

Abstract
Purpose
The purpose of this study was to further expand the mutational spectrum of the Foveal Hypoplasia, Optic Nerve Decussation defect, and Anterior segment abnormalities (FHONDA syndrome), to describe the phenotypic spectrum, and to compare it to albinism.
Subjects and Methods
We retrospectively collected molecular, ophthalmic, and electrophysiological data of 28 patients molecularly confirmed with FHONDA from the Netherlands (9), Israel (13), France (2), and the United States of America (4). We compared the data to that of 133 Dutch patients with the 3 most common types of albinism in the Netherlands: oculocutaneous albinism type 1 (49), type 2 (41), and ocular albinism (43).
Results
Patients with FHONDA had a total of 15 different mutations in SLC38A8, of which 6 were novel. Excluding missing data, all patients had moderate to severe visual impairment (median visual acuity [VA] = 0.7 logMAR, interquartile range [IQR] = 0.6–0.8), nystagmus (28/28), and grade 4 foveal hypoplasia (17/17). Misrouting was present in all nine tested patients. None of the patients had any signs of hypopigmentation of skin and hair. VA in albinism was better (median = 0.5 logMAR, IQR = 0.3–0.7, P 0.006) and the phenotypes were more variable: 14 of 132 without nystagmus, foveal hypoplasia grades 1 to 4, and misrouting absent in 16 of 74.
Conclusions
Compared to albinism, the FHONDA syndrome appears to have a more narrow phenotypic spectrum, consisting of nonprogressive moderately to severely reduced VA, nystagmus, severe foveal hypoplasia, and misrouting. The co-occurrence of nystagmus, foveal hypoplasia, and misrouting in the absence of hypopigmentation implies that these abnormalities are not caused by lack of melanin, which has important implications for understanding the pathogenesis of these features.
Keywords: SLC38A8, FHONDA, foveal hypoplasia, misrouting, melanin
Until 2006, misrouting of the visual pathways was only described in albinism and believed to be secondary to the lack of ocular pigmentation.1–4 In 2006, van Genderen et al. first reported the combination of misrouting and foveal hypoplasia in patients without albinism.5 The patients described in this paper showed similarities to a family described by Pal et al. with foveal hypoplasia and anterior segment dysgenesis.6 In 2013, the term FHONDA was introduced for the disorder, which is short for Foveal Hypoplasia, Optic Nerve Decussation defects and Anterior segment dysgenesis.7 Poulter et al. discovered that mutations in the SLC38A8 gene were responsible for the FHONDA syndrome and demonstrated that in embryonic Medaka fish, knockdown of both orthologs of SLC38A8 did not result in any pigmentation defect of the eye or tegumen.8 Perez et al. described 9 patients with homozygous SLC38A8 mutations who had a combination of foveal hypoplasia and nystagmus also without any signs of hypopigmentation.9
FHONDA and albinism share the clinical features of nystagmus, foveal hypoplasia, and misrouting, but because of the lack of pigmentation defect in Medaka fish, and in earlier reported patients with FHONDA it seems that albinism and FHONDA are distinct disorders.
The phenotypic spectrum of albinism has been extensively investigated and appears to be very broad.10–15 Albinism is a heterogeneous condition, and even patients who have mutations in the same gene also show variable clinical features. Visual acuity in albinism ranges from very poor to normal and foveal hypoplasia of all grades has been described, from absent (grade 0) to severe (grade 4). None of the clinical features is universal for all patients with albinism, with nystagmus being absent in at least 7% and misrouting in 16%.10 FHONDA appears to be rare, and seems more homogeneous than albinism. Since the identification of the first patient, only a few cases have been reported.5–9,14,16–20 The disorder is unknown to most clinicians and therefore is not adequately recognized. The purpose of this study is to further define the phenotypic and genetic spectrum of FHONDA, and to compare its presentation to albinism.
Methods
The study was approved by the Medical Ethics Committee of Leiden University Medical Center and adhered to the tenets of the Declaration of Helsinki.
Patients With FHONDA
We included 28 patients with FHONDA from 4 countries, 12 male patients and 17 female patients, aged 1 to 71 years (median age 24 years). All patients, or their affected siblings, had two likely disease-causing variants in SLC38A8, and had records on their pigmentation status and ophthalmic findings.
Patients with FHONDA from the Netherlands (9 patients and 5 families) were diagnosed at the Bartiméus Institute. Additional patients came from tertiary referral centers in Israel (13 patients, 7 families), France, (2 patients, 1 family), and the United States of America (4 patients, 3 families). Five patients from the Netherlands, nine from Israel, and one patient from France were previously reported, however, several clinical features were not described, for instance, grading of foveal hypoplasia.5,7,8,14 Data were obtained through medical record review for best-corrected visual acuity (VA), refractive error, pigmentation of eyes, skin, and hair, slit lamp examination, ophthalmoscopy, fundus photography, optical coherence tomography (OCT) scans, and multichannel Visual Evoked Potentials (VEPs) tests. We used a grading scheme for foveal hypoplasia previously described by Thomas et al., with grade 0 indicating normal foveal structure, grades 1 and 2 not having incursion of the inner retinal layers, and grades 3 and 4 also affecting the photoreceptor differentiation (Supplementary Table S1).21 Multichannel VEPs were obtained with Espion E2 or E3 (Diagnosys LLC, Cambridge, UK), according to ISCEV standards.22 A chiasm coefficient was calculated and the cutoff values were used from the study of Kruijt et al.23
FHONDA Versus Albinism
We compared the phenotypic spectrum of FHONDA to that of 133 patients with genetically confirmed albinism, 99 male patients and 34 female patients, aged 0 to 77 years (median age 10 years). These patients had either two mutations in TYR (49 patients) or OCA2 (41 patients), or a mutation in GPR143 (43 patients). Mutations in TYR and OCA2 cause oculocutaneous albinism (OCA) type 1 and 2, respectively, and those in GPR143 cause ocular albinism (OA1), the 3 most common types of albinism in the Netherlands. We used IBM SPSS Statistics software version 22 to perform statistical analysis. Data were not normally distributed, and therefore we used nonparametric tests.
