

Abstract
CD8+ T cells can potentiate long-lived immunity against COVID-19. We screened longitudinally-sampled convalescent human donors against SARS-CoV-2 tetramers and identified a participant with an immunodominant response against residues 322 to 311 of nucleocapsid (Nuc322–331), a peptide conserved in all variants of concern reported to date. We conducted 38-parameter cytometry by time of flight on tetramer-identified Nuc322–331–specific CD8+ T cells and on CD4+ and CD8+ T cells recognizing the entire nucleocapsid and spike proteins, and took 32 serological measurements. We discovered a coordination of the Nuc322–331–specific CD8+ T response with both the CD4+ T cell and Ab pillars of adaptive immunity. Over the approximately six month period of convalescence monitored, we observed a slow and progressive decrease in the activation state and polyfunctionality of Nuc322–331–specific CD8+ T cells, accompanied by an increase in their lymph node–homing and homeostatic proliferation potential. These results suggest that following a typical case of mild COVID-19, SARS-CoV-2–specific CD8+ T cells not only persist but continuously differentiate in a coordinated fashion well into convalescence into a state characteristic of long-lived, self-renewing memory.
The uncertainty about the longevity of the immune response elicited by prior SARS-CoV-2 infection or vaccination has been a major area of concern as the world tries to exit from the ongoing COVID-19 pandemic. Studies at the start of the pandemic that suggested a short-lived SARS-CoV-2 Ab response (1) brought about widespread concern, but follow-up studies now suggest that infected individuals exhibit a prolonged and evolving humoral immune response (2, 3). Furthermore, SARS-CoV-2–specific memory T cells, a second arm of adaptive immunity, can be detected more than six months into convalescence, and these cells can self-renew in response to the homeostatic proliferation cytokine IL-7 (4–6). Encouragingly, memory T cells against the nucleocapsid protein from the closely related SARS-CoV-1 virus can be detected 17 years postinfection (7), suggesting the potential for durable T cell immunity against pathogenic β-coronaviruses. Importantly, relative to Abs, T cells are less prone to evasion by the variants of concern emerging worldwide (A. Tarke, J. Sidney, N. Methot, Y. Zhang, J. Dan, B. Goodwin, P. Rubiro, A. Sutherland, R. da Silva Antunes, A. Frazier, et al., manuscript posted on bioRxiv, DOI: 10.1101/2021.02.27.433180), suggesting a potentially important role for these immune effectors in long-term population-based immunity in the years ahead.
Characterizing the memory T cells responding to SARS-CoV-2 will improve our understanding of the features defining long-lived immunity and of the ability of T cells to protect against reinfection. Although the breadth of the SARS-CoV-2–specific response during convalescence has been extensively examined (8, 9), much less is known about the phenotypes of SARS-CoV-2–specific memory T cells. To phenotype SARS-CoV-2–specific T cells, most studies rely on stimulating T cells with SARS-CoV-2–specific Ags/peptides and examining the cells that respond by expressing activation-induced markers or cytokines (5, 8, 10–12). These studies likely underestimate the phenotypic complexity of Ag-specific T cells because of the limited number of activation-induced markers or cytokine markers that can be used to identify responsive cells. These assays are also limited because they do not capture Ag-specific T cells in their original, unstimulated states. Detecting Ag-specific unstimulated cells requires other, more technically-involved approaches, such as the use of T cell multimers/tetramers. Tetramers, which consist of four linked peptide-MHC complexes that specifically bind epitope-specific T cells, are one of the only ways to examine the original phenotypes of Ag-specific T cells. A handful of studies have incorporated the use of SARS-CoV-2 MHC class I multimers to examine CD8+ T cell responses (13–17). Because of small numbers of multimer+ cells of a single specificity, most of these studies examined the combined phenotypes of multimer+ cells recognizing different epitopes and/or pre-enriched for multimer+ cells (to increase detectability), which can bias the resulting collection of Ag-specific cells. One of the studies (16) conducted a longitudinal analysis of multimer+ cells from one patient at six time points (two during acute infection and four at convalescence) by examining by FACS the levels of five phenotyping parameters on pre-enriched multimer+ cells. Although these studies together have revealed multimer+ cells to be distributed among multiple canonical subsets and pinpointed a handful of surface markers expressed by these cells, the inability to identify enough epitope-specific cells for high-parameter phenotypic analysis has made it challenging to perform a comprehensive analysis of how SARS-CoV-2–specific CD8+ T cells against a defined specificity evolve over the course of convalescence.
To fill this void, we screened banked longitudinal specimens from the University of California San Francisco (UCSF) COVID-19 Host Immune Response Pathogenesis (CHIRP) cohort against a collection of SARS-CoV-2 tetramers to try to identify an immunodominant response that can be captured by tetramer analysis. This screen identified one individual with a particularly robust response detectable by one of the tetramers harboring a nucleocapsid peptide. The immunodominance of this response enabled us to perform a longitudinal analysis without the need to pre-enrich for tetramer+ cells or combine tetramer+ cells of multiple specificities. By combining 38-parameter cytometry by time of flight (CyTOF) phenotyping with detection of these tetramer+ cells, we established an in-depth view of epitope-specific T cell responses at five longitudinal time points from early to late (greater than six months) COVID-19 convalescence. Effector responses by these epitope-specific T cells were examined by treating cells with cognate peptide and examining by CyTOF the cytokine and cytolytic effector mechanisms of these cells. All longitudinal specimens were also phenotyped by the same effector CyTOF panel for total nucleocapsid- and spike-specific CD4+ and CD8+ T cell responses and assessed for the levels of 32 different isotype-specific SARS-CoV-2 Abs. Altogether, we measured nearly 400 different SARS-CoV-2–specific parameters for each of the five time points and analyzed them in association with features of the tetramer+ response to provide an integrated and comprehensive overview of the immunological context surrounding the epitope-specific CD8+ T cell response. Although this study focuses on only one individual, this person exhibited a typical mild course of infection that was very well defined clinically and that fully resolved in a timely manner. We therefore consider the response we have characterized to potentially reflect a common one in individuals that have recovered from mild COVID-19.
Materials and Methods
Human subjects
This study was approved by the UCSF (Institutional Review Board number 20–30588). Informed consent was obtained from all subjects. The study used specimens from the UCSF acute CHIRP longitudinal cohort. Five longitudinal specimens were collected from acute COVID-19–infected individuals, the first within 31 d of symptom onset or SARS-CoV-2 exposure (week 0, baseline visit), followed by collections at 1, 3, 10, and 24 wk from baseline visit. Whole blood was collected in EDTA tubes, and PBMCs were isolated by Ficoll, as previously described (5), and cryopreserved in 10% DMSO diluted in FBS. Plasma from the same specimens were collected and cryo-preserved. A total of 21 CHIRP participants with confirmed SARS-CoV-2 infection as assessed by RT-PCR were screened by FACS for specific binding to allophycocyanin-labeled MHC class I tetramers, as described in the following section. PID4103 was identified as a donor with an immunodominant response against the HLA-B*40:01/residues 322 to 311 of nucleocapsid (Nuc322–331) tetramer (Table I).
Table I.
PID4103 participant characteristics
| Patient Identifier | Gender | Age (y) | Date of Symptom Onset | Date of First PCR+ Test | Date of Blood Draw(s) | Time Between Symptom Onset and Analysis (d) | Time Between First PCR+ Test and Analysis (d) |
|---|---|---|---|---|---|---|---|
| PID4103 | Female | 42 | 3/13/20 | 04/09/20 | 04/29/20 (PCR+) | 46 | 20 |
| 05/06/20 (PCR−) | 53 | 27 | |||||
| 05/20/20 (PCR−) | 67 | 41 | |||||
| 07/08/20 (PCR−) | 117 | 90 | |||||
| 10/07/20 (PCR−) | 207 | 181 |
Flow cytometry
Cryopreserved PBMCs from 21 CHIRP participants were thawed and cultured overnight to enable Ag recovery and then screened by FACS for specific binding to allophycocyanin-labeled MHC class I tetramers (Supplemental Table I) obtained from the National Institutes of Health Tetramer Core Facility. These tetramers harbor predicted MHC class I epitopes from SARS-CoV-2. For tetramer staining, 1 million cells were transferred into 96-well, V-bottom polystyrene plates and washed once with FACS buffer (PBS supplemented with 2% FBS and 2 mM EDTA), stained with 15 μg/ml allophycocyanin-labeled MHC class I tetramer for 1 h at room temperature. Cells were then washed twice with FACS buffer and stained for 30 min on ice with a mixture of Abs diluted in a 1:1 mixture of FACS buffer and the Brilliant Stain Buffer (BD Biosciences). The Ab mixture consisted of allophycocyanin/Cy7-CD3 (SK7; BioLegend), BD Horizon BV650-CD8 (RPA-T8; BD Biosciences), BD Horizon BUV737-CD4 (SK3; BD Biosciences), and the LIVE/DEAD Zombie Aqua Fixable Viability Kit (BioLegend). After three additional washes with FACS buffer, cells were fixed and analyzed on an LSRFortessa (BD Biosciences).
Tetramerization of biotinylated MHC class I monomers with metal-labeled streptavidin
Streptavidin was labeled with metal, as previously described (18). Biotinylated HLA-B*40:01 monomers with SARS-CoV-2 Nuc322–331 were obtained from the National Institutes of Health Tetramer Core Facility. Tetramerization was performed as previously described (18). Briefly, the biotinylated monomers and metal-labeled streptavidin were each diluted to 50 μg/ml in PBS. A total of 10 μl of metal-labeled streptavidin was then transferred to 100 μl of peptide–MHC class I monomer. The solution was then mixed and incubated for 10 min at room temperature. After repeating the process twice (resulting in a total of 30 μl of metal-labeled streptavidin being transferred to 100 μl of the monomer solution), CyFACS buffer (metal contaminant-free PBS [Rockland] supplemented with 0.1% BSA and 0.1% sodium azide) was added to reach a final volume of 500 μl. For each specimen containing up to 6 × 106 cells, 100 μl of metal-labeled tetramer was used.
Preparation of specimens for CyTOF
Cryopreserved PBMCs were thawed and cultured overnight. Baseline specimens were stained directly in the absence of ex vivo stimulation. For identification of Ag-specific T cells through intracellular cytokine staining, 6 × 106 cells were stimulated with 0.5 mg/ml anti-CD49d clone L25 (BD Biosciences) and 0.5 mg/ml anti-CD28 clone L293 (BD Biosciences) in the absence or presence of peptides for 4 h in RP10 media (RPMI 1640 [Corning] supplemented with 10% FBS [VWR International], 1% penicillin [Life Technologies], and 1% streptomycin [Life Technologies]) in the presence of 3 μg/ml brefeldin A solution (eBioscience). The peptides used were 1 mM PepMix SARS-CoV-2 peptide (spike glycoprotein) (JPT Peptide Technologies), 1 mM PepMix SARS-CoV-2 Peptide (NCAP) (JPT Peptide Technologies), or 1 mM PepMix SARS-CoV-2 Peptide (MEVTPSGTWL) (custom synthesized by JPT Peptide Technologies).
