

Abstract
The developing human brain is uniquely vulnerable to methylmercury (MeHg) resulting in lasting effects especially in developing cortical structures. Here we assess by single-cell RNA sequencing (scRNAseq) persistent effects of developmental MeHg exposure in a differentiating cortical human-induced pluripotent stem cell (hiPSC) model which we exposed to in vivo relevant and non-cytotoxic MeHg (0.1 and 1.0 μM) concentrations. The cultures were exposed continuously for 6 days either once only during days 4–10, a stage representative of neural epithelial- and radial glia cells, or twice on days 4–10 and days 14–20, a somewhat later stage which includes intermediate precursors and early postmitotic neurons. After the completion of MeHg exposure the cultures were differentiated further until day 38 and then assessed for persistent MeHg-induced effects by scRNAseq. We report subtle, but significant changes in the population size of different cortical cell types/stages and cell cycle. We also observe MeHg-dependent differential gene expression and altered biological processes as determined by Gene Ontology analysis. Our data demonstrate that MeHg results in changes in gene expression in human developing cortical neurons that manifest well after cessation of exposure and that these changes are cell type-, developmental stage-, and exposure paradigm-specific.
Keywords: Cortex, Glutamatergic, Neuron, Human-induced pluripotent stem cells, Methylmercury, single cell RNA sequencing
1. Introduction
The developing brain is uniquely vulnerable to environmental insult (Rice and Barone, 2000) and its sensitivity to methylmercury (MeHg) toxicity became evident with the Minamata disease tragedy, where an epidemic of spasticity, blindness and profound mental retardation was seen in infants born to mothers who had consumed seafood from the heavily contaminated bay water (Harada, 1995; Harada, 1968). Similar outbreaks of profound neurodevelopmental disorders in infants were ascribed to the consumption of seed grain treated with MeHg fungicides in Iraq (Clarkson et al., 1976).. In many of the MeHg contamination incidences reported, it was observed that mothers giving birth to significantly affected infants, were themselves only minimally affected, if at all (Harada, 1995; Myers and Davidson, 2000). Despite extensive research the threshold levels for neurotoxic effects on human fetal brains are still not clear. Several recent studies have focused on prenatal exposures to lower concentrations of MeHg (Castoldi et al., 2008; Grandjean et al., 1999; Grandjean et al., 1997; Huang et al., 2018; Kjellstroem et al., 1989; Oken et al., 2008; Sanfeliu et al., 2003; Trasande et al., 2005). The threshold level for neurologic effects in adults (50 ppm in maternal hair) was estimated to be about five times higher than for prenatal effects (10 ppm in maternal hair) (Bakir et al., 1973; Cox et al., 1989; Swedish Expert Group, 1971). The fetal brain dose has been estimated to be 20-fold lower than that of maternal hair, suggesting that lowest observable effect level occurs at fetal brain doses of 0.3–1 ppm (Burbacher et al., 1990; Cox et al., 1989; Kjellstrom et al., 1986; Marsh, 1987). Based on these studies and the fact that the major source of MeHg is fish consumption, the National Research Council concluded that MeHg poses a public health threat and suggested MeHg threshold levels of blood and hair at 0.5 ppb and 1 ppm, respectively (NRC, 2000). An EPA study report to Congress states that 8% of US women of child bearing age have blood Hg levels in excess of the recommended limits emphasizing the urgency of this environmental threat (EPA, 1997).
MeHg exposure of the adult human brain is primarily localized to the granule layer of the cerebellum and the visual cortex of the cerebrum (Aschner and Syversen, 2005; Choi, 1989; Clarkson, 2002). The damage resulting from exposure during human CNS development is less localized, and the earlier the exposure during development, the more generalized the damage (Choi, 1989; Clarkson, 2002; Costa et al., 2004). Subacute MeHg exposures cause neurological symptoms with latency periods of years in infants, showing persistent and long-lasting, even life-long consequences (Amin-Zaki et al., 1979; Debes et al., 2006). This observation, combined with the finding that MeHg-induced pathological changes are not specific to MeHg intoxication but are seen in a variety of neurodevelopmental disorders (Choi, 1989), suggest that MeHg interferes with fundamental neurodevelopmental processes.
CNS development involves intricate and developmental time dependent cell-cell interactions. The interference with any of these manifold and intricate cell-communications may result in aberrant neurodevelopment and thus, have long-term persistent consequences that may not result in immediate overt toxicity but become manifest at a later time in development as abnormal tissue structure and/or impaired cellular functions (Kraft et al., 2016; Stolp et al., 2011). Prolonged pre and/or perinatal MeHg exposure even at moderate doses interferes with development of neurons and glial cells through diverse cellular mechanisms, including cell proliferation, cell migration, cellular oxidative metabolism, cellular aggregation and process extension and synapse formation (Antunes Dos Santos et al., 2016; Choi, 1986; Choi et al., 1978; Peckham and Choi, 1988). Indeed, histological postmortem observations of brains from in utero exposed infants indicate abnormal brain development and ectopic cell masses in the cortex, disorganization of cortical cell layers, smaller than normal cortical pyramidal neurons, white matter astrogliosis and the persistence of periventricular germinal zone (matrix) cells forming a thin layer of large round cells in the periventricular region around the third ventricle (Burbacher et al., 1990; Choi, 1989; Clarkson, 2002; Matsumoto et al., 1965).
Cortical development begins with the induction of the neuroepithelium which proliferates and forms the ventricular zone (VZ). At the start of neurogenesis these neuroepithelial cells (NE) give rise to the apical progenitors, including the apical (inner, ventricular) radial glial cells (aRG) in the VZ. Their derivatives the basal progenitors, which include the basal (outer) RG (bRG) and intermediate progenitor cells (IPC) divide in subventricular zone. RG cells produce similar proportions of neurons, astrocytes and oligodendrocytes, while IPCs produce mainly neurons (Bystron et al., 2008; Florio and Huttner, 2014; Namba and Huttner, 2017). Thus, during cortical development there is a co-existence and interaction of different cell types including NE, RG, IPCs, migrating neural precursors (NP) and postmitotic neurons at different stages of differentiation (Greig et al., 2013).
Human-induced pluripotent stem cell (hiPSC) technology provides us with an in vitro model to study human cortical development. Differentiating hiPSC-derived cortical cultures display a similar heterogeneity of different stages of developing neurons as has been reported for fetal cortical tissue (Bhaduri et al., 2020; Burke et al., 2020; Camp et al., 2015; Handel et al., 2016). In addition, the time frame of the in vivo and in vitro hiPSC-derived early cortical neuron development is quite similar. Thus, in vivo embryonic day 30 (postconceptional week (PCW) 5) marks the start of neurogenesis with the closure of the neural tube and neuroepithelial cells forming the ventricular zone (Bystron et al., 2008; Kostović et al., 2019). On embryonic day 33 there is a switch to asymmetrical cell proliferation which results in the first postmitotic cells and radial glial cells expressing the transcription factors PAX6 and FOXG1and about another 30 days later (PCW 8–9) early neurons are present (Kostović et al., 2019). In our in vitro differentiation dual SMAD inhibition is used to induced hiPSCs to differentiate into neural epithelial cells and similar to the in vivo time frame 3–4 days later the expression of radial glial markers PAX6 and FOXG1 is observed (Neely et al., 2012). As we report here about 30 days later,on day 38, we see expression of markers defining early cortical neurons. Thus, overall at least for the early stages of human cortical development, the time frame for in vivo and in vitro development is comparable.
Single-cell transcriptomics offers a method of assessing the effects of MeHg on neurodevelopment on a cell type-specific level by eliminating the complexity of this cellular heterogeneity and thus allows identification of cell-specific vulnerability (Romito-DiGiacomo et al., 2007). Much of our understanding regarding the effects of MeHg on the developing human CNS comes from observations of prenatal exposures to relatively high doses, while the effects of sub-cytotoxic exposures on the developing brain are less clear. In vitro observations suggest that mechanisms underlying MeHg toxicity are different at higher and lower doses (Choi and Kim, 1984; Jebbett et al., 2013). Thus, observations from in vivo and in vitro studies suggest that levels, duration and time of exposure during development likely differentially affect different cell populations and play crucial roles in the eventual consequences of the exposure. Given the reported toxicity of MeHg on NP proliferation, migration of early cortical neurons, and the resulting disorganization of the cortical layers (Choi, 1989; Clarkson, 2002) we assessed the effects of MeHg exposure at a very early stage (e.g. NE, RG) MeHg (E-exposure, Days 4–10) only and a combined exposure paradigm that included an early and a somewhat later (e.g. IPC, NP) stage (E+L-exposure, Days 4–10 and 14–20) at lower (0.1 μM) and higher (1 μM) MeHg levels. We then performed single cell RNA sequencing (scRNAseq) at a subsequent time point (Day 38) to examine potential persistent effects of different MeHg exposure paradigms on population size, changes in cell cycle and gene expression on different neuronal cell populations. The delay in assessment to a time period when post-mitotic neurons are known to be present enabled us to more easily detect and differentiate the consequences of the early only (E-exposure) and the combined early and later (E+L-exposure) developmental exposures.
2. Materials and Methods:
2.1. Derivation, validation and differentiation of hiPSCs
The hiPSC line used for this study (CC3) was derived and validated from a control subject according to our established protocol (Neely et al., 2017). In brief, dermal fibroblasts were obtained by skin biopsy after appropriate patient consent/assent under the guidelines of an approved IRB protocol (Vanderbilt No. 080369). About 6 × 105 fibroblasts were reprogrammed by electroporation with the CXLE plasmid vectors using the Neon Transfection System (Life Technologies, Carlsbad, CA) following published methods (Okita et al., 2011) and then plated at 5 × 104 cells/well into matrigel coated 6 well plates. Two days later the cells were transferred into TeSR-E7 medium (#05919, #05914; StemCell Technologies, Vancouver, Canada) and maintained until hiPSC colonies were ready to be manually isolated (about 4 weeks) and propagated in mTeSR medium (#85851, #85852; StemCell Technologies). The lack of plasmid integration into the genomic DNA was demonstrated by qPCR, karyotype analyses performed using standard protocols with at least 20 metaphase spreads (Genetics Associates, Nashville, TN) and pluripotency validated by Pluritest (Muller et al., 2011), and by the capacity of the hiPSCs to differentiate into neural lineages (Kumar et al., 2014; Neely et al., 2017; Tidball et al., 2016). Starting on day 0 cortical NPs were derived from hiPSCs via an 11 day dual-SMAD inhibition as previously described except that we used 0.4 μm LDN in place of noggin or DMH1 (Chambers et al., 2009; Neely et al., 2012). Starting on day 11 the cultures were further differentiated in cortical differentiation medium consisting of 50% Neurobasal medium (#211030490, ThermoFisher Scientific, Waltham, MA) supplemented with B27 (#17504044, ThermoFisher Scientific) and 50% DMEM/F12 medium with glutamax (Invitrogen #10565018) containing N2 supplement (Invitrogen #17502–048), MEM non-essential amino acid mixture (Invitrogen #M7145), Penicillin/Streptomycin (2x; Mediatech, CellGro-Corning, Manassas, VA #30002Cl) and β-mercaptoethanol (100 μM, Sigma, St. Louis, MO # M3148) as described (Joshi et al., 2019). On day 21 of differentiation the cells were passaged by incubating them with accutase (STEMCELL Technologies, Cambridge, MA; #01–0006) for 18 minutes and reseeding them into Matrigel (BD Bioscience #354277)-coated 6 well plates at 1 × 105 cells/cm2 in cortical differentiation medium containing 10 μM Rock-inhibitor (Tocris, Minneapolis, MN #1254) which was removed after 24 hours. The cultures were maintained in cortical differentiation medium until day 38 of differentiation when the cells were harvested for scRNAseq.
2.2. Quantitative reverse transcription PCR (RT-qPCR)
For validation of hiPSC-lines, assessment of neural differentiation and quantification of TTR expression on day 38, total RNA was prepared using a RNeasy Plus Mini kit (#74134, Qiagen, Valencia, CA) according to the manufacturer’s instructions. Isolated RNA was reverse transcribed into cDNA on a MyCycler Thermal Cycler (Bio-Rad, Hercules, CA) using SuperScriptIII First-Strand Synthesis System with oligo(dT)20 (#18080051, ThermoFisher Scientific) according to the protocol provided by manufacturer. Primer sequences used for RT-qPCR are provided in suppl. Table 1. The expression of housekeeping genes GAPDH, PGK1, UBC, and ACTIN were assessed and ACTIN and UBC expression found to be the most consistent between the different developmental stages. All mRNA signals were normalized to the expression of actin. RT-qPCR was performed using a Power SYBR Green Master Mix (#4367659, Applied Biosystems, Carlsbad, CA) on an ABI 7900HT fast real-time PCR detection system (Applied Biosystems) at the Vanderbilt University Medical Center VANTAGE Core facility.
2.3. Immunofluorescence
For immunofluorescence analysis cells were plated into 96-well plates (Greiner Bio-One, Monroe, North Carolina, μclear) and immunofluorescence performed as described (Neely et al., 2017; Neely et al., 2012). Briefly, the cells were fixed in PBS containing 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PZ) for 30 min at room temperature, permeabilized with 0.2% Triton X-100 for 20 min at room temperature and then incubated in PBS containing 5% donkey serum (Jackson ImmunoResearch, West Grove, PA) and 0.05% Triton X-100 overnight at 4oC. The following primary antibodies were applied for 24 hours at 4oC: rabbit anti-PAX6 (# PRB-278P, Covance, Emeryville, CA, dilution 1:200), mouse anti-β3-tubulin antibody (# MA1–19187, ThermoFisher Scientific, 1:500), rabbit anti-glutamate (#G6642, Sigma, 1:2000), mouse anti-MAP2 (#13500, ThermoFisher Scientific, 1:200), rabbit anti-VGLUT1 (#135 302, Synaptic Systems, Mill Bay, Canada, 1:500). Secondary antibodies conjugated to AlexaFluuor 488 (#715–545-151, 1:800) or Cy3 (1:800, #711–165-152, 1:800, both from Jackson ImmunoResearch, West Grove, PA) were applied for 24 hours at 4oC. Cells were counterstained with the nuclear dye Hoechst 33258 (#B1155, Sigma) and images were obtained with a Zeiss ObserverZ1 microscope and AxioVs40 software (version 4.7.2).
2.4. MeHg exposures
Exposures to 1 μM and 0.1 μM MeHg (Alfa Aesar, Ward Hill, MA; #33553) were performed continuously either only early during differentiation on days 4–10 (E exposure) or early and later, on days 4–10 and days 14–20 (E+L exposure) of differentiation. Thus, the four exposure paradigms are referred to as E 0.1 μM, E 1 μM, E+L 0.1 μM, and E+L 1 μM.
2.5. Single cell RNA sequencing:
Cell harvest:
To harvest the cells the cultures were washed twice with DMEM/F12 medium to remove dead cells and cell debris, dissociated with accutase for 12 minutes and collected in ice-cold DMEM/F12 containing 10 μM Rock-inhibitor. The cells were pelleted at 300 x g for 5 minutes at 4oC, resuspended in ice-cold DMEM/F12 containing glutamax, 0.04% ultrapure BSA (#AM2616, ThermoFisher Scientific) and 10 μM Rock-inhibitor and gently triturated using a 5 ml glass pipette (#13–678-36B, ThermoFisher Scientific). The cell suspensions were strained through a 40 μm filter (#C4040, MTCBio, Sayreville, NJ), the viability of the harvested cells determined with trypan blue exclusion assay using an automated cell counter (Cellometer AutoT4 from Nexcelome Bioscience, Lawrence, MA); it was approximately 90% for all samples.
Capturing of single cells and preparation of cDNA:
The cell concentrations were adjusted to 550 cells/μl and approximately 10,000 cells per channel (to give estimated recovery of 5,000–8,000 cells per channel) were loaded onto a Chromium Single Cell 3′ Chip (10x Genomics, PN-2000168) and processed through the Chromium controller to generate single-cell gel beads in emulsion (GEMs). scRNAseq libraries were prepared with the Chromium Single Cell 3′ Library & Gel Bead Kit v.2 (10x Genomics, PN-1000075). Each sample was ligated and barcoded prior to performing library QC. The Agilent Bioanalyzer and QuantStudio 12K Flex were used to assess the library quality prior to sequencing. Libraries from different samples were pooled based on molar concentrations and sequenced on a NovaSeq 6000 instrument (Illumina) with 151 bases for read 1, 151 bases for read 2 and 8 bases for index 1. After the first round of sequencing, libraries were re-pooled on the basis of the actual number of cells in each and re-sequenced to give an equal number of reads per cell in each sample and to reach a sequencing saturation of at least 50%.
2.6. Single cell RNAseq bioinformatics analysis:
Reads were processed and aligned to the GRCh38 human reference genome using Cell Ranger (v 3.1.0) pipeline (https://github.com/10XGenomics/cellranger). For each sample, a digital gene expression matrix was generated containing the raw unique molecular identifier (UMI) counts for each cell in a given sample. Downstream analysis was performed using various functions in Seurat package (v3.1.1) (Butler et al., 2018; Satija et al., 2015; Stuart et al., 2019). Cells with fewer than 1000 detected genes or more than 7000 detected genes or more than 10% mitochondria reads were excluded. ‘Normalized Data’ function was used to normalize the count data. Highly variable genes were then extracted using ‘FindVariableFeatures’ function. We used ‘ScaleData’ function to scale the expression of each gene, so that the mean expression across cells is 0 and the variance across cells is 1 to provide equal weight to all genes.
