

Abstract
Nicotinic acetylcholine receptors (nAChRs) are pentameric ligand-gated ion channels that play a central role in neuronal and neuromuscular signal transduction. Here, we have developed FANG ligands, fibronectin antibody-mimetic nicotinic acetylcholine receptor-generated ligands, using mRNA display. We generated a 1 trillion-member primary e10FnIII library to target a stabilized α1 nicotinic subunit (α211). This library yielded 270000 independent potential protein binding ligands. The lead sequence, α1-FANG1, represented 25% of all library sequences, showed the highest-affinity binding, and competed with α-bungarotoxin (α-Btx). To improve this clone, a new library based on α1-FANG1 was subjected to heat, protease, binding, off-rate selective pressures, and point mutations. This resulted in α1-FANG2 and α1-FANG3. These proteins bind α211 with KD values of 3.5 nM and 670 pM, respectively, compete with α-Btx, and show improved subunit specificity. α1-FANG3 is thermostable (Tm = 62 °C) with a 6 kcal/mol improvement in folding free energy compared with that of the parent α1-FANG1. α1-FANG3 competes directly with the α-Btx binding site of intact neuromuscular heteropentamers [(α1)2β1γδ] in mammalian culture-derived cellular membranes and in Xenopus laevis oocytes expressing these nAChRs. This work demonstrates that mRNA display against a monomeric ecto-domain of a pentamer has the capability to select ligands that bind that subunit in both a monomeric and a pentameric context. Overall, our work provides a route to creating a new family of stable, well-behaved proteins that specifically target this important receptor family.
Graphical Abstract
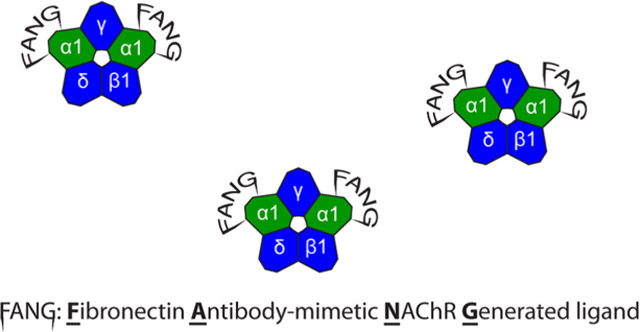
Nicotinic acetylcholine receptors (nAChRs) make up a superfamily of ligand-gated ion channels located at both pre- and postsynaptic junctions.1 nAChRs are a subset of the Cys-loop receptor family that also includes GABAA, serotonin, and glycine receptors.2,3 nAChRs are composed of homo- or heteropentamers. One distinction among heteropentameric nAChRs is neuronal type and muscle type. The muscle-derived nAChR is composed of two α1, β1, γ, and δ or ε subunits, while the neuronal heteropentamers contain only α (α2–10) and β (β2–4) subunits.4,5 In all nAChRs, acetylcholine (ACh) binds at the interface between an α subunit and an adjacent non-α subunit. The α subunit forms most of the contacts with ACh and is termed the “primary” contributor to the binding interface. ACh binding activates the gate of the receptor, revealing the open channel on the axis of the pentamer.6 At the muscle receptor, several existing drugs target the α1-containing interface and suppress spasms during anesthesia.7–9
The standard reagent for binding and recognition of the extracellular domain of α1 is α-bungarotoxin. α-Btx is an 8 kDa toxin isolated from the Taiwanese banded krait (Bungarus multicinctus), and similar toxins occur in other elapid snakes. These toxins have a three-finger tertiary structure that uses four disulfide bonds to maintain the toxin structure.10 The tip of one finger inserts into the agonist binding site of α1.11 This toxin stabilizes an inactive conformation of the α1 subunit, blocking responses to the agonist, blocking nerve–muscle transmission, and paralyzing the snake’s prey.5,12–14 α-Btx is highly stable and has a high affinity for α1 (KD = 0.2 nM), rendering it one of the most useful reagents for studying α1.15 α-Btx was crucial in obtaining the crystal structure of the extracellular domain of mouse α1 because the toxin stabilized the normally dynamic C loop, which is vital for obtaining resolvable crystals of the α1/α-Btx complex.16 The elapid toxins appear to have evolved in the snake salivary gland from members of the lynx/Ly6 family.17
Despite its widespread use in research, α-Btx is an imperfect tool for the study of α1. α-Btx binds α1 but also binds to paralogous nAChR subunits α7 and α9. The affinity of α-Btx for α7 (KD = 1.8 nM18) is similar to that for α1 (KD = 0.27 nM16), while the level of binding of α-Btx to α9 (KD = 66 nM19) is lower but still significant.20 Additionally, there are no publications about convenient α-Btx synthesis or expression in a renewable system due to the disulfide-rich nature of α-Btx. Other strategies to alter the toxin, such as excision of the loops in α-Btx responsible for contacting α1, produce peptides with specificities and affinities lower than those of the intact toxin.21 Also problematic is the fact that the study of nAChR subunits in vivo with α-Btx is complicated by its nAChR blockade, which paralyzes test organisms and requires tedious maintenance on a respirator.22 The limitations of α-Btx render it an undesirable starting point for the generation of new α1 binding reagents and argue for the development of new ligands.
Several previous efforts tried to find alternative reagents to α-Btx with improved properties. These approaches included using rabies virus glycoprotein (RVG) and even regions of the gp120 protein of the human immunodeficiency virus (HIV).21,23 Unfortunately, these attempts generated only peptides and proteins with specificities similar to and affinities lower than those of α-Btx;21,23 consequently, α-Btx has remained the standard reagent for α1 research for several decades.
Here, we used mRNA display to generate novel ligands for α1. We used human fibronectin,24 a scaffold containing variable loops, for our library (Figure 1b). mRNA display enables the design of proteins and peptides via in vitro selection and directed evolution experiments using high-complexity libraries (≤1014 independent sequences) in a strictly monomeric format, with precise control over both binding conditions and library sequence content.24,25 Our prior work demonstrates that libraries may be constructed using a well-defined protein domain bearing variable loops similar to antibodies.24–28 Our goal was to create an α1 reagent (1) with selectivity for the α1 nAChR subunit, (2) with a binding affinity similar to that of α-Btx, (3) that could be genetically encoded for protein expression and in vivo experiments,29 and (4) to help determine if in vitro-developed ligands would alter the ACh-induced current in the muscle nAChR.
Figure 1.
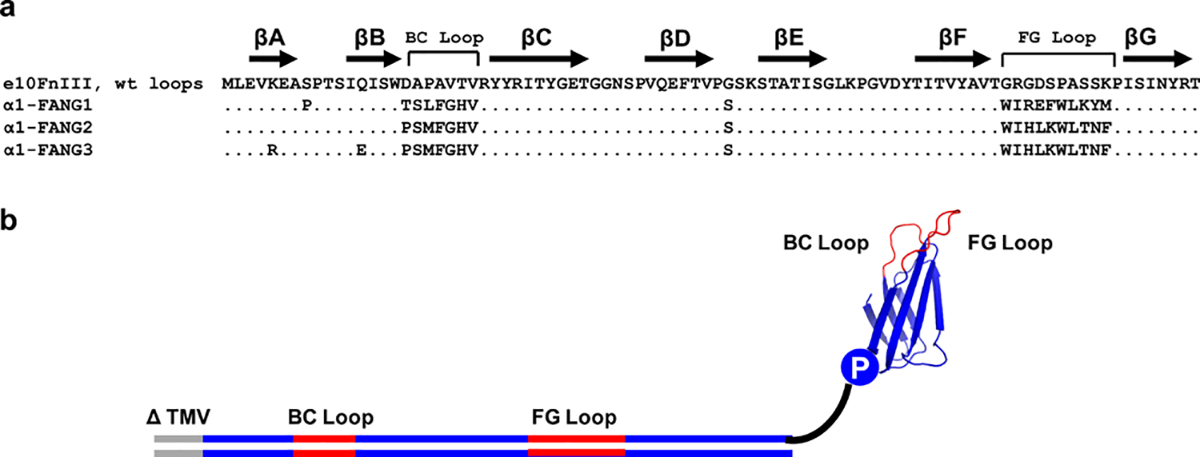
α1-FANG protein sequences and library design. (a) α1-FANG protein sequences. (b) An mRNA display fibronectin selection library contains a promoter sequence (ΔTMV), a constant scaffold with two randomized regions (BC loop and FG loop), and a covalent linkage via puromycin to the nascent fibronectin protein.
