

Abstract
Colorectal cancer is one of the most common malignant tumors and, hence, has become one of the most important public health issues in the world. Treatment with immune checkpoint inhibitors (ICIs) successfully improves the survival rate of patients with melanoma, non‐small‐cell lung cancer, and other malignancies, and its application in metastatic colorectal cancer is being actively explored. However, a few patients develop drug resistance. Predictive molecular markers are important tools to precisely screen patient groups that can benefit from treatment with ICIs. The current article focused on certain important predictive molecular markers for ICI treatment in colorectal cancer, including not only some of the mature molecular markers, such as deficient mismatch repair (d‐MMR), microsatellite instability‐high (MSI‐H), tumor mutational burden (TMB), programmed death‐ligand‐1 (PD‐L1), tumor immune microenvironment (TiME), and tumor‐infiltrating lymphocytes (TILs), but also some of the novel molecular markers, such as DNA polymerase epsilon (POLE), polymerase delta 1 (POLD1), circulating tumor DNA (ctDNA), and consensus molecular subtypes (CMS). We have reviewed these markers in‐depth and presented the results from certain important studies, which suggest their applicability in CRC and indicate their advantages and disadvantages. We hope this article is helpful for clinicians and researchers to systematically understand these markers and can guide the treatment of colorectal cancer.
Keywords: colorectal cancer, immune checkpoint inhibitors, immunotherapy, molecular markers
Predictive molecular markers are important tools to precise screen of patient groups who can benefit from immune checkpoint inhibitors treatment. This article focused on some important predictive molecular markers for ICI treatment in colorectal cancer, which included not only some mature molecular markers such as deficient mismatch repair (d‐MMR) or microsatellite instability‐high (MSI‐H), tumor mutational burden (TMB), programmed death‐ligand 1(PD‐L1), and tumor immune microenvironment (TiME) or tumor‐infiltrating lymphocytes (TILs), but also some novel molecular markers, such as POLE or POLD1, circulating tumor DNA and consensus molecular subtypes (CMS). We have made in‐depth reviews for these markers, which included some important studies in the past, possibility of their application in CRC, the advantages and disadvantages of their application as molecular markers, and the difficulties they are currently facing.

1. INTRODUCTION
Colorectal cancer (CRC) is one of the most common malignant tumors in the world. The mortality rate of CRC is increasing in India, China, and other countries, and CRC is one of the main causes of death in cancer patients. 1 The 5‐year survival rate of metastatic CRC (mCRC) is only 13%–15%. 2 , 3 The treatments for CRC face certain challenges, such as no option of surgery, poor effects of radiotherapy, and chemotherapy, and only a small group of patients’ benefits from targeted therapy. Therefore, it is urgent to explore new treatments to improve the survival rate of mCRC. With in‐depth research on tumor immunity and the increasing emphasis on immunotherapy, treatment with immune checkpoint inhibitors (ICIs) is being considered for mCRC. However, in clinical studies, only a small number of unscreened CRC patients was treated with ICIs and these patients responded to the treatment, 4 , 5 , 6 suggesting that appropriate molecular markers need to be identified to evaluate treatment effects of ICIs. The current article summarized the available molecular markers to help clinicians make better decisions.
2. MOLECULAR MARKERS FOR ICI TREATMENT IN CRC PATIENTS
2.1. Deficient mismatch repair (d‐MMR) and microsatellite instability‐high (MSI‐H)
Microsatellites are highly polymorphic repetitive sequences in the eukaryotic genome comprising 1–6 bases. During DNA replication, irremediable deletion, insertion, and translocation may result in microsatellite instability‐high (MSI‐H). 3 Mismatch repair genes (MMR) are evolutionarily conserved housekeeping genes that can accurately identify base mismatches during DNA replication and transcription and repair them in time. 7 MMR is an important factor in determining the status of microsatellites. When patients have proficient MMR (p‐MMR), the errors can be repaired in time and microsatellite stability (MSS) is maintained. However, when patients have deficient MMR (d‐MMR), where the errors are not repaired in time, MSI‐H is observed. 8 , 9 MMR includes four main genes, mutS homolog 6 (MSH6), mutS homolog 2 (MSH2), mutL homolog 1 (MLH1), and PMS1 homolog 2 (PMS2), and d‐MMR occurs when at least one of these four genes has heterozygous germline mutations or there is hypermethylation of the MLH1 promoter. 10 , 11 , 12 , 13
Studies have suggested that in sporadic CRC, d‐MMR/MSI‐H was associated with the prognosis of the patients. 14 , 15 , 16 With the development of immunotherapy, the predictive values of d‐MMR/MSI‐H tumors in immunotherapy have been identified. The d‐MMR/MSI‐H tumors have high somatic mutations, resulting in high neoantigen loads. Compared to p‐MMR/MSS, d‐MMR/MSI‐H tumors have nearly 20 times more mutations, 17 , 18 , 19 , 20 which are processed and presented by antigen‐presenting cells (APCs), thereby enhancing the infiltration by various immune cells. 21 , 22 , 23 In addition, d‐MMR/MSI‐H tumor microenvironments are more likely to express inhibitory immune checkpoint ligands, such as programmed cell death‐ligand‐1 (PD‐L1), and such patients showed a higher response rate to ICI treatment. 24 Therefore, d‐MMR/MSI‐H is a key factor in ICI treatment, which can be used as a predictive marker of response to ICIs and a prognostic indicator of the efficacy of ICIs in mCRC patients.
As an important molecular marker for ICI treatment, the occurrence rate of d‐MMR/MSI‐H tumors has attracted much attention. In 2016, the American Association for Cancer Research (AACR) reviewed the occurrence rate of MSI‐H in different cancers (MSI‐H was defined as the variation in at least 2 out of 5 or 30% of the tested microsatellite loci): The occurrence rate of MSI‐H exceeded by 10% in patients with CRC, endometrial cancer, gastric cancer, and liver cancer. In ovarian cancer, cervical cancer, esophageal adenocarcinoma, soft tissue tumor, kidney cancer, Ewing's sarcoma, and head and neck cancers, the occurrence rate of MSI‐H was between 2% and 10%. In squamous cell skin cancer, basal cell skin cancer, prostate cancer, lung cancer, and breast cancer, it was <2%. 25 About 10%–20% of the CRC patients have MSI‐H tumors of which about 3% are diagnosed with Lynch syndrome (LS) and about 5% are detected having mCRC. 11 , 26 , 27 LS and sporadic CRC are different in terms of their pathogenesis and treatment. The pathogenesis of LS involves heterozygous germline mutations in MMR. 11 , 28 However, the most common cause of MSI‐H in sporadic CRC is the epigenetic silencing of the MLH1 promoter caused by the abnormal methylation of the CpG islands, which is related to the somatic cell B‐Raf proto‐oncogene pV600E (BRAF pV600E) mutation. 12 , 26 Therefore, testing for BRAF pV600E mutation is the best way to distinguish patients with LS from those with sporadic CRC.
Several studies have shown that compared to p‐MMR/MSS mCRC, d‐MMR/MSI‐H mCRC has a higher response to ICIs. KEYNOTE 016 initially enrolled 10 d‐MMR/MSI‐H mCRC, 18 p‐MMR/MSS mCRC, and 9 d‐MMR/MSI‐H non‐CRC patients. The results of this study showed that the average number of somatic mutations and the mutation‐associated neoantigens in d‐MMR/MSI‐H patients were 1782 and 578, respectively, while those in p‐MMR patients were 73 and 21, respectively. The objective response rate (ORR) and the progression‐free survival (PFS) rate in the d‐MMR/MSI‐H mCRC patients were 40% and 78%, respectively, while those in p‐MMR/MSS mCRC patients were 0% and 11%, respectively. The response in d‐MMR/MSI‐H non‐mCRC patients was similar to that in d‐MMR/MSI‐H mCRC patients. 29 The 124 d‐MMR/MSI‐H mCRC patients included in KEYNOTE 164 also showed a higher response rate when treated with Pembrolizumab. 30 KEYNOTE 177 further confirmed that using ICIs as first‐line treatment for d‐MMR/MSI‐H mCRC patients significantly increased the PFS. 31 The results of NCT03026140 supported the treatment of d‐MMR CRC patients with ICIs as neo‐adjuvants. 32
In May 2017, based on the results of KEYNOTE 016, KEYNOTE 164, and other studies, the Food and Drug Administration (FDA)‐approved Pembrolizumab for advanced and metastatic solid d‐MMR/MSI‐H tumors in adults and children. 33 This was the first time that the FDA classified the patients based on specific biomarkers without considering the source of the tumor, making it a landmark event in the field of immunotherapy. In August of the same year, the FDA‐approved Nivolumab as the second‐line treatment drug for d‐MMR/MSI‐H mCRC patients. 34 In June 2020, based on the latest results of KEYNOTE 177, the FDA‐approved Pembrolizumab as the first‐line treatment drug for D‐MMR/MSI‐H mCRC patients. 35 These pieces of evidence are sufficient to demonstrate the importance of d‐MMR/MSI‐H as a major predictive molecular marker for the treatment of CRC patients with ICIs. Therefore, experts recommend the detection of MMR function or microsatellites status in CRC to predict the prognosis of early‐stage patients and guide the treatment of late‐stage patients. 36 , 37
Since the MMR function and microsatellites status have high consistency, d‐MMR is equivalent to MSI‐H in clinical applications. Common MMR and microsatellites detection methods include immunohistochemistry (IHC), polymerase chain reaction (PCR), next‐generation sequencing (NGS), and radiomics, 38 all of which have their advantages and disadvantages. In general, IHC is used to detect the MMR function and PCR to detect microsatellites status, and the results of the two tests showed over 90% similarity. 39 However, there is evidence that using only one detection method may lead to misdiagnosis. For example, MMR function is usually evaluated using IHC. When one or more MMR proteins are missing, it is diagnosed as d‐MMR. This method is simple and feasible, and can accurately detect specific proteins. However, the results of IHC may be affected by the quality of the samples, human errors, and other factors, and MSI‐H cases caused by non‐MMR mutations may be missed. 38 The gold standard for microsatellites detection is based on the Bethesda Guidelines proposed by the National Cancer Institute of the United States, which adopted the PCR method and involves five types of microsatellites. MSI‐H was defined as alterations in at least two of the five microsatellite loci or 30% or more loci when larger panels are tested. An alteration in one locus was defined as microsatellite instability‐low (MSI‐L), which behaves similarly to MSS. 40 Although PCR is more precise and can identify MSI‐H caused by any genetic mutations, certain limitations exist. For example, due to partially redundant functions of MSH6 and mutL homolog 3 (MSH3) proteins, patients with MSH6 mutations will not be classified as MSI‐H, but PCR ignores this, and PCR results cannot directly confirm which MMR protein is functionally deficient. Also, the detection is expensive, limiting its use further. 38 , 41 NGS can be used to detect MSI‐H in a large number of microsatellites using a small number of nucleic acids and determine the tumor mutation burden (TMB). However, this technology is currently mainly used in biomedical research. It is necessary to further standardize the NGS workflow to make it suitable for use in clinical practice. 38 In conclusion, the Colorectal Cancer Committee of the Chinese Society of Clinical Oncology recommended using IHC to test the MMR function universally and PCR routinely to test microsatellites status in institutions with available platforms to perform PCR. A third diagnostic approach (such as NGS) is considered when the results are inconsistent or drug resistance is suspected. 42
2.2. Tumor mutational burden (TMB)
As mentioned above, d‐MMR/MSI‐H phenotype increases the neoantigen loads and immune cell infiltration, and it is easier to trigger the immune response. Therefore, tumor mutational burden (TMB) has become a preferable marker in recent years. TMB is a measure of the number of mutations in the tumor genome. It is defined as the total number of non‐synonymous somatic mutations in each coding region of the tumor genome, expressed as the number of mutations per trillion bases.