Cell clusters were identified using ‘FindNeighbors’ and ‘FindClusters’ functions with a value of “resolution” parameter of 0.1 and with the first 20 principal components calculated by ‘RunPCA’ function. We visualized our results in a two-dimensional space using Uniform Manifold Approximation and Projection (UMAP) plots. Potential markers were identified by applying ‘FindAllMarkers’ function to each of cluster, compared to cells in all other clusters, genes identified first had to pass the preliminary filtering thresholds (only.pos = TRUE & min.pct = 0.1 & logfc.threshold = 0.25), and then were included if FDR <0.05.
Cell cycle analysis was performed using ‘CellCycleScoring’ function with the parameter “set.ident = TRUE” to set the identity of the Seurat object to the cell-cycle phase. This function uses the mean expression of S phase marker genes and G2M phase marker genes to compute standardized scores for S and G2M for each cell and then assigns each cell to the phase with the highest score. If both scores are negative, then cell is assigned to G1 phase.
Differentially expressed genes (DEG) for clusters and MeHg treatments were detected using the R package edgeR (Robinson et al., 2010) on the corresponding read matrixes. Public repository: The raw data and processed data for scRNAseq analysis are available on NCBI GEO with accession number GSE169751.
2.7. Gene Ontology (GO) analysis:
Analysis was performed for cluster markers using FDR<1e-20 gene lists from the ‘FindAllMarkers’ analysis, in which each cluster was compared against all other clusters. Analysis was performed for MeHg differentially expressed genes using FDR<0.05 for the DEG gene lists for each exposure paradigm compared to the matched vehicle for that exposure (E 0.1 μM, E 1.0 μM, E+L 0.1 μM, and E+L 1.0 μM). Gene lists were further refined and GO analysis was performed using the http://geneontology.org/ server running PANTHER 16.0 release and the human reference genome; with the GO Biological Processes Complete analysis applying the Fisher’s Exact test, and a calculated FDR set at 0.05. Processes were sorted hierarchically.
2.8. quantitative RT-PCR confirmation of MeHg-induced changes in TTR gene expression.
To determine which changes may be detectable at the level of total RNA extracted from the entire population of cells we performed a pseudo-replicate analysis of our scRNAseq data. The pseudo-replicate analysis randomly divided all the cells into an n=3, n=6 (randomize three different times as well), or n=9 data sets that Differential Expression analysis was performed on. Across all exposure group comparisons only TTR would survive FDR for the strongest effects in the E+L 1μM exposure), data not shown. Quantitative RT-PCR (qPCR) was performed as previously described (Prince et al., 2021). In brief, we prepared total RNA from both a duplicate CC3 sample collected on day 38 as well as an independent biological replicate of another control human stem cell line (CD10) that had cells collected at Day 40 (E) and Day 43 (E+L). cDNA was synthesized using SuperScript III First-Strand kit (Invitrogen, 18080051). The amount of RNA utilized was based on final RNA yield within a biological replicate and was matched between treated and control samples within a biological replicate. Oligo(dT)s were used as primers for these reactions and cDNA was stored at −20oC. For qPCR experiments, SsoAdvanced Universal Probes Supermix (Biorad, 1725280), PrimePCR Primers, and PrimePCR Assays (Biorad) were used. qPCR was performed using a CFX384 Optics Module (Biorad), with Actin used as a reference gene. ANOVA of the technical replicates across the two differentiations confirmed our scRNAseq data and revealed a significant increase in TTR expression for 1.0μM exposure vs vehicle for both, the E and the E+L exposure paradigms (data not shown).
2.9. Statistical analysis:
Changes in cluster cell populations and in cell cycle expressed as a % of the total population of all cells were analyzed by binomial analysis calculating a 95% confidence interval of the count data by the Wilson/Brown method using Prism version 9.0.2. The same method was used to calculate binomial based 95% confidence intervals for the number of genes differentially expressed between clusters and exposure paradigms using statistical cutoffs calculated by the edgeR program as noted above.
3. Results
3.1. Neural differentiation and exposure:
hiPSCs were differentiated into cortical glutamatergic neurons (Fig. 1) for a total of 38 days. Differentiating cultures were exposed to MeHg at 0.1 μM or 1 μM either at an early stage of differentiation only (E-exposure; continuously from days 4–10 of differentiation) or at the early and a slightly later time point (E+L-exposure, continuously from days 4–10 and again days 14–20). Cell counts 24 hours after the completion of the exposure on days 11 and 21 (suppl. Fig. 2) indicated no acute loss of viability, confirming our previous observations (Prince et al., 2021). Our previous assessment of the expression of neuronal lineage markers on days 12 and 22 revealed no gross interference of MeHg treatment on cortical glutamatergic differentiation on day 22 for cultures that underwent the same MeHg exposure paradigms, thus the immediate toxicological impact was either just at or below the lowest observed adverse effect concentration (LOAEC) (Prince et al., 2021). Given the known in vivo latency and persistence of developmental MeHg exposures, we sought here to test the hypothesis that more significant toxicological effects might manifest themselves at a later time point of differentiation and that subsets of developing neurons may be selectively and/or differentially affected.
Fig. 1. Differentiation of hiPSC into cortical glutamatergic neurons.
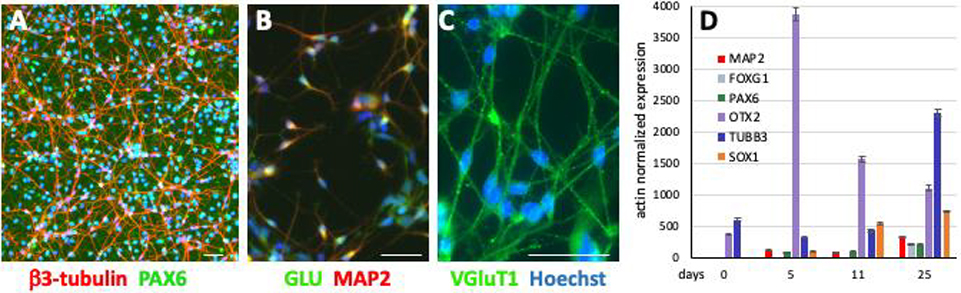
hiPSCs were differentiated into cortical glutamatergic neurons for 30 days. β3-tubulin positive neurites and PAX6 positive nuclei are abundant in these cultures (A). In addition, these neurons are also positive for MAP2 and glutamate (B), as well as VGluT1 (C). The temporal expression of the genes PAX6, FOXG1, OTX2 and SOX1 (encode neural precursor cell markers) and TUBB3 and MAP2 (encode neuronal markers) were quantified in day 25 neuronal cultures by RT-qPCR (D). (A-C: scale bars = 50 μm); (D = mean ± 95% confidence intervals, n = 6, technical replicates). Similar gene expression patterns were observed in several other hiPSC lines from control subjects as well as patients with different disease-specific mutations, an additional example of another control line is provided in suppl. Fig. 1.
3.2. Identification of cellular subtypes (clusters) in cortical cultures at day 38 of differentiation:
Cellular diversity in the developing cortex follows a specific temporal sequence (Bystron et al., 2008; Greig et al., 2013; Lodato and Arlotta, 2015). Therefore, we set out to examine if there might be persistent or latent MeHg-induced effects, if certain cellular subpopulations within the cultures might be selectively affected, or if new (aberrant) developmental lineages would emerge. In order to address these questions, we performed scRNAseq analysis on day 38 of differentiation, thus, 28 days or 18 days after completion of MeHg E- and E+L-exposures, respectively. The viabilities of cells harvested on day 38 ranged from 83.5% to 90.5%, and were not different between vehicle and MeHg treated cultures, regardless of the exposure paradigm.
Unsupervised clustering was performed on the gene expression profiles and visualized using Uniform Manifold Approximation and Projection (UMAP) plots which reveal the presence of the same 9 distinct clusters of cells (clusters 0–8) for all 6 treatment conditions with no new cell types (clusters) emerging (Fig. 2).
Fig. 2. UMAP clustering identifies 9 cell clusters.
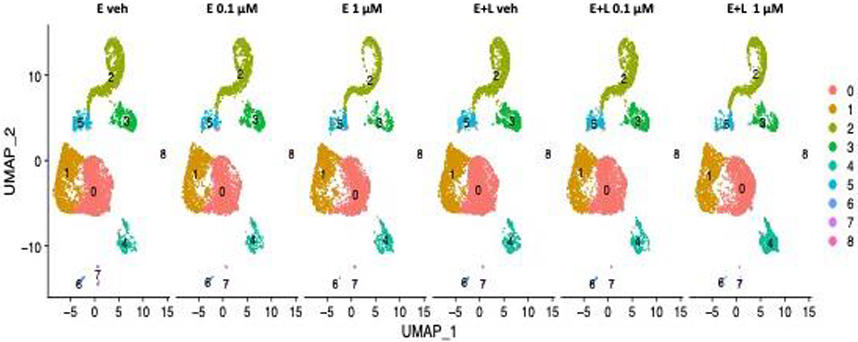
Unsupervised clustering on the gene expression profiles visualized using Uniform Manifold Approximation and Projection (UMAP) plots revealed 9 distinct clusters of cells (clusters 0–8) for all 6 treatment conditions. We identified clusters 0 and cluster 1 as radial glial cells (RG) and cluster 5 as intermediate progenitor cells (IPCs). We assigned clusters 2 and 3 immature postmitotic excitatory neurons (EN) identity of cortical (cluster 2) and thalamic (cluster 3) origin. Cluster 4 we ascribed choroid plexus precursor identity. Clusters 6–8 were predicted to be other RG and mural cells.
Marker identification (using Wilcoxon Rank Sum Test) and differential gene expression analysis (using edgeR (Robinson et al., 2010)) (suppl. Fig. 3; suppl. Table 2) combined with recently published marker enrichment analysis based on scRNAseq of human fetal cortex and human cortical organoids (Bhaduri et al., 2020; Burke et al., 2020; Camp et al., 2015; Mayer et al., 2019; Nowakowski et al., 2017; Pollen et al., 2015; Velasco et al., 2019) were used to assign identifies to the cell types represented across the 9 clusters. Table 1 provides relative expression values across all 9 clusters for key markers of cortical neurodevelopment.
Table 1. Relative expression of selected genes representing cell type markers across clusters.
Gene expression levels indicated in green (higher) to red (lower) shades are expressed on ‘ScaleData’ units so that the mean expression across cells is 0 and the variance across cells is 1 to provide equal weight to all genes. Key: RG, radial glia; IPC, intermediate progenitor cell; EN, excitatory neuron; CP, choroid plexus progenitor; EP, ependymal; VF, ventral forebrain.
| gene_id | C0 | C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | Marker type | Alt Gene Name |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PAX6 | 0.19 | 0.50 | −0.92 | −0.79 | −0.22 | 0.16 | 1.45 | −0.62 | −1.19 | RG | |
| NES | 0.43 | 0.35 | −0.82 | −0.86 | 0.85 | −0.09 | −0.82 | −0.83 | 0.46 | RG | Nestin |
| OTX1 | 0.25 | 0.22 | −0.50 | −0.58 | −0.26 | −0.16 | 0.14 | −0.33 | −0.63 | RG | |
| SOX1 | 0.14 | 0.25 | −0.45 | −0.38 | 0.04 | 0.09 | −0.46 | −0.47 | −0.45 | RG | |
| SOX2 | 0.56 | 0.65 | −1.33 | −1.21 | 0.37 | −0.66 | −1.21 | −1.13 | −1.29 | RG, EP | |
| GLI3 | 0.56 | 0.41 | −1.00 | −0.93 | −0.67 | −0.35 | −0.43 | −0.82 | −0.73 | RG | |
| GFAP | −0.02 | 0.00 | −0.05 | −0.05 | 0.27 | −0.05 | −0.05 | −0.05 | −0.05 | RG | |
| EMX2 | 0.30 | 0.32 | −0.91 | −0.83 | −0.83 | −0.26 | −1.34 | −0.79 | −1.33 | RG | |
| SLC1A3 | 0.22 | 0.19 | −0.47 | −0.46 | 0.28 | −0.33 | −0.43 | 0.89 | 1.96 | RG | |
| HES1 | 0.37 | 0.53 | −1.23 | −1.20 | −0.18 | −0.92 | −0.56 | −0.62 | −0.21 | RG | |
| VIM | 0.56 | 0.57 | −1.48 | −1.53 | 0.52 | −0.42 | 0.66 | −1.51 | 0.63 | RG | |
| FOXP1 | 0.31 | 0.27 | −0.58 | −0.61 | −0.33 | −0.25 | 0.58 | 0.16 | 0.97 | RG | |
| MKI67 | −0.31 | 1.25 | −0.61 | −0.62 | −0.30 | 0.51 | −0.37 | −0.51 | −0.45 | RG | |
| DLL1 | −0.21 | −0.06 | 0.05 | −0.31 | −0.29 | 2.94 | −0.35 | −0.36 | −0.35 | IPC | |
| EOMES | −0.25 | −0.23 | 0.25 | 0.02 | −0.21 | 2.68 | −0.23 | −0.23 | −0.27 | IPC | TBR2 |
| NEUROD1 | −0.32 | −0.31 | 1.02 | −0.08 | −0.31 | 0.91 | −0.30 | 0.86 | −0.33 | IPC | |
| NEUROD4 | −0.18 | −0.18 | 0.03 | −0.18 | −0.14 | 3.40 | −0.18 | 0.16 | −0.19 | IPC | |
| CCND1 | 0.42 | 0.31 | −0.98 | −0.96 | 0.77 | −0.53 | 0.93 | −0.35 | 0.23 | IPC | |
| NEUROG1 | −0.16 | −0.14 | 0.00 | −0.15 | −0.11 | 2.69 | −0.11 | 0.59 | −0.19 | IPC | |
| NEUROG2 | −0.17 | 0.33 | 0.10 | −0.79 | −0.79 | 1.62 | −0.79 | −0.76 | −0.80 | IPC | |
| ASCL1 | −0.08 | 0.21 | −0.28 | −0.20 | −0.05 | 1.42 | −0.37 | −0.40 | −0.38 | IPC | |
| NEUROD4 | −0.18 | −0.18 | 0.03 | −0.18 | −0.14 | 3.40 | −0.18 | 0.16 | −0.19 | IPC | |
| ASCL1 | −0.08 | 0.21 | −0.28 | −0.20 | −0.05 | 1.42 | −0.37 | −0.40 | −0.38 | IPC | |
| FOXG1 | 0.04 | −0.03 | 0.57 | −0.24 | −1.22 | −0.20 | −1.02 | −0.88 | −1.44 | EN | |
| NEUROD2 | −0.42 | −0.41 | 1.76 | −0.29 | −0.43 | −0.23 | −0.41 | −0.37 | −0.44 | EN | |
| NEUROD6 | −0.47 | −0.46 | 1.78 | −0.32 | −0.48 | 0.09 | −0.44 | −0.41 | −0.49 | EN | |
| SLC17A6 | −0.27 | −0.27 | 0.47 | 1.48 | −0.26 | −0.14 | −0.28 | −0.13 | −0.28 | EN | VGLUT2 |
| SLC17A7 | −0.30 | −0.29 | 1.28 | −0.23 | −0.30 | −0.21 | −0.30 | −0.19 | −0.31 | EN | VGLUT1 |
| MAP2 | −0.57 | −0.50 | 1.24 | 1.31 | −0.01 | 0.31 | −1.02 | −0.83 | −1.10 | EN | |
| MAPT | −0.45 | −0.47 | 1.03 | 1.98 | −0.45 | −0.47 | −0.44 | 0.16 | −0.49 | EN | |
| TUBB3 | −0.29 | −0.30 | 0.98 | 0.55 | −0.46 | 0.13 | −0.61 | −0.11 | −0.56 | EN | |
| DCX | −0.57 | −0.60 | 1.41 | 1.76 | −0.39 | 0.04 | −0.73 | −0.06 | −0.76 | EN | |
| NCAM1 | −0.34 | −0.19 | 0.70 | 1.44 | −0.73 | −0.19 | −0.86 | −0.07 | −0.82 | EN | |
| NCAM2 | −0.26 | −0.24 | 0.81 | 0.26 | −0.18 | −0.22 | 0.03 | −0.01 | 0.05 | EN | |
| FEZF1 | −0.13 | −0.12 | 0.14 | 0.44 | 0.23 | 0.16 | −0.17 | 1.21 | −0.17 | EN | |
| FEZF2 | −0.02 | 0.00 | 0.33 | −1.31 | −0.41 | 0.70 | −1.29 | −1.32 | −1.38 | EN | |
| CALB2 | −0.30 | −0.29 | 0.63 | 0.69 | −0.26 | −0.28 | −0.31 | 1.14 | −0.32 | EN | Calretinin |
| BCL11B | −0.37 | −0.32 | 1.27 | 0.00 | −0.46 | −0.10 | −0.50 | 0.27 | −0.57 | EN | |
| NFIA | −0.31 | −0.31 | 1.35 | −1.32 | 0.08 | 0.18 | −0.95 | −1.00 | −0.95 | EN | |
| NFIB | −0.27 | −0.23 | 1.33 | −1.21 | −0.70 | 0.63 | −0.77 | −0.09 | −0.64 | EN | |
| SATB2 | 0.02 | 0.01 | −0.03 | −0.14 | −0.05 | −0.01 | −0.06 | 0.00 | −0.09 | EN | |
| SYN1 | −0.25 | −0.25 | 0.60 | 1.16 | −0.22 | −0.24 | −0.21 | −0.09 | −0.17 | EN | |
| HOMER1 | −0.18 | −0.25 | 0.38 | 0.50 | 0.03 | −0.03 | 0.16 | 0.02 | 0.19 | EN | |
| TBR1 | −0.44 | −0.44 | 1.52 | 0.48 | −0.42 | −0.24 | −0.42 | −0.43 | −0.41 | EN | |
| DLG4 | −0.28 | −0.22 | 0.74 | 0.53 | −0.19 | −0.24 | −0.20 | −0.27 | −0.41 | EN | |
| MAP2 | −0.57 | −0.50 | 1.24 | 1.31 | −0.01 | 0.31 | −1.02 | −0.83 | −1.10 | EN | |
| BDNF | −0.07 | −0.07 | 0.00 | 0.14 | 0.36 | −0.08 | 0.01 | 0.29 | 0.59 | EN | |
| DLX2 | −0.06 | −0.06 | −0.03 | 0.60 | −0.07 | −0.07 | −0.08 | 0.09 | −0.08 | VF | |
| FOXP2 | −0.05 | −0.05 | −0.11 | 0.66 | −0.16 | −0.09 | 0.63 | 0.11 | 0.44 | VF | |
| GBX2 | −0.04 | −0.04 | −0.04 | 0.09 | 0.17 | 0.00 | 0.04 | −0.04 | −0.04 | VF | |
| PBX3 | −0.15 | −0.03 | −0.42 | 2.61 | −0.19 | −0.30 | 0.14 | 0.39 | −0.07 | VF | |
| MEIS2 | 0.18 | 0.02 | −0.52 | 1.73 | −0.67 | −0.63 | 0.17 | 0.24 | −0.67 | VF | |
| PCP4 | −0.20 | −0.21 | −0.10 | 0.06 | 2.04 | −0.22 | 0.78 | 0.32 | −0.09 | CP | |
| NEAT1 | −0.11 | −0.20 | −0.35 | −0.10 | 1.77 | −0.20 | 4.22 | 0.81 | 1.90 | CP | |
| RSPO1 | −0.17 | −0.16 | −0.17 | −0.16 | 2.44 | −0.16 | −0.13 | −0.15 | −0.17 | CP | |
| RSPO2 | −0.20 | −0.21 | −0.21 | −0.19 | 2.90 | −0.19 | −0.18 | −0.21 | −0.21 | CP | |
| RSPO3 | −0.22 | −0.29 | 0.25 | −0.15 | 2.59 | −0.28 | −0.36 | −0.37 | −0.34 | CP | |
