

Abstract
The idea that tumour microenvironment (TME) is organised in a spatial manner will not surprise many cancer biologists; however, systematically capturing spatial architecture of TME is still not possible until recent decade. The past five years have witnessed a boom in the research of high‐throughput spatial techniques and algorithms to delineate TME at an unprecedented level. Here, we review the technological progress of spatial omics and how advanced computation methods boost multi‐modal spatial data analysis. Then, we discussed the potential clinical translations of spatial omics research in precision oncology, and proposed a transfer of spatial ecological principles to cancer biology in spatial data interpretation. So far, spatial omics is placing us in the golden age of spatial cancer research. Further development and application of spatial omics may lead to a comprehensive decoding of the TME ecosystem and bring the current spatiotemporal molecular medical research into an entirely new paradigm.
Keywords: cancer ecology, single‐cell RNA‐seq, spatial omics, tumour microenvironment
Spatial omics is transforming our understanding of the cancer ecosystem at the systemic level.
The integration of spatial omics and single‐cell omics can fundamentally improve our understanding of tumourigenesis and cancer microenvironment.
Generating the spatial atlas of human cancers across multiple omics and timescales will potentially pioneer the revolution of spatiotemporal molecular medicine.
1. BACKGROUND
One of the central issues that hinders successful anti‐cancer treatment is the heterogeneity of tumour microenvironment (TME). 1 Spatially, TMEs in distinct tumours are diversely organised and hierarchically structured. 2 , 3 . Shapes of TMEs are critical for cancer cell fate determination and development, which are coordinated by precise tumour intrinsic transcriptional regulation and intercellular crosstalk. 4 , 5 , 6 In response to external stimuli (e.g., chemotherapy), spatial reprogramming will be initiated including anti‐tumour immunity renaissance and stromal cell relocation. 7 Understanding the spatial structure of TME assembly is hence essential for discovering tumourigenesis mechanisms and designing novel therapeutic strategies.
A challenge in decoding tumour spatial structure is how to capture the high‐throughput spatial profile of TME at the genome‐wide level. Solving this issue requires the ability to record the transcriptional information and spatial coordinates simultaneously. An applicable way is high‐dimensional imaging (i.e., imaging mass cytometry, IMC, and multiplex immunohistochemistry), 8 , 9 ; however, those approaches can only quantify the low‐throughput profile of TME (designed gene sets) rather than the whole transcriptome/proteome.
By contrast, the state‐of‐the‐art spatial omics has now made the whole transcriptome or even epigenome measurable. 10 , 11 , 12 In light of this, we review the technological advances plus computational strategies of spatial omics and discuss how they may accelerate spatiotemporal oncological research. The clinical relevance of spatial omics will potentially extend into novel clinical‐relevant biomarker discovery, novel immunotherapy designing, and precision medicine. The ultimate spatial tumour atlas will be an essential resource uncovering the black box of cancers across space and time.
2. SPATIAL OMICS TECHNOLOGIES
2.1. Laser capture microdissection‐based approaches
The first attempt to dissert the high‐throughput spatial tissue structure can be traced to laser capture microdissection (LCM)‐based strategies 13 , 14 (Figure 1A). This approach utilises LCM to dissect tissues into small segments which are subsequently profiled using high‐throughput technologies such as RNA‐seq. For example, LCM‐seq 14 , 15 , 16 combines single‐cell RNA‐seq (scRNA‐seq) and LCM to trace the spatial transcriptome at the single‐cell level. This technology allows the accurate quantification of compartment of tissue structures and the discovery of diversified cell subpopulation distribution within tissues. Using a similar strategy, topographic single cell sequencing (TSCS) is designed to capture the genomic copy number profile of single tumour cells spatially. 17 By utilising TSCS in breast cancer samples, the results show a direct genomic lineage of breast cancer cell progression. Interestingly, the authors observe that most mutations and copy number aberrations evolved prior to invasion, indicating that cancer cells are well prepared before progression. This technique allows the unbiased discovery of copy number variations at the 2D level. Geographical position sequencing (GEO‐seq), 18 , 19 another method combining the two technologies (LCM and scRNA‐seq), can capture the spatial transcriptome based on a relatively small number of cells. However, compared with LCM‐seq, each spot of GEO‐seq captures more number of cells. Similarly, Tomo‐seq enables the cryosectioning of the region of interest and allows the RNA‐seq on individual sections. 20 , 21 , 22 In the context of oncology, only a few spatial omics studies utilised LCM‐based approaches. 14 , 23 To summarise, LCM‐based high‐throughput technologies can quantify the transcriptome at the cellular level, however, those technologies failed to reach higher resolution and can merely trace the regional location information. Laser microdissection is also time‐consuming, posing challenges for capturing the high‐throughput profile of complex tissues without spending a lot of time.
FIGURE 1.
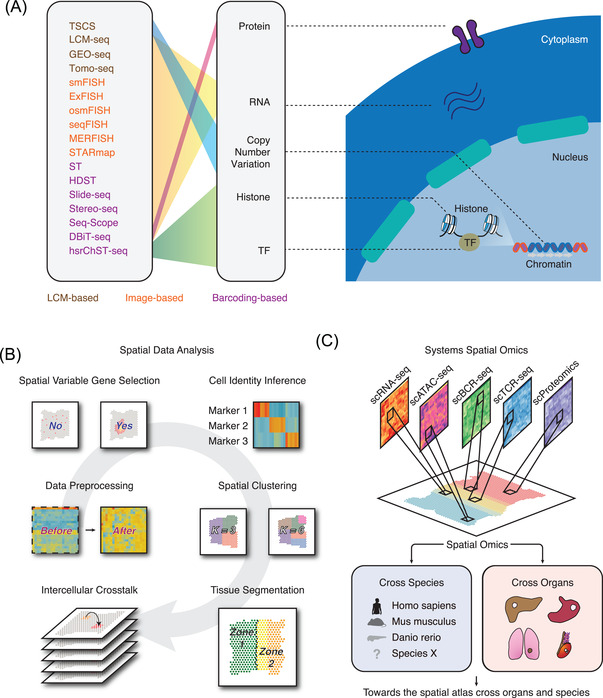
Spatial omics can decode the three‐dimensional structure of tumour microenvironment. (A) Summary of published spatial omics technologies. The brown text represents the LCM‐based technologies. The orange text represents the imaging‐based technologies. The purple text represents the barcoding‐based technologies. TF, transcription factor; LCM, laser capture microdissection. (B) The data analysis strategies which can be adopted in spatial omics data treatment. (C) Spatial omics can be utilised to study cancer samples across different species and distinct organs. Integrating of spatial omics and other omics techniques can systematically decode the structure of tumour microenvironment. scRNA‐seq, single‐cell RNA‐seq; scATAC‐seq, single cell assay for transposase‐accessible chromatin‐seq; scBCR‐seq, single cell B‐cell receptor‐seq; scTCR‐seq, single‐cell T‐cell receptor‐seq; mIHC, multiplex immunohistochemistry
2.2. Image‐based in situ transcriptomics
Another strategy to capture the spatial architecture of tissue transcripts is image‐based in situ transcriptomics technology (Figure 1A). Single‐molecule fluorescence in situ hybridization (smFISH) 24 , 25 , 26 , 27 , 28 can detect several RNAs at the same time. On the basis of smFISH, expansion FISH (ExFISH), ouroboros single‐molecule FISH (osmFISH), and sequential FISH+ (seqFISH+) are designed to increase the number of detected genes (up to 10 000). 29 , 30 , 31 , 32 , 33 , 34 Multiplexed error‐robust fluorescence in situ hybridization (MERFISH), a robust single‐molecule imaging approach, can capture 100–1000 distinct RNA species in hundreds of individual cells, 35 which evolves the gene throughput to ∼10000 in 2019. 36 Similarly, Spatially‐resolved Transcript Amplicon Readout mapping (STARmap), integrating hydrogel‐tissue chemistry, targeted signal amplification, and in situ sequencing, is tested to capture 160–1020 genes simultaneously. 37 Those imaged‐based technologies identified specific RNAs enriched in cellular compartments or even high‐order chromatin structure. 36 , 38 In a nutshell, most image‐based in situ transcriptomics technologies cannot capture the whole transcriptome profile but can offer single‐cell or even subcellular resolution within tissues, hence enabling the discovery of complex cellular states of cancer cells. 39 , 40
2.3. Spatial barcoding‐based transcriptomics
Distinct from image‐based in situ transcriptomics, spatial barcode‐based approaches allow the unbiased sequencing of RNA species at the whole transcriptome level (Figure 1A). Spatial transcriptomics (ST, also named as Visium), one of the most widely used spatial omics technologies, enables the sequencing of 6 mm × 6 mm tissues with each spot at the resolution of ∼100 μm containing 2–10 cells. 41 One of the advantages of such a method is that it can capture thousands of genes with low‐level transcript expression and even from formalin‐fixed paraffin embedding (FFPE) tissues. 42 The upgraded method named high‐definition spatial transcriptomics (HDST) was then developed with resolution at 2 μm. 43 This approach opens the avenue of spatial quantification of complex tissues at the single‐cell resolution. Slide‐seq 44 and Slide‐seqV2 45 , another high‐resolution spatial sequencing technology, can reach the ∼50% RNA capture efficiency of scRNA‐seq and successfully characterise the spatiotemporal developing trajectory of mouse neocortex. Spatio‐Temporal Enhanced REsolution Omics‐sequencing (Stereo‐seq), which combines DNA nanoball chips and in situ RNA capture, can reach the resolution ∼0.5 μm of each bin. 46 Seq‐Scope, newly‐developed spatial transcriptome sequencing technique based on a solid‐phase amplification of randomly barcoded single‐molecule oligonucleotides, can also reach the sub‐cellular resolution (0.5–1 μm). Those methods put us in a unique position in exploring the new functions of organelles and may lead to a major advance of our understandings of spatiotemporal molecular medicine. 47 , 48
Rather than placing the spatial barcodes onto chips, a new class of quantifying spatial coordination of RNAs, named microfluidic deterministic barcoding, was recently developed in Rong Fan's lab. Using a unique barcode delivering method, deterministic barcoding in tissue for spatial omics sequencing (DBiT‐seq) does not require sophisticated steps of tissue lysis to release mRNAs but presents high resolution at ∼10 μm. 49 , 50 On the basis of this work, they developed spatial epigenomics technique for histone modifications quantification such as H3K27me3, H3K4me3 and H3K27ac. 51 This fantastic method enables the discovery of spatially key regulatory elements controlling identity (i.e., spatial enhancer prediction) and brings the epigenetics research into the spatial era. This microfluidic deterministic barcoding based approach promises to extend single omics to spatial multi‐omics sequencing and will open exciting opportunities in quick and straightforward profiling of complex cells and tissues. The revolution of the above spatial omics will pave the way for spatiotemporal molecular medical research 47 , 48 and refresh our understanding of the single‐cell heterogeneity and spatial diversity in TME (Figure 1A).
