

Abstract
Recessive gene mutations underlie many developmental disorders and often lead to disabling neurological problems. Here, we report identification of a homozygous c.170G>A (p.Cys57Tyr or C57Y) mutation in the gene coding for protein disulfide isomerase A3 (PDIA3, also known as ERp57), an enzyme that catalyzes formation of disulfide bonds in the endoplasmic reticulum, to be associated with syndromic intellectual disability. Experiments in zebrafish embryos show that PDIA3C57Y expression is pathogenic and causes developmental defects such as axonal disorganization as well as skeletal abnormalities. Expression of PDIA3C57Y in the mouse hippocampus results in impaired synaptic plasticity and memory consolidation. Proteomic and functional analyses reveal that PDIA3C57Y expression leads to dysregulation of cell adhesion and actin cytoskeleton dynamics, associated with altered integrin biogenesis and reduced neuritogenesis. Biochemical studies show that PDIA3C57Y has decreased catalytic activity and forms disulfide‐crosslinked aggregates that abnormally interact with chaperones in the endoplasmic reticulum. Thus, rare disease gene variant can provide insight into how perturbations of neuronal proteostasis can affect the function of the nervous system.
Keywords: actin cytoskeleton, cell adhesion, integrins, intellectual disability, protein disulfide isomerase
Subject Categories: Molecular Biology of Disease, Neuroscience
Study of a rare intellectual disability‐linked variant in ER enzyme PDIA3 links cellular proteostasis defects directly to cytoskeleton and adhesion pathways required for neuronal morphogenesis and connectivity.

Introduction
Congenital malformations affect 2–5% of children depending on the ethnic origin, with consanguinity representing a major risk factor for one‐tenth of the world population (Bittles & Black, 2010; Sheridan et al, 2013; Ravindranath et al, 2015). Recessive gene mutations in consanguineous populations underlie 30% of the developmental disorders, which often include disabling neurological problems (Martin et al, 2018). Intellectual disability (ID) encompasses a broad range of conditions with reduced cognitive capacity and adaptive behaviors, preventing the individual from developing the functional and social skills of everyday life (Schalock, 2011). Genetic heterogeneity of these conditions poses a challenge for understanding the underlying pathogenic mechanisms and designing therapeutic interventions (Vissers et al, 2016). The estimated number of genes linked to recessive ID surpasses thousands (Ropers, 2010), indicating that most genes have not yet been identified despite the important progress made by massive genome sequencing (Vissers et al, 2016; Riazuddin et al, 2017; Hu et al, 2019). Given this complex genetic landscape, functional studies of mutant genes are paramount for the elucidation of molecular networks associated with abnormal development and malfunctioning of the nervous system.
Here, we performed a clinical and genetic study on a large consanguineous family presenting syndromic ID and identified a homozygous variant in PDIA3 segregating with the disease. PDIA3 encodes protein disulfide isomerase family A member 3 (PDIA3, also known as ERp57 or Grp58), an oxidoreductase containing thioredoxin‐like domains that catalyzes the formation and isomerization of disulfide bonds in the endoplasmic reticulum (ER) (Ellgaard & Ruddock, 2005). PDIA3 has been associated with the pathogenesis of neurodegenerative diseases involving abnormal protein aggregation (Erickson et al, 2005; Hetz et al, 2005; Woehlbier et al, 2016), but not to neurodevelopmental disorders. Despite available evidence suggesting the protective role of PDIA3 against protein misfolding causing neurodegeneration (Perri et al, 2015), the physiological role of the protein in the nervous system remains elusive.
The ER supports essential cellular functions by promoting the synthesis and folding of membrane and secreted proteins. Disturbance to ER proteostasis has emerged as a molecular signature of myriad diseases including cancer, neurodegeneration, metabolic syndromes, fibrosis, and inflammatory conditions (Oakes & Papa, 2015; Hetz & Saxena, 2017; Hetz et al, 2020). However, defining the contribution of the ER folding machinery to the function of the nervous system remained a challenge (Martínez et al, 2018). We functionally addressed the consequences of expressing ID‐linked PDIA3 using various model systems to establish pathogenic mechanisms linking ER proteostasis to neurodevelopmental disorders. Our results uncovered a previously unanticipated pathway for the development of ID, where perturbations to specific components of the ER folding machinery have adverse effects to neuronal connectivity and synaptic function.
Results
Identification of homozygous p.Cys57Tyr in PDIA3 as candidate variant causing a neurodevelopmental syndrome
We performed clinical and genetic analysis on four affected subjects born to consanguineous marriages (Fig 1A). A boy and his three first‐cousins once removed (siblings) are afflicted with a syndrome marked by neurodevelopmental impairment including ID, delayed developmental milestones, and behavioral problems as well as skeletal anomalies, growth retardation, and mild facial dysmorphism (Fig 1B and C, Appendix Fig S1, Appendix Tables S1 and S2, Appendix Supplementary Text). Cranial MRI of a patient did not show any gross structural brain defect (Fig 1D). The condition is non‐progressive and distinguished by severe ID based on the criteria of the American Psychiatric Association from 2013. To identify the locus of the genetic defect underlying the familial trait, we performed linkage mapping. SNP genotype‐based linkage analysis was applied as a multistep approach because the high inbreeding in the kindred and the large pedigree size exceeded the capacity of the programs employed. Additionally, we launched exome sequencing of patient 502. Regions where the homozygous genotype was not shared exclusively by affected individuals or where exome data revealed heterozygosity were excluded. We mapped the disease gene with a maximal multipoint LOD score of 3.85 and identified gene locus as an approximately 2.2‐Mb region at 15q15.2‐21.1 between rs1197547 (nucleotide 42905340) and rs17518970 (nucleotide 45095902; see Materials and Methods, Appendix Fig S2 and Dataset EV1). The locus did not harbor any known genes related to ID.
Figure 1. Identification of a missense variant in PDIA3 causing intellectual disability with developmental impairment.
- Family pedigree. Filled symbols indicate subjects suffering from severe intellectual disability with developmental impairment, crossed symbols deceased subjects, horizontal lines subjects physically examined, * subjects who participated in genetic studies, and + subjects with SNP genotype data.
- Pictures of affected individuals showing mild facial dysmorphism such as protruding ears and broad nose. From left to right, subjects 401, 502, and 504.
- Palmar creases of subject 502. Distal and proximal creases indicated by black dashed lines are normal while thenar and median creases are missing.
- Cranial MRI of subject 502 showing no gross structural brain defects.
- DNA sequence electropherograms showing the substitution c.170G>A (p.Cys57Tyr) in PDIA3.
- Domain structure of PDIA3. Catalytic (a and a′, with active site motif CGHC) and non‐catalytic (b and b′) domains are shown in green and blue, respectively. The C57Y mutant is depicted, with Y57 indicated by a red arrowhead. The region shown in gray corresponds to the X‐linker between domains b′ and a′.
- Structural modeling of substitution of cysteine‐57 with tyrosine in the N‐terminal PDIA3 active site.
To identify the putative gene defect, we investigated exome sequence data of an affected individual and identified at the disease region homozygous c.170G>A (p.Cys57Tyr or C57Y) variant in exon 2 of PDIA3 (Fig 1E and Dataset EV2). Furthermore, the exome file listed 20 other rare (frequency < 0.01) exonic and possibly homozygous variants, but none of them were in the sole region of homozygosity shared exclusively by affected individuals (Dataset EV3). Sanger sequencing validated homozygosity of PDIA3 c.170G>A in patients 401 and 502 while identifying unaffected subject 406 as a heterozygote (Appendix Fig S3). Thus, we concluded that the homozygous variant in PDIA3 is linked to the neurodevelopmental impairment in the kindred. The substituted amino acid residue is a catalytic cysteine in PDIA3 completely conserved across species (Appendix Fig S4A). This variant is novel (not listed in the public databases for genetic variation), fully segregates with the disease and is predicted to be pathogenic by computational algorithms (Appendix Fig S4B and Appendix Supplementary Text).
PDIA3C57Y is pathogenic
PDIA3 is a main component of the ER proteostasis network that cooperates with the lectin‐like chaperones calnexin (CNX) and calreticulin (CRT) in the folding and quality control of glycosylated substrates (Ellgaard & Ruddock, 2005; Jessop et al, 2007) (Appendix Fig S4C). It is ubiquitously expressed and composed of four thioredoxin‐like domains abb′a′, where both the N‐terminal a and the C‐terminal a′ domains contain a catalytic CGHC motif (Fig 1F). The mutated cysteine‐57 (C57) residue directly participates in catalysis of disulfide bond formation (Frand et al, 2000). In addition, C57 is involved in the formation of a heterodimer between PDIA3 and tapasin, which serves as scaffold to assist peptide loading of the major histocompatibility complex class I (MHC‐I) (Garbi et al, 2006). Despite the death of one patient due to recurrent infections (Appendix Supplementary Text), the other cases did not present signs of compromised immunity as would be expected if the MHC‐I pathway was disrupted. The recessive pattern of inheritance suggests that PDIA3 variant is pathogenic due to loss of function. Of note, ubiquitous Pdia3 deletion is embryonic lethal in mice (Garbi et al, 2006), indicating a vital role of the enzyme during development. The substitution of C57 with tyrosine in our patients is expected to compromise thiol‐oxidoreductase activity of the N‐terminal active site (Frand et al, 2000), in addition to impact its geometry due to the proximity of C57 and W56 aromatic side chains (Fig 1G). We reason that PDIA3C57Y may retain sufficient catalytic activity to support viability of offspring, but not normal development. Thus, we investigated PDIA3C57Y pathogenicity in vivo and in vitro to understand the relationship between ER proteostasis and neurodevelopmental impairment.
To determine the consequences of PDIA3C57Y expression on vertebrate development, we performed experiments in zebrafish embryos by injection of mRNA at the one‐cell stage (Fig EV1A and B). While wild‐type (WT) PDIA3 expression had no adverse effects, PDIA3C57Y caused significant morphological abnormalities in approximately 50% of the embryos, such as shortening of the anterior–posterior axis, tail curvature, and cyclopia (Figs 2A and EV1C). PDIA3C57Y expression was not lethal and did not induce cell death in embryos as measured by acridine orange staining (Fig EV1D). Rather, the phenotype was correlated with deficient morphogenetic movements of epiboly and convergent extension during gastrulation of embryos (Fig 2B). Since patients presented skeleton abnormalities, we performed cartilage staining with Alcian blue and observed that embryos expressing PDIA3C57Y developed craniofacial defects including abnormal pharyngeal arch cartilages with a pronounced compression of the anterior–posterior axis, in addition to the presence of kinks in some cartilage elements (Fig 2C). To explore whether PDIA3C57Y affects the nervous system of zebrafish, we carried out immunofluorescent staining of acetylated tubulin to label axonal tracts in the brain. PDIA3C57Y expression led to axonal disorganization detected even in embryos of normal appearance, marked by fused axon tracts in the optic tectum and at the midline of the hindbrain (Fig 2D).
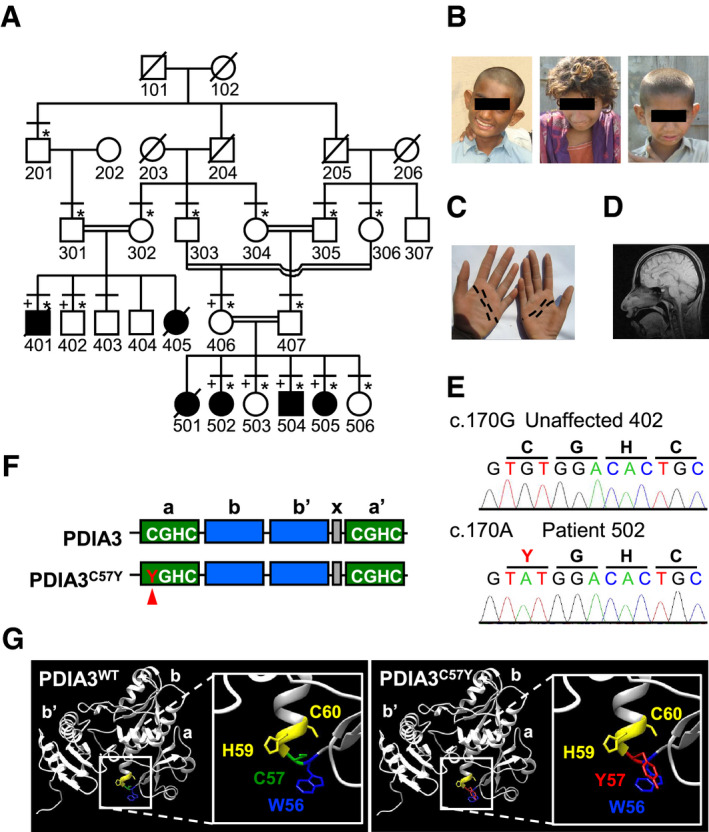
Figure EV1. Effects of PDIA3C57Y expression in zebrafish.
-
A–DZebrafish embryos at the one‐cell stage were injected with sense mRNA coding for wild‐type PDIA3‐V5 or PDIA3C57Y‐V5. (A) PDIA3 mRNA detection in embryos at 24 hpf by conventional PCR. Actin mRNA was employed as loading control. (B) PDIA3 protein levels at 24 hpf determined by Western blot analysis. β‐actin was employed as loading control. (C) Quantification of abnormal morphologies in embryos expressing PDIA3C57Y presented in Fig 2A. (D) Analysis of apoptosis in transgenic embryos with pan‐neuronal expression of mCherry in the central nervous system at 72 hpf. Acridine orange staining (Acridine) shown in green and mCherry in red. BF, bright field. Scale bar 100 μm. Graph shows quantification of acridine orange staining (number of positive spots). Mock, 2; PDIA3, 2; PDIA3C57Y, 5. Data are shown as mean ± s.e.m.
Source data are available online for this figure.
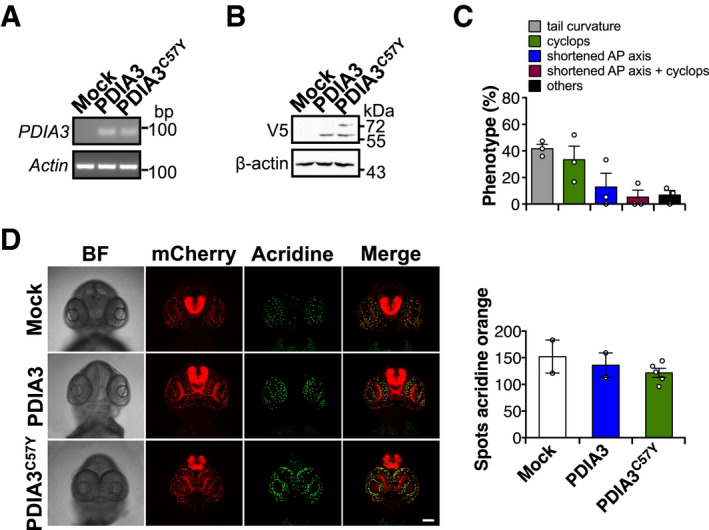
Figure 2. Expression of PDIA3C57Y affects vertebrate development.
