

Abstract
A protein binding-induced supramolecular dissociation strategy is developed with the ratio of monomer and excimer fluorescence as the tool for protein sensing and quantification. Due to the “lock-and-key” strategy based on specific ligand-protein binding, the probe exhibits excellent selectivity and quantification accuracy to the protein of interest. The ratiometric approach is immune to interference from extrinsic quenchers, while preserving the opportunity to be protein specific.
Aberrant expression of proteins is often considered as symptoms of many diseases.1 Thus, developing rapid, convenient and accurate methods for protein sensing and quantification is of significant importance for diagnosis and the understanding of many diseases.2 An ideal protein sensing method should meet several critical criteria: (i) good selectivity and accuracy, (ii) excellent sensitivity, (iii) rapid signal read-out, and (iv) easy operations. Fluorescent methods have been especially popular for this purpose.3 Among these, array-based4,5 and ligand-directed6 sensing are two main categories. The former approach usually relies on the signal pattern from nonspecific protein-sensor interactions,7–12 while the latter utilizes specific intermolecular interactions.13–15 Most of these methods are based on a change in the fluorescence intensity when fluorophores interact with targets.16–18 Such signal changes are vulnerable to the interference from other components in the system. Thus, developing alternate methods that avoid the dependence on direct fluorescent signals may be a potential way to exclude environmental interference.
Excimer and monomer fluorescence features are available in many fluorophores, such as pyrene and perylene.19–23 These have been utilized for the study of sensing,24 imaging,25 and dynamics of self-assemblies26. Ratiometric signal changes of excimer and monomer intensities, instead of a direct fluorescence intensity, make them promising alternatives for sensing.27,28,29 We recently utilized 7-diethylaminocoumarin as a probe to study the dynamics of amphiphiles in temperature-responsive supramolecular assemblies.30 The interruption of hydrophobic and lipophilic balance (HLB) of assemblies by the cleavage of covalent bonds or temperature alterations led to changes in the fidelity of assemblies which were reflected by the monomer and excimer fluorescence. In 2015, we reported that when proteins interacted with supramolecular assemblies which were modified with the specific binding ligands (“lock and key” pairs), the supramolecular dissociation of assemblies led to a direct fluorescence intensity change of pyrene.15 The non-covalent interaction of bulky proteins with relatively small amphiphilic macromolecules interrupt the HLB within the assemblies, destabilizing the system and causing the supramolecular dissociation.31 Because the disassembly was based on protein-ligand binding, it should be a concentration-dependent process, offering an opportunity for application in protein sensing and quantification. However, as aforementioned, no matter using the dye release or fluorophore-quencher separation strategies, they were both based on direct fluorescence changes and easily interfered by the microenvironment. In this context, we herein incorporate excimer-monomer ratio-based fluorescence with the protein-induced supramolecular disassociation strategy and develop a new protein sensing and quantification method. We hypothesize that the different extent of supramolecular dissociation upon ligand and protein binding could lead to distinct monomer and excimer fluorescence ratios, providing quantitative information about protein concentrations.
To test our hypothesis, bovine carbonic anhydrase (bCA) and phenyl sulfonamide were chosen as the model protein and ligand system respectively. Carbonic anhydrase is an important series of proteins, the abnormal expression of which has been implicated as a biomarker for cancers.32,33 Besides, the binding of bCA with sulfonamide ligands has been extensively studied, providing substantial confidence in their selective interaction.34 To start this study, we first designed and synthesized a dendritic molecule D1 which was modified with a phenyl sulfonamide ligand and 7-diethylaminocoumarin. As shown in Fig. 2d, the amphiphilic molecule has pentaethylene glycol moieties (EG5) as the hydrophilic chain and decyl group (C10) as the hydrophobic chain. Coumarin was incorporated into the hydrophobic part of the amphiphile, which in turn buries this functionality into hydrophobic core of the amphiphilic nanoassembly, generating an excimer fluorescence. As the designed binding ligand, phenylsulfonamide is incorporated on the hydrophilic face of the amphiphile, which in turn presents the ligand on the solvent-exposed shell of the nanoassembly, ensuring accessibility to protein binding.
Fig. 2.

(a) Particle size and (b) fluorescence change of D1 assemblies in response of 1 eq. of bCA. (c) Schematic representative of the ligand-bCA displacement assay. (d) Structure of molecule D1. (e) Fluorescence change with/without ligand displacement.
