

Summary
Asthenoteratozoospermia, defined as reduced sperm motility and abnormal sperm morphology, is a disorder with considerable genetic heterogeneity. Although previous studies have identified several asthenoteratozoospermia-associated genes, the etiology remains unknown for the majority of affected men. Here, we performed whole-exome sequencing on 497 unrelated men with asthenoteratozoospermia and identified DNHD1 bi-allelic variants from eight families (1.6%). All detected variants were predicted to be deleterious via multiple bioinformatics tools. Hematoxylin and eosin (H&E) staining revealed that individuals with bi-allelic DNHD1 variants presented striking abnormalities of the flagella; transmission electron microscopy (TEM) further showed flagellar axoneme defects, including central pair microtubule (CP) deficiency and mitochondrial sheath (MS) malformations. In sperm from fertile men, DNHD1 was localized to the entire flagella of the normal sperm; however, it was nearly absent in the flagella of men with bi-allelic DNHD1 variants. Moreover, abundance of the CP markers SPAG6 and SPEF2 was significantly reduced in spermatozoa from men harboring bi-allelic DNHD1 variants. In addition, Dnhd1 knockout male mice (Dnhd1‒/‒) exhibited asthenoteratozoospermia and infertility, a finding consistent with the sperm phenotypes present in human subjects with DNHD1 variants. The female partners of four out of seven men who underwent intracytoplasmic sperm injection therapy subsequently became pregnant. In conclusion, our study showed that bi-allelic DNHD1 variants cause asthenoteratozoospermia, a finding that provides crucial insights into the biological underpinnings of this disorder and should assist with counseling of affected individuals.
Keywords: male infertility, DNHD1, sperm flagella, whole-exome sequencing, mitochondrial sheath
Graphical abstract
Introduction
Infertility has become a global health problem, affecting 10%–15% of couples trying to conceive.1, 2, 3 Male factors are suspected to account for approximately half of all infertility cases,2,4,5 which mainly manifest as azoospermia, oligozoospermia, asthenozoospermia, teratozoospermia, or a combination of the above, such as asthenoteratozoospermia and oligoasthenoteratozoospermia. Asthenoteratozoospermia, which is defined as reduced sperm motility and abnormal sperm morphology,6,7 accounts for approximately 19% of male infertility.2,7
It has been widely recognized that genetic factors are most likely involved in a large proportion of asthenoteratozoospermia with sperm flagellum defects.8,9 Recently, a growing number of genes responsible for asthenoteratozoospermia have been identified. Briefly, variants in a series of cilia and flagella associated protein (CFAP) genes, dynein axonemal heavy chain (DNAH) family member genes, and other genes such as USP26 (MIM: 300309), WDR19 (MIM: 609151), and DZIP1 (MIM: 608761) have been reported to cause asthenoteratozoospermia.10, 11, 12 Interestingly, this kind of asthenoteratozoospermia with sperm flagellum defects caused by the above-mentioned genes is usually described as multiple morphological abnormalities of the sperm flagella (MMAF), and the phenotypes of sperm tails with different variants show inconsistent and heterogeneity. For example, some individuals have normal sperm concentrations, while others exhibit reduced sperm counts. Moreover, except for pure flagellum abnormalities, MMAF caused by variants in CFAP58 (MIM: 619129) and CFAP65 (MIM: 614270) also show abnormalities with mitochondrial sheath (MS).13,14 Therefore, identifying candidate genes of asthenoteratozoospermia and their specific clinical phenotypes will promote the understanding of the etiology of asthenoteratozoospermia and assist the development of related treatment strategies.
In this study, we performed whole-exome sequencing (WES) on a cohort of 497 unrelated infertile men with asthenoteratozoospermia and identified bi-allelic deleterious DNHD1 variants in eight individuals. Further, we generated Dnhd1‒/‒ mice and found that they exhibited similar asthenoteratozoospermia features and male infertility. Our findings strongly suggest that bi-allelic variants of DNHD1 cause asthenoteratozoospermia and male infertility in humans and mice.
Material and methods
Study subjects
In this study, 314 infertile men with asthenoteratozoospermia were recruited between June 2014 and March 2021 in the Reproductive and Genetic Hospital of CITIC-Xiangya (Changsha, Human, China), as previously described.14 The individuals were recruited according to the guidelines of the World Health Organization (WHO) Laboratory Manual for the Examination and Processing of Human Semen.15 All individuals were idiopathic with a normal 46, XY karyotype and no Y chromosome microdeletion. Other possible causes of infertility, such as iatrogenic injury, reproductive tract infection, testicular inflammation, and drug exposure, were excluded. Physical examination of these individuals showed normal results, including height, weight, hair distribution, mental state, testis size, and external genital organs. Additionally, 392 fertile Han Chinese men with a similar genetic background were used as ethnically matched controls, and all of them had normal semen parameters and had fathered at least one healthy child.
In addition, we analyzed the WES data obtained from 183 male infertile individuals affected with asthenoteratozoospermia, which were previously described by the First Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China) and the Xiamen Maternity and Child Care Hospital of Xiamen University (Xiamen, Fujian, China).16, 17, 18 The criteria for all selected individuals were the same as the Reproductive and Genetic Hospital of CITIC-Xiangya.
The genetic study was approved by the corresponding ethics committees of the Reproductive and Genetic Hospital of CITIC-Xiangya (LL-SC-2017-025 and LL-SC-2019-034), the First Affiliated Hospital of Anhui Medical University (20150345), and the Xiamen Maternity and Child Care Hospital (KY-2019-060). All individuals enrolled in this study signed a written informed consent.
Whole-exome sequencing and in-silico bioinformatics analysis
Genomic DNA was extracted from whole peripheral blood samples with the QIAamp DNA Blood Midi Kit (QIAGEN, 51106) according to the manufacturer’s protocol. Genomic DNA was prepared with the Agilent SureSelect Human All Exon V6 Kit and sequenced with the Illumina HiSeq 2000 or HiSeq X-TEN platform, as described previously.16,19,20 Original data were mapped to the human genome assembly GRCh37/hg19 with the Burrows-Wheeler Aligner (BWA) software. The identification of candidate pathogenic variants has been detailed previously.20 The pathogenicity of the variants was evaluated according to the standards and guidelines established by the American College of Medical Genetics and Genomics (ACMG) for the interpretation of the variants.
The suspected variants were confirmed via polymerase chain reaction (PCR) and Sanger sequencing for the probands and their available family members with the Green Mix kit (Promega, M7123). PCR primer sequences and conditions have been listed in Table S1.
Semen parameter analysis
Semen analysis was conducted in the source laboratories during routine biological examination of individuals according to WHO guidelines. Semen samples from men harboring DNHD1 variants were collected through masturbation after 2–7 days of sexual abstinence and evaluated after liquefaction for 30 min at 37°C. Analysis of semen volume, sperm concentration, and motility were conducted during routine examination. The morphology of sperm cells was assessed via H&E staining. For each subject, we examined at least 300 spermatozoa to evaluate the percentage of morphologically abnormal spermatozoa.
Semen samples from mice were collected from the cauda epididymides obtained through dissection of adult male mice. The samples were then diluted in a 1 mL solution for capacitation for 30 min at 37°C. Semen characteristics were analyzed with the computer-assisted sperm analysis (CASA) system and H&E staining. At least three 8-week-old male C57BL/6 mice were analyzed for each group.