CyTOF staining
We designed a 38-parameter CyTOF panel that allows assessment of the phenotypes, differentiation states, effector functions, and activation status of T cells, as well as homing receptors and transcription factors (see Table II). Abs that required in-house conjugation were conjugated to their corresponding metal isotopes using X8 Antibody Labeling Kits according to manufacturer’s instructions (Fluidigm). For CyTOF staining, 6 × 106 cells were blocked for 15 min on ice with sera from mouse (Thermo Fisher Scientific), rat (Thermo Fisher Scientific), and human (AB serum; Sigma-Aldrich) in Nunc 96 DeepWell Polystyrene Plates (Thermo Fisher Scientific). Cells were washed twice with CyFACS buffer, then stained with tetramer for 1 h at room temperature in the presence of 50 nM dasatinib (Sprycel) to reduce TCR internalization and improve tetramer staining. Cells were then washed twice with CyFACS buffer and stained for 45 min on ice with the mixture of CyTOF surface staining Abs (see Table II) in a total volume of 100 μl/well. Cells were then washed three times with CyFACS buffer and stained with Maleimide DOTA (Macrocyclics) for 30 min on ice. Cells were then washed twice with CyFACS and fixed overnight at 4°C with 2% paraformaldehyde (Electron Microscopy Sciences) in metal contaminant-free PBS (Rockland). On the next day, cells were permeabilized by incubation for 30 min at 4°C with Intracellular Fixation & Permeabilization Buffer (eBioscience) and then washed twice with Permeabilization Buffer (eBioscience). Cells were then blocked for 15 min on ice with sera from mouse (Thermo Fisher Scientific) and rat (Thermo Fisher Scientific). Cells were then washed twice with Permeabilization Buffer (eBioscience) and stained for 45 min on ice with the mixture of CyTOF intracellular staining Abs (see Table II). Cells were next washed once with CyFACS and incubated for 20 min at room temperature with 250 nM Cell-ID DNA Intercalator-Ir (Fluidigm) in 2% paraformaldehyde diluted in PBS. As a final step, cells were washed twice with CyFACS, once with Maxpar Cell Staining Buffer (Fluidigm), once with Maxpar PBS (Fluidigm), and once with Maxpar Cell Acquisition Solution (Fluidigm). Immediately prior to acquisition, cells were resuspended to a concentration of 7 × 105/ml in EQ Four Element Calibration Beads (Fluidigm) diluted in Maxpar Cell Acquisition Solution. Cells were acquired at a rate of 250–350 events/sec on a CyTOF2 instrument (Fluidigm) at the UCSF Flow Cytometry Core (Table II).
Table II.
List of CyTOF staining Abs
| Ab | Metal Label | Clone | Vendor |
|---|---|---|---|
| CD49d (α4) | 141Pr | 9F10 | Fluidigm |
| CD19 | 142Nd | HIB19 | Fluidigm |
| CCR5 | 144Nd | NP6G4 | Fluidigm |
| CD8 | 146Nd | RPAT8 | Fluidigm |
| CD7 | 147Sm | CD76B7 | Fluidigm |
| ICOS | 148Nd | C398.4A | Fluidigm |
| CCR4 | 149Sm | L291H4 | Fluidigm |
| CD62L | 153Eu | DREG56 | Fluidigm |
| TIGIT | 154Sm | MBSA43 | Fluidigm |
| CCR6 | 155Gd | G034E3 | In-house |
| CD29 (β1) | 156Gd | TS2/16 | Fluidigm |
| OX40 | 158Gd | ACT35 | Fluidigm |
| CCR7 | 159Tb | G043H7 | Fluidigm |
| CD28 | 160Gd | CD28.2 | Fluidigm |
| CD45RO | 161Dy | UCHL1 | In-house |
| CD69 | 162Dy | FN50 | Fluidigm |
| Tetramer | 163Dy | N/A | In-house |
| PD1 | 164Dy | EH12.1 | In-house |
| CD127 | 165Ho | A019D5 | Fluidigm |
| CXCR5 | 166Er | RF8B2 | In-house |
| CD27 | 167Er | L128 | Fluidigm |
| CD45RA | 169Tm | HI100 | Fluidigm |
| CD3 | 170Er | UCHT1 | Fluidigm |
| CD57 | 171Yb | HCD57 | In-house |
| CD38 | 172Yb | HIT2 | Fluidigm |
| Tetramer | 173Yb | N/A | In-house |
| CD4 | 174Yb | SK3 | Fluidigm |
| CD25 | 176Yb | M-A251 | In-house |
| HLADR | 112Cd | Tu36 | Invitrogen |
| NFAT1a | 143Nd | D43B1 | Fluidigm |
| BIRC5a | 145Nd | 91630 | In-house |
| TNF-αa | 150Nd | MAb11 | In-house |
| CD107aa | 151Eu | H4A3 | Fluidigm |
| Granzyme Ba | 152Sm | O94E6/GZMB | In-house |
| CTLA4a | 157Gd | 14D3 | In-house |
| IFN-γa | 168Er | B27 | Fluidigm |
| Perforina | 175Lu | B-D48 | Fluidigm |
| IL-6a | 209Bi | MQ2–13A5 | In-house |
Intracellular Abs.
CyTOF data analysis
The CyTOF datasets were normalized to EQ Four Element Calibration Beads using Fluidigm’s CyTOF Software, exported as Flow Cytometry Standard files, and imported into FlowJo (BD Biosciences) and Cytobank for gating and downstream analysis. Total T cells were identified using a sequential gating strategy based on DNA content, viability, cell length, and a CD3+CD19− gate. SARS-CoV-2–specific T cells were then identified by subgating on CD4+ or CD8+ T cells, as appropriate, followed by tetramer+ or IFN-γ+ gating. FlowJo (BD Biosciences) was used for gating, generation of histogram plots, and mapping of defined populations onto the t-distributed stochastic neighbor embedding (tSNE) plots. Cytobank was used to calculate the median signal intensity of cell populations based on standard two-dimensional dot plots. Cytobank was also used to generate tSNE plots and FlowSOM plots using the default settings (with a modification of total metaclusters from 10 to 5 for FlowSOM analysis). All of the phenotyping markers were used in tSNE and FlowSOM analysis, except for CD19, which was a parameter used in the upstream gating strategy. Line graphs were generated using ggplot2 in R. The raw CyTOF datasets generated from this study are available for download through the public repository Dryad via the following link: https://doi.org/10.7272/Q6D21VVD.
Serology
The flow cytometry–based serological assay, based off previously validated methods (19), was used to quantitate the relative levels of IgA1, IgA2, IgE, IgG1, IgG2, IgG3, IgG4, and IgM against the nucleocapsid and various domains of the spike protein (S1, S2, and the receptor binding domain [RBD] domain of S1). This assay for measuring serum Ab levels uses biotin-conjugated Ags coupled to streptavidin-coated microspheres (beads). Incubation of Ag–bead complexes with patient sera and subsequent staining by fluorescently conjugated, isotype-specific Abs produces a flow cytometric readout of bead fluorescence that reveals the levels of Ag-specific Abs and their isotypes. The assay was calibrated using mouse monoclonal (IgG2B) Abs raised against the RBD/S1/S2/NP Ags. The calibration revealed high specificity and no cross-reactivity between Ags, with the exception of cross-reactivity of anti-RBD Abs against S1, which was expected, as RBD is contained within S1. To assess isotype usage of RBD/S1/S2/NP–specific Abs from PID4103, we incubated Ag-coated beads with plasma from the five time point specimens of PID4103 (1:2 dilution in HBSS plus 0.1% BSA media). Nonspecific Ab binding was assessed by incubation of plasma with uncoated (Ag-free) beads. The beads were washed with HBSS supplemented with 0.1% BSA and then stained with an isotype-specific, fluorescently conjugated mixture of Abs at a concentration empirically determined to have minimal background binding to both Ag-coated and uncoated beads. Processed beads were analyzed using a BD FACSymphony flow cytometer. Raw values were normalized by subtraction of nonspecific signal as determined by the signal from the Ag-free beads. The dotted lines (Fig. 2H) mark low-confidence signals where the mean fluorescence intensity difference between specific and nonspecific signal was less than 100 U.
FIGURE 2.
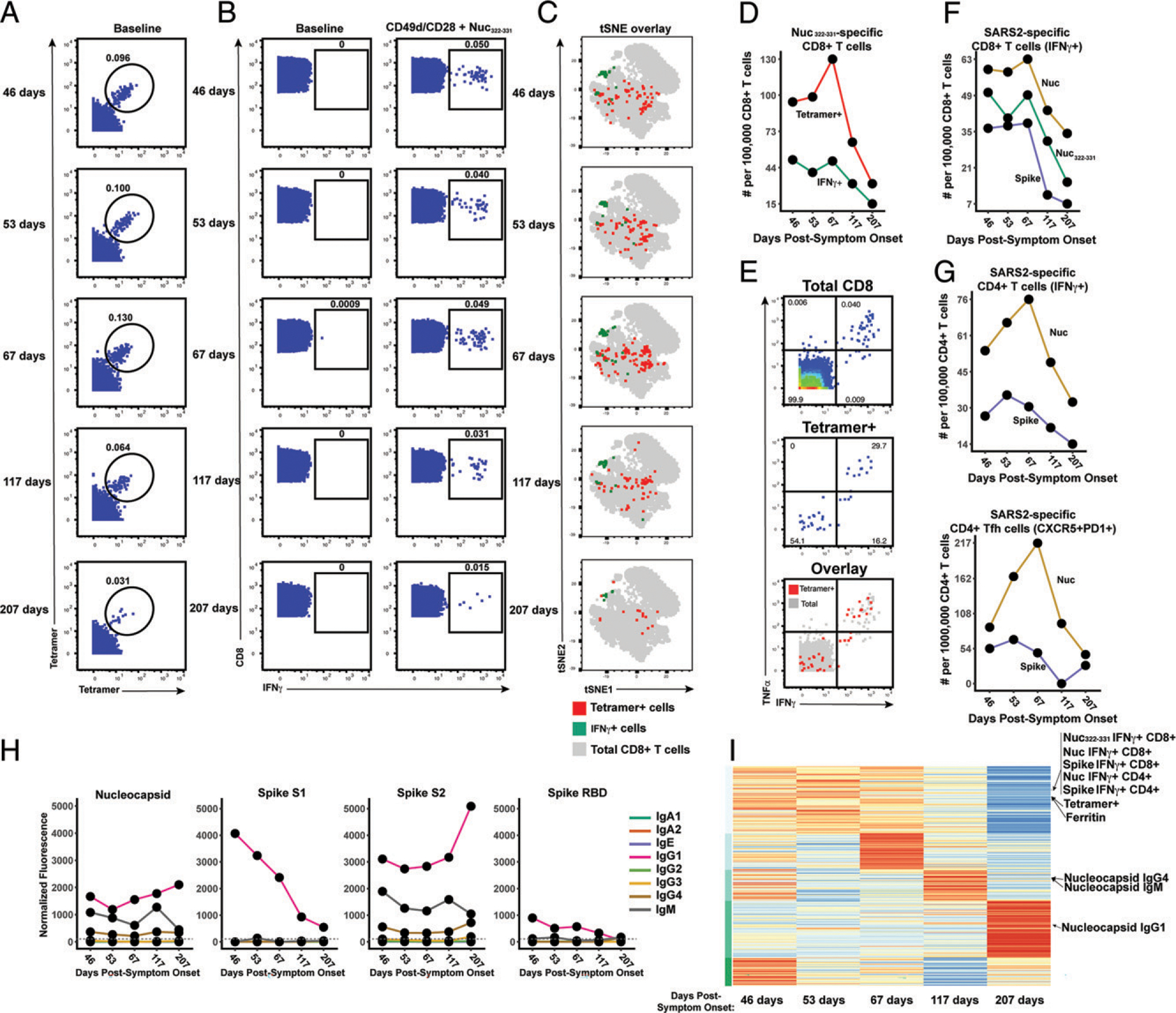
Longitudinal assessment of Nuc322–331–specific CD8+ T cell responses in PID4103 reveals coordination with other components of Ag-specific adaptive immunity. (A) Identification of Nuc322–331–specific CD8+ T cells by CyTOF. Baseline specimens that never underwent any stimulation were stained with HLA-B*40:01/Nuc322–331 tetramers detectable on two different CyTOF channels. The timeline refers to days since symptom onset. Numbers correspond to the percentage of cells within the gate. Results are gated on live, singlet CD3+CD8+ cells. (B) CD8+ T cells specifically producing IFN-γ in response to Nuc322–331 stimulation were detected at all five timepoints. Numbers correspond to the percentage of cells within the gate. Results are gated on live, singlet CD3+CD8+ cells. (C) Tetramer+ and IFN-γ+ cells responding to Nuc322–331 treatment reside in unique regions of the tSNE, suggesting phenotypic changes elicited by cognate peptide recognition. tSNE plots of total CD8+ T cells (gray), tetramer+ (red) from the baseline samples, and IFN-γ+ (green) cells from the peptide-stimulated samples over the course of convalescence of PID4103. Datasets correspond to those extracted from the data presented in (A) and (B). (D) The tetramer+ response is higher in magnitude than the IFN-γ+ response but exhibits similar kinetics, peaking 67 d postsymptom onset. Datasets correspond to those extracted from the data presented in (A) and (B). (E) Approximately half of tetramer+ cells in Nuc322–331–stimulated samples do not secrete IFN-γ or TNF-α. PBMCs from PID4103 were stimulated with Nuc322–331, stained with HLA-B*40:01/Nuc322–331 tetramers, and analyzed by CyTOF. A total of 54.1% of tetramer+ cells expressed neither IFN-γ nor TNF-α, suggesting that approximately half of tetramer+ cells are not identified using the cytokine secretion assay. (F) The responses of CD8+ T cell to Nuc322–331, the entire nucleocapsid protein (Nuc), and the entire spike protein are coordinated. Note that the IFN-γ+ response to Nuc322–331 is greater than the response to the entire spike proteins and less than the response to the entire nucleocapsid protein. (G) The total and Tfh CD4+ T cell responses against nucleocapsid peaks 67 d postsymptom onset, whereas the response to spike peaks slightly earlier. Total (left) or Tfh (CD4+CD45RO+CD45RA−PD1+CXCR5+) (right) CD4+ T cells responding to overlapping peptides spanning the entire nucleocapsid or spike proteins were assessed. (H) Titers of different Ab types against nucleocapsid, and the S1, S2, and RBD domains of spike monitored at the five timepoints and expressed as normalized fluorescence values (see Materials and Methods). The dotted line indicates the limit of detection. (I) Unsupervised k-means clustering of cells, Abs, and other biomarkers based on their abundance in PID4103’s blood across five time points. For each biomarker, abundance is normalized across time points and colored from red (highest) to blue (lowest). The CD4+ and CD8+ T cell against Nuc322–331, nucleocapsid, and spike clustered together. Interestingly, ferritin levels clustered close to them. In contrast, Ab responses against nucleocapsid were delayed and occurred after the peaks of the T cell responses. The green bars on the left correspond to clustering as determined by k-means.