The accumulation of TMB was significantly correlated with the continuous accumulation of somatic gene mutations, deletion of MMR, and mutations in tumor protein p53 (TP53), apolipoprotein B mRNA editing enzyme, catalytic polypeptide 3 (APOBEC), DNA polymerase epsilon (POLE) exonuclease domain, and polymerase delta 1 (POLD1). 43 A TMB high (TMB‐H) indicates an increased formation of new antigens and increasing tumor cell immunogenicity. Because of the relationship between TMB and d‐MMR, TMB is considered an independent marker for ICI treatment. Zang et al. found a significant correlation between TMB‐H and d‐MMR in CRC. 44 Chalmers et al. found that a majority of MSI‐H patients had TMB‐H and 97% had TMB ≥ 10 mut/Mb. However, only 16% of the samples with TMB‐H were classified as MSI‐H. 45 Moreover, there were few patients with both low TMB and d‐MMR/MSI‐H phenotypes. 7 , 45 , 46 Therefore, researchers believed that MSI‐H was a subset of TMB‐H (TMB‐H is defined as >20 mut/Mb). 45 Studies also found that MSS/TMB‐H tumors were more common than MSI‐H/ TMB‐H tumors, which may benefit from immunotherapy. 47 In summary, the use of TMB as a molecular marker is independent of MMR, which means that TMB has a wider application and more TMB‐H patients may benefit from ICI treatment. The relationship between TMB‐H and MSI‐H is shown in Figure 1.
FIGURE 1.
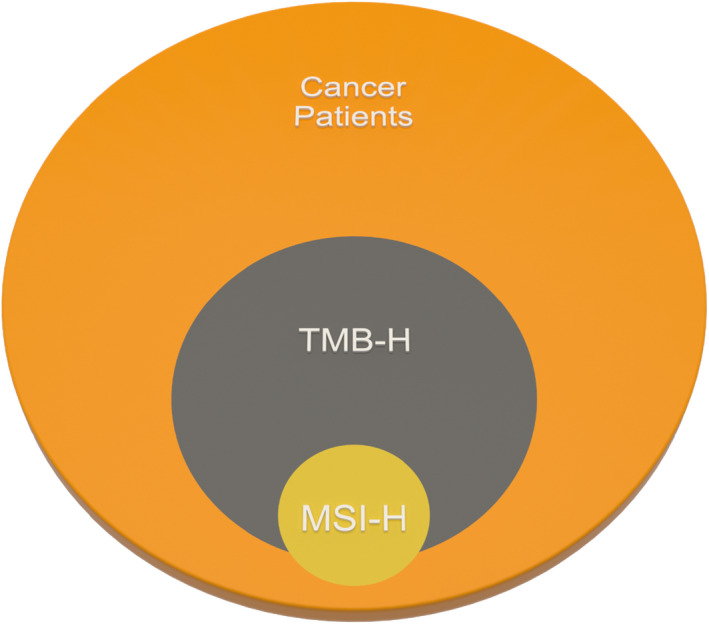
The relationship between TMB‐H and MSI‐H
The idea of TMB as an independent molecular marker for ICI treatment was demonstrated in a variety of tumors. The feasibility of TMB as a biomarker was initially evaluated in melanoma. The study showed that TMB‐H was associated with a high response rate to ICI treatment in melanoma patients. 48 Subsequently, researchers found that in non‐small‐cell lung cancer (NSCLC) patients with TMB > 10 mut/Mb, combined programmed cell death protein 1 (PD‐1) and cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) inhibitor treatment led to an increased PFS. 49 Samstein et al. investigated the relationship between TMB and overall survival (OS) rate after ICI treatment in 1662 patients with advanced malignant tumors and found that higher TMB was associated with improved OS rate. 50 A study published in 2017 including 1638 pan‐cancer patients also proved the effectiveness of TMB as a molecular marker for different types of immunotherapy in patients with different tumors. 51 More importantly, some researchers found that non‐MSI‐H patients with TMB‐H also benefited from ICI treatment, 46 , 47 which suggested that TMB can be used as an independent molecular marker for ICI treatment. In KEYNOTE 158, there was a correlation between the anti‐tumor activity of Pembrolizumab and TMB. The ORR of TMB‐H tumors was 29%, while that of non‐TMB‐H tumors was 6%. Patients with TMB‐H were able to achieve higher ORR. 52 These results prompted the FDA to approve Pembrolizumab for the treatment of refractory advanced solid tumors in adults and children with TMB‐H (TMB ≥ 10 mut/Mb), which confirmed the importance of TMB as an independent molecular marker for ICI treatment. CHECKMATE‐227 trial showed that the PFS in NSCLC patients with TMB‐H was significantly higher with Nivolumab and Ipilimumab combination therapy than chemotherapy. The results validated the benefit of Nivolumab and Ipilimumab combination therapy and the role of TMB as a molecular marker for patient selection. 49 Evaluating TMB helps in screening better responders for ICI treatment in MSI‐H CRC and identifying beneficial ICIs in MSS CRC. Fabrizio et al. found that the ability of TMB to identify CRC subgroups that may be responsive to ICIs surpassed that of microsatellite status. 46 The REGONIVO trial included 25 CRC patients. They were divided into TMB‐H and TMB‐L groups using 22.55 mut/Mb as the cutoff value. It was found that when Regorafenib and Nivolumab combination therapy was applied, the OS of the TMB‐H group was higher than that of the TMB low (TMB‐L) group (12.5 vs. 7.9 months, respectively). 53 In conclusion, TMB is a useful molecular marker for CRC.
Tumor mutational burden is defined as a pan‐cancer marker. However, researchers found that in some genetically unstable germ cell tumors, the relationship between TMB and ICI treatment was unclear, which suggested that the scope of application of TMB needs to be defined. 54 , 55 In addition, as a pan‐cancer marker, a fixed cutoff of TMB that can be applied to different tumors was difficult to identify. 50 In a meta‐analysis, the researchers summarized the most common cutoff values of TMB, which were 10, 16, and 20 mut/Mb. The CHECKMATE 227 study supported the application of the cutoff of 10 mut/Mb. 49 , 56 Another study classified the cutoff values of TMB into 3 groups: low (1–5 mut/Mb), intermediate (6–19 mut/Mb), and high (≥20 mut/Mb). They found that cancers with a higher TMB, measured by comprehensive genomic profiling, have a higher likelihood of responding to immunotherapy. 51 In CRC, Schrock et al. studied 22 d‐MMR/MSI‐H mCRC patients who were treated with PD‐1 or PD‐L1 inhibitors. They not only proved that TMB‐H can be used as an independent molecular marker but also found that when the cutoff value of TMB was between 37 and 41 mu/Mb, a better prediction was achieved. 57 In short, no reliable evidence exists that supports the use of a universal cutoff value to define TMB‐H and TMB‐L. On the contrary, it seems more feasible to determine individual cutoff values for specific tumors.
