

Abstract
Background:
Inflammation plays a major role in the development and progression of cardiovascular disease (CVD) morbidity and mortality. The well-established relationship between periodontal disease (PD) and CVD may be causal. Left untreated, PD can lead to high systemic inflammation, thus contributing to inflammatory CVD, such as atherosclerosis. Multiple mechanisms have been proposed to elucidate the causal relationship between PD and its contribution to CVD.
Objective:
This review article highlights the current evidence supporting the role of PD in the development and progression of atherosclerosis.
Methods:
After creating a list of relevant Medical Subject Heading (MeSH) terms, a systematic search within PubMed in English for each MeSH term between 2000–2019 was used to generate evidence for this review article.
Conclusion:
There is overwhelming evidence in the current literature that supports an association between PD and CVD that is independent of known CVD risk factors. However, the supporting evidence that PD directly causes CVD in humans continues to remain elusive. Multiple biologically plausible mechanisms have been proposed and investigated, yet most studies are limited to mouse models and in vitro cell cultures. Additional studies testing the various proposed mechanisms in longitudinal human studies are required to provide deeper insight into the mechanistic link between these two related diseases.
Keywords: Periodontal disease, periodontitis, inflammation, atherosclerosis, cardiovascular disease
Introduction
Inflammation and Cardiovascular Disease Overview
Despite major advancements in prevention and treatment, such as lipid control and blood pressure control, cardiovascular disease (CVD) continues to remain the leading cause of death worldwide, including the United States. Emerging evidence has demonstrated that inflammation plays an important role in CVD.1,2 Therefore, targeting residual inflammatory risk may be just as important as targeting residual cholesterol risk for reducing CVD development.1,2 Coronary artery disease (CAD) is now recognized as a complex inflammatory disorder, in which the immune system interacts with metabolic derangements and vascular injury.3,4
Inflammation has been shown to play a crucial role in the pathogenesis of atherosclerosis and atherothrombosis.5 During the initial stages of atherogenesis, oxidized low-density lipoprotein cholesterol (LDL-C), injury, and infection cause monocytes to bind to the site of injury in the endothelial wall.6 Monocytes mature into macrophages and eventually develop into foam cells as they consume oxidized lipids and lipoproteins.6 Macrophages and foam cells release proinflammatory cytokines, activating vascular endothelial cells and recruiting additional leukocytes.7 The increased activity of inflammatory cells contributes to the development of the coronary plaque, consisting of a lipid core, foam cells, and fibrous cap composed of collagen.3 The plaque can then rupture, as macrophages release proteolytic enzymes that break down the collagen in the fibrous cap.3 Plaque rupture releases tissue factors and atherosclerotic debris into circulation, leading to thrombosis and consequent myocardial ischemia and infarction.8
Studies have demonstrated a robust relationship between inflammation and the risk of CAD, as well as major CVD events. An elevated plasma level of high-sensitivity C-reactive protein (hs-CRP), an inflammatory biomarker, has not only been shown to be an independent risk factor for CAD, but also predicts future CVD events, regardless of LDL-C levels.9,10 Clinically, hs-CRP has been shown to be the gold standard for assessing low-grade systemic inflammation, as it captures the upstream activity of the interleukin (IL)-1 to IL-6 inflammatory cytokine axis.11 In intermediate CVD risk patients, the risk of future CVD events was lowered by hs-CRP reduction in addition to standard CVD risk factor control.12 The JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) trial demonstrated that patients without hyperlipidemia but with elevated hs-CRP levels exhibited a reduced incidence of major adverse cardiovascular events (MACE) when treated with a statin.13 Nevertheless, it is important to note in this study that LDL-C was also reduced relative to baseline, and therefore, the inflammatory contribution to MACE could not be completely elucidated.
Thus, targeting inflammation has been an attractive pharmacological strategy to decrease CVD risk. Two major clinical trials have increased our understanding of the value of this approach. The CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcome Study) demonstrated that inhibition of IL-1β with the monoclonal antibody, canakinumab, significantly reduced MACE, independent of lipid lowering, in patients with history of MI and elevated hs-CRP.14 In addition, patients treated with canakinumab had reduced plasma levels of downstream inflammatory cytokines, IL-6 and hs-CRP from baseline.14 Lastly, those who experienced the greatest reduction in hs-CRP also had the most significant reduction in CVD- and total mortality. In contrast, CIRT (Cardiovascular Inflammation Reduction Trial) showed that patients receiving a non-specific anti-inflammatory therapy with low-dose methotrexate did not have reduction in MACE and did not have reduced IL1-β, IL-6, and hs-CRP levels.15 The conclusions of these recent trials suggest that targeted inflammatory therapy, namely inhibition of the IL-1 to IL-6 axis, may provide atherosclerotic protection and thereby help reduce the risk of MACE.
Inflammation as a driver of atherogenesis is evident in patients with chronic inflammatory conditions such as rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, and psoriasis. Patients with chronic systemic inflammatory disease have earlier onset and increased rates of CVD events when compared to that of the healthy population.16–20 Moreover these patients specifically psoriasis on a biologic treatment (e.g. anti-tumor necrosis factor (TNF)-α, anti-IL-12, anti-IL-23, and anti-IL17) had favorable modulation of CAD with reduction of high risk plaque features such as non-calcified coronary plaque burden.21,22 Observational cohorts of patients with rheumatoid arthritis on anti-TNF-α therapy experienced a reduced incidence of MACE.23 These findings suggest that early treatment of systemic chronic inflammatory conditions may reduce the progression of subclinical CVD and potentially reduce the risk of future CVD events.
In addition to the systemic inflammatory conditions mentioned previously, periodontal disease (PD) is a chronic inflammatory disease with a microbiological etiology that increases the overall inflammatory burden. Given its inflammatory nature, PD may potentially contribute to atherosclerosis and CVD events. This review will explore the current mechanistic and clinical evidence supporting the role of PD in the development and contribution to CVD.
Periodontal Disease as a Systemic Inflammatory Condition
PD encapsulates a broad range of chronic inflammatory conditions that affect the gingiva, bone and periodontal ligaments supporting teeth.24 According to the classification set by the American Academy of Periodontology and the European Federation of Periodontology, PD can be divided into three major categories with their respective subcategories:25
- Periodontal health and gingival diseases
- Periodontal Health and Gingival Health
- Gingivitis: Dental Biofilm-Induced
- Gingival Diseases: Non-Dental Biofilm-Induced
- Periodontitis
- Necrotizing Periodontal Diseases
- Periodontitis
- Periodontitis as a Manifestation of Systemic Disease
- Periodontal Abscess and Endodontic-Periodontal Lesions
- Periodontal Manifestations of Systemic Diseases and Developmental and Acquired Conditions
- Systemic Diseases or Conditions Affecting Periodontal Supporting Tissues
- Mucogingival Deformities and Conditions
- Traumatic Occlusal Forces
- Tooth- and Prosthesis-Related Factors
PD is caused by the interaction between the oral flora and host immune system, which results in an inflammatory response and destruction of the supporting structures of the teeth (gingiva, bone and ligament).24 Dental plaque is formed as the oral bacteria produce a microbial biofilm on the teeth.26 Gingivitis ensues as bacteria in the dental plaque induce localized inflammation of the gingiva.26 As gingivitis continues to progress, the loss of gingiva, bone and periodontal ligament leads to the development of deep periodontal pockets that are characteristic of chronic periodontitis, leading to systemic inflammation.26
Immune/Inflammatory Pathway Associated with Periodontal Disease
PD has features of immune dysregulation associated with chronic inflammation, including elevated plasma cytokines, such as IL-1, IL-6, and TNF-α in the gingiva and gingival fluid.27 Inhibiting IL-1/TNF-α activity in animal models of PD resulted in reduced inflammatory cell recruitment and bone loss.28 Clinically, patients with chronic periodontitis exhibit increased systemic inflammatory markers, including TNF-α, IL-1, IL-6, IL-8 and circulating neutrophils.29,30 Treatment of periodontitis in these models led to reduced systemic inflammation, as measured by hs-CRP, over a 6-month period.31 Mice infected with periodontal bacteria displayed alterations in their gut microbiota that were more conducive to systemic inflammation (e.g. decreased tight junctions in the ileum).32 However, it is important to note that oral pathogens were not detected in the gut microbiota of infected mice, and the exact mechanism by which gut microbiota would be altered due to PD was not fully understood. Studies have also suggested that locally produced CRP from PD may modulate systemic CRP.33 However, the direct contribution of the locally increased CRP to increased systemic CRP levels has not been established.
