

SUMMARY
Autotaxin (ATX; ENPP2) produces lysophosphatidic acid (LPA) that regulates multiple biological functions via cognate G protein-coupled receptors LPAR1–6. ATX/LPA promotes tumor cell migration and metastasis via LPAR1 and T cell motility via LPAR2, yet its actions in the tumor immune microenvironment remain unclear. Here, we show that ATX secreted by melanoma cells is chemorepulsive for tumor-infiltrating lymphocytes (TILs) and circulating CD8+ T cells ex vivo, with ATX functioning as an LPA-producing chaperone. Mechanistically, T cell repulsion predominantly involves Gα12/13-coupled LPAR6. Upon anti-cancer vaccination of tumor-bearing mice, ATX does not affect the induction of systemic T cell responses but, importantly, suppresses tumor infiltration of cytotoxic CD8+ T cells and thereby impairs tumor regression. Moreover, single-cell data from melanoma tumors are consistent with intratumoral ATX acting as a T cell repellent. These findings highlight an unexpected role for the pro-metastatic ATX-LPAR axis in suppressing CD8+ T cell infiltration to impede anti-tumor immunity, suggesting new therapeutic opportunities.
In brief
Through LPA production, ATX modulates the tumor microenvironment in autocrine-paracrine manners. Matas-Rico et al. show that ATX/LPA is chemorepulsive for T cells with a dominant inhibitory role for Gα12/13-coupled LPAR6. Upon anticancer vaccination, tumor-intrinsic ATX suppresses the infiltration of CD8+ T cells without affecting their cytotoxic quality.
Graphical Abstract
INTRODUCTION
Efficient infiltration of T cells into tumors is associated with positive outcome in several cancer types and determines the response to immunotherapies (Fridman et al., 2017; Ribas and Wolchok, 2018). Chemokines through their G protein-coupled receptors (GPCRs) are major drivers of T cell migration into tumors, thereby playing a crucial role in the immune response to cancer and influencing tumor fate (Jacquelot et al., 2018; Nagarsheth et al., 2017; Ozga et al., 2021). However, tumors develop various strategies to exclude T cells and suppress T cell-mediated immunogenicity, for example via tumor-intrinsic chemokine silencing and production of immunosuppressive cytokines (Batlle and Massagué, 2019; Joyce and Fearon, 2015; Kerdidani et al., 2019; Spranger et al., 2015; Spranger and Gajewski, 2018). Yet, our understanding of factors that regulate the trafficking of tumor-infiltrating lymphocytes (TILs), either positively or negatively, is incomplete and requires identification of new tractable targets (Anandappa et al., 2020; van der Woude et al., 2017). Here, we explore a role for autotaxin (ATX) in this process.
ATX (encoded by ENPP2) is a unique lysophospholipase D (lysoPLD) that is secreted by diverse cell types to produce the lipid mediator and GPCR agonist lysophosphatidic acid (LPA) from abundantly available extracellular lysophosphatidylcholine (LPC) (Perrakis and Moolenaar, 2014; Tokumura et al., 2002; Umezu-Goto et al., 2002). ATX was originally defined as an “autocrine motility factor” secreted by melanoma cells and characterized as a metastasis-enhancing ecto-phosphodiesterase (Nam et al., 2001; Stracke et al., 1992). The ATX-LPA signaling axis plays a key role in a wide variety of biological and pathophysiological processes, ranging from vascular and neural development (van Meeteren et al., 2006) to lymphocyte homing (Kanda et al., 2008), inflammation, fibrosis, and tumor progression (Benesch et al., 2018; Mills and Moolenaar, 2003). Unfortunately, however, detailed assessment of ATX function in vivo is hampered the embryonic lethality of ATX-deficient mice (van Meeteren et al., 2006).
LPA (mono-acyl-sn-glycero-3-phospate) acts on six specific GPCRs, termed LPAR1–LPAR6 or LPA1–6, showing both unique and shared signaling activities and tissue distributions (Yanagida et al., 2013; Yung et al., 2014). LPAR1–LPAR3 belong to the so-called EDG subfamily of GPCRs alongside the sphingosine 1-phosphate (S1P) receptors, whereas the disparate LPAR4–6 members are related to the P2Y purinergic receptor family (Hisano and Hla, 2019; Yanagida et al., 2013). It is further of note that ATX interacts with cell-surface integrins and/or heparan sulfate proteoglycans thereby facilitating delivery of LPA to its cognate receptors in a highly localized manner (Fulkerson et al., 2011; Hausmann et al., 2011; Houben et al., 2013; Kanda et al., 2008).
Numerous studies have documented a critical role for ATX and/or LPA in stimulating cell migration, tumor cell dispersal, invasion, and metastasis, mediated primarily by LPAR1 (Auciello et al., 2019; David et al., 2010; Lin et al., 2019; Liu et al., 2009; Marshall et al., 2012). LPAR1 also mediates the recruitment and activation of fibroblasts, a prototypic ATX-secreting cell type, and thereby can promote tissue fibrosis (Ledein et al., 2020; Sakai et al., 2019; Tager et al., 2008). Activated fibroblasts constitute a large part of solid tumors, producing cytokines and extracellular matrix to enhance metastasis (Kalluri, 2016; Winkler et al., 2020). Interestingly, LPAR1–LPAR3 commonly mediate enhanced cellular responses, whereas non-EDG receptors LPAR4–LPAR6 can exert counter-regulatory actions in that they suppress the migration and invasion of diverse cell types, depending on their dominant G protein-effector pathways (Jongsma et al., 2011; Lee et al., 2008; Takahashi et al., 2017).
In the immune system, ATX is abundantly expressed in high-endothelial venules (HEVs) that control lymphocyte entry from blood into lymphoid tissue (Kanda et al., 2008; Takeda et al., 2016). Acting predominantly through LPAR2, HEV-secreted ATX promotes the random motility of naive T cells to enhance their transmigration into secondary lymphoid organs and thereby contributes to the control of systemic T cell responses (Bai et al., 2013; Kanda et al., 2008; Knowlden et al., 2014; Takeda et al., 2016). Thus, the ATX-LPA signaling axis regulates the migratory activities of both tumor cells and T cells mainly via LPAR1 and LPAR2, respectively. However, its actions in the tumor immune microenvironment remain unclear, particularly the dominant LPAR signaling pathways and how ATX/LPA may affect antigen-specific T cell responses and effector T cell activity in a tumor context.
Here, we show that ATX/LPA antagonizes the migration of patient-derived TILs and healthy blood-derived CD8+ T cells ex vivo, and define Gα12/13-coupled LPAR6 as a T cell migration inhibitory receptor. By eliciting a robust immune response upon anti-cancer vaccination of tumor-bearing mice, we demonstrate that secreted ATX antagonizes tumor infiltration of cytotoxic CD8+ T cells and thereby impedes tumor control in a therapeutic setting. Concordantly, single-cell analysis of melanoma tumors shows a negative correlation between intratumoral ENPP2 expression and CD8+ T cell infiltration. By revealing ATX as a suppressor of anti-tumor immunity, our findings shed light on its multifaceted actions in the tumor microenvironment.
RESULTS
Through LPA production, ATX secreted by melanoma cells is chemorepulsive for TILs and peripheral CD8+ T cells
Melanoma cells are known for their high ATX expression levels among many human cancer cell lines (Ghandi et al., 2019) and solid tumors (Figures S1A and S1B). This feature is unrelated to genetic changes (https://www.cbioportal.org), but rather reflects high ATX expression in skin melanocytes, a highly motile cell type. We set out to examine how melanoma cell-secreted ATX affects the migration of ex vivo expanded melanoma TILs and peripheral blood CD8+ T cells. Patient-derived TILs constitute a heterogeneous population of T cells in distinct functional states and other immune cells (Li et al., 2019). During their ex vivo expansion driven by anti-CD3 antibody and IL-2 (see STAR Methods), TILs become enriched in CD8+ and CD4+ T cells and are then used for adoptive TIL therapy in patients (Rohaan et al., 2019).
We first analyzed the effects of LPA and ATX/LPC on the transwell migration of TILs (isolated from two patients). As a positive control, we used chemokine CXCL10 that signals via CXCR3 to promote effector T cell migration and is implicated in enhancing cancer immunity (Groom and Luster, 2011; Nagarsheth et al., 2017). Strikingly, LPA strongly suppressed the basal migration rate of TlLs (up to 5-fold in patient 1) when assayed over a period of 2 h, in a concentration-dependent manner (Figures 1A and 1B). LPA was capable of antagonizing TIL migration toward CXCL10 (Figure 1C). LPA was also chemo-repulsive for peripheral blood CD8+ T cells isolated from healthy donors (Figure 1D). When TILs or CD8+ T cells were exposed to recombinant ATX (20 nM) together with its substrate LPC (1–5 μM), their transwell migration was similarly suppressed (Figure 1E).
Figure 1. LPA and ATX/LPC are chemorepulsive for TILs and peripheral CD8+ T cells.
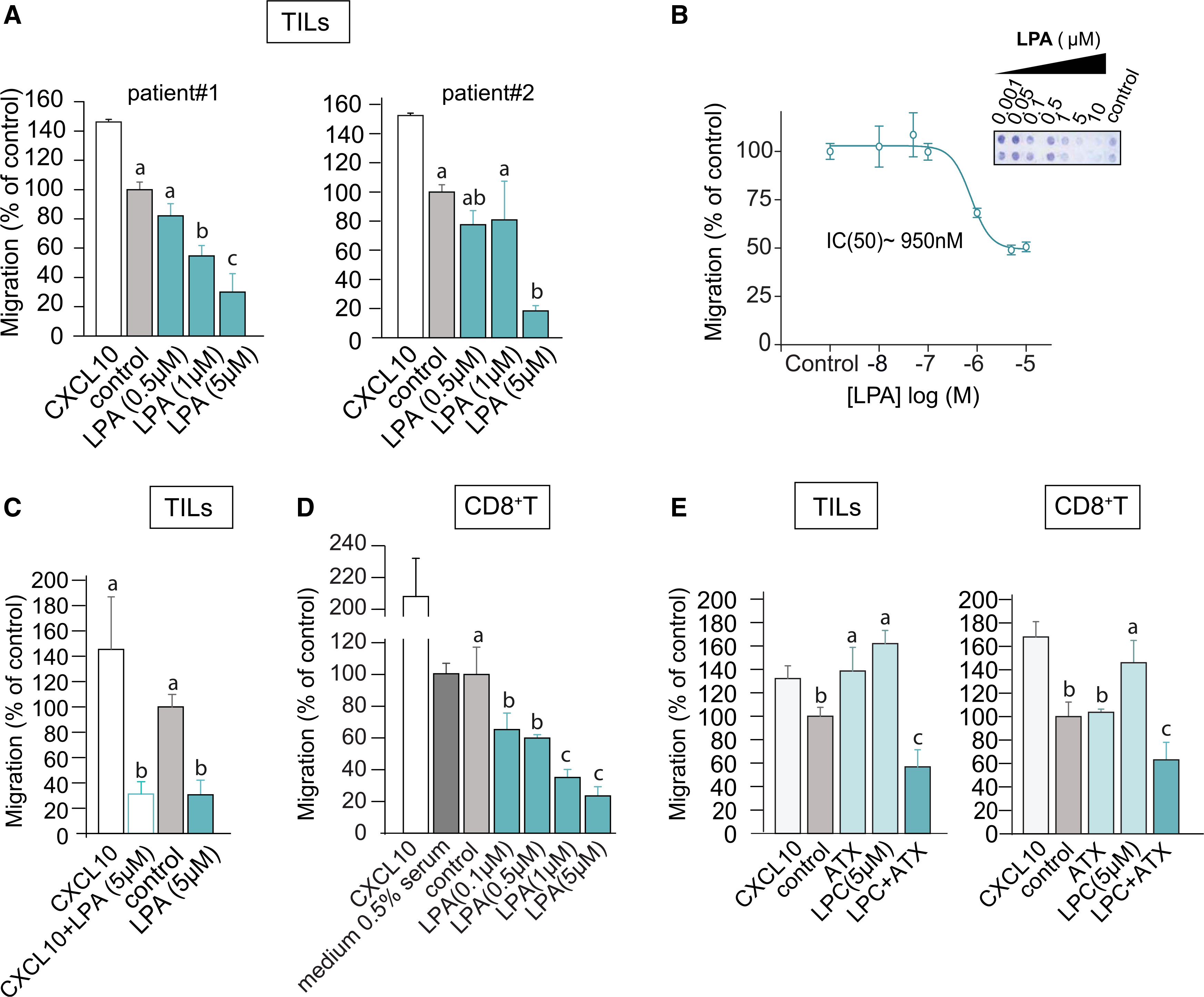
(A) Transwell migration of ex vivo expanded TILs from two melanoma patients stimulated with LPA(18:1) at the indicated concentrations. Chemokine CXCL10 (1 μM) was used as positive control; “control” refers to serum-free medium. Agonists were added to the bottom wells and incubation was carried out for 2 h at 37°C.
(B) LPA dose-dependency of migration. The inset shows a representative transwell filter after staining. Migration was quantified by color intensity using ImageJ.
(C) LPA overrules CXCL10-induced TIL chemotaxis. LPA(18:1) was added together with CXCL10 at the indicated concentrations.
(D) Migration of CD8+ T cells isolated from peripheral blood, measured in the absence (control) and presence of the indicated concentrations of LPA(18:1). Note that the presence of 0.5% serum has no effect.