2.4. Spatial proteomics
The surging of spatial proteomics allows the detection of dozens of proteins without losing the spatial location. Mass spectrometry‐based method is one of the highly multiplex techniques to capture the protein spatial intensity. Multiplexed ion beam imaging (MIBI), using secondary ion mass spectrometry to image labelled antibodies, is able to analyse one hundred markers of the same tissue. 52 This technology yields precise quantification of immune cell subpopulation 53 and their spatial patterns 54 inside the tumour. IMC is another method dependent on metal‐tagged antibodies and enables the imaging of over 100 antibodies. 55 Such method offers unprecedented opportunities to explore regional immunity composition and topological function units of TME. 56 Another technology named CO detection by indexing (CODEX) is capable of profiling up to 50 proteins of single slide based on imaging antibodies conjugated to barcodes. 57 However, all those methods are dependent on the performance of antibodies and are relatively costly. It is still challenging to increase the current throughput to proteome‐wide. Bias may also exist when designing the panel of markers rather than discovering functional proteins from the proteomics data.
2.5. Spatial metabolomics
Identifying the difference of metabolites and the spatial organization of tissues is essential to decipher intra tumour heterogeneity and understand the cancer systems profoundly, however, the spatial metabolic features of tumour s are largely unclear. Matrix‐assisted laser desorption ionization imaging mass spectrometry (MALDI‐IMS) allows the detection of metabolites without losing the spatial information. 58 , 59 Desorption electrospray ionization (DESI)‐IMS is another method to detect the spatial dynamics metabolites without destroying the tissues. 60 , 61 , 62 , 63 Airflow‐assisted desorption electrospray ionization (AFADESI)‐IMS 64 further advances the multiplex capacity and can cover 1,500 metabolites. Applying such method in profiling esophageal squamous cell carcinoma showed the spatial tiny structure of metabolites including pyrroline‐5‐carboxylate reductase 2 (PYCR2) and uridine phosphorylase 1 (UPase1). 65 With the coming of the spatial omics era, spatial metabolomics will become a useful toolkit for identifying novel disease signatures.
2.6. Spatial multi‐omics technologies
Cancer is a multifactorial disorder associated with multiple genetic and environmental factors; hence jointly dissecting spatial multi‐omics profile may enable us to reconstruct the key processes of tumourigenesis. DBiT‐seq supports recording the spatially barcoded mRNA and profiling proteins of interest (a panel of 22 proteins) at the same time. 49 Deterministic barcoding, the underlying key mechanism of this technology, allows the accurate delivering of barcodes containing spatial coordinates without tissue dissociation. The development of spatial multiomics (SM‐Omics) also offers integrated and ST and antibody‐based proteomics profiling without the need for sophisticated infrastructure. 66 This platform enables the automatic profiling of 96 samples and efficiently generates the combined RNA and protein profile in ∼2 days. By testing this platform in mouse brain cortex samples, significant correlation between specific mRNA and protein expression is observed. The authors further develop a computational pipeline and claim its easy deployment to the wide scientific community.
Another strategy to simultaneously decode the spatial and cellular dynamics is to in silico integrating spatial‐omics and single‐cell omics data. Spatially‐resolved Transcriptomics via Epitope Anchoring (STvEA) enables the enrichment of multiplex immunohistochemistry data with scRNA‐seq 67 and is also designed to map CODEX imaging data to scRNA‐seq. As for the epigenomic data, another group imputed single cell multiome (chromatin accessibility and transcriptome multi‐omics) profile from the Visium ST data. 68 Such analysis predicted the spatial epigenetics activity of key genes and linked the opening motif dynamics with phonotypes. These strategies can be potentially adopt to other spatial omics technologies (i.e., spatial metabolomics 65 ). In general, capturing/inferring the spatial multi‐omics profile simultaneously is still in its infancy. The next step is to extend existed single‐omics to multi‐omics and enable the jointly profiling of the complex system of TME.
3. COMPUTATIONAL METHODS FOR SPATIAL OMICS
While the complexity of spatial omics data is what makes it powerful, it also makes them hard to interpret. Usually, these data are collected in batches and generated in large quantities. The large amount of data pose great challenges for computational biologists to digest the big data and construct computational pipelines. Here, we review recent computational methods spanning from data preprocessing, spatial variable gene selection, spatial clustering and tissue segmentation, to spatial inter‐cellular crosstalk (Figure 1B and Table 1).
TABLE 1.
Summary for algorithms designed for spatial omics analysis
| Name | Usage | Environment | URL |
|---|---|---|---|
| SCTransform | Data preprocessing | R | https://github.com/ChristophH/sctransform |
| Giotto | Data preprocessing, spatial variable gene identification, cell identity inference, cell–cell crosstalk modelling, clustering analysis | R | http://spatialgiotto.rc.fas.harvard.edu/giotto.html |
| Seurat | Data preprocessing, spatial variable gene identification, cell identity inference, clustering analysis | R | https://satijalab.org/seurat/vignettes.html |
| SpatialDE | Spatial variable gene identification | Python | https://github.com/Teichlab/SpatialDE |
| trendsceek | Spatial variable gene identification | R | https://github.com/edsgard/trendsceek |
| scGCO | Spatial variable gene identification | Python | https://github.com/WangPeng‐Lab/scGCO |
| SPARK | Spatial variable gene identification | R | https://github.com/xzhoulab/SPARK |
| SOMDE | Spatial variable gene identification | Python | https://pypi.org/project/somde/ |
| BayesSpace | Clustering analysis | R | http://www.bioconductor.org/packages/release/bioc/html/BayesSpace.html |
| SpatialCPie | Clustering analysis | R | https://www.bioconductor.org/packages/release/bioc/html/SpatialCPie.html |
| SPOTlight | Cell identity inference/deconvolution | R | https://github.com/MarcElosua/SPOTlight |
| RCTD | Cell identity inference/deconvolution | R | https://github.com/dmcable/RCTD |
| stereoscope | Cell identity inference/deconvolution | Python | https://github.com/almaan/stereoscope |
| DSTG | Cell identity inference/deconvolution | Python | https://github.com/Su‐informatics‐lab/DSTG |
| STUtility | Data preprocessing, spatial variable gene identification, clustering analysis, tissue segmentation, image processing | R | https://ludvigla.github.io/STUtility_web_site/index.html |
| Squidpy | Data preprocessing, spatial variable gene identification, cell identity inference, cell–cell crosstalk modelling, clustering analysis, tissue segmentation | Python | https://github.com/theislab/squidpy |
| Baysor | Tissue segmentation | Linux | https://github.com/kharchenkolab/Baysor |
| SPATA | Tissue segmentation, trajectory modelling | R | https://themilolab.github.io/SPATA/index.html |
| stLearn | Trajectory modelling; cell–cell crosstalk modelling | Python | https://stlearn.readthedocs.io/en/latest/ |
| GCNG | Cell–cell crosstalk modelling | Python | https://github.com/xiaoyeye/GCNG |
| SpaOTsc | Cell–cell crosstalk modelling | Python | https://github.com/zcang/SpaOTsc |
| MISTy | Cell–cell crosstalk modelling | R | https://github.com/saezlab/mistyR |
3.1. Data preprocessing
The first crucial step for spatial data downstream analysis is normalization against the sequencing depth. The variance of RNA read counts of each spot can be diverse partly due to heterogeneous cell type composition. To resolve this question, several algorithms have been designed for scRNA‐seq, and some of them are still suitable for ST data analysis such as SCTransform. 69 This algorithm integrates regularised negative binomial regression and Pearson residuals which proved to be fit for ST data. 54 SCTransform was embedded in Seurat toolkit and is very easy‐to‐use. 70 Another package supporting spatial RNA read normalization is Giotto. 71 This toolkit supports the all‐in‐one data preprocessing functions including library size adjustment, log transformation and data scaling. Those methods are broadly applicable for spatial barcoding‐based transcriptomics such as ST or Slide‐seq. However, the majority of those computational algorithms do not consider the spatial coordinates and therefore do not measure spatial variability. Spatial normalization and scaling was critical for improving the performance of spatial omics (e.g., normalised expression is more linked with specific morphologies and capture cell types’ relative proportions 72 ). Applying the noise reduction algorithm in image processing such as smoothing or wavelet transform may also enhance the performance of spatial omics.
