

Abstract
Infants with KMT2A-rearranged acute lymphoblastic leukemia (KMT2A-r ALL) have a poor prognosis. KMT2A-r ALL overexpresses FLT3, and the FLT3 inhibitor (FLT3i) lestaurtinib potentiates chemotherapy-induced cytotoxicity in preclinical models. Children’s Oncology Group (COG) AALL0631 tested whether adding lestaurtinib to post-induction chemotherapy improved event-free survival (EFS). After chemotherapy induction, KMT2A-r infants received either chemotherapy only or chemotherapy plus lestaurtinib. Correlative assays included FLT3i plasma pharmacodynamics (PD), which categorized patients as inhibited or uninhibited, and FLT3i ex vivo sensitivity (EVS), which categorized leukemic blasts as sensitive or resistant. There was no difference in 3-year EFS between patients treated with chemotherapy plus lestaurtinib (n = 67, 36 ± 6%) vs. chemotherapy only (n = 54, 39 ± 7%, p = 0.67). However, for the lestaurtinib-treated patients, FLT3i PD and FLT3i EVS significantly correlated with EFS. For FLT3i PD, EFS for inhibited/uninhibited was 59 ± 10%/28 ± 7% (p = 0.009) and for FLTi EVS, EFS for sensitive/resistant was 52 ± 8%/5 ± 5% (p < 0.001). Seventeen patients were both inhibited and sensitive, with an EFS of 88 ± 8%. Adding lestaurtinib did not improve EFS overall, but patients achieving potent FLT3 inhibition and those whose leukemia blasts were sensitive FLT3-inhibition ex vivo did benefit from the addition of lestaurtinib. Patient selection and PD-guided dose escalation may enhance the efficacy of FLT3 inhibition for KMT2A-r infant ALL.
Introduction
KMT2A (MLL) rearrangements (KMT2A-r) occur in 70–80% of infants with acute lymphoblastic leukemia (ALL) and are associated with poor outcomes [1, 2]. Four major cooperative group clinical trials over the past 20 years have reported nearly identical event-free survival (EFS) of 34% [3], 37% [4], 36% [5] and 36% [6] at 4–6 years for KMT2A-r infant ALL. Intensification of chemotherapy has reached the limit of tolerability [7], and stem cell transplantation (SCT) does not improve outcomes for most infants [8–11].
KMT2A-r infant ALL is characterized by a distinct gene-expression profile including striking overexpression of FLT3 [12–14]. FLT3 protein is overexpressed and FLT3 signaling is constitutively activated in these cases, either by activating mutations [15, 16] or, more commonly, by autocrine activation via coexpressed FLT3 ligand [17]. FLT3 overexpression confers especially poor prognosis in KMT2A-r infant ALL [18, 19]. FLT3 inhibitors (FLT3i) selectively kill KMT2A-r ALL cells in vitro and in vivo, and FLT3i synergize with chemotherapy in a sequence-dependent manner [15, 17, 20, 21]. Children’s Oncology Group (COG) AALL0631 tested the hypothesis that the addition of the FLT3i lestaurtinib to post-induction chemotherapy would improve EFS for KMT2A-r ALL.
Methods
Overview of study design
AALL0631 (NCT00557193) was a phase 3 COG clinical trial that enrolled patients from January 2008 to June 2014 from 109 member institutions. For all patients, a parent/guardian provided written informed consent. Eligible patients included infants (age < 366 days at diagnosis) with ALL, acute undifferentiated leukemia, or mixed phenotype acute leukemia with a predominantly lymphoid phenotype (immunophenotype centrally assessed in the Borowitz laboratory at Johns Hopkins University). Central nervous system (CNS) involvement was assessed on cerebrospinal fluid (CSF) obtained prior to starting therapy and classified locally as CNS1 (no blasts in CSF), CNS2 (<5 white blood cells [WBC]/microliter with blasts on cytospin), or CNS3 (≥5 WBC/microliter with blasts). All patients received identical induction chemotherapy. As previously reported, excessive induction toxicity using a modified COG P9407-based regimen for the first 26 patients prompted an amendment changing to an Interfant 99-based induction (Table S1) [7]. After completing induction, patients were stratified into three risk groups based on KMT2A genotype [germline (KMT2A-g) or KMT2A-r] and age at diagnosis: standard risk (SR): KMT2A-g; intermediate risk (IR): KMT2A-r and age 90–365 days; high risk (HR): KMT2A-r and age <90 days. The SR patients received a uniform post-induction chemotherapy regimen (Arm A) without lestaurtinib and will be described separately. For IR and HR patients, AALL0631 proceeded in two phases: a safety/activity phase, designed to identify a safe and biologically active dose of lestaurtinib combined with chemotherapy; and an efficacy phase, designed to determine whether the addition of lestaurtinib to post-induction chemotherapy improved 3-year EFS. Lestaurtinib was supplied by the National Cancer Institute. The backbone of AALL0631 post-induction chemotherapy was based on the predecessor trial COG P9407 [5].
Safety/activity phase
During this phase, IR/HR patients enrolled at one of 44 selected COG sites (based on experience with early phase clinical trials) were non-randomly assigned to receive post-induction chemotherapy plus lestaurtinib (Arm C; Table S2). In parallel, IR/HR patients enrolled at other sites were non-randomly assigned to chemotherapy without lestaurtinib (Arm B; Table S2). Due to expected differences in drug metabolism between younger and older infants, lestaurtinib dose-finding was performed independently in HR and IR patients using different doses at each level (Fig. S1). Dose-limiting toxicity (DLT) was determined during the first two lestaurtinib courses (induction intensification and reinduction; Table S2) and was defined as a lestaurtinib-related adverse event (AE; CTCAEv4) in excess of what was expected with chemotherapy alone (see Supplementary Appendix for detailed DLT definition). Patients without a DLT who received less than 75% of prescribed lestaurtinib doses were deemed inevaluable for DLT assessment and were replaced. Dosing began at dose level 1 (DL1); one dose escalation (DL2) and de-escalation (DL0) were allowed, with dose level cohorts of five evaluable patients. Once a safe dose was identified for IR or HR patients (defined as no more than one of five with DLTs), the randomized efficacy phase for that group commenced only if adequate FLT3i pharmacodynamics (PD) activity was demonstrated at that dose level using FLT3 plasma inhibitory assay [22] (PIA; methods detailed below).
Efficacy phase
During the efficacy phase, patients enrolling at any site were eligible to receive lestaurtinib. The efficacy phase began as a randomized trial, with IR/HR patients randomized 1:1 upon completion of induction to chemotherapy without (Arm B) or with lestaurtinib (Arm C). In April 2013, AALL0631 was amended to non-randomly assign all IR and HR patients to Arm C due to the anticipated expiration of the study supply of lestaurtinib in June 2015 and a desire to optimize the accuracy of the estimated EFS of the IR and HR patients treated with Arm C. Efficacy phase schemas pre- and post-amendment are shown in Fig. S2.
