

ABSTRACT
Coronavirus disease 2019, caused by SARS-CoV-2, remains an on-going pandemic, partly due to the emergence of variant viruses that can “break-through” the protection of the current vaccines and neutralizing antibodies (nAbs), highlighting the needs for broadly nAbs and next-generation vaccines. We report an antibody that exhibits breadth and potency in binding the receptor-binding domain (RBD) of the virus spike glycoprotein across SARS coronaviruses. Initially, a lead antibody was computationally discovered and crystallographically validated that binds to a highly conserved surface of the RBD of wild-type SARS-CoV-2. Subsequently, through experimental affinity enhancement and computational affinity maturation, it was further developed to bind the RBD of all concerning SARS-CoV-2 variants, SARS-CoV-1 and pangolin coronavirus with pico-molar binding affinities, consistently exhibited strong neutralization activity against wild-type SARS-CoV-2 and the Alpha and Delta variants. These results identify a vulnerable target site on coronaviruses for development of pan-sarbecovirus nAbs and vaccines.
KEYWORDS: Computational antibody discovery, Sars-CoV-2, emerging variants, broadly neutralizing antibody, broad-spectrum vaccine
Introduction
Zoonotic coronaviruses have caused three major disease outbreaks in the past two decades: the SARS outbreak caused by severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) in 2002–2004, the Middle East respiratory syndrome (MERS) outbreaks caused by MERS-CoV since 2012, and coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 since December 2019.1,2 The three viruses that caused these outbreaks are positive-sense single-stranded RNA viruses3 belonging to the genera Betacoronavirus. SARS-CoV-1 and SARS‑CoV‑2 are members of the subgenus Sarbecovirus (lineage B), while MERS-CoV belongs to the subgenus Merbecovirus (lineage C).1,4 SARS‑CoV‑2 is divergent from SARS‑CoV‑1, residing in its own distinct phylogenetic clade.5
As of October 2021, the ongoing global COVID-19 pandemic has caused more than 240 million infections and claimed at least 4.8 million lives worldwide. With unprecedented speed, several vaccines were developed and licensed for use in a massive global vaccination program.6 Since the beginning of the pandemic, however, SARS-CoV-2 has undergone significant antigenic drift, resulting in the emergence of several more virulent variants. The Alpha variant was first found in the United Kingdom, but it quickly became a dominant strain.7 Alpha was then outcompeted by the Delta variant, which arose in India and spread to become the most prevalent strain in the world.8 Mutations can confer increased infectivity, resistance to neutralizing antibodies (nAbs), and higher replication efficiency.9–11 As an example of this process, the key mutations (K417N and E484K) that distinguish the Beta and Gamma variants were found to abrogate binding to and neutralization by the two receptor-binding domain (RBD)-binding antibody families most frequently elicited by the spike glycoprotein or RBD derived from the Wuhan strain, thus reducing the effectiveness of current vaccines.12 Rising concern about viral escape triggered efforts to develop a more broadly protective second-generation of coronavirus vaccines13,14 or another novel vaccination strategy.15
While the deployment of preventive vaccines has been the most effective approach to curb the spread of SARS-CoV-2, nAbs can also provide immediate solutions for the treatment of COVID-19 patients. To date, more than 10 monoclonal antibodies (mAbs) have been developed for COVID-19 therapy or prophylaxis, and five of them are approved for clinical use.16,17 Most of these mAbs prevent the RBD from binding the ACE2 receptor, thus blocking SARS-CoV-2 entry into cells. These nAbs were based on the original virus strain, and four clinical-stage nAbs (etesevimab, bamlanivimab, casirivimab, and regdanvimab) show reduced neutralization potency against SARS-CoV-2 variants of concern (VOCs) (Alpha/B.1.1.7, Beta/B.1.351, Gamma/P.1, Delta/B.1.617.2, DeltaPlus/AY.1, AY.2, AY.3).16–19 This underscores the need for new nAbs that protect against all currently circulating variants, ideally all sarbecovirus clades. Several broadly neutralizing mAbs were discovered in blood samples from COVID-19 convalescent individuals or vaccinated individuals.15,19–21
Herein, we report a computational approach with additional experimental affinity enhancement that we used to develop a highly potent nAb that broadly targets all currently circulating SARS-CoV-2 variants, as well as SARS-CoV-1 and pangolin coronavirus.
Results
Computational design approach to discover an antibody against the wild-type SARS-CoV-2 RBD
We initially aimed to discover nAbs against wild-type SARS-CoV-2 (Wuhan strain) by computational design of mAbs that bind to the SARS-CoV-2 RBD. We used four available crystal structures of the SARS-CoV-1 RBD bound to the antigen-binding fragment (Fab) of four nAbs against different surfaces of the RBD of SARS-CoV-1 (m396,22 F26G19,23 80 R,24 S23025) (Figure 1(a)). By superposing the SARS-CoV-2 RBD in complex with human angiotensin-converting enzyme 2 (hACE2)26 on these structures, we were able to extract four models of the variable fragment (Fv) bound to the SARS-CoV-2 RBD. Of note, none of these four antibodies showed any cross-reactivity with the SARS-CoV-2 spike glygcoprotein.27,28 Assuming it was possible, with these Fab docking positions, to shift the antigen-binding specificity to the SARS-CoV-2 RBD, we performed a sequence design on the complementarity-determining regions (CDRs) using the Rosetta software suite.29,30 From ~1000 outputs, we selected 55 designs based on criteria including shape-complementarity, buried solvent-accessible surface area, and number of unsatisfied polar atoms. We then reformatted the selected designs into their full IgG1 form and produced them in Chinese hamster ovary (CHO) cells. One mAb, designated D27, exhibited outstanding binding with the SARS-CoV-2 RBD in a biolayer interferometry (BLI) experiment (Figure 1(b)). When we quantified their interaction, however, we measured a weak dissociation constants (KD) of only 177 nM (Figure 1(c)), which is even weaker than the interaction between hACE2 and the RBD (KD < 100 nM), as reported in the literature.31,32 Of note, D27 was derived from m396, and contains 16 amino acid substitutions relative to m396, all in the CDRs.
Figure 1.
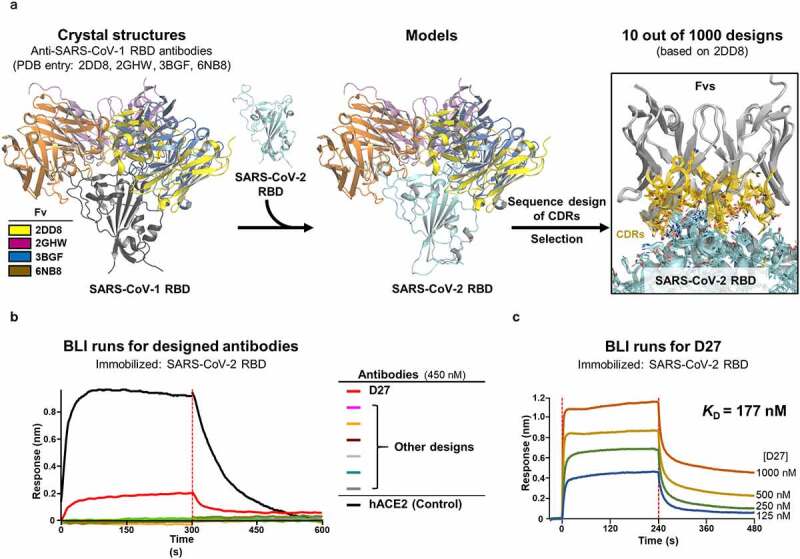
Computational design approach to discover mAbs against the SARS-CoV-2 RBD. (a) The anti-SARS-CoV-1 RBD Fvs derived from the four indicated structures were modeled on the SARS CoV-2 RBD and then subjected to a sequence design protocol. Only CDR sequences were designed, with 55 out of 1000 generated Fv designs ultimately selected for experimental validation. (b) Screening. 55 Fvs were converted into their full IgG format, produced in culture, and tested for binding to the SARS-CoV-2 RBD by BLI. One antibody, D27, showed outstanding binding signal. (c) Quantification of the binding affinity of D27. The SARS-CoV-2 RBD concentration ranged from 126–1000 nM. The BLI experiment was technically duplicated with similar results, and representative sensorgrams are shown.
