

Here, Singh et al. show extensive but selective recruitment of intestinal enhancers by CDX2 in gastric cells and that HNF4A-mediated ectopic CDX2 expression in the stomach occurs through a conserved shadow cis-element. These findings identify mechanisms for TF-driven intestinal metaplasia and a likely pathogenic TF hierarchy.
Keywords: gastric intestinal metaplasia, Barrett's esophagus, transcriptional control of cell identity, CDX2, HNF4A
Abstract
Barrett's esophagus (BE) and gastric intestinal metaplasia are related premalignant conditions in which areas of human stomach epithelium express mixed gastric and intestinal features. Intestinal transcription factors (TFs) are expressed in both conditions, with unclear causal roles and cis-regulatory mechanisms. Ectopic CDX2 reprogrammed isogenic mouse stomach organoid lines to a hybrid stomach–intestinal state transcriptionally similar to clinical metaplasia; squamous esophageal organoids resisted this CDX2-mediated effect. Reprogramming was associated with induced activity at thousands of previously inaccessible intestine-restricted enhancers, where CDX2 occupied DNA directly. HNF4A, a TF recently implicated in BE pathogenesis, induced weaker intestinalization by binding a novel shadow Cdx2 enhancer and hence activating Cdx2 expression. CRISPR/Cas9-mediated germline deletion of that cis-element demonstrated its requirement in Cdx2 induction and in the resulting activation of intestinal genes in stomach cells. dCas9-conjugated KRAB repression mapped this activity to the shadow enhancer's HNF4A binding site. Altogether, we show extensive but selective recruitment of intestinal enhancers by CDX2 in gastric cells and that HNF4A-mediated ectopic CDX2 expression in the stomach occurs through a conserved shadow cis-element. These findings identify mechanisms for TF-driven intestinal metaplasia and a likely pathogenic TF hierarchy.
Differential enhancer usage defines cell identities. Enhancer activity requires nucleosome displacement (Felsenfeld 1992; Polach and Widom 1995), and “pioneer” TFs “open” inaccessible chromatin (Iwafuchi-Doi and Zaret 2016). A few tissue-restricted TFs, including pioneer factors, together regulate any cell's unique complement of cis-elements, commonly co-occupy active enhancers, and regulate each other's expression (Saint-André et al. 2016). Other “shadow” enhancers lie within open chromatin and become active only in special circumstances (Cannavò et al. 2016). Aberrant enhancer access or ectopic TF expression can cause developmental defects, cancer, and other disorders (Lee and Young 2013; Lin et al. 2016).
The absence of CDX2 disrupts intestinal specification in mouse embryos (Stringer et al. 2008; Gao et al. 2009) and causes intestinal failure in adults (Verzi et al. 2010, 2013). TFs such as HNF4α, HNF4γ, and GATA4 cooperate with CDX2 within a core intestinal network (Beuling et al. 2011; San Roman et al. 2015; Chen et al. 2019). Barrett's esophagus (BE) and gastric intestinal metaplasia (GIM), closely related human disorders that precede gastric and esophageal adenocarcinomas (Giroux and Rustgi 2017), almost always express CDX2 and other intestinal TFs (Eda et al. 2003; Groisman et al. 2004; Lee et al. 2012; Colleypriest et al. 2017; Rogerson et al. 2019; Nowicki-Osuch et al. 2021). Recent evidence affirms the gastric origins of BE (Nowicki-Osuch et al. 2021; Singh et al. 2021) and reinforce the view that BE and GIM are fundamentally similar conditions arising in the gastric cardia or antrum, respectively. Both BE (Lavery et al. 2014; McDonald et al. 2015) and GIM (Tsukamoto et al. 2006; Correa et al. 2010; Singh et al. 2021) display mixed gastric and intestinal features, reflecting concomitant activation of stomach and intestinal enhancers with incomplete intestinal differentiation (Singh et al. 2021). Although ectopic CDX2 expression induces intestinal genes in mouse fetal stomachs (Silberg et al. 2002; Mutoh et al. 2004), when endoderm fate is plastic (Banerjee et al. 2018), adult foregut epithelia are reported to resist CDX2-mediated intestinalization (Stairs et al. 2008; Kong et al. 2011; Simmini et al. 2014). It is therefore unknown how CDX2 is first expressed and may then access and activate intestinal enhancers in adult stomach cells.
Here we report that CDX2 reprograms adult mouse stomach organoids to a hybrid gastric–intestinal state, with activation of selected intestinal enhancers and partial attenuation of stomach-specific enhancers. These effects phenocopy human intestinal metaplasia (IM), and organoids derived from squamous esophageal epithelium are resistant. Metaplasia follows from pioneer-like CDX2 activity at thousands of previously inaccessible intestinal enhancers. HNF4A, a core intestinal TF implicated in IM (Kojima et al. 2006; Nowicki-Osuch et al. 2021; Wang et al. 2021), intestinalizes stomach organoids by activating Cdx2, a locus otherwise silent in the stomach (James et al. 1994), through a 3′ shadow enhancer. Deletion of this cis-element preserved intestinal development and structure, but failed to activate Cdx2 or to intestinalize mutant stomach organoids. These findings shed new mechanistic light on enhancer accessibility as a safeguard for adult stomach identity and suggest a model in which chemical or biological triggers for IM might first increase the low basal levels of stomach HNF4A. This TF's action at a Cdx2 shadow enhancer then drives ectopic CDX2 expression, which directly activates a substantial fraction of the intestinal cistrome.
Results
Intestinal TFs induce cellular metaplasia and intestinal enhancers in mouse stomach organoids
The adult mouse gastric antrum is histologically similar to the mouse cardia (Barker et al. 2010) and to the source of human BE (Lavery et al. 2014). In RNA analyses of single human cells, the cardia and antrum/pylorus (Busslinger et al. 2021b; Nowicki-Osuch et al. 2021) show overlapping profiles (Supplemental Fig. S1A,B), especially among undifferentiated stem-like cells (Supplemental Fig. S1C,D), and a recent study also identified strong similarities between the cardia and antrum (Busslinger et al. 2021a). Organoids cultured from adult mouse antrum (Mahe et al. 2013; Miyoshi and Stappenbeck 2013) can therefore serve as a model to replicate IM and, unlike the cardia, the antrum avoids the risk of contamination from adjoining squamous epithelium.
CDX2 is abundantly expressed in BE and GIM (Liu et al. 2007; Lee et al. 2012; Singh et al. 2021), and HNF4A, a TF expressed at low levels in adult stomachs, is up-regulated in both conditions (Kojima et al. 2006; Nowicki-Osuch et al. 2021; Wang et al. 2021). Transcriptional profiling of BE identifies CDX2 and HNF4A as prominent candidate regulators (Supplemental Fig. S2A,B), similar to findings in recent single-cell RNA analyses (Nowicki-Osuch et al. 2021). Moreover, CDX2 specifies the mouse embryonic intestine (Stringer et al. 2008; Gao et al. 2009), and some of its adult functions occur in conjunction with HNF4A (Verzi et al. 2010; San Roman et al. 2015; Chen et al. 2019). To ask whether these TFs intestinalize stomach epithelium, we generated isogenic antral organoid lines with stable expression of GFP (controls), HNF4A, or CDX2. Lentivirus-driven CDX2 levels were similar to those in intestinal epithelium; HNF4A levels were lower than those in the intestine but increased above basal gastric expression (Fig. 1A,B).
Figure 1.
