

In this study, Bryll et al. found that inactivation of the RNA exosome leads to global reduction of nascent mRNA transcripts, and that this defect is accentuated by loss of deposition of histone variant H2A.Z. They identify the mRNA for the sirtuin deacetylase Hst3 as a key target for the RNA exosome that mediates communication between RNA degradation and transcription machineries.
Keywords: transcription, RNA decay, chromatin, H2A.Z, sirtuin, Hst3
Abstract
Eukaryotic cells maintain an optimal level of mRNAs through unknown mechanisms that balance RNA synthesis and degradation. We found that inactivation of the RNA exosome leads to global reduction of nascent mRNA transcripts, and that this defect is accentuated by loss of deposition of histone variant H2A.Z. We identify the mRNA for the sirtuin deacetylase Hst3 as a key target for the RNA exosome that mediates communication between RNA degradation and transcription machineries. These findings reveal how the RNA exosome and H2A.Z function together to control a deacetylase, ensuring proper levels of transcription in response to changes in RNA degradation.
Eukaryotic cells rely on dynamic, multifaceted regulation at each step of RNA biogenesis to maintain mRNA pools and ensure normal protein synthesis. Loss of regulation at any step of this process can lead to aberrant gene expression and deleterious effects on cell fitness. Studies in S. cerevisiae suggest a buffering phenomenon that maintains global mRNA transcript levels through the balance of nuclear transcriptional control and cytoplasmic RNA decay (Haimovich et al. 2013; Sun et al. 2013). For instance, global decreases in transcription caused by depletion of general transcription factors are associated with a compensatory increase in mRNA half-lives, such that steady-state RNA levels remain largely unaffected (Rodríguez-Molina et al. 2016; Baptista et al. 2017). It has been proposed that expression of the cytoplasmic ribonuclease Xrn1 may play a key role in regulating RNA half-lives in response to decreases in transcription (Sun et al. 2013), but it remains unclear how a decrease in RNA degradation efficiency feeds back to control transcription.
Although previous work focused on cytoplasmic RNA degradation pathways (Haimovich et al. 2013; Sun et al. 2013), inactivation of the nuclear RNA exosome also leads to increased RNA half-lives as well as decreases in RNA synthesis, indicating that nuclear RNA decay can also activate a buffering mechanism (Sun et al. 2013). The RNA exosome is a 3′-to-5′ exonuclease that functions within the nucleus or cytoplasm depending on which catalytic subunit is incorporated; yeast Rrp6 functions within the nuclear RNA exosome, while Dis3 is the nuclease for the cytoplasmic enzyme (Liu et al. 2006). The RNA exosome is highly conserved across eukaryotes and is considered a key RNA quality control pathway that regulates cryptic unstable transcripts (CUTs), noncoding RNAs, and abnormal coding transcripts (Mitchell et al. 1997; Torchet et al. 2002; Kiss and Andrulis 2010; Schneider et al. 2012; Delan-Forino et al. 2017). In S. pombe, the RNA exosome also functions with RNA polymerase II (RNAPII) to facilitate transcription termination (Lemay et al. 2014).
Recently, we found that the RNA exosome masks changes in the steady state pools of both coding and noncoding transcripts following dysregulation of key promoter-proximal, nucleosomal features. For instance, loss of histone H3-K56 acetylation (H3-K56Ac) through inactivation of the Rtt109 acetylase and chaperone Asf1 results in a global decrease in nascent RNAPII transcripts, while hyperacetylation of H3-K56, following depletion of the Hst3 sirtuin deacetylase, leads to a global increase in nascent transcripts. However, in both cases, changes in the steady-state pools are only detected following depletion of the RNA exosome (Rege et al. 2015; Feldman and Peterson 2019; Topal et al. 2019). In addition to H3-K56Ac, nucleosomes flanking RNAPII gene promoters are enriched for the histone variant H2A.Z (Albert et al. 2007; Rufiange et al. 2007). Similar to H3-K56Ac, histone H2A.Z functions with the RNA exosome to regulate divergent noncoding RNAs in both yeast and mouse embryonic stem cells, and yeast strains lacking both the nuclear RNA exosome and H2A.Z show synthetic growth phenotypes (Rege et al. 2015).
We found that the sirtuin histone deacetylase Hst3 plays a key role in a feedback circuit in which nuclear RNA degradation is coupled to RNA synthesis. Using native elongating transcript sequencing (NET-seq) (Churchman and Weissman 2011), we confirmed that conditional depletion of the nuclear RNA exosome leads to a global reduction in RNAPII nascent transcription. Interestingly, depletion of both the RNA exosome and an essential component of the histone H2A.Z deposition machinery, the Swr1 ATPase, leads to a greater decrease in nascent transcription, suggesting two partially redundant mechanisms that buffer transcript levels. Analysis of steady-state mRNA pools shows that the RNA exosome regulates levels of the Hst3 mRNA, and overexpression of Hst3 in wild-type cells is sufficient to drive a global decrease in nascent transcription. Thus, these findings support a novel model in which the sirtuin deacetylase Hst3 is a key player in a regulatory circuit containing both the nuclear RNA exosome and the histone variant H2A.Z that connects RNA degradation and transcription initiation.