Structural validation of the computational design
We next determined the crystal structure of the Fab of D27 (D27-Fab) bound to the SARS-CoV-2 RBD at 2.2 Å resolution (Table 1, PDB ID: 7VYR). This structure was closely superposable onto the D27 design, validating the design was largely correct (Figure 2(a)). In comparison to the original m369, a number of amino acid residues substituted in the sequence design are involved in favorable interactions. For example, S92FL and S93LL on light-chain CDR 3 (CDRL3) are involved in hydrophobic interactions with V407 and Y506, as well as the aliphatic portions of T376 and K378 on the RBD (Figure 2(b) and Figure S1b; Region 1). Likewise, S31RH on CDRH1 approaches a cavity of the RBD and forms a hydrogen-bonded network involving backbone atoms of Y369, S371, and F374 (Figure 2(b) and Figure S1b; Region 2).
Table 1.
X-ray data collection and structure refinement statistics
| Data Collection | D27-Fab‒SARS-CoV-2 RBD (PDB ID: 7VYR) |
|---|---|
| Space group | P21 |
| Unit cell dimensions | |
| a, b, c (Å) | 61.80, 191.68, 65.90 |
| α, β, γ (°) | 90.0, 93.99, 90.0 |
| Wavelength (Å) | 1.0000 |
| Resolution (Å) | 47.92–2.2 (2.279–2.2)a |
| R-merge | 0.087 (0.267)a |
| I/σ(I) | 20.04 (7.35)a |
| Completeness (%) | 95.37 (90.13)a |
| Multiplicity | 6.3 |
| Refinement | |
| No. of reflections | 74,394 (6,968)a |
| Rwork/Rfree (%) | 19.2/23.5 |
| R.m.s deviations | |
| Bond (Å)/Angle (º) | 0.009/1.22 |
| Average B-values (Å2) | 29.07 |
| Ramachandran plot (%) | |
| Favored/Additional allowed | 94.78/4.40 |
| Outliers | 0.82 |
| aThe numbers in parentheses are the statistics from the highest resolution shell. | |
Figure 2.
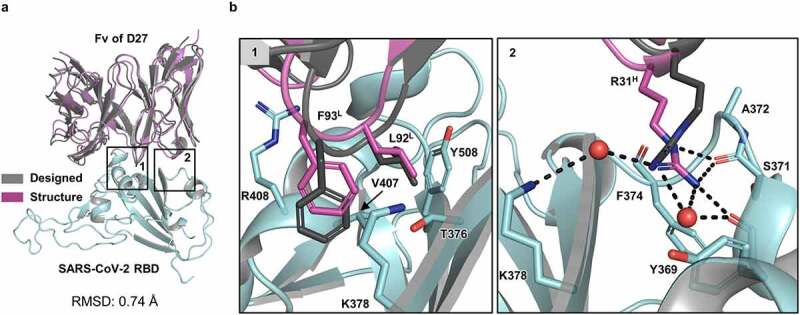
Structural validation of the D27 design. (a) The computational design model (gray) aligns well with the crystal structure of D27-Fab‒SARS-CoV-2 RBD (magenta). For clarity, only the Fv portion of the Fab in the crystal structure is shown. The RMSD value for all aligned C⍺ atoms is 0.74 Å. (b) Zoomed-in views show selected intermolecular interactions in two regions (1 and 2) shown in (a). Region 1 shows key hydrophobic interactions of L92 and F93 (on CDRL3) with a hydrophobic pocket formed by the side chains of the RBD residues T376, K378, V407, R408, and Y508. Region 2 shows key hydrogen-bonding interactions of the guanidine group of R31H (on CDRH1) inserted into a pocket formed by backbone atoms Y369, S371, and F374, plus two water molecules. This hydrogen-bonded network in the crystal structure was not recapitulated by the sequence design because it does not include explicit water molecules.
Structure-based CDR3 loop extension enhanced the binding affinity by 25 fold
With the high-resolution D27-Fab‒SARS-CoV-2 RBD structure in hand, we sought to improve the binding affinity between the two proteins. One way to accomplish this is saturation mutagenesis either of all CDR residues or at least those in contact with the target antigen. We chose not to take this approach because closer examination of the structure revealed that the CDRL3 and the CDRH3 loops are rather loosely packed against the RBD (Figure 3(a)). Thus, extension of the tips of these loops by one and two residues, respectively, would lead to better occupation of the interfacial space between D27 and the RBD, likely strengthening their intermolecular interactions. To find the best possible sequences, we used random DNA libraries in the form of two sets of oligonucleotides to convert F93L (at the tip of CDRL3) into two randomized residues, as well as E101H and Q102H (at the tip of CDRH3) into four randomized residues (Figure 3(a)). Subsequently, these oligonucleotide libraries were incorporated into template DNA encoding the single-chain Fv (scFv) of D27. The resulting scFv library (~6.4 x 107 diversity) was expressed and displayed on the surface of yeast (Figure 3(b)). We performed two rounds of magnetic-activated cell sorting (MACS) and three rounds of fluorescence-activated cell sorting (FACS) with increasing stringency. This allowed us to enrich and select scFvs with increased binding affinity for the RBD (Figure 3(c)). Of the nine scFvs we selected, the one with strongest binding affinity was reformatted in its full IgG form (Figure 3(d) and Table 2). We then found that this mAb, designated D27LE, bound the SARS-CoV-2 RBD with a KD of 7.12 nM (Figure 3(e)), representing a binding affinity roughly 25-fold tighter than that of D27.
Figure 3.
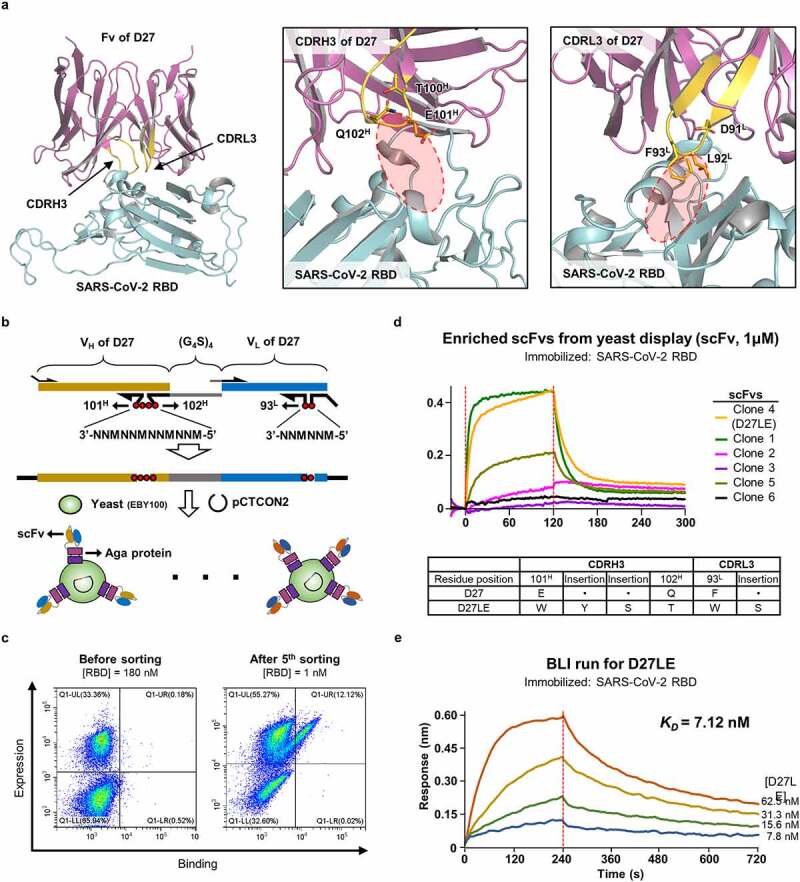
Affinity enhancement by extending the CDR3 loops. (a) Room for improving intermolecular contacts. Left: The positions of the CDRH3 and CDRL3 loops (yellow) are indicated on the crystal structure. Middle, Right: Unoccupied space near the tips of CDRL3 and CDRH3 are encircled. (b) Library construction. A synthetic DNA oligomer library covering the indicated positions was cloned into a vector, and the resulting D27-scFv library was displayed on the surface of yeast. (c) Cell sorting. Yeast cells displaying high-affinity D27-scFvs were successively enriched and selected with increasing stringency ([RBD] from 1 μM to 1 nM). The initial and final rounds are shown. (d) Six scFvs from the final enriched yeast cells were evaluated individually by BLI at fixed concentrations to select D27LE. The amino acid sequences of the randomized segments of D27 and D27LE are compared. (e) Quantification of the binding affinity of D27LE was performed in triplicate at the four indicated concentrations.