Disease response assessments and off-protocol criteria
Disease response assessment was required after induction (week 6), induction intensification (week 10), and consolidation (week 20). Patients without morphologic remission (<5% marrow blasts, resolution of extramedullary disease) by the end of induction intensification (week 10) were deemed treatment failures (event), removed from protocol therapy, and followed for survival. Patients who achieved remission and then relapsed (event) were removed from protocol therapy and followed for survival. Patients removed from protocol therapy for other reasons were followed for events (relapse, second malignancy, and death in remission) and survival. Bone marrow (BM) minimal residual disease (MRD) testing was performed centrally in the Borowitz laboratory as an optional research assay. MRD results were not provided to investigators, not used for risk stratification, response evaluation, or treatment assignment, and will be reported separately.
Correlative laboratory assays
FLT3i PD
Blood for FLT3i PD was drawn during induction intensification (before the first dose on day 20, and at trough time points on days 24 and 27) and reinduction (before the first dose on day 5, and at trough time points on days 9, 12, and 19; Fig. S3). Samples were sent fresh to the Brown laboratory at Johns Hopkins University; plasma was isolated by centrifugation and cryopreserved. The FLT3i PD was determined using the PIA assay as described [22]. Briefly, a model cell line with activated FLT3 is incubated for 1 h with patient plasma from ≥1 pre-treatment and ≥1 post-treatment trough time points, then lysed and assessed for inhibition of FLT3 phosphorylation relative to the pre-treatment plasma using Western blotting and densitometry. As shown in Fig. S4, PIA was measured at five lestaurtinib trough time points relative to the pretreatment samples. Values ranged from 0% (no FLT3 inhibition) to 100% (full inhibition). For the purposes of proceeding from the safety/activity phase to the efficacy phase, adequate FLT3i PD was defined as achieving at least 90% FLT3 PIA in at least 3 of 5 trough time points for at least 3 of 5 evaluable patients. For the purposes of correlating FLT3i PD with EFS, the mean PIA of 5 troughs was calculated for each patient, and categorized based on pre-defined protocol criteria as “inhibited” (≥85%) or “uninhibited” (<85%). The threshold of 85% inhibition for correlation with efficacy was based on prior lestaurtinib studies in adult acute myeloid leukemia (AML) demonstrating that patients achieving trough PIA of at least 85% had better outcomes [23].
FLT3i ex vivo sensitivity (EVS)
Marrow (or peripheral blood, in patients with high circulating blast counts) collected from patients at study entry was sent fresh to the Brown laboratory. Leukemia blasts were enriched by Ficoll centrifugation. FLT3i EVS was determined using WST-1 cell proliferation assay [48 h cytotoxicity of diagnostic blasts exposed to 50 nM lestaurtinib vs. vehicle control; mean of triplicate wells; ranging from 0% (no cytotoxicity) to 100% (all cells killed)].
Statistical methods
Data frozen on March 31, 2019, are included in this report. AALL0631 was originally designed as a randomized trial with the primary endpoint to compare 3-year EFS from the time of randomization for Arm B vs. Arm C. Goal accrual was 162 IR/HR patients (81 per arm), providing 80% power to detect a hazard ratio of 0.62 with the addition of lestaurtinib (1-sided alpha = 0.15) by log-rank test. The impending expiration of the lestaurtinib supply prompted the modification of the AALL0631 design (April 2013) to non-randomly assign all newly-enrolled IR/HR patients to Arm C. The primary objective was changed to estimate 3-year EFS from the beginning of post-induction therapy for patients treated on Arm C at DL2, with an accrual goal of 80 evaluable Arm C patients allowing estimation of 3-year EFS with a maximum standard error of 5.6%. In the modified design, a secondary objective was to compare 3-year EFS of patients treated on Arms B and C. With 55 patients accrued to Arm B at the time of amendment and 80 patients on Arm C, there was 82% power to detect a hazard ratio of 0.62 with the addition of lestaurtinib (1-sided alpha = 0.15). EFS was defined as the time from post-induction treatment assignment to first event (treatment failure, relapse, second malignancy, death) or censored at the date of the last follow-up for event-free patients. Overall survival (OS) was defined as the time from post-induction treatment assignment to death or censored at the date of the last follow-up. Survival rates were estimated using the Kaplan–Meier method with standard errors [24, 25]. Survival curves were compared using the log-rank test. Cumulative incidence (CI) rates were computed using the CI function for competing risks, and comparisons were made using the K-sample test [26]. Continuous descriptive variables were compared using Wilcoxon Rank Sum tests and frequencies compared using Fisher’s exact or chi-square tests. All analyses were conducted using SAS® software version 9.4 (SAS Institute, Cary, NC), and R (http://www.R-project.org, version 2.13.1).
Results
Enrollment, risk assignment, and baseline characteristics
Of 218 patients enrolled, 210 were eligible (Fig. 1). Reasons for ineligibility included misdiagnosis (central review showed AML, n = 4) and improper informed consent (n = 4). Risk assignment for the eligible patients included 64 SR (30%), 111 IR (53%), and 35 HR (17%). Table 1 shows baseline characteristics for eligible IR/HR patients. Baseline characteristics and outcomes for SR patients will be described separately. Other than age (which defined risk group), there were no significant differences in baseline characteristics between the IR and HR patients, with the exception of CNS involvement; 17% of HR patients were CNS1 compared to 50% of IR. The most common KMT2A fusion partners were AFF1 (38%), MLLT1 (34%), MLLT3 (5%) and MLLT10 (4%), with partner unknown for 12%. As in prior studies [3–6], we noted a female pre-dominance in infant KMT2A-r ALL. Infant KMT2A-g ALL and non-infant ALL, by contrast, have a slight male pre-dominance [27].
Fig. 1. CONSORT diagram: the flow of patients from enrollment to treatment assignment.

Risk assignment followed induction (Supplemental Table S1) for eligible patients. Post induction treatment assignment was non-random during the safety/activity (S/A) phase, including 25 patients to Arm B (19 IR, 6 HR), 11 to Arm C-DL1 (5 IR, 6 HR), and 11 to Arm C-DL2 (6 IR, 5 HR). In addition, 28 patients (22 IR, 6 HR) were non-randomly assigned to Arm C-DL2 after the amendment shown in Supplemental Fig. S1.
Table 1.