Results and Discussion
Depletion of the RNA exosome stabilizes RNA pools and reduces transcription
Strains harboring a deletion of RRP6, encoding the catalytic subunit of the nuclear RNA exosome, exhibit a global increase in mRNA half-lives and decreased mRNA synthesis rates compared with a wild-type strain (Sun et al. 2013). Given this global impact on the transcriptome, we worried that growth of rrp6 deletion cells for many generations may lead to indirect effects that either mask or augment the impact on mRNA biogenesis. To investigate more immediate impacts of RNA exosome loss on transcription, the anchor away system (Haruki et al. 2008) was used to conditionally deplete the Rrp6 catalytic subunit from the nucleus.
To evaluate the effectiveness of the nuclear depletion strategy, RNA-seq was used to monitor changes in the steady-state mRNA pool following a 1-h treatment of RRP6-FRB cells with rapamycin, which triggers Rrp6 nuclear depletion. For library normalization, a S. pombe spike-in control was added to each sample. Previous studies reported varying impacts of the RNA exosome on protein-coding transcripts, though loss of the RNA exosome has a large impact on the abundance of small structural RNAs and noncoding RNAs, such as cryptic unstable transcripts (CUTs) (Gudipati et al. 2012; Schneider et al. 2012). CUTs are 5′ capped and polyadenylated noncoding RNAs that harbor multiple Nrd1–Nab1–Sen1 (NNS) binding motifs. Binding of the NNS complex leads to the recruitment of the RNA exosome and subsequent degradation prior to capping and polyadenylation (Arigo et al. 2006; Thiebaut et al. 2006; Vasiljeva and Buratowski 2006; Schulz et al. 2013). Importantly, a 1-h depletion of Rrp6 was sufficient to cause an increase for 369 of the 929 CUTs by >1.5-fold (Fig. 1A). By comparison, we previously found that a 3-h rapamycin treatment led to up-regulation of 868 CUTs (Feldman and Peterson 2019), nearly identical to the impact of an rrp6 deletion allele, as assayed by microarray analyses (765 CUTs increased 1.5× or more) (Rege et al. 2015). In addition to changes in CUT expression, nearly 1600 coding transcripts were significantly increased compared with wild-type (WT) cells following the 1-h depletion (Fig. 1A). This result is similar to our previous study, where we found that 985 coding transcripts were increased in an rrp6 deletion strain, and 780 coding transcripts were increased following a 3-h depletion. Furthermore, many of the same coding transcripts are observed in all three data sets (Supplemental Fig. S1A). These results indicate that although a 1-h depletion of Rrp6 is not equivalent to a null allele, it is sufficient to cripple RNA exosome function and globally disrupt RNA pools.
Figure 1.

Loss of the RNA exosome stabilizes RNAs and reduces nascent transcripts. (A) RNA-seq analyses comparing coding (left panel) and CUT (right panel) RNAs; RRP6-FRB to WT. Density scatter plots of mean log2 RNA abundance of two biological replicates of WT and two biological replicates of RRP6-FRB normalized with S. pombe. Black line denotes x = y (no change). FDR was determined by edgeR. Log2(fold change mutant/WT) is denoted as LFC. Average fold change ± standard error and standard deviation (stdev) are displayed. (B) NET-seq analyses of asynchronous WT and RRP6-FRB strains treated with rapamycin for 1-h and normalized with S. pombe spike-in. Density scatter plots of coding nascent transcripts show the log2 mean intensity for three biological replicates of RRP6-FRB cells plotted against six WT replicates. The black line indicates x = y (no change). P-value was determined by Mann-Whitney U-test. Average fold change ± standard error and standard deviation (stdev) are displayed. (C) Normalized RNAPII density for all replicates of WT and RRP6-FRB cells. NET-seq reads for each gene, scaled to 500 bp, are normalized to total reads. Shaded region represents 95% confidence interval.
To investigate the impact of RNA exosome depletion on transcription, we performed nascent elongation transcript sequencing (NET-seq) on asynchronous WT and RRP6-FRB strains following 1 h of rapamycin treatment (Fig. 1B, schematic). For library normalization, a S. pombe spike-in control was added to each sample. Nuclear depletion of Rrp6 led to a global decrease in nascent, coding transcripts with a mean fold change of >1.25 (Fig. 1B). In order to evaluate whether this decrease in transcription spans the entire length of the coding region, genes were sorted by length, and log2 fold changes (LFCs) of nascent transcripts were plotted across each gene. Depletion of Rrp6 results in a decrease in nascent transcription across the entire length of coding genes, irrespective of gene size (Supplemental Fig. S1B,C). In contrast to Rrp6, nuclear depletion of the Dis3 exosome subunit had no apparent impact on transcription, consistent with its reported cytoplasmic localization (Supplemental Fig. S1D).
