

Abstract
The conversion of soluble tau protein to insoluble, hyperphosphorylated neurofibrillary tangles (NFTs) is a major hallmark leading to neuronal death observed in neurodegenerative tauopathies. Unlike NFTs, the involvement of monomeric tau in the progression of tau pathology has been less investigated. Using live-cell confocal microscopy and flow cytometry, we demonstrate that soluble 0N4R monomers were rapidly endocytosed by SH-SY5Y and C6 glioma cells via actin-dependent macropinocytosis. Further, cellular endocytosis of monomeric tau has been demonstrated to be HSPG-dependent, as shown in C6 glial cells with genetic knockouts of xylosyltransferase-1—a key enzyme in HSPG synthesis—with a reduced level of tau uptake. Tau internalization subsequently triggers ERK1/2 activation and therefore, the upregulation of IL-6 and IL-1β. The role of ERK1/2 in regulating the levels of pro-inflammatory gene transcripts was confirmed by inhibiting the MEK-ERK1/2 signaling pathway, which led to the attenuated IL-6 and IL-1β expressions but not that of TNF-α. Moreover, as a key regulator of tau internalization, LRP1 (low-density lipoprotein receptor-related protein 1) levels were downregulated in response to monomeric tau added to C6 cells, while it was upregulated in HSPG-deficient cells, suggesting that the involvement of LRP1 in tau uptake depends on the presence of HSPGs on the cell surface. The subsequent LRP1 knockdown experiment we performed shows that LRP1 deficiency leads to an attenuated propensity for tau uptake and further elevated IL-6 gene expression. Collectively, our data suggest that tau has multiple extracellular binding partners that mediate its internalization through distinct mechanisms. Additionally, this study demonstrates the important role of both HSPGs and LRP1 in regulating cellular immune responses to tau protein monomers, providing a novel target for alleviating the neuroinflammatory environment before the formation of neurofibrillary tangles.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12031-021-01943-2.
Keywords: Heparan sulfate, Endocytosis, Tauopathies, Alzheimer’s disease, 0N4R tau, Lipoprotein receptor
Introduction
More than 25 neurological disorders feature filamentous aggregates of hyperphosphorylated tau protein in the brain and are collectively known as neurodegenerative tauopathies (Lee et al. 2001). Progressive deposition of tau-containing neurofibrillary tangles (NFTs) throughout various brain regions is a well-characterized hallmark of tauopathies; however, the mechanism behind NFT formation and spread remains poorly understood. Tau binds and stabilizes axonal microtubules (MTs) but can be hyperphosphorylated and lose its affinity for MTs (Bramblett et al. 1993). MAPT gene mutation has also been proposed to explain the impaired ability of tau to promote the binding of tau to MTs (Zhang et al. 2004), leaving it available to interact with other cellular components. Indeed, in the past few decades many additional binding partners, subcellular locations, and physiological roles of tau have been identified (Wang and Mandelkow 2016).
Increasing evidence suggests that tau can be physiologically released into the extracellular space, both as monomers and aggregate forms. Tau has been found in both the interstitial fluid (ISF) and cerebrospinal fluid (CSF) in both humans and wild-type rodents, suggesting that tau release can occur in the absence of neurodegeneration (Dujardin et al. 2014; Cummings et al. 2018; Barten et al. 2011; Wang et al. 2017; Furman et al. 2017; Yamada et al. 2011). Many species of tau have been characterized outside of cells including free monomer, C-terminal truncations, low molecular weight aggregates, fibrils, or membrane-encapsulated tau (Sengupta et al. 2017; Saman et al. 2012; Kanmert et al. 2015; Karch et al. 2012). Interestingly, endogenous tau was released in an activity-dependent manner from primary cortical cultures (Pooler et al. 2013). Several cell culture studies show that tau monomer and oligomer in various phosphorylation states are constitutively released into the extracellular space (Saman et al. 2012; Merezhko et al. 2018; Katsinelos et al. 2018). Tau monomer in the ISF may eventually have pathogenic consequences as tau monomers are able to adopt both an inert and a seed-competent conformation, while some seed-competent monomers can form multiple unique pathological strains (Mirbaha et al. 2018; Sharma et al. 2018). Furthermore, full-length, soluble tau monomer added externally to culture media eventually forms aggregates inside cells (Michel et al. 2014). Taken together, these findings suggest that cellular uptake of physiologically released tau monomer may represent an initial step in neurodegenerative tauopathies, where cellular uptake provides the initial conditions for aggregation.
Several groups have demonstrated that full-length monomer is readily internalized through clathrin-mediated endocytosis or macropinocytosis and that this internalization depends on heparan sulfate proteoglycans (HSPGs) (Takeda et al. 2015; Mirbaha et al. 2015). In contrast, some studies have shown that only low molecular weight tau trimers or larger are endocytosed while monomer is not (Frost et al. 2009; Wu et al. 2012; Evans et al. 2018; Rauch et al. 2018). HSPGs are emerging as a likely mediator of tau internalization as tau contains several heparin-binding domains (Elbaum-Garfinkle and Rhoades 2012). However, a member of the low-density lipoprotein receptor family, LRP1, also had a significant effect on tau endocytosis by neuronal cells (Elbaum-Garfinkle and Rhoades 2012). Most importantly, astrocytic LRP1 has been implicated in Aβ cleavage during the progression of Alzheimer’s disease, while the mechanism by which LRP1 regulates cellular uptake of tau monomers in glial cells requires further investigation.
Cellular responses following monomeric tau uptake have yet to be widely reported in the field of neurodegeneration research. Phosphorylation or mutation may affect the conformation of wild-type tau protein and could have increased pathogenic consequences on the cell by converting wild-type tau into a more pathological relevant tau protein seed, leading to the subsequent pathological transmission, as discussed in one of our previous papers (Song et al. 2021). This possibility also merits further investigation. Recently, the involvement of astrocytes in the progression of tau pathology has drawn much attention because of their close communication with neuronal cells (Song et al. 2021; Sidoryk-Wegrzynowicz et al. 2017; Leyns and Holtzman 2017). Astrocytes, like microglia, express genes involved in phagocytosis (Cahoy et al. 2008), eliminate synaptic debris (Kim et al. 2019), or protein aggregates as seen by clearance of β-amyloid peptide (Aβ) (Li et al. 2014). For instance, the enhanced cellular uptake of tau fibrils by astrocytes has been related to the intensive autophagy-lysosomal pathway as reported in several tau pathology mouse models. Also, astrocytic tauopathy is common across many tau-related neurodegenerative diseases, including Alzheimer’s disease (AD) (Richetin et al. 2020). However, the cellular mechanisms of tau accumulation in astroglial cells and glial cell responses to different isoforms of pathogenic tau have been less investigated. A neuro-inflammatory hypothesis is one of the dominant postulates describing the progression of AD. It has proposed that pathological hallmarks of AD, amyloid β (Aβ) plaques and neurofibrillary tangles (NFTs), activate glial cells to secrete pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6 (Chiroma et al. 2018). The pro-inflammatory cytokines could then act either through autocrine or paracrine pathways to excite astroglial cells to develop more neurotoxic features by transforming into reactive astrocytes; this would lead to increases in phosphorylated tau, Aβ, and pro-inflammatory molecules in various human neurodegenerative disorders at a more advanced stage (Liddelow et al. 2017).
In this study, cellular endocytosis of tau monomer was examined in several cell lines, as well as neuronal and glial cells. To investigate the molecular contributors to tau endocytosis and altered intracellular activities, we subsequently examined tau uptake propensity, the resulting cellular signaling pathways, and pro-inflammatory gene expression levels in HSPG and LRP1-deficient glial cells. Findings from this study provide novel information for the progression of tau protein-related pathology and shed light on exacerbated pro-inflammatory signaling pathways affected by tau propagation that may provide a therapeutic target.
Materials and Methods
Recombinant Protein Expression and Purification
Full-length human microtubule-associated protein tau was subcloned into pETM-13 (https://www.embl.de/pepcore/pepcore_services/strains_vectors/vectors/bacterial_expression_vectors/) (0N4R isoform described in Morozova et al. (2013)), and GSK-3β (a gift of the Woodgett lab, RRID:Addgene_15898) was subcloned into pBAD (EMBL). Both plasmids were generated using ligation-independent cloning (Flanagan et al. 1997). A triple tau mutant (VPR) containing P243L, V279M, and V348W in the wild-type 0N4R tau isoform was created using site-directed mutagenesis (GeneArt® Site-Directed Mutagenesis System, Thermo Fisher Scientific). P243L, V279M, and V348W in 0N4R correspond to the P301L, V337M, and R406W positions in the full-length 2N4R. The oligonucleotide sequences were as follows: P243L (CCG − > CTG): 5′-GATAATATCAAACACGTCCTGGGAGGCGGCAGTG-3′, V279M (GTG − > ATG): 5′-CAGGAGGTGGCCAGATGGAAGTAAAATCTGAG-3′, and V348W (CGG − > TGG): 5′-GACACGTCTCCATGGCATCTCAGCAATGTCTCC-3′. For expressing wild-type 0N4R and tau variant proteins, the pETM-13-0N4R tau plasmid was transformed by heat shock into chemically competent Rosetta™(DE3) cells (MilliporeSigma, 70,954) using standard techniques (Gómez-Ramos et al. 2008), and selected with 34 µg/mL chloramphenicol and 50 μg/mL kanamycin. For expressing phosphorylated tau, the pETM-13-0N4R tau plasmid was co-transformed with the pBAD-GSK3β plasmid into Rosetta-DE3 cells, and in addition to chloramphenicol and kanamycin, 100 µg/mL ampicillin was added for pBAD-GSK3β plasmid selection. Positive clones containing 0N4R tau were grown in LB media containing 34 µg/mL chloramphenicol and 50 µg/mL kanamycin, and if expressing phospho-tau with 100 µg/mL ampicillin to maintain the GSK-3β plasmid. For protein production, cells were inoculated and grown in Terrific Broth (24 g/L yeast extract, 20 g/L tryptone, 4 mL/L glycerol) at 37 ℃ until the A600 was between 0.5 and 0.9. To induce tau expression, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM, and the culture was grown for 4 h at 37 °C. For phospho-tau expression, the cells co-transformed with pETM-13-0N4R and pBAD-GSK3β also received arabinose at a final concentration of 0.1%, added at the same time as the IPTG. The bacterial cells were pelleted by centrifugation at 10,000 × g and tau directly purified, or the pellet frozen at − 80 °C prior to purification.