Baseline characteristics of KMT2A-rearranged infants by risk group.
| IR n = 111 | HR n = 35 | IR + HR n = 146 | p-valuea IR vs. HR | Arm B n = 54 | Arm C n = 78 | p-valuea Arm B vs. Arm C | |
|---|---|---|---|---|---|---|---|
| Age dx (days) | p = 0.56a | ||||||
| Mean (sd) | 211 (76) | 44 (30) | 171 (98) | 179 (99) | 169 (96) | ||
| Median | 207 | 41 | 169 | 178 | 170 | ||
| Range | 92–360 | 0–88 | 0–360 | 1–353 | 0–360 | ||
| WBC (×109/L) | p = 0.25a | p = 0.29a | |||||
| Mean (sd) | 328 (500) | 406 (488) | 347 (496) | 434 (657) | 286 (351) | ||
| Median | 145 | 213 | 160 | 211 | 132 | ||
| Range | 2–4335 | 12–1800 | 2–4335 | 8–4335 | 2–1800 | ||
| # (%) | # (%) | # (%) | # (%) | # (%) | |||
| CNS status | p = 0.001 | p = 0.92 | |||||
| CNS1 | 55 (50) | 6 (17) | 61 (42) | 22 (41) | 35 (45) | ||
| CNS2 | 38 (34) | 20 (57) | 58 (40) | 23 (43) | 29 (37) | ||
| CNS3 | 16 (14) | 9 (26) | 25 (17) | 8 (15) | 13 (17) | ||
| Unknown | 2 (2) | 0 | 2 (1) | 1 (2) | 1 (1) | ||
| KMT2A Partner | p = 0.07 | p = 0.46 | |||||
| AFF1 | 42 (38) | 13 (37) | 55 (38) | 18 (33) | 31 (40) | ||
| MLLT1 | 35 (32) | 15 (43) | 50 (34) | 18 (33) | 29 (37) | ||
| MLLT3 | 8 (7) | 0 | 8 (5) | 1 (2) | 4 (5) | ||
| MLLT10 | 4 (4) | 2 (6) | 6 (4) | 4 (7) | 2 (3) | ||
| Other | 5 (5) | 4 (11) | 9 (6) | 5 (9) | 3 (4) | ||
| Unknown | 17 (15) | 1 (3) | 18 (12) | 8 (15) | 9 (12) | ||
| Immunophenotype | p = 0.36 | p = 1.0 | |||||
| B-ALL | 107 (96) | 32 (91) | 139 (95) | 52 (96) | 75 (96) | ||
| Otherb | 4 (4) | 3 (9) | 7 (5) | 2 (4) | 3 (4) | ||
| Sex | p = 0.85 | p = 0.11 | |||||
| Female | 63 (57) | 21 (60) | 84 (58) | 35 (65) | 39 (50) | ||
| Male | 48 (43) | 14 (40) | 62 (42) | 19 (35) | 39 (50) | ||
| Race | p = 0.81 | p = 1.0 | |||||
| White | 81 (73) | 27 (77) | 108 (74) | 41 (76) | 58 (74) | ||
| Non-white | 18 (16) | 4 (11) | 22 (15) | 7 (13) | 11 (14) | ||
| Unknown | 12 (11) | 4 (11) | 16 (11) | 6 (11) | 9 (12) | ||
| Ethnicity | p = 0.93 | p = 0.46 | |||||
| Hispanic | 25 (23) | 9 (26) | 34 (23) | 16 (30) | 16 (21) | ||
| Not Hispanic | 81 (73) | 25 (71) | 106 (73) | 36 (67) | 59 (76) | ||
| Unknown | 5 (5) | 1 (3) | 6 (4) | 2 (4) | 3 (4) |
IR intermediate risk, HR high risk, L liter.
Wilcoxon Rank Sum test; all other p-values are based on the Fisher’s Exact test.
Other: mixed phenotype B/myeloid (n = 5); Acute undifferentiated leukemia (n = 1); No data (n = 1; centrally reviewed sample inevaluable; local site assessed as B-ALL).
Induction outcomes for IR and HR patients
Among 146 IR/HR patients, 14 (10%) were removed from protocol before post-induction treatment assignment (Fig. 1). Five had infectious induction deaths (3% induction toxic death rate). Four deaths occurred in the first 22 IR/HR patients (18%), prompting changes to induction that significantly reduced induction mortality (2 in 124, 2%) [7]. Nine patients were removed for other reasons [infectious death soon after induction (n = 1), family/physician preference (n = 3), severe toxicity (veno-occlusive disease, n = 1), administrative error (n = 2), and unavailability of lestaurtinib due to international regulatory issues (n = 2)]. Of the 146 patients, 144 were evaluable for morphologic remission at the end of induction (two removed from protocol for non-events prior to evaluation): 113 (78%) achieved remission, 31 (22%) did not achieve remission [25 (17%) with >5% BM blasts, and 6 (4%) died]. These proportions were similar for IR and HR patients.
Safety/activity phase
Five IR patients received lestaurtinib at DL1. All were evaluable and without DLTs. Six IR patients received lestaurtinib at DL2. Five were evaluable and without DLTs. The inevaluable patient discontinued lestaurtinib during reinduction for toxicity unrelated to lestaurtinib. FLT3i PD was assessed for patients treated at DL2 using PIA (Table S3). Four of five IR patients treated at DL2 met the pre-defined criteria for adequate PIA; therefore DL2 (5 mg/kg/day) was deemed to be safe and biologically active, and AALL0631 moved to the efficacy phase for IR patients in January 2011.
Six HR pts received lestaurtinib at DL1. Five were evaluable and without DLTs. The inevaluable patient discontinued lestaurtinib due to parental refusal. Five HR patients received lestaurtinib at DL2. All were evaluable and there was 1 DLT (grade 4 typhlitis during neutropenia following reinduction, possibly related to lestaurtinib). Four of five HR patients treated at DL2 met the pre-defined adequate PIA criteria (Table S3), and therefore DL2 (4.25 mg/kg/day) was deemed to be safe and biologically active, and AALL0631 moved to the efficacy phase for HR patients in February 2012.
Efficacy phase
Adverse event analysis
AE rates were monitored during all post-induction blocks of chemotherapy without (Arm B; 54 patients) or with lestaurtinib (Arm C; 78 patients, 22 from the safety/activity phase and 56 from the efficacy phase: Fig. 1). We prospectively identified 9 specific “target” AEs of interest-based on prior experience with lestaurtinib and 11 AE system organ classes of interest-based on the overall incidence and/or potential seriousness. We compared the proportion of patients with grade 3 or greater AEs for each of these categories between Arms B and C during each post-induction course individually (Table S4) and combined across all reporting periods (Table 2). The only significant difference when combining all reporting periods was the incidence of diarrhea, which was 44% on Arm C and 26% on Arm B (p = 0.04). Thus, adding lestaurtinib to post-induction chemotherapy did not significantly increase the incidence or severity of clinically significant AEs.
Table 2.
Target adverse event and system organ class adverse event rates (grade 3 or greater) by treatment arm, combining all post-induction courses.
| Arm B, N = 54 # (%) | Arm C, N = 78 # (%) | p-valuea | |
|---|---|---|---|
| Target adverse event (AE) | |||
| ALT | 38 (70) | 51 (65) | 0.58 |
| AST | 28 (52) | 37 (47) | 0.72 |
| Neutrophils | 34 (63) | 58 (74) | 0.18 |
| Platelets | 18 (33) | 27 (35) | 1.00 |
| WBC | 11 (20) | 19 (24) | 0.68 |
| Anorexia | 15 (28) | 18 (23) | 0.55 |
| Diarrhea | 14 (26) | 34 (44) | 0.044 |
| Lung infection | 10 (19) | 10 (13) | 0.46 |
| Pneumonitis | 2 (4) | 1 (1) | 0.57 |
| System organ class (SOC) | |||
| Blood/lymphatic | 38 (70) | 56 (72) | 1.00 |
| Cardiac | 2 (4) | 9 (12) | 0.20 |
| Gastrointestinal | 40 (74) | 59 (76) | 0.84 |
| General/administration | 13 (24) | 20 (26) | 1.00 |
| Infections/infestations | 50 (93) | 76 (97) | 0.23 |
| Investigations | 48 (89) | 72 (92) | 0.55 |
| Metabolism/nutrition | 35 (65) | 56 (72) | 0.45 |
| Nervous | 5 (9) | 8 (10) | 1.00 |
| Respiratory/thoracic | 12 (22) | 24 (31) | 0.32 |
| Skin/subcutaneous | 8 (15) | 21 (27) | 0.13 |
| Vascular | 20 (37) | 23 (30) | 0.45 |
Fisher’s Exact test.