k-means cell clustering based on CyTOF profile
We performed unsupervised cell clustering of all the measured parameters using a k-means algorithm implemented using the k-means function in R (https://wwww.R-project.org/). Each kind of measured parameter was centered using the mean of the corresponding levels across the five time points and scaled using the SD of the values before proceeding with unsupervised clustering. To find the optimal number of clusters, the gap statistic, a metric that evaluates clustering efficiency by comparing the sum of within-cluster distance from real data and null data (20), was implemented using the R library factoextra (https://CRAN.R-project.org/package=factoextra). The gap statistic was measured with k values ranging from 1 to 100. Because the gap statistic trend curve increased as k increased, the optimal k value was selected as that within 1 SE from the first local maximum. The optimal k value was determined to be 5, and the validity of cell subtypes was examined by visualizing measured parameter patterns using a heatmap generated using pheatmap (http://CRAN.R-project.org/package=pheatmap).
Results
Identification and description of case study PID4103 with immunodominant Nuc322–331 CD8+ T cell response
The CHIRP study is a prospective longitudinal study designed to understand the evolution of host responses from the acute to convalescent phases of SARS-CoV-2 infection. Individuals within 31 d of symptom onset or SARS-CoV-2 exposure were enrolled, and participants were sampled for 6 mo. Cryopreserved PBMCs from a total of 21 convalescent CHIRP cohort participants, from which specimens from five longitudinal study visits were available and who had mild COVID-19 disease (see Materials and Methods), were screened by flow cytometry against nine MHC class I tetramers harboring predicted CD8+ T cell epitopes from the spike and nucleocapsid proteins of SAR-CoV-2 (Supplemental Table I). We identified one donor, PID4103 (Table I), who harbored an immunodominant response against an HLA-B*40:01-restricted peptide (sequence MEVTPSGTWL) spanning the residues of Nuc322–331 (Fig. 1A). This peptide is 100% conserved in the B.1.1.7, B.1.351, P.1, and B429/CAL20C variants of concern, as well as in the 2002 SARS-CoV-1 virus, but absent from the four common-cold corona-viruses 229E, NL63, OC43, and HKU1 (Supplemental Fig. 1).
FIGURE 1.
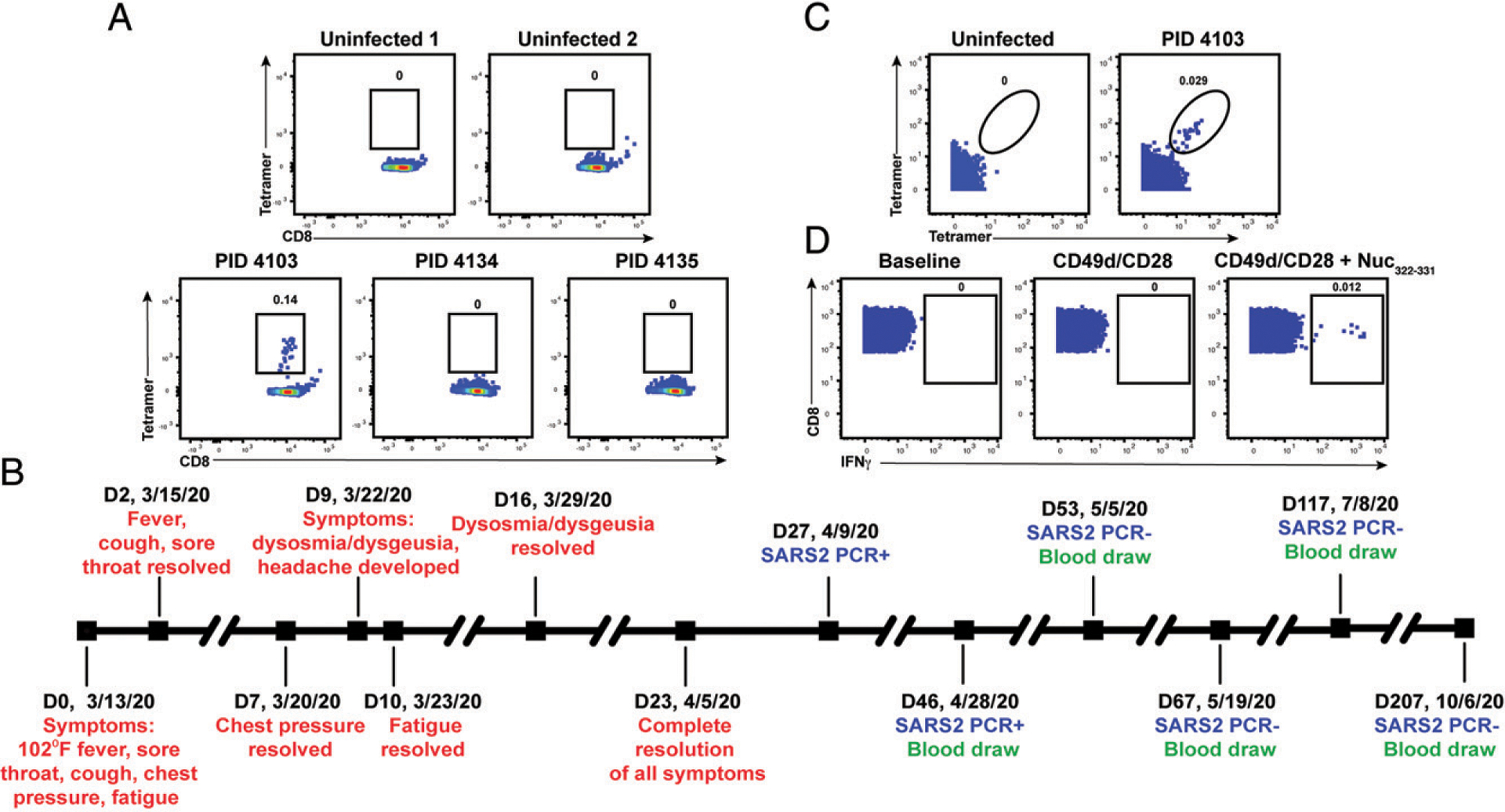
Identification and description of case study PID4103 with immunodominant CD8+ T cell response against Nuc322–331. (A) A distinct population of Nuc322–331–specific CD8+ T cells is detected by FACS tetramer staining in convalescent donor PID4103. Top, PBMCs from uninfected individuals were analyzed by FACS for binding to the HLA-B*40:01/Nuc322–331 tetramer. Results are representative of six independent uninfected donors. Bottom, PBMCs from convalescent COVID-19 individuals from the CHIRP cohort were analyzed by FACS for binding to the HLA-B*40:01/Nuc322–331 tetramer. Participant PID4103 but not participants PID4134 and PID4135 harbors cells binding to the tetramer. Numbers correspond to the percentage of cells within the gates. Results are gated on live, singlet CD3+CD8+ cells. (B) Timeline of clinical course of PID4103’s SARS-CoV-2 infection and sampling. Red indicates the dates of specific symptom initiation and resolution, blue indicates the dates and results of SARS-CoV-2 PCR tests, and green indicates the dates of blood draws. (C) A distinct population of Nuc322–331–specific CD8+ T cells is detected by CyTOF in PID4103 through dual-tetramer staining. PBMCs from one uninfected individual and from PID4103 were stained with two sets of HLA-B*40:01/Nuc322–331 tetramers conjugated to different metal lanthanides, facilitating specific detection of Nuc322–331–specific CD8+ T cells. Numbers correspond to the percentage of cells within the gates. Results are gated on live, singlet CD3+CD8+ cells. (D) Nuc322–331–specific CD8+ T cells can be stimulated by the Nuc322–331 peptide. PBMCs from PID4103 were phenotyped by CyTOF at baseline or following 4 h of costimulation with αCD49d/CD28 Ab in the absence or presence of the Nuc322–331 peptide. Stimulations were conducted in the presence of brefeldin A to enable the detection of IFN-γ. Numbers correspond to the percentage of cells within the gates. Results are gated on live, singlet CD3+CD8+ cells. The PID4103 specimens in (A) were obtained from the day 53 study visit, whereas those from (C) and (D) were obtained from the day 207 study visit.
We decided to focus our study on this participant for three reasons: 1) the presence of an immunodominant response detectable in her blood allowed us to have a sufficient number of cells to perform in-depth CyTOF phenotyping without combining multiple epitope specificities or pre-enrichment for tetramer+ cells, 2) the conservation of the immunodominant epitope among the variants of concern ensured that the memory response we studied would be relevant against the common globally circulating pathogenic strains, and 3) the patient reported symptoms of typical mild COVID-19 disease (as detailed below) and therefore can serve as a model for the typical course of disease experienced by most individuals who become infected with SARS-CoV-2 (21, 22).
Participant PID4103 is a 42-y-old white female whose course of SARS-CoV-2 infection has been extensively characterized (Fig. 1B, Materials and Methods). The participant began experiencing a constellation of mild acute symptoms on March 13, 2020, including a fever of 102°F, sore throat, cough, chest pressure, and fatigue. Fever, cough, and sore throat resolved 2 d later, whereas chest pressure resolved 7 d postsymptom onset. Nine days postsymptom onset, she developed dysosmia/dysgeusia and headache, which lasted approximately 1 wk. Complete resolution of all symptoms did not occur until 23 d from initial symptom onset. The participant tested positive by PCR for SARS-CoV-2 27 d postsymptom onset, and her clinical PCR result was confirmed by nasopharyngeal swab PCR at her baseline study visit 46 d postsymptom onset (407.5 and 161.5 copies/ml for N1 and N2 probes, respectively), which corresponded to 19 d after her first positive PCR test. The participant then attended follow-up visits at 1, 3, 10, and 23 wk after her baseline visit. At all the follow-up study visits, she tested negative by PCR for the virus in specimens from nasopharyngeal swabs, blood, stool, and urine.
PID4103 reported no limitations to her activities of daily living over the course of disease. She had no significant comorbidities other than a prior history of anxiety disorder and hypothyroidism for which she had previously received pharmacology therapy, and she had no concomitant medications during the study period. Her clinical laboratory tests by the time of her baseline visit (46 d postsymptom onset) were within normal limits. However, her ferritin levels, which have previously been shown to strongly correlate with COVID-19 symptoms (23), showed a downtrend over her five study visits (134, 120, 64, 56, 54 ng/ml). Her high sensitivity C-reactive protein (hs-CRP) and erythrocyte sedimentation rate levels were normal by the time she was enrolled in the study (<8.1 mg/l and <20 mm/h, respectively), suggesting lack of systemic inflammation at any of the study visits. PID4103 reported no history of travel in the prior year and no past history of travel that may have coincided with exposure to SARS-CoV-1 (e.g., no 2002 travel to Canada or Asia). Altogether, these clinical features suggest PID4103 to have exhibited a typical case of mild COVID-19 that resolved on its own without medical intervention and that did not result in any long-hauler symptoms.
