

Abstract
The CISD2 (NAF-1) protein plays a key role in regulating cellular homeostasis, aging, cancer and neurodegenerative diseases. It was found to control different calcium, reactive oxygen species (ROS), and iron signaling mechanisms. However, since most studies of CISD2 to date were conducted with cells that constitutively lack, overexpress, or contain mutations in CISD2, the relationships between these different signaling processes are unclear. To address the hierarchy of signaling events occurring in cells upon CISD2 disruption, we developed an inducible system to express CISD2, or the dominant-negative H114C inhibitor of CISD2, in human breast cancer cells. Here, we report that inducible disruption of CISD2 function causes an immediate disruption in mitochondrial labile iron (mLI), and that this disruption results in enhanced mitochondrial ROS (mROS) levels. We further show that alterations in cytosolic and ER calcium levels occur only after the changes in mLI and mROS levels happen and are unrelated to them. Interestingly, disrupting CISD2 function resulted in the enhanced expression of the tumor suppressor thioredoxin-interacting protein (TXNIP) that was dependent on the accumulation of mLI and associated with ferroptosis activation. CISD2 could therefore regulate the expression of TXNIP in cancer cells, and this regulation is dependent on alterations in mLI levels.
Keywords: CISD2, NAF-1, oxidative stress, reactive oxygen species (ROS), iron homeostasis, iron-sulfur cluster [Fe-S], ferroptosis, TXNIP, mitochondria, cancer
Graphical Abstract
Introduction
CISD2 (Nutrient-deprivation autophagy factor-1; NAF-1) is an iron-sulfur [2Fe-2S] membrane-bound homodimeric protein found on the cytosol-facing surface of the endoplasmic reticulum (ER), mitochondria, and mitochondria-associated membranes (MAM) that connect these two compartments [1–4]. It is linked to the regulation of autophagy and apoptosis through interactions with the B-cell lymphoma-2 protein (BCL-2) [5–9], and/or the regulation of apoptosis through interactions with the calcium-activated cysteine protease Calpain2 (CAPN2) and the inhibitor of apoptosis-stimulating protein of p53 (iASPP) proteins [10–12]. CISD2 is also thought to regulate cytosolic and mitochondrial calcium levels through interactions with the calcium channels inositol 1,4,5-trisphosphate receptor (IP3R) [3–5, 13] and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [3, 4, 14, 15]), as well as to regulate mitochondrial iron and reactive oxygen species (ROS) through interactions with CISD1 (mitoNEET) [2, 16], that interacts with and repairs iron regulatory protein 1 (IRP1) [17], as well as anamorsin, a component of the cytosolic Fe-S biogenesis machinery [18]. CISD2 function is critical for the maintenance of cellular homeostasis in different tissues, and proper levels of CISD2 are required for skeletal muscle, heart, nerve, adipocyte, and endocrine cell functions [3, 4]. Accordingly, patients with mutations in, or complete loss of, CISD2 (Wolfram Syndrome Type 2; WFS-T2) suffer from severe diabetes mellitus symptoms, shorter life span, neurodegeneration, upper gastrointestinal tract ulcers, and blood clotting abnormalities [19–23]. CISD2 is also considered a key aging gene, since overexpression or suppression of CISD2 alters life span in mice [4, 14, 15, 24, 25]. In addition to WFS-T2, CISD2 function was recently associated with neurodegenerative diseases such as Alzheimer’s [26–28], as well as with diabetes [20, 23, 29–31], cardiovascular disease [15, 32], and cancer [33–42]. In cancer cells, high levels of CISD2 are thought to prevent mitochondrial labile iron (mLI) and ROS (mROS) overload, inhibit apoptosis, and allow rapid cellular proliferation [33, 35, 37, 38, 40, 42]. Consequentially, CISD2 levels are being used as a prognostic marker for several different cancers, and new treatments for certain cancers were proposed based on CISD2 inhibitors [e.g., 1, 34–43].
Because CISD2 functions as a homodimer, expression of a mutated and/or inactive form of CISD2 in cells can cause a dominant-negative effect in which the mutated/inactive monomer associates with a wild type monomer to generate a homodimer with inhibited function. This approach was used to study the function of the [2Fe-2S] cluster of CISD2 in cancer cells [35]. It was found that expressing a point mutant of CISD2 (H114C) with a significantly higher stability of the CISD2 [2Fe-2S] cluster [35, 44] in human epithelial breast cancer cells impaired CISD2 function and caused the overaccumulation of mLI and mROS, disruption of mitochondrial membrane potential, and activation of apoptosis [35]. This effect also resulted in a significant decrease in the size of xenograft tumors originating from human triple-negative epithelial breast cancer (MDA-MB-231) cells expressing H114C [35].
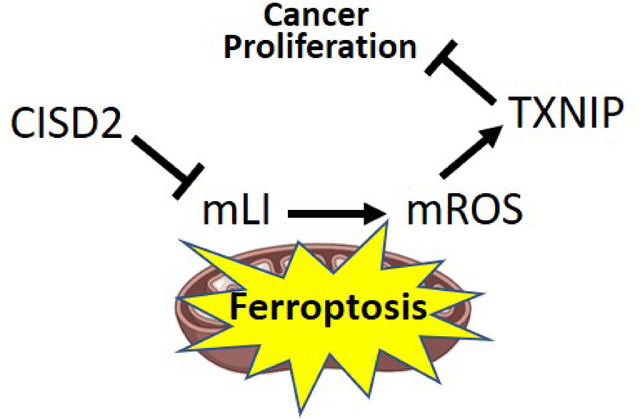
The multiple functions of CISD2 in cells, described above, could result from its role in calcium signaling, iron and/or iron-sulfur homeostasis, and/or ROS signaling/homeostasis [e.g., 3, 4]. The relationship(s) and interdependency between these different signaling and homeostasis processes are however unclear at present, since most studies of CISD2 to date were conducted with cells, tissues or organisms that constitutively lack, overexpress, or contain a mutation(s) in CISD2. Although these experimental systems provided valuable information on CISD2 function, they lack a fine temporal dimension that would allow dissecting the relationship(s) between CISD2 effects on calcium, iron and/or ROS homeostasis and signaling. It is unclear therefore if the primary effect of CISD2 disruption in cancer cells results in alterations in calcium, iron, and/or ROS levels, and whether some of these alterations activate or suppress the other(s). To begin addressing these important questions in cancer cells, we developed an inducible system to express CISD2, or the dominant-negative H114C cluster-stable inhibitor of CISD2, in human epithelial breast cancer cells (MDA-MB-231). Here, we report that inducible disruption of CISD2 function in cancer cells (through inducible expression of H114C) causes an immediate disruption in mLI levels and that this disruption results in altered mROS levels. We further show that alterations in cytosolic and ER calcium levels occur only after the changes in mLI and mROS levels took place, and that H114C-driven changes in calcium levels are unrelated to changes in mLI and mROS. We further reveal that the inducible expression of H114C in cancer cells results in the enhanced accumulation of thioredoxin-interacting protein (TXNIP), that is dependent on the enhanced accumulation of mLI in cancer cells, and that a disruption in CISD2 cluster transfer activity (via inducible expression of H114C) results in the activation of oxidative stress that triggers a ferroptosis-like process of cell death. CISD2 could therefore regulate the expression of the tumor suppressor TXNIP in cancer cells and this regulation is dependent on alterations in mitochondrial function.
Materials and Methods
Cell growth and viability, and inducible expression of CISD2 or H114C
MDA-MB-231 human breast cancer cells were obtained from ATCC (Rockville, MD, USA) and propagated as previously described [33]. The Tet-One Inducible Expression System was purchased from Clontech® Laboratories, Inc. (Cat. No. 634301) and used for the inducible expressing of CISD2 (NAF-1; [35]) or its H114C point mutant [35, 44], according to the manufacturer instructions. MDA-MB-231 cells were transfected with empty, CISD2, or H114C expressing plasmids as previously described [33, 35]. Doxycycline, Hyclate (DOX, EMD Millipore, Cat. No. 324385), was used to induce expression from the Tet-One plasmids, as described previously [45]. For cell growth measurements, cells were seeded in 96-well plates in triplicates at a density of 5000 cells/well, treated with different DOX concentrations (0, 0.5, 1, 1.5, 2, 2.5 μg/ml) and subjected to cell growth assays using an Incucyte® ZOOM system for live-cell imaging and analysis (Essen BioScience, Inc. Michigan, USA) according the manufacturer instructions. Cell growth was determined using the Incucyte® Zoom Software. Cells were subjected to cellular viability assays using Alamar Blue (Thermo Fisher Scientific ™, Cat. No. 00–100) according to the manufacturer instructions and as previously described [33]. Cells were also incubated with or without Ferrostatin-1 (3-Amino-4-cyclohexylaminobenzoic acid ethyl ester; Sigma, Product No. SML0583), using a concentration of 2 μM, and their growth rate and viability in the presence or absence of DOX (0, 0.5, 1, 1.5, 2, 2.5 μg/ml) were determined.