The nanoassemblies of D1 were prepared using the previously developed method.30 First, molecule D1 was dissolved in acetone and then added into deionized water with a 100 μM concentration. Then the mixed solution was stirred in an uncapped vial overnight to evaporate organic solvent. Finally, the solution was diluted to 25 μM for tests. The size of the assembly was measured using dynamic light scattering (DLS) to be ~160 nm. As we planned to use the excimer and monomer ratios as readout-signals, the fluorescence profile of the assemblies was assessed. As shown in Fig. 2b, the assemblies exhibited both the monomer and excimer fluorescence at about 480 nm and 540 nm respectively. After preparing the assemblies of D1, we tested its response to bovine carbonic anhydrase (bCA). However, after adding even one equiv. of the protein (25 μM), the disassembly of the particles was not observed from either the DLS or fluorescence data (Fig. 2a and 2b). There are two potential reasons for the results: First, the hydrophobic interaction in D1 is too strong to be broken and thus the anticipated binding-induced disassembly does not occur. Second, the binding between the phenylsulfonamide ligand in D1 and bCA is too slow or too weak to cause any change to the assemblies during the time scale we studied. To elucidate these possibilities, a competitive protein-binding assay was performed (Fig. 2c).35 Dansyl amide (DNSA) is a pre-fluorophore that exhibits negligible fluorescence by itself, but shows significantly higher fluorescence after binding with bCA.36 Therefore, mixing of DNSA with bCA generates a very strong fluorescence at 460 nm. To this mixture, a solution of the nanoassembly was added. If the phenylsulfonamide from the assembly could compete with DNSA, the latter would be displaced from the bCA binding pocket and its fluorescence would be much lower. As shown in Fig. 2e, after adding 1 equiv. of D1, we did observe a significant decrease of fluorescence. A further decrease was even observed, when an additional 0.5 equiv. of D1 was added. On the other hand, for the control group, without the addition of D1 assembly, there was no significant change in the fluorescence. These results indicate that the particles could indeed bind with bCA, although no disassembly was observed. Therefore, the likely reason for the lack of binding-induced disassembly is the strong hydrophobic interaction in the assemblies.
To solve this problem, a more hydrophilic molecule D2 (Fig. 1) was synthesized. Compared with D1, heptaethylene glycol (EG7) and hexyl group (C6) were installed in D2 as the hydrophilic and hydrophobic chains, respectively. D2 also formed good assemblies in aqueous solution, generating comparable excimer fluorescence as D1 (Fig. 3a), with ~250 nm particle size (Fig. S3). After fabricating assemblies, the possibility for bCA-ligand-binding induced fluorescence change was again examined. To our delight, after mixing with different amount of bCA, we observed a systematic change in monomer and excimer fluorescence (Fig. 3a). As expected, with the increase of protein concentrations, more increase in monomer and decrease in excimer fluorescence were observed. Next, a time dependent study on the monomer and excimer intensity ratio (IM/IE) change was performed. As shown in Fig. 3b, the ratio reached equilibriums in well under 10 min after the addition of bCA, which was a very fast process. Since we previously presumed that protein-binding induced dissociation to be concentration dependent, we were interested in establishing a quantifiable relationship between protein concentrations and the monomer and excimer ratios. To our delight, when plotting the IM/IE ratios versus protein concentrations, a linear relationship was obtained (Fig. 3c). These results demonstrate that this protein binding-induced IM/IE ratio change can be potentially used for protein quantification.
Fig. 1.