CyTOF characterization of Nuc322–331–specific CD8+ T cells through tetramer staining and peptide stimulation
To enable a deep assessment of the phenotypes of Nuc322–331–specific CD8+ T cells, we generated lanthanide-conjugated Nuc322–331 tetramers. This allowed for characterization of Nuc322–331–specific CD8+ T cells by CyTOF, a technique that quantitates up to ~40 proteins simultaneously at the single-cell level through mass spectrometric detection of metal-conjugated Abs (24). To increase the noise-to-signal ratio and improve the specificity of detecting Nuc322–331–specific CD8+ T cells, we conjugated the same tetramer to two different lanthalides and considered only cells binding both sets of tetramers as true positives. A population of Nuc322–331–specific CD8+ T cells could be detected from PID4103 that was absent from uninfected individuals (Fig. 1C). To assess effector function, CD8+ T cells from PID4103 were examined by CyTOF (Table II) in the absence of any stimulation, in the presence of costimulation with anti-CD49d/CD28 for 4 h, and in the presence of costimulation with Nuc322–331 peptide under conditions that enabled detection of peptide-induced cytokines at the single-cell level (5). Only in peptide-stimulated samples did we observe a distinct population of IFN-γ–producing cells (Fig. 1D). These results collectively validate our ability to characterize Nuc322–331–specific CD8+ T cells by CyTOF through tetramer staining as well as through identification of Nuc322–331–specific CD8+ T cells responding to cognate peptide stimulation.
Longitudinal assessment of Nuc322–331–specific CD8+ T cells reveals coordination with other components of Ag-specific adaptive immunity
Having validated the specificity of our reagents, we then characterized by CyTOF the magnitude of the Nuc322–331–specific CD8+ T cell response over the course of convalescence using the PBMCs isolated from PID4103 over the five study visits (Fig. 1B). The magnitude of the response over time was monitored by quantitating both the frequencies of tetramer+ cells in unstimulated specimens (Fig. 2A) and the frequencies of cells inducing IFN-γ in response to Nuc322–331 stimulation (Fig. 2B). Both approaches enabled detection of Nuc322–331–specific CD8+ T cells at all five time points, including the final one, which was >6 mo after symptom onset. The overall phenotypes of the Nuc322–331–specific CD8+ T cells in the tetramer+ versus IFN-γ+ cells differed, as visualized by tSNE (Fig. 2C). This observation is not surprising because the tetramer+ cells are not stimulated, whereas the IFN-γ+ cells are. However, the majority of individually measured CyTOF markers were similar between the tetramer+ and IFN-γ+ cells, and only IFN-γ and TNF-α appeared upregulated on the IFN-γ+ cells relative to the tetramer+ cells (Supplemental Figs. 2, 3). The latter is consistent with the need for ex vivo stimulation to detect cytokine production. Although there were more tetramer+ cells than IFN-γ+ cells, their kinetics were similar over the time course, with a peak 67 d postsymptom onset (Fig. 2D). Our observation that there were approximately twice as many tetramer+ cells as IFN-γ+ cells suggests that approximately half the CD8+ T cells of a given specificity are not captured by the IFN-γ detection method. This was confirmed by examining the tetramer+ cells within the Nuc322–331–stimulated sample, half of which turned out to be IFN-γ+ (Fig. 2E). Interestingly, the majority of these IFN-γ+ cells also produced TNF-α (Fig. 2E), suggesting a polyfunctional response. Importantly, at its maximal peak, the frequency of the Nuc322–331–specific CD8+ T cells as determined by tetramer staining was 1.3 × 10−3 (Fig. 2A), confirming the immunodominance of this epitope because COVID-19 CD8+ T cells with the most dominant epitope reported to date were detected at an average frequency of 6.88 × 10−4 (17).
We then assessed what fraction of the total nucleocapsid-specific CD8+ T cells is accounted for by Nuc322–331–specific cells. To this end, we stimulated samples from all five time points with overlapping peptides covering the entire nucleocapsid protein (Supplemental Fig. 4A) and measured IFN-γ–responding cells by CyTOF. Both cell populations (Nuc322–331 and total nucleocapsid-specific CD8+ T cells) had similar kinetics, as expected (Fig. 2F), and at all five time points, the Nuc322–331–specific CD8+ T cells accounted for the bulk of the nucleocapsid-specific CD8+ T cells (Fig. 2F). For comparison, we also assessed the spike-specific CD8+ T cells from these specimens using overlapping peptides covering the entire spike protein (Supplemental Fig. 4A). These cells also mirrored the kinetics of the Nuc322–331–specific cells but, interestingly, were always less abundant (Fig. 2F). These data reaffirm the immunodominance of Nuc322–331–specific CD8+ T cells, which surpass even that of the spike-specific CD8+ T cells. Furthermore, they demonstrate that the kinetics of the CD8+ T cell response against nucleocapsid and spike in PID4103 is coordinated, peaking 67 d postsymptom onset and decreasing thereafter.
We then assessed to what extent the Nuc322–331-specific CD8+ T cell response is coordinated with the response of CD4+ T cells and Abs. When we assessed by CyTOF total and T follicular helper (Tfh) CD4+ T cells specific to nucleocapsid (Fig. 2G, Supplemental Fig. 4B), we found that both peaked at the third time point (67 d postsymptom onset), just like the Nuc322–331–specific CD8+ T cells did (Fig. 2F, 2G). When we measured eight isotypes (IgM, IgG1, IgG2, IgG3, IgG4, IgA1, IgA2, and IgE) of Abs against full-length nucleocapsid, we found that only IgM, IgG1, and IgG4 nucleocapsid-specific Abs above detectable limits, with IgG1 being the dominant response. All three isotypes of nucleocapsid-specific Abs increased from the third to the fourth time points (Fig. 2H). As a “helped” Ab response would be expected to develop only after a spike in a CD4+ Tfh response (which occurred at the third time point, Fig. 2G), these results suggest a coordinated T cell–dependent Ab response in this individual during convalescence. For comparison, we also assessed the CD4+ T cell and Ab response against spike. Spike-specific CD4+ T cells peaked slightly earlier than the Nuc322–331–specific CD8+ T cells (Fig. 2G). Abs against the spike N-terminal S1 and C-terminal S2 domains, as well as against the RBD domain of S1 that mediates binding to the ACE2 entry receptor, were quantitated. Interestingly, in contrast to the T cell data in which the response to nucleocapsid response dominated over the response to spike, the Ab response to spike was dominant over the response to nucleocapsid (Fig. 2H). Similar to the nucleocapsid data, the dominant Ab isotype against spike was IgG1. Interestingly, however, the Ab response to S1 and RBD progressively decreased over the course of convalescence, whereas the Ab response to S2 more closely mirrored the response to nucleocapsid, increasing from the third to the fourth time points (Fig. 2H). Taken together, these data suggest a synchronous increase in CD4+ and CD8+ T cells preceding the Ab response.
As a complementary way of examining the coordination between these different adaptive immune responses, we conducted an integrated analysis of all the SARS-CoV-2–specific T cell and Ab response measurements from our study. We included the frequencies of all the subsets of Nuc322–331–, Nuc-, and spike-specific T cells that we identified by manual gating as well as FlowSOM clustering (see subsequent sections); the median expression levels of each CyTOF-quantitated Ag on total CD8+ T cells, the tetramer+ cells, and all the CD4+ and CD8+ IFN-γ+ cells responding to Nuc322–331, nucleocapsid, or spike peptide treatments; all 32 Ab measurements (4 proteins times 8 isotypes); and the clinical laboratory measurements. This resulted in a matrix of 393 measured parameters for each of the five time points. When we conducted k-means unsupervised clustering to assess which parameters were closely related, we found the kinetic patterns of the CD4+ and CD8+ T cell responses to Nuc322–331, nucleocapsid, and spike to cluster together (Fig. 2I). Interestingly, the levels of ferritin, which were reported to positively correlate with COVID-19 symptoms (23), clustered right next to the magnitude of the tetramer+ cell response (Fig. 2I). As expected, the nucleocapsid Ab response clustered separately as it was delayed relative to the T cell response (Fig. 2I). Overall, these data suggest that the Nuc322–331–specific CD8+ T cell response is synchronized with CD4+ and CD8+ T cell responses against nucleocapsid and spike, followed by a boosting of the nucleocapsid and S2 IgG1, IgG4, and IgM Abs.
The phenotypes and potential for long-term persistence of Nuc322–331–specific CD8+ T cells evolve during convalescence
We then took advantage of the 38-parameter phenotyping of our tetramer+ cells to characterize the phenotypes of Nuc322–331–specific CD8+ T cells. CD8+ T central memory (Tcm), T effector memory (Tem), T transitional memory (Ttm), Temra, and a mixed population of naive and T stem cell memory cells (Tn/Tscm) were identified through use of various combinations of the phenotyping markers CD8, CD45RO, CD45RA, CCR7, and CD27 (Fig. 3A). In addition, we distinguished, among the memory (CD45RO+) CD8+ T cells, those that were less (CD27+CD28+) or more (CD27−CD28−) terminally differentiated (Fig. 3A). Terminal differentiation and expansion potential were also examined by monitoring expression of CD57 (terminal differentiation marker), CD27 (marker of long-lived cells), and CD127 (α-chain of IL-7R involved in homeostatic proliferation) (Fig. 3B). Cytolytic activity was assessed by monitoring expression of the effector molecules perforin and granzyme B, the degranulation marker CD107a, and CD29, which marks cells with cytolytic activity (25) (Fig. 3C). Tetramer+ cells could be detected among all the aforementioned populations, although in vastly different proportions (Fig. 3A–C).
FIGURE 3.
