

Abstract
Genetically diverse pluripotent stem cells display varied, heritable responses to differentiation cues. Here, we harnessed these disparities through derivation of mouse embryonic stem cells from the BXD genetic reference panel, along with C57BL/6J (B6) and DBA/2J (D2) parental strains, to identify loci regulating cell state transitions. Upon transition to formative pluripotency, B6 stem cells quickly dissolved naïve networks adopting gene expression modules indicative of neuroectoderm lineages, whereas D2 retained aspects of naïve pluripotency. Spontaneous formation of embryoid bodies identified divergent differentiation where B6 showed a propensity toward neuroectoderm and D2 toward definitive endoderm. Genetic mapping identified major trans‐acting loci co‐regulating chromatin accessibility and gene expression in both naïve and formative pluripotency. These loci distally modulated occupancy of pluripotency factors at hundreds of regulatory elements. One trans‐acting locus on Chr 12 primarily impacted chromatin accessibility in embryonic stem cells, while in epiblast‐like cells, the same locus subsequently influenced expression of genes enriched for neurogenesis, suggesting early chromatin priming. These results demonstrate genetically determined biases in lineage commitment and identify major regulators of the pluripotency epigenome.
Keywords: BXD mice, cell fate, epigenetic variability, KRAB zinc‐finger proteins, mESCs
Subject Categories: Chromatin, Transcription & Genomics; Genetics, Gene Therapy & Genetic Disease; Stem Cells & Regenerative Medicine
Extensive mapping highlights a role of genetic background in defining stem cell fate via context‐dependent trans‐regulation of chromatin.
Introduction
The ability to form all somatic and germline tissues, while maintaining the capacity to self‐renew, is defining features of pluripotent stem cells (PSCs) (Evans & Kaufman, 1981; Martin, 1981; Bradley et al, 1984; Buehr & Smith, 2003). Harnessing this potential will transform regenerative medicine. Yet, most studies seeking to identify mechanisms that underlie acquisition of pluripotency and fate determination utilize cells with limited genetic diversity. Historically, successful derivation of naïve mouse embryonic stem cells (ESCs) was achieved for permissive strains (i.e., 129 and C57BL/6 lineages). Nonpermissive strains, such as D2, required inhibition of differentiation pathways (i.e., 2i) (Ying et al, 2008; Nichols et al, 2009; Czechanski et al, 2014), demonstrating that the establishment and maintenance of pluripotency are intrinsic to genetic background. Further, donor genetic background has been identified as the cause of variable efficiencies in derivation and differentiation in human‐induced PSCs (hiPSCs) and ESCs (hESCs) (Allegrucci & Young, 2007; Osafune et al, 2008; Koyanagi‐Aoi et al, 2013; Kyttälä et al, 2016; Li et al, 2018; Volpato & Webber, 2020). Critically, genetic variation within regulatory regions has been attributed with heterogeneity in iPSC differentiation (Nishizawa et al, 2016; Kilpinen et al, 2017). However, these studies have only identified cis regulation of molecular features, such as gene expression and chromatin accessibility, implicated in varied responses to differentiation cues. Identification of trans‐acting factors, enabled by controlled model systems, will provide greater mechanistic insights into genetic control over a complex trait such as capacity to differentiate. While we (Skelly et al, 2020) and others (Ortmann et al, 2020) have recently investigated how genetic variation impacts pluripotency, less is understood about how genetic backgrounds influence cell state transitions.
During development, cells within the epiblast progress along a pluripotent spectrum from pre‐ to post‐implantation (Arnold & Robertson, 2009; Hackett & Surani, 2014), which can be modeled in vitro (Nichols & Smith, 2009). ESCs derived from the preimplantation epiblast (E4.5) capture naïve pluripotency (Evans & Kaufman, 1981; Martin, 1981; Gardner & Beddington, 1988), while epiblast stem cells (EpiSCs) derived post‐implantation (E6.5) represent primed pluripotency (Brons et al, 2007). Between these two states exists formative pluripotency. Transit through formative pluripotency is required for multi‐lineage competency including germ cell induction (Hayashi et al, 2011; Morgani et al, 2017; Smith, 2017). Formative pluripotency can be transiently modeled in vitro by transition of ESCs to epiblast‐like cells (EpiLCs) (Hayashi et al, 2011) and was recently derived directly from the pre‐gastrulation epiblast (Kinoshita et al, 2021). While culture conditions can provide external signaling cues that positions a cell along the pluripotency spectrum (Morgani et al, 2017), variability in acquisition of desired pluripotent states is, in part, dependent on the ESC’s strain of origin (Kawase et al, 1994; Sharova et al, 2007; Schnabel et al, 2012; Czechanski et al, 2014; Garbutt et al, 2018; Ortmann et al, 2020; Skelly et al, 2020). Importantly, differentiation propensity to specific germ layers has been linked to the origin of ESCs within this pluripotency spectrum (Hackett et al, 2017). This demonstrates interactions between genetic background and media conditions may drive cell fate.
Reconfiguration of chromatin accompanies, and often precedes (Bernstein et al, 2006; Bonifer & Cockerill, 2017), transcriptomic changes during cell state transitions (Chen & Dent, 2014; Factor et al, 2014; Novo et al, 2018; Pękowska et al, 2018; Yadav et al, 2018). Regulatory elements, such as enhancers and promoters, largely act in cis to locally control developmentally programmed gene networks (Catarino & Stark, 2018). In concert, DNA‐binding proteins act in trans at many cis‐regulatory elements to either activate or repress gene expression. During development, dramatic alteration of the regulatory landscape can occur even in the absence of changes in transcription factor (TF) expression. For example, when naïve ESCs transition to formative EpiLCs, expression of Pou5f1/Oct4 remains high, while occupancy of POU5F1 at regulatory elements is globally rewired (Buecker et al, 2014; Yang et al, 2019, 3), suggesting the existence of a set of unknown chromatin regulators that influence cell state transitions.
Here, we combine the strength of systems genetics, utilizing extant variation in a diverse genetic reference population, with the power of modeling developmental transitions using PSCs, to identify loci that govern chromatin and gene regulation during exit from pluripotency and determine differentiation propensity.
Results
Position along the pluripotency spectrum is determined by genetic background
To assess genetic control over pluripotency and differentiation, we derived three biological replicate ESCs from independent blastocysts using B6 and D2 mice. Success in derivation of germline‐competent ESCs for these strains required conditions that included serum, feeders, and 2i (Czechanski et al, 2014). Both B6 and D2 ESCs were differentiated to EpiLCs (Fig 1A), and transcript abundance was measured by RNA‐sequencing (‐seq) and chromatin accessibility by ATAC‐seq. While both strains effectively transition from naïve to formative pluripotency, as indicated by expression and accessibility of binding sites of naïve and primed markers, distinct morphological differences were observed (Figs 1A–C and EV1A and B). To determine major features of variation, we performed principal component analysis (PCA) on transcript abundance. While biological replicates closely clustered, demonstrating high reproducibility, PC1 separated ESCs from EpiLCs and PC2 captured strain variation within a cell state (Fig 1B). For ESCs, naïve pluripotency factors (Klf4, Sox2, Esrrb, Nanog, Pou5f1, and Tfcp2l1) were highly expressed but not differentially abundant between B6 and D2 (Fig. 1D). Overall, 1,785 differentially expressed genes (DEGs) in ESCs, and 2,209 DEGs in EpiLCs (FDR < 0.05 and log2FC > 1) were identified between B6 and D2 (Figs 1E and EV1C, Dataset EV1). Of these, only 792 (19.8%) DEGs are shared between cell states, indicating that the strain‐dependent DEGs are largely unique to each cell state.
Figure 1. Genetic background influences position within a pluripotency spectrum through differences in chromatin accessibility.
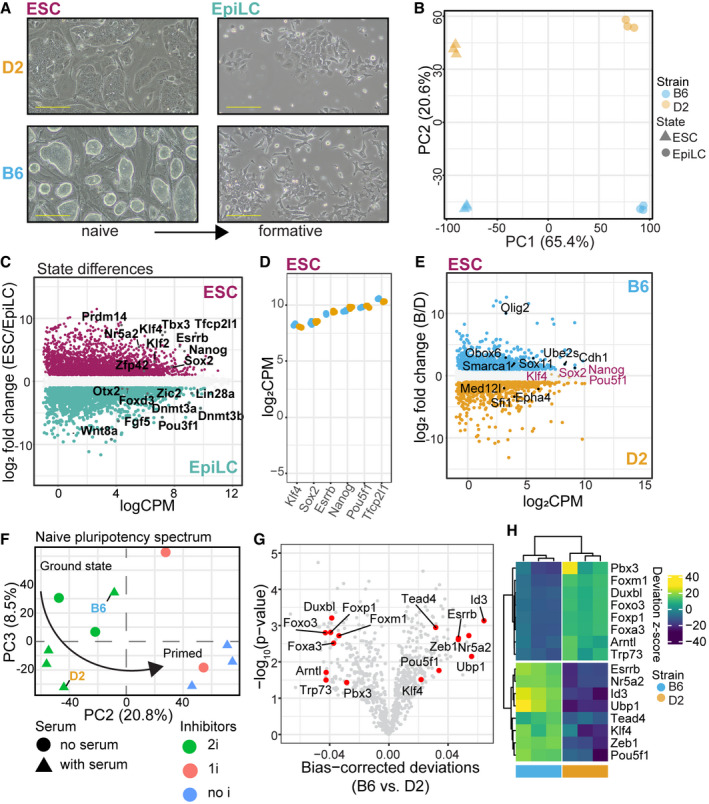
- Phase‐contrast images of representative cultures for B6 and D2 strains maintained in conditions supporting naïve pluripotency and after transitioning to formative EpiLCs. B6 ESC colonies adopt a classical ground state morphology, whereas D2 ESCs grew with a flat morphology and fewer cells per colony. Morphological differences persisted in EpiLCs (scale bar = 50 µm).
- First two principal components show major source of variation in transcript abundance is cell state (PC1, 65.4%) and strain (PC2, 20.6%) for 3 biological replicates of B6 and D2.
- MA plot highlighting genes differentially expressed between state independent of strain (glm state term, false discovery rate‐adjusted P‐value (FDR) < 0.05 and log2FC > 1). Core pluripotent TFs highlighted for each state (maroon = naïve ESCs, teal = formative EpiLCs).
- Scatterplot of expression for core naïve TFs in 3 biological replicate ESCs per strain. Equal high expression of naïve TFs observed in both B6 and D2 ESCs.
- MA plot showing differentially regulated genes between strains within ESCs as determined by pairwise comparison (FDR < 0.05 and logFC > 1). Core naïve TFs are highlighted as not significantly differentially expressed between strains. Genes significantly different between strains within cell state with known roles in promoting naïve pluripotency (B6 = Obox6, Cdh1, Smarca1, Ube2s; D2 = Med12l, Sfi1) and self‐renewal in progenitor cell populations are highlighted (B6 = Olig2, Sox11; D2 = Epha4).
- PCA of full transcript abundance comparing average expression from B6 and D2 ESCs (3 biological replicates) collected here to previously collected RNA‐seq from Hackett et al (2017). PC1 separated experimental origin, indicating batch effects between laboratories, and was not explored further. B6 expression was similar to that of ground state conditions, which exclude serum, whereas D2 expression more closely resembled conditions that include serum. Arrow indicates developmental progression toward primed pluripotency.
- Volcano plot showing the mean difference in bias‐corrected accessibility at TF motifs from ATAC‐seq (3 biological replicates) versus P‐value for that difference. Select TFs critical to pluripotency and differentiation are highlighted.
- Heatmap of motif deviations from 3 biological replicates of B6 and D2 ESCs for TFs highlighted in (G).
Figure EV1. Expression of core naïve TFs does not explain strain‐specific transcriptional programs in ESCs.
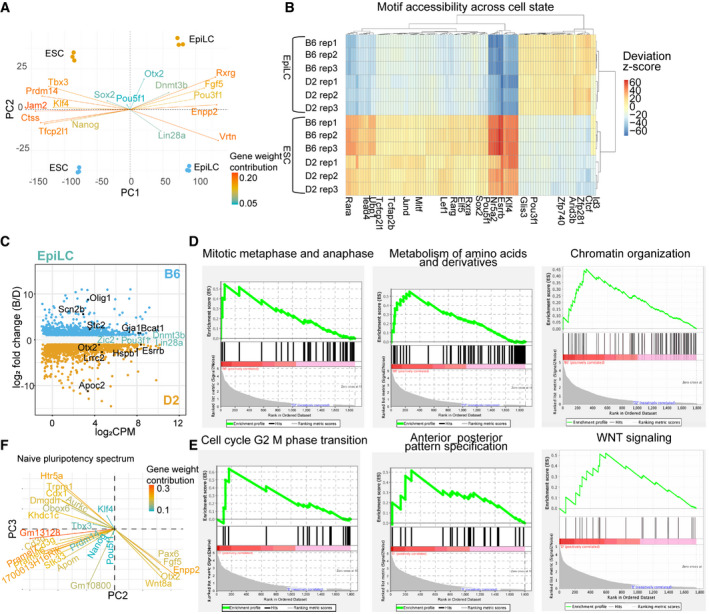
- PCA biplot showing individual gene contributions to the first two principal components. Transcript abundance measured by bulk RNA‐seq from 3 biological replicates for each strain in ESCs and EpiLCs. Core naïve and primed marker genes are indicated, drive separation between cell states, and are largely not differentially expressed between strains. As previously described (Buecker et al, 2014), Pou5f1 expression is unchanged across cell states.
- Heatmap of bias‐corrected deviation z‐scores of chromatin accessibility at TF motifs (n = 3 biological replicates of ESCs and EpiLCs from both B6 and D2 strains).
- MA plot highlighting genes differentially expressed between strain within EpiLCs as determined by pairwise comparison (FDR < 0/05, log2FC > 1). Core primed TFs are highlighted as not significantly differentially expressed between strains.
- GSEA for functional programs curated within MsigDB using DEGs between B6 and D2 ESCs. Upregulated genes in B6 ESCs were enriched for GO terms in biological processes associated with self‐renewal and pluripotency such as regulation of mitotic metaphase and anaphase and metabolism of amino acids and derivatives (significant enrichment in phenotype using 1,000 permutations, FDR < 25%), as well as regulation of chromatin organization (significant for single gene set, nominal P‐value < 0.05).