RESULTS AND DISCUSSION
Primary Selection To Generate α1-FANG1.
The goal of this work was to generate high-affinity, high-specificity, recombinant protein reagents directed at the α1 subunit of the nAChR. To do this, we used an antibody-mimetic library termed e10FnIII in selections against the soluble ecto-domain of α1, which contains three stabilization mutations (V8E/W149R/V155A), termed α211.15,16,27,30 Target-specific binding was observed in round 3, with appreciable binding over background in round 6 (Figure 2a). Once appreciable binding over background appeared in radiolabeled pull-down assays, pool 6 was cloned and sequenced; six clones were assayed for function. Clone 6.4, dubbed α1-FANG1, exhibited the best binding and comprised approximately 25% of the pool sequences (Figures 1a and 2b). Radiolabeled α1-FANG1 bound immobilized α211 with the highest percentage among tested sequences, at 14% of total counts introduced. The initial data we obtained motivated us to focus on characterization of α1-FANG1 and its properties.
Figure 2.
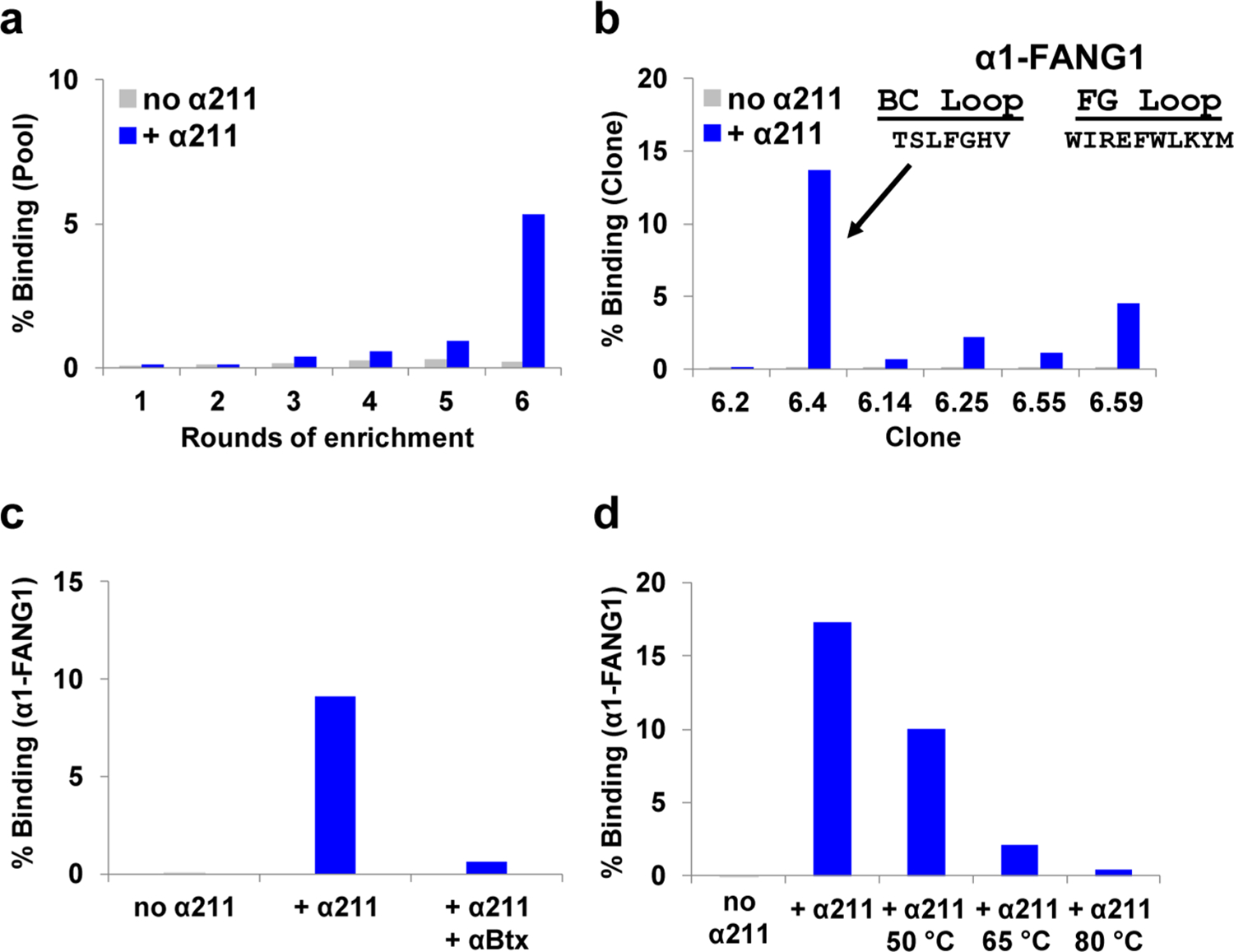
Naïve fibronectin selection against α211. (a) mRNA display selection for the ecto-domain of α211. Binding over background was apparent at pool 3 and reached an appreciable level by pool 6. (b) Radioactive pull down of selected pool 6 fibronectin clones against 100 pmol of α211. We dubbed the clone with the highest level of binding α1-FANG1. (c) Competition of α1-FANG1 with α-Btx. 35S-labeled α1-FANG1 binds beads with immobilized α211; preincubation of α-Btx with α211-immobilized beads causes a decrease in the number of detectable counts of radiolabeled α1-FANG1. (d) α1-FANG1 performance under temperature stress. Pull down of radiolabeled α1-FANG1 against α211 shows α1-FANG1 inactivation at high temperatures.
Preincubation of α-Btx with α211 immobilized on beads greatly reduced the level of binding of α1-FANG1 to α211 (Figure 2c), indicating that α1-FANG1 recognized an epitope similar to α-Btx and had potential as a starting point for generating a new α1 reagent. While α1-FANG1 expressed in reticulocyte lysate seemed to be highly functional in pull-down experiments, expression in Escherichia coli gave rise to aggregation and smeared bands when complexed with α211 in gel shift assays (data not shown). α1-FANG1 is also sensitive to temperature (Figure 2d).
Maturation Selection Generates α1-FANG2 and α1-FANG3.
We sought to improve the affinity, folding stability, and protease resistance of α1-FANG1. Rather than mutating the scaffold, however, our strategy optimized the protein by varying only the loop sequences. The rationale for this approach was to maintain the conserved human-derived scaffold while more fully exploring the remaining sequence space at our randomized positions.
For the maturation selection, we created a doped library based on the α1-FANG1 sequence (Figure 2b) where each position was 58% wild type at the DNA level (19–40% wild type at the amino acid level). We then introduced two pressures: thermal denaturation and proteolytic degradation. First, using the data from Figure 2d, we chose a 10 min incubation step at 65 °C as a thermal challenge. Second, we chose Proteinase K treatment, which degrades unfolded protein across a broad range of amino acid residues and digests unfolded protein more rapidly than folded protein,31,32 which aided our goal of improved protease resistance and stability.
We performed five rounds of mRNA display with the maturation selection library against α211 immobilized on beads (Figure 3a). At round 3, a 10 min, 65 °C incubation step was added. Round 4 added a 1 min exposure to Proteinase K, and round 5 included an off-rate selection by allowing α-Btx to compete with a fibronectin pool already bound to α211. We observed binding over background in round 2 (Figure 3a). In round 3, heat and protease both inactivated the library; by round 5, however, heat and protease treatment resulted in little loss of library binding. Pools 4 and 5 were cloned and sequenced (Figure 3b); the five most represented clones were assayed for function (Figure 3c). One sequence, α1-FANG2, exhibited the best resistance to the temperature and proteolytic challenges from selection (Figure 3c). Further analysis of α1-FANG2 showed a clear binding preference for α211 over the wild-type rat α9 ecto-domain in radioactive pull-down assays (Figure 3d). In this assay, α1-FANG2 bound α211 at 78% and bound α9 at only 0.5%, a 150-fold difference.
Figure 3.