Redundant functional roles of H2A.Z and the RNA exosome in maintaining transcription
Previously, we reported that double mutants lacking both Rrp6 and Swr1, the ATPase subunit of the SWR1 complex (SWR1C), have a synthetic slow-growth phenotype (Rege et al. 2015). Likewise, studies have used epistatic miniarray profiling (E-MAP) to reveal negative genetic interactions between genes encoding Rrp6, multiple subunits of SWR1C, and histone H2A.Z (Collins et al. 2007; Wilmes et al. 2008; Zheng et al. 2010). These genetic interactions led us to investigate the interplay between these two complexes and their influence on transcription by performing RNA-seq and NET-seq analyses for SWR1-FRB and SWR1-FRB RRP6-FRB strains. Consistent with previous deletion allele studies (Rege et al. 2015), a 1-h depletion of Swr1 had no significant impact on the stable coding or noncoding RNA pools (Supplemental Fig. S1E, top panels). Notably, a recent study has found that a 1-h depletion of Swr1 is sufficient to cause the global loss of H2A.Z from chromatin (Ranjan et al. 2020). The codepletion of both Swr1 and Rrp6 led to a similar increase in the stable pool of mRNA transcripts as compared with the sole depletion of Rrp6 (1793 of 5302, LFC > 0.58, edgeR) (Supplemental Fig. S1E bottom left panel). Likewise, the increased levels of noncoding CUT transcripts were nearly identical between the RRP6-FRB and RRP6-FRB SWR1-FRB strains (363 of 920) (Supplemental Fig. S1E, bottom right panel).
Whereas the single depletion of Swr1 had no significant impact on nascent transcription (Fig. 2A), codepletion of Rrp6 and Swr1 resulted in a dramatic decrease (Fig. 2B; see also Supplemental Fig. S1B,C). When further analysis of the data is applied, 3504 nascent transcripts were decreased >1.5-fold (Fig. 2C). In contrast, this same type of analysis revealed that depletion of Rrp6 led to a 1.5-fold decrease of only 15 genes (Fig. 2C; see also Supplemental Fig. S2). Furthermore, the codepletion of Rrp6 and Swr1 had the largest impact on highly expressed genes, specifically the top 25% of genes transcribed in WT (Fig. 2D). Interestingly, nascent transcription of CUTs remained relatively unchanged following codepletion of Rrp6 and Swr1 (Supplemental Fig. S1G). Codepletion of Swr1 and the cytoplasmic RNA exosome component Dis3 resulted in no change in transcription, further confirming a nuclear mechanism of transcriptional homeostasis (Supplemental Fig. S1F). We also analyzed the distribution of RNAPII after normalizing to the overall nascent transcription for a given gene. Whereas depletion of Swr1 had little impact on RNAPII (Fig. 2E), loss of Rrp6 led to an increase in RNAPII distribution past the 3′ end, resulting in readthrough transcripts (Fig. 1C). In the absence of both Swr1 and Rrp6, there is an accentuation of RNAPII distribution at the transcription termination site, suggesting a deficiency in termination, specifically in longer-length genes (Fig. 2E; Supplemental Fig. S1B). These alterations are consistent with previous reports of extended transcript 3′ ends in mutants defective in the RNA exosome, as well as work in fission yeast implicating both H2A.Z and the RNA exosome in transcription termination (Hilleren et al. 2001; Torchet et al. 2002; Zofall et al. 2009; Lemay et al. 2014).
Figure 2.