Tau proteins were purified as described previously (Gómez-Ramos et al. 2009). Briefly, pelleted cells were resuspended in a high-salt buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgSO4, 0.75 M NaCl, 0.02 M NaF, 1 mM PMSF, pH 7.0), disrupted via sonication, and centrifuged at 3200 × g to remove insoluble debris. The supernatant was decanted and then boiled for 10 min, cooled on ice for 15 min, and then centrifuged at 3200 × g for 10 min to remove and discard precipitated protein. The supernatant was decanted and dialyzed overnight at 4 °C in column wash buffer (20 mM PIPES, 10 mM NaCl, 1 mM EGTA, 1 mM MgSO4, 2 mM DTT, 0.1 mM PMSF, pH 6.5). The dialyzed lysate was applied to an SP-sepharose cation-exchange column on a BioLogic DuoFlow chromatography system (Bio-Rad); following application, the column was washed for 10 column volumes and protein eluted with 0.4 M NaCl. Protein-containing fractions were combined, buffer-exchanged into PBS, and concentrated using AmiconⓇ Pro purification concentrators (10 kDa MWCO, ACS501002, MilliporeSigma). The protein concentration was determined from measuring the absorbance at 280 nm (A280) using Beer’s Law with ϵ = 7450 M−1 cm−1 and MW = 40 kDa. A typical final yield was 10–12 mg of tau per liter of culture.
Cell Culture
SH-SY5Y neuroblastoma cells (ATCC CRL-2266™; RRID:CVCL_0019) were cultured in a 1:1 mixture of HAMS and Minimal Essential Media, containing 10% fetal bovine serum (FBS). C6 glioma cells (ATCC CCL-107™; RRID:CVCL_0194) were cultured in HAMS and 15% horse serum (ATCC) and 2.5% FBS. HEK293 cells (ATCC CRL-1573™; RRID:CVCL_0045) were cultured in DMEM and 10% FBS. CHO745 cells and CHO cells (ATCC CRL-9618™; RRID:CVCL_0214) were cultured in 10% FBS and HAMS (ATCC). Rat primary astrocytes (iXCells Biotechnologies 10RA-005) were cultured in astrocyte medium (Thermo Fisher Scientific, USA), 2% FBS and 100 U/mL Penicillin–Streptomycin. Cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C and cultured in CELLSTAR 25-cm2 culture flasks with filters.
CRISPR Plasmid Design and Construction
Primers of single-guide RNAs (sgRNAs) were designed with Benchling online CRISPR Guide RNA Design Tool (https://www.benchling.com/crispr/) that designs sgRNAs with input target sites and provides on- and off-target scores for optimizing higher activity and lowering off-target effects. sgRNAs were designed to target the xylosyltransferase domain; the Exon7-targeting sequence (CCTGTATGGCAACTATCCTG) on the Xylt1 gene (Acc:620,093) was introduced into the pSpCas9(BB)-2A-GFP (RRID:Addgene_48138, a gift of Feng Zhang) using established cloning protocols with BbsI and T7 ligase. The ligation reaction was transformed into Escherichia coli DH5α and the sgRNA expression plasmid obtained via selection of colonies on ampicillin-containing LB plates. Sequences were confirmed using the U6 promoter forward primer: GAGGGCCTATTTCCCATGATT (Integrated DNA Technologies, USA).
C6 Transfection
Twenty-four hours before transfection, C6 cells were seeded at 7 × 105 cells per well in a 6-well plate containing 2 mL of complete growth medium. Cells were grown overnight to approximately 80–90% confluency. On the day of transfection, cells were harvested with 0.05% Trypsin–EDTA and transfected with the sequence-confirmed sgRNA plasmid constructs. For optimizing transfection conditions, 1 × 106 C6 cells were transfected with 10 μg DNA using the SF Cell line 4D-Nucleofector™X kit (V4XC-2012, Lonza) according to the manufacturer’s specifications. Cells were incubated in a humidified 37 °C, 5% CO2 incubator for 48 h.
Fluorescence-Activated Cell Sorting (FACS) of Transfected Cells for Nuclease-Expressing Cells
Two days post-transfection, C6 cell samples were detached with 0.05% Trypsin–EDTA and the top 3% of GFP positive cells were sorted by FACS into prepared 1.5-mL Eppendorf tubes to enrich the cell population with higher levels of nuclease expression. Following a 1-week expansion, enriched cell populations were subjected to a second round of transfection with the sgRNA expression plasmid to improve the level of genome editing, by following the same procedure. Two days post-transfection, transfected cells were harvested and sorted individually into 96-well plates, followed by a 2-week expansion in a humidified incubator. The single-cell clone (CL5) used for experimentation was selected by indel detection by amplicon analysis (IDAA) DNA capillary electrophoresis (Yang et al. 2015).
Alexa Fluor Labeling
Different isoforms of tau protein were labeled with either Alexa Fluor™ 488 (AF488) NHS Ester or Alexa Fluor™ 647 (AF647) NHS Ester (Life Technologies, USA) according to the manufacturer’s instruction by incubating the label and protein (1:10 w/w) in sodium bicarbonate (pH 8.3) for 1 h at room temperature. A Sephadex G-25 column (GE Healthcare) was used to separate labeled protein from the free label and the recovered labeled protein — referred to as tau-AF488 or tau-AF647 — was concentrated using AmiconⓇ Pro purification concentrators (10 kDa MWCO) and quantified via A280 measurement prior to the use.
Live Cell Confocal Microscopy
All cells were plated at 50,000 cells/well in Lab-Tek II confocal imaging chambers (Thermo-Fisher, cat# 155,360) and cultured for 24–48 h in incubators prior to assay. On the day of assay, cell chambers were transferred to a pre-warmed live-cell imaging chamber slide incubator on a Nikon A1 confocal microscope and allowed to equilibrate for approximately 10 min at 37 °C and 5% CO2. All data acquisition was performed with a 488-nM excitation laser with a 60X oil objective attached to a heating element to prevent heat sink from the imaging chamber during acquisition. Thirty seconds after acquisition began, a pre-warmed cell culture media containing tau-AF488 was added to cells in a 2X concentration to ensure complete mixing and to minimize cell surface adherence. For analysis, individual cells were identified at each time point, and the number of tau-containing vesicles was manually counted, using the Nikon Elements software to ensure that the signal did not reach saturation. Three independent biological replicates were performed for all measurements and at least three cells per time point were analyzed. Several alternative methods to quantify tau levels (e.g., total fluorescence, first appearance of vesicles) were examined, and all gave consistent results. Due to cell adherence challenges that caused out of focus changes with time, automated measurements were not able to yield reliable values. To block non-specific endocytosis, cells were pre-incubated on loosely packed wet ice for 30 min. For experiments blocking macropinocytosis via chemical means, culture media was replaced fully with either 1.0 or 0.1 μM cytochalasin-D (MilliporeSigma C8273) for 30 min prior to tau addition to cells.
Tau Internalization Assay and Flow Cytometry
To test the tau uptake propensity by different cell types (HEK293, SH-SY5Y, C6 glioma), single-cell C6 subclone (CL5), LRP1 knockdown C6 cells, and primary rat astrocytes (RA) were seeded at 5 × 105 cells/well in a 24-well plate. After culturing overnight, cells were incubated with tau-AF488 at fixed incremental concentrations of tau proteins (0.25 μM, 0.5 μM, and 1.0 μM). After at least 30 min, the cells were washed with PBS, trypsinized with 0.05% Trypsin–EDTA (Thermo Fisher Scientific, USA), and the fluorescence analyzed with an Accuri flow cytometer (MBIC, Mellon Institute). The relative tau uptake for each condition was calculated by subtracting the non-specific binding of the fluorophore to the cell membrane, which was conducted with identical experimental procedures but at 4 ℃ to limit internalization. For some experiments, C6 glioma, CL5, or primary rat astrocytes (RA) were incubated with unlabeled tau proteins — wild-type 0N4R (tau), phospho-0N4R (ptau), and a triple 0N4R mutant (VPR) — at 1 μM for 48 h before characterization by Western blotting and quantitative real-time PCR analysis. For experiments to investigate the effects of heparin on tau uptake by cells, 0.5 mg/mL heparin (Amsbio, AMS.HEP001) along with the tau protein was added to cell culture, as indicated.
Western Blotting
For analysis of expression or phosphorylation of different proteins present in mammalian cells, the cells were trypsinized and washed with ice-cold PBS prior to suspension in RIPA lysis buffer containing 150 mM NaCl, 0.1% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 50 mM Tri-HCL, and Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, USA) for protein extraction. The cell lysate was maintained at constant agitation for 30 min at 4 °C followed by centrifugation at 16,000 × g for 20 min at 4 °C. Protein concentration was determined by BCA assay (Thermo Fisher Scientific, USA). Prior to gel electrophoresis, cell lysate was mixed with a 4X Laemmli sample loading buffer (Bio-Rad, USA) and boiled at 95 °C for 5 min. Equal amounts of protein were loaded into the wells of a Mini-PROTEAN TGX Precast Gel (Bio-Rad, USA), along with Precision Plus Protein™ WesternC™ standards (Bio-Rad, USA) and then electrophoresed for 30 min at 225 V. Proteins were then transferred to 0.2-µm preassembled PVDF membrane (Bio-Rad, USA) using the Trans-Blot Turbo Transfer System (Bio-Rad, USA). The blots were then subjected to Western analysis as described previously (Bejoy et al. 2020). Briefly, blots were incubated with 5% nonfat dried milk in 1X Tris-buffered saline containing 0.1% (v/v) Tween-20 (TBS-T) buffer for 1 h at room temperature to block non-specific binding, and incubated with the primary antibodies phospho-ERK1/2 (Cell Signaling, 9101S), ERK1/2 (Cell Signaling, 9102S), Xylt1 (Life Technologies, PA5-67,627), LRP1 (Cell Signaling, 64099S), and endogenous control protein, β-actin (Cell Signaling, 3700 T), in TBS-T containing 5% (w/v) BSA overnight at 4 °C. After washing with 1X TBS-T, the blot was incubated with the fluorophore-conjugated secondary antibody (Li-Cor Biosciences, 926–32,211) for 1 h at room temperature. The blots were imaged with ChemiDoc MP Imaging System (Bio-Rad, USA) and the band intensities of target proteins were normalized to the endogenous control protein, β-actin, using ImageJ software.