Table 2.
Binding affinity measurements obtained by BLI for the nine clones enriched in the yeast display/cell sorting procedures
| Name | Sequence in CDRH3 |
Sequence in CDRL3 |
KD (nM) | Analyte concentration (μM)b |
||||
|---|---|---|---|---|---|---|---|---|
| 101 H | Insertion | Insertion | 102 H | Insertion | 93 L | |||
| Clone 1 | W | Y | S | T | W | Q | 677 | 2, 1, 0.5, 0.25 |
| Clone 2 | M | G | T | G | P | K | 1392 | 1 |
| Clone 3 | Q | P | R | . | K | K | No binding | 1. |
| D27LE | W | Y | T | S | W | S | 373 | 2, 1, 0.5, 0.25 |
| Clone 5 | F | Y | T | A | W | V | 4278 | 1 |
| Clone 6 | N | K | T | D | C | W | NMa | . |
| Clone 7 | R | A | R | C | K | S | NM | . |
| Clone 8 | S | P | S | W | C | Q | NM | . |
| Clone 9 | S | Y | N | R | V | T | No binding | 1 |
| D27 (Reference) | E | . | . | Q | . | F | ~4000 | 1 |
aKD was not measured for clones that contain a cysteine residue.
bRBD was immobilized on the biosensor tip and each KD was measured at a single concentration or at four different concentrations.
Computational affinity maturation for the SARS-CoV-2 Alpha variant
By the time we had developed D27LE, the Alpha variant was a serious concern, having spread to many countries. This variant includes the N501Y mutation, which increases RBD binding affinity for the human ACE2 receptor by several fold relative to the wild-type RBD.12,33 On our crystal structure, N501 is close to four residues: one on CDRH1 (S33) and three on CDRH2 (D52, S54, R55) (Figure 4(a)). After modeling the N501Y mutation on the crystal structure, we sought to optimize local interactions with Y501 using a sequence design protocol confined to these four nearby residues in hopes of increasing binding affinity for variants with this mutation. We selected 10 of 100 resulting designs and expressed their scFv forms in E.coli. Eight of the 10 designs exhibited improved binding affinity for the N501Y mutant RBD target, and one design (Y4.2) showed outstanding binding affinity (KD of 4.87 nM) (Figure 4(b-c) and Table 3). Notably, its IgG form bound the N501Y RBD target with ultra-high binding affinity (KD < 1 pM) (Figure 4(c)). Sequencing showed that two of the four designed CDR residues, D52H and S54H, were changed to A52H and G54H. The resulting Ab, named D27LEY, demonstrates that significant affinity maturation against the results of antigenic drift can be achieved quickly by using extensive computational amino acid sampling for a small set of specific residues on a mother antibody. Interestingly, we found D27LE has higher binding affinity for the N501Y mutant RBD (KD = 0.54 nM) than for the wild-type RBD (KD = 7.12 nM) (Figure S2a). Thus, computational affinity maturation for the N501Y mutant RBD enhanced binding affinity > 500 fold. Because N501 is remote from the CDRL3 and CDRH3 loops (extended in D27LE), the affinity enhancement we observed for D27LE must be attributed to the N501Y mutation itself. It is likely that a tyrosine residue at this position provides an even stronger interaction with D27LE than the wild-type asparagine residue because of its hydrophobicity and bulkiness. Of note, the two-residue changes in D27LEY (in comparison with D27LE) that optimized binding to the N501Y mutation increased rather than decreased the binding affinity for the wild-type RBD, with the KD falling from 7.12 nM to 1.14 nM (Figure S2b). These results indicate that the original interface design was suboptimal, at least in part due to inaccuracies in the initial Ab-RBD model. We suspect saturation mutagenesis of CDR residues would have led us to similar results.
Figure 4.
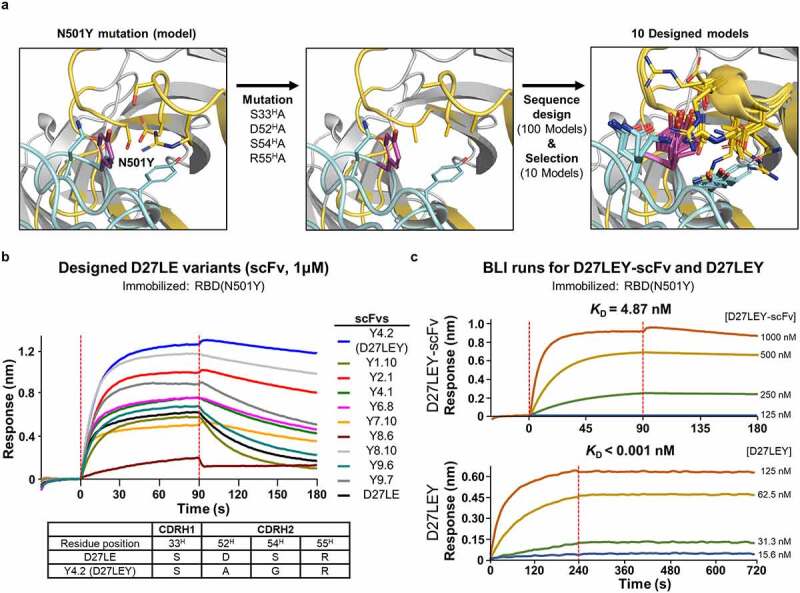
Computational affinity maturation against N501Y mutant RBD. (a) Computational steps. We modeled the Y501 mutation on the crystal structure and replaced four CDR residues (i.e., S33H, D52H, S54H, and R55H) near Y501 with alanine. After subsequent sequence design confined to these four residues, 10 of the resulting 100 models were selected. (b) Screening. These 10 designs in their Fv form evaluated for their binding to the N501Y mutant RBD using BLI. The scFv:RBD(N501Y) concentrations (in nM) were 1000:10. Tabulated below are the amino acid substitutions in the highest-affinity scFv (Y4.2) in comparison with D27LE. (c) The monovalent binding affinity of Y4.2 was measured at the indicated concentrations. Y4.2 was converted to the IgG form (D27LEY), and its bivalent binding affinity was quantified in triplicate at D27LEY concentrations ranging from 15.6–125 nM. The KD value of this interaction is less than 1 pM, which is lower than the instrument sensitivity.
Table 3.