- Similar to (D) showing pathways upregulated in D2. D2 ESCs were associated with regulation of G2/M phase transition and anterior–posterior pattern specification (FDR < 25%) in addition to self‐renewing programs such as WNT signaling (nominal P‐value < 0.05).
- PCA biplot indicating selected gene weights contributing to occupancy of cells along naïve pluripotency spectrum from Fig 1F. Core naïve TFs (such as Klf4, Tbx3, Nanog, Pou5f1, and Prdm14) do not contribute to separation of cells grown in different culture conditions or differential occupancy of pluripotency continuum by B6 and D2 ESCs. Cells grown in conditions lacking 2i have higher expression of primed TFs (i.e., Fgf5, Otx2, Wnt8a). While B6 and D2 both associate with conditions that include 2i, D2 is separated from B6 ESCs by expressing transcription profiles similar to conditions that include serum. Gene weights selected are in top 100 genes contributing to separation along PC2 and PC3; only genes driving separation between B6 and D2 along PC3 are plotted at threshold of 0.15 contribution.
Gene set enrichment analysis in ESCs identified strain‐specific transcriptional programs. B6‐enriched pathways included metabolism of amino acids and chromatin organization (Fig EV1D). Derivatives of amino acid metabolism sustain nucleotide synthesis, required for rapid proliferation (Ito & Suda, 2014) in ESCs, and serve as donors for histone modifications governing the pluripotent epigenetic landscape (Van Winkle & Ryznar, 2019). Additionally, DEGs were enriched for pathways representing different cell cycle phases with B6 enriched for genes regulating M phase and D2 enriched for genes regulating G2/M transition. Cell cycle phase impacts exit from ground state pluripotency as an extended G2 phase delays differentiation (Gonzales et al, 2015). D2 DEGs were paradoxically enriched both for upregulation of anterior–posterior pattern specification (Fig EV1E), typically activated after exit from pluripotency, and for WNT signaling, which prevents exit from ground state (ten Berge et al, 2011; de Jaime‐Soguero et al, 2018).
Next, we compared the transcriptional state of our ESCs with that of published B6 ESCs cultured in nine conditions covering a range of pluripotency (Hackett et al, 2017). PCA of total transcript abundance reconstructed the reported spectrum of pluripotency and showed that B6 and D2 occupied different positions along this continuum (Figs 1F and EV1F). PC2 distinguished ESCs grown in 2i, and PC3 separated conditions containing serum. Along PC2, both B6 and D2 ESCs were transcriptionally similar to conditions that include 2i; however, variation in PC3 suggested that our B6 ESCs were more similar to cells grown in conditions that excluded serum, whereas D2 was transcriptionally similar to cells grown in conditions that included serum.
Since differences in the pluripotent spectrum are not explained by expression of naïve TFs in ESCs, we measured variability in chromatin accessibility as an indicator of TF occupancy. Naïve TFs, including ESRRB, TFCP2L1, KLF4, POU5F1, and NR5A2, all had greater accessibility at their respective motifs in B6 than in D2 (Fig 1G and H). Additionally, the motif for ZEB1, a TF important for neuronal differentiation (Jiang et al, 2018, 1), is more accessible in B6 ESCs. In contrast, D2 ESCs showed greater accessibility of TRP73 motif, p53 family member known to instruct ESC differentiation toward mesoendoderm (Wang et al, 2017). Given that transcript abundance of these TFs is similar between B6 and D2, this suggests the existence of unknown factors that regulate TF accessibility in trans. Together, these data support that differential transcriptional programs, driven by genetic variation, place B6 ESCs in a more naïve position, while D2 ESCs are more primed to exit naïve pluripotency, thus potentially impacting cell fate upon differentiation.
Genetic background dictates activation of distinct biological processes upon exit from naïve pluripotency
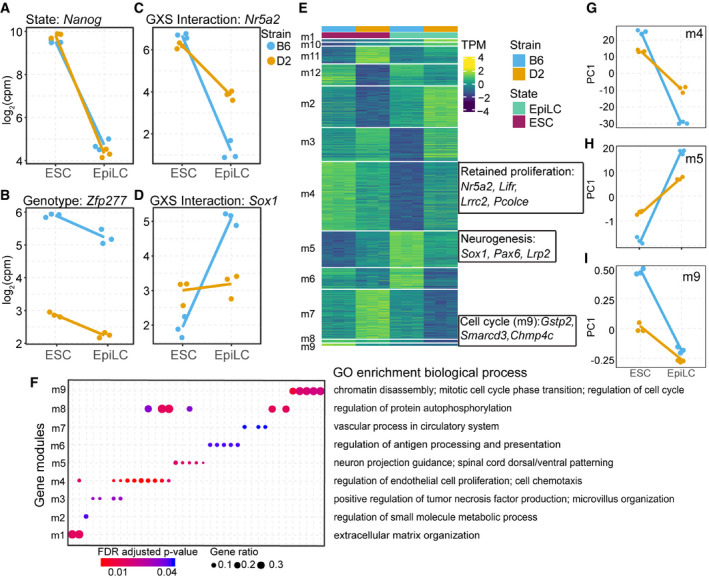
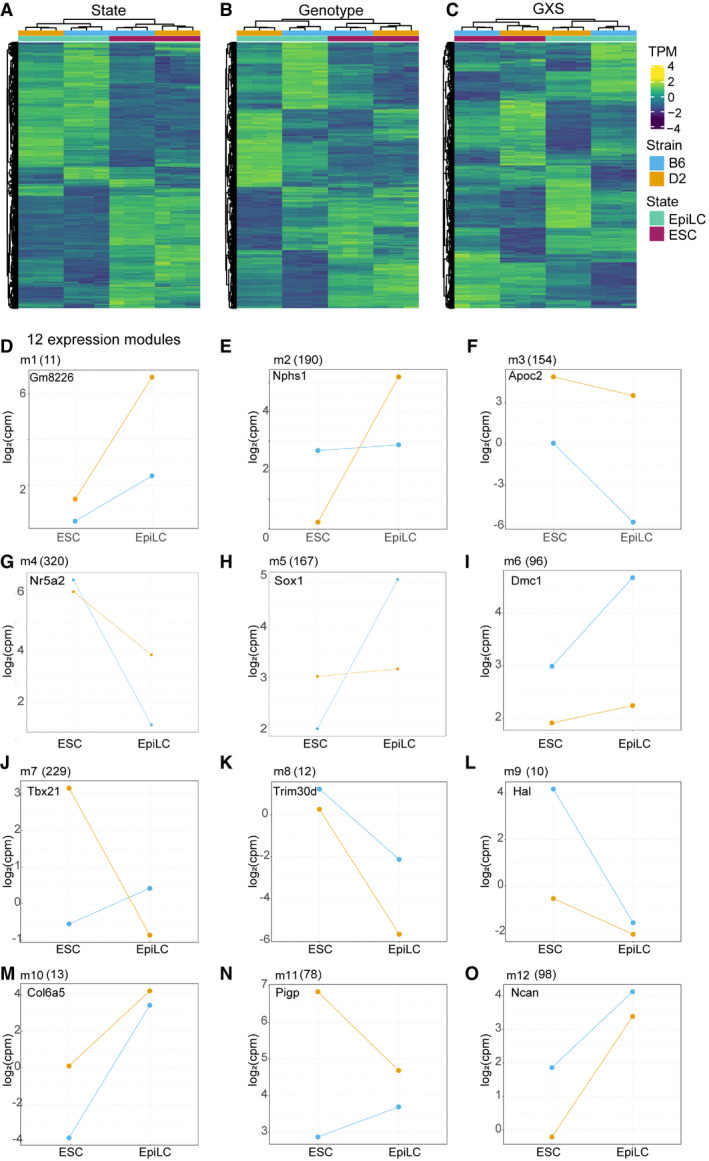
Given observed differences between B6 and D2 in ESCs, we sought to understand how genetic background drives cell state transition. Our linear model identified 4,787 DEGs between cell states, 2,210 DEGs between genetic backgrounds, and 2,083 genes with a significant genotype‐by‐state (GxS) interaction (FDR < 0.05 and log2FC > 1; Figs 2A–D and EV2A–C, Dataset EV2). This GxS interaction is exemplified by Nr5a2 and Sox1, markers for pluripotency and neuronal progenitor cells, respectively (Venere et al, 2012). In ESCs, B6 and D2 show equally high expression of Nr5a2, yet upon exit from naïve state, D2 EpiLCs retain higher expression (Fig 2C). In contrast, Sox1 expression is low in B6 and D2 ESCs, increasing only in B6 EpiLCs (Fig 2D). To functionally characterize GxS interactions, DEGs were divided into modules using a hidden Markov model to classify unique expression paths, followed by filtering for genes with a significant GxS interaction (Fig 2E, Dataset EV3). Example genes demonstrate each module’s unique trajectory upon transition from naïve to formative pluripotency (EV2D‐O). Importantly, different modules represent different biological processes. Nine modules were enriched for GO terms including neurogenesis, proliferation, and cell cycle regulation (Fig 2F and Appendix Fig S1A–E). For example, module 4 genes (m4, n = 320 including Nr5a2) are initially higher expressed in B6 ESCs compared with D2; however, upon transitioning to EpiLCs D2 retains greater expression (Fig 2G). When comparing module 4 genes in EpiLCs between strains, D2 shows enrichment by GSEA (Benporath_ES_H3K27me3; normalized enrichment score = 1.48, nominal P‐value = 0.089) for genes targeted for histone modifications, specifically in ESCs, further suggesting D2 retainment of ESC‐like state in EpiLCs. Supporting the enrichment of M phase of cell cycle regulation in B6 ESCs, m9 (n = 10) was enriched for genes regulating mitotic cell cycle phase transition (Fig 2F and I). Notably, module 5 (m5, n = 167, including Sox1) increased expression in B6 at a higher rate than in D2 and is enriched for GO terms associated with neuronal development (Fig 2F and H). These GxS interactions identify expression modules representing different paths during transition from naïve to formative pluripotency. Additionally, differential upregulation of lineage markers supports genetic biases in cell fate, with B6 primed toward neuronal lineages.
Figure 2. B6 EpiLCs are primed toward neuroectoderm lineage.
-
A–DExample expression patterns for select genes identified by applying a general linear model (glm) including state, genotype, and interaction terms. Dots represent individual biological replicates (N = 3); lines highlight changes in mean values for replicates between each state (log2FC > 1 and FDR < 0.05). (A) State‐dependent expression is exemplified by Nanog, which showed no difference between strains. (B) Expression of Zfp277 exemplifies strain dependence being consistently higher in B6. (C, D) A significant genotype x state (GXS) interaction was identified for Nr5a2 (C, a marker for pluripotency) and Sox1 (D, a marker for neuronal differentiation).
-
EHeatmap of transcript abundances for individual genes (rows) representing 12 expression modules detected by EBseqHMM filtered for genes with a significant GXS glm interaction. Genes with observed functional similarity within modules are highlighted. Nr5a2, Lifr, and other pluripotency genes shared the same expression path (m4) that decreases more significantly in B6, retained expression in D2, when ESCs are differentiated to EpiLCs. Sox1, Pax6, and other neurogenesis genes shared an expression path (m5) that is significantly upregulated in B6 EpiLCs. Gstp2, Smarcd3, and other cell cycle regulation genes shared an expression path (m9) that is significantly upregulated in B6 ESCs.
-
FGene ontology (GO) enrichment of biological processes (FDR‐adjusted P‐value < 0.05) identified for 9 out of 12 expression modules (m1‐9). Each column represents a GO term. Highlighted terms are indicated to the right. GO terms associated with proliferation (m4), neurogenesis (m5), and cell cycle regulation (m9) indicate genetic control of exit from pluripotency and early lineage priming.
-
G–ISummarized differential module behavior between B6 and D2 (3 biological replicates) upon transition from ESCs to EpiLCs represented by PC1 of transcript abundance for all genes within each module. (G) D2 retained proliferation in EpiLCs. (H) Upregulation of neurogenesis in B6 EpiLCs. (I) Increased regulation of cell cycle M phase in B6 ESCs.
Figure EV2. Genetic background induces distinct expression pattern changes across cell state transition.
-
A–CHeatmaps showing expression patterns for genes detected with a significant difference based on general linearized model (glm, state + strain + stain x strain). Hierarchical clustering of samples (columns) results in proper association of cell states and biological replicates within genotype. (A) Genes with significant difference between cellular states independent of genetic background (n = 10,380 at FDR < 0.05). (B) Genes with significant difference between strains independent of state (n = 7,359 at FDR < 0.05). (C) Genes with significant interaction between state and strain (n = 4,912 at FDR < 0.05).
-
D–OCandidate gene expression patterns between genotype and state represent expression modules m1‐12. Modules resulted from intersection between EBseqHMM‐defined expression paths and genes with a significant GxS interaction. Number of genes within each module indicated at top in parentheses.
Genetic background biases differentiation propensity
To determine how genetic differences in pluripotency impact cell fate, we developed an assay for spontaneous differentiation of embryoid bodies (EBs) and profiled these spheroids by single‐cell RNA‐seq (scRNA‐seq) to determine cell composition. Cellular populations within EBs were highly reproducible among biological replicates (Figs 3A and EV3A–E), permitting comparisons between strains. After integration of scRNA‐seq across genetic backgrounds followed by unbiased clustering (Fig 3B), 9 clusters could be annotated with identifiable cellular lineages (Dataset EV4). The remaining clusters were enriched for cellular activities representative of many cell types, such as mitotic cell cycle regulation or chromatin organization, precluding exact lineage identification. Importantly, different genetic backgrounds are biased toward different cell fates (Fig 3C and D). For example, cells in cluster 9 represent primitive erythrocytes and are largely of D2 origin, whereas cells in cluster 5 express vascular endothelium marker genes and are largely B6. Cluster 4, showing equal proportions between strains, lies at the convergence of clusters 5 and 9 and represents yolk sac blood island cells (Fig EV3H), common progenitors of primitive erythrocytes and vascular endothelium (Palis et al, 1999). This suggests that genetic variation modulates trajectories through this developmental bifurcation. In addition to differences in cell fate within the same germ layer (i.e., mesoderm), EBs also identified differentiation bias to primary germ layers. Critically, definitive endoderm (cluster 7) was comprised mainly of D2 cells, whereas neuroectoderm (cluster 11) was comprised mainly of B6 cells.
