

Abstract
Genetic factors and estrogen deficiency contribute to the development of osteoporosis. The single-nucleotide polymorphism (SNP) rs2887571 is predicted from genome-wide association studies (GWASs) to associate with osteoporosis but has had an unknown mechanism. Analysis of osteoblasts from 110 different individuals who underwent joint replacement revealed that the genotype of rs2887571 correlates with WNT5B expression. Analysis of our ChIP-sequencing data revealed that SNP rs2887571 overlaps with an estrogen receptor alpha (ERα) binding site. Here we show that 17β-estradiol (E2) suppresses WNT5B expression and further demonstrate the mechanism of ERα binding at the enhancer containing rs2887571 to suppress WNT5B expression differentially in each genotype. ERα interacts with NFATc1, which is predicted to bind directly at rs2887571. CRISPR-Cas9 and ChIP-qPCR experiments confirm differential regulation of WNT5B between each allele. Homozygous GG has a higher binding affinity for ERα than homozygous AA and results in greater suppression of WNT5B expression. Functionally, WNT5B represses alkaline phosphatase expression and activity, decreasing osteoblast differentiation and mineralization. Furthermore, WNT5B increases interleukin-6 expression and suppresses E2-induced expression of alkaline phosphatase during osteoblast differentiation. We show that WNT5B suppresses the differentiation of osteoblasts via receptor tyrosine kinase-like orphan receptor 1/2 (ROR1/2), which activates DVL2/3/RAC1/CDC42/JNK/SIN3A signaling and inhibits β-catenin activity. Together, our data provide mechanistic insight into how ERα and NFATc1 regulate the non-coding SNP rs2887571, as well as the function of WNT5B on osteoblasts, which could provide alternative therapeutic targets for osteoporosis.
Keywords: ERα, rs2887571, WNT5B, osteoblasts, SNP, estrogen, bone
Graphical abstract
Introduction
Osteoporosis is an age-related metabolic bone disorder characterized by bone mass depletion and microarchitectural deterioration of bone tissue, leading to bone fragility and susceptibility to bone fracture.1 The prototypical osteoporotic fractures are at the hip and spine, which subsequently lead to considerable mobility loss, mortality, and healthcare costs.2
Genetic factors contribute to the development of osteoporosis.3 A genome-wide meta-analysis identified 56 loci associated with bone mineral density (BMD) and the risk of fractures.4 Most of these single-nucleotide polymorphisms (SNPs) are in noncoding regions with no known function. Eight of the SNPs are near WNT signaling genes (WNT3, WNT4, WNT5B, WNT16, CTNNB1, SFRP4, LRP5, and AXIN1), highlighting the importance of WNT signaling in achieving peak bone mass.5
There are 19 evolutionarily conserved WNT ligands in both humans and mice. In the osteoblast lineage, several WNT ligands, including WNT7B and WNT10B, promote osteoblast differentiation through the canonical (β-catenin [CTNNB1]-dependent) signaling pathway, and knockout of these genes in mice leads to decreased BMD.6,7 Most of the eight genes near the implicated SNPs have been shown to have mechanistic roles in bone. Low-density lipoprotein receptor-related protein 5 (LRP5) is a known regulator of bone mineral density and is one of the receptors for the canonical pathway.8 Soluble frizzled-related protein 4 (SFRP4) also binds WNT ligands and modulates their activity.9 WNT16 signals through both the canonical and non-canonical pathways to regulate cortical bone thickness.10,11 In contrast, knockout of the non-canonical Wnt5b slightly increased BMD,12 indicating that WNT5B exhibits negative effects on bone and might promote the development of osteoporosis. However, the mechanism of WNT5B signaling in osteoblasts and the consequence of the SNP near WNT5B were unknown.
In addition to genetic factors, estrogen deficiency in women is a common risk factor for postmenopausal osteoporosis, as estrogens are osteoprotective regulators of bone metabolism by maintaining bone formation and inhibiting bone resorption in an estrogen receptor alpha (ERα)-dependent manner.13,14 ERα can be activated in either a 17β-estradiol (E2)-dependent or -independent mechanism that creates genomic and non-genomic actions. E2 activates ERα to translocate into the nucleus to bind with either an estrogen response element (ERE) or with other transcription factors at estrogen-responsive gene promoters or enhancers.15
To determine whether estrogen signaling is affected by any of the SNPs impacting BMD, we compared the 56 SNPs in the Estrada et al. meta-analysis4 with genome-wide ChIP sequencing for ERα in osteoblast-like cells. Only one of the 56 SNPs is in an ERα binding site: rs2887571, which is near WNT5B. Therefore, we hypothesized that SNP rs2887571 may alter ERα activity to regulate WNT5B expression. We also tested the effects of WNT5B on the osteoblastic lineage and demonstrate mechanistically that the non-canonical WNT5B signaling pathway suppresses ERα activities through its interaction with SIN3A.
Material and methods
Regional association analysis and SNP annotation
Fine mapping of loci 12p13.33 was analyzed by probabilistic identification of causal SNPs (PICS).16 Genome mapping and nucleotide sequence of the region were obtained from the UCSC genome browser.17 The transcription factor binding domains of SNP rs2887571 were annotated by PATCH public 1.0 Pattern Search for Transcription Factor Binding Sites program.
Reagents
17β-Estradiol (E2; Cat. # E2758), Phorbol 12-Myristate 13-Acetate (PMA; Cat. # P8139), and ionomycin calcium salt (Cat. # I3909) were purchased from Sigma-Aldrich. Recombinant mouse WNT5B (rWNT5B; Cat. # 3006-WN) was acquired from R&D Systems. The PPARγ agonist GW1929 hydrochloride (Cat. # 1664) was purchased from Tocris Bioscience. The specific primary antibodies that were used are listed in Table S1.
Primary osteoblast cells
The human study was approved by the Institutional Review Board at The University of Tennessee Health Science Center and all individuals provided informed written consent before participation. All animal work was approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center.
Primary human osteoblasts were isolated from the trabecular bone in the femoral or humeral heads of individuals who underwent total joint replacement surgery using an adaptation of the method previously described.18 The femoral heads were wrapped in sterile gauze with double plastic bags and then the bone was crushed into smaller pieces under sterile conditions. Bone cutters were further used to break the fragments into smaller pieces and separate the trabecular bone from the cortical shell. The bone chips were then transferred to 50 mL tubes containing PBS and washed until no hematopoietic marrow was visible. The bone chips were then incubated for 2 h at 37°C in DMEM (Corning) with 1 mg/mL collagenase IV (Sigma-Aldrich). The bone fragments were seeded into a 75-cm2 tissue culture flask containing DMEM supplemented with 10% FBS (Omega Scientific), 1% Penicillin-Streptomycin-L-Glutamine (Corning), and Normocin (InvivoGen). The cells were left untouched for 5 days in this media, after which the media was changed twice weekly until it reached 90% confluence.
Calvarial osteoblasts were isolated from 2-day-old CD1 mice by sequential collagenase digestion as previously described.19 Calvariae were incubated in MEM (Corning) with 1.0 mg/mL collagenase P (Roche) and 1.25% trypsin (Corning) at 37°C for 40 min. Then cells were washed in MEM and incubated in MEM with 1.0 mg/mL collagenase P and 1.25% trypsin for 1 h at 37°C. Collagenase digestion was stopped by the addition of MEM media containing 10% FBS. The cells from the second digest were obtained and allowed to proliferate in MEM media containing 10% FBS and 1% Penicillin-Streptomycin-L-Glutamine.
Bone marrow-derived mesenchymal stem cells (MSCs) were isolated from tibias and femurs of mice as previously described.20 Cells were flushed with MEM without FBS. Red blood cells were lysed with 3% acetic acid and nucleated cells were counted with a hemocytometer. Total bone marrow was seeded into mesenchymal stem cell media (MesenCult Basal Media, Stem-Cell Technologies Inc.) and left untouched for 5 days, after which the media was changed twice weekly.
For estrogenic activity assays, the media was replaced with phenol-free media containing 5% charcoal-dextran-treated fetal bovine serum (CDT-FBS; Omega Scientific) for 3 days before treatment with 10 nM E2.
Cell line culture
Human osteosarcoma U2OS-ERα cells were kindly provided by Dr. Thomas Spelsberg and were maintained as described.21 Briefly, cells were cultivated in DMEM/F-12 50/50 (Corning) supplemented with 10% FBS, 1% Penicillin-Streptomycin-L-Glutamine, 10 μg/mL Blasticidin S HCL (InvivoGen), and 200 μg/mL Zeocin (Invitrogen). ERα expression was induced by treatment with 100 ng/mL doxycycline (Dox.; Sigma-Aldrich) for 24 h after culture in phenol-red free media containing 5% CDT-FBS for 2 days. Cells were treated with 10 nM E2 or vehicle control (ethanol, EtOH). The cell line was verified each year by STR profiling and tested for mycoplasma.
Osteoblastic differentiation assay
Mouse calvarial osteoblasts were switched to osteogenic induction media (differentiation media, DM), containing 10% FBS, 1% Penicillin-Streptomycin-L-Glutamine, 50 μg/mL L-ascorbic acid 2-phosphate (Sigma-Aldrich), and 2 mM β-glycerophosphate (Sigma-Aldrich). The medium was changed after every 2–3 days. Differentiation was confirmed by quantitation of bone alkaline phosphatase and osteocalcin mRNA.
DNA isolation and genotyping of SNP rs2887571
Genomic DNA was isolated from primary human osteoblasts and U2OS-ERα cells using the DNeasy Blood and Tissue kit (QIAGEN) and genotyped for SNP rs2887571 using the rhAmp SNP Genotyping Assay (IDT).
RNA isolation and qPCR
Total RNA was isolated by TRIzol reagent (Invitrogen) and converted to cDNA using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) for qPCR according to the manufacturer’s instructions. Primers and probes are listed in Table S2. cDNA was quantified using SYBR Green Master Mix (Thermo Fisher Scientific). The qPCR cycling conditions for SYBR Green were initiated at 95°C for 10 min; followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, then detection of the melting curve. β-actin (ACTB or Actb) was used as the internal control for data analysis.
Protein extraction and immunoblotting
Total protein was lysed in EBC lysis buffer (50 mM Tris [pH 8], 120 mM NaCl, 0.5% Nonidet P-40) containing Complete Protease Inhibitor Cocktail (Roche Applied Science). Protein concentrations were quantified using the Bradford method. Aliquots of 10–40 μg protein per lane were loaded onto 5%–12% SDS-polyacrylamide gels and then transferred onto immunoblot polyvinylidene difluoride membrane (Biorad). The membranes were blocked with either 5% milk, 5% BSA, or 5% milk with 1% BSA in TBST at room temperature for 1 h and probed with a specific primary antibody (listed in Table S1) at 4°C overnight, followed by incubation with the secondary antibody conjugated with horseradish peroxide. The reactive bands were visualized using iBright Imaging Systems (ThermoFisher Scientific) and were normalized to β-actin (ACTB).
Immunohistochemistry (IHC)
6-week-old BALB/c mice were sham-operated or ovariectomized and then treated with 50 μg/kg body weight of E2 or sesame oil (as control) by intraperitoneal injection for 24 h.20 ERα (Esr1+/−) heterozygous mice were kindly provided by Dr. Pierre Chambon22 and bred to generate ERα knockout (Esr1−/−) and wild-type littermates. Tibial and femoral bones were collected, de-calcified, formalin-fixed, and embedded in paraffin. Paraffin-embedded samples were deparaffinized and antigen retrieval was performed in 90°C citrate buffer until the temperature reached 55°C. Then non-specific antigens were blocked with 5% normal goat serum and 2.5% bovine serum albumin (BSA) in PBS at pH 7.5 for 30 min at room temperature. A primary WNT5B antibody (PA5-35344) was applied at the dilution of 1:250 at 4°C overnight in a humidified chamber. Samples were incubated with appropriate biotinylated secondary antibodies and visualized by peroxidase/DAB solution (Dako) together with hematoxylin counterstaining.