Screening 10 designs for affinity maturation of D27LE against the N501Y mutation in the RBD
| Name |
Sequence in CDRH1 |
Sequence in CDRH2 |
KD (nM)a |
||
| 33 H |
52 H |
54 H |
55 H |
||
| Y1.10b | T | G | R | Q | 841.9 |
| Y2.1 | S | V | R | D | 30.8 |
| Y4.1 | T | A | T | R | 101.8 |
| Y4.2 (D27LEY) | S | A | G | R | 9.4 |
| Y6.8 | T | A | D | R | 93.5 |
| Y7.10 | T | I | S | R | 41.2 |
| Y8.6 | D | S | G | R | 6627 |
| Y8.10 | S | A | G | Q | 21.3 |
| Y9.6 | T | I | T | I | 262.8 |
| Y9.7 | S | N | S | I | 79.2 |
| D27LE (Reference) | S | D | S | R | 276.1 |
aKD was measured at a single concentration (1 µM) of the N501Y mutant RBD by BLI.
bThe designs were produced in Fv form.
D27LEY potently binds to the RBDs of SARS-CoV-2 variants, SARS-CoV-1 and pangolin CoV
As antigenic drift has continued, new SARS-CoV-2 VOCs have emerged as follows: Beta (B.1.351) and Gamma (P.1) with K417N/T and E484K mutations in addition to N501Y, Delta (B.1.617.2) having key L452R and T478K mutations, and DeltaPlus (AY.1) with an additional K417N mutation. Two SARS-CoV-2 variants of interests (VOIs) (Lambda/C.37, Mu/B.1.621) and a dozen of variants under monitoring (VUMs), including Epsilon (B.1.427) and Kappa (B.617.1), have also emerged. The Lambda variant has key T76I and L452Q mutations. Of note, the K417N and E484K mutations abrogate the RBD binding and neutralization capacity of the two most frequently elicited SARS-CoV-2 antibody families (IGHV3–53/3-66 and IGHV1-2).12 They also reduce the neutralizing activity of sera isolated from convalescent plasma and from vaccinated individuals.34 The T76I and L452Q mutations seem to be responsible for conferring higher infectivity and resistance to nAbs induced by mRNA vaccination.35,36 Remarkably, of all these mutations, only N501Y is located on the D27LEY-binding surface of the RBD (Figure 5(a)). This suggests that D27LEY might not be affected by these key mutations, and thus may bind to the variant RBDs similarly as it binds the wild-type RBD. Indeed, we found D27LEY bound to the RBD of the Beta and Gamma variants (which carry N501Y) with a KD of less than 1 pM (Figure 5(b)). D27LEY bound to the RBDs of the Delta, DeltaPlus, Kappa (B.1.617), Lambda, and Epsilon (B.1.427) variants (which do not carry N501Y) with picomolar affinity (0.01 nM < KD < 0.88 nM) (Figure 5(c) and Figure S2b).
Figure 5.
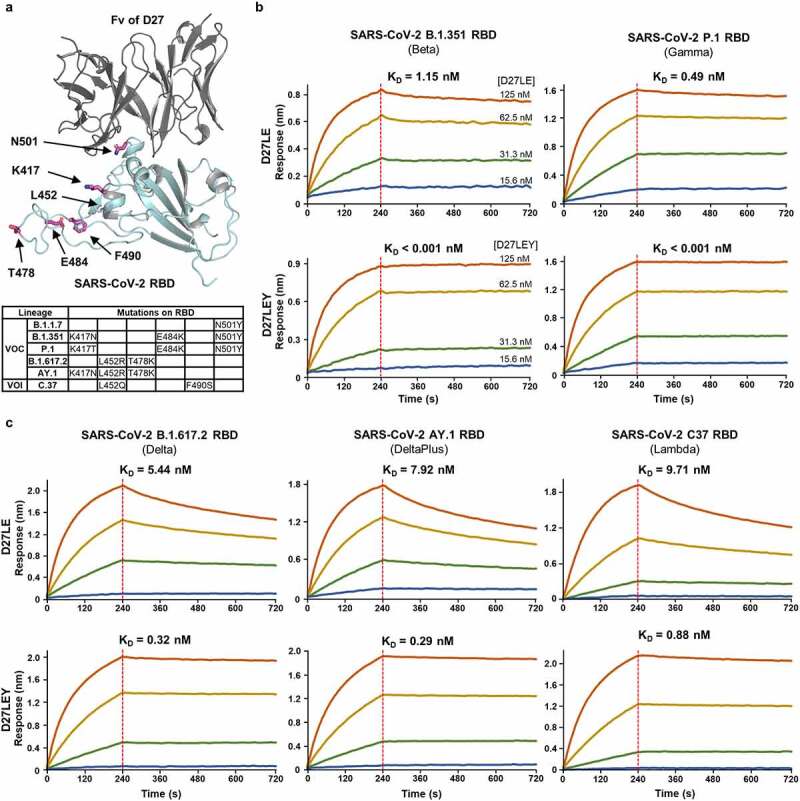
High to ultrahigh binding affinity of D27LEY for the RBDs of SARS-CoV-2 VOCs and a VOI. (a) Apart from N501, key mutations in the existing variants are remote from the antibody binding surface. (b and c) Binding affinity measurements for the RBDs with the N501Y mutation (b) and for those without the N501Y (c). The KD values for each variant are shown. D27LEY showed higher binding affinity than D27LE. In all BLI runs, four different concentrations of the antibody were used (i.e., 15.6, 31.3, 62.5, and 125 nM). The experiments were performed in technical triplicate with each giving similar results. Representative sensorgrams are shown.
We tested whether D27LEY might exhibit cross-reactivity against the RBD of SARS-CoV-1 (Tor 2) and pangolin CoV (PCoV-GD1). Indeed, D27LEY binds to the SARS-CoV-1 RBD with a KD of <1 pM and to the PCoV-GD1 RBD with a KD of 0.66 nM (Figure S3). Thus, D27LEY has RBD-binding potency across sarbecovirus clades Ia and Ib. The sub-picomolar binding affinity of D27LEY for the SARS-CoV-1 RBD is intriguing because its binding interactions were optimized for the SARS-CoV-2 RBD and for its tyrosine substitution of N501, which is a threonine residue in the SARS-CoV-1 RBD. It is possible that the extended CDR3 loops created “hot spot” interactions for the SARS-CoV-1 RBD, and the minor sequence variation in the binding epitopes between the two RBDs, as discussed below, allowed parallel affinity enhancement.
D27LEY strongly or potently neutralizes SARS-CoV-2 variants
The sub-picomolar binding affinity between D27LEY and the N501Y mutant RBD (of the Alpha variant) is at least 10,000-fold higher than the affinity between hACE2 and the N501Y mutant RBD (KD = 22 nM).37 D27LEY also binds the RBD of wild-type SARS-CoV-2 and that of the Delta variant with reduced but still potent affinities (KD = 1.14 nM and 0.32 nM, respectively) (Figure 5(c) and Figure S2b). We examined whether these binding affinities measured in vitro correlate with virus neutralization potency against wild-type SARS-CoV-2 and the SARS-CoV-2 Alpha and Delta variants. In a Vero E6 cell-based focus reduction neutralization test (FRNT), D27LEY exhibited the most potent neutralizing activity against the Alpha variant with a neutralization constant 50 (NC50) of 0.10 nM (Figure 6). D27LEY exhibited similarly strong neutralization activity against the wild-type virus and the Delta variant with NC50 values of 8.3 nM and 12.2 nM, respectively (Figure 6). Given that the Delta variant infects host cells more efficiently than wild-type SARS-CoV-2,38 the neutralization potency of D27LEY correlates nicely with its potency of binding to each respective RBD. Presumably, D27LEY is likely to exhibit similar neutralization activity against most, if not all, circulating SARS-CoV-2 variants and related sarbecoviruses that may emerge in the future.
Figure 6.
