

Abstract
Background and Objective
Posttraumatic stress disorder (PTSD) is a serious and frequently debilitating psychiatric condition that can occur in people who have experienced traumatic stressors, such as war, violence, sexual assault and other life‐threatening events. Treatment of PTSD and traumatic brain injury (TBI) in veterans is challenged by diagnostic complexity, partially due to PTSD and TBI symptoms overlap and to the fact that subjective self‐report assessments may be influenced by a patient's willingness to share their traumatic experiences and resulting symptoms. Corticotropin‐releasing factor (CRF) is one of the main mediators of hypothalamic pituitary adrenal (HPA)‐axis responses in stress and anxiety.
Methods and Results
We analyzed serum CRF levels in 230 participants including heathy controls (64), and individuals with PTSD (53), TBI (70) or PTSD + TBI (43) by enzyme immunoassay (EIA). Significantly lower CRF levels were found in both the PTSD and PTSD + TBI groups compared to healthy control (PTSD vs. Controls: P = 0.0014, PTSD + TBI vs. Controls: P = 0.0011) and chronic TBI participants (PTSD vs. TBI: P < 0.0001, PTSD + TBI vs. TBI: P < 0.0001), suggesting a PTSD‐related mechanism independent from TBI and associated with CRF reduction. CRF levels negatively correlated with PTSD severity on the Clinically Administered PTSD Scale (CAPS‐5) scale in the whole study group.
Conclusions
Hyperactivation of the HPA axis has been classically identified in acute stress. However, the recognized enhanced feedback inhibition of the HPA axis in chronic stress supports our findings of lower CRF in PTSD patients. This study suggests that reduced serum CRF in PTSD should be further investigated. Future validation studies will establish if CRF is a possible blood biomarker for PTSD and/or for differentiating PTSD and chronic TBI symptomatology.
highlights
The HPA axis is activated under acute stress conditions, but an enhanced feedback inhibition may be prevalent in chronic stress conditions such as PTSD.
We observed a reduction in serum CRF levels in veterans with PTSD and PTSD + TBI, but not in veterans with chronic TBI alone.
A serum CRF reduction may be indicative of CNS mechanisms specific to PTSD and should be further evaluated as a possible peripheral biomarker.
Decades of study on the effects of war trauma on the human brain show that post‐traumatic stress disorder (PTSD) has an important and complex relationship with traumatic brain injury (TBI) (1). Both disorders are common in veterans, and while TBI is a risk factor for the development of PTSD, PTSD may be a mediator of TBI pathological consequences (2, 3, 4). Because PTSD and TBI are often comorbid in military personnel, diagnostic complexity due to overlap in PTSD and TBI symptoms, in addition to controversies in PTSD diagnostic criteria (5) represent current challenges for assessment and therapeutic interventions.
The hypothalamic pituitary adrenal (HPA) axis is one of the major neuroendocrine systems in the body. The classic view of the HPA axis involves a modulation of cortisol response under acute stress conditions, which can be altered by physical activity, inflammatory state and the influence of circadian rhythms (6, 7, 8). The general pathway involves integrated brain circuits that control the release of corticotropin‐releasing factor (CRF), also known as corticotropin‐releasing hormone (CRH), from the paraventricular nucleus (PVN) of the hypothalamus, which stimulates the release of adrenocorticotropic hormone (ACTH) from corticotropic cells of the pituitary. ACTH reaches the bloodstream and finally induces the systemic release of glucocorticoids by the adrenal glands and a global peripheral response controlled by the brain. More recent studies show a complex regulation of HPA axis activity in chronic stress and addiction (9, 10).
Early biomarkers studies in humans focused on peripheral cortisol determinations and typically reported increased levels in relation to acute stress. (11, 12) However, neuroendocrine studies consistently suggest an increased negative feedback inhibition of the HPA axis in PTSD (13).
Surprisingly, very few studies have looked at CRF as a possible blood biomarker for PTSD in humans. A study in cerebrospinal fluid (CSF) showed that patients with PTSD with secondary psychotic symptoms had higher CRF compared to PTSD without psychosis, and healthy subjects (14), implicating abnormalities in the secretion of CRF with the production of secondary psychotic symptoms in PTSD. A few studies in PTSD patients reported high levels of CRF in CSF (15), while diurnal plasma cortisol levels on average were decreased (16). One study on plasma CRF showed an increase in PTSD (17). However, the cohorts were of only 30 subjects, and 13 out of 31 PTSD patients were diagnosed with a current depressive episode (16). To our knowledge, no previous studies have proposed serum CRF to have diagnostic potential for the differentiation of PTSD and TBI, and studies on the differentiation of PTSD and TBI have been mostly limited to functional neuroimaging (18, 19).
Diagnosis of PTSD and TBI is most often determined from clinical interviews or self‐report assessments, both of which are prone to distortions and depend on the willingness of patients to discuss their traumatic experiences and related symptoms. Easily accessible blood biomarkers would aid in the understanding, diagnosis and treatment of both PTSD and TBI.
In the Cohen Veterans Center study, which involved many facets including deep phenotyping and imaging (20, 21), US military veterans of the Iraq and Afghanistan Wars were enrolled at NYU Medical Center and Stanford University. All subjects completed an in‐depth clinical interview with licensed psychologists for structured diagnostic screening for psychiatric disorders. In addition to the interviews, over 70% of enrolled subjects completed a series of self‐report questionnaires and agreed to blood draws for biomarker analysis.
Here, we report that serum CRF is significantly lower in a large group of combat veterans with PTSD compared to healthy veterans as well as veterans with evidence of past TBI defined by loss of consciousness (LOC).
Material and Methods
Participants
The individuals included in the study have been deployed to Iraq and/or Afghanistan within the last 10 years. We completed blood biomarker determinations in a total of 230 veterans (7.88% females, mean age 32.54 [s.d. ± 7.23] years) that were selected based on the following categories: healthy controls (n = 64), PTSD (n = 53), chronic TBI (cTBI) (n = 70), PTSD + cTBI (n = 43). Exclusion criteria included current severe medical conditions unrelated to TBI or PTSD and requiring long‐term treatments (e.g., cerebrovascular disorders, multiple sclerosis, cancer, autoimmune conditions and human immunodeficiency virus) or severe psychiatric conditions (i.e., schizophrenia or bipolar disorder, history of psychotic disorder, obsessive‐compulsive disorder and current suicide risk). Due to the nature of the study design, comorbidities that can be found in PTSD veterans exposed to war‐related stress including substance or alcohol use disorder (AUD) were not excluded from the study and included in the analysis. Approximately 50% of the subjects in the PTSD cohort had comorbid AUD. Table 1 shows demographic data for the study groups. All subjects provided written, informed consent to protocols approved by their institutional ethics boards in accordance with the Helsinki Declaration.
