

Abstract
Background:
There is strong evidence that epigenetic age acceleration is associated with increased risk of later-life diseases and all-cause mortality. However, there is currently limited evidence that suggests accelerated epigenetic age is associated with dementia risk.
Objective:
This study aims to clarify whether epigenetic biomarkers of accelerated aging can predict dementia risk, which is an important consideration as aging is the greatest risk factor for the disease.
Methods:
DNA methylation was measured in peripheral blood samples provided by 160 participants from the ASPirin in Reducing Events in the Elderly study, including 73 pre-symptomatic dementia cases and 87 controls matched for age, sex, and smoking and education status. Epigenetic age was calculated using Horvath, Hannum, GrimAge and PhenoAge DNA methylation clocks, and age acceleration (the disparity between chronological age and epigenetic age) was determined.
Results:
There was no difference in age acceleration between dementia cases and controls. In males, only Hannum’s intrinsic epigenetic age acceleration was increased in pre-symptomatic dementia cases compared to controls (Δ +1.8 years, p = 0.03).
Conclusion:
These findings provide no strong evidence that accelerated epigenetic aging measured in peripheral blood can predict dementia risk.
Keywords: Accelerated Aging, dementia, DNA methylation, epigenetic clock, grimAge, hannum, horvath, phenoAge
1. INTRODUCTION
Aging is the primary risk factor for dementia [1], however it remains unclear whether biological markers of aging are also associated with dementia risk. The epigenome is the overarching control of the genome without altering the genetic code by dynamic molecular modifications and includes DNA methylation, histone modifications, and microRNA levels [2–4]. Patterns of DNA methylation, which is a covalent attachment of a methyl group to cytosine by DNA methyltransferases [5], can control gene transcription by blocking or attracting DNA transcription factors [6]. Aberrant epigenetic signatures measured in blood have been associated with disease states [7]. While cancer has been a major focus [8], there is growing evidence that they are implicated in age-related diseases, such as heart disease [9], and neurodegenerative disorders, such as dementia [10]. It is now well accepted that there are age-related changes to epigenetic mechanisms. Indeed, the most promising biomarker of aging is the ‘epigenetic clock,’ in which biological age can be determined by measuring DNA methylation at specific sites across the genome [11].
There are several different epigenetic age measures, or ‘epigenetic clocks’, which are based on the correlation between chronological age and methylation. The two most widely used measures of epigenetic age are Horvath’s and Hannum’s epigenetic clocks, in which methylation levels at specific genomic sites (353 [12] and 71 [13] methylation sites, respectively) are highly correlated with chronological age. The measurable difference between chronological age and epigenetic age, known as age acceleration (AgeAccel), gives an estimation of how much the older (or younger) individual is biologically compared to their actual chronological age. There are also estimates of age acceleration which control blood cell composition, and the ‘intrinsic epigenetic age acceleration’ calculated with Horvath’s clock (IEAA.Horvath), and Hannum’s clock (IEAA.Hannum). A separate measure using Hannum’s clock, ‘extrinsic epigenetic age acceleration’ (EEAA), takes into account age related changes to cell composition by controlling weighted average age-associated cell counts. Measures of age acceleration, and intrinsic and extrinsic age measures, are associated with age-related morbidity and mortality [14], as well as environmental, lifestyle and health factors, such as lower levels of education and higher body mass index [15].
Horvath’s and Hannum’s epigenetic clocks are built directly on the association between DNA methylation and age, whereas more recent epigenetic age measures, such as PhenoAge and GrimAge predict life span by incorporating other age and health-related biological measures. These were created on the premise that clinical biomarkers of all-cause mortality and health-related outcomes are often more accurate at estimating remaining life-span than actual age [16]. PhenoAge is an epigenetic clock that factors in methylation at 513 methylation sites, as well as chronological age, and nine biomarkers of aging, such as levels of albumin, creatinine, and the inflammatory marker C-reactive protein [17]. PhenoAge is a better predictor of long-term mortality risk than both Horvath and Hannum epigenetic clocks. GrimAge is a measure of life-span (time to death), calculated using DNA methylation-based estimations of levels of morbidity and mortality associated plasma-based proteins and smoking pack-years. GrimAge acceleration has been shown to be more accurate in predicting time to disease, such as coronary heart disease and cancer, than accelerated PhenoAge [18].
Not only do measures of epigenetic age have the potential to represent an increased risk of dementia, but studies of the epigenetic clock and dementia could aid in understanding the age-related biological mechanisms that drive dementia risk. Our previous systematic review and meta-analysis, which found that age acceleration was associated with an increased risk of all-cause mortality, included only two studies on dementia [14]. A small study involving only 11 dementia cases found that Horvath’s epigenetic age was associated with incidental dementia [19]. A larger study including 335 Parkinson’s disease participants reported increased age acceleration (AgeAccel Horvath and EEAA) associated with the disease [20]. No study was stratified by sex, which is an important consideration given the clear sex differences in disease prevalence [21, 22] and epigenetic age acceleration [23]. Our systematic review did not include the more recently developed measures of PhenoAge or GrimAge. However, PhenoAge has been positively correlated with amyloid load, neuritic plaques, and neurofibrillary tangles, the primary neuropathology of Alzheimer’s disease [17]. Also, the acceleration of GrimAge has been associated with measures of cognition and brain health, including lower cognitive functioning and vascular lesions in the brain in older age [24].
The primary aim of this study is to determine whether there is an association between accelerated epigenetic aging and pre-symptomatic dementia and to determine whether this differs between males and females. A secondary aim is to assess whether age acceleration changes over time with the manifestation of clinical symptoms by investigating intra-individual change in age acceleration from the pre-symptomatic time-point to dementia diagnosis.
