

Abstract

Hepatocellular carcinoma (HCC) is the second or third leading cause of cancer mortality worldwide (depending on which statistics are used), yet there is no effective treatment. Currently, there are nine FDA-approved drugs for HCC, five monoclonal antibodies and four tyrosine kinase inhibitors. Ornithine aminotransferase (OAT) has been validated as a target in preclinical studies, which demonstrates that it is a potential target to treat HCC. Currently, there are no OAT inactivators in clinical trials for HCC. This Innovation describes evidence to support inhibition of OAT as a novel approach for HCC tumor growth inhibition. After the mechanism of OAT is discussed, the origins of our involvement in OAT inactivation, based on our previous work on mechanism-based inactivation of GABA-AT, are described. Once it was demonstrated that OAT inactivation does lead to HCC tumor growth inhibition, new selective OAT inactivators were designed and their inactivation mechanisms were elucidated. A summary of these mechanistic studies is presented. Inactivators of OAT provide the potential for treatment of HCC, targeting the Wnt/β-catenin pathway.
Keywords: Hepatocellular carcinoma, Ornithine aminotransferase, Wnt/β-catenin, Mechanism-based inactivation, X-ray crystal structure
Hepatocellular carcinoma (HCC) is the sixth or seventh most common cancer and the second or third leading cause of cancer death worldwide (depending on which statistics are used);1 over 800 000 people worldwide contract HCC each year, and about 700 000 die. Various current treatments include surgery, radiation therapy, chemotherapy, immunotherapy, targeted therapy, hormone therapy, stem cell transplant, and precision medicine.2 Despite these numerous approaches available, prognosis for recovery is very poor,3,4 with an overall 5-year survival rate of <10%5 (33% for localized HCC, 11% for spread to nearby structures, and 2% if distant spreading6), and side effects are extensive, largely because HCC is highly chemotherapy-7 and radiation-therapy8-resistant. Although there are nine FDA-approved drugs for hepatocellular carcinoma,9 the HCC market still has a high unmet need because of the lack of efficacy of current treatments. Well over 150 clinical trials for HCC have been terminated or withdrawn, mostly because of liver toxicity, flaws in trial design, and marginal potency.
HCC occurs most frequently in Asia and Africa,10 but because of the spread of hepatitis B and hepatitis C virus infections, which lead to HCC, the incidence of HCC in the United States is rising,11 where about 90% of primary liver cancers are HCC.12 HCC also is part of the natural history of nonalcoholic steatohepatitis (NASH).13 Other causes for the dramatic increase of HCC in the United States include obesity14 and an increase in diabetes.15
Significance of the Wnt/β-Catenin Pathway for Cancer Growth
Activation of the evolutionarily conserved Wnt/β-catenin signaling pathway16 in liver17 correlates with the development of HCC and is essential to normal cellular processes, such as growth, development, survival, and regeneration. The simplified mechanism involves binding of Wnt to co-receptors Frizzled and low-density lipoprotein receptor-related protein5/6 (LRP5/6), which leads to the translocation of β-catenin to the nucleus. Binding of β-catenin to T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors in the nucleus activates transcription of Wnt target genes, such as c-myc, cyclin D1, and AXIN2, important for cell proliferation (Figure 1).18 However, because of mutations in members of the Wnt/β-catenin pathway, β-catenin also is associated with the initiation and progression of cancer.19 The development of HCC in liver correlates with the upregulation of Wnt/β-catenin pathway proteins ornithine aminotransferase (OAT), glutamate transporter GLT-1, and glutamine synthetase.20 OAT converts l-ornithine to l-glutamate, which is transported by GLT-1 to glutamine synthetase to produce l-glutamine. Loss of OAT activity shuts down the formation of its product, l-glutamate, and this leads to the blockage of the synthesis of l-glutamine,21 which is essential for both normal and neoplastic cell growth. Glutamine, however, is taken up more efficiently by tumor cells than normal cells,22 and tumor growth is more accelerated by glutamine than normal cell growth.23
Figure 1.

Binding of Wnt to the Frizzled-LRP5/6 co-receptor allows β-catenin to be translocated into the nucleus, where it binds to TCF/LEF transcription factors, leading to transcription of Wnt target genes.
One important function of glutamine is to maintain tricarboxylic acid cycle intermediates (using the carbon atoms of glutamine) and in the biosyntheses of nucleotides, nonessential amino acids, and hexosamine (from the nitrogen atoms in glutamine).24 Another critical role for glutamine is to suppress oxidative stress by its catabolism to glutathione, an important intracellular antioxidant.25 Consequently, the survival and proliferation of tumor cells depend on a continuous supply of glutamine. As in the case of glucose metabolism, increased glutamine uptake is controlled by oncogenes, such as c-myc,26 and the inhibition of glutamine biosynthesis by oncogenes inhibits the growth of cancer.27 The increased requirement for glutamine by cancer cells is for the purpose of supporting anabolic processes that stimulate proliferation.28 An increase in glutamine supply by enhancement of OAT activity resulting from Wnt/β-catenin activation leads to an increase in tumor cell growth, independent of glutamine supply. Therefore, OAT inhibition to decrease glutamine production has been suggested as a viable mechanism to inhibit tumor growth.29 Also, it has been found that OAT is required to establish spindle formation in cancer cells, again indicating that inhibition of OAT should be an effective cancer treatment.30
Ornithine Aminotransferase
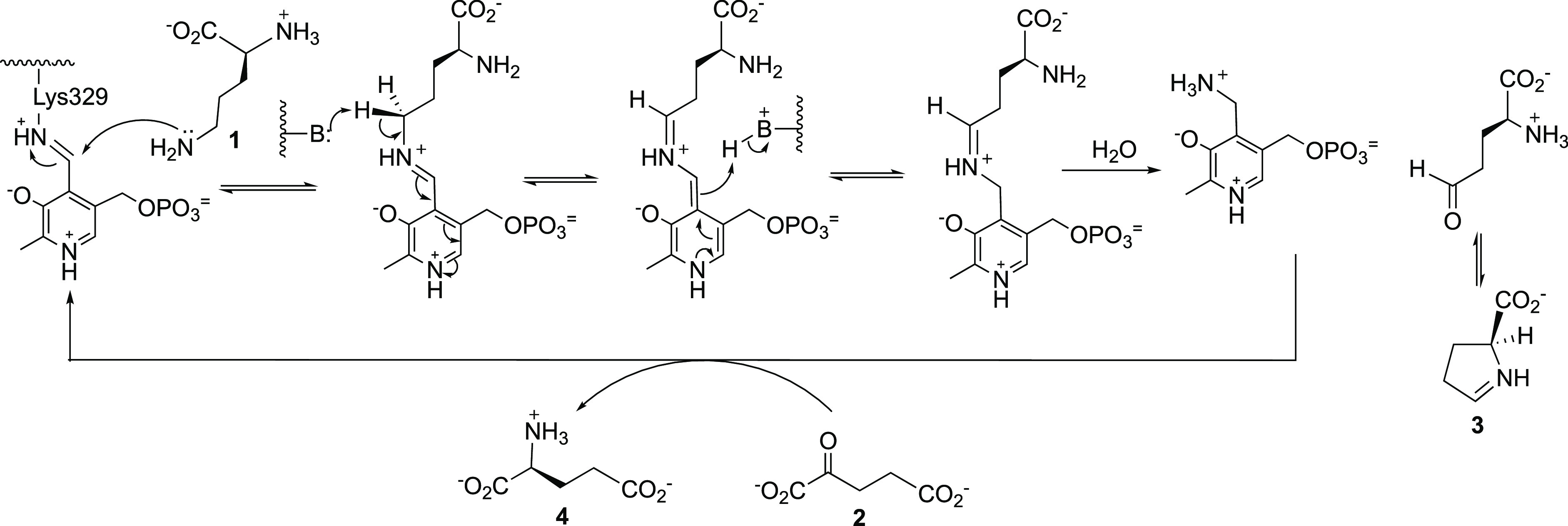
As shown in Scheme 1, OAT, a pyridoxal 5′-phosphate (PLP)-dependent mitochondrial enzyme, catalyzes the reversible interconversion of ornithine (1) and α-ketoglutarate (α-KG, 2) to l-glutamate semialdehyde, which cyclizes to Δ1-pyrroline-5-carboxylate (P5C, 3), and l-glutamate (4). OAT also is involved in proline and hydroxyproline metabolism in cancer cell survival, proliferation, and metastasis.31 Both proline and glutamine are important molecules for tumor growth,32 and they are interconvertible and linked in their metabolism. Under a hypoxic microenvironment proline metabolism is markedly altered in HCC, with accelerated utilization of proline and accumulation of hydroxyproline, which correlates with secretion of alpha-fetoprotein, a biomarker for HCC, and poor prognosis in disease progression. Accumulation of hydroxyproline further promotes HCC growth and resistance to sorafenib, a primary chemotherapeutic agent for HCC.33 There is considerable interplay among ornithine, proline, and glutamine, and their metabolites, α-KG and P5C, in the regulation of OAT and/or proline dehydrogenase/oxidase-dependent proliferation, apoptosis/autophagy, and metastasis.34
Scheme 1. Conversion of l-Ornithine to l-Glutamate Catalyzed by Ornithine Aminotransferase (OAT).