Immunofluorescence (IF)
U2OS-ERα cells or mouse calvarial osteoblasts were fixed with 4% paraformaldehyde. The specific antibodies to non-phospho (active) β-catenin (Ser33/37/Thr42) (1:250), ROR1 (1:100), ROR2 (1:100), and/or WNT5B (1:250) were added overnight at 4°C. Appropriate secondary antibodies conjugated to a fluorophore were incubated for 1 h, followed by mounting with Vectashield Mounting Media with DAPI (H-1200, Vector Laboratories).
Chromatin immunoprecipitation (ChIP) and ChIP-reChIP
U2OS-ERα cells or mouse calvarial osteoblasts were cross-linked in 1% formaldehyde at 37°C for 8 min. Cells were lysed with lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCL) and sonicated using the Bioruptor Pico (Diagenode) for 30 s on/30 s off for 15 cycles. Chromatin was immunoprecipitated with the specific antibody (ERα, NFATc1, SIN3A, H3K27ac, or phospho-c-Fos) at 4°C overnight in dilution buffer (0.1% Triton-X, 0.5M EDTA, 5M NaCl, 1M Tris-HCL). The immune complex was then eluted from beads and reverse cross-linked by incubating in elution buffer (1% SDS, 0.1M NaHCO3) at 65°C overnight. The DNA fragments were purified by QIAquick PCR purification kit (QIAGEN) and used for qPCR using SYBR Green Master Mix. Primers are listed in Table S3. β-actin (ACTB) promoter, hemoglobin (HBB) promoter, or IgG were used as negative controls. For the ChIP-reChIP assay, chromatin was immunoprecipitated with either ERα antibodies, NFATc1 antibodies, or IgG, followed by elution with DTT. The eluate was incubated with NFATc1 antibodies.23
Co-immunoprecipitation (Co-IP)
Cells were lysed in IP lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 10% glycerol, 2 mM EDTA, 50 mM NaF) containing EDTA-free protease inhibitor cocktail (Roche Applied Science). Cell lysates were precleared for 30 min with protein A or protein G. The specific antibody (ERα, SIN3A, WNT5B, or IgG) was incubated with protein A or protein G magnetic beads for 15 min before adding cell lysate. Complex proteins were immunoprecipitated at 4°C overnight and then eluted with 6×SDS at 95°C for 7 min. The supernatants were transferred and analyzed with immunoblotting as described above.
CRISPR-Cas9-mediated gene editing of SNP rs2887571
CRISPR (cr)RNA targeting the SNP rs2887571 (5′-ATTAGAGGGCAAGGTTTCCT-3′; PAM: TGG) was designed by the Alt-R CRISPR HDR Design Tool (Integrated DNA Technologies). The trans-activating CRISPR (tracr)RNA and Cas9 nuclease protein NLS were purchased from Dharmacon. A donor template (5′-A∗GCAAATACCCCACTAATTAGAGGGCAAGGCTTCCTTGGAACTGTGCTGGCTTGGGCCGG∗A-3′) was designed. Asterisk indicates phosphorothioate linkages on the first two and last two nucleotides and the underline indicates the changed nucleotide.
U2OS-ERα cells were transfected with the ribonucleotide complex together with the donor template using DharmaFECT Duo Transfection Reagent (Dharmacon). After 48 h, cells were seeded individually into each well of a 96-well plate and allowed to reach confluence. Genomic DNA was isolated and the genotype of SNP rs2887571 was confirmed as described above.
Dual-luciferase reporter assays
Homozygous GG of SNP rs2887571 was amplified from human genomic DNA with the following primers: 5′-catggtaccGAGCCTGCTTGAAATTTGGCA-3′ and 5′-tacgctagcGTCAGGGAATGGATGGGACC-3′. The DNA fragments were digested with KpnI and Nhel and ligated into the pGL3-promoter-luciferase vector, which produced pGL3-rs2887571. U2OS-ERα cells were transfected with modified plasmid pGL3-rs2887571 and/or EGFPC1-huNFATc1EE-WT together with pRL-SV40 (Renilla) luciferase as an internal control in triplicate using FuGENE 6 Transfection Reagent (Promega). Cells then were treated with EtOH or E2 and after 24 h assayed for luciferase using the Dual-Glo Stop & Glo assay from Promega. EGFPC1-huNFATc1EE-WT was a gift from Jerry Crabtree (Addgene plasmid # 24219).24 U2OS-ERα cells were transfected Super8XTOPFlash (a β-catenin reporter) with pcDNA3-Wnt5B and/or pcDNA-Wnt10B-V5 together with pRenilla-SV40 luciferase. Super8XTOPFlash was a gift from Vitězslav Bryja.25 pcDNA3-Wnt5B (Addgene plasmid #3587526) and pcDNA3-Wnt10B-V5 (Addgene plasmid #3594026) were a gift from Marian Waterman.
Alkaline phosphatase activity assay for osteogenic differentiation
Osteoblasts were differentiated for 7 days and then fixed with 3.7% formaldehyde and stained for alkaline phosphatase using SIGMAFAST BCIP/NBT (Sigma-Aldrich). Cells were collected and split into two parts for alkaline phosphatase activity and total protein. The level of alkaline phosphatase protein activity was measured by SensoLyte pNPP Alkaline Phosphatase Assay Kit (AnaSpec) according to the manufacturer’s instruction. The level of alkaline phosphatase was compared to a standard curve and normalized to total protein.
Alizarin red assay for mineralization
Osteoblasts were differentiated for 2 weeks and then fixed in 50% ethanol for 15 min at 4°C, followed by 30 min incubation with 1% Alizarin Red S (wt./vol. with 0.1% ammonium hydroxide). The stain was then washed with distilled water, dried, and photographed. The amount of mineral content was measured by eluting the Alizarin red stain with 10% cetylpyridinium chloride and the optical density was measured at OD 570 nm.
Adipogenesis assay
MSCs were seeded into a 6-well plate at the density 2 × 106 cells/well and were cultivated in MesenCult Basal Media for 5 days then switched to adipogenic induction medium (MEM supplemented with 10% FBS, 1% Penicillin-Streptomycin-L-Glutamine, and 1 μM of the PPARγ agonist GW1929) with or without 50 ng/mL rWNT5B. Adipocyte differentiation was analyzed by Oil Red O (ORO) staining.27 Lipids were stained with 60% ORO for 20 min at room temperature. The pictures were taken and quantified by ImageJ 1.53a.
CFU-F and CFU-OB
Bone marrow cells were seeded into 6-well plates at a density of 1 × 106 cells/well and were cultivated in MesenCult Basal Media for 5 days, and then were treated with or without 50 ng/mL rWNT5B for 8 days. Undifferentiated cells were fixed and stained with Giemsa (Ricca Chemical Company) and colonies were counted for CFU-F.
Bone marrow cells were plated into 6-well plates at the density of 1 × 106 cells/well and were cultivated in MesenCult Basal Media for 5 days then switched to DM with or without 50 ng/mL rWNT5B for 12 days. Osteoblasts were fixed and stained with Alizarin red as described above.
Knockdown of Wnt5b
Lentivirus expressing a non-mammalian shRNA control (SHC002V) or two different shRNA from The RNAi Consortium (TRC) in pLKO vector for knockdown of mouse Wnt5b (Clone ID TRCN0000226380) was purchased from Sigma Aldrich. Calvarial osteoblasts were plated into 6-well and infected with lentivirus at an MOI of 2.7 in MEM with 8 μg/mL polybrene per well. The plates were centrifuged at 1,400 × g at 30°C for 45 min and left undisturbed for 24 h after which the cells were washed with PBS and treated with or without 50 ng/mL rWNT5B together with 10 μg/mL puromycin (MilliporeSigma). The knockdown of Wnt5b was confirmed by qPCR and immunoblotting.
Overexpression of Sin3a
U2OS-ERα cells were transfected in triplicate with pCS2+MT-mSin3A (Addgene plasmid #30452) together with pGL3-rs2887571, EGFPC1-huNFATc1EE-WT, and pRL-SV40 (Renilla), as an internal control, using FuGENE 6 Transfection Reagent (Promega). The cells were treated with (or without) Dox. to express ERα and with or without 10 nM E2. After 24 h, the luciferase levels were detected by the Dual-Glo Stop & Glo assay from Promega.
Knockdown of SIN3A
ON-TARGETplus Human SIN3A small interfering RNA (siRNA) set of 4 (Dharmacon) was transfected into U2OS-ERα cells using Lipofectamine RNAiMax Reagent (Invitrogen) at 40 nM.28 ON-TARGETplus Non-targeting siRNA (Dharmacon) was used as the negative control.
Giemsa staining
Cells from alkaline phosphatase activity, Alizarin red, and Oil Red O staining were counterstained with Giemsa Stain (Ricca Chemical Company) at the dilution of 1:10 for 30 min and then washed with tap water. The pictures were taken and the number of cells was quantified by ImageJ v. 1.53a.
Linkage Disequilibrium (LD)
HaploReg v4.1 was used to find SNPs in LD with rs2887571 (r2 ≥ 0.8).29
Statistical analysis
All experiments represent biological or both biological and experimental triplicates. Error bars represent the mean with the SD. The normal distribution of data was checked before testing statistics with Student’s t test. p value ≤ 0.05 is considered statistically significant. Analyses were performed with Prism 9 Software. Protein quantification was performed using ImageJ v.1.53a.
As the association between polymorphism SNP rs2887571 and expression levels is not a normal distribution, the graph is shown as the median with the interquartile range and analyzed by Mann-Whitney U-test using SPSS v.27.
Results
SNP rs2887571 is in an ERα binding site
A genome-wide study from Estrada et al. identified 56 SNPs associated with BMD.4 We sought to determine if any of the SNPs associated with BMD might be linked to estrogen signaling, as decreased estrogen signaling leads to osteoporosis. To this end, we analyzed ERα binding sites that were identified by ChIP-sequencing (ChIP-seq) in osteoblast-like U2OS-ERα cells.30 Of the 56 SNPs, only rs2887571 was located within an ERα binding site. As a ChIP-seq signal is larger than the actual binding site, the region around rs2887571 was analyzed by PATCH™ transcription factor binding site software to predict the actual binding site. There is an ERE half-site adjacent to rs2887571 and rs2887571 is predicted to be in a Nuclear Factor of Activated T cells (NFAT) motif (Figure 1A).
Figure 1.

SNP rs2887571 correlates with WNT5B expression in primary human osteoblasts
(A) Nucleotide sequence near SNP rs2887571 with predicted NFAT and ERα binding motifs.
(B) Diagram representing the location between SNP rs2887571 and nearby genes in chromosome 12.
(C–G) Expression of (C) ERC1, (D) LINC00942, (E) LOC107984507, (F) FBXL14, and (G) WNT5B were measured by qPCR and compared with its genotype for SNP rs2887571 in human osteoblasts (AA = 52, AG = 49, and GG = 9). Bars indicate median with interquartile range. Mann-Whitney U-test: ∗∗p = 0.0016.