Measurements of mLI, mROS and calcium levels
Cells were cultured and imaged by Epi-fluorescent Microscopy to determine their mLI with the fluorescent probe RPA (rhodamine B-[(1,10-phenanthrolin-5-yl) aminocarbonyl] benzyl ester) (Squarix biotechnology, ME043.1) as described in [33]. Mitochondrial ROS accumulation was determined using mitoSOX™ Red (Invitrogen™, M36008) according to [11, 35]. Although the mitoSOX™ probe has been extensively used and validated in many publications, care should be taken when interpreting its results [46]. Images were analyzed with Volocity (Quorum Technologies Inc.) and/or with Image-J [33]. Quantification of mLI was performed using 30 different fields (5 cells per field). Quantification of mitoSOX™ fluorescence changes was performed by analyzing 9 different fields (5 cells per field). mLI and mROS levels were averaged from 6–10 independent experiments. Cells were plated onto microscope slides glued to perforated 3 cm diameter tissue culture plates as previously described [11, 33]. For Fluo-4 AM (Invitrogen™, F14201) was used to measure cytosolic Ca2+ following incubation of cells with 4 μM for 30 minutes at 37°C in the presence of 0.01% Pluronic® F-127 (Sigma, P2443). For mitochondrial calcium, cells were treated with Fluo-4 AM, imaged, and then treated with 20 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP, Millipore Sigma, Cat. No. 555602). Following the CCCP treatment, calcium levels were measured again and the increase of Fluo-4 AM fluorescence was calculated as described previously [14]. A similar strategy was used to measure ER calcium levels, but instead of CCCP, cells were treated with 10 μM Thapsigargin (Tg, Sigma-Aldrich, Cat. No. T9033) according to [14]. The measurements described above were conducted using an Olympus FV3000 confocal laser-scanning microscope [47]. Cells were also incubated with or without DFP (3-Hydroxy-1,2-dimethyl-4(1H)-pyridone; Sigma, 379409) at a concentration of 100 μM for 1 hour prior to measuring for mLI, mROS and calcium levels as described in [23, 33].
Protein blots
For protein blot analyses, cells were grown to full confluence, washed twice with 1X PBS, immediately scraped off the plate into a microcentrifuge tube with 1X Laemmli sample buffer for CISD2 and mNT antibodies, and heated to 95°C for 10 minutes. For GPX4, TfR, TXNIP and TRX2, RIPA (Radioimmunoprecipitation assay; Sigma-Aldrich) buffer was used and lysates were kept on ice for 2 hours with vortex every 30 minutes, then centrifugation at 15,000 rpm for 15 minutes, supernatant was used for protein denaturation with Laemmli sample buffer 5X. Protein gels were loaded with equal amounts of protein, determined using The Pierce 660 nm Protein Assay (Thermo Scientific™, Cat. No. 22662) [11], and analyzed using antibodies against CISD2 and mitoNEET [23, 33], TXNIP (Novus Biologicals, NBP2–27095), Thioredoxin 2 (Novus Biologicals, NBP1–92499), β-actin (R&D Systems, MAB8929), GPX4 (R&D Systems, Cat. No. MAB5457), and TfR (Abcam, ab84036). Goat Anti-Rabbit IgG, H & L Chain Specific Peroxidase Conjugate (Sigma, 401315) and Peroxidase-conjugated AffiniPure Goat anti-mouse IgG (H+L) (Jackson ImmunoResearch Laboratories, AB_10015289) were used as secondary antibodies (7, 24). All experiments were repeated at least 3 times.
Lipid Peroxidation
Malondialdehyde (MDA) was measured using QuantiChrome TBARS Assay Kit (DTBA-100, BioAssay Systems, Hayward, CA, USA) according to the manufacturer’s instructions. Briefly, DOX-treated and untreated cells (5X106) of the different lines were harvested 72 hours post treatment, homogenized and sonicated in ice cold PBS buffer. Cell lysates were incubated with ice-cold 10% trichloroacetic acid and centrifuged for 5 minutes at 14,000 rpm. After the neutralization, the clear sample supernatant was mixed with thiobarbituric acid (TBA) solution and incubated at 100°C for 60 minutes. Fluorometric assay was used for quantitative determination of lipid peroxides (thiobarbituric acid reactive substances, TBARS), this was evaluated using the Perkin Elmer EnVision 2104–0020 Multilabel Plate Reader at Ex/Em 530/550nm. Although the MDA TBARS Assay has been extensively used and validated in many publications, care should be taken when interpreting its results [48].
Proteomics Analysis
Cells were grown to 80% confluency and treated with DOX (2 μg/ml), for different times (0, 3, 6, 12, 24 hours). Cells were then sampled and subjected to protein extraction using 0.175 M Tris-HCl, pH 8.8, 5% SDS, 15% glycerol, 0.3M DTT, as described in [49]. Protein samples were precipitated with 100% of Acetone, washed, resuspended in urea buffer (6M urea, 2M thiourea, 100 mM ammonium bicarbonate, pH 8.0) and subjected to protein quantification using EZQ™ Protein Quantitation Kit (Thermo Fisher Scientific ™, Cat. No. R33200). An equal amount of protein (25 μg) from each sample was digested with trypsin, and resulted peptides were cleaned up using C18 100 μL tips (Pierce), lyophilized, resuspended in 25μL of 5% ACN, 0.1% FA and quantified as described in [49]. Peptides were analyzed by liquid chromatography and mass spectrometry (LCMS) using a Bruker nanoElute system attached to a Bruker timsTOF-PRO mass spectrometer via a Bruker CaptiveSpray source (timsToF Pro, Bruker Daltonics, Billerica, MA). One μL injections were made directly onto a 20cm long x 75 μm inner diameter pulled-needle analytical column packed with Waters BEH-C18, 1.7 μm reversed phase resin, and peptides were separated and eluted from the analytical column with a gradient of acetonitrile at 300 nL/min. Total run time was 90 min. MS data were collected in positive-ion data-dependent PASEF mode over an m/z range of 100 to 1700, and an ion-mobility range of 0.6 to 1.6 1/k0. One MS and ten PASEF frames were acquired per cycle of 1.16sec (~1MS and 120 MS/MS). Target MS intensity for MS was set at 10,000 counts/sec with a minimum threshold of 2000 counts/s. An ion-mobility-based rolling collision energy of 20 to 59 eV (1/k0 0.6 to 1.6) was used. An active exclusion/reconsider precursor method with release after 0.4min was used. If the precursor (within mass width error of 0.015 m/z) was >4X signal intensity in subsequent scans, a second MSMS spectrum was collected. Isolation width was set to 2 m/z (<700 m/z) or 3 (800–1500 m/z).
Proteomics data analysis
Reasonable proteome coverage was achieved (~2800 proteins). Raw data was searched using PEAKS (version x+, Bioinformatics Solutions Inc. Canada) with Uniprot human database (UniProt release 2019_11, total 20366 items). Identified proteins were filtered by peptide and protein FDR 1%, for at least 2 spectrum counts per protein. Proteins identified in at least two samples per group were included for further quantitation analysis. Spectrum count was used for differential protein abundance analysis. Each protein spectrum count was normalized to total spectrum counts among the samples. One-way Welch ANOVA was applied for each cell type among five time points. One-way Welch ANOVA was also conducted for each timepoint among three cell types. A protein with p value less than or equal to 0.05 was considered statistically significant. The data were analyzed using the R platform. Proteomics data was deposited in MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp?redirect=auth), under the following accession number: MSV000087436.
Statistics
Statistical significance tests (one-sample t-test, paired t-test, two-way ANOVA followed by a Tukey test, or one-way Welch ANOVA) were used for different data types. GraphPad Prism 8.3.1 and InfoStat Statistical Software were used. Results are presented as mean ± SE of different biological repeat averages, each conducted with 3 technical repeats. Differences were defined statistically significant if the probability value was equal of less than 5% (0.05).