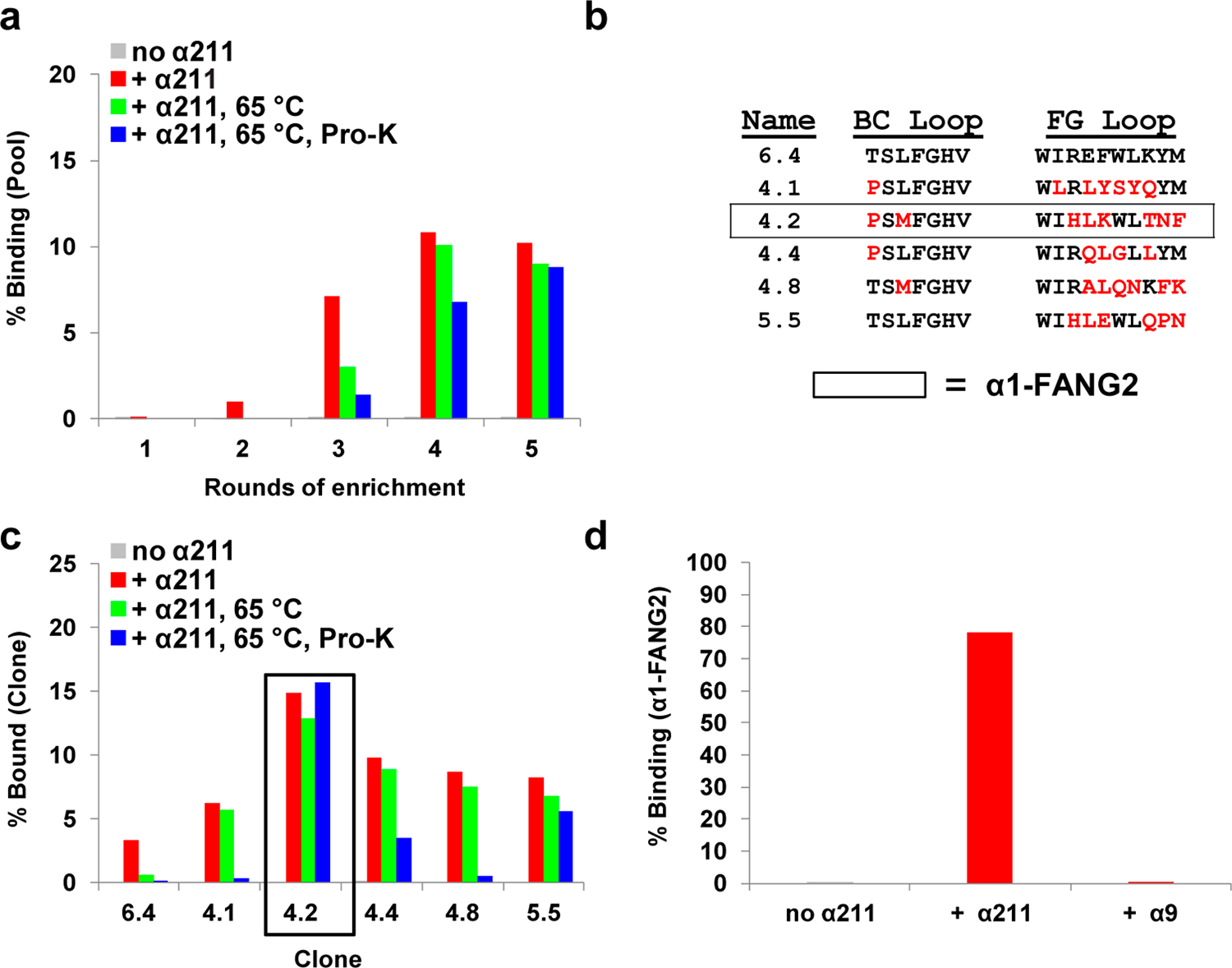
Affinity maturation and stabilization of α1-FANG1. (a) Rounds 1–5 of the maturation selection show increased resistance to introduced pressures as the rounds proceed. Rounds 3–5 were exposed to heat in the form of a 65 °C incubation. Rounds 4 and 5 were exposed to Proteinase K (Pro-K). Round 5 included an off-rate competition with α-Btx. (b) Sequence information for clones derived via maturation selection, with α1-FANG2 highlighted. (c) Behavior of individual clones from α1-FANG1-based maturation selection. Individual clones show a range of resistance to the various selective pressures. (d) Comparative pull downs against α211 and the ecto-domain of rat α9 show that α1-FANG2 is highly selective for α211 over α9.
α1-FANG2 exhibits a stability higher than that of α1-FANG1 but still showed some tendency toward aggregation. To address this, we constructed several scaffold mutants with the intent of improving the stability. Two of the positions we chose to optimize were Lys11 and Gln19, which were previously found to improve clone stability.24,28 Our characterization identified two point mutations that further stabilized these positions in the e10FnIII scaffold, K11R and Q19E (Figure 1a), giving a final protein less prone to aggregation, α1-FANG3.
Tm Determination and ΔG°unfolding,303 K Calculations for α1-FANG Proteins.
With a highly stable protein in hand, we decided to quantify the ΔG°unfolding,303 K for each of our α1-FANG proteins. We measured the change in molar ellipticity at θ222 of purified α1-FANG1, α1-FANG2, and α1-FANG3 protein by circular dichroism (CD) over a range of temperatures to generate melting curves (Figure 4a). We then fit the curves using previously published procedures.33 The melting temperature for α1-FANG1 was 45 °C. The Tm for α1-FANG2 was 69 °C, while α1-FANG3 exhibited a lower Tm of 62 °C (Table 1). Maturation selection helped select for a protein that was 17–24 °C more thermostable than the parent clone. The dramatic increase in Tm for α1-FANG2 and α1-FANG3 indicates that the introduced pressures in selection successfully enhanced stability. Although the Tm of α1-FANG3 is lower than that of α1-FANG2, the profile of the Tm for α1-FANG3 shows a narrower transition zone, characteristic of a larger enthalpy and more cooperative transition.
Figure 4.
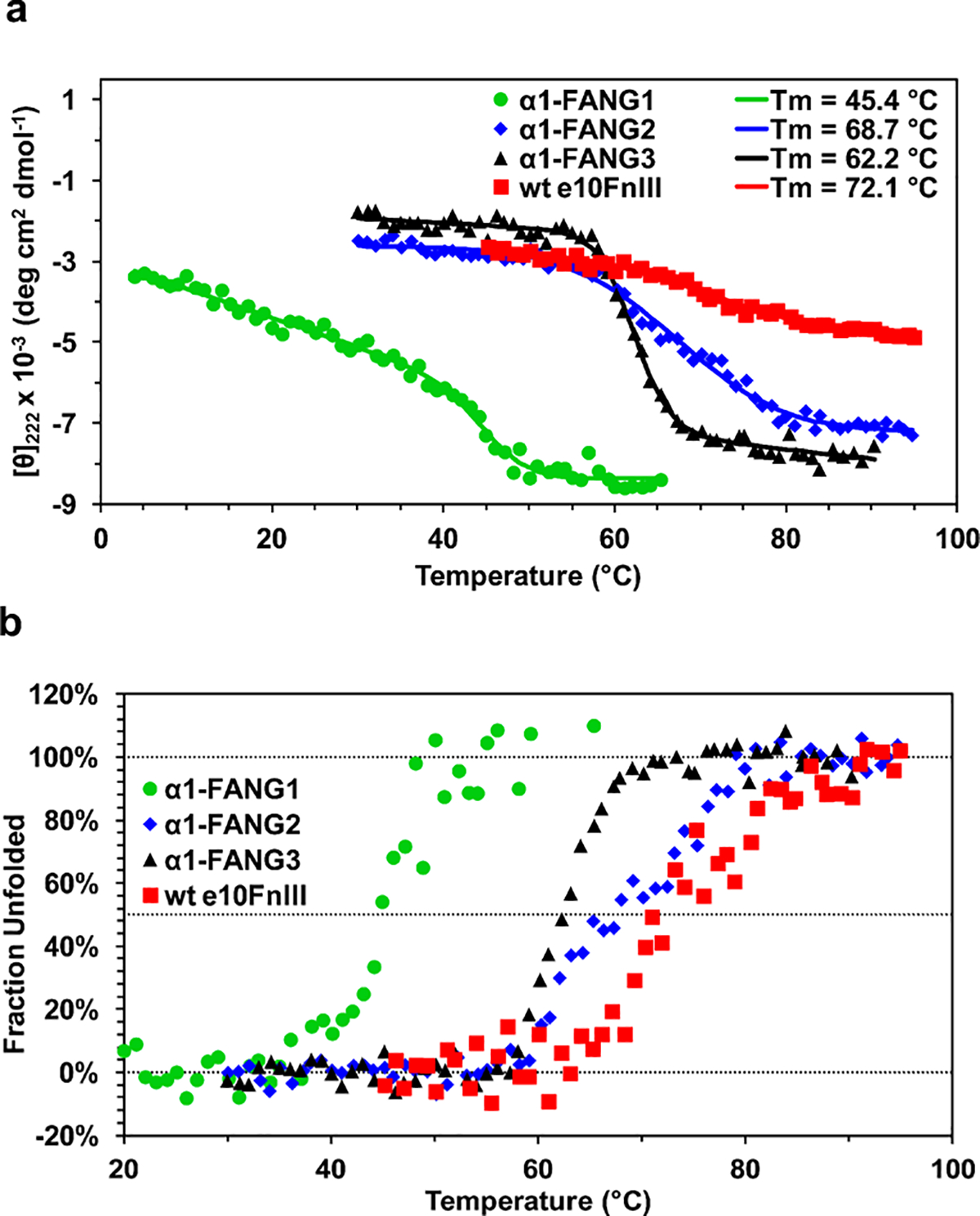
Melting curves of α1-FANG ligands. (a) Melting curves for α1-FANG1, α1-FANG2, and α1-FANG3 scaffolds at θ222. α1-FANG3 shows the most defined transition between two folded states. (b) Folding curves for the clones mentioned above showing the transition from folded to unfolded states.