SNP rs2887571 is associated with WNT5B expression in primary human osteoblasts
SNP rs2887571 is located in a non-coding intergenic region on chromosome 12 near ELKS/RAB6-Interacting/CAST Family Member 1 (ERC1), Long Intergenic Non-Protein Coding RNA 942 (LINC00942), the uncharacterized long non-coding RNA LOC107984507, F-Box and Leucine-Rich Repeat Protein 14 (FBXL14), and WNT Family Member 5B (WNT5B; Figure 1B). To determine whether the SNP correlates with a nearby gene’s expression in osteoblasts, we analyzed DNA and RNA from 110 different individuals (age 33–85 who had undergone joint replacement for degenerative joint disease). Genotyping identified 52 homozygous AA, 49 heterozygous AG, and 9 homozygous GG. The minor allele frequency (MAF) is 0.304 from our mixed population, which is similar to the European MAF of 0.268 from the 1000 Genomes Project Phase 3.31 Among the five nearby genes (Figures 1C–1G), only the level of WNT5B expression was significantly different between homozygous AA and homozygous GG at rs2887571 (∗∗p = 0.0016; Figure 1G).
E2 suppresses WNT5B expression via ERα in osteoblasts
Because SNP rs2887571 shows overlap with an ERα-binding site30 and correlates with WNT5B expression, we hypothesized that E2 might regulate WNT5B through ERα at SNP rs2887571. To test the role of E2 and ERα on WNT5B/Wnt5b expression in osteoblasts, three different osteoblast systems were analyzed: (1) primary human osteoblasts (hOBs), (2) U2OS-ERα cells, and (3) mouse calvarial osteoblasts (mOBs). U2OS-ERα cells are an osteosarcoma cell line that has an inducible expression of ERα. Overexpression of ERα in U2OS cells leads to an osteoblast phenotype that predicts normal osteoblast biology, with the ease of a cell line.19,30 The mRNA and protein levels of WNT5B were decreased by 10 nM E2 in all three osteoblast systems compared to the vehicle (ethanol, EtOH) controls (Figures 2A–2E). The expression levels of alkaline phosphatase (ALPL) were used as a positive control for estrogen activity and differentiation (Figures S1A–S1C). Wnt5b mRNA increased during osteoblast differentiation and E2 suppressed the level of Wnt5b expression only at day 7 (Figure 2E). In contrast, Alpl was upregulated by E2 at the same time point (Figure S1C).
Figure 2.

E2 suppresses WNT5B expression via ERα
(A–D) Human osteoblasts (hOBs) and osteoblast-like U2OS-ERα cells were treated with EtOH or 10 nM E2 for 4 h then levels of (A, C) WNT5B mRNA expression were detected by qPCR. Immunoblotting analysis of WNT5B protein expression after treatment with EtOH or 10 nM E2 for 24 h and normalized to β-actin (BA) and quantified by ImageJ in (B) hOBs and (D) U2OS-ERα cells. Bar = mean with SEM. n = 3, Student’s t test: ∗p<0.05. Three different biological replicates of hOBs and U2OS-ERα cells were tested and a representative picture from one biological replicate is presented.
(E) Mouse osteoblasts (mOBs) were differentiated for 3 and 7 days then treated with EtOH or 10 nM E2 for 24 h, and levels of Wnt5b mRNA expression were detected by qPCR. The level of WNT5B was detected by IHC in mice from sham-treated, ovariectomized (OVX), and OVX then treated with E2.
(F) The number of WNT5B-positive osteoblasts were counted and divided by the total osteoblasts from three different mice in each group. Bar = mean with SEM. n = 3, Student’s t test: ∗∗∗∗p < 0.0001, ∗∗p < 0.01, ∗p < 0.05.
(G) The regions of interest (red square) are shown below at 40× magnification. Black scale bar = 200 μM, red scale bar = 20 μM, red arrow = osteoblast, and asterisk = osteoid.
Next, we analyzed the effect of E2 on WNT5B in bone, in vivo. Mice were ovariectomized (OVX) or sham-operated. They were then treated with vehicle or E2 for 24 h. Immunohistochemistry was performed and the level of WNT5B in osteoblasts, indicated by red arrows, was increased in the OVX group compared to the sham group and was decreased in the OVX+E2 group compared to the OVX group (∗∗∗∗p ≤ 0.0001; ∗p ≤ 0.05; Figures 2F and 2G), demonstrating that E2 suppresses WNT5B in vivo. To confirm this effect was dependent on ERα, we analyzed bones from WT mice and ERα knockout (Esr1−/−) mice. WNT5B is elevated in osteoblasts and bone marrow cells of Esr1−/− mice compared to WT mice (∗∗∗∗p ≤ 0.0001; Figure S1D). Bone marrow-derived MSCs were isolated from each genotype and were differentiated to osteoblasts, before detecting the mRNA levels of Wnt5b, Alpl, and Esr1 (Figures S1E–S1G). Wnt5b was increased in Esr1−/− compared to WT mice (∗p ≤ 0.05; Figure S1E), indicating that ERα is required to suppress the level of WNT5B.
ERα functions as a transcriptional repressor at SNP rs2887571 via SIN3A recruitment
As less than 5% of ERα binding sites are at proximal promoters,19,32 we predicted that SNP rs2887571 was within a WNT5B enhancer and further analyzed the molecular mechanism of the E2-ERα complex at the SNP. E2 activates ERα to bind at targeted gene promoters or enhancers and then recruits other transcriptional co-regulators to control gene expression.19,33
U2OS-ERα cells were treated with or without E2 (10 nM) for 45 min, which is a time point for optimal recruitment of ERα,34 and ChIP-qPCR was performed using an antibody to ERα or IgG. There is a basal level of ERα binding in the absence of E2 and the binding of ERα was significantly increased at SNP rs2887571 in the presence of E2 (∗∗p ≤ 0.01; Figure 3A), compared to the β-actin (ACTB) promoter, which is a negative control region. Although SNP rs35223785, which is in LD with rs2887571, has also been identified as a lead BMD GWAS variant,35, 36, 37 we cannot detect the binding of ERα at this variant compared to IgG (Figure S2A).
Figure 3.

ERα and NFATc1 interact and bind at SNP rs2887571
(A–D) U2OS-ERα cells were treated with or without 10 nM E2 for 45 min, then ChIP was performed using an antibody to (A) ERα, (B) H3K27ac, (C) SIN3A, or (D) NFATc1. qPCR was performed with primers for SNP rs2887571 in comparison with β-actin (ACTB) promoter or hemoglobin (HBB) promoter.
(E) U2OS-ERα cells were treated with 10 nM E2, then ChIP-reChIP was performed with antibodies to ERα, NFATc1 and/or IgG, and primers for SNP rs2887571.
(F) Human genomic DNA around SNP rs2887571 was amplified and inserted into the pGL3 vector, then U2OS-ERα cells were transfected with pGL3-rs2887571, in the presence or absence of EGFPC1-huNFATc1EE-WT. Cells were treated with Dox. to induce ERα. 24 h later, cells were treated with EtOH or 10 nM E2 for an additional 24 h. Relative luciferase activity was measured using dual-luciferase reporter assays.
(G) U2OS-ERα cells were transfected with pGL3-rs2887571 and EGFPC1-huNFATc1EE-WT, together with or without pCS2+MT-mSin3A, before being induced or uninduced to express ERα. Cells were treated with EtOH or 10 nM E2 and the relative luciferase activity was detected at 24 h. Luciferase activity was normalized with pRL-SV40 (Renilla).
(H and I) U2OS-ERα cells were transfected with siRNA directed against SIN3A mRNA, before being treated with or without 10 nM E2. The mRNA levels of (H) SIN3A and (I) WNT5B were detected by qPCR. n = 3, Student’s t test: ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05.
Histone 3 lysine 27 acetylation (H3K27ac) is a chromatin modification that marks open, active, or poised enhancers and promoters, compared with inactive enhancer elements that are marked by H3K27 trimethylation (H3K27me3).38,39 To analyze the chromatin status near SNP rs2887571, ChIP-qPCR was performed with an antibody to H3K27ac or IgG on E2-treated and vehicle control-treated cells. rs2887571 is marked by H3K27ac, indicating that it is an active enhancer. The ACTB promoter is a positive control for H3K27ac. H3K27ac was significantly decreased in the presence of E2 at SNP rs2887571 (∗p ≤ 0.05; Figure 3B) and correlates with a decrease in WNT5B expression after E2 treatment.
We next investigated the mechanism of histone deacetylation at SNP rs2887571 in response to E2. SIN3A, a member of the SIN3 transcription regulator family, has been shown to interact with ERα at the ESR1 promoter and to repress the expression of ESR1 in the presence of E2.40 SIN3 is a scaffold for histone deacetylases (HDACs) and acts as a co-regulator complex for genes involved in mammalian development and diseases. SIN3 lacks a DNA binding motif and it requires other transcription factors for DNA binding.41 Therefore, U2OS-ERα cells were treated in the presence or absence of 10 nM E2, and ChIP-qPCR was performed using an antibody to SIN3A or normal IgG. The binding of SIN3A was significantly increased at SNP rs2887571 in the presence of E2 (∗p ≤ 0.05; Figure 3C). These results indicate that the E2-ERα complex is a repressor of WNT5B via SIN3A at the enhancer containing SNP rs2887571.
SNP rs2887571 is also predicted to be part of an NFAT binding site42 (Figure 1A). The NFAT transcription factor family consists of five members (NFAT1-5), which bind to the highly conserved DNA binding domain, 5′-GGAAA-3′. NFATs show a weak DNA binding capacity and frequently interact with other transcription factors, including ERα.43 As NFAT signaling regulates bone mass by increasing osteoblast proliferation,44 we hypothesized that ERα and NFATs might work together to suppress WNT5B. First, we identified that NFATC1 significantly had the highest expression among other NFATs in human osteoblasts and U2OS-ERα cells (Figures S2B–S2C), suggesting the specific function of NFATc1 in osteoblasts. Indeed, NFATc1 binds at SNP rs2887571 by ChIP-qPCR assay; however, the binding of NFATc1 at rs2887571 was not changed by the presence or absence of E2 in U2OS-ERα cells (Figure 3D). The ACTB promoter and the hemoglobin (HBB) promoter were used as negative controls for NFATc1 binding sites (∗∗p ≤ 0.01; Figure 3D). In addition, we found that ERα and NFATc1 bound at rs2887571 at the same time and place in the presence of E2, as determined by ChIP-reChIP assays. ChIP with an antibody to ERα, followed by reChIP with an antibody to NFATc1 showed enrichment over the normal IgG control (∗∗p ≤ 0.01; Figure 3E).
To further confirm that ERα and NFATc1 interact, co-immunoprecipitations were performed in U2OS-ERα cells. 10 nM E2 was used to activate ERα, and the combination of 2.5 nM PMA and 1 μM ionomycin was used to activate NFATc1.45 The combination of E2, PMA and ionomycin (“EPI,” E2, PMA, and ionomycin) was used to simultaneously activate both ERα and NFATc1. Then, immunoprecipitations were performed with an antibody to ERα or IgG before blotting with a specific antibody to NFATc1. ERα and NFATc1 interact in U2OS-ERα cells (lane 1 Figure S2D), and the interaction increased after EPI treatment (lane 2 Figure S2D).
We further elucidated the effect of ERα and NFATc1 on SNP rs2887571 in luciferase reporter assays. A 401-base pair region of DNA around rs2887571 was cloned into the pGL3-promoter vector (pGL3-rs2887571), which was transfected into U2OS-ERα cells, in the presence of Dox. to induce ERα and in the presence or absence of an NFATC1-expressing plasmid. Then cells were treated with or without 10 nM E2 before detecting luciferase activity. NFATc1 increased luciferase activity by over 4-fold compared to the empty vector and E2 significantly suppressed this activity in the presence of NFATc1 (∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001; Figure 3F), suggesting differential functions at the SNP.