Results
Inducible expression of CISD2 or its H114C dominant-negative inhibitor alters the viability and growth rate of cancer cells
To study the impact of altering CISD2 function in cancer cells using a time-course design, we expressed the CISD2 protein (NAF-1) or its H114C dominant-inhibitor point mutant (H114C) in human epithelial breast cancer (MDA-MB-231) cells using a Doxycycline (DOX)-inducible expression system. As control (Control) we used an empty DOX-inducible expression vector. Treatment of CISD2 or H114C cells with DOX resulted in the accumulation of CISD2 protein (CISD2, H114C; Figures 1B, 1C). In contrast, the level of the endogenous CISD2 protein was unchanged upon DOX treatment of control cells transformed with the empty DOX-inducible expression vector (Control; Figure 1A). The level of CISD2 or H114C was also unchanged in mock-treated cells in the absence of DOX (Supplementary Figure S1). The enhanced (1.2–1.4-fold) expression of the CISD2 protein was evident at 24 hours post DOX application and remained high for up to 72 hours (Figures 1B, 1C). Constitutive overexpression of CISD2 was previously reported to result in enhanced cancer cell proliferation and tumor growth, while constitutive overexpression of H114C had the opposite effect [35]. We therefore tested the effect of DOX-induced CISD2 or H114C expression on cell viability and growth rate of MDA-MB-231 cells. For this purpose, we induced CISD2 or H114C expression using different concentrations of DOX and determined cellular growth and viability at 72 hours following DOX application. In agreement with previous studies [35], DOX-induced expression of CISD2 resulted in enhanced cellular viability and growth, while DOX-induced expression of H114C had the opposite effect (Figures 1D and 1E). The results shown in Figure 1 demonstrate that the DOX-inducible system we developed can enhance the expression of CISD2 or H114C and affect cancer cell growth and viability in a similar manner to constitutive overexpression of CISD2 or H114C [35]. This system can therefore be used to study the impacts of a disruption in CISD2 function (by expressing H114C) on different cellular functions of MDA-MB-231 cells.
Figure 1.

Inducible expression of H114C suppresses the growth rate of human epithelial breast cancer cells. A-C. Protein blot analysis of CISD2 expression in Control (A), CISD2 (B) and H114C (C) cells upon induction of the Tet-One inducible expression system using Doxycycline (DOX) (2μg/ml). Representative protein blots are shown on top and β-actin-normalized expression graph are shown on bottom. Results in (A-C) are shown as mean ± SE of 5 biological control averages, each conducted with 3 technical repeats. ***P<0.001; N=5; one sample t-test. D-E. Cellular viability (D) and growth rate (E) of Control-, CISD2- and H114C-expressing cells 72 hours following the induction of protein expression with DOX (0, 0.5, 1.0, 1.5, 2.0, 2.5 μg/ml). Cell viability was measured using Alamar blue (D), and cell growth was measured with an Incucyte® Live-Cell Imaging apparatus (E). Results in (D-E) are shown as mean ± SE of 4 biological control averages, each conducted with 3 technical repeats. N=4; two-way ANOVA followed by a Tukey test; ns, not significant.
Alterations in mLI and mROS upon DOX-induced expression of H114C
After establishing that the DOX-induced system to study CISD2 function overexpresses the CISD2 and H114C proteins, and that these proteins had the expected effect on cellular growth (Figure 1) [35], we measured changes in mLI and mROS in cancer cells following DOX-inducible expression of CISD2 or H114C. Inducible expression of H114C, but not CISD2, caused a significant increase in the amount of mLI (evident by quenching of the RPA fluorescence probe; Figure 2A). This increase was evident as early as 12 hours post DOX application and continued for up to 72 hours post DOX application (Figure 2A). Inducible expression of H114C, but not CISD2, also caused a significant increase in mROS accumulation (evident by the increased fluorescence of the mitoSOX™ probe; Figure 2B). This increase was also evident as early as 12 hours post DOX application and continued for up to 72 hours post DOX application (Figure 2B). The findings presented in Figure 2 directly link a disruption in CISD2 function (caused by expression of the H114C dominant-negative inhibitor of CISD2) to impairments in mLI and mROS. Because both mLI and mROS accumulation initiated together at 12 hours post DOX application (Figure 2) it is likely that these two processes are linked to each other. Nevertheless, their hierarchy (i.e., who came first, iron or ROS) could not be deduced from the experiments shown in Figure 2.
Figure 2.

Accumulation of mitochondrial labile iron (mLI) and mitochondrial ROS (mROS) following Doxycycline (DOX)-induced expression of H114C in cancer cells. A. Quantitative analysis of mLI accumulation in H114C cells upon DOX (2μg/ml)-induced protein expression in Control, CISD2 and H114C cells. Quenching of RPA fluorescence indicates mLI accumulation. B. Quantitative analysis of mROS accumulation in Control, CISD2 and H114C cells, upon DOX (2μg/ml)-induced protein expression. Results in A and B are mean ± SE of N=10 (A), or N=6 (B), biological control averages, each conducted with 3 technical repeats; two-way ANOVA followed by a Tukey test; ns, not significant.
Alterations in cytosolic and ER calcium levels upon DOX-induced expression of H114C
To determine whether changes in mROS and mLI levels proceeded or preceded changes in calcium levels in MDA-MB-231 cells upon DOX-inducible expression of CISD2 or H114C, we measured cytosolic, ER and mitochondrial calcium levels following DOX application in the different lines. Inducible expression of H114C, but not CISD2, caused a significant decrease in cytosolic calcium levels (Figure 3A), that was accompanied by a significant increase in ER calcium levels (Figure 3B). In contrast, mitochondrial calcium levels remained unaffected (Figures 3C). In contrast to changes in mLI and mROS, that initiated at 12 hours, changes in cytosolic and ER calcium levels initiated at 72 hours post DOX application. These findings suggest that, at least in epithelial breast cancer cells, within the time-course samples studied, alterations in CISD2 function (caused by DOX-induced expression of the H114C CISD2 dominant-negative inhibitor) primarily resulted in changes in mLI and mROS levels (Figure 2) and that these changes occurred before the changes in cytosol or ER calcium levels (Figure 3).
Figure 3.

Alterations in cytosolic and ER calcium levels following Doxycycline (DOX) (2μg/ml) -induced expression of H114C in cancer cells. A. Decreased cytosolic calcium levels in H114C cells upon DOX (2μg/ml)-induced protein expression in control, CISD2 and H114C cells. B. Enhanced accumulation of ER calcium levels in H114C cells upon DOX (2μg/ml)-induced protein expression in Control, CISD2 and H114C cells. C. Levels of mitochondrial calcium levels upon DOX (2μg/ml)-induced protein expression in Control, CISD2 and H114C cells. Results are shown as mean ± SE of 5 biological control averages, each conducted with 3 technical repeats. N=5; two-way ANOVA followed by a Tukey test; ns, not significant.
Inhibiting H114C-mediated changes in mLI levels suppresses the accumulation of mROS, but does not affect changes in calcium levels
In previous studies we reported that suppressing the accumulation of mLI, that resulted from shRNA suppression of CISD2 expression, suppressed the accumulation of mROS levels [11, 33]. These studies suggested that accumulation of mLI directly impacts enhanced mROS levels in CISD2 deficient cells. Because changes in mLI and mROS co-occurred at 12 hours post DOX application and changes in calcium levels occurred at 72 hours (Figures 2, 3), we tested whether suppressing the DOX-induced H114C-mediated accumulation of mLI accumulation, in our inducible experimental system, would also suppress the accumulation of mROS, and whether this suppression will further affect changes in calcium levels. For this purpose, we used the mitochondria-permeable iron chelator Deferiprone (DFP) that was shown to suppress mLI accumulation in cancer cells with suppressed levels of CISD2 [33]. As expected [33], application of DFP to cancer cells, following DOX application, suppressed the accumulation of mLI upon H114C induction (Figure 4A). This application also suppressed mROS accumulation (Figure 4B) but had no significant effect on H114C-induced changes in ER and cytosolic calcium levels (Figures 4C, 4D). The results presented in Figure 4 suggest that inducible expression of H114C primarily caused an induction in mLI accumulation and that this process triggered a rise in mROS levels. In contrast, at least at the time resolution we tested, the accumulation of mLI and mROS were not associated with H114C-induced changes in ER or cytosolic calcium levels.
Figure 4.