TABLE 1.
Demographic data
| Characteristic | TBI | PTSD | PTSD + TBI | Control | P Value |
|---|---|---|---|---|---|
| Age, mean (SD), y | 32.24 (7.65) | 32.54 (5.97) | 33.82 (7.95) | 31.58 (7.33) | 0.159 |
| Female, no. (%) | 3 (3.85%) | 5 (8.20%) | 3 (6.82%) | 8 (10.81%) | 0.411 |
| Race, no. (%) | 0.398 | ||||
| White | 40 (51.95%) | 32 (53.33%) | 20 (45.45%) | 41 (55.41%) | |
| African American | 5 (6.49%) | 8 (13.33%) | 8 (18.18%) | 11 (14.86) | |
| Native American or Hawaiian | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.35%) | |
| Asian | 5 (6.49%) | 6 (10.0%) | 1 (2.27%) | 9 (12.16) | |
| Hispanic | 19 (24.68%) | 11 (18.33%) | 11 (25%) | 9 (12.16) | |
| Others | 8 (10.39%) | 3 (5.0%) | 4 (9.09%) | 3 (4.05%) | |
| Time since last deploy. (months), mean (sd) | 27.78 (30.97) | 32.63 (38.61) | 43.01 (45.54) | 26.85 (39.60) | 0.136 |
| TBI group characteristics, mean (sd) | |||||
| CSI current score | 4.22 (7.94) | 6.81 (11.82) | 11.10 (12.39) | 0.74 (1.82) | 0.0013 |
| CSI lifetime score | 13.32 (13.48) | 13.27 (14.30) | 24.39 (19.84) | 5.42 (6.67) | 0.0007 |
| Loss of consciousness, minutes median | 0.75 | 0.75 | 0.900 | ||
| Time since last event, months mean (sd) | 75.62 (70.28) | 74.05 (46.31) | 0.507 | ||
Note: The significance is evaluated across four groups when the data of four groups are available, between TBI and TBI‐PTSD groups when the data of these two groups only are available.
Abbreviations: CSI, Concussion Symptoms Inventory; PTSD, posttraumatic stress disorder; SD, standard deviation; TBI, traumatic brain injury.
PTSD and TBI Diagnosis
PTSD diagnosis was established according to DSM‐5 criteria using the Clinically Administered PTSD Scale (CAPS‐5). Briefly, CAPS‐5 is a 30‐item interview, which provides information on frequency and severity of PTSD symptoms and functioning, used to make current (past month) and lifetime diagnosis of PTSD. PTSD status was based on the CAPS‐5, with additional follow‐up questions included in the assessment from the CAPS‐IV (i.e., measures of intense fear/helplessness/horror at the time of exposure and sense of foreshortened future), in order to score data and obtain diagnoses based on both DSM‐IV and DSM‐5 criteria. Beck Depression Inventory (BDI), Pittsburgh Sleep Quality Index (PSQI), Pain Rating Scale and Recent Medication were also ascertained for every subject.
To be considered a TBI case, participants reported a history of previous TBI (1–40 years before the blood draw, with mean = 12.9 years) based on closed head injury with LOC. Ohio State University TBI Identification Method–Short Form was used for assessment of TBI. Concussion Symptoms Inventory (CSI) was used for the assessment of post concussive symptoms. LOC time and time since last LOC to blood draw were included as variables. Controls and PTSD cases followed the same inclusion and exclusion criteria showing no history of TBI. Alcohol use was considered positive if SCID lifetime or current alcohol abuse was moderate or severe.
Biofluid Samples
Blood samples were collected between 9 and 10 a.m. under fasting conditions and following blood processing standardized guidelines for biomarkers studies (22). Upon arrival at the clinical site, subjects were asked to rest in a supine position for 30 min prior to venipuncture. Nurses completed blood draws using 21G butterfly needles. For serum extraction, 10 mL of blood were collected into BD Vacutainer Serum Separation Tubes (Thermo Fisher), allowed to clot for 30 min prior to centrifugation (10 min, 2000 g, 4°C). All samples were always kept at 4°C during processing, and stored at −80°C within the first 2 h after the blood draw. All samples were aliquoted into low binding profile tubes (1 ml) labeled with barcodes and stored at −80°C until use. To avoid batching effects, once sufficient samples were collected, experiments were pre‐designed including a similar number of individuals from all study groups. To avoid multiple freeze/thawing cycles, samples were thawed once, aliquoted into smaller fractions (0.050 ml) and stored at −80°C until use.
Determination of CRF Levels
Experiments were performed at the Biofluid Biomarkers Core of the Cohen Veteran Center in Dr. Fossati's lab at NYULMC. Operators performing assays were blind to study group. Barcode readers were used to allow for sample blinding. CRF was measured in serum samples by an enzyme immunoassay (EIA) (Phoenix Pharmaceuticals Inc.). Absorbance was detected using a Spectra Max i3 system (Molecular Devices). Limit of detection (LOD) for the assay is 0.33 ng/ml, Intra‐assay variation is below 10%, and Inter‐assay variation, below <15%. The assay is specific for human CRF, with no cross‐reactivity for human Prepro‐CRF (125‐151), PACAP‐38, LH‐RH, ACTH or [Arg8]‐Vasopressin. All serum samples were well above the LOD and within the levels expected from the assay specifications. All assays were run in duplicate. Previously obtained serum aliquots from healthy donors were included in all experiments and used for batch normalization. Samples showing coefficients of variation higher than 20% were excluded and measured again.