(E) Recombinant ATX (20 nM) added together with the indicated concentrations of LPC(18:1) recapitulates the inhibitory effects of LPA(18:1) on TILs and CD8+ T cells.
(A and C–E) Results are representative of three independent experiments each performed in technical triplicates and expressed as means ± SEM; bars annotated with different letters were significantly different according to Fisher’s least significant difference (LSD) test (p ≤ 0.05) after ANOVA.
We next analyzed melanoma cell supernatants for their modulatory activity on T cell migration. In addition, we measured concurrently secreted ATX protein and lysoPLD activity. Culture media (containing 0.5% serum) conditioned by melanoma cells (MDA-MB-435 and A375) for 24 h markedly suppressed the basal migration and CXCL10-induced chemotaxis of TILs and peripheral CD8+ T cells (Figure 2A). Secreted ATX protein was readily detected by immunoblotting (Figure 2B), whereas lysoPLD activity was detected simultaneously (Figure 2C). By contrast, conditioned media from either ATX knockdown melanoma cells (Figure 2D) or ATX-deficient MDA-MB-231 breast carcinoma cells (Figure 2F) lacked chemorepulsive activity (Figures 2E and 2F). TIL migration could be rescued by incubating melanoma media with established ATX inhibitors, notably PF-8380 and IOA-289 (formerly CRT750) (Shah et al., 2016) (Figure 2G). Together, these results show that LPA-producing ATX released from melanoma cells is a major T cell repellent.
Figure 2. ATX secreted by melanoma cells repels TILs and peripheral CD8+ T cells.
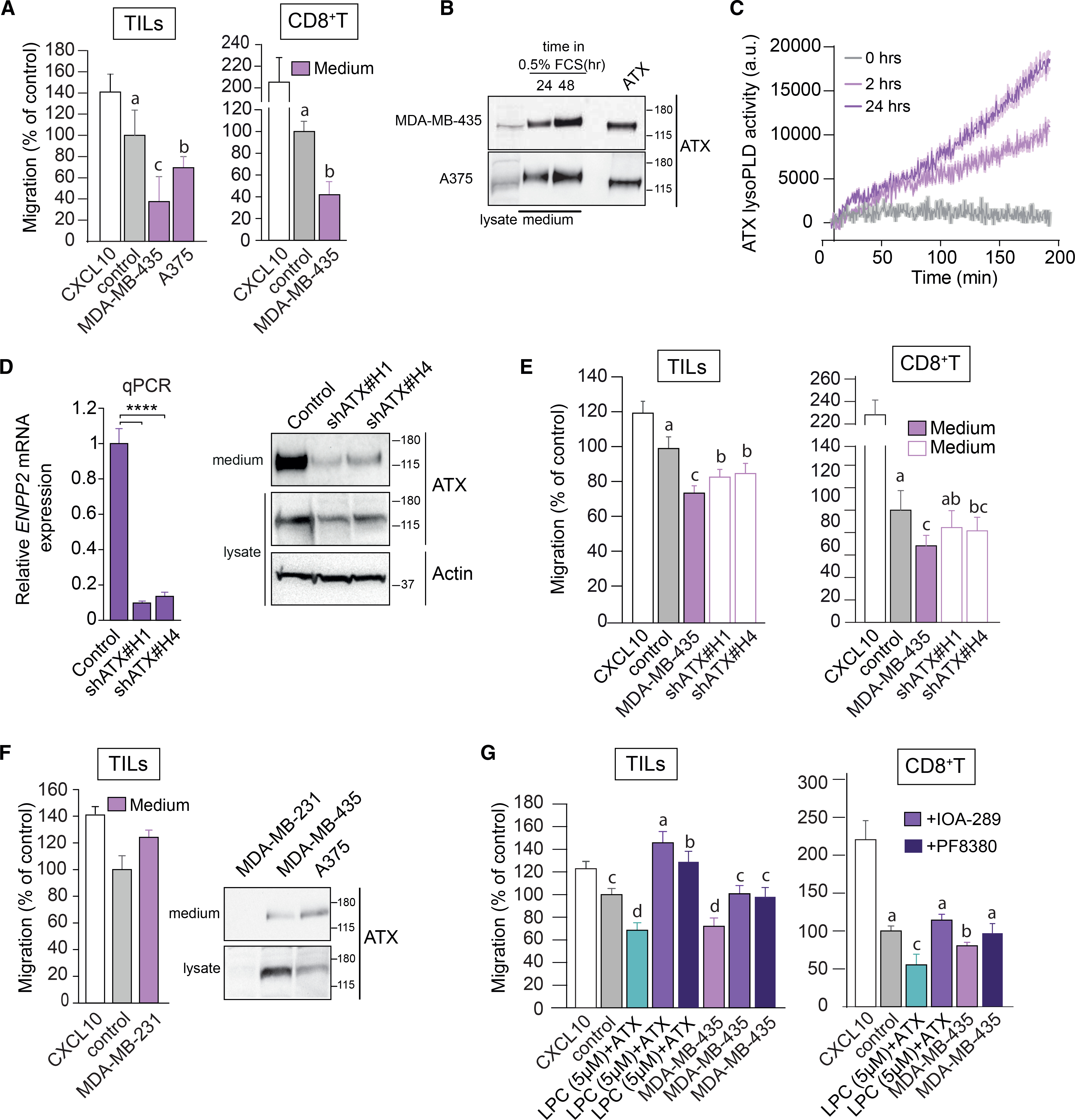
(A) Melanoma-conditioned medium from MDA-MB-435 and A375 cells (collected after 24 h) is chemorepulsive for TILs and blood-derived CD8+ T cells. Experimental conditions as in Figure 1.
(B) Immunoblot showing ATX expression in medium and cell lysates of MDA-MB-435 and A375 melanoma cells. Cells were incubated in DMEM with 0.5% FCS for 24 or 48 h. Recombinant ATX (20 nM) was used as positive control (right lane).
(C) LysoPLD activity accumulating in melanoma-conditioned media over time. Medium from MDA-MB-435 cells was collected after 2 and 24 h, and lysoPLD activity was measured as choline release from added LPC(18:1).
(D) ATX (ENPP2) mRNA expression (relative to cyclophilin) in control and ENPP2-depleted MDA-MB-435 cells stably expressing short hairpin RNAs (shRNAs) against ATX. Maximal ENPP2 knockdown was obtained with shRNA 1 and 4 (of 5 different hairpins). Data represent the mean ± SEM of three independent experiments using triplicate samples; ****p < 0.0001 (unpaired Student’s t test). Right: immunoblot analysis of ATX expression using shRNA 1 and 4. Actin was used as loading control.
(E) Melanoma-conditioned medium from ATX knockdown MDA-MD-435 cells (collected after 24 h) lacks chemorepulsive activity for CD8+ T cells and TILs.
(F) Conditioned media from ATX-deficient MDA-MB-231 breast carcinoma cells lack chemo-repulsive activity for TILs compared to media from ATX-expressing melanoma cells (MDA-MB-435 and A375; cf. A). Right panel: ATX immunoblots from the indicated media and cell lysates.
(G) ATX inhibition restores the migration TILs and CD8+ T cells exposed to melanoma cell-conditioned media. Cells were plated at day 0 in medium containing 10% FCS. After 16 h, cells were exposed to medium containing 0.5% FCS and ATX inhibitors (PF-8380 or IOA-289). Conditioned media were collected after 24 h.
(A and D–G) Representative data of three independent experiments each performed in triplicate. Values are expressed as mean ± SEM; bars annotated with different letters were significantly different according to Fisher’s least significant difference (LSD) test (p ≤ 0.05) after ANOVA.
ATX as an LPA-producing chaperone
We investigated the relationship between ATX-mediated T cell repulsion and extracellular LPA levels. It is well established that LPA in freshly isolated plasma increases to high levels due to constitutive ATX-catalyzed LPC hydrolysis (Aoki et al., 2008). Extracellular LPA comprises distinct molecular species that differ in their acyl chain composition and binding affinity for individual LPA receptors (Yung et al., 2014). We measured LPA species in media from melanoma cells conditioned at 0, 24, and 48 h using liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Kraemer et al., 2019) (Figure 3A). LPA(12:0), LPA(16:0), LPA(18:0), LPA(18:1), and LPA(20:4) were the predominant species in media containing 0.5% serum (Figure 3B). Remarkably, total LPA in TIL-repulsive media declined to very low levels within 24 h, despite the fact that ATX activity increased concurrently (Figures 2B, 2C, 3C, and 3E); by contrast, the corresponding LPC species in these media remained constant or increased over time (Figure 3D). Hence, the loss of LPA in the face of ATX activity is not due to substrate depletion. Depletion of extracellular LPA by melanoma cells has been reported previously (Muinonen-Martin et al., 2014) and is due to its degradation by cell-associated lipid phosphate phosphatases (Sciorra and Morris, 2002). That ATX is fully bioactive at near-zero steady-state LPA levels can be explained by the fact that ATX binds LPA in its “exit tunnel” where it is protected from degradation (Keune et al., 2016; Moolenaar and Perrakis, 2011; Nishimasu et al., 2011; Salgado-Polo et al., 2018). These results thus support the notion that ATX both produces and “chaperones” LPA for local delivery to its receptors at the cell surface.
Figure 3. Lysolipid species and secreted lysoPLD activity in conditioned media from melanoma cells.
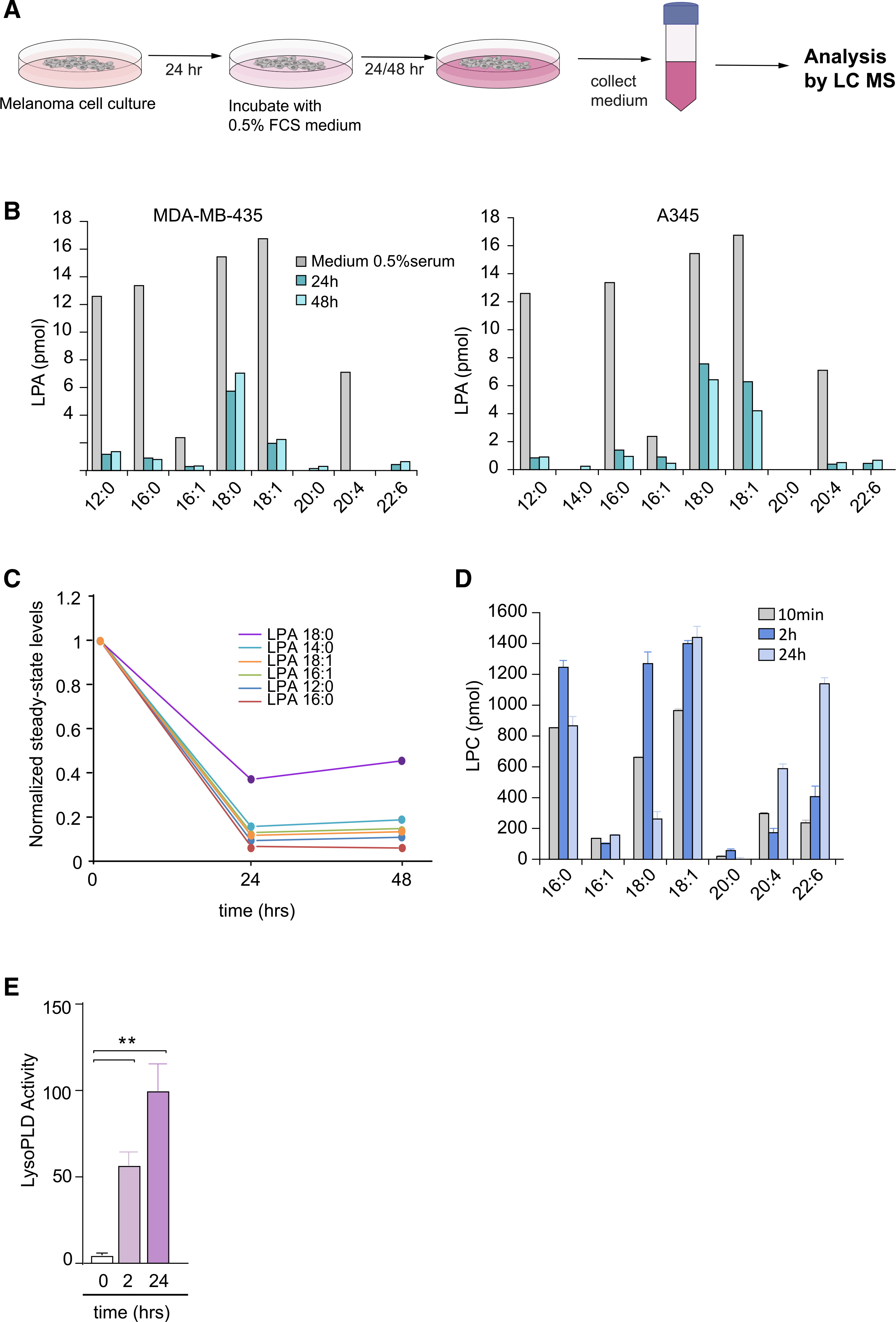
(A) Preparation of cell-conditioned media. Melanoma cells in 10-cm dishes were cultured for 24 h, washed, and then incubated in DMEM containing 0.5% FCS. Media were harvested after 24 and 48 h, and centrifuged to remove cell debris. LPA species were measured using LC/MS/MS.
