

Abstract
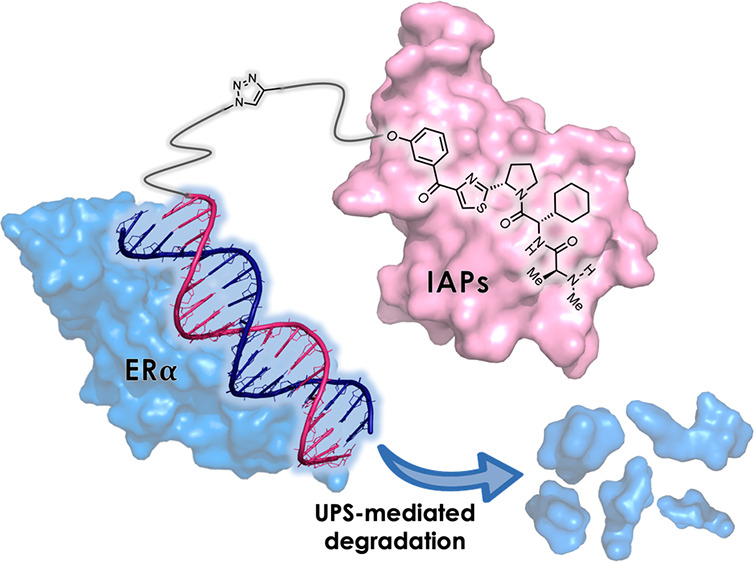
Targeted protein degradation using chimeric small molecules, such as proteolysis-targeting chimeras (PROTACs) and specific and nongenetic inhibitors of apoptosis protein (IAP)-dependent protein erasers (SNIPERs), has attracted attention as a method for degrading intracellular target proteins via the ubiquitin-proteasome system (UPS). These chimeric molecules target a variety of proteins using small molecules that can bind to the proteins. However, it is difficult to develop such degraders in the absence of suitable small-molecule ligands for the target proteins, such as for transcription factors (TFs). Therefore, we constructed the chimeric molecule LCL-ER(dec), which consists of a decoy oligonucleotide that can bind to estrogen receptor α (ERα) and an IAP ligand, LCL161 (LCL), in a click reaction. LCL-ER(dec) was found to selectively degrade ERα via the UPS. These findings will be applicable to the development of other oligonucleotide-type degraders that target different TFs.
Keywords: ubiquitin-proteasome system, protein knockdown, decoy, transcription factors, estrogen receptor
The identity of many target proteins that are involved in the pathogenesis of various diseases has been elucidated, and various drug discovery techniques have been developed to target these proteins.1,2 In particular, the use of targeted protein degradation has recently been attracting attention. This technology uses chimeric molecules called proteolysis-targeting chimeras (PROTACs) or specific and nongenetic inhibitors of apoptosis protein (IAP)-dependent protein erasers (SNIPERs) to degrade target proteins in cells via the ubiquitin-proteasome system (UPS).3,4 These chimeric molecules consist of a ligand for the target protein (protein of interest (POI)) and an E3 ligand. The proximity of the POI and E3 ligase acts as a molecular mediator, thereby promoting the ubiquitination and subsequent degradation of the POI.2 Small molecules, such as LCL161 (LCL) for IAPs, VH032 (VH) for von Hippel–Lindau protein (VHL), and pomalidomide (POM) for cereblon (CRBN), are often used as ligands for E3 ligases.3 A small molecule that is known to bind to a POI can be used to develop chimeric molecules, and many chimeric degraders using small molecules as POI ligands have been developed to date.3 Because the only activity required for the ligand is for it to be able to bind to the POI, this technology can target proteins that have no enzymatic activity (i.e., for which inhibitors cannot be developed). However, it is difficult to develop small-molecule-based degraders against a POI in the absence of appropriate ligands. An alternative approach is to use molecules, such as peptides, that can bind to the protein surfaces as POI ligands. Several groups have recently reported peptide-based degraders that target transcriptional factors (TFs), such as estrogen receptor α (ERα)5 and neurogenic locus notch homologue protein 1 (NOTCH1),6 by introducing an oligopeptide that interacts with the surface of the POI. This peptide-based protein knockdown strategy is also applicable to several types of POIs that have no specific small-molecule ligands.