Quantitative Real-Time PCR Analysis
Total RNA was isolated from cell samples using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol, followed by treatment with the RNA Clean & Concentrator Kit (Zymo). Reverse transcription of 2 μg of total RNA was carried out using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the protocol of the manufacturer. Primers used in the real-time PCR are listed in Supplementary Table 1. The gene β-actin was used as an endogenous control for the normalization of expression levels. Real-time PCR reactions were performed on a ViiA™ 7 System using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems). The amplification reactions were performed as follows: 15 min at 50 °C, 10 min at 95 °C, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Fold change in gene expression was quantified by means of the comparative Ct method, which is based on the comparison of the target gene expression (normalized to the endogenous control β-actin) between the samples. For experiments designed to inhibit MEK1/2-ERK1/2 signaling, 100 μM MEK1/2-ERK1/2 inhibitor, U0126-EtOH (Selleckchem, S1102), was added to culture media along with tau protein.
Immunocytochemistry
Cell staining was performed as described previously (Song et al. 2019). Briefly, 1 × 106 cells were seeded into 6 well-plate per sample and cultured overnight. The replated cells were then fixed with 4% (w/v) paraformaldehyde in PBS at room temperature for 15 min and washed twice with a staining buffer (2% (v/v) FBS in PBS). For staining intracellular markers, the samples were permeabilized with 0.2–0.5% (v/v) Triton X-100 at room temperature for 15 min. The cells were then blocked with 10% (v/v) FBS at room temperature for 30 min and then incubated with primary antibodies, mouse anti-Heparan Sulfate (HS; USBiologic, H1890), or mouse anti-Glial Fibrillary Acidic Protein (GFAP; Sigma-Aldrich, MAB360-25UL) at room temperature for 4 h. After washing, the cells were incubated with the corresponding secondary antibody: Alexa Fluor® 488 goat anti-Mouse IgM (Abcam, ab150121) and Alexa Fluor® 488 goat anti-Mouse IgG1 (Life Technologies, A21125), at room temperature for 1 h. The samples were counterstained with Hoechst 33,342 (Abcam, ab228551) and visualized using a fluorescent microscope (Keyence BZ-X810).
LRP1 Knockdown by Small Interfering RNA (siRNA)
Double-stranded, rat LRP1 specific siRNAs and non-targeting siRNA were synthesized by IDT. The sequences of LRP1 specific siRNAs were as follows:
LRP1 siRNA1: 5′-GGCGUCACUUACAUCAACAACCGTG-3′,
LRP1 siRNA2: 5′-CCUGAUGUUCUGGACCAAUUGGAAT-3′, and.
LRP1 siRNA3: 5′-GUAAAAAUGAAGGAAUUACUUUUTA-3′.
The 1 × 106 C6 glioma cells were transfected with LRP1-specific siRNAs (300 nM) and non-targeting siRNA using Lonza’s SF Cell line 4D-Nucleofector™X kit (V4XC-2012, Lonza) according to the manufacturer’s specifications. Cells were incubated in a humidified 37 °C, 5% CO2 incubator for 48 h. Gene expression of LRP1 was analyzed by real-time PCR analysis 48 h after transfection.
Statistical Analysis
Statistical significance was determined by unpaired t-tests, one-way analysis of variance (ANOVA), or two-way ANOVA when appropriate using GraphPad Prism 8 software. A 5% cutoff was applied to determine statistical significance, and a p-value of < 0.05 was denoted with one asterisk (*). In all flow cytometry and imaging experiments, at least three independent biological replicates from different days were performed and the standard error of the mean is shown. In all cases, the number of replicates is included in the figure caption.
Results
0N4R Tau Is Rapidly Endocytosed by Immortalized Cell Lines via Actin-Dependent Macropinocytosis Pathway
The primary microtubule-associated protein tau (MAPT) transcript is alternatively spliced into six isoforms, where all six are expressed in mature brains while the shortest isoform (0N3R) is expressed primarily during fetal development (Buée et al. 2000). The 0N4R splice variant has previously been shown to be highly abundant in NFTs (Goedert et al. 1992; Brion et al. 1991), but unlike the 2N4R tau variant, the internalization of 0N4R is not well understood and therefore was the subject of this investigation. The 0N4R isoform was purified as described in the “Methods” section and labeled with Alexa Fluor 488 for fluorescence detection and will be subsequently referred to as tau-AF488. We employed both live-cell confocal microscopy and flow cytometry to give insights into the kinetics of 0N4R tau endocytosis in three immortalized cell lines, HEK293, SH-SY5Y, and rat C6 glioma cells (Fig. 1). HEK293 cells have been previously used for tau uptake studies, while we also examined neuronal and glial cell models of more possible relevance — SH-SY5Y cells provide an effective model of neurodegeneration (Agholme et al. 2010) and rat C6 glioma has been shown to serve as a useful astrocytic model due to its ability to reproduce pro-inflammatory behavior (Galland et al. 2019).
Fig. 1.

Cell type differences in tau uptake. A Representative confocal microscopy images for HEK293, SH-SY5Y, and C6 glioma following addition of 1-µM 0N4R tau monomer labeled with Alexa Fluro 488. Scale bar = 10 µM. B Quantification of live-cell confocal microscopy: C6 glioma cells (triangles) endocytose 0N4R monomer about three to four times faster compared to SH-SY5Y cells (circles) and HEK293 cells (squares) (* < 0.05 between C6 and the other two cell types) C–E The relative uptake of tau-AF488 at indicated concentrations by different cell lines was quantified by flow cytometry, where control (untreated) cells were subtracted from those subjected to incubation with tau-AF488. Percent positive indicates the fraction of the cells incubated with a fluorescence above background. Figure 6C(ii) shows typical histograms obtained. For HEK293 cells (C) reducing the concentration of tau-AF488 to 25 nM (open squares) significantly reduced the tau internalization. For SH-SY5Y cells (D), the internalization was reduced at 25 nM (open circles), while for C6 glioma cells (E), there was no observed difference between the concentrations
For confocal imaging, cells plated on imaging chambers were incubated with tau-AF488 and imaged at 0 min, 5 min, and 15 min. Additionally, cells incubated with tau-AF488 were harvested with 0.05% trypsin–EDTA every 30 min over a 3-h timespan and subjected to flow cytometry characterization. Quantification of live-cell confocal microscopy as number of vesicles per cell showed that rat C6 glioma cells endocytosed tau-AF488 monomer around three to four times as rapidly as HEK293 cells and SH-SY5Y cells (Fig. 1A, B). This result was corroborated by quantitative flow cytometry analysis, as 99% of C6 glioma cells were positive for tau after 15 min compared with 75.2% for SH-SY5Y cells and just 6.8% for HEK293 cells for 1-µM tau addition (Fig. 1C–E). Additionally, the nuclear stain Hoechst was co-imaged with tau-AF488 to determine the spatial distribution of tau relative to the cell nucleus. By 120 min, Alexa 488 signal was detected primarily localized within the cell body for both C6 glioma cells and SH-SY5Y cells (Supplemental Fig. S1). Importantly, free Alexa Fluor 488 label and bovine serum albumin (BSA; 1 μM) labeled with Alexa Fluor 488 did not enter cells over the imaging periods used (Supplemental Fig. S2), suggesting that bulk fluid-phase endocytosis was not responsible for the rapid uptake of 0N4R tau that was observed. Because tau probably exists in lower concentrations in the ISF as compared with the cytosol in living systems (Yamada et al. 2011), we also tested uptake of 25 nM tau-AF488 for all three cell types (Fig. 1C–E). We observed a decrease in tau-AF488 fluorescence in SH-SY5Y and HEK293 cells over the time periods tested; however, no statistically significant difference was observed in C6 glioma cells for the 25-nM versus the 1-µM tau addition (Fig. 1E).
To confirm that this process occurred via cellular endocytosis, we pre-incubated HEK293 cells on ice for 30 min prior to tau addition and found that tau-AF488 monomer did not enter cells at temperatures non-permissive for endocytosis, as determined by confocal microscopy (Fig. 2A, B). Furthermore, flow cytometry confirmed that HEK293 cells preincubated at 4 °C endocytosed approximately seven times less tau-AF488 than cells incubated at 37 °C (Fig. 2C). Similarly, C6 glial cells incubated at 4 °C endocytosed 82% less tau-AF488 than cells incubated at 37 °C (Supplemental Fig. S3). To understand the molecular mechanism responsible for tau endocytosis, SH-SY5Y cells were preincubated with cytochalasin-D as a potent actin-polymerization and macropinocytosis inhibitor. Cytochalasin-D addition reduced tau uptake significantly over the entire observation time (Fig. 2D). Tau pathology is not solely restricted to neuronal cells, as tau inclusions have been found in astrocytes, oligodendrocytes, and even microglia (Ghoshal et al. 2001; Ferrer et al. 2014); therefore, we also sought to validate a similar tau uptake mechanism in an immortalized glial cell line, the rat C6 glioma line. Diminished tau uptake was also observed in C6 glial cells (Fig. 2D) with the addition of cytochalasin-D. Taken together, these data suggest that 0N4R tau monomer is readily endocytosed by all three immortalized cell lines, largely by actin-mediated macropinocytosis.
