

Summary
Mammalian SWI/SNF (BAF) chromatin remodelers play dosage-sensitive roles in many human malignancies and neurologic disorders. The gene encoding the BAF-subunit, ACTL6A, is amplified early in the development of many squamous cell carcinomas (SCCs), but its oncogenic role remains unclear. Here we demonstrate that ACTL6A overexpression leads to its stoichiometric assembly into BAF complexes and drives their interaction and engagement with specific regulatory regions in the genome. In normal epithelial cells, ACTL6A was substoichiometric to other BAF-subunits. However, increased ACTL6A levels by ectopic expression or in SCC cells led to near-saturation of ACTL6A within BAF complexes. Increased ACTL6A occupancy enhanced polycomb opposition genome-wide to activate SCC genes, and also facilitated the co-dependent loading of BAF and TEAD-YAP complexes on chromatin. Both mechanisms appeared to be critical and function as a molecular AND gate for SCC initiation and maintenance, thereby explaining the specificity of the role of ACTL6A amplification in SCCs.
eTOC Blurb:
Chang et al. find ACTL6A plays a dosage-sensitive role underlying squamous cell carcinoma (SCC). Early in the course of the development of SCC, ACTL6A gene amplification increases its normally unsaturated occupancy within BAF complexes, leading to epigenetic de-repression by PRC redistribution and increased chromatin loading of TEAD-YAP.
Graphical Abstract
Introduction
Mammalian SWI/SNF (also known as BAF) complexes belong to a family of ATP-dependent chromatin remodelers, which contain an ATPase motor that binds nucleosomes and acts to distort or disrupt DNA-histone contacts (Clapier et al., 2017; He et al., 2020; Mashtalir et al., 2020). The enzymatic remodeling activity of the BAF complex allows DNA-binding proteins like transcription factors to access their recognition sites for gene regulation as well as other proteins involved in various nuclear processes including DNA repair and decatenation (Barisic et al., 2019; Clapier et al., 2017; Kadoch and Crabtree, 2015). Interestingly, BAF complex composition is dynamic in that various assemblies from its 15 subunits encoded by 29 genes can be formed in a given cell and play distinct roles across the genome and in different cell types (He et al., 2020; Mashtalir et al., 2020; Wang et al., 1996; Wu et al., 2009). While they lack sequence-specific DNA recognition, BAF subunits contain domains involved in binding to diverse histone modifications, AT-rich sequences, cruciform DNA structures as well as chromatin and transcriptional regulators that act in concert to guide BAF complex targeting over the genome (Kadoch and Crabtree, 2015). Unique alterations in BAF complex composition during development and in response to signaling further specialize it for engaging specific transcriptional programs (Wu et al., 2009). And yet, the biochemical and structural properties conferred by individual subunits to diversify the remodeler’s genomic targeting and recruitment of distinct transcriptional regulators remain largely undefined.
The distinct roles of individual subunits in BAF complexes have gained attention as alterations in different subunits cause specific cancers and are found collectively in over 20% of all human cancers (Kadoch et al., 2013; Shain and Pollack, 2013). Frequently, these mutations, such as those found on ARID1A, are heterozygous and loss-of-function, indicating that BAF subunits are dosage-sensitive and that the complex functions as a tumor suppressor. Dosage-sensitive roles for several subunits are also seen in the development of the nervous system and contribute to autism and intellectual disability (Ronan et al., 2013; Wenderski et al., 2020). While the BAF complex is generally considered a tumor suppressor, some cancers bear gain-of-function BAF alterations, as in synovial sarcomas, where a chromosomal translocation at SS18 results in an oncogenic SS18-SSX fusion, which retargets BAF across the genome to reverse polycomb-mediated repression and activate oncogenes including SOX2 (Clark et al., 1994; Kadoch and Crabtree, 2013; McBride et al., 2018). Thus, alterations in individual subunits compromise specific biologic actions of BAF complexes, and identifying the underlying mechanisms holds potential for the development of targeted therapy (Kadoch and Crabtree, 2015; Mashtalir et al., 2020; Wilson and Roberts, 2011).
The BAF-subunit gene encoding actin-like 6a (ACTL6A, originally called BAF53A (Zhao et al., 1998) is located on human chromosomal segment 3q26, an amplification hotspot in multiple SCC types including SCCs in the lung, skin, cervix, and oral mucosa (Ciriello et al., 2013; Heselmeyer et al., 1996; Speicher et al., 1995; Tonon et al., 2005). The amplification event occurs early in the course of lung SCC development and persists to the metastatic stage, and is thus deemed critical to both tumor initiation and progression (Jamal-Hanjani et al., 2017). Several driver genes in this amplicon including PI3KCA, SOX2 and TP63 have been identified (Bass et al., 2009; Keyes et al., 2011; Simpson et al., 2015; Watanabe et al., 2014), but the role ACTL6A plays in SCC oncogenesis is less clear.
SCC tumors arise from epithelial tissues, and in epidermis, ACTL6A expression appears in basal keratinocytes and wanes as cells undergo terminal differentiation (Bao et al., 2013). ACTL6A overexpression leads to an expanded basal layer of the epithelium, and conversely, loss of ACTL6A induces keratinocyte differentiation (Bao et al., 2013). ACTL6A also promotes the proliferation of other adult stem cells including hemopoietic and neural stem cells (Krasteva et al., 2012; Lessard et al., 2007). In head-and-neck SCCs, ACTL6A was found to interact with co-amplified TP63 to co-regulate genes promoting proliferation and suppressing differentiation (Saladi et al., 2017). ACTL6A and β-actin form the actin-related protein (ARP) module in BAF complexes and bind the HSA domain of SMARCA4 (BRG1) or SMARCA2 (BRM) ATPases (He et al., 2020; Mashtalir et al., 2020; Szerlong et al., 2008). Unlike actin, ACTL6A does not have ATPase activity (Zhao et al., 1998). In yeast, homologs of ACTL6A, Arp7/9, increase the efficiency of ATP utilization by the yeast SWI/SNF complex (Szerlong et al., 2008). Nevertheless, the oncogenic mechanism driven by ACTL6A amplification and the roles of BAF complexes in promoting SCCs are still largely unclear and understanding how ACTL6A amplification affects BAF complex composition and interaction surfaces may lead to new treatments for SCCs.
Here we find that amplification and overexpression of ACTL6A, which occurs in ~25% of all SCCs and about 40% of all lung SCCs, increases ACTL6A’s normally unsaturated occupancy within BAF complexes. This prompts polycomb redistribution, leading to the derepression of genes critical for SCC oncogenesis. In addition, increased ACTL6A incorporation directs BAF complex’s interaction with pro-oncogenic TEAD-YAP transcriptional regulators. We find that BAF and TEAD-YAP complexes are co-dependent for chromatin binding, thereby creating a positive-feedback mechanism to maintain open chromatin for transcription. Using structure-guided mutagenesis, we found that mutations of two adjacent hydrophobic amino acids within ACTL6A enhanced the binding between BAF-TEAD/YAP complexes and promoted SCC growth, thereby defining a potential druggable target for SCCs with ACTL6A-overexpression. Importantly, the dosage sensitivity of ACTL6A’s mechanism implies that a small reduction of ACTL6A function would be a viable therapeutic strategy against SCCs.
Results
BAF complex alterations across multiple SCC types
To comprehensively assess the mutational burden to all BAF subunits in SCCs, we quantified the frequencies of SCC tumors with mutations, copy number variations, and mRNA expression alterations in all 29 BAF-subunit genes using available data sets from three SCC tissue types in the cBioPortal for Cancer Genomics (Cerami et al., 2012; Gao et al., 2013). As chromosome 3q26, ACTL6A amplification was prominent as previously reported (Ciriello et al., 2013; Heselmeyer et al., 1996; Saladi et al., 2017; Speicher et al., 1995; Tonon et al., 2005) (41% of lung, 18% of head and neck, and 14% of cervical SCCs; 24.3% on average; Figure 1A). Nearly 50% of SCC tumors had increased ACTL6A expression (69% in the lung, 30% head and neck, and 51% cervical SCCs) (Figure 1A). Another BAF-subunit gene, BRD9, was also amplified (in 10% of combined SCC cases) (Figure 1A), but the amplification of BRD9, located on chromosome 5q15, infrequently overlapped with ACTL6A amplification, suggesting that either might be sufficient (Figure 1B). Surprisingly, the overall point mutation frequencies of BAF-subunit genes were low in all three SCCs despite their prevalence in other cancer types, including in basal cell carcinoma where 26% of cases harbor deleterious point mutations in ARID1A (Bonilla et al., 2016; Kadoch et al., 2013; Shain and Pollack, 2013) (Figure 1A).
Figure 1. ACTL6A amplification and/or overexpression are the most frequent genetic alterations among 29 BAF-subunit genes in lung, head-and-neck and cervical SCCs.

(A) Heat maps for alteration frequencies of 29 BAF-subunit genes in three SCC types. mRNA-high/low: z-score threshold ±2 relative to diploid samples.
(B) Venn diagram between SCC tumors with amplification of ACTL6A and BRD9. Combined cases of lung, cervical and head-and-nect SCCs. chr: chromosome.
(C) Alteration frequencies of 133 genes co-amplified with ACTL6A in SCCs.
(D) As in (B) for SCC tumors with ACTL6A amplification and PIK3CA missense mutations.
(E) Box plots of ACTL6A transcripts-per-million (TPM) in tumors and their paired normal tissues. LUSC: lung SCC. CESC: cervical SCC and endocervical adenocarcinoma. HNSC: head-and-neck SCC. ESCA: esophageal carcinoma. T: tumor samples. N: normal tissue samples. RNA-seq: TCGA and GTEx gene expression data from the GEPIA 2. *P < 0.01.
Known oncogenes upregulated within chromosome 3q25–28 amplicon include SOX2, TP63 and PIK3CA (Bass et al., 2009; Keyes et al., 2011; Simpson et al., 2015; Watanabe et al., 2014). Interestingly, upregulation of ACTL6A was as prevalent as PIK3CA and more prevalent than SOX2 and TP63 across all three SCC types, suggesting ACTL6A upregulation is advantageous to SCC tumors and implying the potential significance of ACTL6A as a SCC biomarker or therapeutic target (Figure 1C). Of note, while mutations in PIK3CA and BAF-subunit gene ARID1A co-occur to promote ovarian cancer (Chandler et al., 2015), PIK3CA mutations in SCCs were frequent but generally exclusive of ACTL6A amplification (Figure 1D). The median expression of ACTL6A in SCCs was 2- to 4-fold higher than in normal matched tissue samples (4.3-fold in the lung, 2.6-fold in head-and-neck, 2.8-fold in cervical SCCs) (Figure 1E). Thus, contrary to most other cancers, ACTL6A amplification and/or overexpression rather than point mutations in BAF complex subunits are the dominant alterations of BAF complexes in SCCs, suggesting that a distinct composition might be important for SCC oncogenesis.
Increased ACTL6A occupancy within BAF complexes in SCC cells
The dosage-sensitive roles of BAF subunits in neurodevelopment and cancers (Kadoch and Crabtree, 2015) led us to investigate how ACTL6A levels affect BAF complex composition in SCC cells. We determined the number of molecules per cell using quantitative Western blotting in three SCC cell lines bearing overexpressed ACTL6A, along with primary normal human keratinocytes (KC; cell type of origin for SCC) (Figures 2A and 2B; Figure S1A). Whole-cell lysates from equal numbers of cells of each cell line were used to quantify the amount of a specific protein from each line using a standard curve generated from purified recombinant proteins, followed by the calculation of protein mass and then the number of molecules per cell (Figure 2A). The total number of BAF complexes was estimated using an antibody that recognizes both SMARCA4 and SMARCA2, which are mutually exclusive catalytic subunits of BAF complexes. Surprisingly, we found that the number of ACTL6A molecules per normal keratinocyte (111,686±9,850) was only half the number of SMARCA4/SMARCA2 (222,311±21,635 per cell), suggesting that ACTL6A is sub-stoichiometric within the complex in normal keratinocytes (Figure 2B). In all three SCC cell lines we examined, however, ACTL6A molecules were ~1.5–2.5 fold more numerous than SMARCA4/SMARCA2, which could result in more ACTL6A-containing complexes (ACTL6A: 539,800±33,426 in FaDu (head and neck SCC cell line), 696,016±50,385 in NCI-H520 (lung SCC cell line), 830,683±116,333 in T.T (esophageal SCC cell line); SMARCA4/SMARCA2: 323,542±25,374 in FaDu, 389,563±9,539 in NCI-H520, 315,344± 20,536 in T.T) (Figure 2B).
Figure 2. Increased expression of ACTL6A in SCCs drives ACTL6A occupancy within BAF complexes.

(A) Outline of method for quantifying the number of molecules of a specific protein per cell from different cell lines.
(B) Quantifications of number of ACTL6A molecules per cell compared to SMARCA4/SMARCA2. SCC cell lines: FaDu (head-and-neck), NCI-H520 (lung), and T.T (esophageal). KC: primary normal human keratinocytes. n=3 experiments. Error bars indicate SEM. *P < 0.05.
(C) Co-immunoprecipitation (Co-IP) experiments using SMARCA4 antibody. Shown are Western blots and quantifications of relative levels of BAF subunits co-IP’d by SMARCA4 in SCC (FaDu) cells versus primary human keratinocytes (KC). n=3 experiments. Error bars indicate SEM. *P < 0.05. n.s.: not significant.
(D) Co-IP experiments using SMARCA4 antibody in primary human keratinocytes (KC) transduced by lentivirus for ACTL6A overexpression and vector control. Shown are Western blots and quantifications of relative levels of co-IP’d BAF subunits normalized to vector control. n=3 experiments. Error bars indicate SEM. *P < 0.05. n.s.: not significant.