Association of Periodontal Disease and Cardiovascular Disease
Multiple epidemiologic and observational studies have consistently demonstrated that PD is independently associated with subclinical, and clinical CVD across diverse populations.34–36 Nevertheless, epidemiologic and observational studies are limited in that they do not support a causal relationship, and there is uncertainty regarding the magnitude of the influence of PD and future risk of atherosclerosis and MACE.37,38 Recently, a prospective observational study attempted to provide a more accurate assessment of the role of PD in CVD.39 In a prospective cohort consisting mainly of middle-aged women, an increased incidence and prevalence of PD was significantly associated with an increased risk of developing future cardiovascular event.39 Furthermore, patients who had acute coronary syndrome (confirmed via coronary angiography) demonstrated worse dental and periodontal health.40 In a most recent case-control study, patients with moderate-to-severe periodontitis demonstrated significantly greater odds of developing myocardial infarction, suggesting a possible relationship between PD severity and CVD state.41 Smoking is also risk factor for both periodontitis and cardiovascular disease.42 Smoking contributes to a chronic inflammatory reaction, impairment in fibroblast function and tissue healing, all of which are involved with both PD and CVD.42
PD is linked to a multitude of cardiac and vascular pathologies. Periodontal bacterial burden was associated with carotid artery intima-media thickness (IMT), a subclinical marker of carotid atherosclerotic disease.43 Treatment and improvement of periodontal status was associated with a reduced progression of carotid IMT at 3-year follow-up.44 Periodontal disease has also been implicated in the progression of abdominal aortic aneurysm (AAA) with periodontopathic bacteria present in the samples of diseased AAA.45,46 A recent systematic review of five studies demonstrated that presence of periodontal bacteria in the bloodstream or in situ in the vascular lesion was associated with AAA.47 Periodontitis was also an independent predictor of arrhythmia-related CVD events in patients with atrial fibrillation.48 A cross-sectional study demonstrated that patients with peripheral artery disease had worsened periodontal health and higher levels of systemic inflammation, including CRP and TNF-α.49 Lastly, a murine study demonstrated that the injection of periodontal pathogens induced myocardial hypertrophy via activation of matrix metalloproteinases.50
Genetic Susceptibility to Periodontal Disease and Cardiovascular Disease
Genetic studies have suggested the existence of shared susceptibility genes that are involved in both the pathogenesis of CVD and PD.51–53 One of the strongest and best replicated genetic CVD risk loci has been identified on human chromosome 9p21.3; this locus has been confirmed by four independent genome-wide association studies and by a subsequent meta-analysis.54–58 Studies have shown that 9p21.3 locus is also associated with PD, suggesting shared susceptibility effects in both CVD and PD.53,59 Nevertheless, the multifaceted and chronic nature of both diseases makes it difficult to establish a definitive causal relationship. Despite the increased evidence for an association between PD and CVD, the confirmation of a genetic link between the two pathologies continues to remain elusive.
Mechanisms of PD Involvement in CVD
Though no study has elucidated the exact underlying biological mechanisms by which PD contributes to atherosclerosis, multiple potential mechanisms link PD to CVD and MACE. These include: 1) endothelial dysfunction; 2) direct microbial injury from periodontal pathogens; and 3) injury and inflammation due to microbial byproducts (namely, lipopolysaccharide and gingipains), and 4) immune cross-reactivity with bacterial antigens.
Periodontal Disease Leads to Endothelial Dysfunction
PD has been implicated in endothelial dysfunction, one of the initial steps of atherogenesis that provide valuable CVD risk stratification.60 Flow-mediated dilation (FMD) is a robust method to assess for endothelial dysfunction in patients with subclinical CVD.61 In one study, patients with severe PD without CVD or known CVD risk factors demonstrated significantly reduced brachial artery FMD when compared to age-matched healthy control subjects.62 Furthermore, FMD in subjects with only mild PD was not significantly different from the control group.62 These findings suggest that impairment of FMD and subsequent endothelial dysfunction may depend on PD severity. Other studies have confirmed the reduction in FMD in PD patients, with impairment of FMD similar to that seen in patients with a history of a MI.63,64 Periodontitis patients with endothelial dysfunction also demonstrated elevated levels of salivary matrix metalloproteinase and tissue inhibitor of metalloproteinase complex.65 Furthermore, patients with severe periodontitis demonstrated improvements in FMD when PD was treated, though the influence of treatment upon subsequent CVD risk reduction and MACE was not investigated.66
Microbes may Directly Induce Inflammation and Atherosclerosis
Bacteremia due to PD resulting in direct vascular injury has been shown to potentially contribute to atherosclerosis. Transient bacteremia can occur due to tooth brushing, chewing, or after any dental procedure and is more frequent in patients with PD.67,68 Chronic infections due to bacteremia may increase inflammatory burden required to accelerate atherogenesis.69 In the setting of PD, gingival lesions and bleeding in periodontal pockets allow periodontal bacteria to enter the systemic circulation.70
Major pathogenic periodontal organisms, such as Pophyromonas gingivalis (P. gingivalis), have been shown to adhere and invade the coronary artery endothelial cells in vitro.71 P. gingivalis, one of the most widely studied periodontal microorganisms, also induces the expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1, and P- and E- selectins in human umbilical vein endothelial cells.72 Apolipoprotein E (ApoE) deficient mice with oral P. gingivalis infection moreover developed periodontitis and demonstrated increased plasma levels of inflammatory cytokines and cholesterol, along with alveolar bone loss and atherosclerotic lesions in the aorta.73,74 In contrast, ApoE deficient mice with oral P. gingivalis infection that were treated with muramyl dipeptide to stimulate nucleotide binding oligomerization domain-containing protein 2 (NOD2) demonstrated a reduction in the same parameters. These results suggest that inhibition of NOD2 may play an important role in triggering a host immune response that is conducive to both periodontitis and atherosclerosis. In mice infected with P. gingivalis, treatment of the oral infection resulted in fewer atheromatous lesions in the aorta and aortic tree compared to those of infected mice not receiving antibiotic treatment.75 Additionally, mice infected with P. gingivalis with deficient fimbria (bacterial component required for endothelial invasion) also had fewer atheromatous lesions compared to mice infected with wild type P. gingivalis.75 These findings suggest that direct bacterial invasion of the vascular endothelium may be involved in accelerated atherosclerosis.