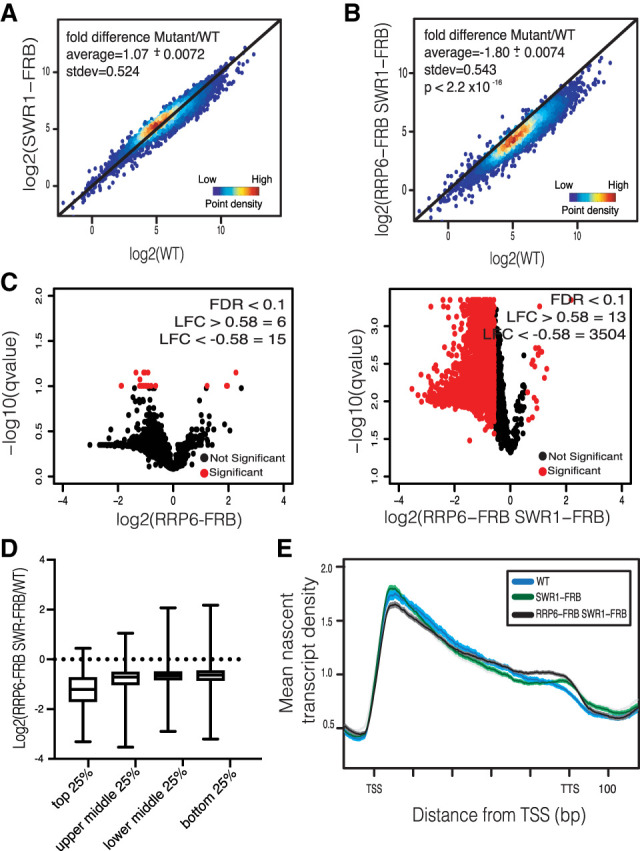
The RNA exosome and H2A.Z globally maintain nascent transcription. (A) Density scatter plots of coding nascent transcripts show the log2 mean intensity for three biological replicates of SWR1-FRB cells plotted against six WT replicates. The black line indicates x = y (no change). Average fold change ± standard error and standard deviation (stdev) are displayed. (B) NET-seq density scatter plots of coding nascent transcripts show the log2 mean intensity for three biological replicates of RRP6-FRB SWR1-FRB cells plotted against six WT replicates. P-value was determined by Mann-Whitney U-test. Average fold change ± standard error and standard deviation (stdev) are supplied on graph. (C) Volcano plots show ORF transcripts that change significantly (denoted in red) by NET-seq for RRP6-FRB SWR1-FRB and RRP6-FRB (FDR ≤ 0.1, LFC ≥ 0.58 or LFC ≤ −0.58). Significant number of genes for LFC ≥ 0.58 or LFC ≤ −0.58 is displayed. (D) Box plot comparing LFC between WT and RRP6-FRB SWR1-FRB mean NET-seq reads of all replicates for the top 25%, top middle 25%, bottom middle 25%, and bottom 25% of genes expressed in WT. (E) Normalized RNAPII density for all replicates of WT (blue), SWR1-FRB (green), and RRP6-FRB SWR1-FRB (black) cells. NET-seq reads for each gene, scaled to 500 bp, are normalized to total reads. Shaded region represents 95% confidence interval.
To investigate whether Rrp6 and Swr1 might also impact expression of an inducible gene set, nascent transcript levels were monitored for genes that are transcriptionally regulated by short exposure to diamide chemical stress (Supplemental Fig. S3A–E; Gasch et al. 2000). In wild-type cells, exposure to diamide leads to the induction of 689 genes and the repression of 644 genes. Similar to constitutively expressed genes, depletion of both Rrp6 and Swr1 led to decreases in nascent transcript levels for both the induced and repressed gene sets.
Our NET-seq analysis indicates that loss of the RNA exosome decreases the number of RNAPII molecules along genes, suggesting that it may alter transcription initiation and elongation. To more directly evaluate how the RNA exosome impacts the levels of actively transcribing RNAPII, we used transient transcriptome sequencing (TT-seq) (Schwalb et al. 2016). In this assay, cells were incubated with 4-thiouridine (4su) for 10 min to label newly transcribed RNAs, followed by biotin capture of labeled RNAs and sequencing library preparation (Fig. 3A). Similar to the results from NET-seq, depletion of Swr1 has little impact on transcription, but loss of Rrp6 leads to a global, ∼1.25-fold reduction in TT-seq signals (Fig. 3B). Furthermore, depletion of both the Rrp6 and Swr1 results in a further decrease in transcription, with reductions nearly 1.5-fold compared with WT (Fig. 3B, right panel; see also Supplemental Fig. S4). Taken together, these findings suggest that the RNA exosome and deposition of the histone variant H2A.Z function together to maintain transcriptional homeostasis.
Figure 3.

Depletion of Rrp6 and Swr1 leads to a decrease in actively transcribing RNAPII. (A) Experimental schematic for TT-seq. (B) Density scatter plots showing coding TT-seq signals between two biological replicates of WT and two biological replicates each of SWR1-FRB (left panel), RRP6-FRB (middle panel), and RRP6-FRB SWR1-FRB (right panel). Black line is x = y. Number of genes significantly decreased >1.5 fold (FDR < 0.1) is displayed. Average fold change ± standard error of the mean and standard deviation (stdev) are included on the graphs.
Hst3 transcript levels are regulated by the RNA exosome
Previous studies suggest that transcriptional buffering involves the cytoplasmic RNA degradation factor Xrn1. In a simple model, decreased transcription produces less Xrn1 mRNA, leading to less cytoplasmic mRNA degradation. However, the mechanism by which decreased mRNA degradation inhibits transcription is less clear. Notably, a nuclease-deficient allele of Xrn1 does not lead to changes in RNA synthesis rates (Sun et al. 2013). Thus, it is not clear that the transcription machinery senses the efficiency of cytoplasmic mRNA decay.