Because the reporter assay suggests that NFATc1 may activate WNT5B transcription, we tested the effect of endogenous NFATc1 on WNT5B protein expression. U2OS-ERα cells were treated with either PBS/DMSO (Ctrl), E2 (10 nM), PMA and ionomycin (2.5 nM and 1 μM, respectively), or the combination of all three (EPI). After 24 h, WNT5B was detected by immunoblotting. Quantification of WNT5B bands (and normalization to β-actin) indicate that WNT5B was elevated after treatment with PMA and ionomycin. In contrast, WNT5B was suppressed by the treatment of E2 and EPI compared to the control treatment (Figure S2E), suggesting that ERα interacts with NFATc1 to repress WNT5B protein expression.
We further evaluated the inhibitory effect of ERα via recruitment of SIN3A. U2OS-ERα cells were transfected with pGL3-rs2887571, NFATC1, Sin3a (or a control plasmid), and pRL-SV40 (Renilla) luciferase. Then, cells were induced to express ERα with Dox. (or uninduced without Dox.) before being treated with or without 10 nM E2 for 24 h. The presence of SIN3A suppressed the levels of luciferase (∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001; Figure 3G). Next, we knocked down SIN3A in U2OS-ERα cells using siRNA directed against SIN3A mRNA. The level of SIN3A mRNA was reduced by more than 2-fold after knockdown (∗∗p ≤ 0.01, Figure 3H). E2 has been shown to suppress SIN3A40 and this is confirmed in Figure 3H (∗∗p ≤ 0.01). Loss of SIN3A did not affect the endogenous level of WNT5B but abolished the inhibitory effect of E2 on WNT5B (Figure 3I).
Taken together, ERα and NFATc1 interact and bind at the WNT5B enhancer containing SNP rs2887571; however, their effects are opposite. NFATc1 induces WNT5B expression, while ERα suppresses it by recruiting SIN3A to deacetylate H3K27.
SNP rs2887571 shows allelic specificities to alter WNT5B levels
Linkage disequilibrium (LD) is the correlation of two alleles that occur together because of their proximity on a chromosome. There are 13 other SNPs that are in LD with rs2887571 and could account for the decreased BMD associated with it. Therefore, to test for LD and verify the specificity of SNP rs2887571 on WNT5B expression, we modified the homozygous AA allele to GG allele in U2OS-ERα cells by using CRISPR-Cas9 technology and homologous recombination with a donor template containing the GG allele. We hypothesized that the cells with the GG allele would express less WNT5B if it is the causative allele.
Immunoblots for WNT5B using protein from parental U2OS-ERα cells (AA) and the CRISPR-modified U2OS-ERα cells (GG) demonstrate that the expression of WNT5B is two times higher in cells with AA at rs2887571 (∗p ≤ 0.05; Figure 4A). The lower levels of WNT5B expression in U2OS-ERα with the GG allele are consistent with the data previously shown in human primary osteoblasts with different genotypes (∗∗p = 0.0016; Figure 1G).
Figure 4.

SNP rs2887571 has allelic specificities to alter WNT5B expression
(A) U2OS-ERα cells with either homozygous AA and GG at SNP rs2887571 were lysed and immunoblotting was performed with an antibody to WNT5B. The level of protein was normalized with β-actin and quantified by ImageJ. Bar = mean with SEM. n = 3, Student’s t test: ∗p < 0.05.
(B–D) Three different biological replicates of U2OS-ERα cells were tested and a representative picture from one biological replicate is presented. ChIP was performed using an antibody to (B) ERα, (C) SIN3A and (D) NFATc1, and qPCR was done with primers for SNP rs2887571 in comparison with the ACTB promoter. n = 3, Student’s t test: ∗∗p < 0.01, ∗p < 0.05.
Next, we further investigated the molecular mechanisms and specificity by performing ChIP for ERα at each allele. We hypothesized that allele G might have more ERα binding than with allele A, which would result in cells with homozygous GG expressing lower WNT5B than cells with homozygous AA (as previously shown in human primary osteoblasts; Figure 1G). U2OS-ERα cells from both genotypes were treated with or without E2 (10 nM) and ChIP-qPCR was performed using an antibody to ERα. E2 significantly increased the binding of ERα in both genotypes, but homozygous GG significantly had more binding than homozygous AA (∗p ≤ 0.05; Figure 4B), suggesting that increased ERα binding in homozygous GG correlates with lower WNT5B expression due to ERα repression of WNT5B.
We also performed ChIP-qPCR using an antibody to SIN3A in cells with each genotype. E2 significantly elevated the binding of SIN3A in homozygous AA; however, there was no change of the binding in homozygous GG with E2 (Figure 4C). Notably, the endogenous binding levels of SIN3A were significantly higher in homozygous GG than those in homozygous AA (∗∗p ≤ 0.01; Figure 4C). The binding of NFATc1 was also different in each genotype. E2 did not affect the binding at homozygous AA but significantly decreased the binding in homozygous GG (∗p ≤ 0.05; Figure 4D). The endogenous binding levels of NFATc1 by ChIP-qPCR were also significantly higher in homozygous GG (∗p ≤ 0.05; Figure 4D).
In summary, SNP rs2887571 showed allelic specificity on WNT5B expression. Homozygous GG had increased binding of ERα, SIN3A, and NFATc1 compared to homozygous AA, which resulted in lower WNT5B levels in homozygous GG. These results also demonstrate that rs2887571 is the causative allele that leads to differential expression of WNT5B protein levels.
WNT5B inhibits osteoblast differentiation and promotes adipogenesis
The mechanism by which WNT5B affects osteoblast differentiation and/or how it regulates gene expression is lacking in the literature. To address this lack of knowledge, we analyzed calvarial osteoblasts and bone marrow-derived mesenchymal stem cells (MSCs) to determine the effects of WNT5B on (1) primary osteoblast differentiation, (2) adipocyte differentiation, and (3) MSCs.
We first determined the expression of WNT5B in calvarial osteoblasts to determine its expression pattern and possible targets. WNT5B mRNA and protein levels increase during osteoblast differentiation by day 7 and through day 18, as demonstrated by both immunoblotting and publicly available RNA-sequencing data (GEO: GSE5446146) (Figures S3A and S3B).
Next, we tested the dose-dependent response to recombinant mouse WNT5B protein (rWNT5B) in mouse calvarial osteoblasts at three different time points of differentiation (day 0 [early], 7 [middle], and 14 [late]). A 24 h treatment of 50 ng/mL rWNT5B did not affect Sp7 (osterix, an early marker of osteoblast differentiation) at any time point (Figure 5A) but significantly decreased Alpl (alkaline phosphatase, a middle marker of osteoblast differentiation) and Bglap (bone gamma-carboxyglutamate protein, also known as osteocalcin, a late marker of osteoblast differentiation) at day 7 and day 14 of differentiation (Figures 5B and 5C). The effects of WNT5B on both osteoblast differentiation and mineralization were confirmed by alkaline phosphatase and Alizarin red staining, respectively, and WNT5B significantly inhibited both differentiation and mineralization (∗∗∗p ≤ 0.001; Figures 5D and 5E). To test whether the inhibitory effect of WNT5B on osteoblast differentiation and mineralization is due to apoptosis, cells from alkaline phosphatase (day 7) and Alizarin red (day14) staining were stained with Giemsa and the number of cells was counted. WNT5B increased the number of cells at day 7 and day 14 compared to the control group (∗∗p ≤ 0.01; Figures S3C and S3D), suggesting that WNT5B inhibits osteoblast differentiation while maintaining cell survival.
Figure 5.

WNT5B suppresses osteoblast differentiation and promotes adipogenesis
(A–C) mOBs were isolated from calvaria then treated with various doses of rWNT5B at day 0 (D0), day 7 after differentiation (D7) or day 14 after differentiation (D14) for 24 h. The levels of (A) Sp7, (B) Alpl, and (C) Bglap were detected by qPCR.
(D) mOBs were treated with or without 50 ng/mL rWNT5B and stained for alkaline phosphatase (ALP) and Alizarin Red.
(E) The levels of Alizarin Red in Ctrl and WNT5B groups were quantified at O.D. 570 nm and shown as % of the control.
(F) mOBs were infected with lentivirus containing sh-Ctrl or sh-Wnt5b, before collecting mRNA or protein. The levels of WNT5B mRNA and protein were normalized with β-actin and quantified by ImageJ.
(G and H) Cells were differentiated for 7 days in the presence or absence of 50 ng/mL rWNT5B, then (G) expression of Bglap was detected by qPCR and (H) alkaline phosphatase (ALP) activity was detected at O.D. 405 nm then was normalized with total protein.
(I) Bone marrow-derived MSCs were treated with or without 50 ng/mL rWNT5B in osteogenic differentiation media for 12 days and stained with Alizarin Red. CFU-OB was reported as the number of colonies.
(J) Bone marrow-derived MSCs were treated with or without 50 ng/mL rWNT5B in adipogenic differentiation medium for 14 days and stained with Oil Red O.
(K) The levels of Oil Red O between Ctrl and WNT5B groups were quantified as % Area by ImageJ.
(L and M) Expression of (L) Pparg and (M) Adipoq in MSC-derived adipocytes treated with or without rWNT5B for 24 h were detected by qPCR. Three different biological replicates were obtained and a representative image or graph from one replicate is presented. n = 3, Student’s t test: ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05.
We next performed a knockdown of Wnt5b in primary murine calvarial osteoblasts to determine the effects of WNT5B on osteoblast differentiation. Wnt5b can be successfully knocked down with lentivirally-expressed short hairpins directed at Wnt5b mRNA. The level of Wnt5b mRNA and protein was reduced by more than 2-fold after knockdown (Figure 5F). Loss of Wnt5b significantly increased the level of Bglap expression compared to the control group, while the addition of 50 ng/mL rWNT5B for 7 days showed the specificity of Wnt5b knockdown and rescued the knockdown (Figure 5G). Furthermore, the level of alkaline phosphatase activity by colorimetric assay supported the negative function of WNT5B on differentiation and the addition of rWNT5B reverses alkaline phosphatase activity in the silenced cells (Figure 5H). Loss of Wnt5b did not affect Sp7 nor Runx2, which are earlier markers of differentiation, thus defining the time of WNT5B action in calvarial osteoblast differentiation (Figures S3E and S3F).
WNT5B also decreased the differentiation of osteoblasts from bone marrow MSCs. The number of CFU-OB was decreased when bone marrow MSCs were treated with 50 ng/mL rWNT5B in osteoblastogenic differentiation media for 12 days compared to the control-treated group (∗p ≤ 0.05; Figure 5I).
We further investigated the effect of WNT5B on MSC-derived adipogenesis. WNT5B has been shown to increase adipogenesis in the 3T3-L1 system,47, 48, 49 but this has not been shown in primary cells. rWNT5B at 50 ng/mL significantly increased the number of adipocytes differentiated in the presence of the PPARγ agonist GW1929 (∗∗∗p ≤ 0.001; Figures 5J and 5K) but did not affect the total cell number, as detected by Giemsa staining (Figure S3G). Mechanistically, rWNT5B had little effect on the expression level of the adipogenic master transcription factor Pparg (PPARγ), when compared to untreated levels at D7 (∗p ≤ 0.05; Figure 5L). More significantly, WNT5B elevated the expression level of other adipocyte markers such as Adipoq (adiponectin), Plin1 (perilipin 1), and Cebpa (CEBPα) (Figures 5M, S3H, and S3I).