Other molecules that can bind to protein surfaces include oligonucleotides, such as aptamers7 and decoys.8 If such oligonucleotides could be used as POI ligands in the development of chimeric degraders, then the number of target proteins that can be degraded would be expanded. DNA-based PROTACs have been developed very recently by several groups. For example, TRAFTAC, by the use of haloPROTAC, d-Cas9-HT7, and dsDNA/CRISPR-RNA chimeras, can induce target protein degradation medicated by the ectopic expression of the Cas9 protein in cells.9 In addition, TF-PROTACs10 (O′PROTACs11) targeting TFs using double-stranded decoy ligands can degrade some TFs. It is particularly advantageous to use a decoy as the ligand when targeting TFs because the decoy can use the known DNA-binding sequence of the target TF, making it easy to design the ligand. In the present study, we developed decoy-based chimeric degraders that target ERα as a model TF (Figure 1). ERα is a member of the nuclear hormone receptor superfamily12 and can bind directly to DNA to act as a TF.13 In addition, ERα homodimers can form transcriptional complexes on the DNA response sequence and regulate the expression of target genes by recruiting coregulators.13 We have previously developed small-molecule-based chimeric degraders against ERα14,15 using the ERα antagonist 4-hydroxytamoxifen (4-OHT) and peptide-based ERα degraders5 using a coactivator motif that interacts with the ERα surface. Therefore, we chose ERα as a model TF as the target for the development of oligonucleotide-type chimeric degraders. Specifically, we designed a thermodynamically stable double-stranded decoy, ER(dec), based on the sequence of the estrogen-responsive element (ERE)16 that is known to bind strongly to ERα. This decoy was conjugated with three different E3 ligase ligands, LCL, VH, and POM, to construct the degraders LCL-ER(dec), VH-ER(dec), and POM-ER(dec), respectively. The structural properties, binding affinities, and ERα degradation activities of these degraders were evaluated.
Figure 1.
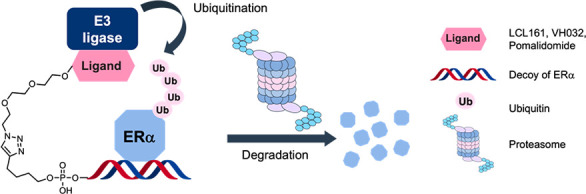
Decoy-type PROTACs degrade ERα via the UPS.
The ERα-binding decoy ER(dec) (21 residues) was designed using information from the DNA sequence and cocrystal X-ray structure of ERα,17 and an ERE from the Xenopus vitellogenin A2 gene.16 The 5′-end of the sense strand 5′-GTCAGGTCACAGTGACCTGAT-3′ of ER(dec) was modified with a hexynyl group. For the construction of the E3 ligase ligands, a PEG3 linker with an azide group was attached to LCL, VH,18 and POM.19 Then, the above sense strand with the alkyne was conjugated to each azide-containing E3 ligase ligand by a copper-catalyzed click reaction. Subsequent hybridization with the antisense strand 5′-ATCAGGTCACTGTGACCTGAC-3′ afforded the desired chimeric molecules LCL-ER(dec), VH-ER(dec), and POM-ER(dec). In addition, three ERα nonbinding decoys were designed with the same base composition as ER(dec): Scrbl-1 with an AT repeat and GC repeat sequence, Scrbl-2 with a completely randomized sequence, and Scrbl-3 with an AGCT repeat sequence. The chimeric molecules LCL-Scrbl-1, LCL-Scrbl-2, and LCL-Scrbl-3 were synthesized in a similar manner as LCL-ER(dec). A nondegradable control NMeLCL-ER(dec) containing an N-methylated analog of LCL161 and the fluorescein (FAM)-labeled decoy FAM-ER(dec) for use in a competitive fluorescence polarization assay were also synthesized (Figure 2). The detailed synthetic protocols of these molecules are described in the Supporting Information.
Figure 2.

Molecules synthesized in this study.
The preferred higher order structures of the synthesized ER(dec) and the chimeric molecules LCL-ER(dec), VH-ER(dec), and POM-ER(dec) were analyzed using CD spectra. Each decoy showed a negative maximum at ∼240 nm and a positive maximum at ∼280 nm, indicating that LCL-ER(dec), VH-ER(dec), and POM-ER(dec) formed typical right-handed B-type structures and the terminal modification of ER(dec) did not affect the higher order structure (Figure S4). The melting temperature (Tm) values of ER(dec), LCL-ER(dec), VH-ER(dec), and POM-ER(dec) were 64.5, 64.3, 66.9, and 62.9 °C, respectively, which indicated that these molecules had similar conformational stability as ER(dec) (Figure S7).
