

Abstract
Increased oxidative stress in glomerular endothelial cells (GEnCs) contributes to early diabetic kidney disease (DKD). While mitochondrial respiratory complex IV activity is reduced in DKD, it remains unclear whether it is a driver or a consequence of oxidative stress in GEnCs. Synthesis of cytochrome C oxidase 2 (SCO2), a key metallochaperone in the electron transport chain, is critical to the biogenesis and assembly of subunits required for functional respiratory complex IV activity. Here, we investigated the effects of Sco2 hypomorphs (Sco2KO/KI, Sco2KI/KI), with a functional loss of SCO2, in the progression of DKD by using a model of type 2 diabetes, db/db mice. Diabetic Sco2KO/KI and Sco2KI/KI hypomorphs exhibited a reduction in complex IV activity but an improvement in albuminuria, serum creatinine, and histomorphometric evidence of early DKD compared with db/db mice. Single-nucleus RNA sequencing using gene set enrichment analysis of differentially expressed genes in the endothelial cluster of Sco2KO/KI;db/db mice demonstrated an increase in genes involved in VEGF-VEGFR2 signaling and reduced oxidative stress compared with db/db mice. These data suggest that reduced complex IV activity as a result of a loss of functional SCO2 might be protective in GEnCs in early DKD.
Introduction
Diabetic kidney disease (DKD) is the leading cause of end-stage renal disease worldwide, developing in ∼40% of patients with diabetes (1). In the early stages, DKD is characterized by glomerular hyperfiltration and microalbuminuria, which eventually progresses to glomerular dysfunction, a rise in albuminuria, and kidney dysfunction in the later stages (1). While glomerular endothelial cell (GEnC) injury, mesangial expansion, and podocyte loss remain hallmark features in DKD, GEnC injury is an early inciting feature that contributes to glomerular hyperfiltration and microalbuminuria (2). Previous studies demonstrated that GEnC dysfunction in early DKD is characterized by an increase in oxidative stress resulting from an imbalance in accumulation and production of reactive oxygen species (ROS), leading to changes in vasoreactivity, coagulation, and inflammation (3). This increase in oxidative stress has also been identified to be more prominent in GEnCs compared with that in podocytes in DKD (4). Furthermore, cross talk between injured GEnCs and podocytes potentiates podocyte loss while exacerbating GEnC injury in DKD, leading to glomerular injury and albuminuria (2,5).
Several studies demonstrated the association between mitochondrial dysfunction in glomeruli and DKD progression, with reduced respiratory complex IV activity along the electron transport chain and an increase in ROS (6,7). Under homeostatic conditions, basal levels of ROS are regulated by antioxidant enzymes, such as superoxide dismutases (SODs) and peroxiredoxins, and low-molecular-weight scavengers, such as cytochrome c, with ROS acting as important second messengers in cellular signaling (6,8). However, excess ROS from overproduction of ROS or a loss of antioxidants is deleterious to mitochondria, leading to mtDNA damage and impaired oxidative phosphorylation (6,9). All respiratory complexes are involved in oxidative phosphorylation, with complex IV being the terminal enzyme in the electron transport chain that catalyzes the reduction of molecular oxygen by transferring electrons from reduced cytochrome c (10).
As the name suggests, synthesis of cytochrome c oxidase (SCO2) is critical to the biogenesis and assembly of subunits required for functional respiratory complex IV, also known as cytochrome c oxidase (COX). SCO2 is one of eight proteins required for the insertion of copper into complex IV (11) and serves as a metallochaperone to transport copper to the copper A site of complex IV as well as a redox sensor under cell stress (10). Furthermore, mutations in the SCO2 gene in humans contribute to devastating mitochondrial diseases, such as fatal infantile cardioencephalomyopathy, because of reduced respiratory complex IV activity (12). The most frequently reported SCO2 mutation is a point mutation at the E140 residue, resulting in a switch from glutamic acid to lysine residue. Mice with knockin (KI) for the homologous human E140 residue exhibit dysfunctional COX assembly with reduced COX activity in the brain, heart, liver, and muscle tissues compared with wild-type mice (13). The addition of exogenous SCO2 cDNA to cancer cells results in a six-fold increase in cellular ROS levels and induction of apoptosis through activation of the apoptosis signal-regulating kinase 1 (ASK1) pathway, whereas silencing of SCO2 leads to a four-fold decrease in cellular ROS in MCF-7 and KB cells (14). Specifically, cell stress induces p53-mediated transcriptional upregulation of SCO2 and leads to increased ROS production and activation of the apoptotic pathway (14). Furthermore, in the absence of p53, reduction in SCO2 contributes to a metabolic switch from oxidative phosphorylation to glycolysis for the generation of ATP (i.e., Warburg effect) in cancer cells (15,16). Conversely, the loss of SCO2 in HCT116 cells shows a reduction in cell respiration that affects intracellular oxygen and redox homeostasis, leading to an increase in ROS production and oxidative DNA damage (17). These data suggest a cell context–dependent role of SCO2 loss and ROS production.
SCO2 is critical to the biogenesis and activity of COX, but an increase in COX activity might exacerbate ROS production during the early phases of DKD, as observed in the liver under early diabetic conditions (18). While several studies reported the increase in glomerular ROS generation under diabetic conditions (3,19,20), it remains unclear whether this is driven by an increase in COX activity in the early stages compared with reduction in activity during the later stages of DKD (21). In addition, the effects of inhibiting complex IV activity on ROS generation and glomerular injury by impairing SCO2 function in early DKD remain unclear. Here, we demonstrate that the loss of complex IV activity in mice expressing Sco2 hypomorph alleles reduces GEnC ROS production and attenuates albuminuria and glomerular injury in a murine model of early DKD.
Research Design and Methods
Animal Models and Genotyping of Sco2 Knockout/KI, Sco2KI/KI, and db/db Mice
Sco2KI/KI and Sco2 knockout/KI (Sco2KO/KI) mice were a gift from the laboratory of Eric Schon (Columbia University, New York, NY), and mice were generated as previously reported (13). To generate Sco2 hypomorph mice under diabetic conditions, Sco2KI/KI and Sco2KO/KI mice were bred with C57BL/6 Lepr db/+ mice to generate Sco2KO/KI db/+ and Sco2KI/KI db/+ mice. The db/+ mice were subsequently bred with the respective genotypes to generate Sco2KO/KI;db/db and Sco2KI/KI;db/db mice. Mice with two wild-type alleles for the Sco2 gene, Sco2KO/KI and Sco2KI/KI, served as respective controls to mice homologous for the leptin receptor mutation (db/db), Sco2KO/KI;db/db and Sco2KI/KI;db/db mice. Male mice at 24 weeks of age were used for the studies. Genotyping by tail biopsy and PCR were performed at 2 weeks of age (Supplementary Table 1) (13). The db/db mice were genotyped using the protocol and primer information from The Jackson Laboratory.
Isolation of Glomeruli From Mice
Mouse glomeruli were isolated as previously described (22). Briefly, mice were perfused with PBS containing 2.5 mg/mL iron oxide and 0.1% BSA. At the end of perfusion, kidneys were removed, decapsulated, minced into 1-mm3 pieces, and digested in PBS containing 1 mg/mL collagenase A and 100 units/mL DNase I at 37°C. Digested tissue was subsequently passed through a 100-μm cell strainer and collected by centrifugation. The pellet was resuspended in 1 mL of PBS, and glomeruli were collected using a magnet. The purity of glomeruli was verified under microscopy.
Measurement of Urine Albumin
The animals were housed in single-mouse metabolic cages (Tecniplast) with free access to food and water for 24-h urine collection. Urine albumin concentration was measured by ELISA kit (Bethyl Laboratories). Twenty-four-hour urine albumin excretion was calculated by multiplying total volume of urine collected and urine albumin concentration.