Results
All but two of the 28 patients with FHONDA were able to cooperate with VA measurement, OCT scans for grading were available in 17 of 28 cases. VEP testing was performed in 9 of 28 patients. VA could be tested in 57 of 90 patients with albinism, in 43 of 90 patients, OCT-scans were obtained, and in 58 of 90 patients VEP tests were done.
FHONDA (N = 28)
Molecular analyses and demographic characteristics of the 28 patients with FHONDA are presented in Table 1. Patients had a total of 15 different mutations in SLC38A8, consisting of missense, (inframe) deletions, frameshift, and truncating mutations located over the entire gene (Table 1, Fig. 1). We report six novel mutations: c.260C > T; p.(Thr87Ile) and c.800T > G; p.(Leu267Arg) in the Dutch family III, c.160G > T; p.(Gly54*) and c.388 + 5G > A; p.(?) in family VII from the United States, c.(805 + 1_806–1)_(1162 + 1_1163–1)del; p.(?) (deletion exon 7 and 8) in the French family VI and family VIII from the United States, respectively, and c.1256G > T; p.(Gly419Val) in family VIII.
Table 1.
Molecular Analyses Patients With FHONDA
| Family | Descent | ID | Mutations SLC38A8 Gene |
|---|---|---|---|
| I* | Afghani | 801 | Homozygous c.1002del; p.(Ser336Alafs*15) |
| I* | Afghani | 802 | Homozygous c.1002del; p.(Ser336Alafs*15) |
| I | Afghani | 828 | Homozygous c.1002del; p.(Ser336Alafs*15) |
| II* | Dutch | 803 | One large deletion and c.1234G > A; p.(Gly412Arg) |
| III | Dutch | 804 | c.260C > T; p.(Thr87Ile) and c.800T > G; p.(Leu267Arg) |
| III | Dutch | 805 | c.260C > T; p.(Thr87Ile) and c.800T > G; p.(Leu267Arg)† |
| IV* | Dutch | 806 | c.598C > T; p.(Gln200*) and c.845_847del; p.(Ala282del) |
| IV* | Dutch | 807 | c.598C > T; p.(Gln200*) and c.845_847del; p.(Ala282del) |
| V | Dutch | 808 | Homozygous c.598C > T; p.(Gln200*) |
| VI | French | 809 | c.697G > A; p.(Glu233Lys) and c.(805 + 1_806–1)_(1162 + 1_1163–1)del; p.(?) deletion exon 7 and 8 |
| VI* | French | 810 | c.697G > A; p.(Glu233Lys) and c.(805 + 1_806–1)_(1162 + 1_1163–1)del; p.(?) deletion exon 7 and 8 |
| VII | Irish/French-Canadian/Puerto Rican | 811 | c.160G > T; p.(Gly54*) and c.388 + 5G > A; p.(?) |
| VIII | Swedish/Italian/Irish/English | 812 | c.(805 + 1_806–1)_(1162 + 1_1163–1)del; p.(?) deletion exon 7 and 8 and c.1256G > T p.(Gly419Val) |
| IX | Ashkenazi-Jewish | 813 | Homozygous c.848A > C; p.(Asp283Ala) |
| IX | Ashkenazi-Jewish | 814 | Homozygous c.848A > C; p.(Asp283Ala) |
| X* | Indian Jewish | 815 | Homozygous c.95T > G; p.(Ile32Ser) |
| X* | Indian Jewish | 816 | Homozygous c.95T > G; p.(Ile32Ser) |
| X* | Indian Jewish | 817 | Homozygous c.95T > G; p.(Ile32Ser) |
| XI* | Indian Jewish | 818 | Homozygous c.95T > G; p.(Ile32Ser) |
| XI* | Indian Jewish | 819 | Homozygous c.95T > G; p.(Ile32Ser) |
| XII* | Indian Jewish | 820 | Homozygous c.95T > G; p.(Ile32Ser) |
| XII* | Indian Jewish | 821 | Homozygous c.95T > G; p.(Ile32Ser) |
| XII* | Indian Jewish | 822 | Homozygous c.95T > G; p.(Ile32Ser) |
| XII* | Indian Jewish | 823 | Homozygous c.95T > G; p.(Ile32Ser) |
| XIII | Jewish; Ashkenazi-Lebanon-Syria/Yemenite-Afghanistan | 824 | c.848A > C; p.(Asp283Ala) and whole gene deletion |
| XIV | Indian Jewish | 825 | Homozygous c.95T > G; p.(Ile32Ser) |
| XIV | Indian Jewish | 826 | Homozygous c.95T > G; p.(Ile32Ser) |
| XV | Ashkenazi-Jewish | 827 | Homozygous c.848A > C; p.(Asp283Ala) |
Figure 1.

Schematic representation of the SLC38A8 gene. The location of the variants found in this study patients is represented on the right and of those retrieved from literature on the left, in grey. Exons are illustrated in orange and blue, transmembrane domains in green. Deletions are depicted at the location of the first deletion coordinate.