Fig. 2.

Monomeric tau is internalized via actin-mediated endocytosis. A Representative confocal microscopy images of HEK293 cells incubated with 1 μM tau-488. Scale bar = 10 µM. B Pre-incubating HEK293 cells on wet ice (4 °C) limits tau uptake as measured by confocal microscopy analysis of mean fluorescence intensity (**p < 0.005, unpaired t-test of 37 °C compared with 4 °C, n > 10/image) at 15 min of incubation. Individual points represent individual cells where bars show mean and SEM. C Similarly, as measured by mean peak intensity via flow cytometry, pre-incubating HEK293 cells on wet ice (4 °C) decreases tau uptake compared with cells at 37 °C (*p < 0.05 for the 37 °C compared to 4 °C; n = 3 independent biological replicates). D 1 μM cytochalasin-D (open symbols, cyto-D) decreases the rate of tau-488 internalization in SH-SY5Y (circles) and C6 glioma cells (triangles) compared with untreated cells (filled symbols; p < 0.05 for 1 µM compared to untreated cells) observed by confocal microscopy
C6 Glioma Cells Efficiently Endocytose 0N4R Monomer in an HSPG-Dependent Manner
HSPGs are highly expressed on the cell surface and have been identified as critical cell-surface endocytosis receptors for 2N4R tau internalization in HEK293 and C17.2 cells (Rauch et al. 2018; Holmes et al. 2013). To confirm the relationship between tau endocytosis and HSPGs on the cell surface, tau uptake was examined in CHO and CHO745 — a CHO-derived cell line deficient in xylosyltransferase, thus incapable of synthesizing all glycosaminoglycans (GAGs) (Lidholt et al. 1992). Quantification of the number of vesicles per cell in live-cell imaging showed that CHO745 cells rapidly endocytosed tau monomer but did so at a significantly slower rate than wild-type CHO cells (Fig. 3A). To further our understanding, heparan sulfate (HS) protein expression on the cell surface was determined via immunolabeling with anti-heparan antibody and quantified by flow cytometry analysis (Fig. 3B). The relative expression levels are consistent with the differential endocytosis rate we observed (Fig. 1), where C6 cells had the highest expression of HS as well as the highest endocytosis rate across all the cell lines we tested, and as expected, CHO showed higher levels of HS relative to the CHO745, consistent with observed tau uptake.
Fig. 3.
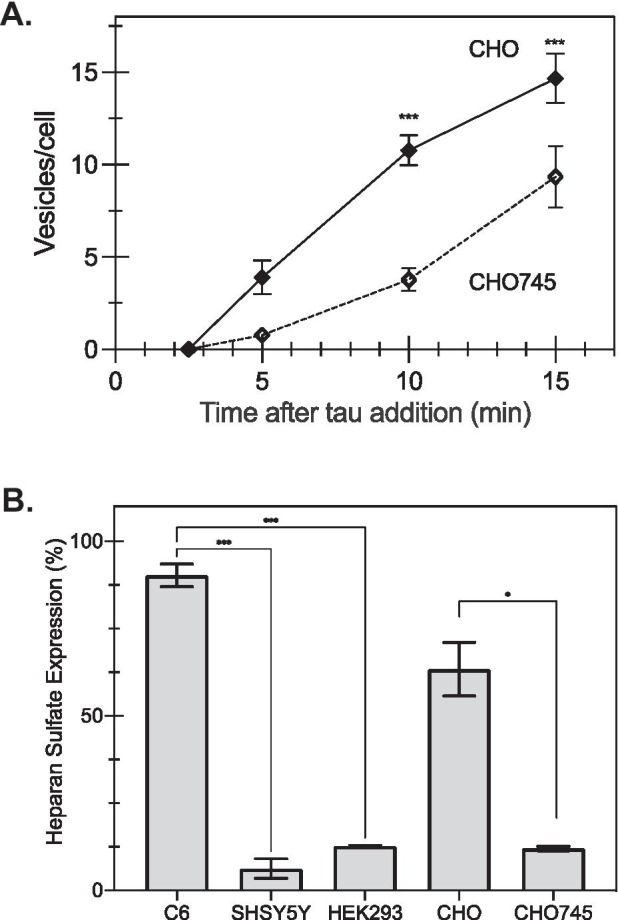
Tau monomer uptake depends on HSPG levels. A When characterized by the number of vesicles per cell observed via live-cell imaging, CHO745 cells had significantly fewer tau-containing vesicles per cell compared with wild-type CHO cells (*p < 0.05 for CHO745 cells compared with wild-type cells; n = 9, where points are the average, and error bars represent the standard error). B Heparan sulfate detection on the cell surface was determined via immunolabeling with anti-heparan sulfate antibodies and detected by flow cytometry, as described in the “Materials and methods” section. (Bars represent the mean for n = 2 independent replicates performed in duplicate, and error bars represent the standard error; *p < 0.05 based on t-test, ***p < 0.001 based on one-way ANOVA)
To validate our hypothesis about the importance of HSPGs as key cell surface receptors for tau, we then constructed an HSPG-deficient C6 cell line by genetic knockdown of xylosyltransferase (Xylt1) using the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system (Cong and Zhang 2014). This approach was informed by the previous approach in CHO cells that showed that Xylt1 deficiency would prevent glycosaminoglycan production and directly disrupt HSPG biosynthesis (Lidholt et al. 1992). Single-guide RNA (sgRNA) was designed to target Exon7 of Xylt1, within the xylosyltransferase domain (Fig. 4A). C6 wild-type cells were transfected with a sequence-verified CRISPR/Cas9-sgRNA construct according to this design. The cell population with a higher level of Cas9 nuclease expression as well as higher sgRNA expression was selected based on the expression of GFP reporter protein. We observed a higher level of GFP expression for transfected cells, while cells lacking the template showed no fluorescence (Fig. 4B). The top 3% of the GFP-positive cell population was subjected to FACS-based single-cell selection (see “Materials and Methods” section), and two clones were characterized following fragment length analysis — clone 3 (CL3) and clone 5 (CL5). As Xylt1 catalyzes the first step in the biosynthesis of HSPG, the levels of heparan sulfate (HS) were quantified by immunostaining to validate the knockdown. Flow cytometry analysis of these stained populations showed that CL5 had about half the HS expression compared with the non-edited C6 parental cells, while CL3 showed a similar level of HS expression compared with the parental cells (Fig. 4C). To confirm the HS expression data, mRNA levels of Xylt1 were quantified, with Xylt1 gene expression of CL5 observed to be about two-fold lower than the non-edited parental C6 (Fig. 4D). Western blot analysis confirmed the absence of Xylt1 expression in CL5, compared with non-edited parental C6 (Fig. 4E). These data confirmed the successful creation of a Xylt1-knockdown C6 cell line that will be referred to as CL5, by using the CRISPR/Cas9 system, which was used for subsequent studies.
Fig. 4.

Construction of a Xylt1 knockdown C6 glioma cell line with CRISPR/Cas9 genome editing technology. A Schematic illustration of Xylt-1- gene of Rattus norvegicus, containing two functional domains and 10 exons. Single-guide RNA (sgRNA) was designed to target Exon7 of Xylt1 within the xylosyltransferase domain as indicated by the red arrow. The sgRNA sequence is highlighted in green. B Overlay of phase-contrast images with fluorescent images of transfected C6 cells with either the control or the pre-constructed CRISPR/Cas9 + sgRNA plasmid which uses GFP as a selection marker to enrich the cell population with higher levels of Cas9 and sgRNA expressions. Scale bar = 100 μm. C Quantification of heparan sulfate (HS) expression by flow cytometry of non-edited C6 (C6), clone 5 (CL5), or clone 3 (CL3), showing highest knockdown for CL5. Shaded histogram: negative control; black curve: parental C6 cells; red curve: either C6 single-cell clone (CL) 5 or 3, as indicated. Xylt1 gene knockdown in clone 5 (CL5) was verified by both quantitative reverse transcription-polymerase reaction (RT-qPCR) (D), where * indicates p < 0.05 between different test conditions, and western blot analysis using Xylt1 antibody (E). F The relative uptake of tau in C6 parental cells (filled circles) compared with CL5 cells (filled triangles) was quantified by flow cytometry after a 30-min incubation at the indicated concentrations. Error bars reflect the standard error, where * represents p < 0.05 for CL5 cells compared with parental (n = 2 independent biological replicates performed in duplicate)
To examine the differential tau uptake capacity, C6 cells and CL5 cells were incubated with tau-AF488 at fixed incremental concentrations of tau proteins (0.25 μM, 0.5 μM, and 1.0 μM) for 30 min. Cells were then washed with PBS, trypsinized with 0.05% Trypsin–EDTA, and the fluorescence was analyzed with a flow cytometer. The relative tau uptake for each condition was calculated by subtracting the non-specific binding of the fluorophore to the cell membrane, which was conducted with identical experimental procedures but at 4 ℃ to limit internalization. Uptake of tau-AF488 by CL5 was compared with parental C6 cells (Fig. 4F and Supplemental Fig. S4). Tau-AF488 was rapidly and efficiently internalized by C6 cells in a concentration-dependent manner, while the HSPG deficiency in CL5 significantly decreased the cellular uptake of tau at every concentration measured during a 30-min incubation. These data indicate that HSPGs indeed play a significant role in the uptake of 0N4R tau monomers by glial cells.