From GABA Aminotransferase Inactivators to OAT Inactivators
About 15 years ago I received an email message from Dr. Yaron Ilan, Professor of Medicine in the Gastroenterology & Liver Units of Hebrew University-Hadassah Medical Center in Jerusalem, asking if I would be willing to send him some of my γ-aminobutyric acid aminotransferase (GABA-AT) inactivators to test in his HCC mouse model. He explained that a DNA microarray analysis was carried out of genes in the liver of a normal rat compared with those in the liver of a rat (Psammomys obesus) that spontaneously develops HCC, and seven genes, including the gene encoding for OAT, were overexpressed.35 He said he wanted to test an OAT inhibitor, but he did not have access to one, and his research assistant had seen my work on GABA-AT inactivators and thought they might work. At first, I thought that it would be futile to test GABA analogue inactivators with OAT because of the low overall sequence identity (17%)36 between the two enzymes. However, a closer look at the active sites revealed that the residues in the two enzymes are quite similar37 and share a high structural homology.38 Also, OAT is a member of the evolutionary aminotransferase subgroup of PLP-dependent enzymes that includes GABA-AT,39 an enzyme found in glial cells and presynaptic neurons;40 all aminotransferases have very similar catalytic mechanisms.41 They both transfer the amino group of their respective substrates (ornithine for OAT and GABA for GABA-AT) to α-ketoglutarate, giving succinic semialdehyde (from GABA)41 or glutamate γ-semialdehyde (from ornithine)42 and l-glutamate.43 But what really got my attention was when I found out that HCC is the most common primary liver malignancy and one of the principal causes of cancer death worldwide.1 However, I thought I should not just send my most potent GABA-AT inactivators because the active sites are not identical between GABA-AT and OAT. Instead, I chose to send a mixture of mediocre potency, low potency, and inactive inhibitors in addition to potent ones (Figure 2).
Figure 2.
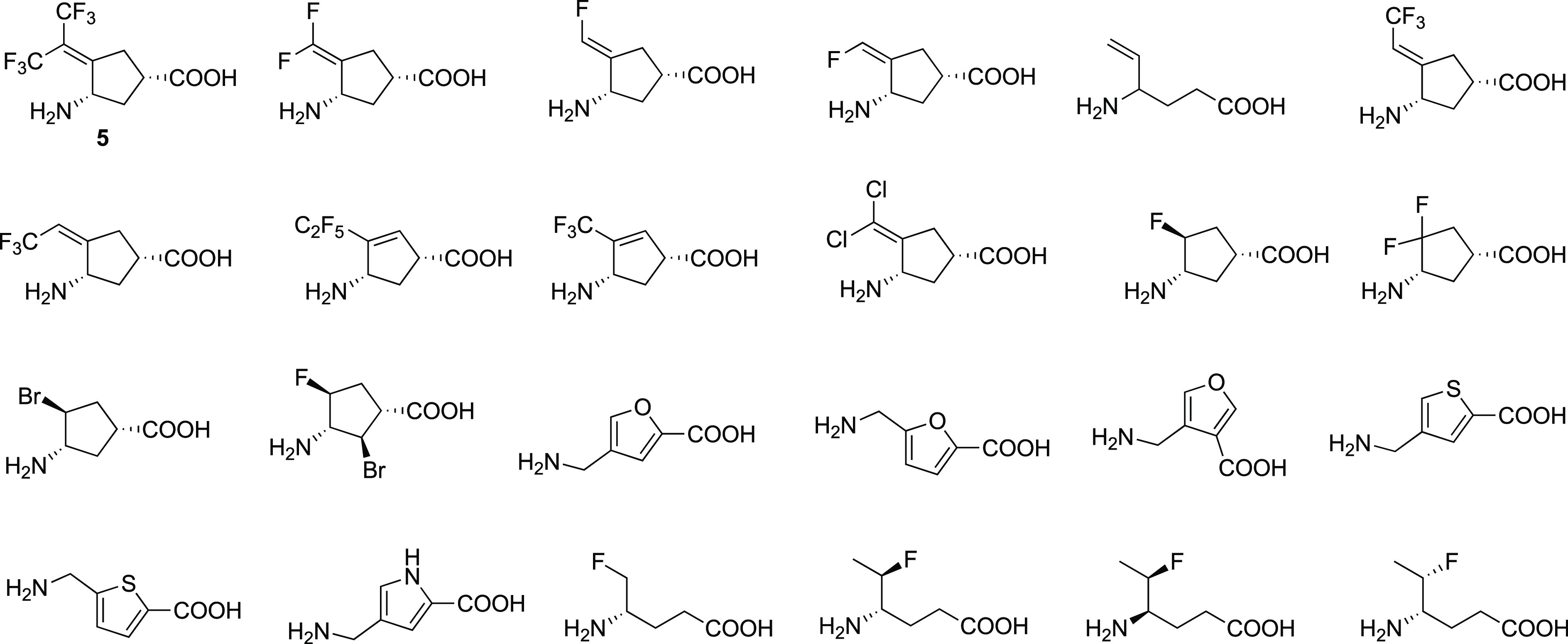
GABA analogues screened for OAT activity. Figure modified from ref (35). Copyright 2015 American Chemical Society.
OAT Inactivators Inhibit HCC Growth
Using these compounds, we showed, for the first time, that OAT inactivators significantly suppress alpha-fetoprotein (AFP) secretion, a biomarker for HCC, in Hep3B and HepG2 cells; the most potent OAT inactivator at that time (5, Figure 2), which was one of the compounds that theoretically could inactivate GABA-AT but did not and, therefore, was OAT selective, significantly suppressed AFP serum levels (Figure 3A) and tumor growth (Figure 3B) in patient-derived HCC-harboring mice, even at 0.1 mg/kg.35 This compound is water-soluble and 42% orally bioavailable, does not inhibit cytochrome P450s or the hERG channel, and is not metabolized by human liver microsomes over 90 min; plasma protein binding is only 62%, maximum tolerated dose is 30 mg/kg (mice), and, in a 7-day repeat study, the no observed adverse effect level (NOAEL) was 10 mg/kg in mice and 30 mg/kg in rats, indicating a therapeutic index of 100–300.