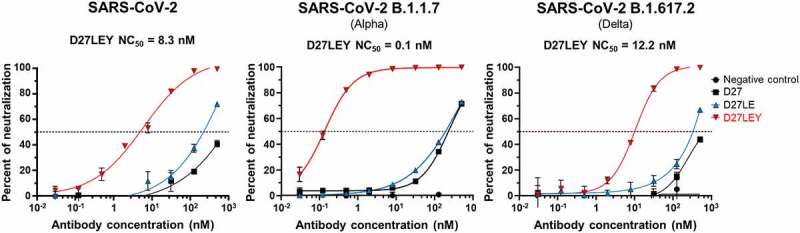
Virus neutralization assessed by FRNT. The neutralization capacity of D27LEY against the wild-type and two variants of SARS-CoV-2 as measured by FRNT (experimental details in the Methods section). The concentration of D27LEY that achieved a > 50% reduction in the number of infectious viral foci (NC50) is shown for each virus strain. These FRNT experiments were performed in technical triplicate with each set showing similar results. A single representative run for each condition is shown.
The epitope of D27LEY is a highly conserved surface on the RBD
Previously, various antigenic sites of the RBD recognized by different nAbs were classified into site Ia, site Ib, site IIa, site IIb, site IIc, site IV, and site V.19,39 Site Ia and site Ib largely overlap with the RBD’s hACE2-binding surface (designated RBM for receptor binding motif). Site IIa is mostly in the RBD core and is accessible only in the open RBD state. All the SARS-CoV-2 RBD mutations that permit the virus to escape recognition by 10 different antibodies were recently mapped and the results showed that a surface patch on the RBD core that includes antigenic site IIa is evolutionarily conserved across sarbecoviruses, and that escape mutations there disrupt proper protein folding or expression.34 In contrast, mutations that appear on the RBM are highly variable across sarbecoviruses.34
Very recently, three potent and broadly nAbs, S2X259, S2X35 and DH1047, were reported. A structural comparison shows that they target the same side on the RBD containing site IIa, which is actually the surface recognized by S2X3539 and is partly shared by D27LEY, S2X259 and DH1047 (Figure 7(a-b)). The D27LEY-binding surface is closer to the RBM than is the other binding surfaces. Consequently, the summed binding surface spans a fairly wide patch on this side of the RBD (Figure 7(b)). This surface does not include the key mutations sites found in many SARS-CoV-2 variants except N501, which is an epitope residue for D27LEY. According to a multiple sequence alignment, apart from N501, the epitope residues recognized by D27LEY are all invariable in the SARS-CoV-2 VOCs, VOIs, and VUMs (Figure 7(c)). This explains the broad and potent binding of D27LEY to currently circulating SARS-CoV-2 variants. The alignment also shows that only three epitope residues for D27LEY are variable in sarbecovirus clade Ia, which includes SARS-CoV, and in clade Ib, which includes pangolin CoV. Moreover, these three residues exhibit limited variation among clades Ia and Ib (Figure 7(c)), explaining the cross-reactivity of D27LEY for their RBDs. Similar conclusions can be drawn to explain the broad neutralization activities of the other three nAbs. Importantly, this comparison of the binding epitopes convincingly defines a vulnerable surface on the RBD that can be targeted for developing broadly protecting nAbs and vaccines.
Figure 7.
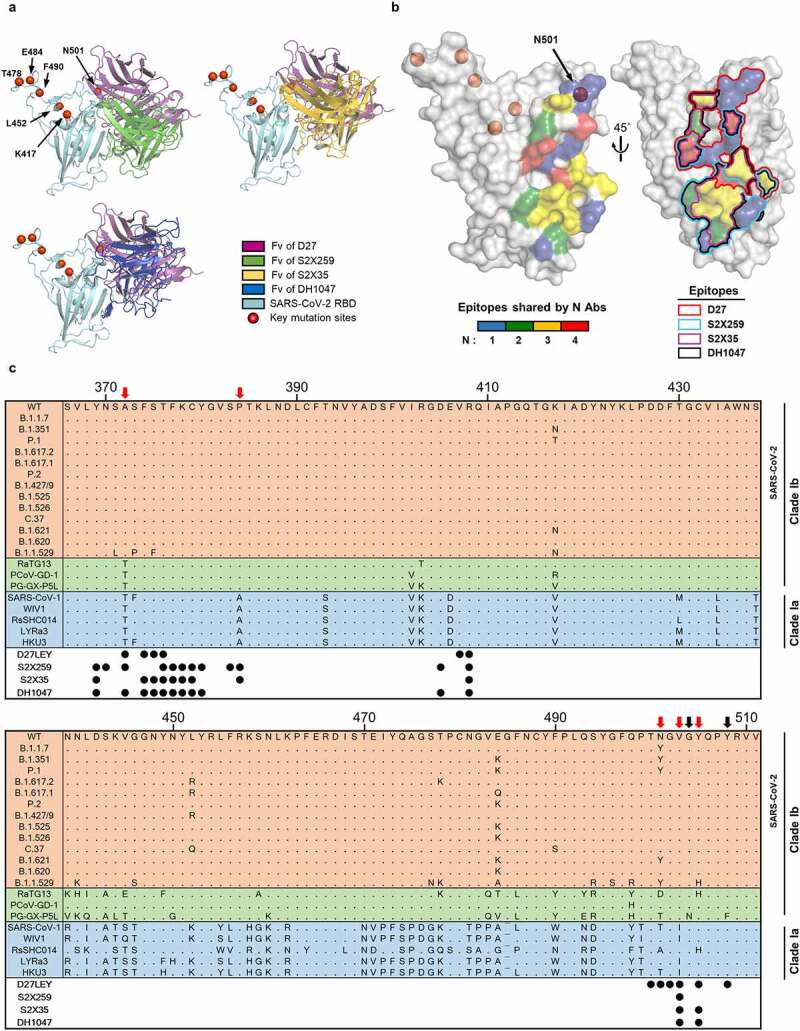
The epitopes recognized by four broadly nAbs. (a) The Fvs of D27, S2X259 (PDB entry: 7RAL), S2X35 (PDB entry: 7R6W) and DH1047 (PDB entry: 7LD1) are shown bound to the RBD (cyan). The red circles indicate key mutation sites shared by many SARS-CoV-2 variants (https://covariants.org/shared-mutations). (b) On the surface of the RBD, the epitopes recognized by the four nAbs are shown, color-coded according to the number of sharing antibodies (Left) and delineated by lines (Right). Epitopes are defined as the RBD residues that contact with ≥ 40 atoms of the bound antibody within 4.5 Å. (c) Alignment of the representative RBD sequences is shown for sarbecovirus clades Ia and Ib, which include the SARS-CoV-2 VOCs, VOIs, and VUMs. The residue positions of the epitopes are indicated by the circles below the alignment. The black arrows indicate epitope residues that are variable in bat and pangolin coronaviruses (RaTG13, PCoV-GD-1, PG-GX-P5L), but not in the SARS-CoV-2 variants. The red arrows indicate epitope residues that are variable across clades Ib and Ia.
Discussion
De novo computational antibody design involves two steps: global docking of scaffold antibodies to antigens, followed by the design of CDR sequences. Yet, the success rate of de novo antibody design is low, with computational approaches remaining mostly limited to affinity maturation based on antibody-antigen co-crystal structures. This indicates that obtaining “plausible” antibody-antigen docking models remains a significant challenge.40 Recently, a computational method using a position-specific structure-scoring method led to the discovery of distinct CDRH3 sequences that exhibit structural homology to the CDRH3 of the original antibody against influenza H1 hemagglutinin. In this approach, the antibody-antigen docking problem is precluded.41 In our study, we constructed docking models for Ab‒SARS-CoV-2 RBD based on four known structures of human mAbs bound to the SARS-CoV-1 RBD. This approach was successful, providing a lead antibody from among only 55 designs. It should be noted, however, that only one model for Ab‒SARS-CoV-2 RBD using m396 gave a hit. In retrospect, the other three mAbs (F26G19, 80 R, S230) used for the initial docking models were in contact with a structurally most variable region between the SARS-CoV-1 and SARS-CoV-2 RBDs. This uncertainty was probably the main reason why the three docking models were not productive, although insufficient sampling might have been responsible for the failure.