Remission induction and treatment failure by risk group
Morphologic remission was assessed at the end of induction intensification (week 10). Of 146 KMT2A-r patients (111 IR/35 HR), 138 (105 IR/33 HR) were evaluable for remission, with 8 (6 IR/2 HR) removed from the protocol for non-events prior to evaluation. Of 138 evaluable, 124 (90%) achieved remission, including 97 of 105 (92%) IR vs. 27 of 33 (82%) HR (p = 0.1). The treatment failure rate was 8 of 138 (6%) overall; 4 of 105 (4%) for IR and 4 of 33 (12%) for HR (p = 0.09). All treatment failures were for lack of BM remission, none for lack of resolution of extramedullary disease.
Survival from study entry by risk group
For 146 KMT2A-r patients, the 5-year EFS and OS (±SE) from study entry was 34 ± 4% and 41 ± 4%, respectively (Fig. 2A), with both 5-year EFS (IR: 40 ± 5% vs. HR: 17 ± 7%, p = 0.003) and OS (IR: 48 ± 5% vs. HR:20 ± 7%, p < 0.001) significantly better for IR compared to HR patients (Fig. 2B). Overall, 96/146 (66%) patients had events (Table 3). In a competing risk analysis of events, the CI of relapse/treatment failure was significantly greater for HR (3-year CI 63 ± 8%) compared to IR patients (44 ± 5%, p = 0.033; Fig. 2C). The site of relapse differed between HR (all 19 relapses included marrow) and IR patients (8/46 (17%) relapses were isolated extramedullary; 7 CNS and 1 cutaneous). Twenty-four deaths occurred as first events: six during induction, and 18 during remission (eight in patients on the protocol, and ten in patients off protocol). Seventeen of 24 (71%) first event deaths were due to infection. Infections were bacterial or fungal in the first year of treatment; late infections were primarily viral and Pneumocystis. There was no difference in the CI of death as the first event between IR and HR patients (3-year CI 20 ± 7% vs. 14 ± 3%, p = 0.48; Fig. 2D).
Fig. 2. Survival and cumulative incidence of events from study entry by risk group.
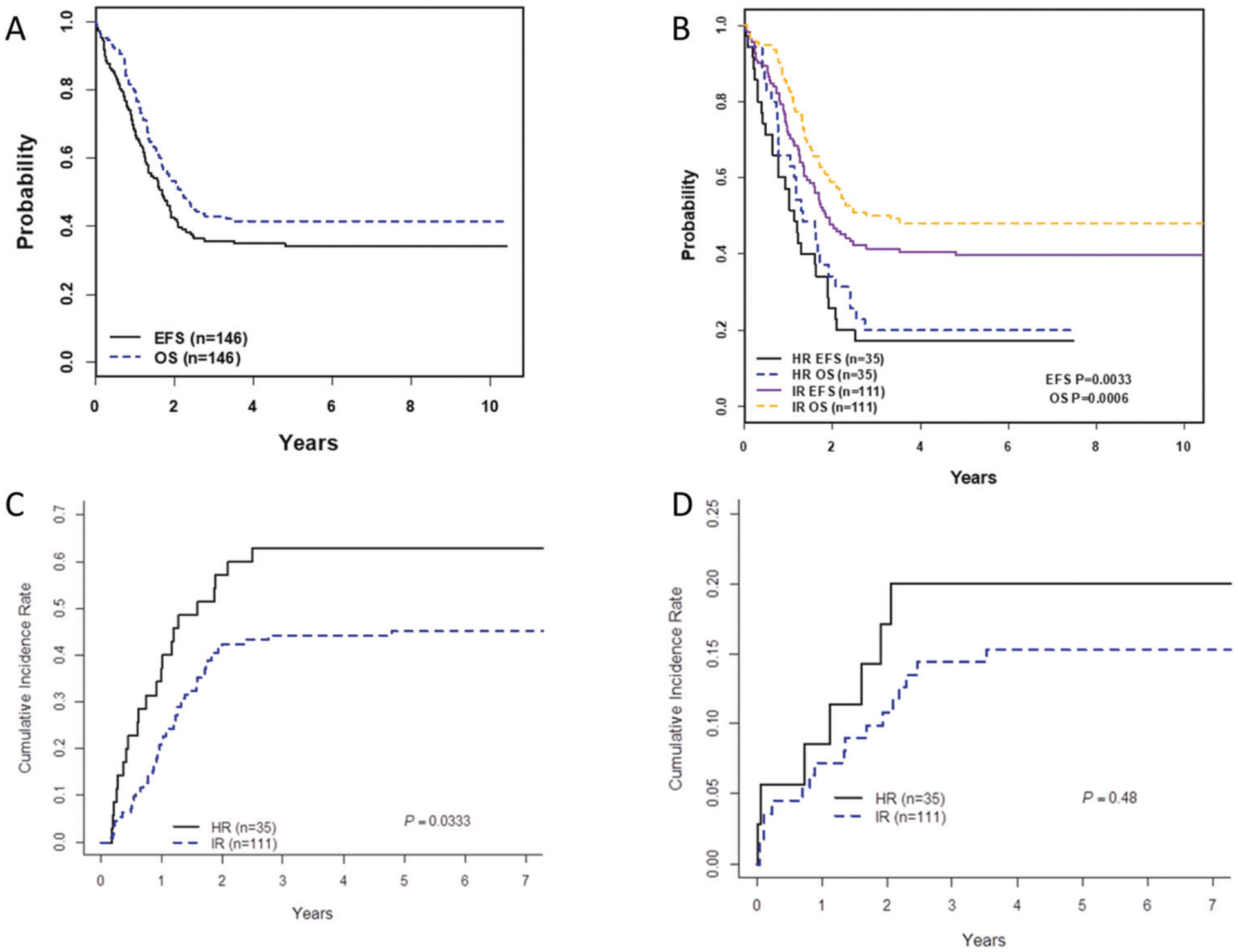
A EFS and OS for all 146 KMT2A-r patients. B EFS and OS for IR patients are superior to EFS and OS for HR patients. C Cumulative incidence of treatment failure or relapse, with death considered a competing event, was significantly higher for HR than IR patients. D Cumulative incidence of death as the first event, with treatment failure and relapse considered competing for events, was similar in HR and IR patients.
Table 3.