Figure 3.
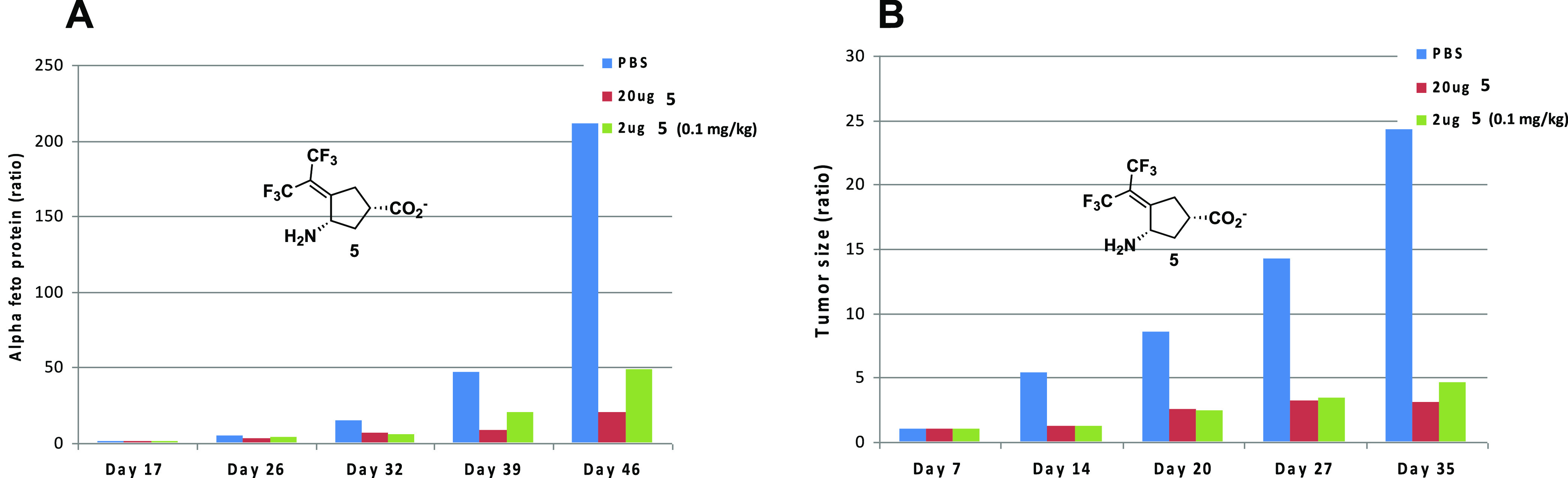
(A) Administration of 5 inhibits serum AFP secretion in vivo. Mice were treated for 27 days, 3 times a week, starting 3 weeks following HCC transplantation with two doses of 1 (0.1 mg/kg [2 μg], green bars or 1 mg/kg [20 μg], red bars), compared with untreated controls (blue bars). Levels are normalized to the starting day of therapy. (B) Compound 5 diminishes tumor volume in both treated groups (0.1 mg/kg [2 μg], green bars and 1.0 mg/kg [20 μg], red bars) compared to untreated controls (blue bars). Figure reproduced from ref (35). Copyright 2015 American Chemical Society.
Inactivation Mechanism for Our First OAT Inactivator
Compound 5 is a mechanism-based inactivator44 of OAT, and the inactivation mechanism, shown in Scheme 2, was elucidated by a combination of intact protein mass spectrometry and crystallography as well as the use of a fluoride ion electrode, partition ratio determination (the number of equivalents of 5 converted to product per inactivation event), and metabolomics.45 Compound 5 forms a Schiff base with coenzyme PLP to give 6. Deprotonation to 7 followed by fluoride ion elimination gives 8. Intermediate 7 does not undergo hydrolysis to 9, but in 92% of the turnovers 8 is hydrolyzed to 10. The remainder of turnovers (8%) results in inactivation from Michael addition of Lys-292 with a loss of two fluoride ions followed by hydrolysis to give covalent adduct 11; hydrolysis of the PLP imine gives 12. Structure 11 was confirmed by intact protein mass spectrometry and the crystal structure shown in Figure 4.
Scheme 2. Inactivation Mechanism for 5 with Human Ornithine Aminotransferase.
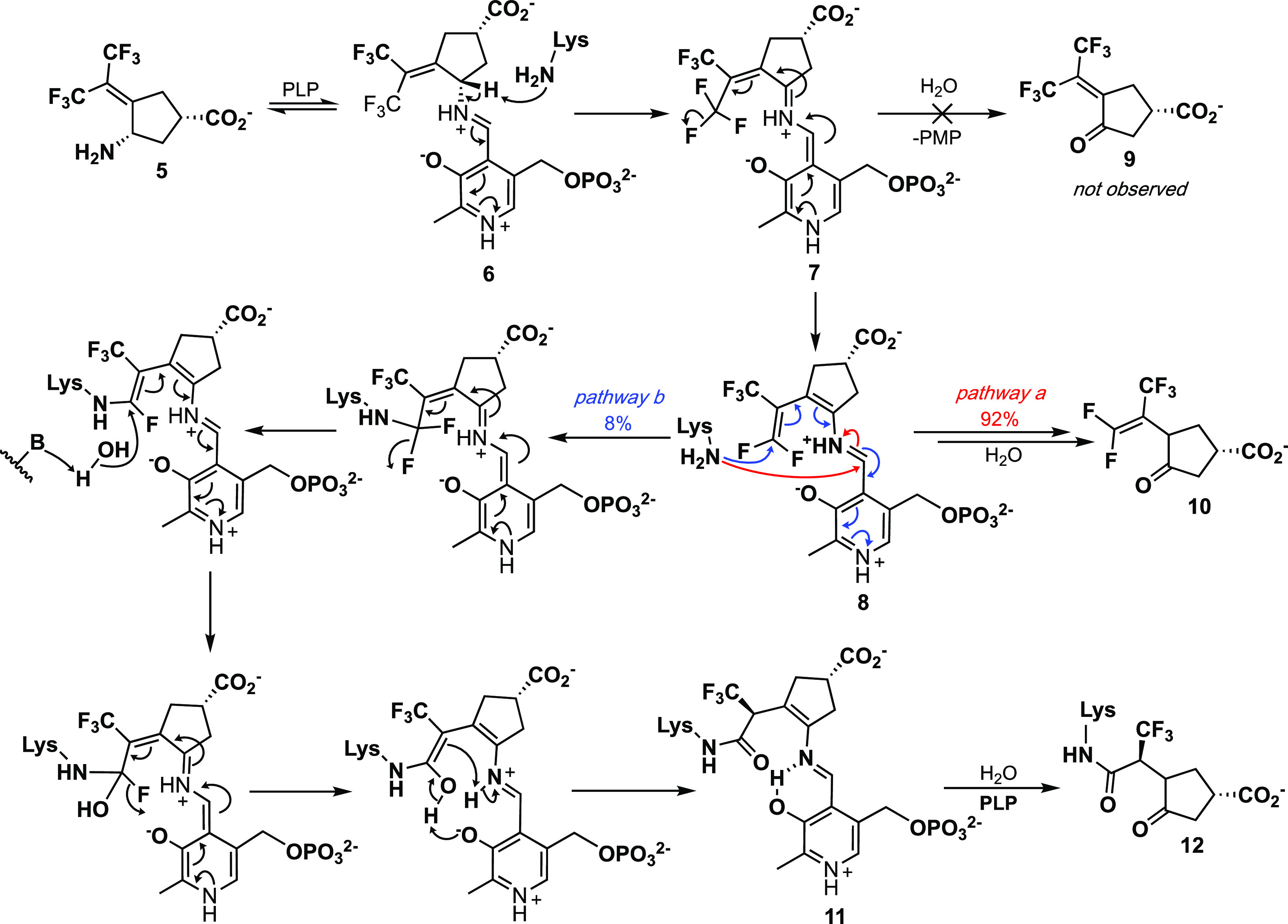
Scheme reproduced from ref (45). Copyright 2019 American Chemical Society.