(E) Quantifications for co-IP experiments by SMARCA4 antibody. Relative levels of co-IP’d ACTL6A in ACTL6A-overexpressing condition normalized to vector control. FaDu: SCC cell line. KC: primary human keratinocytes. HEK293T: human embryonic kidney 293T cells. Error bars indicate SEM. *P < 0.05. n=2–3 experiments.
To compare the occupancy of ACTL6A within BAF complexes in SCC cells to normal keratinocytes, we immunoprecipitated SMARCA4 (Figure 2C) and found the relative levels of ACTL6A (~7:1) in FaDu SCC cells were substantially higher compared to keratinocytes than that of SMARCA4 itself (~2:1), indicating a specific increase in ACTL6A occupancy within BAF complexes in SCC cells (Figure 2C). In contrast, for other BAF subunits including SMARCC1, SMARCE1, and ARID1A, their occupancy within the complexes was unaltered in SCC cells, where the relative co-immunoprecipitated levels of those subunits were similar to that of SMARCA4 (Figure 2C).
To specifically test if ACTL6A expression levels can change its stoichiometry, we reasoned that overexpressing ACTL6A in normal cells should increase ACTL6A incorporation into BAF complexes. Indeed, in keratinocytes transduced by lentivirus expressing ACTL6A, the levels of ACTL6A co-immunoprecipitated with SMARCA4 antibodies were 1.5- to 2-fold higher than in vector-control cells (Figure 2D). SMARCA4 levels remained unaltered, as did the incorporation of other BAF subunits including SMARCC1 (Figure 2D). Overexpressing ACTL6A in another non-SCC line HEK293T (human embryonic kidney 293T) also increased ACTL6A occupancy in BAF complexes (Figure 2E). However, elevating ACTL6A levels in FaDu SCC cells failed to increase its incorporation, indicating the occupancy of ACTL6A within BAF complexes is near saturation in SCC cells (Figure 2E). The increased occupancy was not attributable to ACTL6A polymerization as ectopically expressed ACTL6A-V5 did not bind untagged ACTL6A even though both were incorporated into BAF complexes (Figures S1B and S1C). Density sedimentation analysis of SCC cell nuclear extracts showed most ACTL6A co-migrated with SMARCA4, forming a full BAF complex (Figure S1D). Thus, these results reveal a dynamic occupancy of ACTL6A within BAF complexes in response to ACTL6A dosage. ACTL6A occupancy in BAF complexes is unsaturated in normal keratinocytes and becomes saturated upon its overexpression or in SCCs cells with ACTL6A amplification/overexpression.
ACTL6A regulates the accessibility of specific regulatory regions over the SCC genome in a dosage-dependent manner
To identify accessible chromatin regions in the SCC genomes that are dependent on ACTL6A stoichiometry, we conducted ATAC-seq (assay for transposase-accessible chromatin using sequencing) in SCC cells upon ACTL6A knockdown by small interfering RNA (siRNA). ACTL6A knockdown (siACTL6A) resulted in ~90% reduction of ACTL6A levels and did not affect the levels of other BAF subunits SMARCA4 and SMARCC1, consistent with the notion that ACTL6A is not required for the stability and assembly of BAF complexes (Braun et al., 2021; Krasteva et al., 2012) (Figure S2A). ACTL6A knockdown inhibited SCC cell proliferation (Figure S2B), as previously described (Saladi et al., 2017).
ACTL6A knockdown in SCC cells caused significant accessibility changes in 4,639 regulatory regions, in which 2,053 displayed decreased accessibility and 2,586 displayed increased accessibility (Figure 3A). To see whether ACTL6A’s effect on chromatin accessibility is dosage-dependent and whether its varied dosage would change accessibility to different degrees or at different loci, we reduced ACTL6A siRNA doses to reach an intermediate or 60% ACTL6A reduction in addition to our previous 90% knockdown condition (Figure S2C). Remarkably, the degree of accessibility changes correlated with ACTL6A levels (Figure 3B, 3C and 3F). Across both ACTL6A-promoted and repressed regions, intermediate reduction of ACTL6A resulted in a corresponding intermediate degree of chromatin accessibility decreases or increases, suggesting a dosage-sensitive role of ACTL6A in regulating chromatin accessibility in the SCC genome.
Figure 3. Genome-wide accessible chromatin profiling identifies ACTL6A-dependent regulatory regions in SCC cells.

(A) MA plot for ATAC-seq analysis in FaDu SCC cells 72-hours after transfection with ACTL6A siRNA (siACTL6A) and control siRNA (siControl). Color coded are significantly altered peaks with predicted TEAD-binding motifs (navy) and without TEAD motif (light blue). FDR<0.05. n.s.: not significant. n=2 experiments.
(B and C) Heat maps and metagene plots for ATAC-seq analysis across ACTL6A-promoted (B) or ACTL6A-repressed (C) accessible regions with different levels of ACTL6A reduction by siRNA in FaDu SCC cells. CPM: counts per million.
(D and E) Enrichment for predicted transcription-factor (TF) binding motifs in ACTL6A-promoted (D) or ACTL6A-repressed (E) accessible regions. Matches: number of peaks containing matched TF-binding motifs.
(F) Genome browser tracks showing regions with differential reduction of accessibility upon different ACTL6A knockdown from ATAC-seq. The sites contained TEAD motifs and were also bound by SMARCC1, YAP and TEAD1 identified by CUT&RUN.
(G) Immunofluorescence and quantifications of % of cells with nuclear or cytoplasmic YAP showing unaltered YAP subcellular localization in FaDu SCC cells 72 hours after ACTL6A siRNA (siACTL6A) knockdown versus siRNA control (siControl). Scale bars: 10 μm. n.s.: not significant.
(H) Quantifications for Western blot (WB) signals of phospho-S127 YAP normalized to total YAP levels. Samples including whole cell lysates (WCL), cytoplasmic extracts (cyto) and nuclear extracts (nuclear) from FaDu cells 72 hours after siRNA transfection. n=3 experiments.
(I) RT-qPCR showing YAP/TEAD target genes regulated by ACTL6A. n=3 experiments. Error bars indicate SEM. *P < 0.05.
To identify regulatory elements specifically dependent on ACTL6A for accessibility in SCC cells, we conducted motif enrichment analysis across ACTL6A-promoted and ACTL6A-repressed accessible regions. Interestingly, the top-most significant sequence motifs enriched in ACTL6A-promoted sites were for TEA domain (TEAD1–4) transcription factors (Figure 3D and 3F). 818 out of 2053 ACTL6A-promoted sites contained predicted TEAD motifs, which by contrast were in only 219 out of 2586 ACTL6A-repressed regions (Figure 3A). CEBPA, POU-domain and forkhead-box (FOX) binding motifs were also enriched in ACTL6A-promoted accessible regions, to a lesser extent, whereas CTCF motifs were depleted in these sites, suggesting insulator elements may be refractory to ACTL6A loss (Figure 3D). 92% of ACTL6A-promoted accessible regions were outside gene promoters, implicating ACTL6A’s role in promoting the accessibility of distal regulatory elements (Figure S2D). In contrast, 20% of ACTL6A-repressed regions were within gene promoters (Figure S2E), which were enriched for transcription factor motifs for AP-1 family members FOS and JUN (Figure 3E), likely reflecting the reaction to genotoxic stress characteristic of BAF subunit depletion (Dykhuizen et al., 2013; Smeyne et al., 1993; Wenderski et al., 2020).
The decreased TEAD accessibility by ACTL6A reduction in SCC cells suggests ACTL6A might regulate the oncogenic activity of TEAD-mediated pathways. TEADs, which form complexes with transcriptional co-activators YAP/TAZ, act downstream of the mechano-transduction and Hippo pathway that are involved in tumorigenesis, organ size control, regeneration and cancer resistance to targeted-, immune- and chemo-therapies (Nguyen and Yi, 2019; Yu et al., 2015a; Zanconato et al., 2016). In mammalian skin, YAP/TAZ promotes SCC initiation and progression (Debaugnies et al., 2018; Schlegelmilch et al., 2011; Vincent-Mistiaen et al., 2018). In Drosophila, Hippo signaling has been shown to depend on the Brahma (Brm)-associated proteins (BAP) complex, the fly SWI/SNF complex (Jin et al., 2013; Oh et al., 2013). However, the underlying mechanism remains elusive. Previous studies would suggest ACTL6A loss inhibits YAP activity by upregulating the WWC1 gene, which encodes a scaffold protein in the Hippo pathway and promotes YAP retention in the cytoplasm (Saladi et al., 2017). However, we did not observe increased levels of cytoplasmic YAP by immunostaining in siACTL6A SCC cells relative to siControl cells (Figure 3G). Western blotting also did not show changes in total YAP protein levels (Figure S5G, input lanes) or YAP S127 phosphorylation levels, which promotes its cytoplasmic retention (Yu et al., 2015a) (Figure 3H). WWC1 expression also remained unaltered upon ACTL6A knockdown (Figure S2F). Thus, the accessibility changes at TEAD enhancers upon ACTL6A loss are not caused by YAP translocating to the cytoplasm in these SCC cells. Instead, it implies that ACTL6A-BAF complexes directly promote the remodeling of local chromatin at TEAD enhancers.
The decreases in accessibility at predicted TEAD motifs were accompanied by reduced expression of TEAD/YAP/TAZ target genes in ACTL6A-knockdown SCC cells (Figure 3I; Figure S3A). Using RNA-seq analysis, we identified 188 differentially expressed genes between siACTL6A and siControl conditions in at least two of the three SCC cell lines, which included previously identified TEAD/YAP/TAZ target genes (Zhang et al., 2009) (Figures S3A and S3B). These targets were validated by quantitative PCR (qPCR) of independently prepared samples (Figure 3I). The expression of TEAD 1–4, YAP and TAZ were unaltered in ACTL6A-knockdown SCC cells (Figure S3C). Thus, ACTL6A does not regulate the transcription of TEAD/YAP/TAZ, and the reduced expression of their target genes was not due to reduced expression of TEAD/YAP/TAZ themselves. Of note, although TP63 and SOX2 are co-amplified with ACTL6A in SCCs (Figure 1C), the expression of their target genes (Watanabe et al., 2014) were largely unchanged upon ACTL6A knockdown (Figure S3D), suggesting ACTL6A is not essential for the downstream transcriptional programs of TP63 and SOX2. It is possible that the pioneer factor property of SOX2 which can initiate chromatin opening (Dodonova et al., 2020) or the remodeling activity from residual BAF complexes is sufficient to enable SOX2’s chromatin binding.
BAF complexes and TEAD-YAP co-localize on chromatin and modulate the accessibility of TEAD enhancers through a co-dependent mechanism in SCC cells
If the accessibility of TEAD enhancers is directly modulated by ACTL6A-BAF complexes, ACTL6A-BAF complexes should co-bind with TEAD-YAP across the genome. To test this prediction, we conducted CUT&RUN to profile the distribution of TEAD1, YAP, and BAF complexes (SMARCC1, a DNA-binding subunit of BAF complexes) genome-wide. As expected, the regions bound by TEAD1 and YAP largely overlapped (Figures 4A and 4B). Remarkably, 91% of TEAD1-YAP co-bound regions were also bound by SMARCC1, indicating their co-localization on chromatin (Figures 4A, 4B and 3F). Furthermore, 79% of YAP/TEAD1/SMARCC1 co-bound regions were at active enhancers marked by the histone modifications H3K27Ac and H3K4me1, concordant with earlier observations of TEAD-YAP binding at active enhancers (Stein et al., 2015; Zanconato et al., 2015) (Figure 4B; Figure S4A). A de novo motif search identified TEAD motifs in the 6,251 shared peaks, confirming the specificity of YAP and TEAD1 CUT&RUN profiling (Figure S4B). Notably, while BAF complex-bound regions were enriched most for binding motifs of FOSL2 (JUNB) and SP2 (Figure S4C), the accessibility decreases upon ACTL6A loss were largely at TEAD motifs (Figures 3A and 3D), indicating a specific effect of ACTL6A in the complex on promoting TEAD chromatin binding. The CUT&RUN profiling further confirmed the presence of YAP and TEAD1 at ACTL6A-promoted accessible regions marked by enhancer mark H3K4me1, in contrast to low YAP and TEAD1 levels at ACTL6A-repressed regions, which spanned H3K4me3-marked promoters (Figures S4D). ACTL6A knockdown reduced the binding of YAP-TEAD1 at enhancers that also lost accessibility, accompanied by reduced levels of the active mark H3K27Ac and SMARCC1 (Figures 4C and 4E). SMARCC1 peaks at YAP, TAZ and TEAD1–4 genes themselves were unaltered by ACTL6A loss, supporting the notion that ACTL6A does not affect YAP/TAZ/TEAD transcription (Figure S4E). Together, these results indicate that BAF complexes and TEAD-YAP co-bind across the genome, and ACTL6A functioning within the BAF complex plays a direct and specific role in targeting BAF complexes to TEAD-YAP enhancers and preparing the chromatin landscape to allow TEAD-YAP chromatin binding and transcription activation at their target loci.
Figure 4. Co-dependency between BAF complexes and TEAD-YAP for their chromatin loading.

(A) Venn diagram showing the overlap of peaks of SMARCC1, YAP and TEAD1 CUT&RUN in FaDu SCC cells. n=2 experiments.
(B) Heat maps for CUT&RUN YAP peaks aligned with indicated CUT&RUN peaks.
(C) Metagene plots of SMARCC1, YAP and H3K27Ac CUT&RUN over ACTL6A-promoted accessible sites in siControl and siACTL6A cells.
(D) Heat maps and metagene plots for ATAC-seq and CUT&RUN of SMARCC1, YAP and TEAD1 across regions with reduced YAP binding 48 hours after YAP/TAZ siRNA (siYAP/TAZ) knockdown versus siRNA control (siControl). CPM: counts per million.
(E) Genome browser tracks of ATAC-seq and CUT&RUN of SMARCC1, YAP and TEAD1 in siACTL6A, siYAP/TAZ and siControl cells.