Post-induction events by risk group and treatment arm.
| IR (n = 111) | HR (n = 35) | Total (IR + HR) (n = 146) | Arm B (n = 54) | Arm C-DL2 (n = 67) | Total (Arm B + Arm C-DL2) (n = 121) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | N | % | N | % | |
| Treatment failurea | 4 | 4% | 3 | 9% | 7 | 5% | 3 | 6% | 4 | 6% | 7 | 6% |
| Relapseb | 46 | 41% | 19 | 54% | 65 | 45% | 23 | 43% | 32 | 48% | 55 | 45% |
| BM | 38 | 34% | 19 | 54% | 57 | 39% | 20 | 37% | 29 | 43% | 49 | 40% |
| iBM | 30 | 27% | 17 | 49% | 47 | 32% | 17 | 31% | 24 | 36% | 41 | 34% |
| cBM + CNS | 5 | 5% | 2 | 6% | 7 | 5% | 2 | 4% | 4 | 6% | 6 | 5% |
| cBM + Skin | 1 | 1% | – | – | 1 | 1% | – | – | 1 | 1% | 1 | 1% |
| cBM + Test | 2 | 2% | – | – | 2 | 1% | 1 | 2% | – | – | 1 | 1% |
| iEM | 8 | 7% | – | – | 8 | 5% | 3 | 6% | 3 | 4% | 6 | 5% |
| iCNS | 7 | 6% | – | – | 7 | 5% | 3 | 6% | 2 | 3% | 5 | 4% |
| iSkin | 1 | 1% | – | – | 1 | 1% | – | – | 1 | 1% | 1 | 1% |
| Death as the first event | 17 | 15% | 7 | 20% | 24 | 16% | 8 | 15% | 8 | 12% | 16 | 13% |
| Induction (infx) | 4 | 4% | 2 | 6% | 6 | 4% | – | – | – | – | – | – |
| Remission/on protocolc | 5 | 5% | 3 | 9% | 8 | 5% | 3 | 6% | 5 | 7% | 8 | 7% |
| Infx | 4 | 4% | 3 | 9% | 7 | 5% | 3 | 6% | 4 | 6% | 7 | 6% |
| Neuro | 1 | 1% | – | – | 1 | 1% | – | – | 1 | 1% | 1 | 1% |
| Remission/off protocol | 8 | 7% | 2 | 6% | 10 | 7% | 5 | 9% | 3 | 4% | 8 | 7% |
| Infx | 4 | 4% | – | – | 4 | 3% | 4 | 7% | – | – | 4 | 3% |
| TRM | 2 | 2% | 1 | 3% | 3 | 2% | 1 | 2% | 1 | 1% | 2 | 2% |
| Neuro | 1 | 1% | 1 | 3% | 2 | 1% | – | – | 1 | 1% | 1 | 1% |
| Cardiac aneurysm | 1 | 1% | – | – | 1 | 1% | – | – | 1 | 1% | 1 | 1% |
| Any event | 67 | 60% | 29 | 83% | 96 | 66% | 34 | 63% | 44 | 66% | 78 | 64% |
IR intermediate risk, HR high risk, DL2 dose level 2, BM bone marrow, EM extramedullary, i isolated, c combined, CNS central nervous system, Test testicular, infx infection, neuro neurologic (encephalopathy), TRM transplant-related mortality.
Treatment failure defined as without morphologic remission (<5% BM blasts, resolution of extramedullary disease) by the end of induction intensification.
Combined relapse defined as M2 or M3 marrow at any point after achieving remission with concomitant extramedullary relapse.
Includes two patients who died shortly after going off protocol therapy.
Survival from post-induction treatment assignment without vs. with lestaurtinib
A total of 132 KMT2A-r patients (101 IR/31 HR) completed induction and were randomized or assigned to post-induction therapy on Arm B (chemotherapy alone) or Arm C (chemotherapy plus lestaurtinib). Baseline characteristics (Table 1) were balanced between arms. AALL0631 was designed to compare the 3-year EFS from post-induction risk assignment for Arm B (n = 54) vs. Arm C receiving lestaurtinib at DL2 (n = 67; n = 11 during safety/activity phase, n = 56 during efficacy phase). Arm C patients receiving lestaurtinib at DL1 during the safety/activity phase (n = 11) were excluded from efficacy analysis (Fig. 1). The patient distribution between Arm B (44 IR, 10 HR) and Arm C DL2 (52 IR, 15 HR) was comparable (p = 0.66).
There were no differences in outcome according to the treatment arm. The 3-year EFS from post-induction treatment assignment for all KMT2A-r patients (IR/HR) was 39 ± 7% for Arm B vs. 36 ± 6% for Arm C (p = 0.67; Fig. 3A); and 3-year OS was 46 ± 7% vs. 45 ± 6% (p = 0.95; Fig. 3B). For IR patients, the 3-year EFS was 48 ± 8% Arm B vs. 40 ± 7% Arm C, p = 0.45 (Fig. 3C) and for HR patients were 0% Arm B vs. 20 ± 13% Arm C, p = 0.30 (Fig. 3D). Overall, 78 of 121 (64%) patients had events (Table 3). There were no differences between Arm B vs. Arm C in 3-year CI of relapse/treatment failure (48 ± 7% vs. 52 ± 6%, p = 0.6; Fig. 3E) or death as the first event (13 ± 5% vs. 12 ± 4%, p = 0.69; Fig. 3F). Five patients (2 Arm B, 3 Arm C) received off protocol SCT in first remission. Four had subsequent events and died (two of transplant-related toxicity, two from relapse) and one Arm C patient survived event-free.
Fig. 3. Survival and cumulative incidence of events from post-induction treatment assignment by treatment arm.