-
A–DZebrafish embryos were injected at the one‐cell stage with sense mRNA coding for either wild‐type PDIA3‐V5 or PDIA3C57Y‐V5. (A) Lateral and ventral micrographs of embryos at 48 h post‐fertilization (hpf) showing typical morphological alterations caused by PDIA3C57Y expression such as shortening of anterior–posterior axis, cyclopia, and tail curvature. Scale bar 500 μm. Mock, n = 125; PDIA3, n = 140; PDIA3C57Y, n = 239 embryos injected in three independent experiments. Graph shows quantification of percentage of embryos with altered morphology in each independent experiment. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test. (B) Cell movements during gastrulation. Upper panel: lateral micrographs of embryos undergoing epiboly at 9 hpf. Graph shows quantification of blastoderm displacement. Mock, n = 6; PDIA3, n = 8; PDIA3C57Y, n = 8. Middle panel: dorsal micrograph of embryos at 12.5 hpf showing convergence. Graph shows quantification of somite width. Mock, n = 5; PDIA3, n = 6; PDIA3C57Y, n = 8. Lower panel: lateral micrograph of embryos at 13 hpf showing extension. Graph shows quantification of head to tail angle. Mock, n = 5; PDIA3, n = 6; PDIA3C57Y, n = 8. Scale bar 200 μm. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test. (C) Alcian blue staining of cartilage showing craniofacial features of embryos at 6 days post‐fertilization (dpf). Embryos expressing PDIA3C57Y show reduced distance between Meckel's (m) and ceratohyal (ch) cartilages (double‐headed arrow), kinked palatoquadrate (pq), and additional unidentified pieces of cartilages (?). e, ethmoid plate; bh, basihyal; hs, hyosymplectic; cb, ceratobranchials. Scale bar 200 μm. Mock, n = 6; PDIA3, n = 5; PDIA3C57Y, n = 15. (D) Representative micrographs of acetylated‐tubulin (Ac‐Tub) indirect immunofluorescence in dorsal views of embryos at 96 hpf. White arrowheads and arrows in embryos expressing PDIA3C57Y point to the abnormal organization of axonal tracts in the optic tectum and the fused axons at the dorsal midline of the hindbrain, respectively. BF, bright field. Scale bar 100 μm. Mock, n = 3; PDIA3, n = 3; PDIA3C57Y, n = 11.
Source data are available online for this figure.
Expression of mutant PDIA3 impairs neuronal connectivity and memory consolidation
To study neuronal development in mammals, we performed transient protein expression in primary culture of mouse hippocampus. PDIA3C57Y expression inhibited axonal growth, while no effects on minor neurite length were observed (Fig 3A). Next, we assessed the impact of PDIA3C57Y on brain function by conducting behavioral tests in young mice injected with adeno‐associated virus (AAV) to deliver WT or mutant PDIA3 transgene in the hippocampus. The expression levels of WT and mutant PDIA3 were similar (Fig EV2A), with transduced neurons in CA1 and CA3 regions (Fig EV2B). The locomotion and exploratory behavior of mice injected with AAV‐Mock, AAV‐PDIA3, or AAV‐PDIA3C57Y was comparable in the open‐field test (Fig EV2C). To investigate cognitive function, we employed the Barnes maze to measure spatial learning and memory (Fig 3B). Although the learning curves were similar for the three groups studied (Fig 3B), PDIA3C57Y expression in the hippocampus led to significantly deficient long‐term, and additionally a trend to impaired short‐term, memory acquisition (Fig 3C). Thus, we performed electrophysiological experiments to measure synaptic activity in the hippocampus. Long‐term potentiation (LTP), a long‐lasting form of synaptic plasticity, was monitored by stimulating Schaeffer's collaterals projecting from CA3 and recording field excitatory postsynaptic potentials in the CA1 region (Fig 3D). Consistent with our behavioral results, LTP was impaired in animals expressing PDIA3C57Y in the hippocampus (Fig 3D).
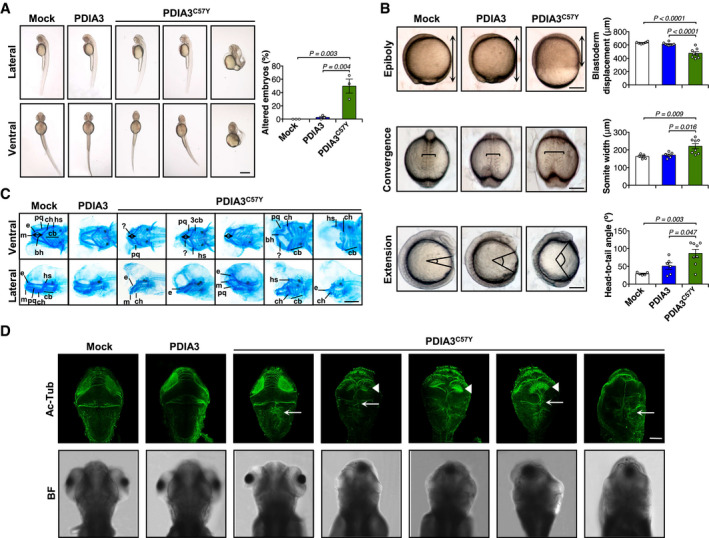
Figure 3. ID‐linked PDIA3C57Y impairs cognitive function.
-
APrimary culture of mouse hippocampal neurons was co‐transfected with constructs for transient expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock), and GFP. Representative micrographs of GFP and indirect immunofluorescence of Tau‐1 to visualize axons at day 3 in vitro. Scale bar 50 μm. Left graph shows quantification of minor neurite length; Mock, n = 175; PDIA3, n = 204; PDIA3C57Y, n = 191 cells quantified in three independent experiments. Right graph shows quantification of axon length; Mock, n = 35; PDIA3, n = 46; PDIA3C57Y, n = 43 cells quantified in three independent experiments. Data are shown as mean ± s.e.m. and statistical analysis performed using Kruskal–Wallis with Dunn's post hoc test.
-
B–DYoung mice at P55 received bilateral stereotaxis injection of adeno‐associated virus serotype 9 (AAV9) to express wild‐type PDIA3‐V5 or PDIA3C57Y‐V5 and GFP or GFP alone (Mock) into the hippocampus for behavioral and electrophysiological analysis. (B) Barnes maze test: mice were placed in a circular arena with 20 holes on the edge and visual cues on the surrounding walls (scheme to the left). The time spent to find the target hole was recorded as latency. The latency from days 1 to 4 is plotted as learning curve (graph to the right); Mock, n = 10; PDIA3, n = 10; PDIA3C57Y, n = 10. Data are shown as mean ± s.e.m. (C) Latency at day 5 is measured as short‐term memory; Mock, n = 10; PDIA3, n = 9; PDIA3C57Y, n = 10. Latency at day 12 is measured as long‐term memory; Mock, n = 9; PDIA3, n = 10; PDIA3C57Y, n = 9. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test. (D) Field excitatory postsynaptic potentials (fEPSPs) were recorded in the stratum radiatum of the CA1 region after stimulation of Schaeffer collateral commissural fibers projecting from CA3 as indicated in the scheme. DG, dentate gyrus. After fEPSP baseline acquisition, long‐term potentiation (LTP) was induced by theta burst stimulation (TBS). Left graph: time course of TBS‐induced LTP in Mock, PDIA3, and PDIA3C57Y mice. Arrow indicates TBS. Inset: representative recordings averaging 10 traces at −10 min (Pre‐TBS, indicated as time “1”) and 50 min (Post‐TBS, indicated as time “2”). Arrow: presynaptic volley. Right graph: LTP quantification during the final 20 min of the recording, presented as the averaged percentage of baseline. Mock, n = 5, 6; PDIA3, n = 7, 8; PDIA3C57Y, n = 5, 7 for number of animals and recorded slices, respectively. Data are shown as mean ± s.e.m. and statistical analysis performed using Wilcoxon–Mann–Whitney two‐sample rank test.
Source data are available online for this figure.
Figure EV2. Analysis of PDIA3C57Y expression in the central nervous system of mice.
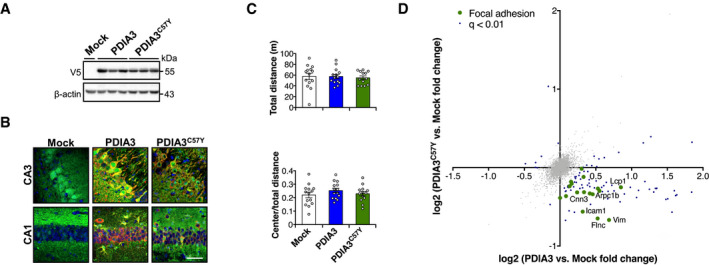
-
A–DYoung mice at P55 received bilateral stereotaxis injection of adeno‐associated virus serotype 9 (AAV9) to express wild‐type PDIA3‐V5 or PDIA3C57Y‐V5 and GFP or GFP alone (Mock) into the hippocampus for behavioral and proteomic analysis. (A) PDIA3 protein levels in hippocampus at P180 detected by Western blot analysis. β‐actin employed as loading control. (B) Representative micrographs of hippocampus analyzed by immunofluorescence to GFP (green) and V5 (red) at P180 showing neuronal transduction in CA3 and CA1 regions. Nucleus stained with Hoechst (blue). Scale bar 50 μm. (C) Open field. Mice were placed in a squared arena and recorded for 15 min. Total distance (upper graph) and center to total distance ratio (lower graph) were measured as locomotor activity and exploratory behavior, respectively. Mock, n = 14; PDIA3, n = 14; PDIA3C57Y, n = 13. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test. No statistically significant differences exist between groups. (D) Proteomic analysis at P180. Mock, n = 3; PDIA3, n = 3; PDIA3C57Y, n = 4. Scatter plot of proteomic analysis of hippocampus tissue of mice expressing wild‐type PDIA3 or PDIA3C57Y. The x‐axis denotes logarithmic fold change in total protein levels in wild‐type PDIA3 mice relative to Mock littermates. The y‐axis denotes logarithmic fold change in total protein levels in PDIA3C57Y mice relative to Mock littermates. Representative hits of focal adhesion are highlighted in green.
Source data are available online for this figure.
PDIA3 dysfunction results in abnormal cell adhesion and actin cytoskeleton
To define possible pathogenic mechanisms associated with PDIA3C57Y expression in the brain, we performed mass spectrometry‐based quantitative proteomics of hippocampus transduced with AAV‐Mock, AAV‐PDIA3, or AAV‐PDIA3C57Y. Gene set enrichment analysis revealed down‐regulation of focal adhesion molecules (e.g., Icam1, Integrin‐β3, Integrin‐αM, Plastin‐2, and Talin‐1), including actin cytoskeleton regulators (e.g., Calponin‐3, Vimentin, and Filamin A, B, and C), in the hippocampus of mice expressing PDIA3C57Y (Figs 4A and B, and EV2D, Datasets EV4 and EV5). In addition, proteins related to the MHC‐I pathway such as tapasin, antigen peptide transporter 1 and 2, β2‐microglobulin, and H2 class I histocompatibility antigen were also downregulated (Datasets EV4 and EV5), consistent with the role of PDIA3 as a scaffold that regulates antigen presentation (Garbi et al, 2006). Quantification of PDIA3 overexpression by measuring shared peptides in mouse and human proteins showed 2.6‐ and 2.1‐fold the endogenous levels for the WT and mutant forms, respectively (Dataset EV6). According to quantification of peptides that are exclusive of the mouse protein, there was minimal variation in the levels of endogenous PDIA3 between AAV‐PDIA3 and AAV‐PDIA3C57Y groups (Dataset EV6). We verified expression levels of selected proteomic hits representing the altered protein clusters using Western blot, confirming decreased levels of Calponin‐3, an actin‐binding protein essential for synaptic function (Ferhat et al, 2003; Junghans & Herzog, 2018), and Psmb10, an immunoproteasome subunit, in the AAV‐PDIA3C57Y group (Fig 4C). Network analysis uncovered meaningful interactions between PDIA3 and actin cytoskeleton components related to focal adhesion (Fig 4D). Based on these results, we performed functional analysis of PDIA3C57Y impact on cell adhesion and actin cytoskeleton dynamics.
Figure 4. Down‐regulation of cell adhesion molecules in brain tissue expressing PDIA3C57Y .
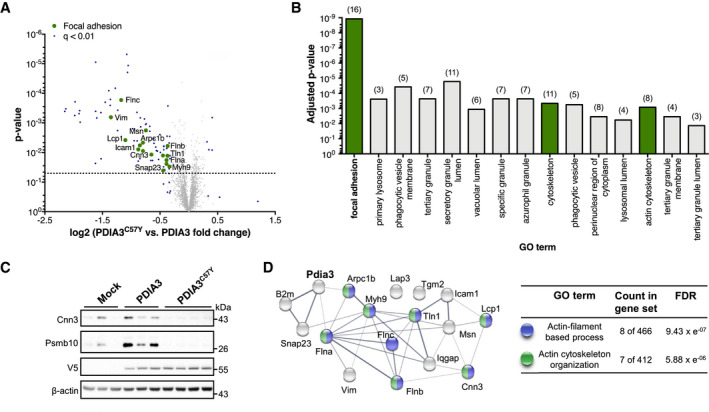
-
A–DYoung mice at P55 received bilateral stereotaxic injection of adeno‐associated virus serotype 9 (AAV9) to express wild‐type PDIA3‐V5 or PDIA3C57Y‐V5 and GFP or GFP alone (Mock) into the hippocampus for proteomic analysis. (A) Volcano plot of proteomic analysis of hippocampus tissue of mice expressing wild‐type PDIA3 or PDIA3C57Y. The x‐axis denotes logarithmic fold change of total protein levels in PDIA3C57Y relative to wild‐type PDIA3 mice. The y‐axis denotes the P‐value of statistical analysis. Dotted line indicates threshold for statistical significance. Statistical analysis performed using two‐tailed Student's t‐test with 95% confidence interval. PDIA3, n = 3; PDIA3C57Y, n = 4. (B) Functional categorization of proteomic hits according to the Gene Ontology (GO) annotation. Graph shows significantly enriched GO terms of cellular component comparing wild‐type PDIA3 to PDIA3C57Y mice. The number of genes associated with each term is indicated in parenthesis. Focal adhesion hits are highlighted in green in the volcano plot presented in A. (C) Western blot validation of the total protein levels of the proteomic hits Calponin‐3 (Cnn3) and Proteasome subunit beta type‐10 (Psmb10; see Dataset EV4). V5 and β‐actin were employed as loading control. (D) Network analysis of Pdia3 interaction with identified genes related to focal adhesion as presented in A and B (see Dataset EV5). The nodes represent the proteins and the edges interactions. The thickness of the edges indicates the extent of interactions, which not necessarily mean physical binding. The table shows GO annotation of biological function of network components. FDR, false discovery rate.
Source data are available online for this figure.
