

Abstract
Plasma tumour DNA (ptDNA) is a potential early noninvasive biomarker of treatment outcome in metastatic castration‐resistant prostate cancer (mCRPC). Herein, we investigated whether pretreatment ptDNA levels reflect metabolic tumour burden in mCRPC and better predict treatment outcome in combination with functional imaging. Targeted next‐generation sequencing was performed to estimate the ptDNA fraction from 102 mCRPC patients receiving abiraterone or enzalutamide. The maximum standardized uptake value (SUVmax), total lesion activity (TLA) and metabolic tumour volume (MTV) were evaluated on 18F‐fluorocholine positron emission tomography/computed tomography. We assessed a Weibull multiple regression model to determine the combined impact of clinical, molecular and imaging characteristics on overall survival (OS) and progression‐free survival (PFS), and to obtain prognostic scores. A significant association was seen between ptDNA and SUVmax, MTV and TLA. For survival analysis, patients were randomly allocated into a training (n = 68) and a validation (n = 34) set. In the training set, multivariable analyses showed that ptDNA, MTV and serum lactate dehydrogenase together with visceral metastasis were independent predictors of both OS and PFS. Prognostic scores were generated, with the identification of three groups of patients with significantly different median OS (29.2, 15.9 and 8.7 months) and PFS (13.3, 7.7 and 3.2 months) probabilities. The differences in median survival between risk groups were confirmed in the validation cohort for both OS and PFS. In our study, we showed that integrating plasma DNA analysis with functional imaging may improve prognostic risk stratification and treatment selection in mCRPC.
Keywords: choline PET/TC, metabolic activity, metastatic castration‐resistant prostate cancer, plasma tumour DNA, prognosis
Plasma tumour DNA (ptDNA) has been recently demonstrated as a potential early noninvasive biomarker in patients affected with metastatic castration‐resistant prostate cancer receiving androgen receptor signalling inhibitors. In our study, we initially showed that ptDNA reflected tumour metabolic activity. Additionally, integrating ptDNA analysis with functional imaging and clinical features showed potential for an improved outcome prediction as well as for a better treatment selection in these patients.
Abbreviations
- ALP
alkaline phosphatase
- AR
androgen receptor
- ARSI
androgen receptor‐signalling inhibitors
- CGA
chromogranin A
- CI
confidence interval
- CT
computed tomography
- CTC
circulating tumour cell
- ECOG
Eastern Cooperative Oncology Group
- HR
hazard ratio
- LDH
lactate dehydrogenase
- mCRPC
metastatic castration‐resistant prostate cancer
- MTV
metabolic tumour volume
- OS
overall survival
- PCWG3
Prostate Cancer Working Group 3
- PET/CT
positron emission tomography/computed tomography
- PFS
progression‐free survival
- PSA
prostate‐specific antigen
- ptDNA
plasma tumour DNA
- SUVmax
maximum standardized uptake value
- TLA
total lesion activity
1. Introduction
Prostate cancer is one of the most frequent malignancies in male adults [1]. Like many cancers, prostate cancer is heterogeneous and reveals a branched evolutionary pattern going from locally advanced to progression of metastatic tumour [2]. Furthermore, clonal evolution continues to happen as a result of systemic therapy, with selection for diverse resistance mechanisms, including reactivation of the androgen–androgen receptor (AR) axis via AR gene aberrations such as amplification, mutations and variants upon challenging of AR signalling inhibitors (ARSI) [3, 4, 5, 6].
In men with prostate cancer receiving a systemic treatment, tumour burden and responses to therapy are commonly assessed through clinical evaluation, prostate‐specific antigen (PSA) levels and radiologic imaging with computed tomography (CT), bone scan, magnetic resonance imaging and positron emission tomography (PET) [7]. Significant efforts have been directed towards developing and enhancing imaging methods for the characterization of prostate tumours. CT/PET offers the opportunity to assess both tumour volume and metabolic disease activity. Despite reliability of current imaging techniques, complementary strategies are warranted to better guide treatment choices.
Liquid biopsy has established itself as method able to overcome the inherent challenges of following treatment resistance using tissue biopsies. Contrary to a tissue biopsy, a liquid biopsy is minimally invasive, capable of real‐time molecular characterization of the disease through the identification of plasma tumour DNA (ptDNA) and able to capture a composite picture of intra‐ and intertumour heterogeneity of the potentially clinically relevant disease [8, 9].
Our group has previously shown that positron emission tomography/computed tomography (PET/CT) with 18F‐fluorocholine (FCH) can be used for early response assessment in metastatic castration‐resistant prostate cancer (mCRPC) patients treated with abiraterone and enzalutamide [10, 11] and that combining PET/CT with AR copy number gain detected in ptDNA associate with outcome in mCRPC [12]. However, the association between ptDNA fraction, as an early noninvasive biomarker of treatment response [3, 13, 14, 15], and tumour burden remains largely unexplored. In this study, we sought to establish the relationship between ptDNA fraction and metabolic tumour burden and if ptDNA in combination with functional imaging more accurately predicts treatment outcome in mCRPC.
2. Materials and methods
2.1. Study design
In this work, we evaluated mCRPC patients with histologically confirmed diagnosis of prostate adenocarcinoma without evidence of neuroendocrine differentiation and small cell histology. Selection criteria included also patients with serum testosterone < 50 ng·dL−1 with androgen deprivation therapy and Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2. Patients received abiraterone 1000 mg with twice‐daily prednisone 5 mg or enzalutamide 160 mg once daily until evidence of progressive disease or unacceptable toxicity.