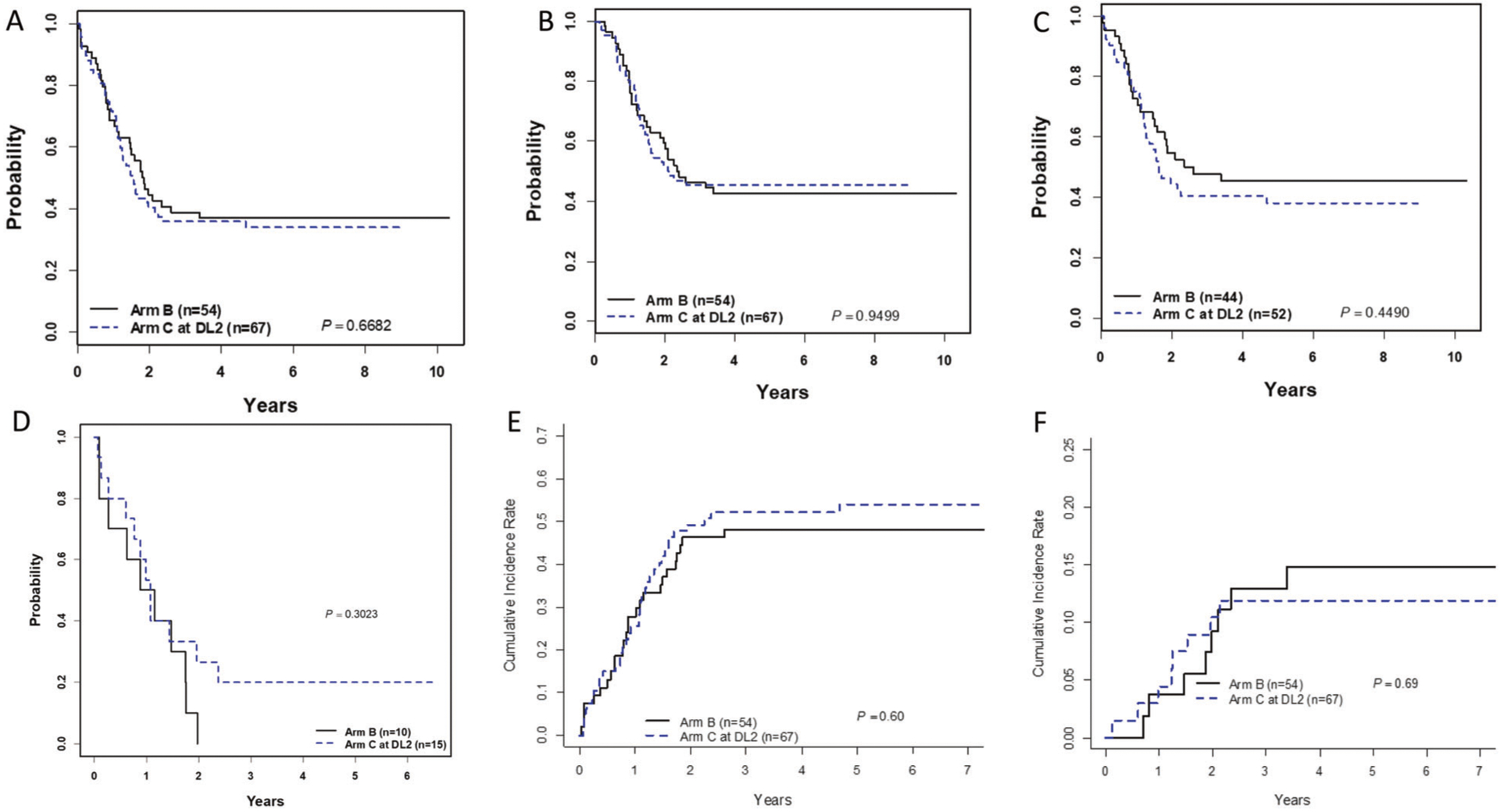
There is no significant difference in EFS (A) or OS (B) for all KMT2A-r patients assigned to Arm B vs. Arm C-DL2. Similarly, there is no significant difference in EFS for IR patients (C) or HR patients (D) between those assigned to Arm B vs. Arm C-DL2. Cumulative incidences of treatment failure/relapse (E) and death as a first event (F) are also not different between Arm B and Arm C-DL2.
FLT3 correlative laboratory studies
We hypothesized that correlative laboratory measures of FLT3i PD and FLT3i EVS would enhance our understanding of clinical responses to lestaurtinib on Arm C. Among 78 IR/HR Arm C patients (n = 67 at DL2 and n = 11 at DL1), data were available for FLT3i PD for 73 (94%) and FLT3i EVS for 62 (79%). For FLT3i PD, of 73 patients with PIA data, 27 (37%) were inhibited and 46 (63%) were uninhibited. Eleven of 20 (55%) HR patients and 16 of 53 (30%) IR patients were inhibited. FLT3i PD significantly correlated with survival (Fig. 4A). The 3-year EFS/OS from post-induction treatment assignment for FLT3-inhibited was 59 ± 10%/67 ± 9% vs. 28%±7/37 ± 7% for FLT3-uninhibited (p = 0.009/0.02). For FLT3i EVS, median cytotoxicity was calculated for all assayed samples on AALL0631 (n = 151, including KMT2A-r and KMT2A-g patients; median cytotoxicity 46%; Fig. S5). Consistent with our prior work [17, 28], samples were categorized as “sensitive” (above median) or “resistant” (below median). Of 62 KMT2A-r Arm C patients with EVS data, 42 (68%) were sensitive and 20 (32%) were resistant. Ten of 17 (59%) HR patients and 32/45 (71%) IR patients were sensitive. FLT3 EVS was significantly correlated with survival (Fig. 4B). The 3-year EFS/OS from post-induction treatment assignment for FLT3i sensitive was 52 ± 8%/62 ± 8% vs. 5 ± 5%/11 ± 7% for FLT3i resistant (p < 0.001 for both). We hypothesized that patients with both inhibited FLT3i PD and sensitive FLT3i EVS would have particularly good clinical outcomes. Of 58 patients with data for both assays, 17 (29%) were both inhibited and sensitive. This cohort had a 3-year EFS/OS of 88 ± 8%/94 ± 6%. The remaining 41 were uninhibited and/or resistant and had 3-year EFS/OS of 20% ± 6/29 ± 7% (p < 0.001 for both; Fig. 4C). Table 4 summarizes the FLT3i PD and EVS results.
Fig. 4. Survival according to results of FLT3 inhibitor laboratory correlative assays for patients treated with lestaurtinib (Arm C).
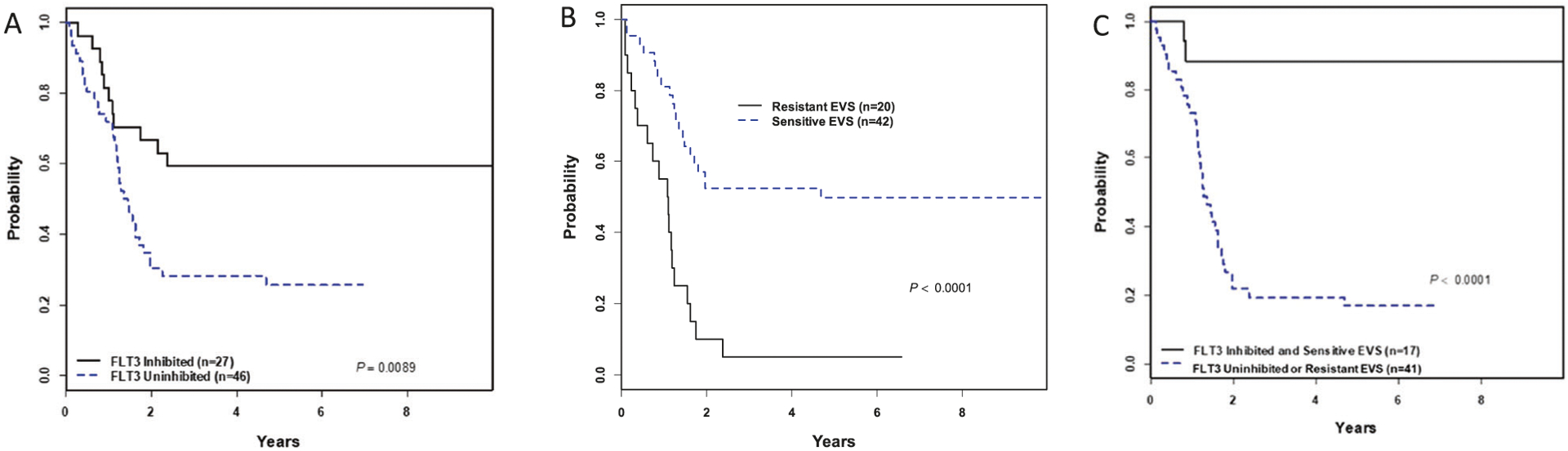
A The mean PIA of five troughs was calculated for each patient, and the patient categorized based on pre-defined protocol criteria as “FLT3 inhibited” (≥85%) or “FLT3 uninhibited” (<85%). Inhibited patients demonstrated significantly higher EFS than uninhibited patients. B Median ex vivo sensitivity (EVS) in lestaurtinib cytotoxicity assays was calculated for all assayed samples on the trial, and patients’ leukemic blasts collected at study entry were categorized as “sensitive EVS” (above median) or “resistant EVS” (below median). Sensitive patients demonstrated significantly higher EFS than resistant patients. C Patients that were both FLT3 inhibited and sensitive EVS had a strikingly higher EFS than patients that were either FLT3 uninhibited or resistant EVS.