The reduced SMARCC1 binding accompanied by reduced YAP binding on chromatin upon ACTL6A loss prompted us to see whether TEAD-YAP/TAZ complexes also facilitate BAF complex’s chromatin recruitment. Knocking down YAP/TAZ (siYAP/TAZ) reduced the expression of their target genes including CTGF and OLR1, as expected, and did not alter ACTL6A levels (Figure S4F). By CUT&RUN, we found substantial regions in siYAP/TAZ cells versus siControl cells with reduced YAP and TEAD1 chromatin binding (Figure 4D). Interestingly, SMARCC1 chromatin binding across these regions was also significantly diminished and corresponded with reduced accessibility as analyzed by ATAC-seq, suggesting TEAD-YAP/TAZ can recruit BAF complexes to chromatin (Figure 4D). Thus, BAF and TEAD-YAP/TAZ complexes are mutually dependent on each other for stable chromatin binding (Figure 4E). The presence of TEAD-YAP/TAZ at enhancers recruits BAF complexes, and meanwhile, ACTL6A in BAF complexes facilitates the chromatin binding of TEAD-YAP/TAZ, creating a positive feedback mechanism to maintain the accessibility of TEAD enhancers and the expression of their target genes.
Increasing ACTL6A levels induces TEAD-YAP binding to BAF complexes
The co-dependency of TEAD cognate motif accessibility on the presence of both ACTL6A-BAF complexes and TEAD-YAP/TAZ suggests an interaction between these two complexes, and that ACTL6A incorporation might modulate this interaction. To test whether ACTL6A assembled into the BAF complex serves to directly recruit TEAD-YAP, we conducted immunoprecipitation experiments. Using TEAD4 and pan-TEAD antibodies, we were able to co-immunoprecipitate SMARCA4 and several other BAF subunits from nuclear extracts of SCC cells (Figure 5A; Figure S5A). Furthermore, the reciprocal immunoprecipitation with SMARCA4 and ACTL6A antibodies yielded TEAD proteins (Figures S5B and S5C). Antibodies to YAP also co-immunoprecipitated BAF subunits (Figure 5B). To further confirm this interaction is direct, we developed an in vitro binding assay (Figure 5C). We noticed that increasing salt concentrations to 500mM (high salt) can disrupt the BAF-YAP interaction (Figure S5D). Hence, we introduced FLAG-tagged YAP into cells and purified FLAG-YAP from the nuclear extract under the high salt conditions, which co-precipitated TEAD but removed most BAF complexes (Figure S5E). We concurrently purified BAF complexes with a SMARCA4 antibody under high salt conditions, which yielded minimal YAP (Figure S5F). After co-incubation under low salt conditions, purified FLAG-YAP and purified BAF complexes were co-immunoprecipitated by the SMARCA4 antibody (Figure 5D), suggesting the interaction is direct.
Figure 5. ACTL6A promotes the direct binding of TEAD-YAP to BAF complexes.

(A-B) Co-IP experiments by TEAD4 (A) and YAP (B) antibodies using nuclear extracts from FaDu SCC cells.
(C) Workflow for in vitro binding experiments. BAF complexes and FLAG-YAP were purified separately under high-salt buffer conditions, and then co-incubated under low-salt buffer condition for in vitro binding examination.
(D) In vitro binding of purified FLAG-YAP and BAF complexes by SMARCA4 antibody-IP.
(E) Co-IP experiments by YAP and IgG antibodies showing decreased binding of SMARCA4 with YAP in ACTL6A CRISPR-knockout (KO) FaDu SCC cells. Quantifications normalized to control. INO80: INO80-complex subunit.
(F) Co-IP experiments by YAP and IgG antibodies in primary human keratinocytes overexpressing ACTL6A and vector-control.
(G) Quantifications for (F) showing increased binding of YAP and BAF subunits upon ACTL6A overexpression. Normalized to vector control. n=3 experiments. Mean ± SEM. *P < 0.05.
(H) The human BAF complex cryo-EM structure (PDB: 6LTJ) showing the position of ACTL6A P373/P374 residues (marked in red) in the nucleosome-bound BAF complex. Cyan: ACTL6A. Red: P373/P374 residues of ACTL6A. Green: HSA domain of SMARCA4. Yellow: Nucleosomal DNA. Olive: histone octamer.
(I) Quantifications for co-IP experiments by YAP antibodies in keratinocytes overexpressing WT, R377G and P373S/P374G ACTL6A. Normalized to vector control. n=3 experiments. Mean ± SEM. *P < 0.05. n.s.: not significant.
(J) Growth curves of FaDu SCC cells transduced with lentiviral constructs for ACTL6A CRISPR-KO and simultaneously reconstituted with KO-resistant WT, R377G, P373S/P374G ACTL6A, or vector-control. n=3 experiments. Error bars indicate SD.
To examine whether the interaction of TEAD-YAP and BAF complexes is dependent on ACTL6A, we conducted ACTL6A loss-of-function and gain-of-function analyses. Reducing ACTL6A levels in SCC cells by siRNA or CRISPR/Cas9 diminished BAF complex binding to YAP (Figure 5E; Figure S5G). Remarkably, overexpressing ACTL6A in normal human keratinocytes, which increased ACTL6A-containing BAF complexes (Figure 2D), enhanced the interaction (Figures 5F and 5G; Figure S5H). We did not observe YAP binding to INO80, another ACTL6A- associated chromatin remodeler (Figure 5E). Thus, ACTL6A is necessary and sufficient for the interaction between BAF complexes and TEAD-YAP.
The WW domains of YAP recognize the PPxY motif on its interaction partners (Chen and Sudol, 1995). While ACTL6A does not contain a PPxY motif, we speculated a proline-rich loop structure (PPSMRLKLI; a.a. 373–381) on ACTL6A that extends toward the nucleosomal DNA bound by the BAF complex (He et al., 2020) may serve as an alternative interaction point (Figure 5H). To examine the effect of ACTL6A P373S/P374G double mutations on the interaction between YAP and BAF complexes, we introduced P373S/P374G ACTL6A in human keratinocytes with WT ACTL6A as control and conducted YAP immunoprecipitation. While WT ACTL6A overexpression is sufficient to enhance the interaction (Figures 5F and 5G), unexpectedly, we found that overexpression of P373S/P374G ACTL6A induced higher levels of YAP binding to BAF complexes than WT ACTL6A (Figure 5I). A nearby R377G mutation did not show this effect on BAF-YAP binding (Figure 5I). To examine whether the increased binding might promote SCC proliferation, we knocked out endogenous ACTL6A using CRISPR/Cas9 and simultaneously reconstituted it with mutated or WT ACTL6A. Reconstituting WT ACTL6A rescued the proliferation defects caused by ACTL6A knockout, and notably, P373S/P374G mutants promoted SCC growth better than WT or R377G ACTL6A (Figure 5J). These mutations neither compromised ACTL6A stability nor altered its incorporation into BAF complexes (Figure S5I). Together, these results indicate that ACTL6A regulates TEAD-YAP activity to drive SCC growth by producing interaction surfaces on BAF complexes for TEAD-YAP binding.
ACTL6A overexpression redistributes polycomb over the genome
The SWI/SNF and polycomb-repressive complexes (PRCs) play opposing roles in epigenetic regulation. In flies, loss-of-function mutations in the SWI/SNF subunits suppress defects that are conferred by mutations in PRC1, indicating a rather dedicated relationship between these two classes of chromatin regulators (Tamkun et al., 1992). In synovial sarcoma, malignant rhabdoid tumors and several other BAF-subunit mutated cancers, PRCs are important primary targets of mammalian SWI/SNF or BAF complexes and distinct mutations in BAF complexes can result in either a gain or loss of its ability to evict PRCs (Bitler et al., 2015; Ho et al., 2011; Kadoch and Crabtree, 2013; Kadoch et al., 2017; Kia et al., 2008; Stanton et al., 2017; Wilson et al., 2010). To determine early consequences of ACTL6A amplification and whether they are attributable to perturbation of the BAF-PRC balance, we examined by CUT&RUN the genome-wide distribution of the PRC2-modified histone mark H3K27me3 in primary normal human keratinocytes following ACTL6A overexpression.
Remarkably, ACTL6A overexpression led to H3K27me3 redistribution over the genome (Figure 6A). We identified gene promoters with altered H3K27me3 deposition, most of which showed decreased H3K27me3 levels, consistent with previous studies which show BAF complexes rapidly and directly evict PRC by an ATP-dependent mechanism (Kadoch et al., 2017; Stanton et al., 2017) (Figure 6B). Affected promoters were primarily bivalent, i.e. marked by both H3K27me3 and H3K4me3, supporting BAF’s role in maintaining bivalent chromatin states (Stanton et al., 2017) (Figures 6B and 6C). Consistent with the effect of ACTL6A overexpression on H3K27me3 domains, knocking down ACTL6A caused increased H3K27me3 levels across the genome accompanied by a few sites with modestly reduced H3K27me3 levels (Figure S6A and S6B). To explore how changes in H3K27me3 levels upon ACTL6A overexpression affected transcription, we conducted RNA-seq analysis. As expected, alterations in H3K27me3 and transcription were largely negatively correlated. Genes with reduced H3K27me3 tended to have increased expression, and vice versa (Figure 6C). Notably, a subset of genes with altered H3K27me3 levels lacked significant changes in their expression, suggesting that H3K27me3 alterations induced by ACTL6A overexpression were likely direct consequences rather than secondary effects from transcriptional changes.
Figure 6. ACTL6A overexpression leads to redistribution of H3K27me3 and activation of SCC genes.

(A) Heat maps for H3K27me3 CUT&RUN differential 5kb-bins between primary human keratinocytes overexpressing ACTL6A and vector-control. CPM : counts per million. n=2 experiments.
(B) Profiles over TSS with decreased (top) and increased (bottom) H3K27me3 levels upon ACTL6A-overexpression versus vector control by CUT&RUN. Right: H3K4me3 ChIP-seq profiles of human keratinocytes (ENCODE). CPM: counts per million.
(C) Scatterplot of H3K27me3 and RNA fold-changes for genes with differential H3K27me3 levels upon ACTL6A-overexpression. Color codes: H3K4me3 levels, high versus low or negative (neg).
(D) Heatmap showing ACTL6A-dependent PRC target genes with corresponding transcriptional changes in SCC tumors. H3K27me3: CUT&RUN as in (A). RNA: HNSC (head-and-neck SCC) and LUSC (lung SCC) tumor versus normal tissue from GEPIA 2.
(E) Genome browser tracks at bivalent WNT7B gene upon ACTL6A-overexpression (+) compared to vector control (−).
(F) WNT7B medium expression levels in tumors and paired normal tissues across various cancers. Data from GEPIA 2. TPM: transcripts per million. HNSC: head-and-neck SCC. LUSC: lung SCC. BLCA: bladder urothelial carcinoma. CESC: cervical SCC and endocervical adenocarcinoma. ESCA: esophageal carcinoma. LUAD: lung adenocarcinoma. CHOL: cholangiocarcinoma. LGG: brain lower grade glioma. PRAD: prostate adenocarcinoma. PAAD: pancreatic adenocarcinoma. BRCA: breast invasive carcinoma. OV: ovarian serous cystadenocarcinoma. THYM: thymoma. THCA: thyroid carcinoma. UCEC: uterine corpus endometrial carcinoma. GBM: glioblastoma multiforme. STAD: stomach adenocarcinoma. SKCM: skin cutaneous melanoma.
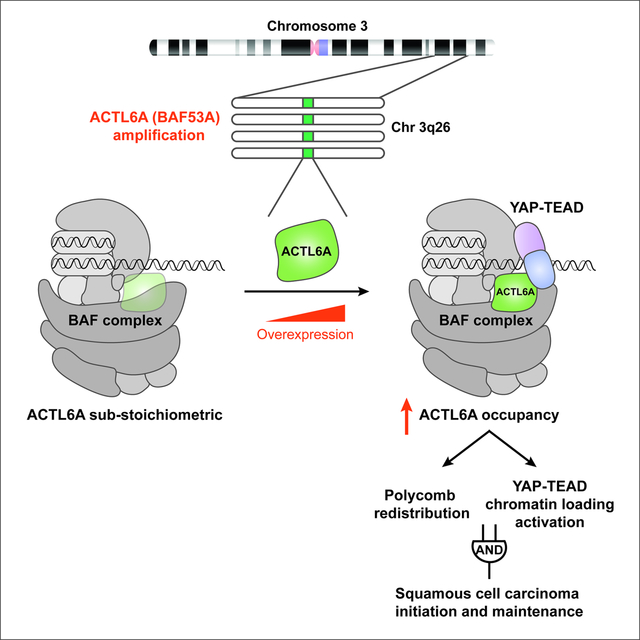
(G) Model for ACTL6A-amplification driven oncogenic mechanism in SCCs. Amplification or overexpression of ACTL6A leads to full occupancy of BAF complexes giving rise to two mechanisms promoting SCC initiation and maintenance.
The decrease of H3K27me3 marks upon ACTL6A overexpression was not due to keratinocyte differentiation, which in contrast leads to a global loss of H3K27me3 and an accompanying decrease in ACTL6A (Bao et al., 2013; Ezhkova et al., 2009). Furthermore, the H3K27me3 levels at polycomb-repressed differentiation genes such as KRT1 and LOR were unaltered (Figure S6C). Also unchanged were the expression of keratinocyte differentiation genes (KRT1, KRT10, IVL, LOR) and progenitor markers (KRT14, KRT5, TP63) (Figure S6D). Consistent with the observation that conditional loss of BAF subunits decreases H3K27me3 levels in HOX clusters (Ho et al., 2011), we observed a gain of H3K27me3 in the HOXB locus upon ACTL6A overexpression (Figure 6C; Figure S6E). Thus, ACTL6A overexpression in normal human keratinocytes leads to a redistribution of H3K27me3 over the genome.