2. MATERIALS AND METHODS
2.1. Study Sub-Sample
Included in this analysis were 160 participants from the ASPirin in Reducing Events in the Elderly (ASPREE) cohort, previously characterised in detail [25]. The ASPREE study was approved by Monash Human Research Ethics Committee (2006/745M), and all participants gave informed written consent. This DNA methylation sub-study was approved by The Alfred Human Ethics Committee (Project 448/16). The study was conducted in accordance with the Declaration of Helsinki 2008 revision, NHMRC Guidelines on Human Experimentation, the federal patient privacy (HIPAA) law, the International Conference of Harmonisation Guidelines for Good Clinical Practice, and the Code of Federal Regulations.
All participants in this sub-sample were self-identified as white Australians and provided blood samples at baseline; for inclusion in ASPREE, the individuals needed to score > 77 out of 100 on the Modified Mini-Mental State Examination (3MS) [26, 27], a screening for general cognition. In addition to the 3MS, other cognitive assessments included Symbol Digit Modalities Test (SDMT) [28], the Controlled Oral Word Association Test (COWAT) [29], and the Hopkins Verbal Learning Test-Revised (HVLT-R) [30, 31]. Participants without serious health complications, such as cardiovascular disease, uncontrolled high blood pressure, or major conditions likely to cause death within five years were included in the study. Participants were aged between 70 and 92 years old.
2.2. Dementia Adjudication
Over the five-year follow-up period, individuals were diagnosed with dementia by a specialist panel of neurologists, neuropsychologists, and geriatricians, who adjudicated on the basis of the Diagnostic and Statistical Manual for Mental Disorders, American Psychiatric Association (DSM-IV) criteria [32]. In the sample of 160 baseline participants, 73 were adjudicated as having dementia within an average of 3.7 years of follow-up, and 87 controls remained cognitively healthy over this period. Cases and controls were matched for age, sex, smoking status, education status, and baseline cognitive function. At 3-years follow-up, 49 of these participants provided further blood samples, including 25 participants with an adjudicated dementia diagnosis and 24 controls who remained cognitively healthy.
2.3. Epigenetic Clock Measures
DNA extracted from peripheral blood samples was run on the Illumina MethylEPIC array at the Australian Genome Research Facility (https://www.agrf.org.au/)). Programming platform ‘R’ version 3.5.1 was used to modify data for use in Horvath’s online “New DNA Methylation Age Calculator” (https://dnamage.genetics.ucla.edu/new), following user manual instructions and suggestions (https://dnamage.genetics.ucla.edu/important-hints). EPIC data was normalized using the preprocessNoob method [33]. Beta values from the EPIC data set were restricted to probes available as measured by 450K array, as expected by the online calculator. Missing probes (33,124 probes, Supplemental File 1) had methylation values filled in as ‘NA’ to complete the data set. To obtain measurements of epigenetic aging, sample beta values, as well as a sample annotation file including sample, age, sex, and tissue type, were uploaded to the calculator. Before submitting the data, options “Normalise data” as well as “Advanced Analysis” were selected, but the “Fast Imputation” option remained unselected. Output from the calculator used in this study included epigenetic age estimated by Horvath, Hannum, PhenoAge, and GrimAge clocks, as well as age acceleration residuals (epigenetic age measures regressed on chronological age) of all clocks and measures of age acceleration adjusted for estimated cell proportions (IEAA) and age-associated cell composition (EEAA).
2.4. Statistical Analysis
STATA 15 was used for all analyses. Correlation between epigenetic age measures and chronological age, as well as the correlation between age acceleration measures, were firstly examined. T-tests were used to compare epigenetic age and age acceleration between pre-symptomatic dementia cases and controls. Logistic regression was used to determine the association between age acceleration and case-control status, adjusting for possible covariates, such as sex, smoking status, education level, and baseline cognitive test scores (3MS, SDMT, COWAT, and HVLT-R). As females have a higher risk of dementia [21], and sex-specific differences in epigenetic aging have been reported previously [23], sex-stratified analysis was also undertaken. Secondary analysis examined the association between age acceleration and dementia cases at the point of diagnosis in a smaller sub-group. Paired t-tests were used to compare age acceleration measured in blood collected during pre-diagnosis at the time of inclusion and blood collected during post-diagnosis at the time of follow up (average 3 years difference).
3. RESULTS
3.1. Age Measure Correlations
Measures of Horvath, Hannum, GrimAge, and PhenoAge age estimates were all correlated with chronological age (r = 0.8 to 0.60, p < 0.0001), with GrimAge being the most highly correlated (Supplementary Table 1). Correlations between baseline chronological age and epigenetic age estimates, stratified by dementia status, can be seen in Fig. (1). GrimAge was most highly correlated with chronological age in both controls (r = 0.79, Fig. 1g) and dementia (r = 0.8, Fig 1h). Most measures of age acceleration were highly correlated with one another, the strongest being between AgeAccel Hannum and EEAA (r = 0.96, p < 0.0001), and all others ranging from r = 0.89, p < 0.0001 to r = 0.21, p = 0.007 (Supplementary Table 2). The only exceptions were from AgeAccelGrim and IEAA.Horvath (r = 0.11, p = 0.19), and AgeAccelGrim and IEAA.Hannum (r = 0.1, p = 0.22).
Fig. (1).