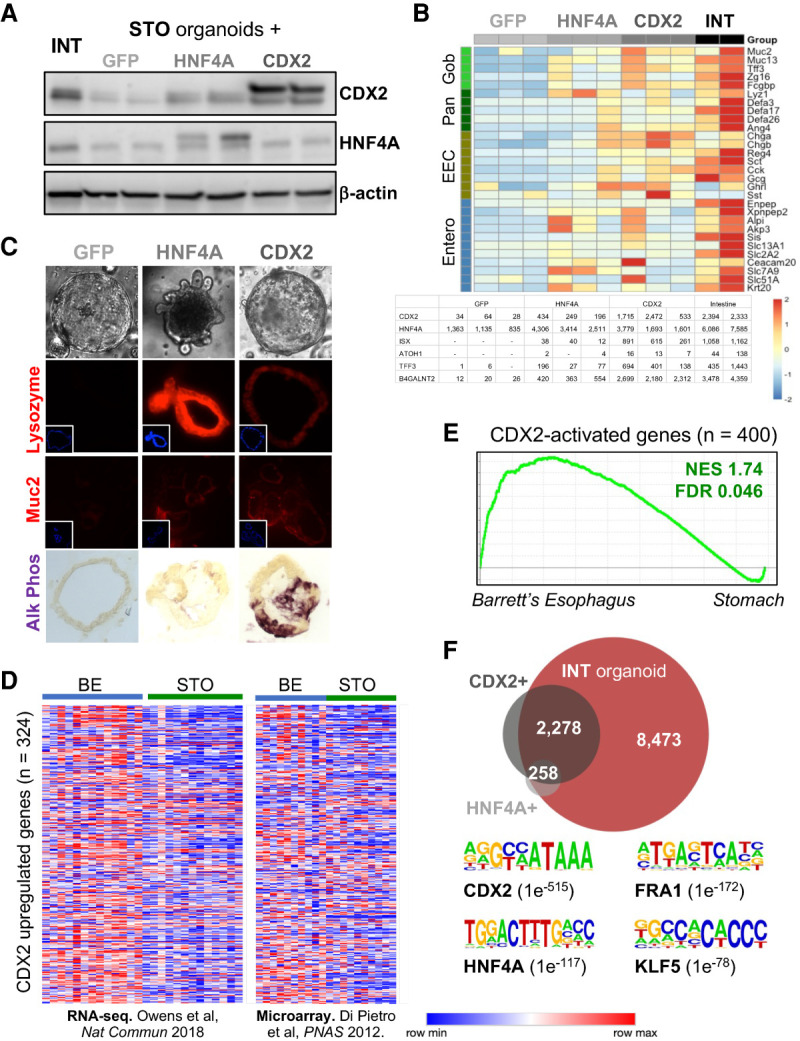
Intestinal TFs induce an intestinal program in mouse stomach organoids. (A) Immunoblots of forced GFP (control), HNF4A, or CDX2 expression in gastric organoids. Each lane represents a distinct organoid line. (B) Expression of intestinal genes representing goblet, enterocyte, enteroendocrine, and Paneth cells in stomach organoids that express GFP, HNF4A, or CDX2, and in intestinal organoids (INT). A table of normalized RNA-seq counts from each organoid line shows CDX2 and HNF4A overexpression and up-regulation of canonical intestinal transcripts. (C, top) Bright-field microscopy of representative stomach organoids, including some with buds that resemble intestinal crypt-like outpouchings. (Bottom) Histochemistry (lysozyme and Muc2 immunostain and alkaline phosphatase) showing intestinal features. (D) Two independent human BE data sets (di Pietro et al. 2012; Owen et al. 2018) show that genes responsive to CDX2 in mouse stomach organoids are up-regulated in BE compared with native stomach epithelium. (E) Gene set enrichment analysis (GSEA) of 400 genes induced in CDX2+ gastric organoids (log2 fold increase >1, q < 0.05, DESeq2), showing resemblance to human BE (data from clinical samples) (Owen et al. 2018). (NES) Normalized enrichment score, (FDR) false discovery rate. (F) Enhancers with increased chromatin access (log2 fold change >1, q < 0.05, DESeq2) in CDX2+ and HNF4A+ stomach organoids overlap significantly with sites selectively accessible in intestinal compared with stomach organoids. Representation factors 7344 (CDX2) and 7748 (HNF4A); P = 0. Motifs for intestinal TFs are highly enriched at enhancers rendered accessible in the presence of CDX2.
Both TFs activated molecular markers specific to the major intestinal cell types, such as alkaline phosphatase (ALPI; enterocytes) and lysozyme (Paneth cells), which are absent or negligible in stomach cells or organoids, and induced crypt-like outpouchings in 1%–10% of stomach organoids (Fig. 1B,C). Compared with GFP+ gastric organoids, independent HNF4A+ and CDX2+ organoid lines activated hundreds of genes (DESeq2, log2 fold difference ≥1, q < 0.05) (Supplemental Table S1) that overlapped significantly with each other (representation factor 29.3; P < 3.575 × 10−193), with genes expressed in intestine-derived organoids, with genes specific to mouse intestines compared with other tissues, and with intestinal microvillus and membrane functions (Supplemental Fig. S2C–E). Small differences in RNA levels of intestinal genes among organoid lines may reflect differences in ectopic TF levels (Fig. 1B), but intestinal features were in every case significant compared with isogenic GFP controls and less robust with HNF4A than with CDX2. Genes induced by CDX2 were highly enriched for functions related to its known transcriptional control of brush border, cytoskeletal, and apico–basal polarity genes (Supplemental Fig. S2E; Gao and Kaestner 2010) and resembled RNA profiles of human BE and GIM (Fig. 1D,E; Supplemental Fig. S2F). Thus, our model system recapitulates significant aspects of these disorders, making it useful to investigate molecular mechanisms of gastric intestinalization likely common to both conditions.
ATAC-seq analysis of CDX2+, HNF4A+, and control (intestinal and GFP+ gastric) organoid lines (Supplemental Table S2) revealed that distant cis-elements (>2 kb upstream of or >1 kb downstream from transcription start sites [TSSs]) specific to intestinal or GFP+ gastric organoids were associated with expression of nearby intestine- or stomach-restricted genes, respectively, and enriched for motifs of TFs corresponding to that tissue (Supplemental Fig. S3A). Thus, ATAC-seq identified bona fide enhancer activity in organoids. Compared with control GFP+ gastric organoids, CDX2+ and HNF4A+ organoids showed increased chromatin accessibility (DESeq2, log2 fold difference ≥1, q < 0.01) at 2898 and 391 enhancers, respectively (Figs. 1F, 2B, 3B). Most HNF4A-induced enhancers (258 of 391) were also induced by CDX2, and nearly all enhancers rendered accessible in CDX2+ gastric organoids were open in intestinal organoids (Figs. 1F 3B). These sites were enriched for CDX2 and other intestinal TF motifs (Fig. 1F) and located near activated genes (Supplemental Fig. S3B). Thus, whereas absence of CDX2 affects developing intestinal epigenomes only until mid-gestation (Banerjee et al. 2018), ectopic CDX2 activates an intestinal program in adult stomach cells by enabling chromatin access at many, but not all, intestinal enhancers.
Direct CDX2 binding at responsive intestinal enhancers
Enhancer activation may reflect direct or indirect TF binding to DNA. We therefore applied CUT&RUN (CnR), a sensitive method by which TF antibodies and protein A-tagged micrococcal nuclease map TF occupancy at nucleotide resolution (Hainer and Fazzio 2019), followed by “footprint” analysis to infer direct and indirect DNA binding (Zhu et al. 2019). Knowing CDX2's role in adult intestinal functions (Gao and Kaestner 2010; Verzi et al. 2010), we first mapped its binding in mouse duodenal villus epithelium. CDX2 occupancy in independent CnR replicates (Supplemental Fig. S3C) overlapped with sites previously identified by ChIP-seq (Saxena et al. 2017), and the absence of CnR signals in Cdx2−/− villus cells confirmed site specificity (Fig. 2A). Motif-centered footprints, indicative of direct DNA binding (Zhu et al. 2019), were present at 10,573 sites. In contrast, 18,626 sites lacked an overt footprint (Fig. 2A), implying that CDX2 does not occupy the latter sites by sequence-specific DNA contacts but indirectly; i.e., through other protein contacts. Chromatin access and H3K27ac marking did not distinguish the two classes of sites (Fig. 2A), indicating that direct or indirect CDX2 occupancy does not define enhancer activity per se.
Figure 2.

Direct CDX2 occupancy at intestinal enhancers in stomach cells. (A) CDX2 binding sites in adult mouse intestinal villus cells, determined by CUT&RUN (CnR), show evidence for direct or indirect DNA occupancy based on the presence of footprints at the CDX2 consensus motif. Cdx2−/− villi lacked signals at CDX2 binding sites. Direct and indirect sites gave similar signals in ATAC-seq and H3K7ac ChIP-seq. (B) In CDX2+ stomach organoids, the TF occupies a large fraction of intestinal cis-elements that bind CDX2 with direct DNA contact (3438 out of 10,573) but few of the 18,626 sites where it binds DNA indirectly in the intestine in vivo. Pre-existing chromatin accessibility is common at indirectly bound CDX2 sites, but chromatin at most direct sites is closed in stomach organoids, and CDX2 expression opens that chromatin. (C) In stomach organoids, CDX2 occupies 3438 intestinal cis-elements that bind CDX2 with direct DNA contact in the native intestine in vivo, whereas most sites that bind CDX2 indirectly in the intestine lack CDX2 binding. All bound sites in stomach organoids, even those that show faint occupancy in the intestine, leave strong footprints at the CDX2 motif (TTTATDR), implying close DNA contact. (D) Schematic showing direct and indirect DNA contact by CDX2 in different contexts.
Of the 10,573 CnR sites bound directly in intestinal cells in vivo, 3438 sites (32.5%) were occupied in CDX2+ gastric organoids (Fig. 2B,C). In contrast, only 831 regions bound indirectly in the intestine were occupied (4.5%; χ2 statistic 44.924; P < 0.00001), even though chromatin was inaccessible at baseline at most directly bound and accessible at most indirectly bound intestinal enhancers (Fig. 2B). Another 7099 CDX2-occupied sites revealed faint intestinal binding (not called as peaks) and equally deep motif-centered footprints in stomach organoids (Fig. 2C), suggesting direct TF contact with DNA. Thus, whereas CDX2 occupies enhancers both indirectly and by direct sequence-specific DNA contact in intestinal cells, all binding in heterologous stomach cells appeared direct, and activation of intestinal enhancers was selective (Fig. 2D).
The stomach epigenome is selectively sensitive to CDX2
To ask whether CDX2-mediated intestinalization is stomach-specific, we generated esophageal organoid lines coexpressing CDX2 and GFP, then used GFP flow cytometry to isolate CDX2-expressing esophageal cells. Open chromatin profiles were nearly identical in CDX2+ and GFP+ esophageal organoids, with only 39 differentially accessible sites (log2 fold difference >1, q < 0.05) (Fig. 3A,B). Whereas highly intestine-restricted loci such as Isx and Atoh1 (Yang et al. 2001; Choi et al. 2006) were extensively reprogrammed in CDX2+ stomach organoids, with proportional increases in mRNA, these and other intestinal genes were unaffected in CDX2+ esophageal organoids (Fig. 3C; Supplemental Fig. S3D). Noting that CDX2 increased expression of several intestinal TFs in gastric organoids (Supplemental Fig. S3E), we asked whether it might activate intestinal genes alone or in concert with other TFs. We revisited motif analysis of open chromatin in CDX2+ gastric organoids, limited to ATAC-seq sites that showed features of direct TF binding: a deep central footprint or increased chromatin accessibility flanking the motif (Baek et al. 2017). CDX1 and CDX2 were the most enriched motifs, with additional significant enrichment only for other homeobox (which resemble that for CDX2, a homeodomain TF) and NFAT/NFATC family motifs (Fig. 3D). Thus, gastric epithelium is uniquely sensitive to the presence of CDX2 and is reprogrammed predominantly by CDX2, with possible contributions from NFAT family and other homeobox TFs such as ISX.