Statistical Analysis
CRF concentration was treated as continuous variables, expressed in nanograms per milliliter (ng/mL) and medians and interquartile ranges were obtained. An analysis of variance (ANOVA) was run to test the null hypothesis that the means of the four groups are equal. Subsequently, after rejecting the null, pairwise comparisons were run to see which of the means differed. Wilcoxon Rank sum tests were used to compare differences between groups in serum CRF values in six pairwise contrasts. To protect the familywise error rate at 0.05, a Bonnferroni correction was applied, that is, p‐values must be divided by 6. To be considered strong evidence of a difference, an adjusted p‐value needs to be less than or equal to 0.0083. The p‐value for tests involving variables other than CRF are reported as unadjusted p‐values. The area under the receiver operating characteristic (ROC) curve (AUC) was determined for serum CRF concentrations based on a logistic regression with 5‐fold cross validation. This function provides a model for the probability of group membership as a function of CRF level. For classification, a threshold was chosen as determined by the Youden index (J), which corresponds to the point on the ROC where sensitivity and specificity are equally important [J = Maxc (Sensitivityc + SpecificityC −1)]. Pearson and Spearman correlations for serum CRF with other study variables were obtained. Unless otherwise indicated, data was reported as mean ± SD. Statistical analyses for demographic and clinical variables were performed using SAS 9.4 (SAS Institute) and R (R Foundation for Statistical Computing). Both R and GraphPad Prism 7 (GraphPad Software Inc.) were used for data visualization.
Results
Demographic and Clinical Characteristics
The demographic characteristics of the 230 participants are shown in Table 1. Participants included: PTSD group (n = 53); cTBI group (n = 70); PTSD + TBI group (n = 43); and control group (n = 64). Basic demographics including age, sex, and race distributions did not differ significantly between study groups. As expected, CSI current and lifetime scores differed between TBI and the other groups (P < 0.05). Because the sample reflects the veteran population, the percentage of female subjects was significantly lower (7.88%) than the males. This represents a limitation common to studies of PTSD and TBI in active duty military and veteran samples and is discussed below.
Relationship Between Serum CRF Levels, PTSD and TBI Measures
The ANOVA test of difference of the four means was highly significant with a p‐value of <0.001. The ANOVA table may be seen in the Supplementary material. Comparing serum CRF levels among the study groups, no differences were found between controls (4.04 ± 0.92 ng/ml) and TBI groups (4.16 ± 0.61 ng/ml) or between PTSD (3.60 ± 0.62 ng/ml) and PTSD + TBI groups (3.59 ± 0.59 ng/ml) (P > 0.05). Significantly lower CRF concentrations were found in both the PTSD and PTSD + TBI groups compared with controls and with the TBI‐only group (P = 0.0014, Effect size [Cohen's d ES] = 0.57 for the comparison between controls and PTSD; P = 0.0011, ES = 0.60 for the comparison between controls and PTSD + TBI; P < 0.0001 and ES of 0.907 for the comparison of PTSD and TBI and P < 0.0001 and ES of 0.934 for the comparison of PTSD + TBI and TBI) (Figure 1). The ROC analyses showed an AUC of 0.73 for the classification of controls versus PTSD (sensitivity = 0.78, specificity = 0.75), of 0.83 for the classification of PTSD versus TBI (sensitivity = 0.82, specificity = 0.75), and 0.80 for the classification of pulled controls and TBI versus pulled PTSD and PTSD + TBI groups (sensitivity = 0.78, specificity = 0.75) (Figure 1B). However, a more conservative estimate from the logistic regression model 5‐fold cross‐validation yielded a mean AUC of 0.676 for control vs. PTSD, and 0.752 for PTSD versus TBI.
FIGURE 1.
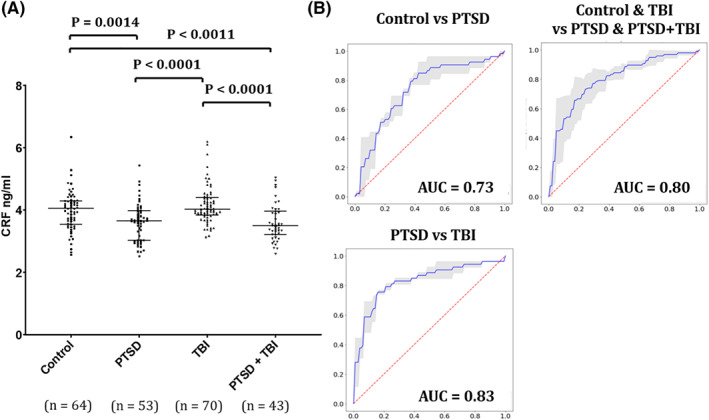
Serum CRF levels differentiate PTSD from controls and TBI patients. (A) Wilcoxon Rank sum test for the group comparisons of CRF serum levels (Y axis = CRF concentration in ng/ml). Bars represent medians and interquartile ranges. (B) Receiver operator curve (ROC) for serum CRF levels separates healthy previously deployed controls from PTSD (AUC = 0.73), PTSD from TBI participants (0.83) and the pulled groups including controls and TBI versus PTSD and PTSD + TBI (AUC = 0.80). AUC, area under the ROC curve (AUC); CRF, corticotropin‐releasing factor; PTSD, posttraumatic stress disorder; SD, standard deviation; TBI, traumatic brain injury
CRF levels were not correlated with duration of LOC (P > 0.05) or time since last LOC within the TBI subjects (P > 0.05).
Because lower serum CRF levels were detected in participants with PTSD, we then explored a potential relationship between CRF levels and the clinician‐administered PTSD scale for DSM‐5 (CAPS‐5). Results showed lower serum CRF levels in subjects with higher CAPS‐5 scores (higher PTSD symptomatology) when subjects were divided in quartiles (Figure 2), both for current CAPS scores (P = 0.0001 for 1st vs. 2nd tertile; p = 0.07 for 1st vs. 3rd) and for lifetime CAPS‐5 evaluation (P = 0.002 for 1st vs. 2nd tertile; p = 0.0007 for 1st vs. 3rd). The correlation between current and lifetime CAPS‐5 and CRF levels was statistically significant in the whole group (Pearson correlation: −0.29, P value < 0.0001; Spearman correlation: −0.31, P value < 0.0001), although it was not maintained in the PTSD group alone. Additionally correlations of individual CAPS‐5 symptom clusters (B,C,D,E) with CRF were not significant in the PTSD group.
