

Abstract
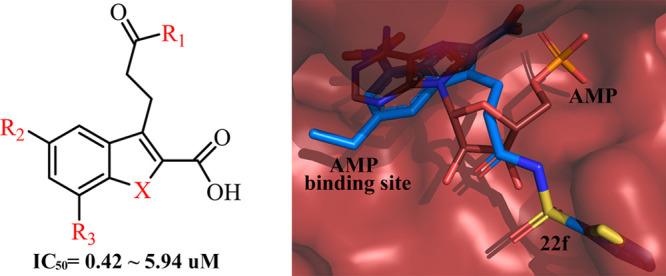
Liver fructose-1,6-bisphosphatase (FBPase) is a key enzyme in the gluconeogenesis, and its inhibitors are expected to be novel antidiabetic agents. Herein, a series of new indole and benzofuran analogues were designed and synthesized to evaluate the inhibitory activity against FBPase. As a result, the novel FBPase inhibitors bearing N-acylsulfonamide moiety on the 3-position of the indole-2-carboxylic acid scaffold (compounds 22f and 22g) were identified with IC50s at the submicromolar levels. Three X-ray crystal structures of the complexes were solved and revealed the structural basis for the inhibitory activity. The chemoinformatics analysis further disclosed the distinct binding features of this class of inhibitors, providing an insight for further modifications to create structurally distinct FBPase inhibitors with high potency and drug-like properties.
Keywords: Fructose-1,6-bisphosphatase; Type 2 diabetes; Allosteric inhibitor; Indole derivatives
As reported by International Diabetes Federation (IDF), approximately 537 million adults (over 81% of adults with diabetes in low- and middle-income countries) are suffering from diabetes worldwide, and this number is estimated to be mounting toward 643 and 784 million by the year of 2030 and 2045.1 One main characteristic symptom of type 2 diabetes is a constant high blood glucose level caused by excessive hepatic glucose production (HGP).2 Patients with long-term hyperglycemia are more likely to suffer from various vascular complications,3 for instance, retinopathy and cataracts,4 nephropathy,5 and cardiovascular diseases,6etc. Glucose in human liver derived generally through two pathways: glycogenolysis and gluconeogenesis (GNG), with GNG being the primary process for liver to output glucose in the fasting state where the three-carbon precursors such as pyruvate, lactate, and glycerol are converted into glucose. For persistent fasting or starvation, gluconeogenesis contributes to more than 50% glucose production, and this contribution is even more remarkable in type 2 diabetes mellitus (T2DM) patients.7,8
In the GNG process, fructose-1,6-bisphosphatase is a key rate-limiting enzyme that catalyzes the conversion of fructose-1,6-bisphosphate into fructose-6-phosphate, where the latter is further converted into glucose under the catalysis of glucose-6-phosphate isomerase and glucose-6-phosphatase.9,10 Human liver FBPase has been considered as a potential therapeutic target for the treatment of T2DM.11 In patients and animal models of type 2 diabetes, the expression levels and activities of FBPase were upregulated in the liver, indicating the importance of this enzyme for the elevation of blood glucose.11,12 FBPase inhibitors have been proven to be effective on reducing hepatic glucose production and lowering blood glucose levels in diabetic animal models.11,13 In particular, two FBPase inhibitors, CS-917 and MB07803, the prodrugs of MB05032 and MB07729 (Figure 1), have been developed and entered the clinical trials.14−17 Therefore, the inhibition of FBPase is a promising strategy for the development of novel antidiabetic agents.
Figure 1.

Chemical structures of several published FBPase inhibitors and structure modifications on the indole-based lead compound.
FBPase is a homotetramer, and every monomer embraces a substrate (fructose-1,6-bisphosphate) binding site and an AMP allosteric binding site.18 Besides many structurally diversified FBPase inhibitors identified by high-throughput screening,19−21 AMP itself was taken as a lead structure to develop highly potent FBPase inhibitors including MB05032 and MB07729 to mimic its negative regulator role.14,22,23 Very recently, novel covalent inhibitors were developed by targeting on Cys128, and that revealed a new cryptic allosteric binding site.24−26 Moreover, a structurally distinct chemotype bearing N-arylsulfonyl-indole-2-carboxamide scaffold was discovered, which exploited both the AMP binding pocket and an interface region between monomers.13,27 Specifically, Cpd118 was disclosed with the pronounced enzymatic activity and glucose lowering efficacy in diabetic animal models28 (Figure 1).
7-Nitro-1H-indole-2-carboxylic acids were disclosed in our continuous efforts to search for novel FBPase inhibitors.29,30 Among them, compound A (Figure 1) was identified as a potent lead structure with low molecular weight. Aiming to further investigate the structure–activity relationships of indole-2-carboxylic acid derivatives, variations of the R1, R2, and R3 substituents and heterocycle replacement were conducted. Herein, we described the synthesis, enzymatic activities, and structure–activity relationships of these compounds as FBPase inhibitors. To probe the binding features of the target molecules, three X-ray cocrystal structures were solved and the chemoinformatics analysis was conducted.
The synthesis of 5,7-disubstituted carboxylic diacids (6a–6i) was illustrated in Schemes 1–3. 7-Nitro and 7-chloro substituted target compounds were prepared as shown in Scheme 1. Under the Japp–Klingemann reaction conditions, 2-nitro-4-bromo aniline or 2-chloro-4-bromo aniline reacted with ethyl 2-oxocyclopentane carboxylate to give rise to phenylhydrazones 2a and 2b in 51% and 88% yields, respectively. Esterification of compounds 2a and 2b afforded 3a (79% yield) and 3b (95% yield), which were converted into indole intermediates 4a and 4b (30% and 48% yields), respectively, under the Fischer indole synthesis conditions.30 Utilizing the Suzuki–Miyaura reaction, the coupling of compounds 4a and 4b with alkylboronic acids yielded 5-alkyl substituted derivatives 5a–5e (64–90% yields), which were hydrolyzed into the carboxylic diacid derivatives 6a–6e (75–90% yields). Compound 6f was obtained by the hydrolysis of compound 4b in 99% yield.
Scheme 1.