Measurement of Serum Creatinine Levels
Serum creatinine levels were measured by isotope dilution liquid chromatography-tandem mass spectrometry at the University of Alabama at the Birmingham O’Brien Core Center.
Measurement of Glomerular Filtration Rate
Glomerular filtration rate (GFR) in mice was determined using the clearance of FITC-sinistrin as previously described (23). Briefly, a transdermal GFR monitor was attached to the shaved back of anesthetized mice, and a baseline reading was obtained for 5 min before retro-orbital injection with a 1.5 mg/20 g dose of 40 mg/mL FITC-sinistrin. After the injection, the mice recovered from anesthesia in a single cage, and the GFR monitor was removed after 2 h of recording. The recorded data were analyzed using MB Lab and MB STUDIO software (MediBeacon, Mannheim, Germany). GFR was calculated from the decrease of fluorescence intensity over time (i.e., plasma half-life of FITC-sinistrin) using a three-compartment model, mice body weight, and an empirical conversion factor.
Blood Pressure Monitoring
Blood pressure was measured noninvasively using the BP-2000 Blood Pressure Analysis System. Mice were initially subjected to an acclimation period of eight cycles before blood pressure assessment. Subsequently, systolic and diastolic blood pressure were measured in each mouse for 10 continuous cycles on 3 consecutive days and expressed as an average as previously described (24).
Cell Culture
Conditionally immortalized mouse GEnCs were derived as previously reported (25). These cells were maintained in RPMI medium with L-glutamine (Invitrogen) containing 10% FBS and 1% penicillin/streptomycin at 33°C with 10 units/mL of interferon-γ in a humidified incubator (5% CO2).
shRNA-Mediated Sco2 Knockdown
Lentivirus Sco2 knockdown in mouse GEnCs was performed using the expression arrest psi-LVRU6GP lentiviral shRNA system (GeneCopoeia, Rockville, MD) as previously described (26). Lentiviral particles were produced by transfecting 293T cells with a combination of lentiviral expression plasmid DNA, pCD/NL-BHΔΔΔ packaging plasmid, and VSV-G–encoding pLTR-G plasmid. For cell infection, viral supernatant was supplemented with 8 μL/mL polybrene and incubated with cells for a 24-h period. Cells expressing shRNA for Sco2 (Sco2-shRNA, MSH089823-LVRU6GP-f, GCAACTACCGCGTATACTACATCAAGAGTGTAGTATACGCGGTAGTTGC) were selected with puromycin for 2–3 weeks before use in all studies. EV-shRNA served as empty vector control. GFP expression and real-time PCR analysis were performed to confirm Sco2 knockdown.
Real-Time PCR
Total RNA was isolated from glomeruli or kidney cortex of mice using the RNeasy Kit (QIAGEN) and from cells using TRIzol. First-strand cDNA was prepared from total RNA using the SuperScript III First-Strand Synthesis Kit (Life Technologies), and cDNA was amplified using SYBR GreenER qPCR Supermix on QuantStudio 3 (Applied Biosystems). Primers for mouse Sco2, vascular endothelial growth factor receptor 2 (Vegfr2), Krüppel-like factor 2 (Klf2), vascular cell adhesion molecule 1 (Vcam1), intercellular adhesion molecule 1 (Icam1), cluster of differentiation 31 (Cd31), EH domain containing 3 (Ehd3), Wilms’ tumor 1 (Wt1), and podocin were designed using National Center for Biotechnology Information Primer-BLAST and validated for efficiency before application (Supplementary Table 2). LightCycler software was used to determine crossing points by the second derivative method. Data were normalized to the housekeeping gene (β-actin) and presented as fold increase compared with RNA isolated from the control group using the 2−ΔΔCT method.
Histopathology and Morphometric Studies by Bright-Field Light Microscopy
Mice were perfused with PBS, and kidneys were fixed in 10% phosphate-buffered formalin overnight and switched to 70% ethanol before processing for histology. Kidney tissue was embedded in paraffin by the histology core facility at Stony Brook University and 4-μm-thick sections were stained with periodic acid-Schiff (PAS) (Sigma-Aldrich) and hematoxylin and eosin (H & E). Liver and heart tissue were fixed and embedded, and 4-μm-thick sections were stained with H and E.
Mesangial expansion, glomerular volume, and average capillary loop area were quantified as previously described (27). In brief, images were scanned, and profile areas were traced using ImageJ. Mean glomerular tuft volume (GV) was determined from mean glomerular cross-sectional area (GA) by light microscopy. GA calculation was based on the average area of 20 glomeruli in each group, and GV calculation was based on the following equation:
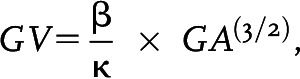 |
where β = 1.38, the shape coefficient of spheres (idealized shape of glomeruli), and κ = 1.1, the size distribution coefficient.
Mesangial expansion was defined as a PAS-positive and nuclei-free area in the mesangium. Quantification of mesangial expansion was based on 20 glomeruli cut at the vascular pole in each group.
Average capillary loop area of a glomerulus was determined by dividing its PAS-negative area by the number of capillary loops. Quantification of average capillary loop area was based on 20 glomeruli cut at the vascular pole in each group.
Quantification of podocyte number per glomerular area was determined using WT1-stained podocytes. Immunostaining for WT1 was initially performed using mouse anti-WT1 (SC-7385; Santa Cruz). Counting of podocytes using WT1+ Hoechst+ cells and measurement of glomerular area was performed using ImageJ, and data were reported as WT1+ Hoechst+ cells per glomerular area.
Immunofluorescence and Immunohistochemistry
All kidney sections from mice were prepared in identical fashion. Immunofluorescence was performed using mouse anti-WT1 (SC-7385; Santa Cruz), rabbit anti-SCO2 (ab115877; Abcam), mouse anti–8-oxoguanine (8-oxoG) (sc130914; Santa Cruz), rabbit anti–translocase of outer mitochondrial membrane 20 (TOMM20) (ab78547; Abcam), and mouse anti–mitochondrially encoded cytochrome c oxidase II (mt-CO2) (ab110258; Abcam). After washing, sections were incubated with a fluorophore-linked secondary antibody (Alexa Fluor 647 donkey anti-mouse, Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 568 donkey anti-rabbit; Life Technologies). After counterstaining with Hoechst (Thermo Fisher Scientific), Ulex europaeus agglutinin I (UEA I), rhodamine (Vector Laboratories), and isolectin (Vector Laboratories), slides were mounted in ProLong Gold Antifade Mountant (Thermo Fisher Scientific) and photographed under a Nikon Eclipse 90i microscope with a digital camera. Quantification of 8-oxoG expression in the glomerulus was determined by measuring the percent area stained in the glomerulus in 20 high-power field (20×) digitized images. For measuring non-glomerular 8-oxoG staining, the image was converted into a binary image, and after subtracting the glomerular area, percent area of 8-oxoG staining in the non-glomerular area per high-power field was quantified using ImageJ. For staining of SCO2, de-identified human kidney biopsy specimens from the Stony Brook University Department of Medicine and University of Utah were categorized into early-stage (<30%) and late-stage (>30%) chronic tubulointerstitial fibrosis by a renal pathologist (M.P.R.). Control kidney biopsy specimens were acquired from the unaffected pole of kidneys that were removed because of renal cell carcinoma. The percentage of glomeruli stained positive for SCO2 was quantified using ImageJ, glomeruli were traced, and positive staining was quantified in the traced area. Quantification of mt-CO2, TOMM20, and SCO2 in the GEnCs was done using ImageJ, the glomeruli were traced, and the threshold was set to trace the isolectin or UEA I staining followed by overlaying the mt-CO2, TOMM20, or SCO2 staining on the previously traced isolectin or UEA I image.