About two thirds of the patients (i.e. 18/28 patients from 8 families, were of south-western Asian origin, Afghani, and Indian, and Ashkenazi Jewish descent). All had homozygous mutations, except the patient from family XIII. The 11 patients from the 4 Indian Jewish families were all homozygous for c.95T > G; p.(Ile32Ser). The c.848A > C variant was found in all Ashkenazi-Jewish patients. The patient from family XIII was of a mixed origin, includingAshkenazi, Lebanon-Syria, and Yemenite-Afghanisti descent, and had a compound heterozygous mutation c.848A > C; p.(Asp283Ala), and a whole gene deletion. In contrast, 9 of 10 (partially) western European patients were compound heterozygotes.
Phenotypic data of patients with FHONDA are presented in Table 2. Patients (25/28) had a median VA of 0.7 logMAR (interquartile range [IQR] = 0.6-0.8). None of the patients was emmetropic, with moderate (1 to 2 diopters) to severe (>2 diopters) astigmatism as the most characteristic refractive error. Only 4 of 28 patients had anterior segment abnormalities (i.e. posterior embryotoxon). All patients had nystagmus (28/28), grade four foveal hypoplasia (17/17), and misrouting of the optic nerve fibers at the chiasm (9/9). All (28/28) patients had normal pigmentation of skin, and/or hair compared to family members and no iris translucency. Fundus imaging showed a lightly pigmented midperiphery in an infant of 6 months of age (VII-811), but some pigmentation was already seen in the macular region. Figures 2A and 2B show examples of foveal hypoplasia and fundus images in patients with FHONDA and patients with albinism. Figure 3 shows misrouting in a patient with FHONDA.
Table 2.
Phenotype Patients With FHONDA
| Family ID | Refraction RE, LE† | VA‡ | Nystagmus Type | Iris Translucency | Foveal Hypoplasia§ | Anterior Segment | Hypopigmentation Fundus | Misrouting |
|---|---|---|---|---|---|---|---|---|
| I*-801 | +1.25D/–1.75Dx179 +1.25D/–1.25Dx179 | 0.7 | Horizontal jerk | No | Grade 4 | Posterior embryotoxon | No | Yes |
| I*-802 | –4.5D/–4.25Dx88 –5.75D/–4.75Dx177 | 0.8 | Horizontal jerk | No | Grade 4 | Posterior embryotoxon | No | Yes |
| I-828 | ND | ND | Horizontal jerk | No | ND | Normal | No | Yes |
| II*-803 | +2.0D/–2.0Dx5 +2.25D/–2.0Dx180 | 0.7 | Horizontal jerk | No | Grade 4 | Posterior embryotoxon | No | Yes |
| III-804 | +4.5D/–2.75Dx4 +3.5D/–3.75Dx174 | 0.8 | Horizontal jerk | No | Grade 4 | Normal | No | Yes |
| III-805 | +4.75D/–2.00x180 +4.25D/–1.50x11 | 0.8 | Horizontal jerk | No | ND | Normal | No | Yes |
| IV*-806 | +6.0D/–3.5Dx175 +6.5D/–4.0Dx15 | 0.7 | Horizontal jerk | No | Grade 4 | Normal | No | Yes |
| IV*-807 | +7.0D/–2.0Dx180 +7.5D/–2.5Dx180 | 1.0 | Horizontal jerk | No | Grade 4 | Posterior embryotoxon | No | Yes |
| V-808 | +10.25D/–1.25Dx17 +10.25D/–2.0Dx176 | 0.8 | Horizontal jerk | No | Grade 4 | Normal | No | Yes |
| VI-809 | +7.0D/–1.0Dx180 +7.25D/–2.0Dx180 | 0.9 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| VI-810 | +2.75D/–1.75Dx180 +3.5D/–1.75Dx20 | 0.7 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| VII-811 | 0.0D/–2.5Dx180 0.0D/–2.5Dx180 | ND | Horizontal jerk | No | Grade 4 | Normal | Blond fundus at six months of age | ND |
| VIII-812 | +7.75D/–4.75Dx2 +7.75D/–4.50Dx2 | 0.7 | Yes, type unknown | No | Grade 4 | Normal | No | ND |
| IX-813 | +4.00D/–1.00Dx166 +6.25D/–2.75Dx25 | 0.6 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| IX-814 | –3.25D/–1.25Dx45 –3.25D/–2.00Dx170 | 0.6 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| X*-815 | +0.25D/–3.5Dx180 +0.25D/–3.75Dx25 | 0.8 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| X*-816 | +4.25D/–2.75Dx8 +5.0D/–3.0Dx180 | 0.8 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| X*-817 | +4.0D/–3.5Dx180 +4.25D/–3.25Dx170 | 0.7 | Horizontal jerk | No | ND | Normal | No | ND |
| XI*-818 | +5.5D/–2.5Dx5 +5.75D/–2.5Dx180 | 0.5 | Periodic alternating | No | ND | Normal | No | ND |
| XI*-819 | +6.5D/–1.75Dx180 +6.25D/–0.5Dx180 | 0.7 | Horizontal jerk | No | Grade 4 | Normal | No | ND |
| XII*-820 | –0.5D/–1.25Dx3 –0.75D/–1.5Dx7 | 1.0 | Horizontal jerk | No | ND | Normal | No | ND |
| XII*-821 | +2.75D/–2.0Dx25 +3.75D/–1.5Dx160 | 0.6 | Horizontal jerk | No | ND | Normal | No | ND |
| XII*-822 | –11.75D/–2.0Dx14 –7.75D/–0.75Dx153 | 0.6 | Horizontal jerk | No | ND | Normal | No | ND |
| XII*-823 | +1.5D/–1.75Dx170 +1.0D/–1.75Dx10 | 0.4 | Periodic alternating | No | ND | Normal | No | ND |
| XIII-824 | –6.5D/–3.25Dx180 –6.5D/–3.25Dx180 | ND | Yes, type unknown | No | ND | Normal | No | ND |
| XIV-825 | ND | 0.7 | Yes, type unknown | No | ND | Normal | No | ND |
| XIV-826 | +1.0D/–1.0Dx180 +2.0D/–0.5Dx180 | 0.5 | Yes, type unknown | No | Grade 4 | Normal | No | ND |
| XV-827 | +3.0D/–3.0Dx10 +4.0D/–2.5Dx10 | 0.8 | Yes, type unknown | No | ND | Normal | No | ND |
Figure 2.