Scanning and transmission electron microscopy
Epididymal sperm samples were prepared for scanning and transmission electron microscopy (SEM and TEM, respectively) as described previously.20 For the SEM assay, spermatozoa were fixed in 2.5% phosphate-buffered glutaraldehyde at 4°C for 2 h. Immobilized spermatozoa were then deposited on poly L-lysine-coated coverslips. The coverslips were washed in distilled water, dehydrated via an ascending gradient of 50%, 70%, 95%, and 100% cold ethanol, and dried at a critical point with a Quorum K850 Critical Point Dryer (East Sussex). Specimens were then attached to specimen holders and coated with gold particles with an ion sputter coater (Quorum Technologies, Q150RS Rotary-Pumped) prior to observation with an S-3400N scanning electron microscope (Hitachi, Japan). For TEM, samples were fixed with glutaraldehyde and osmium tetroxide, which was followed by treatment with OsO4 and sucrose. Subsequently, dehydration was performed with graded ethanol. Then, the samples were embedded in Epon812, dodecenylsuccinic anhydride, methylnadic anhydride, and dimethylaminomethyl phenol. Ultrathin (70–90 nm) sections were contrasted with uranyl acetate and lead citrate. An HT7700 Hitachi electron microscope (Hitachi) and a MegaView III digital camera (Munster) were used for capturing the image.
Immunofluorescence analysis
Sperm cells were washed in 1 phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 30 min at room temperature, and coated on slides pre-coated with 0.1% poly-L-lysine. The slides of sperm cells were washed with 1 PBS, blocked with 5% bovine serum albumin, and incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-DNHD1 (Abnova, PAB15797, 1:100), rabbit polyclonal anti-SPAG6 (Sigma, HPA038440, 1:200), rabbit polyclonal anti-TOMM20 (Proteintech, APR10507G, 1:600), rabbit polyclonal anti-SPEF2 (Sigma, HPA040343, 1:200), rabbit polyclonal anti-AKAP4 (Sigma, HPA020046, 1:500), rabbit polyclonal anti-RSPH1 (Sigma, HPA017382, 1:100), rabbit polyclonal anti-DNAH2 (Novus, NBP2-49506, 1:200), rabbit polyclonal anti-DNAI1 (Bioworld, DNAI1, 1:100), and monoclonal mouse anti-α-tubulin (Sigma, T5168, 1:1,000). Washes were performed with PBST (0.1% Tween-20), followed by 2 h incubation at 37°C with the highly cross-absorbed secondary antibodies: Alexa Fluor 488 anti-mouse IgG (Invitrogen, A21121, 1:1,000) and Alexa Fluor 555 anti-rabbit IgG (Invitrogen, A32732, 1:1,000). Fluorescence images were obtained via an Olympus IX51 fluorescence microscope (Olympus) and analyzed with the VideoTesT-FISH 2.0 software.
Dnhd1-knockout mouse model generation with CRISPR-Cas9
Single-guide RNA (5′-CTAGGGAAAATAATTTAGATAGG-3′) plasmids against exon 22 of Dnhd1 were designed and constructed. The Cas9 messenger RNA (mRNA) and single-guide RNA transcribed by T7 RNA polymerase in vitro were mixed and co-microinjected into fertilized eggs of C57BL/6 mice. Homozygous targeted mice were obtained by intercrossing heterozygous mutant mice. The offspring were genotyped via PCR-Sanger sequencing of mouse genomic DNA with the following primers: Dnhd1-F: 5′-GCTGGTAATAATGCCCTGCTT-3′ and Dnhd1-R: 5′-TCCAGAGATGTCACCGTGTTA-3′. All animals were treated in accordance with the protocols established by the Institutional Animal Care and Use Committee of Central South University (Changsha, China). Adult mice (aged 8 weeks or older) were selected for subsequent experiments.
In vitro fertilization and intracytoplasmic sperm injection in mice
In vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) analysis in mice were conducted, as described previously.21 In brief, wild-type female mice were superovulated by injection of 10 IU of pregnant mare serum gonadotropin (PMSG), followed by injection of 10 IU of human chorionic gonadotropin (HCG, Livzon) 48 h later. After 15–16 h, cumulus-intact oocytes were harvested. For IVF, sperm samples collected from mouse cauda epididymides were added into HTF (EasyCheck, M1150). Next, cumulus-intact oocytes collected from superovulated female mice were transferred into a sperm-containing fertilization drop. After incubation for 5 h, mouse embryos were washed in another fertilization drop and transferred into the M16 medium (Sigma, M7292) for further culture (37°C, 5% CO2). We evaluated the fertilization rates by recording the number of two-cell embryos and blastocysts at 20 h and 96 h. For ICSI, mouse sperm heads were separated from sperm tails and injected into mouse oocytes obtained from superovulated females with a piezo-driven pipette, as described previously.22,23 The injected oocytes were cultured in M16 medium at 37°C under 5% CO2. We counted two-cell embryos and blastocysts at 24 h and 96 h, respectively, to calculate the fertilization rates.
ICSI in DNHD1 variant-associated infertile individuals
ICSI was performed in three hospitals independently with the same protocol and written informed consents were signed by each couple that accepted ICSI treatment. The ICSI protocol was generally descripted as follows: ICSI was performed 3–5 h after oocyte retrieval. The oocyte–cumulus complex (COC) was briefly rinsed in 80 IU/mL hyaluronidase, and cumulus cells were removed by gentle aspiration. Denuded oocytes were washed and transferred into G-MOPS medium (Vitrolife, 10130); only matured oocytes were injected. Sperm were selected for injection on the basis of whether they had good morphology and motility and were then immobilized in polyvinylpyrrolidone (PVP) before injection. The oocyte was held by the holding pipette, and the microinjection needle was pushed through the zona and into the cytoplasm. A small amount of cytoplasm was sucked into the microinjection needle and released back into the oocyte along with the sperm. The injection needle was withdrawn gently, and the oocyte was released from the holding pipette. The injected oocytes were transferred into G-IVF medium and cultured in 6% CO2, 5% O2, and 89% N2 at 37°C in a humidified incubator.
Results
Identification of bi-allelic deleterious variants of DNHD1 in asthenoteratozoospermia-affected men
In this study, we performed WES to analyze the genetic etiology in a cohort of 497 unrelated infertile men with asthenoteratozoospermia. We identified variants in DNHD1 (dynein heavy chain domain 1, GenBank: NM_144666.3) in eight unrelated individuals, representing 1.6% (8/497) of this cohort (Figure 1A, Table 1). Human DNHD1 was highly expressed in the testis in accordance with the NCBI and GTEx databases. Human DNHD1 is located at 11p15.4 with a length of approximately 74.74 kb and encodes a predicted 4,753-amino acid protein that comprises five coiled-coil (CC) domains and two basic and acidic residues domains and includes an acidic residues domain (Figure 1B).
Figure 1.

Identification of bi-allelic DNHD1 variants in men with asthenoteratozoospermia
(A) Pedigree and genetic typing of DNHD1 in eight men with asthenoteratozoospermia. Filled and open signs indicate affected and unaffected individuals, respectively. The eight probands are marked with a black arrow and were screened by WES. Individuals marked with numbers were Sanger sequenced. Abbreviations: F, family; WT, wild type; NC, normal control.
(B) Locations and conservation analysis of the detected variants in DNHD1. The positions of all variants are indicated in the genomic and protein structure of DNHD1, and conservation of the missense mutated amino acids is indicated by the alignment of eight species. All missense variants are conservative according to the sequence alignment. Five red squares in the DNHD1 domain map represent coiled coil domains, the blue square indicates acidic residues domain, and the yellow box shows two basic and acidic residues domains. All domains and regions are described by the UniProt server.
Table 1.