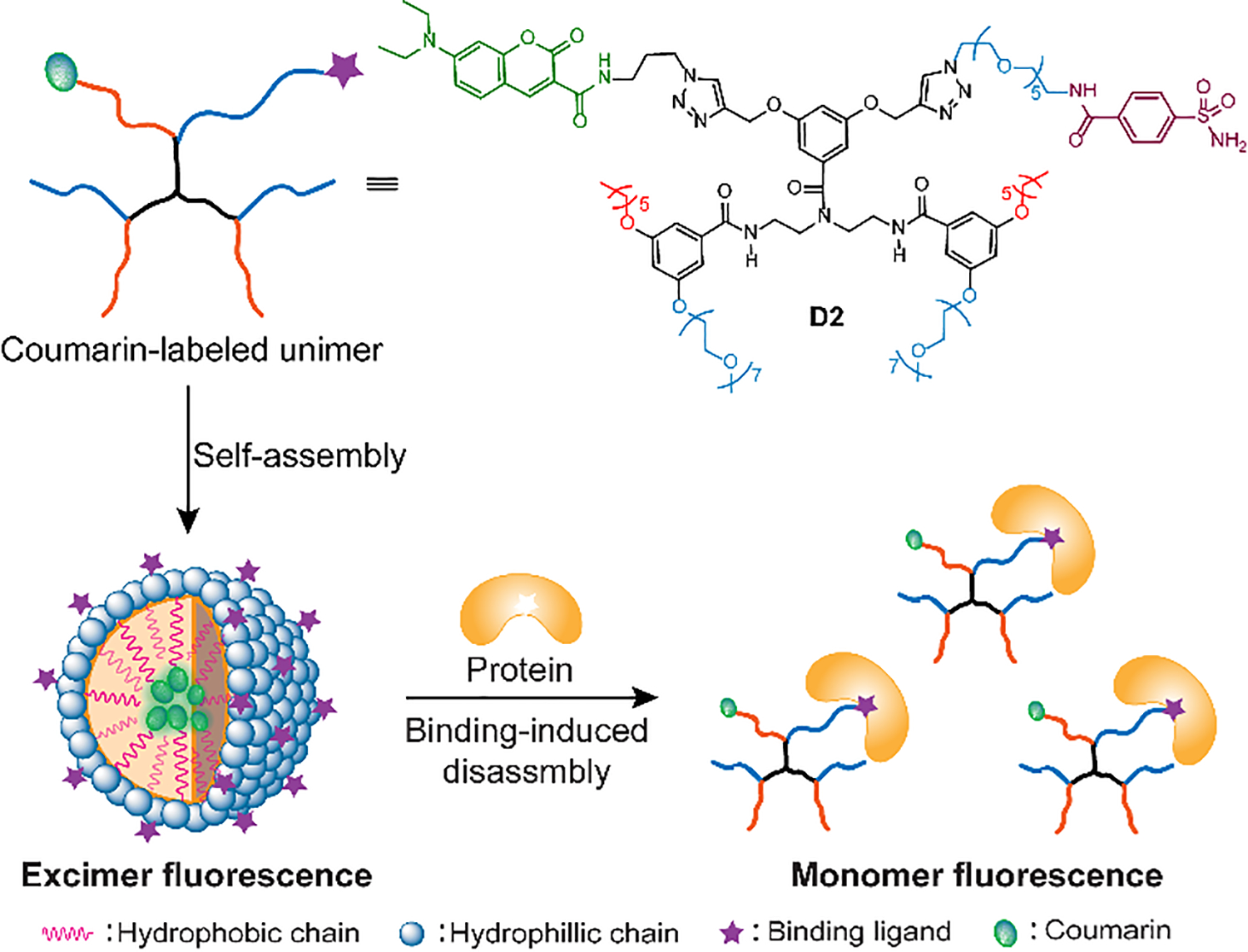
Schematic representative of protein binding-induced disassembly using excimer-monomer transformation
Fig. 3.

(a) Fluorescence change of assemblies D2 (25 μM) in response of different amount of bCA. (b) Time dependent study on the IM/IE ratio change in response to different amount of bCA. (c) The linear plot relationship of IM/IE ratio over bCA concentration. (d) Comparison of IM/IE ratio in response to 1 equiv. of proteins.
To confirm mechanism of IM/IE change is indeed because of protein binding-induced assembly dissociation, we studied the guest molecule release and changes of assembly sizes in response to bCA. As shown in Fig. S4, the accumulated guest molecule release correlates with bCA concentrations: when the amount of bCA changed from 0.2 equiv. to 1 equiv., the accumulated dye release increased from 27% to 55%. Moreover, a burst release of guest molecules was observed in the first a few minutes, consistent with the fast response of IM/IE ratios to bCA. DLS results also show that the addition of protein resulted in the decrease of assembly sizes (Fig. S4). These experiments further support that binding-induced disassembly as the underlying reasons for the observed fluorescence changes.
For sensing and quantification purposes, the specificity and selectivity of the method must be examined. To test for specificity, we examined its response to other seven proteins with different sizes and isoelectric points (Table S1). As shown in Fig. 3d and Fig. S5, compared with other non-specific proteins, only bCA caused a significant change in the IM/IE signal. Bovine serum albumin (BSA) had a weak, but slightly higher response signal among the control proteins, likely due to the relatively stronger interactions with hydrophobic moieties of D2 molecule.37 However, compared with bCA, the response was not significant. Additionally, we also studied whether bCA was specific to the nanoprobe or not. To test this, we synthesized another molecule which phenylsulfonamide ligand was replaced by a phenyl group (D3, Fig. 4a) and prepared the assemblies. As expected, after adding even 1 equiv. of bCA to the D3 system, there was almost no changes in IM/IE ratio over next 1 hour (Fig. 4b and S6), demonstrating that bCA had excellent specificity to the nanoprobe D2. As one problem for the sensing using a direct fluorescence intensity as readout signals is the quenching from other components, we next illustrated the advantage of using the IM/IE signal by dispersing the assemblies in different concentrations of ferric chloride. The fluorescence intensity of assemblies significantly decreased with the increase of Fe3+ concentrations (Fig. S7). However, the IM/IE signal exhibited no significant changes, validating it as an anti-quenching signal for protein sensing.