FIGURE 2.
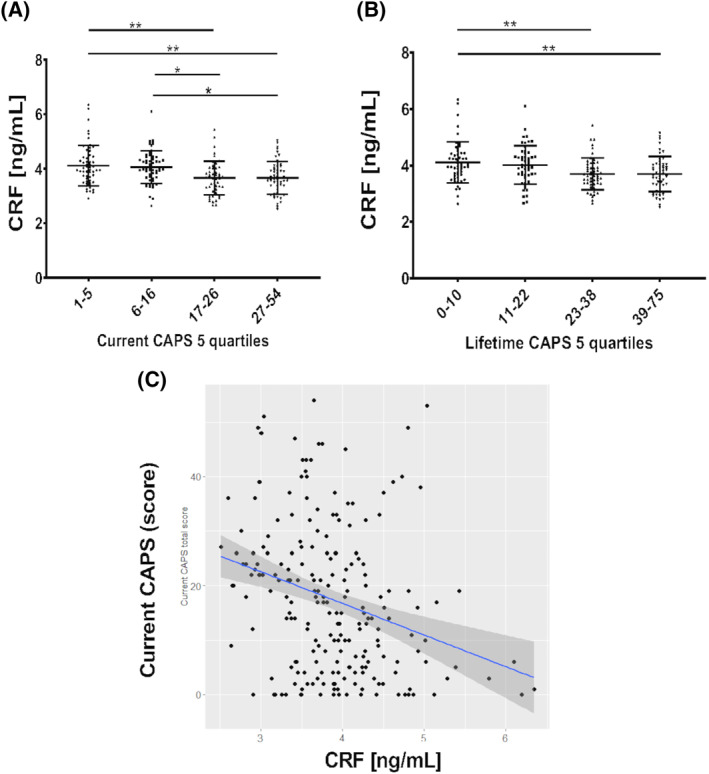
Differences in CRF levels in subjects within quartiles of PTSD symptomatology (measured by clinician‐administered PTSD scale for DSM‐5 [CAPS‐5]). Similar reductions are observed for current (A) and lifetime CAPS‐5 evaluation (B) Each quartile comprises 25% of the participants with available CRF and CAPS measures (total subjects: N = 211 for both current caps and lifetime CAPS). Bars represent mean and standard deviation. * = p < 0.05; ** = p < 0.01 (ANOVA and Tukey's multiple comparison tests). (C) Correlation of current CAPS score with serum CRF in the whole study group is moderate (Spearman r = 0.31; p < 0.0001; Pearson r = −0.29; p < 0.0001). ANOVA, analysis of variance; CAPS, Clinically Administered PTSD Scale; CRF, corticotropin‐releasing factor; PTSD, posttraumatic stress disorder
In order to detect a possible effect of alcohol use within PTSD patients on CRF levels, we separated PTSD subjects with and without comorbid AUD and compared CRF levels between PTSD subjects with AUD, PTSD subjects without AUD and controls. When comparing the two PTSD sub‐groups to healthy controls, both had significantly lower CRF, although the magnitude of the difference between PTSD and controls was more substantial in PTSD subjects with AUD (Wilcoxon p = 0.0038, Cohen's d 0.6) compared to PTSD subjects without AUD (Wilcoxon p = 0.02; Cohen's d = 0.58) (Figure 3). We did not observe significant differences in CRF levels among PTSD subjects with and without AUD, or in PTSD subjects with or without depression.
FIGURE 3.
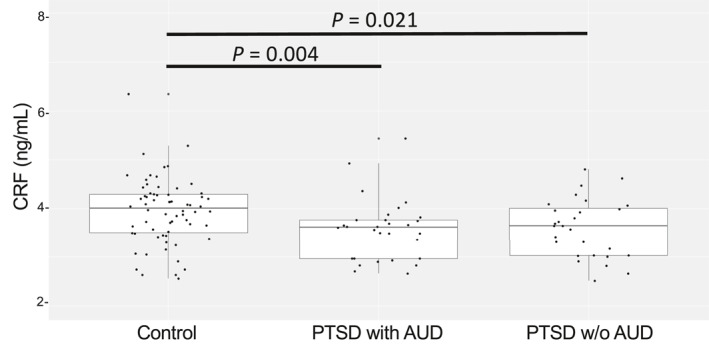
Relationship between alcohol abuse (AUD) and CRF levels. The strength for the differentiation of PTSD from control participants is evident in PTSD subjects with alcohol use (AUD). Wilcoxon p is 0.0038 in PTSD with AUD (N = 26) versus Controls (N = 64) and 0.02 in PTSD without AUD (N = 27) versus Controls. No differences in serum CRF levels are found when comparing PTSD participants with and without AUD. Wilcoxon Rank sum test for the group comparisons. Participants with AUD alone were excluded from the study group. CRF, corticotropin‐releasing factor; PTSD, posttraumatic stress disorder
Discussion
The HPA axis is one of the major neuro‐hormonal systems mediating the stress response in the human body and neuroendocrine studies consistently provide evidence for altered HPA activity in PTSD. (23) The present report points for the first time to reduced serum CRF as a promising biomarker for differentiating PTSD from health individuals and from subjects with chronic TBI. We found that CRF levels were lower in veterans with PTSD and PTSD + TBI, compared to healthy veterans and veterans with a history of TBI, with moderate to large effect sizes. Our findings suggest that, if validated, serum CRF may be considered for membership in future blood biomarker panels for PTSD.
The activity of the HPA axis is regulated by multiple sympathetic and parasympathetic inputs and limbic circuits (e.g., amygdala, hippocampus, and medial prefrontal cortex), directly or indirectly innervating the PVN of the hypothalamus (24). In healthy subjects, integrated brain circuits trigger the parvocellular neurons of the PVN to release infundibular CRF, which stimulates the release of ACTH from corticotropic cells of the pituitary. ACTH reaches the bloodstream and triggers the systemic release of glucocorticoids by the adrenals. The classic view of the HPA axis involves activation of cortisol response under acute stress conditions. However, recent studies show a complex modulation including enhanced feedback inhibitory systems in chronic stress and addiction (9, 10).