Detailed description of the bi-allelic variants in DNHD1 identified in eight infertile men
| Subjects | Gene | cDNA alteration | Amino acid alteration | Mutation zygosity | Function |
Allele frequency |
In-silico bioinformatics prediction |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1000 Genomes | gnomAD | gnomAD-EAS | PolyPhen-2 | MutationTaster | SIFT | CADD | ||||||
| F1-II-1 | DNHD1 | c.5560C>T | p.Arg1854Cys | Het | missense | NAa | 0.000006437 | 0.000 | PD | DC | Del | 23 |
| DNHD1 | c.6498T>G | p.Tyr2166∗ | Het | stop-gain | NAa | 0.00001917 | 0.0001836 | NAb | DC | NAb | 34 | |
| F2-II-1 | DNHD1 | c.8782C>T | p.Arg2928∗ | Hom | stop-gain | 0.000799 | 0.0009866 | 0.000 | NAb | DC | NAb | 36 |
| F3-II-1 | DNHD1 | c.522_525del | p.Arg174Serfs∗3 | Het | frameshift | NAa | NAa | NAa | NAb | DC | NAb | NAb |
| DNHD1 | c.911G>A | p.Arg304Gln | Het | missense | NAa | 0.0001399 | 0.0003020 | D | Poly | Del | 24.8 | |
| F4-II-1 | DNHD1 | c.425_428del | p.Asp142Valfs∗35 | Het | frameshift | NAa | NAa | NAa | NAb | DC | NAb | NAb |
| DNHD1 | c.4072C>T | p.Arg1358Cys | Het | missense | 0.0034 | 0.001054 | 0.009713 | D | DC | Del | 29.9 | |
| F5-II-1 | DNHD1 | c.8909A>G | p.Tyr2970Cys | Hom | missense | 0.0000 | 0.0001438 | 0.001926 | D | DC | Del | 26.7 |
| F6-II-1 | DNHD1 | c. 9649C>T | p.Arg3217∗ | Het | stop-gain | NAa | 0.00005203 | 0.0007224 | NAb | DC | NAb | 36 |
| DNHD1 | c.12473A>G | p.His4158Arg | Het | missense | NAa | NAa | NAa | D | DC | Del | 25.7 | |
| F7-II-1 | DNHD1 | c.12453G>A | p.Trp4151∗ | Het | stop-gain | NAa | NAa | NAa | NAb | DC | NAb | 46 |
| DNHD1 | c.4141C>T | p.Gln1381∗ | Het | stop-gain | NAa | 0.00002538 | 0.0004013 | NAb | DC | NAb | 36 | |
| F8-II-1 | DNHD1 | c.14234T>C | p.Val4745Ala | Het | missense | NAa | 0.001077 | 0.000 | D | Poly | N | 25.2 |
| DNHD1 | c.5347delC | p.Gln1783Serfs∗28 | Het | frameshift | NAa | NAa | NAa | NAb | DC | NAb | NAb | |
The reference transcript of DNHD1 is GenBank: NM_144666.3. Hom, homozygous; Het, heterozygous; D, damaging; PD, possible damaging; DC, disease causing; Poly, polymorphism; Del, deleterious; N, neutral; NAa, not available; NAb, not assessed. CADD (GRCh37-v1.4) score: amino acid substitution is predicted damaging if the score is >4.
Validation and segregation analysis were conducted via PCR-Sanger sequencing, with target site primers in eight families (Table S1). We found that bi-allelic DNHD1 variants detected in seven probands were parentally inherited from heterozygous carriers, which was consistent with an autosomal recessive mode of inheritance (Figure 1A). Briefly, the probands were identified to carry compound heterozygous c.5560C>T (p.Arg1854Cys) and c.6498T>G (p.Tyr2166∗) in family 1 (F1), homozygous c.8782C>T (p.Arg2928∗) in family 2 (F2), compound heterozygous c.522_525del (p.Arg174Serfs∗3) and c.911G>A (p.Arg304Gln) in family 3 (F3), homozygous c.8909A>G (p.Tyr2970Cys) in family 5 (F5), compound heterozygous c. 9649C>T (p.Arg3217∗) and c.12473A>G (p.His4158Arg) in family 6 (F6), and compound heterozygous c.12453G>A (p.Trp4151∗) and c.4141C>T (p.Gln1381∗) in family 7 (F7) (Figure 1A). In family 4 (F4, Figure 1A), the proband was found to carry compound heterozygous c.425_428del (p.Asp142Valfs∗35) and c.4072C>T (p.Arg1358Cys) of DNHD1. Because the c.4072C>T (p.Arg1358Cys) variant was inherited from his mother, the other variant, c.425_428del (p.Asp142Valfs∗35), was suspected to be inherited from his deceased father. In family 8 (F8, Figure 1A), the proband was found to carry heterozygous c.14234T>C (p.Val4745Ala) and heterozygous c.5347delC (p.Gln1783Serfs∗28) of DNHD1. Unfortunately, pedigree analysis was not performed because the samples of his parents were not available.
The detected nonsense and frameshift variants identified in this study were assumed to induce premature termination codons and truncating proteins, suggesting their strong deleterious effects (Figure 1B, Table 1). The remaining six missense variants were highly conserved among species and predicted to be damaging via in-silico bioinformatics tools (Figure 1B, Figure S1, Table 1). Notably, the prevalence analysis from the 1000 Genomes and gnomAD databases revealed that these detected variants were either at low allele frequencies or unavailable in the general population, suggesting their rarity (Table 1). Moreover, none of the detected DNHD1 variants existed in the cohort of 392 fertile controls. Therefore, we speculated that the identified bi-allelic DNHD1 variants might be responsible for the observed infertility phenotypes.
Asthenoteratozoospermia phenotypes in men harboring bi-allelic DNHD1 variants
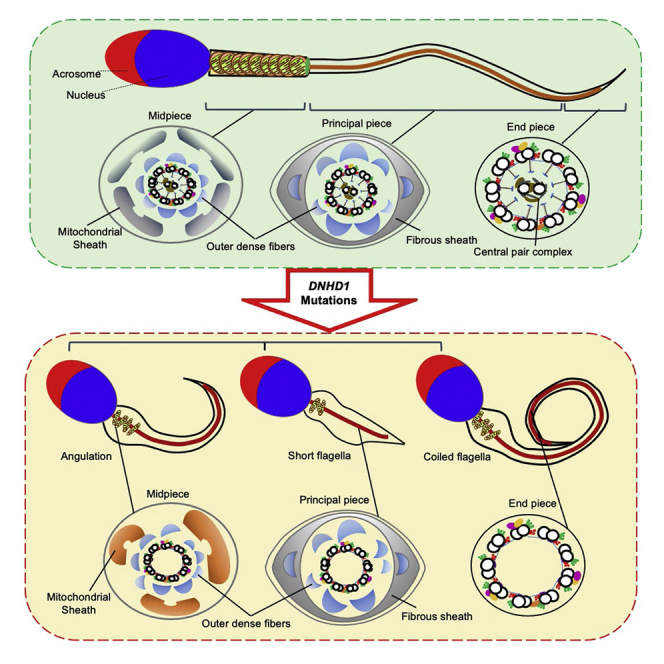
Semen routine assessments were conducted under WHO guidelines, which revealed that sperm motility decreased in eight probands harboring bi-allelic DNHD1 variants (Table 2). Moreover, the progressive motility rate in three of these eight affected individuals decreased dramatically to zero, and the other five had progressive motility rates of 0.5%, 0.8%, 3.2%, 6.3%, and 16.5% (Table 2). The morphology of the sperm cells was assessed with H&E staining and SEM (Figure 2A, Figure S2, Table 2). Spermatozoa from a fertile male control showed normal long and smooth flagella, whereas spermatozoa from men harboring bi-allelic DNHD1 variants displayed multiple morphological abnormalities of the flagella phenotype (including short, coiled, absent, angulation, and irregular caliber flagella) (Figure 2A, Table 2).
Table 2.
