

Abstract
Heart disease remains a significant human health burden worldwide, with a significant fraction of morbidity attributable to environmental exposures. However, the extent to which the thousands of chemicals in commerce and the environment may contribute to heart disease morbidity is largely unknown, because in contrast to pharmaceuticals, environmental chemicals are seldom tested for potential cardiotoxicity. Human induced pluripotent stem cell (iPSC)-derived cardiomyocytes have become an informative in vitro model for cardiotoxicity testing of drugs, with the availability of cells from multiple individuals allowing in vitro testing of population variability. In this study, we hypothesized that a panel of iPSC-derived cardiomyocytes from healthy human donors can be used to screen for the potential cardiotoxicity hazard and risk of environmental chemicals. We conducted concentration-response testing of 1,029 chemicals (drugs, pesticides, flame retardants, PAHs, plasticizers, industrial chemicals, food/flavor/fragrance agents, etc.) in iPSC-derived cardiomyocytes from 5 donors. We used kinetic calcium flux and high-content imaging to derive quantitative measures as inputs into Bayesian population concentration-response modeling of the effects of each chemical. We found that many environmental chemicals pose a hazard to human cardiomyocytes in vitro, with more than half of all chemicals eliciting positive or negative chronotropic, or arrhythmogenic effects. However, most of the tested environmental chemicals for which human exposure and high-throughput toxicokinetics data were available had wide margins of exposure and, thus, do not appear to pose a significant human health risk in a general population. Still, relatively narrow margins of exposure (<100) were estimated for some perfuoroalkyl substances and phthalates, raising concerns that cumulative exposures may pose a cardiotoxicity risk. Collectively, this study demonstrated the value of using a population-based human in vitro model for rapid, high-throughput hazard and risk characterization of chemicals for which little to no cardiotoxicity data is available from guideline studies in animals.
Introduction
Heart disease remains the leading cause of morbidity and mortality in the United States 1 and is a primary contributor to the global burden of disease 2. It is generally recognized that heart disease is the result of a complex interplay of intrinsic 3 and extrinsic 4, 5 factors, the latter being potentially preventable causes of morbidity and mortality 6. Testing of drug candidates for potential adverse effects on the heart is required by regulatory agencies worldwide 7. Such testing occurs in preclinical safety evaluation and during clinical trials, and continues through post-marketing surveillance. By contrast, studies of potential cardiovascular adverse effects are not required for approval of other chemicals in commerce or compounds that persist in the environment 8. The lack of cardiotoxicity testing requirements is concerning, because epidemiological data suggest that about one-third of ischemic heart disease burden can be attributed to environmental exposures 9. In addition, there is emerging evidence from in vitro studies that environmental chemicals have the potential to affect human cardiomyocytes 10–16, or pathways that are known to be important for cardiovascular health 17. Collectively, recent studies that examined dozens of environmental chemicals representative of a number of chemical classes serve as a proof of concept to elevate the concern to various regulatory agencies in the United States, European Union and other countries 18. The lack of information on the extent to which the other thousands of chemicals in commerce and the environment may contribute to human heart disease morbidity is largely because of the lack of medium- to high-throughput toxicity models that can be used to evaluate hazard and variability in cardiac effects 19.
A large number of experimental, pre-clinical and clinical options exist to de-risk potential cardiovascular liabilities of drug candidates 19. Among these, human induced pluripotent stem cell (iPSC)-derived cardiomyocytes have become an attractive model for cardiotoxicity testing; they are an informative tool for characterizing the electrophysiological, contractile, structural, and metabolic effects of drugs and chemicals on cardiomyocytes 19–21. These cells not only have been used to examine the potential cardiac hazard of environmental chemicals 10–16, but also retain the genetic and functional characteristics of the donors from which they were derived, serving as a useful tool to study variability in responses to chemicals 22, 23. Therefore, iPSC-derived cardiomyocytes as a model system in toxicology can be used in several risk characterization contexts 19.
To increase relevance of in vitro studies to human health protection, data on exposure, toxicokinetics, and inter-individual variability is needed to estimate the margin of safety or margin of exposure 12, 13, 23. Our previous studies have shown that using iPSC-derived cardiomyocytes from a population of individuals, rather than a single individual, improves risk characterization 11, 19, 23; however, studies using cells from a large number of donors are far too complex to be used for safety testing on hundreds or thousands of environmental chemicals. Recent work on study size optimization for population-based in vitro testing 24, including iPSC-derived cardiomyocytes 25, provide information on the number of individuals and replicates sufficient for confident conclusions. For example, we recently showed 25 that for iPSC-derived cardiomyocyte studies, a population of 5 unique donors is acceptable to derive a central estimate of population-wide potency and is more accurate and precise than the standard practice of collecting replicates of the same cell line. Moreover, this population size also allows for estimation of risk-based parameters such as margin of exposure 25, 26. Based on these considerations, we hypothesized that a panel of iPSC-derived cardiomyocytes from healthy human donors can be used to screen for the potential cardiotoxicity hazard and risk of environmental chemicals. Specifically, we characterized the potential cardiotoxicity hazard and risk of 1,029 chemicals and drugs from a diverse range of chemical classes in a high-throughput population screening design. We used an optimized population design of iPSC-derived cardiomyocytes (5 donors) and used kinetic calcium flux and high-content imaging to derive quantitative measures as inputs into Bayesian population concentration-response modeling of the effects of each chemical.
Materials and Methods
Materials, chemicals, and plate design.
Cardiomyocyte plating and maintenance media were obtained from Fujifilm Cellular Dynamics (Madison, WI). The EarlyTox Cardiotoxicity Kit (Product number: R8211) was obtained from Molecular Devices (San Jose, CA). Tissue culture-treated 384-well microplates were obtained from Corning (Product number: 3764; Kennebunk, ME). Gelatin from porcine skin (CAS: 9000–70-8) and penicillin/streptomycin (product number: P4333) were obtained from Millipore-Sigma (St. Louis, MO). Trypan Blue (0.4%, product number: 15250–061), Hoechst 33342 (product number: H3570), and MitoTracker Orange (product number: M7510) were from Life Technologies (Grand Island, NY). Phosphate buffered saline (PBS) was obtained from Corning (product number: 21–040-CV). Tissue culture grade dimethyl sulfoxide (DMSO, CAS: 67–68-5) was obtained from Santa Cruz Biotechnology (Dallas, TX). Isoproterenol (CAS: 7683–59-2), propranolol (CAS: 525–66-6), and sotalol (CAS: 959–24-0) were included in the EarlyTox Cardiotoxicity Kit obtained from Molecular Devices. N-acetylprocainamide (CAS: 32795–44-1), sematilide (CAS: 101526–62-9), cisapride (CAS: 260779–88-2), and tetraoctyl-ammonium bromide (CAS: 14866–33-2) were obtained from MilliporeSigma. Lamotrigine (CAS: 84057–84-1), cabazitaxel (CAS: 183133–96-2), and mifepristone (CAS: 84371–65-3) were from ApexBio (Houston, TX).
A total of 1,029 chemicals was tested in this study (a full list of test chemicals with their CASRN, DTXSID, chemical classification, and source is provided in Supplemental Table 1). Test chemicals were chosen (a schematic of the chemical selection workflow is shown in Figure 1A) to represent a broad range of chemical classes (Table 1): pharmaceuticals (including those known to elicit cardiotoxicity), industrial chemicals, pesticides, flame retardants, plasticizers, polycyclic aromatic compounds, solvents, surfactants, dyes, and food/flavor/fragrance agents. All environmental chemicals and most drugs selected for this study are part of the US Environmental Protection Agency (EPA) Toxicity ForeCaster (ToxCast) library 27. Overall, compounds were selected from ToxCast based on the following considerations: (i) chemicals with high-throughput toxicokinetics (httk) data 28 to enable in vitro-to-in vivo extrapolation; (ii) chemicals with hazard and dose-response data in the EPA Integrated Risk Information System (IRIS) 29; (iii) chemicals found to be active for binding of the androgen, estrogen, or thyroid receptors (AR, ER, TR, respectively) in the EPA Endocrine Disruptor Screening Program 30–32; (iv) other chemicals included in the ToxCast library or regulated by the Toxic Substances Control Act (TSCA) that are on the “active” inventory in US commerce; and (v) chemicals tested in previous studies in iPSC-derived cardiomyocytes 10, 11. In addition, a number of pharmaceutical compounds of different risk classifications for Torsades de pointes (TdP) that are tested in the Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative 33 were also included.
Figure 1.

Test chemical selection and characteristics. (A) Schematic showing the selection of the 1029 test chemicals included in this study. Chemicals tested in this study include all pharmaceuticals being validated in the Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative and other pharmaceuticals and environmental chemicals prioritized in existing databases such as ToxCast. (B) Principal component analysis (PCA) plot of the chemical space encompassed in this study, the chemical space tested in the largest screening in a population of iPSC-derived cardiomyocytes to date (Burnett et al., 2019), and the chemical space included in the Collaborative Estrogen Receptor Activity Prediction Project (CERAPP). (C) Histograms comparing the distribution of chemical characteristics in these studies in terms of octanol:water partition coefficient (ALogP), molecular weight (MW), and topological polar surface area (TPSA).
Table 1.
Chemical classes and the number of test chemicals in each class.
| Chemical Class | Number of Chemicals Tested |
|---|---|
| Pharmaceuticals | 125 |
| CiPA Pharmaceuticals | 28 |
| Chemical Intermediates | 179 |
| Herbicides | 147 |
| Pesticides (Non-Herbicide) | 139 |
| Microbiocides | 115 |
| Flame Retardants | 13 |
| Plasticizers | 55 |
| PAH | 14 |
| Solvents | 56 |
| Surfactants | 25 |
| Dyes | 19 |
| Food/Flavor/Fragrances | 100 |
| Other | 14 |
| TOTAL | 1,029 |
Test chemicals (excluding most CiPA compounds) were obtained in microplates from the National Center for Computational Toxicology and Exposure (NCCTE) of the EPA (Research Triangle Park, NC). Quality control of the compounds in the ToxCast library is detailed elsewhere 34. Chemicals were supplied frozen at concentrations of ~20 mM in 100% tissue culture grade DMSO and stored at −80°C until use. CiPA compounds were added to the test panel separately and were obtained from Fisher Scientific (Waltham, MA) or Millipore-Sigma. CiPA compounds were also prepared at concentrations of 20 mM in 100% tissue culture grade DMSO. Each test compound from the chemical master plates was transferred into new tissue culture-treated 384-well microplates using acoustic dispensing (Labcyte Echo 550, Beckman Coulter, Indianapolis, IN) in amounts that would result in 5× concentrations (1, 5, 50, 500 μM) in the working plate in a final volume of 50 μL (as detailed below). These plates were stored at −80°C until the day of the assay. On assay day, the 5× concentrated chemical working plate was prepared by adding 50 μL cardiomyocyte maintenance medium containing 2.5% v/v DMSO to each well of the microplates. Then, 12.5 µL of 5× concentrated test chemical solution was added to the test plate (see “Functional cardiotoxicity screening” section below) to yield final concentrations of 0.2, 1, 10, and 100 µM in 0.5% v/v DMSO.
Chemicals were randomly divided into 16 separate 384-well plates for screening such that the outer-most wells were left empty (308 wells were used on each plate; see a representative plate map in Supplemental Figure 1A). Fifteen of the plates contained unique test chemicals (n = 65 chemicals, 4 concentrations, one replicate each), positive controls (n = 10 chemicals, one concentration, 3 replicates each, see below), vehicle (0.5% DMSO, n = 12), and media-only (n = 6) wells. Plate 16 (Supplemental Figure 1B) contained test chemicals (n = 54 chemicals, 4 concentrations, one replicate each), inter-plate replicates (n = 11 chemicals with replicates, 4 concentrations, one replicate each), and the same design of positive controls and vehicle- and media-only wells as other plates.