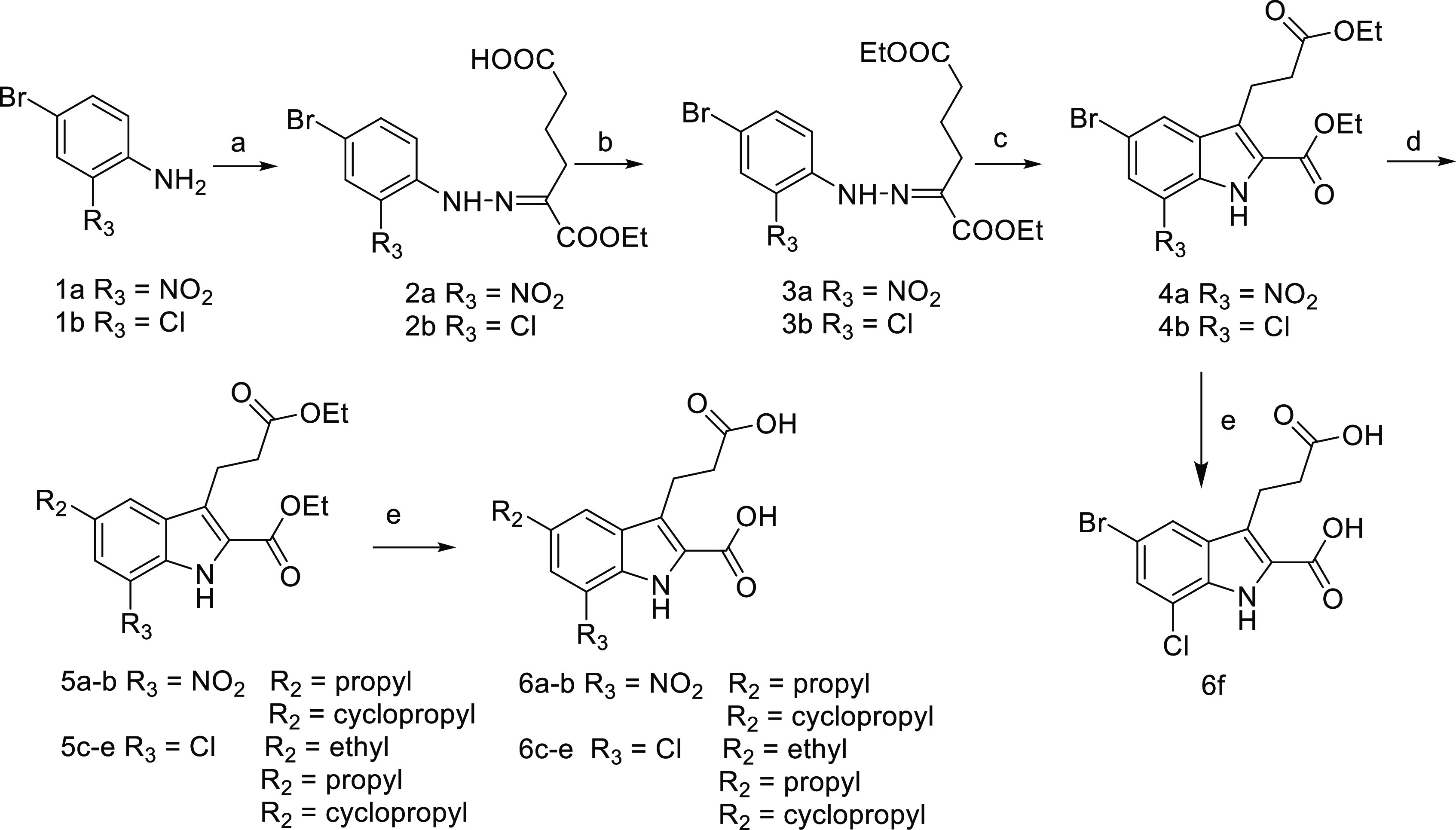
Reagents and conditions: (a) (i) NaNO2, HCl, H2O, 0 °C, (ii) NaOAc, ethyl-2-oxocyclopentane-carboxylate, 15 °C, (iii) Na2CO3, H2O, reflux; (b) conc H2SO4, EtOH, reflux; (c) p-toluene sulfonic acid, 110 °C or PPA, 90 °C; (d) alkylboronic acid, Pd(OAc)2, t-Bu3P·HBF4, K3PO4(aq), toluene, 90 °C; (e) NaOH, THF, EtOH and H2O, rt.
Scheme 3.
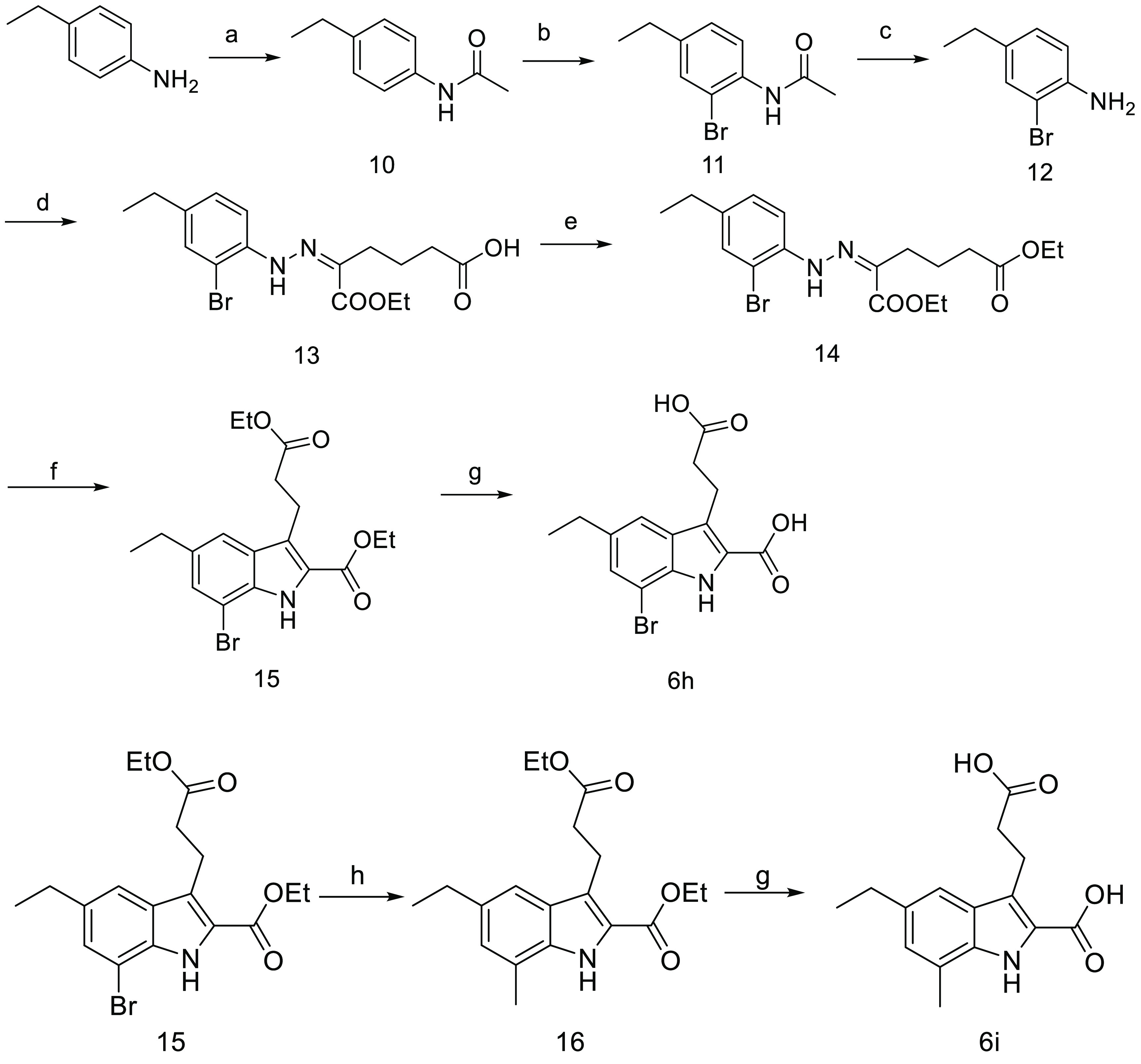
Reagents and conditions: (a) (CH3CO)2O, anhydrous pyridine, EA, rt; (b) Br2, HOAc, 45 °C; (c) KOH, H2O, EtOH, 80 °C; (d) (i) NaNO2, HCl, H2O, 0 °C, (ii) NaOAc, ethyl-2-oxocyclopentane-carboxylate, 15 °C, (iii) Na2CO3, H2O, reflux; (e) conc H2SO4, EtOH, reflux; (f) p-toluene sulfonic acid, 110 °C; (g) NaOH, THF, EtOH, and H2O, rt; (h) CH3B(OH)2, Pd(OAc)2, tricyclohexylphosphonium tetrafluoroborate, K3PO4(aq), toluene, 90 °C.