Peripheral blood samples for plasma DNA analysis, laboratory parameters such as complete blood count, PSA, serum lactate dehydrogenase (LDH), alkaline phosphatase (ALP) and chromogranin A (CGA), were assessed within 1 week of therapy initiation. FCH‐PET/CT imaging was performed within 1 week before starting ARSI therapy. PSA response and toxicity were monthly assessed. Radiographic evaluation was performed with the use of CT scan before treatment and every 3 months thereafter as for institutional clinical practice. Response was retrospectively assessed according to Prostate Cancer Working Group 3 (PCWG3) guidelines [7], and soft tissue disease was evaluated on the basis of Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1
The study was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Conference of Harmonization. The Institutional Review Board approved biomarker study (REC 2192/2013), and patients signed informed consent.
2.2. PET/CT imaging protocol
FCH‐PET/CT scans were carried out on an integrated PET/CT system (Discovery LS camera; General Electric Medical Systems, Waukesha, WI, USA) in 2D acquisition mode for 3 min per bed position. The PET/CT scan takes 45 min after intravenous injection of 18F‐methylcholine (3.7 MBq·kg−1 of body weight, AAA‐Advanced Accelerator Applications, Meldola, Italy). The field of view included the skull to mid‐femurs. Low dose CT (120 kV, 80 mA) without contrast agents was made for attenuation correction and as an anatomical map. The emission data were adjusted for scatter, random coincidence events, and system dead time. Two nuclear medicine physicians with good experience did the reading and interpretation of FCH‐PET/CT scans. Criteria to identify FCH‐PET/CT positivity consisted of the presence of focal areas of raised tracer uptake with or without any underlying lesion found by performing CT. Semiquantitative criteria based on the maximum standardized uptake value (SUVmax) and the target‐to‐background ratio were utilized to aid the visual analysis [16]. The metabolic tumour volume (MTV) parameter was obtained by adding each three‐dimensional volume of interest, and for each lesion volume and SUV mean was multiplied and then summed to have the total lesion activity (TLA). MTV is only a volumetric entity, while TLA takes also into account the metabolic activity of the lesion, so providing an evaluation of the tumour activity. FCH‐PET/CT scans were read sequentially thanks to a Xeleris III Workstation (General Electric Medical Systems). PET, CT and PET/CT fused images supply to provide the scans in axial, sagittal and coronal sections.
2.3. Plasma tumour DNA analysis
Cell‐free DNA was extracted from 1 to 2 mL of plasma with the QIAamp Circulating Nucleic Acid Kit (Qiagen, Santa Clarita, CA, USA) and quantified by spectrophotometric evaluation (NanoDrop® ND‐1000; Celbio, Milan, Italy) or Quant‐iT High Sensitivity PicoGreen Double‐Stranded DNA Assay Kit (Invitrogen, Carlsbad, CA, USA). In plasma and patient‐matched germline DNA, targeted next‐generation sequencing (NGS) was assessed by the PGM Ion Torrent using a 316 or 318 Chip aiming to reach 1000× coverage per target. We estimated the plasma tumour fraction and AR gene copy number for each plasma sample from the study patients by using the approach previously described [3, 4].
2.4. Statistical analysis
Progression‐free survival (PFS) was considered as the time between the first day of ARSI therapy and the date of progression disease or death (whichever came first). Men who had not progressed at database closure were censored at the last tumour assessment or treatment discontinuation for severe adverse events. Overall survival (OS) was considered as the time between the first day of ARSI treatment and the date of death from any cause or the date of the last follow‐up visit.
Categorical variables were summarized using frequency whereas continuous variables were described using median value and interquartile range. The association between categorical variables was determined using the chi‐squared or Fisher's exact test, as appropriate. Spearman correlation was used to assess the association between continuous variables.
Survival curves were estimated by the Kaplan–Meier method, and comparisons were made using the logrank test. Univariable and multivariable Cox regression models were utilized to explore potential factors able to predict PFS and OS and to estimate hazard ratios (HRs) and their 95% confidence interval (CI). All P‐values were two‐sided, and a P < 0.05 was defined as statistically significant. Statistical analyses were done with sas 9.4 software (SAS Institute, Cary, NC, USA).
We used a Weibull multiple regression model to assess the matched impact of molecular, laboratory and imaging characteristics on outcome. From a full model including these factors, we achieved a final parsimonious model by using a backward selection procedure. The prognostic score was built on the final model consisting of four factors for OS and three for PFS. Partial scores were procured by splitting the value of each regression coefficient by the smallest regression coefficient. The total score for each patient resulted from a sum of appropriate partial scores, and three patient groups with different median survival probabilities were recognized. For OS, if the total score was 1 or below, between 1.1 and 2.5, and > 2.5, patients were classified as group I, group II and group III, respectively. For PFS, if the total score was 1 or below, between 1.0 and 2.1, and > 2.1, patients were classified as group I, group II and group III.
3. Results
3.1. Patient cohort characteristics
Between October 2011 and June 2016, 102 plasma samples were collected from CRPC patients at start of treatment with ARSI. Sixty‐six patients were treated with abiraterone, 36 with enzalutamide, 27 (26.5%) were chemotherapy‐naïve, 75 (73.5%) were previously chemotherapy‐treated, and 17 (16.7%) received prior therapy with ARSI. Interactions between ARSI treatment and ptDNA analysed in the Cox models were P = 0.073 for OS and P = 0.213 for PFS.
Patients treated with ARSI were randomly splitted into one training set (n = 68) and one validation set (n = 34). The larger two‐third group of men was utilized as a training set to build the prognostic score, and the remaining one‐third was considered as a validation cohort. Except for lower ALP concentration in the training vs. validation cohort (P = 0.0002), no significant differences in baseline characteristics were seen between training and validation groups (Table 1).
Table 1.