(B) Determination of LPA species in conditioned medium from MDA-MB-435 and A375 melanoma cells, measured at t = 0, 24, and 48 h, using LC/MS/MS. Predominant serum-borne LPA species are (12:0), (16:0), (18:0), (18:1) and (20:4). Note LPA depletion from the medium (within 24 h) upon incubation with ATX-secreting melanoma cells.
(C) Time-dependent decline of the indicated serum-borne LPA species by melanoma cells. Graph shows normalized steady-state LPA levels in conditioned media from MDA-MB-435 cells.
(D) LPC species in conditioned medium from MDA-MB-435 cells, measured at t = 10 min, 2 h and 24 h, using LC/MS/MS. Note that LPC levels tend to increase over time. Values from one experiment performed in triplicate and expressed as mean ± SEM.
(E) Secreted lysoPLD activity increases over time. Medium from MDA-MB-435 cells was collected after 2 and 24 h, and lysoPLD activity was measured as choline release from added LPC(18:1). Values from three independent experiments each performed in triplicate and expressed as mean ± SEM; **p < 0.01 (unpaired Student’s t test).
TIL repulsion involves LPAR6
The T cell repelling activity of ATX/LPA markedly contrasts to its chemotactic activity for tumor cells, strongly suggesting involvement of different LPA receptors. We examined the LPAR expression repertoire in TILs and blood-derived CD8+ T cells using qPCR. Ex vivo expanded melanoma TILs (isolated from eight patients) consistently express high levels of LPAR6 in addition to considerably lower levels of LPAR2; an identical pattern was detected in ovarian carcinoma-derived TILs (Figure 4A, and data not shown). LPAR6 was also the predominant non-EDG LPA receptor in peripheral blood CD8+ T cells alongside LPAR4 and LPAR5 (Figure 4B), in agreement with publicly available data (https://www.immgen.org; http://biogps.org). LPAR4 and LPAR5 may have been lost from TILs during tumorigenesis or their ex vivo expansion, scenarios that warrant further investigation. Incubating TILs with a novel xanthylene-based LPAR6 antagonist, named XAA (Gnocchi et al., 2020), partially overcame T cell repulsion by LPA (Figure 4C). We therefore conclude that repulsion of TILs and peripheral blood CD8+ T cells is primarily mediated by LPAR6, without excluding possible additional anti-migratory roles for LPAR4 and LPAR5.
Figure 4. LPAR expression in TILs and peripheral CD8+ T cells.
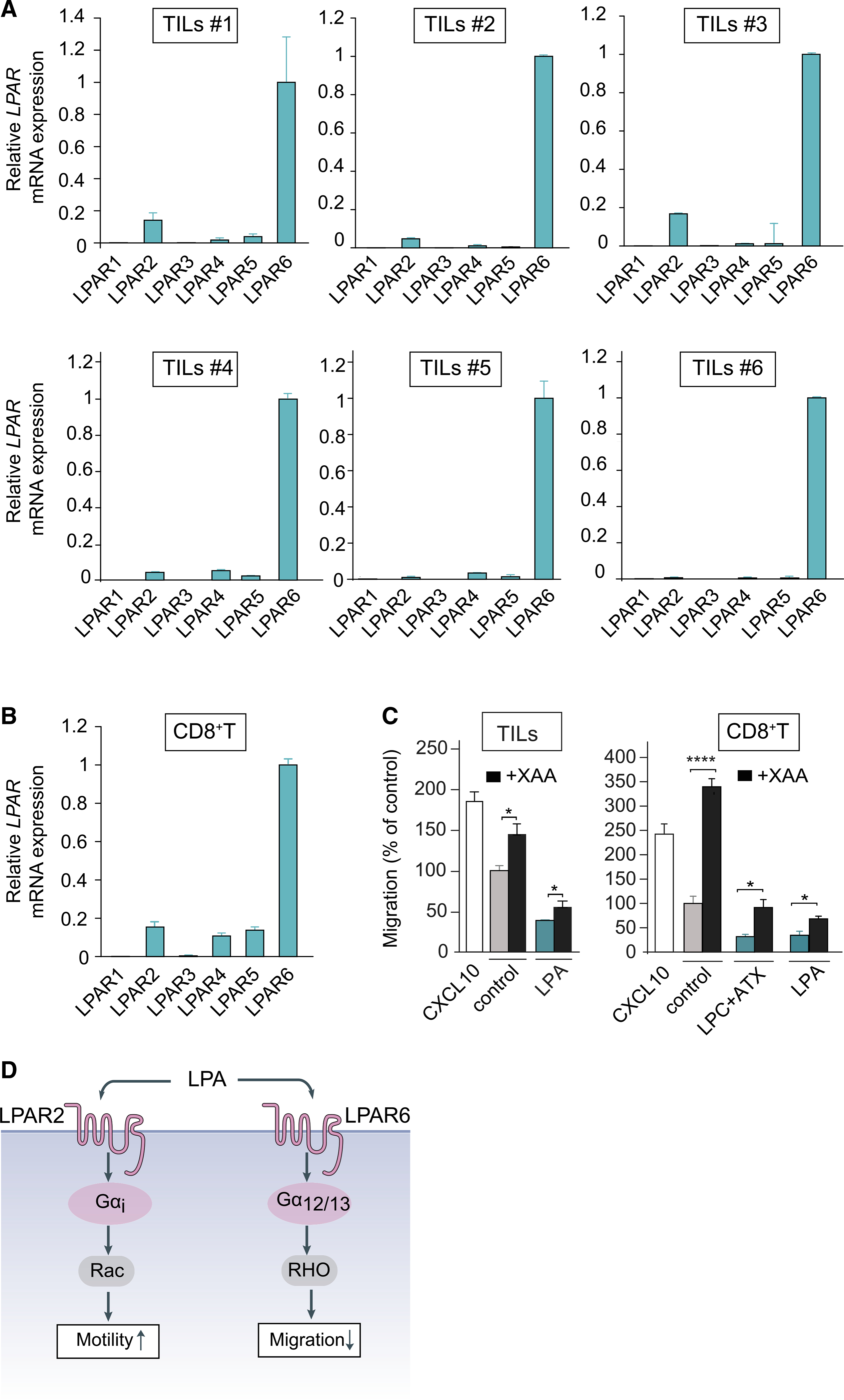
(A) LPAR expression repertoire in ex vivo expanded TILs from six patients (qPCR analysis relative to cyclophilin). TIL values are expressed as mean ± SD.
(B) LPAR expression in peripheral CD8+ T cells from two healthy donors. Values are expressed as mean ± SD.
(C) LPAR6 antagonist XAA restores transwell migration of TILs (left panel) and CD8+ T cells (right panel) in response to LPA or ATX plus LPC. Conditions as in Figure 1. Cells were treated with XAA (10 μM) or vehicle control (0.5% DMSO) for 24 h. Data represent the mean ± SEM of three independent experiments using triplicate samples. Data represent the mean ± SEM of three independent experiments using duplicate samples. *p < 0.05, ****p < 0.0001 (unpaired Student’s t test).
(D) Schematic illustration of dominant G-protein coupling and signaling outcomes of LPAR2 versus LPAR6.
LPAR6 (P2RY5) preferentially couples to the Gα12/13-RhoA pathway that drives cytoskeletal contraction, suppression of cell motility, and other cellular responses (Inoue et al., 2019; Wu et al., 2019; Yanagida et al., 2009; Yung et al., 2014). The function of LPAR6 in T cells has remained largely unexplored despite its high expression in immune cells (http://biogps.org). In contrast to LPAR6, LPAR2 couples to Gi-mediated Rac GTPase activation and other G protein-effector routes and thereby promotes the random motility of T cells (Kanda et al., 2008; Takeda et al., 2016), as schematically illustrated in Figure 4D.
Impact of ATX on the induction of systemic T cell responses and tumor infiltration of cytotoxic CD8+ T cells
Having shown that ATX through generation and protection of LPA repels TILs and blood-derived CD8+ T cells ex vivo, we next investigated how ATX affects the anti-tumor T cell response in vivo. We took advantage of an anti-cancer vaccination model using subcutaneously (s.c.) implanted TC-1 epithelial tumor cells that express the HPV16 E7 oncogene (Lin et al., 1996). TC-1 tumors lack spontaneous T cell infiltration; however, tumor-specific CD8+ T cell infiltration can be induced by vaccination, as we and others previously described (Ahrends et al., 2016, 2017). The DNA vaccine we employed encodes HPV E7 in a gene shuffled configuration to provide a strong MHC class I-restricted CD8+ T cell epitope and HPV-unrelated MHC class II-restricted epitopes that elicit CD4+ T cell “help.” These “helped” CD8+ T cells have optimal cytotoxic and migratory abilities that allow for effective tumor rejection. Specifically, they readily extravasate and infiltrate into the tumor due to upregulation of chemokine receptors and matrix metalloproteases (Ahrends et al., 2017). This therapeutic setting provides a window to examine the impact of ATX on anti-tumor T cell responses and tumor rejection.
Because TC-1 cells were found to lack ATX expression, we generated ATX-expressing TC-1 (TC-1ATX) cells and confirmed that they secrete active ATX (Figures S2A and S2B). Enforced ATX expression did not significantly alter the growth rate of s.c. injected TC-1 tumor cells (Figures S2C and S2D). This agrees with previous tumor implantation studies showing that ATX-LPAR signaling has little effect on primary tumor growth, but does promote metastasis to distant organs mainly through LPAR1 (David et al., 2010; Lee et al., 2015; Marshall et al., 2012).
ATX does not affect induction of systemic T cell responses
We examined how tumor cell-derived ATX may affect the induction of CD8+ and CD4+ T cell responses after vaccination. For this purpose, mice were vaccinated on days 8, 11, and 14 after implantation of wild-type (WT) or ATX-expressing TC-1 tumor cells (Figure 5A). After vaccination, T cells are primed in the vaccine-draining lymph node from where they egress as differentiated effector T cells into the blood and then infiltrate the tumor via chemotaxis (Ahrends et al., 2017). Primed HPV E7-specific CD8+ T cells were detected by flow cytometry using H-2Db/E749–57 MHC tetramers (Tet) (Figure 5B). We monitored vaccine-induced T cell responses in blood over time (Figures 5C–5E). The HPV E7-specific systemic CD8+ T cell response measured in blood was similar in TC-1WT and TC-1ATX tumor-bearing mice (Figures 5C and S3A), as was the frequency of CD8+ T cells with a CD44+ CD62L− effector phenotype (Figures 5D and S3B). Likewise, the frequency of vaccine-induced CD4+ T cells showing a CD44+ CD62L− effector phenotype increased to a similar extent in both groups of tumor-bearing mice (Figures 5E and S3C). Analysis of the spleens (at day 10 after vaccination) showed no differences in the systemic distribution of HPV E7-specific CD8+ T cells (Figure 5F), nor in their differentiation into granzyme B (GZB)- and interferon gamma (IFNγ)-expressing cytotoxic T lymphocytes (CTLs) (Figure 5G). CD4+ T cell responses in the spleen were also similar between both groups of tumor-bearing mice, as measured by the frequency of IFNγ-expressing cells among conventional (FOXP3−) CD4+ T cells (Figures S3D and S3E). Finally, ATX expression did not influence the frequency of FOXP3+ CD4+-regulatory T cells (Tregs) (Figure S3F). Thus, secreted ATX does not affect the induction of systemic CD8+ and CD4+ T cell responses upon vaccination, either in magnitude or quality.
Figure 5. Enforced ATX expression in tumor cells does not affect induction of T cell responses by vaccination.