As shown in Scheme 2, the target molecule 6g was readily prepared starting from compound 7, which was reported in our previous work.29,30 The reduction of nitro group in compound 7 followed by acetylation and hydrolysis produced compound 6g.
Scheme 2.
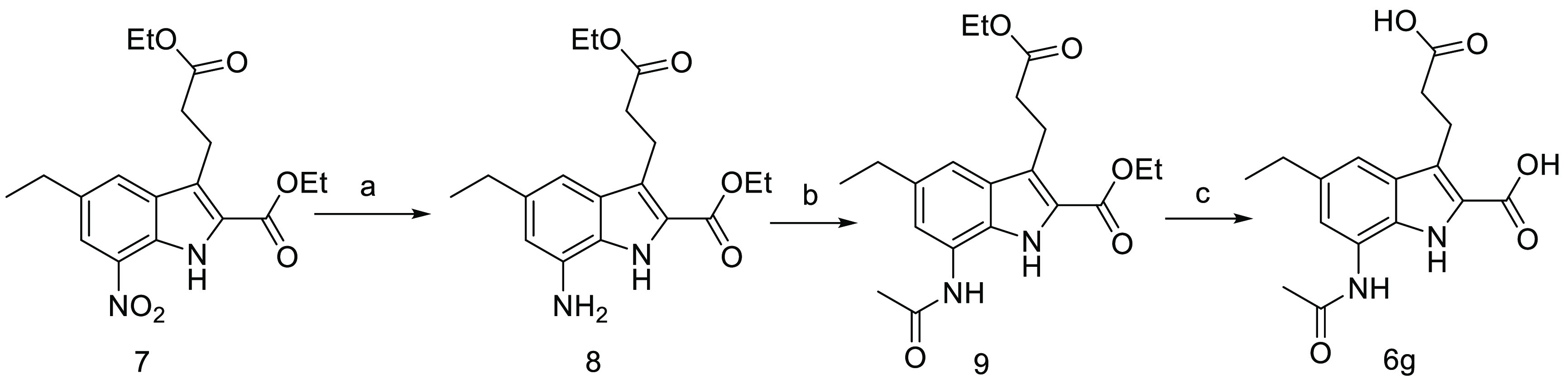
Reagents and conditions: (a) Fe, HOAc, 40 °C; (b) (CH3CO)2O, anhydrous pyridine, rt; (c) NaOH, THF, EtOH and H2O, rt.
The synthesis of 7-bromo and 7-methyl substituted compounds (6h and 6i) was outlined in Scheme 3. In this synthetic route, 4-ethyl aniline as the starting material was successfully converted into the key 2-bromo-4-ethyl aniline 12 upon acetylation, bromination, and deacetylation. Then, by following the same synthetic approach as described in Scheme 1, compound 12 was transformed into the target molecule 6h smoothly. Compound 6i was achieved by consecutive Suzuki coupling of compound 15 with methyl boronic acid and hydrolysis in 55% total yield.
The amide and acyl sulfonamide derivatives 22a–22i were synthesized in a concise manner as shown in Scheme 4. Starting from N-acetyl 4-ethyl aniline, nitration and deacetylation successfully delivered the key compound 18, which was transformed into the desired intermediate 20 by the Japp–Klingemann reaction and Fisher indole synthesis. It is noteworthy to mention that the Fisher indole cyclization was achieved in PPA at 80 °C without esterification of the carboxylic acid group on the 3-position, and therefore this free carboxylic group could couple with amines or sulfonamides to afford compounds 21a–21i (34–81% yields), which were hydrolyzed to give the target compounds 22a–22i.
Scheme 4.
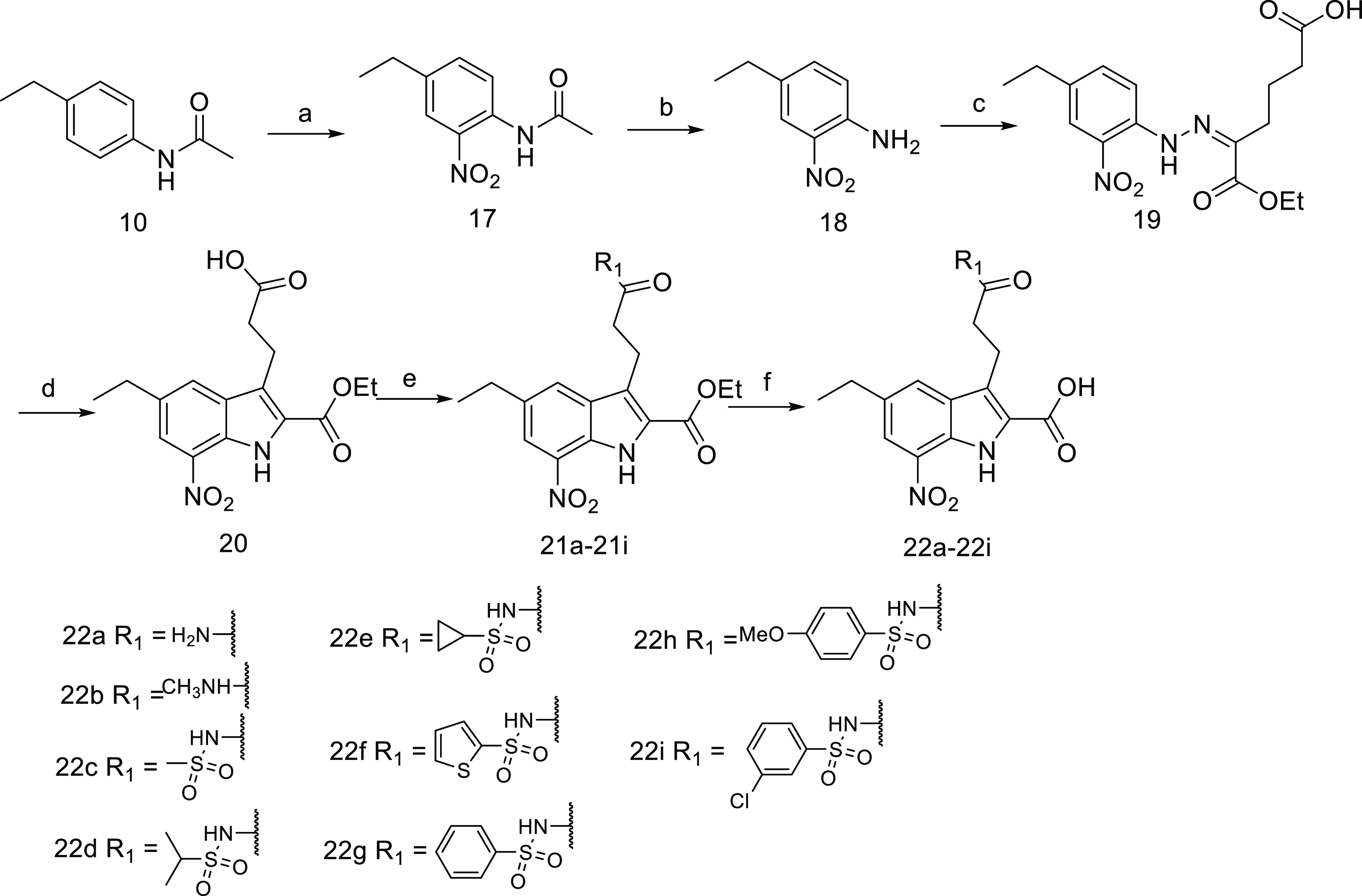
Reagents and conditions: (a) (CH3CO)2O, HOAc, HNO3, rt; (b) KOH, H2O, EtOH, 80 °C; (c) (i) NaNO2, HCl, H2O, 0 °C, (ii) NaOAc, ethyl-2-oxocyclopentane-carboxylate, 15 °C, (iii) Na2CO3, H2O, reflux; (d) PPA, 80 °C; (e) sulfonamide (R′SO2NH2) or amine (R′NH2), HATU, DMAP, Et3N, DCM, rt; (f) NaOH, THF, EtOH, and H2O, rt.