Positive control compounds for [+] and [-] chronotropy were isoproterenol (a β-adrenergic receptor agonist, 10 µM) and propranolol (a β-adrenergic receptor antagonist, 0.5 µM), respectively. Cardioactive pharmaceuticals known to induce clinical QT prolongation were included as positive controls for the phenotype decay-rise ratio, an established in vitro surrogate for clinical QT prolongation which reflects a delayed repolarization of the action potential 23. Positive controls for decay-rise ratio phenotype were cisapride (a serotonin 5-HT4 receptor agonist, 0.1 µM), N-acetylprocainamide (a class III antiarrhythmic agent and K+ channel blocker, 10 µM), sematilide (a class III antiarrhythmic agent and K+ channel blocker, 10 µM), and sotalol (a β-adrenergic receptor antagonist and class III antiarrhythmic agent, 10 µM). Non-cardioactive pharmaceuticals with no known effects on the clinical QT interval were used as negative controls for decay-rise ratio phenotype: mifepristone (a progesterone and glucocorticoid receptor antagonist, 10 µM), cabazitaxel (an antineoplastic chemotherapeutic and microtubule inhibitor, 10 µM), and lamotrigine (an anticonvulsant and Na+ channel blocker, 10 µM). Lastly, tetraoctylammonium bromide (a quaternary ammonium compound, 50 µM) was included as a positive control for cytotoxicity. Concentrations of positive controls were selected based on previous studies in iPSC-derived cardiomyocytes 23, 35, 36.
Calculation of chemical molecular descriptors and diversity in the “chemical space”.
To determine the “applicability domain” of the results from this study based on the chemicals selected for testing, the chemical space occupied by the 1,029 compounds tested herein was compared to that of a panel of chemicals (n = 134) screened in iPSC-derived cardiomyocytes previously 11, 13 and to those in the Collaborative Estrogen Receptor Activity Prediction Project (CERAPP) data set (n = 32,464) 30. The open-source Chemistry Development Kit (CDK) package “rcdk”, as implemented in R (version 3.5) 37, was used to calculate >200 molecular descriptors for each compound. These descriptors are generated based on compound structure and represent features calculated from the chemical structure, such as octanol:water partition coefficient (logP), molecular weight (MW), and topological polar surface area (TPSA). To provide an overview of the diversity of the structures and the overall chemical space, the chemical structures in each list were analyzed using principal component analysis (Figure 1B), as well as using the easily interpretable descriptors AlogP, MW, and TPSA (Figure 1C) as detailed previously 38. The visualizations were performed using the Plotly package in R.
iPSC-derived cardiomyocytes and cell culture.
iPSC-derived cardiomyocytes from 5 human donors were obtained from Fujifilm Cellular Dynamics. Cells were derived from donors with no known or family history of cardiovascular disease and, thus, were meant to represent “healthy” individuals. The selection of 5 donors was based on our previously published study in iPSC-derived cardiomyocytes from 27 individual human donors 36. Specifically, we first performed a clustering analysis and visualized both baseline and drug-induced characteristics of the panel of 27 donors (Figure 2). For baseline cardiomyocyte beating parameters, each cell line was characterized by the median value across control wells for each cell line for beats per minute (BPM), peak amplitude, decay to rise time ratio, peak spacing coefficient of variability (CV), and peak amplitude CV. For treatment-induced effects, we used the effects of positive control compounds isoproterenol (maximum increase in BPM), and propranolol (EC50 in BPM), and cisapride (maximum increase in decay-rise ratio). Additionally, for comparability to previous studies, the “standard” iCell cardiomyocyte donor (Catalog number: CMC-100–010-001) was included. The other 4 donors (donor IDs and their characteristics are listed in Supplemental Table 2) were selected based on maximizing the “distance” from other donors with respect to clustering in both baseline values and positive control treatment effects, while preserving a balance in sexes and ancestry representation. The resulting donor population was comprised of 3 females and 2 males and included individuals of European, African American, and mixed ancestry.
Figure 2.
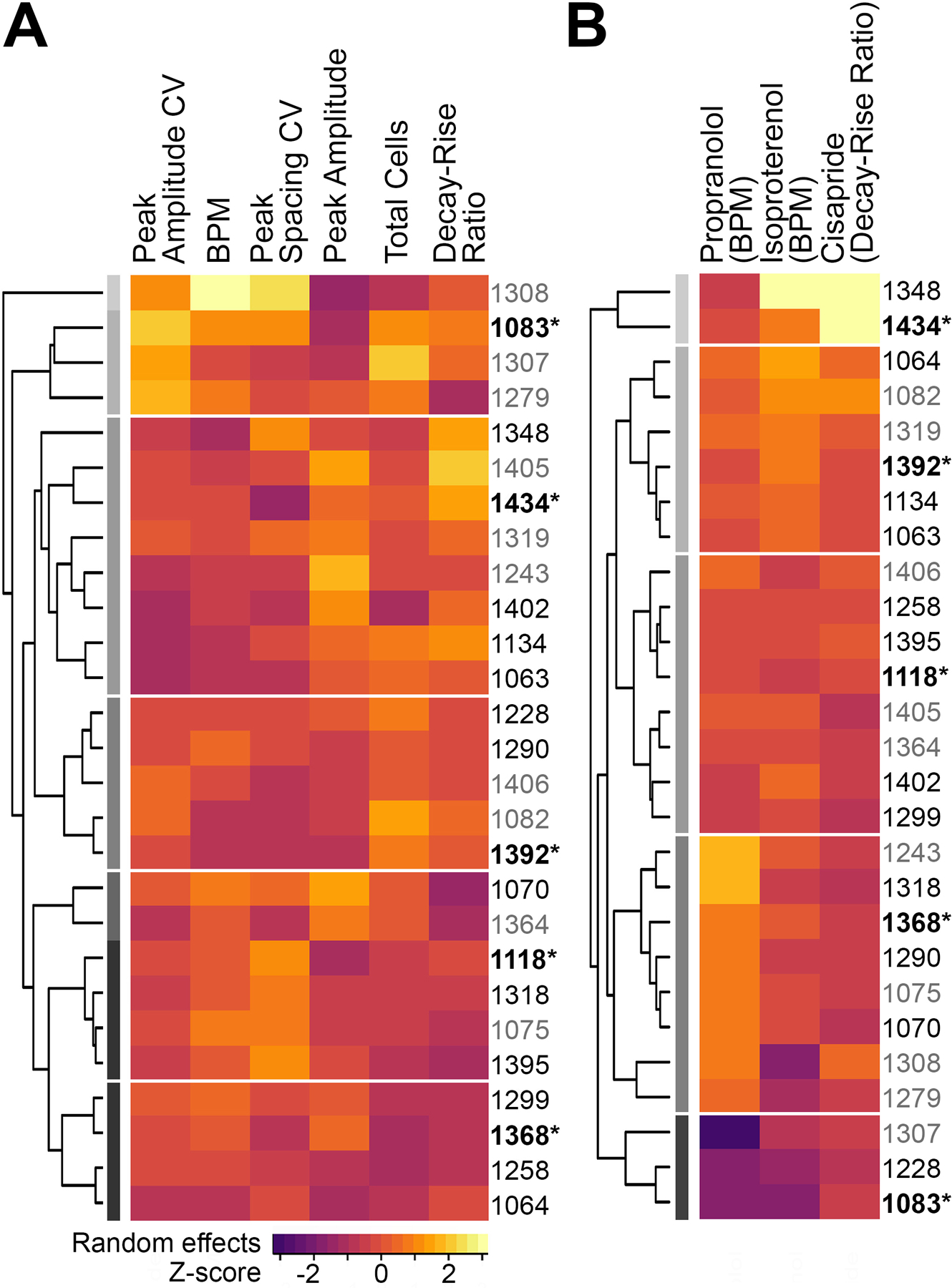
Donor characteristics and selection of the 5 iPSC-derived cardiomyocyte donors included in this study from a pool of 27 donors included in 36. Heat maps show (A) baseline phenotypic characteristics, and (B) chemical-induced characteristics across the donor pool. Donor IDs in grey lettering had insufficient availability from the vendor for testing all chemicals, and were excluded from consideration. Donors selected for testing in this study are designated with bold lettering and an asterisk (*). The donor 1434 is commonly referred to as the “standard donor.” The 5 donors were specifically selected from different clusters to encompass the observed variability in both baseline and chemical-induced characteristics.
iPSC-derived cardiomyocytes from each donor were cultured in multiple batches under identical conditions using an established protocol 11, 36. Cells from each donor were tested in two separate batches (batch 1 = plates 1–8, batch 2 = plates 9–16). Briefly, tissue culture-ready 384-well microplates were treated with 25 µL/well of 0.1% (w/v) gelatin solution (gelatin from porcine skin in 18 MΩ sterile water) and incubated at 37°C and 5% CO2 for 2 hours. Cells were removed from liquid nitrogen storage and thawed in a 37°C water bath for 4 minutes. Cells were then incrementally resuspended in cardiomyocyte plating medium containing 1:500 penicillin/streptomycin solution at room temperature to a final concentration of 2×105 cells/mL, based on manufacturer-provided viability and plating efficiency estimates. Final live cell count in the suspension was confirmed prior to plating using the trypan blue exclusion test. Gelatin solution was decanted from each plate immediately prior to plating cells, and 25 µL cell suspension was dispensed into each well of the microplates (excluding outer wells, which were then filled with an equal amount of sterile phosphate-buffered saline), resulting in a plating density of ~5,000 cells/well. The microplates were kept at room temperature for 30 minutes and then placed for 48 hours in the incubator at 37°C and 5% CO2. At 48 hours post-plating, 17.5 µL plating medium was exchanged with 32.5 µL pre-warmed maintenance medium containing 1:500 penicillin/streptomycin solution, to a final volume of 40 µL/well. Microplates were then incubated at 37°C and 5% CO2 for 13 days post-plating, with 25 µL/well maintenance medium exchanged with fresh pre-warmed maintenance medium containing 1:500 penicillin/streptomycin solution every 48–72 hours. iPSC-derived cardiomyocytes were visually inspected under the light microscope during media changes and started to exhibit spontaneous and synchronous beating in each well from day 7 post-plating. On the evening of day 13 post-plating, the entire volume in each well was carefully aspirated not to disturb the cell monolayer and replaced with 25 µL/well fresh pre-warmed maintenance medium containing 1:500 penicillin/streptomycin solution. On the morning of day 14 post-plating, assays were performed.
Functional cardiotoxicity screening of chemicals using Ca2+ flux assay.
The Ca2+ flux assay (EarlyTox Cardiotoxicity Kit, Molecular Devices) was used to evaluate the functional effects of chemicals on iPSC-derived cardiomyocytes as previously detailed 11, 36. Briefly, this assay measures intracellular Ca2+ flux as a quantitative functional readout based on a fluorescent Ca2+ reporter. Ca2+ flux reads were taken at baseline (before chemical treatment) and after 90 minutes of chemical exposure. The Ca2+ flux assay was performed as follows. Ca2+ dye reagent was first prepared according to manufacturer protocol and equilibrated to 37°C. A volume of 25 µL prepared Ca2+ dye reagent was dispensed into each well of the microplate, resulting in a total volume of 50 µL/well. Plates were incubated at 37°C and 5% CO2 for 2 hours, and then a baseline Ca2+ flux reading was simultaneously recorded in all wells of the microplate using the FLIPR Tetra Cellular Screening System (Molecular Devices). Ca2+ flux was recorded at the speed of 8 frames per second for 100 seconds (n = 800 total reads) with stage temperature = 37°C, λexc = 470–495 nm, λem = 515–575 nm, gain = 2000, and exposure time = 0.05 sec.
Following the baseline Ca2+ flux read, 12.5 µL 5× concentrated test chemical solutions (in 2.5% v/v DMSO and cardiomyocyte maintenance medium) were simultaneously added to all wells of the microplate using the liquid handling robotics of the FLIPR Tetra. FLIPR Tetra settings were optimized to mix and then transfer test chemicals to the microplate at height = 40 µL, speed = 1 µL/sec, and removal speed = 6 mm/sec, resulting in a total volume of 62.5 µL/well with 0.5% v/v DMSO in cardiomyocyte maintenance medium. Immediately after chemical transfer, cells were incubated 37°C and 5% CO2 for 90 minutes, based on optimized protocols from previous reports evaluating functional readouts after different exposure durations 10, 36. Following the 90-minute treatment period, Ca2+ flux was again simultaneously recorded in all wells of the microplate. Data from this 90-minute timepoint was used for further data analysis to evaluate the functional effects of chemicals on cells. The time-point selection was based on the previous studies that examined various intervals and showed that 90 minutes was most representative of the expected effects of positive control compounds that have an effect on ion channels in this model system 35, 36, 39.