Polymicrobial oral infection of major periodontal organisms has demonstrated increased aortic plaque with increased macrophage presence, increased serum cholesterol, and increased triglycerides in ApoE deficient mice.76 Furthermore, several studies have highlighted the presence of periodontal organisms (via DNA, RNA, antigens) in atheromatous plaque samples and vascular walls in patients, which may contribute to the progression of atherosclerosis.77–79 In addition, edentulousness and serum IgG antibodies to periodontal pathogens (Aggregatibacter actinomycetemcomitans [A. actinomycetemcomitans] and P. gingivalis) were associated with CVD in humans.80,81 While the majority of the studies assessed indirect markers of bacterial presence (e.g. serology, presence of dead bacteria, DNA, RNA, antigen), one study demonstrated viable invasive A. actinomycetemcomitans and P. gingivalis in samples of human atherosclerotic plaque.82 Despite mounting evidence suggesting a role for periodontal microbes in inducing atherosclerosis, no study to date has demonstrated that the bacteria themselves are the causative agents of atherosclerosis in humans. These studies suggest that the presence of periodontal bacteria in plaque may accelerate the process of atherogenesis. However, this tentative conclusion requires additional study and more definitive evidence.
Microbiota (including those of the oral cavity) of the human body play important roles in human disease. Probiotics have been suggested to have a positive impact on both the oral cavity and cardiovascular risk.83 There is emerging evidence that probiotics may confer reduction in CVD progression as well as PD.83 In a randomized, double-blind, placebo-controlled study, the use of probiotics demonstrate such has Lactobacillus salivarius WB21 reduced both plaque index and periodontal pocket depth in patients at risk of PD.84 However, the effects of Lactobacillus salivarius WB21 on reducing CVD risk has yet to be established.
Microbial Byproducts may Play a Role in Inducing Systemic Inflammation
Microbial components and byproducts have also been implicated in inducing systemic inflammation and atherosclerosis. Lipopolysaccharide (LPS), also known as endotoxin, is the primary membrane wall component of gram-negative bacteria and is a major factor in sepsis. LPS has also been shown to be an important surface antigen and a potential proinflammatory mediator between PD and CVD.85 High circulating levels of LPS (endotoxemia) is associated with elevated CRP and increased CVD events, when adjusted for traditional risk factors for CVD.86 In the setting of periodontitis, a case-cohort study in the FINRISK 1992 cohort demonstrated that a high antibody response to periodontal pathogens (IgG antibodies to A. actinomycetemcomitans and P. gingivalis) independently predicted incidence of CVD events over a 10-year follow-up period.87 Furthermore, LPS levels correlated positively with periodontal IgG antibody levels.87 However, it is important to note that LPS levels can be derived from a variety other types of chronic infections such as Helicobacter pylori infection or bacterial translocation from the large intestine. A recent follow-up study has confirmed that subgingival microbial burden can associate with and contributes to salivary and serum LPS levels.85 After adjusting for confounding factors, this study found that the association between PD and CVD may be due to elevated LPS levels, suggesting that periodontitis leads to low-grade systemic inflammation that may contribute to a higher risk of CVD.85
LPS is released into the systemic circulation as gram-negative bacteria in the blood circulation are lysed. LPS is recognized by the host immune system by the LPS binding protein (LBP). Once LBP binds to LPS, the antigen is recognized by the CD14 co-receptor present on macrophages, neutrophils, and endothelial cells.88 CD14 then complexes with toll-like receptor 4 (TLR4) and MD2 complex to activate the nuclear factor κB (NF-κB) inflammatory pathway.89 The CD14 and TLR4/NF-κB inflammatory pathway has been associated with the pathogenesis of atherosclerosis and CVD events. Patients with history of a MI have a higher frequency of a single nucleotide polymorphism (C→T) in position −260 in the promoter region of the CD14 receptor gene.90 This polymorphism has been shown to increase the transcriptional activity of the downstream NF-κB inflammatory pathway, which can synergize with CRP in activating the vascular endothelium.91 This finding suggests that certain immune reactions of inflammatory cells to infectious insults may play a role in atherosclerosis.89 Additionally, LPS has been shown to contribute to LDL-C oxidation, foam cell maturation, and thrombogenesis, all of which are involved in the atherosclerotic process.92 In vitro studies in human umbilical vein endothelial cells demonstrated that exposure to P. gingivalis and LPS induced endothelial cell death and increased oxidized LDL-C, and TNF-α levels.93 Thus, the increased systemic levels of LPS due to chronic PD could potentially exert proatherogenic effects.
Periodontal bacteria also produce a wide variety of toxins and virulence factors, such as proteases, lectins, fimbriae, adhesins, and hemagglutinins that can disrupt the host immune response and consequently contribute to CVD. In human monocyte-derived macrophages exposed to P. gingivalis, cytokine antibody arrays demonstrated production of monocyte chemoattractant protein 1, macrophage inflammatory protein 1β, and MIP-3α.94 Furthermore, purified P. gingivalis LPS and fimbrillin, a major fimbrial protein of the bacteria, produced patterns of cytokines suggesting that host immune cells may respond differently to live P. gingivalis compared to its components.95
Gingipains, cysteine proteases produced by P. gingivalis, demonstrate disruptive effects on inflammatory cells, coagulation pathway components, vascular permeability, and the complement system.96 Gingipains reduced LPS-induced IL-8 production in gingival fibroblasts and cleaved ICAM-1 on endothelial cells, thereby allowing immune invasion in periodontal tissues.97,98 Recent studies have also suggested that gingipains may play an important role in lipid dysregulation by cleaving apoB-100, which is the main component of LDL-C that is responsible for binding LDL-C to cell surface receptors.99,100 Disruption of genes involved in gingipain or inhibition of gingipain in P. gingivalis was found to attenuate atherosclerotic lesions in ApoE knockout mice.99 The authors in the study suggest that gingipains, in particular the Arg-gingipain, play an important role in the degradation of apoB-100 in LDL-C particles, and thus are instrumental in development of atherogenesis. Gingipains also demonstrate unfavorable proteolytic effects on lipoproteins by inducing reactive oxygen species and lipid peroxidation.101 Conclusions derived from these study suggest a potential mechanism linking virulence factors from gingival infections to deranged lipid profiles and a pro-inflammatory pathway leading to accelerated atherogenesis .
Molecular Mimicry/Cross Reactivity as Potential Mechanisms of Atherogenesis
Cross-reactive autoantibodies against bacterial antigens are also suggested as a possible mechanism by which periodontal infections promote atherosclerosis, particularly those against heat-shock protein (HSP)60. HSP60 is not only highly conserved with high homology between prokaryotes and eukaryotes, but also is highly immunogenic.102 The proposed mechanism involves endothelial damage from an immune response to bacterial HSP. Cross-reactivity between P. gingivalis and human HSP60 was capable of promoting atherosclerotic changes due to the subsequent autoimmune response in the vascular endothelium.103 This study demonstrated that T-cell immune responses to P. gingivalis HSP60 may be involved with atherosclerosis, since P. gingivalis HSP-specific T-cells were observed in atherosclerotic plaque in the laboratory.104 T-cells recognizing HSP60 were also found in diseased periodontal tissue and peripheral blood from patients.105 In another study, HSP60 antibody levels to were highest in patients with atherosclerosis.106 Clonal analysis demonstrated HSP60-reactive T cells from these patients recognized both human and P. gingivalis HSP60.106 In a cross-sectional study, patients with mild periodontitis had increased levels of anti-serum HSP60 and of small, dense LDL-C when compared to controls matched for age and body mass index.107
Conclusions
There is overwhelming evidence in the current literature that supports an association between PD and CVD. Multiple observational studies have found that PD associates with CVD, independent of known CVD risk factors. However, the supporting evidence that PD directly causes CVD in humans continues to remain elusive. Multiple biologically plausible mechanisms have been proposed and investigated, yet most studies are limited to mouse models and in vitro cell cultures. Additional studies testing the various proposed mechanisms in longitudinal human studies are required to provide deeper insight into the mechanistic link between these two related diseases. Current studies have only assessed how periodontal interventions affect markers of systemic inflammation (e.g. hs-CRP) and markers of subclinical CVD (e.g. endothelial dysfunction). Whether treatment of PD reduces the incidence and/or severity of CVD and MACE remains to be established. A randomized clinical trial involving standardized definitions of PD and standardized periodontal interventions is required to demonstrate that PD treatment is beneficial to CVD risk reduction. In particular, future studies could benefit from expanding on how decreases in markers of systemic inflammation and endothelial function from periodontal therapy affect the incidence of CVD events. With increasing investigation of the role of PD in CVD, PD may be recognized as an important CVD risk factor.