The binding affinities of ER(dec), LCL-ER(dec), VH-ER(dec), and POM-ER(dec) to ERα were evaluated by a competitive fluorescence polarization assay. For the evaluation, the compounds were added to a buffer system containing ERα and FAM-ER(dec), and the inhibitory concentration was calculated as the IC50 value. The IC50 values were 39.4 nM for ER(dec), 31.2 nM for LCL-ER(dec), 43.7 nM for VH-ER(dec), and 52.6 nM for POM-ER(dec). These results indicated that the binding activity of the decoy ER(dec) to ERα was not weakened by the linking of ER(dec) to the E3 ligase ligand, and the compounds had sufficient binding ability to ERα regardless of the type of E3 ligase ligand (Table 1). The binding activities of LCL-Scrbl-1–3 to ERα were considerably weaker than that of ER(dec) composed of the consensus sequence, but some nonspecific binding was observed at high concentrations (Figures S10 and S11).
Table 1. ERα Binding Affinity (IC50) of ER(dec), LCL-ER(dec), VH-ER(dec), POM-ER(dec), LCL-Scrbl-1, LCL-Scrbl-2, and LCL-Scrbl-3a.
| entry | compound | IC50 (nM) |
|---|---|---|
| 1 | ER(dec) | 39.4 ± 12.0 |
| 2 | LCL-ER(dec) | 31.2 ± 5.37 |
| 3 | VH-ER(dec) | 43.7 ± 7.26 |
| 4 | POM-ER(dec) | 52.6 ± 16.9 |
| 5 | LCL-Scrbl-1 | >3000 |
| 6 | LCL-Scrbl-2 | >3000 |
| 7 | LCL-Scrbl-3 | >3000 |
Data represent the mean ± S.D. (n = 3).
To investigate whether LCL-ER(dec), VH-ER(dec), and POM-ER(dec) have degradation activity against ERα, we evaluated the effect of these E3 ligand–decoy conjugates on the protein level of ERα by Western blotting analysis using MCF-7 breast cancer cells. (We confirmed that protein reduction can be observed using a small-molecule ERα degrader, as shown in Figure S13. In addition, we confirmed that decoy-based PROTACs were efficiently taken up by MCF-7 cells by treatment with the transfection reagent, as shown in Figures S15 and S16.) All of the decoys reduced the ERα protein level after 24 h of transfection, and LCL-ER(dec) had the most effect compared with the other decoys (Figure 3A). To investigate the selectivity for the target protein, we examined the effect of these decoys on the levels of different proteins (Figure 3B). None of the decoys effectively reduced the levels of other transcription factors (AR, AhR, and NF-κB p65), transcription-related factors (p300 and BRD4), or proteins not related to transcription (CRABP2, GAPDH, and β-actin), indicating that these decoys selectively reduced the protein level of ERα.
Figure 3.

Degradation of ERα via the UPS by the synthesized decoys. (A,B) Synthesized decoys selectively induced a reduction in the ERα protein level. MCF-7 cells were transiently transfected with the indicated concentrations of LCL-ER(dec), VH-ER(dec), or POM-ER(dec) for 24 h. (C) Effect of UPS inhibitors on the LCL-ER(dec)-induced reduction of ERα levels. MCF-7 cells were transiently transfected with the indicated concentrations of LCL-ER(dec) in the presence or absence of 10 μM MG132 or MLN7243 for 24 h. (D–F) Degradation of the ERα protein by LCL-ER(dec) is IAP-ligand- and DNA-sequence-dependent. MCF-7 cells were transiently transfected with the indicated concentrations of LCL-ER(dec), ER(dec), NMeLCL-ER(dec), LCL-Scramble-1, LCL-Scramble-2, or LCL-Scramble-3 for 24 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies; representative data are shown. The numbers below the ERα panels represent the ER/actin ratios, normalized by designating the expression using the vehicle control (condition without a decoy) as 100%. The changes in protein levels were reproducible between two independent experiments. The data in the bar graph are the mean of two results. Abbreviations: AR, androgen receptor; AhR, aryl hydrocarbon receptor; BRD4, bromodomain-containing protein 4; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; CRABP2, cellular retinoic acid binding protein 2.