Animal studies demonstrate that HPA axis responsivity to acute stressors is altered in animals that have previously been stressed, suggesting a long‐term negative feedback modulation of the HPA axis in response to chronic stress (25). While exposure to severe stressors causes long term dysregulation of resting state and stress‐induced activation of the HPA axis (26, 27, 28), the mechanisms of such modulation are complex and unclear, and both increased and blunted cortisol response are observed depending on the intensity and chronicity of the stressors. (29) Early biomarkers studies in humans typically focused on plasma or saliva cortisol determinations and reported, as a general rule, increased levels in response to acute stress, (11, 12) suggesting that PTSD biology was compatible with a stress response, associated with increased CRF levels, catecholamine depletion within the central nervous system, and reduced hippocampal volume. (30) However, the activation of the HPA axis may vary with duration of PTSD. Cross‐sectional and prospective studies are suggestive of a hypoactive HPA axis in PTSD (31). Indeed, lower urinary and plasma cortisol levels were detected in chronic PTSD patients compared to non‐PTSD trauma survivors and controls. (30) This was confirmed by experiments using dexamethasone as an HPA axis inhibitor in different cohorts. A study by Yehuda and colleagues showed lower post‐DEX ACTH levels in deployed veterans than in a non‐deployed veteran group, suggesting a link between post‐deployment symptoms and chronic stress (32). In our study, we observed a clear reduction in CRF levels in the chronic PTSD group that was also detected in the PTSD + TBI group, but not in the TBI alone, suggesting that the effect may mediated by a biological mechanism related to PTSD and independent from TBI. Our PTSD cohort included both subjects with (n = 42) and without (n = 54) current depression. We didn't observe significant differences in these two subgroups, suggesting that the lower levels of CRF are driven by PTSD and not by the comorbid depression. Additionally, the correlation of serum CRF with BDI was not significant, except for the PTSD + TBI group, in which a positive correlation was observed between BDI and CRF (r = 0.555; P = 0.001). This further confirms that our findings of decreased CRF in PTSD are unrelated to depressive symptoms, as in this group higher CRF levels associated with a higher depression score. To the best of our knowledge, this serum CRF reduction in PTSD compared to controls and TBI subjects has not been described before and is in line with a more recent perspective of PTSD biology that involves a chronic stress state and enhanced negative feedback of the HPA axis. This effect can also be related to the higher sensitivity of glucocorticoid receptors in peripheral tissues that has been previously observed in animal models (30, 33, 34). Our results are also in line with a recent study, which found a reduction in pituitary gland size and altered function in PTSD. That study reported discordant pituitary corticotropin function and pituitary volume in PTSD subjects compared with intact HPA feedback and association of pituitary volume with ACTH levels in healthy control subjects (35).
The observed low serum CRF in chronic PTSD patients may be also consistent with elevated brain CRF utilization, increased CRF metabolism, increased degradation of CRF, or reduction in the CNS secretion of CRF (36).
Alternatively, low serum CRF may represent a risk factor for PTSD. This possibility should be further explored in longitudinal studies with available blood obtained from participants pre‐ and post‐deployment or pre‐ and post‐trauma exposure in civilians.
Since psychotropic medications such as benzodiazepines and antidepressants may influence CRF levels (37), we have compared PTSD subjects (including PTSD + TBI) under benzodiazepines and antidepressant treatments with those without drug treatment. We found that the levels of serum CRF did not differ in the two groups (p = 0.472), confirming that the CRF reduction in PTSD is not due to the use of these medications.
Serum CRF is known to be increased in multiple conditions, including fibromyalgia, mastocytosis, psoriasis, obesity, and pregnancy (released by the placenta) (38, 39, 40, 41). All participants of our study, including the controls, were subjected to a thorough clinical interview and all possible current heath conditions were reported. None of the participants were found to have current fibromyalgia, mastocytosis or psoriasis at the time of the interview and/or blood draw. In addition, we excluded from this study any pregnant subjects.
Low levels of CRF are also observed in tertiary (hypothalamic) adrenal disease (CRF deficiency) (42). Both low and elevated CRF levels have been shown in people with Alzheimer's disease (43, 44), and some scientists suspect that a lack of CRF might cause chronic fatigue syndrome (45). However, further research is needed in both these topics to confirm these findings.
We detected lower CRF levels in subjects within the higher CAPS scores quartiles, for both current and lifetime CAPS (Figure 2), and a moderate negative correlation between CRF and CAPS‐5 in the full group. However, there was no correlation between CAPS‐5 and CRF within the PTSD group, suggesting that the relationship with CRF is likely driven by the differences in CAPS scores between controls and PTSD subjects and is not directly related to the severity of symptoms within the PTSD patients. Further, there was no correlation between CRF and the individual CAPS‐5 symptom clusters (B, C, D or E) in the PTSD group, or between CRF and PSQI.
The reduction in CRF levels had a larger effect size in PTSD patients with reported moderate or severe alcohol use when compared to controls, likely due to the lower variability of CRF levels in the PTSD + AUD group. Alcohol abuse can reach a prevalence of more than 50% in PTSD patients (46, 47, 48) and it is known to be associated with more severe PTSD symptoms and higher levels of cognitive disturbances. Although AUD was not the main focus of this study, which did not include participants with AUD in absence of PTSD, the analysis of HPA axis mediators such as CRF in comorbid PTSD and AUD as well as in AUD alone warrants further attention for a potential biological interaction.
Serum cortisol levels measured in a subset of our subjects (N = 83) did not show significant group differences (Control vs. PTSD: p = 0.46; Control vs. PTSD + TBI: p = 0.33; PTSD vs. PTSD + TBI: p = 0.95), in line with a meta‐analysis in which, across 37 studies, 828 people with PTSD and 800 controls did not differ in cortisol levels. (49) The lack of difference persisted in this meta‐analysis when considering only studies analyzing plasma or serum cortisol. (49).
Although the brain is the main source of CRF production related to stress, CRF is released from the PVN neurons that terminate in the median eminence, a region with fenestrated blood vessels lacking the tight junctions characteristic of the BBB. Here, CRF is released into the peripheral circulation as a hormone. Peripheral CRF (or CRH) plays multiple roles associated to the stress response. For example, CRF released from the brain into the periphery during stress responses induces a colonic response by activating colonic CRF receptors (50). These and other peripheral mechanisms are mediated by central stress, as can be displayed by the “gut wrenching” and nauseous feelings and abdominal pain during or following emotional stress.