In contrast, our data indicate that loss of nuclear RNA decay by the RNA exosome leads to a global decrease in transcription, especially when H2A.Z is also lost from chromatin. One simple model posits that the RNA exosome regulates the stability of a mRNA encoding a global transcriptional repressor. In this model, loss of Rrp6 would lead to increased synthesis of the repressor, leading to inhibition of transcription (Fig. 4A). To test this hypothesis, we evaluated mRNA levels for several well-characterized repressors that impact either preinitiation complex (PIC) assembly or histone modifications. For instance, Mot1 is a member of the Snf2 family of ATPases that removes and relocates TATA-binding protein (TBP) (Auble et al. 1994), while the NC2 heterodimer, containing NC2-β and BUR6, directly inhibits PIC assembly (Cang and Prelich 2002). Rpd3 is a histone deacetylase that has been previously shown to be a transcriptional repressor (Deckert and Struhl 2002), and Hst3 is a sirtuin deacetylase family member that regulates levels of histone H3-K56Ac (Celic et al. 2006). Whereas depletion of Rrp6 had little impact on the levels of Mot1, NC2-β, Bur6, or Rpd3 mRNAs (Supplemental Fig. S5A), steady-state levels of the Hst3 transcript were increased nearly twofold following depletion of the RNA exosome, and levels remained high following codepletion of both Swr1 and Rrp6 (Supplemental Fig. S5A). Similarly, Hst3 protein levels are also increased following depletion of the RNA exosome (Supplemental Fig. S5B). Interestingly, inactivation of the nuclease activity of Xrn1 does not alter HST3 mRNA levels, suggesting that its levels are more sensitive to nuclear decay efficiency (Haimovich et al. 2013). Since Hst3 is cell cycle-regulated, we confirmed there was no significant shift in cell cycle phase following rapamycin treatment and RNA exosome depletion (Supplemental Fig. S5C). The HST3 transcript contains several consensus binding sites for the NNS complex near the 5′ end, and previous work found that the Nrd1 and NAB components of the NNS complex bind to the Hst3 transcript in vivo (Supplemental Fig. S5D; Creamer et al. 2011). A simple model predicts that the RNA exosome regulates HST3 transcript levels by promoting early termination of transcription by the NNS complex. To test this possibility, we analyzed the 5′ to 3′ ratio of TT-seq reads at the HST3 gene for WT, RRP6-FRB, and RRP6-FRB SWR1-FRB strains. In wild-type cells, the 5′ and 3′ read counts are nearly equal (5′ to 3′ ratio of 1.12 ± 0.03), whereas this ratio is decreased in RRP6-FRB and RRP6-FRB SWR1-FRB strains (0.49 ± 0.22 and 0.67 ± 0). This is consistent with less stalling of RNAPII at the 5′ end of HST3 and more productive elongation to the 3′ end. Overall, these data indicate that the RNA exosome regulates HST3 mRNA abundance, likely through NNS-dependent transcription termination.
Figure 4.
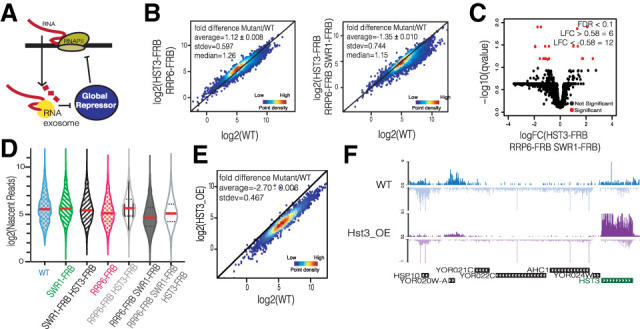
RNA exosome and H2A.Z redundantly regulate global repressor Hst3. (A) Model showing the RNA exosome regulating the transcript of a global repressor. (B) Density scatter plots of coding transcripts show the log2 mean intensity for three biological replicates of HST3-FRB RRP6-FRB (left panel) and four biological replicates of HST3-FRB RRP6-FRB SWR1-FRB (right panel) cells plotted against six WT replicates. The black line indicates x = y (no change). Average fold change ± standard error, standard deviation (SD), and median are displayed. (C) Volcano plot showing ORF transcripts that change significantly (denoted in red) (FDR ≤ 0.1, LFC ≥ 0.58 or LFC ≤ −0.58) in HST3-FRB RRP6-FRB SWR1-FRB strains by NET-seq. Significant number of genes for LFC ≥ 0.58 or LFC ≤ −0.58 is displayed. (D) Violin plot of log2 mean intensity fold change of coding nascent transcripts for all anchor away strains tested in this study. Average of log2 nascent reads is denoted by the red line for each strain. (E) NET-seq density scatter plot for cells in which Hst3 expression was galactose-induced for 1.5 h; data show log2 mean intensity comparing two biological replicates of Hst3 overexpression (HST3_OE) with two biological replicates of galactose-treated WT. Black line is x = y. (F) Representative genome browser views following Hst3 overexpression (HST3_OE) by NET-seq.