Figure 1:
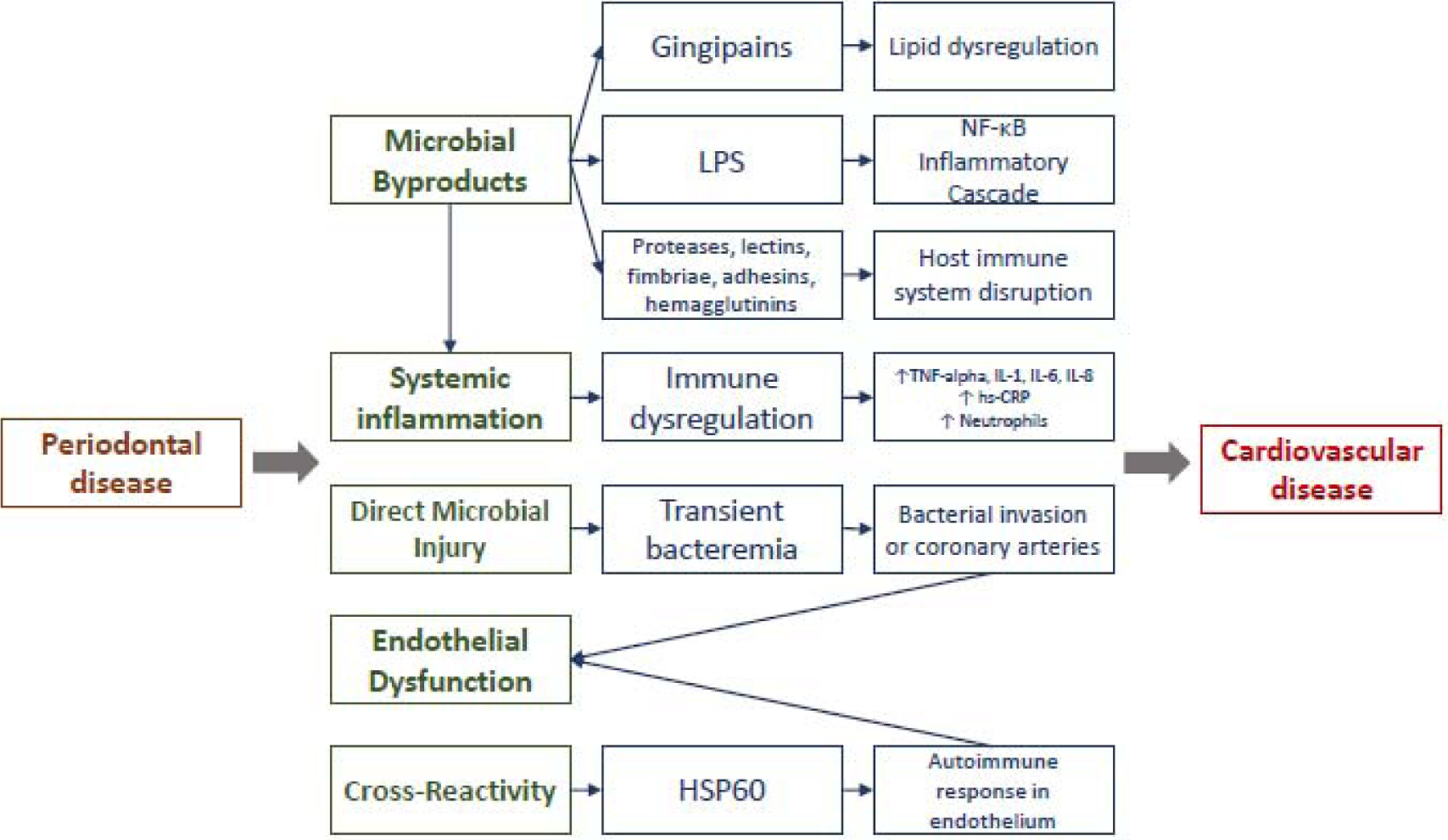
Schematic demonstrating proposed mechanisms linking periodontal disease to cardiovascular disease. Note: proposed mechanisms may overlap.
Table 1.
| Topic | Author | Study design |
|---|---|---|
| Endothelial dysfunction | ||
| Patients with severe PD and no known cardiovascular disease (CVD) or risk factors demonstrated significantly reduced brachial artery in flow-mediated dilation (FMD) when compared to healthy controls, not attributable to vascular smooth muscle cell dysfunction. No significant difference in FMD was found between patients with mild PD and healthy controls. | Salomon et al., 2003 | Cross-sectional study |
| Patients with chronic PD and no known CVD have impaired endothelium-dependent dilatation (EDD) and endothelium-independent dilatation (EID) compared to healthy controls. The reduction in EDD and EID was improved following initial treatment for PD. | Mercanoglu et al., 2004 | Prospective observational study |
| Patients with chronic PD demonstrated reductions in FMD similar to those in patients with a history of a myocardial infarction. | Punj et al., 2017 | Cross-sectional study |
| Periodontitis patients demonstrated impaired FMD as well as higher salivary levels of matrix metalloproteinase-2 complex compared to healthy controls, suggestive of endothelial dysfunction. | Moura et al., 2017 | Cross-sectional study |
| Patients with severe periodontitis demonstrated improvements in FMD, concurrent with treatment of their periodontal disease. The effects on CV risk reduction and events were not investigated. | Seinost et al., 2005 | Prospective observational study |
| Direct microbial injury from periodontal pathogens | ||
| Pathogenic periodontal organisms, P. gingivalis and P. intermedia, invade and adhere to human coronary artery endothelial cells in vitro. | Dorn et al, 1999 | In vitro |
| P. gingivalis induce the expression of surface-associated molecules, including intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and P- and E- selectins in human umbilical vein endothelial cells. | Khlgatian et al., 2002 | In vitro |
| Apolipoprotein E (ApoE) deficient mice subjected to an oral P. gingivalis infection developed periodontitis and demonstrated increased inflammatory cytokines, cholesterol, alveolar bone loss, and atherosclerotic lesions in the aorta, which improved after stimulation of nucleotide binding oligomerization domain-containing protein 2. |
Li et al., 2002
Yuan et al., 2013 |
In vivo |
| Treatment of P. gingivalis in infected mice resulted in fewer atheromatous lesions in the aorta and aortic tree compared to infected mice not receiving treatment. Additionally, mice infected with P. gingivalis with deficient fimbria demonstrated fewer atheromatous lesions compared to those with wild type P. gingivalis. | Amar et al., 2009 | In vivo |
| In ApoE deficient mice, polymicrobial oral infection with major periodontal organisms (P. gingivalis, T. denticola, T. forsythia) led to increased aortic plaque with increased macrophage presence as well as increased serum cholesterol and triglycerides. | Rivera et al., 2013 | In vivo |
| Evidence of periodontal pathogens (DNA, RNA, antigen) identified in atheromatous plaque samples and vascular walls in patients with PD. |
Rath SK et al., 2014
Haraszthy VI et al., 2000 Ott SJ et al., 2006 |
In vitro |
| Edentulousness and serum IgG antibodies to periodontal pathogens (Actinobacillus actinomycetemcomitans and P. Gingivalis) associate with coronary heart disease and stroke. |
Pussinen PJ et al., 2003
Pussinen PJ et al., 2004 |
Prospective observational study |
| Viable invasive A. actinomycetemcomitans and P. gingivalis identified in human atherosclerotic plaque, suggesting periodontitis involvement in CV disease. | Kozarov EV et al,. 2005 | In vitro |
| Injury and inflammation due to microbial byproducts | ||
| Increased circulating levels of LPS associates with elevated cardiometabolic risk factors, including elevated C-reactive protein, diabetes, obesity, as well as CV events. | Kallio KAE et al., 2015 | Prospective observational study |
| High antibody response to periodontal pathogens (IgG angibodies to A. actinomycetemcomitans and P. gingivalis) independently predicts incidence of CV events over a 10-year follow-up period. LPS levels positively correlated with periodontal IgG antibody levels. | Pussinen PJ et al., 2007 | Prospective observational study |
| Subgingival microbial burden associates with and contributes to salivary and serum LPS levels. Additionally, LPS levels associate with stable coronary artery disease. | Liljestrand JM et al.,2017 | Cross-sectional study |
| Patients with history of myocardial infarction have a higher frequency of allele T in position −260 in the promoter region of the CD14 receptor gene of monocytes, which are activated by LPS from Gram-negative bacteria. | Hubacek JA et al., 1999 | In vitro |
| Single nucleotide polymorphism of allele T in position −159 in CD14 increases the transcriptional activity of the downstream NF -κB inflammatory pathway, which can synergize with CRP in activating the vascular endothelium. | LeVan TD et al., 2001 | In vitro |
| LPS shown to contribute to oxidation of low-density lipoprotein, foam cell maturation, and thrombogenesis. | Liao W. et al., 1996 | In vitro |
| Stimulation of P. gingivalis and LPS in human umbilical vein endothelial cells induced endothelial cell death and increased oxidized LDL, and TNF -α levels. | Bugueno IM, et al., 2016 | In vitro |
| P. gingivalis produces toxins and virulence factors, such as proteases, lectins, fimbriae, adhesins, and hemagglutinins that can interfere with signal pathway activation in host immune responses and potentially lead to CVD. |
Zhou Q et al., 2006
Zhou Q et al., 2005 Guo Y et al., 2000 |
In vitro |
| Gingipains shown to reduce LPS-induced IL-8 production in gingival fibroblasts and to cleave ICAM-1 on endothelial cells, allowing for immune invasion in periodontal tissues. Gingipain shown to cleave apoB-100, the main component of LDL that is responsible for allowing the binding of LDL to cell surface receptors. |
Tada H et al., 2002
Tada H., 2003 Hashimoto M et al., 2006 Bengtsson T et al., 2008 |
In vitro |
| Mutant gingipain, lacking either the Rgp- or Kgp gingipains, demonstrated unfavorable proteolytic effects on lipoproteins (LDL, VLDL, and HDL) by inducing reactive oxygen species and lipid peroxidation. | Lonn J et al., 2018 | In vitro |