The benzofuran derivatives were constructed according to the approach depicted in Scheme 5. In the presence of K2CO3, the alkylation of substituted phenol 23 with ethyl bromoacetate produced compound 24 in 62% yield. Upon treatment with DBU, compound 24 cyclized into benzofuran derivative 25 with a yield of 52%. Bromination of 3-methyl group on benzofuran ring using NBS efficiently generated compound 26 (95% yield), which further reacted with diethyl malonate to afford the key intermediate 27 in 54% yield. Under the Suzuki–Miyaura reaction conditions, compound 27 coupled with ethyl boronic acid to give the desired product 28a and a debromo byproduct 28b in 17% and 21% yields, respectively. In the presence of HCl and HOAc, compounds 27, 28a, and 28b were subjected to hydrolysis and decarboxylation to afford benzofuran target molecules 29a–29c with high yields.
Scheme 5.
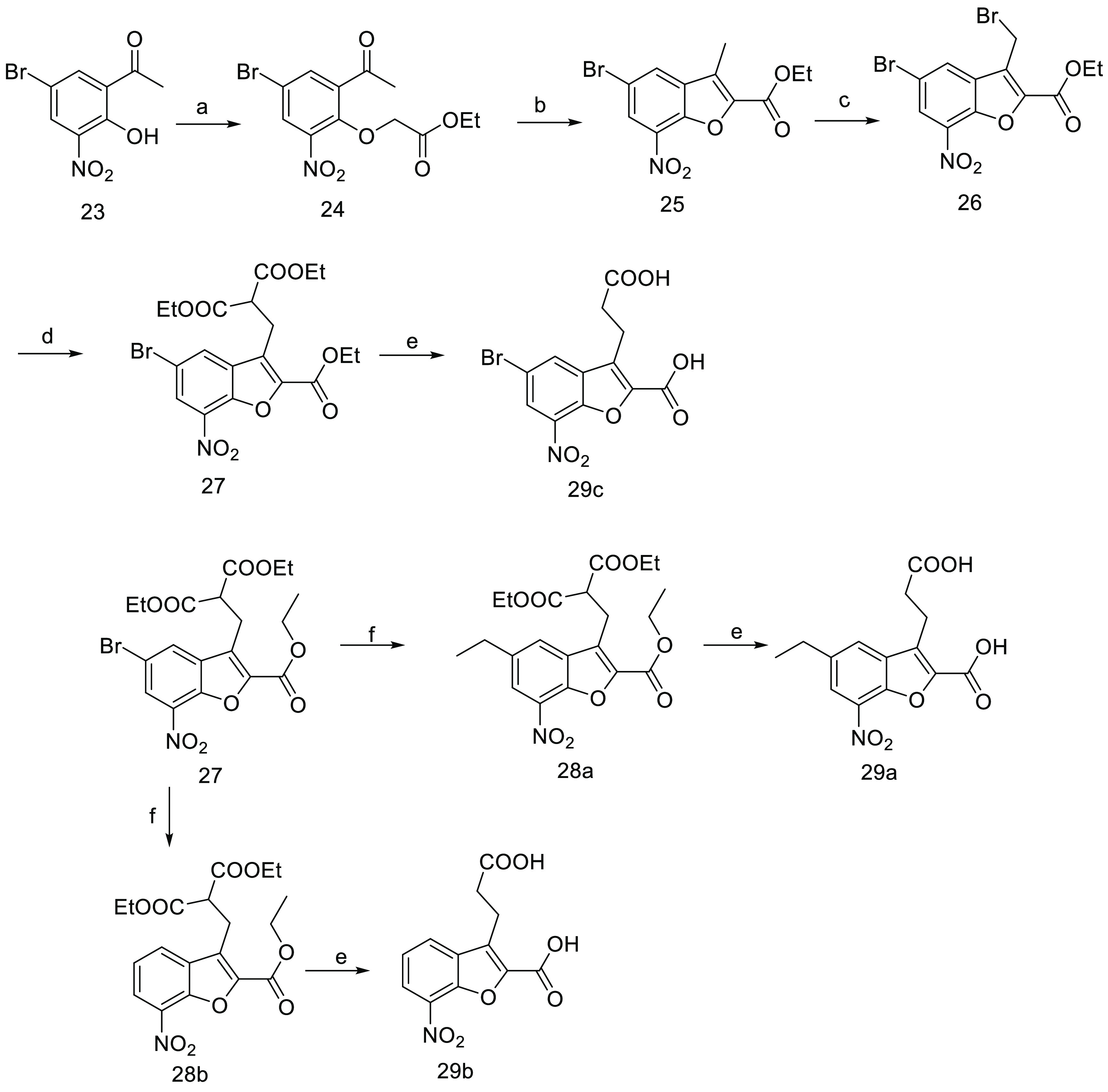
Reagents and conditions: (a) BrCH2COOC2H5, K2CO3, acetone, DMF, 60 °C; (b) DBU, toluene, 80 °C; (c) NBS, AIBN, CCl4, 90 °C; (d) (C2H5OCO)2CH2, NaH, DMF, THF, 70 °C; (e) HCl, HOAc, H2O, 100 °C; (f) C2H5B(OH)2, Pd(OAc)2, t-Bu3PHFP4, K3PO4(aq), toluene, 90 °C.