Immunohistochemistry for COX activity was performed as previously described (24). Mouse kidney cryosections were allowed to dry at room temperature for 1 h. For COX staining, an incubating solution (750 mg of sucrose in 7.5 mL of deionized H2O, 2.5 mL of 0.2 mol/L phosphate buffer, 5 mg of 3,3'diaminobenzidine, 10 mg of cytochrome c, and 20 μg of bovine catalase) was freshly prepared. Slides were incubated at 37°C for 40 min with the incubating solution and subsequently washed twice with 0.1 mol/L PBS. Quantification of the COX staining in the glomeruli and kidney cortex was done using ImageJ. In brief, for glomerular COX staining, the glomeruli were traced and percent area of COX staining in the glomeruli quantified. For measuring nonglomerular COX activity, the image was converted into a binary image, and after subtracting the glomerular area, percent area of COX staining in the nonglomerular area per high-power field was quantified.
Histopathology by Transmission Electron Microscopy
Mice were perfused with PBS and then immediately fixed in 2.5% glutaraldehyde for transmission electron microscopy (TEM). Kidney tissue was embedded, and ultrathin sections were mounted on a copper grid and photographed under a Hitachi H7650 microscope. Briefly, negatives were digitized, and images with a final magnification of approximately ×6,800, ×13,000, and ×23,000 were obtained. Swollen segments of endothelial cytoplasmic rim in which fenestrae were not discernible was identified as glomerular endothelial swelling, as previously described (28). Dysmorphic mitochondria were defined as mitochondria with a focal loss of visible cristae, clustering of residual cristae at the peripheral mitochondrial membrane, and fragmented (<2 μm in length).
The quantification of podocyte effacement was performed as previously described (24). In brief, ImageJ was used to measure the length of the peripheral glomerular basement membrane (GBM), and the number of slit pores overlying this GBM length was counted. Quantification of GBM thickness was performed using ImageJ, as previously described (24). The measurements were from podocyte to endothelial cell membrane at random sites where GBM was displayed in the best cross section.
Single-Nucleus Isolation, Sequencing, Data Processing, and Analysis
Nuclei were isolated from the mouse kidney cortex on the basis of a previously described protocol (29). In brief, mice were perfused with PBS, and kidney cortex was stored in RNAlater solution at −80°C. A 2-mm3 section of tissue was rinsed briefly with PBS and minced before adding 1 mL of lysis buffer containing 20 mmol/L Tris HCl (pH 8), 320 mmol/L sucrose, 5 mmol/L CaCl2, 3 mmol/L MgAc2, 0.1 mmol/L EDTA, 0.1% Triton X-100, 0.1% RNase inhibitor, and 0.1% DAPI. The tissue was initially dissociated by pipetting up and down 10 times with a P1000 tip and then passed through a 25G syringe 10 times. The tissue was incubated on ice for 10 min and then passed through a 30-μm CellTrics filter. Nuclei were pelleted by centrifugation (500g for 5 min at 4°C) and washed with PBS after removal of supernatant. Nuclei were pelleted again and resuspended in 1 mL of PBS containing 0.04% BSA and RNase inhibitor (0.2 units/μL) and then passed through a 40-μm Flowmi filter before generating counts with a hemocytometer. Nuclei were then diluted and prepared for single-nucleus RNA sequencing (snRNA-seq) with the 10x Chromium System according to the manufacturer’s instructions (10x Genomics). Sequencing was performed using a NovaSeqS4 platform.
Raw sequencing data were demultiplexed and aligned to a mouse pre-mRNA reference genome using Cell Ranger on SeaWulf, the High Performance Computing Cluster at Stony Brook University. Quality control, dimensionality reduction, and clustering were performed using the R software package Seurat (30). Genes expressed in a minimum of three cells were retained. Cells expressing <200 or >8,000 genes were excluded. Cells expressing >5% mitochondrial genes were also excluded.
Blue Native PAGE
Mitochondria isolated from perfused kidney were quantified using Bradford assay, and 35 μg of mitochondrial extracts were solubilized with 2% dodecyl maltoside. After incubation on ice for 10 min, mitochondrial extracts were centrifuged at 17,000g for 20 min at 4°C, and the supernatant was loaded on a 3–12% gradient blue native PAGE (Invitrogen) to identify the amount of mitochondrial complexes (31).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 9.0, and P < 0.05 was considered statistically significant. To compare continuous data between two groups and among more than two groups, Student t test and two-way ANOVA with Tukey posttest were used, respectively. Nonparametric statistical tests were performed using the Mann-Whitney test to compare continuous data between two groups and Kruskal-Wallis test with Dunn posttest to compare continuous data among more than two groups. The exact test used for each experiment is denoted in the figure legends, and data are expressed as mean ± SEM. All experiments were repeated a minimum of three times, and representative experiments are shown.
Study Approval
All animal studies conducted were approved by the Stony Brook University Institutional Animal Care and Use Committee. The National Institutes of Health Guide for the Care and Use of Laboratory Animals was followed strictly. The Stony Brook University Institutional Review Board approved the use of archived de-identified human biopsy specimens for immunostaining.
Data and Resource Availability
Raw data from snRNA-seq have been deposited in the Gene Expression Omnibus (under accession no. GSE171722). Reviewer access is available using the token utifiymihfevlmh. R code and the Seurat RDS data object are available through https://github.com/MallipattuLab/MallipattuLab-Diabetes-SCO2-Manuscript.
Results
SCO2 Expression in Human DKD
To assess the role of SCO2 in human DKD, we interrogated a previously reported expression microarray data set using Nephroseq to compare human kidney biopsy specimens from DKD and healthy donor nephrectomies (32). DKD biopsy specimens were grouped on the basis of estimated GFR (eGFR) to represent early- and late-stage DKD (eGFR >60 and <60 mL/min/1.73 m2, respectively). SCO2 expression was significantly increased in microdissected glomeruli in both stages of DKD compared with healthy donor nephrectomy specimens, with no significant changes in tubulointerstitial SCO2 expression (Fig. 1A and B). Immunostaining for SCO2 in healthy donor nephrectomy specimens as well as in kidney biopsy specimens from early-stage (<30% fibrosis) and late-stage patients with DKD (>30% fibrosis) confirmed a significant increase in glomerular SCO2 expression in early- and late-stage DKD compared with healthy donor specimens, with no significant changes in the non-glomerular fraction (Fig. 1C–E). Since GEnC injury is a hallmark feature of early DKD (2,4,33), we immunostained for SCO2 with endothelial-specific lectin (UEA I) to demonstrate that this increase was specific to GEnCs, which was higher in early-stage than in late-stage DKD (Fig. 1C and F). These data suggest that SCO2 in GEnCs is increased in early stages of DKD, which is sustained in the later stages of DKD.
Figure 1.

Increased glomerular SCO2 expression in human kidney biopsy specimens with DKD. A and B: To examine the SCO2 expression, we used previously reported gene expression arrays from Ju et al. (32) collected from microdissected glomerular and tubulointerstitial fractions of kidney biopsy specimens with DKD grouped into eGFR >60 and <60 mL/min/1.73 m2 and healthy living donor biopsy specimens with eGFR >90 mL/min/1.73 m2. C: Immunostaining for SCO2, UEA 1, and DAPI in human kidney biopsy specimens from healthy donor nephrectomies as controls and from patients with DKD grouped into <30% and >30% fibrosis; representative images are shown (n = 5 patients/group). Dashed white line shows the glomerular region. Scale bars = 50 μm. D–F: Glomerular, non-glomerular, and glomerular endothelial SCO2 expression was quantified using ImageJ (10 glomeruli/sample; n = 5 samples/group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Kruskal-Wallis test with Dunn posttest.