Application of the mitochondria-permeable iron chelator Deferiprone (DFP) suppresses mitochondrial labile iron (mLI) and mirochondrial ROS (mROS) accumulation but has no effect on changes in ER and cytosolic calcium levels. A. Application of DFP suppresses the accumulation of mLI levels in DOX (2μg/ml; 48 hour)-treated H114C cells. B. Application of DFP suppresses the accumulation of mROS levels in DOX (2μg/ml; 48 hour)-treated H114C cells. C. Application of DFP does not affect the decrease in cytosolic calcium levels in DOX (2μg/ml; 72 hour)-treated H114C cells. D. Application of DFP does not affect the increase in ER calcium levels in DOX (2μg/ml; 72 hour)-treated H114C cells. Results are shown as mean ± SE of 6, 7 or 12 biological control averages, each conducted with 3 technical repeats. N=6 for (A), N=7 for (B), and N=12 for (C and D); two-way ANOVA followed by a Tukey test; ns, not significant.
Time-course proteomics analysis of cancer cells with disrupted CISD2 function
To further dissect the temporal response of cancer cells to a disruption in CISD2 function, we conducted a time-course proteomics analysis of control, CISD2 and H114C cells following the application of DOX. DOX-induced protein expression in control, CISD2 and H114C cells resulted in the altered expression of 181, 221, and 147 proteins, respectively; with 154, 189, and 118 proteins uniquely expressed in control, CISD2 and H114C, respectively (P < 0.05; Figure 5A; Supplementary Tables S1–S3). Interestingly, the amount of overlap between the different lines was low, with an overlap of 16 (Control and CISD2), 13 (Control and H114C), and 18 (CISD2 and H114C) proteins, indicating that inducible expression of CISD2 or H114C had different effects on cells (Figure 5A). Because inducible expression of H114C caused a disruption in CISD2 function that resulted in suppressed cellular growth (Figure 1), as well as the accumulation of mLI and mROS (Figure 2), we focused our study on the different proteins that were altered in their expression in cancer cells upon expression of H114C (Figure 5A and 5B). Using literature and database searches for each protein, we classified these proteins into iron-, ROS-, and/or calcium-response proteins, and identified 9, 18 and 7, unique iron-, ROS-, and calcium-response proteins altered in cells upon H114C expression, respectively (Figure 5C; Supplementary Tables S4–S6). In accordance with our measurements of mLI and mROS (Figure 2), and our analysis of calcium levels (Figure 3), the expression pattern of the iron- and ROS- response proteins was rapidly altered following DOX application (many peaking in their expression as early as 3 hours post DOX application; Figure 5C), while the expression pattern of the calcium-response proteins was slower (reaching a maximal change at about 6 hours post DOX application; Figure 5C). These results support our findings that measurable changes in mitochondrial iron and ROS levels preceded measurable changes in calcium levels (Figures 2 and 3), and that the changes in mLI and mROS were unrelated to the changes in calcium levels (Figure 4).
Figure 5.

Time-course proteomics analysis of Control-, CISD2- and H114C-expressing cells following application of Doxycycline (DOX; 2μg/ml) for 0, 3, 6, 12, and 24 hours. A. Venn diagram showing the overlap between proteins significantly altered in their expression in Control, CISD2 and H114C cells upon treatment with DOX (2μg/ml) for 0, 3, 6, 12 and 24 hours. B. Heatmap showing the expression pattern of the 118 proteins uniquely expressed following the application of DOX (2μg/ml) for 0, 3, 6, 12, and 24 hours to H114C cells. C. Venn diagram and line graphs showing the expression pattern of iron-, oxidative stress- and calcium- response proteins in H114C cells following the application of DOX (2μg/ml). Please see Supplementary Tables S1–S6, Supplementary Figure 3, and text for additional details. All proteins shown were found to be significantly expressed (P<0.05; N=3) compared to time 0 hour by a one-way Welch ANOVA.
Disrupting CISD2 function in cancer cells results in the enhanced expression of TXNIP and the activation of a ferroptosis-like process
One of the proteins upregulated in H114C expressing cells in response to DOX was TXNIP (Among the 5 proteins responsive to iron, calcium and oxidative stress in Figure 5C). Protein blot analysis of TXNIP expression in H114C cells following DOX application confirm the proteomics results and reveal that the level of TXNIP increased at 24, 48 and 72 hours (Figure 6A). Enhanced accumulation of TXNIP in cells is associated with increased oxidative stress [50, 51], and together with the increased accumulation of mLI and mROS (Figure 2) this expression pattern could suggest that the induced expression of H114C drives cancer cells into ferroptosis. This possibility is supported by prior studies that linked CISD2 expression levels with the activation of ferroptosis in cancer cells [41, 52, 53]. To further examine whether H114C expression in cells is associated with ferroptosis, we tested the expression levels of mitoNEET (mNT; CISD1; another member of the NEET protein family that donates its clusters to CISD2 [16]), a known anti ferroptosis protein [54], in cancer cells upon induction of CISD2 or H114C. In accordance with its proposed functions and interactions with CISD2, mNT expression was enhanced in cancer cells upon induction of CISD2 expression (Figure 6B). In contrast, upon inducible expression of H114C, mNT expression declined (Figure 6B).
Figure 6.

Expression of thioredoxin-interacting protein (TXNIP) and mitoNEET (mNT) following the application of Doxycycline (DOX; 2μg/ml). Expression of the tumor suppressor TXNIP (A) or the anti-ferroptosis ([2Fe-2S] CISD1) protein mNT (B) following the application of Doxycycline (DOX; 2μg/ml) to Control, CISD2 and H114C cells for 0, 3, 6, 12, and 24 hours. Results are shown as mean ± SE of 7 (A) or 5 (B) biological control averages, each conducted with 3 technical repeats. ***P<0.001; N=7 (A); N=5 (B); one sample t-test.
To further examine whether cancer cells enter ferroptosis upon disruption of CISD2 function, we measured the expression of two other markers for ferroptosis, glutathione peroxidase 4 (GPX4) [55, 56] and transferrin receptor (TfR) [56, 57] in H114C following DOX application. Concurring with their proposed role in ferroptosis, the level of GPX4 decreased (Figure 7A and Supplementary Figure S2), while the level of TfR increased (Figure 7B and Supplementary Figure S2) at 72 hours following DOX application. In addition, the level of mitochondrial thioredoxin 2 (TRX2), a key regulator of mitochondrial redox reactions [58, 59], decreased in cancer cells upon the induction of H114C (Figure 7C and Supplementary Figure S2). In addition, the level of lipid peroxidation increased in cancer cells 72 hours following DOX-induced expression of H114C (Figure 7D). Taken together, our findings suggest that at least some aspects of ferroptosis could be triggered in cancer cells upon disruption of CISD2 [2Fe-2S]-related functions and that these could be linked with mNT expression levels.
Figure 7.

Detection of ferroptosis markers in cancer cells following Doxycycline (DOX; 2μg/ml)-induced expression of the H114C protein and suppressed expression of thioredoxin-interacting protein (TXNIP) in H114C cells upon application of the mitochondria-permeable iron chelator Deferiprone (DFP). A-C. Expression of glutathione peroxidase 4 (GPX4; A), transferrin receptor (TfR; B), and mitochondrial thioredoxin 2 (TRX; C) in H114C cells following induction of H114C expression with DOX (2μg/ml) for 0, 24, 48, and 72 hours. Results in (A-C) are shown as mean ± SE of 7 biological control averages, each conducted with 3 technical repeats. **P<0.01, ***P<0.001; N=7; one sample t-test. D. Lipid peroxidation in Control, CISD2 and H114C cells 72 hours following DOX (2μg/ml) application. Results in are shown as mean ± SE of 6 biological control averages, each conducted with 3 technical repeats. *P<0.05, ***P<0.001; N=7; Student’s paired t-test. E. Expression of TXNIP in Control, CISD2 and H114C cells 24 hours following induction with DOX (2μg/ml) in the presence or absence of DFP. Results in are shown as mean ± SE of 6 biological control averages, each conducted with 3 technical repeats. ***P<0.001; N=7; one sample t-test.
The enhanced expression of TXNIP upon disruption in CISD2 function is dependent on mLI accumulation
The disruption in CISD2 cluster-dependent functions resulted in the accumulation of mLI (Figure 2A) and the enhanced expression of TXNIP (Figure 6A). However, it is unknown whether these two processes are linked. To address this question, we studied whether the application of DFP, that inhibits the H114C-dependent accumulation of mLI and mROS (Figure 4), would suppress the enhanced expression of TXNIP (Figure 6A). As shown in Figure 7E, DFP application suppressed the enhanced accumulation of TXNIP following the DOX-induced expression of H114C. In contrast, DFP had no effect on TXNIP expression in control or DOX-induced CISD2 cells (Figure 7E). These findings suggest that the enhanced accumulation of mLI upon disruption of CISD2 function (Figure 2A) not only caused an enhanced accumulation of mROS (Figures 2B and 4) but also caused the enhanced accumulation of TXNIP (Figure 7E), potentially causing an overall cellular enhancement of oxidative stress.