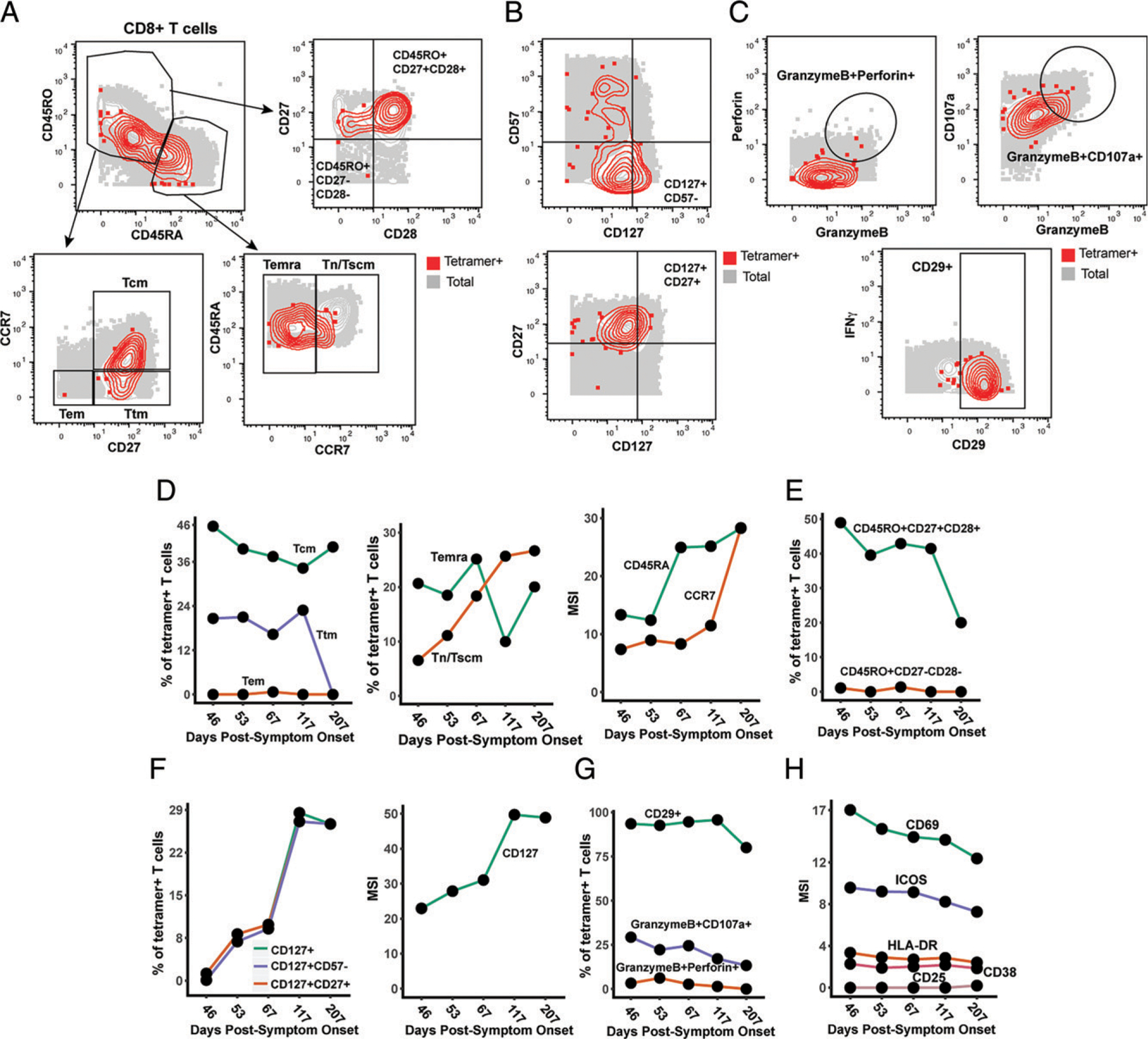
Nuc322–331–specific CD8+ T cells in PID4103 slowly differentiate over the course of convalescence to a less activated state more capable of expanding and migrating to lymph nodes. (A) Gating strategy to identify CD8+ Tcm, Tem, Ttm, Temra, and Tn/Tscm at early and late differentiation stages. The Nuc322–331–specific CD8+ T cells (tetramer+) cells are shown as red contours, whereas total CD8+ T cells are shown in gray. The following gates were used: Tcm (CD45RO+CD45RA−CD27+CCR7+), Ttm (CD45RO+CD45RA−CD27+CCR7−), Tem (CD45RO+CD45RA−CD27−CCR7−), Temra (CD45RO−CD45RA+CCR7−), Tn/Tscm (CD45RO−CD45RA+CCR7+), early differentiation (CD45RO+CD45RA−CD27+CD28+), and late differentiation (CD45RO+CD45RA−CD27−CD28−). (B) Gating strategy to identify different populations of CD127+ cells among total (gray) and Nuc322–331–specific (red) CD8+ T cells. Shown also are gates for less differentiated (CD57−) and Tcm-like (CD27+) CD127+ T cells. (C) Gating strategy to identify cytolytic Nuc322–331–specific CD8+ T cells. Top, Gates defining CD8+ T cells coexpressing granzyme B and perforin, or granzyme and CD107a. Bottom, Gate defining cells expressing high levels of CD29, a marker for cytolytic CD8+ T cells. (D) The proportions of tetramer+ cells belonging to the Tcm, Tem, Ttm, Temra, and Tn/Tscm subsets as defined in (A) are shown in the first two plots. Note the high contribution of Tcm at all timepoints and the progressive increase of the Tn/Tscm subset over time. The panel on the right displays the median expression levels of CD45RA and CCR7 [markers used to define the Tn/Tscm subset (A)] within the tetramer+ population. (E) Early differentiated CD8+ T cells steeply decline in abundance only at the final timepoint, 207 d postsymptom onset. Shown are the proportions of tetramer+ cells belonging to the early (CD45RO+CD27+CD28+) and late (CD45RO+CD27−CD28−)-differentiated subsets over the course of convalescence. (F) Progressive increase in CD127+ Nuc322–331–specific CD8+ T cells over an ~6 mo period of convalescence. Left, Proportions of tetramer+ cells that were CD127+, CD127+CD57−, and CD127+CD27+. The overlapping frequencies of the three populations of cells suggest that most of the CD127+ cells are CD57− and CD27+. Right, Median expression levels of CD127 within the tetramer+ population. (G) Cytolytic Nuc322–331–specific CD8+ T cells slowly decrease over the course of convalescence. The proportions of tetramer+ cells that were CD29+, granzymeB+CD107a+, and granzymeB+perforin+ are shown. (H) The activation state of Nuc322–331–specific CD8+ T cells generally decreases slowly over the course of convalescence. The median expression levels of the indicated activation markers on tetramer+ cells are shown. A gradual decrease was apparent among CD69, ICOS, HLADR, and CD38 but not CD25, whose expression was low at all time points.
We assessed the tetramer+ cells for relative changes in subset distribution over the ~6-mo period analyzed in this study. Among tetramer+ cells, Tem cells were at negligible frequencies throughout the time course, whereas Tcm cells were most common. Tetramer+ Ttm and Temra cells were also abundant, although the Ttm subset frequency dropped precipitously at the final time point. Interestingly, the contribution of the Tn/Tscm subset increased steadily over the course of convalescence, reaching the highest levels at the final time point (Fig. 3D). This increase parallels the increase in expression levels of CD45RA and CCR7 (markers used to define the Tn/Tscm subset) within the tetramer+ population (Fig. 3D). In terms of differentiation state, there was a progressive decrease over time of the early differentiated CD27+CD28+ memory T cell subset among tetramer+ cells (Fig. 3E). This was accompanied by a progressive increase in CD127 positivity, with the CD127+ cells residing almost exclusively among CD57− and CD27+ cells (Fig. 3F). Cytolytic tetramer+ cells decreased over time (Fig. 3G), and this was accompanied by a gradual decrease in the expression levels of some (CD69, ICOS, HLADR, CD38) but not all (CD25) activation markers on these cells (Fig. 3H). Together, these results suggest a continual differentiation of Nuc322–331–specific CD8+ T cells long after resolution of infection. These changes include the evolution of the cells to a state defined by less activation and cytolytic activity and more proliferative and expansion potential.
Clustering of high-dimensional datasets identifies features of convalescence-associated expanding cluster of Nuc322–331–specific CD8+ T cells
The subset identification based on manual gating described above uses only a small fraction of the phenotyping markers examined by our CyTOF panel. Visualization of the phenotypic distribution of the tetramer+ cells by tSNE suggests global changes in phenotypes over time (Fig. 4A) that may not have been captured through manual gating. We next, therefore, implemented a more holistic approach of subset definition by clustering. Total CD8+ T cells in the unstimulated specimens (including the tetramer+ cells) were segregated into five clusters by FlowSOM (26) (Fig. 4B, Supplemental Fig. 5). Although tetramer+ cells were detected among all five clusters, they were not evenly distributed, and the distribution also changed over time (Fig. 4C, 4D). Clusters A2 and A4 were the dominant clusters among tetramer+ cells, but interestingly, whereas cluster A2 increased over time, cluster A4 decreased (Fig. 4D). When we manually gated on a concatenated file corresponding to all of the cluster A2 and A4 cells (which together include most of the tetramer+ cells), we found that these cells collectively harbored Tcm, Tem, Ttm, Temra, and Tn/Tscm subsets (Fig. 4E), suggesting that the dominant population of tetramer+ cells cannot be binned into any single canonical cellular subset. To try to define features of clusters A2 plus A4, we assessed for Ags that were similarly differentially expressed on these two clusters as compared with total CD8+ T cells. Relative to total CD8+ T cells, A2 and A4 cells expressed high levels of CD127 and the transcription factor NFAT1, as well as high levels of the lung-homing molecules CD49d, CD29, and CCR5 (Fig. 4F). We then assessed for differentially expressed markers that were unique to clusters A2 versus A4, to assess the subset features that increase (cluster A2) versus decrease (cluster A4) over the course of convalescence. Cluster A2 expressed high levels of the lymph node–homing receptors CCR7 and CD62L, the checkpoint molecules TIGIT and CTLA4, the costimulatory receptors CD28 and Ox40, and the antiapoptotic protein BIRC5 (Fig. 4G). In comparison, cluster A4 expressed low levels of CCR7 and CD62L, high levels of the activation marker CD69, and high levels of the degranulation marker CD107a (Fig. 4H). These data are consistent with the manual gating results, suggesting a slow expansion of long-lived Nuc322–331–specific CD8+ T cells paired with a decrease in their cytolytic counterparts, but identify additional phenotypic features of the cellular subsets to which these cells belong.
FIGURE 4.
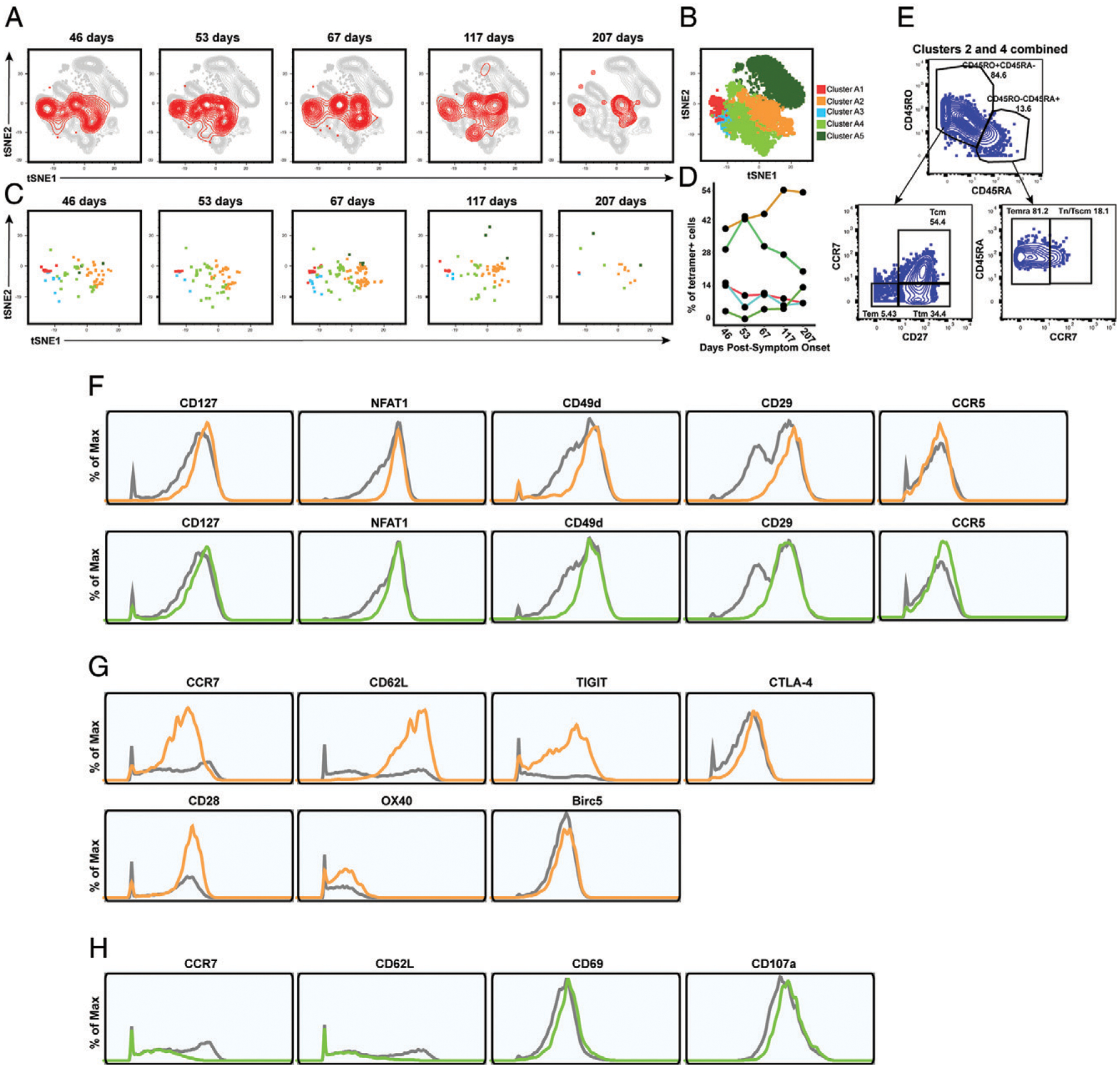
Clusters of Nuc322–331–specific CD8+ T cells from PID4103 exhibit different expansion and contraction. (A) The overall phenotypes of Nuc322–331–specific CD8+ T cells change over the course of convalescence. tSNE plots of total (gray) and tetramer+ (red) CD8+ T cells as a function of time since symptom onset. (B) FlowSOM clusters of CD8+ T cells. Total CD8+ T cells (including the tetramer+ cells) were clustered by FlowSOM to identify five clusters. The location of each cluster is mapped onto the tSNE space depicted in (A). (C) Distribution over time of Nuc322–331–specific CD8+ T cells among the five clusters identified in (B). (D) Proportion of Nuc322–331–specific CD8+ T cells in each cluster as a function of time since symptom onset. The dominant clusters, A2 and A4, increase and decrease over time, respectively. (E) Clusters A2 and A4 include multiple cellular subsets. Gating strategy showing the identification of the Tcm, Tem, Ttm, Temra, and Tn/Tscm subsets, all of which were well-represented among the two dominant clusters. (F) Phenotypic features shared by clusters A2 and A4. Relative to total CD8+ T cells, clusters A2 and A4 expressed high levels of CD127, the transcription factor NFAT1, and the lung-homing receptors CD49d, CD29, and CCR5. Total CD8+ T cells are depicted in gray, cluster A2 cells are depicted in orange, and cluster A4 cells are depicted in green. (G) Phenotypic features exhibited by cluster A2 and not A4. Cluster A2, whose contribution among tetramer+ cells increased over the course of convalescence, expressed high levels of the lymph node–homing receptors CCR7 and CD62L, the checkpoint molecules TIGIT and CTLA4, the costimulatory molecules CD28 and Ox40, and the prosurvival factor BIRC5. Total CD8+ T cells are depicted in gray, and cluster A2 cells are depicted in orange. (H) Phenotypic features exhibited by cluster A4 and not A2. Cluster A4, whose contribution among tetramer+ cells decreased over the course of convalescence, expressed low levels of the lymph node–homing receptors CCR7 and CD62L, high levels of the activation marker CD69, and high levels of the degranulation marker CD107a.