Figure 4.
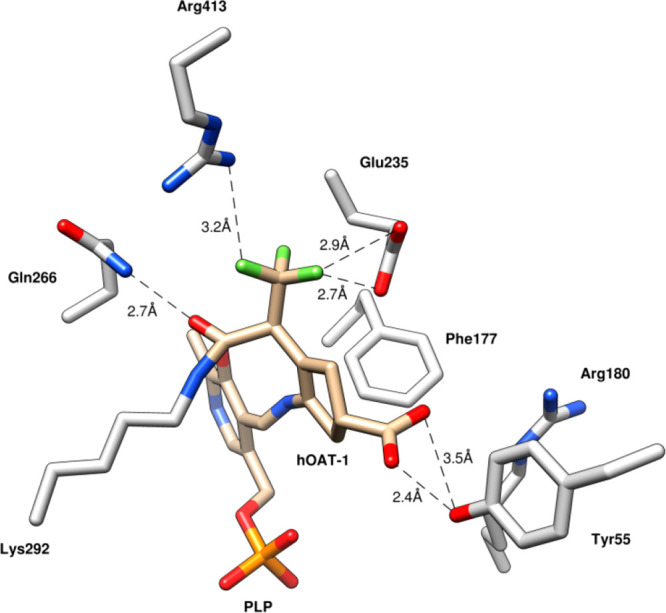
X-ray crystal structure of OAT following inactivation by 5 (PDB code: 6OIA). Figure reproduced from ref (45). Copyright 2019 American Chemical Society.
From the crystal structure it is apparent that only the syn-CF3 group is involved in the inactivation chemistry of 5. The corresponding Z-(13) and E-monotrifluoromethyl (14) analogues, however, had previously been shown to inactivate OAT with comparable inactivation kinetics.35
This was rationalized as the result of interconversion of
the intermediates
derived from 13 and 14 during inactivation.
To prevent rotation and interconversion, the corresponding syn-(15) and anti-monotrifluoromethyl
esters (16) were synthesized (modeling showed that the
ester group could bind with the enzyme to prevent rotation), and,
indeed, 15 was comparable in inactivation efficiency
to 5 and 16 was inactive.
Early Inactivators of OAT
Compound 5 is not
the first compound to inactivate
OAT; l-canaline ((S)-2-amino-4-amino-oxybutyric
acid, 17), a natural product analogue of ornithine,46 and l-fluoromethylornithine (5-FMO; 18)47 were reported in 1965 and
1988, respectively, as inactivators of OAT. However, 17 was shown to strongly inhibit the activity of seven PLP-dependent
enzymes,48 which could account for its
toxicity.49 The alkoxylamine group of 17 forms an oxime with the PLP coenzyme of OAT, causing irreversible
inhibition.50 The crystal structure of 17 with human OAT confirmed the oxime structure.51 5-FMO (18) also irreversibly inhibits
OAT, but selectively, and long-term inhibition of OAT does not produce
apparent detrimental effects in mice,52 possibly because 10–20% of OAT is refractory to inactivation
by 18.53 Of the four possible
stereoisomers of 5-FMO, only the (2S,5S) isomer inactivates OAT.54 The crystal
structure of human OAT inactivated by 5FMO55 shows that the PLP is modified to give the expected product of mechanism-based
inactivation by an enamine mechanism (19, Scheme 3).54 The enamine inactivation mechanism was first introduced by Metzler
and co-workers for two other PLP-dependent enzymes, glutamate decarboxylase56 and aspartate aminotransferase,57 both inactivated by l-serine O-sulfate.
Scheme 3. Mechanism of Inactivation of OAT by 5-Fluoromethylornithine (18).

Newer OAT Inactivators and Inactivation Mechanisms
We have designed
several new OAT mechanism-based inactivators on
the basis of organic chemical intuition and computer modeling, including 20–23. (S)-3-Amino-4,4-difluorocyclopent-1-enecarboxylic
acid (20)58 is an irreversible
inhibitor of OAT, comparable in inactivation efficiency to 5. The corresponding cyclopentane analogue is a very weak inactivator,
but that was expected on the basis of our earlier studies showing
that incorporation of a double bond into CPP-115 (24)59 to give OV329 (25) enhances inactivation
efficiency 10-fold.60 With the aid of intact
protein mass spectrometry, metabolomics, and protein crystallography,
the turnover and inactivation mechanisms for 20 were
elucidated (Scheme 4).61 Transient-state kinetic measurements,
using rapid-mixing spectrophotometry, were used that supported an
initial direct E2 elimination of HF from the PLP complex to give 27 (pathway a) over the typical E1cB mechanism (26, pathway b). The major pathway (97%) from 27 involves
an attack by Lys292 to release, after hydrolysis of the enamine, 28 (pathway c); only 3% of turnovers proceed by water attack
to give 29 (pathway d). The loss of a fluoride ion from 29 gives 30, which, based on transient-state
spectrophotometry and intact protein mass spectrometry, undergoes
tautomerization to give noncovalent adducts 31/32 84% of the turnovers (pathway e) and attack by Lys292 to
give covalent adducts 33/34 16% of the turnovers
(pathway f). In the conversion of 27 to 28, evidence for Lys292 attack (pathway c) was obtained by soaking 20 into OAT crystals and determining the X-ray crystal structure
(Figure 5A); the co-crystallization
of 20 with OAT gave the structures of 31/32 (Figure 5B).
Scheme 4. Mechanism Proposed for the Inactivation of OAT by 20.

Scheme modified from ref (61). Copyright 2021 American Chemical Society.
Figure 5.

(A) Crystal structure of OAT resulting from soaking of 20 over 1 h, shown in two alternate conformations (beige): one in which the carboxylate group interacts with Tyr55 (conformation A) and the other in which the carboxylate forms a salt bridge with Arg413 (conformation B); PDB code: 7LK1. (B) Co-crystal structure of OAT with 20; PDB code: 7LK0. Carbon atoms in the residues are colored gray, nitrogen in dark blue, and oxygen in red; the water molecule is shown as a red sphere. Hydrogen bonding distances between atoms are in Ångstroms (Å) and are shown as black dashed lines. Figure reproduced ref (61). Copyright 2021 American Chemical Society.