3.2. Spatial variable gene identification
When data normalization is completed, a question naturally arises: are there spatially variable genes linked with the well‐organised tissue structures? For example, some genes show extremely variable spatial expression patterns (i.e., tumour and normal edge specific genes), while some genes are ubiquitously expressed (i.e., cancer housekeeping genes). Computationally, quantifying the spatial variable genes is hence expected to be fundamental to discovering the molecular basis of TME architecture. 73 The direct way is to compute the differentially expressed genes according to clusters/anatomical structures 70 or select genes with high variance. 71 Those methods partly depend on prior knowledge (e.g., Pathologist's annotation) and supervised clustering results. On the contrary, spatial‐patterned‐based methods do not rely on supervised annotation and is broadly applicable for spatial data. Seurat 70 utilised variogram models and measures the distance between two spots. SpatialDE builds on Gaussian process regression and decomposes gene expression into spatial and non‐spatial elements. 73 Another group reported the marked point process‐based statistical framework which proved to be robust for simulated and real data 74 This algorithm is non‐parametric and does not depend on any prior knowledge. scGCO, a method based on Markov Random Fields with graph cuts, is able to process millions of cells in hours but does not require consuming large memory. Interestingly, scGCO is widely applicable to a full range of spatial data even image‐based in situ transcriptomics (e.g., seqFISH and MERFISH). 75 SPARK is also a time‐saving algorithm 76 . The authors use the generalised linear spatial models and provide efficient control of type 1 errors. Recently, Hao et al. proposed an artificial neural network (ANN) based clustering method named SOMDE and utilised Gaussian process to conduct feature selection. 77 This method is also featured with super‐fast speed (∼5 min for 20 K spots) and low memory consumption. In summary, a wide spectrum of algorithms are focused on spatial variable gene identification, partly due to its importance for selecting features for downstream spot clustering and dimensional reduction analysis. Most of them are designed for Visium. Hence, their performance on other techniques (e.g., DBiT‐seq and MERFISH) still remain unknown due to the completely different read count distributions and spot resolutions.
3.3. Clustering the spots
Defining the clusters in a given tissue is fundamental for cell‐type identification and downstream functional annotation. In the context of scRNA‐seq, filtering the pure clusters, the population exhibiting identical functions, requires manual gene signature checking and tricky parameter adjustment. A robust method is to compute the entropy metric (ROGUE algorithm 78 ) or Gini index (GiniClust algorithm 79 , 80 ) of given clusters. As for spatial data treatment, simultaneously considering the locational information and mixed cell types pose challenges for accurate spot clustering. The direct way is to provide a user‐friendly package for users to manually select the parameters such as resolution. 81 More complicatedly, Zhao et al. utilised the Bayesian model with a Markov random field and successfully enhanced the clustering efficacy. 82 This algorithm has been demonstrated to perform well on multiple datasets (i.e., squamous cell carcinoma and prefrontal cortex) generated by distinct spatial omics technologies. Other computational methods utilising graph convolutional network or deep learning also appeared to perform well for ST data. 83 , 84 . To summarise, the existing clustering methods are mostly designed for scRNA‐seq and do not consider the neighbourhood spot information or spatially patterned structure, pressing the need for developing more algorithms. Robust spatial clustering algorithms can generate the precise subpopulation maps of cancer cells and correlate the cellular states with their spatial distributions.
3.4. Cell identity inference
A lot of strategies have emerged to link gene expression with cell identity, although most of them are designed for single cell transcriptomics. 85 Some of them are based on the correlation between test data and reference data, while others require supervised classification and classifier training. 85 Given the spatial omics data usually contain a mixture of cells such as ST, new algorithms are needed for optimising the automatic analysis pipeline. One of the most used approaches is deconvolution, such as SPOTlight (using non‐negative matrix factorization regression), 86 RCTD (using non‐negative least‐squares regression), 87 DSTG (using graph‐based convolutional networks) 88 and stereoscope (a probabilistic model based on the negative binomial distribution). 89 Another practical way is to modify the scRNA‐seq integration algorithm. For example, Seurat 70 enabled the ‘anchor’‐based workflow and transferred labels from scRNA‐seq data to ST data. This algorithm was previously demonstrated effective for cross‐species integration and has now shown robustness in ST. The above methods are mostly designed for spatial omics of near‐single‐cell resolution such as Visium and can effectively infer the cellular composition of a spot mixture. Also, those algorithms largely rely on matched scRNA‐seq data to compute the cell‐type probability. As a result, such analysis is largely dependent on single‐cell clustering analysis and the uncertain level of resolution parameter. Annotating higher resolution data (e.g., Stereo‐seq) without prior knowledge is still challenging.
3.5. Single‐cell segmentation
Accurately defining the tissue borders is critical for understanding how cells locally interact with others (i.e., cancer cells interact with immune cells). For example, robust segmentation is able to increase the number of detected cells and allow the inference of the real cell states. 90 To resolve this question, Baysor enables cellular segmentation based on the likelihood of transcriptional composition, size and shape of the cell. 90 A pipeline named Squidpy allows the extraction of image features and nuclei segmentation. 91 This pipeline is largely based on image processing and provides options of different deep learning algorithms such as Stardist 92 and Cellpose. 93 However, a lot of questions regarding tissue segmentation still remain unexplored. How to computationally model the cellular spatial patterns such as immune cells (sparse) and endothelial cells (linear)? How to segment the organelle structure (e.g., nuclei and cytoplasm) and trace the RNA origin in subcellular spatial omics data? A possible way is to train a machine learning model by using the spatial coordinates of existed multiplexed immunofluorescence imaging, given that spatial distribution patterns of spatial omics is similar to immunofluorescence. New algorithms supporting high spatial resolution segmentation need to be developed and will allow its broad application in the spatial omics community.
3.6. Cell–cell communication analysis
In the context of scRNA‐seq data analysis, decoding cell–cell communication is crucial for understanding how cells interact with each other and how such crosstalk networks are changed under specific disease conditions such as cancer. 94 Decoding the global cell–cell communication network will help design specific targeting strategies. 6 , 95 Existed algorithms 96 , 97 , 98 , 99 are mostly designed for scRNA‐seq and do not consider spatial information. To trace the spatial cellular communication dynamics, several algorithms have been proposed. For example, SpaOTsc can infer intercellular gene–gene regulatory information flows between genes by using a machine learning model. 100 Another intercellular communication quantification package named stLearn can compute the morphological similarity between spots and then perform the cell–cell communication analysis. 101 GCNG encodes the spatial location as a graph, integrates it with an expression profile using supervised training, and infers the cell–cell interactive scores. 102 MISTyis a flexible and scalable machine learning framework for capturing the cell–cell communication score and performs well on multiple datasets. 103 Well‐established ST pipelines including Giotto 71 and Squidpy 91 also support the spatial intercellular crosstalk analysis. By utilising those methods, 71 , 100 spatial interactive cells such as interacting astrocytes and inhibitory neurons were observed. Taking the advantage of high‐resolution spatial technologies such as Slide‐seq or Stereo‐seq, it may be possible to computationally model the dynamics of intercellular gene regulation and fundamental biological processes (i.e., phase separation).
4. SPATIAL OMICS IN CANCER RESEARCH: FROM BENCH TO BEDSIDE
Cancer can be viewed as a complex system. 104 The evolution of tumour cells is like a dynamic interaction between microenvironmental/therapeutic selective forces and their intrinsic adaptive strategies to survival. 105 With the help of the state‐of‐the‐art spatial omics, it is now possible to tackle a range of basic and fundamental questions of oncology at the systemic level. Current spatial research is spanning from multi‐omics (e.g., transcriptomics, 106 epigenomics 50 ), and multiple organs (e.g., breast cancer, 43 melanoma 107 ), to multiple species (e.g., Homo sapiens, 54 Danio rerio 108 ) (Figure 1C).
4.1. Spatially decoding the heterogeneity of TME
The core problem hindering patients’ long‐term survival is cancer heterogeneity. 1 This heterogeneity was featured as mixed cell types with spatial differences in gene expression. 4 , 109 As a consequence, diverse cellular populations exist within the TME, making any gene‐ or pathway‐specific therapy less effective. 1 Existed single‐cell omics techniques have decoded the intra tumour heterogeneity at the systems level, 110 , 111 , 112 , 113 however, such a method does not retain the spatial coordinates of each cell. In 2018, Lundeberg's lab first utilised ST to explore the prostate TME diversity in the cancer research. 106 They precisely computed the dynamic gene expression changes during cancer progression and demonstrated the cancer cell state difference between the TME periphery and centre. In stage III metastatic melanoma samples, they found that immune‐related genes such as HLA genes and CD74 are highly active in cancer regions. 107 Surprisingly, a recent study indicates that cancer hallmark pathways are specifically activated even in specific regions of tumours. 114
Another grand challenge in tumour ecology research is the spatial quantification of immune cells. Andersson et al. used the deconvolution algorithm 89 and inferred the region‐specific enrichment/depletion of B cells. They further developed a gene signature of tertiary lymphoid structure (TLS) and observed the activation of cell activation/differentiation pathway in TLS 115 . By using the SPOTlight deconvolution algorithm, 86 the authors spatially trace the T cell subsets in glioblastoma patient samples and infer the trajectory of those T cells. 116 We recently reported the spatiotemporal immune profile of colon cancer liver metastasis and observed the increased infiltration of MRC1+ CCL18+ macrophages in the metastatic sites. 95 All these studies are partly based on ST which cannot directly profile the transcriptome of single immune cell but the combined profiles of cancer/immune cell mixture. In the future, more data generated by high‐resolution spatial omics (e.g., DBiT‐seq) will identify the mechanisms that are shared among varied cell‐type compositions and will help develop new therapeutic approaches targeting the spatial TME organization.