Table 4.
Summary of correlation between correlative laboratory assays and survival for patients treated with lestaurtinib.
| FLT3i PD | FLT3i EVS | Combined | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibited (n = 27) | Uninhibited (n = 46) | p-value | Sensitive (n = 42) | Resistant (n = 20) | p-value | Inhibited and Sensitive (n = 17) | Uninhibited and/or Resistant (n = 41) | p-value | |
| 3 year EFS | 59% | 28% | 0.009 | 52% | 5% | <0.001 | 88% | 20% | <0.001 |
| 3 year OS | 67% | 37% | 0.02 | 62% | 11% | <0.001 | 94% | 29% | <0.001 |
FLT3i FLT3 inhibitor, PD pharmacodynamics, EVS ex vivo sensitivity, EFS event-free survival, OS overall survival.
Discussion
COG AALL0631 was an innovative trial for several reasons. First, it was the first to incorporate a molecularly targeted agent into the frontline treatment of KMT2A-r infant ALL. Second, it established a standard for how to define DLT for a molecularly targeted agent added to an intensive and toxic chemotherapy backbone in a vulnerable patient population, spurring an international publication of consensus guidelines [29]. Third, it was the first to prospectively incorporate a pharmacodynamic measure of biologic activity (FLT3 PIA) into pre-defined criteria for proceeding to an efficacy evaluation. Lestaurtinib had to be both tolerable and meet a validated pharmacodynamic benchmark [22] associated with clinical activity in prior adult trials [23, 30]. Fourth, the strategy of adding lestaurtinib sequentially after each block of chemotherapy was based on extensive preclinical modeling demonstrating maximal synergy with that schedule, compared with simultaneous dosing or the opposite sequence [18, 31]. This chemotherapy synergy was particularly important due to the lack of expected efficacy with single-agent FLT3i [32]. Finally, we incorporated correlative laboratory assays prospectively designed to test the hypothesis that a combination of ex vivo leukemic blast sensitivity and pharmacodynamic FLT3 inhibition would correlate with clinical responses. Together, these provided crucial insights into the observed outcomes.
Unfortunately, no overall benefit was observed with the addition of lestaurtinib (Fig. 3). This was attributable to two factors. The first was pharmacologic. Patients with good FLT3i PD benefited from lestaurtinib, but only 37% of patients met this criterion. Notably, FLT3i PD was better in the 20 patients treated during the safety/activity phase (overall mean 82.5%; mean for DL1 (n = 10) 75.8%; mean DL2 (n = 10) 89.2%) than in the 53 patients treated in the efficacy phase (mean 65.2%; p = 0.003). Possible explanations include random variation, the difference in compliance/adherence during the two phases, and a decrease in drug potency as it neared its expiration date. However, site-reported compliance was >95% during both the safety/activity and the efficacy phase. Further, we are unable to test the hypothesis that the potency of the drug supply decreased over the duration of the study, so that remains speculative. These data suggest that real-time intra-patient dose escalation for inadequate PD could enhance the efficacy of FLT3 inhibition for KMT2A-r infant ALL. It is also possible that a more potent FLT3i would have achieved better PD. While “next-generation” FLT3i such as gilteritinib [33] and quizartinib [34, 35] are orders of magnitude more potent against the FLT3 internal tandem duplication (FLT3-ITD) mutant, less than 5% of infant KMT2A-r ALLs harbor this mutation. Rather, wild-type FLT3 (FLT3-wt) is overexpressed and activated by autocrine FLT3 ligand stimulation [17]. The selective potency of gilteritinib and quizartinib for FLT3-ITD would therefore not necessarily translate into improved PD activity for FLT3-wt signaling.
The second factor contributing to the lack of benefit for lestaurtinib was intrinsic FLT3i resistance. Patients whose diagnostic leukemic blasts were ex vivo sensitive to the cytotoxic effects of lestaurtinib clearly benefited from the addition of lestaurtinib, but only 68% of patients met this criterion. Our EVS results (Fig. S5) confirmed prior studies by several laboratories that KMT2A-r ALL patient samples demonstrate greater FLT3i EVS than KMT2A-g ALL patient samples [15, 17, 19]. However, the EVS of the KMT2A-r samples is variable. It is likely that multiple factors contribute to this variability, including the capacity for various compensatory proliferation/survival pathways, the innate balance of expression of various pro- and anti-apoptotic factors, and the relative activity of various drug transport and/or catabolic mechanisms. Our data raise the possibility that using an EVS assay as the basis for pre-selection of patients with FLT3-sensitive leukemia could also enhance the efficacy of FLT3 inhibition for KMT2A-r infant ALL.
The combination of patient selection based on EVS and individualized dosing based on real-time PD could represent a particularly powerful personalized medicine strategy to enhance the efficacy of molecularly targeted therapy. While only 29% of patients treated with lestaurtinib on this study met both sensitivity and inhibition criteria, this small subset had a strikingly better 3-year EFS (88%) than those who did not (20%; p < 0.001). If personalized dosing could increase the proportion of inhibited patients from 37 to 90%, then with pre-selection of EVS sensitive patients, up to two-thirds of KMT2A-r infant ALL patients could be directed towards FLT3i therapy. The remaining one-third could pursue other promising strategies based on other KMT2A-based molecular vulnerabilities (KMT2A-menin interaction inhibitors e.g., [36]).
We recognize the limitations inherent in retrospective correlations between laboratory correlates and outcomes and are appropriately circumspect about them. Furthermore, we acknowledge that AALL0631 clearly failed to improve the overall outcomes of KMT2A-r infants, whose 5-year EFS on this trial (35%) is identical to those in prior trials [3–6], with the exception of a recent smaller Japanese trial that had 5-year EFS of 66% [37]. Most, unfortunately, we have yet again confirmed the dismal outlook for KMT2A-r infants diagnosed in the first 3 months of life, of whom only 20% survived 5 years. It is sobering that despite more than 20 years of logical clinical trial interventions (intensified cytotoxic therapy, SCT, AML-type chemotherapy, and now FLT3i), we remain unable to improve the prognosis for KMT2A-r infant ALL. Despite its limitations and lack of efficacious results, AALL0631 establishes that molecularly targeted agents can feasibly be tested in a rational manner in newly diagnosed HR leukemia populations. We remain hopeful that the lessons learned from this trial will enhance the success of future interventions.