Clinical results. (A) Fundus images and optical coherence tomography scans of the clinical study patient VIII-812. (B) Fundus image and optical coherence tomography scan of clinical study patient III-804. (C) Example of a severely affected albinism patient. Note the completely translucent fundus accompanied by grade four foveal hypoplasia.21 The patient had two mutations in the TYR gene (c.164G > A and c.896G > A), visual acuity was 1.0 logMAR, and patient had nystagmus, and complete iris translucency. (D) Example of an albinism patient without obvious hypopigmentation of the fundus, comparable to patient FIII-804 with FHONDA. Note the minimal foveal hypoplasia grade 1.21 Other manifestations were also mild, with a suboptimal visual acuity of 0.3 logMAR, nystagmus, and absence of iris translucency. The patient was homozygote for mutation c.1037-7T > A in the TYR gene. All optical coherence tomography scans of the left eye and right eye had the same grade of foveal hypoplasia.
Figure 3.

Chiasmal misrouting in FHONDA. (A) Visually evoked pattern onset potentials recordings of a normal subject. Note correlation of responses recorded from right and left eye in the differential signal. (B) Visually evoked pattern onset potentials from clinical study patient V-808. Note the asymmetry in the differential signal recorded from right eye and left eye. The chiasm coefficient was –0.9, indicating chiasmal misrouting.23–25
FHONDA (N = 28) Versus Albinism (N = 133)
VA was poorer and less variable in FHONDA (median = 0.7 logMAR, IQR = 0.6–0.8) compared to the 133 patients with albinism (median = 0.5 logMAR, IQR = 0.3–0.7, Mann-Whitney U test, P = 0.006). Analysis of VA of FHONDA compared to the different subtypes of albinism showed a significantly better VA in OCA1 and OCA2 (median = 0.5 logMAR, IQR = 0.3–0.7, P = 0.004, and median = 0.5 logMAR, IQR = 0.2–0.7, P = 0.006), respectively). Median VA in OA1 was 0.6 logMAR, IQR = 0.5 to 0.8 and was not significantly better than in FHONDA (P = 0.11). Besides VA, all phenotypic characteristics in OCA1, OCA2, and OA1 were more variable than in FHONDA in this study: 11% (14/132) of patients with albinism did not have nystagmus (0% in FHONDA), foveal hypoplasia varied from grades 1 to 4 (only grade 4 in FHONDA), and misrouting was absent in 22% (16/74, 0% in FHONDA; see also Fig. 4) Misrouting was more evident in FHONDA, and was detected with all stimulus types (i.e. pattern onset and flash VEP), regardless of age. In albinism, VEP recordings were much more variable, and when misrouting was confirmed by the method recommended for that age, it was not always present in recordings with other stimuli (Fig. 5).
Figure 4.

Phenotypic spectrum of FHONDA versus oculocutaneous albinism type 1, type 2, and ocular albinism. For the grading of foveal hypoplasia we used a grading scheme according to Thomas et al., with grades 1 and 2 not having incursion of the inner retinal layers, and grades 3 and 4 also affecting the photoreceptor differentiation (see Supplementary Table S1).21 Misrouting was determined by using cutoff values from the study of Kruijt et al. for the calculated chiasm coefficients from the multichannel visually evoked potential recordings.23
Figure 5.
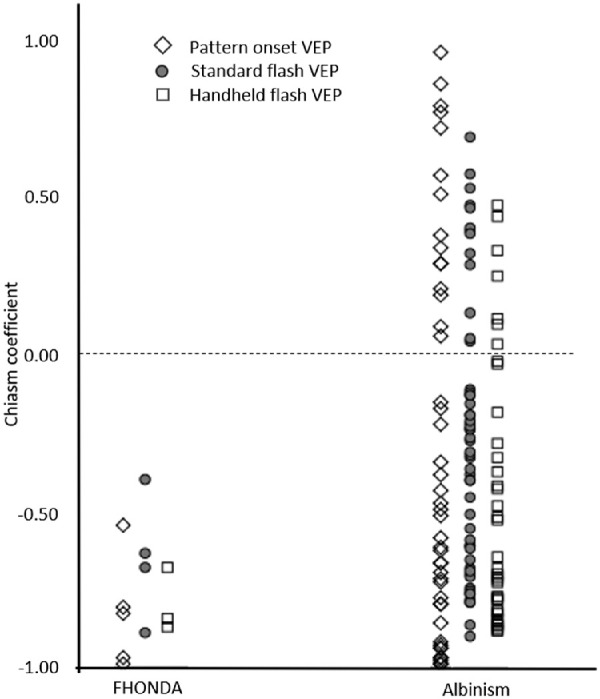
Visually evoked potentials in FHONDA and albinism. The chiasm coefficients were calculated according to Kruijt et al.23 A negative chiasm coefficient indicates misrouting. This figure shows all test results, independently of age. Not all patients were tested with all stimuli, sometimes only the stimulus recommended for the age was used. Note the variability in albinism and the obvious misrouting in patients with FHONDA for all stimuli.