4.2. Tumour invasive margin: the main battlefield in the fight against cancer?
Not all cancer cells are created equal. As for cancer cells located in the boundary or core regions, they exhibit different phenotype states 117 and distinct microenvironmental features. 118 Previous immunohistochemistry‐based low‐throughput studies 119 largely rely on the selection of region of interest and the design of protein panels. By contrast, spatial omics allows the unbiased discovery of key cell types and genes controlling the fate of tumourigenesis. For example, by integrating ST and multiplexed ion beam imaging, Ji et al. identified tumour‐specific keratinocytes (TSKs) which are specifically located in the fibrovascular niche at leading edges. 54 Such TSKs are associated with a high density of intercellular crosstalk with immune cells. Interestingly, this cellular population is featured with an oncogenic transcriptional program sharing the activation of HDAC1 and ETS1, indicating the potential epigenetics reprogramming resides in the leading edge zones. A zebrafish‐based study also reveals that unique edge cell states at the melanoma boundary are associated with upregulated ETS‐family transcription factor activity. 108 These transcriptional states are validated in 10 patient samples, implicating the potential conservation across species. All those pieces of evidence place the tumour leading edges in the limelight and naturally raise the hypothesis TME conditions (i.e., tumour boundary barrier vs. tumour core) may be spatially different (Figure 2A).
FIGURE 2.
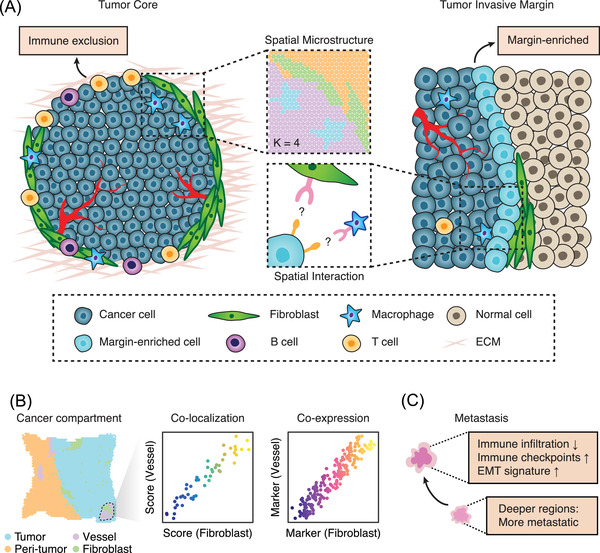
The cancer microenvironment spatial structure and compartment revealed by spatial omics. (A) The distinct microenvironment structure of tumour core and tumour margins. ECM, extracellular matrix. (B) Spatial omics can precisely capture the TME compartment such as CAFs and vessel. CAFs, cancer associated fibroblasts. (C) The microenvironment pf primary and metastatic tumour s are largely different revealed by spatial studies
4.3. The compartment of TME for supporting tumour growth
Significant progress has been made in the field of single‐cell omics and we now know that TME is well‐structured with immune cells, cancer‐associated fibroblasts, and the extracellular matrix, 6 , 120 but the challenges still remain. Importantly, it is difficult to trace back the spatial cellular states and developmental states. The evolution of spatial omics technologies helps to open the black box of TME. For example, the prostate cancer tissues can be divided into compartments with distinct gene expression profiles and pathway activities. 106 A subset of stroma cells are specifically reactive to nearby cancer cells, while the others show inflammatory features. Inside the TME of pancreatic cancer samples, the structure of the stroma is also highly ordered. In particular, the inflammatory fibroblasts and endothelial cells are co‐localised in this region (Figure 2B). As for immune cells, their distribution is closely linked with the compartment zones. 23 , 121 Combined scRNA‐seq and ST revealed the pancreatic cancer subtype‐specific compartment reprogramming in response to neoadjuvant chemotherapy. 7 Collectively, those data explain how the anatomical molecular profile of the TME compartment determines the tumourigenesis fate and how cancer cells spatially respond to therapy.
4.4. Tracing the spatiotemporal evolution of cancer cells
Evolution is the driving force behind cancer cell resistance or metastasis. 122 , 123 Previous research largely relied on multi‐region sample collection, 124 where directly modelling the spatial evolutionary routes cell‐by‐cell was far from applicable. Now, with the help of spatial omics, tracing cancer evolution at different space coordinates and cellular units is now possible. Sundar et al. utilised the NanoString transcriptomics profiling (composed of 770 genes) and found that nearby lymph node metastases may originate from deeper subregions of the primary cancer cells. 125 Another group reported that metastatic tumour s were depleted with immune cell infiltrates but harboured high expression of the immune checkpoints (B7‐H3, TIM‐3, etc.). 126 Interestingly, the epithelial–mesenchymal‐transition (EMT) gradient is specifically enriched in the metastatic tumour but not the primary tumour 127 (Figure 2C). At the genome level, recent data indicated that co‐existed clones surprisingly have different transcriptional and immunological features. Such spatial clonal diversity is deeply impacted by resident tissue structures. TSCS profiling allows the genomic lineage tracing between distinct tumour subpopulations 17 (Figure 3A). These results highlight the spatial rearrangement during cancer evolution, and further multidimensional spatial analysis will allow a broad range of spatiotemporal molecular medicine 47 , 48 problems to be solved.
FIGURE 3.
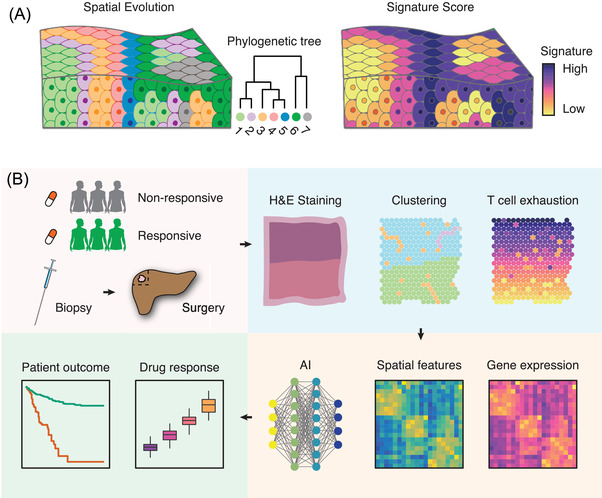
The spatial evolution of cancer cells and the potential clinical application of spatial omics. (A) Spatial genomics and transcriptomics allows the discovery pf evolution and their specific signature of cancer cells. (B) Spatial omics can be potentially used to predict the drug response and the clinical outcomes in the clinical setting. EMT, epithelial‐to‐mesenchymal transition; CAF, cancer‐associated fibroblasts
4.5. Is spatial omics still far from the clinical application?
Initial data showed that spatial arrangements of TME quantified by mIHC perform better in predicting immunotherapy response comparable to existed methods (i.e., PD‐L1 expression, immunohistochemistry, tumour mutational burden and bulk gene expression profiling). 128 These observations indicate that spatial omics can potentially provide innovative solutions for designing precision medicine strategies. Another spatial analysis of the melanoma clinical cohort reveals the spatial interaction between PD‐1/PD‐L1 and IDO‐1/HLA‐DR was tightly linked with anti‐PD‐1 clinical response. 129 All those data revealed that spatially quantifying the TME structure might have better prognostic power than existed biomarkers. This technique may be perfectly suited to discover how TME evolves during therapy (Figure 3B) and has the potential to reveal the targetable structures (e.g., reprogramming the tumour borderlines to engineer the cold TME). However, data from retrospective/prospective clinical cohorts are still lacking.
In the clinical practice, spatial omics technology may pave the way for precision pathology. It is now possible to link the pathological images with spatial gene expression profile by using machine learning or deep learning algorithms. 130 , 131 These studies may enable the prediction of the transcriptomics profile based on existed H&E staining slides which may perform better than existed biomarkers. Generating the spatial atlas of a large sample of tumours will hence not only reveal new ways to improve patients’ outcomes, but also pave the way for spatiotemporal molecular medical research. 47 , 48
5. SPATIAL TUMOUR ECOLOGY: TOWARD A NEW RESEARCH PARADIGM
5.1. Microenvironment is an ecosystem
The cancer microenvironment is similar to an ecological system, a mixture with distinct cellular populations and species (Figure 4A). The establishment and growth of tumours are strikingly similar to an adaptive and evolving ecosystem. 132 For example, the species richness, which can also be referred to intra tumour heterogeneity, is potentially linked with immunotherapy robustness and patients’ long‐term outcome. 133 , 134 The metabolic competition between immune cells and cancer cells, which can be termed as interspecific competition, is also a key determinant of cancer progression. 135 . The behaviour of microenvironmental populations and the structure of TME can be explained by ecological theories, but more high‐dimensional data are needed.
FIGURE 4.