Based on the above, we assayed the role of WNT5B on MSCs, the progenitors of osteoblasts and adipocytes, using the colony-forming unit-fibroblast (CFU-F) assay, which is commonly used to assess the presence of bone marrow-derived MSCs.50,51 We treated MSCs with or without 50 ng/mL rWNT5B in mesenchymal stem cell media for 8 days. WNT5B significantly increased the number of CFU-F and their diameter, compared to the control group (∗p ≤ 0.05; Figure S3J). We determined that the larger size was caused by increased proliferation due to elevated expression levels of several cell cycle genes (Ccne1, Ccne2, Ccna2, Ccnb1, and Ccnb2 but not Ccnd1) (Figure S3K). More importantly, the rWNT5B exposed group had an increased expression level of the stem cell markers Nes (nestin) and Nanog (∗p ≤ 0.05; Figures S3L and S3M) when compared to the control.
Taken together, WNT5B did not affect the early osteogenic differentiation genes but suppressed the middle and late osteogenic differentiation genes, resulting in inhibition of differentiation and mineralization. Moreover, WNT5B increased the number of CFU-F, promoted adipogenic differentiation, and suppressed osteoblast differentiation. Therefore, WNT5B has two roles, one in MSCs promoting “stemness” and one in osteoblasts, blocking differentiation.
WNT5B activates the WNT non-canonical signaling pathway via ROR1/2 and inhibits β-catenin activity in osteoblasts
To gain mechanistic insights and identify the signaling transduction pathway mediated by WNT5B in osteoblasts, we first transfected U2OS-ERα cells with a WNT reporter (TOP-Flash), together with either pcDNA3-WNT5B, pcDNA3-WNT10B, pcDNA3-WNT5B with pcDNA3-WNT10B, or vector alone.52,53 The addition of the canonical WNT ligand WNT10B increased TOP-Flash activity by 5.4-fold. In contrast, WNT5B significantly reduced basal levels of canonical WNT signaling compared to the vector control (∗p ≤ 0.05; Figure 6A). More importantly, WNT5B also inhibited WNT10B-induced β-catenin activity significantly (∗∗p ≤ 0.01; Figure 6A).
Figure 6.

WNT5B activates WNT-PCP signaling pathway via ROR1/2 and inhibits β-catenin activity in osteoblasts
(A) U2OS-ERα cells were transfected with Super8XTOPFlash (a luciferase reporter for β-catenin activity) together with empty vector (pcDNA3), WNT5B, WNT10B, or WNT5B with WNT10B vectors. After 48 h, relative luciferase activity was measured using dual-luciferase reporter assays. Firefly luciferase activity was normalized to pRL-SV40 (Renilla).
(B) mOBs were differentiated for 7 days before treatment with or without 50 ng/mL rWNT5B for 30 min and the levels of active β-catenin (ABC) was detected by immunofluorescence. Scale bar = 10 μM. Three different mOBs were tested in biological replicates and a representative image from one replicate is presented.
(C) Differentiated mOBs at D7 were treated with 50 ng/mL rWNT5B and total cell lysate was collected at different time points before detecting the level of phospho-β-catenin (Ser552), ABC, total β-catenin, and β-actin by immunoblotting.
(D and E) mOBs were differentiated for 7 days before treatment with or without 50 ng/mL rWNT5B for 30 min and localization of WNT5B with (D) ROR1 or (E) ROR2 were detected by immunofluorescence. Scale bar = 10 μM.
(F and G) mOBs were differentiated for 7 days and treated with 50 ng/mL rWNT5B, and protein was collected at 0, 15, 30, and 60 min. The levels of downstream effectors were detected with the indicated antibodies.
(H–J) mOBs (H), MSCs (I), and MSC-derived adipocytes (J) were treated with or without 50 ng/mL rWNT5B for 24 h and qPCR was performed for Il6 mRNA.
(K) mOBs were treated with 50 ng/mL rWNT5B for 1 h and then ChIP was performed using a specific antibody to phospho-c-Fos (Ser32) and qPCR using primers to mouse negative control binding site and the Il6 promoter. n = 3, Student’s t test: ∗∗∗∗p < 0.0001, ∗∗p < 0.01, ∗p < 0.05.
To determine whether WNT5B affects the activity of β-catenin in normal osteoblasts, primary murine calvarial osteoblasts were differentiated for 7 days before treatment with PBS (Ctrl) or 50 ng/mL rWNT5B for 30 min. Immunofluorescence was performed with an antibody to non-phosphorylated (Ser33/37/Thr41), active β-catenin, often referred to as ABC. rWNT5B suppressed the endogenous levels of ABC (Figure 6B). rWNT5B decreased the level of phospho-β-catenin (Ser552) and ABC compared to total β-catenin by immunoblotting (Figure 6C). Total β-catenin protein levels are unchanged compared to β-actin (Figure 6C). Therefore, rWNT5B exposure represses canonical WNT10B/β-catenin-mediated gene transcription.
We further detected the expression level of known non-canonical WNT co-receptors (RYK, ROR1, and ROR2)54 between undifferentiated (at D0) and differentiated (at D7) calvarial osteoblasts. The expression level of ROR1 and ROR2 significantly increased during osteogenesis in both mRNA and protein levels (Figures S4A and S4B), which is similar to the expression pattern of WNT5B (Figures 2E, S3A, and S3B). In contrast, both RYK mRNA and protein levels are unchanged at D7 of differentiation, suggesting that WNT5B signal transduction may be mediated via ROR1/2. Immunofluorescence showed that the co-localization of WNT5B with ROR1 or ROR2 increased in the presence of rWNT5B (50 ng/mL) in mouse osteoblasts (Figures 6D and 6E). To test whether WNT5B physically interacts with either ROR1/2, we used mouse osteoblasts that were differentiated for 7 days and were treated with rWNT5B for 15 min, then co-immunoprecipitations were performed with an antibody to WNT5B or IgG before blotting with a specific antibody to ROR1 or ROR2. WNT5B showed an interaction with ROR2 but not ROR1 (Figure S4C). This result shows that endogenous WNT5B interacts with ROR2.
Next, we investigated the downstream signal transduction effectors of WNT5B. Mouse osteoblasts were differentiated for 7 days before treatment with 50 ng/mL rWNT5B at different time points (0, 15, 30, and 60 min). WNT5B increased the level of dishevelled proteins (DVL) 2 and DVL3 but not DVL1 (Figure 6F). It is often presumed that the higher molecular weight band is an indication that phosphorylation has taken place, and as such, phosphorylated DVL2 and DVL3 are also observed after treatment with WNT5B, suggesting signal transduction has been initiated through these proteins. The rWNT5B signal also activated RAC1/CDC42, phospho-JNK (Thr183/Tyr185), and induced phosphorylation of c-Fos (Ser32) at 15 to 60 min (Figure 6G). In contrast, WNT5B suppressed ROCK2 at 60 min and did not affect the downstream NFATc1. Moreover, c-Jun and phospho-c-Jun (Ser63) were significantly downregulated at 60 min, relative to basal levels (Figure 6G).
c-Fos is a transcription factor that increases the level of the proinflammatory cytokine interleukin 6 (IL6) among other genes.55 IL-6 induces bone loss by inhibiting bone formation56 and increasing bone resorption.57 Moreover, WNT5B was shown to upregulate IL6 in synovial MSCs from individuals with osteoarthritis58 and to correlate with IL6 in people with septic shock.59 We hypothesized that IL6 is a target gene of WNT5B via the activation of c-Fos. We found that WNT5B significantly increased the level of Il6 in mouse osteoblasts, MSCs and MSC-derived adipocytes treated with 50 ng/mL rWNT5B for 24 h (Figures 6H–6J). Mechanistically, ChIP-qPCR with a specific antibody to phospho-c-Fos revealed the binding of phospho-c-Fos at the Il6 promoter, compared to a negative control binding site60 in mouse osteoblasts. The binding of phospho-c-Fos (Ser32) is increased by the addition of rWNT5B (∗p ≤ 0.05; Figure 6K).
These data show that WNT5B colocalized with ROR1/2 then activates signal transduction through DVL2/3 and downstream to WNT non-canonical effectors RAC1/CDC42, phospho-JNK and phospho-c-Fos in osteoblasts, while repressing β-catenin-dependent gene activation.
WNT5B represses E2-induced ALPL expression via SIN3A
Because both ERα and WNT5B differentially regulate alkaline phosphatase (Figures 5B, 5D, and 5H), we further investigated the function of WNT5B on E2-induced ALPL expression. Mouse calvarial osteoblasts were differentiated and treated with or without 10 nM E2 and/or 50 ng/mL rWNT5B for 4 days. In the presence of E2, the level of alkaline phosphatase activity significantly increased; meanwhile, WNT5B inhibited the induction by E2 (Figure 7A). This effect was also confirmed in U2OS-ERα cells (Figure 7B). Furthermore, the mRNA of ALPL was increased by E2 and decreased by WNT5B (Figure 7C). Therefore, U2OS-ERα cells were used to investigate the molecular mechanism of WNT5B on ALPL expression.
Figure 7.

WNT5B inhibits E2-induced ALPL expression via SIN3A
(A and B) mOBs (A) or U2OS-ERα cells (B) were treated with EtOH or 10 nM E2 in the presence or absence of 50 ng/mL rWNT5B for 4 days. Then, alkaline phosphatase (ALP) activity was detected and was normalized with total protein.
(C) Expression of ALPL also was measured in U2OS-ERα cells.
(D) Diagram represents ALPL enhancers in U2OS-ERα cells. U2OS-ERα cells were treated with PBS/EtOH (Ctrl), 10 nM E2, 50 ng/mL rWNT5B, or E2 with rWNT5B for 45 min.
(E and F) ChIP was performed using an antibody to (E) ERα or (F) SIN3A. qPCR was performed with primers to the ALPL enhancers and normalized to the β-actin (ACTB) promoter.
(G) Co-immunoprecipitations were performed with an antibody to SIN3A before blotting with an antibody to ERα or IgG in U2OS-ERα cells.
(H) mOBs were treated with 50 ng/mL rWNT5B for 0 and 15 min, and then were incubated with or without calf intestinal protease (CIP) in vitro before detecting phospho-CDK5 (Tyr15) and SIN3A by immunoblotting and normalized to β-actin (BA) by ImageJ. Three different biological replicates were tested and a representative image from one replicate is presented. n = 3, Student’s t test: ∗∗p < 0.01, ∗p < 0.05.
We have shown previously that ERα binds at two enhancers downstream of ALPL (see schematic, Figure 7D).19 U2OS-ERα cells were treated with either EtOH/PBS (Ctrl), 10 nM E2, 50 ng/mL rWNT5B, or a combination of E2 with rWNT5B (E2/WNT5B), and then ChIP-qPCR was performed with an antibody to ERα. The binding of ERα significantly increased at both enhancers when treated with E2 compared to the control (Figure 7E). Paradoxically, ERα was further enriched when the cells were treated with E2 together with WNT5B compared to only E2 at enhancer 1 (∗∗p ≤ 0.01; Figure 7E). Because the presence of ERα does not match the expression of ALPL, we hypothesized that WNT5B signaling would recruit a repressor complex to these ALPL enhancers. Thus, we performed ChIP-qPCR using an antibody to SIN3A, a member of a histone deacetylase complex (Figure 7F). In the presence of E2, the binding of SIN3A at both enhancers was decreased when compared to the control (Figure 7F), corresponding with the increase in ALPL expression without WNT5B. The addition of WNT5B and E2 increased the recruitment of SIN3A to enhancer 1, compared to E2 alone. We confirmed the deacetylase activity of SIN3A by analyzing for H3K27ac at the ALPL enhancers. Indeed, in the presence of rWNT5B and E2, there is a correlation between SIN3A recruitment and a decrease in H3K27ac compared to E2 alone (Figure S5A).