| IGFBP7 | −0.19 | −0.19 | −0.19 | −0.19 | 1.34 | −0.19 | 5.63 | 0.68 | 3.23 | CP | |
| TTR | −0.20 | −0.20 | −0.18 | −0.18 | 1.57 | −0.20 | −0.19 | −0.24 | −0.23 | CP | |
| CXCL14 | −0.13 | −0.10 | −0.14 | −0.13 | 1.00 | −0.14 | 1.19 | 5.73 | −0.11 | CP | |
| TRPM3 | −0.20 | −0.20 | −0.21 | −0.09 | 1.84 | −0.22 | 4.89 | −0.15 | −0.20 | CP | |
| AQP1 | −0.03 | −0.04 | −0.01 | −0.06 | 0.11 | −0.03 | 0.60 | −0.07 | −0.07 | CP | |
| OTX2 | −0.20 | −0.19 | −0.21 | −0.19 | 2.31 | −0.15 | 4.15 | −0.08 | −0.22 | CP | |
| KCNE2 | −0.03 | −0.03 | 0.01 | −0.04 | 0.01 | −0.05 | 0.03 | −0.05 | −0.05 | CP | |
| FOXJ1 | −0.03 | −0.06 | −0.19 | −0.20 | 1.10 | −0.13 | −0.01 | −0.21 | −0.21 | CP, EP |
In aggregate this analysis suggested that cluster 0 and 1 are radial glia (RG) cells based on their expression of the dorsal telencephalon lineage- and early progenitor markers (GLI3, SOX1, SOX2, OTX1, HES1, VIM, NES) (Table 1). Both clusters also express PROM1, a marker for apical (inner) RG and lacked substantial expression of early basal (outer) RG markers (INSM1, EOMES, HES6) suggesting that cluster 0 and 1 are apical (inner) RG (Camp et al., 2015). GO analysis for biological processes supported our identification of RG for cluster 0, highlighting biological processes such as forebrain radial glial cell differentiation (Enrichment Factor (EF) = 67.6), negative regulation for forebrain neuron differentiation (EF = 67.6), and several processes involved in cell replication (suppl. Table 3A). Comparison of the expression levels of the proliferation marker Ki-67 (MKI67) and the number of cells at G2/M phase of the cell cycle, as well as a cell cycle analysis (Fig. 4) indicates that the cells in cluster 1 proliferate more actively than their counterparts in cluster 0. This was further confirmed by our GO analysis, for which the large majority of the processes identified for cluster 1 relate to mitosis, cell division and proliferation (suppl. Table 3B). A similar observation of two apical RG cell clusters that mainly differ in their proliferation activity has been described for fetal neocortex (Camp et al., 2015). Gene expression of Cluster 5 suggests a basal progenitor identity which include the basal (outer) RG and intermediate progenitor cells (IPCs) based on their high expression of established outer RG/IPC markers (EOMES/TBR2, NEUROD1, NEUROD4, NEUROG1, NEUROG2, ASCL1) with a substantial subset expressing the proliferation marker MKI67 (Table 1). GO analysis supports the suggested identification of cluster 5 cells as IPCs and highlights cell processes such as NE differentiation (EF = 23.5), cerebral cortex neuron differentiation (EF = 13.9) as well as cellular processes indicating on-going mitosis as well as exit from mitosis as expected for this stage of neurodevelopment (suppl. Table 3C). Based on their strong expression of DCX, TUBB3, MAPT, NCAM½ we assigned Clusters 2 and 3 a postmitotic immature glutamatergic excitatory neurons (EN) identity. Cluster 2 cells express VGLUT1, FOXG1, NEUROD2/D6 and are also positive for FEZF2 and marker expression consistent with a layer V/VI cortical identity (e.g. BCL11B) (Table 1). Cluster 3 ENs express the ventral forebrain markers (DLX2, FOXP2), but not FOXG1, NEUROD2 or NEUROD4. This expression pattern in conjunction with the strong expression of VGLUT2, GBX2, PBX3 and FOXP2, thalamic markers (Co et al., 2020) suggests a thalamic (or other subpallial) glutamatergic EN identity for cluster 3 cells (Table 1). Cellular processes indicated by GO analysis include central nervous system maturation (EF = 12.53), positive regulation of synapse maturation (EF = 12.2), axon extension involved in axon guidance (EF = 11.0) as well as several processes involved in cellular transport along microtubules (suppl. Tables 3D, E). Cluster 4 expresses multiple markers consistent with anterior telencephalon choroid plexus progenitors (AQP1, OTX2, SIX3, NES, TRPM3, PCP4, RSPO2, NEAT1, TTR, IGFBP7, KCNJ13) (Bar et al., 2020; Nakamura et al., 1999; Shi et al., 2020; Sivitilli et al., 2020; Trillo-Contreras et al., 2019) (Table 1). GO analysis identified cellular processes such as maintenance of blood-brain barrier (EF = 8.43), establishment of apical/basal cell polarity (EF = 8.1), negative regulation of neural precursor cell proliferation (EF 8.4) and regulation of sodium ion export across pasma membrane (EF = 26.9), all of which support the proposed choroid plexus precursor identity of cluster 4 cells (suppl. Table 3F). Clusters 6, 7 and 8 collectively represent less than 2% of all cells and are predicted to be other radial glia and mural cell types (suppl. Fig. 3; suppl. Table 2). Therefore, scRNAseq allows us to examine gene expression patterns across three distinct stages of cortical development, the apical RG cells (Clusters 0/1), the basal progenitors, outer RG and IPC (cluster 5), and ENs (clusters 2 and 3), as well as co-differentiating choroid plexus progenitors (cluster 4), all of which are co-existing in our differentiations at day 38. Thus, developing cortical glutamatergic neuronal cultures differentiated from hiPSC for 38 days consist of RG (clusters 0 and 1, 63%), IPC (cluster 5, 5%), cortical ENs (cluster 2, 18%), thalamic ENs (cluster 3, 8%), and choroid plexus precursors (4%). Clusters 6–8 made up less than 2% of the overall cell population, and were not further included in the analyses.
Fig. 4. MeHg causes small changes in the cell cycle.
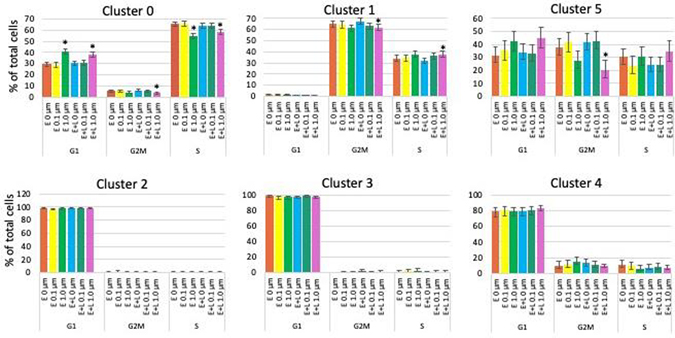
The percentages of cells in G2M, S or G1 phase are plotted. Cells in clusters 2, 3 and 4 are to a large extent postmitotic and not affected by MeHg. E or E+L exposures at 1 μM significantly decreased the percentage of cells in G2M and S phase and increased the population in G1 phase of cluster 0 cells. Clusters 1 and 5 cells were only affected by E+L at 1 μM exposure which caused a decrease in the percentage of G2M phase cells in both clusters and a small increase in the percentage of S phase in cluster 1 cells. Exposures at 0.1 μM did not result in any significant changes. Percent of the total cell population with 95% confidence intervals are shown.
3.3. Effect of developmental MeHg exposure on the population size of different cell types (clusters):
We then examined if MeHg exposure affected one or more of the cell populations (clusters) selectively. We quantified and compared the number of cells in each cluster for each exposure condition by performing binomial analyses calculating a 95% confidence interval of the count data using the Wilson/Brown method. Exposures at the lower (0.1 μM) MeHg concentration did not result in any significant changes in cell numbers for any clusters. E-exposure at 1 μM significantly increased cluster 0 (RG) population, whereas E+L 1 μM decreased the percentage of cells in cluster 1 (RG). E and E+L exposure at 1 μM decreased the cell population of the thalamic ENs (cluster 3) but increased the percentage of choroid plexus precursors (cluster 4) (Fig. 3).
Fig. 3. MeHg causes minor changes in population size in a cell type (cluster)- and exposure paradigm-specific manner.
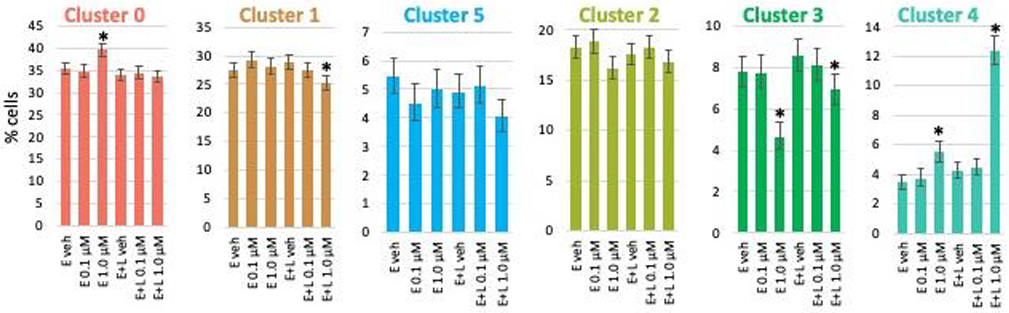
The percentage of cells making up each cluster for each exposure condition was determined. Exposures at the lower (0.1 μM) MeHg concentration did not result in any significant changes of cell numbers for any clusters. E-exposure at 1 μM increased cluster 0 (RG) population, whereas E + L 1 μM decreased cluster 1 (RG) cell percentage. E and E + L exposure at 1 μM decreased the cell population of the thalamic glutamatergic neurons (cluster 3) but increased the percentage of choroid plexus precursors (cluster 4). Percent of the total cell population with 95% confidence intervals are shown.
Thus, prior developmental MeHg exposure at 1 μM (but not 0.1 μM) resulted in small but statistically significant cell type- and exposure paradigm-specific changes in cell populations at day 38 of differentiation, a time point 18 or 28 days after the completion of MeHg exposure, but did not cause a complete (or substantial) loss or the appearance of a new cell type (as would be indicated by identification of a new cluster in the exposed samples). The Seurat software applies modularity optimization technique (Louvain algorithm) to iteratively group cells together based on K-nearest neighbor (KNN) graph structure. While creation of a new cluster or assignment to existing cluster depends on the gene expression of cells rather than the number of cells, presence of a new cell type with expression differences comparable to the other cell types already clustered should be detectable even if the proportion of the cells is very low. The “resolution” parameter is used to control the granularity of the clustering. Based on our domain knowledge of known cell types and marker genes, we used resolution set at 0.1 which was sufficient to detect the 4 cells that cluster 8 has in the E+L 1μM group. Thus, we believe if there was a group of cells whose expression pattern differed by at least a similar degree as the patterns in the identified clusters, that we would have been able to detect such a cluster if it had even as low as 4 cells.
3.4. MeHg-induced apoptosis and effects on glutathione metabolism
MeHg induced apoptosis has been reported in in vivo and in vitro developing neurons (Ceccatelli et al., 2010; Chang et al., 2013). We assessed potential MeHg-indcued changes in apoptotic pathways of day 38 cultures by GO analysis. The most frequently and most prominently enriched apoptotic pathways included apoptotic mechanisms involving: protein insertion into mitochondrial membrane, mitochondrial membrane permeabilization, p53 class mediator and cysteine-type endopeptidase activity (Table 2). These apoptotic pathways were generally modestly enriched with EFs ranging from > 1.4 to <10, with a couple of exceptions that include apoptotic pathways involving p53 class mediator in clusters 0 and 1 exposed to E+L at 0.1 μM and cluster 4 exposed to E+L 1 μM with EFs ranging between 26–35. Apoptotic pathway involving protein insertion into mitochondrial membrane were enriched in clusters 3 and 5 exposed to E at 0.1 μM by an EF of 14. Most importantly, apoptotic pathways were affected in a cluster- and MeHg treatment-specific way, with different apoptotic pathways being implicated in only a subset of clusters by each exposure paradigm.
Table 2: Enrichment of apoptotic pathways is cluster and MeHg-exposure paradigm-specific.
Enrichment of apoptotic pathways in day 38 developing cortical cultures were extracted form our Go analysis (suppl. Tables 5–8) for each cell type (cluster) and at each MeHg treatment paradigm (E at 0.1 μM, E at 1 μM, E+L at 0.1 μM and E+L at 1 μM). The enrichment factor (EF) is listed for each pathway.
| MeHg exposure: E at 0.1 μM | ||
| Cluster | pathway | EF |
| 0 | regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway | 3.44 |
| 0 | regulation of intrinsic apoptotic signaling pathway | 2.28 |
| 1 | release of cytochrome c from mitochondria apoptotic process | 1.73 |
| 1 | positive regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway | 4.15 |
| 1 | regulation of apoptotic process | 1.38 |
| 5 | positive regulation of protein insertion into mitochondrial membrane involved in apoptotic signaling pathway | 14.14 |
| 2 | positive regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway | 4.44 |
| 3 | positive regulation of protein insertion into mitochondrial membrane involved in apoptotic signaling pathway | 14.21 |
| MeHg exposure: E at 1.0 μM | ||
| Cluster | pathway | EF |
| 0 | regulation of oxidative stress-induced intrinsic apoptotic signaling pathway | 4.02 |
| 0 | positive regulation of apoptotic process | 1.73 |
| 0 | apoptotic process | 1.51 |
| 0 | negative regulation of apoptotic process | 1.49 |
| 1 | negative regulation of apoptotic signaling pathway | 2.59 |
| 1 | positive regulation of apoptotic process | 1.95 |
| 1 | apoptotic process | 1.58 |
| 2 | mitochondrial outer membrane permeabilization positive regulation of mitochondrial membrane permeability involved in apoptotic process | 5.04 |
| 2 | negative regulation of intrinsic apoptotic signaling pathway | 3.14 |
| 2 | regulation of cysteine-type endopeptidase activity involved in apoptotic process | 2.14 |
| 2 | positive regulation of apoptotic process | 1.68 |
| MeHg exposure: E+L at 0.1 μM | ||
| Cluster | pathway | EF |
| 0 | positive regulation of intrinsic apoptotic signaling pathway by p53 class mediator | 26.46 |
| 1 | positive regulation of intrinsic apoptotic signaling pathway by p53 class mediator | 35.41 |
| 1 | regulation of intrinsic apoptotic signaling pathway in response to DNA damage | 7.97 |
| MeHg exposure: E+L at 1.0 μM | ||
| Cluster | pathway | EF |
| 0 | regulation of apoptotic process | 1.58 |
| 0 | positive regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway | 3.85 |
| 0 | positive regulation of cysteine-type endopeptidase activity involved in apoptotic process | 2.23 |
| 0 | regulation of neuron apoptotic process | 2.02 |
| 0 | negative regulation of apoptotic signaling pathway | 1.90 |
| 0 | positive regulation of apoptotic process | 1.82 |
| apoptotic process | 1.64 | |
| 1 | negative regulation of striated muscle cell apoptotic process | 5.58 |
| 1 | regulation of cysteine-type endopeptidase activity involved in apoptotic process | 2.21 |
| 1 | positive regulation of apoptotic process | 2.03 |
| apoptotic process | 1.61 | |
| 2 | activation of cysteine-type endopeptidase activity involved in apoptotic process | 5.25 |
| 2 | positive regulation of apoptotic process | 2.48 |
| 4 | positive regulation of intrinsic apoptotic signaling pathway by p53 class mediator | 25.85 |
| 4 | negative regulation of apoptotic process | 1.84 |
MeHg exposures of hiPSC-derived cortical cultures exposed under the exact same MeHg exposure paradigms did not result in acute changes in cellular glutathione (GSH) levels (Prince et al., 2021). Here we assessed for a delayed effect of MeHg on cellular GSH levels by analyzing our data for the enrichment of cellular processes involving glutathione metabolism (GO analysis) and were unable to identify any cellular processes directly involved in cellular glutathione metabolism. In addition we checked for changes in the expression of genes coding for enzymes directly involved in GSH metabolism (GCL, GSR, GSS, GST (all isoforms)) and none of them were significantly changed >2-fold (FDR <0.05). We conclude that MeHg has no significant effect of cellular GSH levels either acutely or delayed in developing hiPSC-derived cortical cultures.