Cell viability screening of chemicals using high-content imaging.
After conclusion of the Ca2+ flux measurements at 90 minutes post-chemical addition, cell viability-related phenotypes were quantitatively assessed through high-content imaging with the ImageXpress Micro Confocal Cellular Imaging System (Molecular Devices) as previously reported 11, 36. The high-content imaging assay was performed as follows. First, the total volume of cell culture medium containing Ca2+ dye was aspirated and replaced with 25 µL/well pre-warmed staining solution with fluorescent probes for nuclei and mitochondria (2.2 µg/mL Hoechst 33342 and 0.2 µM MitoTracker Orange in cardiomyocyte maintenance medium with 1:500 penicillin/streptomycin solution). Microplates were then incubated at standard conditions for 15 minutes, the staining solution was aspirated, and 25 µL/well pre-warmed fresh maintenance medium containing 1:500 penicillin/streptomycin solution was added to each well to wash before proceeding to image acquisition. Images of each well at 10× magnification were acquired in succession using DAPI (Hoechst 33342 for nuclear staining), TRITC (MitoTracker Orange for mitochondrial staining), and FITC (Ca2+ dye) filters.
Image and data processing.
Image and data processing and concentration-response analysis were performed as previously reported 13, 23. Ca2+ flux data were processed and quantified using a custom script 23 in R 40. Four Ca2+ flux-derived phenotypes describing functional cardiotoxicity were analyzed: “[+] chronotropy” (increase in peak frequency), “[-] chronotropy” (decrease in peak frequency), “decay-rise ratio” (ratio of the time from peak maximum to baseline to the time from baseline to peak maximum), and “asystole” (quiescence of beating, where peak frequency goes to zero). Image processing and quantification were performed using the multi-wavelength cell scoring module in the instrument-specific MetaXpress software package (Molecular Devices). The imaging-derived total cells parameter (total number of nuclei in a representative imaging field) was used as a measure of “cytotoxicity”.
Bayesian population concentration-response modeling.
Bayesian population concentration-response modeling was conducted in R on the Texas A&M High Performance Research Computing Core as previously described 13. Briefly, data was preprocessed depending on the endpoint being modeled, and a hierarchical Bayesian random-effects Hill model was used to fit the processed concentration-response data for each chemical and phenotype, consistent with previously published methods 13, 24. For [+] chronotropy and decay-rise ratio (where increases in response are of interest), an “upward” random-effects Hill model was used (Supplemental File 1) and reparametrized at the donor level. An alternate “downward” model was used for the phenotype [-] chronotropy (Supplemental File 2), and a third alternate “zero” model was used for asystole and total cells (Supplemental File 3), as conducted previously 13. Model parameters (including any natural-log transformations), restrictions, and assumptions were previously described 13.
Posterior distribution sampling was conducted using the Markov chain Monte Carlo (MCMC) algorithm in R interfaced with the STAN software package (version 2.17.3) on the Texas A&M University High Performance Research Computing Core as previously described 13. Briefly, simulations were conducted for each chemical and phenotype, consisting of four MC chains of 4,000–36,000 iterations each, the first half of which were warm-up iterations that were discarded prior to analysis. Convergence was assessed at both the inter- and intra-chain levels and was indicated by a potential scale reduction factor R̂ ≤ 1.2 41. For chemicals in which convergence was reached for a given endpoint, a total of 1,000 posterior samples (4 chains, 250 random samples/chain) were saved for subsequent analysis.
Point-of-departure (POD) values were derived for each chemical and phenotype (including replicates, if available). PODs were derived for each donor separately and for a predicted median individual within the population (referred to hereafter as “population median”). PODs for the functional phenotypes were defined as the concentration at which response increased (for the endpoints [+] chronotropy and decay-rise ratio) or decreased (for [-] chronotropy) by 5% from vehicle controls, represented by the EC05. Decay-rise ratio was previously demonstrated to be an accurate in vitro surrogate for in vivo increases in the corrected QT interval (QTc) 23. The POD for the asystole phenotype was represented by the EC95, reflecting a 95% decrease in peak frequency from vehicle controls. The POD for the cytotoxicity phenotype was represented by the EC10, reflecting a 10% decrease in the total number of cells from vehicle controls, consistent with metrics previously reported 24, 42. PODs were restricted to values within the range of tested concentrations (0.2 to 100 μM), and POD values outside of this range were changed to the lowest or highest concentrations tested, respectively. If there was no change in the concentration-response trend at any concentration tested, the POD was defined as the highest concentration tested, 100 μM.
Analysis of assay reproducibility.
Experimental reproducibility was assessed in both an intra- and inter-plate fashion utilizing positive controls and technical replicates, respectively, and in an inter-study fashion by comparing a group of compounds for which PODs were similarly derived for the same 5 donors in previous studies using a larger population of donors 11, 13. For inter-plate and inter-study reproducibility, concordance was assessed by deriving the absolute difference in the log10 transformed POD values across replicates. For example, concordance for technical (inter-plate) replicates was measured as:
where PODorig is the POD derived for the compound in its original plating position (on Plates 1–15), and PODrep is the POD derived for the compound in its replicate plating position (on Plate 16). Similarly, inter-study reproducibility was measured, where PODorig is the POD calculated in this study, and PODrep is the POD previously derived13 for the same compound. A ΔPOD of 0 indicates perfect concordance, and a ΔPOD of 1 indicates concordance within one order of magnitude of concentration. Reproducibility was assessed in this fashion at the population level, using population median estimates of the POD for each compound, and at the individual level, in which a median POD was derived for each individual and compound. Additionally, the effects of positive controls were measured and compared to the effects observed previously 11.
Hazard characterization: Chemical activity calls.
Activity calls were made to inform the cardiotoxicity hazard of each tested chemical, consistent with previously published methods 13. Activity for each chemical and phenotype was determined based on the population median, and chemicals were defined either as “active” or “inactive” for each phenotype. Activity calls were evaluated as follows: (i) convergence was reached, as indicated by R̂ ≤ 1.2; (ii) less than 10% model error in concentration-response fit, as indicated by the model scale parameter for the error (δ) ≤ 0.1 across the four chains; and (iii) the median estimate for the median individual POD < 100 μM (the highest concentration tested in this study) so that the POD was not extrapolated beyond the range of tested concentrations. A chemical was considered “active” in terms of its cardiotoxicity hazard if it fulfilled the criteria for convergence, concentration-response fit, and POD for a given phenotype. The activity calls describe the cardiotoxicity hazard for the population median. A chemical that did not fulfill one of these criteria was considered “inactive” in terms of its cardiotoxicity hazard. Thus, inactivity may reflect inadequate convergence, poor concentration-response fit, or low potency (no response even at the highest tested concentration).
Hazard characterization: Critical endpoint determination.
For each chemical that was active for at least one phenotype (see above), the critical endpoint (the most sensitive phenotype for a chemical) was determined, consistent with previously published methods 13. Briefly, for each chemical and phenotype for which that chemical was active, model parameters were randomly sampled and used to derive a POD. The derived PODs for each active phenotype were compared, and a critical endpoint was selected based on the phenotype with the lowest POD for this random sample. This process was repeated for each of 1000 random posterior samples, and the phenotype most commonly found to be the most sensitive was determined to be the chemical-specific critical endpoint.
Hazard characterization: Ranking of compounds using ToxPi.
The Toxicological Prioritization Index (ToxPi) Graphical User Interface (GUI) (version 2.3) 43 was used to visualize, cluster, and rank the bioactivity profiles of test chemicals. ToxPi charts and ToxPi scores were generated for the “average” chemical in each chemical classification (determined by calculating the average POD value of all chemicals in a given class, for each donor and phenotype). These average POD values for each of the 5 phenotypes (and 5 donors) were formatted into a ToxPi data matrix and inputted into the ToxPi GUI. Data for each phenotype were then normalized and scaled from 0 to 1 using the y = -log10(x) + log10(max(x)) scaling transformation in ToxPi, with a value of 1 representing the lowest relative POD value in the set (the highest relative potency, or bioactivity). In the resultant ToxPi charts, each slice of the ToxPi represents a phenotype, and the area of the slice is proportional to the relative bioactivity for that phenotype. The confidence interval for each slice is represented by the area at the outer edge of the slice that is colored with less intensity. The confidence interval describes the variability in POD values observed across the 5 donors (a larger confidence interval slice area represents a higher degree of variability in response across the donors). The overall ToxPi score represents the relative potency of each chemical class. The ToxPi results were used to rank chemical classes based on their relative overall bioactivity in iPSC-derived cardiomyocytes and to visualize the differences in bioactivity profiles between different chemical classes.
Risk characterization: In vitro-to-in vivo extrapolation (IVIVE).
Cardiotoxicity risk for pharmaceuticals was characterized by deriving margins of safety (MOS), or the ratio of the in vitro POD for the critical (most sensitive) endpoint to the estimated in vivo human exposure, consistent with previously published methods 13. The MOS indicates risk by evaluating the degree of possible overlap between the maximum blood concentration of a drug when administered as intended (Cmax) and the in vitro POD for that drug’s critical endpoint. The lower the MOS, the higher degree of possible overlap between the concentration that induces a response in vitro and the estimated in vivo human exposure. Human Cmax values were sourced and extracted from the PharmaPendium database 44. For each drug with available human pharmacokinetic data and activity in at least one phenotype, the MOS for the median individual was derived by dividing the median individual POD for the critical endpoint by the population median Cmax estimate (with outliers removed).
Cardiotoxicity risk for non-pharmaceutical compounds was characterized by deriving margins of exposure (MOE), an equivalent measurement of risk to the MOS for pharmaceuticals, consistent with previously published methods 13. In contrast to pharmaceuticals, estimated in vivo human exposure values (median exposure predictions, mg/kg body weight/day) for environmental chemicals were sourced and extracted from the EPA’s Exposure Forecasting (ExpoCast) database through the Computational Toxicology (CompTox) Chemicals Dashboard 18. Median exposure estimates were transformed into human steady-state plasma concentrations (Css), assuming median toxicokinetic parameters, using the httk package (version 1.9.2) 28. For each environmental chemical with an available human exposure estimate and activity in at least one phenotype, the MOE for the median individual was derived by dividing the median individual POD for the critical endpoint by the population median Css estimate (the same approach as MOS for pharmaceuticals, as described above). Generally, a margin of ≥100 is considered adequately “protective” in risk assessment for non-genotoxic, non-carcinogenic compounds 45.
Results
Diversity in chemical space and donor characteristics
This study evaluated a large number of diverse chemicals for their potential adverse effects on human iPSC-derived cardiomyocytes, aiming to cover a broad range of environmental compounds. Compound selection criteria (Figure 1A) were based on both chemical class diversity and availability of other information that can enable risk characterization for in vivo exposures. To illustrate the structural diversity of the 1,029 chemicals tested herein, we conducted three-dimensional principal component analysis (PCA, Figure 1B) based on 233 chemical descriptors of the tested compounds as compared to those previously tested (n = 134) in a population of iPSC-derived cardiomyocytes 11, or compounds (n = 32,464) in the CERAPP prediction data set 30. It is evident from comparing the molecular descriptor space that the compounds included in this study are chemically diverse. A similar overlap was evident (Figure 1C) when major physicochemical properties frequently used in the pharmaceutical industry to triage compounds 46 were considered.
An equally important consideration for human health-based interpretation of the outcomes of this study was selection of iPSC-derived cardiomyocyte donors. As shown in Figure 2, the selected 5-donor pool encompassed a diverse range of baseline characteristics and positive control treatment effects. For instance, as compared to the “standard donor” 1434, donor 1083 had a higher BPM, whereas donor 1392 had a lower BPM at baseline. Similarly, propranolol was more potent (lower EC50) in donor 1083 as compared to the standard donor, and less potent (higher EC50) in donor 1392. Interestingly, cisapride had the greatest efficacy (Emax) in the standard donor as compared to the other 4 donors, suggesting that for QT prolongation, the standard donor may be more sensitive than “average.”