Patients with OA1 resemble patients with FHONDA the most, because they have no hypopigmentation of the skin or hair. In addition, they have on average poorer VA and more severe foveal hypoplasia than patients with OCA in our cohort. However, almost all patients with OA1 have ocular hypopigmentation, in contrast to none of the patients with FHONDA. In our cohort, only 3 of 43 patients with OA1 were described with normal ocular pigmentation (no iris translucency AND normal fundus pigmentation). The first patient had grade one hypopigmentation of the posterior pole, according to the scheme of Kruijt et al.,10 which was considered normal in this fair skinned Caucasian patient. However, he had obvious hypopigmentation of the (mid)periphery with choroidal vessels clearly visible through the translucent retinal pigment epithelium. He had VA of 0.5 logMAR, nystagmus, foveal hypoplasia grade 3, and misrouting. The second patient with OA1, from Somalian descent, had high myopia, VA of 1.0 logMAR, nystagmus, and no misrouting. An OCT scan was not obtained. There was possible hypopigmentation of the fundus, but this could also be secondary to the high myopia (S-16/ S-14). The last patient, of mixed Dutch-Indonesian descent, had VA of 0.25 logMAR, nystagmus, foveal hypoplasia grade 2, and misrouting. Ocular pigmentation for FHONDA and the albinism subtypes is shown in Figure 4.
Discussion
In this study, we described the genotypic and phenotypic characteristics of the rare FHONDA syndrome and compared it to the phenotype of the most common types of albinism, OCA1, OCA2, and OA1. To date, only 33 patients with the FHONDA syndrome were reported in the literature.6–8,14,16–20
Genotype
The SLC38A8 gene consists of 10 exons (all coding), spanning almost 32.4 kb of genomic DNA in the region of 16q23.3. This gene encodes the Solute carrier family 38 member 8 protein, a 46.9 kDa putative sodium-coupled neutral amino acid transporter (SNAT) with 11 transmembrane domains, which consists of 435 amino acids. The protein is expressed in the central nervous system and neuronal retina, predominantly in the inner and outer plexiform layers and photoreceptor layers. SLC38A8 has a possible broad substrate profile with high preference for transporting glutamate.8,26
Including all patients from the literature, 15 of the 18 identified missense mutations are localized in, or very near, transmembrane regions (see Fig. 1, Supplementary Table S2). Mutations probably affect the transport function of the protein. The p.(Thr87Ile) and p.(Asp283Ala) are both localized on the extracellular side of the channel and cause a change to a hydrophobic amino acid. Toral et al. postulated that p.(Asp283Ala) missense mutation results in a more positive electrostatic potential at the extracellular side of the channel, potentially disrupting the local concentration of sodium and affecting glutamine transport.17 However, the exact mechanisms of the pathogenicity of the identified missense mutations are still unknown.
The identified nonsense, splice, deletion, and frameshift mutations are spread over the entire gene. They result in translation of a truncated protein or reduced amount of protein due to nonsense mediated decay, thus, affecting localization and/or transport function of the protein. A SLC38A8 functional analysis test is needed to determine the pathological effects of the identified mutations.
It is noteworthy that the c.848A > C; p.(Asp283Ala) mutation was detected in all Ashkenazi Jewish patients reported in this study, and 2 Ashkenazi Jewish patients from the study of Toral et al.17 In total, five patients from three families were homozygous, and one patient of mixed Ashkenazi descent was compound heterozygous. In all Indian Jewish patients from this study, and Indian and Karaite Jewish patients reported in the literature, the mutation c.95T > G; p.(Ile32Ser) was discovered, 17 patients from 8 families were homozygous and one patient was compound heterozygous (see Table 1, Supplementary Table S2). This mutation was not seen in other patients. The six novel SLC38A8 mutations identified in our study significantly extend the genotypic heterogeneity among patients with FHONDA.
Phenotype
All 38 different mutations that are discovered until now in the SLC38A8 gene resulted in similar phenotypes, comprising poor VA, nystagmus, severe foveal hypoplasia (grade 4 in our study, and grade 3 or 4 in previously reported patients), definite chiasmal misrouting, and no signs of any pigmentation defect (skin, hair, iris, or fundus). All newly diagnosed and all but two of the previously reported patients fit this profile.5–9,17–20 These two patients seem to express some amount of hypopigmentation. First, patient P4, one of 990 patients from an albinism study by Lasseaux et al. had iris translucency.14 Further details of this patient were unavailable, so we were unable to determine if this translucency could have other causes. The other patient from this study that had iris translucency was excluded from the series (personal communication). In addition, one patient described by Kuht et al. had mild iris transillumination, but this patient also had a TYR variant which could explain the pigmentation defect of the iris. In three patients, some hypopigmentation of the fundus was described. In all patients, the pigmentation could be considered normal for age, because two patients were only a few months old. The only adult was of white British background and she had no hypopigmentation compared to her family.16,19
It is noteworthy that, even though we detected 6 various novel SLC38A8 mutations among our patients, this did not change the phenotypic homogeneity. The only variable phenotypic characteristic of FHONDA appears to be anterior segment dysgenesis (ASD), mostly consisting of posterior embryotoxon/Axenfeld's anomaly. This was found in only four patients from three families in this study, and seven patients from three families of previously reported patients (see Table 2, Supplementary Table S3). The genetic changes in four families with posterior embryotoxon were solely detected in these patients, and not in any patients without ASD. However, differences in genotype cannot fully explain the presence or absence of ASD, because mutations in the remaining three families with ASD were also observed in patients without any ASD. This means that posterior embryotoxon/Axenfeld's anomaly is not a frequent abnormality in this disorder, occurring in less than 19% of the patients, which is within the range of prevalence in the normal population (7–-32%).27–30