Intracellular Responses Stimulated by Tau Internalization Is HSPG-Dependent
Stancu and colleagues (Stancu et al. 2019) showed previously that aggregated tau was capable of activating the NLRP3 inflammasome, increasing secretion of proinflammatory cytokines. Uptake of fibrillar Aβ also induced the assembly of the NLRP3 inflammasome, which led to caspase 1-dependent release of pro-inflammatory cytokines such as IL-1β and IL-18 (Swanson et al. 2019). Moreover, glial cells play an important role in regulating inflammatory cytokine secretion in the brain (Fleeman and Proctor 2021). Under pathological conditions like the neurodegenerative environment in AD, the pathological hallmarks, Aβ plaques and NFTs, have been thought to activate glial cells to release pro-inflammatory cytokines, such as IL-1β (Blum-Degena et al. 1995), IL-6 (Kálmán et al. 1997), and TNF-α (Fillit et al. 1991). The elevated levels of pro-inflammatory cytokines could then further activate glial cells for additional production of phosphorylated tau and pro-inflammatory cytokines (Chiroma et al. 2018). To identify the inflammatory responses affected by tau monomer endocytosis in C6 glial cells, we next evaluated mRNA transcriptional levels of the pro-inflammatory cytokines, IL-6 and TNF-α. Wild-type 0N4R tau addition significantly upregulated pro-inflammatory gene expression of IL-6 and TNF-α (Fig. 5Ai), suggesting a direct role in activating a pathological cascade in glial cells.
Fig. 5.

The role of HSPGs in regulating the immune response of glial cells to tau endocytosis is mediated by intracellular ERK1/2 signaling. A C6 cells (i and ii) and HSPG-deficient (Xylt-1-) CL5 cells (iii) were incubated with different tau variants for 24 h. RNA was isolated and then subjected to quantitative reverse transcription-polymerase reaction (RT-qPCR) for relative mRNA expression quantification of IL-6 (open bars) and TNF-α (darkly shaded bars). In A.ii, C6 cells were incubated with 1 μM VPR-0N4R along with MEK-ERK1/2 inhibitor or vehicle for 24 h. RNA was extracted and the relative mRNA levels of IL-1β (lightly shaded bars in ii), IL-6, and TNF-α were determined by RT-qPCR. *p < 0.05 between different test conditions; n = 2 independent biological samples performed in replicates. B and C Cell lysates were analyzed by western blotting for p-ERK1/2, total ERK (t-ERK), and β-actin in C6 (B.i) and CL5 (C.i), with a representative western blot shown. Quantification of p-ERK/β-actin ratios in C6 (B.ii) and CL5 (C.ii) from three biological replicates. Dashed line serves as a guide to the eye (*p < 0.05 between different test conditions, ns = not significant; n = 3)
As previous studies demonstrated that the activation of the MEK/ERK pathway was associated with early tau deposition in neurons and glial cells in tauopathies (Ferrer et al. 2001), we recognized that ERK1/2 phosphorylation might lead to cytokine transcription. Internalization of Aβ during the progression of neurofibrillary degeneration in Alzheimer’s disease (AD) has also led to MEK/ERK pathway activation (Pei et al. 2002). To explore the potential mechanism through which tau induced immune responses, the phosphorylation of ERK1/2 was further examined for different experimental conditions (Chua et al. 2004). Intriguingly, the phosphorylation of ERK1/2 increased in glial cells following tau monomer addition as compared with control cells, as determined by Western analysis (Fig. 5B).
To further understand whether cytokine activation was affected by physiological tau differently from pathological tau, several tau variants were examined. A triple mutant VPR (V337M, P301L, R406W) associated with frontotemporal dementia (FTD) that has been shown to hyperphosphorylate and have increased aggregation propensity (Tanemura et al. 2002; Vogelsberg-Ragaglia et al. 2000) and a highly phosphorylated tau protein (ptau) that is expected to drive aggregation (Lim et al. 2014) were created as described in the “Materials and Methods” section, and purified as for wild-type tau. Interestingly, the highest ERK1/2 phosphorylation was concurrent with the highest expression of immune genes in the C6 cells incubated with VPR or ptau (Fig. 5 Ai, B). Addition of the MEK-ERK1/2 inhibitor U0126 abolished the increased levels of IL-1β and IL-6 and enhanced TNF-α mRNA levels that were induced by exogenous VPR addition (Fig. 5Aii). Taken together, we conclude that tau monomer addition induced gene expression changes of pro-inflammatory cytokines, which are regulated by ERK1/2 activation.
HSPGs have been implicated in cellular signaling from a variety of ligands, such as the Wnt, Hedgehog, transforming growth factor-β, and fibroblast growth factor during development (Lin 2004). To investigate the relevance of HSPGs and intracellular signaling activation by tau addition, we further evaluated ERK1/2 phosphorylation and mRNA transcriptional levels of the proinflammatory cytokines IL-6 and TNF-α in the HSPG-deficient C6-derived cell clone CL5. Intriguingly, CL5 cells displayed minimal activation of pro-inflammatory genes upon tau monomer addition compared to parental C6 (Fig. 5Aiii) and the phosphorylation of ERK1/2 was decreased (Fig. 5C). These data suggest that HSPGs regulate the pro-inflammatory gene expression of glial cells in response to monomeric tau via ERK1/2 signaling activation.
LRP1 Receptor Plays a Role in Regulating Intracellular Responses of Glial Cells to Tau Protein
LRP1 has been recently identified as a major regulator of tau internalization, and tau transmission in neurons (Rauch et al. 2020). Ligand binding to LRP1 also activates cytoplasmic signaling pathways including several important intracellular mediators such as JNK and ERK and has been shown to mediate the immune responses of microglia to extracellular signals (Yang et al. 2016). To identify potential interactions between HSPGs and LRP1 upon tau addition, we measured LRP1 expression in both C6 cells and CL5 cells. The addition of VPR protein downregulated LRP1 levels in C6 cells while LRP1 levels in CL5 cells remained at similar levels as the parental C6 cells (Fig. 6A). These data suggests that the involvement of LRP1 in tau uptake depends on the presence of HSPGs on the cell surface, implying the possible interplay between HSPG and LRP1 in tau endocytosis and, therefore, its downstream responses.
Fig. 6.

HSPGs are relevant to LRP-mediated tau endocytosis. A Reduction of LRP1 mRNA was observed for C6 cells but not for CL5 (Xylt1-) after incubation with 1 μM VPR (darkly shaded bars) for 24 h, as analyzed by RT-qPCR (n = 2, *p < 0.05 between different conditions). B Rat C6 glioma cells were transiently transfected with non-targeting siRNA (NC; open bars) or LRP1-specific siRNA1, siRNA2, and siRNA3 for 48 h. LRP1 knockdown was then confirmed by RT-PCR analysis. Darkly shaded bars indicate LRP1-specific siRNA, compared with NC-siRNA (open bars) (n = 2, *p < 0.05 between different conditions). C.i The propensity of AlexaFluor488-labeled monomeric VPR-0N4R (VPR-AF488, green) uptake was examined in C6 cells post-transfection with non-targeting siRNA (NC) or LRP1-specific siRNA3 at 1 μM for 30 min. Scale bar = 100 μm. C.ii Flow cytometry histogram of VPR-AF488 uptake in NC siRNA cells (black curve) compared with LRP1-specific siRNA3 (red curve) shows quantitative decrease in tau uptake following knockdown. Gray filled curve shows distribution with vehicle only. Rat C6 glioma cells were transiently transfected with either non-targeting siRNA (NC) or LRP1-specific siRNA3 for 48, followed by incubation with 1 μM VPR-AF488 or vehicle for 24 h. D RT-qPCR analysis was performed to detect the expression level of IL-6 in C6 cells post-transfection as in C, with β-actin used as an internal control. Open bars, NC-siRNA transfected; gray shaded bars, LRP1-siRNA3 (n = 2, *p < 0.05 between different conditions) E Cell lysates were then analyzed by immunoblotting (i) against p-ERK1/2, total ERK (t-ERK), and β-actin, followed by quantitation (ii) of p-ERK/β-actin ratios from a representative western blot experiment (n = 3, *p < 0.05 between different test conditions)
To validate our observation and identify the role of LRP1 in regulating intracellular responses to monomeric tau, LRP1 gene expression was reduced in C6 glial cells using specific LRP1-siRNAs. LRP1 was successfully knocked down by 20–70% using three different LRP1-siRNA (Fig. 6B), which was further confirmed by western blot at the protein level (Supplemental Fig. S5). LRP1-siRNA3 knockdown cells and control cells were incubated with 1 μM VPR-AF488 at 37 °C for 30 min and then analyzed by fluorescent microscopy (Fig. 6Ci). Tau uptake was slightly decreased in LRP1 knockdown cells, which was subsequently quantified as a 30% reduction by flow cytometry (Fig. 6Cii). LRP1-siRNA3 knockdown exacerbated IL-6 gene expression increases as compared to negative control cells in response to tau (Fig. 6D), suggesting an intermediate regulatory role of LRP1 in response to inflammatory stimuli. Interestingly, ERK appeared unresponsive in LRP1 knockdown cells (Fig. 6E), implying that other possible as yet unknown signaling pathways, regulated by LRP1, may be also responsible for the activation of immune pathways following tau uptake.
Heparin Attenuates Tau Uptake and Mitigates Immune Response to Tau in Primary Astrocytes
In order to ensure that the observations were not limited to immortalized cell lines, experiments in primary astrocytes were performed to determine the effects of heparin in regulating intracellular activities affected by tau endocytosis. The propensity for tau uptake was examined in rat primary astrocytes by fluorescent imaging or flow cytometry to visualize and quantify tau internalization. Here, we selected the VPR-0N4R tau variant, as the most significant changes to cellular immune responses were observed during its uptake in immortalized cells. Monomeric VPR-AF488 was rapidly internalized by rat primary astrocytes (Fig. 7A), and the amount of internalized tau increased over time from 1 to 6 h, while heparin addition attenuated tau uptake, indicating that tau uptake was partially mediated by HSPGs, as in the immortalized cell lines. Phase-contrast images were overlaid with fluorescent images to visualize the spatial localization of tau, where tau protein was observed to be localized primarily within the cell body (Fig. 7A). Additionally, the nuclear stain Hoechst and an astrocyte-specific marker, GFAP, were co-imaged with VPR-AF488 to determine the spatial distribution of tau relative to the cell nucleus (Fig. 7B). Tau protein was evenly distributed outside of the cell nucleus, with negligible overlap with intracellular GFAP protein. In order to quantify this observation, monomeric VPR-AF488 uptake in primary astrocytes was analyzed by flow cytometry (Fig. 7C, D), showing that all cells appear to take up tau following a 6 h incubation.