Semen routine parameters and sperm morphology of men harboring bi-allelic DNHD1 variants
| Human subjects | F1-II-1 | F2-II-1 | F3-II-1 | F4-II-1 | F5-II-1 | F6-II-1 | F7-II-1 | F8-II-1 | Reference valuesa | |
|---|---|---|---|---|---|---|---|---|---|---|
| Semen parameters | ||||||||||
| Sperm volume (mL) | 4.0 | 3.5 | 3.2 | 3.6 | 2.7 | 3.1 | 1.8 | 2.6 | >1.5 | |
| Sperm concentration (106/mL) | 12.1 | 1.8 | 17.6 | 2.7 | 81.4 | 99 | 25.7 | 32.2 | >15.0 | |
| Motility (%) | 0.8 | 20.0 | 0 | 7.3 | 25.0 | 30.0 | 4.5 | 5.8 | >40.0 | |
| Progressive motility (%) | 0.8 | 0 | 0 | 3.2 | 16.5 | 6.3 | 0 | 0.5 | >32.0 | |
| Sperm flagellar morphologyb | ||||||||||
| Absent flagella (%) | 7.1 | 13.8 | 12.5 | 16.0 | 8.6 | 8.0 | N/A | N/A | <5.0 | |
| Short flagella (%) | 27.5 | 15.8 | 38.0 | 22.5 | 6.7 | 8.5 | N/A | N/A | <1.0 | |
| Coiled flagella (%) | 25.0 | 39.2 | 16.9 | 26.6 | 44.5 | 26.9 | N/A | N/A | <17.0 | |
| Angulation (%) | 10.2 | 8.7 | 6.1 | 7.6 | 3.5 | 5.5 | N/A | N/A | <13.0 | |
| Irregular caliber (%) | 27.8 | 20.3 | 24.6 | 24.8 | 16.7 | 12.4 | N/A | N/A | <2.0 | |
| Sperm mid-piece morphology | ||||||||||
| Normal mid-piece (%) | 11.4 | 19.8 | 5.7 | 21.3 | 39.1 | 55.1 | N/A | N/A | >88.6 | |
| Abnormal mid-piece (%) | 88.6 | 80.2 | 94.3 | 78.8 | 60.9 | 44.9 | N/A | N/A | <11.4 | |
Figure 2.

Morphology and DNHD1 deficiency of spermatozoa in the control group and men harboring bi-allelic DNHD1 variants
(A) Morphology of spermatozoa from a fertile male control and men with bi-allelic DNHD1 variants. Spermatozoa from the fertile control showed normal long and smooth flagella, whereas spermatozoa from men harboring bi-allelic DNHD1 variants displayed multiple morphological abnormalities in the flagellar phenotypes (including short, coiled, absent, angulation, and irregular caliber flagella). Scale bars: 5 mm.
(B) Immunofluorescence assay of DNHD1 in the spermatozoa from a fertile control and men with bi-allelic DNHD1 variants. Anti-DNHD1 (red) and anti-a-tubulin (green) antibodies were used. The nuclei of spermatozoa were DAPI labeled (blue). DNHD1 localized at the entire flagella in the fertile control. However, DNHD1 staining was almost absent in the sperm flagella from men with bi-allelic DNHD1 variants. Scale bars: 5 mm.
To examine the pathogenicity of the identified DNHD1 variants, we checked the localization and abundance of DNHD1 in spermatozoa from a fertile control and men harboring bi-allelic DNHD1 variants by using a commercial antibody against DNHD1. Using immunofluorescence staining, we found that DNHD1 is localized in the entire flagella in the fertile control and is predominantly concentrated in the mid-piece of the sperm flagella, while it was almost absent in the sperm flagella in samples obtained from men with DNHD1 variants (Figure 2B). Taken together, our results support causality of bi-allelic DNHD1 variants for flagellum abnormalities and asthenoteratozoospermia.
DNHD1 defects associated with sperm axoneme and mitochondrial sheath malformations
Both cilia and flagella contain an important core component (termed the axoneme), which is an evolutionarily conserved structure comprising nine doublets of microtubules (DMTs) and a central pair of microtubules (CP).26,27 Additionally, the sperm flagellum harbors specific peri-axonemal structures, including a helical mitochondrial sheath (MS) in the mid-piece, a fibrous sheath in the principal piece, and outer dense fibers in the mid-piece and the proximal part of the principal piece (Figure 3A). Sperm flagellar ultrastructure defects were investigated with TEM in men harboring bi-allelic DNHD1 variants. Surprisingly, compared with the sperm flagella from the fertile control, which displayed a typical “9 + 2” microtubule structure (nine DMTs and a CP), more than 95% of the cross-sections of the sperm flagella from men harboring DNHD1 variants were abnormal, including an absence of CP (“9 + 0”), disorganization of the “9 + 2” structure as well as DMTs (Figures 3A and 3B). The main defects included the absence of CP (54.5%, 47.6%, and 66.1% for subjects F1-II-1, F2-II-1, and F3-II-1, respectively), axoneme disorganization (30.1%, 26.2%, and 26.8% for subjects F1-II-1, F2-II-1, and F3-II-1, respectively), and absence of DMTs (10.9%, 23.8%, and 7.1% for subjects F1-II-1, F2-II-1, and F3-II-1, respectively) (Figure 3B).
Figure 3.

DNHD1 deficiency associated with sperm axoneme malformations and central pair loss
(A) Ultrastructure of spermatozoa determined by TEM from a fertile control and men with bi-allelic DNHD1 variants. Cross-sections of the sperm flagella in the control show a typical axoneme and peri-axoneme structure. The axoneme mainly comprises a ‘‘9 + 2’’ structure, including nine pairs of peripheral doublet microtubules (DMTs; green arrows) and a central pair of microtubules (CP; blue arrows). The peri-axoneme structure includes the fiber sheath, nine outer dense fibers (red arrows), and a helical mitochondrial sheath (MS; pink arrows). Scale bars: 200 nm.
(B) Quantification of different categories of flagellar ultrastructural defects. Total cross-section numbers for quantification in the control individual and men harboring bi-allelic DNHD1 variants (subjects F1-II-1, F2-II-1, and F3-II-1) were 135, 156, 84, and 102, respectively. Cross-sectional defects were classified into four categories: absence of CP, absence of DMTs, disorganization, and normal axoneme. The main defective types were an absence of CP (54.5%, 47.6%, and 10.9% for subjects F1-II-1, F2-II-1, and F3-II-1, respectively) and axoneme disorganization (30.1%, 26.2%, and 26.8% for subjects F1-II-1, F2-II-1, and F3-II-1, respectively).
(C and D) SPAG6 and SPEF2 immunofluorescence assays of human sperm. Anti-SPAG6 (red in C) and anti-SPEF2 (red in D) normally localized along the sperm flagella in the control sperm. However, expression of both SPAG6 and SPEF2 was almost absent in sperm obtained from men harboring bi-allelic DNHD1 variants. Anti-a-tubulin (green) marked the sperm flagella. The nuclei of spermatozoa were DAPI-labeled (blue). Scale bars: 5 mm.
Moreover, malformations of the mid-piece in longitudinal sections of the flagella were detected by TEM. Compared with the well-arranged structure of the mid-piece in the control, mutant spermatozoa displayed either absent or abnormal location and/or arrangement, such as a swollen mid-piece with poorly assembled MS, a cytoplasmic mass containing different components, loosely assembled MS, and a completely disorganized mid-piece with a misshapen MS (Figure 4A).
Figure 4.

DNHD1 deficiency associated with mitochondrial sheath malformations in the sperm flagellum
(A) Longitudinal sections of the sperm flagellar mid-piece of a control individual and men harboring bi-allelic DNHD1 variants. The normal sperm had a symmetrical mid-piece with smooth axoneme surrounded by regularly arranged MS. In contrast, DNHD1-deficient sperm displayed either absent or abnormal location and/or arrangement, such as a swollen mid-piece with a poorly assembled MS, a cytoplasmic mass containing different components, loosely assembled MS, and completely disorganized mid-piece with a misshapen MS. Black arrow marks the MS. Scale bars: 1 mm.