Ferrostatin-1 treatment mitigates the negative impacts of CISD2 disruption on cell growth and viability
The findings that disrupting CISD2 function via expression of H114C in cancer cells causes an increase in mLI and mROS, suppresses the expression of GPX4 and TRX2, enhances the expression of TfR, and enhances lipid peroxidation (Figures 2, 6 and 7), strongly suggest that suppressing CISD2 function triggers ferroptosis. However, whether or not the activation of ferroptosis is causing the decrease in cell viability and growth of cancer cells expressing H114C (Figure 1), is unknown. To address this question, we treated Control, CISD2 and H114C cells with DOX at different concentrations (0, 0.5, 1.0, 1.5, 2.0, 2.5 μg/ml), in the presence or absence of the ferroptosis inhibitor Ferrostatin-1 [55–57]. As shown in Figure 8, treatment of DOX-treated H114C cells with Ferrostatin-1 mitigated the decline in cell growth and viability caused by the H114C-driven disruption in CISD2 function. In contrast, Ferrostatin-1 did not affect the growth and viability of DOX-treated Control or CISD2 cells (Figure 8). The findings presented in Figure 8 suggest therefore that a disruption in CISD2 function causes the activation of Ferroptosis and that this process is suppressing the growth and viability of cancer cells.
Figure 8.

Treatment with Ferrostatin-1 mitigates the negative effects of H114 expression on cell viability and growth. A. The effect of Ferrostatin-1 treatment (2μM) on cell viability of Doxycycline (DOX; 0, 0.5, 1.0, 1.5, 2.0, 2.5 μg/ml)-treated Control (Upper), CISD2 (Middle) and H114C (Lower) cells at 72 hours following DOX application (with or without Ferrostatin). Cell viability was measured using Alamar Blue. B. The effect of Ferrostatin-1 treatment (2μM) on cell growth of Doxycycline (DOX; 0, 0.5, 1.0, 1.5, 2.0, 2.5 μg/ml)-treated Control (Upper), CISD2 (Middle) and H114C (Lower) cells at 72 hours following DOX application (with or without Ferrostatin). Cell growth was measured with an Incucyte® Live-Cell Imaging apparatus. Results in A and B are shown as mean ± SE of 7 biological control averages, each conducted with 3 technical repeats. N=7; two-way ANOVA followed by a Tukey test; ns, not significant.
Discussion
The inducible expression system we developed makes use of the H114C point mutant of CISD2 and capitalizes on our previous study that showed this mutant to disrupt CISD2 function in cancer cells [35]. In contrast to CISD2 knock-out or shRNA-suppressed expression systems, that lack or have suppressed levels of the CISD2 protein, the H114C protein expressed in cells functions as a dominant-negative inhibitor with a highly stable (25-fold higher than CISD2) [2Fe-2S] cluster, present in cells in its almost intact form (the crystal structure of H114C is almost indistinguishable from that of CISD2) [44]. Like CISD2, the H114C protein can therefore potentially participate in different protein-protein interactions (e.g., with BCL-2, CAPN2, iASPP and SERCA), however, it is deficient in its [2Fe-2S]-related functions. The [2Fe-2S] clusters of CISD2 were proposed to be primarily involved in two different functions: (i) Electron transfer reactions [1, 60] and (ii) [2Fe-2S] cluster transfer reactions/donation [1, 42, 60]. In the H114C mutant both are disrupted [35, 44]. The use of the H114C mutant of CISD2 in our inducible system is therefore different from studies of complete knockout [9, 13, 15, 24, 61–65] or knock-down [11, 33, 36, 40, 41, 53, 66–68] of CISD2 and primarily focuses on the potential of CISD2 to be involved in [2Fe-2S] cluster and/or electron transfer reactions.
Previous studies of CISD2 function in cells demonstrated that CISD2 deficiency can result in neurodegeneration, damage to skeletal and heart muscles, the development of diabetes and a shorter life span [e.g., 3, 4, 15]. In contrast, overexpression of CISD2 caused longer life span [25], but also promoted cancer cell proliferation and tumor growth [35, 37, 38]. We previously proposed that CISD2 promoted cancer cell and tumor growth by protecting cells from the overaccumulation of mLI and mROS that could result in the activation of apoptosis, and that these processes depended on the function of the [2Fe-2S] clusters of CISD2 [33, 35, 42]. The function of CISD2 in cells was however also proposed to be mediated by different protein-protein interactions and the regulation of ER and mitochondrial calcium signaling [3–16], making it harder to determine what is the hierarchy of events and mode of function of CISD2 in cancer cells. In the current study we used a DOX-inducible system to try and dissect the different events that ensue following a disruption in CISD2 [2Fe-2S] cluster function in cancer cells. As discussed above, using the H114C mutant of CISD2 we attempted to target the impact of a disruption in CISD2 [2Fe-2S] cluster function without causing a significant change in different protein-protein interactions. This was partly achieved by comparing the DOX-induced expression of H114C to that of CISD2 (Figures 1–3) and focusing on H114C-specific events. Using a combination of time-course mLI, mROS and calcium measurements (Figures 2, 3), and use of the mLI attenuator DFP (Figure 4), we were able to determine that the initial effect of disrupting CISD2 [2Fe-2S] cluster function in cancer cells is an increase in mLI and that this process prompted the accumulation of mROS. In contrast, at least at the time resolution studied, changes in ER and cytosolic calcium levels occurred much later (Figure 3) and appeared to be unrelated to changes in mLI (i.e., unaffected by DFP application; Figure 4).
Using a proteomics time-course approach we further identified TXNIP as a protein that increases in its expression upon a disruption in CISD2 [2Fe-2S] cluster function in cancer cells (Figures 5, 6). TXNIP is a thioredoxin-binding protein that triggers oxidative stress by inhibiting the function of the thioredoxin system [69, 70]. It is thought to function as an important regulator for many redox-related signal transduction pathways in cells, and in particular to play a key role in glucose metabolism associated with diabetes [69, 71]. In breast cancer cells, TXNIP is thought to function as a tumor suppressor protein that inhibits the proliferation of cancer cells and activates apoptosis [72, 73]. By altering metabolic reprogramming of cancer cells, TXNIP is also through to affect the invasion and migration of breast cancer cells through the TXNIP-HIF1α-TWIST signaling axis [51]. Our findings that the induced accumulation of TXNIP in cells with induced expression of H114C (Figures 5, 6) was also accompanied by suppressed cellular viability and growth (Figures 1D and 1E), support the proposed role of TXNIP as a tumor suppressor. However, it is unclear why the disrupted function of CISD2 caused the induction of TXNIP. One possibility is that the alterations in mLI and mROS, that specifically occurred in H114C cells upon DOX induction (Figure 2), were responsible for TXNIP induction. Indeed, suppressing the accumulation of mLI and mROS by DFP (Figure 4) suppressed the enhanced expression of TXNIP upon H114C induction (Figure 7E). This finding provides a possible link between enhanced mLI accumulation and enhanced TXNIP expression in cancer cells. In support of this possibility, mROS accumulation (that could result from enhanced mLI; Figures 2 and 4) was shown to cause the activation of the NLRP3/IL-1β pathway through TXNIP during inflammation [74, 75]. A possible link could therefore exist between enhanced mLI, enhanced mROS, accumulation of TXNIP, and suppressed cancer cell proliferation. Our previous findings that suppression of CISD2 protein levels or [2Fe-2S] cluster activity triggered apoptosis of breast cancer cells [11, 35] could further be linked to this pathway, as TXNIP is also thought to be a trigger of apoptosis. In this respect it should be mentioned that at least 59 out of the 118 H114C-specific proteins identified by our proteomics analysis could be linked to apoptosis (Supplementary Table S3). The suppressed [2Fe-2S] cluster function of CISD2 in cancer cells could therefore be triggering apoptosis via TXNIP and this process could depend on the enhanced accumulation of mROS. More studies are of course needed to address this intriguing possibility.