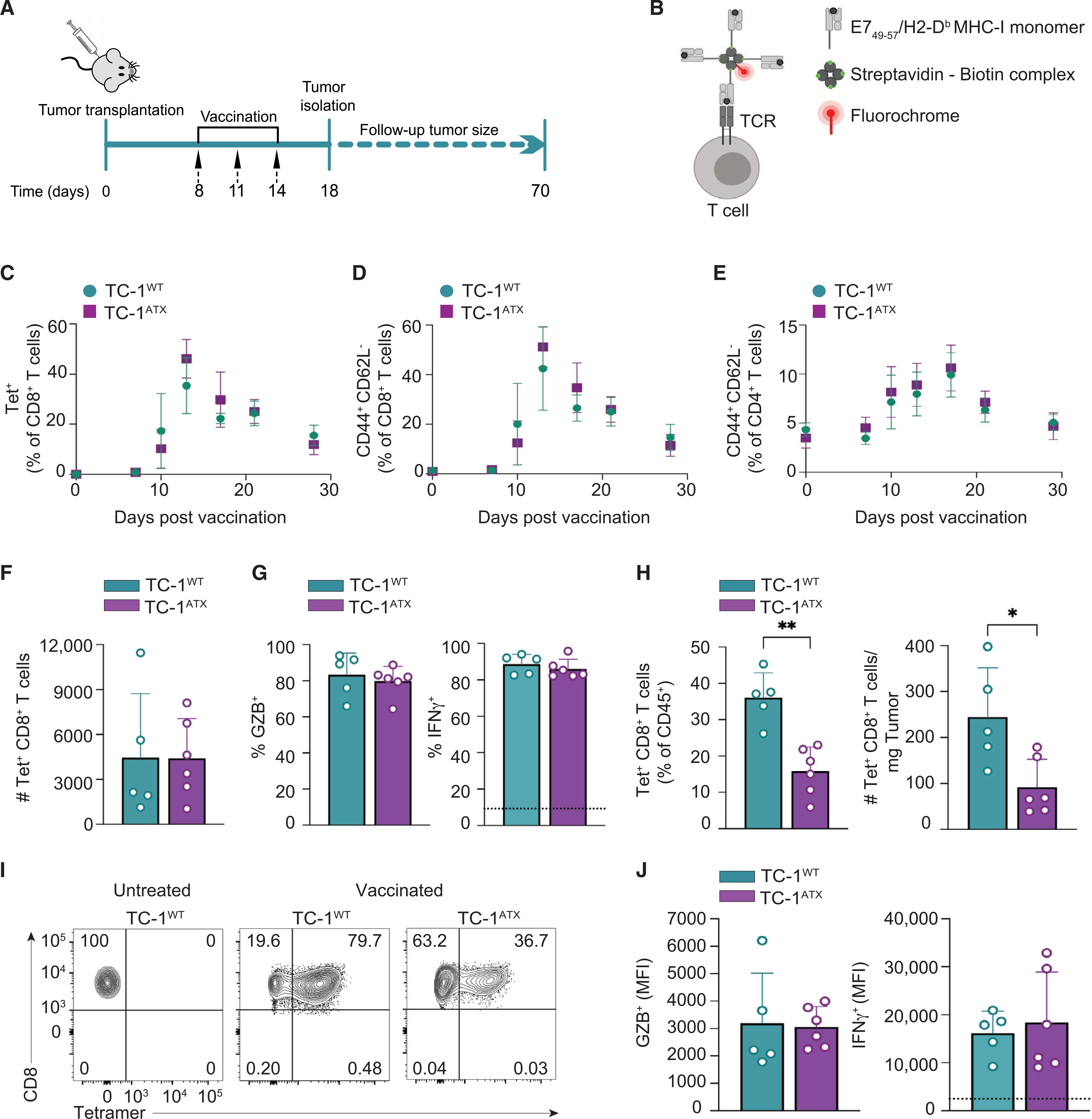
(A) Experimental set-up in the anti-cancer vaccination model. Mice were injected s.c. with wild-type (TC-1WT) or ATX-expressing (TC-1ATX) tumor cells on day 0, vaccinated on days 8, 11, and 14 and were either sacrificed on day 18, or monitored until day 70. Tumor cells were injected into one flank and the vaccine DNA was “tattooed” into the depilated skin of the opposing flank. Data are from one experiment representative of two experiments.
(B) The DNA vaccine encodes HPV-E7 protein together with tumor-unrelated helper epitopes. The CD8+ T cells that have a TCR specific for the immunodominant E749–57 peptide presented in H-2Db can be detected with MHC class I (MHC-I) tetramers. A tetramer is made by folding E749–57 peptide with MHC-I monomer, conjugating this to biotin and multimerizing it with fluorochrome-conjugated streptavidin.
(C–E) Monitoring of the T cell response to vaccination in peripheral blood by flow cytometry in TC-1WT (n = 6) and TC-1ATX (n = 5) tumor-bearing mice.
(C) Frequency of H-2Db/E749–57 tetramer positive (Tet+) cells among total CD8+ T cells. (D and E) Frequency of cells with a CD44+CD62L− effector phenotype among total CD8+ T cells (D) or total CD4+ T cells (E).
(F–J) Analysis of the CD8+ T cell response in spleen (F–J) and tumor (H and J) on day 18 in TC-1WT (n = 5) and TC-1ATX (n = 6) tumor-bearing mice.
(F) Absolute number of tetramer positive (Tet+) CD8+ T cells in spleen.
(G) Frequency of granzyme B (GZB)+ and IFNγ+ cells among Tet+ CD8+ T cells in spleen. IFNγ was measured after ex vivo PMA/ionomycin stimulation. The dotted line indicates IFNγ signal in unstimulated cells.
(H) Frequency among CD45+ hematopoietic cells (left) and absolute number (#, right) of Tet+ CD8+ T cells in TC-1WT and TC-1ATX tumors.
(I) Representative flow cytometry plots indicating the percentage of Tet+ cells among total CD8+ T cells in TC-1WT and TC-1ATX tumors after vaccination and in TC-1WT tumors of non-vaccinated (untreated) mice.
(J) Mean fluorescence intensity (MFI) of GZB+ and IFNγ+ cells within Tet+ CD8+ T cells in TC-1WT and TC-1ATX tumors. IFNγ was measured as in (G).
(C–H and J) Data are expressed as mean ± SD; *p < 0.05, **p < 0.01 (Mann-Whitney U test).
ATX repels cytotoxic CD8+ T cells from the tumor and impairs tumor control
We then investigated how ATX affects anti-tumor immunity and tumor fate after vaccination. Tumor infiltration of vaccine-induced effector CD8+ T cells was analyzed by flow cytometry and immunohistochemistry. Enforced ATX expression significantly reduced the infiltration of HPV E7-specific CD8+ T cells into the tumor, in both absolute numbers and frequency among total hematopoietic (CD45+) cells (Figures 5H and 5I). ATX did not alter the intrinsic cytotoxicity of the infiltrating CD8+ T cells, based on the similar expression levels of GZB and IFNγ in Tet+ CD8+ T cells retrieved from TC-1WT and TC-1ATX tumors (Figure 5J). Tumor-derived ATX did not oppose tumor infiltration by conventional (FOXP3−) CD4+ T cells (Figure S3G), nor did it affect their effector quality as inferred from IFNγ expression levels (Figure S3H). Numbers of infiltrating CD4+ Treg cells were also similar between TC-1WT and TC-1ATX tumors (Figure S3I). In conclusion, ATX expression by TC-1 tumors impaired infiltration of vaccine antigen-specific CD8+ T cells from the blood into the tumor, without affecting CTL quality or infiltration of conventional CD4+ T cells and Treg cells into the tumor.
We verified the flow cytometric data by examining T cell infiltration through quantitative CD8 staining in whole tumor sections by immunohistochemistry. As illustrated in Figure 6A, vaccine-induced CD8+ T cells were less capable of penetrating ATX-expressing tumors compared to parental tumors. In the parental TC-1WT tumors, CD8+ T cells were evenly dispersed throughout the tumor, according to analysis of multiple whole tumor sections. In TC-1ATX tumors, however, CD8+ T cells were detected in separate fields, leaving large parts of the tumor non-infiltrated. Quantitative analysis confirmed reduced CD8+ T cell infiltration in ATX-expressing tumors (Figure 6B). Tumor infiltration of CD4+ T cells and Tregs was not affected by ATX expression (Figures 6C and 6D), in agreement with the flow cytometric data.
Figure 6. Enforced ATX expression in tumor cells inhibits infiltration of effector CD8+ T cells and impedes vaccine-induced tumor control.
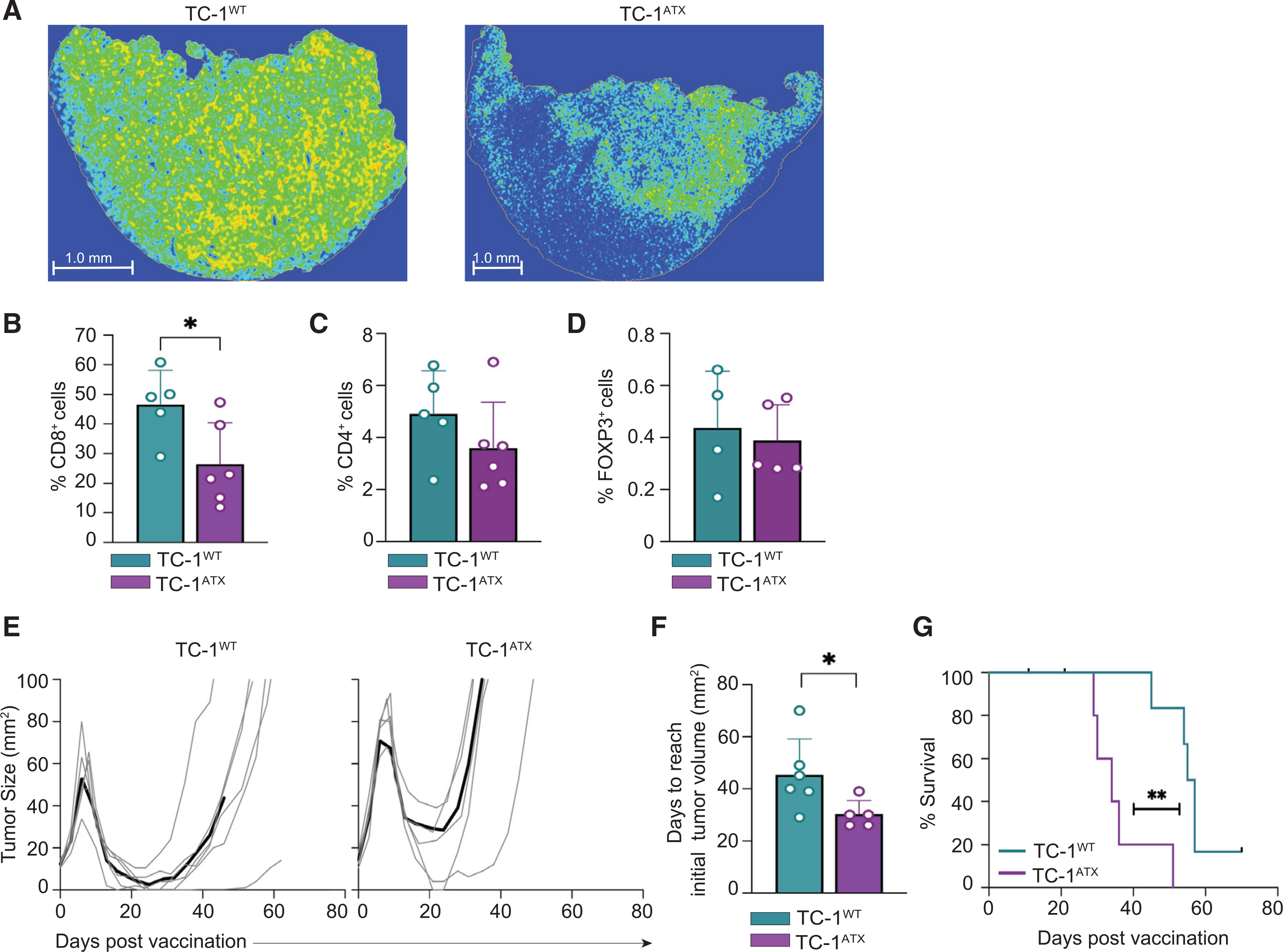
(A–F) Tumor analysis by immunohistochemistry on day 18 in the same mice as analyzed in Figure 5.
(A) Representative heatmaps of CD8+ immunostainings of tumor sections from vaccinated mice bearing TC-1WT or TC-1ATX tumors.
(B–D) Quantification in percentages of CD8+ (B, representative for the data shown in A), CD4+ (C), and FOXP3+ (D) cells out of all nucleated cells as assessed by immunostaining of tumor sections from vaccinated mice bearing TC-1WT or TC-1ATX tumors. Data are depicted as mean + SD, *p < 0.05 (Mann-Whitney U test).
(E–G) TC-1WT (n = 6) and TC-1ATX (n = 5) tumor-bearing mice received vaccination as outlined in Figure 5 and tumor growth was monitored over time up to day 70.
(E) Individual growth curves of TC-1WT and TC-1ATX tumors in vaccinated mice. Black lines represent group average.
(F) Tumor growth delay following vaccination, expressed as number of days required to reach a tumor size corresponding to that at day 7 (see E). Data are depicted as mean + SD, *p < 0.05 (Mann-Whitney U test).
(G) Overall survival curves of tumor-bearing mice. **p < 0.01 (Mantel-Cox analysis).
Data in this figure are from one experiment representative of two independent experiments.
We determined the impact of ATX expression on vaccine-induced TC-1 tumor control, following the experimental protocol of Figure 5A. Vaccination of mice bearing either TC-1WT or TC-1ATX tumors initially resulted in tumor regression (Figure 6E). Importantly, however, vaccine-induced growth delay of ATX-expressing tumors was significantly reduced in comparison to TC-1WT tumors, as was the overall survival rate of mice bearing TC-1ATX tumors (Figures 6F and 6G). Collectively, these findings demonstrate that ATX released by tumor cells impairs cytotoxic CD8+ T cell infiltration and dispersion throughout the tumor and thereby impairs tumor control in a therapeutic setting.
Intratumoral ENPP2 expression in melanoma negatively correlates with CD8+ T cell infiltration
Finally, we sought clinical evidence for intratumoral ATX functioning as a CD8+ T cell repellent in melanoma. Of note, abundant ENPP2 expression is detected not only in melanoma but in virtually all solid tumors (https://www.cbioportal.org), showing remarkably little correlation with ENNP2 expression in the corresponding cancer cell lines (Figures S1A and S1B). This supports the view that a substantial part of the tumor ENPP2 transcripts is derived from non-malignant stromal cells, notably cancer-associated fibroblasts (CAFs) and adipocytes known for their high ATX expression levels, depending on the cancer type (Auciello et al., 2019; Brindley et al., 2020).