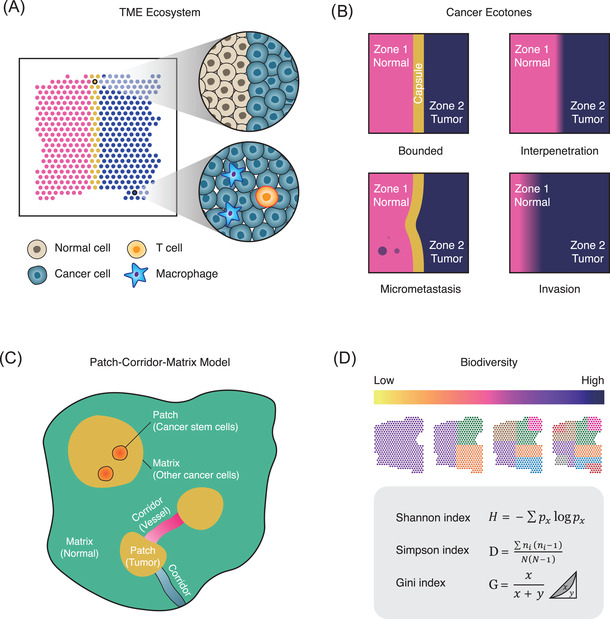
Building up the interdisciplinary link between ecology and oncology. (A) The TME is an ecosystem composed of diversified species such as immune cells and cancer cells. (B) The proposed models for describing different cancer ecotone patterns. The bounded pattern refers to equal and homogeneous interface (capsule) between tumour and adjacent normal tissues. The interpenetration pattern refers to the cancer cell infiltration into the adjacent normal tissues. The micrometastasis pattern refers to the mini cancer cell invasion into the adjacent normal tissues. The invasion pattern refers to the invasive cancer cell infiltration into the adjacent normal tissues. (C) The patch–corridor–matrix model for modelling the spatial tumour heterogeneity. (D) Methods for quantifying the spatial biodiversity inside the tumour microenvironment
5.2. Ecotones of cancer: the transitional zones of cancer‐normal communities
Edge effect refers to a greater diversity of the community at the boundary of habitats. 136 Particularly in TME, the edge effect occurs in tumour ‐normal borders, where two distinct systems meet and mingle. Interestingly, TSKs are specifically located in the edges of skin cancer and those cells harbour frequent intercellular communications with nearby immune cells. 54 A possible explanation is that intratumour environmental conditions drive those species to colonise habitable borders. This observation leads us to hypothesise that the edge effect in the cancer ecosystem widely exists and may be therapeutically targeted to destroy the cancer cell habitats. Those transitional zones are also termed ecotones, referring to the transition area between two biological communities (i.e., interface between forest and grass). 137 At the boundary of TME, how the sharp border is formatted still remains unclear (Figure 4B). Does the formation of cancer ecotone help or prevent the cancer invasion? How does the transitional zone interact with immune system and forms the cold or hot tumour? The study of cancer ecotones by spatial sequencing technologies is still in its infancy. A multimodal and systems‐level spatial atlas of cancer ecotone may fundamentally improve our knowledge of cancer ecology and facilitate the designing of novel anti‐cancer strategies.
5.3. The patch–corridor–matrix landscape model: defining the spatial distribution patterns
The patch–corridor–matrix is an important theory for describing the spatial heterogeneity of ecological landscapes. 138 In general, a patch refers to a spatial unit that harbours distinct features with the surrounding environment and has a certain internal homogeneity. A corridor refers to a linear structure connecting different spatial units; while a matrix refers to a continuous and widely distributed space in space. Recently published ST data reveal that the structure of tissues is in potential accordance with this model. For example, the structure of elongating/elongated spermatids is in consistency with the definition of the patch, while their progenitors such as spermatocytes and spermatogonium are located surrounding elongated spermatids which can be described as the matrix. 139 In the context of oncology, the structure of TME and their adjacent tissues is in line with patch–matrix patterns. We hypothesise that the vascular structure conforms to corridor, which connects the patch (TME) and matrix (adjacent normal tissues). Inside the tumour, the formation of cancer stem cell niche may reside in the TME 140 , 141 , which resembles the patch–matrix model (Figure 4C). Systematically, modelling the TME on the basis of the patch–corridor–matrix may hence provide scientific basis for understanding the size, shape, content and structure of tumour s at microscale (Figure 4C).
5.4. Computationally modelling the TME spatial biodiversity
Biodiversity originally refers to the biological variety of life on the Earth. 142 Similarly, the biodiversity of cancer microenvironment is also a key factor for drug responses and patient outcomes. 134 To accurately trace the TME diversity, statistical models are required to quantify the biodiversity of a given spatial omics data. A widely used equation is the alpha, beta and gamma diversity. 143 Alpha diversity refers to the diversity in a particular ecosystem. This concept is usually used to measure the number of species in a given ecosystem (e.g., the species richness in TME). 143 Beta diversity allows us to compare the biodiversity changes between ecosystems (e.g., metastatic TME vs. primary TME). 143 Gamma diversity refers to the measure of the overall diversity in a large region. 143 Another index assessing biodiversity is Shannon entropy index. 144 This index represents the uncertainty that we can predict which species the individuals randomly selected in the community belong to. If the TME consists of only a single species (e.g., cancer cells), then the randomly selected individual must be that unique species. At this time, the Shannon entropy index is zero. As the number of species in the community increases, the Shannon entropy index will increase. Similarly, the Simpson index 145 and Gini coefficient 146 also represent species richness and evenness. The classic Simpson index represents the probability that two randomly selected individuals in the community belong to the same species. When the species richness of the community increases, this probability decreases, that is, the Simpson index decreases as the species richness increases. 145 With the help of those biodiversity quantification indexes (Figure 4D), it is now possible to link the biodiversity with spatial cancer phenotypes such as immune exclusions.
6. DISCUSSION AND FUTURE PERSPECTIVES
To decode the tumour ecosystem, we need to model how individual cells work and how they interact with each other. Although high‐throughput spatial sequencing technologies coupled with the state‐of‐the‐art computational algorithms have greatly improved our understanding of tumour architecture, many pressing questions still remain to be answered, especially the profiling of the intact tissue structure. Existed experimental protocols merely enable the sequencing of tiny slide(s) of tissues. In fact, such profiling may represent the partial tissue expression profile and cannot fully capture the 3D architecture. An interesting example is the 3D transcriptomics reconstruction of the developing heart, 147 which raises the possibility of generating the 3D model of other tissues such as tumours. Computational strategies originally designed for radiomics, such as 3D Slicer 148 , are expected to reconstruct the spatial molecular organization. We hypothesise that, with the development of advanced spatial omics technologies, a 3D bird's‐eye view of TME that encompasses transcriptomics, proteomics and metabolomics may pave the way towards the comprehensive decoding of the TME ecosystem.
We are now in the golden age for spatiotemporal molecular medicine 47 , 48 research. Spatial omics is transforming our understanding of cancer milieu by offering the precise spatial coordinates of cellular and molecular profiles at the systemic level 149 , 150 , 151 . At the same time, existed methods pose experimental and computational challenges for optimising current protocols and expanding the scope of these models. How to integrate biology data and mathematics model into the same framework, establish the interdisciplinary link between ecology and oncology, digest the booming spatial omics data and develop sophisticated analytical algorithms are still challenging. Achieving those goals are of exceptionally significance for unravelling the biology of TME as well as establishing a framework to explain the aggressive characteristics of malignant cells. Generating the spatial atlas of human cancers across multiple omics and timescales can fundamentally improve our understanding of tumourigenesis, pioneer the revolution of medical research paradigm, and ultimately facilitate the designing of advanced therapeutic strategies in the near future.
CONFLICT OF INTEREST
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (No. 81961128025, 91942313); Program of Shanghai Academic Research Leader (No. 19XD1420700); Shanghai Municipal Key Clinical Specialty; Sanming Project of Medicine in Shenzhen (No. SZSM202003009). The study sponsor did not participate in the study design, collection, analysis or interpretation of data.
Wu Y, Cheng Y, Wang X, Fan J, Gao Q. Spatial omics: Navigating to the golden era of cancer research. Clin Transl Med. 2022;12:e696. 10.1002/ctm2.696
Yingcheng Wu and Yifei Cheng contributed equally.