Reproducibility in chemical-induced effects on iPSC-derived cardiomyocytes
Challenges with reproducibility of the biomedical data are widely acknowledged 47. These concerns are most relevant to the results from large-scale chemical testing studies where test chemical purity/identity 34, data processing methods 48, and genetic variability among donor-derived cells 36, 42 can introduce both uncertainty and variability into the data. Therefore, in accordance with the principles for rigorous science 49, this study’s redundant experimental design included a robust set of positive and negative controls, inter-plate replicates, and a comparison to previous studies with the same chemicals and donor cells.
First, we evaluated technical reproducibility by comparing the concordance of quantitative estimates of the effect (POD) values for 11 test chemicals included in concentration-response as inter-plate replicates (Figure 3A). High concordance was observed between corresponding POD values for all 5 phenotypes, as evidenced by the difference in POD (ΔPOD) between the replicates on two different plates being mostly within a factor of one order of magnitude for every phenotype and donor evaluated. Technical reproducibility was similar among the 5 donors, and the population median also showed high correlation in response to replicate chemicals. The phenotypes cytotoxicity and asystole showed the highest degree of reproducibility (lowest ΔPOD) across donors and replicate chemicals. The functional phenotypes [+] and [-] chronotropy and decay-rise ratio showed a greater degree of variability (higher ΔPOD) in response. Decay-rise ratio was the most variable among phenotypes, but the mean response across the 11 replicate chemicals was still within one order of magnitude for every donor tested.
Figure 3.
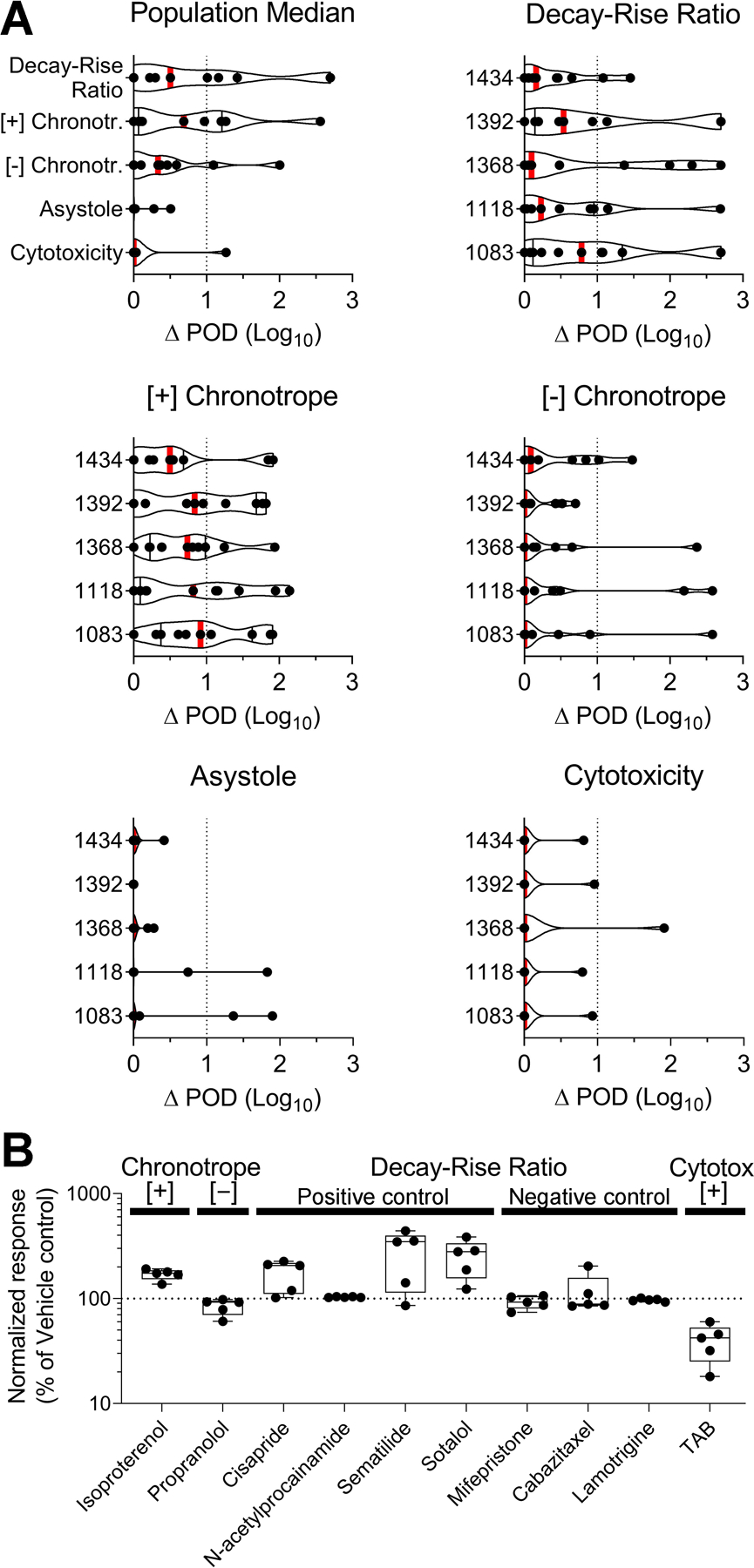
Technical reproducibility. (A) A total of 11 test chemicals were included as technical (inter-plate) replicates. Violin plots showing the difference in point-of-departure (ΔPOD) values between technical replicates. Graphs are separated by phenotype and donor, with the upper left-hand graph describing the population median across the 5 donors. Vertical red bars show the mean response. ΔPOD is plotted on a log scale, and the vertical dotted line at ΔPOD = 1 represents reproducibility within one order of magnitude. (B) A total of 10 positive and negative control compounds were included in box plots showing normalized response (expressed as percent of vehicle control) for each compound across different phenotypes. Boxes include data from 5 donors (dots), and whiskers represent the minimum and maximum values. The horizontal dotted line at normalized response = 100 represents the response of vehicle controls for that phenotype. Test concentrations for positive controls: isoproterenol (10 μM), propranolol (0.5 μM), cisapride (0.1 μM), N-acetylprocainamide (10 μM), sematilide (10 μM), sotalol (10 μM), mifepristone (10 μM), cabazitaxel (10 μM), lamotrigine (10 μM), TAB (50 μM).
A panel of 10 positive and negative control compounds was included on each plate (n = 3 replicates per plate at 1 concentration, Figure 3B). Isoproterenol and propranolol exhibited the expected effects on beat rate, with a wide range of responses among donors, as expected. Cisapride, sematilide, and sotalol increased the decay-rise ratio, again with variability in response among donors. Treatment with sematilide induced the most variability in effects. N-Acetylprocainamide showed no significant effect on decay-rise ratio at the concentration tested. As expected, mifepristone, cabazitaxel, and lamotrigine exhibited no effects on decay-rise ratio. TAB resulted in a decreased cell viability in each donor tested, with differences in sensitivity among donors.
Our study design also allowed for the evaluation of inter-study reproducibility (Figure 4) of a total of 85 test chemicals evaluated herein that were tested in the same donors previously 11, by a similar metric for concordance analysis (ΔPOD between studies). Overall, most of the compound/donor combinations had ΔPOD values (median and inter-quartile ranges) within one order of magnitude. Inter-study reproducibility was similar among the 5 donors, and the population median also showed high correlation in response to replicate chemicals between studies. Again, the phenotype cytotoxicity showed the highest degree of reproducibility between studies (lowest ΔPOD for the population median), with 91% of replicate chemicals exhibiting POD estimates within one order of magnitude of each other. The functional phenotypes showed a greater degree of variability in replicate responses between studies (higher ΔPOD for the population median). For decay-rise ratio, asystole, [-] chronotropy, and [+] chronotropy, 88%, 85%, 81%, and 69% (respectively) of replicate chemicals had ΔPOD between ±1. Though some of the tested chemicals exhibited a greater degree of variability, the mean response for the POD estimate across the 85 overlapping chemicals was still within one order of magnitude for every donor tested.
Figure 4.
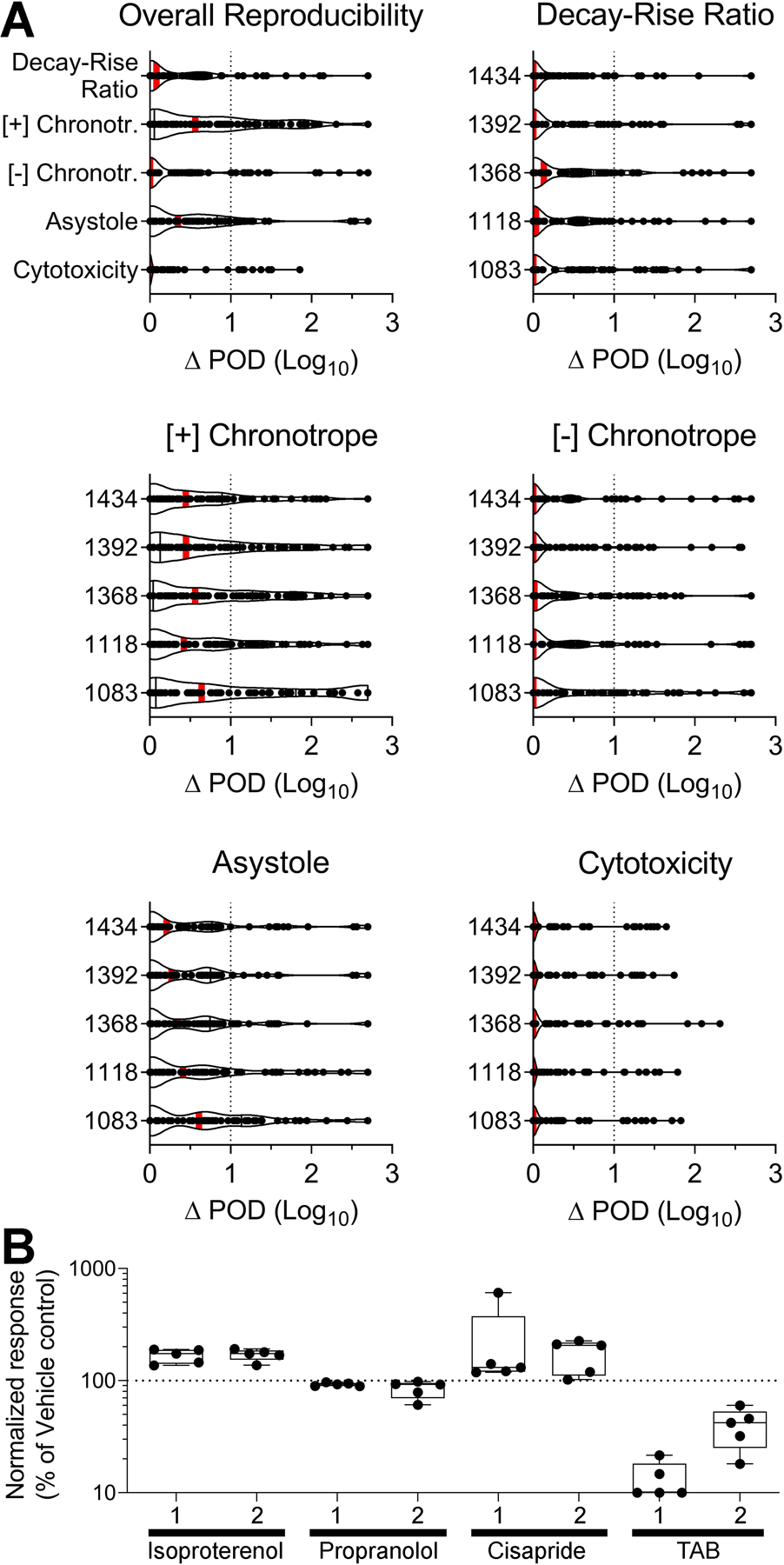
Inter-study reproducibility between this study and previous studies 11, 13 for (A) 85 test chemicals repeated between studies, and (B) 4 positive and negative control compounds repeated between studies. (A) Violin plots showing the difference in point-of-departure (ΔPOD) values between inter-study replicates (n = 85 data points in each box). Graphs are separated by phenotype and donor, with the upper left-hand graph describing the overall reproducibility across the 5 donors. Vertical red bars show the mean response. ΔPOD is plotted on a log scale, and the vertical dotted line at ΔPOD = 1 represents reproducibility within one order of magnitude. (B) Box plots showing normalized response (expressed as percent of vehicle control) for each of 4 positive control compounds repeated between studies (1 = 11, 2 = this study). Isoproterenol and propranolol describe the phenotypes [+] and [-] chronotropy, respectively, cisapride the decay-rise ratio, and TAB cytotoxicity. Boxes are made from 5 donors (n = 5 data points in each box), and whiskers represent the minimum and maximum values. The horizontal dotted line at normalized response = 100 represents the response of vehicle controls for that phenotype. Test concentrations for positive controls: isoproterenol (10 μM), propranolol (0.5 μM), cisapride (0.1 μM), TAB (50 μM).