To study cell adhesion during development, we investigated the clustering of dorsal forerunner cells (DFC) during zebrafish gastrulation (Oteiza et al, 2010). We used the transgenic line Tg(sox17:GFP) to visualize DFC distribution and observed that embryos expressing WT PDIA3 formed a normal oval‐shaped DFC cluster (Fig 5A). In sharp contrast, DFC in embryos expressing PDIA3C57Y organized as a wider cellular aggregate with occasional gaps (Fig 5A), both indicative of impaired cell adhesion in this cell population. We also examined the distribution of the actin cytoskeleton in early zebrafish embryos using a transgenic reporter line for actin (Urra et al, 2018). Deep cells expressing PDIA3C57Y exhibited altered morphology with increased circularity and decreased aspect ratio (Fig 5B). Furthermore, whereas control cells displayed the expected actin peaks along the cell cortex, cells expressing PDIA3C57Y showed a homogeneous distribution pattern of the actin cytoskeleton, suggesting altered filament assembly (Fig 5C). Consistent with these observations, analysis of filamentous actin in skeletal muscle at later developmental stages revealed decreased myotome width and disorganized muscle fibers in animals expressing PDIA3C57Y (Fig 5D).
Figure 5. PDIA3C57Y disturbs cell adhesion and actin cytoskeleton.
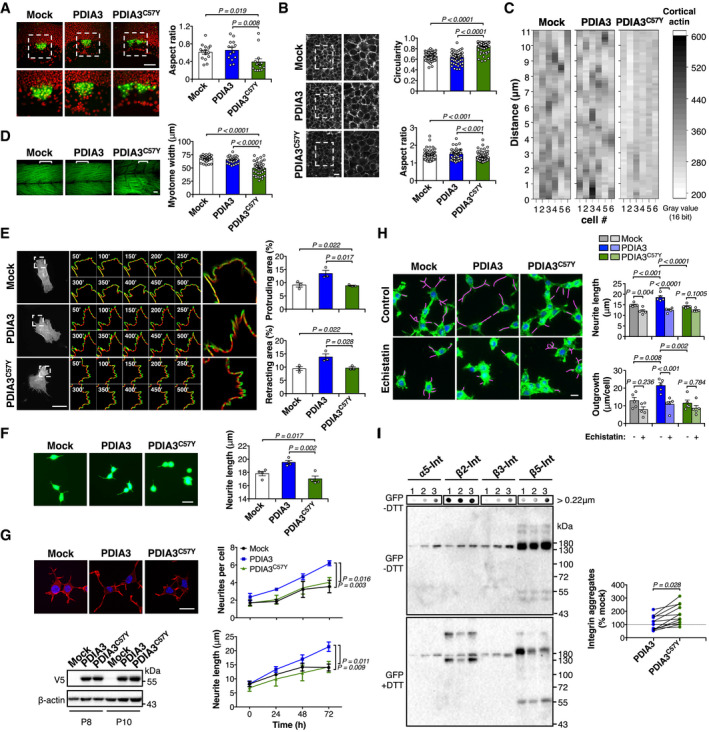
-
ATransgenic zebrafish embryos expressing GFP in dorsal forerunner cells (DFC), Tg(sox17:GFP), were injected at the one‐cell stage with sense mRNA coding for either wild‐type PDIA3‐V5 or PDIA3C57Y‐V5. Fluorescence micrographs of DFC from dorsal views obtained at 8 hpf. Nuclei depicted in red. Right images are digital magnifications of areas delimited by white dashed squares. Scale bar 100 μm. Graph shows quantification of aspect ratio of the DFC cluster. Mock, n = 15; PDIA3, n = 14; PDIA3C57Y, n = 14. Data are shown as mean ± s.e.m. and statistical analysis performed using Kruskal–Wallis with Dunn's post hoc test.
-
B, CTransgenic zebrafish embryos ubiquitously expressing the mCherry‐tagged actin binding domain of utrophin, Tg(actb1:mCherry‐utrCH), were injected at the one‐cell stage with sense mRNA coding for either wild‐type PDIA3‐V5 or PDIA3C57Y‐V5. (B) Fluorescence micrographs of deep cells from the animal pole obtained at 8 hpf. Right images are digital magnifications of areas delimited by white dashed squares. Scale bar 20 μm. Graphs show quantification of cell circularity and aspect ratio. Mock, n = 55; PDIA3, n = 57; PDIA3C57Y, n = 84 cells quantified from at least three embryos per group. Data are shown as mean ± s.e.m. and statistical analysis performed using Mann–Whitney test. (C) Heatmap of fluorescence intensity of mCherry along the cortex of individual deep cells.
-
DPhalloidin staining of skeletal muscle of embryos at 48 hpf. Embryos expressing PDIA3C57Y show decreased myotome width and disorganized muscle fibers. Scale bar 20 μm. Graph shows quantification of myotome width. Mock, n = 40; PDIA3, n = 28; PDIA3C57Y, n = 40 myotomes quantified from at least five embryos. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test.
-
EMouse embryonic fibroblast knock‐out for Pdia3 (MEF Pdia3 KO) was co‐transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock), and EGFP‐LifeAct. Representative micrographs of live cell imaging performed at 48 h after transfection are shown. Segmentation of time‐lapse images was used to obtain protruding (green) and retracting (red) areas. Zoom of representative cell areas is shown. Protruding and retracting area was quantified using Fiji software. Scale bar 50 μm. n = 3 independent experiments. A total of 10 movies per group with 1 or 2 cells per movie were quantified. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test.
-
FNSC‐34 cells cotransfected with constructs for transient expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock), and GFP were differentiated and neurite length measured at 48 h. Scale bar 20 μm. n = 4 independent experiments. Total cells quantified: Mock, 200; PDIA3, 157; PDIA3C57Y, 145. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test.
-
GNSC‐34 neuronal cell lines stably expressing wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock) were differentiated for 72 h and neurite number and length determined over time by high‐content analysis of cells stained with phalloidin. Scale bar 20 μm. n = 3 independent experiments. Total cells quantified: Mock, 343; PDIA3, 388; PDIA3C57Y, 495. Data are shown as mean ± s.e.m. and statistical analysis performed using two‐way ANOVA with Tukey's post hoc test. Western blot analysis of PDIA3 levels in NSC‐34 cell lines employed for the differentiation assay at different passages (P) after initial transfection. β‐actin employed as loading control.
-
HNSC‐34 neuronal cell lines stably expressing wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock) were differentiated for 24 h and treated with 40 nM Echistatin to inhibit integrin signaling. The cells were collected at 72 h of differentiation, stained with phalloidin‐FITC, and scanned by automated microscopy for analysis of neurite length and outgrowth. Magenta traces mark neurite extension. Scale bar 30 μm. n = 5 independent experiments with at least 96 neurites quantified per group in each experiment. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Sidak's post hoc test.
-
IWestern blot and filter‐trap analysis of a panel of integrin paralogs in stable NSC‐34 cell lines overexpressing PDIA3 or PDIA3C57Y, or mock control. The cells were transfected with constructs for overexpression of α5‐integrin (α5‐int) fused to GFP, β2‐integrin (β2‐int) fused to YFP, β3‐integrin (β3‐int) fused to YFP, and β5‐integrin (β5‐int) fused to 2xGFP and analyzed under non‐reducing (−DTT, dithiothreitol) or reducing conditions (+DTT). The anti‐GFP antibody detects both GFP and YFP tags. 1, Mock, 2, PDIA3‐V5, and 3, PDIA3C57Y‐V5. Representative image of three independent experiments. The graph shows quantification of integrin aggregates relative to mock control detected under non‐reducing conditions. Lines connect aggregates quantified in the same experiment. Aggregates from different integrin paralogs were pooled in the statistical analysis using two‐tailed Student's t‐test. Figure EV3C shows quantification of aggregates from each integrin paralog separately.
Source data are available online for this figure.
To further assess the importance of PDIA3 to the regulation of actin cytoskeleton and its possible role in cell adhesion, we used Pdia3 knockout (Pdia3 KO) and control mouse embryonic fibroblasts (MEFs) to investigate the formation and movement of lamellipodia (Urra et al, 2018). We employed expression of the LifeAct reporter coupled to quantitative analysis of live‐cell microscopy to monitor actin filament dynamics (Belin et al, 2014). We found that Pdia3 KO MEFs have reduced lamellipodia index, in addition to diminished protrusion and retraction areas and velocities (Fig EV3A). Importantly, the expression of WT PDIA3, but not PDIA3C57Y, rescued the deficits in actin dynamics observed in Pdia3 KO MEFs (Figs 5E and EV3B). Taken together, these results suggest that PDIA3C57Y alters the actin cytoskeleton and cell adhesion, thereby disturbing tissue morphogenesis.
Figure EV3. Effects of PDIA3C57Y expression on actin cytoskeleton and neuritogenesis.
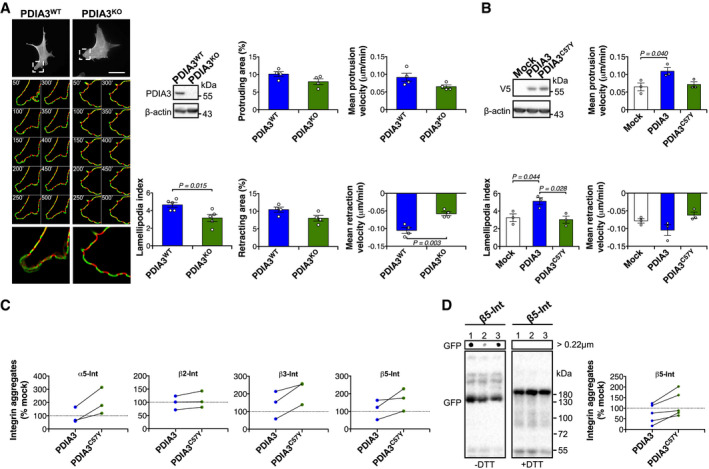
- Mouse embryonic fibroblast (MEF) knock‐out for Pdia3 (Pdia3 KO) and wild‐type MEF (Pdia3 WT) were transfected with construct for expression of EGFP‐LifeAct. Representative micrographs of live cell imaging performed at 48 h after transfection are shown. Segmentation of time‐lapse images was used to obtain protruding (green) and retracting (red) areas. Zoom of representative cell areas is shown. Lamellipodia number and protrusion/retraction movement were quantified using Fiji software. Scale bar 50 μm. n = 4 independent experiments. A total of 9 movies per group with 1 or 2 cells per movie were quantified. Data are shown as mean ± s.e.m. and statistical analysis performed using two‐tailed Student's t‐test. Pdia3 KO MEF was checked by Western blot analysis. β‐actin was employed as loading control.
- Pdia3 KO MEF was co‐transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock), and EGFP‐LifeAct. n = 3 independent experiments. A total of 10 movies per group with 1 or 2 cells per movie were quantified. Graphs show quantification of lamellipodia number and protruding/retracting velocity. Data are shown as mean ± s.e.m. and statistical analysis performed using one‐way ANOVA with Tukey's post hoc test. Wild‐type PDIA3‐V5 or PDIA3C57Y‐V5 levels were checked by Western blot analysis. β‐actin was employed as loading control.
- NSC‐34 neuronal cell lines stably expressing wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock) were transfected with constructs for overexpression of α5‐integrin (α5‐int) fused to GFP, β2‐integrin (β2‐int) fused to YFP, β3‐integrin (β3‐int) fused to YFP, and β5‐integrin (β5‐int) fused to 2xGFP and analyzed by filter trap under non‐reducing (−DTT, dithiothreitol) or reducing (+DTT) conditions as described in Fig 5I. The graphs show quantification of aggregates of each integrin paralog relative to mock control detected under non‐reducing conditions. Lines connect aggregates quantified in the same experiment. n = 3 independent experiments.
- Pdia3 KO MEF was co‐transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock), and β5‐integrin (β5‐int) fused to 2xGFP and analyzed by Western blot and filter‐trap under non‐reducing (−DTT, dithiothreitol) or reducing (+DTT) conditions. 1, Mock, 2, PDIA3‐V5, and 3, PDIA3C57Y‐V5. Representative image of five independent experiments. The graph shows quantification of integrin aggregates relative to mock control detected under non‐reducing conditions. Lines connect aggregates quantification in the same experiment.
Source data are available online for this figure.
PDIA3 regulates neuritogenesis through integrin signaling
We next determined the consequences of PDIA3C57Y expression on neuritogenesis, a relevant process for neural connectivity that depends on actin cytoskeleton remodeling and cell adhesion (da Silva & Dotti, 2002; Kiryushko et al, 2004; Sandi, 2004). Overexpression of PDIA3 in the NSC‐34 neuronal cell line resulted in enhanced neurite length, a phenomenon that was fully abrogated by the ID‐linked variant (Fig 5F). We then examined time course of neuritogenesis in the NSC‐34 cells using phalloidin staining and unbiased automated microscopy. These studies further demonstrated that PDIA3 overexpression, but not the C57Y mutant, enhances neurite number and length (Fig 5G).
Integrins are key adhesion molecules linking the actin cytoskeleton to the extracellular matrix that have been previously identified as PDIA3 substrates (Jessop et al, 2007). Since integrin signaling is important for regulation of neuronal morphogenesis (Park & Goda, 2016), we investigated their possible involvement during neuronal differentiation. Treatment of cells with Echistatin, a potent and selective inhibitor of integrin signaling (Kapp et al, 2017), significantly reduced neurite length in the NSC‐34 cell line, indicating that neuritogenesis in this cellular model depends on integrins (Fig 5H). Moreover, Echistatin totally blunted the positive effects of PDIA3 on neurite length and outgrowth, whereas not exerting a significant action on cells expressing PDIA3C57Y (Fig 5H). This result indicates that integrins are relevant biological effectors of PDIA3 function that may be perturbed in ID.
We then investigated whether PDIA3C57Y expression disturbs integrin folding by examining a panel of four paralogs, α5‐integrin (α5‐int), β2‐integrin (β2‐int), β3‐integrin (β3‐int), and β5‐integrin (β5‐int) fused to either GFP or YFP. Western blot analysis under reducing conditions detected a band between 130 and 180 KDa consistent with the glycosylated monomer of the fusion proteins expressed in the NSC‐34 cell line (Fig 5I). Interestingly, a different band pattern for β2‐int‐YFP and β3‐int‐YFP was observed when comparing cells expressing PDIA3 or PDIA3C57Y. β2‐int‐YFP presented monomer‐like species with faster migration detectable only under reducing conditions that was decreased upon expression of PDIA3, but not PDIA3C57Y (Fig 5I). Similar to β2‐int‐YFP, close inspection of β3‐int‐YFP signal also revealed a faster migrating monomer‐like form upon expression of PDIA3C57Y that is observed exclusively under reducing conditions (Fig 5I).
Western blot analysis under non‐reducing conditions corroborated detection of integrin monomers between 130 and 180 KDa, but it did not reveal any distinct disulfide‐crosslinked oligomeric or aggregated species of the integrin paralogs investigated (Fig 5I). Because the monomer‐like species of β2‐int‐YFP and β3‐int‐YFP are detectable only under reducing conditions, such species may correspond to disulfide‐crosslinked aggregates undetectable by non‐reducing Western blot. Indeed, filter‐trap analysis revealed the existence of integrin aggregates that are sensitive to the thiol reducing agent dithiothreitol (DTT), indicative of disulfide‐crosslinked species. Across the panel of integrins examined, PDIA3C57Y‐expressing cells accumulated higher levels of aggregated species compared to PDIA3‐overexpressing cells (Figs 5I and EV3C). Similar results were obtained in Pdia3 KO MEFs, where higher levels of disulfide‐crosslinked aggregates of β5‐int were detected upon reconstitution with PDIA3C57Y compared to WT PDIA3 (Fig EV3D). These biochemical analyses at steady‐state condition suggest that PDIA3C57Y may alter the folding or impair the quality control of integrins.