Table 1.
Tm and ΔGunfolding,303 K for e10FnIII and α1-FANG Ligands
| fibronectin | Tm (°C) | ΔG°unfolding,303 K (kcal/mol) | KD for α211 |
|---|---|---|---|
| wt10FnIIIΔ1–7 | no data | 7.4 ± 0.327 | not available |
| e10FnIII, wild-type loops | 72 | 6.8 ± 1.1 | not available |
| α1-FANG1 | 45 | 3.4 ± 0.9 | 7.9 nM |
| α1-FANG2 | 69 | 4.7 ± 0.7 | 3.5 nM |
| α1-FANG3 | 62 | 10.4 ± 1.0 | 670 pM |
We also used CD to measure the melting temperature of the enhanced fibronectin scaffold (e10FnIII) that composes our library. For this measurement, we substituted the BC and FG loops of wild-type fibronectin into our enhanced scaffold, which allowed us to have a direct comparison of our clones with something analogous to the native protein. The Tm for e10fnIII is 72 °C. α1-FANG1 has a Tm lower than that of e10FnIII, but α1-FANG2 and α1-FANG3 have melting temperatures within 10 °C of that of the starting scaffold (Table 1).
Using the CD data (Figure 4b), we calculated the ΔG°unfolding,303 K for each α1-FANG protein (Table 1) using an extrapolation of the CD transition zone to 303 K (Supplementary Figure 1). e10FnIII with wild-type loops had a ΔG°unfolding,303 K of 6.8 ± 1.1 kcal/mol, which is in agreement with the published value of 7.4 ± 0.3 kcal/mol for the ΔG°unfolding,303 K of wild-type fibronectin Δ1–7 (wtFnIIIΔ1–7).27 The ΔG°unfolding,303 K for α1-FANG1 is 3.4 ± 0.9 kcal/mol. The ΔG°unfolding,303 K for α1-FANG2 is 4.7 ± 0.7 kcal/mol, showing that on its own, maturation selection improved the folding stability of the protein. The ΔG°unfolding,303 K of α1-FANG2 is 2.1–2.5 kcal/mol lower than that of wtFnIIIΔ1–7 or e10FnIII with wild-type loops. The Tm of 69 °C for α1-FANG2 echoed these data, as it is slightly lower than the Tm of e10FnIII (72 °C) with wild-type loops. Although α1-FANG3 had a Tm lower than those of α1-FANG2 and e10FnIII with wild-type loops, it had a ΔG°unfolding,303 K larger than those of both proteins, with a value of 10.4 ± 1.0 kcal/mol. The introduced point mutation in α1-FANG3 added stability to α1-FANG2, indicating a more stable scaffold. The combination of pressures during the selection process and added mutation directly contributed to the high stability of α1-FANG3.
KD Determination of α1-FANG Proteins.
To test the affinity of α1-FANG2 and α1-FANG3, we determined the forward (k1) and reverse (k−1) rate constants for complex formation. α1-FANG2 bound α211 with a KD of 3.5 nM, and α1-FANG3 bound α211 with a KD of 670 pM (Table 1 and Supplementary Figure 2). The increased stability of the introduced point mutations enhanced the affinity of α1-FANG3 for α211.
Native Gel Assays To Test α1-FANG3 Binding of α211 in Free Solution.
To further characterize α1-FANG3, we tested the ability of α1-FANG3 to bind α211 in a native gel shift assay. There, α1-FANG3 incubated with α211 showed a strong band shift when run on a native gel (Figure 5). The gel shift was similar to that given when the α211/α-Btx complex is run on a native gel, but at a higher level of migration (Figure 5a). After incubation of α1-FANG3 at 37 °C for 30 min, α1-FANG3 was still stable and able to bind to α211 as indicated by a strong gel shift, confirming the temperature stability shown in the maturation selection clone binding assays. Simultaneous incubation of a 1:1 α-Btx:α1-FANG3 molar ratio with α211 resulted in a band shift corresponding to a mixture of the α211/α1-FANG3 complex and the α211/α-Btx complex. α1-FANG3 thus appeared to bind to α211 with an affinity on par with that of α-Btx. An increase of the α-Btx:α1-FANG3 ratio resulted in a decrease in the intensity of the α211/α1-FANG3 complex band and an increase in the intensity of the α211/α-Btx complex band. The gel shift assays also confirmed the high specificity of α1-FANG3 for α211 as α9 did not generate any observable band complex, even at a high α1-FANG3 concentration (Figure 5b).
Figure 5.
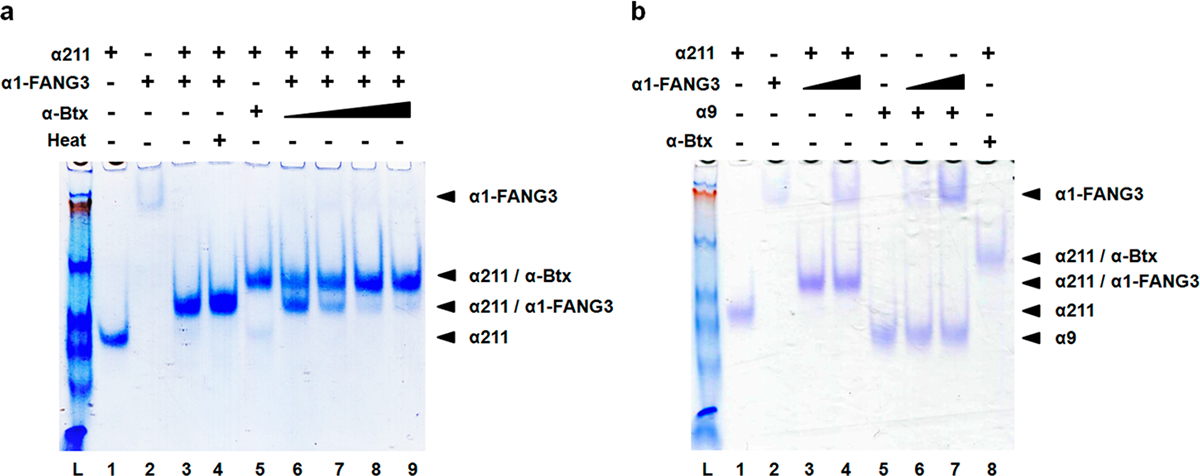
Native gel assays show α1-FANG3 binds α211 in solution. (a) Assay of binding of α1-FANG3 and α-Btx to α211. The α211:α-Btx and α211:α1-FANG3 ratios are 1:1 except in lanes 6–9 [α211:α-Btx:α1-FANG3 ratios of 1:1:1 (lane 6), 1:2:1 (lane 7), 1:5:1 (lane 8), and 1:10:1 (lane 9)]. α211 (lane 1), α1-FANG3 (lane 2), and their complex (lane 3) show distinct mobilities under native gel conditions. Heating at 37 °C for 30 min does not affect the α211/α1-FANG3 complex (compare lanes 3 and 4). The α211/α-Btx complex (lane 5) can also be distinguished from the α211/α1-FANG3 complex on the gel. In direct competition for binding to α211, a 1:1 α-Btx:α1-FANG3 ratio gives a gel shift that corresponds to both the α211/α1-FANG3 complex and the α211/α-Btx complex (lane 6). Increasing the α-Btx:α1-FANG ratio upon co-incubation with α211 decreases the intensity of the α211/α1-FANG3 complex band and increases the intensity of the α211/α-Btx complex band (compare lanes 7–9). (b) α1-FANG3 binds α211 specifically with no binding to related family member α9. The α1-FANG3:α9 and α1-FANG3:α211 ratios are 1:1, except in lanes 4 and 7 (2:1). α9 (lane 5) shows no complex with α1-FANG3 (lane 6), while distinct mobilities for α211 (lane 1), α1-FANG3 (lane 2), and their complex (lane 3) as seen in panel a remain.