To investigate the optimal linker length of the promising LCL-ER(dec) with a PEG3 linker, we designed and synthesized new five chimeric molecules, LCL-ER(dec)-P2 with a PEG2 linker, LCL-ER(dec)-P4 with a PEG4 linker, LCL-ER(dec)-C5 with a pentyl linker, LCL-ER(dec)-C8 with an octyl linker, and LCL-ER(dec)-C11 with an undecyl linker. These five molecules formed stable higher order structures (Figures S6 and S9) and showed strong binding affinities to ERα (Table S2 and Figure S12) similar to LCL-ER(dec). Furthermore, we evaluated the ERα-degrading activities of the six chimeric molecules with different types of linkers and found that LCL-ER(dec) with the PEG3 linker showed the highest activity at lower concentrations (Figure S14).
We next investigated the mechanism of the ERα reduction by focusing on LCL-ER(dec), which showed the highest activity. The LCL-ER(dec)-induced reduction of ERα was abrogated by cotreatment with the proteasome inhibitor MG132 and the ubiquitin-activating inhibitor MLN7243 (Figure 3C), suggesting that LCL-ER(dec) induced UPS-dependent degradation of ERα. Unlike LCL-ER(dec), ER(dec) did not induce degradation of the ERα protein (Figure 3D), indicating that conjugation of the LCL161 ligand is important for the degradation. In addition, the nondegradable control NMeLCL-ER(dec), which contains an N-methylated analog of the LCL161 ligand, did not induce ERα degradation (Figure 3E), suggesting that the ability to bind IAPs is critical for the degradation activity. To examine whether target recognition by the decoy ligands was DNA-sequence dependent, we synthesized LCL161 ligand-decoy chimeras using several different decoys composed of scrambled DNA sequences as the target ligands. These decoy chimeras did not show significant ERα-degradation activity (Figure 3F), which correlated with their weak binding affinity to ERα (Table 1). These results suggested that decoy oligonucleotides can be used in the development of chimeric molecules that induce the DNA sequence-dependent degradation of target proteins.
Because ERα plays an essential role in estrogen signaling, we examined the effect of LCL-ER(dec) on the estrogen-dependent transcriptional activity of ERα. In a luciferase assay using an ERE reporter, LCL-ER(dec) inhibited ERα-dependent transcriptional activation by β-estradiol more effectively than ER(dec), which was in good agreement with the ERα degradation activity (Figure 4). A mixture of the decoy ligand ER(dec) and LCL-PEG3-N3 did not effectively decrease the transcription, indicating that the linking of the two ligands, ER(dec) and LCL-PEG3-N3, is crucial for effective inhibition. In addition, LCL-ER(dec), but not ER(dec), effectively suppressed the proliferation of ERα-positive breast cancer MCF7 cells (Figure S17). These results suggested that degradation of ERα by LCL-ER(dec) leads to effective suppression of estrogen signaling.
Figure 4.
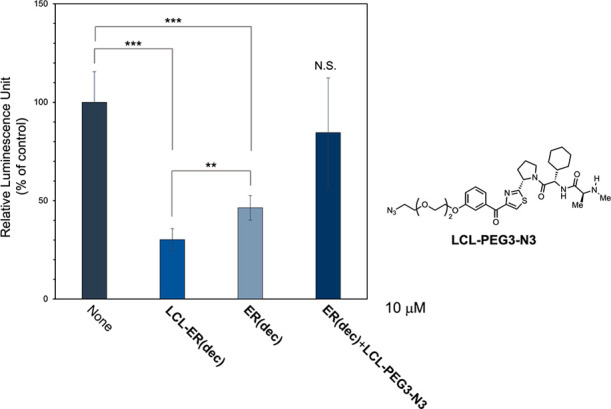
Inhibition of the estrogen-dependent transcriptional activity of ERα by LCL-ER(dec). MCF-7 cells were transiently transfected with a luciferase reporter plasmid containing three tandem copies of the ERE and control Renilla luciferase plasmid-SV40. After 24 h, the cells were further transfected with 10 μM concentrations of the indicated decoys in the presence of 3 nM β-estradiol for 24 h. The ERα-dependent transcriptional activity was evaluated by a luciferase assay, and the relative luciferase activity was normalized by designating the activity of the nontreated control (column 1) as 100%. The data represent the mean ± SD (n = 4). P values were determined using the unpaired two-tailed Student’s t-test. N.S., not significant; ** P < 0.005; *** P < 0.001.