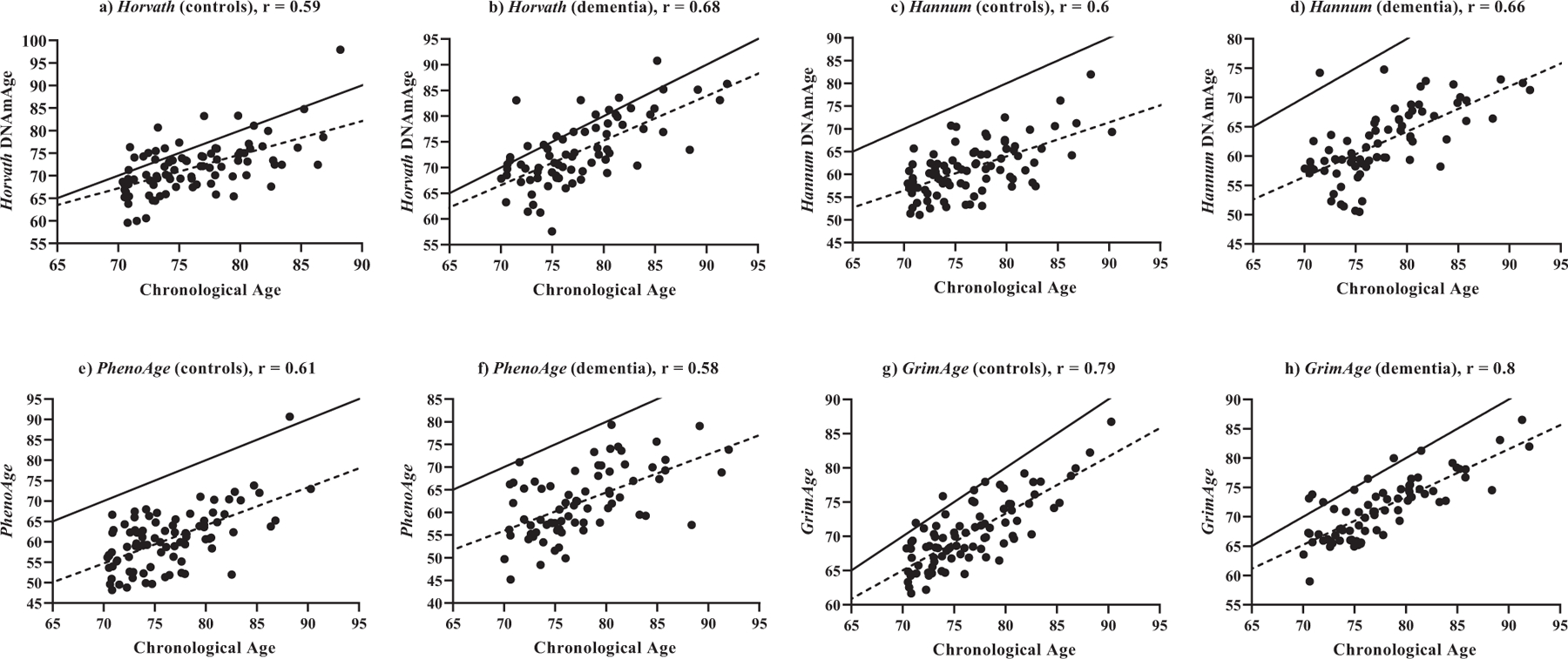
Correlations between estimated epigenetic age (y-axis) and chronological age (x-axis) at baseline. a) Horvath DNAmAge vs. chronological age in control participants (n = 87); b) Horvath DNAmAge vs. chronological age in dementia participants (n = 73); c) Hannum DNAmAge vs. chronological age in control participants (n = 87); d) Hannum DNAmAge vs. chronological age in dementia participants (n = 73); e) PhenoAge vs. chronological age in control participants (n = 87); f) PhenoAge vs. chronological age in dementia participants (n = 73); g) GrimAge vs. chronological age in control participants (n = 87); h) GrimAge vs. chronological age in dementia participants (n = 73); Solid line represents a perfect correlation between age and epigenetic age (r = 1), dotted line represents correlation between age and estimated epigenetic age. All correlations are p < 0.0001. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
3.2. Participant Characteristics
Participant characteristics are shown in Table 1. The only characteristic which differed between cases and controls was baseline COWAT (p = 0.03). Pre-symptomatic dementia cases were on average biologically older than controls (from 1.1 to 1.7 years difference), which is concordant with the difference in chronological age (Table 2).
Table 1.
Baseline characteristics of pre-symptomatic dementia cases and controls.
| - | Dementia (n = 73) | Controls (n = 87) | P |
|---|---|---|---|
| Chronological age (SD) | 77.6 (5.1) | 76.4 (4.6) | 0.11 |
| Female (n, %) | 42, 57.5% | 50, 57.5% | 0.99 |
| Education | - | - | - |
| < = 12 yrs (SD) | 43 (58.9) | 60 (69) | 0.19 |
| > 13 yrs (SD) | 30 (41.1) | 27 (31) | |
| Smoking | - | - | - |
| Current (%) | 0 (0) | 2 (2.3) | 0.42 |
| Past (%) | 32 (43.8) | 36 (41.4) | |
| Never (%) | 41 (56.2) | 49 (56.3) | |
| Mean estimated smoking-packs per year (SD) | 14.1 (7.5) | 13 (8.2) | 0.38 |
| Cognitive tests | - | - | - |
| 3MS (SD) | 92.1 (4.4) | 93.2 (4.8) | 0.14 |
| COWAT (SD) | 14.1 (4.0) | 12.8 (3.6) | 0.03 |
| HVLT-R delayed recall* (SD) | 7.8 (1.9) | 8.2 (1.9) | 0.2 |
| SDMT (SD) | 35.0 (8.4) | 36.2 (9.1) | 0.39 |
Abbreviations: 3MS: Modified Mini-Mental State Examination, COWAT: Controlled-Oral Word Association Test, HVLT-R: Hopkins Verbal Learning Test-Revised, SDMT: Symbol Digit Modalities Test
Data not available for one control participant.