Competition of α1-FANG3 with 125I-Labeled Bungarotoxin in Whole nAChR Pentamer Membranes Derived from a TE671 Cell Line.
We determined whether α1-FANG3 binds intact muscle-type pentamers in addition to in vitro-purified monomers. We assessed the competition between α1-FANG3 and 125I-labeled α-Btx for binding to intact membrane-bound nAChR muscle-type pentamers purified from a muscle-type nAChR-expressing human medulloblastoma cell line (TE671) (Figure 6). α1-FANG3 partially inhibited binding of 125I-labeled α-Btx, to ~70% of total binding, with a Ki of 2.7 nM. A relevant characteristic of the TE671 cell line is that nicotine produces an only 50% reduction in the level of 125I-labeled α-Btx binding, even at a 100000-fold molar excess of nicotine (data not shown). Our inability to reach the expected 50% reduction in the level of nAChR radiolabeling may be due to α-Btx binding other surface proteins besides α1. This idea is consistent with our observation that α1-FANG3 is more specific than α-Btx. On the other hand, α-Btx is known to bind in a 2:1 stoichiometry with the pentamer and α1-FANG3 may not compete equally at both sites.
Figure 6.

α1-FANG3 competes with 125I-labeled α-Btx for binding to the intact pentamer in membranes. An α1-FANG3 competition experiment with 10 nM 125I-labeled α-Btx is fitted by a Ki of 2.9 nM and maximally inhibits to ~70% of the total signal.
Competition of α1-FANG3 with α-Bungarotoxin in Xenopus Oocytes.
It was not obvious whether α1-FANG ligands would affect the gating of the nAChR pentamer, given that there was no selective pressure during the mRNA display process to generate a ligand with a specific gating effect. To assess the effect of α1-FANG3 on intact α1βδγ nAChRs, we studied the well-characterized Xenopus oocyte system. We recorded the currents induced by 10 μM ACh following exposure to various ligand incubations (Figure 7). To test the potential effects of α1-FANG3 on ACh-induced currents, oocytes were preincubated with a 200 nM α1-FANG3 solution for 3 min and then tested with 10 μM ACh. After a 5 min wash, the same oocytes were incubated with a 50 nM α-Btx solution for 3 min, followed by a second test with 10 μM ACh (Figure 7a). The ACh-induced current decreased by only 6% with α1-FANG3 incubation, while incubation with α-Btx decreased the ACh-induced current by 89%, showing that the interaction of α1-FANG3 with the α1βδγ nAChR is functionally silent when compared to that of α-Btx.
Figure 7.
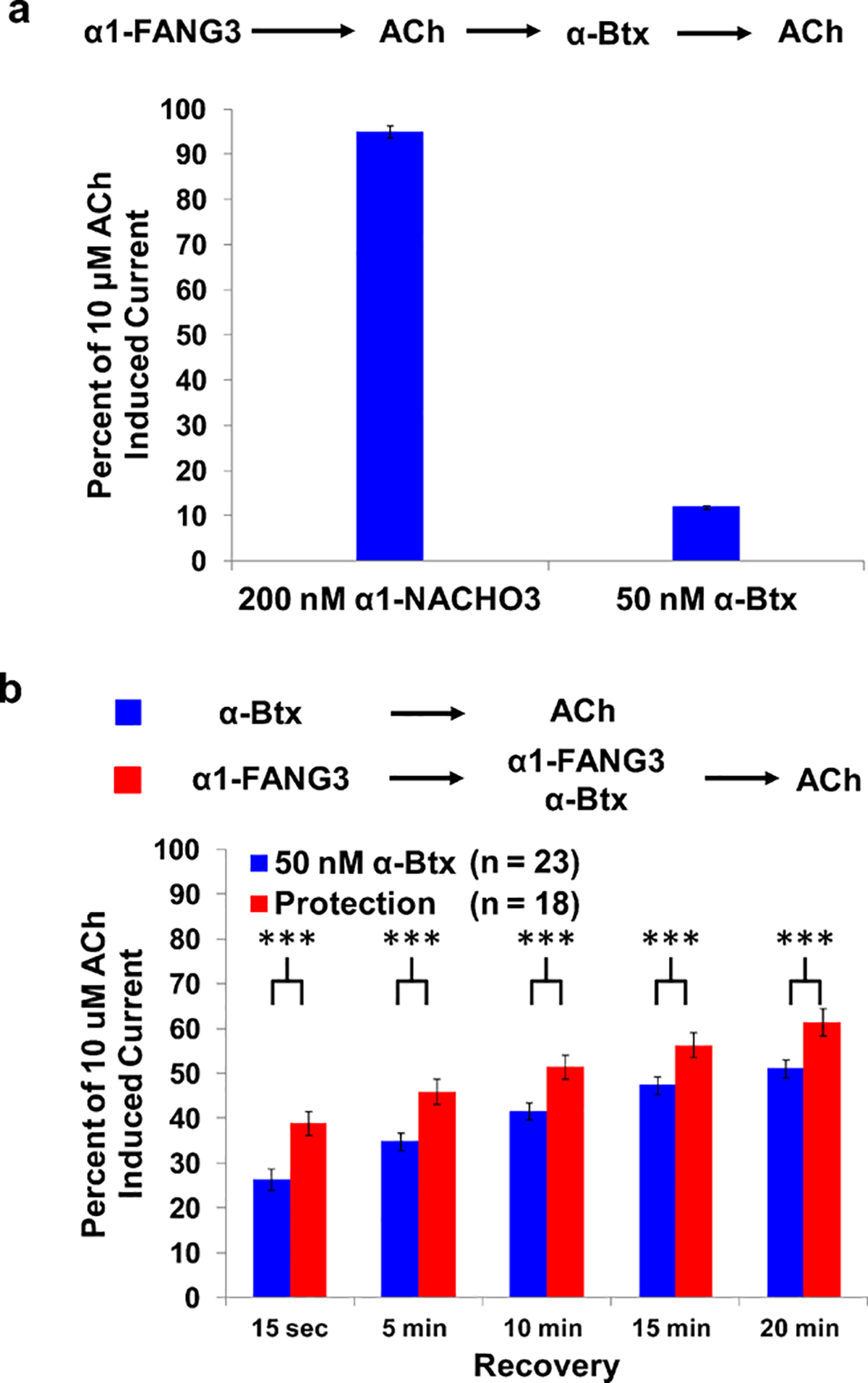
α1-FANG3 protects against α-Btx inhibition of Ach-induced current in the Xenopus laevis oocyte expression system. (a) Preincubation of oocytes (n = 8) with 200 nM α1-FANG3 for 3 min prior to testing with 10 μM ACh reduces the ACh-induced current by ~5%. Preincubation of the same oocytes with 50 nM α-Btx for 3 min prior to additional testing with 10 μM ACh reduces the ACh-induced current by ~90%. (b) Preincubation of oocytes with 200 nM α1-FANG3 for 5 min and then a mixture of 200 nM α1-FANG3 and 50 nM α-Btx for 3 min shows a protection of ACh-induced current compared to incubations with α-Btx alone. Error bars represent the standard error of the mean (SEM). *** = p value ≤ 0.01
To assess competition between α1-FANG3 and α-Btx at α1βδγ nAChRs in the Xenopus oocyte system, two sets of oocytes were recorded for ACh-induced currents. One population was preincubated with a 50 nM α-Btx solution alone for 3 min and then tested with 10 μM ACh. The second set was preincubated with 200 nM α1-FANG3 for 5 min and then with a solution of 200 nM α1-FANG3 and 50 nM α-Btx for 3 min and finally tested with 10 μM ACh. Both populations were tested at 5 min intervals for ACh-induced current over a 20 min recovery period (Figure 7b). Initial ACh-induced currents for the samples treated with α-Btx alone were 26% of the initial value, while the currents for the population preincubated with α1-FANG3 prior to co-incubation with α1-FANG3 and α-Btx were significantly different at 38%. During the washout period, currents increased, as previously noted for the washout of elapid toxins,12 yet the ACh-induced current for the 200 nM α1-FANG3/50 nM α-Btx oocytes remained ~12% greater than for α-Btx-only oocytes, showing that α1-FANG3 had a significant protective effect. Thus, α-Btx blocked 75% of the nAChRs in the absence of α1-FANG3 but only 62% when the nAChRs were protected by α1-FANG3. Direct co-application of α1-FANG3 and α-Btx to oocytes, without α1-FANG3 preincubation, eliminates this protective effect.