Polyfunctional Nuc322–331–specific CD8+ T cells are detected months into convalescence
Although phenotypic analysis of the tetramer+ cells in the unstimulated samples enabled an in-depth assessment of the differentiation states, expansion potential, homing patterns, and cytolytic activity of Nuc322–331–specific CD8+ T cells, they did not allow assessment of the cytokines these cells are capable of producing. We next, therefore, characterized the phenotypes of cells from the specimens stimulated for 4 h with Nuc322–331 peptide. In these specimens, Nuc322–331–specific CD8+ T cells were defined as the IFN-γ+ cells following peptide treatment (Fig. 2B, D), similar to recently implemented methods (5). Characterization of the canonical subsets (Tcm, Tem, Ttm, Temra, Tn/Tscm) revealed that in contrast to the data from unstimulated tetramer+ cells (Fig. 3D), Tcm cells were not the dominant subset among the responding cells (Fig. 5A, 5C). This can be explained by the decreased expression of the Tcm markers CD45RO and CCR7 on the IFN-γ+ as compared with the tetramer+ cells (Supplemental Figs. 2, 3), likely caused by stimulation-induced downregulation of these receptors. Similar to the tetramer+ data, however, the contribution of the Tn/Tscm subset to the IFN-γ+ cells increased over time (Fig. 5C). Cytolytic cells were detected among the IFN-γ+ cells (Fig. 5D). Although these IFN-γ+ cytolytic cells decreased over time, they still represented a sizable proportion of the cells up until the fourth time point, suggesting the existence of polyfunctional (both IFN-γ–producing and cytolytic) Nuc322–331–specific CD8+ T cells well into convalescence (at least 117 d postsymptom onset). To further probe the polyfunctionality of Nuc322–331–specific CD8+ T cells, we assessed to what extent they induced IFN-γ, TNF-α, or IL-6, the latter of which has been implicated in COVID-19 pathogenesis and may be produced by multiple immune cells, including T cells (27). At the first four time points, the vast majority of IFN-γ+ cells also produced TNF-α, whereas at the final time point, responding cells were more equally distributed among IFN-γ+TNF-α+ and IFN-γ+TNF-α− cells (Fig. 5E, 5F). Interestingly, none of the IFN-γ+ cells coproduced IL-6, and overall, the proportion of Nuc322–331–specific IL-6+ T cells was negligible (Fig. 5E). Assessing all three cytokines together, it was apparent that most of the responding cells dually produced IFN-γ and TNF-α (but no IL-6) and that the frequencies of these cells decreased over time, beginning at the third time point 67 d postsymptom onset (Fig. 5F). Not only did the percentage of IFN-γ+TNF-α+ decrease over time, but also there was a decrease in the absolute levels of IFN-γ and TNF-α produced by these cells (Fig. 5G).
FIGURE 5.
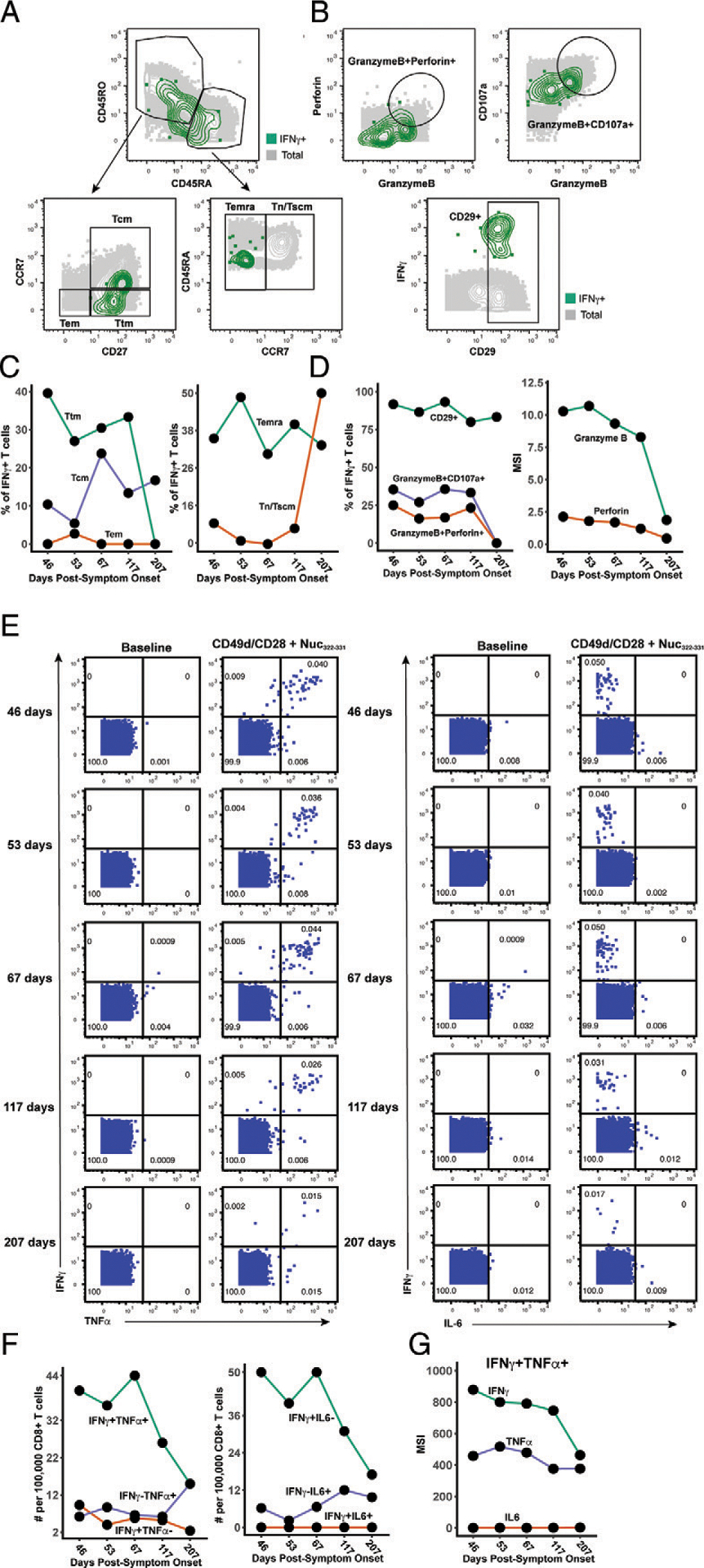
Polyfunctional Nuc322–331–specific CD8+ T cells are detected months into PID4103’s convalescence. (A) Gating strategy to identify the Tcm, Tem, Ttm, Temra, and Tn/Tscm subsets of Nuc322–331–specific CD8+ T cells, identified as those responding to peptide stimulation by producing IFN-γ. The Nuc322–331–specific CD8+ T cells (IFN-γ+) cells are shown as green contours, and total CD8+ T cells are shown in gray. Subset definitions are identical to those used in Figure 3A. (B) Gating strategy to identify cytolytic Nuc322–331–specific CD8+ T cells among those inducing IFN-γ upon cognate peptide stimulation. Top, Gates defining CD8+ T cells coexpressing granzyme B and perforin or granzyme and CD107a are indicated. Bottom, Gate defining cells expressing high levels of CD29, a marker for cytolytic CD8+ T cells. (C) Proportion of IFN-γ+ Nuc322–331–specific CD8+ T cells belonging to the Tcm, Tem, Ttm, Temra, and Tn/Tscm subsets as defined in (A). The lower contribution of Tcm at all time points is likely mediated by activation-induced CCR7 downregulation. Similar to the tetramer data (Fig. 3D), an increase in the contribution of the Tn/Tscm subset was observed over time. (D) Cytolytic Nuc322–331–specific CD8+ T cells slowly decrease over the course of convalescence. Left, Proportion of IFN-γ+ cells that were CD29+, granzymeB+CD107a+, and granzymeB+perforin+. Right, Median expression levels of the indicated cytolytic activity markers on the IFN-γ+ cells. (E) Most Nuc322–331–specific CD8+ T cells responding to peptide stimulation secrete multiple cytokines. Dot plots showing the expression of IFN-γ and TNF-α (left) or IFN-γ and IL-6 (right) on CD8+ T cells among baseline or peptide-stimulated samples. Numbers correspond to the percentage of cells within the gates. Results are gated on live, singlet CD3+CD8+ cells. Most responding Nuc322–331–specific CD8+ T cells were IFN-γ+TNF-α+IL-6−. (F) The proportion of IFN-γ+TNF-α+IL-6−CD8+ T cells responding to Nuc322–331 stimulation decreases over the course of convalescence. The cell populations are taken from the gates shown in (E). (G) The level of IFN-γ and TNF-α produced by Nuc322–331–specific CD8+ T cells decreases over the course of convalescence, as shown by median signal intensity of the IFN-γ+TNF-α+ cells.
Finally, to further probe the effector features of these polyfunctional responding cells, we implemented FlowSOM clustering. As we had conducted CyTOF phenotyping of CD8+ T cells responding not only to Nuc322–331 but also to overlapping peptides from the entire nucleocapsid and spike proteins (Fig. 2E), we clustered all of these responding cells together to compare their effector functions and overall phenotypes (Supplemental Fig. 6). The overall phenotypes of the Nuc322–331–specific CD8+ T cells were similar to those of the nucleocapsid-specific CD8+ T cells (Fig. 6A), consistent with the response to this immunodominant peptide being representative of the response to its parent protein. Interestingly, however, the phenotypes of the spike-specific CD8+ T cell cells differed as reflected by their different distribution within the tSNE plot (Fig. 6A). Furthermore, whereas the majority of the Nuc322–331–specific and Nuc-specific CD8+ T cells belonged to cluster B1, the phenotypes of the spike-specific CD8+ T cells were more evenly distributed, although cluster B1 was also the dominant cluster for the spike-specific cells (Fig. 6B). Cluster B1 included T cells expressing cytolytic markers (granzyme B, CD107a) and cytokines (IFN-γ, TNF-α) (Fig. 6C) consistent with the polyfunctionality of SARS-CoV-2–specific CD8+ T cells during convalescence. Cluster B1, however, also included cells with low levels of cytolytic effectors, and these became more prevalent at the later time points (Fig. 6D), consistent with the diminishing polyfunctionality of SARS-CoV-2–specific CD8+ T cells over the course of convalescence and their differentiation into long-lived memory cells.