FHONDA Versus Albinism
It is interesting that FHONDA appears to have a more narrow phenotypic spectrum compared to albinism, especially with regard to nystagmus, grade of foveal hypoplasia, and chiasmal misrouting. Mutations in several genes can cause different subtypes of albinism, but within one genetic subtype, the phenotypic spectrum is still broad.10,11,13,14 A previous study on albinism concluded that absence of photoreceptor specialization (grades 3 and 4 foveal hypoplasia) was associated with worse VA in albinism, with grade 4 associated with the poorest VA.10 Photoreceptor differentiation was affected in all patients with FHONDA, which may explain the significantly poorer VA than in albinism. Besides poorer VA and more severe foveal hypoplasia, misrouting was detected with all VEP test stimuli with chiasm coefficients not higher than −0.45. This could be explained by, on average, more crossing of the optic nerve fibers at the chiasm than in albinism, so that noise is not affecting the signal as much (noise causes a shift of the chiasm coefficient towards zero).23 This theory is also supported by the findings of Ahmadi et al., who demonstrated that in a patient with FHONDA, all temporal retinal fibers project to the contralateral hemisphere, instead of excessive crossing only in most patients with albinism.31
Despite the recently described diagnostic criteria for albinism, the clinical distinction between patients with FHONDA of Caucasian descent and patients with OA1 without evident ocular hypopigmentation might still be difficult. For instance, mild hypopigmentation of the (mid)peripheral fundus can be normal in a lightly pigmented Caucasian family, as is seen in the patient with FHONDA VII-811 and in proband 2 described by Campbell et al. In our OA1 cohort, we identified only three patients without ocular hypopigmentation. One patient was mildly affected, in contrast to FHONDA, with better VA, and grade 1 or 2 foveal hypoplasia. In the other two patients, fundus hypopigmentation could be related to an overall light skinned Caucasian phenotype and high myopia, respectively. Of these two patients, one resembled FHONDA closely (VA 0.5 logMAR, nystagmus, foveal hypoplasia grade 3, and misrouting), whereas the other patient did not have misrouting, which has not yet been reported in patients with FHONDA. Therefore, only in 1 of 43 patients with OA1 the differentiation with FHONDA could not be clearly made. To further differentiate the two disorders, in a recent study describing the phenotypic spectrum of albinism, poor visual acuity, grade 4 foveal hypoplasia, and misrouting were always accompanied by iris translucency or other obvious signs of hypopigmentation.10 This means that in a severely affected patient but without evident hypopigmentation the possibility of FHONDA is most likely (see Fig. 2). It should be noted, however, that this conclusion is based on a small number of patients with FHONDA. The identification of only 3e patients with mutations in SLC38A8 in a series of 990 presumed patients with albinism, and only 61 patients with FHONDA reported to date confirm that this is a rare entity.14 We hope that this study will raise physicians’ awareness to this uncommon disease and that SLC38A8 will be added to gene panels for nystagmus.
This study confirms that, in consent with the findings of Poulter et al., FHONDA and albinism appear to be different entities, and the combination of foveal hypoplasia and misrouting of the optic nerve fibers can occur independently of abnormal melanin synthesis. It is therefore plausible that a common pathway exists that causes both foveal hypoplasia and misrouting. The uniformly severe phenotype of FHONDA suggests that SLC38A8 might be located at the end of this pathway, without the possibility of a partial rescue.
Although Ehprin-B1 was previously identified as playing a key role in ipsilateral guidance in species with a small part of the ventral retina projecting ipsilaterally, Hoffmann et al. did not detect an association with visual pathway abnormalities in humans with Ephrin-B1 deficiency.32–35 We hypothesize that other components of the Eph system may play an important role in this pathway. Ephrins are a family of membrane-tethered proteins that serve as ligands of the Eph receptor. Eph/ephrin signaling regulates many developmental processes, including the guidance of axonal growth of cones, and formation of tissue boundaries.36–38
The formation of a foveal avascular zone (FAZ) is necessary for normal foveal development.39 Kozulin et al. showed involvement of pigment epithelium derived factor (PEDF) and EphA6 in the definition of the FAZ.40,41 During formation of the FAZ, EphA6 levels start to rise in the macular region and continue to rise after birth while cone differentiation and elongation of the most central cones occurs, a process that appears not to take place in patients with grade four foveal hypoplasia.39–41 During axon guidance, EphA6 expression is highest in the temporal retina and gradually drops around the central retina, implying an important role of EphA6 in retinal ipsilateral axon guidance.34,39 Therefore, EphA6 might be a promising candidate for a common pathway causing foveal hypoplasia and excessive crossing of the optic nerve fibers at the chiasm.
In conclusion, we describe the mutational spectrum, including six novel mutations, and narrow phenotypic spectrum of FHONDA, consisting of poor VA, nystagmus, severe foveal hypoplasia (grade 3 or 4), and chiasmal misrouting. Our study confirms that lack of pigmentation is not essential to cause the combined occurrence of foveal hypoplasia and misrouting. This, in turn, may have important implications for current research on the pathogenesis of these abnormalities and future therapeutic options for alleviation of ocular anomalies leading to vision loss in albinism.42,43
Supplementary Material
Acknowledgments
The authors thank Herman Talsma for his insightful help with the creation of the illustrations. The authors would also like to thank Herman Talsma, Frans Riemslag, and Frank Hoeben for performing many of the visually evoked potentials recordings.