REFERENCES
- 1. Dagogo‐Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. 10.1038/nrclinonc.2017.166 [DOI] [PubMed] [Google Scholar]
- 2. Yuan Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med. 2016;6(8):a026583. 10.1101/cshperspect.a026583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith EA, Hodges HC. The spatial and genomic hierarchy of tumor ecosystems revealed by single‐cell technologies. Trends Cancer. 2019;5(7):411–425. 10.1016/j.trecan.2019.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin MZ, Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5(1):166. 10.1038/s41392-020-00280-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ren X, Kang B, Zhang Z. Understanding tumor ecosystems by single‐cell sequencing: promises and limitations. Genome Biol. 2018;19(1):211. 10.1186/s13059-018-1593-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hwang WL, Jagadeesh KA, Guo JA, et al. Single‐nucleus and spatial transcriptomics of archival pancreatic cancer reveals multi‐compartment reprogramming after neoadjuvant treatment. bioRxiv 2020. 10.1101/2020.08.25.267336 [DOI] [Google Scholar]
- 8. Hartmann FJ, Bendall SC. Immune monitoring using mass cytometry and related high‐dimensional imaging approaches. Nat Rev Rheumatol. 2020;16(2):87–99. 10.1038/s41584-019-0338-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ali HR, Jackson HW, Zanotelli VRT, et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nature Cancer. 2020;1(2):163–175. 10.1038/s43018-020-0026-6 [DOI] [PubMed] [Google Scholar]
- 10. Liao J, Lu X, Shao X, Zhu L, Fan X. Uncovering an organ's molecular architecture at single‐cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 2021;39(1):43–58. 10.1016/j.tibtech.2020.05.006 [DOI] [PubMed] [Google Scholar]
- 11. Crosetto N, Bienko M, van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat Rev Genet. 2015;16(1):57–66. 10.1038/nrg3832 [DOI] [PubMed] [Google Scholar]
- 12. Waylen LN, Nim HT, Martelotto LG, Ramialison M. From whole‐mount to single‐cell spatial assessment of gene expression in 3D. Commun Biol. 2020;3(1):602. 10.1038/s42003-020-01341-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nichterwitz S, Benitez JA, Hoogstraaten R, Deng Q, Hedlund E. LCM‐Seq: a method for spatial transcriptomic profiling using laser capture microdissection coupled with polyA‐based RNA sequencing. Methods Mol Biol. 2018;1649:95–110. 10.1007/978-1-4939-7213-5_6 [DOI] [PubMed] [Google Scholar]
- 14. Nichterwitz S, Chen G, Aguila Benitez J, et al. Laser capture microscopy coupled with Smart‐seq2 for precise spatial transcriptomic profiling. Nat Commun. 2016;7:12139. 10.1038/ncomms12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Civita P, Franceschi S, Aretini P, et al. Laser capture microdissection and RNA‐Seq analysis: high sensitivity approaches to explain histopathological heterogeneity in human glioblastoma FFPE archived tissues. Front Oncol. 2019;9:482. 10.3389/fonc.2019.00482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nichterwitz S, Nijssen J, Storvall H, et al. LCM‐seq reveals unique transcriptional adaptation mechanisms of resistant neurons and identifies protective pathways in spinal muscular atrophy. Genome Res. 2020;30(8):1083–1096. 10.1101/gr.265017.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Casasent AK, Schalck A, Gao R, et al. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell. 2018;172(1‐2):205–217. 10.1016/j.cell.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng G, Suo S, Chen J, et al. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid‐gastrula mouse embryo. Dev Cell. 2016;36(6):681–697. 10.1016/j.devcel.2016.02.020 [DOI] [PubMed] [Google Scholar]
- 19. Chen J, Suo S, Tam PP, Han JJ, Peng G, Jing N. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo‐seq. Nat Protoc. 2017;12(3):566–580. 10.1038/nprot.2017.003 [DOI] [PubMed] [Google Scholar]
- 20. Junker JP, Noel ES, Guryev V, et al. Genome‐wide RNA tomography in the zebrafish embryo. Cell. 2014;159(3):662–675. 10.1016/j.cell.2014.09.038 [DOI] [PubMed] [Google Scholar]
- 21. Kruse F, Junker JP, van Oudenaarden A, Bakkers J. Tomo‐seq: a method to obtain genome‐wide expression data with spatial resolution. Methods Cell Biol. 2016;135:299–307. 10.1016/bs.mcb.2016.01.006 [DOI] [PubMed] [Google Scholar]
- 22. Wu CC, Kruse F, Vasudevarao MD, et al. Spatially resolved genome‐wide transcriptional profiling identifies BMP Signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev Cell. 2016;36(1):36–49. 10.1016/j.devcel.2015.12.010 [DOI] [PubMed] [Google Scholar]
- 23. Massalha H, Bahar Halpern K, Abu‐Gazala S, et al. A single cell atlas of the human liver tumor microenvironment. Mol Syst Biol. 2020;16(12):e9682. 10.15252/msb.20209682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of single RNA transcripts in situ. Science. 1998;280(5363):585–590. 10.1126/science.280.5363.585 [DOI] [PubMed] [Google Scholar]
- 25. Fan Y, Braut SA, Lin Q, Singer RH, Skoultchi AI. Determination of transgenic loci by expression FISH. Genomics. 2001;71(1):66–69. 10.1006/geno.2000.6403 [DOI] [PubMed] [Google Scholar]
- 26. Levsky JM, Shenoy SM, Pezo RC, Singer RH. Single‐cell gene expression profiling. Science. 2002;297(5582):836–840. 10.1126/science.1072241 [DOI] [PubMed] [Google Scholar]
- 27. Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4(10):e309. 10.1371/journal.pbio.0040309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5(10):877–879. 10.1038/nmeth.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen F, Wassie AT, Cote AJ, et al. Nanoscale imaging of RNA with expansion microscopy. Nat Methods. 2016;13(8):679–684. 10.1038/nmeth.3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagendran M, Riordan DP, Harbury PB, Desai TJ. Automated cell‐type classification in intact tissues by single‐cell molecular profiling. Elife. 2018;7:e30510. 10.7554/eLife.30510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single‐cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11(4):360–361. 10.1038/nmeth.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shah S, Lubeck E, Zhou W, Cai L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. 2016;92(2):342–357. 10.1016/j.neuron.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eng CL, Lawson M, Zhu Q, et al. Transcriptome‐scale super‐resolved imaging in tissues by RNA seqFISH. Nature. 2019;568(7751):235–239. 10.1038/s41586-019-1049-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Codeluppi S, Borm LE, Zeisel A, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods. 2018;15(11):932–935. 10.1038/s41592-018-0175-z [DOI] [PubMed] [Google Scholar]
- 35. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging: spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348(6233):aaa6090. 10.1126/science.aaa6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia C, Fan J, Emanuel G, Hao J, Zhuang X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle‐dependent gene expression. Proc Natl Acad Sci U S A. 2019;116(39):19490–19499. 10.1073/pnas.1912459116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Allen WE, Wright MA, et al. Three‐dimensional intact‐tissue sequencing of single‐cell transcriptional states. Science. 2018;361(6400):eaat5691. 10.1126/science.aat5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su JH, Zheng P, Kinrot SS, Bintu B, Zhuang X. Genome‐scale imaging of the 3D organization and transcriptional activity of chromatin. Cell. 2020;182(6):1641–1659 e26. 10.1016/j.cell.2020.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Polonsky M, Round K, Seewaldt V, Cai L. Abstract B117: analysis of luminal breast cancer tumor microenvironment using seqFISH. Cancer Epidemiol Biomark. Prev. 2020;29(6 Suppl 1):B117–B117. 10.1158/1538-7755.Disp18-b117 [DOI] [Google Scholar]
- 40. Rowland TJ, Dumbovic G, Hass EP, Rinn JL, Cech TR. Single‐cell imaging reveals unexpected heterogeneity of telomerase reverse transcriptase expression across human cancer cell lines. Proc Natl Acad Sci U S A. 2019;116(37):18488–18497. 10.1073/pnas.1908275116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stahl PL, Salmen F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78–82. 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- 42. Villacampa EG, Larsson L, Kvastad L, Andersson A, Carlson J, Lundeberg J. Genome‐wide spatial expression profiling in FFPE tissues. bioRxiv 2020. 10.1101/2020.07.24.219758 [DOI] [Google Scholar]
- 43. Vickovic S, Eraslan G, Salmen F, et al. High‐definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16(10):987–990. 10.1038/s41592-019-0548-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodriques SG, Stickels RR, Goeva A, et al. Slide‐seq: a scalable technology for measuring genome‐wide expression at high spatial resolution. Science. 2019;363(6434):1463–1467. 10.1126/science.aaw1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stickels RR, Murray E, Kumar P, et al. Highly sensitive spatial transcriptomics at near‐cellular resolution with Slide‐seqV2. Nat Biotechnol. 2021;39(3):313–319. 10.1038/s41587-020-0739-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen A, Liao S, Cheng M, et al. Large field of view‐spatially resolved transcriptomics at nanoscale resolution. bioRxiv 2021. 10.1101/2021.01.17.427004 [DOI] [Google Scholar]
- 47. Wang X, Fan J. Spatiotemporal molecular medicine: a new era of clinical and translational medicine. Clin Transl Med. 2021;11(1):e294. 10.1002/ctm2.294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X, Fan J. Spatiotemporal molecular imaging is a critical part of spatiotemporal molecular medicine. Clin Transl Med. 2021;11(3):e347. 10.1002/ctm2.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu Y, Yang M, Deng Y, et al. High‐spatial‐resolution multi‐omics sequencing via deterministic barcoding in tissue. Cell. 2020;183(6):1665–1681. 10.1016/j.cell.2020.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Y, Enninful A, Deng Y, Fan R. Spatial transcriptome sequencing of FFPE tissues at cellular level. bioRxiv. 2020. 10.1101/2020.10.13.338475 [DOI] [Google Scholar]