(B) TOMM20 localization in the sperm from a control individual and men harboring bi-allelic DNHD1 variants. TOMM20 (red) localized at the flagellar mid-piece in which the MS can be visualized well. However, TOMM20 showed misshapen, uneven distribution and swelling of the flagellar mid-piece in men with bi-allelic DNHD1 variants. The nuclei of sperm were DAPI labeled (blue). Anti-a-tubulin (green) marked the sperm flagella. Scale bars: 5 mm.
To further investigate the ultrastructural defects revealed by TEM, we performed immunofluorescence assays to examine the presence and localization of several proteins belonging to different substructures of the axoneme, including SPAG6 (component of CP), SPEF2 (component of sperm flagella assembly), TOMM20 (component of MS), AKAP4 (component of fibrous sheath), DNAI1 (component of outer dynein arms), DNAH2 (component of inner dynein arms), and RSPH1 (component of radial spoke). We observed that SPAG6 and SPEF2 were almost absent in the spermatozoa of men harboring bi-allelic DNHD1 variants (Figures 3C and 3D). Moreover, TOMM20 showed misshapen, uneven distribution and swelling of the flagellar mid-piece in men with bi-allelic DNHD1 variants (Figure 4B). In contrast, the results for AKAP4, DNAI1, DNAH2, and RSPH1 were comparable to those observed in the controls (Figure S3), suggesting that the fibrous sheath, outer dynein arms, inner dynein arms, and radial spoke were not directly affected by the presence of DNHD1 variants.
Asthenoteratozoospermia phenotypes in Dnhd1‒/‒ male mice
To investigate how DNHD1 affects spermatogenesis and flagellogenesis in mice, we generated Dnhd1‒/‒ mice with 31 bases deletion in exon 22 of Dnhd1, which induced the truncating protein c.5572_5602del (p.1858Trpfs∗24), by using CRISPR-Cas9 technology (Figures S4A and S4B). Reverse-transcription quantitative PCR, which was performed according to the manufacturer’s instructions (Promega, A5000), showed that the expression of Dnhd1 mRNA was undetectable in the testes of Dnhd1‒/‒ male mice compared with that in Dnhd1+/+ male mice (Figure S4C, Table S2).
To investigate the reproductive habits and fertility rate of Dnhd1‒/‒ mice, Dnhd1+/+ and Dnhd1‒/‒ male mice (8 weeks old) were mated with fertile Dnhd1+/+ female mice, and the number of pups per litter was counted. Notably, Dnhd1‒/‒ male mice had an average of zero offspring per litter during the 3-month reproductive period, whereas the average number of offspring per litter for the Dnhd1+/+ male mice was nine (p < 0.001) (Figure 5A). Further comparison between Dnhd1+/+ and Dnhd11‒/‒ male mice revealed that Dnhd1 deficiency did not cause a significant reduction in the size or weight of testes (Figures 5B and 5C). H&E staining was performed on the testis of Dnhd1+/+ and Dnhd1‒/‒ male mice, and no significant differences were observed (Figure S4D). Sperm was collected from the cauda epididymis of the Dnhd1+/+ and Dnhd11‒/‒ male mice, and sperm parameters were analyzed by CASA. Compared with those of Dnhd1+/+ male mice, the sperm concentration, motility rate, and progressive motility of Dnhd1‒/‒ mice were all significantly decreased, which is generally consistent with the results obtained for human individuals (Figures 5D, 5E, and 5F, Table S3). H&E staining indicated that Dnhd1‒/‒ male mice exhibited more than 95% abnormalities of the sperm flagella, including coiled flagella (39.4%), absent flagella (23.6%), bent caliber (21.9%), short flagella (5.5%), and irregular flagella (5.5%) (Figure 5G, Table S3), but the morphologies of sperm head analysis showed low abnormality rate and exhibited no significant difference between Dnhd1+/+ and Dnhd1‒/‒ male mice. Furthermore, TEM analysis showed the abnormality rate of the sperm flagella in Dnhd1‒/‒ male mice were increased significantly compared with Dnhd1+/+ male mice (53.3%, 32/60 versus 4.8%, 2/42) (Figure S5).
Figure 5.

Effect of Dnhd1 deficiency on typical multiple morphological abnormalities of the flagella phenotypes and infertility in male mice
(A) The mean number of pups per litter was nine in Dnhd1+/+ male mice, whereas all Dnhd1‒/‒ male mice were completely infertile. ∗∗∗ indicates p < 0.001. n = 3.
(B) Testis sizes were comparable between Dnhd1+/+ and Dnhd1‒/‒ male mice at 8 weeks of age.
(C) Testis weights were comparable between Dnhd1+/+ and Dnhd1‒/‒ male mice at 8 weeks of age. NS, not significant. n = 3.
(D) Sperm concentration comparison between Dnhd1+/+ and Dnhd1‒/‒ male mice (63.7 × 106/mL and 20.3 × 106/mL, respectively. ∗∗ indicates p < 0.01). n = 3.
(E) Sperm motility comparison between Dnhd1+/+ and Dnhd1‒/‒ male mice (70.5% and 28%, respectively. ∗∗ indicates p < 0.01). n = 3.
(F) Sperm progressive motility comparison between Dnhd1+/+ and Dnhd1‒/‒ male mice (40% and 4%, respectively. ∗∗∗ indicates p < 0.001). n = 3.
(G) Morphology of spermatozoa from Dnhd1+/+ and Dnhd1‒/‒ male mice. When compared with the normal morphology of spermatozoa obtained from Dnhd1+/+ male mice, Dnhd1‒/‒ male mice spermatozoa exhibited normal head morphologies, but aberrant flagellar morphologies, which were consistent with the clinical phenotypes observed in men harboring bi-allelic DNHD1 variants.
(H) Dnhd1 localization in spermatozoa from Dnhd1+/+ and Dnhd1‒/‒ male mice revealed by immunofluorescence staining. Anti-DNHD1 (red) localized at the sperm axoneme from Dnhd1+/+ male mice, whereas DNHD1 (red) staining was almost absent in the sperm flagella of Dnhd1‒/‒ male mice. Anti-a-tubulin indicated the flagella (green) and the nuclei of spermatozoa were DAPI labeled (blue). Scale bars: 5 mm.
Immunofluorescence assays were performed with sperm cells from Dnhd1+/+ and Dnhd1‒/‒ male mice. Consistent with the findings in humans, DNHD1 localized to the entire sperm flagella in Dnhd1+/+ male mice, whereas DNHD1 staining was almost absent in the sperm flagella of Dnhd1‒/‒ male mice (Figure 5H). Overall, these data suggested that DNHD1 deficiency could result in male infertility in both humans and mice.
ICSI treatment of DNHD1-associated male infertility
To evaluate whether male infertility induced by DNHD1 variants could be overcome with assisted reproductive technology (ART), we performed IVF to explore the fertility of Dnhd1‒/‒ male mice. We calculated the two-cell embryo and blastocyst rates after we used caudal epididymal sperm to fertilize oocytes collected from superovulated Dnhd1+/+ females. Unsurprisingly, the rates of two-cell embryos and blastocysts were markedly lower in Dnhd1‒/‒ male mice than that in Dnhd1+/+ male mice (Figure S6A and Table S4). Subsequently, ICSI, which is an efficient treatment for most asthenoteratozoospermia-affected individuals, was conducted with sperm from Dnhd1+/+ and Dnhd1‒/‒ male mice. The results showed that the rates of two-cell embryos and blastocysts in Dnhd1‒/‒ male mice were almost equivalent to those in Dnhd1+/+ male mice (Figure S6B and Table S4).