Supported by the following foundations in the form of unrestricted funds: ODAS, Vereniging Bartiméus Sonneheerdt and Landelijke Stichting Blinden en Slechtzienden (via contributions through UitZicht). The funding organizations had no role in the design or conduct of this research.
Disclosure: C.C. Kruijt, None; L. Gradstein, None; A.A. Bergen, None; R.J. Florijn, None; B. Arveiler, None; E. Lasseaux, None; X. Zanlonghi, None; L. Bagdonaite-Bejarano, None; A.B. Fulton, None; C. Yahalom, None; A. Blumenfeld, None; Y. Perez, None; O.S. Birk, None; G.C. de Wit, None; N.E. Schalij-Delfos, None; M.M. van Genderen, None
References
- 1. Kinnear PE, Jay B, Witkop CJ.. Albinism. Surv Ophthalmol. 1985; 30(2): 75–101. [DOI] [PubMed] [Google Scholar]
- 2. Summers CG. Vision in albinism. Trans Am Ophtalmol Soc. 1996; 94: 1095–1155. [PMC free article] [PubMed] [Google Scholar]
- 3. Charles SJ, Green JS, Grant JW, Yates JRW, Moore AT.. Clinical features of affected males with X linked ocular albinism. Br J Ophthalmol. 1993; 77(January): 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jeffery G, Brem G, Montoliu L.. Correction of retinal abnormalities found in albinism by introduction of a functional tyrosinase gene in transgenic mice and rabbits. Dev Brain Res. 1997; 99(1): 95–102. [DOI] [PubMed] [Google Scholar]
- 5. van Genderen MM, Riemslag FC, Schuil J, Hoeben FP, Stilma JS, Meire FM.. Chiasmal misrouting and foveal hypoplasia without albinism. Br J Ophthalmol. 2006; 90(9): 1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pal B, Mohamed MD, Keen TJ, et al.. A new phenotype of recessively inherited foveal hypoplasia and anterior segment dysgenesis maps to a locus on chromosome 16q23.2-24.2. J Med Genet. 2004; 41(10): 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al-Araimi M, Pal B, Poulter J a, et al.. A new recessively inherited disorder composed of foveal hypoplasia, optic nerve decussation defects and anterior segment dysgenesis maps to chromosome 16q23.3-24.1. Mol Vis. 2013; 19(October): 2165–2172. [PMC free article] [PubMed] [Google Scholar]
- 8. Poulter JA, Al-Araimi M, Conte I, et al.. Recessive mutations in SLC38a8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013; 93: 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez Y, Gradstein L, Flusser H, et al.. Isolated foveal hypoplasia with secondary nystagmus and low vision is associated with a homozygous SLC38A8 mutation. Eur J Hum Genet. 2014; 22(5): 703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kruijt CC, de Wit GC, Bergen AA, Florijn RJ, Schalij-Delfos NE, van Genderen MM.. The Phenotypic Spectrum of Albinism. Ophthalmology. 2018; 125(12): 1953–1960. [DOI] [PubMed] [Google Scholar]
- 11. Grønskov K, Ek J, Sand A, et al.. Birth Prevalence and Mutation Spectrum in Danish Patients with Autosomal Recessive Albinism. Invest Ophthalmol Vis Sci. 2009; 50(3): 1058–1064. [DOI] [PubMed] [Google Scholar]
- 12. Mauri L, Manfredini E, Del Longo A, et al.. Clinical evaluation and molecular screening of a large consecutive series of albino patients. J Hum Genet. 2017; 62(2): 277–290. [DOI] [PubMed] [Google Scholar]
- 13. Marti A, Lasseaux E, Ezzedine K, et al.. Lessons of a day hospital: Comprehensive assessment of patients with albinism in a European setting. Pigment Cell Melanoma Res. 2018; 31(2): 318–329. [DOI] [PubMed] [Google Scholar]
- 14. Lasseaux E, Plaisant C, Michaud V, et al.. Molecular characterization of a series of 990 index patients with albinism. Pigment Cell Melanoma Res. 2018; 31(4): 466–474. [DOI] [PubMed] [Google Scholar]
- 15. Von Dem Hagen EAH, Houston GC, Hoffmann MB, Morland AB.. Pigmentation predicts the shift in the line of decussation in humans with albinism. Eur J Neurosci. 2007; 25(2): 503–511. [DOI] [PubMed] [Google Scholar]
- 16. Kuht HJ, Han J, Maconachie GDE, et al.. SLC38A8 mutations result in arrested retinal development with loss of cone photoreceptor specialization. Hum Mol Genet. 2020; 29(18): 2989–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Toral MA, Velez G, Boudreault K, et al.. Structural modeling of a novel SLC38A8 mutation that causes foveal hypoplasia. Mol Genet Genomic Med. 2017; 5(3): 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vincent A, Kemmanu V, Shetty R, Anandula V, Madhavarao B, Shetty B.. Variable expressivity of ocular associations of foveal hypoplasia in a family. Eye. 2009; 23(8): 1735–1739. [DOI] [PubMed] [Google Scholar]
- 19. Campbell P, Ellingford JM, Parry NRA, et al.. Clinical and genetic variability in children with partial albinism. Sci Rep. 2019; 9(1): 16576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiner C, Hecht I, Rotenstreich Y, Guttman S, Or L.. The pathogenicity of SLC38A8 in five families with foveal hypoplasia and congenital nystagmus. Exp Eye Res. 2020; 193: 107958. [DOI] [PubMed] [Google Scholar]