Fig. 7.

Rat primary astrocytes readily take up tau in an HPSG-dependent manner. A The amount of VPR-0N4R labeled with AlexaFluor488 (+ VPR, green) internalized by primary astrocytes increases over time from 1 to 6 h, while the presence of heparin has attenuated the tau uptake rate in astrocytes, as observed by fluorescence microscopy. Scale bar = 100 μm B Representative fluorescent images of GFAP (red) and Hoechst (blue) for primary astrocytes under different conditions, as indicated, where VPR indicates the addition of VPR-488 with (middle panel) or without (bottom panel) heparin to the cell as observed via fluorescence microscopy. Scale bar = 100 μm. C To determine population behavior, VPR-AF488 endocytosed by primary astrocytes was determined by flow cytometry with and without heparin addition. Grey filled line: negative control, black line: cells without heparin, red line: cells with heparin. D The propensity of VPR-0N4R uptake was quantified in primary astrocytes by flow cytometry (n = 2) E An RT-qPCR analysis was performed to detect the expression levels of IL-1β (lightly shaded bars), IL-6 (open bars), and TNF-α (filled bars) in primary astrocytes affected by incubation with VPR and heparin, as indicated. β-Actin was used as an internal control. *p < 0.05 between different test conditions; n = 2
A body of evidence has shown that the presence of HSPGs is required for tau internalization. The rapid internalization of 2N4R monomer by neuronal cells relies on the presence of 6-O-sulfation modification on the HSPGs (Rauch et al. 2018). Similarly, the heparin-induced tau fibrils from 2N4R monomer are internalized by primary neurons in a HSPG-dependent manner. HSPGs are also found to be crucial for tau intracellular seeding activity (Holmes et al. 2013; Barbara Stopschinski et al. 2018). The important role of 3-O-sulfation on HSPGs in regulating tau internalization has been recently reported in a non-neuronal cellular model (Zhao et al. 2020). In fact, tau-HS affinity is mediated by various factors such as the HS modifications and the conformational structure of tau (Song et al. 2021). Different cell types have different HS-modifying enzyme patterns, while the physical structure of tau changes with mutations or post-translational modifications of tau. In our study, the interaction between HSPGs and tau monomers in glial cells was investigated by treating cells with heparin, a glycosaminoglycan, that has been used to competitively inhibit binding of tau to HSPGs (Murakami et al. 2017). We observed that heparin addition led to a 50% decrease in 0N4R tau uptake compared to the control group without heparin addition. A similar effect of heparin reduction of wild-type tau and ptau internalized by astrocytes was observed (Supplemental Fig. S6), suggesting that heparin competes with tau for binding with HSPGs at the cell surface, regardless of physiological tau state. Further, the presence of heparin also ameliorated the immune activation of astrocytes upon the addition of unlabeled monomeric VPR by driving the IL-6 and IL-1β gene expression back to baseline levels, though TNFα levels remained elevated (Fig. 7E).
Discussion
Rapid HSPG-Dependent Endocytosis of Tau Monomers
Extracellular tau is present in brain cerebrospinal fluid (CSF) and interstitial fluid (ISF) of AD patients either as free tau or within vesicles (Merezhko et al. 2018). The majority of extracellular tau consists of soluble oligomers and monomers, while a minority of tau species are in a truncated form from cleavage by various pro-inflammatory cytokines released by reactive astrocytes and microglia in AD brains (Dal Prà et al. 2015). The cellular internalization of extracellular tau is a major part of neurodegenerative disease progression, which is still under investigation (Holmes and Diamond 2014). HSPGs, highly expressed on the cell surface, have been identified as an important cell-surface endocytosis receptor of tau, as reported in various studies (Holmes et al. 2013; Christianson and Belting 2014). Ranch and coworkers reported that knockouts of extension enzymes of the HSPG biosynthetic pathway, such as extension enzymes exostosin 1 (EXT1), exostosin 2 (EXT2), and exostosin-like 3 (EXTL3), sufficiently reduced the uptake of tau oligomers in HEK293 cells (Rauch et al. 2018). Primary neurons and mouse models with heparin pretreatment showed diminished tau aggregate uptake, suggesting that HSPGs are required for tau aggregate internalization in vivo (Holmes et al. 2013). We have observed a similar effect of HSPGs on regulating monomeric tau protein uptake, using CNS-relevant cell lines. In our study, reduced tau uptake was observed in CHO745 and Xylt-knockout C6 glial cell lines, as well as in primary astrocytes, and validated a role for HSPGs in tau uptake. The efficient uptake of tau (2N4R isoform) protein by astrocytes was observed in a previous study (Perea et al. 2019), where tau-containing cells were detected as early as 30 min. However, the authors showed that heparin pre-treatment exhibited little effect on monomeric tau internalization, which is inconsistent with our findings on 0N4R uptake by astrocytes. The interaction between tau monomers and HSPGs has been characterized previously at the structural level by surface plasmon resonance and NMR mapping. Interestingly, tau was found to contain a specific heparin-binding domain; residues in both the proline-rich region (PRR) and the second repeat motif (R2) were specifically recognized by negatively charged sulfo groups on heparin sulfate (Zhao et al. 2020). We believe the discrepancy between the research of Perea et al. (2019), and our results may arise from conformational differences of the tau isoforms, 0N4R and 2N4R, used for the studies that result in differential tau-HSPG interaction. Taken together, the data suggest a complex regulation of tau interactions with HSPG and heparin that merits more attention in further studies.
ERK1/2 Signaling Events Are Regulated by Cell Surface Heparan Sulfate Proteoglycans
Cell surface HSPGs have been implicated in a variety of receptor signaling pathways via the binding of extracellular molecules and entities, including tau (Christianson and Belting 2014; Song et al. 2021). HSPGs act as internalizing receptors of macromolecular cargo such as FGF2, lipoproteins, and ApoE in a receptor-independent mechanism via the clathrin-coated pit endocytosis pathway. There are two major classes of cell surface HSPGs defined by the type of core protein — the glypican (GPC) protein family and the syndecan protein family (Lin 2004). Most relevant to our study, the binding of ligands to cell surface syndecan HSPG activates ERK1/2 signaling to facilitate actin-dependent endocytosis (Christianson and Belting 2014). Similarly, our study showed that monomeric tau uptake activates ERK1/2 signaling in an HSPG-dependent way, suggesting a regulatory role of HSPGs between tau and intracellular ERK1/2 signaling (Fig. 5A). Additionally, HSPG, as a co-receptor, activates receptor signaling following binding of cytokines to their corresponding receptors, including the binding between IL-6 and IL-6R (Lin 2004). A recent study demonstrated the inflammatory effects of low molecular weight heparin in attenuating IL-6 release in patients with COVID-19 by competing with HSPGs for the binding of IL-6 (Shi et al. 2020). Moreover, the transcriptional level of IL-6 was significantly decreased after the addition of a heparinase enzyme inhibitor. The underlying mechanism behind these effects was that cell surface HSPGs regulate IL-6 ligand-receptor interaction and IL-6 secretion via facilitating the activation of JAK associated with the IL-6 receptor. Subsequently, downstream ERK 1/2 signaling was activated to mediate the transcriptional activation of IL-6 (Maddahi et al. 2012). Analogously, the addition of ERK1/2 inhibitor in our study reduced the transcriptional level of IL-6 significantly, which validates the role of the ERK pathway in regulating IL-6 transcription and therefore, promoting subsequent IL-6 secretion. Additionally, the mild responses of HSPG-deficient cells to tau addition are concurrent with inactive ERK1/2 signaling (Fig. 5A); this result further indicates that HSPGs play a critical role in regulating inflammatory gene expression in glial cells following the addition of exogenous monomeric tau via intracellular ERK1/2 signaling activation (Fig. 8). However, further study is needed to identify the primary contributor to ERK1/2 phosphorylation, HSPG-dependent endocytosis of tau or positive-feedback regulation of IL-6 secretion, or both.
Fig. 8.

Schematic illustration of the intracellular responses affected by monomeric tau endocytosis. Monomeric tau is rapidly internalized by both neuronal and glial cells, mediated by the actin-dependent macropinocytosis pathway. In addition, an important role of HSPG has been demonstrated in regulating inflammatory gene expression, including IL-6 and IL-1β, partially via ERK1/2 activation following tau uptake. LRP1 modulates IL-6 responses to tau endocytosis, and this interaction is HSPG-dependent but not ERK-dependent. Solid arrows represent the pathways that have been demonstrated in this study. Dashed arrows represent the pathways speculated to be responsible for subsequent cellular responses based on other relevant publications, as described in the “Discussion” section
Differential Tau-HSPG Interaction Defined by Specific Tau Variants
Previous work describes a “paperclip-like” conformation for tau in solution, where the N- and C-terminal ends fold over in proximity to the center of the repeat domains (Jeganathan et al. 2006). Site-specific phosphorylation and mutations have been found to directly impact the conformation of monomeric tau and affect the stability of a folded conformation, contributing to the propensity for tau protein to aggregate (Tenreiro et al. 2014). A recent study reported that FTD-associated mutations enhanced tau oligomer internalization in SH-SY5Y cells, presenting in vitro evidence that mutation-induced conformational changes may lead to changes in tau binding to HSPGs on the cell surface (Karikari et al. 2019). Indeed, site-specific modifications of tau monomer may cause conformational changes to tau structure that facilitate conversion of inert wild-type tau into seed-competent tau states that have a high propensity to form aggregates and filaments (Mirbaha et al. 2018). In our study, no significant difference was observed in monomeric tau uptake between different protein variants within 30 min. Nevertheless, differential pro-inflammatory gene expression and ERK1/2 phosphorylation was observed with these variants. This result implies that differential tau-HSPG interaction may be responsible for intracellular change; further investigation of tau protein variant binding to heparan sulfate proteoglycans may uncover differential affinities. Alternatively, changes to the tau variants following uptake — such as aggregation or phosphorylation — could alter intracellular pathways. To our knowledge, our study is the first that finds different tau variants’ behavior in activating downstream cellular pathways in a CNS-relevant cellular system, which argues that the tau-HSPG interaction may be also epitope and tau variant-dependent.