Finally, we evaluated inter-study reproducibility in response to four of the control compounds discussed above which were also used as positive controls in the same donors previously 11 (Figure 4B). Isoproterenol and propranolol exhibited the expected effects on beat rate in both studies, with comparable responses across donors. Cisapride increased the decay-rise ratio in both studies, again with comparable effects across donors. Of all control compounds, cisapride exhibited the most variability in response among the donors, with some donors exhibiting a larger effect than others. TAB decreased cell viability in both studies, with some differences in sensitivity observed among donors.
Cardiotoxicity hazard characterization
We characterized the cardiotoxicity hazard of chemicals by evaluating their activity for each donor and designating chemicals as either “active” or “inactive” based on their POD for the population median (Figure 5A). Most chemicals (n = 743 out of 1,029 tested) were deemed “active” for at least one phenotype (Supplemental Figure 2), the most common of which was [+] chronotropy (n = 519 active compounds, or about half of the tested chemicals). In order of decreasing activity, [+] chronotropy was followed by decay-rise ratio (n = 256 active), [-] chronotropy (n = 199 active), and asystole (n = 42 active). Cytotoxicity had the smallest number of active compounds (n = 29), as expected based on the selection criteria for non-cytotoxic compounds.
Figure 5.
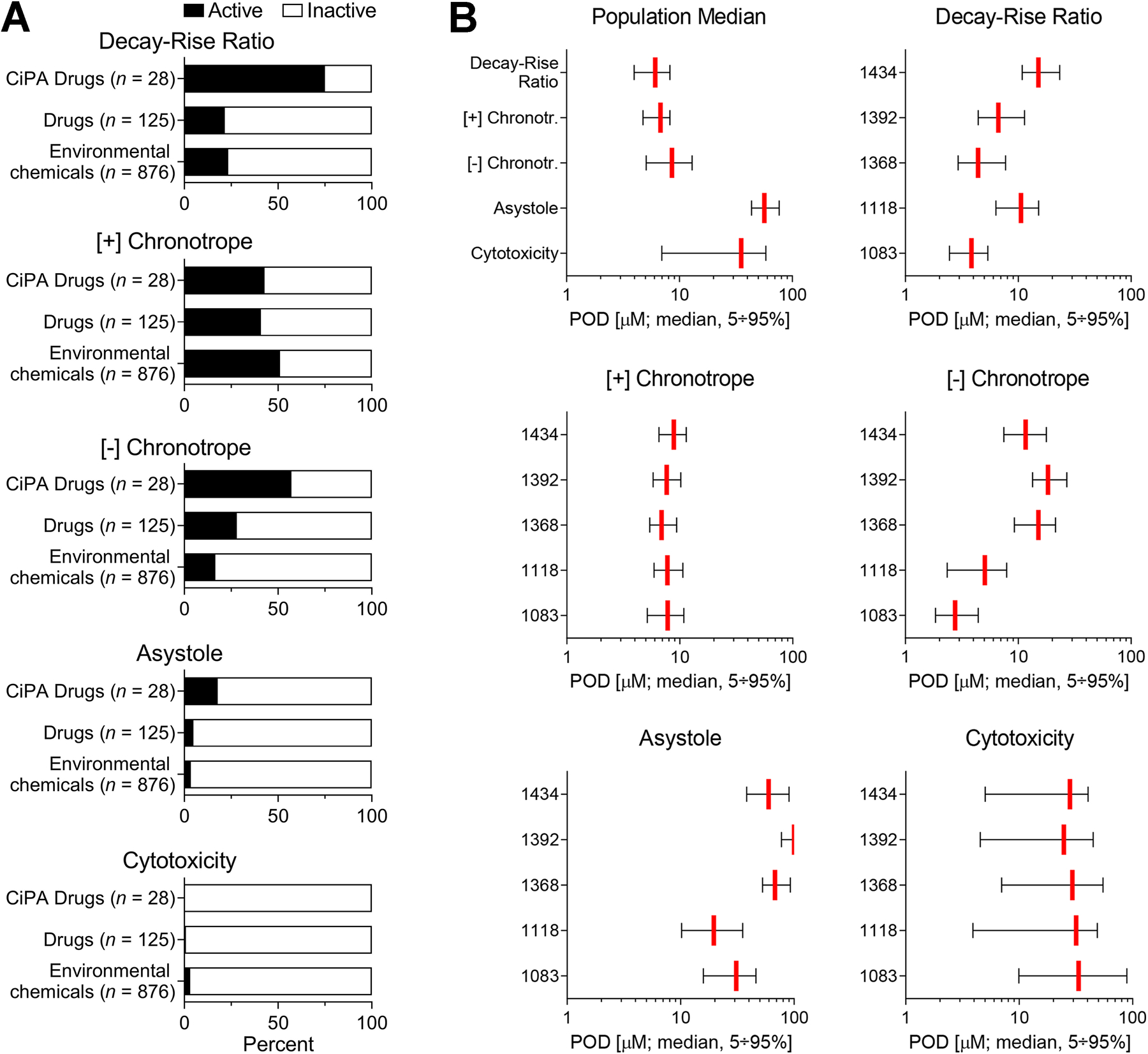
Cardiotoxicity hazard characterization: chemical activity calls. Activity calls across the 1,029 test chemicals. (A) The distribution of activity calls, separated for each phenotype and divided by chemical class. Activity describes whether a chemical is “active” or “inactive” for a phenotype at the population median. Bars show the percentage of chemicals in each class found to be active for that phenotype. (B) Bar plots (median ± 95% confidence intervals) showing the distribution of POD values for all chemicals found to be active at the population median for that phenotype (n = 256 compounds for decay-rise ratio, n = 519 for [+] chronotropy, n = 199 for [-] chronotropy, n = 42 for asystole, and n = 29 for cytotoxicity). Graphs are separated by phenotype and donor, with the upper left-hand graph describing the population median across the 5 donors. Vertical red bars show the median response, and the black bars represent the 95% confidence intervals across all active chemicals.
Most CiPA drugs (75%) were active for decay-rise ratio, a much greater proportion than for non-CiPA drugs or environmental chemicals. Of the CiPA drugs inactive for decay-rise ratio (n = 7 out of 28 compounds), most are classified as “low” risk for drug-induced TdP on the CiPA list. Only one “high”-risk CiPA compound, vandetanib, was classified as inactive for decay-rise ratio in this study. Many CiPA drugs (57%) were also active for the phenotype [-] chronotropy, a much higher proportion than was found for non-CiPA drugs or environmental chemicals. This result was expected, as a longer decay-rise ratio (delayed repolarization) often correlates with a simultaneously decreased beat rate.
For environmental chemicals, [+] chronotropy was the most common active phenotype, for which 51% of all tested non-pharmaceuticals exhibited activity, a higher proportion than observed for CiPA or other pharmaceutical compounds. A smaller number of environmental chemicals (of various subclasses) were active for decay-rise ratio and [-] chronotropy (23% and 17%, respectively). Only 28 (3%) environmental chemicals exhibited activity for cytotoxicity, many of which were herbicides and other non-herbicide pesticides.
The distribution of POD values for chemicals found to be active at the population median for each phenotype is shown in Figure 5B. Chemicals that were active for asystole and cytotoxicity exhibited effects at relatively high concentrations (larger POD). For [+] and [-] chronotropy and decay-rise ratio, active chemicals exhibited effects at lower concentrations (smaller POD), indicating that functional phenotypes are on average more sensitive for chemical-induced effects, and that functional effects are unlikely to be secondary to cytotoxicity. There were no statistically significant differences in sensitivity between donors across all endpoints combined (p>0.05). However, statistically significant differences between endpoints were detected in a two-way ANOVA (p<2e-16, ad hoc Tukey Honest Significant Differences test), with significant differences observed between all endpoint pairs except for [+] chronotropy-asystole, [-] chronotropy-decay-rise ratio, and cytotoxicity-decay-rise ratio.
We also characterized the cardiotoxicity hazard of chemicals by evaluating their critical endpoint (most sensitive phenotype, or phenotype with the lowest POD for the population median) (Figure 6A). The primary critical endpoint for most CiPA drugs (39%) was decay-rise ratio, a much higher proportion than was observed for non-CiPA drugs (13%) or any subclass of environmental chemicals. Some CiPA drugs (29%) had [-] chronotropy as the critical endpoint, and only a few (7%) were inactive for all phenotypes. The most common critical endpoint for non-CiPA drugs was [+] chronotropy (37%). While the majority of environmental chemicals exhibited effects and were designated a critical endpoint, a considerable number (20–40%) of chemicals in each subclass were inactive for all phenotypes, even when tested at up to 100 μM. For all subclasses of environmental chemicals, the most common critical endpoint was consistently [+] chronotropy, comprising between 36% (surfactants) and 55% (herbicides) of chemicals in each class. These are similar or higher proportions than observed for CiPA drugs or other pharmaceuticals. Of all subclasses, microbiocides had the highest proportion of chemicals with decay-rise ratio as the critical endpoint (21%), second only to CiPA drugs. Dyes had the highest proportion of chemicals with asystole as the critical endpoint (5%).
Figure 6.