Loss of Functional SCO2 Attenuates Early DKD
To ascertain the role of SCO2 in DKD, we initially used mice with a point mutation at residue 129 in the functional domain of mouse Sco2, which is analogous to human disease mutation at residue 140, resulting in a switch from glutamic acid to lysine (E129K) (13). Since mice with homozygous KO of Sco2 are embryonically lethal (13), we used heterozygous Sco2KO/KI and homozygous (E129K) Sco2KI/KI mice. These mice have been previously reported to exhibit reduced COX activity with dysfunctional complex IV assembly in other tissues (13). In the kidney, we demonstrate that Sco2 expression, COX activity, and complex IV assembly are significantly reduced in Sco2KO/KI and Sco2KI/KI mice compared with wild-type mice (Supplementary Fig. 1A–C). However, these Sco2 hypomorphs (Sco2KO/KI and Sco2KI/KI) were viable and fertile, with no significant increase in albuminuria or histological evidence of kidney injury at 24 weeks of age (Supplementary Fig. 1D and E).
Leprdb (db/db) mouse is a widely used representative model of type 2 diabetes as a result of leptin receptor mutation (34). Since these db/db mice have been previously reported to exhibit GEnC injury with histological features consistent with early DKD (35,36), we sought to interrogate the role of Sco2 hypomorphs in this murine model of early DKD. Sco2KO/KI and Sco2KI/KI mice were bred with db/+ mice to generate Sco2KO/KI;db/db and Sco2KI/KI;db/db by the second filial generation. All diabetic mice (db/db, Sco2KO/KI;db/db, and Sco2KI/KI;db/db) exhibited a significant increase in blood glucose and body weight compared with the nondiabetic mice (wild type, Sco2KO/KI, and Sco2KI/KI) (Table 1). We also observed that all diabetic mice exhibited a significant increase in kidney weight compared with the nondiabetic mice. Since Sco2 is expressed in other cell types (37) and db/db mice are known to exhibit hepatosteatosis (38,39), we measured liver weight. While liver weight was increased in all diabetic mice compared with nondiabetic mice, no significant changes were noted among the db/db, Sco2KO/KI;db/db, and Sco2KI/KI;db/db mice. We observed mild changes in liver and heart histology at baseline (Supplementary Fig. 1E), and Sco2KO/KI mice have been previously shown to develop liver hepatosteatosis with aging (40). No changes were observed in heart weight or blood pressure across the groups (Table 1).
Table 1.
Blood pressure, blood glucose, and kidney, heart, liver, and body weight measurements
| Wild type | Sco2KO/KI | Sco2KI/KI | db/db | Sco2KO/KI;db/db | Sco2KI/KI;db/db | |
|---|---|---|---|---|---|---|
| Blood glucose (mg/dL) | 111 ± 21 | 107 ± 15 | 126 ± 20 | 377 ± 134** | 319 ± 64** | 333 ± 93** |
| Body weight (g) | 31.2 ± 1.8 | 32.6 ± 2.8 | 33.6 ± 5.6 | 57.6 ± 5.1* | 62.7 ± 7.6*** | 64.8 ± 4.5*** |
| Kidney weight (g) | 0.20 ± 0.02 | 0.19 ± 0.01 | 0.20 ± 0.03 | 0.29 ± 0.07* | 0.26 ± 0.02** | 0.28 ± 0.02*** |
| Heart weight (g) | 0.20 ± 0.02 | 0.19 ± 0.04 | 0.19 ± 0.07 | 0.20 ± 0.03 | 0.19 ± 0.03 | 0.18 ± 0.03 |
| Liver weight (g) | 1.5 ± 0.14 | 1.5 ± 0.14 | 1.5 ± 0.2 | 4.1 ± 1.5* | 3.8 ± 0.9* | 4.3 ± 1.1** |
| Blood pressure (mmHg) | ||||||
| Systolic | 111 ± 15 | 112 ± 15 | 109 ± 8 | 107 ± 23 | 105 ± 10 | 110 ± 12 |
| Diastolic | 57 ± 13 | 57 ± 14 | 59 ± 12 | 44 ± 27 | 53 ± 11 | 47 ± 5 |
Data are mean ± SD. n = 4–6 mice/group.
P < 0.05,
P < 0.01,
P < 0.001 compared with the corresponding genotype by Kruskal-Wallis test.
While the db/db mice exhibited an increase in albuminuria compared with the wild-type mice, this increase was abrogated in the Sco2KO/KI;db/db and Sco2KI/KI;db/db mice (Fig. 2A). Although hyperfiltration is a feature of early DKD, serum creatinine was not decreased in db/db mice, despite the increase in albuminuria compared with wild-type mice, indicating a progression in DKD (Fig. 2B). However, both the Sco2KO/KI;db/db and the Sco2KI/KI;db/db mice exhibited a significant decrease in serum creatinine with a corresponding increase in GFR compared with the db/db mice, suggesting that the loss of functional Sco2 contributes to continued hyperfiltration under diabetic conditions (Fig. 2B and C). We also observed that Sco2KO/KI;db/db and Sco2KI/KI;db/db mice exhibited an increase in overall survival compared with db/db mice (Fig. 2D). Subsequent staining with PAS and H & E showed an increase in mesangial expansion and glomerular volume in all diabetic mice compared with their respective controls (Fig. 3A–C). However, the Sco2KO/KI;db/db and Sco2KI/KI;db/db mice exhibited less mesangial expansion and glomerular hypertrophy than the db/db mice (Fig. 3A–C). Interestingly, there was a significant increase in glomerular capillary dilation in the Sco2KO/KI;db/db and Sco2KI/KI;db/db mice compared with the db/db mice and their respective controls (Fig. 3A and D), which was further confirmed upon blinded review by the renal pathologist (M.P.R.). We also immunostained for WT1, a podocyte-specific marker, to assess podocyte loss between the groups. While all diabetic mice had fewer podocytes per glomerular area than nondiabetic mice, we observed an improvement in podocyte number in Sco2KO/KI;db/db and Sco2KI/KI;db/db mice compared with db/db mice (Fig. 3E). Interestingly, nondiabetic Sco2KO/KI and Sco2KI/KI mice had a slight reduction in podocyte number compared with wild-type mice (Fig. 3E). Furthermore, we investigated the ultrastructural changes using TEM and found that the db/db mice had an increase in GBM thickness and foot process effacement compared with wild-type mice, which was mitigated in the Sco2KO/KI;db/db and Sco2KI/KI;db/db mice (Fig. 4A and Supplementary Fig. 2). In addition, db/db mice exhibited endothelial swelling, with a significant loss of endothelial fenestrations compared with the Sco2KO/KI;db/db and Sco2KI/KI;db/db mice (Fig. 4A). While we observed more dysmorphic mitochondria in GEnCs in the Sco2KO/KI and Sco2KI/KI mice than in the wild-type mice, these changes were attenuated in Sco2KO/KI;db/db and Sco2KI/KI;db/db mice compared with db/db mice (Fig. 4A).
Figure 2.

Diabetic Sco2 hypomorph (Sco2KO/KI;db/db, Sco2KI/KI;db/db) mice exhibit reduced albuminuria and serum creatinine and improved GFR and overall survival compared with db/db mice. A: Urine albumin was measured using 24-h urine collected in metabolic cages at 24 weeks of age in all six groups. B: Serum creatinine was measured at 24 weeks of age. Data are mean ± SEM (n = 4–6 mice/group). **P < 0.01, ***P < 0.001 by Kruskal-Wallis test with Dunn posttest. C: GFR in the db/db mice (n ≥3 mice/group). *P < 0.5, **P < 0.01 by Welch t test. D: Survival curves for all groups until age 24 weeks (n = 6 mice/group). *P < 0.05 by log-rank (Mantel-Cox) test.
Figure 3.