Hence, although due to the nature of our samples we cannot demonstrate that serum CRF levels correlate with those in the brain in our cohort, there is evidence indicating that peripheral CRF levels can be indicative of CNS perturbations, and associated to emotional disturbances and HPA axis responses. Previous research also suggests that a major component of plasma CRF is of hypothalamic origin, however, other extrahypothalamic tissues cannot be ruled out as a minor source of plasma CRF (51). Other studies have shown lower levels of peripheral CRF (plasma) and an increase in PTSD (17). However, the study did not control for depression, and patients with severe depression were almost half of the PTSD group in this study. We have observed an opposite direction of the correlation in depression, with an increased CRF correlating with increased depression scores (BDI) in the PTSD + TBI group. In addition, this study used an RIA assay, while we use EIA. Differences in apparent concentrations are conceivable due to the inherent technical differences of these assays. The higher overall concentration found with the commercial EIA assay we used, compared to other previously used RIA and Mass spectrometry assays for CRF is a limitation of our study, and the results should be confirmed in the future employing mass spectrometry, RIA, or other quantitative types of analysis to confirm concentrations. High backgrounds of the EIA assay, as well as possible detection of both bound and unbound CRF are possible reasons. Nevertheless, the strong effect size of our group differences in serum CRF (in controls vs PTSD and PTSD + TBI groups) reinforces the confidence that these results are biologically important, independently from possible assay‐dependent variability. We believe that the present data need to be reported, so that additional studies can focus on serum CRF determinations in PTSD and PTSD + TBI populations using multiple different assays.
The present report has some limitations that will need to be explored in future studies. First, due to the veteran nature of our study sample, to validate CRF as a biomarker for PTSD results will need to be confirmed in separate cohorts of civilian subjects. Second, our study included relatively young veterans with a homogeneous age (average 32.54) and future studies should explore if results hold in elderly subjects and if CRF levels are related to age. Third, it is important to consider that sex can affect HPA axis activity, as established by multiple animal and human studies which highlight an effect of sex hormones on stress responses (52, 53). Our sample of Iraq and Afghanistan veterans consisted of 7.88% female subjects. Therefore, results will need to be confirmed in validation cohorts comprising a balanced number of men and women and in studies of female veterans and civilians with PTSD and TBI. Additional limitations are the lack of cortisol level measurments in all patients and the lack of a group of AUD participants without PTSD. The relationships between alcohol use and serum CRF levels as well as circadian rhythm and CRF levels should be explored in future studies. Additionally, our study did not account for potential binding of CRF to CRF‐binding protein in plasma which may impact CRF soluble levels. Future studies will be planned to assess these molecular events. Finally, longitudinal studies analyzing CRF levels before, immediately after trauma, and in chronic PTSD within the same patients, would help understanding if the lower CRF levels found in PTSD subjects are the result of a chronic stress state, or if lower CRF is an inherent characteristic of certain individuals, which constitutes a risk factor for PTSD. Studies will need to further confirm if the observed results are due, at least in part, to changes in CRF of peripheral origin, since CRF is known to be produced by other peripheral sources including immune and endothelial cells, the adrenal medulla, testes, ovaries, GI tract, pancreas, and the myometrium.
Conclusion
We report for the first time reduced serum CRF levels in PTSD participants compared to TBI and healthy controls in our Iraq and Afghanistan veteran cohort. We propose that future studies should evaluate peripheral CRF as a possible mechanistic mediator and a potential diagnostic biomarker to differentiate chronic PTSD from healthy and TBI subjects. Although it is still premature to consider CRF as a valuable PTSD biomarker, this study supports the need for a validation of serum CRF reduction in different cohorts including women and civilians with PTSD, in those with AUD comorbid with PTSD, and in prospective longitudinal studies, using multiple assays.
Supporting information
Supporting information 1
The project described was made possible with support by grants from the Steven A. and Alexandra M. Cohen Foundation, Inc. and Cohen Veterans Bioscience, Inc. (CVB) to CM and NYU School of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Foundation or CVB. In addition, this work was supported in part by the American Heart Association Grant 13SDG16860017, the Leon Levy Fellowship in Neuroscience, the NYU CTSA grant UL1TR001445 from the National Center for Advancing Translational Sciences (NCATS), NIH, NIH R01NS104127 awarded to SF, and NIAAA P01AA027057 awarded to CM. AE was supported by the Sierra‐Pacific Mental Illness Research, Education and Clinical Center (MIRECC) at the Palo Alto VA.
The authors declare no conflicts of interest related to this work. AE received stock options at Mindstrong Health and Akili Interactive for unrelated consulting. CRM received unrelated support from the NIAAA, Department of Defense, Cohen Veterans Network, Robin Hood Foundation, McCormick Foundation, Home Depot Foundation, and the City of New York.
SF and JRC designed the study, analyzed data and wrote the manuscript. JRC and LD performed biomarkers experiments. CRM and AE provided the clinical cohorts and gave important contributions to study design and manuscript editing. AG, DAA, NM and EB contributed to clinical and biomarker data collection, organization and retrieval, biofluid banking and project/data management. MQ, ML, EL and CS contributed with statistical data analysis and statistical advice. EL and CS helped in manuscript revision. JN and CM provided guidance on clinical phenotyping of subjects.