| Cross-reactivity due to bacterial antigen | ||
| T-cells observed in atherosclerotic plaque, suggesting involvement of T-cell immune response to P. gingivalis HSP60 in atherosclerosis. | Choi J-I et al., 2002 | In vitro |
| Cross-reactive T-cells recognizing HSP60 found in diseased periodontal tissue and peripheral blood. | Ford P et al., 2005 | In vitro |
| HSP60 antibody levels to both human and P. gingivalis were highest in patients with atherosclerosis. Clonal analysis demonstrated HSP60-reactive T cells that recognized both human and P. gingivalis HSP60 in these patients. | Yamazaki K et al., 2004 | Prospective observational study |
| Patients with mild periodontitis had increased levels of serum HSP60 and small, dense LDL when compared to controls matched for age and body mass index. | Rizzo et al., 2012 | Prospective observational study |
Role of the Funder:
The funding source had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Abbreviations:
- PD
periodontal disease
- CVD
cardiovascular disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest relevant to this work: All authors declare no potential conflicts of interest related to this article.
References
- 1.Ridker PM. How Common Is Residual Inflammatory Risk? Circ Res 2017;120:617–619. [DOI] [PubMed] [Google Scholar]
- 2.Aday AW, Ridker PM. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front Cardiovasc Med 2019;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansson GK. Inflammation, Atherosclerosis, and Coronary Artery Disease. N Engl J Med 2005;352:1685–1695. [DOI] [PubMed] [Google Scholar]
- 4.Ridker Paul M Anticytokine Agents. Circ Res 2019;124:437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willerson James T, Ridker Paul M Inflammation as a Cardiovascular Risk Factor. Circulation 2004;109:II–2. [DOI] [PubMed] [Google Scholar]
- 6.Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation 1994;90:775–778. [DOI] [PubMed] [Google Scholar]
- 7.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res 2015;107:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation 2001;103:1718–1720. [DOI] [PubMed] [Google Scholar]
- 9.Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GDO, Pepys MB, Gudnason V. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004;350:1387–1397. [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N Engl J Med 2000;342:836–843. [DOI] [PubMed] [Google Scholar]
- 11.Ridker PM. A Test in Context: High-Sensitivity C-Reactive Protein. J Am Coll Cardiol 2016;67:712–723. [DOI] [PubMed] [Google Scholar]
- 12.Emerging Risk Factors Collaboration, Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P, Walker M, Thompson A, Sarwar N, Caslake M, Butterworth AS, Amouyel P, Assmann G, Bakker SJL, Barr ELM, Barrett-Connor E, Benjamin EJ, Björkelund C, Brenner H, Brunner E, Clarke R, Cooper JA, Cremer P, Cushman M, Dagenais GR, D’Agostino RB, Dankner R, Davey-Smith G, Deeg D, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med 2012;367:1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein | NEJM. https://www.nejm.org/doi/full/10.1056/NEJMoa0807646 (3 April 2019) [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 15.Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, Mam V, Hasan A, Rosenberg Y, Iturriaga E, Gupta M, Tsigoulis M, Verma S, Clearfield M, Libby P, Goldhaber SZ, Seagle R, Ofori C, Saklayen M, Butman S, Singh N, Le May M, Bertrand O, Johnston J, Paynter NP, Glynn RJ. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N Engl J Med 2019;380:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aviña-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum 2008;59:1690–1697. [DOI] [PubMed] [Google Scholar]
- 17.Mehta NN, Yu Y, Pinnelas R, Krishnamoorthy P, Shin DB, Troxel AB, Gelfand JM. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med 2011;124:775.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gelfand JM, Dommasch ED, Shin DB, Azfar RS, Kurd SK, Wang X, Troxel AB. The risk of stroke in patients with psoriasis. J Invest Dermatol 2009;129:2411–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006;296:1735–1741. [DOI] [PubMed] [Google Scholar]
- 20.Gu M-M, Wang X-P, Cheng Q-Y, Zhao Y-L, Zhang T-P, Li B-Z, Ye D-Q. A Meta-Analysis of Cardiovascular Events in Systemic Lupus Erythematosus. Immunol Invest 2019;1–16. [DOI] [PubMed] [Google Scholar]
- 21.Elnabawi YA, Dey AK, Goyal A, Groenendyk JW, Chung JH, Belur AD, Rodante J, Harrington CL, Teague HL, Baumer Y, Keel A, Playford MP, Sandfort V, Chen MY, Lockshin B, Gelfand JM, Bluemke DA, Mehta NN. Coronary artery plaque characteristics and treatment with biologic therapy in severe psoriasis: results from a prospective observational study. Cardiovasc Res 2019;115:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lerman JB, Joshi AA, Chaturvedi A, Aberra TM, Dey AK, Rodante JA, Salahuddin T, Chung JH, Rana A, Teague HL, Wu JJ, Playford MP, Lockshin BA, Chen MY, Sandfort V, Bluemke DA, Mehta NN. Coronary Plaque Characterization in Psoriasis Reveals High-Risk Features That Improve After Treatment in a Prospective Observational Study. Circulation 2017;136:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnabe C, Martin B-J, Ghali WA. Systematic review and meta-analysis: anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res 2011;63:522–529. [DOI] [PubMed] [Google Scholar]
- 24.Kinane DF, Stathopoulou PG, Papapanou PN. Periodontal diseases. Nat Rev Dis Primer 2017;3:17038. [DOI] [PubMed] [Google Scholar]
- 25.Caton JG, Armitage G, Berglundh T, Chapple ILC, Jepsen S, Kornman KS, Mealey BL, Papapanou PN, Sanz M, Tonetti MS. A new classification scheme for periodontal and peri-implant diseases and conditions - Introduction and key changes from the 1999 classification. J Periodontol 2018;89 Suppl 1:S1–S8. [DOI] [PubMed] [Google Scholar]
- 26.Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000 2014;64:57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebersole JL. Humoral immune responses in gingival crevice fluid: local and systemic implications. Periodontol 2000 2003;31:135–166. [DOI] [PubMed] [Google Scholar]
- 28.Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol Baltim Md 1950 1998;160:403–409. [PubMed] [Google Scholar]
- 29.Loos BG, Craandijk J, Hoek FJ, Dillen PMEW, Velden UVD. Elevation of Systemic Markers Related to Cardiovascular Diseases in the Peripheral Blood of Periodontitis Patients. J Periodontol 2000;71:1528–1534. [DOI] [PubMed] [Google Scholar]