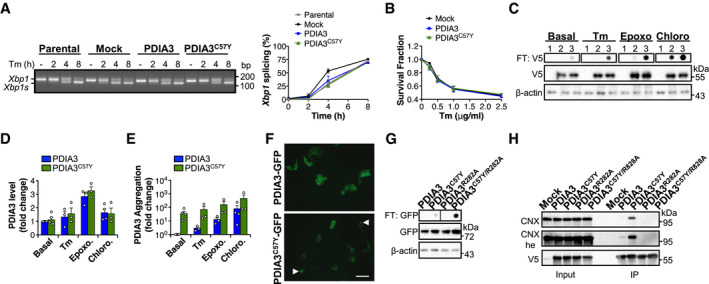
Since PDIA3 is a central component of the folding machinery in the secretory pathway, we also examined the possible occurrence of ER stress and toxicity upon expression of PDIA3C57Y. Consistent with our previous studies in Pdia3 gain‐ and loss‐of‐function models in mice and cell lines (Torres et al, 2015), PDIA3C57Y expression in NSC‐34 cells neither enhanced UPR signaling nor decreased cell viability under basal or under ER stress induced with the pharmacological agent tunicamycin (Fig EV4A and B).
Figure EV4. Characterization of PDIA3C57Y expression in cell culture.
-
AKinetics of unfolded protein response activation measured by splicing of X‐box binding protein 1 (Xbp1) mRNA in NSC‐34 cell lines stably expressing wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock) treated with 1 μg/ml of the ER stressor tunicamycin (Tm). Xbp1s, spliced form of Xbp1 mRNA. Parental, non‐transfected NSC‐34. Graph shows quantification of percentage of Xbp1 mRNA splicing in three independent experiments. Data are shown as mean ± s.e.m.
-
BNSC‐34 cells were transfected with constructs for transient expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock). Viability was determined after treatment with the indicated dose of Tm for 24 h in three independent experiments. Data are shown as mean ± s.e.m.
-
C–ENSC‐34 cells were transfected with constructs for transient expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock). (C) Filter‐trap analysis of PDIA3‐V5 aggregates under non‐reducing conditions at 24 h after treatment with Tm (1 μg/ml), the proteasome inhibitor epoxomicin (Epoxo, 250 nM), or the lysosome inhibitor chloroquine (Chloro, 30 μM). β‐actin was employed as loading control. 1, Mock, 2, PDIA3‐V5, and 3, PDIA3C57Y‐V5. Representative image of three independent experiments. (D) Quantification of total levels of PDIA3 by Western blot under different stress conditions. PDIA3 levels expressed as fold change relative to the wild‐type protein under basal condition. Data are shown as mean ± s.e.m. (E) Quantification of PDIA3 aggregates by filter trap under different stress conditions. PDIA3 aggregates levels expressed as fold change relative to the wild‐type protein under basal condition. Data are shown as mean ± s.e.m.
-
FNSC‐34 cells were transfected with constructs for transient expression of wild‐type PDIA3‐GFP or PDIA3C57Y‐GFP. Fluorescence micrographs show live cells at 48 h after transfection. Arrowheads point to puncta of PDIA3C57Y‐GFP. Scale bar 50 μm. The image is representative of a single experiment performed for construct validation.
-
GNSC‐34 cells were transfected with constructs for transient expression of wild‐type PDIA3‐GFP, PDIA3C57Y‐GFP, PDIA3R282A‐GFP, or PDIA3C57Y/R282A‐GFP. Filter‐trap analysis under non‐reducing conditions was performed at 48 h after transfection. β‐actin was employed as loading control. The image is representative of a single experiment performed for construct validation.
-
HHEK cell line was transfected with constructs for transient expression of wild‐type PDIA3‐V5, PDIA3C57Y‐V5, PDIA3R282A‐V5, or PDIA3C57Y/R282A‐V5, or empty vector (Mock). Immunoprecipitation of V5‐tag was carried out at 48 h after transfection and PDIA3 interaction with calnexin (CNX) measured by Western blot; he, high exposure. The image is representative of a single experiment performed for construct validation.
Source data are available online for this figure.
Mutant PDIA3 forms aggregates and abnormally interacts with chaperones in the ER
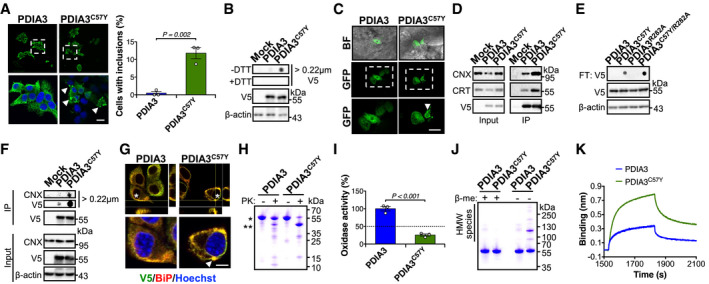
We examined intrinsic properties of PDIA3 to define possible toxic effects generated by variant C57Y. Surprisingly, analysis of the cellular distribution of PDIA3 using immunofluorescence indicated that a small fraction of the cells expressing PDIA3C57Y presented a puncta pattern reminiscent of protein inclusions (Fig 6A). To determine whether variant C57Y causes protein aggregation in cell culture, we performed filter‐trap analysis. This approach demonstrated that PDIA3C57Y forms aggregates that are solubilized under reducing conditions, indicating the occurrence of aberrant disulfide protein crosslinks (Fig 6B). Treatment of cells with lysosome inhibitor chloroquine preferentially enhanced the accumulation of PDIA3 aggregates when compared to the ER stressor tunicamycin or the proteasome inhibitor epoxomicin (Fig EV4, EV5), suggesting that degradation of these disulfide‐crosslinked species occurs through the lysosomal pathway. We also observed the aggregation of PDIA3C57Y in vivo using mosaic overexpression of GFP‐tagged PDIA3 in zebrafish embryos, detecting clusters of PDIA3C57Y‐GFP in epidermal cells (Fig 6C) similar to the protein inclusions observed in cell culture using the same construct (Fig EV4F and G).
Figure 6. Abnormal biochemical properties of PDIA3C57Y .
-
A, BNSC‐34 cells were transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock). (A) Fluorescence micrographs of V5 immunostaining at 48 h after transfection. Bottom images are digital magnifications of areas delimited by white dashed squares. Arrowheads point to PDIA3C57Y‐V5 puncta. Scale bar 15 μm. Graph shows quantification of percentage of cells containing PDIA3 puncta. n = 3 independent experiments. Total cells quantified: PDIA3, 365; PDIA3C57Y, 301. Data are shown as mean ± s.e.m. and statistical analysis performed using two‐tailed Student's t‐test. (B) Filter‐trap analysis of PDIA3‐V5 aggregates under non‐reducing (−DTT, dithiothreitol) and reducing (+DTT) conditions at 48 h after transfection. V5 and β‐actin Western blot analysis was employed for loading control. Representative image of five independent experiments.
-
CZebrafish embryos were injected at the four‐cell stage with plasmid DNA for expression of wild‐type PDIA3‐GFP or PDIA3C57Y‐GFP. Representative fluorescence micrographs of embryos at 48 hpf show mosaic expression of PDIA3‐GFP and PDIA3C57Y‐GFP in epithelial cells. Bottom images are digital magnifications of areas delimited by white dashed squares. Arrowhead points to PDIA3C57Y‐GFP puncta. BF, bright field. Scale bar 50 μm. n = 3 embryos in each group.
-
DHEK cell line was transiently transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock). Western blot analysis of immunoprecipitation of V5‐tag to measure PDIA3 interaction with calnexin (CNX) and calreticulin (CRT) at 48 h after transfection. Representative image of three independent experiments.
-
ENSC‐34 cells were transiently transfected with constructs for expression of wild‐type PDIA3‐V5, PDIA3C57Y‐V5, PDIA3R282A‐V5, or PDIA3C57Y/R282A‐V5. Filter‐trap analysis under non‐reducing conditions was performed at 48 h after transfection. β‐actin was employed as loading control. Representative image of three independent experiments.
-
FHEK cell line was transiently transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock). Native filter‐trap analysis of immunoprecipitation of V5‐tag to measure CNX interaction with PDIA3 aggregates at 48 h after transfection. Representative image of three independent experiments.
-
GNSC‐34 cells were transfected with constructs for expression of wild‐type PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (Mock). Fluorescence micrographs with XZ and YZ orthogonal views from Z‐stacks of seven confocal planes show co‐localization of V5 and BiP immunostaining. Bottom images are digital magnifications of cells indicated with white asterisks. Arrowhead points to PDIA3C57Y‐V5 puncta positive for BiP. Scale bar 5 μm. Representative image of three independent experiments.
-
HPurified recombinant wild‐type PDIA3 and PDIA3C57Y were treated with proteinase K and analyzed by SDS–PAGE with Coomassie blue staining. *, undigested protein. **, main fragment of PDIA3C57Y digestion. Representative image of four independent reactions.
-
IRelative thiol oxidase activity of recombinant wild‐type PDIA3 and PDIA3C57Y using NRCSQGSCWN as substrate peptide. n = 3 independent reactions. Data are shown as mean ± s.e.m. and statistical analysis performed using two‐tailed Student's t‐test.
-
JSDS–PAGE analysis of recombinant wild‐type PDIA3 and PDIA3C57Y under reducing (+β‐me, β‐mercaptoethanol) and non‐reducing (−β‐me) conditions with Coomassie blue staining.
-
KRepresentative traces for binding of 400 nM wild‐type PDIA3 or PDIA3C57Y to immobilized CRT‐P domain using Bio‐Layer Interferometry. A total of five independent measurements were performed.
Source data are available online for this figure.
Figure EV5. Biophysical and biochemical properties of PDIA3C57Y .
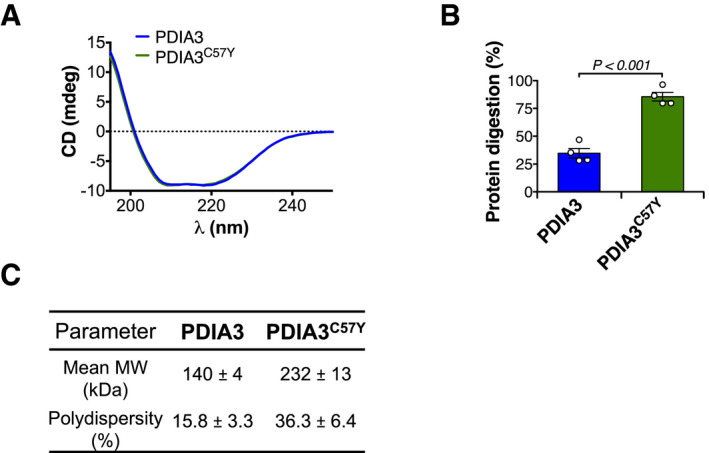
- Circular dichroism (CD) analysis of recombinant wild‐type PDIA3 and PDIA3C57Y. Average traces of CD spectroscopic scans are shown.
- Purified recombinant wild‐type PDIA3 and PDIA3C57Y were treated with proteinase K and analyzed by SDS–PAGE with Coomassie blue staining as shown in Fig 6H. n = 4 independent reactions. Bar graph shows quantification of protein digestion. Data are shown as mean ± s.e.m. and statistical analysis performed using two‐tailed Student's t‐test.
- Dynamic light scattering determination of mean molecular weight (MW) and percent polydispersity of wild‐type PDIA3 and PDIA3C57Y. Data are shown as mean ± s.d.
Source data are available online for this figure.
Since PDIA3 function is tightly coupled to CNX/CRT (Appendix Fig S4C) (Jessop et al, 2007), we assessed protein interaction using co‐immunoprecipitation and found augmented association of PDIA3C57Y with both chaperones (Fig 6D). To address the possible relationship between PDIA3C57Y aggregation and its increased interaction with CNX/CRT, we introduced point mutation p.Arg282Ala (R282A) to PDIA3, previously shown to abolish the binding of the protein to these chaperones (Fig EV4H) (Jessop et al, 2009). R282A variant greatly increased the levels of PDIA3C57Y aggregates while having no impact on the WT protein (Figs 6E and EV4G), suggesting that CNX/CRT may participate in the quality control of PDIA3C57Y aggregates in the ER. Coimmunoprecipitation followed by native filter‐trap corroborated interaction of CNX with PDIA3C57Y aggregates (Fig 6F). Moreover, immunofluorescence analysis against BiP showed that PDIA3C57Y puncta colocalizes with ER chaperones (Fig 6G).
Biochemical analysis of mutant PDIA3
We then expressed and purified recombinant proteins to analyze the structural and biochemical properties of PDIA3C57Y. Circular dichroism analysis revealed no gross alterations of secondary structure of PDIA3C57Y (Fig EV5A). However, PDIA3C57Y was more susceptible to proteolysis with proteinase K, indicating increased flexibility of its tertiary structure (Figs 6H and EV5B). We measured the enzymatic activity and found that PDIA3C57Y retains only around 25% of the disulfide oxidase activity of the WT form (Fig 6I). To test whether purified PDIA3C57Y forms aggregates, we performed non‐reducing SDS–PAGE analysis of the recombinant proteins. Remarkably, PDIA3C57Y formed disulfide‐crosslinked HMW oligomers that were absent in the WT PDIA3 preparation (Fig 6J), implying an intrinsic propensity of the mutant protein to aggregate. Dynamic light scattering (DLS) revealed that WT PDIA3 was mainly a homodimer, whereas the mutant protein was predominantly present as HMW species with higher polydispersity (Fig EV5C). Furthermore, PDIA3C57Y physically associated with the P‐domain of CRT in vitro with increased stoichiometric ratio as compared to the WT protein (Fig 6K), indicating that mutant PDIA3 HMW oligomers may interact with CRT.
Discussion
ER proteostasis is maintained by a highly interconnected network of chaperones, quality control checkpoints, and degradative mechanisms that ensure the proper folding and maturation of proteins and disposal of irreversibly misfolded polypeptides (Calì et al, 2008). The continuous accumulation of misfolded proteins in the ER lumen leads to chronic ER stress and sustained activation of the UPR, constituting a signature of age‐related pathologies such as type 2 diabetes, cancer, and neurodegenerative diseases (Hetz & Saxena, 2017; Hetz et al, 2020). Despite the strong association of disturbed ER proteostasis with human diseases, direct evidence for its involvement in developmental disorders is scarce. Variants in genes of the glycosylation pathway are linked to congenital disorders, but underlying pathogenic mechanisms remain poorly defined (Freeze et al, 2015). Other nodes of the proteostasis network such as autophagy and protein translation control have been also associated with neurodevelopmental disorders (Hetz, 2021). Interestingly, the activity of the UPR has been recently suggested to contribute to normal nervous system development, highlighting the regulation of brain architecture through the control of neurogenesis, cortical neurons migration, and neuronal differentiation (Martínez et al, 2018; Urra et al, 2018).