In addition, the detection methods of TMB were also an important factor in determining its value. Whole‐exome sequencing (WES) was the gold standard for TMB detection, but it was costly, and the detection standards lacked uniformity. 43 , 58 NGS was the most widely used because of its convenience and low cost, although it introduced errors that were dependent on the panel size. 59 , 60 Studies reported a correlation between TMB predicted by NGS and that by WES but the methods used to calculate TMB in NGS and WES also affect the TMB results. 61 , 62 For example, WES involved only missense mutations. MSK‐IMPACT's NGS sequencing method considered only non‐synonymous mutations but the F1CDx NGS sequencing method not only counted non‐synonymous mutations but also the synonymous mutations, 63 , 64 suggesting that the detection methods also have an impact on TMB.
In the selection of biological specimens, tissue specimens were used for tissue‐based TMB (t‐TMB) detection, which had high detection accuracy but the sampling requirements were also high. 43 Blood‐based TMB (b‐TMB) was a valuable substitute for t‐TMB because of its ease of sampling and high consistency with t‐TMB in the predicted results. 65 , 66 , 67
In conclusion, TMB, as an independent molecular marker for ICI treatment, has a high application value. However, it is urgent to define the cutoff of TMB and optimize its detection method.
3. PROGRAMMED CELL DEATH‐LIGAND‐1 (PD‐L1)
Programmed cell death protein 1 (PD‐1) is currently one of the most widely known inhibitory immune checkpoints, which has two main ligands: programmed cell death‐ligand‐1 (PD‐L1) and ligand‐2 (PD‐L2). PD‐L1 is usually expressed on activated T‐ and B‐lymphocytes and macrophage subsets induced by inflammatory cytokines, 68 and plays a role in protecting tissues from excessive inflammation and autoimmune responses. 69 In tumor cells, PD‐L1 was overexpressed as a carcinogenic factor. 70 In a study involving 1000 Chinese cancer patients, PD‐L1 amplification occurred most frequently in lung squamous cell carcinoma (14.3%), human epidermal growth factor receptor‐2 (HER2)‐positive breast cancer (8.8%), and breast cancer with unknown HER2 status (5.8%), while in lung adenocarcinoma and CRC, the amplification rate of PD‐L1 was low. 44
As the main ligands of PD‐1, PD‐1 and PD‐L1 combined to transmit co‐inhibitory signals to limit the proliferation of tumor‐infiltrating lymphocytes and peripheral lymphocytes, stimulate effector T cells to transform into regulatory T cells, and induce T‐cell exhaustion. 71 , 72 , 73 Previous studies have shown that there is a potential connection between PD‐L1 levels and the ICI treatment, prompting researchers to consider PD‐L1 as a molecular marker for ICI treatment. 74 , 75 , 76
Studies have found that PD‐L1‐positive patients benefit from ICI treatment more than PD‐L1‐negative patients. In an earlier study, in patients with NSCLC, melanoma, kidney cancer, prostate cancer, and CRC, using 5% of the tumor cells expressing PD‐L1 as the threshold, it was found that PD‐L1‐positive patients treated with Nivolumab showed a significantly higher objective response than the negative patients. 5 The KEYNOTE 001 study showed that when the tumor proportion score (TPS) of PD‐L1 reached at least 50% in NSCLC, the efficacy of ICI was better, and when TPS < 50%, the higher the TPS, the higher was the response rate. 77 KEYNOTE 042 suggested that when TPS ≥ 50%, ≥20%, and ≥1%, patients showed better OS with Pembrolizumab treatment compared to chemotherapy. 78 Similarly, KEYNOTE 024 also showed that when Pembrolizumab was used to treat patients with advanced NSCLC and PD‐L1 expression in at least 50% of the tumor cells, the PFS and OS were significantly higher and fewer adverse events occurred than those administered platinum‐based chemotherapy. 79 The results of this study led the FDA to approve the use of Pembrolizumab in the treatment of untreated metastatic NSCLC (without epidermal growth factor receptor (EGFR) or anaplastic Lymphoma Kinase (ALK) abnormalities) and propose that when >50% of tumor cells expressed PD‐L1, Pembrolizumab can be used as the first‐line drug. When >1% of tumor cells express PD‐L1, Pembrolizumab can be used for second‐line treatment. 80
It is worth noting that although numerous experiments have proved the feasibility of PD‐L1 as a molecular marker, several reasons limit its use as a molecular marker for ICI treatment. Firstly, the expression of PD‐L1 shows poor association with that of the other molecular markers such as d‐MMR and TMB. A systematic review using pan‐cancer analysis showed that the co‐expression ratio of PD‐L1, TMB‐H, and MSI‐H was 2.9%, but those of PD‐L1 and MSI‐H and PD‐L1 and TMB were only 0.59% and 8.5%, respectively. In CRC, the co‐expression ratio of PD‐L1, TMB‐H, and MSI‐H was 12.8%, but those of MSI‐H and PD‐L1 and PD‐L1 and TMB‐H were only 0.6% and 1.2%, respectively. 7 Other studies found that the expression of PD‐L1 showed great variability across tumors with different microsatellite states not only in d‐MMR/MSI‐H or TMB‐H but also in p‐MMR/MSS or TMB‐L. 7 , 81 , 82 Secondly, the expression of PD‐L1 showed temporal and spatial heterogeneity, which may be affected by changes in the tumor microenvironment, different stages of the tumor, chosen treatment, sampling time, and sampling site. Differences in the detection methods, antibodies, standards, and thresholds of the various detection platforms of PD‐L1 are also factors affecting its accuracy. 81 , 83 , 84 , 85
Therefore, the use of PD‐L1 as a molecular marker to guide ICI treatment is not validated, but it is also true that PD‐L1 is one of the most mature molecular markers available. Actively optimizing, standardizing, and achieving reproducibility in PD‐L1 detection and developing the combined application of molecular markers may promote the use of PD‐L1 as a stable molecular marker for ICI treatment in CRC patients.
4. TUMOR IMMUNE MICROENVIRONMENT (TIME) AND TUMOR‐INFILTRATING LYMPHOCYTES (TILS)
Tumor microenvironment (TME) refers to the environment in which the cancer cells interact with their surroundings, which consist of a variety of cells (immune cells, fibroblasts, endothelial cells, etc.) and extracellular components (growth factors, cytokines, extracellular matrix, hormones, etc.). 86 Tumor progression and the therapeutic effect of anti‐tumor drugs are not only regulated by genes in the tumor cells but also by the immune factors in TME. 86 , 87 One of the most important TME factors is the tumor‐infiltrating lymphocytes (TILs). TILs play an important role in anti‐tumor immunity and can be used as a marker of patient prognosis. Galon et al. proved that the level of TILs in the primary tumor was a strong independent predictor of recurrence and OS, and high TILs level was a positive prognostic factor. 88 Also, in CRC, researchers generally believed that the high level of TILs was positively correlated with prognosis. 89 , 90 , 91 TILs have a high predictive value in response to ICI treatment and play an important role in tumor immunotherapy. In mCRC, patients with high levels of TILs treated with ICIs had better response rates, PFS, and OS than patients with low levels of TILs. 92
In recent years, studies have found that high TILs were significantly correlated with the expression of MSI‐H, TMB, and PD‐L1. Therefore, it is proposed that TILs can be used as a molecular marker for ICI treatment in CRC patients. Smyrk et al. found that the level of TILs was related to the patient's microsatellites status. When the level was 5, the sensitivity of predicting the MSI‐H status in colorectal cancer was 93%, and the specificity was 62%. 93 Fotios Loupakis et al. found that there was a significant correlation between the levels of TILs and TMB‐H. 92 Mara Kitsou et al. studied the relationship between immune checkpoint molecules and the level of TILs in CRC and found that in colon cancer, it was positively correlated with the expression of CD8, adenosine A2a receptor (ADORA2A), CTLA‐4, hepatitis A virus cellular receptor‐2 (HAVCR2), Lymphocyte activation gene 3 protein (LAG3), PD‐L1, PD‐L2, T‐cell immunoreceptor with Ig and ITIM domains (TIGIT), and V‐domain Ig suppressor of T‐cell activation (VISTA). 94 Based on these results, it is reasonable to suggest that TILs have the potential to be used as molecular markers for ICI treatment.
At present, researchers generally believe that the evaluation and intervention of TILs was one of the important means to predict or mediate the resistance to immunotherapy. 95 To achieve this goal, researchers put forward the concept of “Immunoscore (I).” The Immunoscore assessed the density of CD45RO and CD8 immune cells in the core of the tumor (CT) and invasive margin (IM) of the tumor tissue, and the patients were scored (0–4 points) based on that. “I0” meant that the density of the two types of cells was low in both the regions; “I4” meant that the two types of cells had high density in both the regions; and “I1‐I3” represented the intermediate densities. 96 , 97 Studies have found that in stage I‐III CRCs, the higher the density of immune cell infiltration, the higher was the patient score, and the better was the prognosis. 96 , 98 , 99 In mCRC patients, MSI‐H was confirmed to be associated with a high Immunoscore. 100 The expression of PD‐1/PD‐L1 was also significantly higher in I3 and I4 tumor patients in CRC, who might potentially benefit from ICI treatment. 98 , 101 In summary, researchers suggested that Immunoscore is a novel marker independent of microsatellites. 102 , 103 However, the current evidence to support Immunoscore as a predictive marker is insufficient and further research is needed.