We found that SIN3A expression was increased by rWNT5B in U2OS-ERα cells (lane 1 versus 3, Figure 7G). Next, we tested for the interaction of ERα and SIN3A by co-immunoprecipitation. ERα and SIN3A interact, and their interaction increased when WNT5B was added (lane 5 versus 8, Figure 7G). As a negative control, ERα was not immunoprecipitated with normal IgG (lane 9–12, Figure 7G). Phosphorylated CDK5 at tyrosine 15 (Tyr15) is active61 and can phosphorylate SIN3A to induce proteasome degradation.62 Due to the lack of a phospho-specific SIN3A antibody, we analyzed SIN3A phosphorylation using calf intestinal phosphatase (CIP) assays and a phospho-specific CDK5 antibody (Tyr15). Mouse osteoblasts were treated with 50 ng/mL rWNT5B for 0 and 15 min, and protein was obtained. The protein lysates were treated with or without CIP in vitro, before detecting the level of phospho-specific CDK5 (Tyr15) and SIN3A by immunoblotting. Phospho-CDK5 (Tyr15) was decreased and, in contrast, non-phosphorylated SIN3A was increased by the activation of WNT5B (lane 1 versus 3, Figure 7H). Moreover, ImageJ quantification of the immunoblots confirms our results. Therefore, WNT5B suppresses CDK5 activity, which prevents phosphorylation of SIN3A and blocks protein degradation that ultimately allows SIN3A to activate gene suppression (e.g., ALPL).
Interestingly, we found that the expression of ALPL in U2OS-ERα homozygous GG cells, that has lower WNT5B expression, was higher than homozygous AA by qPCR (∗∗∗∗p ≤ 0.0001; Figure S5B), further demonstrating the role of WNT5B in the suppression of ALPL expression.
Taken together, WNT5B suppresses E2-induced ALPL expression by protecting SIN3A degradation via the inhibition of CDK5 activity and then recruiting SIN3A to interact with ERα, resulting in loss of acetylation at H3K27 at the ALPL enhancer.
Discussion
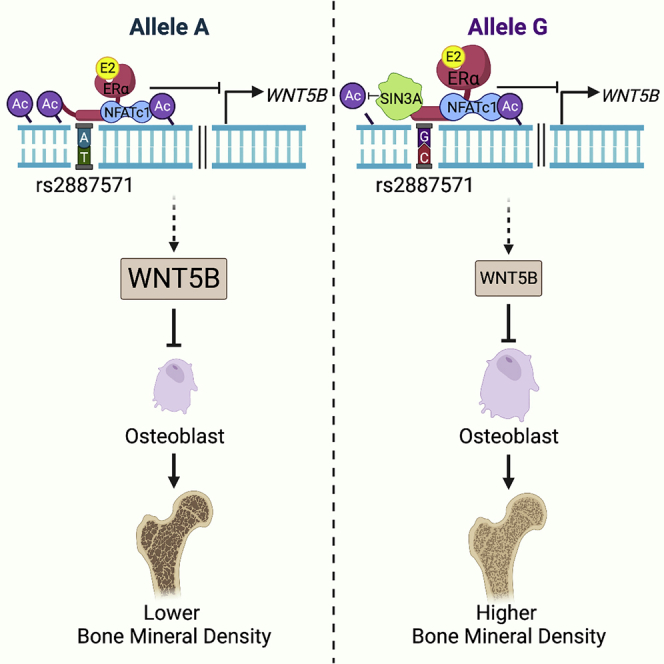
Many genetic factors significantly influence osteoporosis.3 The non-coding intergenic SNP rs2887571 at 12p13.33 is predicted to affect BMD;4,63 however, the mechanism of rs2887571 in bone was not known. Here we show that SNP rs2887571 is a binding site for ERα and NFATc1 and they regulate the expression of WNT5B (Figure 8A). Increased expression of WNT5B with the AA genotype correlates with decreased bone mineral density, as WNT5B inhibits osteoblast differentiation. WNT5B signals through ROR1/2 signaling pathways to inhibit osteoblast differentiation genes such as alkaline phosphatase. Mechanistically, we also reveal that SIN3A is a downstream effector of WNT5B to suppress gene expression (see model, Figure 8B).
Figure 8.

Model of the regulation of WNT5B expression at SNP rs2887571 and the signaling pathway of WNT5B in osteoblasts
(A) The molecular mechanisms of ERα and NFATc1 at SNP rs2887571 in cells with homozygous AA (right) or homozygous GG (left) at rs2887571. (Top) Homozygous GG has increased ERα, NFATc1 and SIN3A compared to homozygous AA, which results in lower endogenous WNT5B expression due to the repressive effects of SIN3A. (Bottom) The presence of E2 recruits SIN3A to suppress the expression of WNT5B in both genotypes, with increased suppression in cells with GG due to increased recruitment of SIN3A.
(B) WNT5B binds with ROR1/2 then activates DVL2/3, RAC1/CDC42, p-JNK, and p-c-Fos. Phosphorylation of SIN3A by CDK5 results in proteasome degradation. WNT5B inhibits phosphorylation of CDK5 (Tyr15), resulting in accumulation of SIN3A. WNT5B also inhibits β-catenin activity in osteoblasts. Solid lines indicate the signaling pathways described in this study. The dashed line indicates the signaling that has been previously reported. P indicates phosphorylation. Created with BioRender.com
GWASs have discovered hundreds of variants associated with bone density64 and the trait-associated variants are mostly far from protein-coding regions.65 The effects of non-coding causal variants can be highly cell-type-, context- and disease-specific; therefore, the greatest challenge after a GWAS is to understand the underlying biological mechanisms of the variants on each trait.66 The intergenic variant SNP rs2887571 is reported to connect with WNT5B in silico by the Gene Relationships Across Implicated Loci (GRAIL) text-mining algorithm4 and is predicted to associate with bone density and osteoporotic fractures.42,63,67 We identified that SNP rs2887571 targets WNT5B, but not ERC1, LINC00942, LOC107984507, nor FBXL14, in primary human osteoblasts. We also provide evidence that this was not due to another SNP in linkage disequilibrium, as shown by CRISPR substitutions of the AA allele for the GG allele altering the expression of WNT5B.
Several WNT proteins, including WNT10B and WNT5A, have been shown to promote bone mass by activating osteoblastogenesis and inhibiting adipogenesis from MSCs.7,68, 69, 70 Although WNT5A and WNT5B have a high amino acid identity in mice (87%) and humans (80.5%),71,72 they show non-redundant functions in most tissues.73 The distinct effects of WNT5A and WNT5B are not only in bone12,74 and adipose tissue47,48,69 but also in the nervous system,75,76 immune system,77,78 and mammary gland.79 Moreover, our results elucidate that WNT5B affects both bone marrow-derived MSCs and calvarial differentiating osteoblasts. WNT5B promotes MSC proliferation and induces the commitment to the adipocyte lineage rather than osteoblast lineage. WNT5B may increase progenitor cell exhaustion as it does in hematopoietic stem cells.77 The role of WNT5B in stem cells, osteoblasts, bone metabolic diseases, and osteosarcoma is still limited and should be investigated in future studies.
WNTs function through either a β-catenin-dependent (canonical) or a β-catenin-independent (non-canonical) WNT signaling pathway.54,80 We find that WNT5B suppresses β-catenin activity and activates non-canonical WNT signaling via DVL2/3, RAC1/CDC42, JNK, and CDK5/SIN3A in osteoblasts. ROR1 and ROR2 show dimerization to trigger non-canonical WNT signal transduction in chronic lymphocytic leukemia,81 and potentially could do the same thing in osteoblasts, as our mechanistic results also show that WNT5B is activating non-canonical signaling via ROR1 and/or ROR2. During osteoblast differentiation (D7), both levels of mRNA and protein expression pattern for ROR1/2 are elevated, similar to WNT5B, yet the expression is more prominent for ROR1. Colocalization by immunofluorescence suggests that ROR1/2 are WNT5B co-receptors. WNT ligands use a two-component system of receptors to create a WNT-ligand-specific signalosome and ligand-selective WNT signaling.82 Further work will be required using conditional KOs or CRISPR-Cas9 technique for ROR1/2 and/or other co-receptors in osteoblasts to delineate the recruitment of the co-receptors and WNT5B interactions.
We have demonstrated the mechanisms of WNT5B on suppression or stimulation of several target genes in osteoblasts. WNT5B increases Il6, an osteoporotic-induction cytokine,56,57 via phospho-c-Fos (Ser32). We also reveal that SIN3A is a WNT5B effector to suppress gene expression via CDK5. SIN3A is a global transcription repressor and phosphorylation by CDK5 leads to proteasome degradation.62 WNT5B decreases CDK5 Tyr15 phosphorylation, thus decreasing active CDK5,61 resulting in the accumulation and activation of SIN3A. Finally, we show that WNT5B inhibits the function of β-catenin by decreasing the levels of non-phospho (active) β-catenin (Ser33/37/Thr41) and phospho-β-catenin (Ser552). Although phosphorylation at Ser33/37/Thr41 leads to proteasome degradation,83 phosphorylation of β-catenin at Ser552 induces transcriptional activity which is mediated by protein kinase B (AKT).84 The PI3K/AKT signaling pathway is reported to inhibit osteoporosis by promoting osteoblast proliferation, differentiation, and bone formation.85 WNT5B may suppress AKT/β-catenin transcriptional activity via JNK. A high-fat diet in mice increases the level of insulin, resulting in the upregulation of Wnt5b,86 while decreasing the level of AKT signaling.87 JNK attenuates the activity of AKT through a p70 ribosomal S6 kinase alpha (RPS6KB1)-dependent mechanism.87 However, the crosstalk between JNK and AKT signaling pathway is complex and requires further investigation.
Estrogens, via ERα, are important in the development and maintenance of bone mineral density.13 Estrogens regulate osteoblast differentiation and induce osteoclast apoptosis.14,20 Here, we show that E2 additionally protects bone by suppressing WNT5B. E2 activates the ERα/SIN3A complex to bind at SNP rs2887571. A 3D interaction study, such as Hi-C or HiChIP, could identify specific promoter and enhancer gene interactions between ERα, WNT5B, and rs2887571.66
We find that ERα and NFATc1 interact with each other and bind at SNP rs2887571. However, the effects of ERα and NFATc1 on WNT5B are in opposition. The E2/ERα complex suppresses WNT5B and NFATc1 increases WNT5B. Which NFAT interacts with ERα is cell type specific. In breast cancer cells, NFATc2 interacts with ERα and enhances ERα-mediated transcription activity.88 In contrast, E2 and ERα have also been shown to inhibit NFATc2 transcriptional activity.89 Our results clearly show that NFATC1 has the highest expression compared to other NFAT members in osteoblasts and activation of NFATc1 represses osteoblastogenesis by activating WNT5B expression. These results support the previous studies that show that NFATc1 suppresses osteoblast differentiation by inhibiting ERα and osteocalcin expression.90,91 Nevertheless, the effects of NFATs on osteoblasts are still controversial92 and need further investigation.