Table 2.
Epigenetic age and age acceleration in pre-symptomatic dementia cases compared to controls.
| - | Dementia (n = 73) | Controls (n = 87) | P |
|---|---|---|---|
| Epigenetic age measure | Mean epigenetic age in years (SD) | - | |
| Horvath | 73.2 (6.6) | 72 (5.8) | 0.22 |
| Hannum | 62.3 (6.0) | 61.2 (5.8) | 0.24 |
| PhenoAge | 62.4 (7.5) | 60.7 (7.1) | 0.14 |
| GrimAge | 71.4 (5.2) | 70.3 (4.9) | 0.75 |
| Age acceleration measure | Mean age acceleration in years (SD) | - | |
| AgeAccel Horvath | +0.32 (4.9) | +0.30 (4.5) | 0.98 |
| AgeAccel Hannum | +0.32 (4.6) | +0.20 (4.5) | 0.86 |
| IEAA.Horvath | +0.13 (4.5) | +0.06 (4.1) | 0.91 |
| IEAA.Hannum | +0.30 (3.9) | -0.10 (3.6) | 0.50 |
| EEAA | +0.32 (5.8) | +0.39 (6.3) | 0.94 |
| AgeAccel Pheno | +0.47 (6.2) | +0.02 (5.5) | 0.62 |
| AgeAccel Grim | +0.09 (3.1) | +0.01 (3.0) | 0.87 |
Abbreviations: AgeAccel Grim: age acceleration calculated using epigenetic GrimAge, AgeAccelPheno: age acceleration calculated using epigenetic PhenoAge, AgeAccel Hannum: age acceleration calculated using Hannum epigenetic age, AgeAccel Horvath: age acceleration calculated using Horvath epigenetic age, EEAA: extrinsic epigenetic age acceleration, IEAA.Hannum: intrinsic epigenetic age acceleration calculated using Hannum epigenetic age, IEAA.Horvath: intrinsic epigenetic age acceleration calculated using Horvath epigenetic age; ‘+’ indicates increased epigenetic age compared to chronological age.
3.3. Age Acceleration
Compared to controls, individuals with pre-symptomatic dementia did not have accelerated epigenetic aging for any of the measures examined. Similar results were observed in cases at the time of dementia diagnosis compared with controls (Supplementary Table 3). When stratifying by sex, there was a significant association with Hannum’s intrinsic epigenetic age acceleration and pre-symptomatic dementia in males (n = 31 cases (IEAA.Hannum = +1.12), n = 37 controls (IEAA.Hannum = −0.70), Δ +1.82 years, p = 0.03), but not in females (Fig. 2., Supplementary Table 4). This remained significant in logistic regression analysis after adjusting for smoking status, batch effects (methylation array chip), education level, and baseline 3MS, SDMT, COWAT, and HVLT-R (OR:1.21, SE:0.11, Adj. p = 0.035).
Fig. (2).
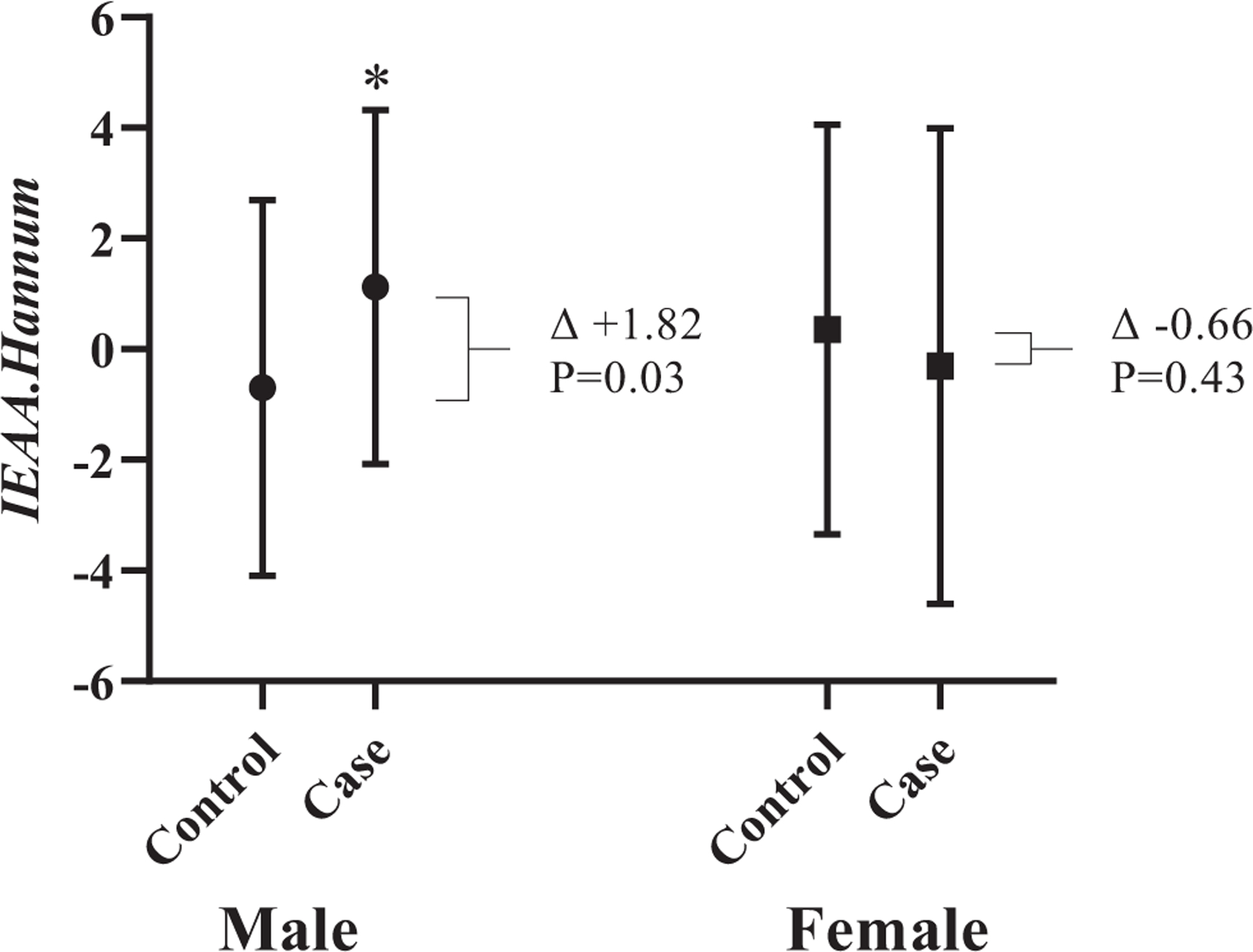
Hannum’s intrinsic epigenetic age acceleration in controls vs. pre-symptomatic dementia, stratified by sex; Left: Male controls (n = 37, IEAA.Hannum = −0.70) vs. pre-symptomatic dementia participants (n = 31, IEAA.Hannum = +1.12), p = 0.03; Right: Female controls (n = 50, IEAA.Hannum = +0.35) vs. pre-symptomatic dementia participants (n = 42, IEAA.Hannum = −0.31), p = 0.43; Error bars represent standard deviation.
The change in age acceleration within individuals was then compared between pre-symptomatic dementia cases (at baseline) and at the time of diagnosis (3-years follow-up) (Supplementary Table 5). Three age acceleration measures, AgeAccel.Horvath, AgeAccel.Hannum and EEAA, in dementia participants, were significantly different between baseline at pre-symptomatic and post dementia diagnosis (three years after). All showed an increased epigenetic age acceleration at baseline and a decreased epigenetic age acceleration at diagnosis, reflecting a decrease in age acceleration over time (AgeAccel.Horvath: Δ−2.10, p = 0.003, AgeAccel.Hannum: Δ−1.41, p = 0.01, and EEAA: Δ−1.71, p = 0.03). Only AgeAccel.Horvath (Δ−1.61, p = 0.03) differed between controls at baseline and the same control participants at follow-up, which also decreased over time (Δ−1.61, p = 0.03).