5. DNA POLYMERASE EPSILON (POLE) OR POLYMERASE DELTA 1 (POLD1)
In addition to chromosomal instability (CIN) and d‐MMR, DNA damage response (DDR) system dysfunction caused by polymerase epsilon (POLE) or polymerase delta 1 (POLD1) mutations is also one of the important mechanisms underlying colorectal cancer. 104 POLE/POLD1 are the key genes of the DDR pathway, encoding the catalytic subunit of DNA polymerase epsilon and the 125‐kDa catalytic subunit of DNA polymerase delta, respectively, which contribute to the fidelity of DNA replication. 105 Regarding the use of POLE/POLD1 as molecular markers for ICI treatment, the prevailing view was that mutations in POLE/POLD1 may lead to TMB‐H and neoantigen load. 106 , 107 , 108 Some researchers have also proposed that during DDR dysfunction, the stimulator of the interferon gene (STING) pathway may be activated to mediate natural anti‐tumor immunity. 109 , 110 It is worth noting that POLE/POLD1 carry the exonuclease domains, which correct errors during the copying process and ensure high fidelity. When mutations occur in exonuclease domains (exonuclease domain mutations (EDMs), also referred to as proofreading domain mutations), they generate a high number of mutant phenotypes in tumor cells. 111 , 112 Therefore, carrying EDMs are called pathogenic mutations, which are more closely related to tumor immune response.
Some studies found that POLE/POLD1 mutation analysis may help determine which patients can benefit from ICI treatment, and this ability is independent of d‐MMR/MSI‐H. It was found that POLE/POLD1 mutations were associated with an improved response rate, PFS, and OS in patients with metastatic uroepithelial carcinoma, NSCLC, melanoma, CRC, and other malignant tumors treated with PD‐1 inhibitors. 108 , 113 , 114 Wang et al. analyzed the incidence of POLE/POLD1 mutations in 47,721 patients with different tumors. The mutation rates of POLE/POLD1 were 2.79% and 1.37%, respectively, and the mutations were higher in skin tumors (16.59%), endometrial cancer (14.85%), melanoma (14.73%), and CRC (7.37%). Also, in most cancers, the TMB of patients who carried POLE/POLD1 mutations was significantly higher than that of non‐carriers. Among patients treated with ICIs, the OS of patients who carried POLE/POLD1 mutations was significantly better than that of non‐carriers. This study also found that 26% of the patients who had POLE/POLD1 mutations also showed MSI‐H. After removing this subset of patients, the remaining patients with MSS were found to benefit significantly from ICI treatment. Multivariate Cox regression analysis showed that POLE/POLD1 mutation was an independent factor determining which solid tumor patients may benefit from ICI treatment. 108
In CRC patients, the application of POLE/POLD1 mutation as a molecular marker for ICI treatment is being researched. In 2017, Gong et al. reported a case of an MSS colon cancer patient with POLE mutation who showed a significant response rate after receiving Pembrolizumab. This patient not only had a high level of TILs but also showed a hypermutated tumor profile, with TMB reaching 122 mut/Mb, and the expression of PD‐1 and PD‐L1 as high as in 90% and 99% of the tumor cells, respectively. 115 Subsequently, researchers found that nearly 1.8%‐3.1% of CRC patients were diagnosed with pathogenic POLE mutation and showed higher TMB and PD‐L1. 111 , 112 , 116 Another group of researchers found that patients with POLE pathogenic mutation had similar immunogenicity as d‐MMR patients, with higher CD8+ lymphocytes, cytotoxic T‐cell markers, TMB, neoantigen load, and effector cytokines. PD‐1, PD‐L1, CTLA‐4, LAG3, and indoleamine 2,3‐dioxygenase 1 (IDO1) were highly expressed, too. 117 At present, there have been more and more clinical studies focusing on POLE/POLD1 mutation and ICI treatment, and more evidence supporting the use of POLE/POLD1 mutation as molecular markers is expected. The clinical trials studying the efficacy of ICI in treating CRC patients with POLE/POLD1 mutation are summarized in Table 1.
TABLE 1.
The clinical trials of ICI treatment of CRC patients with POLE/POLD1 mutations
| Clinical Trials.gov Identifier | Title | Condition or Disease | Treatment | Phase | Actual Enrollment | Primary Outcome Measures |
|---|---|---|---|---|---|---|
| NCT03150706 131 | A Phase II Study of Avelumab in Patients with Mismatch Repair Deficient or POLE Mutated Metastatic Colorectal Cancer | Metastatic Colorectal Cancer | Avelumab | Phase 2 | 33 participants | Serum CEA; TSH; T3; free T4; EKG; CT (or MRI) scans of evaluable/measurable lesions by RECIST 1.1 |
| NCT03435107 132 | A Phase II Study of Durvalumab in Patients with Mismatch Repair Deficient or POLE Mutated Metastatic Colorectal Cancer | Metastatic Colorectal Cancer | Durvalumab | Phase 2 | 33 participants | Objective Response Rates; CT (or MRI) scans of evaluable/measurable lesions by RECIST 1.1 |
| NCT04969029 133 | Randomized, Controlled Phase II Study of Immunotherapy Versus Standard Chemotherapy as Adjuvant Therapy After Surgery for Colon Cancer With MSI‐H or POLE/ POLD1 Mutations | Immunotherapy, Adjuvant Therapy, Colon Cancer, MSI‐H | Tirelizumab | Phase 2 | 30 participants | 3‐year Relapse‐Free Survival |
| NCT03810339 134 | A Phase II Open‐Label Study of Toripalimab, a PD−1 Antibody, in Participants with POLE or POLD1 Mutated and Non‐MSI‐H Advanced Solid Tumors | Solid Tumor, Advanced Cancer | Toripalimab | Phase 2 | 35 participants | Objective Response Rates; Overall Survival; Progression‐Free Survival; Adverse Events |
| NCT03461952 135 | A Phase II Open‐Label, Randomized Non‐Comparative Trial of Nivolumab Alone or in Combination with Ipilimumab for the Treatment of Patients with Advanced Hypermutated Solid Tumors Detected by a Blood‐Based Assay | Advanced Solid Tumors with POLE and/or POLD1 mutation | Nivolumab Ipilimumab | Phase 2 | 4 participants | Objective Response Rate by RECIST 1.1 |
| NCT03767075 136 | Basket of Baskets: A Modular, Open‐label, Phase II, Multicentre Study to Evaluate Targeted Agents in Molecularly Selected Populations with Advanced Solid Tumors | Advanced Solid Tumor (Arm 1C: tumors with POLE mutation, POLD1 mutation) | Arm 1: Atezolizumab | Phase 2 | 1000 participants | Overall response rate by RECIST 1.1 |
In conclusion, the use of POLE/POLD1 pathogenic mutation as molecular markers for ICI treatment has been confirmed in several types of tumors. Although the incidence of POLE/POLD1 pathogenic mutations in CRC is low, these patients have unique immuno‐phenotypes and extraordinary responses to ICI treatment, which are worthy of further study. More importantly, the POLE/POLD1 pathogenic mutation has a higher application value in MSS patients that may enable more patients to benefit from ICI treatment.
6. CIRCULATING TUMOR DNA (CTDNA)
Circulating tumor DNA (ctDNA), a classic short‐stranded DNA fragment having an average length of 120–160 bp, is present in the serum and is released due to necrosis and apoptosis of circulating tumor cells. 118 , 119 Previous studies showed that ctDNA was widely used to detect minimal residual diseases, predict patient prognosis, assess tumor burden, assess mutational burden, predict microsatellite status, monitor the effects of chemotherapy and targeted therapy, detect drug resistance, and identify novel mutations to predict the efficacy of treatment. 118 , 120 In recent years, ctDNA has been widely explored and much progress has been made in predicting the response rate of ICI treatment in tumor patients.
A study including 1,000 patients with locally advanced or metastatic tumors treated with ICIs showed that on‐treatment ctDNA dynamics appear to be predictive of the long‐term benefit of immunotherapy across tumor types. ctDNA dynamics can help differentiate patients who will ultimately derive benefit from immunotherapy from those who are unlikely to derive further benefit. 121 A prospective clinical trial involving 5 different tumor types also revealed the correlation between the level of ctDNA and the efficacy of ICI treatment. The trial measured the ctDNA level in 73 patients after 3 cycles of Pembrolizumab treatment and found that 33 patients had a ctDNA level lower than the baseline. These patients showed better clinical efficacy during the treatment, and 14 patients achieved an objective response. The ctDNA level of 40 patients increased compared to the baseline. Most of these patients experienced disease progression or poor survival, and only one patient experienced objective response. When the ctDNA was eliminated due to the treatment, the patient's OS was 100%. 122 The results indicated that ctDNA may be used as a molecular marker independent of PD‐L1 and TMB.