Figure 3.

Intestinal TFs open intestinal enhancers in stomach but not esophageal organoids (A) Principal component analysis (PCA) of the 5000 most variable enhancers by ATAC-seq. Ectopic expression of CDX2 in stomach, but not in esophageal, organoids shifts the global enhancer profile toward that of intestinal epithelium. (B) ATAC-seq data at 2898 unique enhancers accessible in CDX2+ compared with GFP+ gastric organoids. These regions are closed in esophageal CDX2+ organoids. Each column shows an independent biological replicate, and all signals are quantile-normalized. (C) IGV data tracks of open chromatin at the intestine-restricted Isx locus in replicate samples. CDX2+ stomach organoids show increased access at multiple enhancers, in contrast to CDX2+ esophageal organoids, which lack these or other chromatin changes. Chromatin access was accompanied by gene activation (RNA-seq counts, mean ± SD) in gastric CDX2+ organoids. (D) Bagfoot analysis (Baek et al. 2017) of motif enrichment at sites that display metrics of TF binding; i.e., altered footprint depth or increased accessibility flanking ATAC peaks. CDX1/2 and HOX family motifs are significantly enriched in CDX2+ stomach organoids. (E) Hierarchical clustering of RNA-seq data from organoid lines (DESeq2, likelihood ratio test, q < 0.001) showing major groups of differentially expressed genes in GFP+ (n = 826) or intestinal (INT; n = 737) organoids. Exemplary genes within the major differential clusters are listed.
Intestinal TF-mediated stomach reprogramming occurs independent of SOX2
On a background of increased intestinal genes, the presence of intestinal TFs was associated with reduced expression of stomach-specific transcripts (Fig. 3E). In agreement with this finding, chromatin access was reduced (log2 fold change ≥1, q < 0.05) at 924 gastric enhancers, and classic gastric loci showed extensive attenuation of enhancers in CDX2+ lines that were variably attenuated in HNF4A+ lines (Fig. 4A; Supplemental Fig. S4A). Partial gains and losses of intestinal and gastric enhancer activity, respectively, in antral organoids mirror the mixed stomach–intestinal states described in human BE (Van De Bovenkamp et al. 2003; Lavery et al. 2014) and GIM (Tsukamoto et al. 2006; Correa et al. 2010; Singh et al. 2021). Notably, CDX2 CnR revealed no binding at the gastric enhancers attenuated in stomach organoids (Fig. 4A). This finding implies that, unlike CDX2 activity at intestinal enhancers, loss of stomach enhancers is a secondary effect, likely reflecting suppression of gastric TFs. Indeed, among TFs whose DNA motifs were enriched at attenuated gastric enhancers, mRNA encoding SOX2, a foregut master TF (Que et al. 2007; Arnold et al. 2011), was notably reduced in CDX2+ gastric organoids (Fig. 4B).
Figure 4.

CDX2 and HNF4A reprogram stomach organoids independent of SOX2. (A) ATAC-seq signals on stomach-specific cis-elements that lose chromatin access in CDX2+ stomach organoids. Access is reduced to lesser degrees in HNF4A+ organoids. ATAC-seq data tracks at the stomach-restricted Muc6 locus show reduced chromatin access at the promoter and 5′ enhancers (asterisks) in CDX2+ organoids. CDX2 (CnR, red) does not bind to gastric enhancers that lose access (ATAC-seq, blue) in CDX2+ stomach organoids, indicating that suppression of the stomach state occurs indirectly. (B) Sox2 mRNA is reduced and the SOX2 motif is significantly enriched at stomach-specific enhancers that show diminished accessibility in CDX2+ stomach organoids. (C) Comparative analysis of superenhancer (SE) and motif enrichment between normal tissues identifies SOX2 and CDX2 among candidate master TFs for stomach and intestinal epithelia, respectively. SE-bearing TF loci were identified, enrichment of the respective motifs at their own SEs (inward binding) and all other SEs (outward binding) were calculated, and the graph depicts the difference between stomach and intestinal scores for inward and outward binding for each TF. Unexpressed TFs are not scored; e.g., inward and outward binding scores = 0 for CDX2 in the stomach. (D) TF and control (Actin) immunoblots of GFP+, HNF4A+, and CDX2+ stomach organoid lines expressing (WT) or lacking (null) SOX2. Exposure of organoids to adenoviral Cre depleted SOX2. Of note, SOX2 is reduced in CDX2+ organoids. (E) SOX2 depletion does not impact global mRNA profiles of control, HNF4A+, or CDX2+ antral organoids, as judged by sample–sample correlations between RNA-seq data. (F) ATAC-seq profiles of SOX2-proficient and SOX2-null control, HNF4A+, and CDX2+ stomach organoid lines showing no change in stomach- or intestine-specific enhancers, defined and arrayed as in Supplemental Figure S3A.
In the stomach mucosa, the Sox2 locus carries a superenhancer (Supplemental Fig. S4B), as expected from its defining role in foregut tissue identities (Que et al. 2007). Moreover, the SOX2 motif is enriched in the Sox2 and other stomach-restricted superenhancers (Fig. 4C), a characteristic of master TF genes (Saint-André et al. 2016), and a previous study proposed that the balance between SOX2 and CDX2 dictates fetal stomach versus intestinal identity (Raghoebir et al. 2012). To test whether loss of SOX2 accentuates TF-mediated gastric reprograming, we cultured antral organoids from conditional Sox2fl/fl mice (Sarkar et al. 2016), expressed intestinal TFs using lentiviral delivery, and then depleted SOX2 by treating organoids with adenoviral CRE delivery over two sequential passages. Immunoblots confirmed SOX2 loss (Fig. 4D), which elicited no material effect on global organoid RNA profiles (Fig. 4E) or on CDX2- or HNF4A-responsive genes (Supplemental Fig. S4C). ATAC-seq revealed no changes in open chromatin at intestine- or stomach-restricted enhancers compared with SOX2-proficient CDX2+ or HNF4A+ stomach organoids (only 33 regions were significantly increased in SOX2-null CDX2+ organoids) (Fig. 4F). Thus, the absence of SOX2 does not augment intestinal TF effects on gastric reprogramming.
Pioneer-like CDX2 activity in stomach organoids
Although CDX2 is intestine-restricted in adults, other tissues express it during development and have different transcriptional targets. CDX2 occupancy in gastric organoids correlated uniquely with intestine-specific sites, compared with its binding in epiblast (Amin et al. 2016), neuronal (Mazzoni et al. 2013), or embryonic endodermal (Mahony et al. 2014) cells (Fig. 5A); binding at a 3′ Cdx2 and intronic Mapk14 enhancers illustrate the specificity. Thus, the native stomach epigenome distinctly favors CDX2 binding at intestinal sites. Critical examination of organoid ATAC-seq data at the 3438 binding sites shared with normal intestine revealed previously accessible chromatin at <1000 sites (28%); this group was enriched for promoters (Fig. 5B), which in contrast to enhancers are generally accessible across tissues (Heintzman et al. 2009). Importantly, >71% of sites lacked prior chromatin access, became accessible in the presence of CDX2, and were associated with increased expression of nearby intestinal genes (Fig. 5B). In contrast, the ∼7100 sites that CDX2 occupies in gastric organoids but not in normal intestines (Fig. 2C) showed minimally increased chromatin access, comparable with the low levels seen in intestinal organoids (Supplemental Fig. S4D). Overall, chromatin access increased most at intestine-restricted elements (Fig. 5C). Together, these findings imply that in stomach cells CDX2 contacts DNA directly to activate ∼2500 intestinal enhancers where chromatin was previously closed. As these enhancers are not affected in squamous esophageal organoids (Fig. 3B), only select cell types accommodate this pioneer-like CDX2 activity.
Figure 5.