Because ACTL6A amplification is a very early event in the pathogenesis of SCC (Jamal-Hanjani et al., 2017), we reasoned that overexpressing it in normal keratinocytes might initiate a program of SCC gene expression. If PRC redistribution is a major driving mechanism, then these SCC genes should be distinguished by PRC loss upon ACTL6A overexpression. To determine whether the polycomb target genes affected by ACTL6A dosage are also misregulated in SCC tumors in vivo, we examined their transcripts in SCC tumors versus normal tissues using TCGA/GTEx data sets available in GEPIA (Tang et al., 2017) (Figure 6D). Interestingly, we found 64 of the PRC targets displayed corresponding changes in their RNA levels in either lung SCC (LUSC) or head-and-neck SCC (HNSC) tumors compared to paired normal tissues with p-value<0.05. 47 genes that lost H3K27me3 upon ACTL6A overexpression were preferentially upregulated in LUSC or HNSC tumors, whereas 17 genes that gained H3K27me3 by ACTL6A overexpression were downregulated in LUSC or HNSC tumors (Figure 6D). The derepressed genes included WNT7B (Figures 6E and 6F). WNT7B encodes a Wnt ligand and has been found to contribute to skin carcinogenesis (Krimpenfort et al., 2019) and promote proliferation and invasion of oral SCC cells (Shiah et al., 2014). In pancreatic adenocarcinoma, WNT7B promotes tumors’ anchorage-independent growth and sphere formation (Arensman et al., 2014). ACTL6A overexpression in primary keratinocytes induced WNT7B upregulation and correspondingly reduced H3K27me3 levels at its bivalent promoter (Figure 6E). Two SMARCC1 CUT&RUN peaks near the H3K27me3 domain and within the WNT7B gene body were unaltered, suggesting ACTL6A incorporation did not affect BAF chromatin binding but instead affected its activity in antagonizing PRCs (Figure 6E). Upregulation of WNT7B occurred in several types of SCCs including head-and-neck SCC (HNSC) and lung SCC (LUSC), as well as cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) and esophageal carcinoma (ESCA) (Figure 6F).
Besides WNT7B, other identified ACTL6A-dependent PRC targets known to play roles in SCC oncogenesis included TWIST1, which is associated with epithelial–mesenchymal transition (EMT) in esophageal and head-and-neck SCCs (Jouppila-Matto et al., 2011; Lee et al., 2012); and SATB2, which drives carcinogenesis of oral SCC as well as other cancers and promotes the survival and chemoresistance of head-and-neck SCCs in part by interacting with ΔNp63α (Chung et al., 2010; Ge et al., 2020b; Seong et al., 2015; Yu et al., 2017) (Figure 6D). Several other ACTL6A-dependent PRC target genes belong to the forkhead box (FOX) family, members of which are often repressed by polycomb and poised for activation (Golson and Kaestner, 2016). FOXD1 upregulation induces EMT and chemoresistance of oral SCC cells (Chen et al., 2020); and FOXL2 is upregulated in SCC tumors and a driver of granulosa-cell tumors (Ge et al., 2020a; Shah et al., 2009). Another ACTL6A-dependent PRC target, CDKN2A, is considered a tumor suppressor; however, overexpression of CDKN2A has been noted in several tumors including SCC tumors (Romagosa et al., 2011). In sum, our results suggest that before malignant transformation, early ACTL6A overexpression in epithelial cells is sufficient to reduce polycomb-mediated repression of genes necessary for SCC oncogenesis by perturbing chromatin architecture and BAF-PRC opposition.
Discussion
Our studies reveal that ACTL6A gene amplification and/or overexpression leads to its increased occupancy within BAF complexes that then facilitates the establishment of an altered chromatin state for SCC development (Figure 6G). BAF subunits are highly dosage-sensitive (Kadoch and Crabtree, 2015). Mutations of BAF subunit genes implicated in human cancers and neurological disorders such as autism and intellectual disability are commonly heterozygous, indicating that a half-normal level is biologically significant (Kadoch and Crabtree, 2015). Thus, the 2- to 4-fold increase of ACTL6A levels in SCCs and the consequent increase in ACTL6A occupancy within the complex are consistent with the dosage-sensitive role for BAF complexes in human diseases. Indeed, our studies show that reductions of ACTL6A levels result in alterations in chromatin accessibility of the SCC genome and that altering ACTL6A dosage has two consequences. First, complexes having stoichiometric occupancy of BAF are more effective at evicting polycomb from bivalent promoters; and secondly, ACTL6A-BAF’s function prepares enhancers to receive signals mediated by TEAD-YAP (Figure 6G). These findings are consistent with previous studies showing that BAF complexes are required for the activation of both enhancers and polycomb-regulated bivalent promoters (Hodges et al., 2018; Nakayama et al., 2017). Our studies indicate that both mechanisms are critical and likely to function as an epigenetic AND gate for SCC initiation and maintenance. The requirement of both ACTL6A-dependent mechanisms likely explains the oncogenic specificity of ACTL6A amplification.
In the development of the mammalian nervous system, ACTL6A exchanges with ACTL6B to generate neuron-specific BAF complexes that coordinate gene expression underlying cell cycle exit and the initiation of neural differentiation (Braun et al., 2021; Lessard et al., 2007). In epithelial cells, ACTL6A levels fall as the cells differentiate. This reduction triggers a programmatic switch from proliferation to keratinocyte differentiation with the activation of keratinocyte differentiation genes including KLF4 (Bao et al., 2013; Krasteva et al., 2012; Lu et al., 2015). ACTL6A is also essential for proliferation and maintaining stem cell potency (Bao et al., 2013; Krasteva et al., 2012). Thus, we propose that the degree of occupancy of ACTL6A in chromatin remodelers is regulatory and therefore its dosage acts as a decisive signal underlying the transition between chromatin states during the initiation of SCCs.
Intriguingly, in contrast to ACTL6A amplification in SCCs, basal cell carcinoma (BCC), another cancer originating from basal epithelial cells, has a high frequency of heterozygous loss-of-function mutations in the BAF-subunit gene ARID1A (Bonilla et al., 2016), while such mutations are rare in SCCs (Figure 1A). This indicates different epidermal lineages (Sanchez-Danes and Blanpain, 2018) are specifically susceptible to distinct BAF complex alterations and illustrates the biologic specificity of their functions. Recent structural studies (He et al., 2020; Mashtalir et al., 2020) suggest that this specificity emerges from combinatorial assembly of the products of 29 genes encoding the 15 subunits creating composite surfaces at their interfaces available to interact with proteins such as TEADs and YAP.
Considerable effort has been dedicated to developing TEAD/YAP/TAZ small molecule inhibitors, with limited success (Calses et al., 2019). We find that ACTL6A incorporation promotes TEAD-YAP binding to BAF complexes, and the mutual dependency of BAF and TEAD-YAP creates a positive feedback mechanism to enhance their chromatin co-binding and promote transcriptional activity, suggesting a new therapeutic approach. The activating effects of ACTL6A as a BAF subunit on the TEAD-YAP pathway are consistent with genetic studies in flies, in which mutations in Brahma, the fly homolog of the SWI/SNF ATPase, hinder transcriptional activation by Yorkie (YAP homolog) (Jin et al., 2013; Oh et al., 2013). Our findings also support studies in breast epithelial lineage commitment where BAF complexes interact with the Hippo pathway component TAZ and positively regulate TAZ-induced transcription (Skibinski et al., 2014). Our CUT&RUN genome-wide mapping of the BAF complex and TEAD-YAP elucidate their co-occupancy across the genome and reveal the co-dependency of TEAD-YAP and ACTL6A-containing BAF complexes to bind to enhancers, which hence prepare the chromatin landscape to allow TEAD-YAP mediated transcription at target loci. How P373S/P374G mutations facilitate the interaction between BAF complexes and TEAD-YAP is unclear, and we speculate that perhaps mutating the rigid proline-proline motif might make the loop structure more flexible and create more room for the interaction. While ACTL6A reduction decreases the accessibility of TEAD enhancers, we find overexpression of ACTL6A alone in keratinocytes is not sufficient to significantly create new TEAD accessible sites or further enhance the accessibility of TEAD elements (data not shown). We did observe genome-wide chromatin accessibility changes, particularly at FOX binding motifs, upon ACTL6A overexpression (data not shown), but further investigation will be needed to decipher the mechanisms underlying the specificity of ACTL6A overexpression-induced effects, as well as possible consequences of overexpression of other BAF-subunit genes. Our data suggest already-accessible TEAD elements in keratinocytes might not be further increased by ACTL6A overexpression and additional signals or co-alterations of BAF and TEAD-YAP might be required to make new TEAD sites accessible.
Of note, others (Saladi et al., 2017) have reported ACTL6A activates YAP rather by an indirect mechanism, wherein ACTL6A, in collaboration with TP63, controls YAP nuclear localization by repressing genes including WWC1 that modulate YAP nuclear-cytoplasm shuttling. However, we did not detect changes in YAP subcellular localization upon ACTL6A loss; instead, we find BAF complexes directly interact with YAP and TEAD, and the interaction is dependent on ACTL6A.
Besides TEAD-YAP modulation, we find ACTL6A overexpression is sufficient to induce polycomb redistribution, resulting in the activation of genes known to have roles in SCCs. Although the exact mechanism of BAF-PRC antagonism remains unknown, the effects could be rooted in altered SMARCA4/SMARCA2 ATPase activity, which is required for BAF complexes to evict PRC1 (Kadoch et al., 2017; Stanton et al., 2017) and in yeast is promoted by the ACTL6A homologs Arp7/9 (Szerlong et al., 2008). Interestingly, the outcomes of ACTL6A-induced polycomb redistribution are rather selective for bivalent genes such as WNT7B, whose role in tumor initiation merits further investigation given the early occurrence of ACTL6A amplification during SCC development (Jamal-Hanjani et al., 2017), as does the role of ACTL6A in the interplay between BAF and PRC complexes. Interestingly, despite its high mutation rate, CDKN2A is overexpressed in some SCC tumors (Romagosa et al., 2011) and the reduction of polycomb repression at CDKN2A upon ACTL6A overexpression might contribute to SCC oncogenesis. In line with this, polycomb removal and CDKN2A activation have also been found upon SMARCB1 re-expression in SMARCB1-deficient tumor cells (Kia et al., 2008).
In summary, our studies demonstrate that altering subunit stoichiometry within a chromatin regulatory complex can be oncogenic, and that the dynamics of ACTL6A occupancy in BAF complexes may play roles in normal development by enabling protein-protein interactions with key regulators engaged in proliferation and stem cell function. Our studies indicate that both polycomb redistribution and TEAD-YAP facilitation are essential downstream mechanisms for the initiation and maintenance of SCCs. Therefore, therapeutic efforts might be directed towards reducing ACTL6A function or stoichiometry. The discovery that mutations of two adjacent residues in ACTL6A enhance TEAD-YAP binding to BAF complexes and SCC proliferation suggests a precise therapeutic target.
Limitations of the Study
This study primarily employs an in vitro cell culture system. Thus, further exploration of the link between ACTL6A dosage and TEAD-YAP/TAZ activation using in vivo models could better define the relevance of this mechanism in human cancers and how and when it contributes to SCC etiology. Additionally, many hallmarks of cancer such as metastasis, cellular signaling in the tumor microenvironment, immune cell infiltration and angiogenesis are absent from the cell culture systems and the potential role(s) of ACTL6A in regulating the genes involved in these processes are outside the reach of this study. The depletion of ACTL6A and YAP/TAZ by siRNA- and CRISPR-based methods took 48 to 72 hours, and it would be necessary to use auxin-inducible degron (AID) or other protein degradation technologies that allow rapid ACTL6A degradation to separate immediate from secondary effects. Direct recruitment of ACTL6A-containing versus ACTL6A-absent BAF complexes to polycomb-bound domains could also provide minute-by-minute kinetic analysis addressing the underlying mechanism of ACTL6A’s role in BAF-polycomb antagonism that is absent from this study.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Gerald R. Crabtree (crabtree@stanford.edu)
Materials availability
Plasmids generated in this study will be available upon request.
Data and code availability.