Consistent with the above observations in mice, ICSI treatment was performed with the sperm extracted from the seven men exhibiting DNHD1 variants, and four treatments resulted in successful pregnancy (Table S5).
Discussion
In this study, we identified bi-allelic variants of DNHD1 in eight unrelated asthenoteratozoospermia-affected men (1.6%, 8/497) from a cohort built from three different hospitals. The WES results showed that all eight men harbored bi-allelic DNHD1 variants and no other known pathogenic gene variants that were associated with sperm flagellar development or function. All DNHD1 variants had extremely rare frequencies or were absent in public databases, such as gnomAD and 1000 Genomes, and are predicted to be potentially pathogenic by in-silico bioinformatics. Moreover, bi-allelic DNHD1 variants co-segregated with asthenoteratozoospermia in the pedigree analysis, indicating an autosomal recessive inheritance pattern. The evidence obtained from functional experiments combined with analysis with Dnhd1‒/‒ male mice consistently supported the finding that DNHD1 is an asthenoteratozoospermia-associated gene.
Dynein heavy chain domain 1 (DNHD1), also known as coiled-coil domain-containing 35 (CCDC35), is an evolutionarily conserved protein with five CC domains. The CC domain is a highly conserved superhelical protein motif characterized by the presence of one or more α-helical peptides that twist around each other to form a supercoil.28 Previous studies have shown that CCDC proteins are implicated in sperm flagellum formation and movement, and several CCDC gene defects result in male infertility due to the abnormalities in flagella assembly.13,14,29 For example, testis-enriched CFAP58 (also termed CCDC147) plays a vital role in sperm flagellogenesis, and CFAP58 deficiency causes infertility in humans and mice with abnormalities in both axonemes and peri-axonemes.13 Therefore, DNHD1, which belongs to the CCDC family and is highly expressed in the testis, is thought to be essential for the normal function of flagellar axonemes, and defects in DNHD1 may be responsible for severe asthenoteratozoospermia.
The flagellar axoneme is a motility apparatus of spermatozoa and has a “9 + 2” structure that contains a CP surrounded by nine DMTs (each of which has type-A and type-B microtubules).13 Previous studies have shown that defects in proteins involving the axoneme and peri-axoneme might lead to asthenoteratozoospermia.30,31 In our study, TEM revealed that approximately 95% of the axoneme cross-sections exhibited serious ultrastructural defects in men with bi-allelic DNHD1 variants, wherein the absence of CP and axoneme disorganization were the prominent defect types. Further immunofluorescence analysis showed that both SPAG6 and SPEF2 were almost absent in the sperm from men harboring bi-allelic DNHD1 variants, indicating that DNHD1 might function synergistically with axoneme molecules and is an essential protein for normal flagella assembly.
Notably, we also found severe MS malformations in the sperm from DNHD1-associated individuals, including lost, completely disorganized, and misshapen of MS. MS is an essential component of sperm flagella, and its abnormality presents a type of flagellar defect. Until now, at least twenty genes related to sperm flagella morphological malformation have been identified, but only a few genes are simultaneously associated with MS abnormalities. For example, CFAP58 and CFAP65 defects13,14 can cause asthenoteratozoospermia with flagellar defects, including MS abnormalities in the mid-piece. We noticed that these two genes as well as DNHD1 contain the CC domain, suggesting that the CC domain plays an important role in MS assembly and defects of the CC domain might be related to MS abnormality.
It is worth noting that CP loss in sperm flagella is often observed in individuals with CCDC family proteins deficiency, but MS defect is rarely reported, and its underlying molecular mechanism is unclear. In this study, protein-protein interaction network analysis with STRING showed that DNHD1 may strongly interact with multiple cytoplasmic dynein 1 components (such as DYNLT1, DYNLL1, DYNLL2, and DYNC1LI2) (Figure S7A), and these cytoplasmic dynein components can be used as motors in the retrograde intraflagellar transport (IFT) processes.32, 33, 34, 35 IFT is a highly evolutionarily conserved bidirectional transport platform and uses motors for the trafficking of cargo-related transport complexes into the tail,36 such as structural components of the axoneme, the fiber sheath, and the MS.36,37 Therefore, it is suggested that a protein transport defect (e.g., IFT defect) might explain the defect in accessory structure assembly. For example, our previous study revealed that CFAP65 (CCDC108) deficiency resulted in an abnormal morphology of sperm flagella, accompanied by a defect in the flagellar axoneme and mitochondria sheath.14 Further functional study showed a decrease of IFT-related protein IFT20 and cytoplasmic dynein 1 (DYNLT1, a motor for anterograde IFT) in Cfap65 mutant testis, indicating that the defects in CP and MS might be involved in IFT process.38 In this study, defects in CP and MS were frequently observed in the sperm from DNHD1-mutant individuals. We speculate that functional defect of DNHD1 may disrupt the interaction with proteins involved in the IFT process or affect the expression of IFT-related proteins, leading to abnormal flagellar assembly.
Our phenotypic analysis revealed that men harboring bi-allelic DNHD1 variants displayed typical asthenoteratozoospermia phenotypes, including reduced sperm motility and abnormalities of the sperm flagella. The sperm concentrations and motility vary a lot among males with the DNHD1 variation. Interestingly, similar differences in sperm phenotype were also reported for other asthenoteratozoospermia-related genes, such as DNAH10 (MIM: 605884), CFAP47, DNAH8 (MIM: 603337), TTC29 (MIM: 618735), and DNAH17 (MIM: 610063).16,39,40,41,42 The cause of variable sperm concentration and motility may be partially related to variation types and variation sites, individual heterogeneity, phenotypic heterogeneity, and/or differences in environment and lifestyle among males with the DNHD1 variation.
In this study, we constructed a Dnhd1‒/‒ mouse model to further explore whether DNHD1 is required for sperm flagella assembly and normal sperm motility. Consistent with the clinical presentation of men harboring bi-allelic DNHD1 variants, Dnhd1‒/‒ male mice were sterile and displayed various abnormal flagella as well as sperm immobility. In morphology analysis, only 6.0% sperm with flagellar defects were found in Dnhd1+/+ mice, while more than 95.5% sperm presented flagellar abnormalities in Dnhd1‒/‒ mice. For motility analysis, semen parameters revealed a reduced motility and progressive motility rate of sperm in Dnhd1‒/‒ male mice compared with that in Dnhd1+/+ mice. Surprisingly, sperm concentration was reduced in Dnhd1‒/‒ mice, which was also observed in several gene-specific mouse models, such as those for Cfap58, Cfap47, and Dnah10.13,16,39 This variability in sperm concentrations may reflect the evolutionarily divergent protein regulatory networks of DNHD1 between humans and mice (Figure S7). Our observations in the mouse model further supported the finding that DNHD1 is an essential gene for maintaining normal sperm flagellar morphology and motility.
For individuals with asthenoteratozoospermia, no empirical medicinal treatment can effectively improve semen parameters, and ICSI is the only method for conception.43 However, in previous studies, different clinical outcomes were observed in men whose asthenoteratozoospermia was caused by variants in different genes. For example, individuals with asthenoteratozoospermia associated with USP26, CFAP58, and CFAP47 (MIM: 301057) variants exhibited good clinical outcomes,10,13,39 while pregnancy failure was experienced by those with bi-allelic variants of CEP135 (MIM: 611423) and CFAP65.14,44 In this study, in order to evaluate the efficacy of ICSI, we conducted ICSI with sperm from Dnhd1+/+ and Dnhd1‒/‒ male mice and found that the rates of two-cell embryos and blastocysts showed no differences between these two groups, suggesting that male infertility in Dnhd1‒/‒ mice could be overcome by ICSI. Based on the results of mouse experiments, we conducted ICSI treatment in seven subjects with bi-allelic DNHD1 variants, four of whom successfully achieved clinical pregnancy. Therefore, our findings suggest that ICSI should be recommended for men with DNHD1-associated asthenoteratozoospermia.