- 51. Deng Y, Zhang D, Liu Y, et al. Spatial epigenome sequencing at tissue scale and cellular level. bioRxiv. 2021. 10.1101/2021.03.11.434985 [DOI] [Google Scholar]
- 52. Angelo M, Bendall SC, Finck R, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20(4):436–442. 10.1038/nm.3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Keren L, Bosse M, Marquez D, et al. A structured tumor‐immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. 2018;174(6):1373–1387. 10.1016/j.cell.2018.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ji AL, Rubin AJ, Thrane K, et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell. 2020;182(2):497–514. 10.1016/j.cell.2020.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Giesen C, Wang HA, Schapiro D, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11(4):417–422. 10.1038/nmeth.2869 [DOI] [PubMed] [Google Scholar]
- 56. Sheng J, Zhang J, Wang L, et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut. 2021. 10.1136/gutjnl-2021-324339 [DOI] [PubMed] [Google Scholar]
- 57. Goltsev Y, Samusik N, Kennedy‐Darling J, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 2018;174(4):968–981. 10.1016/j.cell.2018.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kompauer M, Heiles S, Spengler B. Atmospheric pressure MALDI mass spectrometry imaging of tissues and cells at 1.4‐mum lateral resolution. Nat Methods. 2017;14(1):90–96. 10.1038/nmeth.4071 [DOI] [PubMed] [Google Scholar]
- 59. Maier SK, Hahne H, Gholami AM, et al. Comprehensive identification of proteins from MALDI imaging. Mol Cell Proteomics. 2013;12(10):2901–2910. 10.1074/mcp.M113.027599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306(5695):471–473. 10.1126/science.1104404 [DOI] [PubMed] [Google Scholar]
- 61. Cooks RG, Ouyang Z, Takats Z, Wiseman JM. Detection technologies: ambient mass spectrometry. Science. 2006;311(5767):1566–1570. 10.1126/science.1119426 [DOI] [PubMed] [Google Scholar]
- 62. Wiseman JM, Ifa DR, Zhu Y, et al. Desorption electrospray ionization mass spectrometry: imaging drugs and metabolites in tissues. Proc Natl Acad Sci U S A. 2008;105(47):18120–18125. 10.1073/pnas.0801066105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stopka SA, Rong C, Korte AR, et al. Molecular imaging of biological samples on nanophotonic laser desorption ionization platforms. Angew Chem Int Ed Engl. 2016;55(14):4482–4486. 10.1002/anie.201511691 [DOI] [PubMed] [Google Scholar]
- 64. He J, Sun C, Li T, et al. A sensitive and wide coverage ambient mass spectrometry imaging method for functional metabolites based molecular histology. Adv Sci (Weinh). 2018;5(11):1800250. 10.1002/advs.201800250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun C, Li T, Song X, et al. Spatially resolved metabolomics to discover tumor‐associated metabolic alterations. Proc Natl Acad Sci U S A. 2019;116(1):52–57. 10.1073/pnas.1808950116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vickovic S, Lötstedt B, Klughammer J, Segerstolpe Å, Rozenblatt‐Rosen O, Regev A. SM‐Omics: an automated platform for high‐throughput spatial multi‐omics. bioRxiv 2020. 10.1101/2020.10.14.338418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Govek KW, Troisi EC, Miao Z, Aubin RG, Woodhouse S, Camara PG. Single‐cell transcriptomic analysis of mIHC images via antigen mapping. Sci Adv. 2021;7(10):eabc5464. 10.1126/sciadv.abc5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Foster DS, Januszyk M, Chinta MS, et al. Integrated spatial multi‐omics reveals fibroblast fate during tissue repair. bioRxiv. 2021. 10.1101/2021.04.02.437928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hafemeister C, Satija R. Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. 10.1186/s13059-019-1874-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dries R, Zhu Q, Dong R, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021;22(1):78. 10.1186/s13059-021-02286-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saiselet M, Rodrigues‐Vitoria J, Tourneur A, et al. Transcriptional output, cell‐type densities, and normalization in spatial transcriptomics. J Mol Cell Biol. 2020;12(11):906–908. 10.1093/jmcb/mjaa028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Svensson V, Teichmann SA, Stegle O. SpatialDE: identification of spatially variable genes. Nat Methods. 2018;15(5):343–346. 10.1038/nmeth.4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Edsgard D, Johnsson P, Sandberg R. Identification of spatial expression trends in single‐cell gene expression data. Nat Methods. 2018;15(5):339–342. 10.1038/nmeth.4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang K, Feng W, Wang P. Identification of spatially variable genes with graph cuts. bioRxiv. 2018. 10.1101/491472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sun S, Zhu J, Zhou X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat Methods. 2020;17(2):193–200. 10.1038/s41592-019-0701-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hao M, Hua K, Zhang X. SOMDE: a scalable method for identifying spatially variable genes with self‐organizing map. bioRxiv. 2021. 10.1101/2020.12.10.419549 [DOI] [PubMed] [Google Scholar]
- 78. Liu B, Li C, Li Z, Wang D, Ren X, Zhang Z. An entropy‐based metric for assessing the purity of single cell populations. Nat Commun. 2020;11(1):3155. 10.1038/s41467-020-16904-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang L, Chen H, Pinello L, Yuan GC, GiniClust: detecting rare cell types from single‐cell gene expression data with Gini index. Genome Biol. 2016;17(1):144. 10.1186/s13059-016-1010-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tsoucas D, Yuan GC. GiniClust2: a cluster‐aware, weighted ensemble clustering method for cell‐type detection. Genome Biol. 2018;19(1):58. 10.1186/s13059-018-1431-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bergenstrahle J, Bergenstrahle L, Lundeberg J. SpatialCPie: an R/Bioconductor package for spatial transcriptomics cluster evaluation. BMC Bioinform. 2020;21(1):161. 10.1186/s12859-020-3489-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhao E, Stone MR, Ren X, et al. BayesSpace enables the robust characterization of spatial gene expression architecture in tissue sections at increased resolution. bioRxiv. 2020. 10.1101/2020.09.04.283812 [DOI] [Google Scholar]
- 83. Chang Y, He F, Wang J, et al. Define and visualize pathological architectures of human tissues from spatially resolved transcriptomics using deep learning. bioRxiv. 2021. 10.1101/2021.07.08.451210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hu J, Li X, Coleman K, et al. Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. bioRxiv. 2020. 10.1101/2020.11.30.405118 [DOI] [PubMed] [Google Scholar]
- 85. Pasquini G, Rojo Arias JE, Schafer P, Busskamp V. Automated methods for cell type annotation on scRNA‐seq data. Comput Struct Biotechnol J. 2021;19:961–969. 10.1016/j.csbj.2021.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Elosua‐Bayes M, Nieto P, Mereu E, Gut I, Heyn H, SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single‐cell transcriptomes. Nucleic Acids Res. 2021;. 49:e50. 10.1093/nar/gkab043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cable DM, Murray E, Zou LS, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2021. 10.1038/s41587-021-00830-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Song Q, Su J. DSTG: deconvoluting spatial transcriptomics data through graph‐based artificial intelligence. Brief Bioinform. 2021;22:bbaa414. 10.1093/bib/bbaa414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Andersson A, Bergenstrahle J, Asp M, et al. Single‐cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun Biol. 2020;3(1):565. 10.1038/s42003-020-01247-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Petukhov V, Xu RJ, Soldatov RA, et al. Cell segmentation in imaging‐based spatial transcriptomics. Nat Biotechnol. 2021. 10.1038/s41587-021-01044-w [DOI] [PubMed] [Google Scholar]
- 91. Palla G, Spitzer H, Klein M, et al. Squidpy: a scalable framework for spatial single cell analysis. bioRxiv. 2021. 10.1101/2021.02.19.431994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schmidt U, Weigert M, Broaddus C, Myers G. Cell detection with star‐convex polygons. Springer International Publishing; 2018:265–273. [Google Scholar]
- 93. Stringer C, Wang T, Michaelos M, Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nat Methods. 2021;18(1):100–106. 10.1038/s41592-020-01018-x [DOI] [PubMed] [Google Scholar]
- 94. Armingol E, Officer A, Harismendy O, Lewis NE. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet. 2021;22(2):71–88. 10.1038/s41576-020-00292-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wu Y, Yang S, Ma J, et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single‐cell level. Cancer Discov. 2022. 10.1158/2159-8290.CD-21-0316 [DOI] [PubMed] [Google Scholar]
- 96. Wang S, Karikomi M, MacLean AL, Nie Q. Cell lineage and communication network inference via optimization for single‐cell transcriptomics. Nucleic Acids Res. 2019;47(11):e66. 10.1093/nar/gkz204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kumar MP, Du J, Lagoudas G, et al. Analysis of single‐cell RNA‐Seq identifies cell–cell communication associated with tumor characteristics. Cell Rep. 2018;25(6):1458–1468. 10.1016/j.celrep.2018.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jin S, Guerrero‐Juarez CF, Zhang L, et al. Inference and analysis of cell–cell communication using CellChat. Nat Commun. 2021;12(1):1088. 10.1038/s41467-021-21246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Efremova M, Vento‐Tormo M, Teichmann SA, Vento‐Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi‐subunit ligand‐receptor complexes. Nat Protoc. 2020;15(4):1484–1506. 10.1038/s41596-020-0292-x [DOI] [PubMed] [Google Scholar]
- 100. Cang Z, Nie Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat Commun. 2020;11(1):2084. 10.1038/s41467-020-15968-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pham D, Tan X, Xu J, et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell–cell interactions and spatial trajectories within undissociated tissues. bioRxiv. 2020. 10.1101/2020.05.31.125658 [DOI] [Google Scholar]