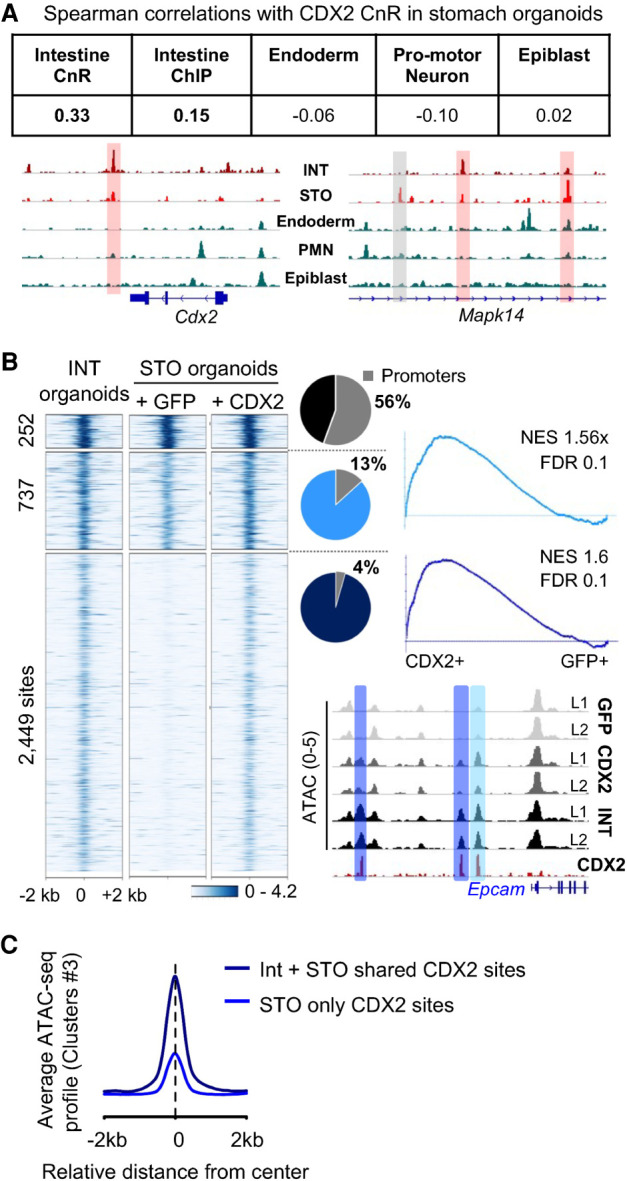
Pioneer-like activity of CDX2 at select intestinal enhancers. (A) CDX2 binding determined by CnR in gastric organoids overlapped most with binding sites in adult intestines and poorly with binding in embryonic tissues. Data tracks for CDX2 binding show shared binding sites with adult intestines (red boxes) and others unique to stomach organoids (gray box). In embryonic tissues, CDX2 binds at sites distinct from these. (B) K-means clustering of ATAC data at the 3438 cis-elements with direct CDX2 binding shared with native intestines. Sites with the highest pre-existing access in stomach organoids are mainly promoters. CDX2 increased access at 737 and opened the chromatin at 2449 enhancers. Gene set enrichment analysis shows that both types of increased chromatin access are linked to genes that increase expression in CDX2+ organoids. The Epcam locus exemplifies increased chromatin access at previously open (light blue) and closed (dark blue) sites. (C) Summation of ATAC-seq signals over cis-elements previously inaccessible in stomach organoids: 2449 sites where CDX2 binding is shared with normal intestines (black curve; cluster 3 in Fig. 2C) and 5546 sites unique to gastric organoids (blue curve; cluster 3 in Supplemental Fig. S4D). Shared sites gained more access than unique sites.
HNF4A and CDX2 in mouse stomach organoids and human tissues
In light of ectopic CDX2 expression (Eda et al. 2003; Phillips et al. 2003; Rogerson et al. 2019; Singh et al. 2021) and likely functions in human IM, we asked how stomach cells, which normally lack CDX2, might activate this TF gene. Mouse Cdx2 is controlled in part by an upstream (−8.5-kb) enhancer that binds HNF4A and other intestinal TFs (Benahmed et al. 2008; Saandi et al. 2013); a downstream repressive element has been described in the liver (Watts et al. 2011), without evidence for a role in the luminal gut. Although the basis for aberrant CDX2 expression in heterologous tissues is unknown, several observations together nominate HNF4A as a candidate activator. First, CDX2 levels increase in HNF4A+ mouse (Figs. 1A,B, 6A) and human (Nowicki-Osuch et al. 2021) stomach organoids. Second, HNF4A is expressed in normal mouse (see Fig. 1A,B) and human (Fig. 6B) stomachs at considerably lower levels than the intestine, and is substantially increased in BE (Fig. 6B). To assess whether this increase is associated with CDX2 expression, we immunostained human BE and GIM (n = 5 specimens each) for both TFs. Low baseline HNF4A levels in normal stomach epithelium were considerably increased in both metaplasias, and CDX2 was present only in cells with elevated HNF4A (Fig. 6C,D; Supplemental Fig. S5). Compared with normal stomach mucosa, ChIP-seq analysis of human BE specimens (Singh et al. 2021) also showed markedly increased levels of the active histone mark H3K4me2 across both the CDX2 and HNF4A loci (Supplemental Fig. S6A,B). In contrast, GATA4 and GATA6 showed similar enhancer features in normal stomach and BE specimens (Supplemental Fig. S6C). Third, recent evidence points to HNF4A-driven transcription in human BE (Nowicki-Osuch et al. 2021). CnR identified significantly more binding in HNF4A+ than in GFP+ organoids (5401 vs. 312 called peaks) (Supplemental Table S3), but these sites barely overlapped with known intestinal enhancers (Supplemental Fig. S6D). The marked contrast to CDX2-bound enhancers (Fig. 2C) suggests that HNF4A might intestinalize stomach cells by activating Cdx2.
Figure 6.

Tightly correlated HNF4A and CDX2 expression in gastric organoids and tissue. (A) Forced HNF4A expression in gastric organoids induces Cdx2, as shown by normalized RNA read counts. Protein levels (see Fig. 1A) also match this induction. (B, left) HNF4A expression in human endoderm-derived tissues (highest in the liver, lowest in the stomach) taken from the genotype-tissue expression (GTEX) consortium (Melé et al. 2015). (Right) HNF4A is up-regulated in human BE compared with normal stomach (data from Owen et al. 2018). (C,D) Sequential tissue sections from primary human BE (C) and GIM (D) reveal perfect concordance of HNF4A up-regulation with CDX2 expression. Nonmetaplastic stomach epithelium (dotted area in C; asterisk in D) shows low basal HNF4A and no CDX2, whereas areas of IM with goblet cells (arrowheads; see also the corresponding hematoxylin and eosin-stained sections in Supplemental Fig. S5A) show high HNF4A and CDX2. Additional examples are in Supplemental Figure S5. (E, top) ATAC-seq data tracks on isogenic organoid lines showing increased ATAC activity at the Cdx2 promoter in HNF4A+ compared with control GFP+ gastric organoids. HNF4A binding, assessed by CUT&RUN (CnR), is not detected at the promoter but at a previously accessible area immediately downstream from the 3′ untranslated region (blue box); GFP+ organoids lack HNF4A binding at this candidate cis-element. (Bottom) ATAC-seq tracks from primary mouse gastric and intestinal Lgr5+ stem cells and H3K27ac ChIP-seq tracks from mouse stomach and intestinal epithelium. The 3′ Cdx2 enhancer is accessible (ATAC+) but inactive (H3K27ac−) in the stomach and encompassed within a superenhancer in the intestine. (F) The novel 3′ Cdx2 shadow enhancer is homologous in mice and humans; a representative 42-bp region within the HNF4A CnR peak (red) shows one base mismatch and shared HNF4A motifs. Pooled (pseudo-bulk) scATAC-seq data from all epithelial cells in an intestinal and two independent human stomach samples (Singh et al. 2021) show chromatin accessibility in both tissues, centered on the HNF4A motif (asterisk).
Although ATAC signals were reproducibly increased at the Cdx2 promoter in HNF4A+ gastric organoids, in keeping with increased mRNA levels, CnR did not reveal promoter HNF4 occupancy; instead, HNF4A occupied a downstream region that is accessible in control gastric organoids but bound only when HNF4A is overexpressed, even modestly (Fig. 6E). This region is 92% similar to the homologous human region, including an identical HNF4A consensus motif, and the chromatin is accessible in human stomach epithelium (Fig. 6F). Furthermore, ATAC-seq analysis of Lgr5+ stem cells freshly isolated from mouse duodenum or stomach antrum (Supplemental Table S4) showed accessible chromatin in stem cells from both sources; however, H3K27ac indicative of enhancer activity was absent in stomach epithelium and restricted to intestinal cells, where the element is part of a locus-wide superenhancer (Fig. 6E). Together, these findings identify a conserved candidate shadow enhancer (Cannavò et al. 2016) in the Cdx2 locus.
HNF4A activates ectopic Cdx2 expression through a shadow 3′ enhancer
As increased HNF4A dosage in gastric organoids results in ectopic CDX2 expression and binding to this 3′ cis-element, we postulated that HNF4A may mediate IM by activating Cdx2. To test this hypothesis, we used CRISPR/Cas9 editing to generate a mouse strain (ΔEnh) with a 1447-bp deletion of the shadow enhancer while preserving coding sequences, including the full 3′ untranslated region (Fig.7A). Heterozygote crosses yielded wild-type, heterozygous, and ΔEnh/ΔEnh mice in Mendelian ratios and homozygotes were overtly normal, with preserved intestinal expression of CDX2 (Fig. 7B,C; Supplemental Fig. S7A,B). To test for Cdx2 induction, we forced HNF4A expression in cultured stomach antrum organoids from wild-type, +/ΔEnh, and ΔEnh/ΔEnh adult littermates. Cdx2 mRNA and protein were readily induced in wild-type organoids, as expected, but were attenuated in +/ΔEnh and totally abrogated in ΔEnh/ΔEnh organoids, despite comparable levels of HNF4A expression in all cases (Fig. 7D,E; Supplemental Fig. S7C). Compared with HNF4A+ organoids with wild-type enhancer, +/ΔEnh and ΔEnh/ΔEnh stomach organoids had reduced expression of 119 and 155 genes, respectively (log2 fold change >1, q < 0.05) (Supplemental Table S5), with an overlap representation factor of 73.2 (P < 5.8 × 10−171) (Supplemental Fig. S7D). Down-regulated genes were enriched for those that constitute the HNF4A transcriptional response in wild-type stomach organoids (Supplemental Fig. S7E); thus, enhancer-deleted organoids have a muted transcriptional response to HNF4A overexpression (Supplemental Fig. S7F), including key intestinal genes and likely direct CDX2 targets (Supplemental Fig. S7G). These findings establish the necessity of the 3′ shadow enhancer in HNF4A-mediated ectopic CDX2 activation and, because genetic perturbation was confined to that cis-element, they indicate that intestinalization by HNF4A occurs through Cdx2.