We previously identified dominant variants in PDIA3 as risk factors for amyotrophic lateral sclerosis (Gonzalez‐Perez et al, 2015). Here, we report a rare homozygous variant that causes severe syndromic ID in a single family. To validate PDIA3 as the culprit gene, we performed various biochemical, cellular, and whole organism experiments that contributed to further understand the biological significance of a major ER oxidoreductase in the nervous system. We provide compelling evidence supporting pathogenicity of the variant, which may act by both loss‐ and gain‐of‐function mechanisms.
Haploinsufficiency or dominant negative effects resulting from the C57Y variant are unlikely since heterozygous individuals are unaffected. The substitution of a catalytic cysteine strongly suggested loss of function as the pathogenic mechanism. On the other hand, PDIA3C57Y may acquire deleterious features that surpass a toxicity threshold for clinical manifestation upon expression of two mutant PDIA3 alleles. Loss‐ and gain‐of‐function mechanisms are not mutually exclusive. Indeed, our data show that the C57Y variant causes PDIA3 aggregation and abnormal interactions with CNX/CRT while greatly compromising enzymatic activity. Overexpression experiments both in vivo and in vitro also corroborate this notion. Zebrafish development and cognitive function of mice may be affected by gain‐of‐toxic properties of PDIA3C57Y. Alternatively, when overexpressed the mutant protein may outcompete endogenous PDIA3 for substrates and chaperones in the ER milieu, generating non‐productive enzymatic cycles that disturb folding of specific clients.
The accumulation of mutant PDIA3C57Y aggregates may be limited by interactions with calnexin or calreticulin directing their lysosomal degradation. Since alterations of PDIA3/CNX/CRT network may affect only a subset of ER cargo, no overt ER stress appears to result from mutant PDIA3 expression. Instead, the pathogenicity of PDIA3C57Y is associated with alteration in the expression of a defined cluster of proteins involved in cell adhesion, in addition to regulators of actin cytoskeleton organization. Remarkably, expression of PDIA3C57Y led to down‐regulation of members of the integrin family, a class of cell adhesion proteins that are obligate substrates for the PDIA3/CNX/CRT pathway in the ER (Jessop et al, 2007). Accordingly, inhibition of integrin signaling suppressed the effects of PDIA3 expression on neuritogenesis. Moreover, biochemical analysis indicates that mutant PDIA3 may disturb the folding and/or quality control of integrins. We reason that the failure to induce signaling events initiated by these adhesion molecules results in altered actin cytoskeleton dynamics, impacting neuronal connectivity and function. As a result, cells expressing PDIA3C57Y could lose the capacity to support tissue morphogenesis and synaptic activity, which are predicted to underlie the neurodevelopmental impairment observed in patients (Fig 7).
Figure 7. Proposed model of PDIA3C57Y role in ID.
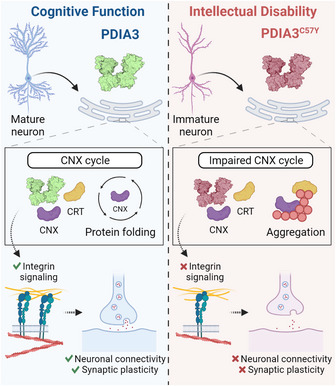
Dysfunction of protein folding and quality control by PDIA3/CNX/CRT disturbs integrin signaling, neuronal connectivity and synaptic plasticity that support cognitive function.
The malfunctioning of the PDIA3/CNX/CRT pathway disturbs the folding and quality control of glycosylated proteins (Jessop et al, 2007). In the nervous system, synaptic activity influences the glycoproteome in neuronal processes where the ER supports local protein synthesis and sorting (Jeyifous et al, 2009; Hanus et al, 2016). Moreover, disturbance of synaptogenesis and synaptic plasticity is emerging as a relevant pathological mechanism causing ID (van Bokhoven, 2011). Our findings provide mechanistic grounds on the contribution of ER proteostasis alterations to the etiology of human neurodevelopmental disorders. The MHC‐I pathway has been shown to regulate synaptic plasticity, promoting signaling from post‐ to presynaptic terminal (Shatz, 2009). MHC‐I has been also suggested to mediate crosstalk between immune cells and neurons, contributing to synaptic pruning by microglia, a possibility not yet proven (Elmer & McAllister, 2012). Whether mutant PDIA3 impairs MHC‐I activity in the nervous system leading to altered synaptic refinement and cognitive dysfunction in ID is an important point to be addressed in future studies. We are currently generating mutant Pdia3 knockin mice to explore this concept. Since mutant PDIA3‐linked ID involves synaptic dysfunction instead of untreatable brain malformations, our study offers a genetic and biological framework to disease modeling and discovery of novel therapeutic targets.
Materials and Methods
Family and data collection
The consanguineous family is from a rural area in Southern Punjab, Pakistan, and was initially ascertained in 2010. A total of six affected family members (two males and four females) were reported in two branches of the kinship. A detailed physical and medical examination of the surviving four affected individuals (a boy and his three first‐cousins once removed—siblings) at ages 12–18 years and 11 unaffected relatives was carried out with the help of physicians including neurologists at Nishtar Medical Hospital, Multan. Detailed phenotypic data of affected individuals and cranial MRI and roentgenograms of one of them were acquired. Anthropometric measurements of affected individuals and three unaffected relatives were obtained.
Affected individuals were evaluated for ID by the criteria proposed by the American Psychiatric Association (2013). The classification into four types, namely mild, moderate, severe, and profound, is according to increasing severity and based on limited mental ability and extent of inadequacy and support required in conceptual, social, and practical domains of daily life.
The study protocol was approved by the Ethical Review Committee of Quaid‐i‐Azam University and the Boğaziçi University Institutional Review Board for Research with Human Participants. Informed consent was obtained from the family. All material was acquired according to the Helsinki II declaration.
Gene localization
SNP genome scan data were generated for siblings 401 and 402 (affected and unaffected, respectively) plus their unaffected multiple‐second‐cousin 406 and her three affected and one unaffected sib (502, 503, 504, and 505; Fig 1A) using Illumina Human OmniExpress‐24 BeadChip. Multipoint LOD score calculations were performed to search for a unique region where the homozygous SNP genotype was shared by affected individuals only, and thus homozygosity was possibly due to identity by descent. Autosomal recessive inheritance, full penetrance, and a disease allele frequency of 0.001 were assumed. The analysis was in three steps (Linkages A, B, and C; details are given below) and assuming simplified partial pedigrees in each, as using all available SNP data together would have exceeded the capacity of the programs. Firstly, analysis A was performed and detected 10 regions yielding maximal LOD scores > 2.8, on chromosomes 1, 6, 7, 8, 13, 15, 17, 19, and 21. In analysis B, the regions on chromosomes 1, 8, 13, 17, and 21 were eliminated due to decreased LOD scores. Region on chromosome 7 was also eliminated due to small size. We investigated the genotypes in the remaining homozygous regions and found that in the two regions on chromosome 15, but not in regions on chromosomes 6 and 19, exclusively patients shared the homozygosity. We performed detailed linkage analysis C for those regions (15q15.1‐21.1 and 15q22.31) using all SNP markers and a simplified pedigree that included all participants with SNP genotype data but assumed closer kinship. Both regions yielded high LOD scores, but the latter region was excluded because exome data were not homozygous. We thus identified 15q15.1‐21.1, the only region where patients exclusively shared the homozygosity, as the disease gene locus.
Linkage analysis A
Markers were selected with 0.01‐Mb spacing, and sets of 30 markers were used in Allegro implemented in easyLINKAGE (v.5.08; Appendix Fig S2A). Regions yielding LOD scores > 2.8 and with sizes > 500 kb were evaluated further.
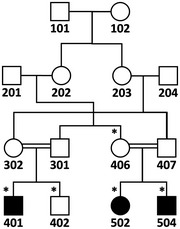
Simplified pedigree used in linkage analysis A. SNP genotype data of individuals marked with asterisks were used.
Linkage analysis B
This analysis was performed for the larger core family for chromosomes that yielded high multipoint LOD scores (> 2.8) in linkage analysis A, i.e., chromosomes 1, 6, 7, 8, 13, 15, 17, 19, and 21 (Appendix Fig S2B), again using easyLINKAGE. Chromosomes 1, 8, 13, 17, and 21 were eliminated because the LOD scores were lower than those in linkage analysis A. The remaining regions on chromosomes 6, 7, 15, and 19 were investigated on MS Excel for possible identity by descent from a recent ancestor and the extent of shared homozygosity in affected individuals. The only region on chromosome 7 and four of the six regions on chromosome 15 were eliminated due to small size (< 500 kb) of the shared homozygosity in patients, hypothesizing that they were due to common haplotypes in the village. For the genotypes in the remaining shared homozygosity regions, in the two regions on chromosome 15, but not in the regions on chromosomes 6 and 19, only patients shared the homozygosity. As a result, all regions were eliminated except for the two at 15q15.1‐21.1 and 15q22.31.
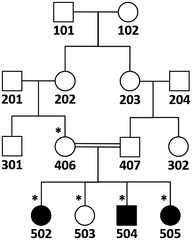
Simplified pedigree used in linkage analysis B. SNP genotype data of individuals marked with asterisks were used.
Linkage analysis C
This final linkage analysis was performed for regions 15q15.1‐21.1 and 15q22.31 not eliminated in linkage analysis B, including all available SNP data and with 10 marker sets (Appendix Fig S2C). Program SimWalk was used so that all samples could be included. The program can handle a larger family albeit with very slow pace. Both regions yielded maximal LOD scores > 3.8. At 15q15.1‐21.16, maximal homozygosity was approximately 2.2 Mb, between rs17767270 (nucleotide 42898612) and rs1288092 (nucleotide 45090821). At 15q22.31, maximal homozygosity was 665 kb. Exome sequence data of affected individual 502 generated later revealed that all three rare variants in the latter region were heterozygous, indicating that in the region, the SNPs used for genotyping were non‐informative and the homozygosity was not due to identity by descent from a recent ancestor. Hence, we eliminated the latter locus and identified the disease locus as the 2.2‐Mb region at 15q15.1‐21.16. The maximal LOD score was 3.85 for the simplified pedigree.
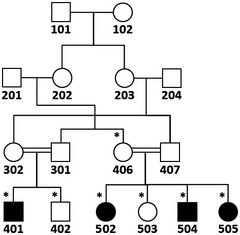
Simplified pedigree used for linkage analysis C. SNP genotype data of individuals marked with asterisks were used.
Sequence analyses
DNA sample of affected individual 502 was subjected to exome sequencing. The captured exome library was created using the Agilent SureSelect Target Enrichment System and sequenced on the Illumina HiSeq2000 platform. Raw reads generated by sequencing were aligned to the GRCh37/hg19 using BWA‐0.7.12‐r1039 (Burrows‐Wheeler alignment; http://bio‐bwa.sourceforge.net/), resulting in a mean depth of 42X for disease locus and 33X for all targeted regions. Variant calling was performed with SAMtools‐0.1.14 (Sequence Alignment/Map tools; http://samtools.sourforge.net) and variant annotation with ANNOVAR (Annotate Variation; http://www.openbioinformatics.org/annovar/). Of the exonic variants in the exome file, we selected those with frequencies < 0.01 in all populations reported in public databases and allele read to total read ratios > 0.6 (considered to be homozygous). We evaluated for shared homozygosity the loci of the genes harboring those variants. PDIA3 was the only gene in a region where homozygous genotype was shared exclusively by all affected individuals (Dataset EV2). The variant was validated by Sanger sequencing using primers 5′‐GGAAGTGTCTACTAGCTCAAAGG‐3′ and 5′ ‐CCATGTAACAAAGCTGAGACAAC‐3′. Its segregation with the disease was ascertained by single‐strand conformational polymorphism analysis and its novelty was interrogated in databases dbSNP and gnomAD that contain 10,000 Pakistani samples. The variant has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) with Submission ID SUB3488668. Online computational algorithms PolyPhen2 (Polymorphism Phenotyping‐2; http://genetics.bwh.harvard.edu/pph), Mutation Taster (http://www.mutationtaster.org/), SIFT (Sorting Intolerant From Tolerant; http://blocks.fhcrc.org/sift/SIFT.html), PROVEAN (Protein Variation Effect Analyzer; http://provean.jcvi.org/index.php) and UMD‐Predictor (http://umd‐predictor.eu/), M‐CAP (http://bejerano.stanford.edu/mcap/), REVEL (https://sites.google.com/site/revelgenomics/downloads), and CADD (https://cadd.gs.washington.edu/snv) were used to predict whether the single‐candidate variant (PDIA3 c.170G>A; NM_005315) possibly underlying the disease is damaging to the protein.
No deletion or duplication shared by patients only was detected with cnvPartition (v3.2.0) CNV Analysis Plug‐in for Genome Studio (http://support.illumina.com/downloads/cnvpartition plug‐in_v320_for_genomestudio.ilmn) (Hoffmann & Lindner, 2005).
Molecular modeling
Structural analysis was performed using the SWISSMODEL server (http://swissmodel.expasy.org/) and Chimera1.11 imaging software. The PDB structure 3f8u (Dong et al, 2009) was used to model the structure of mutant PDIA3C57Y.
Plasmid construction and site‐directed mutagenesis
pcDNA3.1/V5 encoding human WT PDIA3 was previously described (Jessop et al, 2007). Constructs for expression of PDIA3C57Y, PDIA3R282A, and PDIA3C57Y/R282A were generated by the QuikChange XL Site‐Directed Mutagenesis Kit (Agilent Technologies) using the protocol recommended by the manufacturer. Constructs for expression of GFP‐tagged PDIA3 were generated by restriction‐based cloning. All variants were validated by Sanger sequencing using the following primers: for PDIA3 (p.Cys57Tyr), 5′‐GTGTGGCGCTGCTTCTT‐3′, and 5′‐TGGCAGTGCAATCAACCTTT‐3′; for PDIA3 (p.Arg282Ala), 5′‐TGGTATCTGCCCTCACATGAC‐3′ and 5′‐GCTGAACTTGTGGCCGTTTA‐3′. The constructs for expression of integrin paralogs were obtained from Addgene: α5‐integrin‐GFP was a gift from Rick Horwitz (Laukaitis et al, 2001), β2‐integrin‐YFP from Timothy Springer (Kim et al, 2003), β3‐integrin‐YFP from Jonathan Jones (Tsuruta et al, 2002), and β5‐integrin‐2xGFP from Staffan Stromblad (Lock et al, 2018).
Zebrafish experiments
Embryos of zebrafish (Danio rerio) were obtained by natural spawning, raised at 28°C in embryo medium, and staged according to morphology (Kimmel et al, 1995) and age (hours and days post‐fertilization; hpf and dpf). Zebrafish lines used were WT Tübingen, Tg(Huc:mCherry) to visualize neurons in the central nervous system, Tg(sox17:GFP) to visualize dorsal forerunner cells (DFC), and Tg(actb1:mCherry‐utrCH) to visualize actin in the whole embryo. All animal protocols were approved by the Bioethics Committee of the Faculty of Medicine, University of Chile.