In summary, we synthesized LCL-ER(dec) with the aim of developing a chimeric degrader using a decoy ligand that can be applied to a variety of TFs. For the target protein, we selected ERα, which has previously been shown to be degraded by chimeric molecules containing small molecules or peptides. The three chimeric molecules LCL-ER(dec), VH-ER(dec), and POM-ER(dec) were synthesized by conjugating the ERα decoy ER(dec) with LCL161, VH032, and POM, E3 ligase ligands commonly used in PROTACs and SNIPERs. Among these three chimeric molecules, LCL-ER(dec) showed the highest ERα degradation-inducing activity, as assessed by Western blotting. In contrast, the ERα-nonbinding chimeric molecules, LCL-Scrbl-1–3, did not reduce the protein levels of ERα, suggesting that LCL-ER(dec) binds to ERα in a sequence-specific manner to induce the degradation. In addition, the nondegradable control, NMeLCL-ER(dec), did not induce ERα degradation, suggesting that the ability to bind IAPs is critical for the degradation activity. Studies using UPS inhibitors suggested that LCL-ER(dec) degraded ERα via the UPS. Thus decoy-type PROTACs are also useful as a new type of POI inducer. In the future, we aim to apply the methods developed in this study to the design of degradation inducers for transcription-related factors that are difficult to target.
Acknowledgments
We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Glossary
Abbreviations
- PROTACs
proteolysis targeting chimeras
- IAP
inhibitor of apoptosis protein
- SNIPERs
specific and nongenetic IAP-dependent protein erasers
- UPS
ubiquitin proteasome system
- TF
transcription factor
- ERα
estrogen receptor α
- POI
protein of interest
- VHL
von Hippel–Lindau protein
- NOTCH1
neurogenic locus notch homologue protein 1
- 4-OHT
4-hydroxytamoxifen
- ERE
estrogen-responsive element
- FAM
fluorescein
- CD
circular dichroism
- Tm
melting temperature
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00629.
Synthetic procedures for all compounds listed in this Letter and the protocols for the in vitro assays (binding and protein degradation assays) (PDF)
Author Contributions
M. Naganuma, N.O., G.T., and H.T. performed the experiments and analyzed results. M. Naganuma, N.O., T.I., M. Naito, and Y.D. designed the research and wrote the paper. All authors discussed the results and commented on the manuscript.
This study was supported in part by grants from AMED under grant numbers JP21mk0101197 (to Y.D. and N.O.) and JP21ak0101073 (to N.O., T.I., and Y.D.), 21mk0101187 (to N.O. and T.I.), and JP21am0401003 (to T.I.), the Japan Society for the Promotion of Science (KAKENHI, grants JP21K05320 to Y.D., JP18H05502 to M. Naito and Y.D., JP18K06567 and JP21K06490 to N.O., and JSPS Fellows 21J23036 to M. Naganuma), the Terumo Foundation for Life Sciences and Arts (to Y.D.), the Takeda Science Foundation (to Y.D.), the Naito Foundation (to Y.D.), the Sumitomo Foundation (to Y.D.), the Novartis Foundation (Japan) for the Promotion of Science (to Y.D.), the Japan Foundation of Applied Enzymology (to Y.D.), the Kobayashi Foundation for Cancer Research (to Y.D.), the Foundation for Promotion of Cancer Research in Japan (to Y.D.), and the Scholarship of the Tokyo Biochemical Research Foundation (to M. Naganuma).
The authors declare no competing financial interest.