Figure 7.

HNF4A activates endogenous Cdx2 in gastric organoids through the shadow 3′ enhancer. (A) Design and validation of genetically modified mice with enhancer deletion by CRISPR–Cas9. Location of the enhancer downstream from the Cdx2 3′ UTR, targeting short guide (sg) RNAs, and primers used for PCR validation are shown; a maroon box delineates the 1447-bp enhancer deletion in the principal founder strain, confirmed by Sanger sequencing. (Bottom) Genotyping PCR for the enhancer-deleted (Δenh) strain. (Left) Differentiation of monoallelic from biallelic deletion (primers: CDX2e_F and CDX2e_WT_Rv). (Right) Enhancer deletion (primers: CDX2e_F and CDX2e_Del_Rv). (WT) Wild type. (B,C) Heterozygous and homozygous Δenh mice showed no loss of intestinal CDX2 by qRT-PCR (B) or changes in intestinal histology (C; representative images of duodenum) in three independent mice of each genotype. (D,E) HNF4A overexpression failed to induce CDX2 in gastric organoids cultured from enhancer-deleted homozygous mice, as shown by RNA-seq (D; confirmed by qRT-PCR) (Supplemental Fig. S7C) and immunoblots (E). CDX2 induction was also blunted in heterozygote organoids. (F) Design of two guide RNAs, e1 and e6, compatible with a repressive dCas9-KRAB. e1 is located at +363 bp, within the enhancer but outside the CnR-defined HNF4A binding site (Fig. 6D), whereas e6 is located at +781 bp within the HNF4A binding site (red box). (G) HNF4A+ organoids with an intact CDX2 enhancer (+/+) were transduced with lentivirus encoding dCas9-KRAS and e6, e1, or a nontargeting guide. Compared with the latter, the guide targeting the HNF4A binding site (e6) reduced Cdx2 mRNA expression, while the control e1 guide did not. (H) Model for increased HNF4A levels activating the 3′ shadow Cdx2 enhancer, hence inducing expression of CDX2, the TF that activates intestinal genes in stomach cells.
HNF4A binding occurs at a single site within an ∼200-bp region with a consensus HNF4A motif in the center of this 3′ enhancer (Fig. 7F). To investigate this subregion, we directed enzymatically inactive CAS9 fused with the KRAB transcriptional repressor (dCAS9-KRAB) (Rosenbluh et al. 2017) to the target site using guide RNAs complementary to the HNF4A binding site (e6), a control guide (e1) that targets the enhancer outside this HNF4A binding site, or a nontargeting control guide complementary to extraneous regions (Fig. 7F). In HNF4A+ stomach organoids with the shadow 3′ enhancer intact, we used lentiviruses to generate isogenic organoid lines. Compared with each control guide RNA, the e6 guide reduced Cdx2 selectively and significantly (Fig. 7G). Thus, HNF4A-mediated Cdx2 induction in stomach epithelium depends on the HNF4A binding site within the 3′ shadow enhancer (Fig. 7H).
Discussion
IM arises on backgrounds of chronic tissue injury from acid and bile (proximal stomach) or H. pylori infection (distal stomach), progressing in some cases to dysplasia and cancer. Like the cancers they may spawn (Cancer Genome Atlas Research Network 2017), BE and GIM are phenotypically similar (Piazuelo et al. 2004; Singh et al. 2021) and both metaplasias arise from native stomach cells (Nowicki-Osuch et al. 2021; Singh et al. 2021). Neoplastic evolution has been studied extensively (Stachler et al. 2015; Huang et al. 2018), but it is unclear how adult stomach cells first adopt intestinal properties. Both metaplasias express intestinal TFs (Kojima et al. 2006; Liu et al. 2007; Lee et al. 2012; Rogerson et al. 2019; Wang et al. 2021), but it is unclear whether HNF4A and CDX2 are simply lineage markers or drive altered cell identity. CDX2-induced intestinal heterotopia in developing mouse stomachs (Silberg et al. 2002; Mutoh et al. 2004) likely reflects stage-limited fetal plasticity (Banerjee et al. 2018), and previous expression of CDX2 did not intestinalize adult gastric organoids (Simmini et al. 2014), possibly owing to low CDX2 levels. We show that forced CDX2 activity at physiologic levels in organoids cultured from adult gastric antrum induces the partial intestinalization and hybrid gastric–intestinal identity characteristic of BE and GIM. Although a previous study showed that CDX2 could intestinalize transitional epithelial cells at the squamo–columnar junction (Jiang et al. 2017), esophageal squamous organoids resisted CDX2-mediated intestinalization. Transitional junction cells are one postulated source for BE, but recent data strongly favor a gastric origin (Nowicki-Osuch et al. 2021; Singh et al. 2021). Our antral organoid model represents both partial intestinalization and suppressed gastric states seen in clinical metaplasia.
In stomach organoids, CDX2 did not occupy its indirect intestinal target sites, even though chromatin was accessible at most of them, nor did it bind the sites it engages in embryonal tissues. Rather, it bound a fraction of the sites it normally occupies in adult mouse intestinal cells, where chromatin was inaccessible at baseline. This pioneer-like CDX2 activity in stomach cells reflected direct DNA contact, activated intestine-restricted genes, and was not evident in esophageal squamous cells, which are also foregut-derived. Together, these findings imply that a cofactor or some epigenome feature other than accessible chromatin helps direct CDX2 to intestinal enhancers in stomach cells and that such factors or features are absent in esophageal cells. Further investigation may uncover the basis for differential sensitivity of foregut epithelia to CDX2-driven metaplasia and for only partial activation of the intestinal enhancer repertoire.
Modest overexpression of HNF4A in stomach organoids activated endogenous Cdx2 and a CDX2-dependent transcriptional program, revealing a TF network that underlies adult foregut plasticity. Less-robust intestinalization in HNF4A+ versus CDX2+ stomach organoids may reflect less CDX2 induction than we achieved with lentiviral CDX2 expression. We identified a conserved shadow 3′ Cdx2 enhancer that is dispensable for normal intestinal expression but is bound and aberrantly activated by HNF4A in stomach cells; accessible chromatin at this region in murine and human stomach and intestinal epithelia may reflect their shared developmental origin. Previous identification of FOXA1 and repressive RFX1 binding at this site in the liver (Watts et al. 2011) raises the possibility that repressive TFs restrain ectopic Cdx2 expression in other endoderm derivatives and may explain why HNF4A, which is present at low levels in the stomach, binds the shadow enhancer only when expressed at the higher levels found in the intestine. Aberrant CDX2 expression in cancers of other endoderm-derived organs, such as the pancreas and lung (Moskaluk et al. 2003; Cowan et al. 2016), may also occur from activity at this shadow enhancer. Last, studies in mice and cell lines reveal Cdx2 autoregulation (Barros et al. 2011), suggesting that its ectopic activation in stomach epithelium may be self-sustaining.
We suggest that increased HNF4A expression is a limiting factor in IM, followed by CDX2 activation and resulting intestinalization. Human BE and GIM show heterogeneous and subclonal intestinal identity: The tissues and even presumptive clonal BE or GIM glands carry cells that express and cells that lack CDX2 and other intestinal markers (Singh et al. 2021). Thus, CDX2 expression is not inherent in the metaplastic state, but a dynamic and regulated variable, with intestinal markers present only in CDX2+ cells (Singh et al. 2021) and CDX2 expressed only in HNF4A+ cells. Our findings provide a framework to regard cell state changes in IM as governed by this hierarchy of lineage-determining TFs. Future studies might address whether acid and bile reflux or H. pylori colonization triggers ectopic CDX2 expression via HNF4A and whether evolution of TF-defined cell states contributes to pathogenesis of gastric and distal esophageal dysplasia and adenocarcinoma.
Materials and methods
Generation and culture of mouse gastric and esophageal organoids
Organoids from the gastric antrum were generated as described previously (Mahe et al. 2013). After dissection, the antrum was rotated for 25 min at room temperature in phosphate-buffered saline (PBS) plus 10 mM EDTA. Glandular epithelium was separated from the stroma and muscularis by gentle scraping, washed in PBS, and plated in Matrigel (Corning 356234) supplemented with Glutamax, HEPES, N2, B27, and 65% conditioned media containing Wnt3, Rspo1, and Noggin (Miyoshi and Stappenbeck 2013). The esophagus was opened and rinsed, first with Hank's balanced salt solution (HBSS) supplemented with antibiotic–antimycotic (Gibco 15240112) and then with ice-cold PBS. The epithelium was isolated with tweezers and minced with fine scissors, and tissue fragments were digested in 1 mL of collagenase solution (Life Technologies 17100017) for 10 min at 37°C. Culture and passage details are in the Supplemental Material.