- 21. Thomas MG, Kumar A, Mohammad S, et al.. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011; 118(8): 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robson AG, Nilsson J, Li S, et al.. ISCEV guide to visual electrodiagnostic procedures. Doc Ophthalmol. 2018; 136(1): 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kruijt CC, De Wit GC, Talsma HE, Schalij-Delfos NE, Van Genderen MM.. The detection of misrouting in albinism: Evaluation of different VEP procedures in a heterogeneous cohort. Investig Ophthalmol Vis Sci. 2019; 60(12): 3963–3969. [DOI] [PubMed] [Google Scholar]
- 24. Jansonius NM, van der Vliet TM, Cornelissen FW, Pott JW, Kooijman AC.. A girl without a chiasm: electrophysiologic and MRI evidence for the absence of crossing optic nerve fibers in a girl with a congenital nystagmus. J Neuroophthalmol. 2001; 21(1): 26–29. [DOI] [PubMed] [Google Scholar]
- 25. Pott JWR, Jansonius NM, Kooijman AC.. Chiasmal coefficient of flash and pattern visual evoked potentials for detection of chiasmal misrouting in albinism. Doc Ophthalmol. 2003; 106: 137–143. [DOI] [PubMed] [Google Scholar]
- 26. Hägglund MGA, Hellsten SV, Bagchi S, et al.. Transport of l-glutamine, l-alanine, l-arginine and l-histidine by the neuron-specific slc38a8 (SNAT8) in CNS. J Mol Biol. 2015; 427(6): 1495–1512. [DOI] [PubMed] [Google Scholar]
- 27. Forsius H, Eriksson AFJ.. Embryotoxon corneae posterius in an isolated population. Acta Ophthamol. 1964; 42: 42–49. [DOI] [PubMed] [Google Scholar]
- 28. Ozeki H, Shirai S, Majima A, Sano MIK.. Clinical evaluation of posterior embryotoxon in one institution. Jpn J Ophthalmol. 1997; 41(6): 422–425. [DOI] [PubMed] [Google Scholar]
- 29. Rennie CA, Chowdhury S, Khan J, Rajan F, Jordan K, Lamb RJVA.. The prevalence and associated features of posterior embryotoxon in the general ophthalmic clinic. Eye (Lond). 2005; 19(4): 396–399. [DOI] [PubMed] [Google Scholar]
- 30. Hashemi H, Khabazkhoob M, Emamian MH, Shariati M, Yekta A, Fotouhi A.. The frequency of occurrence of certain corneal conditions by age and sex in Iranian adults. Contact Lens Anterior Eye. 2015; 38(6): 451–455. [DOI] [PubMed] [Google Scholar]
- 31. Ahmadi K, Fracasso A, van Dijk JA, et al.. Altered organization of the visual cortex in FHONDA syndrome. Neuroimage. 2019; 190: 224–231. [DOI] [PubMed] [Google Scholar]
- 32. Nakagawa S, Brennan C, Johnson KG, Shewan D, Harris WA, Holt CE.. Ephrin-B Regulates the Ipsilateral Routing of Retinal Axons at the Optic Chiasm. Neuron. 2000; 25: 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams SE, Mann F, Erskine L, et al.. Ephrin-B2 and EphB1 Mediate Retinal Axon Divergence at the Optic Chiasm. Neuron. 2003; 39: 919–935. [DOI] [PubMed] [Google Scholar]
- 34. Lambot M, Depasse F, Noel J, Vanderhaeghen P.. Mapping Labels in the Human Developing Visual System and the Evolution of Binocular Vision. J Neurosci. 2005; 25(31): 7232–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoffmann MB, Thieme H, Liedecke K, Meltendorf S, Zenker M, Wieland I.. Visual Pathways in Humans With Ephrin-B1 Deficiency Associated With the Cranio-Fronto-Nasal Syndrome. Invest Ophthalmol Vis Sci. 2015; 56(12): 7427–7437. [DOI] [PubMed] [Google Scholar]
- 36. Egea J, Klein R.. Bidirectional Eph-ephrin signaling during axon guidance. Trends Cell Biol. 2007; 17(5): 230–238. [DOI] [PubMed] [Google Scholar]
- 37. Rohani N, Canty L, Luu O, Fagotto F, Winklbauer R.. EphrinB/EphB signaling controls embryonic germ layer separation by contact-induced cell detachment. PLoS Biol. 2011; 9(3): 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fagotto F, Rohani N, Touret AS, Li R.. A Molecular Base for Cell Sorting at Embryonic Boundaries: Contact Inhibition of Cadherin Adhesion by Ephrin/Eph-Dependent Contractility. Dev Cell. 2013; 27(1): 72–87. [DOI] [PubMed] [Google Scholar]
- 39. Provis JM, Dubis AM, Maddess T, Carroll J.. Adaptation of the central retina for high acuity vision: Cones, the fovea and the a vascular zone. Prog Retin Eye Res. 2013; 35: 63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kozulin P, Natoli R, Madigan MC, Brien KMBO, Provis JM.. Gradients of Eph-A6 expression in primate retina suggest roles in both vascular and axon guidance. Mol Vis. 2009; 15: 2649–2662. [PMC free article] [PubMed] [Google Scholar]
- 41. Kozulin P, Natoli R, Brien KMBO, Madigan MC, Provis JM.. Differential expression of anti-angiogenic factors and guidance genes in the developing macula. Mol Vis. 2009; 15: 45–59. [PMC free article] [PubMed] [Google Scholar]
- 42. Onojafe F, Chan C, Onojafe IF, et al.. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism. J Clin Invest. 2011; 121(10): 3914–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dolinska MB, Kovaleva E, Backlund P, Wingfield PT, Brooks BP, Sergeev YV. Albinism-causing mutations in recombinant human tyrosinase alter intrinsic enzymatic activity. PLoS One. 2014; 9(1): e84494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.