In conclusion, we identified a gene causing male infertility due to asthenoteratozoospermia. The genetic and experimental evidence from DNHD1-associated men and Dnhd1‒/‒ male mice strongly suggests that DNHD1 plays a vital role in sperm flagellogenesis. DNHD1 deficiency causes abnormalities in both axonemes and peri-axonemes, leading to asthenoteratozoospermia. Our study provides insights into the understanding of asthenoteratozoospermia and the counseling of individuals with asthenoteratozoospermia.
Acknowledgments
We would like to thank all individuals who participated in this study. We thank the Department of Medical Ultrastructure, School of Basic Medical Science, and the Lab of Biomedical Electron Microscopy of Higher Research Center, Central South University for assistance with SEM work, and we would be grateful to Dr. Xiaoying Wu and Dr. Junpu Wang for their help of taking images. We also thank Zhejiang University for technical support of transmission electron microscopy. This work was supported by grants from the National Key Research and Developmental Program of China (2018YFC1004901), The National Natural Science Foundation of China (81971447, 81971441, 82071705, 82171608), the Key Grant of Prevention and Treatment of Birth Defect from Hunan Province (2019SK1012), Hunan Provincial Grant for Innovative Province Construction (2019SK4012), the Research Grant of CITIC-Xiangya (YNXM-202006, YNXM-202004), and the Graduate Research and Innovation Projects of Central South University (512191019).
Declaration of interests
The authors declare no competing interests.
Published: December 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2021.11.022.
Contributor Information
Ge Lin, Email: linggf36@hotmail.com.
Yue-Qiu Tan, Email: tanyueqiu@csu.edu.cn.
Data and code availability
The WES datasets supporting the current study have not been deposited in a public repository because of privacy and ethical restrictions but are available from the corresponding authors on request.
Web resources
1000 Genomes Project, https://www.internationalgenome.org/
Database of Genomic Variations, http://dgv.tcag.ca/dgv/app/home
MutationTaster, https://www.mutationtaster.org/
National Center for Biotechnology Information (NCBI), https://www.ncbi.nlm.nih.gov/
OMIM, http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
String, https://www.string-db.org/
UCSC Genome Browser, http://genome.ucsc.edu
UniProt, https://www.uniprot.org
Supplemental information
References
- 1.Cocuzza M., Cocuzza M.A., Bragais F.M., Agarwal A. The role of varicocele repair in the new era of assisted reproductive technology. Clinics (São Paulo) 2008;63:395–404. doi: 10.1590/s1807-59322008000300018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal A., Mulgund A., Hamada A., Chyatte M.R. A unique view on male infertility around the globe. Reprod. Biol. Endocrinol. 2015;13:37. doi: 10.1186/s12958-015-0032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharlip I.D., Jarow J.P., Belker A.M., Lipshultz L.I., Sigman M., Thomas A.J., Schlegel P.N., Howards S.S., Nehra A., Damewood M.D., et al. Best practice policies for male infertility. Fertil. Steril. 2002;77:873–882. doi: 10.1016/s0015-0282(02)03105-9. [DOI] [PubMed] [Google Scholar]
- 4.Tournaye H., Krausz C., Oates R.D. Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. 2017;5:544–553. doi: 10.1016/S2213-8587(16)30040-7. [DOI] [PubMed] [Google Scholar]
- 5.Mascarenhas M.N., Flaxman S.R., Boerma T., Vanderpoel S., Stevens G.A. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9:e1001356. doi: 10.1371/journal.pmed.1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shahrokhi S.Z., Salehi P., Alyasin A., Taghiyar S., Deemeh M.R. Asthenozoospermia: Cellular and molecular contributing factors and treatment strategies. Andrologia. 2020;52:e13463. doi: 10.1111/and.13463. [DOI] [PubMed] [Google Scholar]
- 7.Krausz C., Riera-Escamilla A. Genetics of male infertility. Nat. Rev. Urol. 2018;15:369–384. doi: 10.1038/s41585-018-0003-3. [DOI] [PubMed] [Google Scholar]
- 8.Tu C., Wang W., Hu T., Lu G., Lin G., Tan Y.Q. Genetic underpinnings of asthenozoospermia. Best Pract. Res. Clin. Endocrinol. Metab. 2020;34:101472. doi: 10.1016/j.beem.2020.101472. [DOI] [PubMed] [Google Scholar]
- 9.Coutton C., Escoffier J., Martinez G., Arnoult C., Ray P.F. Teratozoospermia: spotlight on the main genetic actors in the human. Hum. Reprod. Update. 2015;21:455–485. doi: 10.1093/humupd/dmv020. [DOI] [PubMed] [Google Scholar]
- 10.Liu C., Shen Y., Shen Q., Zhang W., Wang J., Tang S., Wu H., Tian S., Cong J., He X., et al. Novel Mutations in X-Linked, USP26-Induced Asthenoteratozoospermia and Male Infertility. Cells. 2021;10:1594. doi: 10.3390/cells10071594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni X., Wang J., Lv M., Liu C., Zhong Y., Tian S., Wu H., Cheng H., Gao Y., Tan Q., et al. A novel homozygous mutation in WDR19 induces disorganization of microtubules in sperm flagella and nonsyndromic asthenoteratospermia. J. Assist. Reprod. Genet. 2020;37:1431–1439. doi: 10.1007/s10815-020-01770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lv M., Liu W., Chi W., Ni X., Wang J., Cheng H., Li W.Y., Yang S., Wu H., Zhang J., et al. Homozygous mutations in DZIP1 can induce asthenoteratospermia with severe MMAF. J. Med. Genet. 2020;57:445–453. doi: 10.1136/jmedgenet-2019-106479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He X., Liu C., Yang X., Lv M., Ni X., Li Q., Cheng H., Liu W., Tian S., Wu H., et al. Bi-allelic Loss-of-function Variants in CFAP58 Cause Flagellar Axoneme and Mitochondrial Sheath Defects and Asthenoteratozoospermia in Humans and Mice. Am. J. Hum. Genet. 2020;107:514–526. doi: 10.1016/j.ajhg.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W., Tu C., Nie H., Meng L., Li Y., Yuan S., Zhang Q., Du J., Wang J., Gong F., et al. Biallelic mutations in CFAP65 lead to severe asthenoteratospermia due to acrosome hypoplasia and flagellum malformations. J. Med. Genet. 2019;56:750–757. doi: 10.1136/jmedgenet-2019-106031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Alvarez J., Cano-Corres R., Fuentes-Arderiu X. A Complement for the WHO Laboratory Manual for the Examination and Processing of Human Semen (First Edition, 2010) EJIFCC. 2012;23:103–106. [PMC free article] [PubMed] [Google Scholar]
- 16.Tu C., Cong J., Zhang Q., He X., Zheng R., Yang X., Gao Y., Wu H., Lv M., Gu Y., et al. Bi-allelic mutations of DNAH10 cause primary male infertility with asthenoteratozoospermia in humans and mice. Am. J. Hum. Genet. 2021;108:1466–1477. doi: 10.1016/j.ajhg.2021.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Y., Tian S., Sha Y., Zha X., Cheng H., Wang A., Liu C., Lv M., Ni X., Li Q., et al. Novel bi-allelic variants in DNAH2 cause severe asthenoteratozoospermia with multiple morphological abnormalities of the flagella. Reprod. Biomed. Online. 2021;42:963–972. doi: 10.1016/j.rbmo.2021.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Li Y., Sha Y., Wang X., Ding L., Liu W., Ji Z., Mei L., Huang X., Lin S., Kong S., et al. DNAH2 is a novel candidate gene associated with multiple morphological abnormalities of the sperm flagella. Clin. Genet. 2019;95:590–600. doi: 10.1111/cge.13525. [DOI] [PubMed] [Google Scholar]