Because of the larger active site pocket of OAT relative to that of GABA aminotransferase (GABA-AT), CPP-11535 and OV32960a are nonselective inactivators of both enzymes. By increasing the size of the fluorine substituents on these inactivators to trifluoromethyl groups (to give 5), selective OAT inactivation was realized. Another approach to gain selectivity over GABA-AT based on size was taken by increasing the ring size from cyclopentene to cyclohexene (21/22).62 Kinetic analyses revealed that 21 was 2.9 times more efficient as an inactivator of OAT than 5 and 22 is 23.4 times more efficient than 5. Although 21 and 22 also inactivate GABA-AT, they do so much less efficiently: 65.6 times and 13.3 times less efficiently, respectively. With the aid of fluoride ion release studies, intact protein mass spectrometry, and protein crystallography, inactivation mechanisms were proposed for 21 and 22. Three potential mechanisms are shown in Scheme 5. They all are initiated by Schiff base formation with the active site PLP followed by deprotonation and elimination of a fluoride ion to give 35. Pathway a is an enamine mechanism; when X = H, enamine adduct 36 results, and when X = F enamine addition is followed by elimination of HF and aromatization (37). Pathway b results in aromatization without loss of X (38). Pathway c is a Michael addition mechanism, leading to either covalent adduct 39 when X = H or aromatized adduct 40 when X = F. On the basis of intact protein mass spectrometry, 21 formed a covalent adduct with OAT with an additional mass (369.29) that corresponds to either 36a in pathway a or 39 (pathway c, Scheme 5); 22 added an additional 366.34 mass units to OAT, which corresponds to either 37 (pathway a) or 40 (pathway c). Protein crystallography clarified the structures, clearly showing adduct 36a (pathway a) from 21 and adduct 40 (pathway c) from 22 (Figure 6). A difference of a single fluorine atom made the difference between an enamine mechanism (21) and a Michael addition/aromatization mechanism (22). Furthermore, shrinkage of the ring from cyclohexene (22) to cyclopentene (20) was sufficient to again change the inactivation mechanism (Scheme 4), this time to Michael addition by water followed by tautomerization (31/32) or attack by Lys292 (33/34). The major pathway (97%) for 20, however, was the release of metabolite 28 (pathway c).
Scheme 5. Possible Inactivation Mechanisms by 21 and 22.

Scheme reproduced from ref (62). Copyright 2020 American Chemical Society.
Figure 6.

Crystal structures of 21 (PDB code: 6 V8D) and 22 (PDB code: 6 V8C) cocrystallized with OAT. Amino acid residues are in silver; compounds bound to PLP are in gold; oxygen is red, and nitrogen is blue. Figure reproduced from ref (62). Copyright 2020 American Chemical Society.
A chimera of 22 and OV329 (25) resulted in 23, which was expected, if it were an inactivator, to have an inactivation mechanism similar to that for 22 or 25. Compound 23 is 22 times more efficient as an inactivator of OAT than 5 and comparable to 22; however, another completely different inactivation mechanism was revealed.63 On the basis of native mass spectrometry (to preserve noncovalent substrate binding and protein quaternary structure), intact protein mass spectrometry (to identify covalent adducts), top-down tandem mass spectrometry (to fragment the intact protein), protein crystallography, and metabolomics, the most plausible mechanisms for turnover and inactivation are shown in Scheme 6. The methods used to identify each of the intermediates and final adducts are given next to the structures. Schiff base formation of 23 with the active site PLP gives 41. Deprotonation to 42 leads to partitioning of the pathway, with 63% (pathway a) leading to inactivation intermediate 43 and 37% (pathway b) leading to turnover intermediate 44. Attack by Lys292 on 44 gives 45, which eliminates F– to give 46; deprotonation leads to aromatic metabolite 47. Inactivation intermediate 43 undergoes bond rotation because of steric hindrance with the difluoromethylenyl group to give 48. Tautomerization results in reactive intermediate 49, which first is attacked by Thr322, displacing a F– to give 50, whose crystal structure was obtained by soaking 23 into crystalline OAT (Figure 7A). Attack by Lys292 gives intermediate 51; protonation leads to the final double covalent adduct (52, Figure 7B), the first example of a mechanism-based inactivator leading to a single adduct with two covalent bonds from two different active site residues, one of which is not catalytic.
Scheme 6. Plausible turnover and Inactivation Mechanisms for the Inactivation of OAT by 23.
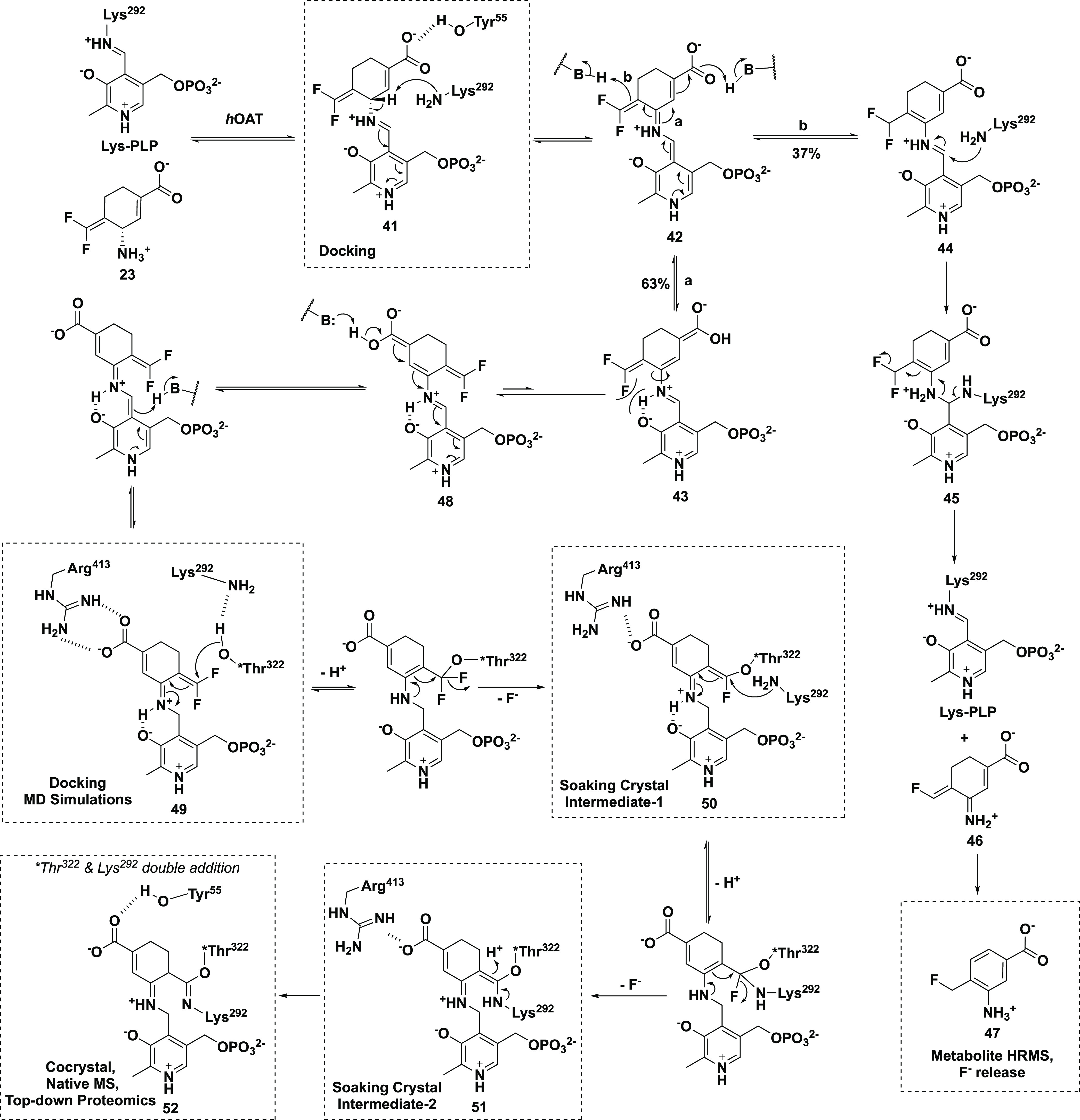
Scheme modified from ref (63). Copyright 2021 American Chemical Society.
Figure 7.