3.5. Effect of developmental MeHg exposure on the cell cycle:
Based on previous observations (Bose et al., 2012; Burke et al., 2006; Vogel et al., 1986; Yuan et al., 2018) we examined the persistent effects of MeHg on the cell cycle at day 38 using the same statistical methods described above. In control cultures more than 95 % of ENs (clusters 2 and 3) are in G1 phase of the cell cycle and therefore no longer undergoing mitosis (Fig. 4). This is in line with their identification based on gene expression analysis as early postmitotic neurons. Approximately 70% of the IPCs (cluster 5) are in G2M/S phase of the cell cycle and thus still proliferative, with the rest being in G1 phase. GO analysis also highlights cell processes involved in both, on-going mitosis as well as exit from mitosis (suppl. Table 3C). Interestingly the proliferation activities of the two RG cell populations are quite distinct, with cluster 1 showing >99% of the cells proliferating, whereas only 70% cluster 0 are in G2M/S phase, an observation also highlighted by the preponderance of cellular processes indicated by GO analysis involved in cell proliferation, mitosis, and cell division in cluster 1 (suppl. Table 3B).
Only the higher MeHg concentration (1 μM) resulted in cell cycle changes. Cluster 0 cells are the only cell type affected by both, E and E+L exposures. In these cells MeHg slightly but significantly decreased the percentage of cells in G2M and S phases and increased the number of cells in G1 phase, indicating a decreased proliferative activity (Fig. 4). For cluster 1 cells we observed a decrease in G2M phase, and concomitant increase in S-phase, possibly indicating an elevated rate of cell cycle progression. Cluster 5 cells show reduced percentage of cells in G2M phase only (Fig. 4). Cells in clusters 2, 3 and 4 were to a large extent postmitotic and not affected by MeHg. Thus, MeHg had only minor effects on the cell cycle for the remaining proliferative cell types and only at the higher (1 μM) exposure concentration.
In addition to changes in the percentage of cells in different stages of the cell cycle we also observed subtle but significant changes in a subset of cell cycle regulatory genes that have previously been reported to be affected by MeHg in other model systems (Fujimura and Usuki, 2015; Ou et al., 1999; Xu et al., 2010). Thus, in cluster 1 cells we observed a small, but significant 1.3-fold down regulation of p21 (CDKN1A) after E and E+L exposure at 1 μM with a concomitant small but significant 1.2-fold upregulation of CDK2 after E+L at 1 μM (suppl. Table 4). Cluster 0 cells exposed under the E+L at 1 μM paradigm responded with a 1.2-fold decrease in cyclin E1 (CCNE1) expression, a gene not affected in cluster 0 cells (suppl. Table 4). Thus, consistent cell type-specific changes in the expression of cell cycle related genes were present in the most proliferative cell types following developmental MeHg exposure conditions where we also saw changes in the percentage of cells in different phases of the cell cycle.
3.6. MeHg effects on gene expression:
In order to obtain more detailed insight into the effects MeHg on in vitro cortical neurodevelopment, we compared gene expression patterns between clusters and MeHg exposure paradigms. We determined the number of genes from a total of 33538 genes identified in the scRNAseq, with significant changes in expression levels (FDR < 0.05) and observed cluster- and MeHg exposure paradigm-specific differences (Fig. 5, suppl Fig. 4 and 5).
Fig. 5. MeHg causes cluster- and exposure paradigm-specific changes in gene expression.
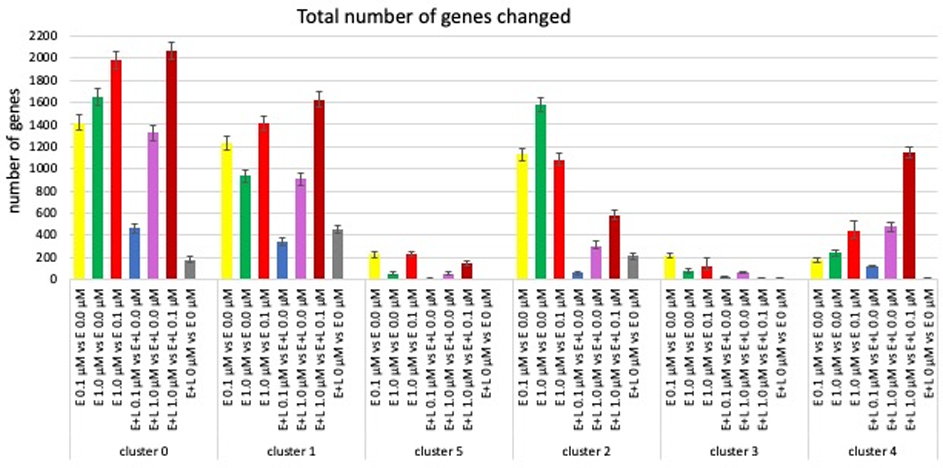
The number of genes with significantly (FDR < 0.05) changed expression from a total of 33538 genes assessed are shown. The grey bars indicate differences in gene expression between two control cultures. The red bars show number of genes differentially expressed between a low (0.1 μM) and high (1.0 μM) exposure. (Error bars represent 95% confidence levels).
Thus, as might be expected exposure E+L at 1 μM MeHg changed the expression of significantly more genes than E+L at 0.1 μM in all cell types (clusters), whereas such a concentration-dependent effect for E exposures at 0.1 and 1.0 μM was not observed, rather the relative size of the response there varied among clusters. The largest number of genes differentially expressed is observed in a comparison between exposures at 1 μM versus 0.1 μM (for E or E+L exposures) illustrating that MeHg at 0.1 μM or 1.0 μM largely impacts different sets of genes (Fig. 5, red bars; suppl Fig. 5). Most strikingly, a one-time exposure early in development, E at 0.1 μM affected a significantly larger number of genes at day 38 of differentiation than a repeated exposure E+L at 0.1 μM in all clusters, with the changes in expression levels at E+L at 0.1 μM in clusters 1, 2 and 5 not significantly higher than the number of genes differentially expressed between the two vehicle-treated cultures (Fig. 5, blue vs grey bars). This suggests that a moderate second exposure (0.1 μM) not only does not cause further changes, but might actually reverse already initiated changes, a phenomenon observed by others (Dahl and Balfour, 1964; Dhodda et al., 2004; Stenzel-Poore et al., 2007; Zeiger et al., 2010). The effect of MeHg on gene expression also appears to be cell type-specific, with clusters 0 and 2 showing the largest overall changes in gene expression at exposure E 1 μM, clusters 1, 3 and 5 at exposure E 0.1 μM while cluster 4 is most most significantly affected by E+L at 1 μM. (Fig. 5; suppl. Fig. 4).
Using a more stringent criteria we examined the number and identity of genes with changes of expression >2 fold up or down (FDR <0.05) (Fig. 6). Exposures E at 0.1 μM or 1.0 μM resulted in a comparable number of gene expression changes amongst all clusters, except for the choroid plexus precursors (cluster 4) which showed a significantly 10-fold higher number of genes with changed expression levels > 2-fold at E at 1.0 μM than any of the other clusters. E+L-exposure at 0.1 μM did not result in any gene expression changes >2-fold in either direction for clusters 0, 1 and 2, only 2 changes in clusters 3 and 5, and cluster 4 again with significantly higher number of genes (14) being affected. The number of genes with expression changes due to the E+L 1 μM exposure was more homogenous among the clusters. More striking were the differences in the number of genes altered depending on the exposure paradigm within each cluster (Fig. 6, suppl. Fig. 5, red circles).
Fig. 6. MeHg causes cluster- and exposure paradigm-specific changes in gene expression.
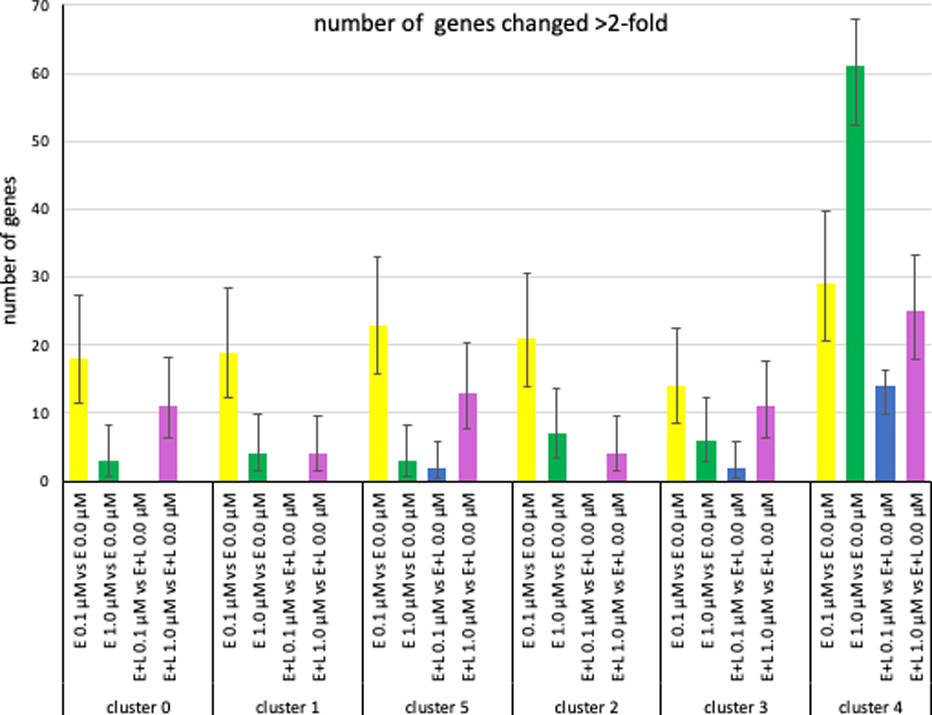
The number genes with changes in expression levels >2-fold (FDR< 0.05) are plotted. (Error bars represent 95% confidence intervals).
When looking at the effect of MeHg concentration, somewhat surprisingly, for all clusters, except clusters 3 and 4, E-exposure at the lower 0.1 μM MeHg concentration resulted in more gene expression changes than E-exposure at 1 μM, while for cluster 3 there was no significant difference between the two and for cluster 4 the number was significantly higher at 1.0 μM than 0.1 μM (Fig. 6, suppl. Fig. 5). A very different outcome was observed for E+L exposure where in all clusters the higher 1 μM concentration resulted in a significantly higher number of genes with > 2-fold expression than the lower 0.1 μM exposure (Fig. 6). Looking at the effect of exposure frequency, we observed that for all clusters a one-time exposure E at 0.1 μM resulted in a significantly higher number of changes than a two-hit E+L-exposure at the same concentration, whereas the equivalent comparison at 1 μm showed a much more varied pattern of response amongst the different clusters (Fig. 6; suppl. Fig. 4, 5).
We then looked at the identity of the genes changing >2-fold (FDR < 0.05). E-exposure at 0.1 μM MeHg changed between 14–23 genes in the neuronal clusters 0 (18 genes), 1 (19 genes), 5 (23 genes), 2 (21 genes) 3 (14 genes) and interestingly, all these genes without exception, were up-regulated (Fig. 6, Table 3A, suppl. Fig. 4A). About 85% of the genes changed in cluster 0 and 1 were identical and contained a high fraction of genes representing ribosomal and mitochondrial components (Table 3A). Out of the 23 genes altered in cluster 5 (IPC), 15 were also changed in clusters 0 and/or 1 and the majority of them encode ribosomal or mitochondrial components. 12 genes showing > 2-fold changes were shared between clusters 2 (12 out of 23) and cluster 3 (12 out of 14), 7 of which encode ribosomal and mitochondrial components. In cluster 4 29 genes showed altered expression with a similar number of genes being up- and down-regulated. Twelve of these genes were shared with at least 3 of the neuronal clusters and 9 of these twelve encode ribosomal and mitochondrial genes. In summary, there is a preponderance of ribosomal and mitochondrial genes whose expression was changed by E 0.1 μM exposure (but not any of the other three exposure paradigms, see below), an observation confirmed by GO analysis which also highlighted a preponderance of cellular processes involving ribosomes/translation and mitochondria for all clusters (suppl. Table 5A–F). In addition, 52% of the genes with changed gene expression in cluster 4 were unique to cluster 4, while the equivalent number of uniquely changed genes is smaller for the neuronal clusters (cluster 0 = 0%; cluster 1 = 0%, cluster 2 = 10%; cluster 3 = 14%, cluster 5 = 35%) (Table 3A).
Table 3A:
E at 0.1 μM Exposure cluster comparison
| Genes | Cluster 0 | Cluster 1 | Cluster 5 | Cluster 2 | Cluster 3 | Cluster 4 |
|---|---|---|---|---|---|---|
| AL355075.4 | 2.04 | |||||
| AL360012.1 | 2.09 | |||||
| ANKRD1 | 0.44 | |||||
| ARHGAP11B | 2.05 | |||||
| ATF3 | 0.41 | |||||
| ATP5ME | 2.23 | 2.27 | 2.21 | 2.26 | 2.18 | 2.11 |
| C11orf98 | 2.002 | |||||
| CEBPB | 0.49 | |||||
| COX16 | 2.13 | 2.15 | 2.05 | |||
| CYTOR | 0.48 | |||||
| DHX8 | 2.37 | |||||
| DLK1 | 4.36 | |||||
| DLX1 | 2.08 | |||||
| EMX2 | 2.08 | |||||
| GPM6A | 2.19 | |||||
| GPNMB | 0.41 | |||||
| HES1 | 2.004 | 2.13 | ||||
| HIST1H4C | 2.01 | 2.25 | 2.39 | |||
| HYOU1 | 2.02 | |||||
| IER3 | 0.45 | |||||
| IQCH-AS1 | 2.08 | |||||
| MEG3 | 3.56 | |||||
| MRPS24 | 2.50 | 2.63 | 2.14 | 2.34 | 2.78 | |
| MT-ATP8 | 2.09 | |||||
| MT-ND3 | 2.24 | 2.32 | 2.31 | 2.32 | 2.12 | 2.03 |
| MT-ND4L | 2.40 | 2.29 | 2.55 | 2.32 | 2.44 | 2.18 |
| NDUFA3 | 2.09 | 2.1 | 2.07 | 2.18 | 2.11 | |
| NDUFB1 | 2.17 | 2.29 | 2.32 | 2.13 | ||
| NME2 | 4.23 | 4.12 | 3.16 | 3.6 | 2.37 | 4.36 |
| NPPB | 0.32 | |||||
| NUDCD3 | 2.07 | |||||
| PAM16 | 2.06 | 2.4 | 3.03 | 2.55 | 2.17 | 2.71 |
| RNASEK | 2.04 | 2.15 | 2.18 | 2.68 | 2.51 | 2.86 |
| RPL17 | 2.12 | 2.31 | 2.15 | 2.18 | 2.03 | |
| RPL37A | 2.04 | |||||
| RPL38 | 2.03 | 2.06 | 2.07 | 2.08 | ||
| RPP21 | 2.01 | |||||
| RPS27 | 2.11 | 2.22 | 2.13 | 2.19 | 2.07 | 2.02 |
| RPS29 | 2.41 | 2.49 | 2.36 | 2.55 | 2.47 | 2.37 |
| S100A6 | 0.44 | |||||
| SNHG25 | 2.17 | 2.25 | 2.50 | |||
| SNHG9 | 2.03 | 2.35 | 2.01 | 2.05 | ||
| SPTY2D1OS | 2.07 | |||||
| TLN1 | 2.08 | |||||
| TOMM5 | 2.09 | 2.08 | 2.13 | |||
| TXNDC16 | 2.02 | |||||
| UBE2V1 | 2.18 | 2.31 | 2.47 | 2.55 | 2.16 | 2.50 |
| ZSCAN16-AS1 | 2.14 | 2.04 | ||||
| total | 18 | 19 | 23 | 21 | 14 | 29 |
| % up | 100 | 100 | 100 | 100 | 100 | 72.4 |
| % down | 0 | 0 | 0 | 0 | 0 | 27.6 |
| % cluster specific genes | 0 | 0 | 34.8 | 9.5 | 14.3 | 51.7 |
E exposure at 1.0 μM resulted in significantly fewer genes showing a >2 fold (up or down) change in expression in the neuronal clusters (between 3–7 genes), but an increased number in cluster 4 (61 genes) when compared to E at 0.1 μM exposure (Fig. 6, Table 3B, suppl. Fig. 4, 5). Except for the one unique gene in cluster 1, all genes differentially expressed were shared between clusters 0 and 1 and were changed to the same degree and in the same direction and most importantly they were all different from the genes changed by E at 0.1 μM exposure (Table 4). GO analysis further suggested different cellular processes are affected by E-exposures at 0.1 μM and 1.0 μM for clusters 0 and 1, with cerebral cortex regionalization and radial glial cell differentiation processes being the most affected by 1.0 μM (suppl. Tables 6A, B), rather than the ribosome/translation and mitochondria involving cellular processes affected by E at 0.1 μM mentioned above (suppl. Tables 5A, B). Of the 3 genes affected in cluster 5 cells two are involved in forebrain neuron development (ID1 and NR2FR) (Table 3B). GO analysis for cluster 5 shows enrichment in forebrain regionalization as well as ribosome/translation processes (suppl. Table 6C). The genes affected in the two excitatory neuron clusters 2 and 3 are distinct from each other, except for one gene (NTS), but include several genes involved in forebrain neuronal differentiation (Table 3B). GO analysis highlights mitochondrial electron transport for cluster 2 and mitochondrial ATP synthesis coupled proton transport and ribosomal processes/translation for cluster 3 (suppl. Table 6 D, E). Most importantly, all of the genes affected by E at 1.0 μM in the neuronal clusters and the large majority (60 out of 61 genes) in cluster 4 are different from the genes affected by E at 0.1 μM (Table 4). Thus, E at 0.1 μM and 1.0 μM exposure paradigms lead to drastically different changes in gene expression patterns in all six cell types (clusters) assessed.
