK, Guo MG, Ji AL, and Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nature Reviews Genetics. 2021. [DOI] [PMC free article] [PubMed]
- 38.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, Mclndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. [DOI] [PubMed] [Google Scholar]
- 40.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Twigger A-J, and Khaled WT. Mammary gland development from a single cell ‘omics view. Seminars in Cell & Developmental Biology. 2021;114:171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561 ):486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen H, Lareau C, Andreani T, Vinyard ME, Garcia SP, Clement K, et al. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biology. 2019;20(1):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung CY, Ma Z, Dravis C, Preissl S, Poirion O, Luna G, et al. Single-Cell Chromatin Analysis of Mammary Gland Development Reveals Cell-State Transcriptional Regulators and Lineage Relationships. Cell Rep. 2019;29(2):495–510.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell. 2018;172(1–2):205–17.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nichterwitz S, Chen G, Aguila Benitez J, Yilmaz M, Storvall H, Cao M, et al. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat Commun. 2016;7:12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods. 2017;14(9):865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nature Biotechnology. 2017;35(10):936–9. [DOI] [PubMed] [Google Scholar]
- 49.Bandura DR, Baranov VI, Ornatsky Ol, Antonov A, Kinach R, Lou X, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–22. [DOI] [PubMed] [Google Scholar]
- 50.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (New York, NY). 2011;332(6030):687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spitzer MH, and Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165(4):780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Reveaied by Multiplexed Ion Beam Imaging. Cell. 2018;174(6): 1373–87 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A 2013;110(29):11982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin JR, Fallahi-Sichani M, and Sorger PK. Highly multiplexed imaging of single cells using a high-throughputcyclic immunofluorescence method. Nat Commun. 2015;6:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014; 11 (4):417–22. [DOI] [PubMed] [Google Scholar]
- 56.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nature Medicine. 2014;20(4):436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science (New York, NY). 2014;343(6177):1360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alon S, Goodwin DR, Sinha A. Wassie AT, Chen F, Daugharthy ER, et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science (New York, NY) 2021;371(6528):eaax2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science (New York, NY). 2015;348(6233):aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (New York, NY). 2016;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 61.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science (New York, NY). 2019;363(6434):1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maynard KR, Collado-Torres L, Weber LM, Uytingco C, Barry BK, Williams SR, et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nature Neuroscience. 2021;24(3):425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16(10):987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caswell-Jin JL, Lorenz C, and Curtis C. Molecular Heterogeneity and Evolution in Breast Cancer. Annual Review of Cancer Biology. 2021;5(1):79–94. [Google Scholar]
- 65.Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174(5):1293–308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. Embo j. 2020;39(19):e104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell. 2019;177(5):1330–45.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature. 2020;578(7796):615–20. [DOI] [PubMed] [Google Scholar]
- 70.Ali HR, Jackson HW, Zanotelli VRT, Danenberg E, Fischer JR, Bardwell H, et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nature Cancer 2020;1 (2):163–75. [DOI] [PubMed] [Google Scholar]
- 71.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell. 2018;174(6):1373–87.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. The Journal of clinical investigation. 2019;129(4):1785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Almendro V, Marusyk A, and Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol. 2013;8:277–302. [DOI] [PubMed] [Google Scholar]
- 74.Black JRM, and McGranahan N. Genetic and non-genetic clonal diversity in cancer evolution. Nat Rev Cancer. 2021. [DOI] [PubMed]
- 75.Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, and Swanton C. Cancer: Evolution Within a Lifetime. Annual Review of Genetics. 2014;48(1):215–36. [DOI] [PubMed] [Google Scholar]
- 76.Davis A, Gao R, and Navin N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim Biophys Acta Rev Cancer. 2017;1867(2):151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marusyk A, and Polyak K. Tumor heterogeneity: Causes and consequences. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2010;1805(1):105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davis A, Gao R, and Navin N. Tumor evolution: Linear, branching, neutral or punctuated? Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2017;1867(2):151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, and Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514(7520):54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cleary AS, Leonard TL, Gestl SA, and Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508(7494):113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang M, Tsimelzon A, Chang CH, Fan C, Wolff A, Perou CM, et al. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer Discov. 2015;5(5):520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Janiszewska M, Tabassum DP, Castano Z, Cristea S, Yamamoto KN, Kingston NL, et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nature Cell Biology. 2019;21(7):879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nature genetics. 2016;48(10):1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cross W, Kovac M, Mustonen V, Temko D, Davis H, Baker AM, et al. The evolutionary landscape of colorectal tumorigenesis. Nat Ecol Evol. 2018;2(10):1661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153(3):666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature. 2020;578(7793):122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Minussi DC, Nicholson MD, Ye H, Davis A, Wang K, Baker T, et al. Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature. 2021;592(7853):302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mazor T, Pankov A, Song JS, and Costello JF. Intratumoral Heterogeneity of the Epigenome. Cancer cell. 2016;29(4):440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nature genetics. 2019;51(6):1060–6. [DOI] [PubMed] [Google Scholar]
- 90.Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell. 2018;34(6):939–53.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yuan S, Norgard RJ, and Stanger BZ. Cellular Plasticity in Cancer. Cancer Discovery. 2019;9(7):837–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bakir B, Chiarella AM, Pitarresi JR, and Rustgi AK. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020;30(10):764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nam AS, Chaligne R, and Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 2021;22(1):3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan C-L, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nature Communications. 2018;9(1):2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marusyk A, Janiszewska M, and Polyak K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell. 2020;37(4):471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Janiszewska M, Liu L, Almendro V, Kuang Y, Paweletz C, Sakr RA, et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nature genetics. 2015;47(10):1212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. [DOI] [PubMed] [Google Scholar]
- 99.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell. 2018; 173(4):879–93.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]