Hst3 and Hst4 are two yeast paralogs of mammalian Sirt6 that target the deacetylation of H3-K56 (Celic et al. 2006). Previously, we showed that depletion of both Hst3 and Hst4 resulted in hyperacetylation of H3-K56 and a global, ∼2× increase in nascent RNA transcription (Maas et al. 2006; Feldman and Peterson 2019). In contrast to HST3, the HST4 mRNA was not significantly stabilized by loss of Rrp6, and thus this sirtuin does not appear to be involved in this feedback mechanism. These data are consistent with a model in which increased levels of Hst3, due to Rrp6 depletion, might lead to a global decrease in RNA synthesis. To test this idea, we performed NET-seq analyses in triple depletion (HST3-FRB RRP6-FRB SWR1-FRB) and double depletion (HST3-FRB RRP6-FRB) strains. Strikingly, whereas depletion of both Rrp6 and Swr1 led to a global reduction in nascent transcripts, the additional depletion of Hst3 restored RNA synthesis and RNAPII distribution to wild-type levels (Fig. 4B, Supplemental Fig. S6A). Specifically, whereas ∼3500 genes were significantly decreased following depletion of both Rrp6 and Swr1 (Fig. 2C), the codepletion of Rrp6, Swr1, and Hst3 results in only 12 genes significantly decreased (Fig. 4C). A violin plot of nascent transcripts, with the mean denoted in red, shows this shift in nascent transcription to WT levels (Fig. 4D).
If deacetylation of H3-K56 is key for the role of the RNA exosome in transcriptional regulation, then decreases in transcription due to loss of H3-K56Ac should be epistatic with RNA exosome depletion. Asf1 is a histone chaperone that functions with the Rtt109 acetyltransferase to catalyze H3-K56Ac (Han et al. 2007; Tsubota et al. 2007), and nuclear depletion of Asf1 leads to a global decrease in transcription (see also Supplemental Fig. S6B; Topal et al. 2019). However, codepletion of both Asf1 and Rrp6 did not result in a further reduction in nascent transcription, suggesting that these two factors function within the same pathway (Supplemental Fig. S6B).
To test whether increased levels of Hst3 are sufficient to induce a global decrease in transcription, Hst3 was transiently overexpressed from a galactose-inducible promoter, followed by NET-seq analysis. Consistent with the model, Hst3 overexpression is sufficient to reduce nascent transcription of nearly all genes >1.5-fold (Fig. 4E). A representative genome browser view reveals the increase in Hst3 transcription and the global reduction in transcription across several genes (Fig. 4F). These findings are consistent with a feedback model whereby the RNA exosome regulates the levels of the Hst3 mRNA, which in turn impacts RNA synthesis.
The cellular mechanism that maintains transcriptional homeostasis appears to be dependent on the appropriate regulation of histone acetylases and deacetylases. Our previous results suggest that H3-H56Ac may function as a transcriptional rheostat in which loss of this mark leads to an approximately twofold decrease in global transcription while hyperacetylation of H3K56 causes an approximately twofold increase in transcription (Feldman and Peterson 2019; Topal et al. 2019). Here, our data indicate that smaller modulations in H3K56Ac, mediated by changes in Hst3 mRNA abundance, also cause global transcriptional defects. These transcriptional changes are further augmented by loss of H2A.Z deposition, even though loss of H2A.Z by itself has little impact on transcription. This suggests a new, key role for H2A.Z in buffering promoters against changing levels of histone deacetylases, such as Hst3. Given that H2A.Z is often enriched at the same promoter-proximal nucleosomes that harbor H3-K56Ac, one attractive possibility is that nucleosomes harboring H2A.Z impact the deacetylation activity of Hst3. Together, our work uncovers new roles for H2A.Z and uncovers a feedback circuit in which nuclear mRNA decay is linked to transcription initiation.
Materials and methods
Strains and media
The S. cerevisiae strains used here were derived from MATa tor1-1 fpr1::loxP-LEU2-loxP RPL13A-2xFKBP12::loxP (HHY221) bar1Δ::HISG RPB3-FLAG:NAT. Unless noted otherwise, cells were cultivated in YPD (10% yeast extract, 20% bacterial peptone, 2% glucose). Diamide stress analysis was performed in YPD media with treatment for 30 min of diamide at 1.5 mM final concentration. For Hst3 overexpression, cells were grown in SLGg (0.6% yeast nitrogen base without amino acids, 2% lactic acid, 3% glycerol, 0.05% glucose at pH 6.6) treated with 2% galactose for 1.5 h.
RNA-seq
Asynchronous cells were grown in 50 mL of YPD media at 30°C to midlog phase, treated with rapamycin at a final concentration of 8 µg/mL for 1 h, and harvested, and total RNA was isolated through hot acid phenol extraction. Strand-specific RNA-seq library preparation was done as described previously with a S. pombe spike-in for normalization (Feldman and Peterson 2019; Topal et al. 2019). Briefly, cells were collected and spiked with S. pombe at a 1:6 ratio. Following hot phenol extraction and ethanol precipitation, RNA was further purified with RNAeasy miniprep kit, DNA was digested, and then rRNA was removed using RiboZero magnetic beads (Illumina) from 3 µg of RNA. Two biological replicate libraries for WT and each anchor away mutant were then prepared as described by Zhang et al. (2012) and sequenced using paired-end sequencing on an Illumina NextSeq 500 with read length of 75 bp. Data analysis is described in the Supplemental Material.