Patient characteristics. n, number; NLR, neutrophil–lymphocyte ratio; PS, performance status; SUV, standardized uptake value.
| Total (n = 102) | Training (n = 68) | Validation (n = 34) | P | |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | ||
| Age, years | ||||
| ≤ 74 a | 54 (52.9) | 40 (58.8) | 14 (41.2) | |
| > 74 | 48 (47.1) | 28 (41.2) | 20 (58.8) | 0.092 |
| Prostatectomy | ||||
| No | 60 (58.8) | 40 (58.8) | 20 (58.8) | |
| Yes | 42 (41.2) | 28 (41.2) | 14 (41.2) | 1.000 |
| Radical radiotherapy | ||||
| No | 58 (56.9) | 42 (61.8) | 16 (47.1) | |
| Yes | 44 (43.1) | 26 (38.2) | 18 (52.9) | 0.157 |
| Gleason score | ||||
| 6–7 | 41 (45.6) | 28 (45.9) | 13 (44.8) | |
| 8–10 | 49 (54.4) | 33 (54.1) | 16 (55.2) | 0.924 |
| Site of metastasis | ||||
| Bone | 91 (89.2) | 63 (92.7) | 28 (82.3) | 0.173 |
| Lymph nodes | 53 (52.0) | 37 (54.4) | 16 (47.1) | 0.483 |
| Liver | 5 (4.9) | 3 (4.4) | 2 (5.9) | 0.746 |
| Lung | 9 (8.8) | 8 (11.8) | 1 (2.9) | 0.265 |
| ECOG PS | ||||
| 0–1 | 99 (97.1) | 66 (97.1) | 33 (97.1) | |
| ≥ 2 | 3 (2.9) | 2 (2.9) | 1 (2.9) | 1.000 |
| Presence of pain | ||||
| No | 94 (92.2) | 62 (91.2) | 32 (94.1) | |
| Yes | 8 (7.8) | 6 (8.8) | 2 (5.9) | 0.602 |
| Type of treatment | ||||
| Abiraterone | 66 (64.7) | 46 (67.7) | 20 (58.8) | |
| Enzalutamide | 36 (35.3) | 22 (32.3) | 14 (41.2) | 0.379 |
| Chemotherapy‐naive | ||||
| No | 75 (73.5) | 51 (75.0) | 24 (70.6) | |
| Yes | 27 (26.5) | 17 (25.0) | 10 (29.4) | 0.634 |
| Prior therapeutic lines | ||||
| 1–2 | 65 (63.7) | 45 (66.2) | 20 (58.8) | |
| > 2 | 37 (36.3) | 23 (33.8) | 14 (41.2) | 0.466 |
| Serum LDH, U·L−1 | ||||
| < 225 b | 78 (76.5) | 50 (73.5) | 28 (82.3) | |
| ≥ 225 | 24 (23.5) | 18 (26.5) | 6 (17.7) | 0.322 |
| ALP, U·L−1 | ||||
| < 129 b | 67 (65.7) | 53 (77.9) | 14 (41.2) | |
| ≥ 129 | 35 (34.3) | 15 (22.1) | 20 (58.8) | 0.0002 |
| NLR | ||||
| < 3 b | 54 (52.9) | 35 (51.5) | 19 (55.9) | |
| ≥ 3 | 48 (47.1) | 33 (48.5) | 15 (44.1) | 0.674 |
| Serum CGA, ng·mL−1 | ||||
| < 120 b | 48 (47.1) | 29 (42.7) | 19 (55.9) | |
| ≥ 120 | 54 (52.9) | 39 (57.3) | 15 (44.1) | 0.207 |
| Haemoglobin, g·dL−1 | ||||
| > 12.5 b | 40 (39.2) | 25 (36.8) | 15 (44.1) | |
| ≤ 12.5 | 62 (60.8) | 43 (63.2) | 19 (55.9) | 0.473 |
| Serum albumin, g·dL−1 | ||||
| > 4 b | 50 (52.6) | 32 (50.8) | 18 (56.2) | |
| ≤ 4 | 45 (47.4) | 31 (49.2) | 14 (43.8) | 0.615 |
| Unknown/missing | 7 | 5 | 2 | |
| Serum PSA, ng·dL−1 | ||||
| < 23.24 a | 50 (49.5) | 32 (47.8) | 18 (52.9) | |
| ≥ 23.24 | 51 (50.5) | 35 (52.2) | 16 (47.1) | 0.623 |
| Unknown/missing | 1 | 1 | 0 | |
| Number of lesions on FCH‐CT/PET | ||||
| < 12 a | 51 (50.0) | 33 (48.5) | 18 (52.9) | |
| ≥ 12 | 51 (50.0) | 35 (51.5) | 16 (47.1) | 0.674 |
| SUVmax | ||||
| < 83.60 a | 50 (49.5) | 33 (49.2) | 17 (50.0) | |
| ≥ 83.60 | 51 (50.5) | 34 (50.8) | 17 (50.0) | 0.943 |
| Unknown/missing | 1 | 1 | 0 | |
| MTV | ||||
| < 102.79 a | 51 (50.0) | 35 (51.5) | 16 (47.1) | |
| ≥ 102.79 | 51 (50.0) | 33 (48.5) | 18 (52.9) | 0.674 |
| TLA | ||||
| < 391343 a | 51 (50.0) | 34 (50.0) | 17 (50.0) | |
| ≥ 391343 | 51 (50.0) | 34 (50.0) | 17 (50.0) | 1.000 |
| ptDNA | ||||
| ≤ 0.188 a | 51 (50.0) | 33 (48.5) | 18 (52.9) | |
| > 0.188 | 51 (50.0) | 35 (51.5) | 16 (47.1) | 0.674 |
| AR copy number | ||||
| Normal | 75 (73.5) | 51 (75.0) | 24 (70.6) | |
| Gain | 27 (26.5) | 17 (25.0) | 10 (29.4) | 0.634 |
Median value.