FIGURE 6.
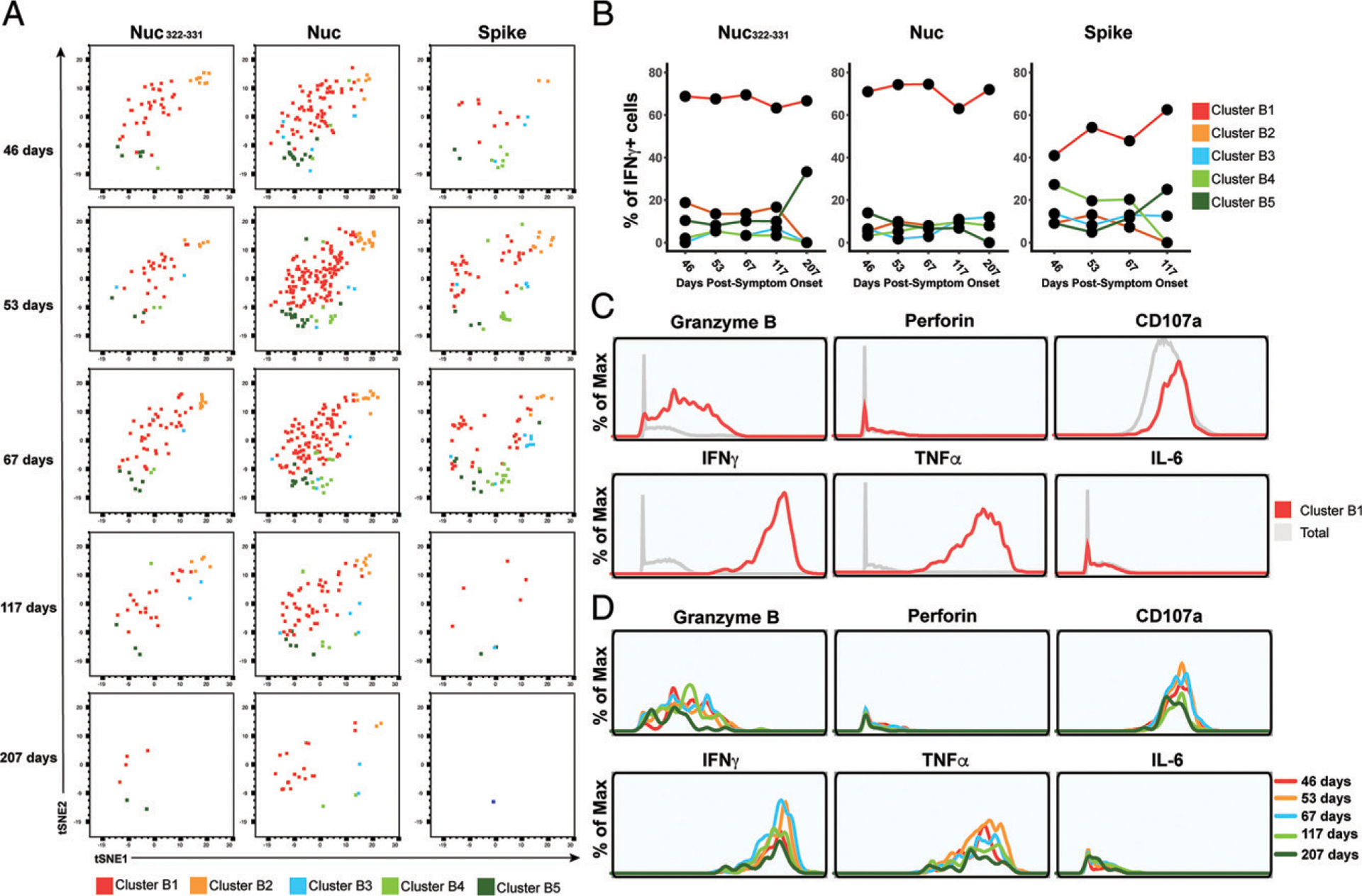
PID4103’s CD8+ T cells responding to Nuc322–331 stimulation are more similar to those responding to nucleocapsid than to spike peptides. (A) Cluster distribution of CD8+ T cells responding to Nuc322–331 or to peptides spanning the entire nucleocapsid or spike proteins. IFN-γ+ CD8+ T cells from the Nuc322–331−, nucleocapsid-, or spike-stimulated specimens were split into five clusters (B1–B5) by FlowSOM. The responding cells are shown as dot plots and colored according to their cluster membership. Note the higher similarity of cells in the tSNE among the Nuc322–331–and Nuc-specific cells, relative to the spike-specific ones. (B) Cluster B1 is dominant among CD8+ T cells with all three specificities but more prominent among the Nuc322–331 and Nuc-specific cells. (C) Cluster B1 cells, to which most cells responding to Nuc322–331, Nuc, and Spike stimulation CD8+ T cells belong, are characterized by high expression levels of the cytolytic markers granzyme B and CD107a and the cytokines IFN-γ and TNF-α. (D) The subpopulations of cluster B1 cells expressing higher levels of effector cytokines and cytolytic molecules decrease over the course of convalescence. Shown are histogram plots depicting cluster B1 cells colored according to time point. Although all the cells shown belong to cluster B1, those from the later time points expressed lower levels of granzyme B, CD107a, IFN-γ, and TNF-α.
Discussion
An important role for CD8+ T cells in effective host response against SARS-CoV-2 was implicated early during the COVID-19 pandemic, when it was observed that lymphopenia, particularly of CD8+ T cells, associates with disease severity (28). Moreover, the CD8+ T cell response directed against the nucleocapsid protein of SARS-CoV-2 may be particularly important, as it appears to be more common than the response directed against spike and other nonstructural proteins (13, 16, 29). Interestingly, CD8+ T cell responses against SARS-CoV-1 also appear to be nucleocapsid-focused, and SARS-CoV-1–specific CD8+ T cells still detectable 17 years after the 2002 SARS outbreak were found to be reactive against nucleocapsid (7). These prior observations implicate nucleocapsid-specific CD8+ T cells not only as important guardians during acute infection but also as a reservoir of long-lived memory cells.
In this study, we screened both spike and nucleocapsid MHC class I tetramers against convalescent COVID-19 participants from the CHIRP cohort, and identified Nuc322–331 as an immunodominant epitope in one of the participants. The maximal proportion of Nuc322–331–specific CD8+ T cells was 0.13%, which exceeds the ~0.0688% that was recently reported as the most immunodominant CD8+ T cell response known to date in COVID-19 (17) and is about two orders of magnitude higher than the proportion of SARS-CoV-2 CD8+ T cells against other epitopes (15). Further supporting its immunodominance is our observation that at all five time points, the proportion of Nuc322–331–specific cells exceeded that of the CD8+ T cells directed against the entire spike protein. Other groups have also reported Nuc322–331–specific CD8+ T cells at frequencies similar to ours [0.093% (12)] or lower [< 0.01% (16)]. The lower frequency detected in the latter study may be donor-dependent or could reflect a frequency calculation based off measurements of pre-enriched tetramer+ cells. Of note, the relatively high frequency of Nuc322–331–specific CD8+ T cells is not due to prior clonal amplification elicited by common-cold coronaviruses, as this peptide is not conserved in those strains. It is also unlikely to have been previously primed by SARS-CoV-1 in PID4103 because, despite 100% conservation of this peptide between the two SARS strains, the participant’s travel history suggest that she could not have been previously exposed to the 2002 virus. Whether the immunodominance of Nuc322–331–specific CD8+ T cells is due to high frequencies of precursors in the naive TCR repertoire, the molecular features of this peptide interacting with MHC, and/or the kinetics of its presentation by APCs in vivo, remains to be determined. Importantly, as the Nuc322–331 sequence is 100% identical in all of the current SARS-CoV-2 variants of concern, the Nuc322–331 responses characterized in this study can be presumed to be as effective against the variants as they are against the original strain.
Extensive CyTOF phenotyping of tetramer+ CD8+ T cells of the longitudinal samples from PID4103 revealed that, among the canonical subsets, Tcm were most common. These results are consistent with recent reports that SARS-CoV-2 nucleocapsid-derived tetramer+ cells were commonly of the CD45RA−CD27+ phenotype (17), characteristic of both the Tcm and Ttm subsets. We further observed that one of the surface markers used to define Tcm cells, CCR7, increased over the course of convalescence among tetramer+ cells. Furthermore, unbiased clustering of the datasets identified a dominant cluster (A2) of tetramer+ cells that increased over the course of convalescence, and this cluster was characterized by high levels of both CCR7 and CD62L expression. As both of these receptors direct immune cells into lymph nodes, the data suggest that over the course of ~6 mo of convalescence, Nuc322–331–specific CD8+ T cells continuously differentiate toward a state more likely to home to lymph nodes. Tcm cells, a canonical lymph node–homing subset, are thought to be relatively long-lived compared with other memory subsets such as Tem and Ttm (30). Another long-lived subset expressing CCR7 are T stem cell memory (Tscm) cells, which have long telomeres and are maintained by ongoing proliferation (31). Although, in our CyTOF panel, we could not distinguish between Tn and Tscm cells, we suspect that the CD45RA+CD45RO−CCR7+ subset (comprising both Tn and Tscm cells) that we detected among tetramer+ cells were predominantly of the Tscm phenotype because naive T cells recognizing Nuc322–331 are expected to be very rare. These Tn/Tscm cells increased steadily over the course of convalescence, plateauing at 117 days postsymptom onset. A recent cross-sectional study, in which convalescents were binned into early versus late convalescence, also found CD45RA+CCR7+ tetramer+ cells to be higher in the latter phase (14), consistent with our longitudinal data. That study also found that, among tetramer+ cells, the percentage of CD127+ cells increased as a function of days since symptom onset out to 120 d. This again mirrors our longitudinal analysis, in which we found that both the percentage of CD127+ cells and the median expression levels of CD127 on the cells steadily increased from 43 to 207 days postsymptom onset. Interestingly, the CD127+ cells were almost exclusively CD27+ and CD57−, suggesting a high expansion potential for these cells. Indeed, our recent study demonstrated that SARS-CoV-2–specific T cells expressing CD127 were capable of homeostatically expanding ex vivo (5). Together with prior demonstrations that CD127 expression identifies CD8+ T cell memory precursors, giving rise to long-lived memory cells (32), these data suggest a slow differentiation of SARS-CoV-2–specific CD8+ T cells to lymph node–homing, long-lived memory cells with expansion potential in the months following recovery from mild COVID-19.
In contrast, other features of the Nuc322–331–specific CD8+ T cells tended to decrease over time. Within the first four months of convalescence, Nuc322–331–specific CD8+ T cells were largely polyfunctional, with most producing both IFN-γ and TNF-α, and a substantial fraction of these cells additionally expressing cytolytic markers. This changed at the final time point (>six months convalescence), when the cytolytic activity of the cells dropped and cells capable of producing both IFN-γ and TNF-α decreased. Interestingly, the decrease in polyfunctionality was mirrored by a decrease in the activation state of the cells, as reflected by a decrease in CD69 and ICOS and to a lesser extent HLADR and CD38. This was in some aspect surprising, as we would have expected the activation state of the cells to have returned back to normal by the second time point, which was almost two months after symptom onset and 30 days after the complete resolution of all symptoms. Together, these results suggest slow changes in the features and functional responses of SARS-CoV-2–specific CD8+ T cells long after full recovery from mild COVID–19.