In melanoma patients, researchers found that patients with low ctDNA (≤20 copies/ml) before commencing therapy who received first‐line ICI treatment showed a higher PFS. When ctDNA was elevated, patients who received CTLA‐4 and PD‐1 inhibitors showed better PFS than patients treated only with PD‐1 inhibitors. 123 The IMvigor010 (NCT02450331) study tested the ctDNA of patients with infiltrating urothelial carcinoma who received Atezolizumab and analyzed the relationship between the level of ctDNA and the treatment effect of Atezolizumab, showing that the treatment with Atezolizumab improved the disease‐free survival (DFS) and OS of ctDNA‐positive patients but showed no change in ctDNA‐negative patients. The ctDNA clearance occurred in 18.2% of ctDNA‐positive patients treated with Atezolizumab who showed better OS and DFS. In the observation group, this phenomenon was observed in only 3.8% of the patients. 124 This result showed that the level and clearance rate of ctDNA can be used as predictors of the response and effect of ICI treatment in tumor patients. To further explore the mechanism, researchers performed exploratory transcriptional analysis to assess the expression of other immune‐related biomarkers and found that compared to ctDNA‐negative patients, ctDNA‐positive patients showed a higher expression of cell cycle and keratin genes, suggesting that these tumors may be more aggressive. In ctDNA‐positive patients, when TMB and PD‐L1 were overexpressed or pan‐fibroblast TGFβ response (F‐TBRS) signature and angiogenesis genes were under‐expressed, Atezolizumab significantly improved patient prognosis, but there was no such relationship in ctDNA‐negative patients. 124 Other researchers also found that it is feasible to evaluate TMB, PD‐L1, and microsatellites states using ctDNA 118 , 119 ; therefore, it is a potential molecular marker for ICI treatment and worthy of further research. Kim et al. found that, in metastatic gastric cancer, the total effective rate and PFS of patients with a high ctDNA burden who were treated with PD‐1 inhibitor significantly improved compared to patients with low ctDNA burden. 125 These results provided support for ctDNA as a promising molecular marker for ICI treatment. In CRC, it was found that ctDNA can be used for the detection of initial tumor burden, patient prognosis assessment, minimal residual disease detection, recurrence monitoring, chemotherapy or targeted therapy response prediction, drug resistance monitoring, etc. 119 However, studies suggesting the possibility of ctDNA as a molecular marker for ICI treatment in CRC patients are not available.
In addition to b‐TMB, ctDNA can be detected in liquid biopsies. Compared to obtaining specimens from tissues, liquid biopsy has a profound impact on the precision treatment of tumors due to its advantages of non‐invasiveness, high safety, fast detection speed, cost‐effectiveness, ease of sampling, and strong reproducibility. Common detection methods include digital PCR, BEAMing (Bead, Emulsion, Amplification, and Magnetic), WES, and NGS. These different technologies have their advantages and disadvantages, requiring comprehensive consideration by researchers. 119 In recent years, with the advancements in NGS technology, more and more researchers have adopted NGS for ctDNA detection. Sequencing Quality Control Phase 2 (SEQC2) project studied the performance of the NGS‐based ctDNA detection method. The study used a cross‐platform blind verification method to comprehensively evaluate the sensitivity, specificity, accuracy, and reproducibility of ctDNA liquid biopsy products. The results of this study may help establish industry standards, provide technical guidelines for liquid biopsies, and promote the development and transformation of tumor and clinical studies. 126
In conclusion, ctDNA is a potential novel molecular marker for ICI treatment for CRC.
7. CONSENSUS MOLECULAR SUBTYPES (CMS)
Using genome expression and cell phenotypes, Guinney et al. proposed the consensus molecular subtypes (CMS) of CRC, which included CMS1 (MSI Immune, 14%), CMS2 (Canonical, 37%), CMS3 (Metabolic, 13%), and CMS4 (Mesenchymal, 23%). 127 CMS1 is mainly composed of d‐MMR/MSI‐H tumors, with TMB‐H, BRAF mutation, and high immune infiltration characteristics.
CMS1 patients showed highly infiltrated cytotoxic lymphocytes, CD8+ T cells, CD4+ memory T cells, T helper cells 1, follicular helper T cells, γδ T cells, activated dendritic cells, natural killer cells (NK cells), and M1 macrophages and active immune microenvironment and good prognosis. 127 , 128 , 129 At the same time, PD‐1, PD‐L1, CTLA‐4, etc., were highly expressed in CMS1. 127 , 128 , 129 , 130 TMB and neoantigen loads were high, too, 103 suggesting that the CMS1 patients may benefit from ICI treatment.
High chromosome instability, “WNT and MYC activation—APC mutation—adenoma—TP53 mutation—adenocarcinoma” model were characteristics of CMS2. 127 , 130 CMS2 was also known as the “immune desert.” There were lesser infiltrating immune cells in tumor tissues and were mainly composed of static NK cells and naive CD4+ T cells or B cells. 128 Therefore, CMS2 patients cannot acquire active anti‐tumor immunity. Among all CMS groups, the CMS2 group had the least MSI‐H patients and the expression of PD‐1/PD‐L1 in tumor tissues was lower. Therefore, it showed a weak response to ICI treatment. 127 , 128 , 129
CMS3 was characterized by high Kirsten rat sarcoma viral oncogene (KRAS) mutation, metabolic dysregulation, and impaired lipid oxidation. 127 , 130 About 16% of the CMS3 group population was composed of MSI‐H patients, 103 and some patients expressed T helper cell 17, naive B cells, naive T cells, resting T cells, and PD‐1+ T cells. Same as CMS2, lymphocytes, monocytes, and myeloid cells were less among the infiltrated cells, suggesting that the immune microenvironment of CMS3 was in a dormant state. 127 , 128 , 129
CMS4 was characterized by epithelial‐mesenchymal transition, stromal remodeling, interstitial activation, angiogenesis, and immune cell infiltration. MSI‐H tumors accounted for 6% of CMS4. CMS4 had an activated complement pathway and was rich in lymphocytes and macrophages, showcasing its inflammatory characteristics. However, due to fewer CD8+ and CD4+ T cells and more Treg cells and M2 macrophages, CMS4 presented an immune mechanism different from CMS1. 127 , 128 , 129 Characteristics of consensus molecular subtypes are summarized in Table 2.
TABLE 2.
Characteristics of Consensus molecular subtypes
| CMS1 | CMS2 | CMS3 | CMS4 | |
|---|---|---|---|---|
| Percentage (%) | 14 | 37 | 13 | 23 |
| Microsatellite | Mainly composed of MSI‐H (76%) | MSI‐L or MSS, MSI‐H (2%) | Mixed MSI status, MSI‐H (16%) | MSI‐L or MSS,MSI‐H (6%) |
| CIMP | High | – | Low | – |
| SCNA | – | High | Low | High |
| CIN | – | High | Low | Middle |
| APC and TP53 | APC low and TP53 low | APC high and TP53 high | – | – |
| Characteristics | Immune infiltration and activation, BRAF mutation | WNT and MYC activation | Metabolic dysregulation, KRAS mutation | EMT, angiogenesis, stromal infiltration, TGF‐β activation |
| TiME | Immune activated, high immune checkpoint inhibitor expression, TMB‐H, and neoantigen loads | Immune desert, less infiltrating immune cells, poor immune checkpoint inhibitor expression | Immune excluded, less infiltrating immune cells | Immune inflamed, high complement, Treg cells, and M2 macrophages |
Abbreviations: BRAF, B‐Raf proto‐oncogene; CIMP, CpG Island Methylator Phenotype; CIN, chromosomal instability; EMT, Epithelial‐Mesenchymal Transition; KRAS, Kirsten rat sarcoma viral oncogene; MSI‐H, microsatellite instability‐high; MSI‐L, microsatellite instability‐low; MSS, microsatellite stability; P53, tumor protein p53; SCNA, somatic copy number alterations; TGF‐β, transforming growth factor‐β.
TiME, tumor immune microenvironment.
CMS1 and CMS4 tumors were also known as “hot tumors.” They are immunoreactive and the main targets of ICI treatment. CMS2 and CMS3 tumors, known as “cold tumors,” had a low response rate to ICI treatment. Therefore, methods to make them “hot” to increase the response rate to ICI treatment need to be identified.
8. CONCLUSION
Precision medicine began with targeted therapy and thriving in immunotherapy. The original intention of immunotherapy was to use anti‐tumor immunity to develop new treatment methods and achieve more precise treatment. As the main method of immunotherapy, ICI treatment has been used to treat a variety of malignant tumors, and it has become one of the main treatment methods for mCRC. In the ICI treatment, the concept of applying molecular markers to guide the treatment of different diseases helps clinicians develop an open mind and is an important step to bringing precision medicine into practice. However, there are no perfect molecular markers at present. It is difficult to identify a molecular marker that shows a consistently accurate performance to predict ICI treatment response and curative effect. Therefore, we hope that better molecular markers are identified or the existing ones are developed further to promote the standardization of ICI treatment so that more patients can benefit from it. An ideal molecular marker should have high specificity and sensitivity, a wide range of applications, ease of sampling and measurement, and standardized detection methods. However, the aforementioned molecular markers do not meet these requirements currently. Therefore, we hope that better markers are identified or the value of existing markers can be fully tapped through joint applications and improved detection methods. In short, the development of ideal molecular markers has a long way to go.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Fenqi Du contributed to the writing of the manuscript; Yanlong Liu conceived and finalized the content of the manuscript.
Du F, Liu Y. Predictive molecular markers for the treatment with immune checkpoint inhibitors in colorectal cancer. J Clin Lab Anal.2022;36:e24141. doi: 10.1002/jcla.24141
DATA AVAILABILITY STATEMENT
The datasets generated in the current study will be made available on request by the corresponding author.