For in vitro synthesis of capped RNA with the T7 mMESSAGE mMACHINE system (Ambion), pcDNA3.1/V5 constructs coding for human PDIA3 and PDIA3C57Y were linearized with StuI. For overexpression experiments, zebrafish embryos were microinjected at one‐cell stage with sense mRNA diluted in water using a Picospritzer III microinjector. After testing different doses, the final quantity of mRNA used was 70 pg/embryo. Mock group corresponds to non‐injected embryos. Viability was evaluated at 48 hpf and the global overexpression phenotypes were analyzed at 48 and 96 hpf in a SMZ‐1000 Nikon stereomicroscope as described (Rojas‐Rivera et al, 2012; Woehlbier et al, 2016).
For determination of the extent of cell movements during gastrulation, mRNA‐injected WT Tübingen embryos were imaged using a SMZ‐1000 Nikon stereomicroscope and the progression of the deep blastoderm, the head‐to‐tail angle, and the width of the first three somites were measured at 9, 12.5, and 13 hpf as previously described (Urra et al, 2018).
For immunofluorescent staining of axons, mRNA‐injected WT Tübingen embryos were fixed at 96 hpf in TCA for 3 h and washed three times in phosphate‐buffered saline (PBS) for 10 min. After 8 min of 1× trypsin treatment, larvae were incubated with 1:1,000 dilution of mouse anti‐tubulin acetylated (Sigma), followed by incubation with 1:200 dilution of Alexa 488‐conjugated goat anti‐mouse antibody (Invitrogen). Fluorescence micrographs of fixed embryos were taken using a Leica LSI confocal microscope. Images were processed with ImageJ (http://rsb‐web.nih.gov/ij/) and Adobe Photoshop CS3 software (Adobe).
For in vivo detection of apoptotic cells, Tg(Huc:mCherry) embryos were injected at one‐cell stage with 70 pg of mRNA coding for PDIA3 or PDIA3C57Y, and maintained for 72 h in E3 medium at 28.5°C. Then, larvae were incubated with E3 medium containing 2 mg/ml acridine orange for 30 min at 28.5°C using 24‐well plates (6 embryos per well). After eight 5‐min washes with E3 medium, embryos were anesthetized with tricaine (Sigma) and fluorescence micrographs taken with Leica LSI confocal microscope using exactly the same exposure, gain, and magnification. Images were processed with ImageJ (http://rsb‐web.nih.gov/ij/) and Adobe Photoshop CS3 software (Adobe).
For evaluation of DFC clustering, Tg(sox17:GFP) was injected at one‐cell stage with 70 pg of mRNA coding for PDIA3 or PDIA3C57Y and fixed in 4% paraformaldehyde in PBS at 75% epiboly as previously described (Oteiza et al, 2010). Fixed embryos were incubated for 2 h in Hoechst 33342 to stain nuclei. Fluorescence micrographs of fixed embryos were taken using a Leica TCS LSI macro zoom confocal microscope. Images were processed and analyzed with ImageJ (http://rsb‐web.nih.gov/ij/).
For measurement of actin cytoskeleton in vivo, Tg(actb1:mCherry‐utrCH) embryos were injected at one‐cell stage with 70 pg of mRNA coding for PDIA3 or PDIA3C57Y, placed in custom‐made chambers 8 hpf, and imaged on a Volocity ViewVox spinning disc (Perkin Elmer) coupled to a Zeiss Axiovert 200 confocal microscope using a Pan‐Apochromatic 40×/1.2W objective. To quantify circularity, aspect ratio, and pixel signal intensity, cells were manually segmented using Fiji software.
For mosaic expression of PDIA3, blastomeres of WT embryos at four‐cell stage were microinjected with 80 pg of constructs for expression of GFP‐tagged WT PDIA3 or PDIA3C57Y using a Picospritzer III microinjector. Fluorescence signal was analyzed in embryos at 48 hpf and the micrographs were acquired using a Zeiss Axiovert 200 confocal microscope.
For skeletal staining, mRNA‐injected larvae were fixed at 6 dpf with 4% formaldehyde in 0.1 M sodium phosphate buffer for 2 h and stored in 70% methanol. The protocol for simultaneous bone and cartilage staining was adapted from that previously described (Walker & Kimmel, 2007). Briefly, larvae were rinsed in 50% ethanol and subsequently stained with 0.2 mg/ml Alcian Blue 8 GX (Sigma) in 70% ethanol/80 mM MgCl2. After washes in 0.02% Triton X‐100, embryos were bleached in 1% H2O2/1% KOH for 30 min, washed in a saturated sodium tetraborate solution, and digested for 1 h in 1 mg/ml trypsin (Sigma) in 60% saturated sodium tetraborate. Bones were stained with 0.04 mg/ml Alizarin Red S (Sigma) in 1% KOH. Destaining was carried out in an increasing glycerol series (10, 30, and 70%) and specimens were stored at 4°C in 70% glycerol. Micrographs were acquired using MZ‐1000 Nikon stereomicroscope.
Primary culture of hippocampal neurons
Hippocampal neuronal cultures were done as previously described (Kaech & Banker, 2006). Briefly, hippocampi were dissected from Mus musculus embryos at E18.5 and cells disaggregated with trypsin and plated on coverslips coated with poly‐l‐lysine. Neurons were maintained in Neurobasal medium with B27 and N2 supplements and co‐transfected on day 0 in vitro (DIV) with constructs for expression of green fluorescent protein (pEGFP‐N1) and PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5) in 1:4 ratio (0.8 μg of total DNA) using Lipofectamine 2000 according to manufacturer's instructions (Invitrogen). For axonal length measurements, neurons were fixed and stained on 3 DIV with 1:500 dilution of mouse anti‐Tau‐1 (Millipore) followed by incubation with 1:500 dilution of Alexa 546‐conjugated donkey anti‐mouse antibody (Invitrogen). Minor neurite length was measured using GFP signal. Fluorescence micrographs were acquired using Zeiss LSM 710 confocal microscope. Quantifications were performed with ImageJ (http://rsb‐web.nih.gov/ij/) software as previously described (Villarroel‐Campos et al, 2016).
Preparation of adeno‐associated virus
Adeno‐associated virus (AAV) vectors carry a transgene cassette composed of the chicken β‐actin (CBA) promoter driving human PDIA3 or mutant PDIA3 (p.Cys57Tyr) expression followed by an IRES‐EGFP cassette and a bovine growth hormone (BGH) polyadenylation signal. The open reading frame is preceded by a Kozak sequence. The PDIA3 protein contains a C‐terminus V5 tag. All constructs were generated by PCR amplification and validated by Sanger sequencing prior to production. A control vector for expression of EGFP only (Mock) was included. The AAV serotype 9 vector stocks were produced at the University of Massachusetts Medical School Horae Gene Therapy Center Vector Core as previously described (Ayuso et al, 2010). Vector titers were determined by quantitative PCR for viral genomes using the following primers and a probe specific for the BGH polyA: Primer1: 5′‐GCCAAAAATTATGGGGACAT‐3′; Primer2: 5′‐ATTCCAACACACTATTGCAATG‐3′; and Probe: 6FAM‐ATGAAGCCCCTTGAGCATCTGACTTCT‐TAMRA.
Mouse experiments
Male C57BL/6 mice were employed, maintained in a facility with 12 h light/dark cycle at 25°C with food and water provided ad libitum. All animal procedures were approved by the Bioethics Committee of the Faculty of Medicine, University of Chile. Young mice at P55 received bilateral stereotaxic injections of AAV9 (8.0 × e + 009 particles/injection) in the hippocampi using the coordinates anterior–posterior: −1.8, dorsal–ventral: +1.8, and medial–lateral: ±1.8. The mice were injected with either control AAV9 vector (Mock) or AAV9 vectors for expression of PDIA3 or PDIA3C57Y.
The injected mice were submitted to behavioral evaluation at 4 months of age by open‐field and Barnes maze. In the open field, mice were placed on a 40 × 40 cm arena with 40‐cm‐high acrylic glass walls and spontaneous activity recorded for 15 min as previously described (Chew et al, 2015). The arena and walls were cleaned between measurements of different animals. The recorded video was analyzed for quantification of multiple parameters such as total distance traveled and total distance spent on the center using ANY‐maze software (Stoelting Co). In the Barnes maze, mice were placed on a circular arena (92 cm of diameter and 72.5 cm above the floor) containing 20 equally spaced holes on the edge (5 cm diameter and 7.5 cm apart from each other). Visual cues were placed around the arena for spatial navigation. Before each trial, the animal was placed for 10 s inside a cylindrical container in the center of arena. Upon removal of the cylinder, the mice were exposed to an aversive sound (beep of a timer). The time the animal took to find and enter the target hole leading to an escape box was recorded as primary and total latency, respectively. The protocol consisted of three phases: adaptation, acquisition, and test. During adaptation, the mice were gently guided into the escape box by the experimenter and allowed to stay there for 2 min. The acquisition phase consisted of four trials per day, for four consecutive days (day 1–4). During the acquisition phase, each animal was allowed to explore the maze for 3 min or until entering the escape box. After the mouse entered the box, the sound was immediately turned off and the animal allowed to stay there for 1 min. If the mouse did not enter the escape box within 3 min, the experimenter guided it into the box, allowing it to stay there for 1 min. Each mouse was then placed in its cage until the next trial (15 min of interval between trials). The arena and walls were cleaned between measurements of different animals. Primary and total latency were measured. The test was conducted at days 5 and 12 to assess short‐ and long‐term spatial memory, respectively. The target hole without the escape box was maintained in the same position used in the acquisition period. The test was performed for 90 s and the latency recorded. The primary latency from days 1 to 4 was measured as learning curve, at day 5 as short‐term memory, and at day 12 as long‐term memory.
For histological and biochemical analysis of AAV9 transduction and PDIA3 expression, brain tissue was collected at 6 months of age. Briefly, mice were anesthetized with ketamine/xylazine and perfused with cold saline. The brain was removed from the skull and separated into two hemispheres. The hippocampus of the left hemisphere was dissected out and stored frozen at −80°C until analysis. The right hemisphere was post‐fixed in 4% paraformaldehyde in PBS overnight at 4°C, followed by cryopreservation in 30% sucrose in PBS and inclusion in freezing medium (OCT, TissueTek). Fifty‐µm‐thick sagittal sections were obtained free floating in a Leica cryostat for immunostaining using anti‐GFP and anti‐V5 tag antibodies. Five serial sections every 200 µm were stained per animal. Briefly, the tissue was washed three times in PBS, blocked using Mouse‐on‐Mouse kit according to the protocol recommended by the manufacturer (Vector Laboratories), and incubated free‐floating overnight at RT with 1:1,000 dilution of rabbit anti‐GFP (abcam) and 1:250 dilution of mouse anti‐V5 (Invitrogen) antibodies. Then, sections were washed three times with PBS and incubated for 2 h at RT with 1:1,000 dilution of Alexa 488‐conjugated goat anti‐rabbit (Invitrogen) and Alexa 568‐conjugated goat anti‐mouse (Invitrogen) secondary antibodies using Hoechst 33342 for nuclear staining. Fluorescence micrographs were acquired using a Nikon C2+ confocal microscope.
Preparation of hippocampal slices for electrophysiology
Three‐hundred‐μm‐thick transversal slices were prepared from injected mice at 6 months of age. Animals were deeply anesthetized with isoflurane and transcardially perfused with ice‐cold oxygenated (95% O2, 5% CO2) N‐methyl‐d‐glucamine (NMDG)‐based solution for a better cell preservation in slices (Ting et al, 2014). After perfusion, the brain was quickly removed and submerged in ice‐cold oxygenated (95% O2, 5% CO2) NMDG solution containing (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 sodium ascorbate, 3 Na‐pyruvate, 0.5 CaCl2, 10 MgSO4, pH 7.4 adjusted with HCl. Slices were cut in this solution at a thickness of 300 μm using a Leica vibratome. After slicing, the tissue was incubated for 10 min at 33°C in NMDG buffer solution. Then, slices were transferred into a storage chamber kept at RT in artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 glucose, 2 CaCl2 and 2 MgSO4, pH 7.4 (95% O2, 5% CO2).
Field excitatory postsynaptic potentials
Field excitatory postsynaptic potentials (fEPSPs) were recorded as previously described (Rozas et al, 2012, 2015). Briefly, the fEPSPs were evoked by applying electrical stimulations from Schaeffer collateral commissural fibers and recorded on the stratum radiatum of the CA1 region. Test pulses were applied every 15 s. A baseline was established with test pulses adjusted to evoke 50% of the maximal response. After recording a stable baseline for at least 20 min, long‐term potentiation (LTP) was induced with theta burst stimulation (TBS, consisting of five trains of 10 bursts at 5 Hz each, one burst, and four pulses at 100 Hz). In all experiments, the fEPSP recordings were continued for 60 min after applying TBS. Electrophysiological data are presented as mean ± s.e.m. and normalized relative to the baseline (average slope of fEPSPs measured before the TBS protocol). LTP was measured during the final 20 min of the recording and presented as the averaged percentage of baseline. Statistical significance for mean differences between experimental groups was assessed using Wilcoxon–Mann–Whitney two‐sample rank test.