Table 3B:
E at 1 μM Exposure cluster comparison
| Genes | Cluster 0 | Cluster 1 | Cluster 5 | Cluster 2 | Cluster 3 | Cluster 4 |
|---|---|---|---|---|---|---|
| ACTA2 | 0.24 | |||||
| ADAMTS1 | 0.45 | |||||
| AL138826.1 | 0.24 | |||||
| ALDH1A1 | 0.36 | |||||
| ANKRD1 | 0.40 | |||||
| ATP2B2 | 2.09 | |||||
| CALB2 | 2.38 | |||||
| CCK | 3.92 | |||||
| CCL2 | 0.27 | |||||
| CCNO | 4.12 | |||||
| CDC20B | 2.08 | |||||
| CFI | 0.39 | |||||
| COL4A1 | 0.50 | |||||
| COL9A3 | 2.03 | 2.03 | 2.07 | |||
| CRH | 0.29 | |||||
| DCT | 0.24 | |||||
| DLK1 | 4.47 | |||||
| ECEL1 | 2.07 | |||||
| EFNA5 | 0.42 | |||||
| ERICH5 | 0.43 | |||||
| FAM69C | 0.47 | |||||
| FBN2 | 0.48 | |||||
| FSTL5 | 2.14 | |||||
| GALNT5 | 2.23 | |||||
| GPNMB | 0.38 | |||||
| HECW1 | 2.1 | 2.1 | ||||
| ID1 | 2.04 | 2.11 | ||||
| IGF1 | 2.48 | |||||
| KCNJ13 | 2.23 | |||||
| KCTD8 | 2.26 | |||||
| KLHDC8A | 2.00 | |||||
| LGALS3 | 0.46 | |||||
| LHX1 | 2.23 | |||||
| LINC00982 | 2.05 | |||||
| LINGO1 | 2.11 | |||||
| LRRC17 | 0.40 | |||||
| LUM | 0.31 | |||||
| LYPD1 | 0.39 | |||||
| MAB21L2 | 0.49 | |||||
| MEG3 | 2.06 | |||||
| MYL9 | 0.35 | |||||
| NEFM | 0.38 | |||||
| NEUROD6 | 2.63 | |||||
| NEUROG1 | 2.47 | |||||
| NPPB | 0.30 | |||||
| NR2F2 | 0.4 | 0.42 | ||||
| NRN1 | 2.62 | |||||
| NTN1 | 0.48 | |||||
| NTS | 3.61 | 3.7 | 0.29 | |||
| NUPR1 | 0.37 | |||||
| OTP | ||||||
| PDZRN3 | 2.23 | |||||
| PLAC9 | 0.48 | |||||
| PLK2 | 0.45 | |||||
| PLS3 | 2.26 | |||||
| PMCH | 7.35 | |||||
| PMEL | 0.13 | |||||
| PPP1R14C | 0.43 | |||||
| RBP1 | 0.33 | |||||
| S100A11 | 0.47 | |||||
| SEMA6D | 0.48 | |||||
| SFRP2 | 0.26 | |||||
| SIX3 | 0.39 | 0.41 | 0.25 | |||
| SLIT1 | 0.48 | |||||
| SNTG1 | 2.72 | |||||
| SST | 0.07 | |||||
| TAGLN | 0.19 | |||||
| TPM1 | 0.38 | |||||
| TPM2 | 0.50 | |||||
| TTR | 4.72 | |||||
| TWIST1 | 0.32 | |||||
| UNC5A | 2.96 | |||||
| WLS | 2.01 | |||||
| XIST | 2.41 | |||||
| ZFYVE16 | 2.04 | |||||
| total | 3 | 4 | 3 | 7 | 6 | 61 |
| % up | 67 | 75 | 67 | 71 | 50 | 37.7 |
| % down | 33 | 25 | 33 | 14 | 50 | 62.3 |
| % cluster specific genes | 0 | 25 | 0 | 71.4 | 50.0 | 98.4 |
Table 4: Comparison of gene expression changes induced by the different MeHg exposure paradigms within each cluster.
The data contained in this table is the same as presented in Tables 2A–D, but presented in a fashion that allows the easy comparison of the differential effect of the four exposure paradigms within each cluster. All genes with expression changes > 2-fold (FDR 0.05) are listed for the four different exposure paradigms. Numbers indicated represent fold change, values > 0 indicate up-regulation, values < 0 indicate down regulation. Five groups of genes particularly relevant to this analysis are highlighted in colors as indicated. The assignment of those genes to the particular groups is based on information from several different sources including https://medlineplus.gov/genetics/gene/; https://www.ncbi.nlm.nih.gov/gtr/genes/; https://www.genecards.org/; www.uniprot.org/uniprot/; https://pathcards.genecards.org/card/; and the literature cited in the text.
| ribosomal component | actin dynamics/muscle tissue | |||
| mitochondrial component | cell cycle | |||
| forebrain/neuron development | ||||
| Exposure paradigm comparison by cluster | ||||
| Gene | E 0.1 μM | E 1 μM | E+L 0.1 μM | E+L 1.0 μM |
| Cluster 0 | ||||
| ACTC1 | 5.38 | |||
| ATP5ME | 2.23 | |||
| COL9A3 | 2.03 | 2.05 | ||
| COX16 | 2.13 | |||
| HECW1 | 2.1 | |||
| ID3 | 2.18 | |||
| MRPS24 | 2.50 | |||
| MSX2 | 2.15 | |||
| MT-ND3 | 2.24 | |||
| MT-ND4L | 2.40 | |||
| MYH3 | 3.57 | |||
| MYL1 | 2.06 | |||
| MYLPF | 3.85 | |||
| NDUFA3 | 2.09 | |||
| NDUFB1 | 2.17 | |||
| NME2 | 4.23 | |||
| PAM16 | 2.06 | |||
| RNASEK | 2.04 | |||
| RPL17 | 2.12 | |||
| RPL38 | 2.03 | |||
| RPS27 | 2.11 | |||
| RPS29 | 2.41 | |||
| SIX3 | 0.39 | |||
| SNHG25 | 2.17 | |||
| SNHG9 | 2.03 | |||
| TNNC1 | 2.42 | |||
| TNNI1 | 3.02 | |||
| TOMM5 | 2.09 | |||
| TPM2 | 2.19 | |||
| TTR | 2.33 | |||
| UBE2V1 | 2.18 | |||
| Total number of genes | 18 | 3 | 0 | 11 |
| Cluster 1 | ||||
| Gene | E 0.1 μM | E 1 μM | E+L 0.1 μM | E+L 1.0 μM |
| ATP5ME | 2.27 | |||
| COL9A3 | ||||
| DHRS3 | 0.49 | |||
| HECW1 | 2.1 | |||
| HES1 | 2.004 | |||
| HIST1H4C | 2.01 | |||
| ID3 | 2.12 | |||
| KLHDC8A | 2.0002 | |||
| MRPS24 | 2.63 | |||
| MSX2 | 2.08 | |||
| MT-ND3 | 2.32 | |||
| MT-ND4L | 2.29 | |||
| NDUFA3 | 2.1 | |||
| NDUFB1 | 2.29 | |||
| NME2 | 4.12 | |||
| PAM16 | 2.4 | |||
| RNASEK | 2.15 | |||
| RPL17 | 2.31 | |||
| RPL38 | 2.06 | |||
| RPS27 | 2.22 | |||
| RPS29 | 2.49 | |||
| SIX3 | 0.41 | |||
| SNHG25 | 2.25 | |||
| TOMM5 | 2.08 | |||
| TTR | 2.28 | |||
| UBE2V1 | 2.31 | |||
| ZSCAN16-AS1 | 2.14 | |||
| Total | 19 | 4 | 0 | 4 |
| Cluster 5 | ||||
| Gene | E 0.1 μM | E 1 μM | E+L 0.1 μM | E+L 1.0 μM |
| AL355075.4 | 2.04 | |||
| AL360012.1 | 2.09 | |||
| ARHGAP11B | 2.05 | |||
| ATP5ME | 2.21 | |||
| BUB1 | 0.46 | |||
| C11orf98 | 2.002 | |||
| C2orf40/ECRG4 | 2.36 | |||
| CDC20 | 0.41 | |||
| CENPE | 0.41 | |||
| COL9A3 | 2.07 | |||
| COX16 | 2.15 | |||
| DHRS3 | 0.39 | |||
| DHX8 | 2.37 | |||
| ECT2 | 0.49 | |||
| GPM6A | 2.19 | |||
| HMMR | 0.46 | |||
| HYOU1 | 2.02 | |||
| ID1 | 2.04 | |||
| MRPS24 | 2.14 | |||
| MT-ND3 | 2.31 | |||
| MT-ND4L | 2.55 | |||
| NDUFA3 | 2.07 | |||
| NME2 | 3.16 | |||
| NR2F2 | 0.4 | |||
| NRN1 | 2.28 | |||
| NSUN5 | 2.12 | |||
| PAM16 | 3.03 | |||
| PRR11 | 0.48 | |||
| RNASEK | 2.18 | |||
| RPL17 | 2.15 | |||
| RPL38 | 2.07 | |||
| RPS27 | 2.13 | |||
| RPS29 | 2.36 | |||
| SNHG9 | 2.35 | |||
| STMN4 | 0.39 | |||
| TRH | 0.10 | 0.13 | ||
| TTR | 2.72 | |||
| TXNDC16 | 2.02 | |||
| UBE2V1 | 2.47 | |||
| VEGFA | 2.02 | |||
| Total number of genes | 23 | 3 | 13 | |
| Cluster 2 | ||||
| Gene | E 0.1 μM | E 1 μM | E+L 0.1 μM | E+L 1.0 μM |
| AL138826.1 | 0.24 | |||
| ATP5ME | 2.26 | |||
| CALB2 | ||||
| COX16 | 2.05 | |||
| HES1 | 2.13 | |||
| HIST1H4C | 2.25 | |||
| MEG3 | 2.06 | 0.33 | ||
| MRPS24 | 2.34 | |||
| MT-ND3 | 2.32 | |||
| MT-ND4L | 2.32 | |||
| NDUFA3 | 2.18 | |||
| NDUFB1 | 2.32 | |||
| NME2 | 3.6 | |||
| NR2F2 | 0.42 | |||
| NTS | 3.61 | 4.4 | ||
| PAM16 | 2.55 | |||
| RNASEK | 2.68 | |||
| RPL17 | 2.18 | |||
| RPL37A | 2.04 | |||
| RPL38 | 2.08 | |||
| RPS27 | 2.19 | |||
| RPS29 | 2.55 | |||
| SNHG9 | 2.01 | |||
| TAC3 | 0.48 | |||
| TLN1 | 2.08 | |||
| TOMM5 | 2.13 | |||
| TTR | 4.72 | 2.5 | ||
| UBE2V1 | 2.55 | |||
| WLS | 2.01 | |||
| Total number of genes | 21 | 7 | 0 | 4 |
| Cluster 3 | ||||
| Gene | E 0.1 μM | E 1 μM | E+L 0.1 μM | E+L 1.0 μM |
| ATP5ME | 2.18 | |||
| CARTPT | 2.22 | |||
| EMX2 | 2.08 | |||
| FST | 2.2 | |||
| FSTL5 | 2.14 | |||
| GAL | 3.31 | |||
| HIST1H4C | 2.39 | |||
| ID1 | ||||
| ID2 | 2.18 | |||
| ID3 | 2.43 | |||
| ISL1 | 2.18 | |||
| MEG3 | 3.56 | 2.94 | ||
| MT-ND3 | 2.12 | |||
| MT-ND4L | 2.44 | |||
| NDUFA3 | 2.11 | |||
| NME2 | 2.37 | |||
| NTS | 3.7 | |||
| OTP | 0.39 | 0.41 | ||
| PAM16 | 2.17 | |||
| RASL11B | 2.31 | |||
| RELN | 2.16 | |||
| RGS16 | 2.28 | |||
| RNASEK | 2.51 | |||
| RPS27 | 2.07 | |||
| RPS29 | 2.47 | |||
| RSPO3 | 2.35 | |||
| SIX3 | 0.25 | |||
| SNHG9 | 2.05 | |||
| SST | 0.07 | 0.32 | ||
| TTR | 2.27 | |||
| UBE2V1 | 2.16 | |||
| Total number of genes | 14 | 6 | 2 | 12 |
| Cluster 4 | ||||
| Gene | E 0.1 μM | E 1 μM | E+L 0.1 μM | E+L 1.0 μM |
| ACTA2 | 0.24 | 0.31 | ||
| ACTG2 | 0.4 | |||
| ADAMTS1 | 0.45 | |||
| AL139246.5 | ||||
| ALDH1A1 | 0.36 | |||
| ANKRD1 | 0.44 | 0.40 | 0.44 | |
| ASS1 | 2.02 | |||
| ATF3 | 0.41 | |||
| ATP2B2 | 2.09 | |||
| ATP5ME | 2.11 | |||
| BBOX1 | 2.54 | |||
| BCAN | 0.39 | |||
| C9orf24 | 2.06 | |||
| CAPS | 2.07 | |||
| CCK | 3.92 | |||
| CCL2 | 0.27 | 4.23 | ||
| CCNO | 4.12 | |||
| CDC20B | 2.08 | |||
| CEBPB | 0.49 | |||
| CFI | 0.39 | |||
| COL3A1 | 0.33 | |||
| COL4A1 | 0.50 | |||
| CRH | 0.29 | 0.28 | 0.18 | |
| CYTOR | 0.48 | |||
| DCN | 0.41 | |||
| DCT | 0.24 | |||
| DLK1 | 4.36 | 4.47 | ||
| DLX1 | 2.08 | |||
| ECEL1 | 2.07 | |||
| EDNRB | 2.27 | |||
| EFNA5 | 0.42 | |||
| ERICH5 | 0.43 | |||
| FABP7 | 0.39 | |||
| FAM107A | 2.22 | |||
| FAM69C | 0.47 | |||
| FBN2 | 0.48 | |||
| FIBIN | 2.21 | |||
| FN1 | 2.50 | |||
| FOLR1 | 2.66 | |||
| GALNT5 | 2.23 | |||
| GPNMB | 0.41 | 0.38 | ||
| HES6 | 0.26 | |||
| HMX1 | 0.44 | |||
| HOPX | 0.42 | |||
| IER3 | 0.45 | |||
| IFIT2 | 2.29 | |||
| IGF1 | 2.48 | 2.01 | ||
| IGFBP7 | 2.52 | |||
| IQCH-AS1 | 2.08 | |||
| KCNJ13/Kir7.1 | 2.12 | |||
| KCTD8 | 2.26 | |||
| LAMA4 | 2.03 | |||
| LGALS3 | 0.46 | |||
| LHX1 | 2.23 | |||
| LINC00982 | 2.05 | |||
| LINGO1 | 2.11 | |||
| LRRC17 | 0.40 | 0.33 | ||
| LUM | 0.31 | |||
| LYPD1 | 0.39 | 0.4 | ||
| MAB21L2 | 0.49 | |||
| MEG3 | 0.42 | |||
| MRPS24 | 2.78 | |||
| MT-ATP8 | 2.09 | |||
| MT-ND3 | 2.03 | |||
| MT-ND4L | 2.18 | |||
| MYL9 | 0.35 | |||
| NDP | 0.495 | |||
| NDUFB1 | 2.13 | |||
| NEFM | 0.38 | |||
| NEUROD6 | 2.63 | |||
| NEUROG1 | 2.47 | |||
| NME2 | 4.36 | |||
| NPPB | 0.32 | 0.30 | ||
| NRN1 | 2.62 | |||
| NTN1 | 0.48 | |||
| NTS | 0.29 | 0.45 | ||
| NUDCD3 | 2.07 | |||
| NUPR1 | 0.37 | |||
| PAM16 | 2.71 | |||
| PDZRN3 | 2.23 | |||
| PLAC9 | 0.48 | |||
| PLK2 | 0.45 | |||
| PLS3 | 2.26 | |||
| PMCH | 7.35 | |||
| PMEL | 0.13 | 0.29 | ||
| PPP1R14C | 0.43 | |||
| PTN | 2.22 | |||
| RBP1 | 0.33 | |||
| RGS16 | 0.46 | |||
| RNASEK | 2.86 | |||
| RPL17 | 2.03 | |||
| RPP21 | 2.01 | |||
| RPS27 | 2.02 | |||
| RPS29 | 2.37 | |||
| S100A11 | 0.47 | |||
| S100A6 | 0.44 | |||
| SEMA6D | 0.48 | |||
| SFRP2 | 0.26 | 0.38 | ||
| SLIT1 | 0.48 | |||
| SNHG25 | 2.50 | |||
| SNTG1 | 2.72 | |||
| SPTY2D1OS | 2.07 | |||
| TAGLN | 0.19 | 0.23 | ||
| TPM1 | 0.38 | |||
| TPM2 | 0.50 | |||
| TTR | 2.87 | |||
| TWIST1 | 0.32 | |||
| UBE2V1 | 2.50 | |||
| UNC5A | 2.96 | |||
| XIST | 2.41 | |||
| ZFYVE16 | 2.04 | |||
| ZSCAN16-AS1 | 2.04 | |||
| Total number of genes | 29 | 61 | 14 | 25 |
To our surprise E+L exposure at 0.1 μM resulted in very few changes of gene expression. No gene expression changes >2-fold were observed for clusters 0, 1, and 2 and for cluster 5 the expression of only two genes was changed. Thus, we observed a 2.5-fold down regulation of STMN4, a gene which affects microtubule function, is highly expressed in the fetal human cortex and known to play a role in neuronal development (Chauvin and Sobel, 2015; Nakao et al., 2004; Ohkawa et al., 2007; Tao et al., 2016) and a 10-fold drop in the expression of Thyrotropin Releasing Hormone (TRH) (Table 3C). Cluster 3 neurons also showed 2 genes whose expression was affected, a 3.3-fold increase in GAL, a gene encoding Galanin, a neuropeptide that appears to have neuroprotective properties (Cordero-Llana et al., 2014) and a 3-fold decrease in SST, coding for somatostatin, which has been implicated to play a role in the modulation of neuronal differentiation (Bulloch, 1987; Laquerrière et al., 1992). Indeed, subtle changes of somatostatin levels during neurodevelopment may result in long-term alterations of the neuronal circuitry (Sakai et al., 1995; Volk and Lewis, 2014). As observed for the E-exposure paradigms, cluster 4 was significantly more affected with 14 genes showing a >2-fold change in expression, with all these genes uniquely affected in cluster 4 (Table 3C). GO analysis for E+L at 0.1 μM exposure indicated the same cellular processes affected in all clusters, including SRP-dependent co-translational protein targeting to membrane, nuclear-transcribed mRNA catabolic process nonsense-mediated decay, translation initiation and rRNA processing (suppl. Tables 7A–F)
Table 3C:
E+L at 0.1 μM Exposure cluster comparison
| Genes | Cluster 0 | Cluster 1 | Cluster 5 | Cluster 2 | Cluster 3 | Cluster 4 |
|---|---|---|---|---|---|---|
| AL139246.5 | 2.46 | |||||
| ASS1 | 2.02 | |||||
| BBOX1 | 2.54 | |||||
| CCL2 | 4.23 | |||||
| CRH | 0.28 | |||||
| DCN | 0.41 | |||||
| EDNRB | 2.27 | |||||
| FAM107A | 2.22 | |||||
| FIBIN | 2.21 | |||||
| FN1 | 2.50 | |||||
| GAL | 3.31 | |||||
| IFIT2 | 2.29 | |||||
| LAMA4 | 2.03 | |||||
| MEG3 | 0.42 | |||||
| PTN | 2.22 | |||||
| SST | 0.32 | |||||
| STMN4 | 0.39 | |||||
| TRH | 0.10 | |||||
| total | 0 | 0 | 2 | 0 | 2 | 14 |
| up % | N/A | N/A | 0 | N/A | 50 | 79 |
| down % | N/A | N/A | 100 | N/A | 50 | 21 |
| cluster specific genes | N/A | N/A | 100 | N/A | 100 | 100 |