Fig. 4.

(a) Structure of molecule D3. (b) Comparison of IM/IE ratios of assemblies D3 with or without 1 equiv. of bCA.
We next tested whether this supramolecular dissociation-based method can be used for the quantification of proteins in complex systems where multiple proteins may present. To test this, different amount of bCA was mixed with various proteins and then quantified using the designed nanoprobe (Fig. S8). As shown in Table 1, when bCA was dispersed 1:1 ratio with α-chymotrypsin (Chy), the recovery ratio was 103.6 ± 4.4 %. We next tested the quantification in more complex system with multiple different proteins: When 7.5 μM bCA was mixed with α-chymotrypsin, lysozyme (Lys) and β-lactoglobulin A (Lac A) (7.5, 7.5, 2.5 μM respectively), the spike recovery was 103.6 ± 4.4 %. These experiments further demonstrate the specificity and accuracy of this method. To explore the dynamic range of protein sensing, we assessed the protein concentration range in which a linear relationship with IM/IE is maintained (Fig. S9); the maximum bCA concentration for an accurate quantification is 30 μM, i.e. 1.2 equiv. of the probe concentration.
Table 1.
bCA spiked recovery experiment in complex system
| entry | protein system | CA spiked (μM) | CA found (μM) | recovery (%) |
|---|---|---|---|---|
|
| ||||
| 1 | 5.0 μM Chy | 5.0 | 5.18 | 103.6 ± 4.4 |
| 2 | Chy, Lys, Lac A (7.5, 7.5, 2.5 μM) | 7.5 | 7.74 | 103.2 ± 6.2 |
In summary, we designed an amphiphilic nanoprobe that could be used for protein sensing and quantification via a supramolecular disassembly mechanism. This method has a few notable features: i) the sensing and quantification was based on the protein binding-induced supramolecular disassembly, extending the application of this interesting supramolecular observation; ii) the readout signal was the monomer and excimer fluorescence ratio instead of fluorescence intensity, exhibiting better resistance to interferences, such as from metal quenching; iii) the sensing was a “lock and key” type recognition, ensuring the specificity and accuracy of the quantification; and iv) the introduction of binding ligand at the last step of the synthetic scheme makes this method a convenient one for broadly adapting this strategy for developing sensors for other proteins.
Supplementary Material
Acknowledgements
We thank U.S. Army Research Office (W911NF-15-1-0568) for supporting this work.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Footnotes relating to the title and/or authors should appear here.
References
- 1.Rifai N, Gillette MA and Carr SA, Nat. Biotechnol, 2006, 24, 971–983. [DOI] [PubMed] [Google Scholar]
- 2.Nettikadan S, Radke K, Johnson J, Xu J, Lynch M, Mosher C and Henderson E, Mol. Cell. Proteomics, 2006, 5, 895–901. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Wang H, Li J, Zheng J, Xu X and Yang R, Anal. Chem, 2011, 83, 1268–1274. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, Askim JR and Suslick KS, Chem. Rev, 2019, 119, 231–292. [DOI] [PubMed] [Google Scholar]
- 5.Wright AT and Anslyn EV, Chem. Soc. Rev, 2006, 35, 14–28. [DOI] [PubMed] [Google Scholar]
- 6.Ferreira De Freitas R and Schapira M, Medchemcomm, 2017, 8, 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.González DC, Savariar EN and Thayumanavan S, J. Am. Chem. Soc, 2009, 131, 7708–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savariar EN, Ghosh S, González DC and Thayumanavan S, J. Am. Chem. Soc, 2008, 130, 5416–5417. [DOI] [PubMed] [Google Scholar]
- 9.Sandanaraj BS, Demont R and Thayumanavan S, J. Am. Chem. Soc, 2007, 129, 3506–3507. [DOI] [PubMed] [Google Scholar]
- 10.Geng Y, Peveler WJ and Rotello VM, Angew. Chem., Int. Ed, 2019, 58, 5190–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Bender M, Seehafer K and Bunz UHF, Angew. Chem., Int. Ed, 2016, 55, 7689–7692. [DOI] [PubMed] [Google Scholar]