Next-generation sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Pan-TEAD (D3F7L) | Cell Signaling Technology | Cat# 13295S |
| Mouse anti-TEAD4 | Abcam | Cat# ab58310 |
| Mouse anti-TEAD1 | BD Biosciences | Cat# 610922 |
| Mouse anti-BAF53A (ACTL6A)(5H3L6) | Invitrogen | Cat# 702414 |
| Mouse anti-ARID1A (PSG3) | Santa Cruz Biotechnology | Cat# sc-32761 |
| Rabbit anti-BAF57 (SMARCE1) | Bethyl Laboratories | Cat# A300-810A |
| Rabbit anti-Phospho-YAP (Ser127) (D9W2I) | Cell Signaling Technology | Cat# 13008 |
| Mouse anti-Brg1 (SMARCA4) (H-10) | Santa Cruz Biotechnology | Cat# sc-374197 X |
| Rabbit anti-INO80 | Bethyl Laboratories | Cat# A303-371A |
| Mouse anti-EZH2 | BD Biosciences | Cat# 612666 |
| Rabbit anti-GAPDH (D16H11) XP | Cell Signaling Technology | Cat# 5174S |
| Mouse anti-V5 tag | Invitrogen | Cat# R960-25 |
| Mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025 |
| Rabbit IgG | MilliporeSigma | Cat# 12-370 |
| Mouse anti-YAP | Abnova | Cat# H00010413-M01 |
| Rabbit anti-BAF53A (ACTL6A) | Novus Biologicals | Cat# NB100-61628 |
| Mouse anti-FLAG (M2) | Sigma-Aldrich | Cat# F1804 |
| Donkey anti-mouse IgG | Invitrogen | Cat# A-21202 |
| Rabbit anti-TEAD1(D9X2L) | Cell Signaling Technology | Cat# 12292S |
| Rabbit anti-histone H3K4me1 | Abcam | Cat# ab8895 |
| Rabbit anti-histone H3K4me3 | Active motif | Cat# 39159 |
| Rabbit anti-histone H3K27me3 | Cell Signaling Technology | Cat# 9733 |
| Rabbit anti-histone H3K27Ac | Abcam | Cat# ab4729 |
| Rabbit anti-BAF53A (ACTL6A) | The Crabtree laboratory | N/A |
| Rabbit anti-Brg1/BRM (SMARCA4/SMARCA2) (J1) | The Crabtree laboratory | N/A |
| Rabbit anti-BAF155 (SMARCC1) | The Crabtree laboratory | N/A |
| Rabbit anti-YAP (D8H1X) XP | Cell Signaling Technology | Cat# 14074S |
| Bacterial and Virus Strains | ||
| One-Shot Stbl3 chemically competent E. coli | Invitrogen | Cat# C7373-03 |
| BL21(DE3) Competent E.coli | New England Biolabs | Cat# C2527I |
| Rosetta 2(DE3) competent cells | EMD Millipore | Cat# 71397-3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polybrene | Santa Cruz Biotechnology | Cat# sc-134220 |
| Polyethylenimine Max (PEI MAX) (MW 40,000) | Polysciences | Cat# 24765 |
| Digitonin | Millipore | Cat# 300410-250MG |
| Digitonin | Promega | Cat# G9441 |
| Spermidine trihydrochloride | Sigma-Aldrich | Cat# S2501-1G |
| Benzonase Nuclease | Sigma-Aldrich | Cat# E1014-25KU |
| Amylose resin | New England Biolabs | Cat# E8021L |
| Glutathione-superflow resin | Clontech | Cat# 635608 |
| Glutathione reduced | Sigma-Aldrich | Cat# G-4251 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10002D |
| 3X FLAG Peptide | MilliporeSigma | Cat# F4799-4MG |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10009D |
| Geltrex | Thermo Fisher Scientific | Cat# A1413302 |
| Transferrin | Roche | Cat# 10652202001 |
| EGF | Thermo Fisher Scientific | Cat# PHG0311 |
| Insulin | Sigma-Aldrich | Cat# I5500 |
| Forskolin | Tocris Bioscience | Cat# 1099 |
| VX-745 | Tocris Bioscience | Cat# 3915 |
| RO4929097 | Cellagen Technology | Cat# C7649 |
| Dexamethasone | Tocris Bioscience | Cat# 1126 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140-163 |
| Deoxyribonuclease I | Worthington Biochemical Corporation | Cat# LS002058 |
| TRIsure | Bioline | Cat# BIO-38033 |
| Plasmocin | InvivoGen | Cat# ant-mpp |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Fibronectin | Sigma-Aldrich | Cat# F1141-1MG |
| DharmaFECT 1 transfection reagent | Horizon Discovery | Cat# T-2001-01 |
| 5X siRNA Buffer | Horizon Discovery | Cat# B-002000-UB-100 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833-100mg |
| ON-TARGETplus | Horizon Discovery | Cat# L-008243-00 |
| Human ACTL6A siRNA | ||
| ON-TARGETplus Non-targeting Pool | Horizon Discovery | Cat# D-001810-10 |
| Blasticidin S HCl | Thermo Fisher Scientific | Cat# R21001 |
| Critical Commercial Assays | ||
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490S |
| NEBNext MultiplexOligos for Illumina (Dual Index Primers Set 1) | New England Biolabs | Cat# E7600S |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541S |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32854 |
| ZymoPURE II Plasmid Midiprep Kit | Zymo Research | Cat# D4200 |
| DNA Clean & Concentrator-5 | Zymo Research | Cat# D4013 |
| Direct-zol RNA Miniprep Kits | Zymo Research | Cat# R2050 |
| High Sensitivity D1000 Reagents (Sample Buffer & Ladder) | Agilent Technologies | Cat# 5067-5585 |
| High Sensitivity D1000 ScreenTapes | Agilent Technologies | Cat# 5067-5584 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| Bio-Rad Protein Assay Dye (Bradford) | Bio-Rad | Cat# 500-0006 |
| Lenti-X GoStix | Clontech | Cat# 631281 |
| PhiX Control v3 | Illumina | Cat# FC-110-3001 |
| SensiFAST SYBR Lo-ROX Kit | Bioline | Cat# BIO-94020 |
| SensiFAST cDNA Synthesis Kit | Bioline | Cat# BIO-65054 |
| KLD Enzyme Mix | New England Biolabs | Cat# M0554S |
| In-Fusion HD Cloning Kit | Clontech | Cat# 639650 |
| NEBNext Ultra II DNA Library Prep with Sample Purification Beads | New England Biolabs | Cat# E7103S |
| NEBNext Ultra II Directional RNA Library Prep Kit | New England Biolabs | Cat# E7760S |
| Deposited Data | ||
| Structure of nucleosome-bound human BAF complex | Protein Data Bank | ID# 6LTJ |
| Deep sequencing datasets from this study | Gene Expression Omnibus | GSE156788 |
| H3K4me3 ChIP-seq (keratinocytes) | ENCODE | ENCSR075OQB |
| H3K27me3 ChIP-seq | ENCODE | ENCSR377MRR |
| Experimental Models: Cell Lines | ||
| Human Epidermal Keratinocytes, adult (HEKa) | Thermo Fisher Scientific | Cat# C0055C |
| HEK293T | Takara | Cat# 632180 |
| FaDu | ATCC | Cat# HTB-43 |
| NCI-H520 | ATCC | Cat# HTB-182 |
| T.T | JCRB Cell Bank | Cat# JCRB0262 |
| KYSE70 | Sigma | Cat# 94072012 |
| Oligonucleotides | ||
| Primers | See Table S1 | N/A |
| Recombinant DNA | ||
| pQCXIH-Myc-YAP | Addgene | Cat# 33091 |
| CYC244-Flag-NLS-hYAP | This study | N/A |
| N106-hACTL6A | This study | N/A |
| CYC103-hACTL6A | This study | N/A |
| pGSTag | Addgene | Cat# 21877 |
| pMAL-c2X | Addgene | Cat# 75286 |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| Software and Algorithms | ||
| Adobe Creative Cloud | Adobe | https://www.adobe.com/creativecloud.html |
| Rstudio | RStudio | https://www.rstudio.com/ |
| Image Studio Lite | LI-COR | https://www.licor.com/bio/ |
| SnapGene | Insightful Science | https://www.snapgene.com/ |
| Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SeqPurge | Sturm et al., 2016 | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| deeptools | Ramirez et al., 2016 | N/A |
| macs2 | Zhang et al., 2008 | N/A |
| Homer | Heinz et al., 2010 | N/A |
| DEseq2 | Love et al., 2014 | N/A |
| edgeR | Robinson et al., 2010 | N/A |
| DiffBind | Ross-Innes et al., 2012 | N/A |
| chromVar | Schep et al., 2017 | N/A |
| ChiPseeker | Yu et al., 2015b | N/A |
| ChrAccR | https://github.com/GreenleafLab/ChrAccR | N/A |
| PyMOL v2.4.2 | Schrodinger | https://pymol.org |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell lines and culture conditions.
FaDu, a pharyngeal squamous cell carcinoma cell line, was purchased from ATCC (HTB-43) and cultured in ATCC-formulated Eagle’s Minimum Essential Medium supplemented with 10% fetal bovine serum (FBS; Omega Scientific) and antibiotics (100 units/mL Penicillin and 100 μg/mL Streptomycin; Gibco). NCI-H520 lung squamous cell carcinoma cells were purchased from ATCC (HTB-182) and cultured in RPMI-1640 medium (ATCC modification, Gibco) supplemented with 10% FBS and antibiotics. T.T esophageal squamous cell carcinoma cells were purchase from JCRB Cell Bank (JCRB0262) and cultured in medium Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12; Gibco, catalog no. 10565) supplemented with 10% FBS, and antibiotics. KYSE70 esophageal squamous cell carcinoma cell line was purchased from Sigma (94072012) and cultured in medium RPMI-1640 (Gibco, catalog no. 21870092) supplemented with 10% FBS, 10 mM HEPES (Gibco), 1 mM sodium pyruvate (Gibco), 2mM GlutaMax (Gibco) and antibiotics. Primary normal human epidermal keratinocytes (KC) were purchased from Gibco (C0055C) and cultured on Geltrex (Gibco) coated plates with EpiLife basal medium with 60 μM calcium (Gibco) plus 5 μg/mL insulin, 15 μg/mL transferrin, 10ng/mL epidermal growth factor, 10 μM forskolin, 500nM VX-745, 250nM RO4929097, 100nM dexamethasone and antibiotics. HEK293T cells were purchased from Takara Bio USA (632180) and cultured in high glucose DMEM (GIBCO) medium supplemented with 10% FBS (GIBCO), 10 mM HEPES (Gibco), 1 mM sodium pyruvate (Gibco), 2mM GlutaMax (Gibco) and antibiotics. Cells were maintained in a humidified incubator at 37 °C in the presence of 5% CO2 and passaged every 2–3 days. Cell lines were routinely tested for mycoplasma and immediately tested upon suspicion. None of the cell lines used in the reported experiments tested positive.
METHOD DETAILS
Estimate of protein molecules.
For the preparation of whole-cell lysates, cells were lysed in RIPA buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 20 mM NaF, 1 mM DTT, 1 mM sodium orthovanadate, 0.25 mM PMSF and protease inhibitors) supplemented with 5 mM MgCl2 and 0.5 U/ul of Benzonase (Sigma). After the samples were kept on ice for 30 minutes, LDS sample buffer with final 2.5% β-mercaptoethanol was added, followed by boiling for 5 minutes. The extracts from 300,000 cells were subjected to SDS–PAGE and western blot analysis together with 1.25 ng, 5 ng, 10 ng, and 20 ng of purified ACTL6A or SMARCA4 recombinant proteins. Odyssey CLx LI-COR was used to analyze and quantify the Western blot signals. The standard curves of signal to mass from the recombinant proteins were applied for estimating the amount of ACTL6A or SMARCA4/SMARCA2 from the cell lysates, which was further divided by the cell number (300,000) to obtain the mass (g) per cell, and then the number of molecules (N) per cell was calculated using the following formula: N= mass (g) / (molecular weight (kDa) ×103) × Avogadro constant (6.022×1023)
For producing recombinant proteins, the DNA fragment encoding human ACTL6A amino acid 43–119, the region used to raise anti-ACTL6A antibodies (Crabtree laboratory), was inserted between the BamHI and HindIII sites of pGSTag (Addgene 21877); and the fragment expressing human SMARCA4 amino acid 1086–1307, used to raise the J1 antibodies (Crabtree laboratory) that recognize both SMARCA4 and SMARCA2, was inserted between the BamHI and HindIII sites of pMAL-c2X (Addgene 75286). After 0.4mM IPTG induction for 2 hours at 37°C, the bacteria were collected and resuspended in PBS with 1mM EDTA, 1mM PMSF, 0.02% (~3mM) β-mercaptoethanol, and protease inhibitors. The cells were lysed by Diagenode Bioruptor for 15 min, high output. After 3,500 rpm spin for 10 mins, the supernatants were rotated with amylose resin (New England Biolabs) for MBP tag, or glutathione-superflow resin (Clontech) for GST tag overnight at 4°C. The resins were washed four times by PBS supplemented with 350mM NaCl, 0.1% triton-X-100, 1mM EDTA, 1mM PMSF, 0.02% (~3mM) β-mercaptoethanol, and protease inhibitors. The GST-tagged proteins were eluted by 10mM reduced glutathione (Sigma, 100mM stock made in 50mM Tris, pH7.6) in PBS (containing 1mM PMSF and 0.02% (~3mM) β-mercaptoethanol), and MBP-tagged proteins were eluted by 10mM maltose.
ACTL6A and YAP/TAZ knockdown.
siRNA transfections were performed using DharmaFECT 1 transfection reagents (Horizon Discovery) in antibiotics-free medium according to the manufacturer’s instructions. The siRNA reagents were purchased from Horizon Discovery (ON-TARGETplus Human ACTL6A siRNA (Dharmacon L-008243–00); control ON-TARGETplus Non-targeting Pool (Dharmacon D-001810–10); custom YAP siRNA (GACATCTTCTGGTCAGAGA); custom TAZ siRNA (ACGTTGACTTAGGAACTTT)). siRNAs were resuspended in siRNA buffer (Horizon Discovery). Cells were collected either 48 or 72 hours after transfection, as specified in the figure legends.
CRISPR gRNAs were cloned into vector lentiCRISPR v2 (Addgene 52961). The sequences of human ACTL6A targeting gRNAs are: TAATGCTCTGCGTGTTCCGA, ATGAGCGGCGGCGTGTACGG, GCGTGTTCCGAGGGAGAATA, AGATGACGGAAGCACATTAA. For producing lentiviral particles, 2 lentiviral vectors (18 μg per 15 cm dish) together with packaging vectors pMD2.G (4.5 μg) and psPAX2 (13.5 μg) were delivered into lenti-X 293T cells (Clontech) using 108–144 μg PEI MAX 40K (Polysciences, cat. 24765; stock 1 μg /ul) mixed in 1.8 ml Opti-MEM (Gibco) according to the manufacturer’s instructions. 12–16 hours after transfection, the medium was replaced by viral production medium (UltraCULTURETM serum-free cell culture medium (Lonza) supplemented with 10mM HEPES (Gibco), 1 mM sodium pyruvate (Gibco), 2mM GlutaMax (Gibco) and antibiotics (Gibco)). 72 hr post-transfection, lentiviral particles were collected by centrifugation of 0.45 μm pore size-filtered cell culture supernatants at 20,000 rpm for 2 hours at 4 °C, followed by PBS resuspension. Lentiviral transduction was conducted by spinfection methods in the presence of 10 μg/ml polybrene at 1,100 ×g for 30 min at 37°C. 48 hours post-infection, infected cells were selected by 2μg/ml puromycin (Sigma), followed by 10μg/ml Blasticidin (Gibco) in ACTL6A reconstitution experiments.