Figure 3. Embryoid bodies confirm genetic background influences differentiation propensity with B6 trajectory toward neuroectoderm.
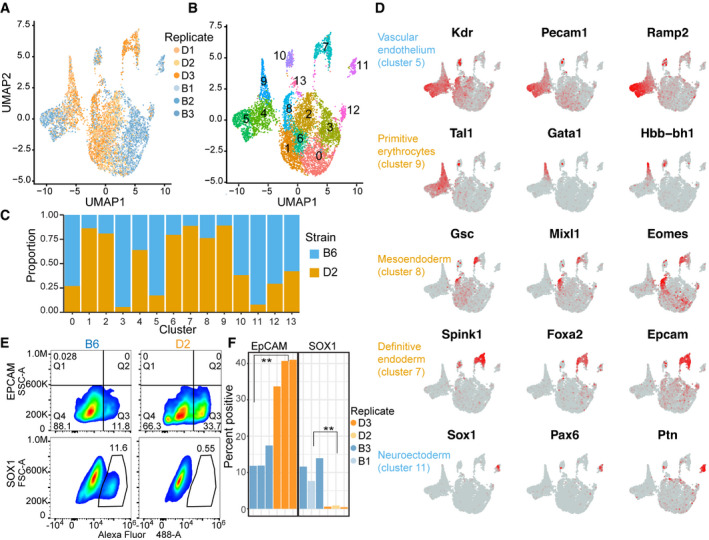
- UMAP embeddings of transcriptional profiles from spontaneously differentiated embryoid bodies (EBs). Each point represents a cell colored by genetic background and shaded by biological replicate (N = 3).
- Similar to (A) indicating 13 major cell populations based on unsupervised clustering. Each cell is colored based on cluster.
- Proportion of cells in a given cluster from (B) represented by either B6 or D2 genetic background.
- Feature plot of expression gradients for indicated gene overlaid on UMAP from (A). Three lineage markers that help to distinguish cellular identity are shown for five cell clusters.
- Pseudocolor plots of FACS analysis on cells from EBs for both B6 and D2 strains. Cells were labeled with anti‐EpCAM (top) or biological replicates with anti‐SOX1 antibody (bottom). Gated population indicates percentages of EpCAM+ (Q3) or SOX1+ cells.
- Bar chart showing percent positive cells gated for EpCAM+ population (left) or SOX1+ population (right). As predicted by scRNA‐seq, more D2 EB cells express EpCAM compared with B6 and more B6 EB cells express SOX1 compared with D2 (N = 3, two‐sided t‐test, EpCAM P‐value = 0.0012, and SOX1 P‐value = 0.0048).
Figure EV3. Spontaneous differentiation of ESCs to EBs identifies divergent developmental trajectories between B6 and D2 strains.
-
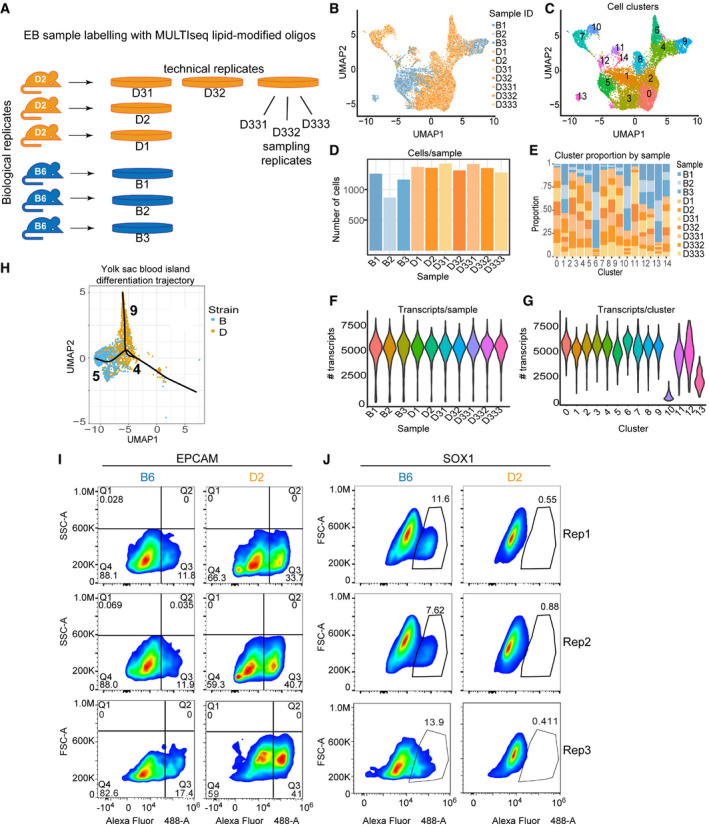
AExperimental design to assess reproducibility of EB formation and scRNA‐seq characterization of cell proportions. Reproducibility was accessed by growing (i) three biological replicates (originating from independently derived ESC lines), (ii) one ESC cell line grown for three technical replicates, and (iii) sampling replicates (pool of disaggregated EBs sampled three times). Each replicate was barcoded using lipid‐modified oligonucleotides, pooled, and loaded onto a single 10X Chromium microfluidic lane.
-
BUMAP embeddings for all 10 scRNA‐seq libraries colored by individual replicate. Each dot represents a single cell. Cells from replicate EBs clustered in proximity demonstrating reproducibility.
-
CSimilar to (B), UMAP embeddings of cell clustering based on transcript abundance for all 10 samples.
-
DNumber of cells per sample shown for each sample in (B).
-
EProportion of cells within each cluster color coded by sample in (B). Overall, biological, technical, and sample replicates made up similar proportions of clusters. Because technical and sampling replicates were collected from D2 ESCs, downstream analysis for Fig 3 was performed with only 3 biological replicates for each genotype in order to allow comparisons between similar cell numbers.
-
FViolin plot of all 10 samples in (B) indicating the distribution of number of transcripts per sample.
-
GViolin plot indicating number of transcripts per cluster from 6 samples (3 biological replicates per strain) represented in UMAP shown in Fig 3B. Clusters 10 and 13 had fewer transcripts compared with all other clusters. Attempts at annotation of these two clusters mostly identified cellular processes instead of obvious cellular lineages.
-
HUMAP embeddings showing individual cells for clusters 4, 5, and 9 from Fig 3B. Trajectory of pseudotime analysis based on gene expression, using Slingshot, is overlaid on UMAP. Cluster 4 represents yolk sac blood island cells, which are common progenitors of vascular endothelium (cluster 5) and primitive erythrocytes (cluster 9).
-
I, JPseudocolor plots for replicate FACS analysis on cells from EBs for both B6 and D2 strains. Cells from technical and biological replicates were labeled with anti‐EpCAM (I) and anti‐SOX1 antibody (J). Gated population indicates percentages of EpCAM+ (Q3) or SOX1+ cells. Replicate 1 is redisplayed here from Fig 3E to show with the other replicates.
To validate transcriptional measurements of differentiation bias, FACS analysis was performed on protein abundance using antibodies against cell lineage markers. Compared with B6, a greater proportion of D2 EB cells expressed EpCAM, indicating definitive endoderm differentiation (13.7% versus 38.5%, respectively; Figs 3E and F, and EV3I and J). Likewise, a greater proportion of cells from B6 EBs expressed SOX1 compared with D2 (11.04% versus 0.61%), indicating neuroectoderm bias. In summary, differentiation propensity is predominantly driven by genetic background. Importantly, B6 bias in neuroectoderm differentiation discovered in EBs is consistent with lineage priming of neuronal development evident in EpiLCs.
Cellular systems genetics identifies trans‐regulation of chromatin accessibility and gene expression
To identify loci influencing differences between strains, we took a cellular systems genetics approach by deriving ESCs from 33 individual BXD recombinant inbred mice, each representing a unique homozygous mosaic of the B6 and D2 founders (Peirce et al, 2004). RNA‐ and ATAC‐seq were performed for both ESCs and EpiLCs. PCA of total RNA found that PC1 separated cell state (57.6% of total variance, Fig 4A), while PC2‐9 captured variance in genetic background (22.2% total variance, Fig EV4A–C), supporting genetic governance over position within cell state.
Figure 4. Trans‐acting quantitative trait loci co‐regulate chromatin accessibility and gene expression in ESCs and EpiLCs.
-
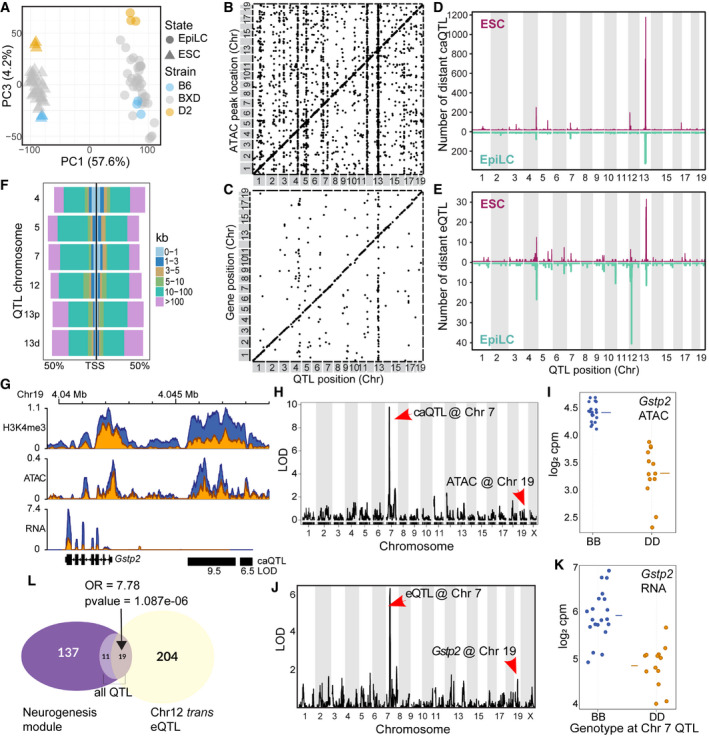
APCA of total transcript abundance in ESCs and EpiLCs from parental (3 biological replicates) and 33 distinct recombinant inbred BXD strains generated from crosses between B6 and D2. PC1 captures variation between cell state (57.6%), and PC3 captures transcript variation within cell states.
-
BScatterplot of genomic locations of individual chromatin accessibility (ca)QTL (x‐axis, LOD > 5) versus location of ATAC peak being regulated (y‐axis) for ESCs. Genetic effects that act locally (n = 3,973), i.e., in cis, fall on the diagonal line, while distal‐acting QTL (n = 13,767) lie off the diagonal. Several prominent distal‐QTL hotspots appear as vertical lines.
-
CSimilar to (B), plotting expression (e)QTL position versus gene location (LOD > 5, n = 701 local‐eQTL, n = 357 distal‐eQTL).
-
DNumber of distal‐caQTL in 1 Mb windows versus genomic location mapped in ESCs (maroon) and EpiLCs (teal).
-
ESimilar to (D) comparing eQTL location and number between ESCs and EpiLCs.
-
FDistribution of caQTL hotspot targets in relation to nearest transcription start site in ESCs as percentage of total targets.
-
GCoverage profile for Gstp2 locus on Chr 19 showing H3K4me3, ATAC, and RNA read depth from merged replicates. Locations of Chr 7 trans‐caQTL targets are indicated with black bars and LOD scores listed below.
-
HLOD score plot from QTL scan in ESCs for chromatin accessibility at the putative enhancer at Gstp2. Genetic variation at Chr 7 QTL distally regulates chromatin accessibility on Chr 19.
-
IPhenotype by genotype plot of the Chr 7 caQTL from h. A B6 haplotype at Chr 7 is associated with increased chromatin accessibility on Chr 19 (horizontal line represents mean).
-
J, KSimilar to H&I showing QTL mapping and haplotype effect for expression of Gstp2 in ESCs (horizontal line represents mean).
-
LEuler diagram showing overlap between genes in the neurogenesis module (m5 from Fig 2H) and Chr 12 trans‐eQTL (LOD > 4) in EpiLCs (enrichment compared to all eQTL in neurogenesis module, Fisher’s exact test—odds ratio (OR) = 7.78, P‐value = 1.087e‐06).
Figure EV4. QTL mapping chromatin accessibility and gene expression in EpiLCs.
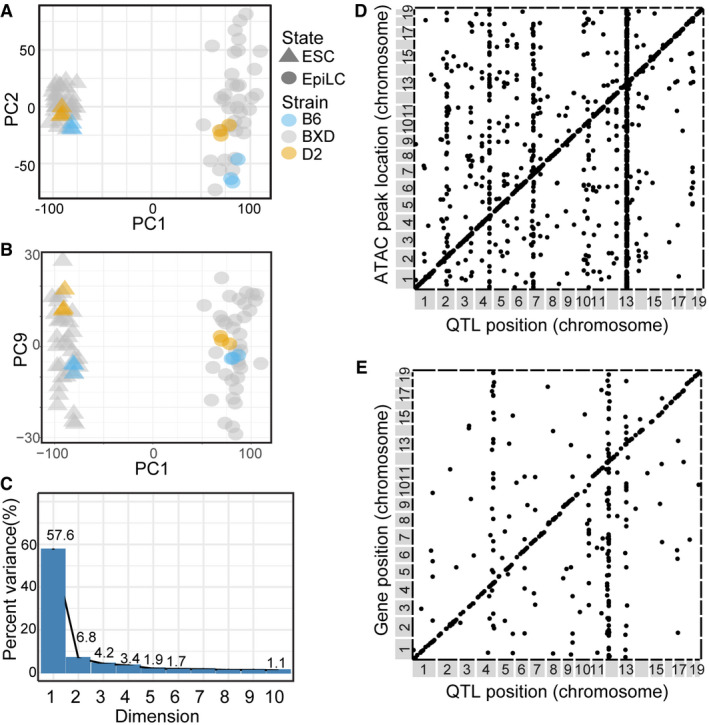
- Scatterplot of PC1 and PC2 showing variance of total transcript abundance from RNA‐seq for 33 distinct strains of BXDs and 3 biological replicates each for B6 and D2 strains in ESCs and EpiLCs.
- Similar to (A) showing variance in PC1 versus PC9.