The computational sequence design of CDRs involving the Monte Carlo search for amino acid sequence is analogous to the experimental phage display biopanning. It is notable that recent experimental approach led to the discovery of two nAbs against the SARS-CoV-2 RBD by using a ~ 109-diversity phage display library constructed based on the anti-SARS-CoV-1 mAbs (m396, 80 R, CR3022).42 In this study, experimental affinity enhancement based on structural validation at high-resolution increased the binding affinity for the wild-type RBD by more than 20-fold, while computational affinity maturation for the N501Y mutant RBD increased affinity much further. This was all accomplished while also enhancing the antibody’s affinity for the wild-type RBD. Whereas D27LEY exhibits ultrapotent binding affinity for SARS-CoV-2 variants with the N501Y mutation, its binding affinities for other variants lacking this mutation are lower, but still potent. It is likely that saturation mutagenesis at other selected sites could further enhance binding affinity for all variants.
In conclusion, D27LEY appears to be a strong candidate for development into a therapeutic or prophylactic mAb for COVID-19. Our work here also reveals a rational path toward vaccine design aimed at broad and effective protection against SARS-CoV-2 and other related viruses. While all the broadly protective nAbs were discovered from COVID-19 patients or vaccinated individuals, D27LEY was discovered and developed via a computational antibody design. This suggests our approach can also be used as a general method for developing mAbs with ultrapotent binding affinities for a given antigen.
Materials and methods
Sequence design of the binding interface
All computations were performed with Rosetta.29 Four pseudo-complexes between the SARS-CoV-2 RBD and a Fv derived from four human nAbs isolated from SARS-CoV-1 convalescent plasma were constructed by aligning the hACE2‒SARS-CoV-2 RBD structure (PDB entry: 6LZG) with each of four Fab‒SARS-CoV-1 RBD structures (PDB entry: 2DD8, 2GHW, 3BGF, 6NB8). These four complex models were first refined with the Rosetta FastRelax protocol and then subjected to a sequence design protocol in which the CDR residues within 7 Å of the RBD were allowed to change and those within 7–9 Å of the RBD were restricted to repacking in the PackRotamersMover and FastDesign protocols in Rosetta (Supplementary data 1). Water molecules were placed at the Fv-RBD interface in the output design models using ExplicitWaterMover43 so they could be used for counting buried unsatisfied polar atoms (buried_unsat). Finally, 55 of 1000 design models were selected based on multiple filtering criteria, such as buried_unsat, buried solvent accessible surface area, pack score and shape complementarity.
Production and purification of designed antibodies
The DNA fragments encoding CL of the қ-type light chain or CH1-CH2-CH3 of the gamma heavy chain were inserted into the pCEP4 vector (Invitrogen). The resulting vectors were designated pCEP4(k-light) and pCEP4(heavy), respectively. For each design, DNA fragments encoding VL and VH were synthesized (IDT) and cloned into either pCEP4(k-light) or pCEP4(heavy), respectively. Each resulting pair of vectors was introduced into CHO-S cells (Gibco) at a density of 6 × 106 cells/ml using ExpiFectamine (Gibco) to express full-length mAbs. The cells were grown in ExpiCHO expression medium (Gibco) for 12 d. Then, the culture supernatant was collected by centrifugation at 4,000 g for 30 min at 4°C, filtered through a 0.45 µm filter (Millipore), diluted by half with the addition of protein A binding buffer (150 mM NaCl, 20 mM Na2HPO4, pH 7.0), loaded onto an open column containing Protein A resin (Sino Biological), and eluted with Protein A elution buffer (0.1 M glycine, pH 3.0). The eluent was then immediately neutralized with Protein A neutralizing buffer (1 M Tris-HCl, pH 8.5). The antibodies were then further purified using a HiLoad 26/60 Superdex 200 gel-filtration column (Cytiva) equilibrated with buffer A (20 mM Tris-HCl, 150 mM NaCl, pH 7.5).
Biolayer interferometry
BLI experiments were performed using an Octet QKe384 system (ForteBio). Biotinylated or 6x(His)-tagged RBD and its variants (Acrobio Systems) at 5 nM was loaded onto streptavidin or Ni-NTA biosensor tips (ForteBio), respectively, in Kinetics Buffer (ForteBio) for 40 s. For screening, the designed mAbs at 450 nM went through an association step for 120 s and a dissociation step for 120 s before their binding kinetics were analyzed with the BLItz Pro software (ForteBio). For measuring KD, the designed mAbs at four to five different concentrations were subjected to BLI runs with 240-s association steps and 480-s dissociation steps before the binding kinetics were analyzed with the Octet DataAnalysis 10.0 software package (ForteBio).
Production and purification of D27-Fab
DNA fragments encoding VL and CL or VH and CH1 with an 8xHis tag were cloned separately into the pCEP4 vector. CHO-S cells were then co-transformed with the two resulting vectors. The expression and purification procedures we followed were virtually identical to those used for full-length mAbs, except that we used HisPur Ni-NTA resin (Thermo Scientific) instead of Protein A resin.
Production of the wild-type SARS-CoV-2 RBD and truncated hACE2
The SARS-CoV-2 RBD and the N-terminal hACE2 peptidase domain were expressed using the Bac-to-Bac® Baculovirus system (Invitrogen, USA). A DNA fragment encoding the RBD (residues R319-F541) with an N-terminal gp67 signal peptide and a C-terminal HRV3C cleavage site, a 10x(His) tag, and a FLAG tag was cloned into the pFASTbac1 vector (GenScript, USA). The resulting construct was expressed in Spodoptera frugiperda (Sf9) insect cells at 27°C for 3 days. The culture medium was collected, filtered, and loaded onto a HisTrap excel column (Cytiva, USA). The column was then washed with buffer A containing 20 mM imidazole. Bound RBD was eluted with buffer A containing 250 mM imidazole. After cleaving the C-terminal tags with HRV3C protease, the RBD was further purified using a HiLoad® 26/600 Superdex 200 pg column (Cytiva) equilibrated with buffer A containing 1 mM dithiothreitol. The N-terminal hACE2 peptidase domain (residues S19-N615) was cloned, expressed, and purified according to the same protocol used for the SARS-CoV-2 RBD.
Production and purification of RBDs
DNA fragments encoding the RBD of the Alpha, Delta, DeltaPlus, or Epsilon variants were prepared by site-directed mutagenesis and cloned into the pCEP4 vector. The DNA fragment encoding the RBD of PG-GD1 was synthesized (IDT) and cloned into the pCEP4 vector. The protein production and purification procedures were virtually identical to those used for D27-Fab. The RBDs of the Beta and Gamma variants, however, were purchased from Acrobio Systems and the RBD of SARS-CoV-1 was purchased from R&D Systems.