(A) X-ray crystal structure of first intermediate (50) from soaking 23 into OAT; PDB code: 7LOM. (B) X-ray cocrystal structure of double covalently bonded 23 (52) with Thr322 and Lys292 in OAT; PDB code: 7LON. Figure reproduced from ref (63). Copyright 2021 American Chemical Society.
The compounds described above have been designed to allow for a variety of potential inactivation mechanisms to occur. Inactivators with related structures demonstrate how minor structural differences can have a major impact on inactivation mechanisms. Enzymes evolve to catalyze highly specific chemistry on specific molecules, but when enzymes are presented with compounds having unnatural structures, unexpected catalytic chemistry can result. This exemplifies the ongoing struggle between Man and Nature, each trying to outwit the other. With the assistance of mechanism-based inactivators, it is sometimes possible to get the better of Mother Nature.
Currently, there are nine FDA-approved drugs for HCC. Five are monoclonal antibodies that either act as checkpoint inhibitors to trigger the immune system to fight the cancer or act by binding to VEGF, leading to a reduction in microvascular growth of tumor blood vessels and limiting the blood supply to the tumor. The other four drugs are tyrosine kinase inhibitors. OAT is a novel target for drugs to treat HCC. Inactivators of OAT provide the promise of treatment for this dreaded disease by targeting a yet untapped point of a new pathway, the Wnt/β-catenin pathway. Possibly a combination of drugs acting on different pathways will be the way forward to successfully inhibit progression of this disease.
Acknowledgments
I am grateful for financial support from the National Institutes of Health (R01 CA260250 and R01 DA030604) and for my outstanding graduate students (GS), postdocs (PD), and collaborators (C) involved in the OAT inactivator project, who all make me look good: Hyunbeom Lee (GS), Jose I. Juncosa (PD), Hoang V. Le (PD), Matthew J. Moschitto (PD), Wei Zhu (PD), Pathum Weerawarna (PD), Sida Shen (PD), Timothy A. Dwight (PD), Hejun Lu (PD), Yaron Ilan (C), Ehud Zigmond (C), Ami Ben Ya’acov (C), Yoav Lichtenstein (C), Zvi Shalev (C), Yoav Smith (C), Lidya Zolotarov (C), Ehud Ziv (C), Rony Kalman (C), Neil L. Kelleher (C), Peter F. Doubleday (C), Rafael D. Melani (C), Zdzislaw Wawrzak (C), Dali Liu (C), Daniel S. Catlin (C), Arseniy Butrin (C), Brett A. Beaupre (C), Noel Kadamandla (C), Peidong Zhao (C), Graham R. Moran (C), Mauricio T. Tavares (C), and Glaucio M. Ferreira (C).
Glossary
Abbreviations
- AFP
alpha-fetoprotein
- α-KG
α-ketoglutarate
- 5-FMO
l-fluoromethylornithine
- GABA-AT
gamma-aminobutyric acid aminotransferase
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- hERG
human ether-à-go-go-related gene
- NASH
nonalcoholic steatohepatitis
- NOAEL
no observed adverse effect level
- OAT
ornithine aminotransferase
- PLP
pyridoxal 5′-phosphate
- P5C
Δ1-pyrroline-5-carboxylate
- VEGF
vascular endothelial growth factor
The author declares no competing financial interest.
References
- a Tang A.; Hallouch O.; Chernyak V.; Kamaya A.; Sirlin C. B. Epidemiology of hepatocellular carcinoma: target population for surveillance and diagnosis. Abdom. Radiol. 2018, 43, 13–25. 10.1007/s00261-017-1209-1. [DOI] [PubMed] [Google Scholar]; b Bray F.; Ferlay J.; Soerjomataram I.; Siegel R. L.; Torre L. A.; Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 2018, 68, 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]; (c) https://www.bluefaery.org/statistics/.
- https://www.cancer.gov/about-cancer/treatment/types.
- de Lope C. R.; Tremosini S.; Forner A.; Reig M.; Bruix J. Management of HCC. J. Hepatol. 2012, 56 (Suppl. 1), S75. 10.1016/S0168-8278(12)60009-9. [DOI] [PubMed] [Google Scholar]
- Xie B.; Wang D. H.; Spechler S. J. Sorafenib for treatment of hepatocellular carcinoma: a systematic review. Dig. Dis. Sci. 2012, 57, 1122. 10.1007/s10620-012-2136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarveazad A.; Agah S.; Babahajian A.; Amini N.; Bahardoust M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J. Res. Med. Sci. 2019, 24, 86. 10.4103/jrms.JRMS_1017_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.emedicinehealth.com/survival_rate_for_hepatocellular_carcinoma/article_em.htm.
- a Marin J. J. G.; Castano B.; Martinez-Becerra P.; Rosales R.; Monte M. J. Chemotherapy in the treatment of primary liver tumours. Cancer Ther. 2008, 6, 711–728. [Google Scholar]; b Hong L.; Han Y.; Zhang H.; Zhao Q.; Wu K.; Fan D. Drug resistance-related miRNAs in hepatocellular cancer. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 283–288. 10.1586/17474124.2014.881713. [DOI] [PubMed] [Google Scholar]
- Han S.-X.; Zhu Q.; Ma J.-L.; Lv Y.; Zhao J.; Huang C.; Jia X.; Ou W.; Guo H.-T. Apoptin sensitizes radiation-induced cell death via classic mitochondrial, caspase and p53-dependent signaling in HepG2 elss. Mol. Med. Rep. 2010, 3, 59–63. 10.3892/mmr.2010.391. [DOI] [PubMed] [Google Scholar]
- https://www.cancer.gov/about-cancer/treatment/drugs/liver.