In conclusion, this work shows a functional mechanism of how the SNP rs2887571 regulates WNT5B and the inhibitory effect of WNT5B on osteoblastogenesis. In our study, we find that homozygous GG at SNP rs2887571 correlates with lower WNT5B expression than homozygous AA in primary human osteoblasts. We propose that SNP rs2887571 is an enhancer for WNT5B (Figure 8A) and that ERα represses WNT5B to protect osteoblasts. Using the CRISPR-Cas9 technique to alter SNP rs2887571 in U2OS-ERα cells reveals that allele G leads to higher suppression of endogenous WNT5B via recruitment of SIN3A, leading to increased osteoblastogenesis. These findings reveal the importance of GWAS-identified signals in the non-coding region, as well as the biological effects of WNT5B in osteoblasts, for the pathophysiology of bone metabolic diseases such as osteoporosis that may lead to the development of alternative therapies for bone disease.
Acknowledgments
The research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number AR-064354 to S.A.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.S. was supported by a Development and Promotion of Science and Technology Talents Project scholarship from the Royal Government of Thailand.
Declaration of interests
The authors declare no competing interests.
Published: December 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2021.11.018.
Data availability
Data for ERα ChIP sequencing in U2OS-ERα cells have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE28918.
Web Resources
Supplemental information
References
- 1.Imel E.A., DiMeglio L.A., Burr D.B. In: Basic and Applied Bone Biology, Chapter 16. Burr D.B., Allen M.R., editors. Academic Press; 2014. Metabolic Bone Diseases; pp. 317–344. [DOI] [Google Scholar]
- 2.Compston J.E., McClung M.R., Leslie W.D. Osteoporosis. Lancet. 2019;393:364–376. doi: 10.1016/S0140-6736(18)32112-3. [DOI] [PubMed] [Google Scholar]
- 3.Ralston S.H., Uitterlinden A.G. Genetics of osteoporosis. Endocr. Rev. 2010;31:629–662. doi: 10.1210/er.2009-0044. [DOI] [PubMed] [Google Scholar]
- 4.Estrada K., Styrkarsdottir U., Evangelou E., Hsu Y.H., Duncan E.L., Ntzani E.E., Oei L., Albagha O.M., Amin N., Kemp J.P., et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012;44:491–501. doi: 10.1038/ng.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moorer M.C., Riddle R.C. Regulation of Osteoblast Metabolism by Wnt Signaling. Endocrinol. Metab. (Seoul) 2018;33:318–330. doi: 10.3803/EnM.2018.33.3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maupin K.A., Droscha C.J., Williams B.O. A Comprehensive Overview of Skeletal Phenotypes Associated with Alterations in Wnt/β-catenin Signaling in Humans and Mice. Bone Res. 2013;1:27–71. doi: 10.4248/BR201301004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevens J.R., Miranda-Carboni G.A., Singer M.A., Brugger S.M., Lyons K.M., Lane T.F. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J. Bone Miner. Res. 2010;25:2138–2147. doi: 10.1002/jbmr.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norwitz N.G., Mota A.S., Misra M., Ackerman K.E. LRP5, Bone Density, and Mechanical Stress: A Case Report and Literature Review. Front. Endocrinol. (Lausanne) 2019;10:184. doi: 10.3389/fendo.2019.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haraguchi R., Kitazawa R., Mori K., Tachibana R., Kiyonari H., Imai Y., Abe T., Kitazawa S. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci. Rep. 2016;6:25198. doi: 10.1038/srep25198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wergedal J.E., Kesavan C., Brommage R., Das S., Mohan S. Role of WNT16 in the regulation of periosteal bone formation in female mice. Endocrinology. 2015;156:1023–1032. doi: 10.1210/en.2014-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Movérare-Skrtic S., Henning P., Liu X., Nagano K., Saito H., Börjesson A.E., Sjögren K., Windahl S.H., Farman H., Kindlund B., et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 2014;20:1279–1288. doi: 10.1038/nm.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brommage R., Liu J., Hansen G.M., Kirkpatrick L.L., Potter D.G., Sands A.T., Zambrowicz B., Powell D.R., Vogel P. High-throughput screening of mouse gene knockouts identifies established and novel skeletal phenotypes. Bone Res. 2014;2:14034. doi: 10.1038/boneres.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khalid A.B., Krum S.A. Estrogen receptors alpha and beta in bone. Bone. 2016;87:130–135. doi: 10.1016/j.bone.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krum S.A., Brown M. Unraveling estrogen action in osteoporosis. Cell Cycle. 2008;7:1348–1352. doi: 10.4161/cc.7.10.5892. [DOI] [PubMed] [Google Scholar]
- 15.Cui J., Shen Y., Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol. Med. 2013;19:197–209. doi: 10.1016/j.molmed.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farh K.K., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J., Shishkin A.A., et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zweig A.S., Karolchik D., Kuhn R.M., Haussler D., Kent W.J. UCSC genome browser tutorial. Genomics. 2008;92:75–84. doi: 10.1016/j.ygeno.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Beresford J.N., Gallagher J.A., Poser J.W., Russell R.G.G. Production of osteocalcin by human bone cells in vitro. Effects of 1,25(OH)2D3, 24,25(OH)2D3, parathyroid hormone, and glucocorticoids. Metab. Bone Dis. Relat. Res. 1984;5:229–234. doi: 10.1016/0221-8747(84)90064-X. [DOI] [PubMed] [Google Scholar]
- 19.Krum S.A., Miranda-Carboni G.A., Lupien M., Eeckhoute J., Carroll J.S., Brown M. Unique ERalpha cistromes control cell type-specific gene regulation. Mol. Endocrinol. 2008;22:2393–2406. doi: 10.1210/me.2008-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krum S.A., Miranda-Carboni G.A., Hauschka P.V., Carroll J.S., Lane T.F., Freedman L.P., Brown M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27:535–545. doi: 10.1038/sj.emboj.7601984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monroe D.G., Getz B.J., Johnsen S.A., Riggs B.L., Khosla S., Spelsberg T.C. Estrogen receptor isoform-specific regulation of endogenous gene expression in human osteoblastic cell lines expressing either ERalpha or ERbeta. J. Cell. Biochem. 2003;90:315–326. doi: 10.1002/jcb.10633. [DOI] [PubMed] [Google Scholar]
- 22.Dupont S., Krust A., Gansmuller A., Dierich A., Chambon P., Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 23.Truax A.D., Greer S.F. ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol. Biol. 2012;809:175–188. doi: 10.1007/978-1-61779-376-9_12. [DOI] [PubMed] [Google Scholar]
- 24.Beals C.R., Clipstone N.A., Ho S.N., Crabtree G.R. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- 25.Veeman M.T., Slusarski D.C., Kaykas A., Louie S.H., Moon R.T. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 2003;13:680–685. doi: 10.1016/S0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 26.Najdi R., Proffitt K., Sprowl S., Kaur S., Yu J., Covey T.M., Virshup D.M., Waterman M.L. A uniform human Wnt expression library reveals a shared secretory pathway and unique signaling activities. Differentiation. 2012;84:203–213. doi: 10.1016/j.diff.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wend K., Wend P., Drew B.G., Hevener A.L., Miranda-Carboni G.A., Krum S.A. ERα regulates lipid metabolism in bone through ATGL and perilipin. J. Cell. Biochem. 2013;114:1306–1314. doi: 10.1002/jcb.24470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das T.K., Sangodkar J., Negre N., Narla G., Cagan R.L. Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene. 2013;32:3184–3197. doi: 10.1038/onc.2012.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward L.D., Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miranda-Carboni G.A., Guemes M., Bailey S., Anaya E., Corselli M., Peault B., Krum S.A. GATA4 regulates estrogen receptor-alpha-mediated osteoblast transcription. Mol. Endocrinol. 2011;25:1126–1136. doi: 10.1210/me.2010-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R., 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carroll J.S., Meyer C.A., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 33.Palaniappan M., Nguyen L., Grimm S.L., Xi Y., Xia Z., Li W., Coarfa C. The genomic landscape of estrogen receptor α binding sites in mouse mammary gland. PLoS ONE. 2019;14:e0220311. doi: 10.1371/journal.pone.0220311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Métivier R., Penot G., Hübner M.R., Reid G., Brand H., Kos M., Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 35.Morris J.A., Kemp J.P., Youlten S.E., Laurent L., Logan J.G., Chai R.C., Vulpescu N.A., Forgetta V., Kleinman A., Mohanty S.T., et al. 23andMe Research Team An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 2019;51:258–266. doi: 10.1038/s41588-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S.K. Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture. PLoS ONE. 2018;13:e0200785. doi: 10.1371/journal.pone.0200785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kichaev G., Bhatia G., Loh P.R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellison-Zelski S.J., Solodin N.M., Alarid E.T. Repression of ESR1 through actions of estrogen receptor alpha and Sin3A at the proximal promoter. Mol. Cell. Biol. 2009;29:4949–4958. doi: 10.1128/MCB.00383-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams G.E., Chandru A., Cowley S.M. Co-repressor, co-activator and general transcription factor: the many faces of the Sin3 histone deacetylase (HDAC) complex. Biochem. J. 2018;475:3921–3932. doi: 10.1042/BCJ20170314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin L., Liu Y., Wang Y., Wu G., Chen J., Ye W., Yang J., Huang Q. Computational Characterization of Osteoporosis Associated SNPs and Genes Identified by Genome-Wide Association Studies. PLoS ONE. 2016;11:e0150070. doi: 10.1371/journal.pone.0150070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mognol G.P., Carneiro F.R., Robbs B.K., Faget D.V., Viola J.P. Cell cycle and apoptosis regulation by NFAT transcription factors: new roles for an old player. Cell Death Dis. 2016;7:e2199. doi: 10.1038/cddis.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winslow M.M., Pan M., Starbuck M., Gallo E.M., Deng L., Karsenty G., Crabtree G.R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell. 2006;10:771–782. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 45.Fromigué O., Haÿ E., Barbara A., Marie P.J. Essential role of nuclear factor of activated T cells (NFAT)-mediated Wnt signaling in osteoblast differentiation induced by strontium ranelate. J. Biol. Chem. 2010;285:25251–25258. doi: 10.1074/jbc.M110.110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kemp J.P., Medina-Gomez C., Estrada K., St Pourcain B., Heppe D.H., Warrington N.M., Oei L., Ring S.M., Kruithof C.J., Timpson N.J., et al. Phenotypic dissection of bone mineral density reveals skeletal site specificity and facilitates the identification of novel loci in the genetic regulation of bone mass attainment. PLoS Genet. 2014;10:e1004423. doi: 10.1371/journal.pgen.1004423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanazawa A., Tsukada S., Kamiyama M., Yanagimoto T., Nakajima M., Maeda S. Wnt5b partially inhibits canonical Wnt/beta-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2005;330:505–510. doi: 10.1016/j.bbrc.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Kanazawa A., Tsukada S., Sekine A., Tsunoda T., Takahashi A., Kashiwagi A., Tanaka Y., Babazono T., Matsuda M., Kaku K., et al. Association of the gene encoding wingless-type mammary tumor virus integration-site family member 5B (WNT5B) with type 2 diabetes. Am. J. Hum. Genet. 2004;75:832–843. doi: 10.1086/425340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Tienen F.H., Laeremans H., van der Kallen C.J., Smeets H.J. Wnt5b stimulates adipogenesis by activating PPARgamma, and inhibiting the beta-catenin dependent Wnt signaling pathway together with Wnt5a. Biochem. Biophys. Res. Commun. 2009;387:207–211. doi: 10.1016/j.bbrc.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Castro-Malaspina H., Gay R.E., Resnick G., Kapoor N., Meyers P., Chiarieri D., McKenzie S., Broxmeyer H.E., Moore M.A. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood. 1980;56:289–301. [PubMed] [Google Scholar]