4. DISCUSSION
We found no evidence of accelerated epigenetic aging measured in blood in individuals with pre-symptomatic dementia compared to controls. This suggests that despite age being the greatest risk factor for dementia, it is not directly associated with DNA methylation markers of biological aging. The only significant finding was a slight increase in IEAA.Hannum for males with dementia versus controls, but given the lack of consistency, this could be a chance finding. While correlations were significant between all epigenetic age measures and chronological age, the correlation coefficients of Horvath, Hannum, and PhenoAge were low relative to GrimAge (r = 0.80). This suggests that GrimAge measures may more reliably predict age than the other epigenetic clocks in this population comprised of participants aged over 70 years old who were relatively healthy, without cardiovascular diseases. However, we found no evidence that GrimAge was predictive of dementia. Indeed, GrimAge is considered a measure of time-to-death rather than chronological age per se and uses DNA methylation-based estimations of plasma-based proteins (adrenomedullin, beta-2-microglobulim, cystatin C, growth differentiation factor 15, leptin, plasminogen activation inhibitor 1, and tissue inhibitor metalloproteinase 1) and smoking pack-years [18]. This contrasts with the Horvath’s and Hannum’s clocks, which use the correlation between DNA methylation at specific probes and age.
Only a couple of studies have investigated whether dementia is associated with increased epigenetic age or age acceleration. The first measured AgeAccel Horvath, IEAA. Horvath and EEAA in 508 Caucasians and 84 Hispanics, including 335 with Parkinson’s disease [20]. They observed that AgeAccel Horvath and EEAA were associated with disease status after adjusting for exposure to organophosphate (pesticide), cumulative smoking pack-years, education, coffee intake, and ethnicity. Almost all individuals in our study had probable or possible Alzheimer’s Disease [34], which could help account for the divergent findings, given these are two different diseases with differing etiology. A smaller study compared Horvath’s epigenetic age between participants with dementia (n = 11) and controls (number unclear) [19]. After controlling for chronological age and gender, this study observed that epigenetic age was associated with dementia status (p = 0.03). However, our findings are in agreement with the most current study of epigenetic aging and late-life dementia [35]. The study by Sibbett et al. measured age acceleration at a baseline time point comparing differences between incidental dementia (n = 109, average time to dementia 9.7 years) and controls (n =379) who either remained without dementia before death (average time to death = 7.8 years) or survived beyond the last date of data collection (average time = 17.8 years). That study also found no evidence of age acceleration associated with dementia incidence after adjusting for age, sex, smoking status, as well as diseases, such as cardiovascular disease, cerebrovascular disease, hypertension, and diabetes, and APOE ɛ4 status [35].