All target compounds (6a–6i, 22a–22i, 29a–29c) were tested for their inhibitory activities against recombinant human FBPase according to the method in our previous work.29,30 The results were summarized in Tables 1–3. MB05032 and AMP were used as reference molecules.
Table 1. Chemical Structures and Inhibitory Activities against FBPase of Compounds 6a–6fa.

| compd | R2 | R3 | IC50 (μM) |
|---|---|---|---|
| compd A | ethyl | NO2 | 0.10 |
| 6a | propyl | NO2 | 0.42 ± 0.9 |
| 6b | cyclopropyl | NO2 | 0.47 ± 0.5 |
| 6c | ethyl | Cl | 1.90 ± 0.2 |
| 6d | propyl | Cl | 1.30 ± 1.4 |
| 6e | cyclopropyl | Cl | 1.40 ± 0.3 |
| 6f | bromo | Cl | 36.4%b |
AMP and MB05032 were used as reference molecules. IC50 for AMP was 3.3 ± 0.1 μM. IC50 for MB05032 was 0.044 ± 0.012 μM.
The percentage inhibition at the concentration of 10 μM of tested compounds.
Table 3. Chemical Structures and Inhibitory Activities against FBPase of Compounds 22a–22ia.


AMP and MB05032 were used as reference molecules. IC50 for AMP was 3.3 ± 0.1 μM. IC50 for MB05032 was 0.044 ± 0.012 μM.
As shown in Table 1, the placement of ethyl, propyl, and cyclopropyl groups on the 5-position of indole ring resulted in the comparable FBPase inhibitions in either 7-chloro series or 7-nitro series of derivatives, while the incorporation of a bromine atom led to a marked reduction in potency. These results suggested that alkyl groups were favorable for this position, and presumably they were located in a hydrophobic pocket. Compared with 7-chloro substituted target compounds (6c–6e), their 7-nitro substituted counterparts (compound A, 6a, and 6b) exhibited more potent inhibitory activity, with IC50 values ranging from 0.10–0.47 μM. It was indicated that 7-substituents on the indole scaffold play an important role in the binding with FBPase. Therefore, further variations were accomplished on the 7-position as shown in Table 2. When an acetylamino or a methyl moiety was utilized as substitutes for the nitro or chloro group, the inhibitory activity of compounds 6g and 6i was completely lost. Compound 6h with a bromine atom also showed weak inhibition as well. Taken together, the nitro group was the most favorable fragment on the 7-position, presumably due to its electron-withdrawing ability and/or the capability of acting as an H-bond acceptor.
Table 2. Chemical Structures and Inhibitory Activities against FBPase of Compounds 6d–6ia.

AMP and MB05032 were used as reference molecules. IC50 for AMP was 3.3 ± 0.1 μM. IC50 for MB05032 was 0.044 ± 0.012 μM.
The percentage inhibition at the concentration of 10 μM of tested compounds.
Generally recognized, molecules with more than two carboxylic acid groups might bring about poor permeability. In our previous work, we found that 2-carboxylic acid was critical for the binding affinity by mimicking the phosphate group of AMP and the carboxylic group on the side chain extended into a solvent exposure region. Therefore, we tentatively replaced this carboxylic group with a bioisosteric group, and a number of amides and acyl sulfonamides were prepared. As shown in Table 3, all of the compounds exhibited inhibitory activities with IC50 values ranging from 0.50 to 5.94 μM. In comparison with amide analogues (22a–22b), the acyl sulfonamide derivatives (22c–22i) had stronger inhibition against FBPase, and among which, benzenesulfonamide and thiophene sulfonamide derivatives were the most potent inhibitors (22f and 22g), with IC50 values of 0.66 and 0.50 μM, respectively. These results suggested that acyl sulfonamide derivatives could serve as novel FBPase inhibitors.
Three benzofuran derivatives (29a–29c, Scheme 5) were also constructed by keeping the characteristic groups on the 2-, 3-, and 7-positions of the scaffold. Interestingly, all three compounds, bearing a hydrogen atom, a bromine atom, or an ethyl group on the 5-position, had no inhibitory effects at all, suggesting that the NH group on the indole ring made a critical interaction with FBPase.
Collectively, the 7-nitro group and the indole NH moiety were vital for the binding affinity, probably constructing critical H-bond interactions with the key amino acids. An alkyl substituent on the 5-position was beneficial to the inhibition. The acyl sulfonamide fragment was a favorable isostere for the carboxylic group on the side chain, offering a new opportunity to discover potent and drug-like FBPase inhibitors.
To reveal the binding behavior of indole-2-carboxylic acid derivatives, we cocrystallized small molecules with FBPase, obtaining three FBPase-inhibitor complexes, including *14b(30) (Figure 1), 6c (this work), and 22f (this work).
In general, the newly solved FBPase structures presented as a homotetramer, and one subunit bound to one inhibitor in the AMP allosteric site and one fructose-1,6-bisphosphate in the substrate site (Figure 2). Three inhibitors were oriented with a similar binding pose (Figure 3a,c,e,g). The indole scaffold was situated in the purine pocket and interacted with Glu20, Ala24, and Leu30 via hydrophobic interactions. The 2-COOH constructed the extensive hydrogen bond interactions with Thr27, Gly28, and Thr31 via partly mimicking the phosphate group of AMP and contributed significantly to the binding affinity. The indole NH formed an H-bond with the side chain OH group of Thr31, and this H-bond is critical as well, as the corresponding benzofuran analogue lost activity completely. In addition, the 7-NO2 was able to form a hydrogen bond with Thr31, while the 7-Cl was trapped in a hydrophobic cavity lined with Val17, Leu30, Leu34, and Met177. This result further supported that incorporation of an H-bond acceptor on the 7-position would be desirable for enhancing the binding affinity, and this was consistent with the SAR results. The 5-alkyl fragments interacted with Met177 and Cys179 via hydrophobic interactions, and that was an additional binding feature for this class of inhibitors. Of note, as observed from Figure 3b,d,f, the bound same inhibitors in four monomers were overlapped, and their detailed binding orientations of 3-substituents were somewhat different, although these side chains extended into the similar solvent exposure region. The side chains built up interactions with Lys112, Tyr113, and/or Arg140 to simulate the ribose moiety of AMP. We speculated that this phenomenon was the result of flexibility of both 3-subsituents and the side chain of Lys112, Tyr113, and Arg140. In addition, the acyl sulfonamide side chain preferred to interact with Lys112, Tyr113, and Arg140 via the cation−π or H-bond interactions, providing the structural basis for its application to replace the ribose or the acidic moiety to improve the drug-like property.
Figure 2.