REFERENCES
- 1. Zou X, Jia M, Wang X, et al. Interpretation of the World Cancer Report 2020. Chin J Clin Thorac Cardiovasc Surg. 2021;28(1):8. doi: 10.7507/1007-4848.202010033 [DOI] [Google Scholar]
- 2. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177‐193. doi: 10.3322/caac.21395 [DOI] [PubMed] [Google Scholar]
- 3. Thomas J, Leal A, Overman MJ. Clinical development of immunotherapy for deficient mismatch repair colorectal cancer. Clin Colorectal Cancer. 2020;19(2):73‐81. doi: 10.1016/j.clcc.2020.02.002 [DOI] [PubMed] [Google Scholar]
- 4. Chung KY, Gore I, Fong L, et al. Phase II study of the anti‐cytotoxic T‐lymphocyte‐associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010;28(21):3485‐3490. doi: 10.1200/jco.2010.28.3994 [DOI] [PubMed] [Google Scholar]
- 5. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443‐2454. doi: 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167‐3175. doi: 10.1200/jco.2009.26.7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luchini C, Bibeau F, Ligtenberg M, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD‐1/PD‐L1 expression and tumour mutational burden: a systematic review‐based approach. Ann Oncol. 2019;30(8):1232‐1243. doi: 10.1093/annonc/mdz116 [DOI] [PubMed] [Google Scholar]
- 8. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248‐5257. [PubMed] [Google Scholar]
- 9. Jácome AA, Eng C. Role of immune checkpoint inhibitors in the treatment of colorectal cancer: focus on nivolumab. Expert Opin Biol Ther. 2019;19(12):1247‐1263. doi: 10.1080/14712598.2019.1680636 [DOI] [PubMed] [Google Scholar]
- 10. Funkhouser WK, Lubin IM, Monzon FA, et al. Relevance, pathogenesis, and testing algorithm for mismatch repair‐defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn. 2012;14(2):91‐103. doi: 10.1016/j.jmoldx.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 11. Koopman M, Kortman GAM, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100(2):266‐273. doi: 10.1038/sj.bjc.6604867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870‐6875. doi: 10.1073/pnas.95.12.6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ward R, Meagher A, Tomlinson I, et al. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut. 2001;48(6):821‐829. doi: 10.1136/gut.48.6.821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim CG, Ahn JB, Jung M, et al. Effects of microsatellite instability on recurrence patterns and outcomes in colorectal cancers. Br J Cancer. 2016;115(1):25‐33. doi: 10.1038/bjc.2016.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang S, Na Y, Joung SY, et al. The significance of microsatellite instability in colorectal cancer after controlling for clinicopathological factors. Medicine. 2018;97(9):e0019. doi: 10.1097/md.0000000000010019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gavin PG, Colangelo LH, Fumagalli D, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18(23):6531‐6541. doi: 10.1158/1078-0432.Ccr-12-0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568‐571. doi: 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rooney M, Shukla S, Wu C, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48‐61. doi: 10.1016/j.cell.2014.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kreiter S, Vormehr M, van de Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692‐696. doi: 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McGranahan N, Furness AJS, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463‐1469. doi: 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science. 2017;357(6349):409‐413. doi: 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michael‐Robinson JM, Biemer‐Hüttmann A, Purdie DM, et al. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut. 2001;48(3):360‐366. doi: 10.1136/gut.48.3.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin Z, Yoon HH. The promise of PD‐1 inhibitors in gastro‐esophageal cancers: microsatellite instability vs. PD‐L1. J Gastrointest Oncol. 2016;7(5):771‐788. 10.21037/jgo.2016.08.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter‐inhibitory checkpoints. Cancer Discov. 2015;5(1):43‐51. doi: 10.1158/2159-8290.Cd-14-0863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dudley JC, Lin M‐T, Le DT, et al. Microsatellite instability as a biomarker for PD‐1 blockade. Clin Cancer Res. 2016;22(4):813‐820. doi: 10.1158/1078-0432.Ccr-15-1678 [DOI] [PubMed] [Google Scholar]
- 26. Sun BL. Current microsatellite instability testing in management of colorectal cancer. Clin Colorectal Cancer. 2021;20(1):e12‐e20. doi: 10.1016/j.clcc.2020.08.001 [DOI] [PubMed] [Google Scholar]
- 27. Damato A, Iachetta F, Antonuzzo L, et al. Phase II study on first‐line treatment of NIVolumab in combination with folfoxiri/bevacizumab in patients with Advanced COloRectal cancer RAS or BRAF mutated ‐ NIVACOR trial (GOIRC‐03‐2018). BMC Cancer. 2020;20(1):822. doi: 10.1186/s12885-020-07268-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuismanen SA, Moisio A‐L, Schweizer P, et al. Endometrial and colorectal tumors from patients with hereditary nonpolyposis colon cancer display different patterns of microsatellite instability. Am J Pathol. 2002;160(6):1953‐1958. doi: 10.1016/s0002-9440(10)61144-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. doi: 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Le DT, Kim TW, Van Cutsem E, et al. Phase II Open‐Label Study of Pembrolizumab in Treatment‐Refractory, Microsatellite Instability‐High/Mismatch Repair‐Deficient Metastatic Colorectal Cancer: KEYNOTE‐164. J Clin Oncol. 2020;38(1):11‐19. doi: 10.1200/jco.19.02107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. André T, Shiu K‐K, Kim TW, et al. Pembrolizumab in microsatellite‐instability‐high advanced colorectal cancer. N Engl J Med. 2020;383(23):2207‐2218. doi: 10.1056/NEJMoa2017699 [DOI] [PubMed] [Google Scholar]
- 32. Chalabi M, Fanchi LF, Dijkstra KK, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR‐proficient and MMR‐deficient early‐stage colon cancers. Nat Med. 2020;26(4):566‐576. doi: 10.1038/s41591-020-0805-8 [DOI] [PubMed] [Google Scholar]
- 33. FDA . FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017. Accessed September 20, 2021. https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐grants‐accelerated‐approval‐pembrolizumab‐first‐tissuesite‐agnostic‐indication
- 34. FDA . FDA grants nivolumab accelerated approval for MSI‐H or dMMR colorectal cancer. 2017. Accessed September 20, 2021. https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐grants‐nivolumab‐accelerated‐approval‐msi‐h‐or‐dmmr‐colorectal‐cancer
- 35. FDA . FDA approves pembrolizumab for first‐line treatment of MSI‐H/dMMR colorectal cancer. 2020. Accessed September 20, 2021. https://www.fda.gov/drugs/drug‐approvals‐and‐databases/fda‐approves‐pembrolizumab‐first‐line‐treatment‐msi‐hdmmr‐colorectal‐cancer
- 36. NCCN . NCCN Clinical Practice Guidelines in Colon Cancer. 2021. Accessed September 20, 2021. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf
- 37. NCCN . NCCN Clinical Practice Guidelines in Rectal Cancer. 2021. Accessed September 20, 2021. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf
- 38. Damilakis E, Mavroudis D, Sfakianaki M, et al. Immunotherapy in metastatic colorectal cancer: could the latest developments hold the key to improving patient survival? Cancers. 2020;12(4):889. doi: 10.3390/cancers12040889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cohen R, Hain E, Buhard O, et al. Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of microsatellite instability or mismatch repair deficiency status. JAMA Oncol. 2019;5(4):551‐555. doi: 10.1001/jamaoncol.2018.4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rodriguez‐Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89(23):1758‐1762. doi: 10.1093/jnci/89.23.1758 [DOI] [PubMed] [Google Scholar]
- 41. Svrcek M, Lascols O, Cohen R, et al. MSI/MMR‐deficient tumor diagnosis: which standard for screening and for diagnosis? Diagnostic modalities for the colon and other sites: differences between tumors. Bull Cancer. 2019;106(2):119‐128. doi: 10.1016/j.bulcan.2018.12.008 [DOI] [PubMed] [Google Scholar]
- 42. Wang F, Wang Z‐X, Chen G, et al. Expert opinions on immunotherapy for patients with colorectal cancer. Cancer Commun. 2020;40(10):467‐472. doi: 10.1002/cac2.12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zeng Z, Yang B, Liao Z. Biomarkers in immunotherapy‐based precision treatments of digestive system tumors. Front Oncol. 2021;11: 650481. doi: 10.3389/fonc.2021.650481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zang Y‐S, Dai C, Xu X, et al. Comprehensive analysis of potential immunotherapy genomic biomarkers in 1000 Chinese patients with cancer. Cancer Med. 2019;8(10):4699‐4708. doi: 10.1002/cam4.2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. doi: 10.1186/s13073-017-0424-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fabrizio DA, George Jr TJ, Dunne RF, et al. Beyond microsatellite testing: assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol. 2018;9(4):610‐617. doi: 10.21037/jgo.2018.05.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goodman AM, Sokol ES, Frampton GM, et al. Microsatellite‐stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol Res. 2019;7(10):1570‐1573. doi: 10.1158/2326-6066.Cir-19-0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 2014;371(23):2189‐2199. doi: 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hellmann MD, Ciuleanu T‐E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093‐2104. doi: 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samstein RM, Lee C‐H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202‐206. doi: 10.1038/s41588-018-0312-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598‐2608. doi: 10.1158/1535-7163.Mct-17-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open‐label, phase 2 KEYNOTE‐158 study. Lancet Oncol. 2020;21(10):1353‐1365. doi: 10.1016/s1470-2045(20)30445-9 [DOI] [PubMed] [Google Scholar]