ACTL6A overexpression.
Human ACTL6A cDNA were cloned between Not1 and Mlu1 restriction enzyme sites downstream of the EF1α promoter in Crabtree lentiviral vector CYC103 (puromycin selection) and N106 (blasticidin selection), which harbor a second promoter PGK driving drug resistance gene for selection. See the “ACTL6A knockdown” section for lentiviral production and infection. For site-directed mutagenesis, ACTL6A cDNA were cloned into pUC-19 vector between HindIII and Kpn1, and the construct was used as template in PCR with primers- for P373S/P374G: TCAGAAAACTTCTGGAAGTATGCGG, GACAGCTCTCTATTCAAC; for R377G: TCCAAGTATGGGCTTGAAATTGATTGC, GGAGTTTTCTGAGACAGC. The PCR products were treated by kinase, ligase and Dpn1 (KLD) enzyme mix (New England Biolabs), followed by bacterial transformation, clone selection by Sanger DNA sequencing, and subcloning back to lentiviral vector CYC103 (puromycin selection) and N106 (blasticidin selection) between Not1 and Mlu1 sites. For generating ACTL6A expressing constructs that are resistant to ACTL6A-CRISPR KO, the following silent mutations marked by underlines were introduced to block ACTL6A gRNA binding: atgTCTggAggAgtCtaTgg, GgaCgaTggCTCTacCttGa, CaaCgcCctCAGGgtCccTCgCgaAaata.
Immunoprecipitation and Western blot.
For protein-protein interaction studies, cells reaching 80–90% confluence on the culture plates were washed once by cold PBS and lysed in cold hypotonic lysis buffer A, ~0.5ml/10cm2 growth area (Buffer A: 25 mM HEPES pH 7.5, 25 mM KCl, 0.05 mM EDTA, 5mM MgCl2, 10% glycerol, 0.1% NP-40, 1 mM DTT, 1 mM sodium orthovanadate, 0.25 mM PMSF and protease inhibitors). After incubation on ice for 5 minutes, cells were scraped from plates by cell lifter, harvested, and spun down at 1500 rpm for 5 mins at 4 °C. Then, the nuclei were washed by buffer A twice. For each wash, cold 5ml buffer A per 20-million cells were added to the pellet, followed by 5-minute incubation on ice, centrifugation at 1,500 rpm for 5 mins, and discarding the supernatants. The nuclei were lysed in immunoprecipitation (IP) buffer, ~1 ml per 20-million cells (IP buffer: 20 mM HEPES pH7.5, 100 mM KCl, 2.5 mM MgCl2, 5% glycerol, 1% Triton X-100, 0.5% NP-40, 1 mM dithiothreitol (DTT), 1mM sodium orthovanadate, 0.25 mM phenylmethylsulphonylfluoride (PMSF) and protease inhibitors). The nuclei were further passed through a 1 ml 27G-needle syringe 5 times and sonicated for three cycles of 10 sec-ON and 1 minute-OFF, high output (Diagenode Bioruptor). After centrifugation at 15,000 rpm for 10 min to collect the supernatants, the concentrations of the nuclear extracts were measure by Bradford assay (Bio-Rad) and adjusted to 1–1.5 μg/μl by IP buffer. For each IP, 500–1000ug protein extracts were incubated under rotary agitation overnight at 4 °C with antibodies against SMARCA4 (Santa Cruz Biotechnology, sc-374197 X; 4 μg), YAP (Cell Signaling Technology, 14074S; 5 μl), Pan-TEAD (Cell Signaling Technology, 13295S; 5 μl), TEAD4 (Abcam, ab58310; 3ug), ACTL6A (Invitrogen, 702414; 2.5 μg), or mouse/rabbit IgG control (Santa Cruz Biotechnology sc-2025; MilliporeSigma, 12–370). After additional one hour incubation with 40ul Protein A or G dynabeads (Invitrogen), the beads were washed four times by 1 ml IP buffer and resuspended in 20 μl 1x LDS sample buffer (Invitrogen) containing 2.5% β-mercaptoethanol and then boiled at 95 °C for 5 min. The samples were subjected to SDS–PAGE and western blot analysis. For ACTL6A perturbations, cells were collected 72 hours after siRNA transfection or 5 days after infection by lentiCRISPR, or 1–2 weeks after infection by lentivirus carrying ACTL6A.
Antibodies used for Western blotting included those against SMARCC1 (Crabtree laboratory), SMARCA4 (Santa Cruz Biotechnology, sc-374197 X), ACTL6A (Novus Biologicals, NB100–61628; or homemade in the Crabtree laboratory), J1 SMARCA4/SMARCA2 (Crabtree laboratory), ARID1A (Santa Cruz Biotechnology, sc-32761), BAF57 (Bethyl Laboratories, A300–810A), YAP (Abnova, H00010413-M01; Cell Signaling Technology, 14074S), Phospho-YAP Ser127 (Cell Signaling Technology, 13008), Pan-TEAD (Cell Signaling Technology, 13295S), TEAD1 (BD Biosciences, 610922), TEAD4 (Abcam, ab58310), INO80 (Bethyl Laboratories, A303–371A), EZH2 (BD Biosciences, 612666), GAPDH (Cell Signaling Technology, 5174S), V5 (Invitrogen, R960–25).
In vitro binding.
To produce FLAG-YAP proteins in mammalian cells, doxycycline-inducible lentiviral plasmid CYC244-FLAG-NLS-hYAP was generated by cloning FLAG-NLS-hYAP DNA into Not1 restriction enzyme site downstream of the tetracycline response element (TRE) in Crabtree lentiviral vector CYC244, which harbors a second promoter PGK driving puromycin-resistance gene and transactivator rtTA. FLAG-NLS-hYAP DNA were amplified from pQCXIH-Myc-YAP (Addgene# 33091). See the “ACTL6A knockdown” section for lentiviral production and infection. 24 hours after doxycycline (0.5μg/ml) addition, nuclear extracts from HEK293T cells were prepared as described in the “Immunoprecipitation and Western blot” section with modification of using 500mM KCl in the IP buffer (high-salt IP buffer), followed by immunoprecipitation using anti-FLAG antibody (Sigma-Aldrich, F1804) or mouse/rabbit IgG as control. After high-salt IP buffer wash, FLAG-YAP proteins were eluted by 3xFlag peptide (Sigma-Aldrich, F4799) in the IP buffer on the Thermomixer at 400 rpm for 10min at RT and the dynabeads were discard after magnetic seperation. To purify BAF complexes, nuclear extracts from HEK293T cells were prepared as above and a SMARCC1 antibody (Crabtree laboratory) was used for immunoprecipitation using high-salt IP buffer. After 5 time washes by high-salt IP buffer, the dynabeads bound by BAF complexes were resuspended in the IP buffer, incubated with the purified FLAG-YAP overnight, followed by 5-time IP buffer washing step. The dynabeads were resuspended in 1x LDS sample buffer (Invitrogen) containing 2.5% β-mercaptoethanol and then boiled at 95 °C for 5 min. The samples were subjected to SDS–PAGE and western blot analysis.
Density gradient sedimentation analysis.
A detailed description has been published elsewhere (Lessard et al., 2007). In brief, 30–40 million cells were washed by PBS once, lysed in 10 ml cold hypotonic lysis buffer A (see Immunoprecipitation), and then incubated on ice for 7 minutes. After centrifugation at 1500 rpm for 5 mins at 4 °C, nuclei were further washed by 10 ml buffer A twice and re-suspended in 700 μl buffer C (10 mM HEPES, pH7.5, 100 mM KCl, 0.1 mM EDTA, 3mM MgCl2, 10% glycerol, 1 mM DTT, 1mM sodium orthovanadate, 0.25 mM PMSF and protease inhibitors). Chromatin proteins were extracted with 0.3M ammonium sulfate (pH 7) by adding 1/9 volume of 3M ammonium sulfate stock and incubated under rotary agitation for 1–2 hours at 4 °C. Nuclear extracts were collected after ultracentrifugation at 100,000 rpm for 15 minutes at 4 °C (TLA 120.2 rotor), and proteins were precipitated with 0.33 mg/μl ammonium sulfate on ice for 20 min.
Precipitated proteins were pelleted by another ultracentrifugation at 100,000 rpm for 15 minutes at 4 °C and re-suspended in 200 μl HEMG-0 buffer (25 mM HEPES, pH7.9, 100 mM KCl, 0.1 mM EDTA, 12.5mM MgCl2 supplemented with 1 mM DTT, 1mM sodium orthovanadate, 0.25 mM PMSF and protease inhibitors). Protein concentration was measured by Bradford assay (Bio-Rad) and adjusted accordingly for glycerol gradient analyses. 200 μl of the solution with 500–1000 μg total protein was overlaid on a 10-ml density-gradient liquid column with 10 to 30% glycerol (in HEMG buffer) and placed in a SW-40 swing bucket rotor for centrifugation at 40,000 rpm for 16 h at 4 °C. A series of 0.5ml fractions were then recovered top-down and subsequently subjected to SDS–PAGE and western blot analysis.
Subcellular fractionation.
Cells were first lysed in cold hypotonic lysis buffer A (Buffer A: 25 mM HEPES pH 7.5, 25 mM KCl, 0.05 mM EDTA, 5mM MgCl2, 10% glycerol, 0.1% NP-40, 1 mM DTT, 1 mM sodium orthovanadate, 0.25 mM PMSF and protease inhibitors). After incubation on ice for 5 minutes, cells were spun down at 1500 rpm for 5 mins at 4 °C. The supernatants were collected as cytoplasmic fraction. Nuclei were washed by buffer A twice, lysed (in equal volume to the cytoplasmic fractions) with RIPA buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 20 mM NaF, 1 mM DTT, 1 mM sodium orthovanadate, 0.25 mM PMSF and protease inhibitors) supplemented with 5 mM MgCl2 and 0.5 U/ul of Benzonase (Sigma), and then incubated on ice for 30 minutes. Both cytoplasmic fractions and nuclei solutions were subjected to centrifugation at 15,000 rpm for 10 min at 4 °C to remove the cell debris. Samples were diluted with 4x LDS sample buffer (Invitrogen)/β-mercaptoethanol, boiled for 5 minutes, and analyzed by Western blot.
Immunofluorescence.
24 hours after siRNA transfection, cells were re-seeded onto chamber slides coated with fibronectin (coating: 20 μg/ml fibronectin (Sigma) at 37 °C overnight). 72 hours post-transfection, cells were fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature (RT). For immunostaining, cells were permeabilized in PBS with 0.3% Triton X-100 for 20 min and blocked for 1h at RT in blocking buffer (PBS containing 2.5 % normal donkey serum, 2.5 % normal goat serum, 1% BSA and 0.1% Triton X-100) supplemented with M.O.M. blocking reagent (Vector Laboratories). Primary YAP antibody (Abnova, H00010413-M01) was diluted in blocking buffer supplemented with M.O.M. Protein Concentrate (Vector Laboratories) and applied to cells, followed by overnight incubation at 4 °C. After washing with PBS+ 0.1% Triton X-100 three times at RT, cells were incubated for 1 hour at RT with secondary antibodies conjugated to Alexa-488 (Invitrogen). The slides were mounted with ProLong Diamond Antifade Mountant with DAPI (Invitrogen). Images were captured on the Keyence BZ-X700 microscope, and formatted using ImageJ, Adobe Photoshop, and Illustrator CS6.
RNA extraction, RT-qPCR, and RNA-seq analysis.
48 or 72 hours after siRNA transfection or one week after lentiviral transduction and drug selection, cells were lysed directly in culture plates with TRIsure reagents (Bioline), and RNA was extracted using Direct-zol RNA MiniPrep kits (Zymo Research) with in-column DNase I digestion to remove residual genomic DNA. For RT-qPCR, complementary DNA was synthesized using the SensiFAST cDNA synthesis kit (Bioline). cDNAs were mixed with indicated primers and SensiFAST SYBR lo-ROX reagents (Bioline), and quantitative PCR (qPCR) was performed on a Applied Biosystems QuantStudio 6 Flex Real-Time PCR System. Primer specificity was confirmed by subsequent melting curve analysis or gel electrophoresis. Levels of PCR products were expressed as a function of peptidylprolyl isomerase B (PPIB). Primers were designed through Primer 3 or from previous reports, and amplified products encompass exon/intron boundaries. The primer sequences of primers used in this study are listed in Table S1.
RNA-seq libraries were generated using the NEBNext ultra II directional RNA library prep kit coupled with NEBNext multiplex oligos for Illumina (New England Biolabs) and following the manufacturer’s directions. PE75 sequencing was performed on a NextSeq 550 sequencing system (Illumina). Alignment of RNA-sequencing reads was performed with STAR (Dobin et al., 2013) with ENCODE standard options to GENCODE v19, and read counts were generated using eXpress (Roberts and Pachter, 2013). Differential gene expression was determined using DEseq2 (Love et al., 2014).
CUT&RUN and data analysis.
CUT&RUN was performed as previously described (Skene et al., 2018). Cells were collected 48 or 72 hours after siRNA transfection or one week after lentiviral transduction and drug selection. 250,000 cells were bound to 10 μl concanavalin-A beads and collected into a 0.2 ml PCR tube. Antibody binding was conducted in the volume of 50 μl on thermomixer for 96-well PCR plates (Eppendorf) in 4 °C cold room for 2 hours, and Protein A-MNase binding for 1 hour. Wash buffer volume was adjusted to 150 μl. The digestion was performed under high Ca2+/low salt condition on ice for 15 minutes, followed by incubation at 37 °C for 30 min to release CUT&RUN fragments. Antibodies were diluted 1/100 and included: YAP (Cell signaling, 14074T), TEAD1 (Cell signaling, 12292S), SMARCC1 (Crabtree laboratory), H3K4me1 (Abcam, ab8895), H3K4me3 (Active motif, 39159), H3K27Ac (Abcam, ab4729), H3K27me3 (cell signaling, 9733). For histone marks profiling, CUT&RUN fragments were purified by spin column-based methods (Zymo DNA clean & concentrator-5 kit). For transcription factors and BAF complex profiling, CUT&RUN fragments were extracted by phenol-chloroform to recover small fragments.