Generation of TF-expressing organoids
We used Gibson modular assembly (Akama-Garren et al. 2016) to modify the lentiviral vector lentiCas9-Blast (Addgene 52962) to replace the Cas9 module with a PuroR cassette, GFP, and TF cDNAs, separated by P2A and T2A cleavage sites, respectively. We cloned the EF1a-KRAB-dCas9-HA cassette from pLX_311-KRAB-dCas9 (Addgene 96918) into a lentiviral backbone containing a U6-gRNA-tracr_v2 cassette and a P2A-BlastR cassette. Virus was harvested 1 and 2 d after transfection of 293T cells and concentrated by ultracentrifugation at 76,755g for 2.5 h at 4°C. Viral pellets were resuspended in 150 µL of Opti-MEM medium (Gibco), stored overnight at 4°C, and frozen in liquid nitrogen. Organoids dissociated to near-single cells by digestion in trypsin were exposed to lentivirus by spinfection at 600g for 1 h at 32°C in the presence of 10 µg/mL polybrene. Infected cells were selected 48 h later in 1.5 µg/mL puromycin, and TF-expressing and control organoid lines obtained from the lowest viral titer were expanded. To avoid cells that may have silenced the TFs, we isolated RNA and nuclei from GFP+ cells collected by flow cytometry.
Organoid histology
We embedded Matrigel drops containing organoids directly into Tissue-Tek OCT compound (Sakura Products) and froze the blocks, and then cut 10-µm thick tissue sections. Alkaline phosphatase staining (Reprocell 000055) was performed using the manufacturer's protocol. Lysozyme immunostaining used polyclonal rabbit Ab (1:100; Dako A0099) and Alexa Fluor 546-conjugated goat antirabbit IgG (Invitrogen A11035).
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections (4 μm) were baked overnight at 37°C, deparaffinized, and rehydrated (100% xylene four times for 3 min each, 100% ethanol four times for 3 min each, and running water for 5 min). Sections were treated with 1.5% H2O2 in methanol for 10 min, washed under running water for 5 min, and placed in a pressure cooker (Biocare Medical) at 120°C in target retrieval solution (pH 6.1 citrate buffer) (DAKO). After cooling and transfer to Tris-buffered saline, consecutive sections were incubated for 40 min at room temperature with anti-CDX2 mouse monoclonal antibody (mAb; 1:150; BioGenex clone CDX2-88) or anti-HNF4A rabbit mAb (1:250; Cell Signaling clone C11F12), followed by secondary antibody (Envision+ mouse or rabbit [DAKO]) for 30 min. Stains were developed using 3,3′-diaminobenzidine (brown product) and counterstained with Mayer's hematoxylin.
RNA-seq and ATAC-seq
Organoids were removed from Matrigel using cell recovery solution (Corning 354253) and treated with Trizol (Thermo 10296010) to isolate RNA or dissociated to single cells in 0.05% trypsin with manual trituration for ATAC-seq. RNA was extracted using the Qiagen RNA minikit with on-column DNase treatment. Libraries were prepared using a PolyA mRNA library preparation kit (New England Biolabs). For ATAC-seq, 35,000–50,000 single viable cells were sorted on a Sony SH800z cell sorter using DAPI as a viability marker, followed by OMNI-ATAC (Corces et al. 2017). Briefly, a crude nuclear preparation was made by pelleting FACS-sorted cells at 1000g for 5 min at 4°C and resuspending them in 50 µL of ice-cold ATAC resuspension buffer (10 mM Tris-HCl at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween-20, 0.01% digitonin) for 3–5 min. Cells were washed in the same buffer containing 0.1% Tween-20 and nuclei were isolated by centrifugation at 1000g for 10 min at 4°C. Nuclear pellets were resuspended in 50 µL of transposition mix (25 µL of 2× TD buffer [Illumina 15027866], 2.5 µL of transposase [Illumina 15027865], 16.5 µL of PBS, 0.5 µL of 1% digitonin, 0.5 µL of 10% Tween-20, 5 µL of water) and incubated for 30 min at 37°C, followed by elution of DNA using the Mini-Elute PCR purification kit (Qiagen 28004). Libraries were prepared as described (Buenrostro et al. 2013). RNA and ATAC libraries were sequenced on a HiSeq-X instrument (Illumina) to generate paired-end 150-bp reads (Novogene). Computational analysis of ATAC-seq and RNA-seq data is described in the Supplemental Material.
CUT&RUN
Single-cell suspensions were prepared from intestinal villi as described (Saxena et al. 2017) and from organoids as described above. Single viable cells (3 × 105 to 5 × 105) were isolated on a Sony SH800z cell sorter using DAPI as a viability marker and CUT&RUN was performed as described (Hainer and Fazzio 2019). Briefly, cells were pelleted at 1000g for 5 min at 4°C and resuspended in nuclear extraction buffer followed by immobilization of nuclei on Concanavalin A beads (Polysciences 86057-3). Immobilized nuclei were incubated with blocking buffer containing 2 mM EDTA to prevent aberrant MNase activation, then rotated overnight with CDX2 Ab (1:50; Cell Signaling Technology D11D10) at 4°C, and finally rotated with protein A-conjugated micrococcal nuclease (pA-MNase; 143 µg/mL stock diluted 1:500; a kind gift from S. Henikoff) for 1 h at 4°C. Samples were equilibrated in an ice bath for 5 min and pA-MNase was activated using 2 mM CaCl2 for 1 h, followed by enzyme inactivation and extraction of DNA with phenol and chloroform. Libraries were made using Next Ultra II kit (New England Biolabs E7645S) with minor modifications to enrich for small fragments likely bound to a TF (Liu et al. 2018; Liu 2019). Libraries were sequenced on a HiSeq-X instrument (Illumina) to generate paired-end 150-bp reads (Novogene). Computational analysis is described in the Supplemental Material.
Cdx2 enhancer-deleted mice
Animal experiments were performed under a protocol approved by the Boston Children's Hospital Animal Care and Use Committee. To generate Cdx2ΔEnh mice, we synthesized two sgRNAs flanking the targeted enhancer. Two overlapping oligonucleotides carrying the T7 RNA polymerase promoter, the target sequence, and the sgRNA scaffold were annealed and used as a template for in vitro transcription (MEGAshortscript T7 transcription kit, Ambion), and reaction products were purified using Nucaway spin columns (Seruggia et al. 2015, 2020; Sher et al. 2019). Prior to use, 200 ng/μL SpCas9 protein (PNA Bio) and 200 ng/μL each sgRNA were mixed in 50 μL of Opti-MEM (Gibco). Batches of 20 one-cell-stage C57BL/6/N/Crl mouse embryos were electroporated using a square-wave electroporator (seven pulses, 30 V, 100-msec intervals) and transferred into CD1 foster mothers. Founder mice were screened by PCR, and genotypes were confirmed by Sanger sequencing. Primers and sgRNA sequences are listed in Supplemental Table S6.
Quantitative RT-PCR
RNA was converted to cDNA using SuperScript III first strand kit (Invitrogen 18080051). Up to 2 µg of RNA was used for each reverse transcriptase reaction using oligo-dT primers. RT-PCR for Cdx2 was performed using Power SYBR Green Master mix (Applied Biosystems 4367659) and the primers 5′-TAAACTCCACTGTCACCCA-3′ (forward) and 5′-AGCGTCCATACTCCTCAT-3′ (reverse). Results were quantified using the Δ−ΔCt method with normalization against 0 on CFX Maestro software (Bio-Rad).
Access to Data
All ChIP, ATAC, and mRNA data are deposited in the Gene Expression Omnibus (GEO) under accession number GSE160264.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01DK082889 and P50CA127003, the Dana-Farber/Novartis Drug Discovery Program (DDP17026), and a generous gift from the Sarah Rhodes Fund for Cancer Research (to R.A.S.). H.S. is a William Raveis Charitable Fund Physician-Scientist of the Damon Runyon Cancer Research Foundation (PST-15-18).
Author contributions: H.S. and R.A.S. conceived the study. H.S., D.S., M.S., A.K.N., Z.W., J.Z., A.J.H., J.W., and A.M. performed experiments. H.S., S.M., J.L.H., and R.A.S. analyzed data. K.H., S.H.O., A.J.B., and R.A.S. supervised various elements of the study. H.S. and R.A.S. drafted the manuscript with input from all authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348983.121.
Competing interest statement
M.S. is currently an employee of Bristol Myers Squibb. The other authors declare no competing interests.