- 30.Noack B, Genco RJ, Trevisan M, Grossi S, Zambon JJ, De Nardin E. Periodontal infections contribute to elevated systemic C-reactive protein level. J Periodontol 2001;72:1221–1227. [DOI] [PubMed] [Google Scholar]
- 31.Offenbacher S, Beck JD, Moss K, Mendoza L, Paquette DW, Barrow DA, Couper DJ, Stewart DD, Falkner KL, Graham SP, Grossi S, Gunsolley JC, Madden T, Maupome G, Trevisan M, Dyke TEV, Genco RJ. Results From the Periodontitis and Vascular Events (PAVE) Study: A Pilot Multicentered, Randomized, Controlled Trial to Study Effects of Periodontal Therapy in a Secondary Prevention Model of Cardiovascular Disease. J Periodontol 2009;80:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, Gotoh K, Motooka D, Nakamura S, Iida T, Yamazaki K. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep 2014;4:4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maekawa T, Tabeta K, Kajita-Okui K, Nakajima T, Yamazaki K. Increased expression of C-reactive protein gene in inflamed gingival tissues could be derived from endothelial cells stimulated with interleukin-6. Arch Oral Biol 2011;56:1312–1318. [DOI] [PubMed] [Google Scholar]
- 34.Kebschull M, Demmer RT, Papapanou PN. ‘Gum bug, leave my heart alone!’-- epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res 2010;89:879–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, Taubert KA, Newburger JW, Gornik HL, Gewitz MH, Wilson WR, Smith SC, Baddour LM, American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, Council on Epidemiology and Prevention, Council on Peripheral Vascular Disease, and Council on Clinical Cardiology. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association?: a scientific statement from the American Heart Association. Circulation 2012;125:2520–2544. [DOI] [PubMed] [Google Scholar]
- 36.Tonetti MS, Van Dyke TE, Working group 1 of the joint EFP/AAP workshop. Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol 2013;40 Suppl 14:S24–29. [DOI] [PubMed] [Google Scholar]
- 37.Dorn JM, Genco RJ, Grossi SG, Falkner KL, Hovey KM, Iacoviello L, Trevisan M. Periodontal disease and recurrent cardiovascular events in survivors of myocardial infarction (MI): the Western New York Acute MI Study. J Periodontol 2010;81:502–511. [DOI] [PubMed] [Google Scholar]
- 38.Holmlund A, Holm G, Lind L. Number of teeth as a predictor of cardiovascular mortality in a cohort of 7,674 subjects followed for 12 years. J Periodontol 2010;81:870–876. [DOI] [PubMed] [Google Scholar]
- 39.Yu Y-H, Chasman DI, Buring JE, Rose L, Ridker PM. Cardiovascular Risks Associated with Incident and Prevalent Periodontal Disease. J Clin Periodontol 2015;42:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stryjewska K, Pytko-Polonczyk J, Sagbraaten S, Sagbraaten SVSV, Stryjewski PJ. The oral health of patients with acute coronary syndrome confirmed by means of coronary angiography. Pol Merkur Lek Organ Pol Tow Lek 2020;48:23–26. [PubMed] [Google Scholar]
- 41.Gomes-Filho IS, Coelho JMF, Miranda SS, Cruz SS, Trindade SC, Cerqueira EMM, Passos-Soares JS, Costa M da CN, Vianna MIP, Figueiredo ACMG, Hintz AM, Coelho AF, Passos LCS, Barreto ML, Scannapieco F. Severe and moderate periodontitis are associated with acute myocardial infarction. J Periodontol 2020; [DOI] [PubMed] [Google Scholar]
- 42.Mozos I, Stoian D. Oral Health and Cardiovascular Disorders. Underst Mol Crosstalk Biol Process IntechOpen; 2019; [Google Scholar]
- 43.Desvarieux Moïse Demmer Ryan T., Tatjana Rundek, Bernadette Boden-Albala, Jacobs David R., Sacco Ralph L., Papapanou Panos N. Periodontal Microbiota and Carotid Intima-Media Thickness. Circulation American Heart Association; 2005;111:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desvarieux M, Demmer RT, Jacobs DR, Papapanou PN, Sacco RL, Rundek T. Changes in clinical and microbiological periodontal profiles relate to progression of carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology study. J Am Heart Assoc 2013;2:e000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paraskevas KI, Mikhailidis DP, Giannoukas AD. Periodontitis and abdominal aortic aneurysms: a random association or a pathogenetic link? Int Angiol J Int Union Angiol 2009;28:431–433. [PubMed] [Google Scholar]
- 46.Kurihara N, Inoue Y, Iwai T, Umeda M, Huang Y, Ishikawa I. Detection and localization of periodontopathic bacteria in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg Off J Eur Soc Vasc Surg 2004;28:553–558. [DOI] [PubMed] [Google Scholar]
- 47.Salhi L, Rompen E, Sakalihasan N, Laleman I, Teughels W, Michel J-B, Lambert F. Can Periodontitis Influence the Progression of Abdominal Aortic Aneurysm? A Systematic Review. Angiology 2019;70:479–491. [DOI] [PubMed] [Google Scholar]
- 48.Im SI, Heo J, Kim BJ, Cho K-I, Kim HS, Heo JH, Hwang JY. Impact of periodontitis as representative of chronic inflammation on long-term clinical outcomes in patients with atrial fibrillation. Open Heart 2018;5:e000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kure K, Sato H, Aoyama N, Izumi Y. Accelerated inflammation in peripheral artery disease patients with periodontitis. J Periodontal Implant Sci 2018;48:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sekinishi A, Suzuki J-I, Aoyama N, Ogawa M, Watanabe R, Kobayashi N, Hanatani T, Ashigaki N, Hirata Y, Nagai R, Izumi Y, Isobe M. Periodontal pathogen Aggregatibacter actinomycetemcomitans deteriorates pressure overload-induced myocardial hypertrophy in mice. Int Heart J 2012;53:324–330. [DOI] [PubMed] [Google Scholar]
- 51.Mucci LA, Hsieh C-C, Williams PL, Arora M, Adami H-O, Faire U de, Douglass CW, Pedersen NL. Do genetic factors explain the association between poor oral health and cardiovascular disease? A prospective study among Swedish twins. Am J Epidemiol 2009;170:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ernst FD, Uhr K, Teumer A, Fanghänel J, Schulz S, Noack B, Gonzales J, Reichert S, Eickholz P, Holtfreter B, Meisel P, Linden GJ, Homuth G, Kocher T. Replication of the association of chromosomal region 9p21.3 with generalized aggressive periodontitis (gAgP) using an independent case-control cohort. BMC Med Genet 2010;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaefer AS, Richter GM, Groessner-Schreiber B, Noack B, Nothnagel M, El Mokhtari N-E, Loos BG, Jepsen S, Schreiber S. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet 2009;5:e1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science 2007;316:1488–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007;316:1491–1493. [DOI] [PubMed] [Google Scholar]