Although our primary findings differed, something of note in the Degerman study mentioned above was the disparity between epigenetic age and chronological age in older individuals [19]. Participants’ chronological age (average 57.9 years) and epigenetic age (average 57.1 years) were correlated at baseline (r = 0.69). After 15 years of follow-up, however, when participants were between 70 to 80 years (average 72.8), the average epigenetic age was considerably younger (average 69.5 years) than chronological age. This trend is similar to that observed in our paired analysis of age acceleration, where AgeAccel.Horvath, AgeAccel.Hannum and EEAA were significantly decreased at 3-years follow-up compared to the same participants at baseline. This has also been reported previously in another study, including both blood and brain tissue from Alzheimer’s Disease (n = 61) and controls (n = 31) [36]. They observed that age acceleration slows down or reverses in older individuals. Indeed, epigenetic age algorithms are not as linear in older populations, with epigenetic age appearing to be younger in comparison to chronological age in individuals above 80 years old [37]. As our sample of participants was between 70 and 92 years old, it could also partly explain our findings. A further explanation could be that DNA methylation at Horvath and Hannum clock-specific probes respond to the pathophysiology associated with the clinical presentation of the disease, similar to what is seen in the prenatal environment, where epigenetic age will become decelerated at birth in response to adverse conditions [38].
A source of possible variation between other epigenetic age studies and ours is in the cell estimation used in the calculation of IEAA measures, which adjust for estimated cell type. The IEAA measures calculated in this study used cell estimation based on the older 450K methylation array, however, updated cell estimations for EPIC, the array used in this study, differ, as they are derived using cell sorted DNA run on the EPIC array [39]. This may also affect IEAA.Hannum, which was observed in our study to be slightly increased in men pre-symptomatic for dementia compared to controls. Furthermore, epigenetic age measures by Horvath and Hannum are generated using an older epigenetic array than used in this study. The Illumina EPIC array used in this study measures ~850,000 methylation probes across the epigenome [40]. Although most EPIC probes overlap (93.3%) with the 450K array (~450,000 probes) was used to calculate Horvath and Hannum epigenetic age measures [41], there were still 33,124 probes missing from the EPIC array that are present in the 450 K array. This results in 19 missing probes from Horvath’s 353 methylation site clock and six missing from Hannum’s 71 site clock. However, a previous study found that the lack of probes generally does not affect Horvath and Hannum clock outcomes [42]. These differences, however, do not affect measures of GrimAge and PhenoAge. GrimAge was purposefully built using probes that were available on both the EPIC and 450K arrays to ensure future compatibility [18], and PhenoAge builds on probes available on the three most recent generations of Illumina Arrays (Epic, 450K and 27K) [17]. This highlights the need for updated epigenetic age algorithms based on the EPIC, which outperform older clock algorithms, as studies will increasingly start to use the EPIC array.
4.1. Study limitation
A potential limitation of this study is the small sample size, which may have limited power to detect associations if they were present. One recent study suggests that a small sample size is subject to confounding by estimated blood cell compositions when compared to larger studies [43], however, this study is similar in size relative to the aforementioned studies in the field. As previously mentioned, a general limitation to epigenetic aging studies, particularly using older populations, is that the correlation between age and epigenetic age does not have a linear relationship with chronological age in those past 80 years old [37]. This suggests the need for ‘older aging’ epigenetic clocks to be developed. Another limitation is that although some methylation sites are strongly conserved across brain and blood tissue [44], the sites used in measuring the epigenetic clocks do not necessarily reflect age-related changes in the brain. Thus, for use in age-associated brain diseases, future efforts should be made to specifically compare brain aging and associated methylation sites. Progress is being made in this field, for example, the recently published ‘cortical DNAm age clock,’ which can accurately predict brain age based on 347 cortical methylation sites [45, 46]. However, this signature still needs to be measurable in blood, if it is to be used as an easy to access biomarker of brain aging.
CONCLUSION
There is no evidence that peripheral blood-based accelerated epigenetic aging, measured using Horvath, Hannum, GrimAge, and PhenoAge epigenetic clocks, is predictive of dementia risk. Furthermore, our study provides an important addition to the current evidence. Together, this suggests that biological aging does not drive dementia risk. Updated clock algorithms for specific use in observing biological age differences associated with dementia should be considered. More generally, blood-based DNA methylation biomarkers for dementia remain a promising area of research, with the potential for early diagnostics and better risk prediction.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge the dedicated and skilled staff in Australia and the U.S. involved in the ASPREE cohort. The authors also are most grateful to the ASPREE participants, who so willingly volunteered for this study and the general practitioners and staff of the medical clinics who cared for the participants.
FUNDING
ASPREE was supported by grants from the National Institute on Aging and the National Cancer Institute at the U.S. National Institutes of Health (U01AG029824 and U19AG062682); the National Health and Medical Research Council of Australia (334047 and 1127060); Monash University (Australia) and the Victorian Cancer Agency (Australia). JR is funded by a NHMRC Dementia Research Leader Fellowship (1135727). PDF was gratefully funded by RTP stipend PhD scholarship, awarded by Monash University and the Australian Government.