In addition to TXNIP, our protein expression analysis shown in Figure 7 revealed that the expression of GPX4 and transferrin receptor (TfR), two known ferroptosis markers [56, 76], was altered following H114C induction in cancer cells. While the expression of GPX4 decreased, the expression of TfR increased, at 72 hours, suggesting the activation of ferroptosis. In addition, lipid peroxidation increased at 72 hours further supporting the involvement of ferroptosis. Moreover, treatment of DOX-induced H114C cells with the ferroptosis inhibitor Ferrostatin-1 recovered the decline in cell growth and viability caused by the disruption in CISD2 function of cancer cells (Figure 8). The suppression of cancer cell proliferation upon disruption of CISD2 cluster-related functions could therefore be mediated by a ferroptosis-like mechanism and this process could involve TXNIP. To the best of our knowledge, TXNIP was not previously linked to the activation of ferroptosis in cancer cells and this aspect of its function should be examined in future studies.
In contrast to the deleterious effects of H114C expression on cancer cells growth and viability, overexpression of CISD2 in cancer cells improved cellular growth and viability (Figures 1D, 1E, and 8). The enhanced expression of CISD2 was also associated with suppressed expression of TXNIP (Figure 7E), a decrease in lipid peroxidation (Figure 7D), a decrease in the expression of TfR (Supplementary Figure S2E), and an increase in the levels of TRX2 (Supplementary Figure S2F). These findings suggest that overexpression of CISD2 enhances the antioxidative capacity of cancer cells, presumably through improved flux of iron in the form of [2Fe-2S] clusters from the mitochondria to the cytosol [42]. This finding is in agreement with our previous analysis of xenograft tumors with enhanced expression of CISD2 [35]. Indeed, at least 38 proteins identified by our current proteomics analysis were also identified in xenograft tumors with enhanced expression of CISD2 (Supplementary Table S7).
Taken together, our findings suggest that disrupting the function of the [2Fe-2S] clusters of CISD2 primarily results in enhanced mLI levels that trigger mROS accumulation, enhance the expression of TXNIP, and activate ferroptosis. At least in human epithelial breast cancer cells, CISD2 function could therefore be linked to the enhanced expression of the tumor suppressor TXNIP, as well as to the activation of ferroptosis, and this role should be explored in other cellular systems such as nerve, cardiovascular and endocrine cells, as well as serve as a guide to the development of future therapies for different diseases associated with CISD2 function. Due to its involvement in diabetes and cancer [e.g., 1, 3, 15, 23], the NEET protein CISD2 could also serve as an interesting target for drugs that target different diabetes-associated cancers [3, 77].
Supplementary Material
Highlights.
An inducible system that disrupts CISD2 function in cancer cells was developed.
Inducible disruption in CISD2 function resulted in the accumulation of mitochondrial labile iron and reactive oxygen species.
Alterations in cytosolic and ER calcium levels followed the changes in mitochondrial labile iron and reactive oxygen species.
Disrupting CISD2 function resulted in the enhanced expression of the tumor suppressor thioredoxin-interacting protein (TXNIP) and the activation of ferroptosis.
The induction of TXNIP was dependent on the enhanced accumulation of mitochondrial labile iron.
Acknowledgments
O.K. acknowledges The Annual Golda Meir Fellowship and The Hebrew University Rector for the Emergency Scholarship recipients for Postdoctoral fellows, 2020-2021.
Funding
This work was supported by the NSF-Binational Science Foundation (BSF) Grant NSF-MCB 1613462 (to R.M.), BSF Grant 2015831 (to R.N.), and GM111364 (to R.M.). We also acknowledge the Charles W. Gehrke Proteomics Center, Research Core Facilities, University of Missouri, Columbia.
Abbreviations Used:
- BCL-2
B-cell lymphoma-2 protein
- CAPN2
Calpain2
- CCCP
Carbonyl cyanide 3-chlorophenylhydrazone
- DFP
Deferiprone
- DOX
Doxycycline
- ER
Endoplasmic reticulum
- GPX4
Glutathione Peroxidase 4
- iASPP
inhibitor of apoptosis-stimulating protein of p53
- IP3R
Inositol 1,4,5-triphosphate receptor
- IRP1
Iron regulatory protein 1
- MAM
Mitochondria-ER-associated membranes
- MDA
Malondialdehyde
- mLI
Mitochondrial labile iron
- mROS
Mitochondrial reactive oxygen species
- NAF-1
Nutrient-deprivation Autophagy Factor-1
- OMM
Outer mitochondrial membrane
- RIPA
Radioimmunoprecipitation assay
- ROS
Reactive oxygen species
- RPA
Rhodamine B-[(1,10-phenanthrolin-5-yl) aminocarbonyl] benzyl ester
- SERCA2
Sarco/endoplasmic reticulum Ca2+-ATPase
- TBARS
Thiobarbituric acid reactive substances
- TfR
Transferrin Receptor
- Tg
Thapsigargin
- TRX
Thioredoxin
- TXNIP
Thioredoxin-interacting protein
- WFS-T2
Wolfram syndrome type 2
Footnotes
Author disclosure statement
No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Tamir S, Paddock ML, Darash-Yahana-Baram M, Holt SH, Sohn YS, Agranat L, Michaeli D, Stofleth JT, Lipper CH, Morcos F, Cabantchik IZ, Onuchic JN, Jennings PA, Mittler R, Nechushtai R, Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease, Biochim Biophys Acta 1853(6) (2015) 1294–315. [DOI] [PubMed] [Google Scholar]
- [2].Karmi O, Marjault H-B, Pesce L, Carloni P, Onuchic JN, Jennings PA, Mittler R, Nechushtai R, The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease, JBIC Journal of Biological Inorganic Chemistry (2018) 23(4):599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nechushtai R, Karmi O, Zuo K, Marjault H-B, Darash-Yahana M, Sohn Y-S, King SD, Zandalinas SI, Carloni P, Mittler R, The balancing act of NEET proteins: Iron, ROS, calcium and metabolism, Biochimica et Biophysica Acta (BBA)-Molecular Cell Research (2020) 118805. [DOI] [PubMed] [Google Scholar]
- [4].Shen Z-Q, Huang Y-L, Teng Y-C, Wang T-W, Kao C-H, Yeh C-H, Tsai T-F, CISD2 maintains cellular homeostasis, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research (2021) 118954. [DOI] [PubMed] [Google Scholar]
- [5].Chang NC, Nguyen M, Germain M, Shore GC, Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1, EMBO J 29(3) (2010) 606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang NC, Nguyen M, Shore GC, BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis?, Autophagy 8(5) (2012) 856–7. [DOI] [PubMed] [Google Scholar]
- [7].Chang NC, Nguyen M, Bourdon J, Risse PA, Martin J, Danialou G, Rizzuto R, Petrof BJ, Shore GC, Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle, Hum Mol Genet 21(10) (2012) 2277–87. [DOI] [PubMed] [Google Scholar]
- [8].Tamir S, Rotem-Bamberger S, Katz C, Morcos F, Hailey KL, Zuris JA, Wang C, Conlan AR, Lipper CH, Paddock ML, Mittler R, Onuchic JN, Jennings PA, Friedler A, Nechushtai R, Integrated strategy reveals the protein interface between cancer targets Bcl-2 and NAF-1, Proc Natl Acad Sci U S A 111(14) (2014) 5177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang CH, Kao CH, Chen YF, Wei YH, Tsai TF, Cisd2 mediates lifespan: is there an interconnection among Ca(2)(+) homeostasis, autophagy, and lifespan?, Free Radic Res 48(9) (2014) 1109–14. [DOI] [PubMed] [Google Scholar]
- [10].Lu S, Kanekura K, Hara T, Mahadevan J, Spears LD, Oslowski CM, Martinez R, Yamazaki-Inoue M, Toyoda M, Neilson A, Blanner P, Brown CM, Semenkovich CF, Marshall BA, Hershey T, Umezawa A, Greer PA, Urano F, A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome, Proc Natl Acad Sci U S A 111(49) (2014) E5292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Holt SH, Darash-Yahana M, Sohn YS, Song L, Karmi O, Tamir S, Michaeli D, Luo Y, Paddock ML, Jennings PA, Onuchic JN, Azad RK, Pikarsky E, Cabantchik IZ, Nechushtai R, Mittler R, Activation of apoptosis in NAF-1-deficient human epithelial breast cancer cells, J Cell Sci 129(1) (2016) 155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Iosub-Amir A, Bai F, Sohn YS, Song L, Tamir S, Marjault HB, Mayer G, Karmi O, Jennings PA, Mittler R, Onuchic JN, Friedler A, Nechushtai R, The anti-apoptotic proteins NAF-1 and iASPP interact to drive apoptosis in cancer cells, Chem Sci 10(3) (2019) 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang CH, Chen YF, Wu CY, Wu PC, Huang YL, Kao CH, Lin CH, Kao LS, Tsai TF, Wei YH, Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis, Hum Mol Genet 23(18) (2014) 4770–85. [DOI] [PubMed] [Google Scholar]