Diabetic Sco2 hypomorph (Sco2KO/KI;db/db, Sco2KI/KI;db/db) mice exhibit less glomerular injury compared with db/db mice. A: Representative images of paraffin-embedded sections stained with PAS (magnification ×10 and ×20) and H & E (magnification ×20) are shown to demonstrate mesangial expansion, glomerular volume, and tubulointerstitial changes in each group. Scale bars = 50 μm. B–E: Quantification of mesangial expansion, glomerular volume, capillary dilation, and podocyte number (defined as the number of WT1+ cells per glomerular area) (20 glomeruli/mouse, n = 4–6 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001 by Kruskal-Wallis test with Dunn posttest; #P < 0.01 by Mann-Whitney U test.
Figure 4.

Diabetic Sco2 hypomorph (Sco2KO/KI;db/db, Sco2KI/KI;db/db) mice exhibit less glomerular injury compared with db/db mice. A: Ultrastructural changes are shown at original magnification by TEM to demonstrate changes in podocyte structure, GEnC structure, and GBM. Arrowheads indicate foot process effacement, asterisks indicate GEnC swelling and injury, and arrows indicate dysmorphic mitochondria in GEnCs. Scale bars = 2 μm (magnification ×6,800) and 500 nm (magnification ×13,000 and ×23,000). B–E: mRNA expression quantified from real-time PCR for Vegfr2, Klf2, Icam1, and Vcam1 in isolated glomeruli (n = 4–6 mice/group). *P < 0.05, **P < 0.01 by Kruskal-Wallis test with Dunn posttest.
To assess whether these changes in GEnCs were mediated by changes in endothelial markers involved in angiogenesis, cell adhesion, and inflammation and are known to be dysregulated in DKD (36,41), we measured the mRNA expression of Vegfr2, Klf2, Icam1, and Vcam1 in isolated glomeruli from all groups. We observed a significant increase in Vegfr2 and Klf2 (anti-inflammatory, pro-angiogenesis) and a decrease in Icam1 and Vcam1 (cell adhesion, pro-inflammatory) in Sco2KO/KI;db/db and Sco2KI/KI;db/db mice compared with db/db mice (Fig. 4B–E). Collectively, these data suggest that the loss of functional SCO2 attenuated GEnC injury and progression of DKD, with an improvement in functional and histological markers of glomerular injury consistent with early DKD.
Loss of Functional SCO2 Attenuates Oxidative Stress in GEnCs Under Diabetic Conditions
While we observed no significant changes in kidney injury between the Sco2 hypomorphs and wild-type mice at baseline, the loss of functional SCO2 significantly increased the glomerular capillary dilation and attenuated the loss of endothelial fenestrations and podocyte injury under diabetic conditions (Figs. 3D and 4A). To assess the cellular pathways that might mediate the renoprotective effects of SCO2 loss in early DKD at the single-cell level, we initially performed snRNA-seq on kidney cortex from wild-type, Sco2KO/KI, db/db, and Sco2KO/KI;db/db mice. Our rationale for choosing snRNA-seq instead of single-cell RNA sequencing was based on studies in the kidney (42). Using the 10x Genomics platform, we successfully generated transcriptomes for 16,196 nuclei that passed all quality control checks (Supplementary Fig. 3A and B). With use of the R software package Seurat, 21 clusters were generated using unsupervised clustering analysis (Supplementary Fig. 3C). To assign cell-type identity to the clusters, we manually compared the top significant marker genes with established cell-type markers from several published data sets (29,43), and clusters with similar cell-type identity were combined to generate 18 unique clusters (Fig. 5A). Annotated clusters include many of the most abundant cell types in the kidney, including proximal tubule (PT) segment 1 (S1) and S2, PT S3, endothelial cells, mesangial cells, podocytes, connecting tubule, distal convoluted tubule (CD-PC), intercalated cells A and B, loop of Henle (ascending and descending loops), collecting duct principal cell, and macrophages (Fig. 5A and B). We also report the relative abundance of each cell type as a percentage of nuclei in each cluster with respect to each group (Fig. 5C). Since we observed an improvement in markers of GEnC injury in Sco2KO/KI;db/db mice compared with db/db mice, we identified 99 differentially expressed genes (DEGs) between these groups and ranked the genes on the basis of log2 fold change (Supplementary File 1). To differentiate the subpopulation of endothelial cells, we isolated and subclustered the endothelial cluster. Subclustering of the endothelial cluster revealed six subclusters with higher expression of previously reported GEnC-specific markers, such as kinase insert domain receptor (Kdr), plasmalemma vesicle associated protein (Plvap), and Eps15 homology domain-containing 3 (Ehd3) in subclusters 1 and 4 (Supplementary Fig. 4A) (44–46). We highlight the key upregulated DEGs, such as Kdr, glutathione peroxidase 3 (Gpx3), Zinc finger and BTB domain containing 16 (Zbtb16), heparan sulfate proteoglycan 2 (Hspg2), Plvap, and insulin-like growth factor binding protein 5 (Igfbp5) (Fig. 5D). We subsequently performed enrichment analysis with Enrichr (47,48) on the list of genes differentially upregulated in Sco2KO/KI;db/db compared with db/db endothelial clusters. Enrichment analysis using WikiPathways (49), Reactome (50), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (51) revealed that these DEGs encompassed key pathways, such as VEGF-VEGFR signaling, detoxification of ROS signaling, and oxidative stress (Fig. 5E). To assess the level of specificity of these pathways to the endothelial cluster, we demonstrate the changes in gene expression and the percentage of cells in all identified clusters between the Sco2KO/KI;db/db and db/db mice for genes involved in antioxidative stress and VEGF-VEGFR2 signaling (Fig. 5F). In addition to the endothelial cluster, we observed that other clusters, such as PT clusters, also demonstrate an increase in genes countering oxidative stress. Furthermore, enrichment analysis for all other clusters that had a minimum of 40 DEGs between Sco2KO/KI;db/db and db/db mice showed enrichment of oxidative stress pathways, suggesting that the global loss of SCO2 function plays a role in reducing oxidative stress in other cell types in the kidney (Supplementary Figs. 5 and 6). Conversely, enrichment analysis of significantly downregulated DEGs in the endothelial cell cluster showed pathways involved in regulation of focal adhesion assembly, regulation of cell matrix adhesion, complement, and coagulation cascade (Supplementary Fig. 7A and B). These changes in the endothelial cluster are reflective of GEnCs as shown in the subclusters of the endothelial cluster (Supplementary Fig. 4B and C).
Figure 5.

snRNA-seq with enrichment analysis demonstrates an increase in anti-oxidative stress and VEGF-VEGFR2 signaling genes in Sco2KO/KI;db/db compared with db/db mice. A: Uniform Manifold Approximation and Projection (UMAP) plot illustrates all 16,158 nuclei, mapping to 17 clusters from wild-type, Sco2KO/KI, Sco2KO/KI;db/db, and db/db mice. B: Cluster identities aligned to canonical cell types in the adult mouse kidney on the basis of a variety of cell-type–specific marker genes. C: Relative cell-type abundance compared among all four groups. D: Violin plot of key statistically significant DEGs in the endothelial cluster between Sco2KO/KI;db/db and db/db mice. E: Reactome, WikiPathways, and KEGG enrichment analyses in upregulated genes in the Sco2KO/KI;db/db mice compared with db/db mice in the endothelial cluster. F: Expression and percentage of nuclei of key anti-oxidative stress and VEGF-VEGFR2 signaling genes in all cell clusters from snRNA-seq. Cat, catalase; CD-PC, collecting duct principal cell; CNT, connecting tubule; DCT, distal convoluted tubule; Endo, endothelial; Fib, fibroblast; Flt1, Fms-related receptor tyrosine kinase 1; Gclc, glutamate-cysteine ligase catalytic subunit; Gsr, glutathione S-reductase; Gstt2, glutathione S-transferase theta 2; IC, intercalated cell; LH(AL), loop of Henle (ascending loop); LH(DL), loop of Henle (descending loop); MAPK, mitogen-activated protein kinase; Mes, mesangial; Mø, macrophage; Pod, podocyte; p.val, P value; TGF, transforming growth factor; Txnrd1, thioredoxin reductase 1.