LIST OF ABBREVIATIONS
- 3MS
Modified Mini-Mental State Examination
- AgeAccel
Age Acceleration
- AgeAccelGrim
Age Acceleration Calculated Using GrimAge Epigenetic Clock
- AgeAccel Hannum
Age Acceleration Calculated Using Hannum’s Epigenetic Clock
- AgeAccel Horvath
Age Acceleration Calculated Using Horvath’s Epigenetic Clock
- AgeAccelPheno
Age Acceleration Calculated Using PhenoAge Epigenetic Clock
- APOE
Apolipoprotein E
- ASPREE
ASPirin in Reducing Events in the Elderly Cohort
- COWAT
Controlled Oral Word Association Test
- DSM-IV
Diagnostic and Statistical Manual for Mental Disorders version 4
- EEAA
Extrinsic Epigenetic Age Acceleration
- EPIC
Illumina MethylEPIC array
- HVLT-R
Hopkins Verbal Learning Test Revised
- IEAA.Horvath
Intrinsic Epigenetic Age Acceleration Calculated Using Horvath’s Epigenetic Clock
- IEAA.Hannum
Intrinsic Epigenetic Age Acceleration Calculated Using Hannum’s Epigenetic Clock
- SDMT
Symbol Digit Modalities Test
Footnotes
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The ASPREE study was approved by Monash Human Research Ethics Committee (2006/745M). This DNA methylation sub-study was approved by the Alfred Human Ethics Committee (Project 448/16).
HUMAN AND ANIMAL RIGHTS
No animals were used for this research. The study on humans was conducted in accordance with the Declaration of Helsinki 2008 revision, NHMRC Guidelines on Human Experimentation, the federal patient privacy (HIPAA) law, the International Conference of Harmonisation Guidelines for Good Clinical Practice, and the Code of Federal Regulations.
CONSENT FOR PUBLICATION
Written informed consent was given by all ASPREE participants.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the ASPREE principal investigators, but restrictions apply to the availability of these data, which were used under license for the current study, therefore, they are not publicly available. Data are, however, available from the authors upon reasonable request and with permission of the ASPREE principal investigators through the website (www.ASPREE.org). Data will be shared by the researchers upon request for use in the epigenetic analysis; if approved, data will be made available through a web-based data portal safe haven at Monash University, Australia.
CONFLICT OF INTEREST
AM reports receiving consulting fees from Alkahest, Inc. and reports grants from National Institute on Aging. RCS reports grants for clinical research regarding dementia and Alzheimer’s disease from National Institutes of Health, the Centers for Medicare and Medicaid Services, the Department of Defence, and the Illinois Department of Public Health, being a non-compensated board member of the Alzheimer’s Association - Illinois Chapter and, being the site principal investigator or sub-investigator for clinical trials for which his institution (Rush University Medical Center) is compensated [Amylyx Pharmaceuticals, Inc., Eli Lilly & Co., Inc., Genentech, Inc., Merck & Co, Inc., Navidea Biopharmaceuticals, Novartis Pharmaceuticals, Inc., Roche Holdings AG, and Takeda Development Center Americas, Inc.]. PF, PL, RS, RV, RLW and JR report no disclosures relevant to the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material for this paper is available on the publisher’s website along with the published article.
REFERENCES
- [1].Hickman RA, Faustin A, Wisniewski T. Alzheimer disease and its growing epidemic: Risk factors, biomarkers, and the urgent need for therapeutics. Neurol Clin 2016; 34(4): 941–53. 10.1016/j.ncl.2016.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Booth LN, Brunet A. The aging epigenome. Mol Cell 2016; 62(5): 728–44. 10.1016/j.molcel.2016.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang W, Qu J, Liu G-H, Belmonte JCI. The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol 2020; 21(3): 137–50. 10.1038/s41580-019-0204-5 [DOI] [PubMed] [Google Scholar]
- [4].Campbell RR, Wood MA. How the epigenome integrates information and reshapes the synapse. Nat Rev Neurosci 2019; 20(3): 133–47. 10.1038/s41583-019-0121-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lyko F The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat Rev Genet 2018; 19(2): 81–92. 10.1038/nrg.2017.80 [DOI] [PubMed] [Google Scholar]
- [6].Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacol 2013; 38(1): 23–38. 10.1038/npp.2012.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis 2018; 5(1): 1–8. 10.1016/j.gendis.2018.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: Moving forward. PLoS Genet 2018; 14(6): e1007362. 10.1371/journal.pgen.1007362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Soler-Botija C, Gálvez-Montón C, Bayés-Genís A. Epigenetic biomarkers in cardiovascular diseases. Front Genet 2019; 10(950): 950. 10.3389/fgene.2019.00950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fransquet PD, Ryan J. The current status of blood epigenetic biomarkers for dementia. Crit Rev Clin Lab Sci 2019; 56(7): 435–57. 10.1080/10408363.2019.1639129 [DOI] [PubMed] [Google Scholar]
- [11].Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 2018; 19(6): 371–84. 10.1038/s41576-018-0004-3 [DOI] [PubMed] [Google Scholar]
- [12].Horvath S DNA methylation age of human tissues and cell types. Genome Biol 2013; 14(10): R115. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49(2): 359–67. 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fransquet PD, Wrigglesworth J, Woods RL, Ernst ME, Ryan J. The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clin Epigenetics 2019; 11(1): 62. 10.1186/s13148-019-0656-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ryan J, Wrigglesworth J, Loong J, Fransquet PD, Woods RL. A systematic review and meta-analysis of environmental, lifestyle, and health factors associated with DNA methylation age. J Gerontol A Biol Sci Med Sci 2020; 75(3): 481–94. 10.1093/gerona/glz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Levine ME. Modeling the rate of senescence: Can estimated biological age predict mortality more accurately than chronological age? J Gerontol A Biol Sci Med Sci 2013; 68(6): 667–74. 10.1093/gerona/gls233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Levine ME, Lu AT, Quach A, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 2018; 10(4): 573–91. 10.18632/aging.101414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 2019; 11(2): 303–27. 10.18632/aging.101684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Degerman S, Josefsson M, Nordin Adolfsson A, et al. Maintained memory in aging is associated with young epigenetic age. Neurobiol Aging 2017; 55: 167–71. 10.1016/j.neurobiolaging.2017.02.009 [DOI] [PubMed] [Google Scholar]