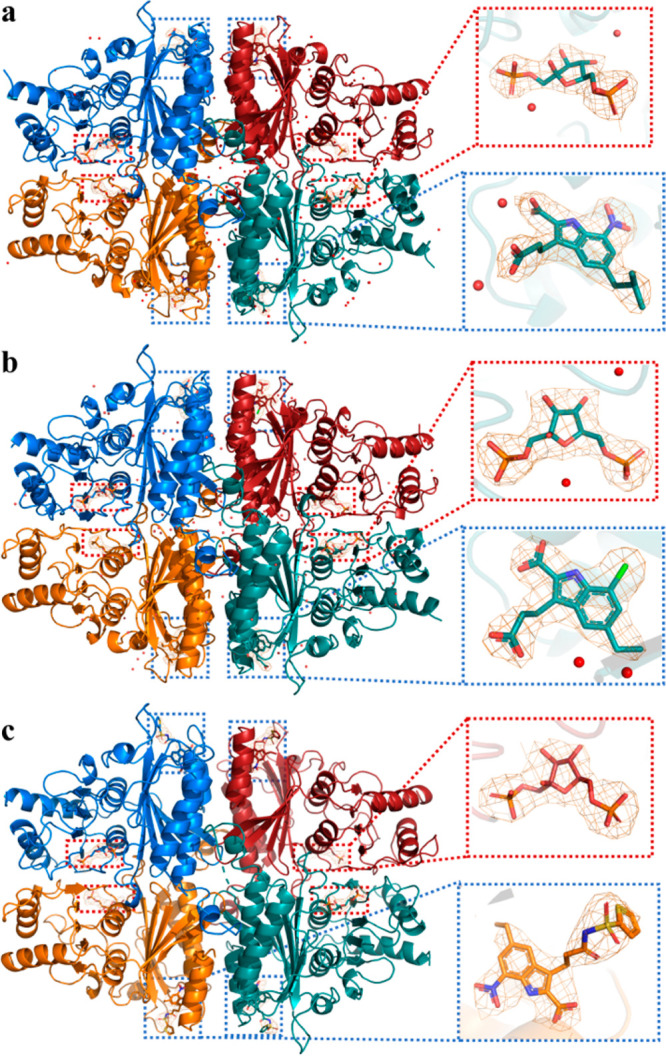
Overviews and the ligand close-views of solved FBPase crystal structures: (a) *14b (PDB 7EZP), (b) 6c (PDB 7EZF), (c) 22f (PDB 7EZR). The 2Fo–Fc electron density maps were contoured at 1σ.
Figure 3.

Detailed noncovalent interactions of receptor–ligand and binding pose comparison: (a) *14b; (b) 3-position side chain of *14b; (c) 6c; (d) 3-position side chain of 6c; (e) 22f; (f) 3-position side chain of 22f. (a–f) Marine for chain A, orange for chain B, firebrick for chain C, deep teal for chain D, green dash for hydrogen bond, magentas dash for hydrophobic interaction, blue dash for water hydrogen bond, and olive dash for salt bridge. (g) The superimposition of PDB: firebrick for AMP (PDB 1FTA, chain A), deep teal for *14b (PDB 7EZP, chain A), orange for 6c (PDB 7EZF, chain B), and marine for 22f (PDB 7EZR, chain A). The images were generated with PyMol-open-source.
To further compare the binding mode profiles, PLEC interaction fingerprint31 and Tanimoto factor were applied to generate similarities for the solved protein complexes in this work and those from PDB database, and all small molecules harbored in PDB were targeting AMP allosteric site.
In general, the binding modes of all PDB structures are approximately classified into two clusters (Figure 4), namely, cluster 1 containing PDB from 2VT5 to 2FIE, and cluster 2 including PDB from 7EZP (*14b) to 3A29. Notably, the ligands (SI, Figure S-1a) in cluster 1 bind to part of AMP allosteric site and the interface between two subunits; in contrast, the ligands (SI, Figure S-1b) in cluster 2 only occupy the AMP allosteric site. Obviously, the binding modes in cluster 1 are markedly different from those in cluster 2, suggesting that capitalizing on different binding subpockets is an efficient approach to generate structurally diversified inhibitors with distinct binding modes.
Figure 4.

Clustered heatmap of crystal structures from this work and Protein Data Bank.
Compounds *14b, 22f, and 6c were all bound to AMP allosteric site and belong to cluster 2 (Figure 4; SI, Figure S-1b). Interestingly, the detailed binding features of compound 6c are somewhat different from those of compounds *14b and 22f, even though they are in the same cluster. Compound 6c with a 7-chloro atom could form hydrophobic interactions with Val17 and Met177 and presents more similar binding interactions to the tricyclic inhibitors (3KC0, 3KC1, 3KBZ, 3A29).22,32 Nonetheless, compounds *14b and 22f with a 7-NO2 group tend to interact via hydrogen bonds. Of note, compared with that of AMP, more hydrophobic interactions were involved in the binding of these three inhibitors due to the fact that the 5-alkyl groups on the indole ring are able to create interactions with Met177 and Cys179. These results provide some insights for the discovering of nonphosphate-based inhibitors with high potency and drug-like properties by fully taking advantage of the AMP binding pocket.
Overall, two remarkably distinct binding modes were identified for the known FBPase inhibitors according to the PLEC interaction fingerprint, and they exploited different binding subpockets and represented two distinctive pharmacophores. The indole-2-carboxylic acid derivatives showed significantly dissimilar binding behavior to the dual-site inhibitors (cluster 1) and resembled the AMP in the binding mode (cluster 2). The variation on the indole-2-carboxylic acid scaffold to rationally explore the AMP pocket could lead to promising FBPase inhibitors.
In summary, on the basis of compound A and the known SAR of the indole-based FBPase inhibitors, a series of new indole analogues and benzofuran derivatives were synthesized and evaluated against FBPase. The SAR results demonstrated that the incorporation of an H-bond acceptor on the 7-position and an alkyl group on the 5-postion of the indole ring would benefit the inhibition. The bioisosteric replacement of carboxylic acid moiety on the 3-position with N-acyl sulfonamide group achieved new FBPase inhibitors with IC50 values at the submicromolar levels. Three FBPase complex structures provided informative evidence for the SAR results. Two key interactions could be recognized: the hydrogen bonds formed by the indole NH, 2-COOH, and 7-NO2 group, and the hydrophobic interactions of the indole ring and 5-alkyl substituent. The N-acyl sulfonamide side chain extended into a solvent exposure region, and that could be used to improve the drug-like properties. Furthermore, the chemoinformatics analysis by using PLEC further outlined the binding modes and features of the known FBPase inhibitors. Altogether, this work provided new chemical template and structural insights for developing diversified FBPase inhibitors with improved potency and drug-like properties.
Acknowledgments
We appreciate the X-ray crystallography facility platform at the National Protein Research Facility Base (Tsinghua) and Shanghai Synchrotron Radiation Facility for the support in the protein crystallographic experiment. Financial support from National Natural Science Foundation of China (81502933), Beijing Natural Science Foundation (no. 7132138), CAMS Initiative for Innovative Medicine (CAMS-I2M-2-004, 2020-I2M-1-003), and Beijing Outstanding Young Scientist Program (BJJWZYJH01201910023028) is gratefully acknowledged.