Crosstalk Between LRP1 and HSPGs in Regulating Intracellular Responses of Glial Cells to Tau Monomer
Apart from HSPG-dependent endocytosis, tau internalization is also regulated by specific receptor-mediated endocytosis, as suggested by several previous reports (Evans et al. 2018; Rauch et al. 2020). LRP1 has recently been recognized as a master regulator of tau uptake; LRP1 knockdown abolished uptake of various forms of tau including monomers, oligomers, and fibrils in H4 neuroglioma cells (Rauch et al. 2020). Our study showed consistent results as observed in neuronal cells, demonstrating the involvement of LRP1 in tau endocytosis by glial cells. In addition, the cooperative interplay between HSPGs and LRP1 has been found to regulate neuronal Aβ uptake (Kanekiyo et al. 2011). Interestingly, LRP1 has been shown to facilitate the endocytosis and lysosomal degradation of the HSPG-hedgehog complex (Capurro et al. 2012), suggesting a possible LRP1-based regulation of HSPG-dependent endocytosis.
In our study, downregulation of LRP1 mRNA was observed in the glial cells upon monomeric tau addition, while LRP1 remained at a similar level in HSPG-deficient cells compared to control cells upon tau addition. Further, LRP1 knockdown exaggerated IL-6 mRNA expression, while no ERK activation was observed. Thus, we suggest that IL-6 changes likely were altered via the JNK pathway (Fig. 8). Alternatively, upregulated IL-6 expression may be the outcome of cooperation between LRP1 and HSPG via its binding to monomeric tau. Further studies are needed to identify the intracellular signaling pathways involved in the immune response of glial cells to tau, with a special focus on the MAPK and JNK pathways, which have been previously identified as the key protein kinases regulated by Aβ-LRP1 interaction in microglial cells (Yang et al. 2016).
Implications of Efficient Monomeric Tau Endocytosis for Tau Pathogenesis
Extensive studies suggest that the progression of tau pathologies, including FTD, Pick disease, progressive supranuclear palsy, and corticobasal degeneration, rely on the effective spreading of pathogenic tau, known as “seeds,” along both synaptic and non-synaptic pathways (Holmes and Diamond 2014; Hock and Polymenidou 2016). This propagation of tau pathology involves the transfer of pathogenic tau seeds from “donor cell” to “recipient cell” and templated-seeding of aggregation via recruitment of normal tau and formation of new tau seeds in donor cells (Mudher et al. 2017). Soluble tau oligomers have been shown to be the main toxic seeds inducing endogenous tau misfolding (Shafiei et al. 2017) and transneuronal propagation (Pooler et al. 2013). However, the role of monomeric tau in the pathogenesis of tauopathies is still under investigation. Until this report, there has been only one publication that successfully showed the capacity of exogenous monomeric tau for seeding intracellular tau aggregates and its subsequent propagation (Michel et al. 2014). Rapid endocytosis of monomeric tau protein provides an excess of tau protein available for seeding intracellular tau aggregates, though the templating-seeding properties of tau seeds mostly rely on the recruitment of endogenous tau protein (Holmes and Diamond 2014). It remains more plausible that this seeding occurs under pathological conditions where tau seeds facilitate intracellular tau aggregate formation (Leyns and Holtzman 2017; Guo and Lee 2011). Apart from the role of monomeric tau in tau pathogenesis, how astrocytic tau endocytosis affects tau pathology needs further discussion. In our study, abundant and rapid accumulation of tau protein in astrocytic-like cells was observed when compared with neuronal cells. This observation is consistent with prior work demonstrating that astrocytes exhibit greater avidity for oligomeric tau than neurons (Piacentini et al. 2017) primarily due to the enriched phagocytosis pathways in astrocytes (Martini-Stoica et al. 2018). Astrocytic internalization of tau mainly disrupted intracellular Ca2+ signaling, leading to a significant reduction in the levels of released gliotransmitters and synaptic vesicles (Piacentini et al. 2017). Informed by these previous studies, rapid endocytosis of monomeric tau by astrocytes observed in our study would likely alter astrocytic neurotransmission, causing synaptic degeneration. We suggest that the seeding propensity for aggregation and potential alterations in neurotransmission following uptake of monomeric tau may have an overlapping role in the progression of tau pathogenesis. Future studies will focus on further investigating the functional alterations in CNS cell lines following monomeric tau internalization.
Conclusions
Our study shows that monomeric tau is rapidly endocytosed by multiple CNS-relevant cell lines, and the fastest tau endocytosis rate was observed in glial cells among all the cell types tested. In addition, we have demonstrated that tau endocytosis is facilitated by multiple pathways including actin-dependent macropinocytosis as well as HSPG- and LRP1-dependent pathways. Moreover, HSPG-mediated tau endocytosis in glial cells also triggered the downstream upregulation of pro-inflammatory gene expression, in part via mediating ERK1/2 signaling activation. Further, ERK1/2 phosphorylation also displayed a tau variant-dependent activation, where disease-relevant tau variants induced the highest levels of ERK1/2 phosphorylation. Further investigation also revealed the pivotal role of LRP1 in regulating the pro-inflammatory gene expression and tau internalization via altering the expression level of LRP1. The study has provided experimental evidence to advance the understanding of the important role of monomeric tau uptake in glial cells in the progression of tau pathology.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Dr. Deborah Grimm (Tulane University) for the assistance with confocal microscopy experiments and analysis.
Abbreviations
- AD
Alzheimer’s disease
- HSPG
Heparan sulfate proteoglycans
- NFT
Neurofibrillary tangles
- Xylt1
Xylosyltransferase-1
- FTD
Frontotemporal dementia
- CNS
Central nervous system
- Aβ
β-Amyloid peptide
- ISF
Interstitial fluid
- CSF
Cerebrospinal fluid
- HSPG
Heparan sulfate proteoglycans
- HS
Heparan sulfate
- LRP1
Low-density lipoprotein receptor-related protein 1
- MT
Microtubules
Author contribution
All authors contributed to the overall study conception and design, and Anne S. Robinson was responsible for overall supervision. Material preparation, data collection, and analysis were performed by Liqing Song, Daniel E. Oseid, and Evan A. Wells. The first draft of the manuscript was written by Liqing Song, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported in part by the Marko Spark Innovation fund (Tulane University Brain Institute) and by Carnegie Mellon University.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Nothing to declare.
Declarations
Additional declarations for articles in life science journals that report the results of studies involving humans and/or animals
Nothing to declare.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Liqing Song and Daniel E. Oseid contributed equally to this work.
References
- Agholme L, Lindström T, Kgedal K, et al. An in vitro model for neuroscience: differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J Alzheimer’s Dis. 2010;20:1069–1082. doi: 10.3233/JAD-2010-091363. [DOI] [PubMed] [Google Scholar]
- Barbara Stopschinski XE, Holmes BB, Miller GM, et al. Specific Glycosaminoglycan Chain Length and Sulfation Patterns Are Required for Cell Uptake of Tau versus-Synuclein and-Amyloid Aggregates. 2018 doi: 10.1074/jbc.RA117.000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barten DM, Cadelina GW, Hoque N, et al. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimer’s Dis. 2011;24:127–141. doi: 10.3233/JAD-2011-110161. [DOI] [PubMed] [Google Scholar]
- Bejoy J, Bijonowski B, Marzano M, et al. Wnt-notch signaling interactions during neural and astroglial patterning of human stem cells. Tissue Eng - Part A. 2020;26:419–431. doi: 10.1089/ten.tea.2019.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum-Degena D, Müller T, Kuhn W, et al. Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, et al. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-X. [DOI] [PubMed] [Google Scholar]
- Brion JP, Hanger DP, Couck AM, Anderton BH. A68 proteins in Alzheimer’s disease are composed of several tau isoforms in a phosphorylated state which affects their electrophoretic mobilities. Biochem J. 1991;279:831. doi: 10.1042/BJ2790831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, et al. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/S0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capurro MI, Shi W, Filmus J. LRP1 mediates Hedgehog-induced endocytosis of the GPC3-Hedgehog complex. J Cell Sci. 2012;125:3380–3389. doi: 10.1242/jcs.098889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiroma SM, Baharuldin MTH, Taib CNM et al (2018) Inflammation in Alzheimer’s disease: a friend or foe? Biomed Res Ther 5:2552–2564. 10.15419/bmrat.v5i8.464
- Christianson HC, Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014;35:51–55. doi: 10.1016/j.matbio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Chua CC, Rahimi N, Forsten-Williams K, Nugent MA. Heparan sulfate proteoglycans function as receptors for fibroblast growth factor-2 activation of extracellular signal-regulated kinases 1 and 2. Circ Res. 2004;94:316–323. doi: 10.1161/01.RES.0000112965.70691.AC. [DOI] [PubMed] [Google Scholar]
- Cong L, Zhang F (2014) Genome engineering using crispr-cas9 system. In: Chromosomal Mutagenesis: Second Edition 197–217
- Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement Transl Res Clin Interv. 2018;4:195–214. doi: 10.1016/j.trci.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Prà I, Chiarini A, Gui L, et al. Do astrocytes collaborate with neurons in spreading the “infectious” aβ and Tau drivers of Alzheimer’s disease? Neuroscientist. 2015;21:9–29. doi: 10.1177/1073858414529828. [DOI] [PubMed] [Google Scholar]
- Dujardin S, Bégard S, Caillierez R, et al. Ectosomes: a new mechanism for non-exosomal secretion of Tau protein. PLoS ONE. 2014 doi: 10.1371/journal.pone.0100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S, Rhoades E. Identification of an aggregation-prone structure of tau. J Am Chem Soc. 2012;134:16607–16613. doi: 10.1021/ja305206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans LD, Wassmer T, Fraser G, et al. Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. 2018;22:3612–3624. doi: 10.1016/j.celrep.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, et al. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy an. Brain Pathol. 2001;11:144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, López-González I, Carmona M, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol. 2014 doi: 10.1097/NEN.0000000000000030. [DOI] [PubMed] [Google Scholar]
- Fillit H, Ding W, Buee L, et al. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-K. [DOI] [PubMed] [Google Scholar]