Upper normal value.
3.2. Associations of ptDNA with clinical variables and functional imaging
Median fraction of ptDNA before starting treatment was 0.188 (0.014–0.96). We evaluated the distribution of tumour content fraction in plasma among patients with different number and types of metastasis. A significant correlation was reported between ptDNA and number of lesions (rs = 0.53, P < 0.0001) (Fig. 1A). However, ptDNA did not significantly associate with the number of different types of sites of metastasis (Fig. 1B) nor the specific type of metastasis a patient harboured although there was a trend for patients with liver metastasis (P = 0.058) or bone metastasis (P = 0.104) to have a higher ptDNA fraction (Fig. S1).
Fig. 1.
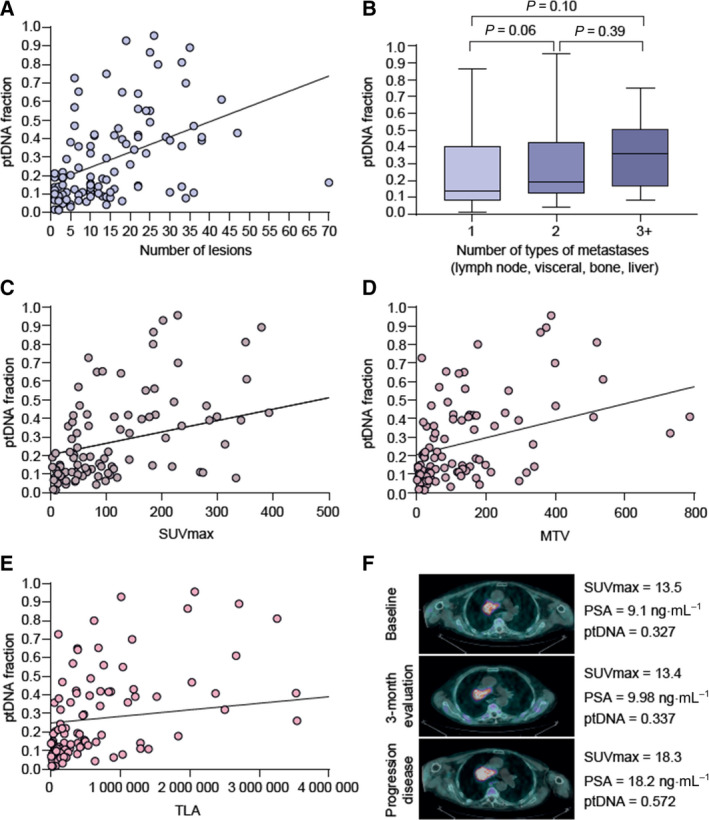
Association of ptDNA fraction with metastatic sites and metabolic activity. (A) Correlation of the number of metastatic sites and ptDNA. The outcome was the relationship between quantitative variables that was examined using the linear correlation coefficient (Pearson product moment correlation coefficient), r = 0.46, P < 0.0001. (B) Association of median ptDNA fraction and the number of types of metastases (66 patients had only one metastatic site, 54 had two metastatic sites, and five had more than two metastatic sites of disease). Box␣plot error bars show the range of the data set. All reasonably ‘extreme’ data are contained between the two ends of the error bars. Error bars are typically extended to be 1.5 times the interquartile range beyond the first and third quartiles if outlier values are present. We used Wilcoxon–Mann–Whitney test for comparison of ptDNA fraction considered as continuous data and number of types of metastases (two independent groups, P = 0.06 and P = 0.39) or Kruskal–Wallis test (three independent groups, P = 0.10). SUVmax (C), MTV (D) and TLA (E) associated with ptDNA fraction. The outcome was the relationship between quantitative variables that was examined using the Pearson linear correlation coefficient: (C) r = 0.48, P < 0.0001; (D) r = 0.47, P < 0.0001; (E) r = 0.37, P < 0.0001. (F) Representative case of association of metabolic activity and ptDNA fraction. MTV, metabolic tumour activity; TLA, tumour lesion activity.
In addition, we correlated ptDNA and clinical variables at plasma collection and observed that ptDNA levels were significantly associated with LDH, ALP and PSA (Fig. S2). We also correlated AR copy number gain, detected in cell‐free DNA from 27.3% of overall patients, with ptDNA level showing a significant correlation (rs = 0.45, P < 0.0001) (Fig. S2).
Furthermore, we investigated the association between choline uptake measured as median SUVmax, MTV, TLA and ptDNA levels. As expected, FCH‐PET/CT parameters (SUVmax, MTV and TLA) were correlated with each other (Fig. S3). As it is known that FCH‐PET/CT is not an appropriate method for the evaluation of the liver status because of the normal tracer distribution [17], we excluded five patients harbouring hepatic metastases from all analyses of the current study (Table S1).
We reported a meaningful correlation between ptDNA fraction and choline uptake measured by SUVmax (rs = 0.544, P < 0.0001), MTV (rs = 0.488 P < 0.0001) and TLA (rs = 0.538 P < 0.0001) (Fig. 1C–E). A representative case, of a mCRPC postdocetaxel patient treated with abiraterone, illustrates this direct relationship between ptDNA and choline uptake on FCH‐PET/CT (Fig. 1F).
3.3. Ability of combining ptDNA analysis and functional imaging to predict clinical outcome
Median follow‐up was 49 months (range 1–50). The median OS and PFS for the overall population were 15.3 months (95% CI 11.4–17.5) and 5.7 months (95% CI 5.0–6.7), respectively. Recognizing the need for a prognostic tool that mirrors outcomes from currently available treatments, we examined variables associated with OS and PFS. Thus, we developed index models based on clinical, molecular and imaging factors.