- [20].Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY) 2015; 7(12): 1130–42. 10.18632/aging.100859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Beam CR, Kaneshiro C, Jang JY, Reynolds CA, Pedersen NL, Gatz M. Differences between women and men in incidence rates of dementia and Alzheimer’s disease. J Alzheimers Dis 2018; 64(4): 1077–83. 10.3233/JAD-180141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Miller IN, Cronin-Golomb A. Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov Disord 2010; 25(16): 2695–703. 10.1002/mds.23388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Horvath S, Gurven M, Levine ME, et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol 2016; 17(1): 171–93. 10.1186/s13059-016-1030-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hillary RF, Stevenson AJ, Cox SR, et al. An epigenetic predictor of death captures multi-modal measures of brain health. Mol Psychiatry 2019. 10.1038/s41380-019-0616-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McNeil JJ, Woods RL, Nelson MR, et al. Baseline characteristics of participants in the ASPREE (ASPirin in Reducing Events in the Elderly) study. J Gerontol A Biol Sci Med Sci 2017; 72(11): 1586–93. 10.1093/gerona/glw342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ryan J, Woods RL, Britt C, et al. Normative performance of healthy older individuals on the Modified Mini-Mental State (3MS) examination according to ethno-racial group, gender, age, and education level. Clin Neuropsychol 2019; 33(4): 779–97. 10.1080/13854046.2018.1488996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jones TG, Schinka JA, Vanderploeg RD, Small BJ, Graves AB, Mortimer JA. 3MS normative data for the elderly. Arch Clin Neuropsychol 2002; 17(2): 171–7. 10.1093/arclin/17.2.171 [DOI] [PubMed] [Google Scholar]
- [28].Smith A Symbol digit modalities test: Manual. Los Angeles: Western Psychological Services; 1982. [Google Scholar]
- [29].Ruff RM, Light RH, Parker SB, Levin HS. Benton controlled oral word association test: Reliability and updated norms. Arch Clin Neuropsychol 1996; 11(4): 329–38. 10.1093/arclin/11.4.329 [DOI] [PubMed] [Google Scholar]
- [30].Ryan J, Woods RL, Murray AM, et al. Normative performance of older individuals on the Hopkins Verbal Learning Test-Revised (HVLT-R) according to ethno-racial group, gender, age and education level. Clin Neuropsychol 2020; 1–17. 10.1080/13854046.2020.1730444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Benedict RHB, Schretlen D, Groninger L, Brandt J. Hopkins Verbal Learning Test – Revised: Normative data and analysis of interform and test-retest reliability. Clin Neuropsychol 1998; 12(1): 43–55. 10.1076/clin.12.1.43.1726 [DOI] [Google Scholar]
- [32].First MB, Frances A, Pincus HA. DSM-IV-TR handbook of differential diagnosis. Arlington, VA, US: American Psychiatric Publishing, Inc. 2002. 10.1176/appi.books.9781585622658 [DOI] [Google Scholar]
- [33].Triche TJ Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res 2013; 41(7): e90–0. 10.1093/nar/gkt090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ryan J, Storey E, Murray AM, et al. Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology 2020; 95(3): e320–31. 10.1212/WNL.0000000000009277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sibbett RA, Altschul DM, Marioni RE, Deary IJ, Starr JM, Russ TC. DNA methylation-based measures of accelerated biological ageing and the risk of dementia in the oldest-old: A study of the Lothian Birth Cohort 1921. BMC Psychiatry 2020; 20(1): 91. 10.1186/s12888-020-2469-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].El Khoury LY, Gorrie-Stone T, Smart M, et al. Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol 2019; 20(1): 283. 10.1186/s13059-019-1810-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bell CG, Lowe R, Adams PD, et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol 2019; 20(1): 249. 10.1186/s13059-019-1824-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Palma-Gudiel H, Eixarch E, Crispi F, Morán S, Zannas AS, Fañanás L. Prenatal adverse environment is associated with epigenetic age deceleration at birth and hypomethylation at the hypoxia-responsive EP300 gene. Clin Epigenetics 2019; 11(1): 73. 10.1186/s13148-019-0674-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Salas LA, Koestler DC, Butler RA, et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol 2018; 19(1): 64. 10.1186/s13059-018-1448-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Illumina Inc. Illumina Infinium MethylationEPIC Array. Illumina Inc; 2020. [Google Scholar]
- [41].Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 2016; 17(1): 208–08. 10.1186/s13059-016-1066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McEwen LM, Jones MJ, Lin DTS, et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. Clin Epigenetics 2018; 10(1): 123. 10.1186/s13148-018-0556-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang Q, Vallerga CL, Walker RM, et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med 2019; 11(1): 54. 10.1186/s13073-019-0667-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Braun PR, Han S, Hing B, et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry 2019; 9(1): 47. 10.1038/s41398-019-0376-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shireby GL, Davies JP, Francis PT, et al. Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain 2020; 143(12): 3763–75. 10.1093/brain/awaa334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Grodstein F, Lemos B, Yu L, Iatrou A, De Jager PL, Bennett DA. Characteristics of epigenetic clocks across blood and brain tissue in older women and men. Front Neurosci 2021; 14: 555307–07. 10.3389/fnins.2020.555307 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.