We analyzed ENPP2 expression patterns and CD8+ T cell infiltration using single-cell RNA-sequencing (scRNA-seq) results from 32 melanoma tumors (prior to immunotherapy) in which diverse cell subsets can be distinguished (Jerby-Arnon et al., 2018). ENPP2 expression in individual cells (n = 7,186) and its association with CD8+ T cell infiltration was examined in all subsets, namely malignant cells, CD8+ and CD4+ T cells, B cells, natural killer (NK) cells, CAFs, tumor-associated macrophages, and endothelial cells. Figure 7A shows the melanoma samples grouped by individual cell types. Whereas lymphocytes do not express ATX, significant ENPP2 expression was detected not only in malignant cells and CAFs but also in macrophages and endothelial cells (Figure 7B). Tumors with the highest intratumoral ENPP2 expression—in both cancer and stromal cells—contained significantly fewer CD8+ T cells, whereas low ENPP2 expression correlated with enhanced CD8+ T cell infiltration, as quantified by Pearson’s correlation analysis (r = 0.4; p = 0.01) (Figure 7C).
Figure 7. Single-cell analysis of ENNP2 expression in melanoma tumors and its inverse correlation with CD8+ T cell accumulation.
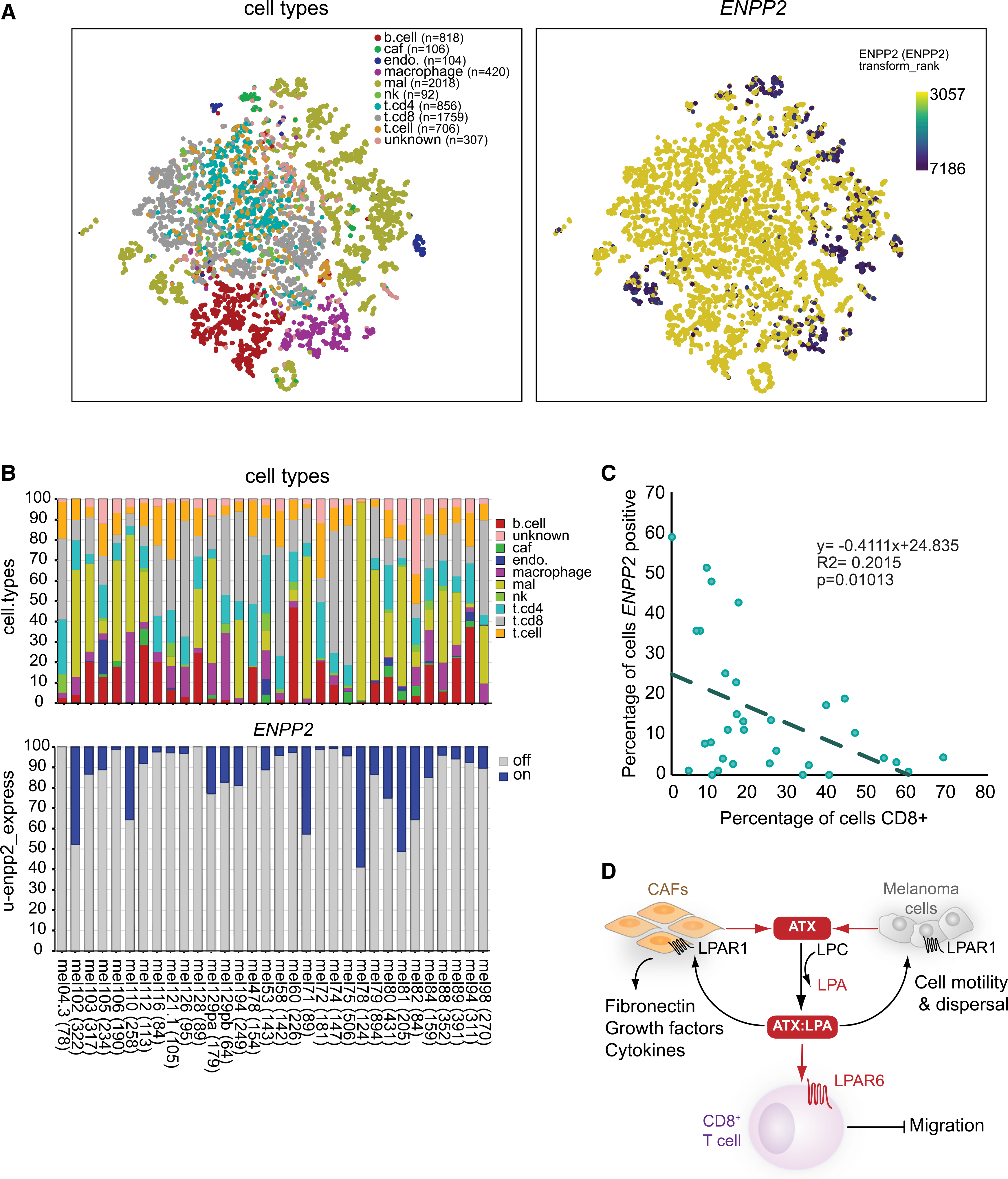
(A) t-distributed stochastic neighbor embedding (tSNE) embedding of 7,186 single cells (complexity = 5) from 32 melanoma patients as described (Jerby-Arnon et al., 2018). Data were used to project patients, inferred cell types, and log2 ENPP2 expression values, respectively, as described in STAR Methods. Right panel shows ENPP2 expression (blue/purple dots high expression) as overlay on single cells presented in the left panel. Intratumoral ENPP2 expression is detected in malignant cells (mal), cancer-associated fibroblasts (caf), macrophages, and endothelial cells (endo), but not in lymphocytes (T, B, and NK cells).
(B) Stacked bar graph showing the percentages of inferred cell type per individual patient sample (top), and the percentage of ENPP2-expressing cell types (bottom).
(C) Inverse correlation between intratumoral ENPP2 expression and CD8+ T cell accumulation. Pearson correlation between the percentage of inferred ENPP2-expressing cells and CD8+-positive cells (R = 0.4; p = 0.01).
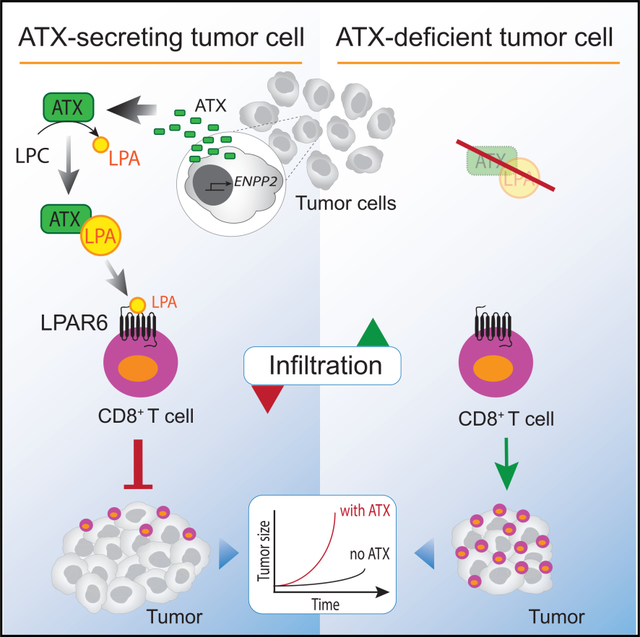
(D) Model of the melanoma immune microenvironment. In this model, ATX is secreted by melanoma cells and diverse stromal cells, particularly fibroblasts (CAFs), to convert extracellular LPC into LPA. ATX functions as an LPA-producing chaperone (ATX:LPA) that carries LPA to its GPCRs and exerts dual actions: it suppresses T cell infiltration through G12/13-coupled LPAR6, while it promotes melanoma cell dispersal and activates CAFs via LPAR1 (mainly via GI). Activated CAFs release growth factors and produce extracellular matrix. Random T cell motility mediated by LPAR2 is not illustrated (see Figure 4D). See text for further details.
Elevated ENPP2 expression in melanoma samples was also associated with reduced CD4+ T cell infiltration, but not with macrophage accumulation (Figures S4A and S4B). Although ATX-mediated repulsion of CD4+ T cells was not observed in the above vaccination model, differences in the functional state of the respective CD4+ T cell populations or species-specific receptor expressions might account for this discrepancy. Despite some caveats concerning the interpretation of scRNA-seq results, as will be discussed below, the single-cell transcriptomics analysis is consistent with our in vivo findings, namely that intratumoral ATX repels CD8+ T cells from the tumor. Collectively, our findings support a model of intratumoral ATX/LPAR signaling (Figure 7D) to be discussed below.
DISCUSSION
The signaling mechanisms that contribute to the exclusion of CD8+ T cells from tumors remain poorly understood, which hampers progress in improving immunotherapy efficacy (Anandappa et al., 2020; Joyce and Fearon, 2015; van der Woude et al., 2017). Tumor-intrinsic mechanisms underlying T cell exclusion involve, for example, transcriptional chemokine silencing (Spranger et al., 2015; Spranger and Gajewski, 2018) and production of immunosuppressive cytokines such as transforming growth factor β (TGF-β) (Batlle and Massagué, 2019; Mariathasan et al., 2018). However, secreted factors and T cell GPCRs that counteract T cell infiltration remain to be identified.
Here, we demonstrate that LPA-producing ATX secreted by tumor cells is a major repellent for human TILs and healthy CD8+ T cells under ex vivo conditions, with a dominant anti-migratory role for Gα12/13-coupled LPAR6. Moreover, we show that secreted ATX repels cytotoxic CD8+ T cells from s.c. engrafted tumors to impede anti-tumor immunity and tumor regression in a therapeutic setting.
ATX/LPA is widely known for its chemotactic activities toward both normal and tumor cells, acting mainly via LPAR1, and to enhance the random motility of T cells via LPAR2 (Kanda et al., 2008; Knowlden et al., 2014). Unexpectedly, we initially observed T cell chemo-repulsive effects of exogenous ATX/LPA and melanoma cell-secreted ATX of TILs and peripheral blood CD8+ T cells ex vivo, with ATX/LPA antagonizing the migration toward chemokine CXCL10 (Figures 1 and 2). Whereas CD8+ T cells express multiple LPA receptors, the unique LPAR expression pattern in TILs and the use of a novel LPAR6 antagonist allowed us to define LPAR6 as the predominant T cell anti-migratory receptor (Figure 4). In this respect, it should be emphasized that biological outcome is determined by the balance in expression of GPCRs that signal mainly via Gi (i.e., chemokine and chemotactic EDG receptors LPAR1–LPAR3) versus those that couple predominantly to the Gα12/13-RhoA pathway, notably anti-chemotactic non-EDG receptors LPAR4–LPAR6, as exemplified by the present findings.
Contrary to prevailing notions, secreted ATX failed to raise extracellular LPA levels as its lysoPLD activity was outperformed by cell-associated LPA-degrading activity (Figure 3). By binding LPA in its “exit tunnel,” presumably at a 1:1 ratio, ATX protects bioactive LPA from degradation (Keune et al., 2016; Moolenaar and Perrakis, 2011; Nishimasu et al., 2011; Salgado-Polo et al., 2018) and, as such, functions as an LPA-producing “chaperone.” Based on its calculated lifetime (Saunders et al., 2011), the ATX:LPA complex can diffuse over a relatively long distance in the extracellular milieu (Keune et al., 2016) and hence may shape an ATX/LPA gradient and its paracrine signaling range. Precisely how ATX releases LPA to its cognate receptors upon interaction with the cell surface awaits further functional and structural studies.
LPAR6 (P2RY5) now joins the few select GPCRs that counteract T cell chemotaxis through the Gα12/13-RhoA pathway. Among them, EDG-family sphingosine-1-phosphate receptor S1PR2 is arguably the best characterized member (Baeyens et al., 2015; Laidlaw et al., 2019), but a role for S1PR2 in immuno-oncology has not been documented to date. LPAR6 (P2RY5) is of special interest as it displays its highest expression in immune cells and is strongly induced upon activation of chicken T cells through as-yet-unknown mechanisms (Kaplan et al., 1993; Webb et al., 1996). Furthermore, LPAR6 prefers 2-acyl- rather than 1-acyl-LPA species as ligand (Yung et al., 2014), which may explain the relatively high IC50 value for 1-oleyl-LPA observed in T cell migration assays (Figure 1B). Although its non-EDG relatives LPAR4 and LPAR5 were not detected in ex vivo expanded TILs (Figure 4A), the latter receptor is nonetheless of immuno-oncological importance since its genetic deletion in mice enhances T cell receptor activity and anti-tumor responses (Mathew et al., 2019). To what extent LPAR6 and LPAR5 may act redundantly or synergistically in T cell signal transmission remains to be investigated.
Building on our in vitro findings, we pursued the impact of tumor-intrinsic ATX on T cell responses in the mouse TC-1 tumor model that is often used in anti-cancer vaccination studies (Ahrends et al., 2017; Borst et al., 2018). For this purpose, we stably expressed ATX in TC-1 cells that lack endogenous Enpp2 expression and confirmed their LPA-producing activity. Vaccination induces the simultaneous activation of CD4+ and CD8+ T cells to optimize the cytotoxic T cell response in magnitude and quality (Ahrends et al., 2017). “Helped” CD8+ T cells acquire chemokine receptors to increase their migration capacity and enhanced metalloprotease activity that enables them to invade tumor tissue to promote tumor regression (Ahrends et al., 2017; Borst et al., 2018). We established that tumor-intrinsic ATX has no effect on vaccine-induced CD4+ and CD8+ T cell responses (Figure 5). The cytotoxic CD8+ T cells thus displayed optimal effector capacity independent of ATX activity. Importantly, despite the robust anti-tumor immune response, tumor-intrinsic ATX was capable of significantly impeding tumor infiltration of cytotoxic CD8+ T cells and suppressing tumor rejection (Figure 6). These findings highlight a key role for LPA-producing ATX in suppressing anti-tumor immunity in a therapeutic setting. By inference, LPAR6 most likely plays a dominant role in mediating ATX-induced T cell repulsion in vivo, possibly in concert with LPAR5, but this needs further investigation. Further development of specific LPAR6 antagonists would enable a robust pharmacological characterization and help dissect the ATX-LPAR immune signaling network in further detail.