E+L exposure at 1 μM had different effects on different cell types. Thus, for the radial glial cells, 11 genes for cluster 0, but only 4 in cluster 1 showed > 2-fold change in expression (Table 3D). Strikingly, 8 of 13 upregulated genes in cluster 0 were genes involved in actin dynamics and typically expressed in muscle tissue. Except for MSX2, which was similarly upregulated in cluster 0 and 1, none of these genes were affected in any other neuronal clusters. GO analysis highlighted effects of E+L at 1 μM exposure on cellular processes involving actin dynamics in skeletal, cardiac and smooth muscle for cluster 0, but not any of the other clusters (suppl. Table 8A–F). These observations suggest a possible cluster 0-specific neural to myogenic shift in lineage specification. A similar observation has been reported for another model of developmental neurotoxicity, where the effects of valproate on mouse hES neuronal differentiation resulted in a similar neural-myogenic shift (Smirnova et al., 2014). The largest change for neuronal cell types was seen for cluster 5 (IPC) with a total of 13 genes changed, 4 of which play a role in cell cycle/cell division and 4 are related to neuronal development (Table 3D). These same processes were identified via GO analysis (suppl Table 8C). Clusters 2 and 3, both representing ENs, also were differentially affected, with 11 changes for cluster 3, but only 4 for cluster 2 (Table 3D). With very few exceptions these genes were all upregulated and more than half of these genes are involved in neuronal development (Table 3D), a cellular process also identified by GO analysis (suppl. Table 8D, E). Such an up-regulation of genes involved in neurodevelopment has also been observed in mouse embryonic stem cell-derived neuronal cultures exposed to continuous low MeHg concentrations over a 24–96 hour period of time (Theunissen et al., 2011). As seen in all other exposure paradigms, cluster 4 (choroid plexus precursor cells) showed the largest number (25) genes with > 2-fold expression change with the majority (18 of 25 genes) being down regulated. Interestingly, the expression of TTR (transthyretin, a key functional marker of choroid plexus) was elevated to a very similar degree in all 6 clusters. In addition, GO analysis indicated that E+L at 1 μM, but not any of the other exposure paradigms, affects cholesterol metabolism in all six clusters (suppl. Tables 8A–F). However, overall, the vast majority of all genes affected in all 6 clusters (50 out of 68 genes) by E+L at 1 μM are cluster-specific (Table 3D).
Table 3D:
E+L at 1 μM Exposure cluster comparison
| Genes | Cluster 0 | Cluster 1 | Cluster 5 | Cluster 2 | Cluster 3 | Cluster 4 |
|---|---|---|---|---|---|---|
| ACTA2 | 0.31 | |||||
| ACTC1 | 5.38 | |||||
| ACTG2 | 0.4 | |||||
| AL138826.1 | ||||||
| ANKRD1 | 0.44 | |||||
| BCAN | 0.39 | |||||
| BUB1 | 0.46 | |||||
| C2orf40/ECRG4 | 2.36 | |||||
| C9orf24 | 2.06 | |||||
| CAPS | 2.07 | |||||
| CARTPT | 2.22 | |||||
| CDC20 | 0.41 | |||||
| CENPE | 0.41 | |||||
| COL3A1 | 0.33 | |||||
| COL9A3 | 2.05 | |||||
| CRH | 0.18 | |||||
| DHRS3 | 0.49 | 0.39 | ||||
| ECT2 | 0.49 | |||||
| FABP7 | 0.39 | |||||
| FOLR1 | 2.66 | |||||
| FST | 2.2 | |||||
| HES6 | 0.26 | |||||
| HMMR | 0.46 | |||||
| HMX1 | 0.44 | |||||
| HOPX | 0.42 | |||||
| ID2 | 2.18 | |||||
| ID3 | 2.18 | 2.12 | 2.43 | |||
| IGF1 | 2.01 | |||||
| IGFBP7 | 2.52 | |||||
| ISL1 | 2.18 | |||||
| KCNJ13/Kir7.1 | 2.12 | |||||
| LRRC17 | 0.33 | |||||
| LYPD1 | 0.4 | |||||
| MEG3 | 0.33 | 2.94 | ||||
| MSX2 | 2.15 | 2.08 | ||||
| MYH3 | 3.57 | |||||
| MYL1 | 2.06 | |||||
| MYLPF | 3.85 | |||||
| NDP | 0.495 | |||||
| NRN1 | 2.28 | |||||
| NSUN5 | 2.12 | |||||
| NTS | 4.4 | 0.45 | ||||
| OTP | 0.41 | |||||
| PMEL | 0.29 | |||||
| PRR11 | 0.48 | |||||
| RASL11B | 2.31 | |||||
| RELN | 2.16 | |||||
| RGS16 | 2.28 | 0.46 | ||||
| RSPO3 | 2.35 | |||||
| SFRP2 | 0.38 | |||||
| TAC3 | 0.48 | |||||
| TAGLN | 0.23 | |||||
| TNNC1 | 2.42 | |||||
| TNNI1 | 3.02 | |||||
| TPM2 | 2.19 | |||||
| TRH | 0.13 | |||||
| TTR | 2.33 | 2.28 | 2.72 | 2.5 | 2.27 | 2.87 |
| VEGFA | 2.02 | |||||
| total | 11 | 4 | 13 | 4 | 11 | 25 |
| up % | 100 | 75 | 38 | 50 | 91 | 28 |
| down % | 0 | 25 | 62 | 50 | 9 | 72 |
| % unique genes | 72.7 | 0.0 | 84.6 | 25.0 | 72.7 | 88.0 |
In conclusion, not only are MeHg-induced changes in the number and identity of genes vastly different between different cell types (clusters), but the different MeHg exposure paradigms also affect gene expression within each cluster in radically different ways (Table 4).
4. Discussion:
4.1. Changes on cluster population size
Exposure to MeHg, regardless of the exposure paradigm, did not cause loss of a major fraction of cells in any of the clusters nor did it result in new clusters (e.g. new cell types). Overall population size changes were only observed at the higher 1 μM MeHg concentration, and generally these changes were small, with by far the biggest change observed in cluster 4 cells (choroid plexus precursors), where we observed a 3-fold increase from 4% to 12% in the total cell population in E+L exposed cultures. Thus, we conclude that none of the four MeHg exposure paradigms caused major changes in the culture cell composition, suggesting that the developmental trajectory in these differentiating cortical cultures remains unchanged by the implemented exposures.
4.2. MeHg effect on cell cycle/proliferation
Several lines of evidence support the hypothesis that the reduced cell number observed in the in vivo MeHg-exposed developing CNS is a result of the inhibition of cell proliferation, especially mitosis (Burke et al., 2006; Faustman et al., 2002; Fujimura and Usuki, 2015; Rodier et al., 1984), and a differentiation stage-specific effect of MeHg on the cell cycle has been reported (Kim et al., 2007). Thus, we examined if MeHg similarly affects the cell cycle of in vitro differentiating human neurons. We observed subtle effects at the higher MeHg concentration (1 μM) and only in proliferating cells (clusters 0, 1 and 5). But as we saw for the changes in relative cell populations, the cell cycle response pattern was different amongst different cell types. Thus, for cluster 0 cells, we observed a decrease in the number of cells in the S-phase suggesting a persistent MeHg-induced inhibition of DNA synthesis and a consequential decrease and increase in the number of cells in the G2M- and G1 phase, respectively, for both, E and E+L exposures. Whereas in clusters 1 and 5, we saw an increase in the number of cells in S-phase accompanied with a decrease in cells in the G2M phase only for the E+L exposure, suggesting that MeHg persistently affects the cell cycle via a different mechanism in cells of cluster 1 and 5, versus cluster 0. As expected, non-proliferative (or less proliferative) cell types (clusters 2, 3 and 4) were not significantly affected by MeHg. Diverging results regarding the effects of MeHg on the cell cycle of neuronal cells have been reported, with some studies reporting increases or decreases in number of cells in G2M-, S and G1 phases of the cell cycle (Miura et al., 1999; Rodier et al., 1984; Sager et al., 1984; Sudo et al., 2019; Zhang et al., 2009). These differences are likely at least partially due to differences in the model systems and MeHg exposure paradigms used. Indeed, the effects of MeHg on the cell cycle have been shown to depend on the MeHg concentration and exposure length (Ponce et al., 1994; Vogel et al., 1986). In vivo observations suggest that these cell cycle changes might be mostly mediated by an acute antimitotic action of MeHg (Rodier et al., 1984). MeHg, mostly at exposures higher than what we have used here, is known to bind to tubulin interfering with the function of microtubules, the main component of the mitotic spindle apparatus (Abe et al., 1975; Choi, 1989; Choi, 1991; Sager, 1988; Sager and Matheson, 1988). The fact that in this study the cell cycle was persistently and/or latently affected 18 or 28 days after MeHg had been removed suggests a mechanism other than tubulin modification by MeHg. This conclusion is supported by our previous observation, that in these same cortical cultures cellular Hg content is reduced > 90% 10 days after exposure when compared to a 24 hour after exposure time point (Prince et al., 2021).
Aside from directly inhibiting microtubule function, MeHg has been shown to cause cell cycle arrest via changes in the expression of genes related to cell cycle regulation such as p21, cyclin E, and CDK2 (Burke et al., 2006; Fujimura and Usuki, 2015; Ou et al., 1999; Xu et al., 2010). Most importantly in the context of our observations, Bose and collaborators observed that some of these changes in cell cycle gene expression were inherited by daughter cells never directly exposed to MeHg, and are thus by definition persistent (Bose et al., 2012). We observed a small, but significant down regulation of p21 (CDKN1A, 1.3 fold) resulting from both E and E+L exposures at 1.0 μM for cluster 1 cells with a small (1.2-fold) concomitant and significant upregulation of CDK2 in cluster 1 cells exposed to 1 μM MeHg (E+L only). Cluster 0 cells responded with a 1.2-fold decrease in cyclin E1 (CCNE1) expression. Thus, cell type-specific changes in cell cycle related gene expression were present in the most proliferating cell types under MeHg exposure conditions where we also noted changes in the number of cells in different cell cycle phases.
4.3. Gene expression changes:
Here we looked at persistent, and potentially latent, gene expression effects (day 38 of differentiation, 18 or 28 days after removal of the last MeHg exposure) in developing neurons exposed to MeHg (0.1 μM or 1.0 μM) either only during the very early NP stage (E, days 4–10) or early and later (E+L, days 4–10 and 14–20). The second exposure coincided with a developmental stage when early postmitotic cells are first being produced. The two MeHg concentrations used here result in cellular MeHg levels in these hiPSC-derived differentiating neuronal cultures that are comparable to MeHg levels measured in human brains of individuals that show mild to severe neurological effects, respectively (Aschner, 2012; Burbacher et al., 1990; Choi et al., 1978; Lapham et al., 1995; Prince et al., 2021).
The most surprising and profound observations from this study are: (1) the differential response of the varying cell populations (clusters) to the developmental MeHg exposures; (2) the different effects of the different MeHg exposure paradigms (concentration and timing of exposure); and (3) an apparent greater sensitivity of cluster 4 (choroid plexus precursors) compared to the neuronal cell types of clusters 0, 1, 2, 3, and 5.
Somewhat surprising to us is that not only was the number of genes affected by the two different MeHg concentrations (within E or E+L exposure) significantly different, but more importantly the set of genes were also mostly different. Thus, a tenfold difference in MeHg concentration resulted in a very different gene-expression response. Similar observations have been reported for other in vitro model systems where low and high mercury concentrations resulted in different cellular responses suggesting the mechanisms underlying the cellular responses to low and high doses of MeHg are different (Choi and Kim, 1984; Jebbett et al., 2013). MeHg binds strongly to proteins via sulfhydryl groups (Ajsuvakova et al., 2020), and we postulate that different levels and combinations of protein modifications result in diverse effects on gene expression. We further postulate, based on our cluster-specific responses that such responses are cell-type specific.
Interesting also was our observation that the “two-hit” exposure E+L at 0.1 μM resulted in significantly fewer genes showing expression changes than the “single-hit” E at 0.1 μM exposure in all clusters and the genes affected were different in the two exposure paradigms (Fig. 5, Fig. 6). Our observations suggest that a mild (0.1 μM) ‘second hit” not only does not affect additional genes, but protects from further cellular damage and might actually reverse already initiated cellular responses. Indeed, a similar observation was made for MeHg toxicity in another in vitro model system although in those studies the initial insult was elicited by a stressor different from MeHg (Usuki et al., 2013; Usuki et al., 2017). This phenomenon is not unique to MeHg toxicity, but has already been described in other insult paradigms such as in brains of rats repeatedly subjected to episodes of anoxia (Dahl and Balfour, 1964). These authors and later others concluded that the initial insult resulted in a preconditioning of the cells, which provided protection for a later insult (Calabrese and Kozumbo, 2021; Dahl and Balfour, 1964; Dhodda et al., 2004; Stenzel-Poore et al., 2007; Zeiger et al., 2010).