- 53. Fukuoka S, Hara H, Takahashi N, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an Open‐Label, Dose‐Escalation, and Dose‐Expansion Phase Ib Trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38(18):2053‐2061. doi: 10.1200/jco.19.03296 [DOI] [PubMed] [Google Scholar]
- 54. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD‐1 inhibition. N Engl J Med. 2017;377(25):2500‐2501. doi: 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Labriola MK, Zhu J, Gupta R, et al. Characterization of tumor mutation burden, PD‐L1 and DNA repair genes to assess relationship to immune checkpoint inhibitors response in metastatic renal cell carcinoma. J Immunother Cancer. 2020;8(1):e000319. doi: 10.1136/jitc-2019-000319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krieger T, Pearson I, Bell J, et al. Targeted literature review on use of tumor mutational burden status and programmed cell death ligand 1 expression to predict outcomes of checkpoint inhibitor treatment. Diagn Pathol. 2020;15(1):6. doi: 10.1186/s13000-020-0927-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schrock AB, Ouyang C, Sandhu J, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI‐high metastatic colorectal cancer. Ann Oncol. 2019;30(7):1096‐1103. doi: 10.1093/annonc/mdz134 [DOI] [PubMed] [Google Scholar]
- 58. Garofalo A, Sholl L, Reardon B, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8(1):79. doi: 10.1186/s13073-016-0333-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Buchhalter I, Rempel E, Endris V, et al. Size matters: dissecting key parameters for panel‐based tumor mutational burden analysis. Int J Cancer. 2019;144(4):848‐858. doi: 10.1002/ijc.31878 [DOI] [PubMed] [Google Scholar]
- 60. Budczies J, Allgäuer M, Litchfield K, et al. Optimizing panel‐based tumor mutational burden (TMB) measurement. Ann Oncol. 2019;30(9):1496‐1506. doi: 10.1093/annonc/mdz205 [DOI] [PubMed] [Google Scholar]
- 61. Cyriac G, Gandhi L. Emerging biomarkers for immune checkpoint inhibition in lung cancer. Semin Cancer Biol. 2018;52(Pt 2):269‐277. doi: 10.1016/j.semcancer.2018.05.006 [DOI] [PubMed] [Google Scholar]
- 62. Roszik J, Haydu LE, Hess KR, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016;14(1):168. doi: 10.1186/s12916-016-0705-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251‐264. doi: 10.1016/j.jmoldx.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Endris V, Buchhalter I, Allgäuer M, et al. Measurement of tumor mutational burden (TMB) in routine molecular diagnostics: in silico and real‐life analysis of three larger gene panels. Int J Cancer. 2019;144(9):2303‐2312. doi: 10.1002/ijc.32002 [DOI] [PubMed] [Google Scholar]
- 65. Chen Y‐T, Seeruttun SR, Wu X‐Y, et al. Maximum somatic allele frequency in combination with blood‐based tumor mutational burden to predict the efficacy of atezolizumab in advanced non‐small cell lung cancer: a Pooled Analysis of the Randomized POPLAR and OAK Studies. Front Oncol. 2019;9:1432. doi: 10.3389/fonc.2019.01432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang Z, Duan J, Cai S, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non‐small cell lung cancer with use of a next‐generation sequencing cancer gene panel. JAMA Oncol. 2019;5(5):696‐702. doi: 10.1001/jamaoncol.2018.7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen XI, Fang L, Zhu Y, et al. Blood tumor mutation burden can predict the clinical response to immune checkpoint inhibitors in advanced non‐small cell lung cancer patients. Cancer Immunol Immunother. 2021;70(12):3513‐3524. doi: 10.1007/s00262-021-02943-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mazanet MM, Hughes CCW. B7–H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol. 2002;169(7):3581‐3588. doi: 10.4049/jimmunol.169.7.3581 [DOI] [PubMed] [Google Scholar]
- 69. Duffy MJ, Crown J. Biomarkers for predicting response to immunotherapy with immune checkpoint inhibitors in cancer patients. Clin Chem. 2019;65(10):1228‐1238. doi: 10.1373/clinchem.2019.303644 [DOI] [PubMed] [Google Scholar]
- 70. Kythreotou A, Siddique A, Mauri FA, et al. PD‐L1. J Clin Pathol. 2018;71(3):189‐194. doi: 10.1136/jclinpath-2017-204853 [DOI] [PubMed] [Google Scholar]
- 71. Nikolova M, Lelievre J‐D, Carriere M, et al. Regulatory T cells differentially modulate the maturation and apoptosis of human CD8+ T‐cell subsets. Blood. 2009;113(19):4556‐4565. doi: 10.1182/blood-2008-04-151407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069‐1086. doi: 10.1158/2159-8290.Cd-18-0367 [DOI] [PubMed] [Google Scholar]
- 73. Han Y, Liu D. Li L PD‐1/PD‐L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727‐742. [PMC free article] [PubMed] [Google Scholar]
- 74. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847‐856. doi: 10.1158/1535-7163.Mct-14-0983 [DOI] [PubMed] [Google Scholar]
- 75. Gu L, Chen M, Guo D, et al. PD‐L1 and gastric cancer prognosis: a systematic review and meta‐analysis. PLoS One. 2017;12(8):e0182692. doi: 10.1371/journal.pone.0182692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen Q, Li T, Yue W. Drug response to PD‐1/PD‐L1 blockade: based on biomarkers. Onco Targets Ther. 2018;11:4673‐4683. doi: 10.2147/ott.S168313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Garon EB, Hellmann MD, Rizvi NA, et al. Five‐year overall survival for patients with advanced non‐small‐cell lung cancer treated with pembrolizumab: results from the Phase I KEYNOTE‐001 Study. J Clin Oncol. 2019;37(28):2518‐2527. doi: 10.1200/jco.19.00934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mok TSK, Wu Y‐L, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD‐L1‐expressing, locally advanced or metastatic non‐small‐cell lung cancer (KEYNOTE‐042): a randomised, open‐label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819‐1830. doi: 10.1016/s0140-6736(18)32409-7 [DOI] [PubMed] [Google Scholar]
- 79. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375(19):1823‐1833. doi: 10.1056/NEJMoa1606774 [DOI] [PubMed] [Google Scholar]
- 80. FDA . FDA expands pembrolizumab indication for first‐line treatment of NSCLC (TPS ≥1%). 2019. Accessed September 20, 2021. https://www.fda.gov/drugs/fda‐expands‐pembrolizumab‐indication‐first‐line‐treatment‐nsclc‐tps‐1
- 81. Wang X, Teng F, Kong L, et al. PD‐L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016;9:5023‐5039. doi: 10.2147/ott.S105862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Llosa NJ, Luber B, Tam AJ, et al. Intratumoral adaptive immunosuppression and type 17 immunity in mismatch repair proficient colorectal tumors. Clin Cancer Res. 2019;25(17):5250‐5259. doi: 10.1158/1078-0432.Ccr-19-0114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Huyghe N, Baldin P, Van den Eynde M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch‐repair tumours? Gastroenterol Rep. 2020;8(1):11‐24. doi: 10.1093/gastro/goz061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aguiar PN, Santoro IL, Tadokoro H, et al. The role of PD‐L1 expression as a predictive biomarker in advanced non‐small‐cell lung cancer: a network meta‐analysis. Immunotherapy. 2016;8(4):479‐488. doi: 10.2217/imt-2015-0002 [DOI] [PubMed] [Google Scholar]
- 85. Marginean EC, Melosky B. Melosky b is there a role for programmed death ligand‐1 testing and immunotherapy in colorectal cancer with microsatellite instability? Part II‐the challenge of programmed death ligand‐1 testing and its role in microsatellite instability‐high colorectal cancer. Arch Pathol Lab Med. 2018;142(1):26‐34. doi: 10.5858/arpa.2017-0041-RA [DOI] [PubMed] [Google Scholar]
- 86. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61‐68. doi: 10.1016/j.canlet.2016.01.043 [DOI] [PubMed] [Google Scholar]
- 87. Barker HE, Paget JTE, Khan AA, et al. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15(7):409‐425. doi: 10.1038/nrc3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mlecnik B, Bindea G, Pagès F, et al. Tumor immunosurveillance in human cancers. Cancer Metastasis Rev. 2011;30(1):5‐12. doi: 10.1007/s10555-011-9270-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rozek LS, Schmit SL, Greenson JK, et al. Tumor‐infiltrating lymphocytes, Crohn's‐like lymphoid reaction, and survival from colorectal cancer. J Natl Cancer Inst. 2016;108(8):djw027. doi: 10.1093/jnci/djw027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kwak Y, Koh J, Kim D‐W, et al. Immunoscore encompassing CD3+ and CD8+ T cell densities in distant metastasis is a robust prognostic marker for advanced colorectal cancer. Oncotarget. 2016;7(49):81778‐81790. doi: 10.18632/oncotarget.13207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xie QK, He WZ, Hu WM, et al. Tumor‐infiltrating lymphocyte as a prognostic biomarker in stage IV colorectal cancer should take into account the metastatic status and operation modality. Cancer Manag Res. 2018;10:1365‐1375. doi: 10.2147/cmar.S162147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Loupakis F, Depetris I, Biason P, et al. Prediction of benefit from checkpoint inhibitors in mismatch repair deficient metastatic colorectal cancer: role of tumor infiltrating lymphocytes. Oncologist. 2020;25(6):481‐487. doi: 10.1634/theoncologist.2019-0611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Smyrk TC, Watson P, Kaul K, et al. Tumor‐infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91(12):2417‐2422. [PubMed] [Google Scholar]