References
- Akama-Garren EH, Joshi NS, Tammela T, Chang GP, Wagner BL, Lee DY, Rideout WM 3rd, Papagiannakopoulos T, Xue W, Jacks T. 2016. A modular assembly platform for rapid generation of DNA constructs. Sci Rep 6: 16836. 10.1038/srep16836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S, Neijts R, Simmini S, van Rooijen C, Tan SC, Kester L, van Oudenaarden A, Creyghton MP, Deschamps J. 2016. Cdx and T brachyury co-activate growth signaling in the embryonic axial progenitor niche. Cell Rep 17: 3165–3177. 10.1016/j.celrep.2016.11.069 [DOI] [PubMed] [Google Scholar]
- Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, Hochedlinger K. 2011. Sox2+ adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9: 317–329. 10.1016/j.stem.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S, Goldstein I, Hager GL. 2017. Bivariate genomic footprinting detects changes in transcription factor activity. Cell Rep 19: 1710–1722. 10.1016/j.celrep.2017.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee KK, Saxena M, Kumar N, Chen L, Cavazza A, Toke NH, O'Neill NK, Madha S, Jadhav U, Verzi MP, et al. 2018. Enhancer, transcriptional, and cell fate plasticity precedes intestinal determination during endoderm development. Genes Dev 32: 1430–1442. 10.1101/gad.318832.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, et al. 2010. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6: 25–36. 10.1016/j.stem.2009.11.013 [DOI] [PubMed] [Google Scholar]
- Barros R, da Costa LT, Pinto-de-Sousa J, Duluc I, Freund JN, David L, Almeida R. 2011. CDX2 autoregulation in human intestinal metaplasia of the stomach: impact on the stability of the phenotype. Gut 60: 290–298. 10.1136/gut.2010.222323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benahmed F, Gross I, Gaunt SJ, Beck F, Jehan F, Domon-Dell C, Martin E, Kedinger M, Freund JN, Duluc I. 2008. Multiple regulatory regions control the complex expression pattern of the mouse Cdx2 homeobox gene. Gastroenterology 135: 1238–1247.e3. 10.1053/j.gastro.2008.06.045 [DOI] [PubMed] [Google Scholar]
- Beuling E, Baffour-Awuah NY, Stapleton KA, Aronson BE, Noah TK, Shroyer NF, Duncan SA, Fleet JC, Krasinski SD. 2011. GATA factors regulate proliferation, differentiation, and gene expression in small intestine of mature mice. Gastroenterology 140: 1219–1229.e2. 10.1053/j.gastro.2011.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger GA, de Barbanson B, Oka R, Weusten BLA, de Maat M, van Hillegersberg R, Brosens LAA, van Boxtel R, van Oudenaarden A, Clevers H. 2021a. Molecular characterization of Barrett's esophagus at single-cell resolution. Proc Natl Acad Sci 118: e2113061118. 10.1073/pnas.2113061118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger GA, Weusten BLA, Bogte A, Begthel H, Brosens LAA, Clevers H. 2021b. Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep 34: 108819. 10.1016/j.celrep.2021.108819 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. 2017. Integrated genomic characterization of oesophageal carcinoma. Nature 541: 169–175. 10.1038/nature20805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavò E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, Ciglar L, Korbel JO, Furlong EE. 2016. Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol 26: 38–51. 10.1016/j.cub.2015.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toke NH, Luo S, Vasoya RP, Fullem RL, Parthasarathy A, Perekatt AO, Verzi MP. 2019. A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat Genet 51: 777–785. 10.1038/s41588-019-0384-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MY, Romer AI, Hu M, Lepourcelet M, Mechoor A, Yesilaltay A, Krieger M, Gray PA, Shivdasani RA. 2006. A dynamic expression survey identifies transcription factors relevant in mouse digestive tract development. Development 133: 4119–4129. 10.1242/dev.02537 [DOI] [PubMed] [Google Scholar]
- Colleypriest BJ, Burke ZD, Griffiths LP, Chen Y, Yu WY, Jover R, Bock M, Biddlestone L, Quinlan JM, Ward SG, et al. 2017. Hnf4α is a key gene that can generate columnar metaplasia in oesophageal epithelium. Differentiation 93: 39–49. 10.1016/j.diff.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. 2017. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962. 10.1038/nmeth.4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P, Piazuelo MB, Wilson KT. 2010. Pathology of gastric intestinal metaplasia: clinical implications. Am J Gastroenterol 105: 493–498. 10.1038/ajg.2009.728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan ML, Li QK, Illei PB. 2016. CDX-2 Expression in primary lung adenocarcinoma. Appl Immunohistochem Mol Morphol 24: 16–19. 10.1097/PAI.0000000000000250 [DOI] [PubMed] [Google Scholar]
- di Pietro M, Lao-Sirieix P, Boyle S, Cassidy A, Castillo D, Saadi A, Eskeland R, Fitzgerald RC. 2012. Evidence for a functional role of epigenetically regulated midcluster HOXB genes in the development of Barrett esophagus. Proc Natl Acad Sci 109: 9077–9082. 10.1073/pnas.1116933109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eda A, Osawa H, Satoh K, Yanaka I, Kihira K, Ishino Y, Mutoh H, Sugano K. 2003. Aberrant expression of CDX2 in Barrett's epithelium and inflammatory esophageal mucosa. J Gastroenterol 38: 14–22. 10.1007/s005350300001 [DOI] [PubMed] [Google Scholar]
- Felsenfeld G. 1992. Chromatin as an essential part of the transcriptional mechanism. Nature 355: 219–224. 10.1038/355219a0 [DOI] [PubMed] [Google Scholar]
- Gao N, Kaestner KH. 2010. Cdx2 regulates endo-lysosomal function and epithelial cell polarity. Genes Dev 24: 1295–1305. 10.1101/gad.1921510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, White P, Kaestner KH. 2009. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell 16: 588–599. 10.1016/j.devcel.2009.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux V, Rustgi AK. 2017. Metaplasia: tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat Rev Cancer 17: 594–604. 10.1038/nrc.2017.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman GM, Amar M, Meir A. 2004. Expression of the intestinal marker Cdx2 in the columnar-lined esophagus with and without intestinal (Barrett's) metaplasia. Mod Pathol 17: 1282–1288. 10.1038/modpathol.3800182 [DOI] [PubMed] [Google Scholar]
- Hainer SJ, Fazzio TG. 2019. High-Resolution chromatin profiling using CUT&RUN. Curr Protoc Mol Biol 126: e85. 10.1002/cpmb.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KK, Ramnarayanan K, Zhu F, Srivastava S, Xu C, Tan ALK, Lee M, Tay S, Das K, Xing M, et al. 2018. Genomic and epigenomic profiling of high-risk intestinal metaplasia reveals molecular determinants of progression to gastric cancer. Cancer Cell 33: 137–150.e5. 10.1016/j.ccell.2017.11.018 [DOI] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Zaret KS. 2016. Cell fate control by pioneer transcription factors. Development 143: 1833–1837. 10.1242/dev.133900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James R, Erler T, Kazenwadel J. 1994. Structure of the murine homeobox gene cdx-2. Expression in embryonic and adult intestinal epithelium. J Biol Chem 269: 15229–15237. 10.1016/S0021-9258(17)36596-1 [DOI] [PubMed] [Google Scholar]
- Jiang M, Li H, Zhang Y, Yang Y, Lu R, Liu K, Lin S, Lan X, Wang H, Wu H, et al. 2017. Transitional basal cells at the squamous-columnar junction generate Barrett's oesophagus. Nature 550: 529–533. 10.1038/nature24269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Kishimoto T, Nagai Y, Tanizawa T, Nakatani Y, Miyazaki M, Ishikura H. 2006. The expression of hepatocyte nuclear factor-4α, a developmental regulator of visceral endoderm, correlates with the intestinal phenotype of gastric adenocarcinomas. Pathology 38: 548–554. 10.1080/00313020601024011 [DOI] [PubMed] [Google Scholar]
- Kong J, Crissey MA, Funakoshi S, Kreindler JL, Lynch JP. 2011. Ectopic Cdx2 expression in murine esophagus models an intermediate stage in the emergence of Barrett's esophagus. PLoS One 6: e18280. 10.1371/journal.pone.0018280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery DL, Nicholson AM, Poulsom R, Jeffery R, Hussain A, Gay LJ, Jankowski JA, Zeki SS, Barr H, Harrison R, et al. 2014. The stem cell organisation, and the proliferative and gene expression profile of Barrett's epithelium, replicates pyloric-type gastric glands. Gut 63: 1854–1863. 10.1136/gutjnl-2013-306508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. 2013. Transcriptional regulation and its misregulation in disease. Cell 152: 1237–1251. 10.1016/j.cell.2013.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Kim N, Lee HS, Kang JM, Park HK, Jo HJ, Shin CM, Lee SH, Park YS, Hwang JH, et al. 2012. The role of CDX2 in intestinal metaplasia evaluated using immunohistochemistry. Gut Liver 6: 71–77. 10.5009/gnl.2012.6.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al. 2016. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530: 57–62. 10.1038/nature16546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N. 2019. Library prep for CUT&RUN with NEBNext® Ultra™ II DNA library prep kit for Illumina® (E7645). protocolsio. https://protocols.io/view/library-prep-for-cut-amp-run-with-nebnext-ultra-ii-wvgfe3w.html; 10.17504/protocols.io.wvgfe3w [DOI] [Google Scholar]