- 102. Yuan Y, Bar‐Joseph Z. GCNG: graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 2020;21(1):300. 10.1186/s13059-020-02214-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tanevski J, Gabor A, Flores ROR, Schapiro D, Saez‐Rodriguez J. Explainable multi‐view framework for dissecting intercellular signaling from highly multiplexed spatial data. bioRxiv. 2020. 10.1101/2020.05.08.084145 [DOI] [Google Scholar]
- 104. Du W, Elemento O. Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies. Oncogene. 2015;34(25):3215–3225. 10.1038/onc.2014.291 [DOI] [PubMed] [Google Scholar]
- 105. Gatenby RA, Brown JS. Integrating evolutionary dynamics into cancer therapy. Nat Rev Clin Oncol. 2020;17(11):675–686. 10.1038/s41571-020-0411-1 [DOI] [PubMed] [Google Scholar]
- 106. Berglund E, Maaskola J, Schultz N, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018;9(1):2419. 10.1038/s41467-018-04724-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Thrane K, Eriksson H, Maaskola J, Hansson J, Lundeberg J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018;78(20):5970–5979. 10.1158/0008-5472.CAN-18-0747 [DOI] [PubMed] [Google Scholar]
- 108. Hunter MV, Moncada R, Weiss JM, Yanai I, White RM. Spatially resolved transcriptomics reveals the architecture of the tumor/microenvironment interface. bioRxiv. 2020. 10.1101/2020.11.05.368753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Qian J, Olbrecht S, Boeckx B, et al. A pan‐cancer blueprint of the heterogeneous tumor microenvironment revealed by single‐cell profiling. Cell Res. 2020;30(9):745–762. 10.1038/s41422-020-0355-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhou Y, Yang D, Yang Q, et al. Single‐cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. 2020;11(1):6322. 10.1038/s41467-020-20059-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Levitin HM, Yuan J, Sims PA. Single‐cell transcriptomic analysis of tumor heterogeneity. Trends Cancer. 2018;4(4):264–268. 10.1016/j.trecan.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cheng S, Li Z, Gao R, et al. A pan‐cancer single‐cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184(3):792–809. 10.1016/j.cell.2021.01.010 [DOI] [PubMed] [Google Scholar]
- 113. Duan M, Hao J, Cui S, et al. Diverse modes of clonal evolution in HBV‐related hepatocellular carcinoma revealed by single‐cell genome sequencing. Cell Res. 2018;28(3):359–373. 10.1038/cr.2018.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nagasawa S, Kuze Y, Maeda I, et al. Genomic profiling reveals heterogeneous populations of ductal carcinoma in situ of the breast. Commun Biol. 2021;4(1):438. 10.1038/s42003-021-01959-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Andersson A, Larsson L, Stenbeck L, et al. Spatial deconvolution of HER2‐positive breast tumors reveals novel intercellular relationships. bioRxiv. 2020. 10.1101/2020.07.14.200600 [DOI] [Google Scholar]
- 116. Ravi VM, Neidert N, Will P, et al. Lineage and spatial mapping of glioblastoma‐associated immunity. bioRxiv. 2021. 10.1101/2020.06.01.121467 [DOI] [Google Scholar]
- 117. Bastola S, Pavlyukov MS, Yamashita D, et al. Glioma‐initiating cells at tumor edge gain signals from tumor core cells to promote their malignancy. Nat Commun. 2020;11(1):4660. 10.1038/s41467-020-18189-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zheng B, Wang D, Qiu X, et al. Trajectory and functional analysis of PD‐1(high) CD4(+)CD8(+) T cells in hepatocellular carcinoma by single‐cell cytometry and transcriptome sequencing. Adv Sci (Weinh). 2020;7(13):2000224. 10.1002/advs.202000224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Halama N, Michel S, Kloor M, et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011;71(17):5670–5677. 10.1158/0008-5472.CAN-11-0268 [DOI] [PubMed] [Google Scholar]
- 120. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor‐infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–821. 10.1038/s41423-020-0488-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Romanens L, Chaskar P, Tille J‐C, et al. Spatial transcriptomics of tumor microenvironment in formalin‐fixed paraffin‐embedded breast cancer. bioRxiv. 2020. 10.1101/2020.01.31.928143 [DOI] [Google Scholar]
- 122. Birkbak NJ, McGranahan N. Cancer genome evolutionary trajectories in metastasis. Cancer Cell. 2020;37(1):8–19. 10.1016/j.ccell.2019.12.004 [DOI] [PubMed] [Google Scholar]
- 123. Amirouchene‐Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. 2017;7(8):805–817. 10.1158/2159-8290.CD-17-0343 [DOI] [PubMed] [Google Scholar]
- 124. Zhao Y, Fu X, Lopez JI, et al. Selection of metastasis competent subclones in the tumour interior. Nat Ecol Evol. 2021;5:1033‐1045. 10.1038/s41559-021-01456-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sundar R, Liu DH, Hutchins GG, et al. Spatial profiling of gastric cancer patient‐matched primary and locoregional metastases reveals principles of tumour dissemination. Gut. 2020;70:1823‐1832. 10.1136/gutjnl-2020-320805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Brady L, Kriner M, Coleman I, et al. Inter‐ and intra‐tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun. 2021;12(1):1426. 10.1038/s41467-021-21615-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang DY‐T, Tan TZ, Tai Y‐T, et al. Case study: digital spatial profiling of metastatic clear cell carcinoma reveals intra‐tumor heterogeneity in epithelial‐mesenchymal gradient. bioRxiv. 2021. 10.1101/2021.05.27.445912 [DOI] [Google Scholar]
- 128. Lu S, Stein JE, Rimm DL, et al. Comparison of biomarker modalities for predicting response to PD‐1/PD‐L1 checkpoint blockade: a systematic review and meta‐analysis. JAMA Oncol. 2019;5:1195‐1204. 10.1001/jamaoncol.2019.1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Johnson DB, Bordeaux J, Kim JY, et al. Quantitative spatial profiling of PD‐1/PD‐L1 interaction and HLA‐DR/IDO‐1 predicts improved outcomes of anti‐PD‐1 therapies in metastatic melanoma. Clin Cancer Res. 2018;24(21):5250–5260. 10.1158/1078-0432.CCR-18-0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Levy‐Jurgenson A, Tekpli X, Kristensen VN, Yakhini Z. Spatial transcriptomics inferred from pathology whole‐slide images links tumor heterogeneity to survival in breast and lung cancer. Sci Rep. 2020;10(1):18802. 10.1038/s41598-020-75708-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yoosuf N, Navarro JF, Salmen F, Stahl PL, Daub CO. Identification and transfer of spatial transcriptomics signatures for cancer diagnosis. Breast Cancer Res. 2020;22(1):6. 10.1186/s13058-019-1242-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Reynolds BA, Oli MW, Oli MK. Eco‐oncology: applying ecological principles to understand and manage cancer. Ecol Evol. 2020;10(16):8538–8553. 10.1002/ece3.6590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021;27(2):212–224. 10.1038/s41591-021-01233-9 [DOI] [PubMed] [Google Scholar]
- 134. Ma L, Hernandez MO, Zhao Y, et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell. 2019;36(4):418–430. 10.1016/j.ccell.2019.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Chang CH, Qiu J, O'Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. 10.1016/j.cell.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ries L, Fletcher RJ, Battin J, Sisk TD. Ecological responses to habitat edges: mechanisms, models, and variability explained. Annu Rev Ecol Evol Syst. 2004;35(1):491–522. 10.1146/annurev.ecolsys.35.112202.130148 [DOI] [Google Scholar]
- 137. Kent M, Gill WJ, Weaver RE, Armitage RP. Landscape and plant community boundaries in biogeography. Prog Phys Geogr. 2016;21(3):315–353. 10.1177/030913339702100301 [DOI] [Google Scholar]
- 138. Forman RTT. Some general principles of landscape and regional ecology. Landsc Ecol. 1995;10(3):133–142. 10.1007/bf00133027 [DOI] [Google Scholar]
- 139. Chen H, Murray E, Laumas A, et al. Dissecting mammalian spermatogenesis using spatial transcriptomics. bioRxiv. 2020. 10.1101/2020.10.17.343335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Melzer C, von der Ohe J, Lehnert H, Ungefroren H, Hass R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol Cancer. 2017;16(1):28. 10.1186/s12943-017-0595-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–238. 10.1016/j.stem.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gaston KJ. Global patterns in biodiversity. Nature. 2000;405(6783):220–227. 10.1038/35012228 [DOI] [PubMed] [Google Scholar]
- 143. Whittaker RH. Evolution and measurement of species diversity. Taxon. 2019;21(2‐3):213–251. 10.2307/1218190 [DOI] [Google Scholar]
- 144. Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27(3):379–423. 10.1002/j.1538-7305.1948.tb01338.x [DOI] [Google Scholar]
- 145. Simpson EH. Measurement of diversity. Nature. 1949;163(4148):688–688. 10.1038/163688a0 [DOI] [Google Scholar]
- 146. Gini C. On the measure of concentration with special reference to income and statistics. Colorado College Publ Gen Ser. 1936;208(1):73–79. [Google Scholar]
- 147. Asp M, Giacomello S, Larsson L, et al. A spatiotemporal organ‐wide gene expression and cell atlas of the developing human heart. Cell. 2019;179(7):1647–1660. 10.1016/j.cell.2019.11.025 [DOI] [PubMed] [Google Scholar]
- 148. Fedorov A, Beichel R, Kalpathy‐Cramer J, et al. 3D slicer as an image computing platform for the quantitative imaging network. Magn Reson Imaging. 2012;30(9):1323–1341. 10.1016/j.mri.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lewis SM, Asselin‐Labat ML, Nguyen Q, et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods. 2021;18(9):997–1012. 10.1038/s41592-021-01203-6 [DOI] [PubMed] [Google Scholar]
- 150. Rao A, Barkley D, Franca GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596(7871):211–220. 10.1038/s41586-021-03634-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single‐cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Rev Genet. 2021;22(10):627–644. 10.1038/s41576-021-00370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]