Supplementary Material
Acknowledgements
The correlative laboratory studies were funded by grants NIH/NCI (K23 CA111728; PAB), Damon Runyon Cancer Research Foundation (CIA 30-06; PAB), Leukemia and Lymphoma Society (SCOR 7327-07 to DS and PAB, and LLS Scholar 2365-12 to PAB), and Children’s Cancer Foundation (PAB). The clinical trial was funded by grants to the children’s oncology group from the National Institutes of Health/National Cancer Institute (U10CA098543, U10CA098413, U10CA180886, and U10CA180899) and the St. Baldrick’s Foundation. Lestaurtinib was provided to study participants by Teva via a Collaborative Research and Development Agreement with the National Institutes of Health/National Cancer Institute/Cancer Therapy and Evaluation Program.
Conflict of interest
The authors report the following potential conflicts of interest: PAB: Scientific Advisory Committees for Novartis, Servier, Jazz, and Janssen. LG: Consultancy for Amgen, Novartis, Roche/Genentech; Equity Ownership for Amgen, Anchiano, Blueprint Medicines, Celgene, Clovis, Mirati, Sanofi Paris; Honoraria for Amgen, Roche/Genentech; Scientific Advisory Committee for Amgen; Data Safety and Monitoring Committee member for Novartis and Celgene. EAR: Research Funding from Pfizer; Data Safety and Monitoring Committee member for Celgene. MJB: Honoraria from Beckman Coulter. MLL: Scientific Advisory Committee for Medisix Therapeutics. SPH: Consultancy for Amgen, Bristol Myers Squibb,, and Novartis; Equity Ownership for Amgen; Honoraria for Jazz.
Footnotes
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-021-01177-6.
References
- 1.Brown P, Pieters R, Biondi A. How I treat infant leukemia. Blood. 2019;133:205–14. [DOI] [PubMed] [Google Scholar]
- 2.Brown P Treatment of infant leukemias: challenge and promise. Hematol Am Soc Hematol Educ Program. 2013;2013:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, Heerema NA, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the children’s oncology group. Blood. 2006;108:441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370: 240–50. [DOI] [PubMed] [Google Scholar]
- 5.Dreyer ZE, Hilden JM, Jones TL, Devidas M, Winick NJ, Will-man CL, et al. Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (cohort 3). Pediatr Blood Cancer. 2015;62:419–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pieters R, De Lorenzo P, Ancliffe P, Aversa LA, Brethon B, Biondi A, et al. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the Interfant-06 protocol: results from an international phase III randomized study. J Clin Oncol. 2019;37:2246–56. [DOI] [PubMed] [Google Scholar]
- 7.Salzer WL, Jones TL, Devidas M, Dreyer ZE, Gore L, Winick NJ, et al. Decreased induction morbidity and mortality following modification to induction therapy in infants with acute lymphoblastic leukemia enrolled on AALL0631: a report from the children’s oncology group. Pediatr Blood Cancer. 2015;62:414–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sison EA, Brown P. Does hematopoietic stem cell transplantation benefit infants with acute leukemia? Hematol Am Soc Hematol Educ Program. 2013;2013:601–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dreyer ZE, Dinndorf PA, Camitta B, Sather H, La MK, Devidas M, et al. Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the children’s oncology group. J Clin Oncol. 2011;29:214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mann G, Attarbaschi A, Schrappe M, De Lorenzo P, Peters C, Hann I, et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 study. Blood. 2010;116: 2644–50. [DOI] [PubMed] [Google Scholar]
- 11.Van der Velden VHJ, Corral L, Valsecchi MG, Jansen MWJC, De Lorenzo P, Cazzaniga G. et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia. 2009;23: 1073–9. [DOI] [PubMed] [Google Scholar]
- 12.Kang H, Wilson CS, Harvey RC, Chen IM, Murphy MH, Atlas SR, et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a children’s oncology group study. Blood. 2012;119:1872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stam RW, Schneider P, Hagelstein JAP, Van der Linden MH, Stumpel DJPM, de Menezes RX. et al. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood. 2010;115:2835–44. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7. [DOI] [PubMed] [Google Scholar]
- 15.Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3:173–83. [DOI] [PubMed] [Google Scholar]
- 16.Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, et al. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004;103:1085–8. [DOI] [PubMed] [Google Scholar]
- 17.Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. 2005;105:812–20. [DOI] [PubMed] [Google Scholar]
- 18.Chillon MC, Gomez-Casares M, Lopez-Jorge C, Rodriguez-Medina C, Molines A, Sarasquete ME, et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia. 2012;26:2360–6. [DOI] [PubMed] [Google Scholar]
- 19.Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, den Boer ML, Pieters R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood. 2007;110:2774–5. [DOI] [PubMed] [Google Scholar]
- 20.Brown P, Levis M, McIntyre E, Griesemer M, Small D. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia. 2006;20: 1368–76. [DOI] [PubMed] [Google Scholar]
- 21.Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005;106: 2484–90. [DOI] [PubMed] [Google Scholar]
- 22.Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 25.Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br J Cancer. 1977;35:1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–54. [Google Scholar]
- 27.Schultz KR, Pullen DJ, Sather HN, Shuster JJ, Devidas M, Borowitz MJ, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the pediatric oncology group (POG) and children’s cancer group (CCG). Blood. 2007;109:926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown P, Meshinchi S, Levis M, Alonzo TA, Gerbing R, Lange B, et al. Pediatric AML primary samples with FLT3/ITD mutations are preferentially killed by FLT3 inhibition. Blood. 2004;104:1841–9. [DOI] [PubMed] [Google Scholar]
- 29.Horton TM, Sposto R, Brown P, Reynolds CP, Hunger SP, Winick NJ, et al. Toxicity assessment of molecularly targeted drugs incorporated into multiagent chemotherapy regimens for pediatric acute lymphocytic leukemia (ALL): review from an international consensus conference. Pediatr Blood Cancer. 2010;54:872–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knapper S, Russell N, Gilkes A, Hills RK, Gale RE, Cavenagh JD, et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood. 2017;129:1143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104:1145–50. [DOI] [PubMed] [Google Scholar]
- 32.Zwaan CM, Soderhall S, Brethon B, Luciani M, Rizzari C, Stam RW, et al. A phase 1/2, open-label, dose-escalation study of midostaurin in children with relapsed or refractory acute leukaemia. Br J Haematol. 2019;185:623–7. [DOI] [PubMed] [Google Scholar]
- 33.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381:1728–40. [DOI] [PubMed] [Google Scholar]
- 34.Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20:984–97. [DOI] [PubMed] [Google Scholar]
- 35.Cooper TM, Cassar J, Eckroth E, Malvar J, Sposto R, Gaynon P, et al. A phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: a therapeutic advances in childhood leukemia & lymphoma (TACL) study. Clin Cancer Res. 2016;22:4014–22. [DOI] [PubMed] [Google Scholar]
- 36.Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell. 2019;36:660.673.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomizawa D, Miyamura T, Imamura T, Watanabe T, Moriya Saito A, Ogawa A, et al. A risk-stratified therapy for infants with acute lymphoblastic leukemia: a report from the JPLSG MLL-10 trial. Blood. 2020;136:1813–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.