- a Petrick J. L.; Florio A. A.; Znaor A.; Ruggieri D.; Laversanne M.; Alvarez C. S.; Ferlay J.; Valery P. C.; Bray F.; McGlynn K. A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. 10.1002/ijc.32723. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McGlynn K. A.; Petrick J. L.; El-Serag H. B. Epidemiology of hepatocellular carcinoma. Hepatology 2021, 73, 4–13. 10.1002/hep.31288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altekruse S. F.; McGlynn K. A.; Reichman M. E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London W. T.; McGlynn K. A.. Liver cancer, In Cancer Epidemiology and Prevention, 3rd ed.; Schottenfeld D., Fraumeni J. F. Jr., Eds.; Oxford University Press: New York, 2006; pp 763–786. [Google Scholar]
- a Ong J. P.; Younossi Z. M. Epidemiology and natural history of NAFLD and NASH. Clin. Liver Dis. 2007, 11, 1–16. 10.1016/j.cld.2007.02.009. [DOI] [PubMed] [Google Scholar]; b Bugianesi E. Non-alcoholic steatohepatitis and cancer. Clin. Liver Dis. 2007, 11, 191–207. 10.1016/j.cld.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Nishikawa H.; Fukunishi S.; Asai A.; Nishiguchi S.; Higuchi K. Obesity and liver cancer in Japan: a comprehensive review. Anticancer Res. 2021, 41, 2227–2237. 10.21873/anticanres.14999. [DOI] [PubMed] [Google Scholar]
- Kim H.; Lee D. S.; An T. H.; Park H.-J.; Kim W. K.; Bae K.-H.; Oh K. J. Metabolic spectrum of liver failure in type 2 diabetes and obesity: from NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22 (9), 4495. 10.3390/ijms22094495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson M. D.; Monga S. P. S. WNT//-catenin signaling in liver health and disease. Hepatology 2007, 45, 1298–1305. 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- Cadoret A.; Ovejero C.; Terris B.; Souil E.; Levy L.; Lamers W. H.; Kitajewski J.; Kahn A.; Perret C. New targets of -catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- a Li Q.; Sun M.; Wang M.; Feng M.; Yang F.; Li L.; Zhao J.; Chang C.; Dong H.; Xie T.; Chen J. Dysregulation of Wnt//-catenin signaling by protein kinases in hepatocellular carcinoma and its therapeutic application. Cancer Sci. 2021, 112, 1695–1706. 10.1111/cas.14861. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang Y.; Wang X. Targeting the Wnt//-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. 10.1186/s13045-020-00990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucero O. M.; Dawson D. W.; Moon R. T.; Chien A. J. A re-evaluation of the ″oncogenic″ nature of Wnt/beta-catenin signaling in melanoma and other cancers. Curr. Oncol. Rep. 2010, 12, 314–318. 10.1007/s11912-010-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cadoret A.; Ovejero C.; Terris B.; Souil E.; Levy L.; Lamers W. H.; Kitajewski J.; Kahn A.; Perret C. New targets of -catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]; b Zucman-Rossi J; Benhamouche S; Godard C; Boyault S; Grimber G; Balabaud C; Cunha A S; Bioulac-Sage P; Perret C Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 2007, 26, 774–780. 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]
- Sekine S.; Lan B. Y-A.; Bedolli M.; Feng S.; Hebrok M. Liver-specific loss of -catenin blocks glutamine synthesis pathway activity and cytochrome P450 expression in mice. Hepatology 2006, 43, 817–825. 10.1002/hep.21131. [DOI] [PubMed] [Google Scholar]
- Souba W. W. Glutamine and cancer. Ann. Surg. 1993, 218, 715–728. 10.1097/00000658-199312000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina M. A. Glutamine and cancer. J. Nutr. 2001, 131 (9 Suppl), 2539S–2542S. 10.1093/jn/131.9.2539S. [DOI] [PubMed] [Google Scholar]
- a Dang C. V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DeBerardinis R. J.; Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shanware N. P.; Mullen A. R.; DeBerardinis R. J.; Abraham R. T. Glutamine: pleiotropic roles in tumor growth and stress resistance. J. Mol. Med. 2011, 89, 229–236. 10.1007/s00109-011-0731-9. [DOI] [PubMed] [Google Scholar]; b DeBerardinis R. J.; Mancuso A.; Daikhin E.; Nissim I.; Yudkoff M.; Wehrli S.; Thompson C. B. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19345–19350. 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gao P.; Tchernyshyov I.; Chang T.-C.; Lee Y.-S.; Kita K.; Ochi T.; Zeller K. I.; De Marzo A. M.; Van Eyk J. E.; Mendell J. T.; Dang C. V. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Le A.; Lane A. N.; Hamaker M.; Bose S.; Gouw A.; Barbi J.; Tsukamoto T.; Rojas C. J.; Slusher B. S.; Zhang H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu W.; Le A.; Hancock C.; Lane A. N.; Dang C. V.; Fan T. W-M; Phang J. M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (23), 8983–8988. 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tong X.; Zhao F.; Thompson C. B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009, 19 (1), 32–37. 10.1016/j.gde.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise D. R.; Thompson C. B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Amadasi A.; Bertoldi M.; Contestabile R.; Bettati S.; Cellini B.; di Salvo M. L.; Bori-Voltattorni C.; Bossa F.; Mozzarelli A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007, 14, 1291–1324. 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]; b Dekaney C. M.; Wu G.; Yin Y. L.; Jaeger L. A. Regulation of ornithine aminotransferase gene expression and activity by all-transretinoic acid in Caco-2 intestinal epithelial cells. J. Nutr. Biochem. 2008, 19, 674–681. 10.1016/j.jnutbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Wang G.; Shang L.; Burgett A. W.; Harran P. G.; Wang X. Diazonamide toxins reveal an unexpected function for ornithine delta-aminotransferase in mitotic cell division. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2068–2073. 10.1073/pnas.0610832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Phang J. M. Proline metabolism in cell regulation and cancer biology: Recent advances and hypotheses. Antioxid. Redox Signaling 2019, 30, 635–649. 10.1089/ars.2017.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu W.; Le A.; Hancock C.; Lane A. N.; Dang C. V.; Fan T. W. M.; Phang J. M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8983–8988. 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu W.; Hancock C. N.; Fischer J. W.; Harman M.; Phang J. M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. 10.1038/srep17206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang J. M.; Liu W.; Hancock C. N.; Fischer J. W. Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. 10.1097/MCO.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L.; Zeng J.; Geng P.; Fang C.; Wang Y.; Sun M.; Wang C.; Wang J.; Yin P.; Hu C.; Guo L.; Yu J.; Gao P.; Li E.; Zhuang Z.; Xu G.; Liu Y. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin. Cancer Res. 2018, 24, 474–485. 10.1158/1078-0432.CCR-17-1707. [DOI] [PubMed] [Google Scholar]
- Tanner J. J.; Fendt S. M.; Becker D. F. The proline cycle as a potential cancer therapy target. Biochemistry 2018, 57, 3433–3444. 10.1021/acs.biochem.8b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond E.; Ben Ya’acov A.; Lee H.; Lichtenstein Y.; Shalev Z.; Smith Y.; Zolotarov L.; Ziv E.; Kalman R.; Le H. V.; Lu H.; Silverman R. B.; Ilan Y. Suppression of hepatocellular carcinoma by inhibition of overexpressed ornithine aminotransferase. ACS Med. Chem. Lett. 2015, 6, 840–844. 10.1021/acsmedchemlett.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici P.; Capitani G.; Muller R.; Schirmer T.; Jansonius J. N. Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol. 1999, 285 (1), 297–309. 10.1006/jmbi.1998.2289. [DOI] [PubMed] [Google Scholar]
- Markova M.; Peneff C.; Hewlins M. J. E.; Schirmer T.; John R. A. Determinants of substrate specificity in ω-sminotransferases. J. Biol. Chem. 2005, 280 (43), 36409–36416. 10.1074/jbc.M506977200. [DOI] [PubMed] [Google Scholar]