NET-seq
NET-seq libraries were prepared as described in Churchman and Weissman (2011) for three biological replicates for each anchor away mutant (unless stated otherwise) and three biological replicates and three technical replicates for WT. A comparison of replicates is in Supplemental Figure S7. As previously described, we implemented a 1:10 S. pombe spike-in, which contained a Flag-tagged Rpd3 subunit (JY741), to normalize reads (Feldman and Peterson 2019; Topal et al. 2019). Overnight cultures were diluted to OD 0.05 in 1 L of YPD unless stated otherwise. Once cultures reached OD 0.45, cells were treated for 1 h with rapamycin (final concentration 8 μg/mL), S. pombe was added, and cells were collected. RNA Pol II IP, RNA purification, and library construction were carried out as described (Churchman and Weissman 2011); 3′ end sequencing was performed on an Illumina NextSeq 500 with a read length of 75 bp. Detailed analysis is described in the Supplemental Material.
TT-seq
TT-seq was performed as described (Schwalb et al. 2016) with an ERCC spike-in (three unlabeled and three 4sU-labeled) for normalization (Topal et al. 2019). Two biological replicates for each strain were grown to 1.5 × 107 cells and labeled with 2.5 mM 4-thiouracil (4su) (Sigma-Aldrich) for 10 min. Cells were harvested by centrifugation at 3000g for 2 min and RNA was extracted by hot acid phenol. RNA was fragmented to <1.5 kb and 4sU-labeled RNA was purified by streptavidin beads. Library prepareation and sequencing were completed as described for RNA-seq. TT-seq data were processed and analyzed as described in the Supplemental Material.
Western blot and flow cytometry are described in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank Jessica Feldman (University of Massachusetts Medical School), Salih Topal (University of Massachusetts Medical School), and Ozkan Aydemir (University of Massachusetts Medical School) for help with bioinformatics analyses, and other members of the Peterson laboratory for helpful discussions. This work was supported by a grant from the National Institutes of Health (R35-GM122519) to C.L.P., and a University of Massachusetts Medical School General Medical Sciences Medical Scientist Training Program Training Grant (MSTP T32GM107000) to A.R.B.
Author contributions: A.R.B. and C.L.P. designed the experiments. A.R.B. performed all experiments and analyzed the data. A.R.B. and C.L.P. wrote and edited the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348923.121.
Competing interest statement
The authors declare no competing interests.
References
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. 2007. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576. 10.1038/nature05632 [DOI] [PubMed] [Google Scholar]
- Arigo JT, Eyler DE, Carroll KL, Corden JL. 2006. Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell 23: 841–851. 10.1016/j.molcel.2006.07.024 [DOI] [PubMed] [Google Scholar]
- Auble DT, Hansen KE, Mueller CG, Lane WS, Thorner J, Hahn S. 1994. Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev 8: 1920–1934. 10.1101/gad.8.16.1920 [DOI] [PubMed] [Google Scholar]
- Baptista T, Grünberg S, Minoungou N, Koster MJE, Timmers HTM, Hahn S, Devys D, Tora L. 2017. SAGA is a general cofactor for RNA polymerase II transcription. Mol Cell 68: 130–143.e5. 10.1016/j.molcel.2017.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang Y, Prelich G. 2002. Direct stimulation of transcription by negative cofactor 2 (NC2) through TATA-binding protein (TBP). Proc Natl Acad Sci 99: 12727–12732. 10.1073/pnas.202236699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, Verreault A. 2006. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol 16: 1280–1289. 10.1016/j.cub.2006.06.023 [DOI] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373. 10.1038/nature09652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, et al. 2007. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446: 806–810. 10.1038/nature05649 [DOI] [PubMed] [Google Scholar]
- Creamer TJ, Darby MM, Jamonnak N, Schaughency P, Hao H, Wheelan SJ, Corden JL. 2011. Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet 7: e1002329. 10.1371/journal.pgen.1002329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert J, Struhl K. 2002. Targeted recruitment of Rpd3 histone deacetylase represses transcription by inhibiting recruitment of Swi/Snf, SAGA, and TATA binding protein. Mol Cell Biol 22: 6458–6470. 10.1128/MCB.22.18.6458-6470.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delan-Forino C, Schneider C, Tollervey D. 2017. Transcriptome-wide analysis of alternative routes for RNA substrates into the exosome complex. PLoS Genet 13: e1006699. 10.1371/journal.pgen.1006699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Peterson CL. 2019. Yeast sirtuin family members maintain transcription homeostasis to ensure genome stability. Cell Rep 27: 2978–2989.e5. 10.1016/j.celrep.2019.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. 10.1091/mbc.11.12.4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, Jacquier A, Libri D. 2012. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell 48: 409–421. 10.1016/j.molcel.2012.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimovich G, Medina DA, Causse SZ, Garber M, Millan-Zambrano G, Barkai O, Chávez S, Pérez-Ortín JE, Darzacq X, Choder M. 2013. Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell 153: 1000–1011. 10.1016/j.cell.2013.05.012 [DOI] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. 2007. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315: 653–655. 10.1126/science.1133234 [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. 2008. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932. 10.1016/j.molcel.2008.07.020 [DOI] [PubMed] [Google Scholar]