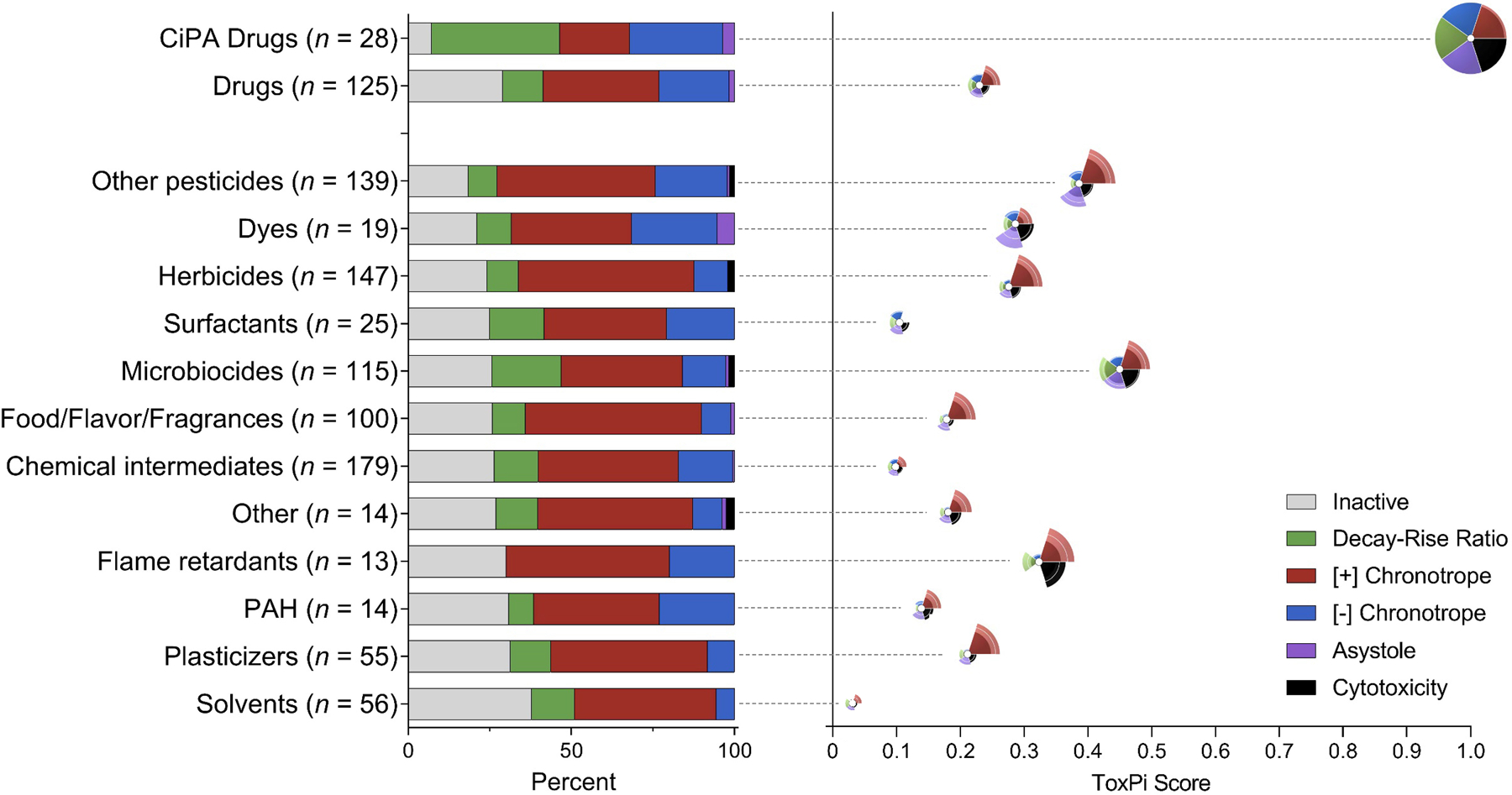
Cardiotoxicity hazard characterization: critical endpoint calls and ToxPi bioactivity visualization. (A) The distribution of critical endpoint calls for each chemical class with B) the corresponding ToxPi score and bioactivity profile of the average chemical in that class, ordered by increasing percent inactivity. Critical endpoint is defined as the most sensitive phenotype (the phenotype with the lowest point-of-departure for the population median). Bars are colored by phenotype and show the percentage of chemicals in each class having each phenotype as its critical endpoint. (B) The ToxPi bioactivity profile visualizations are positioned such that the white dot at the center of the ToxPi denotes the ToxPi score for that class. A higher ToxPi score indicates a higher relative bioactivity than other chemical classes. Slices are colored by endpoint and indicate potency for that phenotype, with a larger slice size indicating higher relative bioactivity (note that there is no slice for “inactive,” as potency cannot be described for chemicals that were inactive for every phenotype). Each slice has a corresponding confidence interval, represented by the area at the outer edge of each slice that is colored with less intensity. A larger confidence interval indicates a higher degree of variability in POD values among the 5 donors.
Next, we visualized the bioactivity profile for the average chemical in each class using ToxPi (Figure 6B). CiPA drugs were the most bioactive class (largest slices) and exhibited the highest potency across phenotypes (lowest POD, indicated by the highest overall ToxPi score of 1). Non-CiPA drugs were most bioactive for [+] chronotropy but exhibited lower relative potency in effects (ToxPi score of 0.24). Similarly, most subclasses of environmental chemicals exhibited the highest bioactivity for [+] chronotropy, at varying degrees of potency. Microbiocides were the most potent subclass, having the highest ToxPi score of any environmental chemical class (0.45), second only to CiPA drugs. In addition, microbiocides exhibited the highest bioactivity for decay-rise ratio among subclasses (a larger slice than for any other chemical class except CiPA drugs). Solvents were the least bioactive (smallest slice across phenotypes) and least potent (lowest overall ToxPi score of 0.03) class. Most chemical classes showed little to no bioactivity for cytotoxicity or asystole.
Cardiotoxicity risk characterization
In addition to characterizing hazard, we also characterized the cardiotoxicity risk of chemicals by evaluating margins of safety (MOS, for pharmaceuticals) or margins of exposure (MOE, for environmental chemicals) for the population median (Figure 7). MOS was derived for all tested pharmaceuticals that were active for at least one phenotype and had available human pharmacokinetic data in PharmaPendium (n = 46 out of 125 drugs, and n = 23 out of 28 CiPA drugs). Similarly, MOE was derived for all tested environmental chemicals that were active for at least one phenotype and had available exposure estimates in ExpoCast (n = 346 out of 876 non-pharmaceutical compounds). For pharmaceuticals, margins were relatively low (mostly below 100), indicating a higher chance of overlap between the dose that causes an effect in vitro and the normal human exposure level. 76% of all tested drugs (n = 35) and 91% of all tested CiPA drugs (n = 21) had margins below 100. Both CiPA drugs with margins above 100 were low-risk CiPA compounds (tamoxifen = 522 and metoprolol = 226) and did not exhibit decay-rise ratio as the critical endpoint. Of all drugs, propofol and tolcapone had the lowest MOS (less than 1).
Figure 7.
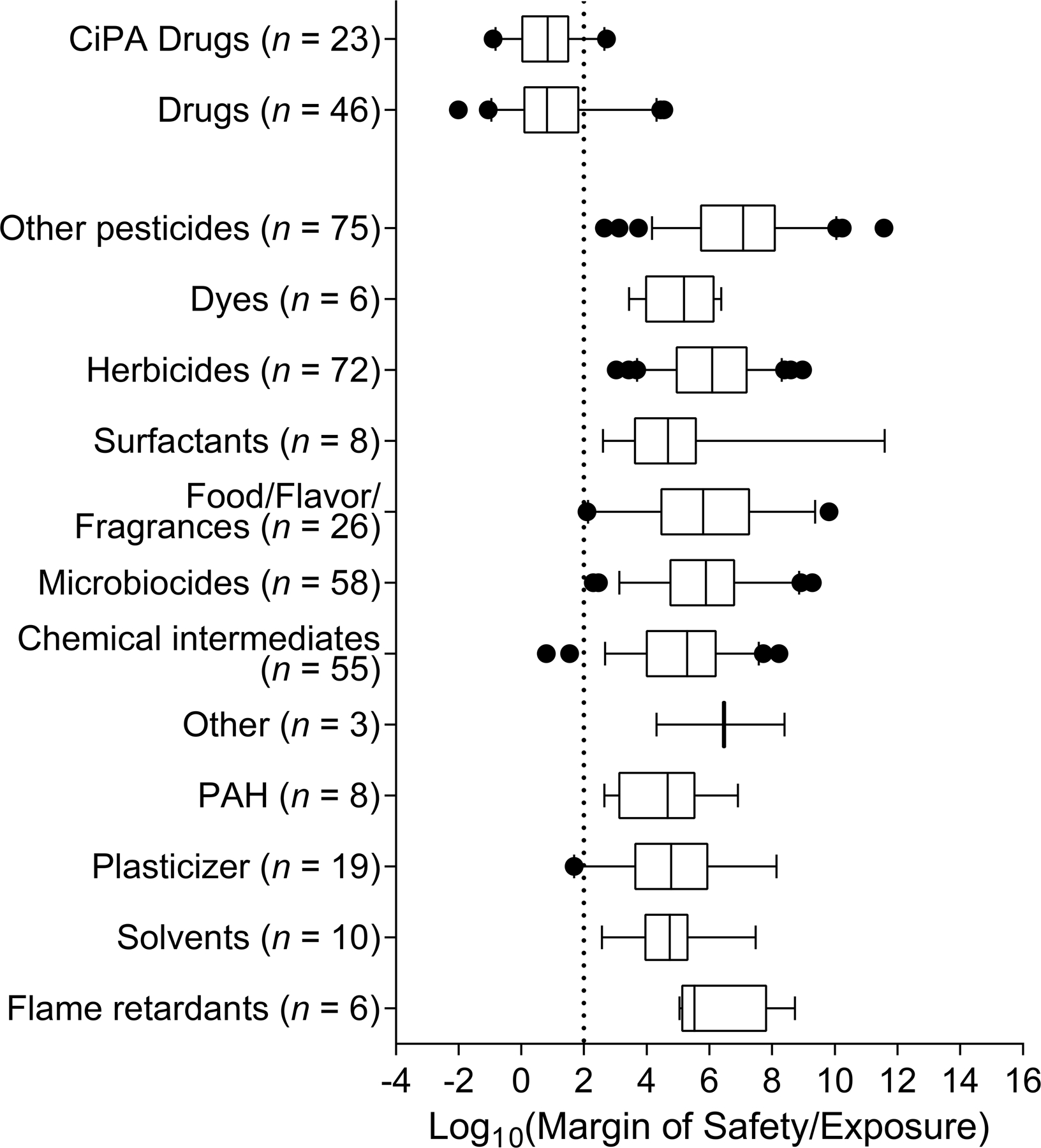
Cardiotoxicity risk characterization. Margins of safety (MOS, for pharmaceuticals) or margins of exposure (MOE, for environmental chemicals) for each chemical class. The MOS/MOE is calculated by dividing the point-of-departure for the population median in vitro by the exposure level for the population median in vivo. Lower margins represent higher chance of overlap between the concentration which causes an effect in vitro and the exposure level normally encountered by the population. Boxes are made of all chemicals with available exposure data in that class, whiskers show the 95% confidence intervals for MOS/MOE, and dots represent outliers. Margins are plotted on a log scale, and the vertical dotted line at MOS/MOE = 2 represents a margin of 100, a commonly used indicator as an adequately “protective” margin for non-genotoxic, non-carcinogenic compounds in chemical risk assessment.
By contrast, margins were relatively high (mostly above 100) for all classes of environmental chemicals. Overall, non-herbicide pesticides exhibited the highest margins of any chemical class, with a median of 5 orders of magnitude above the protective risk level. Surfactants and PAHs exhibited the lowest median margins across environmental chemical subclasses, but margins were still 2–3 orders of magnitude above the protective risk level. Interestingly, plasticizers and food/flavor/fragrance agents had the lowest 95% confidence intervals for the margin across chemical subclasses. Only three non-drug compounds had margins below 100, ammonium perfluorooctanoate (MOE = 6), 2,5-di-tert-butylbenzene-1,4-diol (MOE = 35), and dihexyl phthalate (MOE = 49), with one additional food/flavor/fragrance agent, coumarin, having a margin near 100 (MOE = 125).
Discussion
This study demonstrated how a human population-based in vitro model can be used to characterize the cardiotoxicity hazard and risk of drugs and chemicals in a high-throughput manner. We used iPSC-derived cardiomyocytes from 5 human donors to assess both hazard and risk for cardiotoxicity of more than 1,000 diverse chemicals in terms of their (i) activity in iPSC-derived cardiomyocytes, (ii) critical endpoint (most sensitive phenotype affected), and (iii) margin of safety/margin of exposure. We integrated in vitro data derived from calcium flux and high-content imaging with Bayesian population concentration-response modeling to characterize chemical-induced effects on 5 functional and viability-related phenotypes. Tested compounds encompassed diverse classes of pharmaceutical and environmental chemicals and included pesticides, flame retardants, plasticizers, PAHs, dyes, and food/flavor/fragrance agents, many of which lack adequate information on their potential human cardiotoxicity hazard.
Importantly, we found high technical reproducibility in chemical effects for all phenotypes and donors used in this study (Figure 3), as well as high inter-study reproducibility between this and our previous study 11 (Figure 4). Generally, phenotypes describing cell viability effects were less variable than those of functional effects, highlighting the need to include multiple replicates of each test condition, multiple concentrations, and adequate positive controls in study designs to more confidently understand the concentration-response of functional effects. Nonetheless, the high observed concordance in derived point-of-departure (POD) values within and across studies adds confidence that this model is a valuable and reliable screening tool for quantitatively characterizing the effects of chemicals of a broad diversity.
We characterized the cardiotoxicity hazard of chemicals by defining chemical activity (i.e. either “active” or “inactive” for the population median, Figure 5A). Overall, CiPA drugs exhibited the most activity across phenotypes, it was predominantly effects on decay-rise ratio and [-] chronotropy, with the former most commonly being the critical endpoint (Figure 6). This finding was similar to observations from our previous study on a subset of about half of CiPA drugs, in which about 90% showed activity for decay-rise ratio 13. These results were expected, because this phenotype is an in vitro surrogate for QT prolongation, and the CiPA drugs were specifically chosen because of their association with this and related arrhythmias. Additionally, in a previous study it was shown that using a population of iPSC-derived cardiomyocytes enables accurate recapitulation of clinical QTc prolongation following exposure to drugs 23. Together, these and other studies support the use of iPSC-derived cardiomyocytes as a platform of choice for assessing the proarrhythmic liabilities of drugs during preclinical development 50, 51, which can be integrated with other lines of evidence to improve the workflow and efficiency of regulatory decision making 52, 53.