- 19.Zhu F., Wang F., Yang X., Zhang J., Wu H., Zhang Z., Zhang Z., He X., Zhou P., Wei Z., et al. Biallelic SUN5 Mutations Cause Autosomal-Recessive Acephalic Spermatozoa Syndrome. Am. J. Hum. Genet. 2016;99:942–949. doi: 10.1016/j.ajhg.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan Y.Q., Tu C., Meng L., Yuan S., Sjaarda C., Luo A., Du J., Li W., Gong F., Zhong C., et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet. Med. 2019;21:1209–1217. doi: 10.1038/gim.2017.130. [DOI] [PubMed] [Google Scholar]
- 21.Lyu Q.F., Deng L., Xue S.G., Cao S.F., Liu X.Y., Jin W., Wu L.Q., Kuang Y.P. New technique for mouse oocyte injection via a modified holding pipette. Reprod. Biomed. Online. 2010;21:663–666. doi: 10.1016/j.rbmo.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Gu T.P., Guo F., Yang H., Wu H.P., Xu G.F., Liu W., Xie Z.G., Shi L., He X., Jin S.G., et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 23.Kimura Y., Yanagimachi R. Intracytoplasmic sperm injection in the mouse. Biol. Reprod. 1995;52:709–720. doi: 10.1095/biolreprod52.4.709. [DOI] [PubMed] [Google Scholar]
- 24.Cooper T.G., Noonan E., von Eckardstein S., Auger J., Baker H.W., Behre H.M., Haugen T.B., Kruger T., Wang C., Mbizvo M.T., Vogelsong K.M. World Health Organization reference values for human semen characteristics. Hum. Reprod. Update. 2010;16:231–245. doi: 10.1093/humupd/dmp048. [DOI] [PubMed] [Google Scholar]
- 25.Auger J., Jouannet P., Eustache F. Another look at human sperm morphology. Hum. Reprod. 2016;31:10–23. doi: 10.1093/humrep/dev251. [DOI] [PubMed] [Google Scholar]
- 26.Inaba K. Molecular basis of sperm flagellar axonemes: structural and evolutionary aspects. Ann. N Y Acad. Sci. 2007;1101:506–526. doi: 10.1196/annals.1389.017. [DOI] [PubMed] [Google Scholar]
- 27.Inaba K. Sperm flagella: comparative and phylogenetic perspectives of protein components. Mol. Hum. Reprod. 2011;17:524–538. doi: 10.1093/molehr/gar034. [DOI] [PubMed] [Google Scholar]
- 28.Burkhard P., Stetefeld J., Strelkov S.V. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 29.Priyanka P.P., Yenugu S. Coiled-Coil Domain-Containing (CCDC) Proteins: Functional Roles in General and Male Reproductive Physiology. Reprod. Sci. 2021;28:2725–2734. doi: 10.1007/s43032-021-00595-2. [DOI] [PubMed] [Google Scholar]
- 30.Wang W.L., Tu C.F., Tan Y.Q. Insight on multiple morphological abnormalities of sperm flagella in male infertility: what is new? Asian J. Androl. 2020;22:236–245. doi: 10.4103/aja.aja_53_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rawe V.Y., Hermes R., Nodar F.N., Fiszbajn G., Chemes H.E. Results of intracytoplasmic sperm injection in two infertile patients with abnormal organization of sperm mitochondrial sheaths and severe asthenoteratozoospermia. Fertil. Steril. 2007;88:649–653. doi: 10.1016/j.fertnstert.2006.12.074. [DOI] [PubMed] [Google Scholar]
- 32.Lechtreck K.F. IFT-Cargo Interactions and Protein Transport in Cilia. Trends Biochem. Sci. 2015;40:765–778. doi: 10.1016/j.tibs.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsurumi Y., Hamada Y., Katoh Y., Nakayama K. Interactions of the dynein-2 intermediate chain WDR34 with the light chains are required for ciliary retrograde protein trafficking. Mol. Biol. Cell. 2019;30:658–670. doi: 10.1091/mbc.E18-10-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakayama K., Katoh Y. Ciliary protein trafficking mediated by IFT and BBSome complexes with the aid of kinesin-2 and dynein-2 motors. J. Biochem. 2018;163:155–164. doi: 10.1093/jb/mvx087. [DOI] [PubMed] [Google Scholar]
- 35.Lehti M.S., Zhang F.P., Kotaja N., Sironen A. SPEF2 functions in microtubule-mediated transport in elongating spermatids to ensure proper male germ cell differentiation. Development. 2017;144:2683–2693. doi: 10.1242/dev.152108. [DOI] [PubMed] [Google Scholar]
- 36.Pleuger C., Lehti M.S., Dunleavy J.E., Fietz D., O’Bryan M.K. Haploid male germ cells-the Grand Central Station of protein transport. Hum. Reprod. Update. 2020;26:474–500. doi: 10.1093/humupd/dmaa004. [DOI] [PubMed] [Google Scholar]
- 37.Taschner M., Lorentzen E. The Intraflagellar Transport Machinery. Cold Spring Harb. Perspect. Biol. 2016;8:a028092. doi: 10.1101/cshperspect.a028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang W., Tian S., Nie H., Tu C., Liu C., Li Y., Li D., Yang X., Meng L., Hu T., et al. CFAP65 is required in the acrosome biogenesis and mitochondrial sheath assembly during spermiogenesis. Hum. Mol. Genet. 2021;30:2240–2254. doi: 10.1093/hmg/ddab185. [DOI] [PubMed] [Google Scholar]
- 39.Liu C., Tu C., Wang L., Wu H., Houston B.J., Mastrorosa F.K., Zhang W., Shen Y., Wang J., Tian S., et al. Deleterious variants in X-linked CFAP47 induce asthenoteratozoospermia and primary male infertility. Am. J. Hum. Genet. 2021;108:309–323. doi: 10.1016/j.ajhg.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C., Miyata H., Gao Y., Sha Y., Tang S., Xu Z., Whitfield M., Patrat C., Wu H., Dulioust E., et al. Bi-allelic DNAH8 Variants Lead to Multiple Morphological Abnormalities of the Sperm Flagella and Primary Male Infertility. Am. J. Hum. Genet. 2020;107:330–341. doi: 10.1016/j.ajhg.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu C., He X., Liu W., Yang S., Wang L., Li W., Wu H., Tang S., Ni X., Wang J., et al. Bi-allelic Mutations in TTC29 Cause Male Subfertility with Asthenoteratospermia in Humans and Mice. Am. J. Hum. Genet. 2019;105:1168–1181. doi: 10.1016/j.ajhg.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song B., Liu C., Gao Y., Marley J.L., Li W., Ni X., Liu W., Chen Y., Wang J., Wang C., et al. Novel compound heterozygous variants in dynein axonemal heavy chain 17 cause asthenoteratospermia with sperm flagellar defects. J. Genet. Genomics. 2020;47:713–717. doi: 10.1016/j.jgg.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Chemes H.E., Alvarez Sedo C. Tales of the tail and sperm head aches: changing concepts on the prognostic significance of sperm pathologies affecting the head, neck and tail. Asian J. Androl. 2012;14:14–23. doi: 10.1038/aja.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sha Y.W., Xu X., Mei L.B., Li P., Su Z.Y., He X.Q., Li L. A homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF) Gene. 2017;633:48–53. doi: 10.1016/j.gene.2017.08.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The WES datasets supporting the current study have not been deposited in a public repository because of privacy and ethical restrictions but are available from the corresponding authors on request.