REFERENCES
- 1. Howlett JR, Stein MB. Post‐traumatic stress disorder: relationship to traumatic brain injury and approach to treatment; in Laskowitz D, Grant G, editors. Translational Research in Traumatic Brain Injury. Edited by Laskowitz D, Grant G. Boca Raton, 2016. [PubMed] [Google Scholar]
- 2. Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N Engl J Med 2008;358:453–463. [DOI] [PubMed] [Google Scholar]
- 3. Yurgil KA, Barkauskas DA, Vasterling JJ, Nievergelt CM, Larson GE, Schork NJ, et al. Association between traumatic brain injury and risk of posttraumatic stress disorder in active‐duty Marines. JAMA psychiatry 2014;71:149–157. [DOI] [PubMed] [Google Scholar]
- 4. Shalev A, Liberzon I, Marmar C. Post‐traumatic stress disorder. N Engl J Med 2017;376:2459–2469. [DOI] [PubMed] [Google Scholar]
- 5. Hoge CW, Yehuda R, Castro CA, McFarlane AC, Vermetten E, Jetly R, et al. Unintended consequences of changing the definition of posttraumatic stress disorder in DSM‐5. JAMA Psychiatry 2016;73:750–752. [DOI] [PubMed] [Google Scholar]
- 6. Onakomaiya MM, Porter DM, Oberlander JG, Henderson LP. Sex and exercise interact to alter the expression of anabolic androgenic steroid‐induced anxiety‐like behaviors in the mouse. Horm Behav 2014;66:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neumann A‐M, Schmidt CX, Brockmann RM, Oster H : Circadian regulation of endocrine systems. Auton Neurosci 2019;216:1–8. [DOI] [PubMed] [Google Scholar]
- 8. Bauer ME, Teixeira AL. Inflammation in psychiatric disorders: what comes first? Ann N Y Acad Sci. 2018;1437:57–67. [DOI] [PubMed] [Google Scholar]
- 9. Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol 2014;35:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Logrip ML, Zorrilla EP, Koob GF. Stress modulation of drug self‐administration: implications for addiction comorbidity with post‐traumatic stress disorder. Neuropharmacology 2012;62:552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, et al. Pituitary‐adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. Jama 2000; 284:592–597. [DOI] [PubMed] [Google Scholar]
- 12. Elzinga BM, Schmahl CG, Vermetten E, Van Dyck R, Bremner JD. Higher cortisol levels following exposure to traumatic reminders in abuse‐related PTSD. Neuropsychopharmacology 2003;28:1656–1665. [DOI] [PubMed] [Google Scholar]
- 13. Yehuda R, Hoge CW, McFarlane AC, Vermetten E, Lanius RA, Nievergelt CM, et al. Post‐traumatic stress disorder. Nat Rev Dis Primers. 2015;1:15057. [DOI] [PubMed] [Google Scholar]
- 14. Sautter FJ, Bissette G, Wiley J, Manguno‐Mire G, Schoenbachler B, Myers L, et al. Corticotropin‐releasing factor in posttraumatic stress disorder (PTSD) with secondary psychotic symptoms, nonpsychotic PTSD, and healthy control subjects. Biol Psychiatr 2003;54:1382–1388. [DOI] [PubMed] [Google Scholar]
- 15. Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, et al. Serial CSF corticotropin‐releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatr 1999;156:585–588. [DOI] [PubMed] [Google Scholar]
- 16. Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol Psychiatr 1996; 40:79–88. [DOI] [PubMed] [Google Scholar]
- 17. De Kloet CS, Vermetten E, Geuze E, Lentjes EG, Heijnen CJ, Stalla GK, et al. Elevated plasma corticotrophin‐releasing hormone levels in veterans with posttraumatic stress disorder. Prog Brain Res 2008;167:287–291. [DOI] [PubMed] [Google Scholar]
- 18. Amen DG, Raji CA, Willeumier K, Taylor D, Tarzwell R, Newberg A, et al. Functional neuroimaging distinguishes posttraumatic stress disorder from traumatic brain injury in focused and large community datasets. PloS One 2015;10:e0129659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raji CA, Willeumier K, Taylor D, Tarzwell R, Newberg A, Henderson TA, et al. Functional neuroimaging with default mode network regions distinguishes PTSD from TBI in a military veteran population. Brain Imaging Behav 2015;9:527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Etkin A, Maron‐Katz A, Wu W, Fonzo GA, Huemer J, Vertes PE, et al. Using fMRI connectivity to define a treatment‐resistant form of post‐traumatic stress disorder. Sci Transl Med. 2019;11:eaal3236. 10.1126/scitranslmed.aal3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maron‐Katz A, Zhang Y, Narayan M, Wu W, Toll RT, Naparstek S, et al. Individual patterns of abnormality in resting‐state functional connectivity reveal two data‐driven PTSD subgroups. Am J Psychiatr 2020;177:244–253. [DOI] [PubMed] [Google Scholar]
- 22. O'Bryant SE, Gupta V, Henriksen K, Edwards M, Jeromin A, Lista S, et al. Guidelines for the standardization of preanalytic variables for blood‐based biomarker studies in Alzheimer's disease research. Alzheimer's Dementia 2015;11:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yehuda R, Giller EL, Southwick SM, Lowy MT, Mason JW. Hypothalamic‐pituitary‐adrenal dysfunction in posttraumatic stress disorder. Biol Psychiatr 1991;30:1031–1048. [DOI] [PubMed] [Google Scholar]
- 24. Bellavance MA, Rivest S. The HPA‐immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front Immunol 2014;5:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ladd CO, Owens MJ, Nemeroff CB. Persistent changes in corticotropin‐releasing factor neuronal systems induced by maternal deprivation. Endocrinology 1996;137:1212–1218. [DOI] [PubMed] [Google Scholar]
- 26. Daskalakis NP, Lehrner A, Yehuda R. Endocrine aspects of post‐traumatic stress disorder and implications for diagnosis and treatment. Endocrinol Metab Clin N Am 2013;42:503–513. [DOI] [PubMed] [Google Scholar]
- 27. Golier JA, Caramanica K, Yehuda R. Neuroendocrine response to CRF stimulation in veterans with and without PTSD in consideration of war zone era. Psychoneuroendocrinology 2012;37:350–357. [DOI] [PubMed] [Google Scholar]
- 28. Belda X, Rotllant D, Fuentes S, Delgado Rl, Nadal R, Armario A. Exposure to severe stressors causes long‐lasting dysregulation of resting and stress‐induced activation of the hypothalamic‐pituitary‐adrenal axis. Ann N Y Acad Sci 2008;1148:165–173. [DOI] [PubMed] [Google Scholar]