- Lee H.; Juncosa J. I.; Silverman R. B. Ornithine aminotransferase vs. GABA aminotransferase. Implications for the design of new anticancer drugs. Med. Res. Rev. 2015, 35, 286–305. 10.1002/med.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B.-Y.; Cho B.-K.; Yun H.; Koteshwar K.; Kim B.-G. Revisit of aminotransferase in the genomic era and its application to biocatalysis. J. Mol. Catal. B: Enzym. 2005, 37 (1–6), 47–55. 10.1016/j.molcatb.2005.09.004. [DOI] [Google Scholar]
- Jeremiah S.; Povey S. The biochemical genetics of human gamma-aminobutyric acid transaminase. Ann. Hum. Genet. 1981, 45 (Pt 3), 231–236. 10.1111/j.1469-1809.1981.tb00334.x. [DOI] [PubMed] [Google Scholar]
- a Mehta P. K.; Hale T. I.; Christen P. Evolutionary relationships among aminotransferases. Tyrosine aminotransferase, histidinol-phosphate aminotransferase, and aspartate aminotransferase are homologous proteins. Eur. J. Biochem. 1989, 186 (1–2), 249–253. 10.1111/j.1432-1033.1989.tb15202.x. [DOI] [PubMed] [Google Scholar]; b Jung M. J.; Seiler N. Enzyme-activated irreversible inhibitors of L-ornithine:2-oxoacid aminotransferase. Demonstration of mechanistic features of the inhibition of ornithine aminotransferase by 4-aminohex-5-ynoic acid and gabaculine and correlation with in vivo activity. J. Biol. Chem. 1978, 253 (20), 7431–7439. 10.1016/S0021-9258(17)34520-9. [DOI] [PubMed] [Google Scholar]
- Peraino C.; Bunville L. G.; Tahmisian T. N. Chemical, physical, and morphological properties of ornithine aminotransferase from rat liver. J. Biol. Chem. 1969, 244 (9), 2241–2249. 10.1016/S0021-9258(19)78217-9. [DOI] [PubMed] [Google Scholar]
- Cooper A. J. Glutamate-gamma-aminobutyrate transaminase. Methods Enzymol. 1985, 113, 80–82. 10.1016/S0076-6879(85)13019-3. [DOI] [PubMed] [Google Scholar]
- Silverman R. B.Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology; CRC Press: Boca Raton, FL, 1988; Vols. I and II. [Google Scholar]
- Moschitto M. J.; Doubleday P. F.; Catlin D. S.; Kelleher N. L.; Liu D.; Silverman R. B. Mechanism of inactivation of ornithine aminotransferase by (1S,3S)-3-amino-4-(hexafluoropropan-2-ylidenyl)cyclopentane-1-carboxylic acid. J. Am. Chem. Soc. 2019, 141, 10711–10721. 10.1021/jacs.9b03254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katunuma N.; Okada M.; Matsuzawa T.; Otsuka Y. Studies on ornithine ketoacid transaminase. II. Role in metabolic pathway. J. Biochem. 1965, 57, 445–449. 10.1093/oxfordjournals.jbchem.a128099. [DOI] [PubMed] [Google Scholar]
- Daune G.; Gerhart F.; Seiler N. 5-Fluoromethylornithine, an irreversible and specific inhibitor of L-ornithine:2-oxo-acid aminotransferase. Biochem. J. 1988, 253 (2), 481–488. 10.1042/bj2530481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahiala E. L.; Kekomaki M.; Janne J.; Raina A.; Raiha N. C. Inhibition of pyridoxal enzymes by L-canaline. Biochim, Biophys. Acta 1971, 227 (2), 337–343. 10.1016/0005-2744(71)90065-9. [DOI] [PubMed] [Google Scholar]
- Rosenthal G. A. A mechanism of L-canaline toxicity. Eur. J. Biochem. 1981, 114, 301–304. 10.1111/j.1432-1033.1981.tb05149.x. [DOI] [PubMed] [Google Scholar]
- Kito K.; Sanada Y.; Katunuma N. Mode of inhibition of ornithine aminotransferase by L-canaline. J. Biochem. 1978, 83, 201–206. 10.1093/oxfordjournals.jbchem.a131892. [DOI] [PubMed] [Google Scholar]
- Shah S. A.; Shen B. W.; Brunger A. T. Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure 1997, 5 (8), 1067–1075. 10.1016/S0969-2126(97)00258-X. [DOI] [PubMed] [Google Scholar]
- a Seiler N.; Daune-Anglard G. Endogenous ornithine in search for CNS functions and therapeutic applications. Metab. Brain Dis. 1993, 8, 151–179. 10.1007/BF00996928. [DOI] [PubMed] [Google Scholar]; b Daune-Anglard G.; Bonaventure N.; Seiler N. Some biochemical and pathophysiological aspects of long-term elevation of brain ornithine concentrations. Pharmacol. Toxicol. 1993, 73, 29–34. 10.1111/j.1600-0773.1993.tb01953.x. [DOI] [PubMed] [Google Scholar]
- Daune G.; Seiler N. Interrelationships between ornithine, glutamate and GABA—III. An ornithine aminotransferase activity that is resistant to inactivation by 5-fluoromethylornithine. Neurochem. Int. 1988, 13, 383–391. 10.1016/0197-0186(88)90012-5. [DOI] [PubMed] [Google Scholar]
- Bolkenius F. N.; Knödgen B.; Seiler N. DL-Canaline and 5-fluoromethylornithine. Biochem. J. 1990, 268, 409–414. 10.1042/bj2680409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici P.; Capitani G.; Muller R.; Schirmer T.; Jansonius J. N. Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol. 1999, 285 (1), 297–309. 10.1006/jmbi.1998.2289. [DOI] [PubMed] [Google Scholar]
- Likos J. J.; Ueno H.; Feldhaus R. W.; Metzler D. E. A novel reaction of the coenzyme of glutamate decarboxylase with L-serine O-sulfate. Biochemistry 1982, 21, 4377–4386. 10.1021/bi00261a029. [DOI] [PubMed] [Google Scholar]
- Ueno H.; Likos J. J.; Metzler D. E. Chemistry of the inactivation of cytosolic aspartate aminotransferase by serine O-sulfate. Biochemistry 1982, 21, 4387–4393. 10.1021/bi00261a030. [DOI] [PubMed] [Google Scholar]
- Shen S.; Butrin A.; Doubleday P. F.; Melani R. D.; Beaupre B. A.; Tavares M. T.; Ferreira G. M.; Kelleher N. L.; Moran G. R.; Liu D.; Silverman R. B. Turnover and inactivation mechanisms for (S)-3-amino-4,4-difluorocyclopent-1-enecarboxylic acid, a selective mechanism-based inactivator of human ornithine aminotransferase. J. Am. Chem. Soc. 2021, 143, 8689–8703. 10.1021/jacs.1c02456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman R. B. The 2011 E. B. Hershberg Award for Important Discoveries in Medicinally Active Substances: (1S,3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115), a GABA Aminotransferase Inactivator and New Treatment for Drug Addiction and Infantile Spasms. J. Med. Chem. 2012, 55, 567–575. 10.1021/jm201650r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Juncosa J. I.; Takaya K.; Le H. V.; Moschitto M. J.; Weerawarna P. M.; Mascarenhas R.; Liu D.; Dewey S. L.; Silverman R. B. Design and mechanism of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid, a highly potent GABA aminotransferase inactivator for the treatment of addiction. J. Am. Chem. Soc. 2018, 140, 2151–2164. 10.1021/jacs.7b10965. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weerawarna P. M.; Moschitto M. J.; Silverman R. B. Theoretical and mechanistic studies of the inactivation of GABA aminotransferase by OV329 and CPP-115. Determination of the rate-limiting step and validation of the global kinetic parameters. ACS Chem. Biol. 2021, 16, 615–630. 10.1021/acschembio.0c00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S.; Butrin A.; Doubleday P. F.; Melani R. D.; Beaupre B. A.; Tavares M. T.; Ferreira G. M.; Kelleher N. L.; Moran G. R.; Liu D.; Silverman R. B. Turnover and inactivation mechanisms for (S)-3-amino-4,4-difluorocyclopent-1-enecarboxylic acid, a selective mechanism-based inactivator of human ornithine aminotransferase. J. Am. Chem. Soc. 2021, 143, 8689–8703. 10.1021/jacs.1c02456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Doubleday P. F.; Catlin D. S.; Weerawarna P. M.; Butrin A.; Shen S.; Wawrzak Z.; Kelleher N. L.; Liu D.; Silverman R. B. A remarkable difference that one fluorine atom confers on the mechanisms of inactivation of human ornithine aminotransferase by two cyclohexene analogues of.-aminobutyric acid. J. Am. Chem. Soc. 2020, 142, 4892–4903. 10.1021/jacs.0c00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Doubleday P. F.; Butrin A.; Weerawarna P. M.; Melani R.; Catlin D. S.; Dwight T. A.; Liu D.; Kelleher N. L.; Silverman R. B. Remarkable, unexpected mechanism for (S)-3-amino-4-(difluoromethylenyl)cyclohex-1-ene-carboxylic acid as a selective inactivator of human ornithine aminotransferase. J. Am. Chem. Soc. 2021, 143, 8193–8207. 10.1021/jacs.1c03572. [DOI] [PMC free article] [PubMed] [Google Scholar]