To further validate this change in endothelial markers involved in angiogenesis, cell adhesion, and inflammation with Sco2 knockdown in GEnCs, we used immortalized mouse GEnCs to generate stable knockdown of Sco2. We initially confirmed that previously generated mouse GEnCs express markers specific to GEnCs, such as Cd31 and Ehd3, with no detectable expression of other cellular markers such as podocyte markers, Wt1, and podocin (Supplementary Fig. 8A). We subsequently generated and confirmed stable knockdown of Sco2 using shRNA specific to Sco2 (Sco2-shRNA) with EV-shRNA control GEnCs (Supplementary Fig. 8B). Sco2-shRNA GEnCs exhibited a significant increase in Vegfr2 and Klf2 and a decrease in Vcam1 and Icam1 expression compared with EV-shRNA GEnCs (Supplementary Fig. 8C).
Loss of Functional SCO2 Reduces COX Activity and Oxidative Stress in GEnCs Under Diabetic Conditions
SCO2 is a critical membrane-bound metallochaperone critical for complex IV assembly, and we initially observed that the loss of functional SCO2 results in reduced COX activity and its assembly in the kidney (Supplementary Fig. 1B and C). In snRNA-seq, we also observed an increase in DEGs involved in reduced oxidative stress in the endothelial cluster of Sco2KO/KI;db/db compared with db/db mice (Fig. 5D–F). Furthermore, previous studies demonstrated that reduced COX activity in the setting of SCO2 knockdown might contribute to a decrease in ROS production in other tissues (52). Since GEnC ROS production is a major driver of glomerular injury in DKD (19,25), we sought to determine whether Sco2 hypomorphs contribute to reduced COX activity and oxidative stress under diabetic conditions.
Initially, we observed that the Sco2KO/KI mice exhibited a reduction in COX activity in the glomeruli compared with wild-type mice at basal conditions (Fig. 6A). However, glomerular COX activity was significantly increased in db/db mice compared with wild-type mice, which remained reduced in Sco2KO/KI;db/db and Sco2KI/KI;db/db mice compared with db/db mice (Fig. 6A and Supplementary Fig. 9B). However, we observed no significant differences in non-glomerular COX activity between the groups (Supplementary Fig. 11A). Not surprisingly, glomerular 8-oxoG (marker of oxidative stress from excess ROS) expression was increased in all diabetic mice compared with their respective controls (Fig. 6B and Supplementary Fig. 9C). While Sco2 hypomorphs exhibited a mild increase in glomerular 8-oxoG expression under basal conditions, Sco2KO/KI;db/db and Sco2KI/KI;db/db mice had reduced glomerular 8-oxoG expression compared with db/db mice (Fig. 6B and Supplementary Fig. 9C). We also observed an increase in non-glomerular 8-oxoG expression in db/db mice compared with Sco2KO/KI;db/db and Sco2KI/KI;db/db mice (Supplementary Fig. 11B). We also immunostained and quantified the percentage of TOMM20 expression in GEnCs to demonstrate that reduced glomerular COX activity and 8-oxoG expression were not a result of reduced mitochondria in Sco2KO/KI;db/db and Sco2KI/KI;db/db mice compared with db/db mice (Fig. 6C and Supplementary Fig. 10A). Furthermore, we observed that GEnC TOMM20 was reduced in db/db mice compared with wild-type mice, suggesting that a loss of GEnC mitochondria might be a result of enhanced COX activity leading to oxidative stress from excess ROS (Fig. 6C and Supplementary Fig. 10A).
Figure 6.

Reduced COX activity and ROS production in the diabetic Sco2 hypomorph (Sco2KO/KI;db/db, Sco2KI/KI;db/db) mice compared with db/db mice. Immunohistochemistry for COX activity and immunostaining for 8-oxoG, TOMM20, mt-CO2, and isolectin were performed. A and B: Quantification of in situ glomerular COX activity and 8-oxoG staining. C and D: Quantification of GEnC TOMM20 and mt-CO2 staining (20 glomeruli/mouse, n = 4–6 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001 by Kruskal-Wallis test with Dunn posttest.
SCO2 is essential for transporting copper to the copper A site on mt-CO2 and, therefore, is essential for the synthesis and maturation of cytochrome subunit II (mt-CO2) (53). Immunostaining and quantification of mt-CO2 in GEnCs showed reduced expression in Sco2 hypomorphs compared with wild-type mice, but no changes were observed in db/db mice (Fig. 6D and Supplementary Fig. 10B). While GEnC mt-CO2 expression was unchanged between wild-type and db/db mice, GEnC TOMM20 expression was reduced in db/db mice compared with wild-type mice (Fig. 6C and Supplementary Fig. 10A). These data suggest that mt-CO2 expression is increased in the remaining mitochondria in GEnCs in db/db mice, which contribute to enhanced COX activity leading to excess ROS production. In comparison, GEnC TOMM20 and mt-CO2 expression in Sco2KO/KI;db/db and Sco2KI/KI;db/db mice remains preserved with respect to the nondiabetic Sco2 hypomorphs (Fig. 6C and D). Collectively, these data suggest that the reduction in COX activity due to the loss of functional SCO2 might attenuate GEnC oxidative stress in early DKD.
Discussion
Mitochondrial dysfunction that leads to an increase in mitochondrial ROS (mtROS) contributes to GEnC injury in DKD as well as other glomerular diseases, such as focal segmental glomerulosclerosis (3,5,54–57). mtROS has also been implicated in microvascular dysfunction in patients with chronic kidney disease stages 3–5 (57). In addition, activation of endothelial NAD(P)H oxidase, a major source of vascular ROS, has been reported to exacerbate DKD (56). Mouse GEnCs exposed to high glucose or diabetic sera also contribute to mitochondrial dysfunction with increased mtROS. Conversely, pharmacological inhibition using a mitochondrially targeted antioxidant, mitoTEMPO, attenuated endothelial dysfunction in cultured mouse GEnCs (25). Furthermore, treatment of streptozocin-treated D2 and Akita-D2 mice with mitoTEMPO has been reported to prevent accumulation of 8-oxoG in GEnCs as well as reduce urinary 8-oxodG levels, thereby ameliorating endothelial cell injury in these diabetic mouse models (58). ROS accumulation in GEnCs might occur as a result of enhanced complex IV activity or reduced accessibility of cytoplasmic copper ions by antioxidant SOD1. In this study, we demonstrate that the loss of functional SCO2, an essential assembly factor for complex IV of the electron transport chain, attenuated glomerular injury in our db/db mice model, which in large part is driven by mitigating the increase in COX activity, leading to a reduction in excess ROS production in GEnCs under diabetic conditions. Similar to the studies using mitoTEMPO (58), Sco2 hypomorphs play a protective role in early DKD by attenuating excess ROS production. In addition, we observed that GEnC SCO2 expression is induced in early DKD, suggesting that enhanced COX activity driving ROS production might contribute to the GEnC injury observed in DKD.
Stress conditions such as diabetes can mimic p53-mediated induction of SCO2 expression, which results in increased ROS production and apoptosis in cancer cells (59). SCO2 has been previously reported to activate ASK-1 phosphorylation, triggering the dissociation of the ASK-1-thioredoxin complex, which leads to increased ROS production and eventual apoptosis (14). We postulate that the increase in SCO2 expression in human diabetic kidney might be related to an induction of p53 pathway under diabetic conditions; however, further studies are required to validate these findings. In the presence of diabetes, a significant increase in mtROS production in cardiac myocytes has been reported to be due to increased SCO2 expression (52). The increase in SCO2 expression during the early stages of disease parallels the increase in complex IV activity, and as the disease advances, there is a reduction in SCO2 expression and complex IV activity. Hence, loss of functional SCO2 might play a protective role in conditions such as early-stage DKD, where there is increased COX activity because of increased energy demand, leading to increased ROS production (21). Inhibiting functional SCO2 might counteract the overactivity of complex IV as well as prevent induction of the p53 pathway under diabetic conditions.