- 94. Kitsou M, Ayiomamitis G, Zaravinos A. High expression of immune checkpoints is associated with the TIL load, mutation rate and patient survival in colorectal cancer. Int J Oncol. 2020;57(1):237‐248. doi: 10.3892/ijo.2020.5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Le DT, Hubbard‐Lucey VM, Morse MA, et al. A blueprint to advance colorectal cancer immunotherapies. Cancer Immunol Res. 2017;5(11):942‐949. doi: 10.1158/2326-6066.Cir-17-0375 [DOI] [PubMed] [Google Scholar]
- 96. Angell H, Galon J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013;25(2):261‐267. doi: 10.1016/j.coi.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 97. Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol. 2014;232(2):199‐209. doi: 10.1002/path.4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44(3):698‐711. doi: 10.1016/j.immuni.2016.02.025 [DOI] [PubMed] [Google Scholar]
- 99. Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128‐2139. doi: 10.1016/s0140-6736(18)30789-x [DOI] [PubMed] [Google Scholar]
- 100. Noepel‐Duennebacke S, Juette H, Schulmann K, et al. Microsatellite instability (MSI‐H) is associated with a high immunoscore but not with PD‐L1 expression or increased survival in patients (pts.) with metastatic colorectal cancer (mCRC) treated with oxaliplatin (ox) and fluoropyrimidine (FP) with and without bevacizumab (bev): a pooled analysis of the AIO KRK 0207 and RO91 trials. J Cancer Res Clin Oncol. 2021;147(10):3063‐3072. doi: 10.1007/s00432-021-03559-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yomoda T, Sudo T, Kawahara A, et al. The immunoscore is a superior prognostic tool in stages II and III colorectal cancer and is significantly correlated with Programmed Death‐Ligand 1 (PD‐L1) expression on tumor‐infiltrating mononuclear cells. Ann Surg Oncol. 2019;26(2):415‐424. doi: 10.1245/s10434-018-07110-z [DOI] [PubMed] [Google Scholar]
- 102. Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938‐945. doi: 10.1038/nm.3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Picard E, Verschoor CP, Ma GW, et al. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front Immunol. 2020;11:369. doi: 10.3389/fimmu.2020.00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kreidieh M, Mukherji D, Temraz S, et al. Expanding the scope of immunotherapy in colorectal cancer: current clinical approaches and future directions. Biomed Res Int. 2020;2020:9037217. doi: 10.1155/2020/9037217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230(2):148‐153. doi: 10.1002/path.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mouw KW, Goldberg MS, Konstantinopoulos PA, et al. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675‐693. doi: 10.1158/2159-8290.Cd-17-0226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chae YK, Davis AA, Raparia K, et al. Association of tumor mutational burden with DNA repair mutations and response to anti‐PD‐1/PD‐L1 therapy in non‐small‐cell lung cancer. Clin Lung Cancer. 2019;20(2):88‐96.e86. doi: 10.1016/j.cllc.2018.09.008 [DOI] [PubMed] [Google Scholar]
- 108. Wang F, Zhao Q, Wang YN, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5(10):1504‐1506. doi: 10.1001/jamaoncol.2019.2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING‐mediated T‐cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646‐661. doi: 10.1158/2159-8290.Cd-18-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Härtlova A, Erttmann SF, Raffi FAM, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity. 2015;42(2):332‐343. doi: 10.1016/j.immuni.2015.01.012 [DOI] [PubMed] [Google Scholar]
- 111. Bourdais R, Rousseau B, Pujals A, et al. Polymerase proofreading domain mutations: New opportunities for immunotherapy in hypermutated colorectal cancer beyond MMR deficiency. Crit Rev Oncol Hematol. 2017;113:242‐248. doi: 10.1016/j.critrevonc.2017.03.027 [DOI] [PubMed] [Google Scholar]
- 112. Mo S, Ma X, Li Y, et al. Somatic POLE exonuclease domain mutations elicit enhanced intratumoral immune responses in stage II colorectal cancer. J Immunother Cancer. 2020;8(2):e000881. doi: 10.1136/jitc-2020-000881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ricciuti B, Recondo G, Spurr LF, et al. Impact of DNA Damage Response and Repair (DDR) gene mutations on efficacy of PD‐(L)1 immune checkpoint inhibition in non‐small cell lung cancer. Clin Cancer Res. 2020;26(15):4135‐4142. doi: 10.1158/1078-0432.Ccr-19-3529 [DOI] [PubMed] [Google Scholar]
- 114. Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD‐1/PD‐L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36(17):1685‐1694. doi: 10.1200/jco.2017.75.7740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Gong J, Wang C, Lee PP, et al. Response to PD‐1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Canc Netw. 2017;15(2):142‐147. doi: 10.6004/jnccn.2017.0016 [DOI] [PubMed] [Google Scholar]
- 116. Guo Y, Guo XL, Wang S, et al. Genomic alterations of NTRK, POLE, ERBB2, and microsatellite instability status in chinese patients with colorectal cancer. Oncologist. 2020;25(11):e1671‐e1680. doi: 10.1634/theoncologist.2020-0356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Domingo E, Freeman‐Mills L, Rayner E, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1(3):207‐216. doi: 10.1016/s2468-1253(16)30014-0 [DOI] [PubMed] [Google Scholar]
- 118. Cabel L, Proudhon C, Romano E, et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol. 2018;15(10):639‐650. doi: 10.1038/s41571-018-0074-3 [DOI] [PubMed] [Google Scholar]
- 119. Li L, Zhang J, Jiang X, et al. Promising clinical application of ctDNA in evaluating immunotherapy efficacy. Am J Cancer Res. 2018;8(10):1947‐1956. [PMC free article] [PubMed] [Google Scholar]
- 120. Zhu C, Zhuang W, Chen L, et al. Frontiers of ctDNA, targeted therapies, and immunotherapy in non‐small‐cell lung cancer. Transl Lung Cancer Res. 2020;9(1):111‐138. doi: 10.21037/tlcr.2020.01.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang QU, Luo J, Wu S, et al. Prognostic and predictive impact of circulating tumor DNA in patients with advanced cancers treated with immune checkpoint blockade. Cancer Discov. 2020;10(12):1842‐1853. doi: 10.1158/2159-8290.Cd-20-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bratman SV, Yang SYC, Iafolla MAJ, et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nature Cancer. 2020;1(9):873‐881. doi: 10.1038/s43018-020-0096-5 [DOI] [PubMed] [Google Scholar]
- 123. Marsavela G, Lee J, Calapre L, et al. Circulating tumor DNA predicts outcome from first‐, but not second‐line treatment and identifies melanoma patients who may benefit from combination immunotherapy. Clin Cancer Res. 2020;26(22):5926‐5933. doi: 10.1158/1078-0432.Ccr-20-2251 [DOI] [PubMed] [Google Scholar]
- 124. Powles T, Assaf ZJ, Davarpanah N, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595(7867):432‐437. doi: 10.1038/s41586-021-03642-9 [DOI] [PubMed] [Google Scholar]
- 125. Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD‐1 inhibition in metastatic gastric cancer. Nat Med. 2018;24(9):1449‐1458. doi: 10.1038/s41591-018-0101-z [DOI] [PubMed] [Google Scholar]
- 126. Deveson IW, Gong B, Lai K, et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat Biotechnol. 2021;39(9):1115‐1128. doi: 10.1038/s41587-021-00857-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350‐1356. doi: 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Karpinski P, Rossowska J, Sasiadek MM. Immunological landscape of consensus clusters in colorectal cancer. Oncotarget. 2017;8(62):105299‐105311. doi: 10.18632/oncotarget.22169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Becht E, de Reyniès A, Giraldo NA, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22(16):4057‐4066. doi: 10.1158/1078-0432.Ccr-15-2879 [DOI] [PubMed] [Google Scholar]
- 130. Dienstmann R, Vermeulen L, Guinney J, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17(2):79‐92. doi: 10.1038/nrc.2016.126 [DOI] [PubMed] [Google Scholar]
- 131. ClinicalTrials.gov . A Phase II Study of Avelumab in Patients With Mismatch Repair Deficient or POLE Mutated Metastatic Colorectal Cancer. 2017. Accessed September 20, 2021. https://clinicaltrials.gov/ct2/show/NCT03150706?term=NCT03150706&draw=2&rank=1
- 132. ClinicalTrials.gov . A Phase II Study of Durvalumab in Patients With Mismatch Repair Deficient or POLE Mutated Metastatic Colorectal Cancer. 2018. Accessed September 20, 2021. https://clinicaltrials.gov/ct2/show/NCT03435107?term=NCT03435107&draw=2&rank=1
- 133. ClinicalTrials.gov . Randomized Controlled Phase II Study of Immunotherapy Versus Standard Chemotherapy as Adjuvant Therapy After Surgery for Colon Cancer With MSI‐H or POLE/ POLD1 Mutations. 2021. Accessed September 20, 2021. https://clinicaltrials.gov/ct2/show/NCT04969029?term=NCT04969029&draw=2&rank=1
- 134. ClinicalTrials.gov . A Phase II Open Label Study of Toripalimab, a PD‐1 Antibody, in Participants With POLE or POLD‐1 Mutated and Non‐MSI‐H Advanced Solid Tumors. 2019. Accessed September 20, 2021. https://clinicaltrials.gov/ct2/show/NCT03810339?term=NCT03810339&draw=2&rank=1
- 135. ClinicalTrials.gov . A Phase II Open Label, Randomized Non‐Comparative Trial of Nivolumab Alone or in Combination With Ipilimumab for the Treatment of Patients With Advanced Hypermutated Solid Tumors Detected by a Blood Based Assay. 2018. Accessed September 20, 2021. https://clinicaltrials.gov/ct2/show/NCT03461952?term=NCT03461952&draw=2&rank=1
- 136. ClinicalTrials.gov . Basket of Baskets: A Modular, Open‐label, Phase II, Multicentre Study to Evaluate Targeted Agents in Molecularly Selected Populations with Advanced Solid Tumours. 2018. Accessed September 20, 2021. https://clinicaltrials.gov/ct2/show/NCT03767075?term=NCT03767075&draw=2&rank=1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated in the current study will be made available on request by the corresponding author.