Another interesting observation is that the expression of TTR, a gene encoding transthyretin, was universally upregulated >2-fold in all clusters exposed to E+L at 1 μM. Transthyretin is a thyroid hormone distributor protein, the only one in the human brain (Richardson, 2007) and delivers thyroid hormones to neuronal stem cells and progenitor cells, where these hormones regulate cell division and apoptosis (Lemkine et al., 2005; Richardson, 2007; Richardson et al., 2007). The majority of brain TTR is produced and secreted by the choroid plexus but low levels of neuronal expression have been reported (Buxbaum and Reixach, 2009; Li et al., 2011; Vancamp et al., 2019). TTR has been recognized as a neuronal stress marker and has been shown to be up-regulated by oxidative stress (Sharma et al., 2019). Stress-induced alterations of TTR expression appear to be brain region- and insult specific. Thus, TTR expression was upregulated in the prefrontal cortex, but not hippocampus in a chemical (naltrexone) adult mouse injury model (Yu et al., 2011). In a chronic stress model however, TTR was down regulated in the cortex, but not choroid plexus (Joo et al., 2009). A delayed increase in CSF transthyretin protein in the CSF, but no change in choroid plexus TTR mRNA was observed in adult rats after exposure to MeHg (Nakamura et al., 2011). We are not aware of any studies that have assessed TTR expression during human cortical development under control or stress conditions.
E-exposure at 0.1 μM resulted in the most homogenous response with 9 common genes upregulated > 2-fold in all six clusters. The majority of these genes encode ribosomal or mitochondrial components, a response that was unique to the E at 0.1 μM exposure paradigm. An increase in ribonucleoprotein particles has been reported in brains of adult asymptomatic MeHg treated rats (Brubaker et al., 1973). Mutations in several ribosomal components as well as interference of ribosomal biogenesis by exogenous factors have been shown to disrupt neurodevelopment (Hetman and Slomnicki, 2019). Although it is clear that mitochondrial function is crucial for normal cell function, how the combined changes in several of the mitochondrial genes we observed would affect developing neurons is not clear at this time. In this context however, it is of interest, that combinations of otherwise non-pathogenic mtDNA gene variants have been linked to a hereditary optic neuropathy (Caporali et al., 2018). In addition, our recent analysis of mitochondrial function shortly after MeHg exposure completion (day 22 of differentiation) revealed subtle changes in mitochondrial respiration in cultures subjected to E+L at 0.1 μM or 1 μM, but not single E-exposures (Prince et al., 2021).
Lastly, we report on the effect of MeHg on human choroid plexus precursors. The large number of genes showing >2 -fold changes in expression in these cells (cluster 4), when compared to the neuronal cell types (cluster 0, 1, 2, 3, 5), could be highly relevant to the effects of MeHg on the development of the nervous system. The choroid plexus has been shown to be integral for normal brain development (Liddelow, 2015) with choroid plexus epithelial cells transferring proteins and other molecules important for brain development (Dziegielewska et al., 2001; Liddelow, 2015). Aside from the effects on choroid plexus gene expression, we also observed a significant 3-fold increase in the relative population of these cells (cluster 4) in cultures exposed to MeHg. Significant expansion of the choroid plexus epithelial cells at the expense of cortical neuroepithelium has been reported under conditions of suboptimal BMP signaling (Panchision et al., 2001). To our knowledge, our study reports for the first time on the effects of MeHg on the human developing choroid plexus.
5. Conclusions:
To our knowledge, this is the first assessment of the effects of in-vivo relevant MeHg exposures in an in vitro model of human cortical development assessing persistent effects of different MeHg developmental exposure paradigms performed on a single cell level. We observed small changes in cluster populations but did not observe a significant loss of one cell type (cluster) or appearance of new cell types (clusters), or even a major re-specification of cells from one cluster into another cluster. Changes in cell cycle stage distributions were also relatively small and suggest a subtle inhibition of proliferation. The most profound findings of this study are that the MeHg-induced changes in gene expression were cell type- (cluster) specific and MeHg exposure paradigm specific. Thus, the timing as well as the MeHg concentration itself profoundly impacted the pattern of gene expression changes. Since we assessed gene expression 18 and 28 days after exposures ended, we conclude that the MeHg-induced changes are either persistent or manifest themselves in a delayed manner. Delayed effects resulting from MeHg intoxication have been observed in patients (Weiss et al., 2002). It is of course difficult to determine how exactly changes in these gene expressions alone or especially in combination affect human neural development and what the resulting functional consequences are. To address this question our future studies will look at the consequences of these delayed/latent effects on long term neuronal functions. We conclude that hiPSC-derived developing neuronal cultures provide an excellent human model to study persistent neurodevelopmental toxicity of MeHg in the developing human cortex.
Supplementary Material
Tables 3A-D: Comparison of MeHg-induced gene expression changes between clusters.
All genes with expression changes > 2-fold (FDR 0.05) for clusters 0–5 are listed for the four different exposure paradigms for a comparison between clusters. Numbers indicated represent fold change, values > 0 indicate up-regulation, values < 0 indicate down regulation. Five groups of genes particularly relevant to this analysis are highlighted in colors as indicated. The assignment of those genes to the particular groups is based on information from several different sources including https://medlineplus.gov/genetics/gene/; https://www.ncbi.nlm.nih.gov/gtr/genes/ https://www.genecards.org/; www.uniprot.org/uniprot/; https://pathcards.genecards.org/card/; and the literature cited in the text. Genes changed uniquely in one cluster are highlighted by a diagonally stripped pattern.
| ribosomal component | actin dynamics/muscle tissue |
| mitochondrial component | cell cycle |
| forebrain/neuron development | cluster-specific genes |
6. Acknowledgements:
We thank Bingying Han for technical assistance with the human stem cell modeling. Single cell RNA sequencing was performed at the Vanderbilt University VANTAGE Core Laboratory. Bioinformatics analysis was provided by the Purdue Bioinformatics Core. We thank Morgan Thomas for technical assistant with qPCR. Financial support provided by NIH/NIEHS R01 ES07331 to MA and ABB.
Abbreviations:
- CNS
central nervous system
- E
early exposure
- E+L
early and later combined exposure
- EN
excitatory neurons
- hiPSC
human-induced pluripotent stem cells
- IPC
intermediate progenitor cells
- MeHg
methylmercur
- min
minutes
- NE
neuroepithelial cells
- RG
radial glia
- scRNAseq
single cell RNA sequencing
- NP
neural precursor
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
7. Supplementary Materials: Supplementary Figures and Supplementary Tables are appended to this manuscript. These include Supplementary Figures 1, 2, 3, 4A–F, and 5A–F; supplemental figure legends are incorporated into each file. These also include Supplementary Tables 1, 2, 3A–F, 4, 5A–F, 6A–F, 7A–F, 8A–F; table captions are incorporated into each file.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References:
- Abe T, Haga T, and Kurokawa M 1975. Blockage of axoplasmic transport and depolymerisation of reassembled microtubules by methyl mercury. Brain Res. 86:504–508. [DOI] [PubMed] [Google Scholar]
- Ajsuvakova OP, Tinkov AA, Aschner M, Rocha JBT, Michalke B, Skalnaya MG, Skalny AV, Butnariu M, Dadar M, Sarac I, Aaseth J, and Bjørklund G 2020. Sulfhydryl groups as targets of mercury toxicity. Coord Chem Rev. 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin-Zaki L, Majeed MA, Elhassani SB, Clarkson TW, Greenwood MR, and Doherty RA 1979. Prenatal methylmercury poisoning. Clinical observations over five years. Am J Dis Child. 133:172–177. [PubMed] [Google Scholar]
- Antunes Dos Santos A, Appel Hort M, Culbreth M, López-Granero C, Farina M, Rocha JB, and Aschner M 2016. Methylmercury and brain development: A review of recent literature. J Trace Elem Med Biol. 38:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M 2012. Considerations on methylmercury (MeHg) treatments in in vitro studies. Neurotoxicology. 33:512–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, and Syversen T 2005. Methylmercury: recent advances in the understanding of its neurotoxicity. Ther Drug Monit. 27:278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahahir HI, Clarkson TW, Smith JC, and Doherty RA 1973. Methylmercury poisoning in Iraq. Science. 181:230–241. [DOI] [PubMed] [Google Scholar]
- Bar O, Gelb S, Atamny K, Anzi S, and Ben-Zvi A 2020. Angiomodulin (IGFBP7) is a cerebral specific angiocrine factor, but is probably not a blood-brain barrier inducer. Fluids and barriers of the CNS. 17:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, Pollen AA, Nowakowski TJ, and Kriegstein AR 2020. Cell stress in cortical organoids impairs molecular subtype specification. Nature. 578:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Onishchenko N, Edoff K, Janson Lang AM, and Ceccatelli S 2012. Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicol Sci. 130:383–390. [DOI] [PubMed] [Google Scholar]
- Brubaker PE, Klein R, Herman SP, Lucier GW, Alexander LT, and Long MD 1973. DNA, RNA, and protein synthesis in brain, liver, and kidneys of asymptomatic methylmercury treated rats. Exp Mol Pathol. 18:263–280. [DOI] [PubMed] [Google Scholar]
- Bulloch AG 1987. Somatostatin enhances neurite outgrowth and electrical coupling of regenerating neurons in Helisoma. Brain Res. 412:6–17. [DOI] [PubMed] [Google Scholar]
- Burbacher TM, Rodier PM, and Weiss B 1990. Methylmercury developmental neurotoxicity: a comparison of effects in humans and animals. Neurotoxicol Teratol. 12:191–202. [DOI] [PubMed] [Google Scholar]
- Burke EE, Chenoweth JG, Shin JH, Collado-Torres L, Kim SK, Micali N, Wang Y, Colantuoni C, Straub RE, Hoeppner DJ, Chen HY, Sellers A, Shibbani K, Hamersky GR, Diaz Bustamante M, Phan BN, Ulrich WS, Valencia C, Jaishankar A, Price AJ, Rajpurohit A, Semick SA, Bürli RW, Barrow JC, Hiler DJ, Page SC, Martinowich K, Hyde TM, Kleinman JE, Berman KF, Apud JA, Cross AJ, Brandon NJ, Weinberger DR, Maher BJ, McKay RDG, and Jaffe AE 2020. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat Commun. 11:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke K, Cheng Y, Li B, Petrov A, Joshi P, Berman RF, Reuhl KR, and DiCicco-Bloom E 2006. Methylmercury elicits rapid inhibition of cell proliferation in the developing brain and decreases cell cycle regulator, cyclin E. Neurotoxicology. 27:970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN, and Reixach N 2009. Transthyretin: the servant of many masters. Cell Mol Life Sci. 66:3095–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystron I, Blakemore C, and Rakic P 2008. Development of the human cerebral cortex: Boulder Committee revisited. Nat Rev Neurosci. 9:110–122. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, and Kozumbo WJ 2021. The hormetic dose-response mechanism: Nrf2 activation. Pharmacol Res. 167:105526. [DOI] [PubMed] [Google Scholar]
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Bräuninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, Knoblich JA, Lachmann R, Pääbo S, Huttner WB, and Treutlein B 2015. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A. 112:15672–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali L, Iommarini L, La Morgia C, Olivieri A, Achilli A, Maresca A, Valentino ML, Capristo M, Tagliavini F, Del Dotto V, Zanna C, Liguori R, Barboni P, Carbonelli M, Cocetta V, Montopoli M, Martinuzzi A, Cenacchi G, De Michele G, Testa F, Nesti A, Simonelli F, Porcelli AM, Torroni A, and Carelli V 2018. Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber’s hereditary optic neuropathy. PLoS Genet. 14:e1007210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi AF, Johansson C, Onishchenko N, Coccini T, Roda E, Vahter M, Ceccatelli S, and Manzo L 2008. Human developmental neurotoxicity of methylmercury: impact of variables and risk modifiers. Regul Toxicol Pharmacol. 51:201–214. [DOI] [PubMed] [Google Scholar]
- Ceccatelli S, Daré E, and Moors M 2010. Methylmercury-induced neurotoxicity and apoptosis. Chem Biol Interact. 188:301–308. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotech. 27:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Lee HJ, Kang B, Yu KN, Minai-Tehrani A, Lee S, Kim SU, and Cho MH 2013. Methylmercury induces caspase-dependent apoptosis and autophagy in human neural stem cells. J Toxicol Sci. 38:823–831. [DOI] [PubMed] [Google Scholar]
- Chauvin S, and Sobel A 2015. Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog Neurobiol. 126:1–18. [DOI] [PubMed] [Google Scholar]
- Choi BH 1986. Methylmercury poisoning of the developing nervous system: I. Pattern of neuronal migration in the cerebral cortex. Neurotoxicology. 7:591–600. [PubMed] [Google Scholar]
- Choi BH 1989. The effects of methylmercury on the developing brain. Prog Neurobiol. 32:447–470. [DOI] [PubMed] [Google Scholar]
- Choi BH 1991. Effects of methylmercury on neuroepithelial germinal cells in the developing telencephalic vesicles of mice. Acta Neuropathol. 81:359–365. [DOI] [PubMed] [Google Scholar]
- Choi BH, and Kim RC 1984. The comparative effects of methylmercuric chloride and mercuric chloride upon DNA synthesis in mouse fetal atrocytes in vitro. Experimental and Molecular Pathology. 41:371–376. [DOI] [PubMed] [Google Scholar]
- Choi BH, Lapham LW, Amin-Zaki L, and Saleem T 1978. Abnormal neuronal migration, deranged cerebral cortical organization, and diffuse white matter astrocytosis of human fetal brain: a major effect of methylmercury poisoning in utero. J Neuropathol Exp Neurol. 37:719–733. [DOI] [PubMed] [Google Scholar]
- Clarkson TW 2002. The three modern faces of mercury. Environ Health Perspect. 110 Suppl 1:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, Amin-Zaki L, and Al-Tikriti SK 1976. An outbreak of methylmercury poisoning due to consumption of contaminated grain. Federation proceedings. 35:2395–2399. [PubMed] [Google Scholar]
- Co M, Anderson AG, and Konopka G 2020. FOXP transcription factors in vertebrate brain development, function, and disorders. Wiley Interdiscip Rev Dev Biol. 9:e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Llana O, Rinaldi F, Brennan PA, Wynick D, and Caldwell MA 2014. Galanin promotes neuronal differentiation from neural progenitor cells in vitro and contributes to the generation of new olfactory neurons in the adult mouse brain. Exp Neurol. 256:93–104. [DOI] [PubMed] [Google Scholar]
- Costa LG, Aschner M, Vitalone A, Syversen T, and Soldin OP 2004. Developmental neuropathology of environmental agents. Annual review of pharmacology and toxicology. 44:87–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C, Clarkson TW, Marsh DO, Amin-Zaki L, Tikriti S, and Myers GG 1989. Dose-response analysis of infants prenatally exposed to methyl mercury: an application of a single compartment model to single-strand hair analysis. Environ Res. 49:318–332. [DOI] [PubMed] [Google Scholar]
- Dahl NA, and Balfour WM 1964. Prolonged anoxic survival due to anoxia pre-exposure: Brain ATP, lactate, and pyruvate. The American journal of physiology. 207:452–456. [DOI] [PubMed] [Google Scholar]
- Debes F, Budtz-Jørgensen E, Weihe P, White RF, and Grandjean P 2006. Impact of prenatal methylmercury exposure on neurobehavioral function at age 14 years. Neurotoxicol Teratol. 28:536–547. [DOI] [PubMed] [Google Scholar]
- Dhodda VK, Sailor KA, Bowen KK, and Vemuganti R 2004. Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis. J Neurochem. 89:73–89. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Ek J, Habgood MD, and Saunders NR 2001. Development of the choroid plexus. Microsc Res Tech. 52:5–20. [DOI] [PubMed] [Google Scholar]
- EPA, U. 1997. Health effects of mercury and mercury compounds. In Mercury Study Report to Congress, Office of Air Quality Planning & Standards and Office of Research Development. Vol. V. EPA, Washington, DC. [Google Scholar]
- Faustman EM, Ponce RA, Ou YC, Mendoza MA, Lewandowski T, and Kavanagh T 2002. Investigations of methylmercury-induced alterations in neurogenesis. Environ Health Perspect. 110 Suppl 5:859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, and Huttner WB 2014. Neural progenitors, neurogenesis and the evolution of the neocortex. Development. 141:2182–2194. [DOI] [PubMed] [Google Scholar]
- Fujimura M, and Usuki F 2015. Low concentrations of methylmercury inhibit neural progenitor cell proliferation associated with up-regulation of glycogen synthase kinase 3β and subsequent degradation of cyclin E in rats. Toxicol Appl Pharmacol. 288:19–25. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Budtz-Jørgensen E, White RF, Jørgensen PJ, Weihe P, Debes F, and Keiding N 1999. Methylmercury exposure biomarkers as indicators of neurotoxicity in children aged 7 years. Am J Epidemiol. 150:301–305. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, Murata K, Sørensen N, Dahl R, and Jørgensen PJ 1997. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Teratol. 19:417–428. [DOI] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, and Macklis JD 2013. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. 14:755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Chintawar S, Lalic T, Whiteley E, Vowles J, Giustacchini A, Argoud K, Sopp P, Nakanishi M, Bowden R, Cowley S, Newey S, Akerman C, Ponting CP, and Cader MZ 2016. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum Mol Genet. 25:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M 1995. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 25:1–24. [DOI] [PubMed] [Google Scholar]