- Scree plot shows variance explained for PC1‐10. Visual inspection of PC2‐9 (total variation = 22.2%) identified that, while biological replicates of parental strains tightly cluster, they are separated by variance captured by the PC, suggesting these PCs represent genetic contribution to transcript abundance within cell state. Individual BXDs can have transcriptomes with greater variation compared to the inbred parents, as is often the case in genetic populations with segregating alleles.
- Scatterplot of genomic locations of individual caQTL (x‐axis, LOD > 5) versus location of ATAC peak being regulated (y‐axis) for EpiLCs. Genetic effects that act locally (n = 4,463), i.e., in cis, fall on the diagonal line, while distal‐acting QTL (n = 1,172) lie off the diagonal.
- Similar to (D), plotting eQTL position versus target gene location (n = 318 local‐eQTL, n = 200 distal‐eQTL).
Genetic mapping identified abundant local (cis) and distal (trans) chromatin accessibility (ca) and gene expression (e) quantitative trait loci (QTL) in both ESCs and EpiLCs (Figs 4B–E and EV4D and E, Datasets [Link], [Link], [Link], [Link]). Six prominent caQTL “hotspots” were identified in ESCs, most exhibiting shared distal co‐regulation of chromatin accessibility and gene expression. Interestingly, these QTL hotspots are also active in EpiLCs. Notably, EpiLC hotspots harbored a greater proportion of trans‐regulated gene targets accounting for 42% (84/200) of all distal‐eQTL compared with 23% (82/357) in ESCs, whereas distal‐caQTL were evenly regulated between states, 66.4% (778/1,172) in EpiLC and 62.9% (2,501/3,973) in ESCs. Further, co‐regulation was not confined within a cell state. The Chr 12 QTL hotspot regulates a greater proportion of putative regulatory elements in ESCs, but many more genes in EpiLCs, suggesting state‐dependent trans‐regulation (Fig 4D and E, Appendix Fig S2A).
One causal molecular chain explaining shared regulation of distal chromatin accessibility and gene expression is whether a factor within the QTL regulates chromatin in trans, which then mediates local gene expression in cis. This would be evident by paired targets of caQTL and eQTL mapping to the same locus. Indeed, most distal chromatin targets are 10‐100 kb from the nearest promoter, suggesting putative regulatory elements that may act in cis (Fig 4F). Gstp2, located on Chr 19, is a member of module 9 associated with B6 enrichment of mitotic phase transition. Differential expression of Gstp2 is correlated with differential chromatin accessibility at a nearby putative regulatory element (Fig 4G), with B6 showing higher levels of both. QTL mapping in BXDs identified a single locus on Chr 7, with the B6 haplotype increasing both molecular features in trans (Fig 4H–K). Extending these observations within cell type, about half (46.96%) of all genes regulated by a distal‐eQTL hotspot were associated with a paired caQTL in ESCs, whereas the majority of EpiLC QTL targets were not paired (Appendix Fig S2B, Dataset EV9). Interestingly, the Chr 12 hotspot was unique with a distinct bias toward paired caQTL and eQTL across cell states. While a single Chr 12 target gene matched a nearby caQTL in EpiLCs, 4 of the EpiLC gene targets were associated with chromatin targets in ESCs (Appendix Fig S2C). In EpiLCs, Chr 12 QTL target genes were significantly enriched, over all other eQTL, for developmentally primed genes exhibiting bivalent promoters (Mas et al, 2018) (Fisher’s exact test, OR = 2.95, P‐value = 5.66e‐10) in addition to enrichment for GO terms associated with neuronal development programs (Fisher’s exact test, OR = 7.8, P‐value = 1.087e‐6; Fig 4L). In fact, of the 19 neurogenesis genes distally regulated by Chr 12, 15 were among genes annotated as bivalent and poised for gene activation upon differentiation. Similar to the observed neural bias in B6, the presence of a B6 haplotype in BXD‐derived lines at Chr 12 leads to increased expression of the distal neuronal‐associated genes (Fig EV5A). Summarizing the differential expression of the 19 distal neuronal‐associated genes among the 33 BXD EpiLCs as an eigengene, we performed QTL mapping and found expression of the eigengene maps to the same locus on Chr 12 (Fig EV5B). These data suggest that factors expressed from within the QTL hotspots variably regulate chromatin accessibility distally, ultimately leading to variation in transcript abundance of developmentally important genes.
Figure EV5. B6 haplotype at Chr 12 eQTL increases expression of genes found within the genotype‐by‐state neurogenesis module.
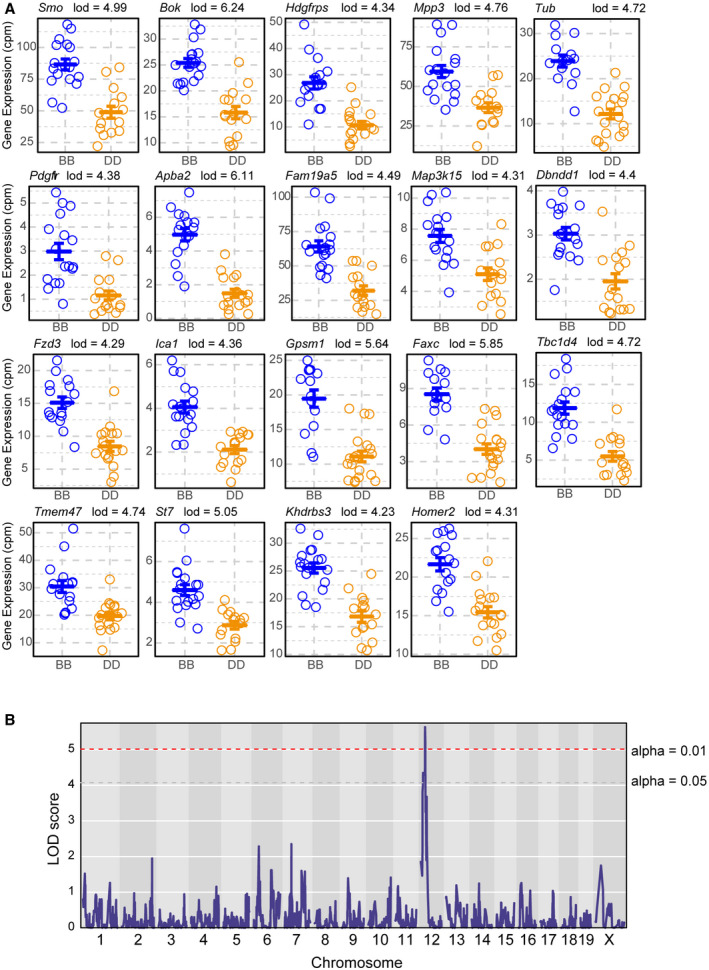
- Phenotype‐by‐genotype plots for all 19 Chr 12 eQTL targets that are found within the GxS neurogenesis module (m5). In each individual case, having the B6 haplotype at Chr 12 resulted in higher expression of the eQTL target gene, consistent with B6’s differentiation propensity toward neuroectoderm (mean ± SEM).
- LOD plot for Chr 12 neurogenesis module eigengene. The eigengene is represented by the first principal component of the expression matrix for the genes indicated in panel (A). The modules maximum LOD score (5.63) maps to the proximal end of Chr 12 with a 1.5 LOD‐drop confidence interval between 14753136 and 29439595, the same interval that the individual gene targets map to. Dashed lines represent empirical P‐value thresholds based on 1,000 permutations (gray < 0.05, red < 0.01).
Genotype of QTL predicts chromatin state at distal targets
The QTL mapping suggests that chromatin state (active versus repressed) at target loci is determined by the genotype at the QTL (trans) and not the local genotype at the peak (cis). To test this prediction, histone H3 lysine 4 trimethylation (H3K4me3) was used as an alternative indicator of active chromatin. Differential H3K4me3 between B6 and D2 parental lines was highly correlated (albeit twofold to fourfold higher) with differential chromatin accessibility between B6 and D2 haplotypes at the QTL in BXD strains (Fig 5A). For example, BXD strains that had high accessibility when genotypically B6 at a QTL also had higher H3K4me3 level at that distal locus in the B6 parent when compared to D2. However, to accurately determine whether the QTL drives chromatin state, the haplotype at the local H3K4me3 site would need to be different from that of the QTL. To accomplish this, H3K4me3 ChIP‐seq data were analyzed from two independent BXD strains (BXD75 and BXD87; Fig 5B–D), for which the genotype at the QTL and target should be different at approximately 50% of targets. As an example, differential chromatin accessibility on Chr 18 at 12.157 Mb was mapped to a QTL on Chr 4, resulting in higher H3K4me3 levels when the haplotype at the QTL is D2. In agreement, the H3K4me3 level at this location is higher in the D2 parent than in B6 and both BXD75 and BXD87 have low levels of H3K4me3. However, BXD87 carries the D2 haplotype at the Chr 18 locus, but carries the B6 haplotype at the Chr 4 QTL, suggesting that the distal haplotype of the QTL determines the chromatin state. To generalize this observation, for all peaks for which haplotypes differ (n = 1,264 distal‐QTL targets) the ratio of H3K4me3 level in the two BXD strains was independently determined for both the parental strain that shared the haplotype at the peak location (cis) and the parental strain that shared haplotype of the QTL (trans). In aggregate, the H3K4me3 level in the BXD strains was consistently closer to that of the parental strain that shared haplotype at the QTL (ratio ~1, Fig 5C and D), validating the prediction that the haplotype of the QTL drives chromatin state.
Figure 5. trans‐QTL regulate chromatin binding of transcription factors required for pluripotency and differentiation.
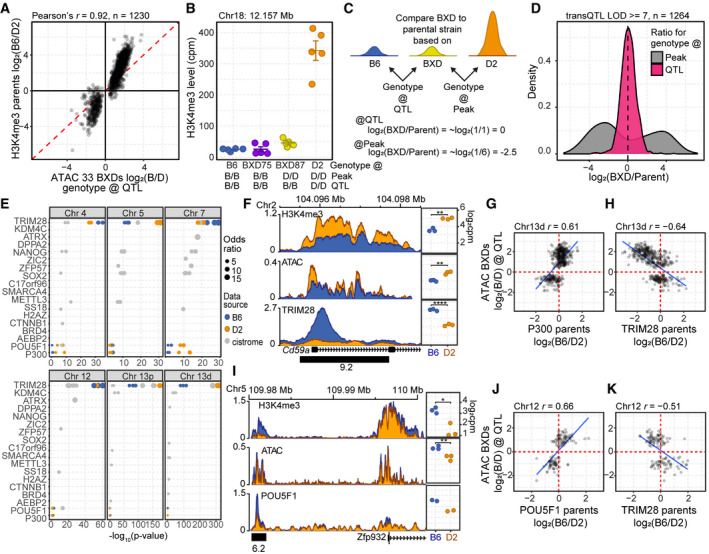
- Scatterplot of difference in chromatin accessibility in the 33 BXD inbred versus difference in H3K4me3 modification in the parental strains for trans‐caQTL (LOD ≥ 8). ATAC‐seq signal represents the log2 ratio of the average signal based on the genotype at the QTL.
- H3K4me3 level is predicted by the genotype at the QTL versus local genotype. H3K4me3 ChIP‐seq for B6 and D2 parental lines compared with two BXD strains (mean ± SEM). Genotypes for both the location of the peak and location of the QTL are indicated below.
- Scheme to determine whether local (peak) or distal (QTL) genotype predicts chromatin state. A ratio of ~1 (log2(ratio) ≈ 0) indicates similar chromatin modification.
- Log2 fold change of H3K4me3 level when comparing BXD75 or BXD87 to the parental genotype either at the location of the peak or based on the genotype at the QTL for all trans‐caQTL for which the local genotype and QTL genotype differ (LOD score ≥ 7).
- Gray—Publicly available ESC ChIP‐seq datasets were evaluated for significant overlap of QTL targets (Fisher’s exact test, top ten factors with q‐value cutoff < 0.01). To validate a subset of these results, ChIP‐seq was performed for TRIM28 (N = 3), POU5F1 (N = 2), and P300 (N = 2) in B6 (blue) and D2 (orange) ESCs. Each circle represents a biological replicate or independent ChIP‐seq accession from Cistrome.
- Left—Coverage profiles (averaged biological replicates, N = 3) for ChIP factor occupancy at Cd59a on Chr 2 under trans‐regulation by the Chr 13d QTL. Distal‐caQTL target region used for quantification is indicated by a black box with LOD score below. Right—Scatterplots for ChIP factors comparing quantitative level of modification/binding for independent replicates between B6 and D2 ESCs.
- The difference in occupancy of P300 in the parental strains is correlated to difference in chromatin accessibility in the 33 BXDs based on the genotype of the BXD at the QTL for Chr 13 caQTL targets (LOD > 7, Pearson’s r = −0.61).
- TRIM28 occupancy at the same targets of Chr 13d caQTL in (F) was negatively correlated with ATAC signal (Pearson’s r = −0.64).
- Similar to (F), coverage profiles for ChIP factor occupancy for a putative regulatory element on Chr 5 near Zfp932, targeted by Chr 12 trans‐caQTL, showed greater occupancy of POU5F1 and increased open chromatin compared with D2. Distal‐caQTL target region used for quantification is indicated by a black box with LOD score below.
- The difference in occupancy of POU5F1 in the parental strains is correlated to difference in chromatin accessibility in the 33 BXDs based on the genotype of the BXD at the QTL for Chr 12 caQTL targets (LOD > 7, Pearson’s r = −0.66).
- Similar to (J) showing negative correlation between TRIM28 occupancy and chromatin accessibility (Pearson’s r = −0.51).
Data information: P‐values in (E, H) represent two‐sided t‐test, *< 0.05, **< 0.01, ****< 0.0001)
Occupancy of pluripotent TFs at regulatory elements is directed by caQTL
Given that expression levels of pluripotent TFs are similar, whereas accessibility of their binding motifs is variable, we hypothesized that causal genes underlying hotspot QTL regulate TF occupancy in trans. We calculated enrichment of overlap between publicly curated ChIP‐seq datasets performed in mESCs and our distal hotspot caQTL intervals (Fig 5E, Dataset EV10). While binding of TRIM28, a chromatin repressor (Friedman et al, 1996), showed the most significant overlap across all caQTL targets, pluripotent TFs, enhancer activators, and chromatin remodelers were all enriched depending on the QTL. For example, targets of QTL on Chrs 4, 5, 7, and 12 are all variably enriched for POU5F1, P300, NANOG, and SOX2.