- 29. McKittrick CR, Caroline Blanchard D, Blanchard RJ, McEwen BS, Sakai RR. Serotonin receptor binding in a colony model of chronic social stress. Biol Psychiatr 1995;37:383–393. [DOI] [PubMed] [Google Scholar]
- 30. Yehuda R. Biology of posttraumatic stress disorder. J Clin Psychiatr 2001;62 (Suppl 17):41–46. [PubMed] [Google Scholar]
- 31. Lehrner A, Daskalakis N, Yehuda R. Cortisol and the hypothalamic‐pituitary‐adrenal axis in PTSD. In: Bremer JD, editor. Posttraumatic stress disorder: from neurobiology to treatment. John Wiley & Sons; 2016. pp. 265–290. 10.1002/9781118356142.ch11 [DOI] [Google Scholar]
- 32. Golier JA, Legge J, Yehuda R. The ACTH response to dexamethasone in Persian Gulf War veterans. Ann N Y Acad Sci 2006; 1071:448–453. [DOI] [PubMed] [Google Scholar]
- 33. Louvart H, Maccari S, Vaiva G, Darnaudéry M. Prenatal stress exacerbates the impact of an aversive procedure on the corticosterone response to stress in female rats. Psychoneuroendocrinology 2009;34:786–790. [DOI] [PubMed] [Google Scholar]
- 34. Faye C, McGowan JC, Denny CA, David DJ. Neurobiological mechanisms of stress resilience and implications for the aged population. Curr Neuropharmacol 2018;16:234–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cooper O, Bonert V, Moser F, Mirocha J, Melmed S. Altered pituitary gland structure and function in posttraumatic stress disorder. J Endocr Soc 2017;1:577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geracioti TD, Jr, Baker DG, Kasckow JW, Strawn JR, Jeffrey Mulchahey J, Dashevsky BA, et al. Effects of trauma‐related audiovisual stimulation on cerebrospinal fluid norepinephrine and corticotropin‐releasing hormone concentrations in post‐traumatic stress disorder. Psychoneuroendocrinology 2008;33:416–424. [DOI] [PubMed] [Google Scholar]
- 37. Wilson MA, Biscardi R, Smith MD, Wilson SP. Effects of benzodiazepine agonist exposure on corticotropin‐releasing factor content and hormonal stress responses: divergent responses in male and ovariectomized female rats. J Pharmacol Exp Therapeut 1996;278:1073–1082. [PubMed] [Google Scholar]
- 38. Tsilioni I, Russell IJ, Stewart JM, Gleason RM, Theoharides TC, Neuropeptides CRH SP. Neuropeptides CRH, SP, HK‐1, and inflammatory cytokines IL‐6 and TNF are increased in serum of patients with fibromyalgia syndrome, implicating mast cells. J Pharmacol Exp Therapeut 2016;356:664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vasiadi M, Therianou A, Sideri K, Smyrnioti M, Sismanopoulos N, Delivanis DA, et al. Increased serum CRH levels with decreased skin CRHR‐1 gene expression in psoriasis and atopic dermatitis. J Allergy Clin Immunol 2012;129:1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Theoharides TC, Petra AI, Stewart JM, Tsilioni I, Panagiotidou S, Akin C. High serum corticotropin‐releasing hormone (CRH) and bone marrow mast cell CRH receptor expression in a mastocytosis patient. J Allergy Clin Immunol 2014;134:1197–1199. [DOI] [PubMed] [Google Scholar]
- 41. Petraglia F, Sawchenko PE, Rivier J, Vale W. Evidence for local stimulation of ACTH secretion by corticotropin‐releasing factor in human placenta. Nature 1987;328:717–719. [DOI] [PubMed] [Google Scholar]
- 42. Fehm H, Voigt K, Lang R, Hetzel W, Pfeiffer E. Adrenal insufficiency secondary to hypothalamic corticotropin releasing factor (CRF) insufficiency with hyperpigmentation: a case report. Horm Metab Res 1976;8:470–474. [DOI] [PubMed] [Google Scholar]
- 43. Florio P, Zatelli MC, Reis FM, degli Uberti EC, Petraglia F. Corticotropin releasing hormone: a diagnostic marker for behavioral and reproductive disorders? Front Biosci 2007;12:551–560. [DOI] [PubMed] [Google Scholar]
- 44. Ishii M, Iadecola C. Metabolic and non‐cognitive manifestations of Alzheimer's disease: the hypothalamus as both culprit and target of pathology. Cell Metabol 2015;22:761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cleare AJ, Miell J, Heap E, Sookdeo S, Young L, Malhi GS, et al. Hypothalamo‐pituitary‐adrenal axis dysfunction in chronic fatigue syndrome, and the effects of low‐dose hydrocortisone therapy. J Clin Endocrinol Metab 2001;86:3545–3554. [DOI] [PubMed] [Google Scholar]
- 46. Blakey SM, Wagner HR, Naylor J, Brancu M, Lane I, Sallee M, et al. Chronic pain, TBI, and PTSD in military veterans: a link to suicidal ideation and violent impulses? J Pain. 2018;19:797–806. 10.1016/j.jpain.2018.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blanco C, Hoertel N, Wall MM, Franco S, Peyre H, Neria Y, et al. Toward understanding sex differences in the prevalence of posttraumatic stress disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatr 2018;79:16m11364. [DOI] [PubMed] [Google Scholar]
- 48. Black AC, Cooney NL, Sartor CE, Arias AJ, Rosen MI. Impulsivity interacts with momentary PTSD symptom worsening to predict alcohol use in male veterans. Am J Drug Alcohol Abuse. 2018;44:524–531. 10.1080/00952990.2018.1454935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meewisse M‐L, Reitsma JB, de Vries G‐J, Gersons BPR, Olff M. Cortisol and post‐traumatic stress disorder in adults. Br J Psychiatry 2007;191:387–392. [DOI] [PubMed] [Google Scholar]
- 50. Taché Y, Million M. Role of corticotropin‐releasing factor signaling in stress‐related alterations of colonic motility and hyperalgesia. J Neurogastroenterol Motil 2015;21:008–024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suda T, Tomori N, Yajima F, Sumitomo T, Nakagami Y, Ushiyama T, et al. Immunoreactive corticotropin‐releasing factor in human plasma. J Clin Invest 1985;76:2026–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stockhorst U, Antov MI. Modulation of fear extinction by stress, stress hormones and estradiol: a review. Front Behav Neurosci 2015;9:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goel N, Workman JL, Lee TT, Innala L, Viau V. Sex differences in the HPA axis. Comp Physiol 2014;4:1121–1155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information 1