Glossary
Abbreviations
- AMP
adenosine-5-monophosphate
- GNG
gluconeogenesis
- HGP
hepatic glucose production
- PLEC
protein–ligand extended connectivity
- PDB
Protein Data Bank
- SAR
structure activity relationship
- T2DM
type 2 diabetes mellitus
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00613.
Experimental procedures and spectra data for intermediates and target compounds; the 1H NMR and 13C NMR spectra for the target compounds; the enzymatic assay for evaluation of FBPase activity; data collection and refinement statistics of cocrystal structures; ligand structures in cocrystal structures of FBPase and their interactions with key amino acids; the IC50 curves of target compounds (PDF). (PDF)
PDB coordinates of the cocrystal structure 7EZF (CIF)
PDB coordinates of the cocrystal structure 7EZP (CIF)
PDB coordinates of the cocrystal structure 7EZR (CIF)
Accession Codes
The PDB coordinates of the cocrystal structures (7EZF, 7EZP, and 7EZR) will be released by Protein Data Bank when this work is published.
Author Contributions
All authors have given approval to the final version of the manuscript. Xiaoyu Wang and Rui Zhao contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- IDF Diabetes Altlas; International Diabetes Federation, 2021; https://diabetesatlas.org (accessed 2021-11-02).
- Yang W. Y.; Lu J. M.; Weng J. P.; Jia W. P.; Ji L. N.; Xiao J. Z.; Shan Z. Y.; Liu J.; Tian H. M.; Ji Q. H.; Zhu D. L.; Ge J. P.; Lin L. X.; Chen L.; Guo X. H.; Zhao Z. G.; Li Q.; Zhou Z. G.; Shan G. L.; He J. Prevalence of Diabetes among Men and Women in China. N. Engl. J. Med. 2010, 362, 1090–1101. 10.1056/NEJMoa0908292. [DOI] [PubMed] [Google Scholar]
- Bonadonna R. C.; Cucinotta D.; Fedele D.; Riccardi G.; Tiengo A. The Metabolic Syndrome is a Risk Indicator of Microvascular and Macrovascular Complications in Diabetes: Results from Metascreen, a Multicenter Diabetes Clinic-based Survey. Diabetes Care 2006, 29 (12), 2701–2707. 10.2337/dc06-0942. [DOI] [PubMed] [Google Scholar]
- Diabetic Retinopathy Guidelines; Scientific Department, The Royal College of Ophthalmologists, 2012; https://www.rcophth.ac.uk/wp-content/uploads/2014/12/2013-SCI-301-FINAL-DR-GUIDELINES-DEC-2012-updated-July-2013.pdf (accessed 2021-11-02).
- Ma Z. J.; Chen R.; Ren H. Z.; Guo X.; Chen J. G.; Chen L. M. Endothelial Nitric Oxide Synthase (eNOS) 4b/a Polymorphism and the Risk of Diabetic Nephropathy in Type 2 Diabetes Mellitus: A Meta-Analysis. Meta Gene 2014, 2, 50–62. 10.1016/j.mgene.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A. D.; Langenberg C.; Rapsomaniki E.; Denaxas S.; Pujades-Rodriguez M.; Gale C. P.; Deanfield J.; Smeeth L.; Timmis A.; Hemingway H. Type 2 Diabetes and Incidence of Cardiovascular Diseases: A Cohort Study in 1.9 Million People. Lancet Diabetes Endocrinol. 2015, 3 (2), 105–113. 10.1016/S2213-8587(14)70219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau B. R.; Wahren J.; Chandramouli V.; Schumann W. C.; Ekberg K.; Kalhan S. C. Contributions of Gluconeogenesis to Glucose Production in the Fasted State. J. Clin. Invest. 1996, 98 (2), 378–385. 10.1172/JCI118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson I.; Rothman D. L.; Katz L. D.; Shulman R. G.; Shulman G. I. Increased Rate of Gluconeogenesis in Type II Diabetes Mellitus. A 13C Nuclear Magnetic Resonance Study. J. Clin. Invest. 1992, 90 (4), 1323–1327. 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrikopoulos S.; Proietto J. The Biochemical Basis of Increased Hepatic Glucose Production in a Mouse Model of Type 2 (Non-Insulin-Dependent) Diabetes Mellitus. Diabetologia 1995, 38, 1389–1396. 10.1007/BF00400598. [DOI] [PubMed] [Google Scholar]
- Kebede M.; Favaloro J.; Gunton J. E.; Laybutt D. R.; Shaw M.; Wong N.; Fam B. C.; Aston-Mourney K.; Rantzau C.; Zulli A.; Proietto J.; Andrikopoulos S. Fructose-1,6-Bisphosphatase Overexpression in Pancreatic β-cells Results in Reduced Insulin Secretion: A New Mechanism for Fat-Induced Impairment of β-cell Function. Diabetes 2008, 57 (7), 1887–1895. 10.2337/db07-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Poelje P. D.; Dang Q.; Erion M. D. Fructose-1,6-Bisphosphatase as a Therapeutic Target for Type 2 Diabetes. Drug Discovery Today: Ther. Strategies 2007, 4 (2), 103–109. 10.1016/j.ddstr.2007.10.003. [DOI] [Google Scholar]
- Li Z. M.; Bie J. B.; Song H. R.; Xu B. L. Recent Advance in the Discovery of Allosteric Inhibitors Binding to the AMP Site of Fructose-1,6-bisphosphatase. Acta Pharm. Sin. 2011, 46 (11), 1291–1300. [PubMed] [Google Scholar]
- Zhou J.; Bie J. B.; Wang X. Y.; Liu Q.; Li R. C.; Chen H. L.; Hu J. P.; Cao H.; Ji W. M.; Li Y.; Liu S. N.; Shen Z. F.; Xu B. L. Discovery of N-Arylsulfonyl-Indole-2-Carboxamide Derivatives as Potent, Selective, and Orally Bioavailable Fructose-1,6-Bisphosphatase Inhibitors—Design, Synthesis, In Vivo Glucose Lowering Effects, and X-ray Crystal Complex Analysis. J. Med. Chem. 2020, 63 (18), 10307–10329. 10.1021/acs.jmedchem.0c00726. [DOI] [PubMed] [Google Scholar]
- Kaur R.; Dahiya L.; Kumar M. Fructose-1,6-Bisphosphatase Inhibitors: A New Valid Approach for Management of Type 2 Diabetes Mellitus. Eur. J. Med. Chem. 2017, 141 (1), 473–505. 10.1016/j.ejmech.2017.09.029. [DOI] [PubMed] [Google Scholar]
- Dang Q.; Liu Y.; Cashion D. K.; Kasibhatla S. R.; Jiang T.; Taplin F.; Jacintho J. D.; Li H.; Sun Z. L.; Fan Y.; DaRe J.; Tian F.; Li W. Y.; Gibson T.; Lemus R.; van Poelje P. D.; Potter S. C.; Erion M. D. Discovery of a Series of Phosphonic Acid-Containing Thiazoles and Orally Bioavailable Diamide Prodrugs That Lower Glucose in Diabetic Animals Through Inhibition of Fructose-1,6-Bisphosphatase. J. Med. Chem. 2011, 54 (1), 153–165. 10.1021/jm101035x. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov; U.S. National Institutes of Health, 2021; https://clinicaltrials.gov/ (accessed 2021-11-30).