While the level of Sco2 expression was reduced in the Sco2 hypomorphs compared with wild-type mice, we did not observe any difference in the Sco2 expression between Sco2KO/KI and Sco2KI/KI mice, which might be due to incomplete penetrance of the KO allele. In addition, there was no evidence of significant kidney injury in these Sco2 hypomorphs, suggesting that degree of residual complex IV activity is sufficient for the energy requirements under basal conditions. Furthermore, in yeast, deletion of Sco2 reduces complex IV activity by 40%, whereas deletion of Sco1 leads to a 90% loss of complex IV activity (60), suggesting that other proteins might be able to compensate for the loss of Sco2 in complex IV assembly. However, under diabetic conditions, the loss of functional SCO2 abrogated albuminuria and serum creatinine compared with db/db mice. The loss of SCO2 might prevent the induction of stress-induced pathways and, therefore, play a protective role in these diabetic conditions. The copper pool in the mitochondrial matrix is shared by complex IV and the mitochondria-localized portion of the antioxidant SOD1, and a balance between the utilization of copper by SOD1 and complex IV might be essential for maintaining oxidative phosphorylation and preventing mitochondrial and cellular damage from increased oxidative stress (61). Under normal physiological conditions, there is no excess of free copper ions, electrons movement is undisturbed, and, therefore, SOD1 uses <2% of the shared copper ion pool to counter any excess ROS production. However, in early diabetes, substrate overload increases mitochondrial activity and ROS production without a change in copper ion concentration, thereby limiting the antioxidant effects of SOD1 or downstream peroxidases to convert the free radicals (O2−) to H2O2. Absence of functional SCO2 under diabetic conditions could help to mitigate this excess ROS production due to reduced complex IV activity (62,63) as well as enhanced antioxidant activity of SOD1 resulting from an increase in the pool of available copper ions (37). Therefore, the decrease in GEnC ROS production we observed in the diabetic Sco2 hypomorphs might be a result of reduced complex IV activity or an increase in accessible copper ions by antioxidant SOD1. Interestingly, the deletion of SCO2 or SCO1 does not affect the growth of yeast in the presence of ROS conditions, but deletion of SCO2 and SOD1 concurrently leads to a decrease in yeast growth (60). Additional studies are required to determine the role of SOD1 in Sco2 hypomorphs under these diabetic conditions.
Surfeit locus protein 1 (Surf1) is another key factor required for the assembly of complex IV (10). Loss of Surf1 in mice initiates mitochondrial stress response and improves insulin sensitivity as a result of reduced complex IV assembly and activity (64,65). Despite this reduced complex IV assembly and activity, Surf1−/− mice exhibit improved survival compared with wild-type mice (66). While we did not observe similar effects on survival at baseline in our Sco2 hypomorphs, loss of functional SCO2 did restore overall survival in mice under diabetic conditions.
In the presence of DKD, ROS has been reported to be detrimental in multiple cell types in the kidney. High-glucose–mediated ROS production in podocytes from mitochondrial and non-mitochondrial sources has been reported to induce apoptosis and depletion of podocytes at the onset of diabetes (67). ROS also plays a critical role in the differentiation of resident interstitial fibroblasts to myofibroblasts (68). Proximal tubules have higher mitochondrial content, which might reduce their threshold for ROS production and subsequent tubular damage. Interestingly, the excess copper ion pool resulting from a loss of functional SCO2 can be used by SOD1, which is highly expressed in kidney PTs (69). As such, in our mouse model, the reduction in oxidative stress pathways in other cell clusters in the snRNA-seq might also be due to a loss of functional SCO2 and reduced COX activity. Furthermore, inhibition of increased superoxide production in the kidney cortex mitochondria isolated from db/db mice attenuated ROS-induced mitochondrial dysfunction (70).
An improvement in early glomerular injury under diabetic conditions might not be restricted to only a reduction in complex IV activity. Interestingly, inhibition of complex I by rotenone has been previously reported to have renoprotective effects in a murine model of DKD (42). While several studies have reported a reduction in complex IV, as well as others, in later stages of DKD (21), it remains unclear whether this is a consequence of reduced activity at later stages of DKD or a result of fewer or damaged mitochondria resulting from substrate overload and ROS production. Furthermore, persistent inhibition of complex IV activity might be detrimental at later stages of DKD because of a reduction in overall mitochondrial content. Additional studies are necessary to delineate the precise stage at which inhibition of complex IV activity might play a protective role in the progression of DKD.
To our knowledge, this study is the first to demonstrate that the loss of functional SCO2 might be protective by attenuating glomerular injury in early DKD. Specifically, we observed that these renoprotective effects under diabetic conditions might be a result of reduced complex IV activity, leading to a decrease in oxidative stress in GEnCs. These data suggest that inhibiting SCO2 might be a potential therapeutic target in the early stages of DKD.
Article Information
Acknowledgments. The authors thank Dr. Eric Schon (Columbia University) for the gift of Sco2KO/KI and Sco2KI/KI mice and the Northport Veterans Affairs Medical Center-Stony Brook University Single Cell Genomic Core for single nuclei library generation.
Funding. This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants DK-112984 and DK-121846 and U.S. Department of Veterans Affairs grants 1I01BX003698 and 1I01BX005300 to S.K.M.
Duality of Interest. J.C.H. declares a ShangPharma research grant of $250,000 per year in 2019–2020 and is a scientific advisory board member for Renalytix. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. N.A.G., A.R.L., and S.K.M. conducted the study, analyzed the data, and prepared the manuscript. J.M.V., Y.G., B.O.F., E.F., and M.P.R. participated in analyzing the data. I.S.D., J.C.H., and D.B. assisted in the preparation of the manuscript. All authors reviewed and approved the final version of the manuscript. N.A.G. and S.K.M. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.16828219.
N.A.G. and A.R.L. contributed equally to this work.