To validate binding of TFs at QTL targets and test for differential occupancy between strains, we performed TRIM28, P300, and POU5F1 ChIP‐seq in B6 and D2 mESCs and correlated changes in occupancy in the parents with our chromatin data from BXDs (Fig 5E–K, Appendix Fig S3). For example, a Chr 13d eQTL and caQTL target at the Cd59a locus showed higher TRIM28 binding in B6 correlated with reduced chromatin accessibility, H3K4me3 level, and gene expression in D2 (Fig 5F, Dataset EV11). This example can be generalized to most Chr 13 QTL targets. Distal targets of Chr 13d showed positive correlation between differential P300 binding in the parents and differential chromatin accessibility in the BXDs when grouped by the genotype at the QTL (Fig 5G), whereas differential TRIM28 binding is negatively correlated with chromatin accessibility at the same loci (Fig 5H). At a Chr 12 caQTL target, increased POU5F1 occupancy in B6 coincided with greater open chromatin near Zfp932 on Chr 5 (Fig 5I). Chr 12 distal‐caQTL targets show a positive correlation between differential POU5F1 binding and chromatin accessibility (Fig 5J) and negative correlation with TRIM28 occupancy (Fig 5K). Reanalyzing overlap of our ChIP‐seq data with QTL targets, we found that generally when one parental background had higher TRIM28 repressor binding, the other parent exhibited higher TF binding (Fig 5E colored points, Dataset EV11). Together, these data show that trans‐regulation of chromatin in ESCs has a significant impact on regulatory elements bound by factors critical for establishment and maintenance of pluripotency and suggest that this might be due to differential binding of the TRIM28 repressor.
KRAB zinc‐finger proteins are implicated as trans‐acting factors underlying QTL
The enrichment of TRIM28 binding at the caQTL targets suggests that repression in one strain may drive differential accessibility between strains. To test this, we measured chromatin accessibility and gene expression using ESCs derived from heterozygous (B6xD2)F1 hybrid mice. The locus near the genes Riok3 and Rmc1 (3110002H16Rik) provided an example of co‐regulation for both chromatin accessibility and gene expression by the same QTL (Fig 6A). A prominent TRIM28 binding site showed differential occupancy with increased binding in B6 strain, whereas P300, ATAC, and H3K4me3 levels were reciprocally higher in D2. Similarly, there are twelve chromatin accessibility targets in this region all regulated by the distal Chr 4 QTL, which showed reduced accessibility when the QTL is B6 (Dataset EV5). In (B6xD2)F1 hybrids, both expression of Riok3/Rmc1 and chromatin accessibility at the TRIM28 binding site were reduced to similar levels found in the B6 parent (Fig 6B). These data suggest that the B6 allele of Chr 4 QTL dominantly suppresses gene expression and chromatin accessibility on both B6 and D2 chromosomes. To generalize this observation, we used the approach outlined in Tian et. al (2016) to characterize QTL effects. When the additive effect (half the distance between each parental mean) equals the dominant effect (F1 minus the combined parental mean), the effect of the QTL is considered dominant, and when these two values are plotted against each other, the results should fall along a diagonal line. If the dominant effect is repressive, these values will be negative (parental mean is greater than F1). The majority of Chr 4 caQTL targets showed a pattern consistent with being dominantly repressed (Fig 6C), with approximately equal numbers being more open when the haplotype at the QTL is B6 or D2.
Figure 6. Validation of Chr 4 QTL on gene expression and chromatin accessibility implicate KRAB‐ZFPs in trans‐regulation.
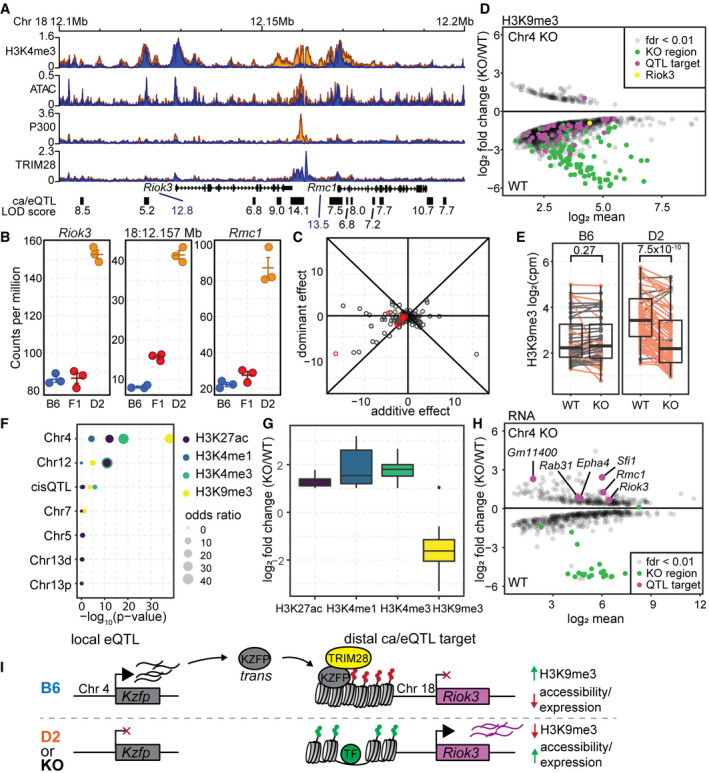
- Coverage profile for H3K4me3, ATAC, P300, and TRIM28 ChIP at a target locus for the QTL on Chr 4 for both chromatin accessibility (black boxes) and gene expression (Riok3 and Rmc1). LOD scores are listed under each target locus.
- Gene expression (Riok3 and Rmc1) or chromatin accessibility (18:12.157 Mb) level measured in B6, D2, and (BxD)F1 hybrid mESCs. For this locus, both gene expression and chromatin accessibility are dominantly repressed in the F1 (mean ± SEM).
- Scatterplot of the additive versus dominant effect for each Chr 4 caQTL target locus. Points along the diagonal lines indicate dominant effects (red—caQTL targets found at the Riok3/Rmc1 locus.
- MA plot of significant (FDR < 0.01) differential H3K9me3 level between wild‐type (B6) mESCs and those with a 2.47‐Mb deletion encompassed by the Chr 4 QTL (n = 2 replicates). Regions that overlap Chr 4 caQTL targets are indicated in magenta (Chr18:12.157 Mb locus from (A) in yellow).
- Box‐and‐whisker plot indicating change in H3K9me3 levels between WT and Chr 4 KO mESCs grouped by whether chromatin accessibility is higher when the QTL is B6 (left) or D2 (right). Dots represent mean values from biological duplicate experiments, and lines connect means. Orange indicates loci for which H3K9me3 signal decreases in the KO (P‐value indicated on top, two‐sided paired Wilcoxon’s test).
- LOLA analysis of significant enrichment between sites that change between WT and Chr 4 KO mESCs and overlap QTL hotspot targets. For H3K4me3, H3K4me1, and H3K27ac, there was only one ChIP experiment performed, and therefore, all sites with a log2 fold change ≥ 1 were used for overlap enrichment.
- Box‐and‐whisker plot indicating direction of change for histone modifications between WT and Chr 4 KO cells.
- MA plot similar to (D) showing change in gene expression between WT and Chr 4 KO mESCs (n = 2). Chr 4 QTL targets are indicated in magenta and increase expression in the KO.
- Model of trans‐regulation of chromatin state for the Chr 4 QTL. Local variation at the Chr 4 QTL results in differential expression of KZFPs, leading to decreased expression of distal genes through recruitment of TRIM28 and formation of heterochromatin (H3K9me3).
Data information: In the boxplot, the central lines represent medians, the box represents the first and third quartile, and the whiskers extend 1.5 times the interquartile range.
The Chr 4 QTL was mapped to an approximate 5.6 Mb region (Chr 4: 143,302,047–148,864,661 bp) that encompasses a cluster of genes encoding a class of chromatin repressors known as KRAB zinc‐finger proteins (KZFPs). Canonically, KZFPs provide sequence‐specific DNA binding through their zing‐finger domains and recruit TRIM28 to repress transposable element expression (Friedman et al, 1996; Rowe et al, 2010; Bruno et al, 2019). To study transposable element reactivation, a group recently engineered a B6 ESC line to contain an approximate 2.47‐Mb deletion (~ Chr 4: 145,383,917–147,853,435 bp) (Wolf et al, 2020) that removed a cluster of 21 genes encoding KZFPs within the Chr 4 QTL interval. In order to validate trans‐regulation between B6 and D2, RNA‐ and ChIP‐seq data collected from the Chr 4 deletion (KO) and matched wild‐type (WT) mESCs were reanalyzed in context of the QTL targets mapped here. KZFPs/TRIM28 recruit a complex that establishes heterochromatin marked by the histone modification H3K9me3. H3K9me3 loci that were identified to be differentially modified between WT and KO ESCs (FDR < 0.01, n = 1,992) showed significant overlap with Chr 4 QTL targets (Fisher’s exact test, P‐value = 1.7 × 10−47, odds ratio = 64.4), including the TRIM28 binding site at Riok3/Rmc1 locus (Fig 6D). All but one of these QTL targets showed higher H3K9me3 modification in WT cells, suggesting repression was attenuated in the KO, similar to increased open chromatin when the QTL haplotype is D2. The genetic effect of the QTL predicts that only those targets that showed higher accessibility in D2 (repressed in B6) should have decreased H3K9me3 in the KO; indeed, this prediction was found to be true (Fig 6E). Additionally, active histone marks H3K27ac, H3K4me1, and H3K4me1 also showed differential modification (|log2 fold change| > 1) at Chr 4 QTL targets compared with other QTL (Fig 6F) and predicted increases due to derepression in the KO (Fig 6G). Finally, of the seven distal gene targets controlled by Chr 4 QTL that are higher expressed in D2 and detected in the KO dataset, six were expressed significantly higher in the KO (Fig 6H, Fisher’s exact test, P‐value = 1.36 × 10−7, odds ratio = 109.2), including Riok3 and Rmc1. Additionally, of all the gene targets regulated by other QTL hotspots mapped in this study (i.e., Chrs 5, 7, 12, or 13) only one gene (Cd59a, regulated by Chr 13 distal‐QTL) showed differential expression in the Chr 4 KO highlighting the QTL specificity to Chr 4 targets. Importantly, expression of four of the genes that encode KZFPs within the deleted region was regulated by local, cis‐acting, eQTL (FDR < 0.05), resulting in higher expression in B6 than in D2 (log2 fold change > 1, Dataset EV6). This suggests that one or more KZFPs, that are higher expressed when the Chr 4 QTL is B6, represses distal chromatin and gene expression. Alternatively, the D2 haplotype lacks or reduces the expression of the KZFPs; therefore, the QTL targets, such as Riok3, are expressed (Fig 6I). While a greater number of genes were differentially expressed after deletion of the Chr 4 locus compared with the number of genes regulated by the Chr 4 QTL, many more KZFPs are deleted in the KO compared with the number that are differentially expressed between B6 and D2, potentially explaining the increased impact of the KO. Together, these lines of evidence suggest that the underlying molecular identity of the Chr 4 QTL is due to the activity of rapidly evolving KZFPs with either expression or structural differences between B6 and D2.
Discussion
Here, we provide a rationally designed genetic framework to critically assess genetic contribution to differentiation potential. Our data suggest that ESCs from B6 and D2 genetic backgrounds (i) reside at different locations along a pluripotent spectrum, (ii) exhibit divergence in gene networks during transition from naïve to formative pluripotency, and (iii) harbor at least 6 loci that distally co‐regulate chromatin accessibility, transcription factor binding, and gene expression; all of which are consistent with outcomes that alter cell‐fate specification.
Modeling in vivo developmental progression in vitro has captured critical intermediate states between naïve and primed pluripotency (Morgani et al, 2017). Formulation of culture conditions permitting propagation of intermediate states, such as peri‐implantation rosettes, has revealed aspects of pluripotent gene networks important for balancing maintenance of self‐renewal with competency for differentiation (Bedzhov & Zernicka‐Goetz, 2014; Neagu et al, 2020). Here, we show ESCs derived from different genetic backgrounds, B6 and D2 strains of mice, yet cultured in identical conditions achieve occupancy of discrete states along the pluripotent spectrum. B6 ESCs acquire transcriptional profiles closer to ground state pluripotency, even in undefined media conditions, and upon transitioning to formative pluripotency rapidly, they dissolve the naïve program along with apparent priming toward neuroectoderm. In contrast, D2 ESCs exit pluripotency more slowly, as evidenced by stable expression of naïve TFs with little overt lineage bias. We observe conflicting behavior of ESCs as they exit naïve pluripotency and establish the capacity for multi‐lineage differentiation in EpiLCs. The position of B6 ESCs in gold standard ground state does not guarantee responsiveness to differentiation cues; instead, B6 EpiLCs appear biased toward neuronal lineages. In comparison, D2 ESCs are positioned in a more developmentally progressed state of naïve pluripotency, showing expression of contradicting programs representing WNT signaling alongside early developmental patterning. This may suggest D2 cells are primed to exit naïve but are prevented by WNT signaling. Despite D2 ESC status, EpiLCs were able to retain higher expression of naïve markers when prompted to exit. Whether WNT signaling continues to play a role in slowing the transition and preventing early priming toward lineage commitment is an outstanding question. This highlights the utility of naturally occurring genetic variation to reveal critical stages of development that allow for multi‐lineage commitment. Further investigation into pathways allowing for a slower transit while creating an enhanced opportunity to receive differentiation cues may provide applications to better control cell‐fate choice in vitro.
Even though B6 and D2 ESCs occupy different positions along the pluripotent spectrum, they express similar levels of naïve markers, providing a conundrum as to where differences arise. To reconcile this, we adopted a multi‐omics approach, measuring chromatin accessibility as a gauge for regulatory element activity. This approach showed that chromatin accessibility at pluripotent TF motifs is higher in B6 than in D2, indicating other trans‐acting chromatin regulation may determine pluripotent spectrum. We identified 6 QTL hotspots that alter binding of naïve TFs in trans, and ultimately differentially regulate proximal genes pertinent to cell fate. Our study additionally suggests chromatin priming in ESCs influences gene expression in EpiLCs, for example, instructing commitment to neuroectoderm. The fact that a larger proportion of caQTL and eQTL are paired in ESCs but not EpiLCs could suggest that the primary effect of QTL on influencing chromatin accessibility resides in ESCs, ultimately leading to secondary or tertiary gene expression changes after differentiation. In EpiLCs, these downstream expression changes would still map back to the same distal locus identified in ESCs but may no longer be accompanied by local changes in chromatin directly. This finding supports recent observations of chromatin priming preceding cell‐fate decisions during gastrulation (Argelaguet et al, 2019) and highlights the potential for early events in development to have differential cascading effects on adult phenotypes (Maurano et al, 2012).