- van Poelje P. D.; Potter S. C.; Erion M. D.. Fructose-1,6-bisphosphatase Inhibitors for Reducing Excessive Endogenous Glucose Production in Type 2 Diabetes. In Diabetes–Perspectives in Drug Therapy; Handbook of Experimental Pharmacology; Schwanstecher M., Eds.; Springer, 2011; Vol. 203, pp 279–301 10.1007/978-3-642-17214-4_12. [DOI] [PubMed] [Google Scholar]
- Ke H. M.; Zhang Y. P.; Lipscomb W. N. Crystal Structure of Fructose-1,6-Bisphosphatase Complexed with Fructose-6-Phosphate, AMP, and Magnesium. Proc. Natl. Acad. Sci. U. S. A. 1990, 87 (14), 5243–5247. 10.1073/pnas.87.14.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng S.; Harris K. M.; Kantrowitz E. R. Designing Inhibitors against Fructose-1,6-Bisphosphatase: Exploring Natural Products for Novel Inhibitor Scaffolds. Eur. J. Med. Chem. 2010, 45 (4), 1478–1484. 10.1016/j.ejmech.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng S.; Gryncel K. R.; Kantrowitz E. R. A Library of Novel Allosteric Inhibitors against Fructose-1,6-Bisphosphatase. Bioorg. Med. Chem. 2009, 17 (11), 3916–3922. 10.1016/j.bmc.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnitskaya A.; Borkin D. A.; Huynh K.; Török B.; Stieglitz K. Rational Design, Synthesis, and Potency of N-Substituted Indoles, Pyrroles, and Triarylpyrazoles as Potential Fructose-1,6-Bisphosphatase Inhibitors. ChemMedChem. 2010, 5 (3), 384–389. 10.1002/cmdc.200900493. [DOI] [PubMed] [Google Scholar]
- Tsukada T.; Takahashi M.; Takemoto T.; Kanno O.; Yamane T.; Kawamura S.; Nishi T. Synthesis, SAR, and X-ray Structure of Tricyclic Compounds as Potent FBPase Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19 (20), 5909–5912. 10.1016/j.bmcl.2009.08.081. [DOI] [PubMed] [Google Scholar]
- Erion M. D.; van Poelje P. D.; Dang Q.; Kasibhatla S. R.; Potter S. C.; Reddy M. R.; Reddy K. R.; Jiang T.; Lipscomb W. N. MB06322 (CS-917): A Potent and Selective Inhibitor of Fructose-1,6-Bisphosphatase for Controlling Gluconeogenesis in Type 2 Diabetes. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (22), 7970–7975. 10.1073/pnas.0502983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. Y.; Wei L.; Han X. Y.; Chen H. F.; Ren Y. L.; Xu Y. H.; Song R. R.; Rao L.; Su C.; Peng C.; Feng L. L.; Wan J. Discovery of Novel Allosteric Site and Covalent Inhibitors of FBPase with Potent Hypoglycemic Effects. Eur. J. Med. Chem. 2019, 184 (15), 111749–111762. 10.1016/j.ejmech.2019.111749. [DOI] [PubMed] [Google Scholar]
- Huang Y. Y.; Xu Y. X.; Song R. R.; Ni S. S.; Liu J. Q.; Xu Y. H.; Ren Y. L.; Rao L.; Wang Y. J.; Wei L.; Feng L. L.; Su C.; Peng C.; Li J.; Wan J. Identification of the New Covalent Allosteric Binding Site of Fructose-1,6-Bisphosphatase with Disulfiram Derivatives toward Glucose Reduction. J. Med. Chem. 2020, 63 (11), 6238–6247. 10.1021/acs.jmedchem.0c00699. [DOI] [PubMed] [Google Scholar]
- Xu Y. X.; Huang Y. Y.; Song R. R.; Ren Y. L.; Chen X.; Zhang C.; Mao F.; Li X. K.; Zhu J.; Ni S. S.; Wan J.; Li J. Development of Disulfide-Derived Fructose-1,6-Bisphosphatase (FBPase) Covalent Inhibitors for the Treatment of Type 2 Diabetes. Eur. J. Med. Chem. 2020, 203, 112500–112518. 10.1016/j.ejmech.2020.112500. [DOI] [PubMed] [Google Scholar]
- Bie J. B.; Wang X. Y.; Liu S. N.; Zhou J.; Liu Q.; Shen Z. F.; Xu B. L. Design, Synthesis, Biological Evaluation and Binding Mode Analysis of 7-Nitro-indole-N-acylarylsulfonamide-based Fructose-1,6-Bisphosphatase Inhibitors. Chin. J. Med. Chem. 2020, 30 (12), 713–724. 10.14142/j.cnki.cn21-1313/r.2020.12.001. [DOI] [Google Scholar]
- Hu J. P.; Zhou J.; Sun Y. H.; Tan H. X.; Wang F. H.; Liu S. N.; Xu B. L.; Li Y.; Shen Z. F. Quantitative Determination of Cpd118, a Novel FBPase Inhibitor, in Dog Plasma by HPLC–MS/MS. Bioanalysis 2021, 13 (11), 865–873. 10.4155/bio-2021-0035. [DOI] [PubMed] [Google Scholar]
- Bie J. B.; Liu S. N.; Zhou J.; Xu B. L.; Shen Z. F. Design, Synthesis and Biological Evaluation of 7-Nitro-1H-Indole-2-Carboxylic Acid Derivatives as Allosteric Inhibitors of Fructose-1,6-Bisphosphatase. Bioorg. Med. Chem. 2014, 22 (6), 1850–1862. 10.1016/j.bmc.2014.01.047. [DOI] [PubMed] [Google Scholar]
- Bie J. B.; Liu S. N.; Li Z. M.; Mu Y. Z.; Xu B. L.; Shen Z. F. Discovery of Novel Indole Derivatives as Allosteric Inhibitors of Fructose-1,6-Bisphosphatase. Eur. J. Med. Chem. 2015, 90, 394–405. 10.1016/j.ejmech.2014.11.049. [DOI] [PubMed] [Google Scholar]
- Wójcikowski M.; Kukiełka M.; Stepniewska-Dziubinska M. M.; Siedlecki P. Development of a Protein–Ligand Extended Connectivity (PLEC) Fingerprint and Its Application for Binding Affinity Predictions. Bioinformatics 2019, 35 (8), 1334–1341. 10.1093/bioinformatics/bty757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada T.; Takahashi M.; Takemoto T.; Kanno O.; Yamane T.; Kawamura S.; Nishi T. Structure-Based Drug Design of Tricyclic 8H-Indeno[1,2-d][1,3]Thiazoles as Potent FBPase Inhibitors. Bioorg. Med. Chem. Lett. 2010, 20 (3), 1004–1007. 10.1016/j.bmcl.2009.12.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.