References
- 1. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol 2017;12:2032–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol 2015;308:F287–F297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Daehn I, Casalena G, Zhang T, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 2014;124:1608–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fu J, Wei C, Lee K, et al. Comparison of glomerular and podocyte mRNA profiles in streptozotocin-induced diabetes. J Am Soc Nephrol 2016;27:1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daehn IS. Glomerular endothelial cell stress and cross-talk with podocytes in early [corrected] diabetic kidney disease. Front Med (Lausanne) 2018;5:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol 2014;306:F367–F378 [DOI] [PubMed] [Google Scholar]
- 7. Granata S, Dalla Gassa A, Tomei P, Lupo A, Zaza G. Mitochondria: a new therapeutic target in chronic kidney disease. Nutr Metab (Lond) 2015;12:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem 2001;276:23357–23361 [DOI] [PubMed] [Google Scholar]
- 9. Gujarati NA, Vasquez JM, Bogenhagen DF, Mallipattu SK. The complicated role of mitochondria in the podocyte. Am J Physiol Renal Physiol 2020;319:F955–F965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Timón-Gómez A, Nývltová E, Abriata LA, Vila AJ, Hosler J, Barrientos A. Mitochondrial cytochrome c oxidase biogenesis: recent developments. Semin Cell Dev Biol 2018;76:163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shoubridge EA. Cytochrome c oxidase deficiency. Am J Med Genet 2001;106:46–52 [DOI] [PubMed] [Google Scholar]
- 12. Papadopoulou LC, Sue CM, Davidson MM, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet 1999;23:333–337 [DOI] [PubMed] [Google Scholar]
- 13. Yang H, Brosel S, Acin-Perez R, et al. Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum Mol Genet 2010;19:170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Madan E, Gogna R, Kuppusamy P, Bhatt M, Mahdi AA, Pati U. SCO2 induces p53-mediated apoptosis by Thr845 phosphorylation of ASK-1 and dissociation of the ASK-1-Trx complex. Mol Cell Biol 2013;33:1285–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science 2006;312:1650–1653 [DOI] [PubMed] [Google Scholar]
- 16. Wang S, Peng Z, Wang S, et al. KRAB-type zinc-finger proteins PITA and PISA specifically regulate p53-dependent glycolysis and mitochondrial respiration. Cell Res 2018;28:572–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sung HJ, Ma W, Wang PY, et al. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat Commun 2010;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alimujiang M, Yu XY, Yu MY, et al. Enhanced liver but not muscle OXPHOS in diabetes and reduced glucose output by complex I inhibition. J Cell Mol Med 2020;24:5758–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lassén E, Daehn IS. Molecular mechanisms in early diabetic kidney disease: glomerular endothelial cell dysfunction. Int J Mol Sci 2020;21:9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Østergaard JA, Cooper ME, Jandeleit-Dahm KAM. Targeting oxidative stress and anti-oxidant defence in diabetic kidney disease. J Nephrol 2020;33:917–929 [DOI] [PubMed] [Google Scholar]
- 21. Mise K, Galvan DL, Danesh FR. Shaping up mitochondria in diabetic nephropathy. Kidney360 2020;1:982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takemoto M, Asker N, Gerhardt H, et al. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol 2002;161:799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scarfe L, Schock-Kusch D, Ressel L, et al. Transdermal measurement of glomerular filtration rate in mice. J Vis Exp 2018;(140):58520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horne SJ, Vasquez JM, Guo Y, et al. Podocyte-specific loss of Krüppel-like factor 6 increases mitochondrial injury in diabetic kidney disease. Diabetes 2018;67:2420–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Casalena GA, Yu L, Gil R, et al. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun Signal 2020;18:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mallipattu SK, Horne SJ, D’Agati V, et al. Krüppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest 2015;125:1347–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mallipattu SK, Gallagher EJ, LeRoith D, et al. Diabetic nephropathy in a nonobese mouse model of type 2 diabetes mellitus. Am J Physiol Renal Physiol 2014;306:F1008–F1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lafayette RA, Druzin M, Sibley R, et al. Nature of glomerular dysfunction in pre-eclampsia. Kidney Int 1998;54:1240–1249 [DOI] [PubMed] [Google Scholar]
- 29. Lake BB, Chen S, Hoshi M, et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 2019;10:2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wittig I, Braun H-P, Schägger H. Blue native PAGE. Nat Protoc 2006;1:418–428 [DOI] [PubMed] [Google Scholar]
- 32. Ju W, Greene CS, Eichinger F, et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res 2013;23:1862–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fu J, Wei C, Zhang W, et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int 2018;94:326–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev 2014;10:131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haku S, Wakui H, Azushima K, et al. Early enhanced leucine-rich α-2-glycoprotein-1 expression in glomerular endothelial cells of type 2 diabetic nephropathy model mice. BioMed Res Int 2018;2018:2817045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gil CL, Hooker E, Larrivée B. Diabetic kidney disease, endothelial damage, and podocyte-endothelial crosstalk. Kidney Med 2020;3:105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leary SC, Kaufman BA, Pellecchia G, et al. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum Mol Genet 2004;13:1839–1848 [DOI] [PubMed] [Google Scholar]
- 38. Sahai A, Malladi P, Pan X, et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol 2004;287:G1035–G1043 [DOI] [PubMed] [Google Scholar]
- 39. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 2006;87:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hill S, Deepa SS, Sataranatarajan K, et al. Sco2 deficient mice develop increased adiposity and insulin resistance. Mol Cell Endocrinol 2017;455:103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng H, Harris RC. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc Hematol Disord Drug Targets 2014;14:22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 2019;30:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu M, Li S, Yu X, et al. Mitochondrial activity contributes to impaired renal metabolic homeostasis and renal pathology in STZ-induced diabetic mice. Am J Physiol Renal Physiol 2019;317:F593–F605 [DOI] [PubMed] [Google Scholar]
- 44. Dumas SJ, Meta E, Borri M, et al. Single-cell RNA sequencing reveals renal endothelium heterogeneity and metabolic adaptation to water deprivation. J Am Soc Nephrol 2020;31:118–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karaiskos N, Rahmatollahi M, Boltengagen A, et al. A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 2018;29:2060–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He B, Chen P, Zambrano S, et al. Single-cell RNA sequencing reveals the mesangial identity and species diversity of glomerular cell transcriptomes. Nat Commun 2021;12:2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44(W1):W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Slenter DN, Kutmon M, Hanspers K, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 2018;46(D1):D661–D667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jassal B, Matthews L, Viteri G, et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2020;48(D1):D498–D503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000;28:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nakamura H, Matoba S, Iwai-Kanai E, et al. p53 promotes cardiac dysfunction in diabetic mellitus caused by excessive mitochondrial respiration-mediated reactive oxygen species generation and lipid accumulation. Circ Heart Fail 2012;5:106–115 [DOI] [PubMed] [Google Scholar]
- 53. Horn D, Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 2008;60:421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ebefors K, Wiener RJ, Yu L, et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int 2019;96:957–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh A, Ramnath RD, Foster RR, et al. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS One 2013;8:e55852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagasu H, Satoh M, Kiyokage E, et al. Activation of endothelial NAD(P)H oxidase accelerates early glomerular injury in diabetic mice. Lab Invest 2016;96:25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kirkman DL, Muth BJ, Ramick MG, Townsend RR, Edwards DG. Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am J Physiol Renal Physiol 2018;314:F423–F429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qi H, Casalena G, Shi S, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes 2017;66:763–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2011;2:948–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ekim Kocabey A, Kost L, Gehlhar M, Rödel G, Gey U. Mitochondrial Sco proteins are involved in oxidative stress defense. Redox Biol 2019;21:101079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bourens M, Fontanesi F, Soto IC, Liu J, Barrientos A. Redox and reactive oxygen species regulation of mitochondrial cytochrome c oxidase biogenesis. Antioxid Redox Signal 2013;19:1940–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jaksch M, Paret C, Stucka R, et al. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum Mol Genet 2001;10:3025–3035 [DOI] [PubMed] [Google Scholar]
- 63. Rebelo AP, Saade D, Pereira CV, et al. SCO2 mutations cause early-onset axonal Charcot-Marie-Tooth disease associated with cellular copper deficiency. Brain 2018;141:662–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pulliam DA, Deepa SS, Liu Y, et al. Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem J 2014;462:359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Deepa SS, Pulliam D, Hill S, et al. Improved insulin sensitivity associated with reduced mitochondrial complex IV assembly and activity. FASEB J 2013;27:1371–1380 [DOI] [PubMed] [Google Scholar]
- 66. Deepa SS, Pharaoh G, Kinter M, et al. Lifelong reduction in complex IV induces tissue-specific metabolic effects but does not reduce lifespan or healthspan in mice. Aging Cell 2018;17:e12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006;55:225–233 [PubMed] [Google Scholar]
- 68. Jha JC, Banal C, Chow BSM, Cooper ME, Jandeleit-Dahm K. Diabetes and kidney disease: role of oxidative stress. Antioxid Redox Signal 2016;25:657–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 2015;26:2669–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sourris KC, Harcourt BE, Tang PH, et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic Biol Med 2012;52:716–723 [DOI] [PubMed] [Google Scholar]