- 56.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann H-E, Barrett JH, König IR, Stevens SE, Szymczak S, Tregouet D-A, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, et al. Genomewide association analysis of coronary artery disease. N Engl J Med 2007;357:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schunkert H, Götz A, Braund P, McGinnis R, Tregouet D-A, Mangino M, Linsel-Nitschke P, Cambien F, Hengstenberg C, Stark K, Blankenberg S, Tiret L, Ducimetiere P, Keniry A, Ghori MJR, Schreiber S, El Mokhtari NE, Hall AS, Dixon RJ, Goodall AH, Liptau H, Pollard H, Schwarz DF, Hothorn LA, Wichmann H-E, König IR, Fischer M, Meisinger C, Ouwehand W, Deloukas P, et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation 2008;117:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ernst FD, Uhr K, Teumer A, Fanghänel J, Schulz S, Noack B, Gonzales J, Reichert S, Eickholz P, Holtfreter B, Meisel P, Linden GJ, Homuth G, Kocher T. Replication of the association of chromosomal region 9p21.3 with generalized aggressive periodontitis (gAgP) using an independent case-control cohort. BMC Med Genet 2010;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reriani MK, Lerman LO, Lerman A. Endothelial function as a functional expression of cardiovascular risk factors. Biomark Med 2010;4:351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi H, Uceda DE, Dey AK, Mehta NN. Application of Non-invasive Imaging in Inflammatory Disease Conditions to Evaluate Subclinical Coronary Artery Disease. Curr Rheumatol Rep 2019;22:1. [DOI] [PubMed] [Google Scholar]
- 62.Salomon Amar, Noyan Gokce, Sonia Morgan, Mariana Loukideli, Van Dyke Thomas E., Vita Joseph A. Periodontal Disease Is Associated With Brachial Artery Endothelial Dysfunction and Systemic Inflammation. Arterioscler Thromb Vasc Biol 2003;23:1245–1249. [DOI] [PubMed] [Google Scholar]
- 63.Mercanoglu F, Oflaz H, Oz O, Gökbuget AY, Genchellac H, Sezer M, Nişanci Y, Umman S. Endothelial dysfunction in patients with chronic periodontitis and its improvement after initial periodontal therapy. J Periodontol 2004;75:1694–1700. [DOI] [PubMed] [Google Scholar]
- 64.Punj A, Shenoy SB, Subramanyam K. Comparison of Endothelial Function in Healthy Patients and Patients With Chronic Periodontitis and Myocardial Infarction. J Periodontol 2017;88:1234–1243. [DOI] [PubMed] [Google Scholar]
- 65.Moura MF, Navarro TP, Silva TA, Cota LOM, Soares Dutra Oliveira AM, Costa FO. Periodontitis and Endothelial Dysfunction: Periodontal Clinical Parameters and Levels of Salivary Markers Interleukin-1β, Tumor Necrosis Factor-α, Matrix Metalloproteinase-2, Tissue Inhibitor of Metalloproteinases-2 Complex, and Nitric Oxide. J Periodontol 2017;88:778–787. [DOI] [PubMed] [Google Scholar]
- 66.Seinost G, Wimmer G, Skerget M, Thaller E, Brodmann M, Gasser R, Bratschko RO, Pilger E. Periodontal treatment improves endothelial dysfunction in patients with severe periodontitis. Am Heart J 2005;149:1050–1054. [DOI] [PubMed] [Google Scholar]
- 67.Lockhart PB, Brennan MT, Thornhill M, Michalowicz BS, Noll J, Bahrani-Mougeot FK, Sasser HC. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J Am Dent Assoc 1939 2009;140:1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hajishengallis G Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 2015;15:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Libby P, Loscalzo J, Ridker PM, Farkouh ME, Hsue PY, Fuster V, Hasan AA, Amar S. Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week. J Am Coll Cardiol 2018;72:2071–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Friedewald VE, Kornman KS, Beck JD, Genco R, Goldfine A, Libby P, Offenbacher S, Ridker PM, Van Dyke TE, Roberts WC, American Journal of Cardiology, Journal of Periodontology. The American Journal of Cardiology and Journal of Periodontology Editors’ Consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol 2009;104:59–68. [DOI] [PubMed] [Google Scholar]
- 71.Dorn BR, Dunn WA, Progulske-Fox A. Invasion of human coronary artery cells by periodontal pathogens. Infect Immun 1999;67:5792–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khlgatian M, Nassar H, Chou H-H, Gibson FC, Genco CA. Fimbria-Dependent Activation of Cell Adhesion Molecule Expression in Porphyromonas gingivalis-Infected Endothelial Cells. Infect Immun 2002;70:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yuan H, Zelkha S, Burkatovskaya M, Gupte R, Leeman SE, Amar S. Pivotal role of NOD2 in inflammatory processes affecting atherosclerosis and periodontal bone loss. Proc Natl Acad Sci U S A 2013;110:E5059–E5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li L, Messas E, Batista EL, Levine RA, Amar S. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 2002;105:861–867. [DOI] [PubMed] [Google Scholar]
- 75.Amar S, Wu S, Madan M. Is Porphyromonas gingivalis cell invasion required for atherogenesis? Pharmacotherapeutic implications. J Immunol Baltim Md 1950 2009;182:1584–1592. [DOI] [PubMed] [Google Scholar]
- 76.Rivera MF, Lee J-Y, Aneja M, Goswami V, Liu L, Velsko IM, Chukkapalli SS, Bhattacharyya I, Chen H, Lucas AR, Kesavalu LN. Polymicrobial infection with major periodontal pathogens induced periodontal disease and aortic atherosclerosis in hyperlipidemic ApoE(null) mice. PloS One 2013;8:e57178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rath SK, Mukherjee M, Kaushik R, Sen S, Kumar M. Periodontal pathogens in atheromatous plaque. Indian J Pathol Microbiol 2014;57:259. [DOI] [PubMed] [Google Scholar]
- 78.Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of Periodontal Pathogens in Atheromatous Plaques. J Periodontol 2000;71:1554–1560. [DOI] [PubMed] [Google Scholar]
- 79.Ott SJ, El Mokhtari NE, Musfeldt M, Hellmig S, Freitag S, Rehman A, Kühbacher T, Nikolaus S, Namsolleck P, Blaut M, Hampe J, Sahly H, Reinecke A, Haake N, Günther R, Krüger D, Lins M, Herrmann G, Fölsch UR, Simon R, Schreiber S. Detection of diverse bacterial signatures in atherosclerotic lesions of patients with coronary heart disease. Circulation 2006;113:929–937. [DOI] [PubMed] [Google Scholar]
- 80.Pussinen PJ, Jousilahti P, Alfthan G, Palosuo T, Asikainen S, Salomaa V. Antibodies to periodontal pathogens are associated with coronary heart disease. Arterioscler Thromb Vasc Biol 2003;23:1250–1254. [DOI] [PubMed] [Google Scholar]