In the training cohort, we performed univariate (Tables S2 and S3) and multivariable (Tables 2 and 3) Cox proportional hazards regression analyses of OS and PFS, respectively, to assess the associations between functional imaging, clinical features and molecular biomarkers and to generate prognostic scores.
Table 2.
Multivariable analysis of OS after backward stepwise procedure in the training cohort. Total score ranges from 1 to 5.2.
| Factor estimate (standard error) | Standard error | P | HR (95% CI) | Partial score | |
|---|---|---|---|---|---|
| MTV | 0.599 | 0.268 | 0.026 | 1.82 (1.08–3.08) | 1.00 |
| ptDNA | 0.848 | 0.289 | 0.003 | 2.34 (1.32–4.12) | 1.40 |
| Visceral metastasis | 1.033 | 0.383 | 0.007 | 2.81 (1.33–5.95) | 1.70 |
| Serum LDH, U·L−1 | 1.239 | 0.331 | 0.0002 | 3.45 (1.81–6.60) | 2.10 |
| Risk groups | No. pts (%) | Total score |
|---|---|---|
| I | 22 (33.9) | < 1.4 |
| II | 24 (36.9) | 1.4–2.8 |
| III | 19 (29.2) | ≥ 2.8 |
Table 3.
Multivariable analysis of PFS after backward stepwise procedure in the training cohort. Total score ranges from 0 to 5.85.
| Factor estimate (standard error) | Standard error | P | HR (95% CI) | Partial score | |
|---|---|---|---|---|---|
| MTV | 0.586 | 0.271 | 0.031 | 1.80 (1.06–3.06) | 1.00 |
| ptDNA | 0.645 | 0.266 | 0.015 | 1.91 (1.13–3.21) | 1.10 |
| Visceral metastasis | 0.997 | 0.424 | 0.019 | 2.71 (1.18–6.22) | 1.70 |
| Serum LDH, U·L−1 | 1.204 | 0.323 | 0.0002 | 3.33 (1.77–6.27) | 2.05 |
| Risk groups | No. pts (%) | Total score |
|---|---|---|
| I | 15 (23.1) | < 1.0 |
| II | 34 (52.3) | 1.0–2.1 |
| III | 16 (24.6) | > 2.1 |
Multivariable analysis of OS after a backward stepwise procedure demonstrated that the presence of MTV, ptDNA, visceral metastasis and pretreatment serum LDH were significantly associated with OS [HR 1.82, 95% CI 1.08–3.08, P = 0.026; HR 2.34, 95% CI 1.32–4.12, P = 0.0003; HR 2.81, 95% CI 1.33–5.95, P = 0.007; and HR 3.45, 95% CI 1.81–6.60, P = 0.0002, respectively]. These variables were used to establish a prognostic index model of OS in mCRPC.
Table 2 presents the partial score value for category, originating from the subdivision of each regression coefficient by the smallest one as described in Statistical analysis. The total score for each patient was resulted from each patient's appropriate partial scores, and individuals with different median survival probabilities were classified into three groups. If the total score was equal to 1.4 or lower, the patient was allocated in group I, group II was characterized by a score between 1.4 and 2.8, and group III required a score > 2.8. The choice of time and number of groups with a different survival was more than 70% (group II, 15 months: OS was 30–70%; group III, 15 months: OS was < 30%). In the training set, 22 patients (33.9%) were assigned to risk group I, 24 (36.9%) to risk group II and 19 (29.2%) to risk group III.
The survival experience of the three patient categories was established by the score. Survival probabilities were assessed by the exponential model and by the Kaplan–Meier method. In the training set, we observed a different median OS among the three risk groups (risk group I, 29.2 months [95% CI, 18.3–37.0 months]; risk group II, 14.8 months [95% CI, 9.8–24.0 months]; and risk group III, 9.4 months [95% CI, 6.3–17.4 months]; P < 0.0001) (Fig. 2A). Similarly, in the validation set, we showed risk group I, 23.4 months (95% CI, 8.1–38.5 months); risk group II, 11.2 months (95% CI, 6.0–15.8 months); and risk group III, 9.0 months (95% CI, 3.4–12.9 months); P = 0.009 (Fig. 2B).
Fig. 2.
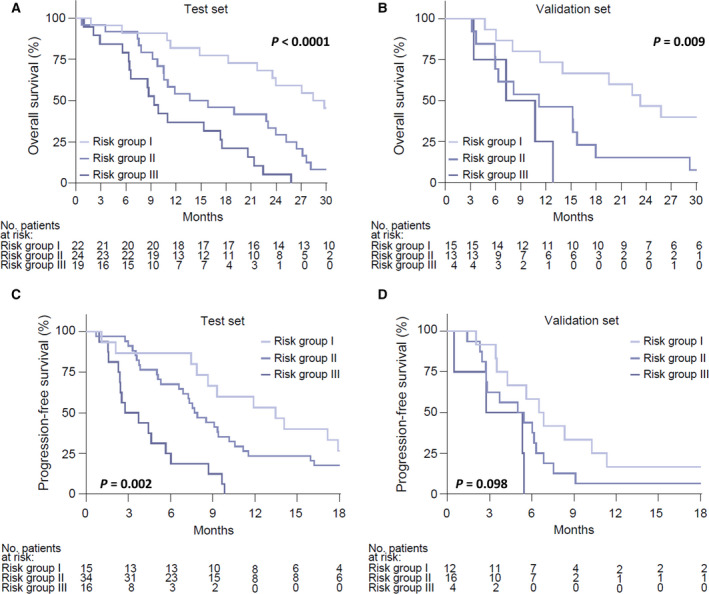
Risk group survival probabilities. Kaplan–Meier curves for OS by OS risk groups in the training set (A) and validation set (B) and PFS by PFS risk groups in the training set (C) and validation set (D). Survival curves were compared using Logrank test.panel. Pearson linear correlation coefficient: (A) P < 0.0001; (B) P = 0.009; (C) 0.002; (D) P = 0.098.