- Liu Q, Teh M, Ito K, Shah N, Ito Y, Yeoh KG. 2007. CDX2 expression is progressively decreased in human gastric intestinal metaplasia, dysplasia and cancer. Mod Pathol 20: 1286–1297. 10.1038/modpathol.3800968 [DOI] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. 2018. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173: 430–442.e17. 10.1016/j.cell.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe MM, Aihara E, Schumacher MA, Zavros Y, Montrose MH, Helmrath MA, Sato T, Shroyer NF. 2013. Establishment of gastrointestinal epithelial organoids. Curr Protoc Mouse Biol 3: 217–240. 10.1002/9780470942390.mo130179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Edwards MD, Mazzoni EO, Sherwood RI, Kakumanu A, Morrison CA, Wichterle H, Gifford DK. 2014. An integrated model of multiple-condition ChIP-Seq data reveals predeterminants of Cdx2 binding. PLoS Comput Biol 10: e1003501. 10.1371/journal.pcbi.1003501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Peljto M, Patel T, Thornton SR, McCuine S, Reeder C, Boyer LA, Young RA, Gifford DK, et al. 2013. Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat Neurosci 16: 1191–1198. 10.1038/nn.3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SA, Graham TA, Lavery DL, Wright NA, Jansen M. 2015. The Barrett's gland in phenotype space. Cell Mol Gastroenterol Hepatol 1: 41–54. 10.1016/j.jcmgh.2014.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. 2015. Human genomics. The human transcriptome across tissues and individuals. Science 348: 660–665. 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Stappenbeck TS. 2013. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 8: 2471–2482. 10.1038/nprot.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskaluk CA, Zhang H, Powell SM, Cerilli LA, Hampton GM, Frierson HF Jr. 2003. Cdx2 protein expression in normal and malignant human tissues: an immunohistochemical survey using tissue microarrays. Mod Pathol 16: 913–919. 10.1097/01.MP.0000086073.92773.55 [DOI] [PubMed] [Google Scholar]
- Mutoh H, Sakurai S, Satoh K, Osawa H, Hakamata Y, Takeuchi T, Sugano K. 2004. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: comparative study with Cdx2 transgenic mice. Gut 53: 1416–1423. 10.1136/gut.2003.032482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicki-Osuch K, Zhuang L, Jammula S, Bleaney CW, Mahbubani KT, Devonshire G, Katz-Summercorn A, Eling N, Wilbrey-Clark A, Madissoon E, et al. 2021. Molecular phenotyping reveals the identity of Barrett's esophagus and its malignant transition. Science 373: 760–767. 10.1126/science.abd1449 [DOI] [PubMed] [Google Scholar]
- Owen RP, White MJ, Severson DT, Braden B, Bailey A, Goldin R, Wang LM, Ruiz-Puig C, Maynard ND, Green A, et al. 2018. Single cell RNA-seq reveals profound transcriptional similarity between Barrett's oesophagus and oesophageal submucosal glands. Nat Commun 9: 4261. 10.1038/s41467-018-06796-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RW, Frierson HF Jr., Moskaluk CA. 2003. Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am J Surg Pathol 27: 1442–1447. 10.1097/00000478-200311000-00006 [DOI] [PubMed] [Google Scholar]
- Piazuelo MB, Haque S, Delgado A, Du JX, Rodriguez F, Correa P. 2004. Phenotypic differences between esophageal and gastric intestinal metaplasia. Mod Pathol 17: 62–74. 10.1038/modpathol.3800016 [DOI] [PubMed] [Google Scholar]
- Polach KJ, Widom J. 1995. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol 254: 130–149. 10.1006/jmbi.1995.0606 [DOI] [PubMed] [Google Scholar]
- Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL. 2007. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134: 2521–2531. 10.1242/dev.003855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghoebir L, Bakker ER, Mills JC, Swagemakers S, Kempen MB, Munck AB, Driegen S, Meijer D, Grosveld F, Tibboel D, et al. 2012. SOX2 redirects the developmental fate of the intestinal epithelium toward a premature gastric phenotype. J Mol Cell Biol 4: 377–385. 10.1093/jmcb/mjs030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson C, Britton E, Withey S, Hanley N, Ang YS, Sharrocks AD. 2019. Identification of a primitive intestinal transcription factor network shared between esophageal adenocarcinoma and its precancerous precursor state. Genome Res 29: 723–736. 10.1101/gr.243345.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Xu H, Harrington W, Gill S, Wang X, Vazquez F, Root DE, Tsherniak A, Hahn WC. 2017. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat Commun 8: 15403. 10.1038/ncomms15403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saandi T, Baraille F, Derbal-Wolfrom L, Cattin AL, Benahmed F, Martin E, Cardot P, Duclos B, Ribeiro A, Freund JN, et al. 2013. Regulation of the tumor suppressor homeogene Cdx2 by HNF4α in intestinal cancer. Oncogene 32: 3782–3788. 10.1038/onc.2012.401 [DOI] [PubMed] [Google Scholar]
- Saint-André V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, Bradner JE, Young RA. 2016. Models of human core transcriptional regulatory circuitries. Genome Res 26: 385–396. 10.1101/gr.197590.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Roman AK, Aronson BE, Krasinski SD, Shivdasani RA, Verzi MP. 2015. Transcription factors GATA4 and HNF4A control distinct aspects of intestinal homeostasis in conjunction with transcription factor CDX2. J Biol Chem 290: 1850–1860. 10.1074/jbc.M114.620211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Huebner AJ, Sulahian R, Anselmo A, Xu X, Flattery K, Desai N, Sebastian C, Yram MA, Arnold K, et al. 2016. Sox2 suppresses gastric tumorigenesis in mice. Cell Rep 16: 1929–1941. 10.1016/j.celrep.2016.07.034 [DOI] [PubMed] [Google Scholar]
- Saxena M, Roman AKS, O'Neill NK, Sulahian R, Jadhav U, Shivdasani RA. 2017. Transcription factor-dependent ‘anti-repressive’ mammalian enhancers exclude H3K27me3 from extended genomic domains. Genes Dev 31: 2391–2404. 10.1101/gad.308536.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seruggia D, Fernández A, Cantero M, Pelczar P, Montoliu L. 2015. Functional validation of mouse tyrosinase non-coding regulatory DNA elements by CRISPR-Cas9-mediated mutagenesis. Nucleic Acids Res 43: 4855–4867. 10.1093/nar/gkv375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seruggia D, Fernández A, Cantero M, Fernández-Minan A, Gomez-Skarmeta JL, Pelczar P, Montoliu L. 2020. Boundary sequences flanking the mouse tyrosinase locus ensure faithful pattern of gene expression. Sci Rep 10: 15494. 10.1038/s41598-020-72543-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher F, Hossain M, Seruggia D, Schoonenberg VAC, Yao Q, Cifani P, Dassama LMK, Cole MA, Ren C, Vinjamur DS, et al. 2019. Rational targeting of a NuRD subcomplex guided by comprehensive in situ mutagenesis. Nat Genet 51: 1149–1159. 10.1038/s41588-019-0453-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberg DG, Sullivan J, Kang E, Swain GP, Moffett J, Sund NJ, Sackett SD, Kaestner KH. 2002. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology 122: 689–696. 10.1053/gast.2002.31902 [DOI] [PubMed] [Google Scholar]
- Simmini S, Bialecka M, Huch M, Kester L, van de Wetering M, Sato T, Beck F, van Oudenaarden A, Clevers H, Deschamps J. 2014. Transformation of intestinal stem cells into gastric stem cells on loss of transcription factor Cdx2. Nat Commun 5: 5728. 10.1038/ncomms6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Ha K, Hornick JL, Madha S, Cejas P, Jajoo K, Singh P, Polak P, Lee H, Shivdasani RA. 2021. Hybrid stomach-intestinal chromatin states underlie human Barrett's metaplasia. Gastroenterology 161: 924–939.e11. 10.1053/j.gastro.2021.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, Davison JM, Nason KS, Loda M, Leshchiner I, et al. 2015. Paired exome analysis of Barrett's esophagus and adenocarcinoma. Nat Genet 47: 1047–1055. 10.1038/ng.3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, Lynch JP, Rustgi AK. 2008. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett's esophagus. PLoS One 3: e3534. 10.1371/journal.pone.0003534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer EJ, Pritchard CA, Beck F. 2008. Cdx2 initiates histodifferentiation of the midgut endoderm. FEBS Lett 582: 2555–2560. 10.1016/j.febslet.2008.06.024 [DOI] [PubMed] [Google Scholar]
- Tsukamoto T, Mizoshita T, Tatematsu M. 2006. Gastric-and-intestinal mixed-type intestinal metaplasia: aberrant expression of transcription factors and stem cell intestinalization. Gastric Cancer 9: 156–166. 10.1007/s10120-006-0375-6 [DOI] [PubMed] [Google Scholar]
- Van De Bovenkamp JH, Korteland-Van Male AM, Warson C, Büller HA, Einerhand AW, Ectors NL, Dekker J. 2003. Gastric-type mucin and TFF-peptide expression in Barrett's oesophagus is disturbed during increased expression of MUC2. Histopathology 42: 555–565. 10.1046/j.1365-2559.2003.01619.x [DOI] [PubMed] [Google Scholar]
- Verzi MP, Shin H, He HH, Sulahian R, Meyer CA, Montgomery RK, Fleet JC, Brown M, Liu XS, Shivdasani RA. 2010. Differentiation-specific histone modifications reveal dynamic chromatin interactions and partners for the intestinal transcription factor CDX2. Dev Cell 19: 713–726. 10.1016/j.devcel.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, Shin H, San Roman AK, Liu XS, Shivdasani RA. 2013. Intestinal master transcription factor CDX2 controls chromatin access for partner transcription factor binding. Mol Cell Biol 33: 281–292. 10.1128/MCB.01185-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Chen M, Ni Z, Li T, Zeng J, Lu G, Wang J, Zhang J, Wu S, Shi Y. 2021. HDAC6/HNF4α loop mediated by miR-1 promotes bile acids-induced gastric intestinal metaplasia. Gastric Cancer 24: 103–116. 10.1007/s10120-020-01108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JA, Zhang C, Klein-Szanto AJ, Kormish JD, Fu J, Zhang MQ, Zaret KS. 2011. Study of foxA pioneer factor at silent genes reveals Rfx-repressed enhancer at Cdx2 and a potential indicator of esophageal adenocarcinoma development. PLoS Genet 7: e1002277. 10.1371/journal.pgen.1002277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. 2001. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294: 2155–2158. 10.1126/science.1065718 [DOI] [PubMed] [Google Scholar]
- Zhu Q, Liu N, Orkin SH, Yuan GC. 2019. CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol 20: 192. 10.1186/s13059-019-1802-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.