For other drugs and environmental chemicals, more than half exhibited activity in [+] chronotropy as the most common phenotype and usually also the most sensitive endpoint (Figure 6). These results are similar to our previous findings in a smaller number (n = 82 total) of environmental chemicals, in which about 60% of tested compounds showed activity for [+] chronotropy 13. Interestingly, microbiocides had the highest proportion of compounds with decay-rise ratio as the critical endpoint and a high average potency, second only to CiPA drugs. There is mechanistic evidence associating some biocides to ion channel dysfunction and altered signaling pathways 17, 54, highlighting the utility of integrating data from multiple lines of evidence to more comprehensively understand chemical hazards and elucidate their mechanisms of cardiotoxicity. Other lines of evidence also support the association between some environmental chemicals and adverse effects on cardiovascular function. For example, exposure to pesticides such as organophosphates and carbamates has been associated with an increased incidence and mortality of myocardial infarction and other arrhythmic effects in humans 55, 56, corresponding cardiovascular adverse effects in animal models 55, and mechanistic evidence of ion channel dysfunction and altered signaling pathways in vitro based on a comprehensive analysis of ToxCast endpoints 17. Additionally, many food/flavor/fragrance agents demonstrated activity, often with [+] chronotropy as the critical endpoint. This finding is consistent with previous studies showing positive chronotropic effects in iPSC-derived cardiomyocytes following acute exposure to compounds such as food and beverage additives 11, 13, 57, 58 and cosmetic ingredients 59.
In contrast to the functional endpoints, very few compounds tested in this study (n = 69 out of 1,029) were active for the phenotypes cytotoxicity or asystole in our tested concentration range (Figure 5). This lack of activity in viability effects is supported by observations from our previous study 13. Moreover, for the few compounds that were active for these viability effects, in most cases, functional effects were also observed, and generally at a lower concentration. Interestingly, previous studies have also shown that functional phenotypes generally exhibit greater measures of population variability than do viability endpoints 13.
Previous studies have demonstrated the utility of integrating in vitro-derived hazard data with estimated human exposure levels to characterize chemical risk by calculating margins of safety (MOS, for pharmaceuticals) or margins of exposure (MOE, for environmental chemicals) 10, 12, 13, 59. Lower margins represent a higher chance of overlap between the concentration which causes an effect in vitro and the exposure level encountered by the population. In our previous study in a population of 43 iPSC-derived cardiomyocytes, most pharmaceuticals (n = 52) had margins below 100, whereas environmental chemicals (n = 82) commonly had margins greater than 10,000 13. Other studies in iPSC-derived cardiomyocytes have also found activity-to-exposure ratios between 1 and 5 orders of magnitude for many environmental chemicals 10, 12. Similarly, in the current study, pharmaceuticals (both CiPA and other drugs) had the lowest margins of any class, with most drugs having a MOS below 100; these results are not unexpected, because drugs are directly administered at doses intended to have pharmacological effects. Low margins for effects on decay-rise ratio (common for CiPA drugs) are generally more concerning than are low margins for chronotropic effects (common for other drugs), as the primary cardiovascular safety liability is the potential to delay cardiac repolarization (proarrhythmia) 26, 60. By contrast, all subclasses of environmental chemicals had high median margins, with most compounds having an MOE multiple orders of magnitude above 100 (commonly 10,000 and above). Only four non-drug chemicals had margins near or below 100: the ammonium salt of PFOA, 2,5-di-tert-butylbenzene-1,4-diol, dihexyl phthalate, and coumarin. The low margins for these chemicals were driven by both high human exposures and low effect concentrations, with all four chemicals having exposure estimates ranked in the top 25 of all environmental compounds in this study and exhibiting effects to peak frequency at concentrations below 0.2 μM. Of particular concern are PFOA and dihexyl phthalate, since these are members of large classes of perfluoroalkyl substances (PFAS) and phthalates, respectively, both of which are recognized to be persistent environmental contaminants. Moreover, there is evidence of cardiovascular effects in humans associated with exposure to these compounds 61, 62. Thus, although most environmental chemicals for which current exposure and TK estimates are available seem to pose little cardiotoxicity risk to the population, the relatively lower margins for PFOA and dihexyl phthalate raise the concern that cumulative exposure to PFAS and phthalates may pose a cardiotoxicity risk given the number of such substances on the market and in the environment and the poor understanding of cumulative exposures to these classes of compounds.
There are a number of considerations why these in vitro-data risk characterization results need to be interpreted with caution. Many exposure predictions in ExpoCast are based on human biomonitoring data (in the form of urine concentrations) from the National Health and Nutrition Examination Survey (NHANES), but the exposure measurements are extrapolated to derive population-wide estimates and, thus, possess inherent uncertainty 63. Further, there is a lack of widespread biomonitoring data across chemicals, so many environmental chemicals lack reliable exposure estimates 64, without which margins cannot be derived. As a result, the cardiotoxicity risk of chemicals with unknown exposures (>600 in this study) cannot be assessed, which limits our ability to broadly generalize our data to the potential cardiotoxicity risk of these chemicals. Additionally, cardiomyocyte assays are not comprehensive as to their coverage of adverse cardiovascular effects, such as effects on mediated by the nervous system or the vasculature.
In conclusion, this study considerably extends the existing body of knowledge describing the bioactivity of environmental chemicals and demonstrates that many environmental chemicals present a hazard to human cardiomyocytes in vitro. Results from this study also suggest that although many environmental chemicals tested do not appear to present a significant human health risk at currently-estimated exposure levels, low margins of exposures for perfluoroalkyl substances and phthalates raise concerns about potential for cardiotoxicity due to cumulative exposure to these large classes of compounds, and highlighting the need for more comprehensive exposure data. More broadly, this research demonstrates the feasibility of this model for high-throughput testing of environmental chemicals that lack data on their potential cardiotoxicity hazard. Thus, this in vitro screening approach is a sensible alternative or supplement to in vivo toxicity testing methodologies and offers a promising avenue to address concerning lack of data on potential cardiotoxicity of a large number of environmental and industrial chemicals.
Supplementary Material
Acknowledgements
The authors wish to thank Drs. Russell Thomas and Ann Richard (US Environmental Protection Agency) for assistance with procuring test chemicals. The authors wish to acknowledge assistance from the scientists and technicians at FujiFilm Cellular Dynamics for enabling these studies by establishing a large panel of iPSC-derived human cardiomyocytes. This work was funded, in part, by a cooperative agreement with the United States Environmental Protection Agency (STAR RD83580201) and grants from the National Institutes of Health (P42 ES027704 and T32 ES026568). The views expressed in this manuscript do not reflect those of the funding agencies. The use of specific commercial products in this work does not constitute endorsement by the authors or the funding agencies.
Footnotes
Conflict of Intertest Statement
The authors declare no relevant conflicts of interest with regard to this study.
References
- (1).Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, American Heart Association Council on, E., Prevention Statistics, C., and Stroke Statistics, S. (2019) Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation, 139, e56–e528. [DOI] [PubMed] [Google Scholar]
- (2).WHO Cvd Risk Chart Working Group. (2019) World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health, 7, e1332–e1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Knowles JW, and Ashley EA (2018) Cardiovascular disease: The rise of the genetic risk score. PLoS Med, 15, e1002546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Page RL 2nd, O’Bryant CL, Cheng D, Dow TJ, Ky B, Stein CM, Spencer AP, Trupp RJ, Lindenfeld J, American Heart Association Clinical, P., Heart, F., Transplantation Committees of the Council on Clinical, C., Council on Cardiovascular, S., Anesthesia, Council on, C., Stroke, N., Council on Quality of, C., and Outcomes, R. (2016) Drugs That May Cause or Exacerbate Heart Failure: A Scientific Statement From the American Heart Association. Circulation, 134, e32–69. [DOI] [PubMed] [Google Scholar]
- (5).Bhatnagar A (2017) Environmental Determinants of Cardiovascular Disease. Circ Res, 121, 162–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Laverty H, Benson C, Cartwright E, Cross M, Garland C, Hammond T, Holloway C, McMahon N, Milligan J, Park B, Pirmohamed M, Pollard C, Radford J, Roome N, Sager P, Singh S, Suter T, Suter W, Trafford A, Volders P, Wallis R, Weaver R, York M, and Valentin J (2011) How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol, 163, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pang L, Sager P, Yang X, Shi H, Sannajust F, Brock M, Wu JC, Abi-Gerges N, Lyn-Cook B, Berridge BR, and Stockbridge N (2019) Workshop Report: FDA Workshop on Improving Cardiotoxicity Assessment With Human-Relevant Platforms. Circ Res, 125, 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Judson R, Richard A, Dix DJ, Houck K, Martin M, Kavlock R, Dellarco V, Henry T, Holderman T, Sayre P, Tan S, Carpenter T, and Smith E (2009) The toxicity data landscape for environmental chemicals. Environ Health Perspect, 117, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pruss-Ustun A, Wolf J, Corvalan C, Neville T, Bos R, and Neira M (2017) Diseases due to unhealthy environments: an updated estimate of the global burden of disease attributable to environmental determinants of health. J Public Health (Oxf), 39, 464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sirenko O, Grimm FA, Ryan KR, Iwata Y, Chiu WA, Parham F, Wignall JA, Anson B, Cromwell EF, Behl M, Rusyn I, and Tice RR (2017) In vitro cardiotoxicity assessment of environmental chemicals using an organotypic human induced pluripotent stem cell-derived model. Toxicol Appl Pharmacol, 322, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Burnett SD, Blanchette AD, Grimm FA, House JS, Reif DM, Wright FA, Chiu WA, and Rusyn I (2019) Population-based toxicity screening in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Appl Pharmacol, 381, 114711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Grimm FA, Klaren WD, Li X, Lehmler HJ, Karmakar M, Robertson LW, Chiu WA, and Rusyn I (2020) Cardiovascular Effects of Polychlorinated Biphenyls and Their Major Metabolites. Environ Health Perspect, 128, 77008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Blanchette AD, Burnett SD, Grimm FA, Rusyn I, and Chiu WA (2020) A Bayesian Method for Population-wide Cardiotoxicity Hazard and Risk Characterization Using an In Vitro Human Model. Toxicol Sci, 178, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen Z, Lloyd D, Zhou YH, Chiu WA, Wright FA, and Rusyn I (2021) Risk Characterization of Environmental Samples Using In Vitro Bioactivity and Polycyclic Aromatic Hydrocarbon Concentrations Data. Toxicol Sci, 179, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen Z, Liu Y, Wright FA, Chiu WA, and Rusyn I (2020) Rapid hazard characterization of environmental chemicals using a compendium of human cell lines from different organs. ALTEX, 37, 623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).House JS, Grimm FA, Klaren WD, Dalzell A, Kuchi S, Zhang SD, Lenz K, Boogaard PJ, Ketelslegers HB, Gant TW, Wright FA, and Rusyn I (2021) Grouping of UVCB substances with new approach methodologies (NAMs) data. ALTEX, 38, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Krishna S, Berridge B, and Kleinstreuer N (2021) High-Throughput Screening to Identify Chemical Cardiotoxic Potential. Chem Res Toxicol, 34, 566–583. [DOI] [PubMed] [Google Scholar]