Crystallization, structure determination, and refinement
Purified RBD (9.8 mg/ml) and D27-Fab (12.3 mg/ml) were mixed in a 1:1.1 molar ratio. Crystals were obtained using the hanging-drop vapor diffusion method at 20°C in a solution containing 20% (v/v) PEG 3350 and 200 mM NH4Cl. For cryoprotection, the crystal was briefly immersed in the mother liquor containing an additional 7.5% (v/v) glycerol. X-ray diffraction data were collected on beamline 11 C at the Pohang Accelerator Laboratory and processed with HKL2000.44 Phases were determined by the molecular replacement protocol in PHENIX45 using the structures of a kappa light chain (PDB ID: 4MIK), SARS-CoV-2 RBD (PDB ID: 7JMO), and the computational design model of the heavy chain as search models. Model building and structure refinement were carried out using COOT46 and PHENIX.45
Construction of the CDR3 loop-extended D27-scFv library
The CDR3 loop-extended D27-scFv library was constructed using DNA oligonucleotides and a standard overlapping polymerase chain reaction (PCR) method. To randomize and extend the E101H and Q102H sequence in D27 to 4 residues and F93L to 2 residues, the synthetic oligonucleotides contained NNMNNMNNMNNM or NNMNNM, respectively, where “N” stands for any base and “M” stands for A or C. A DNA oligomer was prepared to link VH and VL of D27 via a (G4S)4 linker. Its sequence was as follows:
5ʹ-GGCGGAGGTGGTTCAGGTGGCGGCGGTTCTGGAGGAGGGGGCAGTGGTGGCGGAGGTTCA-3ʹ
After PCR, 6 µg of the resulting scFv library and 2 µg of linearized pCTCON2 vector (New England Biolabs) containing a TRP selectable marker were introduced into the S. cerevisiae EBY100 strain using an Electroporator 2510 (Eppendorf) according to a published protocol.47
Yeast surface display and cell sorting
After transformation, the yeast cells were cultured in Trp-deficient SD-CAA medium. Then, 3 × 106 cells/ml were resuspended in galactose-replete SGCAA medium and cultured for another 20 h at 20°C to induce the expression of Aga2p-scFv containing a C-terminal Myc tag. To label the Aga2p-scFv fusion protein produced in yeast, the resulting cells (107 cells/100 µL) were washed two times in phosphate-buffered saline (PBS), resuspended with PBSF (PBS with 1% bovine serum albumin (BSA)), and incubated with 0.2 µL of phycoerythrin (PE)- or PE-Cyanine7 (PE-Cy7)-labeled anti-c-Myc antibody (Cell Signaling Technology; Cat # 3739, Cat # 36271) for 1 h at 4°C. For fluorescence labeling of the RBD, 6x(His)-tagged RBD was incubated with fluorescein isothiocyanate (FITC)-labeled anti-6x(His) antibody (Abcam; Cat # ab1206) at a 1:1 molar ratio for 1 h at 4°C. The cells were then incubated with labeled RBD at different concentrations for 1 h at 4°C and washed two times before cell sorting. Two rounds of MACS were performed with FITC-labeled RBD (5 nanomoles for the 1st round and 1 nanomole for the 2nd round) to deplete the clones with weak target-binding affinity. The cells were then collected using anti-FITC microbeads (Miltenyi Biotec) and subjected to three rounds of FACS using a S3e cell sorter (Bio-Rad) in the presence of 100, 10, and 5 nM FITC-labeled RBD. The resulting sorted cells were plated on an SDCAA plate, and individual plasmids were prepared for 9 clones. The scFv sequences were determined by DNA sequencing (Cosmogenetech, Korea).
Focus reduction neutralization test
Virus neutralization was determined using the FRNT assay.48 In triplicate, mAbs were serially diluted in Eagle’s minimum essential medium (EMEM) containing 2% fetal bovine serum. They were then incubated with 8 × 103 plaque forming units (PFU) of SARS-CoV-2 for 1 h at 37°C in a final volume of 50 μL. The mAb-virus mixture was added to 4 × 104 Vero E6 cells on a 96-well plate. After incubation for 8 h at 37°C, the cells were washed with PBS and fixed with 3.7% formaldehyde. The fixed cells were permeabilized with 100% methanol and incubated with a blocking buffer (1% BSA, 0.5% goat serum, 0.1% tween-20 in PBS) for 30 min. The cells were then stained with a mAb against the nucleocapsid of SARS-CoV-2 (40143-R001, Sino Biological) for 1 h and incubated with a goat anti-rabbit IgG (H + L)-HRP (1706515, Bio-Rad) for 1 h. Using TMB Stabilized Substrate (W4121, Promega), viral infection foci were counted with an ImmunoSpot analyzer (2001726, CTL). NC50s were calculated by nonlinear, dose-response regression analysis using GraphPad Prism 8.
Supplementary Material
Acknowledgments
This work was supported by the KAIST Mobile Clinic Module Project (Grant No. MCM-2021-N11210036 to B.-H.O.), the National Research Council of Science and Technology grant (CRC-16-01-KRICT to K.-D.K.) and the National Research Foundation grant (NRF-2019M3E5D6063903 to H.-S.C.) through the Korean government (MSIP). The X-ray data were collected on the Beamline 11C at the Pohang Accelerator Laboratory, Korea. All figures presenting protein structures were generated using PyMOL 2.0.
Funding Statement
This work was supported by the National Research Council of Science and Technology [CRC-16-01-KRICT]; National Research Foundation of Korea [NRF-2019M3E5D6063903];Korea Advanced Institute of Science and Technology [MCM-2021-N11210036].
List of abbreviations:
nAbs Neutralizing antibodies
mAbs Monoclonal antibodies
hACE2human angiotensin-converting enzyme 2
RBD Receptor-binding domain
SARS-CoV-2Severe acute respiratory syndrome coronavirus-2
VOCs Variants of concern
VOIs Variants of interests
VUMs Variants under monitoring
CDRs Complementarity-determining regions
Author contributions
B.-H.O. directed the work. B.-S.J. further conceptualized the research and performed the computational design, structure determination, affinity maturation, and quantification of binding affinity. H.-S.C., J.S.C. and U.K. performed purification of the SARS-CoV-2 RBD for x-ray crystallography. I.H. performed the virus neutralization assay, as directed by K.-D.K. B.C. developed the ApproximateBuriedUnsatPenalty EnergyMethod and J.A.-B. developed NeighborhoodResidueSelector and ResidueSelector that were used in the Rostta runs. B.-H.O. and B.-S.J. wrote the original draft. All authors reviewed and accepted the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frutos R, Serra-Cobo J, Pinault L, Roig ML, Devaux CA.. Emergence of Bat-Related Betacoronaviruses: hazard and Risks. Front Microbiol. 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Machhi J, Herskovitz J, Senan AM, Dutta D, Nath B, Oleynikov MD, Blomberg WR, Meigs DD, Hasan M, Patel M, et al. The Natural History, Pathobiology, and Clinical Manifestations of SARS-CoV-2 Infections. J Neuroimmune Pharm. 2020;15(3):359–86. doi: 10.1007/s11481-020-09944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu N, Zhang DY, Wang WL, Li XW, Yang B, Song JD, Zhao X, Huang B, Shi W, Lu R, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Engl J Med. 2020;382(8):727–33. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin (vol 579, pg 270, 2020). Nature. 2020;588(7836):E6–E. doi: 10.1038/s41586-020-2951-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fauci AS. The story behind COVID-19 vaccines. Science. 2021;372(6538):109. doi: 10.1126/science.abi8397. [DOI] [PubMed] [Google Scholar]