- Flanagan LA, Cunningham CC, Chen J, et al. The structure of divalent cation-induced aggregates of PIP2 and their alteration by gelsolin and tau. Biophys J. 1997 doi: 10.1016/S0006-3495(97)78176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleeman RM, Proctor EA (2021) Astrocytic propagation of tau in the context of Alzheimer’s disease. Front Cell Neurosci 63. 10.3389/FNCEL.2021.645233 [DOI] [PMC free article] [PubMed]
- Frost B, Jacks RL, Diamond MI. Propagation of Tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Vaquer-Alicea J, White CL, et al. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017;133:91–100. doi: 10.1007/s00401-016-1644-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galland F, Seady M, Taday J, et al. Astrocyte culture models: molecular and function characterization of primary culture, immortalized astrocytes and C6 glioma cells. Neurochem Int. 2019;131:104538. doi: 10.1016/J.NEUINT.2019.104538. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, García-Sierra F, Fu Y, et al. Tau-66: evidence for a novel tau conformation in Alzheimer’s disease. J Neurochem. 2001;77:1372–1385. doi: 10.1046/j.1471-4159.2001.00346.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992 doi: 10.1016/0896-6273(92)90117-V. [DOI] [PubMed] [Google Scholar]
- Gómez-Ramos A, Díaz-Hernández M, Rubio A, et al. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci. 2008 doi: 10.1016/j.mcn.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Gómez-Ramos A, Díaz-Hernández M, Rubio A, et al. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur Neuropsychopharmacol. 2009 doi: 10.1016/j.euroneuro.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Guo JL, Lee VMY. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock E-M, Polymenidou M. Prion-like propagation as a pathogenic principle in frontotemporal dementia. J Neurochem. 2016;138:163–183. doi: 10.1111/jnc.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–E3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, Diamond MI. Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem. 2014;289:19855–19861. doi: 10.1074/jbc.R114.549295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan S, Von Bergen M, Brutlach H, et al. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Kálmán J, Juhász A, Laird G, et al. Serum interleukin-6 levels correlate with the severity of dementia in Down syndrome and in Alzheimer’s disease. Acta Neurol Scand. 1997;96:236–240. doi: 10.1111/J.1600-0404.1997.TB00275.X. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Zhang J, Liu Q, et al. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J Neurosci. 2011;31:1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanmert D, Cantlon A, Muratore CR, et al. C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci. 2015;35:10851–10865. doi: 10.1523/JNEUROSCI.0387-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Jeng AT, Goate AM. Extracellular tau levels are influenced by variability in tau that is associated with tauopathies. J Biol Chem. 2012;287:42751–42762. doi: 10.1074/jbc.M112.380642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikari TK, Nagel DA, Grainger A, et al. Distinct conformations, aggregation and cellular internalization of different tau strains. Front Cell Neurosci. 2019;13:296. doi: 10.3389/fncel.2019.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsinelos T, Zeitler M, Dimou E, et al. Unconventional secretion mediates the trans-cellular spreading of Tau. Cell Rep. 2018;23:2039–2055. doi: 10.1016/j.celrep.2018.04.056. [DOI] [PubMed] [Google Scholar]
- Kim Y, Park J, Choi YK. The role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: a review. Antioxidants. 2019;8:121. doi: 10.3390/antiox8050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017;12:1–16. doi: 10.1186/s13024-017-0192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cheng D, Cheng R, et al. Mechanisms of U87 astrocytoma cell uptake and trafficking of monomeric versus protofibril Alzheimer’s disease amyloid-β proteins. PLoS ONE. 2014;9:e99939. doi: 10.1371/journal.pone.0099939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidholt K, Weinke JL, Kiser CS, et al. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci U S A. 1992;89:2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Haque MM, Kim D, et al. Cell-based models to investigate Tau aggregation. Comput Struct Biotechnol J. 2014;12:7–13. doi: 10.1016/j.csbj.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- Maddahi A, Povlsen G, Edvinsson L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J Neuroinflammation. 2012;9:783. doi: 10.1186/1742-2094-9-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini-Stoica H, Cole AL, Swartzlander DB, et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med. 2018;215:2355–2377. doi: 10.1084/jem.20172158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merezhko M, Brunello CA, Yan X et al (2018) Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep 25. 10.1016/j.celrep.2018.10.078 [DOI] [PubMed]
- Michel CH, Kumar S, Pinotsi D, et al. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem. 2014 doi: 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbaha H, Chen D, Morazova OA et al (2018) Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 7. 10.7554/eLife.36584 [DOI] [PMC free article] [PubMed]
- Mirbaha H, Holmes BB, Sanders DW, et al. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem. 2015;290:14893–14903. doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova OA, March ZM, Robinson AS, Colby DW. Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry. 2013;52:6960–6967. doi: 10.1021/BI400866W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Colin M, Dujardin S, et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun. 2017;5:99. doi: 10.1186/s40478-017-0488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Takenaka-Uema A, Kobayashi T, et al. Heparan sulfate proteoglycan is an important attachment factor for cell entry of Akabane and Schmallenbergviruses. J Virol. 2017 doi: 10.1128/jvi.00503-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, Braak H, An WL, et al. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Mol Brain Res. 2002;109:45–55. doi: 10.1016/S0169-328X(02)00488-6. [DOI] [PubMed] [Google Scholar]
- Perea JR, López E, Díez-Ballesteros JC, Ávila J, Hernández F, Bolós M (2019) Extracellular Monomeric Tau Is Internalized by Astrocytes. Front Neurosci 13:442. 10.3389/fnins.2019.00442 [DOI] [PMC free article] [PubMed]
- Piacentini R, Li Puma DD, Mainardi M, et al. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia. 2017;65:1302–1316. doi: 10.1002/glia.23163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Polydoro M, Wegmann S, et al. Propagation of tau pathology in Alzheimer’s disease: identification of novel therapeutic targets. Alzheimer’s Res Ther. 2013;5:49. doi: 10.1186/alzrt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN, Chen JJ, Sorum AW et al (2018) Tau internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci Rep 8 10.1038/s41598-018-24904-z [DOI] [PMC free article] [PubMed]
- Rauch JN, Luna G, Guzman E, et al. LRP1 is a master regulator of tau uptake and spread. Nature. 2020;580:381–385. doi: 10.1038/s41586-020-2156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetin K, Steullet P, Pachoud M, et al. (2020) Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat Neurosci. 2020;2312(23):1567–1579. doi: 10.1038/s41593-020-00728-x. [DOI] [PubMed] [Google Scholar]
- Saman S, Kim WH, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta U, Portelius E, Hansson O, et al. Tau oligomers in cerebrospinal fluid in Alzheimer’s disease. Ann Clin Transl Neurol. 2017;4:226–235. doi: 10.1002/acn3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiei SS, Guerrero-Muñoz MJ, Castillo-Carranza DL (2017) Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci 9 [DOI] [PMC free article] [PubMed]
- Sharma AM, Thomas TL, Woodard DR, et al. Tau Monomer Encodes Strains Biorxiv. 2018 doi: 10.1101/325043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Wang C, Wang H et al (2020) The potential of low molecular weight heparin to mitigate cytokine storm in severe COVID-19 patients: a retrospective cohort study. Clin Transl Sci 13 10.1111/cts.12880 [DOI] [PMC free article] [PubMed]
- Sidoryk-Wegrzynowicz M, Gerber YN, Ries M, et al. Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol Commun. 2017;5:89. doi: 10.1186/s40478-017-0478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Wells EA, Robinson AS. Critical molecular and cellular contributors to tau pathology. Biomedicines. 2021;9:190. doi: 10.3390/biomedicines9020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Yuan X, Jones Z, et al. Functionalization of brain region-specific spheroids with isogenic microglia-like cells. Sci Rep. 2019;9:1–18. doi: 10.1038/s41598-019-47444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancu IC, Cremers N, Vanrusselt H, et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137:599–617. doi: 10.1007/s00401-018-01957-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Wegmann S, Cho H, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun. 2015;6:1–15. doi: 10.1038/ncomms9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanemura K, Murayama M, Akagi T, et al. Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci. 2002;22:133–141. doi: 10.1523/jneurosci.22-01-00133.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenreiro S, Eckermann K, Outeiro TF (2014) Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci 7 [DOI] [PMC free article] [PubMed]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, et al. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell. 2000;11:4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Balaji V, Kaniyappan S, et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. 2017;12:5. doi: 10.1186/s13024-016-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci [DOI] [PubMed]
- Wu JW, Herman M, Liu L et al (2012) Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. 10.1074/jbc.M112.394528 [DOI] [PMC free article] [PubMed]
- Yamada K, Cirrito JR, Stewart FR, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Liu CC, Zheng H et al (2016) LRP1 modulates the microglial immune response via regulation of JNK and NF-ΚB signaling pathways. J Neuroinflammation 13. 10.1186/s12974-016-0772-7 [DOI] [PMC free article] [PubMed]
- Yang Z, Steentoft C, Hauge C, et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015;43:e59–e59. doi: 10.1093/nar/gkv126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, et al. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci. 2004;24:4657–4667. doi: 10.1523/JNEUROSCI.0797-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhu Y, Song X, et al. 3-O-sulfation of heparan sulfate enhances tau interaction and cellular uptake. Angew Chemie Int Ed. 2020;59:1818–1827. doi: 10.1002/anie.201913029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Nothing to declare.