- 81.Pussinen PJ, Alfthan G, Rissanen H, Reunanen A, Asikainen S, Knekt P. Antibodies to periodontal pathogens and stroke risk. Stroke 2004;35:2020–2023. [DOI] [PubMed] [Google Scholar]
- 82.Kozarov Emil V, Dorn Brian R, Shelburne Charles E, Dunn William A, Progulske-Fox Ann. Human Atherosclerotic Plaque Contains Viable Invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol 2005;25:e17–e18. [DOI] [PubMed] [Google Scholar]
- 83.Ettinger G, MacDonald K, Reid G, Burton JP. The influence of the human microbiome and probiotics on cardiovascular health. Gut Microbes 2014;5:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shimauchi H, Mayanagi G, Nakaya S, Minamibuchi M, Ito Y, Yamaki K, Hirata H. Improvement of periodontal condition by probiotics with Lactobacillus salivarius WB21: a randomized, double-blind, placebo-controlled study. J Clin Periodontol 2008;35:897–905. [DOI] [PubMed] [Google Scholar]
- 85.Liljestrand JM, Paju S, Buhlin K, Persson GR, Sarna S, Nieminen MS, Sinisalo J, Mäntylä P, Pussinen PJ. Lipopolysaccharide, a possible molecular mediator between periodontitis and coronary artery disease. J Clin Periodontol 2017;44:784–792. [DOI] [PubMed] [Google Scholar]
- 86.Kallio KAE, Hätönen KA, Lehto M, Salomaa V, Männistö S, Pussinen PJ. Endotoxemia, nutrition, and cardiometabolic disorders. Acta Diabetol 2015;52:395–404. [DOI] [PubMed] [Google Scholar]
- 87.Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol 2007;27:1433–1439. [DOI] [PubMed] [Google Scholar]
- 88.Role of CD14 in cellular recognition of bacterial lipopolysaccharides. - PubMed - NCBI. https://www.ncbi.nlm.nih.gov/pubmed/10608082 (10 April 2019) [DOI] [PubMed] [Google Scholar]
- 89.Arroyo-Espliguero R, Avanzas P, Jeffery S, Kaski JC. CD14 and toll-like receptor 4: a link between infection and acute coronary events? Heart 2004;90:983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hubacek JA, Rothe G, Pit’ha J, Skodová Z, Stanĕk V, Poledne R, Schmitz G. C(−260)-->T polymorphism in the promoter of the CD14 monocyte receptor gene as a risk factor for myocardial infarction. Circulation 1999;99:3218–3220. [DOI] [PubMed] [Google Scholar]
- 91.LeVan TD, Bloom JW, Bailey TJ, Karp CL, Halonen M, Martinez FD, Vercelli D. A Common Single Nucleotide Polymorphism in the CD14 Promoter Decreases the Affinity of Sp Protein Binding and Enhances Transcriptional Activity. J Immunol 2001;167:5838–5844. [DOI] [PubMed] [Google Scholar]
- 92.Liao W Endotoxin: Possible roles in initiation and development of atherosclerosis. J Lab Clin Med 1996;128:452–460. [DOI] [PubMed] [Google Scholar]
- 93.Bugueno IM, Khelif Y, Seelam N, Morand D-N, Tenenbaum H, Davideau J-L, Huck O. Porphyromonas gingivalis Differentially Modulates Cell Death Profile in Ox-LDL and TNF-α Pre-Treated Endothelial Cells. PloS One 2016;11:e0154590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Q, Amar S. Identification of Proteins Differentially Expressed in Human Monocytes Exposed to Porphyromonas gingivalis and Its Purified Components by High-Throughput Immunoblotting. Infect Immun 2006;74:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun 2005;73:935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guo Y, Nguyen K-A, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000 2010;54:15–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tada H, Sugawara S, Nemoto E, Takahashi N, Imamura T, Potempa J, Travis J, Shimauchi H, Takada H. Proteolysis of CD14 on human gingival fibroblasts by arginine-specific cysteine proteinases from Porphyromonas gingivalis leading to down-regulation of lipopolysaccharide-induced interleukin-8 production. Infect Immun 2002;70:3304–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tada H, Sugawara S, Nemoto E, Imamura T, Potempa J, Travis J, Shimauchi H, Takada H. Proteolysis of ICAM-1 on human oral epithelial cells by gingipains. J Dent Res 2003;82:796–801. [DOI] [PubMed] [Google Scholar]
- 99.Hashimoto M, Kadowaki T, Tsukuba T, Yamamoto K. Selective proteolysis of apolipoprotein B-100 by Arg-gingipain mediates atherosclerosis progression accelerated by bacterial exposure. J Biochem (Tokyo) 2006;140:713–723. [DOI] [PubMed] [Google Scholar]
- 100.Bengtsson T, Karlsson H, Gunnarsson P, Skoglund C, Elison C, Leanderson P, Lindahl M. The periodontal pathogen Porphyromonas gingivalis cleaves apoB-100 and increases the expression of apoM in LDL in whole blood leading to cell proliferation. J Intern Med 2008;263:558–571. [DOI] [PubMed] [Google Scholar]
- 101.Lönn J, Ljunggren S, Klarström-Engström K, Demirel I, Bengtsson T, Karlsson H. Lipoprotein modifications by gingipains of Porphyromonas gingivalis. J Periodontal Res 2018;53:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kiessling R, Gröunberg A, Ivanyi J, Soderströum K, Ferm M, Kleinau S, Nilsson E, Klareskog L. Role of hsp60 during Autoimmune and Bacterial Inflammation. Immunol Rev 1991;121:91–111. [DOI] [PubMed] [Google Scholar]
- 103.Choi J-I, Chung S-W, Kang H-S, Rhim BY, Park Y-M, Kim U-S, Kim S-J. Epitope Mapping of Porphyromonas gingivalis Heat-shock Protein and Human Heat-shock Protein in Human Atherosclerosis. J Dent Res 2004;83:936–940. [DOI] [PubMed] [Google Scholar]
- 104.Choi J-I, Chung S-W, Kang H-S, Rhim BY, Kim S-J, Kim S-J. Establishment of Porphyromonas gingivalis Heat-shock-protein-specific T-cell Lines from Atherosclerosis Patients. J Dent Res 2002;81:344–348. [DOI] [PubMed] [Google Scholar]
- 105.Ford P, Gemmell E, Walker P, West M, Cullinan M, Seymour G. Characterization of Heat Shock Protein-Specific T Cells in Atherosclerosis. Clin Diagn Lab Immunol 2005;12:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamazaki K, Ohsawa Y, Itoh H, Ueki K, Tabeta K, Oda T, Nakajima T, Yoshie H, Saito S, Oguma F, Kodama M, Aizawa Y, Seymour GJ. T-cell clonality to Porphyromonas gingivalis and human heat shock protein 60s in patients with atherosclerosis and periodontitis. Oral Microbiol Immunol 2004;19:160–167. [DOI] [PubMed] [Google Scholar]
- 107.Rizzo M, Cappello F, Marfil R, Nibali L, Marino Gammazza A, Rappa F, Bonaventura G, Galindo-Moreno P, O’Valle F, Zummo G, Conway de Macario E, Macario AJL, Mesa F. Heat-shock protein 60 kDa and atherogenic dyslipidemia in patients with untreated mild periodontitis: a pilot study. Cell Stress Chaperones 2012;17:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]