In addition, MTV, ptDNA fraction, lung metastasis and serum LDH harboured independent value for PFS [HR 1.80, 95% CI 1.06–3.06, P = 0.031; HR 1.91, 95% CI 1.13–3.21, P = 0.015, HR 2.71, 95% CI 1.18–6.22, P = 0.019; and HR 3.33, 95% CI 1.77–6.27, P = 0.0002, respectively] (Table 3). Using Cox coefficient analysis of these parameters, we also established three prognostic groups with differing PFS (median, 13.3 vs. 7.7 vs. 3.2 months; P < 0.0001) (Fig. 2C), which was confirmed in the validation set (median, 6.6 vs. 5.1 vs. 4.0 months; P = 0.037) (Fig. 2D).
4. Discussion
Over the last decades, several prognostic scores have been developed by integrating different clinical variables and associating them with prognosis in mCRPC patients receiving standard chemotherapy and ARSI [18, 19, 20, 21, 22, 23, 24, 25]. The majority of these nomograms were characterized by the absence of predictive discrimination to support treatment selection, often limiting their clinical usefulness. Recent biomarker studies aimed to increase the prognostic and predictive value with the inclusion of AR‐V7 expression in circulating tumour cell (CTC), high circulating tumour content and many genetic aberrations involving AR, TP53, the PI3K/AKT pathway and homologous recombination repair [4, 5, 6, 26, 27]. Their integration with typical clinical variables in future models could increase accuracy and improve outcomes prediction, as highlighted herein through the combination of ptDNA analysis and functional imaging.
In our study, particularly striking was the association of ptDNA fraction with metabolic tumour activity and the number of lesions rather than the specific type of lesion, as shown in previous NGS studies on plasma samples from mCRPC and other clinical variants of prostate cancer [28, 29], suggesting that ptDNA fraction may provide interesting aspects of tumour biology and disease volume that are not captured by common clinical factors. Interestingly, despite these associations, both ptDNA and metabolic tumour activity were independent predictors of clinical outcomes in multivariate regression models. Consequently, metabolic tumour burden in combination with ptDNA analysis has the potential to ameliorate the accuracy of tumour response prediction and prognostication in mCRPC patients. In support of our findings, a recent prospective trial [30] also showed the utility of integrating functional imaging using 18F‐NaF PET/CT scan and the analysis of CTCs in mCRPC patients treated with enzalutamide. The authors showed that individual metastatic sites may show differential AR and AR‐V7 expression as well as neuroendocrine markers, and can be responsible of a heterogeneous response to enzalutamide as assessed by quantitative total bone imaging. Furthermore, De Laere et al. [31] developed a three‐stratum risk stratification system, using both clinical features and TP53‐alteration status in liquid biopsy to stratify patients in good and poor prognostic subgroups treated with ARSI. The current work builds on our previous discoveries describing AR copy number detected in plasma, by using digital droplet PCR, as a possible biomarker to generate a novel survival model [32]. In the study, circulating AR copy number, FCH‐PET parameters and other clinical factors were significant predictors of OS in mCRPC treated with ARSI and similar to this study could classify men into three risk groups thanks to the independent prognostic value of these biomarkers. In the present study, we included 65 patients previously investigated [32] with updated survival data and only those with plasma DNA suitable for tumour content detection. Altogether, these findings show that liquid biopsy analysis represents a noninvasive complementary tool to functional imaging, able to reflect changes in tumour burden and clinical outcomes and with the potential to identify predictive biomarkers.
The main limitations of this work were a single‐institution retrospective design and the small number of patients, and the inclusion of a very heterogeneous patient population, regardless of treatment line. In addition, with growing interest in novel tracers such as prostate‐specific membrane antigen, with higher sensitivity and specificity than other lipid metabolism tracers [33], the use of FCH‐PET/CT is debated. Nevertheless, FCH‐PET/CT is still utilized in clinical practice and has greatly improved the management of prostate cancer patients providing both anatomical information and metabolic information.
5. Conclusions
The current prognostic model was built and validated using data routinely collected in mCRPC treated with ARSI in combination with molecular and metabolic data, leading to the identification of patient subsets with very different survival outcomes. This model may be also useful for clinical trial design of new therapeutic approaches in mCRPC. The inclusion of biomarkers with risk assessment models tumour such as hormone‐naive, high‐risk localized cancer. These findings underline the feasibility of combining molecular and functional imaging but require a validation in larger prospective studies leading to future possible standardization and cost‐effectiveness in clinical routine application.
Conflict of interest
V. Conteduca has served as consultant/advisory board member for Janssen, Astellas, Merck, AstraZeneca and Bayer and has received speaker honoraria or travel support from Astellas, Janssen, Ipsen, Bayer and Sanofi. U. De Giorgi reports personal fees from Astellas, Pharma, Bayer, Novartis and MSD; personal fees and nonfinancial support from Bristol Myers‐Squibb, Ipsen, Janssen, Pfizer and grants and personal fees from Sanofi and Roche and grants from AstraZeneca. No potential conflicts of interest were disclosed by the other authors.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.13120.
Author contributions
VC conceptualized and designed the study. VC, PC, CL, GG, NB, GP, AF, AA, GS and FM involved in acquisition of data. VC and ES analysed and interpreted the data. VC, GP and UDG wrote, reviewed and/or revised the manuscript. UDG supervised the study.
Supporting information
Fig. S1. Association of plasma tumour DNA (ptDNA) fraction with metastatic sites.
Fig. S2. Analysis of plasma tumour DNA (ptDNA fraction) and clinical variable associations.