Supplementary Material
References
- Ferguson F. M.; Gray N. S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discovery 2018, 17, 353–376. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Nelson A. L.; Dhimolea E.; Reichert J. M. Development Trends for Human Monoclonal Antibody Therapeutics. Nat. Rev. Drug Discovery 2010, 9, 767–774. 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Small-Molecule Modulation of Protein Homeostasis. Chem. Rev. 2017, 117, 11269–11301. 10.1021/acs.chemrev.7b00077. [DOI] [PubMed] [Google Scholar]
- Itoh Y.; Ishikawa M.; Naito M.; Hashimoto Y. Protein Knockdown Using Methyl Bestatin-Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- Yokoo H.; Ohoka N.; Naito M.; Demizu Y. Design and Synthesis of Peptide-based Chimeric Molecules to Induce Degradation of the Estrogen and Androgen Receptors. Bioorg. Med. Chem. 2020, 28, 115595. 10.1016/j.bmc.2020.115595. [DOI] [PubMed] [Google Scholar]
- Ohoka N.; Misawa T.; Kurihara M.; Demizu Y.; Naito M. Development of a Peptide-based Inducer of Protein Degradation Targeting NOTCH1. Bioorg. Med. Chem. Lett. 2017, 27, 4985–4988. 10.1016/j.bmcl.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. Aptamers as Therapeutics. Nat. Rev. Drug Discovery 2010, 9, 537–550. 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita R.; Higaki J.; Tomita N.; Ogihara T. Application of Transcription Factor ″Decoy″ Strategy as Means of Gene Therapy and Study of Gene Expression in Cardiovascular Disease. Circ. Res. 1998, 82, 1023–1028. 10.1161/01.RES.82.10.1023. [DOI] [PubMed] [Google Scholar]
- Samarasinghe K. T. G.; Jaime-Figueroa S.; Burgess M.; Nalawansha D. A.; Dai K.; Hu Z. Y.; Bebenek A.; Holley S. A.; Crews C. M. Targeted Degradation of Transcription Factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol. 2021, 28, 648–66. 10.1016/j.chembiol.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Chen H.; Kaniskan H. U.; Xie L.; Chen X.; Jin J.; Wei W. Y. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. 10.1021/jacs.1c03852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J.; Yan Y.; Ding D.; Wang D.; He Y.; Pan Y.; Yan W.; Kharbanda A.; Li H. Y.; Huang H. Destruction of DNA-Binding Proteins by Programmable Oligonucleotide PROTAC (O’PROTAC): Effective Targeting of LEF1 and ERG. Adv. Sci. 2021, 8, 2102555 10.1002/advs.202102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak M.; Lefebvre P.; Staels B. General Molecular Biology and Architecture of Nuclear Receptors. Curr. Top. Med. Chem. 2012, 12, 486–504. 10.2174/156802612799436641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Chambon P. The Estrogen-Receptor Binds Tightly to its Responsive Element as a Ligand-induced Homodimer. Cell 1988, 55, 145–156. 10.1016/0092-8674(88)90017-7. [DOI] [PubMed] [Google Scholar]
- Ohoka N.; Okuhira K.; Ito M.; Nagai K.; Shibata N.; Hattori T.; Ujikawa O.; Shimokawa K.; Sano O.; Koyama R.; Fujita H.; Teratani M.; Matsumoto H.; Imaeda Y.; Nara H.; Cho N.; Naito M. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. 10.1074/jbc.M116.768853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N.; Morita Y.; Nagai K.; Shimokawa K.; Ujikawa O.; Fujimori I.; Ito M.; Hayase Y.; Okuhira K.; Shibata N.; Hattori T.; Sameshima T.; Sano O.; Koyama R.; Imaeda Y.; Nara H.; Cho N.; Naito M. Derivatization of Inhibitor of Apoptosis Protein (IAP) Ligands Yields Improved Inducers of Estrogen Receptor α Degradation. J. Biol. Chem. 2018, 293, 6776–6790. 10.1074/jbc.RA117.001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinhitpass L.; Schorpp M.; Wagner U.; Ryffel G. U. An Estorogen-Responsive Element Derived from the 5′ Flanking Region of the Xenopus Vitellogenin A2 Gene Functions in Transfected Human-Cells. Cell 1986, 46, 1053–1061. 10.1016/0092-8674(86)90705-1. [DOI] [PubMed] [Google Scholar]
- Schwabe J. W. R.; Chapman L.; Finch J. T.; Rhodes D. The Crystal Structure of the Estrogen Receptor DNA-binding Domain Bound to DNA: How Receptors Discriminate between their Response Elements. Cell 1993, 75, 567–578. 10.1016/0092-8674(93)90390-C. [DOI] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. Q.; Wu Z. W.; Chen P.; Zhang J.; Wang T.; Zhou J. P.; Zhang H. B. Discovery of a New Class of PROTAC BRD4 Degraders Based on a Dihydroquinazolinone Derivative and Lenalidomide/Pomalidomide. Bioorg. Med. Chem. 2020, 28, 115228. 10.1016/j.bmc.2019.115228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.