In a clinical setting, single-cell analysis of melanoma tumors (Jerby-Arnon et al., 2018) showed significant ENPP2 expression in malignant cells, CAFs, tumor-associated macrophages, and endothelial cells, which further accentuates the complexity of ATX/LPA signaling in the tumor microenvironment (Figures 7A–7C). Consistent with our in vivo findings, intratumoral ENPP2 expression positively correlated with CD8+ T cell exclusion. ENPP2 expression was also associated reduced CD4+ T cell infiltration in these tumors (Figure S4). These findings should be validated in future immuno-histochemical analyses of select patient samples.
Taken together with previous evidence, our findings support a simplified model of the tumor (melanoma) microenvironment illustrated in Figure 7D. In this model, LPA-producing ATX is secreted by both tumor and stromal cells and—complexed with LPA—counteracts tumor infiltration of CD8+ T cells mainly via G12/13-coupled LPAR6, while it activates tumor cells and pro-tumorigenic fibroblasts (CAFs) in autocrine/paracrine loops via LPAR1, which signals predominantly via Gi. ATX/LPA-stimulated tumor cells acquire a pro-metastatic phenotype, whereas activated fibroblasts drive immune escape by generating a physical barrier to TILs and by secreting immunosuppressive molecules (De Jaeghere et al., 2019). Because ATX is abundantly expressed in most solid tumors (Figure S1B), this model obviously extends beyond melanoma to many cancer types. Because the tumor microenvironment is heterogeneous and cancer type-specific, ATX/LPA signaling outcome will critically depend on the composition and LPAR expression repertoire of the immune cell infiltrate, and likely also on the spatial arrangement of ATX-secreting stromal cells within the tumor.
In conclusion, by suppressing anti-tumor immunity while promoting metastasis via different LPA receptors, the ATX-LPAR signaling axis creates a T cell-excluding, pro-tumorigenic microenvironment that is amenable to therapeutic intervention. Our findings pave the way for addressing outstanding questions on the ATX-LPAR axis in other immunotherapeutic settings, such as genetically engineered melanoma models and/or patient-derived xenografts (PDX) engrafted in humanized mouse models (Patton et al., 2021; Rosato et al., 2018). Such clinically relevant models should provide further insight into the dual pro-tumor actions of ATX; furthermore, they will offer an opportunity to evaluate the anti-tumor benefits of pharmacological ATX inhibition, for example in combination with immune checkpoint inhibitors.
Limitations of study
Our study has several limitations. Although LPAR6 acts as a migration-inhibitory receptor for peripheral blood CD8+ T cells, and ex vivo expanded TILs, its role in ATX-mediated T cell repulsion in tumor-bearing mice, as reported here, remains be established by using Lpar6(−/−) mice. Furthermore, our correlative single-cell analysis of ATX expression in melanoma tumors (Figure 7) should be viewed with caution because scRNA-seq studies do not detect all transcripts in every single cell, and intratumoral ENPP2 expression is not necessarily predictive of secreted ATX activity in the tumor microenvironment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wouter H. Moolenaar (w.moolenaar@nki.nl).
Materials availability
Any reagents generated for this study will be made available upon request to the lead contact.
Data and code availability
Raw western blot images have been deposited at Mendeley and are publicly available as of the date of publication.
No new code was written for this study. Analysis of publicly available single-cell RNA-seq data is described in the method details.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Six to eight-week old female C57BL/6JRj (B6) mice were obtained from Janvier Laboratories (Le Genest Saint Isle, France) and maintained in individually ventilated cages (Innovive, San Diego, CA) under specific pathogen-free conditions. All mouse experiments were performed in accordance with institutional and national guidelines and were approved by the Committee for Animal Experimentation at the NKI.
Human cell lines
MDA-MB-435, A375 melanoma cells and MDA-MB-231 breast carcinoma cell line were purchased from ATCC and maintained in low passage numbers according to ATCC guidelines. All cell lines were maintained in master cell banks and undergo routine mycoplasma testing. Any cells displaying abnormal morphological changes or doubling time are discarded and replaced with a new vial. MDA-MB-435, MDA-MB-231 and A375M cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C under 5% CO2. Patient-derived TILs cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% human serum at 37°C under 5% CO2. Human CD8+ T cells were isolated from buffy coats, activated with anti-CD3 and CD28 mAbs that were plate-bound and expanded in RPMI 1640 medium supplemented with 10% human serum and 100 IU/mL IL-2 and 5 ng/ml IL-15 at 37°C under 5% CO2.
Murine TC-1 cells
TC-1 tumor cells are lung epithelial cells engineered to express HPV16 E6 and E7 proteins (Lin et al., 1996). Cells were obtained from the Leiden University Medical Center in 2015, and the authors did not perform further authentication. TC-1 cells stably overexpressing ATX were generated using retroviral vector pBABE-ATX-Myc by retroviral transduction and subsequent selection with puromycin. Phoenix-AMPHO cells were transiently transfected with retroviral vector using calcium phosphate, and virus particles were collected 48 h, thereafter TC-1ATX cells were selected in medium containing 2 mg/ml puromycin ATX overexpression was validated by western blotting (Figure S3A). TC-1 cells were cultured in medium supplemented with 10% fetal calf serum (FCS), 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine, 10 mM HEPES and antibiotics at 37°C, 5% CO2. TC-1 cell stock was tested negative for Mycoplasma by PCR, and cells thawed from this stock were used within 3 passages for in vitro and in vivo experiments.
METHOD DETAILS
Expression vector
Human cDNA ATX was subcloned into a pcDNA3 (-myc) plasmid by amplification with oligos and digestion with BamHI/NotI. The viral plasmid pBABE-ATX-Myc was constructed by subcloning the ATX from pcDNA3-ATX-myc into a pBABE plasmid, pcDNA3-ATX-myc was cut using EcoRI and NotI, and ligated into the pBABE vector, digested with BamHI and SalI.
Isolation and expansion of melanoma-derived TILs
TIL isolation and expansion was started by generation of a single cell suspension by enzymatic digestion of the resected metastatic tumor material obtained by surgery, using collagenase type IV (Sigma Aldrich) and Pulmozyme (Roche). Resulting cell suspensions were cultured in the presence of 6000 IU/ml IL-2 (Proleukin, Novartis) for two to four weeks. During the subsequent Rapid Expansion Protocol of two weeks, T cells were cultured in 50% RPMI/50% AIM-V medium in the presence of 3,000 IU/ml IL-2, 30 ng/ml anti-CD3 antibody (OKT-3, Miltenyi) and irradiated autologous PBMCs (feeder cells in 200-fold excess over TILs).
Isolation of peripheral CD8+ T cells
Human peripheral blood mononuclear cells (PBMCs) were isolated from fresh buffy coats using Ficoll-Paque Plus (GE Healthcare) gradient centrifugation. Total CD8+ T cells were isolated using magnetic sorting with CD8 microbeads (Miltenyi Biotec). Blood samples were obtained from anonymized healthy male donors with written informed consent in accordance to guidelines established by the Sanquin Medical Ethical Committee.
Conditioned media
Conditioned media were collected from MDA-MB435 and A375M cells. Sub-confluent 10-cm dishes of melanoma cells were washed with PBS and incubated in DMEM containing 0.5% FBS. Conditioned medium was harvested after 24 and 48 h, and centrifuged for 30 min at 4000 rpm in a tabletop centrifuge to remove cell debris.
Transwell migration assays
T cell migration was measured using 48-well chemotaxis chambers (Neuro Probe, Inc.) equipped with 5 μm-pore polycarbonate membranes which were coated with fibronectin (1 μg/ml). Cells (1 × 106/ml) were added to the upper chamber. Migration was assessed after 2 h for TILs and CD8+ T cells at 37°C in humidified air containing 5% CO2. Migrated cells were fixed in Diff-Quik Fix and stained using Diff-Quik II. Migration was quantified by color intensity measurements using ImageJ software.
ATX lysoPLD activity
ATX enzymatic activity in conditioned media was measured by steady-state choline release from exogenously added LPC using a coupled reaction, as detailed elsewhere (Salgado-Polo et al., 2018). Briefly, media were centrifuged for 45 min at 4,500 rpm, upon which 75 μL of the supernatants were plated on 96-well plates together with 600 μM LPC(18:1), 1 U ml−1 choline oxidase, 2 U ml−1 horseradish peroxidase (HRP) and 2 mM homovanillic acid (HVA), reaching a final volume of 100 μl. ATX activity was measured by HVA fluorescence at λex/λem = 320/460 nm every 30 s for at least 160 min at 37°C with a Pherastar plate reader (BMG Labtech). Since ATX activity in vitro presents a ~15-min lag phase, the subsequent linear slope (60–160 min) was used to perform all analyses. Triplicate measures were statistically analyzed by an unpaired t test.
Western blotting
Cells were washed in ice-cold PBS (phosphate-buffered saline containing 2 mM Ca2+ and Mg2+), lysed in RIPA buffer with protease inhibitors and spun down. Equal amounts of proteins were determined by BCA protein assay kit (Pierce), separated by SDS-PAGE using pre-cast gradient gels (4%–12% Nu-Page Bis-Tris, Invitrogen) and transferred to nitrocellulose membranes. The membrane was blocked for 1 h at room-temperature in 5% skimmed milk in TBST. Incubation with antibodies was done overnight at 4°C, followed by 1 h incubation with horseradish peroxidase-conjugated secondary antibodies (DAKO, Glostrup, Denmark). Proteins were visualized using ECL western blot reagent (GE Healthcare, Chalfont St. Giles, UK).
qPCR analysis
Expression levels of LPA receptors and ATX/ENPP2 were quantified by RT-qPCR. Total RNA was isolated using GeneJET purification kit (Fermentas). cDNA was synthesized by reverse transcription from 2 mg RNA with oligodT 15 primers and SSII RT enzyme (Invitrogen). qPCR was performed on a 7500 Fast System (Applied Biosystems) as follows: 95°C for 2 min followed by 40 cycles at 95°C for 15 s followed by 60°C for 1 min. 200 nM forward and reverse primers,16 mL SYBR Green Supermix (Applied Biosystems) and diluted cDNA were used in the final reaction mixture. Cyclophilin was used as reference gene and milliQ was used as negative control. Normalized expression was calculated following the equation NE = 2(Ct target-Ct reference). Primers used: LPA1 forward AATCGGGATACCATGATGAGT, reverse CCAGGAGTCCAGCAGATGATAAA; LPA2 forward CGCTCAGCCTGGTCAAAGACT, reverse TTGCAGGACTCACAGCCTAAAC; LPA3 forward AGGACACCCATGAAGCTAATGAA, reverse GCCGTCGAGGAGCAGAAC; LPA4 forward CCTAGTCCTCAGTGGCGGTATT, reverse CCTTCAAAGCAGGTGGTGGTT; LPA5 forward CCAGCGACCTGCTCTTCAC, reverse CCAGTGGTGCAGTGCGTAGT; LPA6 forward AAACTGGTCTGTCAGGAGAAGT, reverse CAGGCAGCAGATTCATTGTCA; ENPP2 forward ATTACAGCCACCAAGCAAGG, reverse TCCCTCAGAGGATTTGTCAT; Cyclophilin forward CATCTGCACTGCCAAGACTGA and reverse TTGCCAAACACCACATGCTT.
ATX knockdown melanoma cells
To generate ATX knockdown melanoma cells, we used five human ENPP2 shRNAs in the lentiviral vector pLKO1: (TRC human shRNA library; TRCN0000048993, TRCN0000048995- TRCN0000048997 and TRCN0000174091). To generate particles for stable infections, HEK293T cells were transfected with single shRNA hairpins using the calcium phosphate protocol; the virus particles were collected at 48 h after transfection. ENPP2 stable knockdown cells were selected and maintained in medium containing 2 μg/ml puromycin.
Lipid extraction and LC-MS/MS measurements of LPA
Extraction of lipids from cell-free conditioned media was done using acidified organic solvents and measurement of seventeen LPA species was accomplished using LC- MS/MS. Quantitation of LPA species was achieved using LPA(17:0) as an internal standard. Experimental details can be found elsewhere (Kraemer et al., 2019).
In vivo tumor growth
On day 0, C57BL/6 mice were anesthetized with isofluorane and injected s.c. with 1 × 105 TC-1 tumor cells in HBSS. Tumor size was measured by calipers in two dimensions and calculated as: area (mm2) = width x length. Mice were monitored twice per week. Mice were sacrificed when the tumor diameter reached 15 mm or when the tumor size reached 100 mm2. In the survival curves, censored events indicate mice were sacrificed before treated tumors reached 100 mm2.
Anti-cancer vaccination
The HELP-E7SH plasmid DNA vaccine was described previously and validated in detail (Ahrends et al., 2017). Intra-epidermal DNA “tattoo” vaccination was performed as described in the same papers. Briefly, the hair on a hind leg was removed using depilating cream (Veet, Reckitt Benckiser) prior to vaccination, mice were anesthetized and 15 μL of 2 mg/ml plasmid DNA solution in 10 mM Tris, 1 m M EDTA, pH 8.0 was applied to the hairless skin with a Permanent Make Up tattoo device (MT Derm GmbH, Berlin, Germany), using a sterile disposable 9-needle bar with a needle depth of 1 mm and oscillating at a frequency of 100 Hz for 45 s.