- 7.Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, Hinsley WR, Laydon DJ, Dabrera G, O’Toole Á, et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature. 2021;593(7858):266-+. doi: 10.1038/s41586-021-03470-x. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira I, Kemp SA, Datir R, Saito A, Meng B, Rakshit P, et al. SARS-CoV-2 B.1.617 Mutations L452R and E484Q Are Not Synergistic for Antibody Evasion. J Infect Dis. 2021;224(6):989–94. doi: 10.1093/infdis/jiab368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bates TA, Leier HC, Lyski ZL, McBride SK, Coulter FJ, Weinstein JB, et al. Neutralization of SARS-CoV-2 variants by convalescent and BNT162b2 vaccinated serum. Nat Commun. 2021;12:12. doi: 10.1038/s41467-020-20168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen RTE, Zhang XW, Case JB, Winkler ES, Liu Y, VanBlargan LA, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. 2021;27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuzmina A, Khalaila Y, Voloshin O, Keren-Naus A, Boehm-Cohen L, Raviv Y, Shemer-Avni Y, Rosenberg E, Taube R. SARS-CoV-2 spike variants exhibit differential infectivity and neutralization resistance to convalescent or post-vaccination sera. Cell Host Microbe. 2021;29(4):522-+. doi: 10.1016/j.chom.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan M, Huang D, Lee CD, Wu NC, Jackson AM, Zhu X, Liu H, Peng L, van Gils MJ, Sanders RW, et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science. 2021;373(6556):818–23. doi: 10.1126/science.abh1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez DR, Schafer A, Leist SR, De La Cruz G, West A, Atochina-Vasserman EN, Lindesmith LC, Pardi N, Parks R, Barr M, et al. Chimeric spike mRNA vaccines protect against Sarbecovirus challenge in mice. Science. 2021;373(6558):991–98. doi: 10.1126/science.abi4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saunders KO, Lee E, Parks R, Martinez DR, Li D, Chen H, Edwards RJ, Gobeil S, Barr M, Mansouri K, et al. Neutralizing antibody vaccine for pandemic and pre-emergent coronaviruses. Nature. 2021;594(7864):553–59. doi: 10.1038/s41586-021-03594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan CW, Chia WN, Young BE, Zhu F, Lim BL, Sia WR, et al. Pan-Sarbecovirus Neutralizing Antibodies in BNT162b2-Immunized SARS-CoV-1 Survivors. N Engl J Med. 2021;385(15):1401–06. doi: 10.1056/NEJMoa2108453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corti D, Purcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184(12):3086–108. doi: 10.1016/j.cell.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, Planchais C, Porrot F, Robillard N, Puech J, et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021;596(7871):276-+. doi: 10.1038/s41586-021-03777-9. [DOI] [PubMed] [Google Scholar]
- 18.McCallum M, Bassi J, De Marco A, Chen A, Walls AC, Di Iulio J, Tortorici Ma, Navarro M-J, Silacci-Fregni C, Saliba C, et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science. 2021;373(6555):648-+. doi: 10.1126/science.abi7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Starr TN, Czudnochowski N, Liu ZM, Zatta F, Park YJ, Addetia A, Pinto D, Beltramello M, Hernandez P, Greaney AJ, et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature. 2021;597(7874):97-+. doi: 10.1038/s41586-021-03807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tortorici MA, Czudnochowski N, Starr TN, Marzi R, Walls AC, Zatta F, Bowen JE, Jaconi S, Di Iulio J, Wang Z, et al. Broad sarbecovirus neutralization by a human monoclonal antibody. Nature. 2021;597(7874):103-+. doi: 10.1038/s41586-021-03817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang LS, Zhou TQ, Zhang Y, Yang ES, Schramm CA, Shi W, Pegu A, Oloniniyi OK, Henry AR, Darko S, et al. Ultrapotent antibodies against diverse and highly transmissible SARS-CoV-2 variants. Science. 2021;373(6556):759-+. doi: 10.1126/science.abh1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prabakaran P, Gan JH, Feng Y, Zhu ZY, Choudhry V, Xiao XD, Ji X, Dimitrov DS. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J Biol Chem. 2006;281(23):15829–36. doi: 10.1074/jbc.M600697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pak JE, Sharon C, Satkunarajah M, Auperin TC, Cameron CM, Kelvin DJ, et al. Structural Insights into Immune Recognition of the Severe Acute Respiratory Syndrome Coronavirus S Protein Receptor Binding Domain. J Mol Biol. 2009;388(4):815–23. doi: 10.1016/j.jmb.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang WC, Lin YQ, Santelli E, Sui JH, Jaroszewski L, Stec B, et al. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J Biol Chem. 2006;281(45):34610–16. doi: 10.1074/jbc.M603275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walls AC, Xiong XL, Park YJ, Tortorici MA, Snijder J, Quispe J, et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion (vol 176, 1026. 1, 2019). Cell. 2020;(183):1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang QH, Zhang YF, Wu LL, Niu S, Song CL, Zhang ZY, Lu G, Qiao C, Hu Y, Yuen K-Y, et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 2020;181(4):894-+. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian XL, Li C, Huang AL, Xia S, Lu SC, Shi ZL, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infec. 2020;9:382–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi CY, Sun XY, Ye J, Ding LF, Liu MQ, Yang Z, Lu X, Zhang Y, Ma L, Gu W, et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol. 2020;17(6):621–30. doi: 10.1038/s41423-020-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, Barth P, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods. 2020;17(7):665–80. doi: 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loffler P, Schmitz S, Hupfeld E, Sterner R, Merkl RR. MSF: a modular framework for multi-state computational protein design. Plos Comput Biol. 2017;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler DS. Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein (vol 180, 281.e1, 2020). Cell. 2020;183(183):1735. doi: 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrapp D, Wang NS, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260-+. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zahradnik J, Marciano S, Shemesh M, Zoler E, Harari D, Chiaravalli J, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol. 2021;6(9):1188-+. doi: 10.1038/s41564-021-00954-4. [DOI] [PubMed] [Google Scholar]
- 34.Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe. 2021;29:44-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura I, Kosugi Y, Wu J, Yamasoba D, Butlertanaka EP, Tanaka YL, et al. SARS-CoV-2 Lambda variant exhibits higher infectivity and immune resistance. bioRxiv. 2021;07.28.454085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carreño JM, Alshammary H, Singh G, Raskin A, Amanat F, Amoako A, et al. Reduced neutralizing activity of post-SARS-CoV-2 vaccination serum against variants B.1.617.2, B.1.351, B.1.1.7+E484K and a sub-variant of C.37. medRxiv. 2021;07.21.21260961. [Google Scholar]
- 37.Collier DA, De Marco A, Ferreira IATM, Meng B, Datir RP, Walls AC, Kemp SA, Bassi J, Pinto D, Silacci-Fregni C, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature. 2021;593(7857):136-+. doi: 10.1038/s41586-021-03412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mlcochova P, Kemp S, Dhar MS, Papa G, Meng B, Ferreira I, Datir R, Collier DA, Albecka A, Singh S, et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021;599(7883):114–19. doi: 10.1038/s41586-021-03944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piccoli L, Park YJ, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, Silacci-Fregni C, Pinto D, Rosen LE, Bowen JE, et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell. 2020;183(4):1024–42 e21. doi: 10.1016/j.cell.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norman RA, Ambrosetti F, Bonvin AMJJ, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2020;21(5):1549–67. doi: 10.1093/bib/bbz095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finn JA, Dong JH, Sevy AM, Parrish E, Gilchuk I, Nargi R, Scarlett-Jones M, Reichard W, Bombardi R, Voss TG, et al. Identification of Structurally Related Antibodies in Antibody Sequence Databases Using Rosetta-Derived Position-Specific Scoring. Structure. 2020;28(10):1124-+. doi: 10.1016/j.str.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rouet R, Mazigi O, Walker GJ, Langley DB, Sobti M, Schofield P, et al. Potent SARS-CoV-2 binding and neutralization through maturation of iconic SARS-CoV-1 antibodies. Mabs-Austin. 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavlovicz RE, Park H, DiMaio F, Wallner B. Efficient consideration of coordinated water molecules improves computational protein-protein and protein-ligand docking discrimination. Plos Comput Biol. 2020;16(9):e1008103. doi: 10.1371/journal.pcbi.1008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–26. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, Mustyakimov M, Afonine PV, Langan P. Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr D. 2009;65(6):567–73. doi: 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D-Biological Crystallography. 2010;66(4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23(4):155–59. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 48.Vanderheiden A, Floyd EVV, Kauffman K, Mantus RC, Anderson E G, Anderson E, Rouphael N, Edupuganti S, Shi P-Y, Menachery VD, et al. Development of a Rapid Focus Reduction Neutralization Test Assay for Measuring SARS-CoV-2 Neutralizing Antibodies. Curr Protoc Immunol. 2020;131(1):e116. doi: 10.1002/cpim.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.