Our mRNA display selection against the soluble ecto-domain of α1, α211, resulted in a ligand with potential as a research tool to further the study of α1. α1-FANG3 is specific for α211 and is the first ligand able to specifically distinguish between the α1 subunit and other nAChR family members. Additionally, α1-FANG3 is superior to a natural product reagent such as α-Btx, because it requires no synthesis and can be expressed in E. coli, while maintaining an affinity similar to that of α-Btx. These properties are a result of the tunable nature of the e10FnIII scaffold; using affinity maturation and thermal and proteolytic challenges, we improved the ΔG°unfolding,303 K of our initially selected ligand by ~7 kcal/mol and the Tm by 17 °C. Additionally, α1-FANG3 targets the α-Btx site of α1 but has no active effect on gating of the nAChR pentamer, making it possible to study the receptor in an unperturbed state. These properties of α1-FANG3 enhance its potential utility for in vivo study of α1 distribution, trafficking, and recycling of α1 at the synaptic junction.
More broadly, our work shows that using a monomeric ecto-domain as a target for in vitro selection can yield ligands that bind a monomer individually and in the context of a fully assembled receptor. In general, expression and purification of a genetically encoded monomer are more straightforward than generation of an entire pentamer; therefore, the process laid out in our study provides a simpler path to target other nAChR subunits, as well as to target the larger Cys-loop family for novel ligand generation. mRNA display-generated fibronectins against this family could have better specificity, higher stability, and different functionality compared to those of existing molecules, generating a new category of reagents with which to study this important family of proteins.
METHODS
α211 Expression and Purification.
The α211 mouse construct was previously published.15 Production of the wild-type rat α9 ECD was identical to that of α211.
Naïve Selection against α211.
A naïve DNA library containing 1 × 1012 unique molecules, encoding an enhanced version of the 10th human fibronectin type III domain (e10FnIII) with randomized BC and FG loops, was constructed and cleared of stop codons as previously described.27,28,34
For the first round of naïve selection, 1120 pmol of ligated RNÄ was translated in vitro, which resulted in ~4 copies of the 1 × 1012 library for the affinity enrichment step. Affinity enrichment was performed with 400 pmol of α211 protein immobilized on M-270 Epoxy Dynabeads (Invitrogen). The selection buffer was phosphate-buffered saline supplemented with 1 mg mL−1 bovine serum albumin (Equitech-Bio), 0.01 mg mL−1 yeast tRNA (Calbiochem), and 0.05% (v/v) Tween 20 (Amresco). In subsequent rounds, 160 pmol of ligated RNA was translated in vitro and 100 pmol of immobilized α211 was used for affinity enrichment.
Binding for naïve selection assays was measured by pull-down assays of RNase (Sigma)-treated, oligo-dT cellulose-purified protein fusions against 100 pmol of immobilized α211, followed by scintillation counting of [35S]methionine (MP Biochemicals). Radiolabeled binding assays for the naïve selection were performed at room temperature for 1 h.
For α1-FANG1 competition assays with α-Btx, 300 pmol of α-Btx (Biotium) was preincubated with 100 pmol of α211 protein immobilized on M-270 Epoxy Dynabeads (Invitrogen) for 1 h prior to incubation with oligo-dT cellulose-purified 35S-labeled α1-FANG1. The rest of the assay was identical to those previously described.
Identification of clones from naïve selection was achieved witḧ Sanger sequencing after insertion of library DNA into the TOPO-TA vector (Life Technologies).
Affinity Maturation Selection.
For affinity maturation selection, a DNA library was prepared as previously described but with the BC and FG loops doped at 58% representation of α1-FANG1 at the nucleotide level.27,28 The affinity enrichment process was the same as that described for naïve selection. For the first round of the affinity matured selection, 960 pmol of ligated RNA was translated in vitro. Affinity enrichment for the first round was performed with 300 pmol of α211 protein immobilized on M-270 Epoxy Dynabeads (Invitrogen). The selection buffer was identical to that used in naïve selection. In subsequent rounds, 180 pmol of ligated RNA was̈ translated in vitro and only 100 pmol of immobilized α211 was used for affinity enrichment. FLAG affinity purification was added in rounds after the first round as previously described. In the third round, a 65 °C incubation at 10 min was added after FLAG purification. In the fourth round, incubation with 0.05 unit of Proteinase K on agarose (Sigma) for 1 min was added after the 65 °C incubation. In the fifth round, an off-rate selection step was introduced by addition of 300 pmol of α-Btx (Biotium) in selection buffer for 10 min to the affinity-enriched beads after the incubation and removal of the library.
Binding was measured by a pull-down assay of RNase (Sigma)-treated, oligo-dT cellulose-purified fusions against 100 pmol of immobilized α211 by scintillation counting of [35S]methionine (MP Biochemicals). Radiolabeled binding assays for the maturation selection were performed at room temperature for 1 h.
For binding assays involving α9, rat α9 was immobilized on M-270 Epoxy Dynabeads (Invitrogen). Assays used 100 pmol of immobilized α9. α1-FANG2 RNA was reticulocyte translated (Green Hectares) in vitro and purified using anti-FLAG M2 agarose beads (Sigma). FLAG-purified α1-FANG2 was used for binding assays similar to that used for the oligo-dT cellulose-purified protein fusions.
Identification of clones from doped selection was achieved by Sanger sequencing after insertion of library DNA into the TOPO-TA vector (Life Technologies).
On- and off-rate calculations were performed as previously published, but with individual clones rather than entire pools of molecules.35 [35S]Methionine-labeled in vitro reticulocyte-translated (Green Hectares) α1-FANG proteins were purified using anti-FLAG M2 agarose beads (Sigma). For off-rate calculations, 1 × 106 radioactive counts were incubated with 100 pmol of α211 immobilized on M-270 Epoxy Dynabeads (Invitrogen) for 1 h. The supernatant was removed, and the beads were washed four times with selection buffer. A 50-fold molar excess of α-Btx (Biotium) in selection buffer was added to the α211-immobilized beads. At designated time points after introduction, aliquots were removed, and the beads and supernatant were separated and individually counted to provide the relative ratio of bound α1-FANG protein. For on-rate calculations, 1 × 106 radioactive counts were incubated with 7 pmol of α211 immobilized on M-270 Epoxy Dynabeads (Invitrogen). At designated time points after introduction, aliquots were removed, and the beads and supernatant were separated and individually counted to provide the relative ratio of bound α1-FANG protein.
Expression of α1-FANG1 and Its Derivatives.
α1-FANG1 was inserted into the pet24a vector (Novagen) containing an N-terminal maltose binding protein (MBP) tag. Sanger sequencing confirmed DNA insertion and the correct reading frame. The fusion protein was expressed using E. coli BL-21 (DE3) cultures induced at an OD of ~0.4 with 0.2 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) for 3 h at 30 °C. The proteins were purified with Ni-NTA beads (Qiagen). The MBP fused proteins were cleaved using HRV3C protease (Thermo Scientific); residual MBP was removed with amylose resin (New England Biolabs), and the protease was removed with GST-Sepharose (GE Healthcare).