- [14].Shen ZQ, Chen YF, Chen JR, Jou YS, Wu PC, Kao CH, Wang CH, Huang YL, Chen CF, Huang TS, Shyu YC, Tsai SF, Kao LS, Tsai TF, CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma, Cell Rep 21(8) (2017) 2198–2211. [DOI] [PubMed] [Google Scholar]
- [15].Yeh CH, Shen ZQ, Hsiung SY, Wu PC, Teng YC, Chou YJ, Fang SW, Chen CF, Yan YT, Kao LS, Kao CH, Tsai TF, Cisd2 is essential to delaying cardiac aging and to maintaining heart functions, Plos Biol 17(10) (2019) e3000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Karmi O, Holt SH, Song L, Tamir S, Luo Y, Bai F, Adenwalla A, Darash-Yahana M, Sohn YS, Jennings PA, Azad RK, Onuchic JN, Morcos F, Nechushtai R, Mittler R, Interactions between mitoNEET and NAF-1 in cells, PLoS One 12(4) (2017) e0175796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huynh N, Ou Q, Cox P, Lill R, King-Jones K, Glycogen branching enzyme controls cellular iron homeostasis via Iron Regulatory Protein 1 and mitoNEET, Nature communications 10(1) (2019) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lipper CH, Paddock ML, Onuchic JN, Mittler R, Nechushtai R, Jennings PA, Cancer-Related NEET Proteins Transfer 2Fe-2S Clusters to Anamorsin, a Protein Required for Cytosolic Iron-Sulfur Cluster Biogenesis, PLoS One 10(10) (2015) e0139699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ajlouni K, Jarrah N, El-Khateeb M, El-Zaheri M, El Shanti H, Lidral A, Wolfram syndrome: identification of a phenotypic and genotypic variant from Jordan, Am J Med Genet 115(1) (2002) 61–5. [DOI] [PubMed] [Google Scholar]
- [20].Amr S, Heisey C, Zhang M, Xia XJ, Shows KH, Ajlouni K, Pandya A, Satin LS, El-Shanti H, Shiang R, A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2, Am J Hum Genet 81(4) (2007) 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rigoli L, Di Bella C, Wolfram syndrome 1 and Wolfram syndrome 2, Curr Opin Pediatr 24(4) (2012) 512–7. [DOI] [PubMed] [Google Scholar]
- [22].Urano F, Wolfram Syndrome: Diagnosis, Management, and Treatment, Curr Diab Rep 16(1) (2016) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Danielpur L, Sohn YS, Karmi O, Fogel C, Zinger A, Abu-Libdeh A, Israeli T, Riahi Y, Pappo O, Birk R, Zangen DH, Mittler R, Cabantchik ZI, Cerasi E, Nechushtai R, Leibowitz G, GLP-1-RA corrects mitochondrial labile iron accumulation and improves beta-cell function in type 2 Wolfram syndrome, J Clin Endocrinol Metab (2016) jc20162240. [DOI] [PubMed] [Google Scholar]
- [24].Chen YF, Kao CH, Kirby R, Tsai TF, Cisd2 mediates mitochondrial integrity and life span in mammals, Autophagy 5(7) (2009) 1043–5. [DOI] [PubMed] [Google Scholar]
- [25].Wu CY, Chen YF, Wang CH, Kao CH, Zhuang HW, Chen CC, Chen LK, Kirby R, Wei YH, Tsai SF, Tsai TF, A persistent level of Cisd2 extends healthy lifespan and delays aging in mice, Hum Mol Genet 21(18) (2012) 3956–68. [DOI] [PubMed] [Google Scholar]
- [26].Annalisa N, Krashia P, D’Amelio M, Cisd2: A promising new target in Alzheimer’s disease, The Journal of Pathology (2020). [DOI] [PubMed] [Google Scholar]
- [27].Chen YF, Chou TY, Lin IH, Chen CG, Kao CH, Huang GJ, Chen LK, Wang PN, Lin CP, Tsai TF, Upregulation of Cisd2 attenuates Alzheimer’s related neuronal loss in mice, J Pathol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hsieh CJ, Weng PH, Chen JH, Chen TF, Sun Y, Wen LL, Yip PK, Chu YM, Chen YC, Sequence variants of the aging gene CISD2 and the risk for Alzheimer’s disease, J Formos Med Assoc 114(7) (2015) 627–32. [DOI] [PubMed] [Google Scholar]
- [29].Mozzillo E, Delvecchio M, Carella M, Grandone E, Palumbo P, Salina A, Aloi C, Buono P, Izzo A, D’Annunzio G, Vecchione G, Orrico A, Genesio R, Simonelli F, Franzese A, A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2, Bmc Med Genet 15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rondinelli M, Novara F, Calcaterra V, Zuffardi O, Genovese S, Wolfram syndrome 2: a novel CISD2 mutation identified in Italian siblings, Acta Diabetol 52(1) (2015) 175–178. [DOI] [PubMed] [Google Scholar]
- [31].Rouzier C, Moore D, Delorme C, Lacas-Gervais S, Ait-El-Mkadem S, Fragaki K, Burte F, Serre V, Bannwarth S, Chaussenot A, Catala M, Yu-Wai-Man P, Paquis-Flucklinger V, A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions, Hum Mol Genet 26(9) (2017) 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Du XH, Xiao RJ, Xiao F, Chen Y, Hua FZ, Yu SC, Xu GH, NAF-1 antagonizes starvation-induced autophagy through AMPK signaling pathway in cardiomyocytes, Cell Biol Int 39(7) (2015) 816–823. [DOI] [PubMed] [Google Scholar]
- [33].Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML, Hochberg A, Cabanchick IZ, Onuchic JN, Jennings PA, Nechushtai R, Mittler R, NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth, Proc Natl Acad Sci U S A 110(36) (2013) 14676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu L, Xia M, Wang J, Zhang W, Zhang Y, He M, CISD2 expression is a novel marker correlating with pelvic lymph node metastasis and prognosis in patients with early-stage cervical cancer, Med Oncol 31(9) (2014) 183. [DOI] [PubMed] [Google Scholar]
- [35].Darash-Yahana M, Pozniak Y, Lu M, Sohn YS, Karmi O, Tamir S, Bai F, Song L, Jennings PA, Pikarsky E, Geiger T, Onuchic JN, Mittler R, Nechushtai R, Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters, Proc Natl Acad Sci U S A (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang L, Hong S, Wang Y, He Z, Liang S, Chen H, He S, Wu S, Song L, Chen Y, A novel prognostic score model incorporating CDGSH iron sulfur domain2 (CISD2) predicts risk of disease progression in laryngeal squamous cell carcinoma, Oncotarget 7(16) (2016) 22720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang L, Ouyang F, Liu XB, Wu S, Wu HM, Xu YD, Wang B, Zhu JR, Xu XH, Zhang L, Overexpressed CISD2 has prognostic value in human gastric cancer and promotes gastric cancer cell proliferation and tumorigenesis via AKT signaling pathway, Oncotarget 7(4) (2016) 3791–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li SM, Chen CH, Chen YW, Yen YC, Fang WT, Tsai FY, Chang JL, Shen YY, Huang SF, Chuu CP, Chang IS, Hsiung CA, Jiang SS, Upregulation of CISD2 augments ROS homeostasis and contributes to tumorigenesis and poor prognosis of lung adenocarcinoma, Sci Rep 7(1) (2017) 11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun Y, Jiang Y, Huang J, Chen H, Liao Y, Yang Z, CISD2 enhances the chemosensitivity of gastric cancer through the enhancement of 5-FU-induced apoptosis and the inhibition of autophagy by AKT/mTOR pathway, Cancer Med 6(10) (2017) 2331–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sun AG, Meng FG, Wang MG, CISD2 promotes the proliferation of glioma cells via suppressing beclin1mediated autophagy and is targeted by microRNA449a, Mol Med Rep 16(6) (2017) 7939–7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim EH, Shin D, Lee J, Jung AR, Roh JL, CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer, Cancer Lett 432 (2018) 180–190. [DOI] [PubMed] [Google Scholar]