One of the most interesting findings from this study was the coordinated response of Nuc322–331–specific CD8+ T cells with other components of the adaptive immune system. The magnitude of the Nuc322–331–specific CD8+ T cell response peaked at 67 days postsymptom onset and then decreased thereafter, as did the total CD4+ and CD8+ T cell response against nucleocapsid and the total CD8+ T cell response against spike. The Ab response also exhibited convalescence spikes, but delayed relative to the T cell peak. The delay of the Ab response is reasonable, given the time needed for B cells to be effectively helped by CD4+ T cells, and our finding that the nucleocapsid-specific CD4+ T cell response peaked at the third time point, which was followed by spikes at the fourth time point in multiple isotypes of Abs against the same protein, suggests a CD4+ T cell helped B cell response in PID4103. An increase in the T cell and Ab response 67 to 117 days postsymptom onset was in fact unexpected, given that this was 30–94 days postsymptom resolution and long after the virus had been cleared. Although this convalescence peak in adaptive immune response could theoretically have been caused by reinfection of this participant, we think that possibility unlikely because 1) she tested negative in PCR tests at all time points, except her baseline visit, 2) we saw no evidence of elevated Nuc322–331–specific CD8+ T cell activation at the spiking time points (in fact activation progressively decreased, as discussed above), which we would have expected upon reinfection, 3) the participant reported no reemergence of COVID-19 symptoms after initial symptom resolution, and 4) reinfection is overall uncommon. Further in-depth analyses of the SARS-CoV-2–specific immune responses in additional individuals are needed to confirm whether the immune response detailed in this study is recapitulated in others who experienced mild COVID-19 disease.
Our study has limitations. It is a longitudinal case study of a single patient but, notably, one that has been extensively well-characterized both clinically and immunologically and in which high-dimensional measurements of SARS-CoV-2–specific CD8+ T cells, CD4+ T cells, and Ab responses were analyzed together. Although Nuc322–331 was identified as an immunodominant epitope from screening of 21 participants, we note that these participants were not HLA typed, and it is unknown to what extent the participants other than PID4103 harbored HLA-B*40:01. We also note that the 21 screened participants were all white. Future studies should investigate, using similar methods, the kinetics and features of the CD8+ T cell response in additional demographic groups. Our study was also limited to examining only three cytokines (IFN-γ, TNF-α, and IL-6); it would be informative in future studies to include IL-2 and chemokines, such as MIP-1β, important for CD8+ T cell effector functions.
In summary, we report an unexpectedly dynamic evolution of Nuc322–331–specific CD8+ T cells during convalescence in PID4103. This evolution was gradual and persistent even up to six months after complete symptom resolution. We observed coordination of this response with the CD4+ T cell and Ab responses directed against the same Ag and found that it was characterized by a progressive diminution of the activation state and polyfunctionality of cells in parallel with increases in their expansion potential. If one assumes similarities to nucleocapsid-specific CD8+ T cells from SARS-CoV-1, then the course of differentiation we describe in this study may be one that leads to SARS-CoV-2–specific memory CD8+ T cells that can persist for up to 17 years, and perhaps even longer.
Supplementary Material
Acknowledgments
We thank Stanley Tamaki and Claudia Bispo for CyTOF assistance at the Parnassus Flow Cytometry Core, Nandhini Raman and Jane Srivastava for assistance in flow cytometry at the Gladstone Flow Cytometry Core, Jeff Milush and Norman Jones for assistance with the specimens at the Core Immunology Lab, Emory Pediatrics/Winship Flow Cytometry Core (access supported in part by Children’s Healthcare of Atlanta) and Emory Children’s Clinical and Translational Discovery Core for their support with flow cytometry-based serological experiments, Heather Hartig for help with recruitment, Min-Gyoung Shin and Reuben Thomas for help with the k-means clustering, Warner Greene for helpful feedback on the project, Françoise Chanut for editorial assistance, and Robin Givens for administrative assistance.
This work was supported by the Van Auken Private Foundation, David Henke, and Pamela and Edward Taft (N.R.R.), philanthropic funds donated to Gladstone Institutes by The Roddenberry Foundation and individual donors devoted to COVID-19 research (N.R.R.), the University of California San Francisco Program for Breakthrough Biomedical Research (N.R.R., E.G., S.A.L.), which is partly funded by Sandler Foundation Fast Grant Awards 2164 (N.R.R.), 2208 (N.R.R.), and 2160 (to S.A.L.), a part of Emergent Ventures at the Mercatus Center, George Mason University. This work was also funded by National Institutes of Health Grant (NIH) R01 AI123127-05S1 (E.G.) and Emory’s Lowance Center for Human Immunology (E.G.). We acknowledge NIH Center Grants P30 DK063720 and S10 1S10OD018040-01, for use of the cytometry by time of flight instrument, and the NIH Tetramer Core Facility (contract number 75N93020D00005), for providing the SARS-CoV-2 tetramers and biotinylated monomers, and support from the Center for AIDS Research (P30AI027763) and the James B. Pendleton Charitable Trust.
Abbreviations used in this article:
- CHIRP
COVID-19 Host Immune Response Pathogenesis
- CyTOF
cytometry by time of flight
- Nuc322–331
residues 322 to 311 of nucleocapsid
- RBD
receptor binding domain
- Tcm
T central memory
- Tem
T effector memory
- Tfh
follicular helper
- Tn/Tscm
mixed population of naive and T stem cell memory cells
- Tscm
T stem cell memory
- tSNE
t-distributed stochastic neighbor embedding
- Ttm
T transitional memory
- UCSF
University of California San Francisco
Footnotes
Disclosures
The authors have no financial conflicts of interest.
The raw cytometry by time of flight datasets presented in this article have been submitted to Dryad (https://doi.org/10.7272/Q6D21VVD).
The online version of this article contains supplemental material.
References
- 1.Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, Agudelo M, Barnes CO, Gazumyan A, Finkin S, et al. 2020. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gudbjartsson DF, Norddahl GL, Melsted P, Gunnarsdottir K, Holm H, Eythorsson E, Arnthorsson AO, Helgason D, Bjarnadottir K, Ingvarsson RF, et al. 2020. Humoral immune response to SARS-CoV-2 in Iceland. N. Engl. J. Med 383: 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, Cho A, Jankovic M, Schaefer-Babajew D, Oliveira TY, et al. 2021. Evolution of antibody immunity to SARS-CoV-2. Nature 591: 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, Grifoni A, Ramirez SI, Haupt S, Frazier A, et al. 2021. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 371: eabf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neidleman J, Luo X, Frouard J, Xie G, Gill G, Stein ES, McGregor M, Ma T, George AF, Kosters A, et al. 2020. SARS-CoV-2-specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Rep. Med 1: 100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuo J, Dowell AC, Pearce H, Verma K, Long HM, Begum J, Aiano F, Amin-Chowdhury Z, Hallis B, Stapley L, et al. 2021. Robust SARS-CoV-2-specific T cell immunity is maintained at 6 months following primary infection. [Published erratum appears in 2021 Nat. Immunol. 22: 928.] Nat. Immunol 22: 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, Chng MHY, Lin M, Tan N, Linster M, et al. 2020. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 584: 457–462. [DOI] [PubMed] [Google Scholar]
- 8.Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, et al. 2020. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181: 1489–1501.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarke A, Sidney J, Kidd CK, Dan JM, Ramirez SI, Yu ED, Mateus J, da Silva Antunes R, Moore E, Rubiro P, et al. 2021. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med 2: 100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, Hippenstiel S, Dingeldey M, Kruse B, Fauchere F, et al. 2020. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 587: 270–274. [DOI] [PubMed] [Google Scholar]
- 11.Sekine T, Perez-Potti A, Rivera-Ballesteros O, Strålin K, Gorin JB, Olsson A, Llewellyn-Lacey S, Kamal H, Bogdanovic G, Muschiol S, et al. ; Karolinska COVID-19 Study Group. 2020. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 183: 158–168.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng Y, Mentzer AJ, Liu G, Yao X, Yin Z, Dong D, Dejnirattisai W, Rostron T, Supasa P, Liu C, et al. 2020. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol 21: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kared H, Redd AD, Bloch EM, Bonny TS, Sumatoh H, Kairi F, Carbajo D, Abel B, Newell EW, Bettinotti MP, et al. 2021. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Invest 131: e145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rha MS, Jeong HW, Ko JH, Choi SJ, Seo IH, Lee JS, Sa M, Kim AR, Joo EJ, Ahn JY, et al. 2020. PD-1-expressing SARS-CoV-2-specific CD8(+) T cells are not exhausted, but functional in patients with COVID-19. Immunity 54: 44–52.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Habel JR, Nguyen THO, van de Sandt CE, Juno JA, Chaurasia P, Wragg K, Koutsakos M, Hensen L, Jia X, Chua B, et al. 2020. Suboptimal SARS-CoV-2-specific CD8+ T cell response associated with the prominent HLA-A*02:01 phenotype. Proc. Natl. Acad. Sci. USA 117: 24384–24391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulien I, Kemming J, Oberhardt V, Wild K, Seidel LM, Killmer S, Sagar FD, Lago MS, Decker A, et al. 2021. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med 27: 78–85. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen THO, Rowntree LC, Petersen J, Chua BY, Hensen L, Kedzierski L, van de Sandt CE, Chaurasia P, Tan HX, Habel JR, et al. 2021. CD8+ T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope display high naive precursor frequency and TCR promiscuity. Immunity 54: 1066–1082.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, and Davis MM. 2013. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat. Biotechnol 31: 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Ghosn EE, Cole LE, Obukhanych TV, Sadate-Ngatchou P, Vogel SN, Herzenberg LA, and Herzenberg LA. 2012. Antigen-specific memory in B-1a and its relationship to natural immunity. Proc. Natl. Acad. Sci. USA 109: 5388–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tibshirani R, Walther G, and Hastie T. 2002. Estimating the number of clusters in a data set via the gap statistic. J. R. Stat. Soc. Series B Stat. Methodol 63: 411–423. [Google Scholar]
- 21.Wu Z, and McGoogan JM. 2020. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 323: 1239–1242. [DOI] [PubMed] [Google Scholar]
- 22.Li LQ, Huang T, Wang YQ, Wang ZP, Liang Y, Huang TB, Zhang HY, Sun W, and Wang Y. 2020. COVID-19 patients’ clinical characteristics, discharge rate, and fatality rate of meta-analysis. J. Med. Virol 92: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed S, Ansar Ahmed Z, Siddiqui I, Haroon Rashid N, Mansoor M, and Jafri L. 2021. Evaluation of serum ferritin for prediction of severity and mortality in COVID-19- A cross sectional study. Ann. Med. Surg. (Lond.) 63: 102163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bendall SC, Nolan GP, Roederer M, and Chattopadhyay PK. 2012. A deep profiler’s guide to cytometry. Trends Immunol. 33: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolet BP, Guislain A, van Alphen FPJ, Gomez-Eerland R, Schumacher TNM, van den Biggelaar M, and Wolkers MC. 2020. CD29 identifies IFN-γ-producing human CD8+ T cells with an increased cytotoxic potential. Proc. Natl. Acad. Sci. USA 117: 6686–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y. 2015. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87: 636–645. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, Sun R, Tian Z, Xu X, and Wei H. 2020. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl. Sci. Rev 7: 998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, Alanio C, Kuri-Cervantes L, Pampena MB, D’Andrea K, et al. ; UPenn COVID Processing Unit. 2020. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369: eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferretti AP, Kula T, Wang Y, Nguyen DMV, Weinheimer A, Dunlap GS, Xu Q, Nabilsi N, Perullo CR, Cristofaro AW, et al. 2020. Unbiased screens show CD8+ T cells of COVID-19 patients recognize shared epitopes in SARSCoV-2 that largely reside outside the spike protein. Immunity 53: 1095–1107.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bacchus-Souffan C, Fitch M, Symons J, Abdel-Mohsen M, Reeves DB, Hoh R, Stone M, Hiatt J, Kim P, Chopra A, et al. 2021. Relationship between CD4 T cell turnover, cellular differentiation and HIV persistence during ART. PLoS Pathog. 17: e1009214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed R, Roger L, Costa Del Amo P, Miners KL, Jones RE, Boelen L, Fali T, Elemans M, Zhang Y, Appay V, et al. 2016. Human stem cell-like memory T cells are maintained in a state of dynamic flux. Cell Rep. 17: 2811–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, and Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol 4: 1191–1198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.