Fig. S3. Linear regression (diagonal line) between maximum standardized total lesion activity (TLA), metabolic tumour volume (MTV), and maximum standardized uptake value (SUVmax).
Table S1. Clinical and molecular features of mCRPC patients included in the training and validation cohorts for OS and PFS models.
Table S2. Univariate analysis of Overall Survival in the training cohort.
Table S3. Univariate analysis of Progression‐Free Survival in the training cohort.
Acknowledgements
We would like to thank Professor Gerhardt Attard and Dr Daniel Wetterskog for their clinical supervision and technical support.
Vincenza Conteduca and Emanuela Scarpi contributed equally to this article
Data accessibility
The data that support the findings of this study are available in the Supporting information of this article and from the corresponding author upon reasonable request.
References
- 1. Siegel RL, Miller KD, Fuchs HE & Jemal A (2021) Cancer statistics, 2021. CA Cancer J Clin 71, 7–33. [DOI] [PubMed] [Google Scholar]
- 2. Davies A, Conteduca V, Zoubeidi A & Beltran H (2019) Biological evolution of castration‐resistant prostate cancer. Eur Urol Focus 5, 147–154. [DOI] [PubMed] [Google Scholar]
- 3. Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, Salvi S, Amadori D, Zafeiriou Z, Rescigno P et al. (2015) Plasma AR and abiraterone‐resistant prostate cancer. Sci Transl Med 312, 312re10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Conteduca V, Wetterskog D, Sharabiani M, Grande E, Fernandez‐Perez MP, Jayaram A, Salvi S, Castellano D, Romanel A, Lolli C et al. (2017) Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration‐resistant prostate cancer: a multi‐institution correlative biomarker study. Ann Oncol 28, 1508–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, Haegert A, Warner EW, Mo F, Brahmbhatt S et al. (2016) Genomic alterations in cell‐free DNA and enzalutamide resistance in castration‐resistant prostate cancer. JAMA Oncol 2, 1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL et al. (2014) AR‐V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, Antonarakis ES, Beer TM, Carducci MA, Chi KN et al. (2016) Trial design and objectives for castration‐resistant prostate cancer: updated recommendations from the prostate cancer clinical trials working group 3. J Clin Oncol 34, 1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. González‐Billalabeitia E, Conteduca V, Wetterskog D, Jayaram A & Attard G (2019) Circulating tumour DNA in advanced prostate cancer: transitioning from discovery to a clinically implemented test. Prostate Cancer Prostatic Dis 22, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, Foye A, Lloyd P, Nykter M, Beer TM et al. (2018) Concordance of circulating tumour DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst 110, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Giorgi U, Caroli P, Scarpi E, Conteduca V, Burgio SL, Menna C, Moretti A, Galassi R, Rossi L, Amadori D et al. (2015) (18)F‐Fluorocholine PET/CT for early response assessment in patients with metastatic castration‐resistant prostate cancer treated with enzalutamide. Eur J Nucl Med Mol Imaging 42, 1276–1283. [DOI] [PubMed] [Google Scholar]
- 11. De Giorgi U, Caroli P, Burgio SL, Menna C, Conteduca V, Bianchi E, Fabbri F, Carretta E, Amadori D, Paganelli G et al. (2014) Early outcome prediction on 18F‐fluorocholine PET/CT in metastatic castration‐resistant prostate cancer patients treated with abiraterone. Oncotarget 5, 12448–12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conteduca V, Scarpi E, Caroli P, Salvi S, Lolli C, Burgio SL, Menna C, Schepisi G, Testoni S, Gurioli G et al. (2017) Circulating androgen receptor combined with 18F‐fluorocholine PET/CT metabolic activity and outcome to androgen receptor signalling‐directed therapies in castration‐resistant prostate cancer. Sci Rep 7, 15541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conteduca V, Wetterskog D, Scarpi E, Romanel A, Gurioli G, Jayaram A, Lolli C, Tandefelt DG, Schepisi G, Casadei C et al. (2020) Plasma tumour DNA as an early indicator of treatment response in metastatic castration‐resistant prostate cancer. Br J Cancer 123, 982–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Du M, Tian Y, Tan W, Wang L, Wang L, Kilari D, Huang CC, Wang L & Kohli M (2020) Plasma cell‐free DNA‐based predictors of response to abiraterone acetate/prednisone and prognostic factors in metastatic castration‐resistant prostate cancer. Prostate Cancer Prostatic Dis 23, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Annala M, Taavitsainen S, Khalaf DJ, Vandekerkhove G, Beja K, Sipola J, Warner EW, Herberts C, Wong A, Fu S et al. (2021) Evolution of castration‐resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clin Cancer Res 27, 4610–4623. [DOI] [PubMed] [Google Scholar]
- 16. Mawlawi O, Podoloff DA, Kohlmyer S, Williams JJ, Stearns CW, Culp RF, Macapinlac H & Association NEM (2004) Performance characteristics of a newly developed PET/CT scanner using NEMA standards in 2D and 3D modes. J Nucl Med 45, 1734–1742. [PubMed] [Google Scholar]
- 17. Ghedini P, Bossert I, Zanoni L, Ceci F, Graziani T, Castellucci P, Ambrosini V, Massari F, Nobili E, Melotti B et al. (2018) Liver metastases from prostate cancer at 11C‐Choline PET/CT: a multicenter, retrospective analysis. Eur J Nucl Med Mol Imaging 45, 751–758. [DOI] [PubMed] [Google Scholar]
- 18. Armstrong AJ, Garrett‐Mayer ES, Yang Y‐CO, de Wit R, Tannock IF & Eisenberger M (2007) A contemporary prognostic nomogram for men with hormone‐refractory metastatic prostate cancer: a TAX327 study analysis. Clin Cancer Res 13, 6396–6403. [DOI] [PubMed] [Google Scholar]