Several lines of evidence support that the QTL discovered in this study are of significant developmental importance and are driven by variable KZFPs. Aside from roles in governing cell state transitions described here, QTL overlapping the same positions on Chrs 4, 12, and 13 have all been implicated in a variety of developmental and disease phenotypes such as limb abnormalities (Johnson et al, 1995), cleft palate (Plamondon et al, 2011), lupus (Treger et al, 2019), and association with differences in priming sites of meiotic recombination (Baker et al, 2019), imprinting stability in PSCs (Swanzey et al, 2020), and stabilization of ground state pluripotency (Skelly et al, 2020). A common family of DNA‐binding proteins, encoded by genes underlying all three of these QTL regions, is the rapidly evolving KZFPs. Here, we show that deletion of a cluster of genes encoding KZFPs within the Chr 4 QTL critical region (Wolf et al, 2020) altered chromatin state and gene expression as predicted. Further, putative regulatory elements targeted by all six QTL hotspots were enriched for binding by TRIM28 (Fig 5E), which is recruited to chromatin through interaction with KZFPs (Friedman et al, 1996). Additionally, the effect of the QTL was found to be dominantly repressive in F1 hybrids, consistent with the function of KZFP/TRIM28 complexes in formation of heterochromatin in trans. Notably, a single KZFP contained within the Chr 13 QTL hotspot interval was shown to be causal in the progression of a lupus phenotype (Treger et al, 2019). In addition, while the other studies outlined above largely have not pinpointed causal factors, the overlapping molecular and physiological QTL harbor clusters of newly emergent murine KZFPs (Kauzlaric et al, 2017; Bruno et al, 2019). This provides exciting future work into assigning causality to a rapidly evolving gene family whose divergence in different strain backgrounds may account for evolution of regulatory function that shapes development and disease (Elmer & Ferguson‐Smith, 2020).
The strength of the cellular systems approach to understand cell state transitions is best exemplified by the B6 trajectory toward neuroectoderm. First, genotype‐by‐state interactions during transition to formative pluripotency uncovered strain divergence during exit from naïve pluripotency and were responsible for installing competency of lineage specification with a predisposition to neuroectoderm in B6. Second, while spontaneous differentiation to EBs confirmed B6 trajectory toward neuroectoderm, genetic background influences differentiation toward multiple lineages, both in the primary germ layers and within the same lineage (i.e., primitive erythrocytes (D2) and vascular endothelium (B6)). The B6 bias toward neuroectoderm, and reduced endoderm formation, was recently reported in another manuscript using a panel of four inbred mouse strains that included B6 (Ortmann et al, 2020). While culture conditions used here contained 2i/LIF/serum, Ortmann et al used 2i conditions lacking serum. This suggests that the effect of genetic background is greater than that of culture conditions. Further, the multi‐omics approach taken here identified increased accessibility of ZEB1 motifs in B6 ESCs, suggesting early establishment of differentiation propensity. Finally, these observational studies in the parental lines are extended through genetic mapping in BXDs. QTL mapping identified a locus on Chr 12 regulating chromatin accessibility distally in ESCs, thereby potentially priming a regulatory landscape directing neuronal development in cells harboring a B6 haplotype at the Chr 12 QTL.
In the absence of inductive signals to direct differentiation, neutralization has been described as the “default” development path (Smukler et al, 2006). In vivo inhibition of neuronal programs is due to extrinsic signaling originating from extraembryonic tissue, of which ESC cultures are typically devoid (Andoniadou & Martinez‐Barbera, 2013). Historically, the study of differentiation in vitro has been limited to ESCs derived from B6 or 129 mouse strains (Lenka & Ramasamy, 2007; Argelaguet et al, 2019). Our study, however, suggests this neuronal default paradigm may not be universal to all genetic backgrounds, supporting other recent work (Ortmann et al, 2020). It is therefore paramount to assess causal factors underlying the Chr 12 QTL, which are likely to play a role in capacitation for multi‐lineage differentiation.
In total, this work demonstrates that ESCs derived from genetically diverse strains do not share equal developmental potential in vitro. Recent experiments have shown that differences in cell‐fate choice during development may be critical in predisposing individuals to complex diseases due to underlying differences in cell‐type composition (The GTEx Consortium atlas of genetic regulatory effects across human tissues, 2020). Clearly, further investigation into genetic governance of differentiation is needed to understand its potential role in complex traits.
Materials and Methods
Derivation of mouse embryonic stem cells
Mouse embryonic stem cells were derived and maintained in conditions previously reported (Czechanski et al, 2014). All mice were obtained from the Jackson Laboratory (Bar Harbor, ME) including C57BL/6J (stock number 000664), DBA/2J (stock number 100006), and BXD recombinant inbred lines (see Table EV1). All animal experiments were approved by the Animal Care and Use Committee of the Jackson Laboratory (summary #04008). Obtaining mESCs derived in this study is achieved through contacting the corresponding author. All cell lines were tested negative for mycoplasma.
Cell culture
Naïve pluripotency supported in ESCs
A vial containing 3 million ESCs (P4–P6) was thawed onto a 60‐mm tissue‐treated culture dish seeded with irradiated mouse embryonic fibroblasts (MEFs) as feeders in DMEM high glucose base medium supplemented with 15% fetal bovine serum (FBS, Lonza, cat. no. 14‐501F lot no. 0000217266), 1X Pen/Strep, 2 mM GlutaMAX, 1 mM sodium pyruvate, 0.1 mM MEM‐NEAA, 0.1 mM 2‐mercaptoethanol, 103 IU LIF, 1 μM PD0325901, and 3 μM CHIR99021. ESCs were expanded for 2–3 days to reach ~70% confluency for molecular and differentiation assays.
Differentiation to formative pluripotency in EpiLCs
EpiLCs were grown from mESCs as previously described (Hayashi et al, 2011; Buecker et al, 2014). Confluent ESCs were washed with 1X PBS, trypsinized (0.05%) to form single‐cell suspension, and filtered through a 40‐µM mesh, and MEFs were excluded by settling on 60‐mm dishes coated with 0.01% gelatin for 30 min. ESCs were seeded at a density of 200,000 cells per 60‐mm dish coated with 5 µg/ml fibronectin in medium containing N2B27 supplemented with 2 mM GlutaMAX, 1 mM sodium pyruvate, 1X Pen/Strep, 0.1 mM MEM‐NEAA, 0.1 mM 2‐mercaptoethanol, 1% KOSR, and 12 ng/ml bFGF. Cells were transitioned to not exceed 48 h. Cells were gently dissociated using TrypLE (Thermo Fisher Scientific, cat. no. 12‐605‐010) for subsequent molecular and differentiation assays.
Spontaneous differentiation to EBs
To form EBs, EpiLCs were washed with 1× PBS and dissociated with TrypLE and resuspended in EB medium containing DMEM high glucose base supplemented with 15% FBS, 2 mM GlutaMAX, 0.1 mM MEM‐NEAA, 1 mM sodium pyruvate, 1× Pen/Strep, 0.1 mM 2‐Mercaptoethanol, and 12 ng/ml bFGF at a seeding density of 750 cells/EB. Uniform and reproducible EBs were achieved using AggreWell 400 plates (STEMCELL Technologies, cat. no. 34415) containing 1,200 microwells. Each well contains 2 ml of single‐cell solution of 900,000 cells per well (750 cells/EB). Preparation of AggreWell 400 was followed per manufacturer’s instructions, with modification to only use middle 8 wells to ensure reproducibility of cultures. Cells were allowed to aggregate in microwells for 48 h undisturbed. After 48 h, each well was filtered through a 40‐µm mesh to discard any unincorporated cells and collect newly formed EBs. Filter was then inverted, and EB medium was passed through filter to collect EBs into 100‐mm Corning Ultra‐low attachment culture dish (MilliporeSigma, cat. no. CLS3262). Dishes were placed on BellyButton rotator and left undisturbed for another 48 h to allow EBs to further develop while limiting aggregation of individual EBs within culture. After 48 h, EBs were filtered through a 40‐µM mesh to discard unincorporated cells. Single‐cell suspensions were achieved by incubating EBs in 0.25% trypsin for 2 min at 37°C followed by manual disaggregation with an Eppendorf P1000 manual pipette. Trypsin was inactivated by resuspending cells in PBS supplemented with 10% FBS, and the cell suspension was filtered through a 40‐µM mesh. Cell suspensions were washed twice with PBS before proceeding with processing for scRNA‐seq or FACS analysis.
RNA‐seq sample preparation and sequencing
For mESC RNA collection, naïve culture was trypsinized and MEFs were removed by plating single‐cell suspension onto gelatin‐coated dishes and allowing MEFs to settle for 30 min. One million enriched mESCs were lysed, and RNA was extracted using the RNeasy (Qiagen) RNA Extraction Kit. EpiLCs were harvested using TrypLE, and 1 million cells were used for RNA extraction. RNA concentration and quality were assessed using the NanoDrop 2000 Spectrophotometer (Thermo Scientific) and the RNA Total RNA Nano Assay (Agilent Technologies). Libraries were constructed using the KAPA mRNA HyperPrep Kit (KAPA Biosystems), according to the manufacturer’s instructions. Library quality and concentration were checked using the D5000 Screen Tape (Agilent Technologies) and quantitative PCR (KAPA Biosystems).
ATAC‐seq and ChIP‐seq sample preparation and sequencing
To measure chromatin accessibility in ESCs and EpiLCs, cells were harvested as described above. For parental ESCs and EpiLCs along with BXD ESCs, the OMNI‐ATAC (Corces et al, 2017) protocol was followed using 100,000 cells. For BXD EpiLCs, the FAST‐ATAC (Corces et al, 2016) protocol was followed using 100,000 cells. Libraries were amplified for a total of 8–10 cycles, and DNA was purified using AMPure XP beads (Beckman Coulter). The quality and concentration of ATAC‐seq libraries were evaluated using the Bioanalyzer DNA High Sensitivity Assay (Agilent Technologies) and quantitative PCR (KAPA Biosystems).
For each biological replicate ESC ChIP‐seq library, 10 million cells were harvested and fixed as previously described (Skelly et al, 2020). Cell lysis, chromatin fragmentation, dialysis, and immunoprecipitation were performed as described (Baker et al, 2015). Immunoprecipitation was performed using antibodies against P300 (12 µl, Bethyl A300‐358A), POU5F1 (20 µl, Cell Signaling Tech Oct‐4A rabbit mAb, 5677S), and TRIM28 (10 µl, Abcam 201C, ab22553). ChIP‐seq libraries were constructed using the KAPA HyperPrep Kit (Roche Sequencing and Life Science). Quality and concentration of libraries were assessed using the High Sensitivity D5000 ScreenTape (Agilent Technologies) and KAPA Library Quantification Kit (Roche Sequencing and Life Sciences).
Single‐cell RNA‐seq sample preparation and sequencing
Three biological replicate EBs, starting from independently derived ESCs, were grown for both B6 and D2 parental strains. Additionally, starting from just one of the derived D2 ESC lines, EBs were grown in triplicate to represent technical replicates within cell line. Finally, 3 aliquots of disaggregated cells from one of the D2 EB cultures were subsampled to determine reproducible representation cell populations within EBs as they represent heterogenous organoids. The MULTIseq protocol (McGinnis et al, 2019) was used to pool single‐cell suspensions from EBs across samples to load onto one 10X Chromium lane to reduce batch effects. Each of the 10 samples outlined above was labeled with a unique lipid‐modified oligo following the published protocol using 500,000 cells per sample. After labeling, cells were counted and adjusted so that the final pool represented 3,000 cells per sample. Pooled cells were washed once in PBS and resuspended in PBS containing 1% BSA supplemented with 1% FBS. A single 10× Chromium microfluidic lane was superloaded with 40,000 cells with the goal of obtaining approximately 1,000 barcoded cells per sample after sequencing, demultiplexing, and filtering. Single‐cell capture, barcoding, and library preparation were performed using the 10X Chromium version 3 chemistry, according to the manufacturer’s protocol (#CG00183). cDNA and barcode libraries were checked for quality on an Agilent 4200 TapeStation, quantified by KAPA qPCR, and sequenced on a single lane (95% transcriptome, 5% barcode) on NovaSeq 6000 (Illumina) to an average depth of 100,000 reads per cell.
Fluorescence‐assisted cell sorting
Spontaneously derived embryoid bodies were independently grown from parental ESC lines to represent technical replicates. Single‐cell suspensions were fixed with 4% paraformaldehyde at a concentration of 1 million cells/ml for 15 min at room temperature. Cells were washed with PBS/1% FBS twice before antibody labeling. Cells were blocked for 15 min in either PBS/1% FBS (EpCAM) or Perm/Wash (SOX1) prior to incubation with primary antibody (α‐EpCAM at 1/10,000, Abcam ab71916; α‐SOX1 at 1/300, R&D Systems AF3369) for 45 min at room temperature. After washing three times for 5 min each in PBS/1% FBS (Perm/Wash for SOX1 samples), cells were incubated with secondary antibody for 45 min at room temperature. After washing three times for 5 min each in PBS/1% FBS (Perm/Wash for SOX1 samples), cells were analyzed on Attune NXT Analyzer (Thermo Fisher). Cells were gated using FlowJoTM software, and density of cell populations was visualized using flowVis package in R (https://bioconductor.org/packages/flowViz/).
Statistical analysis
Data shown represent mean ± SEM unless otherwise indicated. Statistical analysis was performed using R version 3.4.1 for qtl mapping, v.3.5.1 for normalization and transformation of inputs for downstream analysis, and v.3.6 for visualization and analysis (R Core Team, 2018) as outlined below. Tests for statistical significance for QTL mapping, and RNA‐, ChIP‐, and ATAC‐seq are outlined using the indicated R packages below. Significance for differences in distributions in Fig 6E was determined using nonparametric two‐sided paired Wilcoxon’s test with the null hypothesis expecting equal medians. Significance for figure panels 3F, 5F, and 5I was determined using unpaired two‐tailed Student’s t‐test assuming unequal variance.