- Harada Y 1968. Congenital (or fetal) Minamata disease. In Study Group of Minamata Disease. Katsuna M, editor, Kumamoto University, Japan. 93–118. [Google Scholar]
- Hetman M, and Slomnicki LP 2019. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J Neurochem. 148:325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LS, Cory-Slechta DA, Cox C, Thurston SW, Shamlaye CF, Watson GE, van Wijngaarden E, Zareba G, Strain JJ, Myers GJ, and Davidson PW 2018. Analysis of Nonlinear Associations between Prenatal Methylmercury Exposure from Fish Consumption and Neurodevelopmental Outcomes in the Seychelles Main Cohort at 17 Years. Stoch Environ Res Risk Assess. 32:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebbett NJ, Hamilton JW, Rand MD, and Eckenstein F 2013. Low level methylmercury enhances CNTF-evoked STAT3 signaling and glial differentiation in cultured cortical progenitor cells. Neurotoxicology. 38:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo Y, Choi KM, Lee YH, Kim G, Lee DH, Roh GS, Kang SS, Cho GJ, Choi WS, and Kim HJ 2009. Chronic immobilization stress induces anxiety- and depression-like behaviors and decreases transthyretin in the mouse cortex. Neurosci Lett. 461:121–125. [DOI] [PubMed] [Google Scholar]
- Joshi P, Bodnya C, Ilieva I, Neely MD, Aschner M, and Bowman AB 2019. Huntington’s disease associated resistance to Mn neurotoxicity is neurodevelopmental stage and neuronal lineage dependent. Neurotoxicology. 75:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Kim YS, Kim MS, and Ryu JC 2007. The inhibitory mechanism of methylmercury on differentiation of human neuroblastoma cells. Toxicology. 234:1–9. [DOI] [PubMed] [Google Scholar]
- Kjellstroem T, Kennedy P, Wallis S, and et al. 1989. Physical and mental development of children with prenatal exposure to mercury from fish. Stage 2, interviews and psychological tests at age 6 (Report 3642). National Swedish Environmental Protection Board, Stockholm. [Google Scholar]
- Kjellstrom T, Kennedy P, Wallis S, and Mantell C 1986. Physical and mental development of children with prenatal exposure to Hg from fish-Stage 1-preliminary tests at age 4. National Swedish Environmental Protection Board, Report 3080, Solna, Sweden. [Google Scholar]
- Kostović I, Sedmak G, and Judaš M 2019. Neural histology and neurogenesis of the human fetal and infant brain. Neuroimage. 188:743–773. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Aschner M, Cory-Slechta DA, Bilbo SD, Caudle WM, and Makris SL 2016. Unmasking silent neurotoxicity following developmental exposure to environmental toxicants. Neurotoxicol Teratol. 55:38–44. [DOI] [PubMed] [Google Scholar]
- Kumar KK, Lowe EW Jr., Aboud AA, Neely MD, Redha R, Bauer JA, Odak M, Weaver CD, Meiler J, Aschner M, and Bowman AB 2014. Cellular manganese content is developmentally regulated in human dopaminergic neurons. Sci Rep. 4:6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapham LW, Cernichiari E, Cox C, Myers GJ, Baggs RB, Brewer R, Shamlaye CF, Davidson PW, and Clarkson TW 1995. An analysis of autopsy brain tissue from infants prenatally exposed to methymercury. Neurotoxicology. 16:689–704. [PubMed] [Google Scholar]
- Laquerrière A, Leroux P, Gonzalez B, Bodenant C, Tayot J, and Vaudry H 1992. Somatostatin receptors in the human cerebellum during development. Brain Res. 573:251–259. [DOI] [PubMed] [Google Scholar]
- Lemkine GF, Raj A, Alfama G, Turque N, Hassani Z, Alegria-Prévot O, Samarut J, Levi G, and Demeneix BA 2005. Adult neural stem cell cycling in vivo requires thyroid hormone and its alpha receptor. Faseb j. 19:863–865. [DOI] [PubMed] [Google Scholar]
- Li X, Masliah E, Reixach N, and Buxbaum JN 2011. Neuronal production of transthyretin in human and murine Alzheimer’s disease: is it protective? J Neurosci. 31:12483–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA 2015. Development of the choroid plexus and blood-CSF barrier. Front Neurosci. 9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, and Arlotta P 2015. Generating neuronal diversity in the mammalian cerebral cortex. Annu Rev Cell Dev Biol. 31:699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DO 1987. Dose-repsonse relationships in humans-methyl mercury epidemics in Japan and Iraq. In The toxicity of methyl mercury. Eccles CU and A. Z., editors. Johns Hopkins, Baltimore. 45–53. [Google Scholar]
- Matsumoto H, Koya G, and Takeuchi T 1965. Fetal Minamata disease. A neuropathological study of two cases of intrauterine intoxication by a methyl mercury compound. J Neuropathol Exp Neurol. 24:563–574. [PubMed] [Google Scholar]
- Mayer S, Chen J, Velmeshev D, Mayer A, Eze UC, Bhaduri A, Cunha CE, Jung D, Arjun A, Li E, Alvarado B, Wang S, Lovegren N, Gonzales ML, Szpankowski L, Leyrat A, West JAA, Panagiotakos G, Alvarez-Buylla A, Paredes MF, Nowakowski TJ, Pollen AA, and Kriegstein AR 2019. Multimodal Single-Cell Analysis Reveals Physiological Maturation in the Developing Human Neocortex. Neuron. 102:143–158.e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Koide N, Himeno S, Nakagawa I, and Imura N 1999. The involvement of microtubular disruption in methylmercury-induced apoptosis in neuronal and nonneuronal cell lines. Toxicol Appl Pharmacol. 160:279–288. [DOI] [PubMed] [Google Scholar]
- Muller FJ, Schuldt BM, Williams R, Mason D, Altun G, Papapetrou EP, Danner S, Goldmann JE, Herbst A, Schmidt NO, Aldenhoff JB, Laurent LC, and Loring JF 2011. A bioinformatic assay for pluripotency in human cells. Nat Methods. 8:315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers GJ, and Davidson PW 2000. Does methylmercury have a role in causing developmental disabilities in children? Environ Health Perspect. 108 Suppl 3:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Yasutake A, Fujimura M, Hachiya N, and Marumoto M 2011. Effect of methylmercury administration on choroid plexus function in rats. Arch Toxicol. 85:911–918. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Suzuki Y, Sakuta H, Ookata K, Kawahara K, and Hirose S 1999. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+,K+-ATPase. Biochem J. 342 ( Pt 2):329–336. [PMC free article] [PubMed] [Google Scholar]
- Nakao C, Itoh TJ, Hotani H, and Mori N 2004. Modulation of the stathmin-like microtubule destabilizing activity of RB3, a neuron-specific member of the SCG10 family, by its N-terminal domain. J Biol Chem. 279:23014–23021. [DOI] [PubMed] [Google Scholar]
- Namba T, and Huttner WB 2017. Neural progenitor cells and their role in the development and evolutionary expansion of the neocortex. Wiley Interdiscip Rev Dev Biol. 6. [DOI] [PubMed] [Google Scholar]
- Neely MD, Davison CA, Aschner M, and Bowman AB 2017. From the Cover: Manganese and Rotenone-Induced Oxidative Stress Signatures Differ in iPSC-Derived Human Dopamine Neurons. Toxicol Sci. 159:366–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely MD, Litt MJ, Tidball AM, Li GG, Aboud AA, Hopkins CR, Chamberlin R, Hong CC, Ess KC, and Bowman AB 2012. DMH1, a highly selective small molecule BMP inhibitor promotes neurogenesis of hiPSCs: comparison of PAX6 and SOX1 expression during neural induction. ACS Chem Neurosci. 3:482–491 (PMC888888). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, Ounadjela JR, Shuga J, Wang X, Lim DA, West JA, Leyrat AA, Kent WJ, and Kriegstein AR 2017. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 358:1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC NRC 2000. Toxicological effects of methylmercury. National Academy Press (US), Washington (DC). [PubMed] [Google Scholar]
- Ohkawa N, Fujitani K, Tokunaga E, Furuya S, and Inokuchi K 2007. The microtubule destabilizer stathmin mediates the development of dendritic arbors in neuronal cells. J Cell Sci. 120:1447–1456. [DOI] [PubMed] [Google Scholar]
- Oken E, Radesky JS, Wright RO, Bellinger DC, Amarasiriwardena CJ, Kleinman KP, Hu H, and Gillman MW 2008. Maternal fish intake during pregnancy, blood mercury levels, and child cognition at age 3 years in a US cohort. Am J Epidemiol. 167:1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, Shibata T, Kunisada T, Takahashi M, Takahashi J, Saji H, and Yamanaka S 2011. A more efficient method to generate integration-free human iPS cells. Nat Methods. 8:409–412. [DOI] [PubMed] [Google Scholar]
- Ou YC, Thompson SA, Ponce RA, Schroeder J, Kavanagh TJ, and Faustman EM 1999. Induction of the cell cycle regulatory gene p21 (Waf1, Cip1) following methylmercury exposure in vitro and in vivo. Toxicol Appl Pharmacol. 157:203–212. [DOI] [PubMed] [Google Scholar]
- Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, and McKay RD 2001. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev. 15:2094–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham NH, and Choi BH 1988. Abnormal neuronal distribution within the cerebral cortex after prenatal methylmercury intoxication. Acta Neuropathol. 76:222–226. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, Lim DA, Leyrat AA, West JA, and Kriegstein AR 2015. Molecular identity of human outer radial glia during cortical development. Cell. 163:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce RA, Kavanagh TJ, Mottet NK, Whittaker SG, and Faustman EM 1994. Effects of methyl mercury on the cell cycle of primary rat CNS cells in vitro. Toxicol Appl Pharmacol. 127:83–90. [DOI] [PubMed] [Google Scholar]
- Prince LM, Neely MD, Warren EB, Thomas M, Henley MR, Smith KK, Aschner M, and Bowman AB 2021. Environmentally relevant developmental methylmercury exposures alter neuronal differentiation in a human-induced pluripotent stem cell model. Food and Chemical Toxicology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D, and Barone S Jr. 2000. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 108 Suppl 3:511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SJ 2007. Cell and molecular biology of transthyretin and thyroid hormones. Int Rev Cytol. 258:137–193. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Lemkine GF, Alfama G, Hassani Z, and Demeneix BA 2007. Cell division and apoptosis in the adult neural stem cell niche are differentially affected in transthyretin null mice. Neurosci Lett. 421:234–238. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England). 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM, Aschner M, and Sager PR 1984. Mitotic arrest in the developing CNS after prenatal exposure to methylmercury. Neurobehav Toxicol Teratol. 6:379–385. [PubMed] [Google Scholar]
- Romito-DiGiacomo RR, Menegay H, Cicero SA, and Herrup K 2007. Effects of Alzheimer’s disease on different cortical layers: the role of intrinsic differences in Abeta susceptibility. J Neurosci. 27:8496–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager PR 1988. Selectivity of methyl mercury effects on cytoskeleton and mitotic progression in cultured cells. Toxicol Appl Pharmacol. 94:473–486. [DOI] [PubMed] [Google Scholar]
- Sager PR, Aschner M, and Rodier PM 1984. Persistent, differential alterations in developing cerebellar cortex of male and female mice after methylmercury exposure. Brain Res. 314:1–11. [DOI] [PubMed] [Google Scholar]
- Sager PR, and Matheson DW 1988. Mechanisms of neurotoxicity related to selective disruption of microtubules and intermediate filaments. Toxicology. 49:479–492. [DOI] [PubMed] [Google Scholar]
- Sakai K, Maeda K, Chihara K, and Kaneda H 1995. Increases in cortical neuropeptide Y and somatostatin concentrations following haloperidol-depot treatment in rats. Neuropeptides. 29:157–161. [DOI] [PubMed] [Google Scholar]
- Sanfeliu C, Sebastià J, Cristòfol R, and Rodríguez-Farré E 2003. Neurotoxicity of organomercurial compounds. Neurotox Res. 5:283–305. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A 2015. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Khan S, Rahman S, and Singh LR 2019. The Extracellular Protein, Transthyretin Is an Oxidative Stress Biomarker. Front Physiol. 10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sun L, Wang M, Liu J, Zhong S, Li R, Li P, Guo L, Fang A, Chen R, Ge WP, Wu Q, and Wang X 2020. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 18:e3000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivitilli AA, Gosio JT, Ghoshal B, Evstratova A, Trcka D, Ghiasi P, Hernandez JJ, Beaulieu JM, Wrana JL, and Attisano L 2020. Robust production of uniform human cerebral organoids from pluripotent stem cells. Life Sci Alliance. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova L, Block K, Sittka A, Oelgeschläger M, Seiler AE, and Luch A 2014. MicroRNA profiling as tool for in vitro developmental neurotoxicity testing: the case of sodium valproate. PLoS One. 9:e98892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, and Simon RP 2007. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 38:680–685. [DOI] [PubMed] [Google Scholar]
- Stolp HB, Johansson PA, Habgood MD, Dziegielewska KM, Saunders NR, and Ek CJ 2011. Effects of neonatal systemic inflammation on blood-brain barrier permeability and behaviour in juvenile and adult rats. Cardiovasc Psychiatry Neurol. 2011:469046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R 2019. Comprehensive Integration of Single-Cell Data. Cell. 177:1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo K, V.A.N.D. C, Miyamoto A, and Shiraishi M 2019. Comparative analysis of in vitro neurotoxicity of methylmercury, mercury, cadmium, and hydrogen peroxide on SH-SY5Y cells. J Vet Med Sci. 81:828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedish Expert Group. 1971. Methyl mercury in fish. A toxicologic-epidemiologic evaluation of risks. Suppl 4:1–364 pp. [PubMed] [Google Scholar]
- Tao Y, Gao H, Ackerman B, Guo W, Saffen D, and Shugart YY 2016. Evidence for contribution of common genetic variants within chromosome 8p21.2–8p21.1 to restricted and repetitive behaviors in autism spectrum disorders. BMC Genomics. 17:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen PT, Pennings JL, Robinson JF, Claessen SM, Kleinjans JC, and Piersma AH 2011. Time-response evaluation by transcriptomics of methylmercury effects on neural differentiation of murine embryonic stem cells. Toxicol Sci. 122:437–447. [DOI] [PubMed] [Google Scholar]
- Tidball AM, Neely MD, Chamberlin R, Aboud AA, Kumar KK, Han B, Bryan MR, Aschner M, Ess KC, and Bowman AB 2016. Genomic Instability Associated with p53 Knockdown in the Generation of Huntington’s Disease Human Induced Pluripotent Stem Cells. PLoS One. 11:e0150372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasande L, Landrigan PJ, and Schechter C 2005. Public health and economic consequences of methyl mercury toxicity to the developing brain. Environ Health Perspect. 113:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trillo-Contreras JL, Toledo-Aral JJ, Echevarría M, and Villadiego J 2019. AQP1 and AQP4 Contribution to Cerebrospinal Fluid Homeostasis. Cells. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuki F, Fujimura M, and Yamashita A 2013. Endoplasmic reticulum stress preconditioning attenuates methylmercury-induced cellular damage by inducing favorable stress responses. Sci Rep. 3:2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuki F, Fujimura M, and Yamashita A 2017. Endoplasmic reticulum stress preconditioning modifies intracellular mercury content by upregulating membrane transporters. Sci Rep. 7:12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancamp P, Gothié JD, Luongo C, Sébillot A, Le Blay K, Butruille L, Pagnin M, Richardson SJ, Demeneix BA, and Remaud S 2019. Gender-specific effects of transthyretin on neural stem cell fate in the subventricular zone of the adult mouse. Sci Rep. 9:19689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, Paulsen B, Nguyen L, Adiconis X, Regev A, Levin JZ, and Arlotta P 2019. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 570:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel DG, Rabinovitch PS, and Mottet NK 1986. Methylmercury effects on cell cycle kinetics. Cell Tissue Kinet. 19:227–242. [DOI] [PubMed] [Google Scholar]
- Volk DW, and Lewis DA 2014. Early developmental disturbances of cortical inhibitory neurons: contribution to cognitive deficits in schizophrenia. Schizophr Bull. 40:952–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Clarkson TW, and Simon W 2002. Silent latency periods in methylmercury poisoning and in neurodegenerative disease. Environ Health Perspect. 110 Suppl 5:851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Yan C, Tian Y, Yuan X, and Shen X 2010. Effects of low level of methylmercury on proliferation of cortical progenitor cells. Brain Res. 1359:272–280. [DOI] [PubMed] [Google Scholar]
- Yu J, Halder D, Baek MN, Das ND, Choi MR, Oh DY, Choi IG, Jung KH, and Chai YG 2011. Changes in the expression of transthyretin and protein kinase Cγ genes in the prefrontal cortex in response to naltrexone. Neurosci Lett. 488:288–293. [DOI] [PubMed] [Google Scholar]
- Yuan X, Wang J, and Chan HM 2018. Sub-Nanomolar Methylmercury Exposure Promotes Premature Differentiation of Murine Embryonic Neural Precursor at the Expense of Their Proliferation. Toxics. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiger SL, McKenzie JR, Stankowski JN, Martin JA, Cliffel DE, and McLaughlin B 2010. Neuron specific metabolic adaptations following multi-day exposures to oxygen glucose deprivation. Biochim Biophys Acta. 1802:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Xu Y, Sun J, Li X, Wang L, and Jin L 2009. Protection of pyrroloquinoline quinone against methylmercury-induced neurotoxicity via reducing oxidative stress. Free radical research. 43:224–233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.