- Hilleren P, McCarthy T, Rosbash M, Parker R, Jensen TH. 2001. Quality control of mRNA 3′-end processing is linked to the nuclear exosome. Nature 413: 538–542. 10.1038/35097110 [DOI] [PubMed] [Google Scholar]
- Kiss DL, Andrulis ED. 2010. Genome-wide analysis reveals distinct substrate specificities of Rrp6, Dis3, and core exosome subunits. RNA 16: 781–791. 10.1261/rna.1906710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, Larochelle M, Marguerat S, Atkinson S, Bähler J, Bachand F. 2014. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat Struct Mol Biol 21: 919–926. 10.1038/nsmb.2893 [DOI] [PubMed] [Google Scholar]
- Liu Q, Greimann JC, Lima CD. 2006. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127: 1223–1237. 10.1016/j.cell.2006.10.037 [DOI] [PubMed] [Google Scholar]
- Maas NL, Miller KM, DeFazio LG, Toczyski DP. 2006. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell 23: 109–119. 10.1016/j.molcel.2006.06.006 [DOI] [PubMed] [Google Scholar]
- Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. 1997. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′→5′ exoribonucleases. Cell 91: 457–466. 10.1016/S0092-8674(00)80432-8 [DOI] [PubMed] [Google Scholar]
- Ranjan A, Nguyen VQ, Liu S, Wisniewski J, Kim JM, Tang X, Mizuguchi G, Elalaoui E, Nickels TJ, Jou V, et al. 2020. Live-cell single particle imaging reveals the role of RNA polymerase II in histone H2A.Z eviction. Elife 9: e55667. 10.7554/eLife.55667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rege M, Subramanian V, Zhu C, Hsieh TH, Weiner A, Friedman N, Clauder-Münster S, Steinmetz LM, Rando OJ, Boyer LA, et al. 2015. Chromatin dynamics and the RNA exosome function in concert to regulate transcriptional homeostasis. Cell Rep 13: 1610–1622. 10.1016/j.celrep.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Molina JB, Tseng SC, Simonett SP, Taunton J, Ansari AZ. 2016. Engineered covalent inactivation of TFIIH-kinase reveals an elongation checkpoint and results in widespread mRNA stabilization. Mol Cell 63: 433–444. 10.1016/j.molcel.2016.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. 2007. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell 27: 393–405. 10.1016/j.molcel.2007.07.011 [DOI] [PubMed] [Google Scholar]
- Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D. 2012. Transcriptome-wide analysis of exosome targets. Mol Cell 48: 422–433. 10.1016/j.molcel.2012.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P. 2013. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155: 1075–1087. 10.1016/j.cell.2013.10.024 [DOI] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Gagneur J, Cramer P. 2016. TT-seq maps the human transient transcriptome. Science 352: 1225–1228. 10.1126/science.aad9841 [DOI] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Pirkl N, Maier KC, Schenk A, Failmezger H, Tresch A, Cramer P. 2013. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol Cell 52: 52–62. 10.1016/j.molcel.2013.09.010 [DOI] [PubMed] [Google Scholar]
- Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. 2006. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Mol Cell 23: 853–864. 10.1016/j.molcel.2006.07.029 [DOI] [PubMed] [Google Scholar]
- Topal S, Vasseur P, Radman-Livaja M, Peterson CL. 2019. Distinct transcriptional roles for histone H3-K56 acetylation during the cell cycle in yeast. Nat Commun 10: 4372. 10.1038/s41467-019-12400-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchet C, Bousquet-Antonelli C, Milligan L, Thompson E, Kufel J, Tollervey D. 2002. Processing of 3′-extended read-through transcripts by the exosome can generate functional mRNAs. Mol Cell 9: 1285–1296. 10.1016/S1097-2765(02)00544-0 [DOI] [PubMed] [Google Scholar]
- Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD. 2007. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell 25: 703–712. 10.1016/j.molcel.2007.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Buratowski S. 2006. Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol Cell 21: 239–248. 10.1016/j.molcel.2005.11.028 [DOI] [PubMed] [Google Scholar]
- Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, et al. 2008. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell 32: 735–746. 10.1016/j.molcel.2008.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, Zamore PD. 2012. Strand-specific libraries for high throughput RNA sequencing (RNA-seq) prepared without poly(A) selection. Silence 3: 9. 10.1186/1758-907X-3-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Benschop JJ, Shales M, Kemmeren P, Greenblatt J, Cagney G, Holstege F, Li H, Krogan NJ. 2010. Epistatic relationships reveal the functional organization of yeast transcription factors. Mol Syst Biol 6: 420. 10.1038/msb.2010.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, Grewal SI. 2009. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461: 419–422. 10.1038/nature08321 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.