Quantitative proteomic analysis
Hippocampal tissue of mice transduced with AAV9 (Mock, PDIA3, and PDIA3C57Y) was homogenized in TEN buffer (10 mM Tris‐HCl pH 8.0, 1 mM EDTA, 100 mM NaCl, 1% NP‐40, and protein inhibitor cocktail [Roche]). For each sample, 20 μg of lysate was precipitated with chloroform/methanol. Samples for mass spectrometry analysis were prepared as described (Plate et al, 2016). Air‐dried pellets were resuspended in 1% RapiGest SF (Waters) and diluted to final volume in 100 mM HEPES (pH 8.0). Proteins were reduced with 5 mM Tris(2‐carboxyethyl)phosphine hydrochloride (Thermo Fisher) for 30 min and alkylated with 10 mM iodoacetamide (Sigma Aldrich) for 30 min at room temperature in the dark. Proteins were digested for 18 h at 37°C with 0.5 μg trypsin (Promega). After digestion, the peptides from each sample were reacted for 1 h with the appropriate tandem mass tag (TMT) isobaric reagent (Thermo Fisher) in 40% (v/v) anhydrous acetonitrile and quenched with 0.4% ammonium bicarbonate for 1 h. Samples with different TMT labels were pooled and acidified with 5% formic acid. The groups were divided into three mass spectrometry runs to fit the TMT six‐plex channels, with five samples per group and a common reference sample. Acetonitrile was evaporated on a SpeedVac and debris removed by centrifugation for 30 min at 18,000 g. MudPIT microcolumns were prepared as described (Ryno et al, 2014). LC‐MS/MS analysis was performed using a Q‐Exactive HF mass spectrometer equipped with an Ultimate 3000 nLC 1000 (Thermo Fisher). MudPIT experiments were performed by 10 μl sequential injections of 0, 10, 20, 30, …, 100% buffer C (500 mM ammonium acetate in buffer A) and a final step of 90% buffer C/10% buffer B (100% acetonitrile, 0.1% formic acid, v/v/v) and each step followed by a gradient from buffer A (95% water, 5% acetonitrile, and 0.1% formic acid) to buffer B. Electrospray was performed directly from the analytical column by applying a voltage of 2 kV with an inlet capillary temperature of 275°C. Data‐dependent acquisition of MS/MS spectra was performed with the following settings: eluted peptides were scanned from 300 to 1,800 m/z with a resolution of 120,000. The top 15 peaks for each full scan were fragmented by HCD using a normalized collision energy of 30%, isolation window of 2.0 m/z, a resolution of 30,000, ACG target 1e5, maximum IT 60 ms, and scanned from 100 to 1,800 m/z. Dynamic exclusion was set to 10 s. Peptide identification and protein quantification were performed using Proteome Discoverer 2.2 (Thermo Fisher). Spectra were searched using SEQUEST against a UniProt mouse proteome database. The database was curated to remove redundant protein and splice isoforms, and common contaminants were added. Searches were carried out using a decoy database of reversed peptide sequences using Percolator node for filtering and the following settings: 50 ppm peptide precursor tolerance, 6 amino acid minimum peptide length, trypsin cleavage (unlimited missed cleavage events), static Cys modification of 57.0215 (carbamidomethylation), static N‐terminal and Lys modification of 229.1629 (TMT‐6‐plex), FDR 0.01, and two peptide IDs per protein. Normalization of TMT reporter ion intensities was carried out based on total peptide abundance in each channel, and subsequently, TMT ratios for each identified protein were calculated in reference to a common reference sample. An inconsistency was detected in quantification of major differentially regulated proteins in two samples of the PDIA3 group allocated in the first mass spectrometry run, which was then excluded from further analysis. The whole mass spectrometry proteomics dataset has been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with accession number PXD026507. Finally, the reference‐normalized TMT intensities were compared among Mock (n = 3), PDIA3 (n = 3), and PDIA3C57Y (n = 4) transduced samples and significance assessed by a two‐tailed unpaired t‐test using the FDR approach (Benjamini et al, 2006) and Q = 1% in Graphpad Prism. Enrichment analysis of most significant alterations between groups was performed in EnrichR platform using gene ontology (GO) database (Chen et al, 2013; Kuleshov et al, 2016). Protein–protein interaction network was generated in STRING v.11 using Pdia3 and genes related to Focal Adhesion GO term (Szklarczyk et al, 2015).
Cell culture experiments
Mouse embryonic fibroblasts (MEF), NSC‐34 and HEK cells were grown in proliferation medium (Dulbecco's modified Eagle's medium–high glucose [DMEM; Gibco] supplemented with 10% fetal bovine serum [FBS], 100 units/ml penicillin, and 0.1 mg/ml streptomycin [Invitrogen]) at 37°C under 5% CO2. Routinely, cells were transfected with Effectene transfection reagent (Qiagen) using the protocol recommended by the manufacturer. For generation of NSC‐34 stable cell lines expressing WT or mutant PDIA3, cells were transfected with constructs for expression of PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5) and selected with 250 μg/ml G‐418 (Sigma Aldrich).
For neuritogenesis assay, stable NSC‐34 cell lines overexpressing PDIA3 or PDIA3C57Y, or mock control, were seeded on 25‐mm coverslips in Neurobasal medium with 2% B27 supplement, 2 mM glutamine, 100 units/ml penicillin, and 0.1 mg/ml streptomycin (Invitrogen). Cells were fixed every 24 h, stained with rhodamine‐conjugated phalloidin, and imaged by automated microscopy with high‐content screening platform (Cellomics). The software Neuronal Profiling (Cellomics) was used to quantify neurite number and length. For assessing integrin signaling, cells were treated 24 h after seeding with 40 nM Echistatin, fixed at 72 h of differentiation, stained with FITC‐conjugated phalloidin, and scanned by automated microscopy (Cellomics). Neurite length was determined using Simple Neurite Tracer plugin (Fiji software). In another experimental setting, NSC‐34 cells were co‐transfected with constructs for expression of green fluorescent protein (pEGFP‐N1) and PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5) in 1:9 ratio (μg of DNA). Cells were split 24 h post‐transfection. About 50,000 cells were re‐seeded in six‐well format using DMEM medium with 1% FBS. After attachment to the plate for 4 h, cells were washed once with 1 ml PBS pH 7.4, and the medium was changed to Neurobasal with 2% B27 supplement, 2 mM glutamine, 100 units/ml penicillin, and 0.1 mg/ml streptomycin (Invitrogen). Following 48 h of incubation, a minimum of six pictures were taken per condition per experiment using an Olympus IX71 fluorescence microscope. Neuritogenesis was quantified by analyzing the length of the largest neurite of each EGFP‐positive cell per condition using ImageJ software (NIH, Bethesda, Maryland, USA).
For cell viability assay, NSC‐34 cells were transfected with constructs for expression of PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5). Cells were split 24 h post‐transfection. About 10,000 cells per well were re‐seeded in 96‐well format in proliferation medium. Toxicity of ER stress inducer tunicamycin was measured using MTT assay (Promega) after 24 h of drug treatment.
For reverse transcriptase PCR, cDNA was synthesized using SuperScriptIII following manufacturer's instructions (Life Technologies) and conventional PCR performed using GoTaq Green Master Mix (Promega). Actin was employed as housekeeping control. Primer sequences: PDIA3: forward 5′‐GTCATAGCCAAGATGGATGCC‐3′ and reverse 5′‐TTAATTCACGGCCACCTTCATA‐3′; Xbp1: forward 5′‐ ACACGCTTGGGAATGGACAC‐3′ and reverse 5′‐CCATGGGAAGATGTTCTGGG‐3′; and Actin: forward 5′‐CTCAGGAGGAGCAATGATCTTGAT‐3′ and reverse 5′‐TACCACCATGTACCCAGGCA‐3′.
For immunofluorescent staining, NSC‐34 cells were transfected with constructs for expression of PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5). Cells were split 24 h post‐transfection. About 30,000 cells were re‐seeded on glass coverslips coated with poly‐l‐lysine. After 24 h, cells were fixed with 4% paraformaldehyde in PBS, pH 7.4, for 20 min at room temperature (RT), and permeabilized in 0.3% Triton X‐100 in PBS for 10 min at RT. After washing in PBS, cells were incubated in blocking solution (0.5% bovine serum albumin [BSA] and 10% fetal bovine serum [FBS] in PBS) for 15 min at RT. Then, cells were incubated with 1:250 dilution of mouse anti‐V5 antibody (Invitrogen), combined or not with 1:250 dilution of rabbit anti‐BiP (abcam), in 0.5% BSA in PBS overnight at 4°C, followed by incubation with 1:500 dilution of Alexa 488‐conjugated goat anti‐mouse IgG (Invitrogen), and combined or not with 1:500 dilution of Alexa 568‐conjugated goat anti‐rabbit IgG (Invitrogen), for 2 h at RT using Hoechst 33342 for nuclear staining. Fluorescence micrographs were taken using a Nikon C2+ confocal microscope. For imaging of NSC‐34 live cells transfected with constructs for expression of PDIA3‐GFP or PDIA3C57Y‐GFP, micrographs were acquired using an Olympus IX71 fluorescence microscope.
Live cell imaging of actin cytoskeleton
Pdia3 WT and knock‐out (KO) MEF were transfected with a construct for expression of EGFP‐Lifeact, seeded onto fibronectin‐coated 25‐mm coverslips, and imaged in complete medium supplemented with 10 mM HEPES using a Nikon spinning disk confocal microscope with a 40×/1.3 NA oil‐immersion objective lens. Fluorescence micrographs were acquired every 12 s for 10 min. The number of lamellipodia per cell (lamellipodia index) was determined manually as previously described (Urra et al, 2018). For analysis of lamellipodia protrusion and retraction velocity and area, images were automatically analyzed using the ADAPT plugin (Fiji software). The merged images of total protrusion (green) and retraction (red) area and a projection of average slices were shown.
To evaluate the effect of PDIA3C57Y on lamellipodia parameters, MEF Pdia3 KO was co‐transfected with constructs for expression of EGFP‐Lifeact and PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5) in 1:9 ratio (μg of DNA). Lamellipodia index and protrusion/retraction velocity and area were determined as described above.
Biochemical assays
Cells and tissue homogenates were prepared in TEN buffer described above and sonicated for 15 s at 30% amplitude (QSonica). Samples treated or not with dithiothreitol (DTT, final concentration of 0.1 M), diluted in Laemmli sample buffer and loaded onto 10% SDS–polyacrylamide gels, transferred to PVDF membrane, and analyzed by Western blot. The following antibodies and dilutions were used: mouse anti‐V5 1:2,000 (Invitrogen), mouse anti‐β‐actin 1:20,000 (MP Biomedicals), rabbit anti‐calnexin 1:2,000 (Stressgen), mouse anti‐calreticulin 1:2,000 (Stressgen), rabbit anti‐calponin‐3 1:500 (abcam), rabbit anti‐Psmb10 (abcam), mouse anti‐GFP 1:1,000 (Santa Cruz Biotechnology), rabbit anti‐GFP 1:1,000 (abcam), rabbit anti‐PDIA3 1:2,000 (Santa Cruz Biotechnology), goat anti‐rabbit HRP conjugate 1:2,000 (Invitrogen), goat anti‐mouse HRP conjugate 1:2,000 (Invitrogen), and anti‐mouse TrueBlot (Rockland).
Protein extracts were also analyzed by filter trap. Briefly, samples treated or not with DTT were diluted to a final concentration of 0.25 mg/ml in PBS pH 7.4 containing 1% SDS, filtered through a cellulose acetate membrane with a pore size of 0.22 μm using a dot‐blot apparatus (Bio‐Rad) and analyzed by immunoblot as previously described (Medinas et al, 2018).
Immunoprecipitation
Immunoprecipitation was performed as previously described (Woehlbier et al, 2016). HEK cells were transiently transfected with constructs for expression of PDIA3‐V5 or PDIA3C57Y‐V5, or empty vector (pcDNA3.1/V5). After 48 h, cells were collected and washed once with 1 ml PBS, pH 7.4. Cells were resuspended in 500 μl NP‐40 buffer (0.2% NP‐40, 50 mM Tris‐HCl pH 7.5, 150 mM NaCl, and protease inhibitor cocktail [Roche]) and incubated overnight at 4°C. After vortexing for 20 s, cell lysates were centrifuged at 10,000 g for 5 min at 4°C. The supernatant was incubated with anti‐V5 antibody beads (V5‐tagged Protein Purification Kit, MBL International) on a tube rotator for 4 h at 4°C. Then, beads were washed three times with NP‐40 buffer, increasing the salt concentration to 500 mM NaCl in the final wash. Protein complexes were eluted by adding V5 peptide (V5‐tagged Protein Purification Kit, MBL International) for 10 min at RT. Input and eluate were analyzed by Western blot.
Immunoprecipitation was also performed from crude cell lysate for analysis of aggregates in eluate fraction by native filter trap with samples diluted in PBS pH 7.4 containing 0.2% NP‐40, filtered through a cellulose acetate membrane with a pore size of 0.22 μm using a dot‐blot apparatus (Bio‐Rad), and analyzed by immunoblot.
Protein expression and purification
Mature human PDIA3 (ERp57) and Calreticulin P‐domain were expressed and purified as described previously (Woehlbier et al, 2016). Eluted fractions from ion exchange chromatography containing pure protein were pooled, concentrated, and buffer exchanged into 20 mM sodium phosphate pH 7.3 with Amicon stirred cell concentrator using Millipore Ultracel 10 kDa ultrafiltration disc. The concentrations of purified proteins were calculated based on their calculated molar extinction coefficients. Site‐directed mutagenesis was performed according to instructions of the Pfu Turbo DNA polymerase kit (Agilent, Santa Clara, CA, USA) and the constructs were checked by sequencing. Mutant PDIA3C57Y was expressed and purified as per the WT protein.
Biophysical analysis of purified protein
Far UV circular dichroism (CD) spectra were performed as described previously (Woehlbier et al, 2016). Limited proteolysis was also carried out as described previously (Woehlbier et al, 2016) using 10 µM of PDIA3 or PDIA3C57Y in 20 mM sodium phosphate buffer pH 7.3 in the presence of 10 mM β‐mercaptoethanol and 0 or 10 µg/ml Proteinase K.
Dynamic light scattering analysis to determine the molecular weight and polydispersity of PDIA3 and PDIA3C57Y was performed using DynaPro PlateReader II instrument (Wyatt Technology). A sample of the protein (0.5 mg/ml) in 20 mM sodium phosphate buffer pH 7.3 was centrifuged for 30 min at 16,000 g and transferred to four parallel wells on 356‐well plate (30 µl/well). Acquisition time was 5 s with 20 repeats per sample at 22°C.
To examine the potential formation of mixed disulfide bonds, PDIA3 and PDIA3C57Y were incubated with 20 mM N‐ethylmalemide to prevent thiol‐disulfide rearrangement and then analyzed by reducing (with addition of 5% β‐mercaptoethanol) or non‐reducing SDS–PAGE with Coomassie blue staining.
Recombinant PDIA3 activity measurements
The thiol oxidase activity of PDIA3 and PDIA3C57Y was determined as described previously (Ruddock et al, 1996). Time‐dependent changes in the fluorescence intensity (excitation at 280 nm and emission at 350 nm) were monitored on FluoroMax‐4 (HORIBA Instruments, Edison, NJ, USA) in McIlvaine buffer pH 7.0 with 0.2 µM enzyme, 2 mM GSH, 0.5 mM GSSG, and 5 µM of NRCSQGSCWN as substrate peptide for 15 min. Control reactions without addition of enzyme were performed (blank). The catalyzed reaction rate was determined from exponential fit of the data followed by subtraction of blank value. The relative activities of WT and mutant PDIA3 were presented.
The in vitro interaction of PDIA3 and PDIA3C57Y with CRT‐P domain was studied with bio‐layer interferometry (BLI) technology using Octet RED384 system (ForteBio). CRT P‐domain was biotinylated according to manufacturer's instructions (ForteBio). Different concentrations (0–400 nM) of WT or mutant protein were tested in parallel for binding to the biotinylated CRT‐P domain immobilized on Streptavidin (SA) Dip and Read™ Biosensors (ForteBio). All measurements were performed in PBS Kinetics Buffer (ForteBio) at 30°C in 96‐well microplates. Data were analyzed on Octet Data Analysis High Throughput (HT) software 11.0.
Statistical analysis
All experiments were successfully reproduced. Independent biological replicates were used for statistical comparison in mouse, zebrafish, and cell culture studies. Number of animals, individual cells, and replicates are indicated in each figure legend. Results of recombinant protein were obtained from one batch of purification and number of replicates is indicated in each figure legend. For representative data, number of replicates is indicated in each figure legend. If a data point varied two times the standard deviation from the mean, the Grubb's test was performed to identify a possible outlier. Normal distribution of data was checked by D'Agostino & Pearson and/or Shapiro–Wilk normality tests. One‐way and two‐way ANOVA followed by multiple‐comparison post‐test (Tukey's or Sidak's post hoc test), Student's t‐test, Kruskal–Wallis, and Mann–Whitney tests were performed using GraphPad software version 7. Wilcoxon–Mann–Whitney two‐sample rank test for statistical analysis of electrophysiology results was performed using Igor Pro (Wavemetrics, Inc.). Two‐tailed analysis is indicated in each figure legend. P < 0.05 was considered significant. Data are shown as mean. Error bars represent standard error of the mean (s.e.m.). Exact P values are presented in the figures.