The libraries were generated using the NEBNext ultraII DNA library prep kit for Illumina coupled with NEBNext multiplex oligos for Illumina (New England Biolabs) with modifications optimized for small fragments (detailed in dx.doi.org/10.17504/protocols.io.wvgfe3w.) PE75 sequencing was performed on a NextSeq 550 sequencing system (Illumina). Adapter trimming was performed with SeqPurge (Sturm et al., 2016). Demultiplexed fastq files were mapped to the hg19 genome as 2×36 mers using Bowtie2 (Langmead and Salzberg, 2012), and generated BAM files were then filtered for low-quality reads and duplicated reads. TEAD1 and YAP mapped fragments <120bp were used for downstream analysis. Coverage bigwigs were generated using deeptools bamCoverage (Ramirez et al., 2016) with CPM normalization. Peaks were called using macs2 (Zhang et al., 2008) with the nomodel option. Fixed-width peaks from replicates and treatments were combined using the called peaks summits files as described previously (Corces et al., 2018). De novo motif search on all CUT&RUN libraries was done using Homer (v4.11) (Heinz et al., 2010) with default parameters. Counts over peaks were generated using featureCounts (Liao et al., 2014). Differential peaks were determined using DEseq2 (Love et al., 2014) or edgeR (Robinson et al., 2010). To annotate joined YAP/TEAD1/SMARCC1 peaks, we defined the following states based on CUT&RUN datasets: active promoter (high H3K4me3 & low H3K27me3 & TSS distance < 1000), poised promoter (high H3K4me3 & high H3K27me3 & TSS distance < 1000), active enhancer (high H3K4me1 & high H3K27Ac & TSS distance > 1000), poised enhancer (high H3K4me1 & high H3K27me3 & TSS distance > 1000) and repressed sites (H3K27me3 high). High and low were defined by 0.99 percentile.
Differential analysis for H3K27me3 CUT&RUN between ACTL6A -overexpressing and vector-control keratinocytes was performed using DiffBind (Ross-Innes et al., 2012) by binning the genome into 5-kb bins and performing differential analysis (using DESeq2) between the two conditions. We identified 4,035 H3K27me3-differential bins (constituting 2389 broad peaks), of which 1,963 showed decreased H3K27me3 levels upon ACTL6A overexpression. Genes with H3K27me3 differentials in their promoters were defined as genes whose TSS is within the differential H3K27me3 5-kb bin. H3K27me3 CUT&RUN and H3K4me3 ChIP-seq (ENCSR075OQB for keratinocytes from ENCODE (Zhang et al., 2020)) signals around gene promoter with differential H3K27me3 levels was performed using computeMatrix and plotProfile from deeptools (Ramirez et al., 2016). H3K27me3 ChIP-seq for keratinocytes were from ENCODE ENCSR377MRR (Zhang et al., 2020). Heatmaps for CUT&RUN and ATAC-seq data around different genomic features were done using deeptools computeMatrix (v2.5.6) (Ramirez et al., 2016). Plots were generated using deeptools plotHeatmap or plotProfile. Differential gene expression data of RNA-seq for lung squamous cell carcinoma (LUSC) and head-and-neck squamous cell carcinoma (HNSC) versus their normal tissues were from GEPIA2 (Tang et al., 2017) with cutoff p-value < 0.05. HNSC and LUSC datasets were merged by gene name and then merged with genes displaying differential H3K27me3 CUT&RUN signals to identify ACTL6A-dependent polycomb target genes that were preferentially altered in either HNSC or LUSC tumors.
Omni-ATAC-seq and data analysis.
We followed the Omni-ATAC method in Corces MR et al. (Corces et al., 2017). 72 hours after siRNA transfection or one week after lentiviral transduction and drug selection, cells were pretreated with 200 U/ml DNase (Worthington) for 30 min at 37 °C to remove free-floating DNA and DNA from dead cells. Nuclei from 75,000 cells were used in the transposition reactions (Illumina) at 37 °C for 30 min, followed by DNA purification by Zymo DNA clean & concentrator-5 kits, and library preparation. PE75 sequencing was conducted on a NextSeq 550 sequencing system (Illumina). Demultiplexed and trimmed fastq files were mapped to the hg19 genome as 2×36 mers using Bowtie2 (Langmead and Salzberg, 2012). Duplicate reads were removed using picard-tools (v.1.99). Low-quality reads and chrM reads were removed using samtools. Fixed width peaks were called using macs2 (Zhang et al., 2008) with the nomodel option, and summits were called. Peaks from replicates and treatments were combined using the called peaks summits files as described previously (Corces et al., 2018). Read counts over the peaks were done using the ChrAccR package (https://github.com/GreenleafLab/ChrAccR). Differential peaks were determined using DEseq2 (Love et al., 2014) or edgeR (Robinson et al., 2010).
To find transcription factors that are enriched in differential peaks, we used the motifMatcher (part of chromVar (Schep et al., 2017) package to call TF binding sites within the differential peak sets. Using hypergeometric testing, we computed the enrichment of each TF within increasing/decreasing differential peaks with all peaks as background. Heatmaps for CUT&RUN and ATAC-seq data around different genomic features were generated using deeptools computeMatrix (Ramirez et al., 2016) (v2.5.6). Plots were generated using deeptools plotHeatmap or plotProfile. Genomic feature annotation for ATAC-seq peaks that gained or lost accessibility as a result of ACTL6A loss was performed using ChiPseeker (Yu et al., 2015b).
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons were performed in Prism 8 (GraphPad Software) with unpaired two-tailed student’s t-test to determine significance between two groups indicated in figures. The cancer genomic data were pulled from cBioPortal TCGA PanCancer Atlas studies (Cerami et al., 2012; Gao et al., 2013), and the RNA analyses of tumors versus paired normal tissues were pulled from GEPIA 2 (Tang et al., 2017).
Supplementary Material
Highlights:
ACTL6A occupancy in BAF complex is sub-stoichiometric in normal epithelial cells
SCC cells upregulate ACTL6A thus increasing ACTL6A assembly with BAF complex
ACTL6A mediates co-dependent chromatin loading of BAF and TEAD-YAP complexes
ACTL6A upregulation counteracts polycomb-mediated repression at SCC signature genes
Acknowledgments:
We thank all Crabtree laboratory members for intellectual input and suggestions; B. Keyes and W. Lu for critical reading of the manuscript; S. Henikoff for providing protein A-MNase (pA-MN) for CUT&RUN. C.-Y.C. is supported by the GSK Sir James Black fellowship. Z.S. is supported by EMBO Long-Term Fellowship EMBO ALTF 1119–2016 and by Human Frontier Science Program Long-Term Fellowship HFSP LT 000835/2017-L. S.G.L. is supported by NIH grant CA243442. G.R.C. is an investigator of the Howard Hughes Medical Institute. This work was supported by the Howard Hughes Medical Institute, NIH grant CA163915, Department of Defense grant W81XWH-16–1-0083 to G.R.C and the David Korn Professorship. K.M.L. is a Packard Foundation Fellow, Pew Scholar, Human Frontiers Science Program Young Investigator and the Anthony DiGenova Endowed Faculty Scholar; and is supported by the Stanford Ludwig Center for Cancer Stem Cell Research. W.J.G is a Chan Zuckerberg Biohub investigator and acknowledges grants 2017–174468 and 2018–182817 from the Chan Zuckerberg Initiative.
Footnotes
Declaration of Interests: G.R.C is a founder and stockholder in Foghorn Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arensman MD, Kovochich AN, Kulikauskas RM, Lay AR, Yang PT, Li X, Donahue T, Major MB, Moon RT, Chien AJ, et al. (2014). WNT7B mediates autocrine Wnt/beta-catenin signaling and anchorage-independent growth in pancreatic adenocarcinoma. Oncogene 33, 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Tang J, Lopez-Pajares V, Tao S, Qu K, Crabtree GR, and Khavari PA (2013). ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell 12, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisic D, Stadler MB, Iurlaro M, and Schubeler D (2019). Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. (2009). SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 41, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M., Conejo-Garcia JR, et al. (2015). Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 21, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, et al. (2016). Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet 48, 398–406. [DOI] [PubMed] [Google Scholar]
- Braun SMG, Petrova R, Tang J, Krokhotin A, Miller EL, Tang Y, Panagiotakos G, and Crabtree GR (2021). BAF subunit switching regulates chromatin accessibility to control cell cycle exit in the developing mammalian cortex. Genes & development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calses PC, Crawford JJ, Lill JR, and Dey A (2019). Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 5, 297–307. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J, et al. (2015). Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun 6, 6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HI, and Sudol M (1995). The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A 92, 7819–7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yang M, Wang C, Ouyang Y, Chen X, Bai J, Hu Y, Song M, Zhang S, and Zhang Q (2020). Forkhead box D1 promotes EMT and chemoresistance by upregulating lncRNA CYTOR in oral squamous cell carcinoma. Cancer Lett 503, 43–53. [DOI] [PubMed] [Google Scholar]
- Chung J, Lau J, Cheng LS, Grant RI, Robinson F, Ketela T, Reis PP, Roche O, Kamel-Reid S, Moffat J, et al. (2010). SATB2 augments DeltaNp63alpha in head and neck squamous cell carcinoma. EMBO Rep 11, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, and Sander C (2013). Emerging landscape of oncogenic signatures across human cancers. Nat Genet 45, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Iwasa J, Cairns BR, and Peterson CL (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nature reviews. Molecular cell biology 18, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, Gusterson BA, and Cooper CS (1994). Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 7, 502–508. [DOI] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debaugnies M, Sanchez-Danes A, Rorive S, Raphael M, Liagre M, Parent MA, Brisebarre A, Salmon I, and Blanpain C (2018). YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodonova SO, Zhu F, Dienemann C, Taipale J, and Cramer P (2020). Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672. [DOI] [PubMed] [Google Scholar]
- Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, Pfister S, Cho YJ, Zhao K, and Crabtree GR (2013). BAF complexes facilitate decatenation of DNA by topoisomerase IIalpha. Nature 497, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, and Fuchs E (2009). Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136, 1122–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Jiang L, Tian Y, Zheng M, Huang M, and Li J (2020a). FOXL2 expression might be a novel prognostic biomarker in patients with laryngeal squamous cell carcinoma. J Int Med Res 48, 300060520919252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Gao J, Sun QW, Wang CX, Deng W, Mao GY, Li HQ, Guo SS, Cheng J, Wu YN, et al. (2020b). MiR-34a inhibits the proliferation, migration, and invasion of oral squamous cell carcinoma by directly targeting SATB2. J Cell Physiol 235, 4856–4864. [DOI] [PubMed] [Google Scholar]
- Golson ML, and Kaestner KH (2016). Fox transcription factors: from development to disease. Development 143, 4558–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, Li J, Liu B, and Xu Y (2020). Structure of nucleosome-bound human BAF complex. Science 367, 875–881. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heselmeyer K, Schrock E, du Manoir S, Blegen H, Shah K, Steinbeck R, Auer G, and Ried T (1996). Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervix. Proc Natl Acad Sci U S A 93, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, and Crabtree GR (2011). esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol 13, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges HC, Stanton BZ, Cermakova K, Chang CY, Miller EL, Kirkland JG, Ku WL, Veverka V, Zhao K, and Crabtree GR (2018). Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol 25, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. (2017). Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 376, 2109–2121. [DOI] [PubMed] [Google Scholar]
- Jin Y, Xu J, Yin MX, Lu Y, Hu L, Li P, Zhang P, Yuan Z, Ho MS, Ji H, et al. (2013). Brahma is essential for Drosophila intestinal stem cell proliferation and regulated by Hippo signaling. Elife 2, e00999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouppila-Matto A, Narkio-Makela M, Soini Y, Pukkila M, Sironen R, Tuhkanen H, Mannermaa A, and Kosma VM (2011). Twist and snai1 expression in pharyngeal squamous cell carcinoma stroma is related to cancer progression. BMC Cancer 11, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SM, Pulice JL, Chory EJ, and Crabtree GR (2017). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet 49, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina TV, Enikolopov G, et al. (2011). DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 8, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kia SK, Gorski MM, Giannakopoulos S, and Verrijzer CP (2008). SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol 28, 3457–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasteva V, Buscarlet M, Diaz-Tellez A, Bernard MA, Crabtree GR, and Lessard JA (2012). The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood 120, 4720–4732. [DOI] [PubMed] [Google Scholar]
- Krimpenfort P, Snoek M, Lambooij JP, Song JY, van der Weide R, Bhaskaran R, Teunissen H, Adams DJ, de Wit E, and Berns A (2019). A natural WNT signaling variant potently synergizes with Cdkn2ab loss in skin carcinogenesis. Nat Commun 10, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim JH, Han S, Sung CO, Do IG, Ko YH, Um SH, and Kim SH (2012). Twist1 is an independent prognostic factor of esophageal squamous cell carcinoma and associated with its epithelial-mesenchymal transition. Ann Surg Oncol 19, 326–335. [DOI] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, and Crabtree GR (2007). An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Fang L, Ouyang B, Zhang X, Zhan S, Feng X, Bai Y, Han X, Kim H, He Q, et al. (2015). Actl6a protects embryonic stem cells from differentiating into primitive endoderm. Stem Cells 33, 1782–1793. [DOI] [PubMed] [Google Scholar]