DNA inserts containing the sequences of α1-FANG2 and α1-FANG3 were subcloned into the pET 24a vector (Novagen). Sanger sequencing confirmed DNA insertion and the correct reading frame. The constructs were transformed into Rosetta 2(DE3)pLysS competent cells with the heat shock method. The transformants were plated on 2XYT agar plates containing 50 μg mL−1 kanamycin. Plates were incubated at 37 °C overnight. In each case, four colonies were screened for expression on a small scale, followed by larger-scale protein purification. A pre-inoculate was prepared by seeding a single colony in 50 mL of 2XYT medium with kanamycin. The culture was incubated at 37 °C while being shaken (220 rpm) overnight. One liter of inoculate was set using 10 mL of the overnight pre-inoculate, at 37 °C. When the OD600 reached 0.6–0.8 (2.5–3 h), cells were induced at 1 mM IPTG for 3 h at 37 °C. Cells were then harvested by centrifugation at 6000g for 20 min at 4 °C. Cell pellets were stored at −20 °C until they were purified.
Frozen cells were thawed on ice and resuspended with Ni-NTA binding buffer [50 mM NaH2PO4 (pH 7.8), 0.5 M KCl, 10% (v/v) glycerol, 0.1% (v/v) Triton X-100, and 20 mM imidazole] containing protease inhibitors (1 mM PMSF, 1 μg mL−1 leupeptin, and 1 μg mL−1 pepstatin A). Then, cells were lysed with sonication followed by centrifugation at 18000 rpm for 30 min at 4 °C. The supernatant was incubated with Ni-NTA agarose beads (Qiagen) at 4 °C for 2–3 h with end-over-end rotation. The protein was eluted with elution buffer [50 mM NaH2PO4 (pH 7.8), 0.5 M KCl, 10% (v/v) glycerol, and 500 mM imidazole] after being washed with binding buffer to remove loosely bound proteins. The eluted protein was concentrated and diluted with Mono Q buffer A [20 mM HEPES (pH 7.5)] that did not contain any salt to lower the salt concentration for anion exchange chromatography. The final salt concentration was adjusted to 100–120 mM. Some precipitations were observed, and they were removed by centrifugation at 10000g for 10 min or by filtration using a 0.22 μm filter. The sample was subjected to an affinity column (Mono Q HR 5/5, GE Healthcare) to purify proteins using a salt gradient from 0 to 100% buffer B (Mono Q buffer B contains 1 M NaCl). Bound peak fractions and flow-through fractions were separately concentrated and run over a size exclusion column (Superdex 75 10/300 GL, GE Healthcare) using 20 mM HEPES (pH 7.5) and 300 mM NaCl buffer. Each peak was identified and confirmed by OD280 and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels, and fractions of the peaks were pooled and concentrated for future experiments. The final amount of monomer α1-FANG3 was 1–2 mg from a 1 L culture.
Gel Shift Assays.
Mouse α211 was mixed with α1-FANG3 or α-Btx at an equimolar ratio (1 μg of α211 was used), and the mixture incubated on ice for 1 h (except for the competitive assay). For the competitive assay, the amount of α-Btx was increased relative to the amount of α1-FANG3 (α-bungarotoxin:α1-FANG3 ratios of 1:1, 2:1, 5:1, and 10:1), and the samples were allowed to equilibrate for 16 h overnight. Heated samples were prepared by heating α1-FANG3 in a heat block for 37 °C for 30 min, and samples were cooled on ice before the addition of mouse α211. Just before samples were loaded into a gel, 5× loading buffer (containing no dye) was added to each sample tube, and samples were loaded on a 10% native-PAGE gel at 4 °C. The gel was run for 3–3.5 h at 100–120 V and ~15 mA in a 4 °C room with TBE buffer. Bands were detected with Coomassie staining.
Circular Dichroism (CD) Spectroscopy.
Ultraviolet–CD spectra data were acquired with a Jasco J815 spectropolarimeter (at the University of Southern California NanoBiophysics Core Facility) equipped with a Peltier temperature-control device. The buffer consisted of 20 mM sodium phosphate, 1 M NaCl, and 6.3 M urea (pH 7). The E. coli-expressed α1-FANG2, α1-FANG3, and e10FnIII with wild-type loops were measured at a concentration of 25 μM. α1-FANG1 was at a concentration of 12.5 μM. Measurements used a 1 mm path length at 222 nm. The temperature range was 30–95 °C for α1-FANG2, α1-FANG3, and e10FnIII with wild-type loops and 4–95 °C for α1-FANG1. The temperature was increased at a rate of 1 °C min−1. The buffer-only blank was subtracted. Total ellipticities were converted to mean residue molar ellipticities (degrees square centimeters per decimole) before calculation of ΔG°unfolding,303 K and Tmelt using previously published procedures.33
Competition of α1-FANG3 with 125I-Labeled Bungarotoxin in Whole nAChR Pentamer Membranes Derived from a TE671 Cell Line.
Cells of human medulloblastoma clonal line TE671/RD (obtained from G. Crawford, City of Hope Research Institute, Duarte, CA, or from the American Type Culture Collection) have been maintained in our laboratory for years according to established protocols. Cells are cultured in Dulbecco’s modified Eagle’s medium buffered with sodium bicarbonate, containing 4 mM glutamine and 4.5 g L−1 glucose, and supplemented with 10% (v/v) horse serum and 5% (v/v) fetal calf serum, penicillin (100 units mL−1), and streptomycin (100 μg mL−1).
Cells were grown to confluence in 100 mm × 20 mm tissue culture dishes (Sarstedt) and then harvested with a polypropylene spatula into polycarbonate centrifuge tubes. After centrifugation at 4000g, cell culture medium was replaced with 5 mM Tris (pH 7.5) at 0 °C, and cells were homogenized with a Polytron PT2000 for 30 s. The homogenate was centrifuged at 40000g for 10 min, resuspended in Ringer solution with sodium azide (RAZ, 33 mM Tris, 115 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1.3 mM MgSO4, and 0.1 g/L NaN3), centrifuged a second time, and then resuspended in RAZ at a concentration of 0.1 mL. These membrane preparations can be stored at 4 °C for months without degradation of nAChR binding sites.
Reaction mixtures (200 μL) were prepared in RAZ supplemented with 0.1% (w/v) protease-free bovine serum albumin (Jackson ImmunoResearch) with varying concentrations of α1-FANG3 competing with 10 nM [125I]-Tyr54-α-bungarotoxin (PerkinElmer). Nonspecific binding was defined with reaction mixtures containing 1 μM α-cobratoxin. After incubation for 6 h on an orbital shaker at approximately 20 °C, binding reaction mixtures were filtered under partial vacuum through 25 mm GF/C filters (Whatman) pretreated for 30 min in 0.3% (w/v) polyethylenimine (Sigma) and 5% (w/v) powdered skim milk. Borosilicate glass test tubes and GF/C filters were rinsed three times with ice-cold RAZ. Filters were incubated overnight in scintillation cocktail and counted using a Packard TriCarb 2800 TR instrument. Specific counts were normalized to controls, and data were fitted (GraphPad Prism 5.0) to a Ki value assuming single-site binding competition and an α-bungarotoxin KD of 0.27 nM.16
Competition of α1-FANG3 with α-Bungarotoxin in Xenopus Oocytes.
Stage VI oocytes from Xenopus laevis were isolated and maintained at 16 °C in ND96 solution [96 mM NaCl, 2 mM KCl, 1 mM Mg2Cl2, 1.8 mM CaCl2, and 5 mM HEPES (pH 7.5)] supplemented with 2.5 mM sodium pyruvate (Acros Organics), 50 μg/mL gentamicin (Sigma), and 0.6 mM theophylline (Sigma). Oocytes were injected with a 50 nL sample containing 2 ng of a wild-type nAChR α/β/δ/γ mRNA subunit mixture at a 2:1:1:1 ratio.