Murine tissue preparation and flow cytometry
At the indicated days, blood was sampled from live mice or mice were sacrificed and lymphoid tissues and tumors were harvested. Peripheral blood cells were collected by tail bleeding in Microvette CB300 LH tubes (Sarstedt). Red blood cells were lysed in 0.14 M NH4Cl and 0.017 M Tris-HCl (pH 7.2) for 1 min at room temperature and cell suspensions were washed and stained with relevant mAbs, as indicated below. Tumor tissue was mechanically disaggregated using a McIlwain tissue chopper (Mickle Laboratory Engineering), and a single-cell suspension was prepared by digesting the tissue in collagenase type A (Roche) and 25 μg/ml DNase (Sigma) in serum-free DMEM medium for 45 min at 37°C. Enzyme activity was neutralized by addition of DMEM containing 8% FCS, and the tissue was dispersed by passing through a 70-μm cell strainer. Lymphoid tissue was dispersed into single cells by passing it through a 70-μm cell strainer. Single cells were first stained with APC-conjugated E759–57/H-2Db tetramers (Peptide and tetramer facility, Immunology department, Leiden University Medical Center) for 15 min at 4°C in the dark. After tetramer staining, tumor cells were incubated with 2% normal mouse serum with 10 μg/ml DNase for 15 min on ice. For surface staining, cells were incubated for 30 min on ice in the dark with fluorochrome-conjugated antibodies and 0.5 μL anti-APC mAb (clone APC003, BioLegend) per sample in PBS, 0.5% BSA, 0.01% sodium azide to increase intensity of tetramer staining. LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit (1:1000, Invitrogen) was added to exclude dead cells. Intracellular staining of cytokines and transcription factors was performed after cell fixation and permeabilization using the FOXP3 Transcription Factor Staining Buffer Set according to the manufacturer’s protocol (Thermo Fischer Scientific). The following fluorochrome-conjugated mAbs were used for flow cytometry and obtained from BD PharMingen (Breda, the Netherlands) unless otherwise specified: CD45.2-BUV395 (1:200; clone 30-F11), TCRβ-PECy7 (1:100; clone H57–597), CD3-PECy7 (1:100, clone 145–2C11, eBiosciences), CD8-BUV805 (1:300, clone 53–6.7), CD4-BV711 (1:200, clone GK1.5), FOXP3-PECy5.5 (1:100, clone FJK-16 s, eBiosciences), CD44-BV785 (1:200, clone IM7, BioLegend), CD62L-FITC (1:100, clone MEL-14, eBioscience), IFNγ-eF450 (1:200, clone XMG-1.2, eBioscience), Granzyme B-PE (1:200, clone GB11, Sanquin Amsterdam). To detect cytokine production, whole cell preparations from tumor and lymphoid tissue were plated in 100 μl IMDM/8% FCS in a 96-well U-bottom plate. Cells were treated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma Aldrich, Zwijndrecht, the Netherlands) and 1 μM ionomycin (Sigma Aldrich) dissolved in DMSO and diluted in culture medium. Control (unstimulated) cells were treated with an equal volume of DMSO diluted in culture medium. GolgiPlug (1 μg/ml; BD Biosciences) was added to all wells before incubating the cells for 3 h at 37°C, 5% CO2. To determine absolute cell numbers AccuCount Blank Particles (Spherotech) were added to the samples, prior to analysis. For all experiments, cells were analyzed using a BD Symphony A5 flow cytometer with Diva software, and the generated data were analyzed using FlowJo software.
Immuno-histochemical analysis
Harvested tumors were fixed for 24 h in aqueous solution with 50% ethanol, 5% acetic acid and 3.7% formalin, embedded in paraffin, and then sectioned randomly at 5 mm. For immunostaining, sections were rehydrated and incubated with primary antibodies to CD8 (eBioscience; clone 4SM15), CD4 (eBioscience; clone 4SM95) and FOXP3 (eBioscience; clone FJK-16 s). Endogenous peroxidases were blocked with 3% H2O2, and the sections were then incubated with biotin-conjugated secondary antibodies, followed by incubation with HRP-conjugated streptavidin-biotin (DAKO). The substrate was developed using diaminobenzidine (DAB) (DAKO). We included negative controls to determine background staining, which was negligible. For the assessment of immune-cell infiltration in the tumor cross-sections, the immuno-stained slides were scanned and digitally processed using the Pannoramic 1000 scanner (3DHISTECH, Hungary) equipped with a 20x objective. Digital whole slide images of CD8-, CD4- and FOXP3-stained serial tissue sections were registered with the HE sections in HALO image analysis software version (3.2.1851.229) (Indica Labs, Corrales, NM). The tumor area within the stained sections were manually annotated and all nuclei within the tumor area (hematoxylin and/or DAB staining) were automatically segmented with the use of the commercially available Indica Labs – Multiplex IHC v2.3.4 algorithm module. Optimized parameters for the detection of nuclei signal included nuclear weight (1 for hematoxylin and 0.066 for DAB staining), nuclear contrast threshold (0.44), minimum nuclear optical density (0.095), nuclear size (11.3 – 220.7), minimal nuclear roundness (0.033) and nuclear segmentation aggressiveness (0.536). The optimized module parameters for the cytoplasmic and membrane detection included DAB-markup color (198, 163, 122) with the DAB-nucleus positive threshold (0.1105, 2.5, 2.5). The algorithm module parameters were kept constant for the analysis of all the sections across the different lymphocyte stainings. Next, with the utilization of the algorithm the total cell number within the tumor area (per section per staining) was automatically determined along with the equivalent number of each lymphocyte classification as DAB-positive cells. For the quantification analysis, the fraction (percentage) of DAB-positive cells (determined either via nucleus or cytoplasmic/membrane staining) over the total number of cells within the tumor area was used.
Single-cell RNA-seq analysis of human melanoma tumors
Single-cell data from 32 melanoma tumors (Jerby-Arnon et al., 2018) was downloaded from NCBI GEO (gse115978) and exported to the R2 platform (https://hgserver1.amc.nl:443/, Mixed Melanoma SC - Regev - 7186 - tpm - gse115978). tSNE clustering was applied to 7186 cells. A complexity of 5 was chosen to represent the cohort. Inferred cell type information was extracted from the GEO dataset. Expression of ENPP2 and other annotations were projected onto the tSNE embedding. In every patient sample, the percentage of ENPP2-expressing cells was correlated to the percentage of cells inferred to be CD8+-positive. All analyses of the single-cell data were performed in the R2 genomics analysis and visualization platform.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data from in vitro migration assays were analyzed with a Mix Model Analysis of Variance, where treatments were considered as fix factor and repetition of the experiments were included as random factor. Means of each group were compared by Fisher’s LSD post hoc test using IBM-SPSS v. 25. Data from mouse studies were analyzed using GraphPad Prism version 9 (GraphPad Software, La Jolla, CA). Differences between various treatment groups were analyzed using the Mann-Whitney U Test. Differences in survival curves were analyzed using the Log Rank (Mantel-Cox) test. Differences with P values < 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Purified anti-Allophycocyanin (anti-APC;clone APC003) | BioLegend | Cat# 408002; RRID:AB_345358 |
| Anti-mouse CD45 BUV395 (clone 30-F11) | BD Biosciences | Cat# 564279; RRID:AB_2651134 |
| Anti-mouse TCRb PE/Cy7 (clone H57–597) | BioLegend | Cat# 109222; RRID:AB_893625 |
| Anti-mouse CD3 PE/Cy7 (clone 145–2C11) | Thermo Fisher | Cat# 25–0031-81; RRID:AB_469571 |
| Anti-mouse CD8a BUV805 (clone 53–6.7) | BD Biosciences | Cat# 612898; RRID:AB_2870186 |
| Anti-mouse CD8a (clone 4SM15) | Thermo Fisher | Cat# 14–0808-82, RRID:AB_2572861 |
| Anti-mouse CD4 BV711 (clone GK1.5) | BD Biosciences | Cat# 563050; RRID:AB_2737973 |
| Anti-mouse CD4 (clone 4SM95) | Thermo Fisher | Cat# 14–9766-82, RRID:AB_2573008 |
| Anti-mouse/human CD44 BV785 (clone IM7) | BioLegend | Cat# 103059; RRID:AB_2571953 |
| Anti-mouse CD62L FITC (clone MEL14) | Thermo Fisher | Cat# 11–0621-82; RRID:AB_465109 |
| Anti-mouse IFNg eF450 (clone XMG1.2) | Thermo Fisher | Cat# 48–7311-82, RRID:AB_1834366 |
| Anti-human GZMB PE (clone CLB-GB11) | Sanquin, Amsterdam | Cat# M2289; RRID:AB_2114694 |
| Anti-mouse FOXP3 PE/Cyanine5.5 (clone FJK-16 s) | Thermo Fisher | Cat# 35–5773-82; RRID:AB_11218094 |
| Anti-mouse FOXP3 (clone FJK-16 s) | Thermo Fisher | Cat# 14–5773-82, RRID:AB_467576 |
| Anti-human ATX (4F1) | Gift from Junken Aoki | N/A |
|
| ||
| Biological samples | ||
|
| ||
| Human tumor-infiltrating lymphocytes | Melanoma patients; this paper | N/A |
| Human CD8+ T cells | Peripheral blood; this paper | N/A |
| Conditioned media from tumor cells | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| LPA(18:1) | Avanti Polar Lipids | Cat# 857130 |
| LPC(18:1) | Avanti Polar Lipids | Cat# 791643 |
| Near-IR Dead cell stain kit | Life Technologies | Cat# L10119 |
| H-2Db/E749–57 tetramers | This paper | Homemade |
| Collagenase Type A | Roche | Cat# 11088793001 |
| DNase I | Roche | Cat# 10104159001 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# P8139 |
| Ionomycin | Sigma-Aldrich | Cat# 19657 |
| PF-8380 | Sigma-Aldrich | Cat# 1144035–53–9 |
| IOA-289 | iOnctura | https://doi.org/10.1016/j.bmcl.2016.10.036 |
| XAA | This paper | N/A |
| Homovanillic acid | Sigma-Aldrich | Cat# H1252 |
| Horseradish peroxidase (type VI) | Sigma-Aldrich | Cat# P8375 |
| Choline oxidase from Alcaligenes sp. | Sigma-Aldrich | Cat# C5896 |
|
| ||
| Critical commercial assays | ||
|
| ||
| eBioscience Foxp3/transcription factor staining buffer set | Thermo Fisher | 00–5523-00 |
| BD GolgiPlug (brefeldin A) | BD Biosciences | Cat# 555029 |
| HRP-conjugated streptavidin-biotin | DAKO | Cat# P039701–2 |
| AccuCount Blank Particles (7–7.9 μm) | Spherotech | Cat# ACBP-70–10 |
|
| ||
| Deposited data | ||
|
| ||
| Western blot data | Mendeley | https://doi.org/10.17632/c6wjc63frj.1 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| TC-1 | Ramon Arens (LUMC) | N/A |
| MDA-MB-435 | ATCC® | Cat# HTB-129, RRID:CVCL_0622 |
| A375 | ATCC® | Cat# CRL-1619,RRID:CVCL_0132 |
| MDA-MB-231 | ATCC® | Cat# HTB-26, RRID:CVCL_0062 |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse: C57BL/6JRj | Janvier Laboratories | C57BL/6JRj |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for LPAR1–6, ENPP2 and cyclophilin | This paper | N/A |
| shRNA targeting ATX #H1: CCGGCCTGTACCAAATCTGACATATCTCGAGATATGTCAGATTTGGTACAGGTTTTTG | This paper | N/A |
| shRNA targeting ATX #H4: CCGGCCAATCTTCGACTATGACTATCTCGAGATAGTCATAGTCGAAGATTGGTTTTTG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Help-E7SH DNA vaccine | Homemade; this paper | N/A |
| ATX | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo v10 | FlowJo, LLC | RRID:SCR_001456; https://www.bdbiosciences.com/en-us |
| BD FACSDIVA v8.0.1 | BD Biosciences | RRID:SCR_001456; https://www.bdbiosciences.com/en-us |
| Image Lab | Biorad | RRID:SCR_014210; http://www.bio-rad.com/en-us/sku/1709690-image-lab-software |
| GraphPad Prism 8 | Graphpad | RRID:SCR_002798; https://www.graphpad.com/scientific-software/prism/ |
| IBM SPSS Statistics | IBM | RRID:SCR_019096; https://www.ibm.com/products/spss-statistics |
| HALO image analysis software version (3.2.1851) | Indica Labs | RRID:SCR_018350; https://www.indicalab.com |
Highlights.
ATX secreted by tumor cells repels TILs and CD8+ T cells ex vivo
LPAR6 qualifies as a T cell migration inhibitory receptor
Tumor-intrinsic ATX suppresses CD8+ T cell infiltration and tumor control in mice
Melanoma single-cell data provides supporting clinical evidence
ACKNOWLEDGMENTS
We thank Paula Voabil, Jos Urbanus, Yanling Xao, Marjolein Mertz, and Juan Manuel Alba for their help with experiments and data analysis, Kees Franken for making MHC tetramers, Junken Aoki for providing anti-ATX antibodies, and Tomasz Ahrends and Anne van der Leun for helpful discussions and sharing unpublished data. This work was supported by private funding to W.H.M. and grants from the Dutch Cancer Society (NKI 2013–5951 and 10764 to I.V. and NKI 2017–10894 to J.B. and I.V.), the German Research Foundation (DFG) (ME 4924/1–1 to A. Mazzocca), and the NIH (P30 GM127211 to A.J.M.). E.M.-R. is supported by a “Ramón y Cajal” Award (RYC2019–027950-I) from Ministerio de Ciencia e Innovación (MICINN), Spain.