ATAC‐, ChIP‐, and bulk RNA‐seq data processing
Bulk parental, BXD ESC, and EpiLC fastq files were aligned using bowtie (Langmead et al, 2009) to their respective strain‐specific transcriptomes (Ensembl release 84), and transcripts were quantified at gene‐level abundances using EMASE (Raghupathy et al, 2018). RNA‐seq data from Wolf et al (2020) (GEO accession GSE115291) for wild‐type B6 and Chr 4 knockout mESCs (n = 2 replicates each, GSM3173773, GSM3173774, GSM3173775, GSM3173776) were aligned similarly as above to the B6 reference transcriptome and quantified using EMASE. Read counts were filtered for lowly expressed genes (3 counts per million in at least three samples), TMM‐normalized using edgeR (Robinson et al, 2010), and log2‐transformed for differential analysis and QTL mapping.
Illumina adaptors were trimmed from ATAC and ChIP reads using Trimmomatic (version 0.33) and then aligned to mouse reference genome (mm10), modified to incorporate known D2 variants reported in Mouse Genomes Project Database, REL‐1505 (Keane et al, 2011; Yalcin et al, 2011) using hisat2 (Pertea et al, 2016). Duplicate reads were removed using Picard Tools (“Picard Toolkit”, 2019, Broad Institute), and peaks were called for each sample using MACS (Zhang et al, 2008) (version 1.4.2, P = e−5). A comprehensive set of open chromatin locations (i.e., peakome) was generated by combining all ATAC peaks identified across all 33 BXD samples and peaks overlapping by 1 bp merged using bedtools. A similar peakome strategy was used for ChIP samples. H3K9me3 data from Wolf et al (2020) (GEO accession GSE115291 samples GSM3173641, GSM3173642, GSM3173643, GSM3173644) were downloaded from the sequence read archive aligned to mm10 and quantified using combined TRIM28 binding locations identified from ChIP‐seq performed in B6 and D2 in this study. All other locations of histone modification from Wolf et al (H3K4me3—GSM3173670 and GSM3173673; H3K4me1—GSM3173669 and GSM3173672; and H3K27ac—GSM3173671 and GSM3173674) were identified using MACS using paired input DNA (input DNA—GSM3173681 and GSM3173683). ATAC and ChIP read counts were collected using bedtools (Quinlan & Hall, 2010) multicov within respective peakome intervals. Read matrices were TMM‐normalized and log2‐transformed for QTL mapping of ATAC samples in BXDs and downstream differential analysis of chromatin accessibility, histone modification, and ChIP factor occupancy between parental strains.
caQTL and eQTL mapping in ESCs and EpiLCs
Normalized and transformed read counts were used as input for QTL mapping for both transcript abundance and chromatin accessibility. QTL were mapped using the “scan1” function in R/qtl2 (Broman et al, 2019, 2) using a linear mixed model to account for kinship. Processing batch of BXD samples was included in the model as a covariate to account for batch effects (Table EV2). To assess genome‐wide significance in ESC RNA‐seq samples, we calculated empirical P‐values using a permutation strategy (1,000 permutations) and applied a multiple testing correction to obtain FDR values (Benjamini–Hochberg‐adjusted P‐value). Figure 3 reports significant eQTL with LOD cutoff > 5 and caQTL with LOD cutoff > 5 (Datasets [Link], [Link], [Link], [Link]).
Distribution of hotspot caQTL from nearest TSS was determined using ChIPseeker (Yu et al, 2015). Target genes of caQTL were assigned using GREAT (Dataset EV9) (McLean et al, 2010) and further filtered for genes with eQTL mapped to same genomic interval as paired caQTL (within and across cell state).
Differential analysis of gene expression
Differentially expressed transcripts between parental ESCs were determined using pairwise comparisons in edgeR performing quasi‐likelihood F‐test (FDR < 0.05, log2FC > 1). To determine sets of genes enriched in functional programs, we used significantly differentially expressed transcripts between strains (FDR < 0.05) as in input in gene set enrichment analysis (GSEA) (Mootha et al, 2003; Subramanian et al, 2005). Strain‐specific enriched programs were identified using gene sets curated with MSigDB (Subramanian et al, 2005; Liberzon et al, 2011, 2015) and considered significant at FDR < 25% (weighted scoring and 1,000 permutations on phenotypes) and suggestive at nominal P‐value < 0.05 (1,000 permutations on gene set).
Placement of parental ESCs along naïve pluripotency spectrum was determined using previously published RNA‐seq data from ESCs grown in 9 different culture conditions (Hackett et al, 2017). Fastq files were accessed in GEO (Accession: GSE98517, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98517) and uniformly processed to match our RNA‐seq data as described above. Principal components were determined using prcomp function in R on TMM‐normalized and log2‐transformed RNA read counts from the whole transcriptome. Gene contribution to separation of samples along PCs was achieved using factoextra package in R (https://rpkgs.datanovia.com/factoextra/index.html).
To determine differentially expressed transcripts dependent on cell state, strain, and a significant interaction between state and strain, a general linear model (glm; Y = B0 + B1X1 + B2X2 + B3X1X2, where X1 = state, X2 = strain, and X1X2 = interaction term) was applied to normalized RNA read counts from parental ESC and EpiLC samples. Unique expression paths of genes changing similarly across cell state were identified using EBseqHMM (Leng et al, 2015) (following default settings, confidence cutoff = 0.5). Modules were defined by filtering EBseq paths for genes with a significant state by strain interaction (X1X2) from the glm. The principal component analysis was performed separately on each module using the expression of all genes within the module to summarizing the change in expression as an eigenvalue. Gene modules and significantly differentially expressed genes identified in glm were visualized using ComplexHeatmap (Gu et al, 2016) package in R. Gene modules with enriched GO terms were determined using clusterProfiler (Yu et al, 2012) (adjusted P‐value < 0.05).
Differential analysis of chromatin accessibility and ChIP factor occupancy
Differential accessibility of transcription factor motifs between parental ESCs and differences detected across cell states were determined using chromVar (Schep et al, 2017). Unsupervised hierarchical clustering was performed on GC bias‐corrected deviation z‐scores.
To identify differential occupancy of factors at caQTL in ESCs, we tested for overlap of Cistrome (Mei et al, 2017; Zheng et al, 2019) curated ChIP‐seq datasets (mouse factor data downloaded 1/18/19; we extracted mouse ESCs ChIP‐seq for use in this study representing 1,763 ChIP‐seq experiments) along with ChIP‐seq performed in this study (TRIM28, P300, POU5F1) using LOLA (Sheffield & Bock, 2016) package in R. Regions were defined as the collection of chromatin target intervals for each hotspot caQTL; the universe was set as the total ATAC‐seq peakome and LOLA results were filtered by maxRnk (combined score for P‐value and odds ratio). Allele‐dependent ChIP factor binding (P300, TRIM28, POU5F1) correlated with chromatin accessibility at hotspot caQTL was determined using Pearson’s correlation.
Analysis and annotation of single‐cell RNA‐seq
CellRanger was used to align scRNA‐seq fastq files and identify individual cell barcodes from 10X Genomics data retrieving 20,134 individual cell transcriptomes. Individual cells were further classified into MULTIseq lipid hash samples using demultiplex R package (https://github.com/chris‐mcginnis‐ucsf/MULTI‐seq) resulting in 2,150 negative cells (no hash barcode), 3,022 doublets (identified as two hash barcodes), and 14,962 unique cells accounting for ~1,000 cells/sample (Table EV3). Clustering and analysis of scRNA‐seq data was performed using Seurat (Stuart et al, 2019). Cells with > 10% of reads from mitochondrial genes were removed. Preprocess, normalization, and dimensional reduction largely followed default Seurat settings including FindVariableFeatures (nfeatures = 2,000), RunPCA (npcs = 100) with 20 principal components selected for clustering. Because of the large observed differences in clustering between B6 and D2 cells, we used harmony (Nowotschin et al, 2019) to mitigate the impact of cell strain background on clustering with the following settings (theta = 1, dims.use = 1:20, max.iter.harmony = 100). Increasing theta resulted in forced clustering between cells with little biological meaning, while not improving integration between genetic background. Therefore, theta = 1 was chosen to maximize strain integration while preserving cell identity.
Unique and differentially expressed genes in cell clusters were generated from Seurat, and subsequent gene lists were used to annotate cell clusters using MouseMine (Motenko et al, 2015) along with literature searches of top 5 unique and highly expressed genes in each cluster. MouseMine is a public database supporting gene set queries to discover functional relatedness to a biological process, interactions with other genes, and assess spatial expression within relevant anatomical regions of the developing mouse embryo. Nine cell clusters were annotated as a unique cell lineage, and four clusters were enriched for GO terms describing cellular activities with no enrichment in anatomical structures or lineage markers.
Differentiation trajectory of cells in cluster 4 branching to clusters 5 and 9 was inferred using Slingshot (Street et al, 2018). Cells in cluster 4 were selected as starting cluster based on prior knowledge of gene markers indicating yolk sac blood island cells.
Author contributions
CLB and CB conceptualized the study and contributed to methodology. CLB and LGR provided resources. CB, CS, HJF, and AC investigated the study. CB, CS, EIH, DAS, and CLB performed data curation and formal analysis. CB, CS, and CLB performed visualization. CB and CLB wrote the manuscript. CLB, DAS, LGR, and SCM revised and edited the manuscript. CLB performed supervision. CLB, LGR, SCM, and DAS performed funding acquisition.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Acknowledgements
We would like to thank all members of the Baker laboratory for comments and discussion. Additionally, we would like to thank Dr. Martin Pera for constructive feedback on the project and reviewing the manuscript; Dr. Martin Ringwald for assisting with approaches to annotate developmental stages of single‐cell clusters; and Christopher S. McGinnis and the Gartner Laboratory for providing LMOs for MULTIseq. We recognize contributions from The Jackson Laboratory Genome Technologies Services, Flow Cytometry Services, and Single Cell Biology Laboratory for technical assistance and consultation. The Jackson Laboratory scientific services are supported in part through the National Cancer Institute’s Cancer Core Grant P30CA034196. The Special Mouse Strain Resource grant from the National Institute of Health Office of the Director P40OD011102 (L.G.R.) provided support for the BXD recombinant inbred strains used for mESC derivation. This work was supported by the Jackson Laboratory Director’s Innovation Fund to S.C.M., L.G.R., D.A.S., and C.L.B.; C.B was supported by the National Institute of Child Health and Human Development (NICHD) grant T32HD007065; and C.L.B. was supported by the National Institute of General Medical Sciences (NIGMS) grant R35GM133724.
Conflict of interest
The authors declare that they have no conflict of interest.
The EMBO Journal (2022) 41: e109445
Data availability
Original data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE164935 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164935). Along with raw sequencing data, processed data tables in the accession include normalized read counts for gene expression for ESC and EpiLC samples; normalized read counts along with peak intervals for chromatin accessibility and ChIP factor occupancy for ESC and EpiLC samples; and matrices produced by CellRanger for expression libraries for single‐cell RNA‐seq from EBs and single‐cell barcode table displaying EB samples associated with unique lipid‐modified oligos after demultiplexing following MULTIseq pipeline.
H3K4me3 ChIP‐seq data for B6, D2, BXD75, and BXD87 were collected previously (Baker et al, 2019) and are available through GEO accession GSE113192 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113192).
RNA‐seq and histone modification ChIP‐seq data for mESCs from Chr 4 knockout and wild‐type cells lines were published previously (Wolf et al, 2020) and are available through GEO accession GSE115291 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115291).
RNA‐seq data for mESCs grown in different media formulations to access pluripotency spectrum were published previously (Hackett et al, 2017) and are available through GEO accession GSE98517 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98517).
Results in Figs 1 and EV1 for differential gene expression analysis are available as Dataset EV1.
Results in Figs 2 and EV2 and Appendix Fig S1 for defining gene modules are available as Datasets EV2 and EV3.
Results in Figs 3 and EV3 for single‐cell RNA‐seq and FACS analysis are available as Dataset EV4 and Table EV3.
Results in Figs 4 and EV4 for QTL analysis are available as Datasets [Link], [Link], [Link], [Link].
Results in Fig 5E for locus overlap enrichment analysis are available as Dataset EV10.
Results in Fig 5F for ChIP‐seq occupancy are available as Dataset EV11.