- 12.Pérez RL, Cong M, Vaughan SR, Ayala CE, Galpothdeniya WIS, Mathaga JK and Warner IM, ACS Sensors, 2020, 5, 2422–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu L, Huang J, Pu K and James TD, Nat. Rev. Chem, 2021, 5, 406–421. [DOI] [PubMed] [Google Scholar]
- 14.Su D, Teoh CL, Wang L, Liu X and Chang YT, Chem. Soc. Rev, 2017, 46, 4833–4844. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Zhuang J, Raghupathi KR and Thayumanavan S, Chem. Commun, 2015, 51, 17265–17268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miao Y, Sun X, Lv J and Yan G, ACS Appl. Mater. Interfaces, 2019, 11, 2264–2272. [DOI] [PubMed] [Google Scholar]
- 17.Yu WT, Wu TW, Huang CL, Chen IC and Tan KT, Chem. Sci, 2016, 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Zhao X, Zheng K, Guo X, Yan Y and Xu Y, Langmuir, 2019, 35, 5599–5607. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Chen J, Chen Y, Li W and Yu C, Anal. Chem, 2014, 86, 4371–4378. [DOI] [PubMed] [Google Scholar]
- 20.Agafontsev AM, Shumilova TA, Oshchepkov AS, Hampel F and Kataev EA, Chem. - A Eur. J, 2020, 26, 9991–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraskouskaya D, Bancerz M, Soor HS, Gardiner JE and Gunning PT, J. Am. Chem. Soc, 2014, 136, 1234–1237. [DOI] [PubMed] [Google Scholar]
- 22.Kapf A and Albrecht M, J. Mater. Chem. B, 2018, 6, 6599–6606. [DOI] [PubMed] [Google Scholar]
- 23.Paris PL, Langenhan JM and Kool ET, Nucleic Acids Res, 1998, 26, 3789–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bains GK, Kim SH, Sorin EJ and Narayanaswami V, Biochemistry, 2012, 51, 6207–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benni I, Trabuco MC, Di Stasio E, Arcovito A, Boffi A, Malatesta F, Bonamore A, De Panfilis S, De Turris V and Baiocco P, RSC Adv, 2018, 8, 12815–12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seixas De Melo J, Costa T, Francisco A, Maçanita AL, Gago S and Gonçalves IS, Phys. Chem. Chem. Phys, 2007, 9, 1370–1385. [DOI] [PubMed] [Google Scholar]
- 27.Yang CJ, Jockusch S, Vicens M, Turro NJ and Tan W, Proc. Natl. Acad. Sci. U. S. A, 2005, 102, 17278–17283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feiner-Gracia N, Buzhor M, Fuentes E, Pujals S, Amir RJ and Albertazzi L, J. Am. Chem. Soc, 2017, 139, 16677–16687. [DOI] [PubMed] [Google Scholar]
- 29.Rosenbaum I, Harnoy AJ, Tirosh E, Buzhor M, Segal M, Frid L, Shaharabani R, Avinery R, Beck R and Amir RJ, J. Am. Chem. Soc, 2015, 137, 2276–2284. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Lionello C, Westley J, Cardellini A, Huynh U, Pavan GM and Thayumanavan S, Nanoscale, 2021, 11568–11575. [DOI] [PubMed] [Google Scholar]
- 31.Azagarsamy MA, Yesilyurt V and Thayumanavan S, J. Am. Chem. Soc, 2010, 132, 4550–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Türeci Ö, Sahin U, Vollmar E, Siemer S, Göttert E, Seitz G, Parkkila AK, Shah GN, Grubb JH, Pfreundschuh M and Sly WS, Proc. Natl. Acad. Sci. U. S. A, 1998, 95, 7608–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lie M, Mazure NM, Hofman V, Ammadi RE, Ortholan C, Bonnetaud C, Havet K, Venissac N, Mograbi B, Mouroux J, Pouysségur J and Hofman P, Br. J. Cancer, 2010, 102, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB and Whitesides GM, Chem. Rev, 2008, 108, 946–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koutnik P, Shcherbakova EG, Gozem S, Caglayan MG, Minami T and Anzenbacher P, Chem, 2017, 2, 271–282. [Google Scholar]
- 36.Srivastava DK, Jude KM, Banerjee AL, Haldar M, Manokaran S, Kooren J, Mallik S and Christianson DW, J. Am. Chem. Soc, 2007, 129, 5528–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji W, Chen L, Xue X, Guo Z, Yu Z, Zhao B and Ozaki Y, Chem. Commun, 2013, 49, 7334–7336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.