- (18).Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, Baker NC, Patlewicz G, Shah I, Wambaugh JF, Judson RS, and Richard AM (2017) The CompTox Chemistry Dashboard: a community data resource for environmental chemistry. J Cheminform, 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Burnett SD, Blanchette AD, Chiu WA, and Rusyn I (2021) Human induced pluripotent stem cell (iPSC)-derived cardiomyocytes as an in vitro model in toxicology: strengths and weaknesses for hazard identification and risk characterization. Expert Opin Drug Metab Toxicol, in press. [DOI] [PMC free article] [PubMed]
- (20).Anson BD, Kolaja KL, and Kamp TJ (2011) Opportunities for use of human iPS cells in predictive toxicology. Clin Pharmacol Ther, 89, 754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Apati A, Varga N, Berecz T, Erdei Z, Homolya L, and Sarkadi B (2019) Application of human pluripotent stem cells and pluripotent stem cell-derived cellular models for assessing drug toxicity. Expert Opin Drug Metab Toxicol, 15, 61–75. [DOI] [PubMed] [Google Scholar]
- (22).Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmstrom A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, and Wu JC (2016) Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med, 22, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Blanchette AD, Grimm FA, Dalaijamts C, Hsieh NH, Ferguson K, Luo YS, Rusyn I, and Chiu WA (2019) Thorough QT/QTc in a Dish: An In Vitro Human Model That Accurately Predicts Clinical Concentration-QTc Relationships. Clin Pharmacol Ther, 105, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chiu WA, Wright FA, and Rusyn I (2017) A tiered, Bayesian approach to estimating of population variability for regulatory decision-making. ALTEX, 34, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Blanchette A, Burnett SD, Rusyn I, and Chiu WA (2021) A tiered approach to population-based in vitro testing for cardiotoxicity: Balancing estimates of potency and variability Submitted. [DOI] [PMC free article] [PubMed]
- (26).International Conference on Harmonization. (2005) The Clinical Evaluation of QT/QTC Interval Prolongation and Proarrythmic Potential for Non-Antiarrythmic Drugs E14, International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland. [Google Scholar]
- (27).Kleinstreuer NC, Yang J, Berg EL, Knudsen TB, Richard AM, Martin MT, Reif DM, Judson RS, Polokoff M, Dix DJ, Kavlock RJ, and Houck KA (2014) Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat Biotechnol, 32, 583–591. [DOI] [PubMed] [Google Scholar]
- (28).Pearce RG, Setzer RW, Strope CL, Wambaugh JF, and Sipes NS (2017) httk: R Package for High-Throughput Toxicokinetics. J Stat Softw, 79, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).U.S. EPA. (2020) Integrated Risk Information System (IRIS), U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- (30).Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, Zakharov A, Worth A, Richard AM, Grulke CM, Trisciuzzi D, Fourches D, Horvath D, Benfenati E, Muratov E, Wedebye EB, Grisoni F, Mangiatordi GF, Incisivo GM, Hong H, Ng HW, Tetko IV, Balabin I, Kancherla J, Shen J, Burton J, Nicklaus M, Cassotti M, Nikolov NG, Nicolotti O, Andersson PL, Zang Q, Politi R, Beger RD, Todeschini R, Huang R, Farag S, Rosenberg SA, Slavov S, Hu X, and Judson RS (2016) CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ Health Perspect, 124, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Casey WM, Chang X, Allen DG, Ceger PC, Choksi NY, Hsieh JH, Wetmore BA, Ferguson SS, DeVito MJ, Sprankle CS, and Kleinstreuer NC (2018) Evaluation and Optimization of Pharmacokinetic Models for in Vitro to in Vivo Extrapolation of Estrogenic Activity for Environmental Chemicals. Environ Health Perspect, 126, 97001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kleinstreuer NC, Browne P, Chang X, Judson R, Casey W, Ceger P, Deisenroth C, Baker N, Markey K, and Thomas RS (2018) Evaluation of androgen assay results using a curated Hershberger database. Reprod Toxicol, 81, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wallis R, Benson C, Darpo B, Gintant G, Kanda Y, Prasad K, Strauss DG, and Valentin JP (2018) CiPA challenges and opportunities from a non-clinical, clinical and regulatory perspectives. An overview of the safety pharmacology scientific discussion. J Pharmacol Toxicol Methods, 93, 15–25. [DOI] [PubMed] [Google Scholar]
- (34).Richard AM, Huang R, Waidyanatha S, Shinn P, Collins BJ, Thillainadarajah I, Grulke CM, Williams AJ, Lougee RR, Judson RS, Houck KA, Shobair M, Yang C, Rathman JF, Yasgar A, Fitzpatrick SC, Simeonov A, Thomas RS, Crofton KM, Paules RS, Bucher JR, Austin CP, Kavlock RJ, and Tice RR (2021) The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chem Res Toxicol, 34, 189–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sirenko O, Crittenden C, Callamaras N, Hesley J, Chen YW, Funes C, Rusyn I, Anson B, and Cromwell EF (2013) Multiparameter in vitro assessment of compound effects on cardiomyocyte physiology using iPSC cells. J Biomol Screen, 18, 39–53. [DOI] [PubMed] [Google Scholar]
- (36).Grimm FA, Blanchette A, House JS, Ferguson K, Hsieh NH, Dalaijamts C, Wright AA, Anson B, Wright FA, Chiu WA, and Rusyn I (2018) A human population-based organotypic in vitro model for cardiotoxicity screening. ALTEX, 35, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Guha R (2007) Chemical informatics functionality in R. J Stat Softw, 18, 1–16. [Google Scholar]
- (38).Wignall JA, Muratov E, Sedykh A, Guyton KZ, Tropsha A, Rusyn I, and Chiu WA (2018) Conditional Toxicity Value (CTV) Predictor: An In Silico Approach for Generating Quantitative Risk Estimates for Chemicals. Environ Health Perspect, 126, 057008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sirenko O, Cromwell EF, Crittenden C, Wignall JA, Wright FA, and Rusyn I (2013) Assessment of beating parameters in human induced pluripotent stem cells enables quantitative in vitro screening for cardiotoxicity. Toxicol Appl Pharmacol, 273, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).R Core Team. (2015) R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- (41).Gelman A, and Rubin DB (1992) Inference from iterative simulation using multiple sequences. Statistical science, 7, 457–472. [Google Scholar]
- (42).Abdo N, Xia M, Brown CC, Kosyk O, Huang R, Sakamuru S, Zhou YH, Jack JR, Gallins P, Xia K, Li Y, Chiu WA, Motsinger-Reif AA, Austin CP, Tice RR, Rusyn I, and Wright FA (2015) Population-based in vitro hazard and concentration-response assessment of chemicals: the 1000 genomes high-throughput screening study. Environ Health Perspect, 123, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Marvel SW, To K, Grimm FA, Wright FA, Rusyn I, and Reif DM (2018) ToxPi Graphical User Interface 2.0: Dynamic exploration, visualization, and sharing of integrated data models. BMC Bioinf, 19, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Giblin KA, Basili D, Afzal AM, Rosenbrier-Ribeiro L, Greene N, Barrett I, Hughes SJ, and Bender A (2021) New Associations between Drug-Induced Adverse Events in Animal Models and Humans Reveal Novel Candidate Safety Targets. Chem Res Toxicol, 34, 438–451. [DOI] [PubMed] [Google Scholar]
- (45).Scientific Committee on Toxicity Ecotoxicity and the Environment. (2001) Position Paper on Margins of Safety (MOS) in Human Health Risk Assessment Expressed at the 22nd CSTEE Plenary Meeting, Brussels, 06/07 March 2001, European Commission, Brussels, Belgium. [Google Scholar]
- (46).Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug deliv Rev, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (47).Prinz F, Schlange T, and Asadullah K (2011) Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov, 10, 712. [DOI] [PubMed] [Google Scholar]
- (48).Watt ED, and Judson RS (2018) Uncertainty quantification in ToxCast high throughput screening. PLoS One, 13, e0196963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Casadevall A, and Fang FC (2016) Rigorous Science: a How-To Guide. mBio, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ando H, Yoshinaga T, Yamamoto W, Asakura K, Uda T, Taniguchi T, Ojima A, Shinkyo R, Kikuchi K, Osada T, Hayashi S, Kasai C, Miyamoto N, Tashibu H, Yamazaki D, Sugiyama A, Kanda Y, Sawada K, and Sekino Y (2017) A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J Pharmacol Toxicol Methods, 84, 111–127. [DOI] [PubMed] [Google Scholar]
- (51).Blinova K, Dang Q, Millard D, Smith G, Pierson J, Guo L, Brock M, Lu HR, Kraushaar U, Zeng H, Shi H, Zhang X, Sawada K, Osada T, Kanda Y, Sekino Y, Pang L, Feaster TK, Kettenhofen R, Stockbridge N, Strauss DG, and Gintant G (2018) International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep, 24, 3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vargas HM, Rolf MG, Wisialowski TA, Achanzar W, Bahinski A, Bass A, Benson CT, Chaudhary KW, Couvreur N, Dota C, Engwall MJ, Michael Foley C, Gallacher D, Greiter-Wilke A, Guillon JM, Guth B, Himmel HM, Hegele-Hartung C, Ito M, Jenkinson S, Chiba K, Lagrutta A, Levesque P, Martel E, Okai Y, Peri R, Pointon A, Qu Y, Teisman A, Traebert M, Yoshinaga T, Gintant GA, Leishman DJ, and Valentin JP (2021) Time for a Fully Integrated Nonclinical-Clinical Risk Assessment to Streamline QT Prolongation Liability Determinations: A Pharma Industry Perspective. Clin Pharmacol Ther, 109, 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Strauss DG, Wu WW, Li Z, Koerner J, and Garnett C (2021) Translational Models and Tools to Reduce Clinical Trials and Improve Regulatory Decision Making for QTc and Proarrhythmia Risk (ICH E14/S7B Updates). Clin Pharmacol Ther, 109, 319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Xia M, Shahane SA, Huang R, Titus SA, Shum E, Zhao Y, Southall N, Zheng W, Witt KL, Tice RR, and Austin CP (2011) Identification of quaternary ammonium compounds as potent inhibitors of hERG potassium channels. Toxicol Appl Pharmacol, 252, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Georgiadis N, Tsarouhas K, Tsitsimpikou C, Vardavas A, Rezaee R, Germanakis I, Tsatsakis A, Stagos D, and Kouretas D (2018) Pesticides and cardiotoxicity. Where do we stand? Toxicol Appl Pharmacol, 353, 1–14. [DOI] [PubMed] [Google Scholar]
- (56).Bar-Meir E, Schein O, Eisenkraft A, Rubinshtein R, Grubstein A, Militianu A, Glikson M, and Cbrn Medical Branch MCIDF (2007) Guidelines for treating cardiac manifestations of organophosphates poisoning with special emphasis on long QT and Torsades De Pointes. Crit Rev Toxicol, 37, 279–285. [DOI] [PubMed] [Google Scholar]
- (57).Nystoriak MA, Kilfoil PJ, Lorkiewicz PK, Ramesh B, Kuehl PJ, McDonald J, Bhatnagar A, and Conklin DJ (2019) Comparative effects of parent and heated cinnamaldehyde on the function of human iPSC-derived cardiac myocytes. Toxicol In Vitro, 61, 104648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Luo YS, Chen Z, Blanchette AD, Zhou YH, Wright FA, Baker ES, Chiu WA, and Rusyn I (2021) Relationships between constituents of energy drinks and beating parameters in human induced pluripotent stem cell (iPSC)-Derived cardiomyocytes. Food Chem Toxicol, 149, 111979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chaudhari U, Nemade H, Sureshkumar P, Vinken M, Ates G, Rogiers V, Hescheler J, Hengstler JG, and Sachinidis A (2018) Functional cardiotoxicity assessment of cosmetic compounds using human-induced pluripotent stem cell-derived cardiomyocytes. Arch Toxicol, 92, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).FDA US (2005) Guidance for Industry: S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals [PubMed]
- (61).Huang M, Jiao J, Zhuang P, Chen X, Wang J, and Zhang Y (2018) Serum polyfluoroalkyl chemicals are associated with risk of cardiovascular diseases in national US population. Environ Int, 119, 37–46. [DOI] [PubMed] [Google Scholar]
- (62).Mariana M, and Cairrao E (2020) Phthalates Implications in the Cardiovascular System. J Cardiovasc Dev Dis, 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Biryol D, Nicolas CI, Wambaugh J, Phillips K, and Isaacs K (2017) High-throughput dietary exposure predictions for chemical migrants from food contact substances for use in chemical prioritization. Environ Int, 108, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wambaugh JF, Setzer RW, Reif DM, Gangwal S, Mitchell-Blackwood J, Arnot JA, Joliet O, Frame A, Rabinowitz J, Knudsen TB, Judson RS, Egeghy P, Vallero D, and Cohen Hubal EA (2013) High-throughput models for exposure-based chemical prioritization in the ExpoCast project. Environ Sci Technol, 47, 8479–8488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.