References
- Allegrucci C, Young LE (2007) Differences between human embryonic stem cell lines. Hum Reprod Update 13: 103–120 [DOI] [PubMed] [Google Scholar]
- Andoniadou CL, Martinez‐Barbera JP (2013) Developmental mechanisms directing early anterior forebrain specification in vertebrates. Cell Mol Life Sci 70: 3739–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani C‐A, Imaz‐Rosshandler I, Lohoff T, Xiang Y, Hanna CW et al (2019) Multi‐omics profiling of mouse gastrulation at single‐cell resolution. Nature 576: 487–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SJ, Robertson EJ (2009) Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat Rev Mol Cell Biol 10: 91–103 [DOI] [PubMed] [Google Scholar]
- Baker CL, Kajita S, Walker M, Saxl RL, Raghupathy N, Choi K, Petkov PM, Paigen K (2015) PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype‐specific initiation of meiotic recombination. PLoS Genet 11: e1004916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Walker M, Arat S, Ananda G, Petkova P, Powers NR, Tian H, Spruce C, Ji B, Rausch D et al (2019) Tissue‐specific trans regulation of the mouse epigenome. Genetics 211: 831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedzhov I, Zernicka‐Goetz M (2014) Self‐organizing properties of mouse pluripotent cells initiate morphogenesis upon implantation. Cell 156: 1032–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Berge D, Kurek D, Blauwkamp T, Koole W, Maas A, Eroglu E, Siu RK, Nusse R (2011) Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat Cell Biol 13: 1070–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326 [DOI] [PubMed] [Google Scholar]
- Bonifer C, Cockerill PN (2017) Chromatin priming of genes in development: Concepts, mechanisms and consequences. Exp Hematol 49: 1–8 [DOI] [PubMed] [Google Scholar]
- Bradley A, Evans M, Kaufman MH, Robertson E (1984) Formation of germ‐line chimaeras from embryo‐derived teratocarcinoma cell lines. Nature 309: 255–256 [DOI] [PubMed] [Google Scholar]
- Broman KW, Gatti DM, Simecek P, Furlotte NA, Prins P, Sen Ś, Yandell BS, Churchill GA (2019) R/qtl2: software for mapping quantitative trait loci with high‐dimensional data and multiparent populations. Genetics 211: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IGM, Smithers LE, Trotter MWB, Rugg‐Gunn P, Sun B, de Sousa C, Lopes SM, Howlett SK, Clarkson A, Ahrlund‐Richter L et al (2007) Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448: 191–195 [DOI] [PubMed] [Google Scholar]
- Bruno M, Mahgoub M, Macfarlan TS (2019) The arms race between KRAB–zinc finger proteins and endogenous retroelements and its impact on mammals. Annu Rev Genet 53: 393–416 [DOI] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J (2014) Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14: 838–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehr M, Smith A (2003) Genesis of embryonic stem cells. Philos Trans R Soc Lond B Biol Sci 358: 1397–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino RR, Stark A (2018) Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev 32: 202–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Dent SYR (2014) Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet 15: 93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ et al (2016) Lineage‐specific and single‐cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48: 1193–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott‐Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B et al (2017) An improved ATAC‐seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czechanski A, Byers C, Greenstein I, Schrode N, Donahue LR, Hadjantonakis A‐K, Reinholdt LG (2014) Derivation and characterization of mouse embryonic stem cells from permissive and nonpermissive strains. Nat Protoc 9: 559–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer JL, Ferguson‐Smith AC (2020) Strain‐specific epigenetic regulation of endogenous retroviruses: the role of trans‐acting modifiers. Viruses 12: 810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, Kaufman MH (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature 292: 154–156 [DOI] [PubMed] [Google Scholar]
- Factor DC, Corradin O, Zentner GE, Saiakhova A, Song L, Chenoweth JG, McKay RD, Crawford GE, Scacheri PC, Tesar PJ (2014) Epigenomic comparison reveals activation of “seed” enhancers during transition from naive to primed pluripotency. Cell Stem Cell 14: 854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ 3rd (1996) KAP‐1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10: 2067–2078 [DOI] [PubMed] [Google Scholar]
- Garbutt TA, Konneker TI, Konganti K, Hillhouse AE, Swift‐Haire F, Jones A, Phelps D, Aylor DL, Threadgill DW (2018) Permissiveness to form pluripotent stem cells may be an evolutionarily derived characteristic in Mus musculus . Sci Rep 8: 14706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RL, Beddington RS (1988) Multi‐lineage ‘stem’ cells in the mammalian embryo. J Cell Sci Suppl 10: 11–27 [DOI] [PubMed] [Google Scholar]
- Gonzales K, Liang H, Lim Y‐S, Chan Y‐S, Yeo J‐C, Tan C‐P, Gao B, Le B, Tan Z‐Y, Low K‐Y et al (2015) Deterministic restriction on pluripotent state dissolution by cell‐cycle pathways. Cell 162: 564–579 [DOI] [PubMed] [Google Scholar]
- GTEx Consortium (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369: 1318–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849 [DOI] [PubMed] [Google Scholar]
- Hackett JA, Kobayashi T, Dietmann S, Surani MA (2017) Activation of lineage regulators and transposable elements across a pluripotent spectrum. Stem Cell Rep 8: 1645–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Surani MA (2014) Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell 15: 416–430 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146: 519–532 [DOI] [PubMed] [Google Scholar]
- Ito K, Suda T (2014) Metabolic requirements for the maintenance of self‐renewing stem cells. Nat Rev Mol Cell Biol 15: 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jaime‐Soguero A, Abreu de Oliveira WA, Lluis F (2018) The pleiotropic effects of the canonical Wnt pathway in early development and pluripotency. Genes 9: 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Yan L, Xia L, Lu X, Zhu W, Ding D, Du M, Zhang D, Wang H, Hu B (2018) Zinc finger E‐box‐binding homeobox 1 (ZEB1) is required for neural differentiation of human embryonic stem cells. J Biol Chem 293: 19317–19329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Lane PW, Ward‐Bailey P, Davisson MT (1995) Mapping the mouse dactylaplasia mutation, Dac, and a gene that controls its expression, mdac. Genomics 29: 457–464 [DOI] [PubMed] [Google Scholar]
- Kauzlaric A, Ecco G, Cassano M, Duc J, Imbeault M, Trono D (2017) The mouse genome displays highly dynamic populations of KRAB‐zinc finger protein genes and related genetic units. PLoS One 12: e0173746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase E, Suemori H, Takahashi N, Okazaki K, Hashimoto K, Nakatsuji N (1994) Strain difference in establishment of mouse embryonic stem (ES) cell lines. Int J Dev Biol 38: 385–390 [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M et al (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Goncalves A, Leha A, Afzal V, Alasoo K, Ashford S, Bala S, Bensaddek D, Casale FP, Culley OJ et al (2017) Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546: 370–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Barber M, Mansfield W, Cui Y, Spindlow D, Stirparo GG, Dietmann S, Nichols J, Smith A (2021) Capture of mouse and human stem cells with features of formative pluripotency. Cell Stem Cell 28: 453–471.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi‐Aoi M, Ohnuki M, Takahashi K, Okita K, Noma H, Sawamura Y, Teramoto I, Narita M, Sato Y, Ichisaka T et al (2013) Differentiation‐defective phenotypes revealed by large‐scale analyses of human pluripotent stem cells. Proc Natl Acad Sci USA 110: 20569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttälä A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy K, Nakanishi M, Nishimura K, Ohtaka M, Weltner J et al (2016) Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep 6: 200–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Li Y, McIntosh BE, Nguyen BK, Duffin B, Tian S, Thomson JA, Dewey CN, Stewart R, Kendziorski C (2015) EBSeq‐HMM: a Bayesian approach for identifying gene‐expression changes in ordered RNA‐seq experiments. Bioinformatics 31: 2614–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenka N, Ramasamy SK (2007) Neural induction from ES cells portrays default commitment but instructive maturation. PLoS One 2: e1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Miu K‐K, Gu S, Cheung H‐H, Chan W‐Y (2018) Comparison of multi‐lineage differentiation of hiPSCs reveals novel miRNAs that regulate lineage specification. Sci Rep 8: 9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GR (1981) Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA 78: 7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas G, Blanco E, Ballaré C, Sansó M, Spill YG, Hu D, Aoi Y, Le Dily F, Shilatifard A, Marti‐Renom MA et al (2018) Promoter bivalency favors an open chromatin architecture in embryonic stem cells. Nat Genet 50: 1452–1462 [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J et al (2012) Systematic localization of common disease‐associated variation in regulatory DNA. Science 337: 1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS, Werb Z et al (2019) MULTI‐seq: sample multiplexing for single‐cell RNA sequencing using lipid‐tagged indices. Nat Methods 16: 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, Zhu M, Wu J, Shi X, Taing L et al (2017) Cistrome data browser: a data portal for ChIP‐Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res 45: D658–D662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K‐F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E et al (2003) PGC‐1α‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Morgani S, Nichols J, Hadjantonakis A‐K (2017) The many faces of pluripotency: in vitro adaptations of a continuum of in vivo states. BMC Dev Biol 17: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motenko H, Neuhauser SB, O’Keefe M, Richardson JE (2015) MouseMine: a new data warehouse for MGI. Mamm Genome 26: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neagu A, van Genderen E, Escudero I, Verwegen L, Kurek D, Lehmann J, Stel J, Dirks RAM, van Mierlo G, Maas A et al (2020) In vitro capture and characterization of embryonic rosette‐stage pluripotency between naive and primed states. Nat Cell Biol 22: 534–545 [DOI] [PubMed] [Google Scholar]
- Nichols J, Jones K, Phillips JM, Newland SA, Roode M, Mansfield W, Smith A, Cooke A (2009) Validated germline‐competent embryonic stem cell lines from nonobese diabetic mice. Nat Med 15: 814–818 [DOI] [PubMed] [Google Scholar]
- Nichols J, Smith A (2009) Naive and primed pluripotent states. Cell Stem Cell 4: 487–492 [DOI] [PubMed] [Google Scholar]
- Nishizawa M, Chonabayashi K, Nomura M, Tanaka A, Nakamura M, Inagaki A, Nishikawa M, Takei I, Oishi A, Tanabe K et al (2016) Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell 19: 341–354 [DOI] [PubMed] [Google Scholar]
- Novo CL, Javierre B‐M, Cairns J, Segonds‐Pichon A, Wingett SW, Freire‐Pritchett P, Furlan‐Magaril M, Schoenfelder S, Fraser P, Rugg‐Gunn PJ (2018) Long‐range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rep 22: 2615–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Setty M, Kuo Y‐Y, Liu V, Garg V, Sharma R, Simon CS, Saiz N, Gardner R, Boutet SC et al (2019) The emergent landscape of the mouse gut endoderm at single‐cell resolution. Nature 569: 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortmann D, Brown S, Czechanski A, Aydin S, Muraro D, Huang Y, Tomaz RA, Osnato A, Canu G, Wesley BT et al (2020) Naive pluripotent stem cells exhibit phenotypic variability that is driven by genetic variation. Cell Stem Cell 27: 470–481.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz‐Gerald CS, Sato Y, Cowan CA, Chien KR, Melton DA (2008) Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol 26: 313–315 [DOI] [PubMed] [Google Scholar]
- Palis J, Robertson S, Kennedy M, Wall C, Keller G (1999) Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 126: 5073–5084 [DOI] [PubMed] [Google Scholar]
- Peirce JL, Lu L, Gu J, Silver LM, Williams RW (2004) A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 5: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pękowska A, Klaus B, Xiang W, Severino J, Daigle N, Klein FA, Oleś M, Casellas R, Ellenberg J, Steinmetz LM et al (2018) Gain of CTCF‐anchored chromatin loops marks the exit from naive pluripotency. Cell Syst 7: 482–495.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016) Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plamondon JA, Harris MJ, Mager DL, Gagnier L, Juriloff DM (2011) The clf2 gene has an epigenetic role in the multifactorial etiology of cleft lip and palate in the A/WySn mouse strain. Birth Defects Res A Clin Mol Teratol 91: 716–727 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018) R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; [Google Scholar]
- Raghupathy N, Choi K, Vincent MJ, Beane GL, Sheppard KS, Munger SC, Korstanje R, Pardo‐Manual de Villena F, Churchill GA (2018) Hierarchical analysis of RNA‐seq reads improves the accuracy of allele‐specific expression. Bioinformatics 34: 2177–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard‐Liesching H, Verp S, Marquis J et al (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463: 237–240 [DOI] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, Greenleaf WJ (2017) chromVAR: inferring transcription‐factor‐associated accessibility from single‐cell epigenomic data. Nat Methods 14: 975–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel LV, Abratte CM, Schimenti JC, Southard TL, Fortier LA (2012) Genetic background affects induced pluripotent stem cell generation. Stem Cell Res Ther 3: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova LV, Sharov AA, Piao Y, Shaik N, Sullivan T, Stewart CL, Hogan BLM, Ko MSH (2007) Global gene expression profiling reveals similarities and differences among mouse pluripotent stem cells of different origins and strains. Dev Biol 307: 446–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Bock C (2016) LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics 32: 587–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Czechanski A, Byers C, Aydin S, Spruce C, Olivier C, Choi K, Gatti DM, Raghupathy N, Keele GR et al (2020) Mapping the effects of genetic variation on chromatin state and gene expression reveals loci that control ground state pluripotency. Cell Stem Cell 27: 459–469.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A (2017) Formative pluripotency: the executive phase in a developmental continuum. Development 144: 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smukler SR, Runciman SB, Xu S, van der Kooy D (2006) Embryonic stem cells assume a primitive neural stem cell fate in the absence of extrinsic influences. J Cell Biol 172: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S (2018) Slingshot: cell lineage and pseudotime inference for single‐cell transcriptomics. BMC Genom 19: 477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanzey E, McNamara TF, Apostolou E, Tahiliani M, Stadtfeld M (2020) A susceptibility locus on chromosome 13 profoundly impacts the stability of genomic imprinting in mouse pluripotent stem cells. Cell Rep 30: 3597–3604.e3 [DOI] [PubMed] [Google Scholar]
- Tian J, Keller MP, Broman AT, Kendziorski C, Yandell BS, Attie AD, Broman KW (2016) The dissection of expression quantitative trait locus hotspots. Genetics 202: 1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treger RS, Pope SD, Kong Y, Tokuyama M, Taura M, Iwasaki A (2019) The lupus susceptibility locus Sgp3 encodes the suppressor of endogenous retrovirus expression SNERV. Immunity 50: 334–347.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Winkle LJ, Ryznar R (2019) One‐carbon metabolism regulates embryonic stem cell fate through epigenetic DNA and histone modifications: implications for transgenerational metabolic disorders in adults. Frontiers in Cell and Developmental Biology 7: 300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venere M, Han Y‐G, Bell R, Song JS, Alvarez‐Buylla A, Blelloch R (2012) Sox1 marks an activated neural stem/progenitor cell in the hippocampus. Development 139: 3938–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato V, Webber C (2020) Addressing variability in iPSC‐derived models of human disease: guidelines to promote reproducibility. Dis Model Mech 13: dmm042317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, Soh C‐L, Su J, Zhang C, Shu W, Xi Q et al (2017) The p53 family coordinates Wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell 20: 70–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, de Iaco A, Sun M‐A, Bruno M, Tinkham M, Hoang D, Mitra A, Ralls S, Trono D, Macfarlan TS (2020) KRAB‐zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. eLife 9, 10.7554/elife.56337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav T, Quivy J‐P, Almouzni G (2018) Chromatin plasticity: a versatile landscape that underlies cell fate and identity. Science 361: 1332–1336 [DOI] [PubMed] [Google Scholar]
- Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, Nellåker C, Goodstadt L, Nicod J, Bhomra A et al (2011) Sequence‐based characterization of structural variation in the mouse genome. Nature 477: 326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S‐H, Andrabi M, Biss R, Murtuza Baker S, Iqbal M, Sharrocks AD (2019) ZIC3 controls the transition from naive to primed pluripotency. Cell Rep 27: 3215–3227.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q‐L, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L‐G, Han Y, He Q‐Y (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L‐G, He Q‐Y (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31: 2382–2383 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen C‐H, Brown M, Zhang X, Meyer CA et al (2019) Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 47: D729–D735 [DOI] [PMC free article] [PubMed] [Google Scholar]