- Mashtalir N, Suzuki H, Farrell DP, Sankar A, Luo J, Filipovski M, D’Avino AR, St Pierre R, Valencia AM, Onikubo T, et al. (2020). A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 183, 802–817 e824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MJ, Pulice JL, Beird HC, Ingram DR, D’Avino AR, Shern JF, Charville GW, Hornick JL, Nakayama RT, Garcia-Rivera EM, et al. (2018). The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 33, 1128–1141 e1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT, et al. (2017). SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 49, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen CDK, and Yi C (2019). YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 5, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Slattery M, Ma L, Crofts A, White KP, Mann RS, and Irvine KD (2013). Genome-wide association of Yorkie with chromatin and chromatin-remodeling complexes. Cell Rep 3, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids research 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, and Pachter L (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nature methods 10, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, and Ramon y Cajal S (2011). p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 30, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Ronan JL, Wu W, and Crabtree GR (2013). From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 14, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladi SV, Ross K, Karaayvaz M, Tata PR, Mou H, Rajagopal J, Ramaswamy S, and Ellisen LW (2017). ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 31, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Danes A, and Blanpain C (2018). Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer 18, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nature methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, et al. (2011). Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144, 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong BK, Lau J, Adderley T, Kee L, Chaukos D, Pienkowska M, Malkin D, Thorner P, and Irwin MS (2015). SATB2 enhances migration and invasion in osteosarcoma by regulating genes involved in cytoskeletal organization. Oncogene 34, 3582–3592. [DOI] [PubMed] [Google Scholar]
- Shah SP, Kobel M, Senz J, Morin RD, Clarke BA, Wiegand KC, Leung G, Zayed A, Mehl E, Kalloger SE, et al. (2009). Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med 360, 2719–2729. [DOI] [PubMed] [Google Scholar]
- Shain AH, and Pollack JR (2013). The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS one 8, e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiah SG, Hsiao JR, Chang WM, Chen YW, Jin YT, Wong TY, Huang JS, Tsai ST, Hsu YM, Chou ST, et al. (2014). Downregulated miR329 and miR410 promote the proliferation and invasion of oral squamous cell carcinoma by targeting Wnt-7b. Cancer Res 74, 7560–7572. [DOI] [PubMed] [Google Scholar]
- Simpson DR, Mell LK, and Cohen EE (2015). Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol 51, 291–298. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nature protocols 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Skibinski A, Breindel JL, Prat A, Galvan P, Smith E, Rolfs A, Gupta PB, LaBaer J, and Kuperwasser C (2014). The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep 6, 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Vendrell M, Hayward M, Baker SJ, Miao GG, Schilling K, Robertson LM, Curran T, and Morgan JI (1993). Continuous c-fos expression precedes programmed cell death in vivo. Nature 363, 166–169. [DOI] [PubMed] [Google Scholar]
- Speicher MR, Howe C, Crotty P, du Manoir S, Costa J, and Ward DC (1995). Comparative genomic hybridization detects novel deletions and amplifications in head and neck squamous cell carcinomas. Cancer Res 55, 1010–1013. [PubMed] [Google Scholar]
- Stanton BZ, Hodges C, Calarco JP, Braun SM, Ku WL, Kadoch C, Zhao K, and Crabtree GR (2017). Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat Genet 49, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schubeler D, et al. (2015). YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet 11, e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm M, Schroeder C, and Bauer P (2016). SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinformatics 17, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerlong H, Hinata K, Viswanathan R, Erdjument-Bromage H, Tempst P, and Cairns BR (2008). The HSA domain binds nuclear actin-related proteins to regulate chromatin-remodeling ATPases. Nat Struct Mol Biol 15, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, and Kennison JA (1992). brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68, 561–572. [DOI] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research 45, W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonon G, Wong KK, Maulik G, Brennan C, Feng B, Zhang Y, Khatry DB, Protopopov A, You MJ, Aguirre AJ, et al. (2005). High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A 102, 9625–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent-Mistiaen Z, Elbediwy A, Vanyai H, Cotton J, Stamp G, Nye E, Spencer-Dene B, Thomas GJ, Mao J, and Thompson B (2018). YAP drives cutaneous squamous cell carcinoma formation and progression. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, and Crabtree GR (1996). Diversity and specialization of mammalian SWI/SNF complexes. Genes & development 10, 2117–2130. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, Fox C, Francis JM, Pedamallu CS, DeLuca DS, et al. (2014). SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest 124, 1636–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenderski W, Wang L, Krokhotin A, Walsh JJ, Li H, Shoji H, Ghosh S, George RD, Miller EL, Elias L, et al. (2020). Loss of the neural-specific BAF subunit ACTL6B relieves repression of early response genes and causes recessive autism. Proc Natl Acad Sci U S A 117, 10055–10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, and Roberts CW (2011). SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11, 481–492. [DOI] [PubMed] [Google Scholar]
- Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, and Roberts CW (2010). Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, and Crabtree GR (2009). Understanding the words of chromatin regulation. Cell 136, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015a). Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015b). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yu W, Ma Y, Shankar S, and Srivastava RK (2017). SATB2/beta-catenin/TCF-LEF pathway induces cellular transformation by generating cancer stem cells in colorectal cancer. Sci Rep 7, 10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2016). YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, and Guan KL (2009). TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem 284, 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lee D, Dhiman V, Jiang P, Xu J, McGillivray P, Yang H, Liu J, Meyerson W, Clarke D, et al. (2020). An integrative ENCODE resource for cancer genomics. Nat Commun 11, 3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, and Crabtree GR (1998). Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 95, 625–636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next-generation sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Pan-TEAD (D3F7L) | Cell Signaling Technology | Cat# 13295S |
| Mouse anti-TEAD4 | Abcam | Cat# ab58310 |
| Mouse anti-TEAD1 | BD Biosciences | Cat# 610922 |
| Mouse anti-BAF53A (ACTL6A)(5H3L6) | Invitrogen | Cat# 702414 |
| Mouse anti-ARID1A (PSG3) | Santa Cruz Biotechnology | Cat# sc-32761 |
| Rabbit anti-BAF57 (SMARCE1) | Bethyl Laboratories | Cat# A300-810A |
| Rabbit anti-Phospho-YAP (Ser127) (D9W2I) | Cell Signaling Technology | Cat# 13008 |
| Mouse anti-Brg1 (SMARCA4) (H-10) | Santa Cruz Biotechnology | Cat# sc-374197 X |
| Rabbit anti-INO80 | Bethyl Laboratories | Cat# A303-371A |
| Mouse anti-EZH2 | BD Biosciences | Cat# 612666 |
| Rabbit anti-GAPDH (D16H11) XP | Cell Signaling Technology | Cat# 5174S |
| Mouse anti-V5 tag | Invitrogen | Cat# R960-25 |
| Mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025 |
| Rabbit IgG | MilliporeSigma | Cat# 12-370 |
| Mouse anti-YAP | Abnova | Cat# H00010413-M01 |
| Rabbit anti-BAF53A (ACTL6A) | Novus Biologicals | Cat# NB100-61628 |
| Mouse anti-FLAG (M2) | Sigma-Aldrich | Cat# F1804 |
| Donkey anti-mouse IgG | Invitrogen | Cat# A-21202 |
| Rabbit anti-TEAD1(D9X2L) | Cell Signaling Technology | Cat# 12292S |
| Rabbit anti-histone H3K4me1 | Abcam | Cat# ab8895 |
| Rabbit anti-histone H3K4me3 | Active motif | Cat# 39159 |
| Rabbit anti-histone H3K27me3 | Cell Signaling Technology | Cat# 9733 |
| Rabbit anti-histone H3K27Ac | Abcam | Cat# ab4729 |
| Rabbit anti-BAF53A (ACTL6A) | The Crabtree laboratory | N/A |
| Rabbit anti-Brg1/BRM (SMARCA4/SMARCA2) (J1) | The Crabtree laboratory | N/A |
| Rabbit anti-BAF155 (SMARCC1) | The Crabtree laboratory | N/A |
| Rabbit anti-YAP (D8H1X) XP | Cell Signaling Technology | Cat# 14074S |
| Bacterial and Virus Strains | ||
| One-Shot Stbl3 chemically competent E. coli | Invitrogen | Cat# C7373-03 |
| BL21(DE3) Competent E.coli | New England Biolabs | Cat# C2527I |
| Rosetta 2(DE3) competent cells | EMD Millipore | Cat# 71397-3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polybrene | Santa Cruz Biotechnology | Cat# sc-134220 |
| Polyethylenimine Max (PEI MAX) (MW 40,000) | Polysciences | Cat# 24765 |
| Digitonin | Millipore | Cat# 300410-250MG |
| Digitonin | Promega | Cat# G9441 |
| Spermidine trihydrochloride | Sigma-Aldrich | Cat# S2501-1G |
| Benzonase Nuclease | Sigma-Aldrich | Cat# E1014-25KU |
| Amylose resin | New England Biolabs | Cat# E8021L |
| Glutathione-superflow resin | Clontech | Cat# 635608 |
| Glutathione reduced | Sigma-Aldrich | Cat# G-4251 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10002D |
| 3X FLAG Peptide | MilliporeSigma | Cat# F4799-4MG |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10009D |
| Geltrex | Thermo Fisher Scientific | Cat# A1413302 |
| Transferrin | Roche | Cat# 10652202001 |
| EGF | Thermo Fisher Scientific | Cat# PHG0311 |
| Insulin | Sigma-Aldrich | Cat# I5500 |
| Forskolin | Tocris Bioscience | Cat# 1099 |
| VX-745 | Tocris Bioscience | Cat# 3915 |
| RO4929097 | Cellagen Technology | Cat# C7649 |
| Dexamethasone | Tocris Bioscience | Cat# 1126 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140-163 |
| Deoxyribonuclease I | Worthington Biochemical Corporation | Cat# LS002058 |
| TRIsure | Bioline | Cat# BIO-38033 |
| Plasmocin | InvivoGen | Cat# ant-mpp |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Fibronectin | Sigma-Aldrich | Cat# F1141-1MG |
| DharmaFECT 1 transfection reagent | Horizon Discovery | Cat# T-2001-01 |
| 5X siRNA Buffer | Horizon Discovery | Cat# B-002000-UB-100 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833-100mg |
| ON-TARGETplus | Horizon Discovery | Cat# L-008243-00 |
| Human ACTL6A siRNA | ||
| ON-TARGETplus Non-targeting Pool | Horizon Discovery | Cat# D-001810-10 |
| Blasticidin S HCl | Thermo Fisher Scientific | Cat# R21001 |
| Critical Commercial Assays | ||
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490S |
| NEBNext MultiplexOligos for Illumina (Dual Index Primers Set 1) | New England Biolabs | Cat# E7600S |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541S |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32854 |
| ZymoPURE II Plasmid Midiprep Kit | Zymo Research | Cat# D4200 |
| DNA Clean & Concentrator-5 | Zymo Research | Cat# D4013 |
| Direct-zol RNA Miniprep Kits | Zymo Research | Cat# R2050 |
| High Sensitivity D1000 Reagents (Sample Buffer & Ladder) | Agilent Technologies | Cat# 5067-5585 |
| High Sensitivity D1000 ScreenTapes | Agilent Technologies | Cat# 5067-5584 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| Bio-Rad Protein Assay Dye (Bradford) | Bio-Rad | Cat# 500-0006 |
| Lenti-X GoStix | Clontech | Cat# 631281 |
| PhiX Control v3 | Illumina | Cat# FC-110-3001 |
| SensiFAST SYBR Lo-ROX Kit | Bioline | Cat# BIO-94020 |
| SensiFAST cDNA Synthesis Kit | Bioline | Cat# BIO-65054 |
| KLD Enzyme Mix | New England Biolabs | Cat# M0554S |
| In-Fusion HD Cloning Kit | Clontech | Cat# 639650 |
| NEBNext Ultra II DNA Library Prep with Sample Purification Beads | New England Biolabs | Cat# E7103S |
| NEBNext Ultra II Directional RNA Library Prep Kit | New England Biolabs | Cat# E7760S |
| Deposited Data | ||
| Structure of nucleosome-bound human BAF complex | Protein Data Bank | ID# 6LTJ |
| Deep sequencing datasets from this study | Gene Expression Omnibus | GSE156788 |
| H3K4me3 ChIP-seq (keratinocytes) | ENCODE | ENCSR075OQB |
| H3K27me3 ChIP-seq | ENCODE | ENCSR377MRR |
| Experimental Models: Cell Lines | ||
| Human Epidermal Keratinocytes, adult (HEKa) | Thermo Fisher Scientific | Cat# C0055C |
| HEK293T | Takara | Cat# 632180 |
| FaDu | ATCC | Cat# HTB-43 |
| NCI-H520 | ATCC | Cat# HTB-182 |
| T.T | JCRB Cell Bank | Cat# JCRB0262 |
| KYSE70 | Sigma | Cat# 94072012 |
| Oligonucleotides | ||
| Primers | See Table S1 | N/A |
| Recombinant DNA | ||
| pQCXIH-Myc-YAP | Addgene | Cat# 33091 |
| CYC244-Flag-NLS-hYAP | This study | N/A |
| N106-hACTL6A | This study | N/A |
| CYC103-hACTL6A | This study | N/A |
| pGSTag | Addgene | Cat# 21877 |
| pMAL-c2X | Addgene | Cat# 75286 |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| Software and Algorithms | ||
| Adobe Creative Cloud | Adobe | https://www.adobe.com/creativecloud.html |
| Rstudio | RStudio | https://www.rstudio.com/ |
| Image Studio Lite | LI-COR | https://www.licor.com/bio/ |
| SnapGene | Insightful Science | https://www.snapgene.com/ |
| Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SeqPurge | Sturm et al., 2016 | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| deeptools | Ramirez et al., 2016 | N/A |
| macs2 | Zhang et al., 2008 | N/A |
| Homer | Heinz et al., 2010 | N/A |
| DEseq2 | Love et al., 2014 | N/A |
| edgeR | Robinson et al., 2010 | N/A |
| DiffBind | Ross-Innes et al., 2012 | N/A |
| chromVar | Schep et al., 2017 | N/A |
| ChiPseeker | Yu et al., 2015b | N/A |
| ChrAccR | https://github.com/GreenleafLab/ChrAccR | N/A |
| PyMOL v2.4.2 | Schrodinger | https://pymol.org |