- 19. Halabi S, Lin C‐Y, Kelly WK, Fizazi K, Moul JW, Kaplan EB, Morris MJ & Small EJ (2014) Updated prognostic model for predicting overall survival in first‐line chemotherapy for patients with metastatic castration‐resistant prostate cancer. J Clin Oncol 32, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chi KN, Kheoh T, Ryan CJ, Molina A, Bellmunt J, Vogelzang NJ, Rathkopf DE, Fizazi K, Kantoff PW, Li J et al. (2016) A prognostic index model for predicting overall survival in patients with metastatic castration‐resistant prostate cancer treated with abiraterone acetate after docetaxel. Ann Oncol 27, 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smaletz O, Scher HI, Small EJ, Verbel DA, McMillan A, Regan K, Kelly WK & Kattan MW (2002) Nomogram for overall survival of patients with progressive metastatic prostate cancer after castration. J Clin Oncol 20, 3972–3982. [DOI] [PubMed] [Google Scholar]
- 22. Halabi S, Small EJ, Kantoff PW, Kattan MW, Kaplan EB, Dawson NA, Levine EG, Blumenstein BA & Vogelzang NJ (2003) Prognostic model for predicting survival in men with hormone‐refractory metastatic prostate cancer. J Clin Oncol 21, 1232–1237. [DOI] [PubMed] [Google Scholar]
- 23. Stangl‐Kremser J, Mari A, Suarez‐Ibarrola R, D'Andrea D, Korn SM, Pones M, Kramer G, Karakiewicz P, Enikeev DV, Glybochko PV et al. (2020) Development of a prognostic model for survival time prediction in castration‐resistant prostate cancer patients. Urol Oncol 38, 600.e9–600.e15. [DOI] [PubMed] [Google Scholar]
- 24. Armstrong AJ, Lin P, Higano CS, Sternberg CN, Sonpavde G, Tombal B, Templeton AJ, Fizazi K, Phung D, Wong EK et al. (2018) Development and validation of a prognostic model for overall survival in chemotherapy‐naïve men with metastatic castration‐resistant prostate cancer. Ann Oncol 29, 2200–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ryan CJ, Kheoh T, Li J, Molina A, De Porre P, Carles J, Efstathiou E, Kantoff PW, Mulders PFA, Saad F et al. (2017) Prognostic index model for progression‐free survival in chemotherapy‐naïve metastatic castration‐resistant prostate cancer treated with abiraterone acetate plus prednisone. Clin Genitourin Cancer S1558‐7673(17)30211‐2. 10.1016/j.clgc.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson D, Van Allen E, Wu Y‐M, Schultz N, Lonigro R, Mosquera J‐M, Montgomery B, Taplin M‐E, Pritchard C, Attard G et al. (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Conteduca V, Mosca A, Brighi N, de Giorgi U & Rescigno P (2021) New prognostic biomarkers in metastatic castration‐resistant prostate cancer. Cells 10, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Attard G, Gormley M, Urtishak K, Simon JS, Ricci DS, Parekh TV, De Sarkar N, Yu EY, Montgomery RB, Nelson PS et al. (2020) Association of detectable levels of circulating tumour DNA (ctDNA) with disease burden in prostate cancer (PC). J Clin Oncol 38 (15_suppl), 5562. [Google Scholar]
- 29. Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, Orlando F, Fedrizzi T, Ku SY, Dann E et al. (2020) Circulating tumour DNA profile recognizes transformation to castration‐resistant neuroendocrine prostate cancer. J Clin Invest 130, 1653–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyriakopoulos CE, Heath EI, Ferrari A, Sperger JM, Singh A, Perlman SB, Roth AR, Perk TG, Modelska K, Porcari A et al. (2020) Exploring spatial‐temporal changes in 18F‐sodium fluoride PET/CT and circulating tumour cells in metastatic castration‐resistant prostate cancer treated with enzalutamide. J Clin Oncol 38, 3662–3671. [DOI] [PubMed] [Google Scholar]
- 31. De Laere B, Oeyen S, Mayrhofer M, Whitington T, van Dam PJ, Van Oyen P, Ghysel C, Ampe J, Ost P, Demey W et al. (2019) TP53 outperforms other androgen receptor biomarkers to predict abiraterone or enzalutamide outcome in metastatic castration‐resistant prostate cancer. Clin Cancer Res 25, 1766–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conteduca V, Scarpi E, Matteucci F, Caroli P, Ravaglia G, Fantini L, Gurioli G, Schepisi G, Wetterskog D, Menna C et al. (2019) A multimodal approach to outcome prediction in metastatic castration‐resistant prostate cancer by integrating functional imaging and plasma DNA analysis. JCO Precis Oncol 2019. 10.1200/PO.18.00302 [DOI] [PubMed] [Google Scholar]
- 33. Malaspina S, De Giorgi U, Kemppainen J, Del Sole A & Paganelli G (2018) 68Ga‐PSMA‐PET: added value and future applications in comparison to the current use of choline‐PET and mpMRI in the workup of prostate cancer. Radiol Med 123, 952–965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Association of plasma tumour DNA (ptDNA) fraction with metastatic sites.
Fig. S2. Analysis of plasma tumour DNA (ptDNA fraction) and clinical variable associations.
Fig. S3. Linear regression (diagonal line) between maximum standardized total lesion activity (TLA), metabolic tumour volume (MTV), and maximum standardized uptake value (SUVmax).
Table S1. Clinical and molecular features of mCRPC patients included in the training and validation cohorts for OS and PFS models.
Table S2. Univariate analysis of Overall Survival in the training cohort.
Table S3. Univariate analysis of Progression‐Free Survival in the training cohort.
Data Availability Statement
The data that support the findings of this study are available in the Supporting information of this article and from the corresponding author upon reasonable request.