Voltage-clamp currents from oocytes were recorded, ~24 h after injection, using two-electrode circuits (OpusExpress 6000A, Molecular Devices, Axon Instruments). The pipet microelectrodes were filled with 3 M KCl and had resistances of 0.2–10 MΩ. Oocytes were perfused continuously with Ca2+-free ND96, with additions otherwise noted.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Caitlin Nichols for her acronym expertise and Dr. Matthew Mulcahy for his digital prowess.
Funding
Supported by National Institutes of Health Grants R01 AI085583 and R01 DA037161.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00513.
Plot of ΔGunfolding versus inverse temperature of α1-FANG1 and its derivatives (Supplemental Figure 1) and determination of α1-FANG3 Kon and Koff rates via radiolabeled binding assays (Supplemental Figure 2) (PDF)
REFERENCES
- (1).Smart TG, and Paoletti P (2012) Synaptic neurotransmitter-gated receptors. Cold Spring Harbor Perspect. Biol. 4, a009662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lester HA, Dibas MI, Dahan DS, Leite JF, and Dougherty DA (2004) Cys-loop receptors: new twists and turns. Trends Neurosci. 27, 329–336. [DOI] [PubMed] [Google Scholar]
- (3).Sine SM, and Engel AG (2006) Recent advances in Cys-loop receptor structure and function. Nature 440, 448–455. [DOI] [PubMed] [Google Scholar]
- (4).Corringer PJ, Le Novère N, and Changeux JP (2000) Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40, 431–458. [DOI] [PubMed] [Google Scholar]
- (5).Kasheverov IE, Utkin YN, and Tsetlin VI (2009) Naturally occurring and synthetic peptides acting on nicotinic acetylcholine receptors. Curr. Pharm. Des. 15, 2430–2452. [DOI] [PubMed] [Google Scholar]
- (6).Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, and Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411, 269–276. [DOI] [PubMed] [Google Scholar]
- (7).Smith CE, van Miert MM, Parker CJ, and Hunter JM (1997) A comparison of the infusion pharmacokinetics and pharmacodynamics of cisatracurium, the 1R-cis 1’R-cis isomer of atracurium, with atracurium besylate in healthy patients. Anaesthesia 52, 833–841. [DOI] [PubMed] [Google Scholar]
- (8).Motamed C, Menad R, Farinotti R, Kirov K, Combes X, Bouleau D, Feiss P, and Duvaldestin P (2003) Potentiation of mivacurium blockade by low dose of pancuronium: a pharmacokinetic study. Anesthesiology 98, 1057–1062. [DOI] [PubMed] [Google Scholar]
- (9).Hurst R, Rollema H, and Bertrand D (2013) Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol. Ther. 137, 22–54. [DOI] [PubMed] [Google Scholar]
- (10).Changeux JP, Kasai M, and Lee CY (1970) Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc. Natl. Acad. Sci. U. S. A. 67, 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Moise L, Piserchio A, Basus VJ, and Hawrot E (2002) NMR structural analysis of α-bungarotoxin and its complex with the principal α-neurotoxin-binding sequence on the α7 subunit of a neuronal nicotinic acetylcholine receptor. J. Biol. Chem. 277, 12406–12417. [DOI] [PubMed] [Google Scholar]
- (12).Lester HA (1972) Vulnerability of desensitized or curare-treated acetylcholine receptors to irreversible blockade by cobra toxin. Mol. Pharmacol. 8, 632–644. [PubMed] [Google Scholar]
- (13).Arias HR (2000) Localization of agonist and competitive antagonist binding sites on nicotinic acetylcholine receptors. Neurochem. Int. 36, 595–645. [DOI] [PubMed] [Google Scholar]
- (14).Nirthanan S, and Gwee MC (2004) Three-finger α-neurotoxins and the nicotinic acetylcholine receptor, forty years on. J. Pharmacol. Sci. 94, 1–17. [DOI] [PubMed] [Google Scholar]
- (15).Yao Y, Wang J, Viroonchatapan N, Samson A, Chill J, Rothe E, Anglister J, and Wang ZZ (2002) Yeast expression and NMR analysis of the extracellular domain of muscle nicotinic acetylcholine receptor α subunit. J. Biol. Chem. 277, 12613–12621. [DOI] [PubMed] [Google Scholar]
- (16).Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, and Chen L (2007) Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 A resolution. Nat. Neurosci. 10, 953–962. [DOI] [PubMed] [Google Scholar]
- (17).Miwa JM, Lester HA, and Walz A (2012) Optimizing cholinergic tone through lynx modulators of nicotinic receptors: implications for plasticity and nicotine addiction. Physiology 27, 187–199. [DOI] [PubMed] [Google Scholar]
- (18).Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, and Wonnacott S (1999) Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling α7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology 38, 679–690. [DOI] [PubMed] [Google Scholar]
- (19).Baker ER, Zwart R, Sher E, and Millar NS (2004) Pharmacological properties of α9α10 nicotinic acetylcholine receptors revealed by heterologous expression of subunit chimeras. Mol. Pharmacol. 65, 453–460. [DOI] [PubMed] [Google Scholar]
- (20).Hunt SP, and Schmidt J (1978) The electron microscopic autoradiographic localization of α-bungarotoxin binding sites within the central nervous system of the rat. Brain Res. 142, 152–159. [DOI] [PubMed] [Google Scholar]
- (21).Lentz TL, Hawrot E, and Wilson PT (1987) Synthetic peptides corresponding to sequences of snake venom neurotoxins and rabies virus glycoprotein bind to the nicotinic acetylcholine receptor. Proteins: Struct., Funct., Genet. 2, 298–307. [DOI] [PubMed] [Google Scholar]
- (22).Das S, MacDonald K, Chang HY, and Mitzner W (2013) A simple method of mouse lung intubation. J. Visualized Exp, e50318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bracci L, Lozzi L, Rustici M, and Neri P (1992) Binding of HIV-1 gp120 to the nicotinic receptor. FEBS Lett. 311, 115–118. [DOI] [PubMed] [Google Scholar]
- (24).Olson CA, Adams JD, Takahashi TT, Qi H, Howell SM, Wu TT, Roberts RW, Sun R, and Soh HT (2011) Rapid mRNA-display selection of an IL-6 inhibitor using continuous-flow magnetic separation. Angew. Chem., Int. Ed. 50, 8295–8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Roberts RW, and Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. U. S. A. 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Takahashi TT, Austin RJ, and Roberts RW (2003) mRNA display: ligand discovery, interaction analysis and beyond. Trends Biochem. Sci. 28, 159–165. [DOI] [PubMed] [Google Scholar]
- (27).Olson CA, and Roberts RW (2007) Design, expression, and stability of a diverse protein library based on the human fibronectin type III domain. Protein Sci. 16, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Olson CA, Liao HI, Sun R, and Roberts RW (2008) mRNA display selection of a high-affinity, modification-specific phospho-IκBα-binding fibronectin. ACS Chem. Biol. 3, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GC, McGee AW, Sabatini BL, Roberts RW, and Arnold DB (2013) Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang ZZ, Hardy SF, and Hall ZW (1996) Assembly of the nicotinic acetylcholine receptor. The first transmembrane domains of truncated α and δ subunits are required for heterodimer formation in vivo. J. Biol. Chem. 271, 27575–27584. [DOI] [PubMed] [Google Scholar]
- (31).Hilz H, Wiegers U, and Adamietz P (1975) Stimulation of proteinase K action by denaturing agents: application to the isolation of nucleic acids and the degradation of ‘masked’ proteins. Eur. J. Biochem. 56, 103–108. [DOI] [PubMed] [Google Scholar]
- (32).Qasim MA (2014) Specificity of proteinase K at P2 to P3′ sub-sites and its comparison to other serine proteases. Protein Pept. Lett. 21, 164–170. [DOI] [PubMed] [Google Scholar]
- (33).Greenfield NJ (2007) Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 1, 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liao HI, Olson CA, Hwang S, Deng H, Wong E, Baric RS, Roberts RW, and Sun R (2009) mRNA display design of fibronectin-based intrabodies that detect and inhibit severe acute respiratory syndrome coronavirus nucleocapsid protein. J. Biol. Chem. 284, 17512–17520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jalali-Yazdi F, Huong Lai L, Takahashi TT, and Roberts RW (2016) High-Throughput Measurement of Binding Kinetics by mRNA Display and Next-Generation Sequencing. Angew. Chem., Int. Ed. 55, 4007–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.