Footnotes
DECLARATION OF INTERESTS
Z.J. is an employee and shareholder of iOnctura SA, a company developing an ATX inhibitor for use in cancer.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110013.
REFERENCES
- Ahrends T, Bąba1a N, Xiao Y, Yagita H, van Eenennaam H, and Borst J (2016). CD27 Agonism Plus PD-1 Blockade Recapitulates CD4+ T-cell Help in Therapeutic Anticancer Vaccination. Cancer Res. 76, 2921–2931. [DOI] [PubMed] [Google Scholar]
- Ahrends T, Spanjaard A, Pilzecker B, Bąba1a N, Bovens A, Xiao Y, Jacobs H, and Borst J<.au> (2017). CD4+ T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 47, 848–861.e5. [DOI] [PubMed] [Google Scholar]
- Anandappa AJ, Wu CJ, and Ott PA (2020). Directing Traffic: How to Effectively Drive T Cells into Tumors. Cancer Discov. 10, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki J, Inoue A, and Okudaira S (2008). Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 1781, 513–518. [DOI] [PubMed] [Google Scholar]
- Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, et al. (2019). A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 9, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeyens A, Fang V, Chen C, and Schwab SR (2015). Exit Strategies: S1P Signaling and T Cell Migration. Trends Immunol. 36, 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Z, Cai L, Umemoto E, Takeda A, Tohya K, Komai Y, Veeraveedu PT, Hata E, Sugiura Y, Kubo A, et al. (2013). Constitutive lymphocyte transmigration across the basal lamina of high endothelial venules is regulated by the autotaxin/lysophosphatidic acid axis. J. Immunol. 190, 2036–2048. [DOI] [PubMed] [Google Scholar]
- Batlle E, and Massagué J (2019). Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 50, 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch MGK, MacIntyre ITK, McMullen TPW, and Brindley DN (2018). Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers (Basel) 10, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst J, Ahrends T, Bąba1a N, Melief CJM, and Kastenmüller W (2018). CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18, 635–647. [DOI] [PubMed] [Google Scholar]
- Brindley DN, Tang X, Meng G, and Benesch MGK (2020). Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. Int. J. Mol. Sci. 21, 5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Wannecq E, Descotes F, Jansen S, Deux B, Ribeiro J, Serre CM, Grès S, Bendriss-Vermare N, Bollen M, et al. (2010). Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS ONE 5, e9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jaeghere EA, Denys HG, and De Wever O (2019). Fibroblasts fuel immune escape in the tumor microenvironment. Trends Cancer 5, 704–723. [DOI] [PubMed] [Google Scholar]
- Fridman WH, Zitvogel L, Sautès-Fridman C, and Kroemer G (2017). The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 14, 717–734. [DOI] [PubMed] [Google Scholar]
- Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, and Smyth SS (2011). Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 286, 34654–34663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnocchi D, Kapoor S, Nitti P, Cavalluzzi MM, Lentini G, Denora N, Sabbà C, and Mazzocca A (2020). Novel lysophosphatidic acid receptor 6 antagonists inhibit hepatocellular carcinoma growth through affecting mitochondrial function. J. Mol. Med. (Berl.) 98, 179–191. [DOI] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011). CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM, van Meeteren LA, Houben AJ, van Zeijl L, et al. (2011). Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 18, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano Y, and Hla T (2019). Bioactive lysolipids in cancer and angiogenesis. Pharmacol. Ther. 193, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben AJ, van Wijk XM, van Meeteren LA, van Zeijl L, van de Westerlo EM, Hausmann J, Fish A, Perrakis A, van Kuppevelt TH, and Moolenaar WH (2013). The polybasic insertion in autotaxin α confers specific binding to heparin and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 288, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, et al. (2019). Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 177, 1933–1947.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquelot N, Duong CPM, Belz GT, and Zitvogel L (2018). Targeting Chemokines and Chemokine Receptors in Melanoma and Other Cancers. Front. Immunol. 9, 2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongsma M, Matas-Rico E, Rzadkowski A, Jalink K, and Moolenaar WH (2011). LPA is a chemorepellent for B16 melanoma cells: action through the cAMP-elevating LPA5 receptor. PLoS ONE 6, e29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, and Fearon DT (2015). T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80. [DOI] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kanda H, Newton R, Klein R, Morita Y, Gunn MD, and Rosen SD (2008). Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 9, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Smith DI, and Sundick RS (1993). Identification of aG protein coupled receptor induced in activated T cells. J. Immunol. 151, 628–636. [PubMed] [Google Scholar]
- Kerdidani D, Chouvardas P, Arjo AR, Giopanou I, Ntaliarda G, Guo YA, Tsikitis M, Kazamias G, Potaris K, Stathopoulos GT, et al. (2019). Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat. Commun. 10, 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keune WJ, Hausmann J, Bolier R, Tolenaars D, Kremer A, Heidebrecht T, Joosten RP, Sunkara M, Morris AJ, Matas-Rico E, et al. (2016). Steroid binding to Autotaxin links bile salts and lysophosphatidic acid signalling. Nat. Commun. 7, 11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlden SA, Capece T, Popovic M, Chapman TJ, Rezaee F, Kim M, and Georas SN (2014). Regulation of T cell motility in vitro and in vivo by LPA and LPA2. PLoS ONE 9, e101655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer MP, Mao G, Hammill C, Yan B, Li Y, Onono F, Smyth SS, and Morris AJ (2019). Effects of diet and hyperlipidemia on levels and distribution of circulating lysophosphatidic acid. J. Lipid Res. 60, 1818–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Gray EE, Zhang Y, Ramírez-Valle F, and Cyster JG (2019). Sphingosine-1-phosphate receptor 2 restrains egress of γδ T cells from the skin. J. Exp. Med. 216, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledein L, Léger B, Dees C, Beyer C, Distler A, Vettori S, Boukaiba R, Bidouard JP, Schaefer M, Pernerstorfer J, et al. (2020). Translational engagement of lysophosphatidic acid receptor 1 in skin fibrosis: from dermal fibroblasts of patients with scleroderma to tight skin 1 mouse. Br. J. Pharmacol. 177, 4296–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Z, Cheng CT, Zhang H, Subler MA, Wu J, Mukherjee A, Windle JJ, Chen CK, and Fang X (2008). Role of LPA4/p2y9/GPR23 in negative regulation of cell motility. Mol. Biol. Cell 19, 5435–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Fujiwara Y, Liu J, Yue J, Shimizu Y, Norman DD, Wang Y, Tsukahara R, Szabo E, Patil R, et al. (2015). Autotaxin and LPA1 and LPA5 receptors exert disparate functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis. Mol. Cancer Res. 13, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen JBAG, Blank CU, et al. (2019). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, and Wu TC (1996). Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 56, 21–26. [PubMed] [Google Scholar]
- Lin S, Haque A, Raeman R, Guo L, He P, Denning TL, El-Rayes B, Moolenaar WH, and Yun CC (2019). Autotaxin determines colitis severity in mice and is secreted by B cells in the colon. FASEB J. 33, 3623–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, Stephens LC, Cui X, Murrow G, et al. (2009). Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 15, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, Collins JW, Nakayama J, Horak CE, Liewehr DJ, Steinberg SM, Albaugh M, Vidal-Vanaclocha F, Palmieri D, Barbier M, et al. (2012). Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J. Natl. Cancer Inst. 104, 1306–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D, Kremer KN, Strauch P, Tigyi G, Pelanda R, and Torres RM (2019). LPA5 Is an Inhibitory Receptor That Suppresses CD8 T-Cell Cytotoxic Function via Disruption of Early TCR Signaling. Front. Immunol. 10, 1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills GB, and Moolenaar WH (2003). The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 3, 582–591. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH, and Perrakis A (2011). Insights into autotaxin: how to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 12, 674–679. [DOI] [PubMed] [Google Scholar]
- Muinonen-Martin AJ, Susanto O, Zhang Q, Smethurst E, Faller WJ, Veltman DM, Kalna G, Lindsay C, Bennett DC, Sansom OJ, et al. (2014). Melanoma cells break down LPA to establish local gradients that drive chemotactic dispersal. PLoS Biol. 12, e1001966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarsheth N, Wicha MS, and Zou W (2017). Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 17, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SW, Clair T, Kim YS, McMarlin A, Schiffmann E, Liotta LA, and Stracke ML (2001). Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 61, 6938–6944. [PubMed] [Google Scholar]
- Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, Ishitani R, Takagi J, Aoki J, and Nureki O (2011). Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 18, 205–212. [DOI] [PubMed] [Google Scholar]
- Ozga AJ, Chow MT, and Luster AD (2021). Chemokines and the immune response to cancer. Immunity 54, 859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Mueller KL, Adams DJ, Anandasabapathy N, Aplin AE, Bertolotto C, Bosenberg M, Ceol CJ, Burd CE, Chi P, et al. (2021). Melanoma models for the next generation of therapies. Cancer Cell 39, 610–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, and Moolenaar WH (2014). Autotaxin: structure-function and signaling. J. Lipid Res. 55, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohaan MW, Wilgenhof S, and Haanen J (2019). Adoptive cellular therapies: the current landscape. Virchows Arch. 474, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato RR, Dávila-González D, Choi DS, Qian W, Chen W, Kozielski AJ, Wong H, Dave B, and Chang JC (2018). Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res. 20, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Bain G, Furuichi K, Iwata Y, Nakamura M, Hara A, Kitajima S, Sagara A, Miyake T, Toyama T, et al. (2019). The involvement of autotaxin in renal interstitial fibrosis through regulation of fibroblast functions and induction of vascular leakage. Sci. Rep. 9, 7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado-Polo F, Fish A, Matsoukas MT, Heidebrecht T, Keune WJ, and Perrakis A (2018). Lysophosphatidic acid produced by autotaxin acts as an allosteric modulator of its catalytic efficiency. J. Biol. Chem. 293, 14312–14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LP, Cao W, Chang WC, Albright RA, Braddock DT, and De La Cruz EM (2011). Kinetic analysis of autotaxin reveals substrate-specific catalytic pathways and a mechanism for lysophosphatidic acid distribution. J. Biol. Chem. 286, 30130–30141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciorra VA, and Morris AJ (2002). Roles for lipid phosphate phosphatases in regulation of cellular signaling. Biochim. Biophys. Acta 1582, 45–51. [DOI] [PubMed] [Google Scholar]
- Shah P, Cheasty A, Foxton C, Raynham T, Farooq M, Gutierrez IF, Lejeune A, Pritchard M, Turnbull A, Pang L, et al. (2016). Discovery of potent inhibitors of the lysophospholipase autotaxin. Bioorg. Med. Chem. Lett. 26, 5403–5410. [DOI] [PubMed] [Google Scholar]
- Spranger S, and Gajewski TF (2018). Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, and Liotta LA (1992). Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 267, 2524–2529. [PubMed] [Google Scholar]
- Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, et al. (2008). The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Fukushima K, Onishi Y, Inui K, Node Y, Fukushima N, Honoki K, and Tsujiuchi T (2017). Lysophosphatidic acid (LPA) signaling via LPA4 and LPA6 negatively regulates cell motile activities of colon cancer cells. Biochem. Biophys. Res. Commun. 483, 652–657. [DOI] [PubMed] [Google Scholar]
- Takeda A, Kobayashi D, Aoi K, Sasaki N, Sugiura Y, Igarashi H, Tohya K, Inoue A, Hata E, Akahoshi N, et al. (2016). Fibroblastic reticular cell-derived lysophosphatidic acid regulates confined intranodal T-cell motility. eLife 5, e10561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, and Fukuzawa K (2002). Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 277, 39436–39442. [DOI] [PubMed] [Google Scholar]
- Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, and Arai H (2002). Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 158, 227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, and de Vries IJM (2017). Migrating into the Tumor: a Roadmap for T Cells. Trends Cancer 3, 797–808. [DOI] [PubMed] [Google Scholar]
- van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradère JP, Pettit TR, Wakelam MJ, Saulnier-Blache JS, Mummery CL, et al. (2006). Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 26, 5015–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb TE, Kaplan MG, and Barnard EA (1996). Identification of 6H1 as a P2Y purinoceptor: P2Y5. Biochem. Biophys. Res. Commun. 219, 105–110. [DOI] [PubMed] [Google Scholar]
- Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, and Werb Z (2020). Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu V, Yeerna H, Nohata N, Chiou J, Harismendy O, Raimondi F, Inoue A, Russell RB, Tamayo P, and Gutkind JS (2019). Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 294, 11062–11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida K, Masago K, Nakanishi H, Kihara Y, Hamano F, Tajima Y, Taguchi R, Shimizu T, and Ishii S (2009). Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 284, 17731–17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida K, Kurikawa Y, Shimizu T, and Ishii S (2013). Current progress in non-Edg family LPA receptor research. Biochim. Biophys. Acta 1831, 33–41. [DOI] [PubMed] [Google Scholar]
- Yung YC, Stoddard NC, and Chun J (2014). LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 55, 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw western blot images have been deposited at Mendeley and are publicly available as of the date of publication.
No new code was written for this study. Analysis of publicly available single-cell RNA-seq data is described in the method details.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.