- 51.Khalid A.B., Pence J., Suthon S., Lin J., Miranda-Carboni G.A., Krum S.A. GATA4 regulates mesenchymal stem cells via direct transcriptional regulation of the WNT signalosome. Bone. 2021;144:115819. doi: 10.1016/j.bone.2020.115819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fatima I., El-Ayachi I., Taotao L., Lillo M.A., Krutilina R.I., Seagroves T.N., Radaszkiewicz T.W., Hutnan M., Bryja V., Krum S.A., et al. The natural compound Jatrophone interferes with Wnt/β-catenin signaling and inhibits proliferation and EMT in human triple-negative breast cancer. PLoS ONE. 2017;12:e0189864. doi: 10.1371/journal.pone.0189864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wend P., Runke S., Wend K., Anchondo B., Yesayan M., Jardon M., Hardie N., Loddenkemper C., Ulasov I., Lesniak M.S., et al. WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol. Med. 2013;5:264–279. doi: 10.1002/emmm.201201320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niehrs C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 55.McKay S., Bromhaar M.M., de Jongste J.C., Hoogsteden H.C., Saxena P.R., Sharma H.S. Pro-inflammatory cytokines induce c-fos expression followed by IL-6 release in human airway smooth muscle cells. Mediators Inflamm. 2001;10:135–142. doi: 10.1080/09629350124155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hughes F.J., Howells G.L. Interleukin-6 inhibits bone formation in vitro. Bone Miner. 1993;21:21–28. doi: 10.1016/s0169-6009(08)80117-1. [DOI] [PubMed] [Google Scholar]
- 57.Kurihara N., Bertolini D., Suda T., Akiyama Y., Roodman G.D. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J. Immunol. 1990;144:4226–4230. [PubMed] [Google Scholar]
- 58.Huang J., Chen C., Liang C., Luo P., Xia G., Zhang L., Wang X., Wen Z., Cao X., Wu S. Dysregulation of the Wnt Signaling Pathway and Synovial Stem Cell Dysfunction in Osteoarthritis Development. Stem Cells Dev. 2020;29:401–413. doi: 10.1089/scd.2019.0260. [DOI] [PubMed] [Google Scholar]
- 59.Gatica-Andrades M., Vagenas D., Kling J., Nguyen T.T.K., Benham H., Thomas R., Körner H., Venkatesh B., Cohen J., Blumenthal A. WNT ligands contribute to the immune response during septic shock and amplify endotoxemia-driven inflammation in mice. Blood Adv. 2017;1:1274–1286. doi: 10.1182/bloodadvances.2017006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khalid A.B., Slayden A.V., Kumpati J., Perry C.D., Osuna M.A.L., Arroyo S.R., Miranda-Carboni G.A., Krum S.A. GATA4 Directly Regulates Runx2 Expression and Osteoblast Differentiation. JBMR Plus. 2018;2:81–91. doi: 10.1002/jbm4.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zukerberg L.R., Patrick G.N., Nikolic M., Humbert S., Wu C.L., Lanier L.M., Gertler F.B., Vidal M., Van Etten R.A., Tsai L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 62.Lam E., Pareek T.K., Letterio J.J. Cdk5 controls IL-2 gene expression via repression of the mSin3a-HDAC complex. Cell Cycle. 2015;14:1327–1336. doi: 10.4161/15384101.2014.987621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitchell J.A., Chesi A., Elci O., McCormack S.E., Roy S.M., Kalkwarf H.J., Lappe J.M., Gilsanz V., Oberfield S.E., Shepherd J.A., et al. Physical Activity Benefits the Skeleton of Children Genetically Predisposed to Lower Bone Density in Adulthood. J. Bone Miner. Res. 2016;31:1504–1512. doi: 10.1002/jbmr.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu X., Bai W., Zheng H. Twelve years of GWAS discoveries for osteoporosis and related traits: advances, challenges and applications. Bone Res. 2021;9:23. doi: 10.1038/s41413-021-00143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hardison R.C. Genome-wide epigenetic data facilitate understanding of disease susceptibility association studies. J. Biol. Chem. 2012;287:30932–30940. doi: 10.1074/jbc.R112.352427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Broekema R.V., Bakker O.B., Jonkers I.H. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. 2020;10:190221. doi: 10.1098/rsob.190221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y., Wactawski-Wende J., Sucheston-Campbell L.E., Preus L., Hovey K.M., Nie J., Jackson R.D., Handelman S.K., Nassir R., Crandall C.J., Ochs-Balcom H.M. The influence of genetic susceptibility and calcium plus vitamin D supplementation on fracture risk. Am. J. Clin. Nutr. 2017;105:970–979. doi: 10.3945/ajcn.116.144550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baron R., Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 69.Okamoto M., Udagawa N., Uehara S., Maeda K., Yamashita T., Nakamichi Y., Kato H., Saito N., Minami Y., Takahashi N., Kobayashi Y. Noncanonical Wnt5a enhances Wnt/β-catenin signaling during osteoblastogenesis. Sci. Rep. 2014;4:4493. doi: 10.1038/srep04493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wend P., Wend K., Krum S.A., Miranda-Carboni G.A. The role of WNT10B in physiology and disease. Acta Physiol. (Oxf.) 2012;204:34–51. doi: 10.1111/j.1748-1716.2011.02296.x. [DOI] [PubMed] [Google Scholar]
- 71.Gavin B.J., McMahon J.A., McMahon A.P. Expression of multiple novel Wnt-1/int-1-related genes during fetal and adult mouse development. Genes Dev. 1990;4(12B):2319–2332. doi: 10.1101/gad.4.12b.2319. [DOI] [PubMed] [Google Scholar]
- 72.Saitoh T., Katoh M. Molecular cloning and characterization of human WNT5B on chromosome 12p13.3 region. Int. J. Oncol. 2001;19:347–351. doi: 10.3892/ijo.19.2.347. [DOI] [PubMed] [Google Scholar]
- 73.Suthon S., Perkins R.S., Bryja V., Miranda-Carboni G.A., Krum S.A. WNT5B in Physiology and Disease. Front. Cell Dev. Biol. 2021;9:667581. doi: 10.3389/fcell.2021.667581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maeda K., Kobayashi Y., Udagawa N., Uehara S., Ishihara A., Mizoguchi T., Kikuchi Y., Takada I., Kato S., Kani S., et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 2012;18:405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- 75.Wu M., Li Z., Liang L., Ma P., Cui D., Chen P., Wu G., Song X.J. Wnt signaling contributes to withdrawal symptoms from opioid receptor activation induced by morphine exposure or chronic inflammation. Pain. 2020;161:532–544. doi: 10.1097/j.pain.0000000000001738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gupta K., Schnell E. Neuronal network remodeling and Wnt pathway dysregulation in the intra-hippocampal kainate mouse model of temporal lobe epilepsy. PLoS ONE. 2019;14:e0215789. doi: 10.1371/journal.pone.0215789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Rezende M.M., Ng-Blichfeldt J.P., Justo G.Z., Paredes-Gamero E.J., Gosens R. Divergent effects of Wnt5b on IL-3- and GM-CSF-induced myeloid differentiation. Cell. Signal. 2020;67:109507. doi: 10.1016/j.cellsig.2019.109507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Louwette S., Labarque V., Wittevrongel C., Thys C., Metz J., Gijsbers R., Debyser Z., Arnout J., Van Geet C., Freson K. Regulator of G-protein signaling 18 controls megakaryopoiesis and the cilia-mediated vertebrate mechanosensory system. FASEB J. 2012;26:2125–2136. doi: 10.1096/fj.11-198739. [DOI] [PubMed] [Google Scholar]
- 79.Kessenbrock K., Smith P., Steenbeek S.C., Pervolarakis N., Kumar R., Minami Y., Goga A., Hinck L., Werb Z. Diverse regulation of mammary epithelial growth and branching morphogenesis through noncanonical Wnt signaling. Proc. Natl. Acad. Sci. USA. 2017;114:3121–3126. doi: 10.1073/pnas.1701464114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perkins R.S., Suthon S., Miranda-Carboni G.A., Krum S.A. WNT5B in cellular signaling pathways. Semin. Cell Dev. Biol. 2021 doi: 10.1016/j.semcdb.2021.09.019. S1084-9521(21)00262-7. [DOI] [PubMed] [Google Scholar]
- 81.Yu J., Chen L., Cui B., Widhopf G.F., 2nd, Shen Z., Wu R., Zhang L., Zhang S., Briggs S.P., Kipps T.J. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J. Clin. Invest. 2016;126:585–598. doi: 10.1172/JCI83535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eubelen M., Bostaille N., Cabochette P., Gauquier A., Tebabi P., Dumitru A.C., Koehler M., Gut P., Alsteens D., Stainier D.Y.R., et al. A molecular mechanism for Wnt ligand-specific signaling. Science. 2018;361:eaat1178. doi: 10.1126/science.aat1178. [DOI] [PubMed] [Google Scholar]
- 83.Yost C., Torres M., Miller J.R., Huang E., Kimelman D., Moon R.T. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 84.Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G.B., Kobayashi R., Hunter T., Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xi J.C., Zang H.Y., Guo L.X., Xue H.B., Liu X.D., Bai Y.B., Ma Y.Z. The PI3K/AKT cell signaling pathway is involved in regulation of osteoporosis. J. Recept. Signal Transduct. Res. 2015;35:640–645. doi: 10.3109/10799893.2015.1041647. [DOI] [PubMed] [Google Scholar]
- 86.Kurita Y., Ohki T., Soejima E., Yuan X., Kakino S., Wada N., Hashinaga T., Nakayama H., Tani J., Tajiri Y., et al. A High-Fat/High-Sucrose Diet Induces WNT4 Expression in Mouse Pancreatic β-cells. Kurume Med. J. 2019;65:55–62. doi: 10.2739/kurumemedj.MS652008. [DOI] [PubMed] [Google Scholar]
- 87.Miller W.P., Ravi S., Martin T.D., Kimball S.R., Dennis M.D. Activation of the Stress Response Kinase JNK (c-Jun N-terminal Kinase) Attenuates Insulin Action in Retina through a p70S6K1-dependent Mechanism. J. Biol. Chem. 2017;292:1591–1602. doi: 10.1074/jbc.M116.760868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang H., Xie X., Zhu X., Zhu J., Hao C., Lu Q., Ding L., Liu Y., Zhou L., Liu Y., et al. Stimulatory cross-talk between NFAT3 and estrogen receptor in breast cancer cells. J. Biol. Chem. 2005;280:43188–43197. doi: 10.1074/jbc.M506598200. [DOI] [PubMed] [Google Scholar]
- 89.Qin X., Wang X.H., Yang Z.H., Ding L.H., Xu X.J., Cheng L., Niu C., Sun H.W., Zhang H., Ye Q.N. Repression of NFAT3 transcriptional activity by estrogen receptors. Cell. Mol. Life Sci. 2008;65:2752–2762. doi: 10.1007/s00018-008-8273-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Choo M.K., Yeo H., Zayzafoon M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone. 2009;45:579–589. doi: 10.1016/j.bone.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Penolazzi L., Lisignoli G., Lambertini E., Torreggiani E., Manferdini C., Lolli A., Vecchiatini R., Ciardo F., Gabusi E., Facchini A., et al. Transcription factor decoy against NFATc1 in human primary osteoblasts. Int. J. Mol. Med. 2011;28:199–206. doi: 10.3892/ijmm.2011.701. [DOI] [PubMed] [Google Scholar]
- 92.Sitara D., Aliprantis A.O. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 2010;233:286–300. doi: 10.1111/j.0105-2896.2009.00849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for ERα ChIP sequencing in U2OS-ERα cells have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE28918.