- [42].Mittler R, Darash-Yahana M, Sohn YS, Bai F, Song L, Cabantchik IZ, Jennings PA, Onuchic JN, Nechushtai R, NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer, Antioxid Redox Signal (2018) 30(8):1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Geldenhuys WJ, Skolik R, Konkle ME, Menze MA, Long TE, Robart AR, Binding of thiazolidinediones to the endoplasmic reticulum protein nutrient-deprivation autophagy factor-1, Bioorganic & medicinal chemistry letters 29(7) (2019) 901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tamir S, Eisenberg-Domovich Y, Conlan AR, Stofleth JT, Lipper CH, Paddock ML, Mittler R, Jennings PA, Livnah O, Nechushtai R, A point mutation in the [2Fe-2S] cluster binding region of the NAF-1 protein (H114C) dramatically hinders the cluster donor properties, Acta Crystallogr D Biol Crystallogr 70(Pt 6) (2014) 1572–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dhanesuan N, Sharp JA, Blick T, Price JT, Thompson EW, Doxycycline-inducible expression of SPARC/Osteonectin/BM40 in MDA-MB-231 human breast cancer cells results in growth inhibition, Breast cancer research and treatment 75(1) (2002) 73–85. [DOI] [PubMed] [Google Scholar]
- [46].Zielonka J, Kalyanaraman B, Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med. 48(8) (2010) 983–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhou Z, Fan D, Wang J, Sohn YS, Nechushtai R, Willner I, Triggered Dimerization and Trimerization of DNA Tetrahedra for Multiplexed miRNA Detection and Imaging of Cancer Cells, Small 17(6) (2021) 2007355. [DOI] [PubMed] [Google Scholar]
- [48].Meagher EA, FitzGerald GA, Indices of lipid peroxidation in vivo: strengths and limitations. Free Radic Biol Med. 28(12) (2000) 1745–1750. [DOI] [PubMed] [Google Scholar]
- [49].Yang B, Li R, Liu PN, Geng X, Mooney BP, Chen C, Cheng J, Fritsche KL, Beversdorf DQ, Lee JC, Quantitative proteomics reveals docosahexaenoic acid-mediated neuroprotective effects in lipopolysaccharide-stimulated microglial cells, Journal of proteome research 19(6) (2020) 2236–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhou J, Chng W-J, Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer, Mitochondrion 13(3) (2013) 163–169. [DOI] [PubMed] [Google Scholar]
- [51].Chen Y, Ning J, Cao W, Wang S, Du T, Jiang J, Feng X, Zhang B, Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers, Frontiers in Oncology (2020) 10:568574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen X, Yu C, Kang R, Tang D, Iron metabolism in ferroptosis, Frontiers in Cell and Developmental Biology (2020) 8:590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shao F, Du J, Li Y, Yu J, Wu H, Ying K, Downregulation of CISD2 has prognostic value in non-small cell lung cancer and inhibits the tumorigenesis by inducing mitochondrial dysfunction, Frontiers in Oncology 10 (2020) 3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yuan H, Li X, Zhang X, Kang R, Tang D, CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation, Biochem Biophys Res Commun 478(2) (2016) 838–44. [DOI] [PubMed] [Google Scholar]
- [55].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR, Regulation of ferroptotic cancer cell death by GPX4, Cell 156(1–2) (2014) 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jiang X, Stockwell BR, Conrad M, Ferroptosis: Mechanisms, biology and role in disease, Nature Reviews Molecular Cell Biology (2021) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka JM, Transferrin receptor is a specific ferroptosis marker, Cell reports 30(10) (2020) 3411–3423. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang H, Go Y-M, Jones DP, Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress, Archives of biochemistry and biophysics 465(1) (2007) 119–126. [DOI] [PubMed] [Google Scholar]
- [59].Kameritsch P, Singer M, Nuernbergk C, Rios N, Reyes AM, Schmidt K, Kirsch J, Schneider H, Müller S, Pogoda K, The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity, Proceedings of the National Academy of Sciences 118(7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE, Jennings PA, MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone, Proc Natl Acad Sci U S A 104(36) (2007) 14342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chen YF, Kao CH, Chen YT, Wang CH, Wu CY, Tsai CY, Liu FC, Yang CW, Wei YH, Hsu MT, Tsai SF, Tsai TF, Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice, Genes Dev 23(10) (2009) 1183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chen YF, Wu CY, Kirby R, Kao CH, Tsai TF, A role for the CISD2 gene in lifespan control and human disease, Ann N Y Acad Sci 1201 (2010) 58–64. [DOI] [PubMed] [Google Scholar]
- [63].Tsai PH, Chien Y, Chuang JH, Chou SJ, Chien CH, Lai YH, Li HY, Ko YL, Chang YL, Wang CY, Liu YY, Lee HC, Yang CH, Tsai TF, Lee YY, Chiou SH, Dysregulation of Mitochondrial Functions and Osteogenic Differentiation in Cisd2-Deficient Murine Induced Pluripotent Stem Cells, Stem Cells Dev 24(21) (2015) 2561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Huang YL, Shen ZQ, Wu CY, Teng YC, Liao CC, Kao CH, Chen LK, Lin CH, Tsai TF, Comparative proteomic profiling reveals a role for Cisd2 in skeletal muscle aging, Aging Cell 17(1) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gaines CH, Snyder AE, Ervin RB, Farrington J, Walsh K, Schoenrock SA, Tarantino LM, Behavioral characterization of a novel Cisd2 mutant mouse, Behavioural Brain Research 405 (2021) 113187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen B, Shen S, Wu J, Hua Y, Kuang M, Li S, Peng B, CISD2 associated with proliferation indicates negative prognosis in patients with hepatocellular carcinoma, Int J Clin Exp Pathol 8(10) (2015) 13725–38. [PMC free article] [PubMed] [Google Scholar]
- [67].Yang Y, Bai YS, Wang Q, CDGSH iron sulfur domain 2 activates proliferation and EMT of pancreatic cancer cells via Wnt/beta-catenin pathway and has prognostic value in human pancreatic cancer, Oncol Res (2016) 25(4):605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shen Z-Q, Huang Y-L, Tsai T-F, Cisd2 haploinsufficiency: A driving force for hepatocellular carcinoma, Molecular & cellular oncology 5(3) (2018) e1441627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chong C-R, Chan WPA, Nguyen TH, Liu S, Procter NE, Ngo DT, Sverdlov AL, Chirkov YY, Horowitz JD, Thioredoxin-interacting protein: pathophysiology and emerging pharmacotherapeutics in cardiovascular disease and diabetes, Cardiovascular drugs and therapy 28(4) (2014) 347–360. [DOI] [PubMed] [Google Scholar]
- [70].Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J, Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases, Frontiers in immunology 4 (2014) 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Singh LP, Thioredoxin interacting protein (TXNIP) and pathogenesis of diabetic retinopathy, Journal of clinical & experimental ophthalmology 4 (2013) 4: 10.4172/2155-9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Shen L, O’Shea JM, Kaadige MR, Cunha S, Wilde BR, Cohen AL, Welm AL, Ayer DE, Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP, Proceedings of the National Academy of Sciences 112(17) (2015) 5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Park JW, Lee SH, Woo G-H, Kwon H-J, Kim D-Y, Downregulation of TXNIP leads to high proliferative activity and estrogen-dependent cell growth in breast cancer, Biochemical and biophysical research communications 498(3) (2018) 566–572. [DOI] [PubMed] [Google Scholar]
- [74].Wang X, Jiang M, He X, Zhang B, Peng W, Guo L, N-acetyl cysteine inhibits the lipopolysaccharide-induced inflammatory response in bone marrow mesenchymal stem cells by suppressing the TXNIP/NLRP3/IL-1β signaling pathway, Molecular Medicine Reports 22(4) (2020) 3299–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yan YR, Zhang L, Lin YN, Sun XW, Ding YJ, Li N, Li HP, Li SQ, Zhou JP, Li QY, Chronic intermittent hypoxia-induced mitochondrial dysfunction mediates endothelial injury via the TXNIP/NLRP3/IL-1β signaling pathway, Free Radical Bio Med 165 (2021) 401–410. [DOI] [PubMed] [Google Scholar]
- [76].Li J, Cao F, Yin H.-l., Huang Z.-j., Lin Z.-t., Mao N, Sun B, Wang G, Ferroptosis: past, present and future, Cell Death & Disease 11(2) (2020) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Samuel SM, Varghese E, Varghese S, Büsselberg D, Challenges and perspectives in the treatment of diabetes associated breast cancer, Cancer treatment reviews 70 (2018) 98–111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.