Author contributions
DBM, SMa, RHB, CH, and AT designed the study. SMa, MA and SMu performed clinical evaluation. EY‐B, SMu, and AT performed genetic analysis. DBM, PR, UW, HU, FB, and CG‐B performed cell culture experiments. JB, EP, and MLC performed zebrafish experiments. DBM, RD, GQ, FC‐M, and VV performed mouse studies. MTW and LP performed proteomic analysis. DC, RP, CR, and BM performed electrophysiological studies. MJS and LWR performed studies with recombinant proteins. OU, CR, MS‐E, and RHB contributed reagents. DBM, SMa, CH, and AT wrote the manuscript. All authors revised and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
SDataEV
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank family members for their collaboration. This work was funded by FONDECYT 1191538 (DBM), FONDECYT 1200459 (UW), FONDECYT 11180825 (HU), FONDECYT 11140430 and DICYT 021843RS (CR), FONDECYT 1161524, DICYT 021843MM and 021943MM_POSTDOC (BM), FONDECYT 1180419 (CG‐B), CONICYT PIA ACT 192015, FONDEQUIP EQM130051 and Nucleus Physics of Active Matter (MLC), FONDECYT 1140549 (CH), FONDAP program 15150012 (CG‐B, MLC, CH), Millennium Institute P09‐015‐F (MLC, CH), European Commission R&D MSCA‐RISE 734749 (CH), URF‐QAU Pakistan (SMalik), AOF 318182 and Biocenter Oulu (LWR), and Boğaziçi University Research Fund project 7695 (AT). We also thank the support from Muscular Dystrophy Association 575897 and ALS Association 19‐IIA‐456 (DBM), FONDEF ID16I10223, FONDEF ID11E1007, Michael J Fox Foundation for Parkinson's Research—Target Validation grant 9277, US Office of Naval Research‐Global N62909‐16‐1‐2003, US Air Force Office of Scientific Research FA9550‐16‐1‐0384, and Department of Defense W81XWH2110960 (CH). Figure 7 and the synopsis figure were created with BioRender.com.
The EMBO Journal (2022) 41: e105531
Contributor Information
Claudio Hetz, Email: chetz@uchile.cl, Email: chetz@buckinstitute.org.
Aslıhan Tolun, Email: tolunasli@itu.edu.tr.
Data availability
Variant (PDIA3 c.170G>A; NM_005315) has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) with Submission ID SUB3488668. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with accession number PXD026507 (http://www.ebi.ac.uk/pride/archive/projects/PXD026507). Tables with data quantification and uncropped blots and gels are provided as supplemental material. Raw data are available from the corresponding authors upon reasonable request. All materials will be made available upon request after execution of Material Transfer Agreement between institutions.
References
- American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. Arlington, VA: American Psychiatric Association; [Google Scholar]
- Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, Edmonson SA, Africa L, Zhou S, High KA et al (2010) High AAV vector purity results in serotype‐ and tissue‐independent enhancement of transduction efficiency. Gene Ther 17: 503–510 [DOI] [PubMed] [Google Scholar]
- Belin BJ, Goins LM, Mullins RD (2014) Comparative analysis of tools for live cell imaging of actin network architecture. Bioarchitecture 4: 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Krieger AM, Yekutieli D (2006) Adaptive linear step‐up procedures that control the false discovery rate. Biometrika 93: 491–507 [Google Scholar]
- Bittles AH, Black ML (2010) Consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA 107: 1779–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H (2011) Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet 45: 81–104 [DOI] [PubMed] [Google Scholar]
- Calì T, Vanoni O, Molinari M (2008) The endoplasmic reticulum crossroads for newly synthesized polypeptide chains. Prog Mol Biol Transl Sci 83: 135–179 [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang Y‐J, Castanedes‐Casey M, Lee CW, Jansen‐West K, Kurti A, Murray ME et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science (New York, NY) 348: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM (2009) Insights into MHC class I peptide loading from the structure of the tapasin‐ERp57 thiol oxidoreductase heterodimer. Immunity 30: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Ruddock LW (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep 6: 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer BM, McAllister AK (2012) Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci 35: 660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RR, Dunning LM, Olson DA, Cohen SJ, Davis AT, Wood WG, Kratzke RA, Holtzman JL (2005) In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind beta‐amyloid. Biochem Biophys Res Comm 332: 50–57 [DOI] [PubMed] [Google Scholar]
- Ferhat L, Esclapez M, Represa A, Fattoum A, Shirao T, Ben‐Ari Y (2003) Increased levels of acidic calponin during dendritic spine plasticity after pilocarpine‐induced seizures. Hippocampus 13: 845–858 [DOI] [PubMed] [Google Scholar]
- Frand AR, Cuozzo JW, Kaiser CA (2000) Pathways for protein disulphide bond formation. Trends Cell Biol 10: 203–210 [DOI] [PubMed] [Google Scholar]
- Freeze HH, Eklund EA, Ng BG, Patterson MC (2015) Neurological aspects of human glycosylation disorders. Annu Rev Neurosci 38: 105–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbi N, Tanaka S, Momburg F, Hämmerling GJ (2006) Impaired assembly of the major histocompatibility complex class I peptide‐loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol 7: 93–102 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Perez P, Woehlbier U, Chian R‐J, Sapp P, Rouleau GA, Leblond CS, Daoud H, Dion PA, Landers JE, Hetz C et al (2015) Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 566: 158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus C, Geptin H, Tushev G, Garg S, Alvarez‐Castelao B, Sambandan S, Kochen L, Hafner A‐S, Langer JD, Schuman EM (2016) Unconventional secretory processing diversifies neuronal ion channel properties. eLife 5: e20609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C (2021) Adapting the proteostasis capacity to sustain brain healthspan. Cell 184: 1545–1560 [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis‐Carneiro M, Wälchli S, Carboni S, Vial‐Knecht E, Maundrell K, Castilla J, Soto C (2005) The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci 25: 2793–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13: 477–491 [DOI] [PubMed] [Google Scholar]
- Hetz C, Zhang K, Kaufman RJ (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21: 421–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann K, Lindner TH (2005) easyLINKAGE‐Plus–automated linkage analyses using large‐scale SNP data. Bioinformatics (Oxford, England) 21: 3565–3567 [DOI] [PubMed] [Google Scholar]
- Hu H, Kahrizi K, Musante L, Fattahi Z, Herwig R, Hosseini M, Oppitz C, Abedini SS, Suckow V, Larti F et al (2019) Genetics of intellectual disability in consanguineous families. Mol Psychiatry 24: 1027–1039 [DOI] [PubMed] [Google Scholar]
- Jessop CE, Chakravarthi S, Garbi N, Hämmerling GJ, Lovell S, Bulleid NJ (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J 26: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop CE, Tavender TJ, Watkins RH, Chambers JE, Bulleid NJ (2009) Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J Biol Chem 284: 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyifous O, Waites CL, Specht CG, Fujisawa S, Schubert M, Lin EI, Marshall J, Aoki C, de Silva T, Montgomery JM et al (2009) SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat Neurosci 12: 1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junghans D, Herzog S (2018) Cnn3 regulates neural tube morphogenesis and neuronal stem cell properties. FEBS J 285: 325–338 [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1: 2406–2415 [DOI] [PubMed] [Google Scholar]
- Kapp TG, Rechenmacher F, Neubauer S, Maltsev OV, Cavalcanti‐Adam EA, Zarka R, Reuning U, Notni J, Wester H‐J, Mas‐Moruno C et al (2017) A comprehensive evaluation of the activity and selectivity profile of ligands for RGD‐binding integrins. Sci Rep 7: 39805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Carman CV, Springer TA (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science (New York, NY) 301: 1720–1725 [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310 [DOI] [PubMed] [Google Scholar]
- Kiryushko D, Berezin V, Bock E (2004) Regulators of neurite outgrowth: role of cell adhesion molecules. Ann N Y Acad Sci 1014: 140–154 [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukaitis CM, Webb DJ, Donais K, Horwitz AF (2001) Differential dynamics of alpha 5 integrin, paxillin, and alpha‐actinin during formation and disassembly of adhesions in migrating cells. J Cell Biol 153: 1427–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock JG, Jones MC, Askari JA, Gong X, Oddone A, Olofsson H, Göransson S, Lakadamyali M, Humphries MJ, Strömblad S (2018) Reticular adhesions are a distinct class of cell‐matrix adhesions that mediate attachment during mitosis. Nat Cell Biol 20: 1290–1302 [DOI] [PubMed] [Google Scholar]
- Martin HC, Jones WD, McIntyre R, Sanchez‐Andrade G, Sanderson M, Stephenson JD, Jones CP, Handsaker J, Gallone G, Bruntraeger M et al (2018) Quantifying the contribution of recessive coding variation to developmental disorders. Science (New York, NY) 362: 1161–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez G, Khatiwada S, Costa‐Mattioli M, Hetz C (2018) ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci 41: 610–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medinas DB, Rozas P, Martínez Traub F, Woehlbier U, Brown RH, Bosco DA, Hetz C (2018) Endoplasmic reticulum stress leads to accumulation of wild‐type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 115: 8209–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Papa FR (2015) The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 10: 173–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oteiza P, Köppen M, Krieg M, Pulgar E, Farias C, Melo C, Preibisch S, Müller D, Tada M, Hartel S et al (2010) Planar cell polarity signalling regulates cell adhesion properties in progenitors of the zebrafish laterality organ. Development (Cambridge, England) 137: 3459–3468 [DOI] [PubMed] [Google Scholar]
- Park YK, Goda Y (2016) Integrins in synapse regulation. Nat Rev Neurosci 17: 745–756 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri ER, Thomas CJ, Parakh S, Spencer DM, Atkin JD (2015) The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front Cell Dev Biol 3: 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L, Cooley CB, Chen JJ, Paxman RJ, Gallagher CM, Madoux F, Genereux JC, Dobbs W, Garza D, Spicer TP et al (2016) Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 5: e15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath V, Dang H‐M, Goya RG, Mansour H, Nimgaonkar VL, Russell VA, Xin Y (2015) Regional research priorities in brain and nervous system disorders. Nature 527: S198–S206 [DOI] [PubMed] [Google Scholar]
- Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL, Song Y, van Beusekom E, Khan AA, Tomas‐Roca L et al (2017) Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry 22: 1604–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas‐Rivera D, Armisén R, Colombo A, Martínez G, Eguiguren AL, Díaz A, Kiviluoto S, Rodríguez D, Patron M, Rizzuto R et al (2012) TMBIM3/GRINA is a novel unfolded protein response (UPR) target gene that controls apoptosis through the modulation of ER calcium homeostasis. Cell Death Differ 19: 1013–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH (2010) Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 11: 161–187 [DOI] [PubMed] [Google Scholar]
- Rozas C, Loyola S, Ugarte G, Zeise ML, Reyes‐Parada M, Pancetti F, Rojas P, Morales B (2012) Acutely applied MDMA enhances long‐term potentiation in rat hippocampus involving D1/D5 and 5‐HT2 receptors through a polysynaptic mechanism. Eur Neuropsychopharmacol 22: 584–595 [DOI] [PubMed] [Google Scholar]
- Rozas C, Carvallo C, Contreras D, Carreño M, Ugarte G, Delgado R, Zeise ML, Morales B (2015) Methylphenidate amplifies long‐term potentiation in rat hippocampus CA1 area involving the insertion of AMPA receptors by activation of β‐adrenergic and D1/D5 receptors. Neuropharmacology 99: 15–27 [DOI] [PubMed] [Google Scholar]
- Ruddock LW, Hirst TR, Freedman RB (1996) pH‐dependence of the dithiol‐oxidizing activity of DsbA (a periplasmic protein thiol:disulphide oxidoreductase) and protein disulphide‐isomerase: studies with a novel simple peptide substrate. Biochem J 315(Pt 3): 1001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryno LM, Genereux JC, Naito T, Morimoto RI, Powers ET, Shoulders MD, Wiseman RL (2014) Characterizing the altered cellular proteome induced by the stress‐independent activation of heat shock factor 1. ACS Chem Biol 9: 1273–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C (2004) Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci 5: 917–930 [DOI] [PubMed] [Google Scholar]
- Schalock RL (2011) The evolving understanding of the construct of intellectual disability. J Intellect Dev Disabil 36: 227–237 [DOI] [PubMed] [Google Scholar]
- Shatz CJ (2009) MHC class I: an unexpected role in neuronal plasticity. Neuron 64: 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan E, Wright J, Small N, Corry PC, Oddie S, Whibley C, Petherick ES, Malik T, Pawson N, McKinney PA et al (2013) Risk factors for congenital anomaly in a multiethnic birth cohort: an analysis of the Born in Bradford study. Lancet (London, England) 382: 1350–1359 [DOI] [PubMed] [Google Scholar]
- da Silva JS, Dotti CG (2002) Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat Rev Neurosci 3: 694–704 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al (2015) STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G (2014) Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol (Clifton, NJ) 1183: 221–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Medinas DB, Matamala JM, Woehlbier U, Cornejo VH, Solda T, Andreu C, Rozas P, Matus S, Muñoz N et al (2015) The protein‐disulfide isomerase ERp57 regulates the steady‐state levels of the prion protein. J Biol Chem 290: 23631–23645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruta D, Gonzales M, Hopkinson SB, Otey C, Khuon S, Goldman RD, Jones JCR (2002) Microfilament‐dependent movement of the beta3 integrin subunit within focal contacts of endothelial cells. FASEB J 16: 866–868 [DOI] [PubMed] [Google Scholar]
- Urra H, Henriquez DR, Cánovas J, Villarroel‐Campos D, Carreras‐Sureda A, Pulgar E, Molina E, Hazari YM, Limia CM, Alvarez‐Rojas S et al (2018) IRE1α governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat Cell Biol 20: 942–953 [DOI] [PubMed] [Google Scholar]
- Villarroel‐Campos D, Henríquez DR, Bodaleo FJ, Oguchi ME, Bronfman FC, Fukuda M, Gonzalez‐Billault C (2016) Rab35 functions in axon elongation are regulated by p53‐related protein kinase in a mechanism that involves Rab35 protein degradation and the microtubule‐associated protein 1B. J Neurosci 36: 7298–7313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LELM, Gilissen C, Veltman JA (2016) Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17: 9–18 [DOI] [PubMed] [Google Scholar]
- Walker MB, Kimmel CB (2007) A two‐color acid‐free cartilage and bone stain for zebrafish larvae. Biotech Histochem 82: 23–28 [DOI] [PubMed] [Google Scholar]
- Woehlbier U, Colombo A, Saaranen MJ, Pérez V, Ojeda J, Bustos FJ, Andreu CI, Torres M, Valenzuela V, Medinas DB et al (2016) ALS‐linked protein disulfide isomerase variants cause motor dysfunction. EMBO J 35: 845–865 [DOI] [PMC free article] [PubMed] [Google Scholar]