

Abstract
The hypocretins (Hcrts, also known as orexins) are two peptides, both synthesized by a small group of neurons, most of which are in the lateral hypothalamic and perifornical regions of the hypothalamus. The hypothalamic Hcrt system directly and strongly innervates and potently excites noradrenergic, dopaminergic, serotonergic, histaminergic, and cholinergic neurons. Hcrt also has a major role in modulating the release of glutamate and other amino acid transmitters. Behavioral investigations have revealed that Hcrt is released at high levels in active waking and rapid eye movement (REM) sleep and at minimal levels in non-REM sleep. Hcrt release in waking is increased markedly during periods of increased motor activity relative to levels in quiet, alert waking. Evidence for a role for Hcrt in food intake regulation is inconsistent. I hypothesize that Hcrt’s major role is to facilitate motor activity tonically and phasically in association with motivated behaviors and to coordinate this facilitation with the activation of attentional and sensory systems. Degeneration of Hcrt neurons or genetic mutations that prevent the normal synthesis of Hcrt or of its receptors causes human and animal narcolepsy. Narcolepsy is characterized by an impaired ability to maintain alertness for long periods and by sudden losses of muscle tone (cataplexy). Administration of Hcrt can reverse symptoms of narcolepsy in animals, may be effective in treating human narcolepsy, and may affect a broad range of motivated behaviors.
Keywords: REM sleep, hypothalamus, narcolepsy, cataplexy, feeding, motor, alertness
THE DISCOVERY OF HYPOCRETIN (OREXIN)
The discovery of hypocretin (Hcrt) and of its clinical and biological significance is a landmark in recent neuroscience. In fewer than three years, the discovery led to the unraveling of the cause of narcolepsy, which had been a mystery since the identification of the disorder more than 120 years ago. We now know that most explanations prior to 2000, including the ideas that narcolepsy was a psychosomatic illness, was caused by excessive masturbation, was a form of schizophrenia (Douglass et al. 1993, Passouant 1976), or was a symptom of a subtle neurochemical imbalance were far off the mark. Now, five years after the identification of Hcrt, we know that the peptide has a major role in behavioral control through its regulation of aminergic, cholinergic, and amino acid transmitter systems in relation to sleep and waking behaviors. The Hcrt story is notable as an example of how basic research can lead to completely unanticipated insights into human disease.
In the late 1990s, Gregor Sutcliffe’s group in San Diego was interested in identifying peptides that were linked to the control of food intake and the regulation of weight (De Lecea et al. 1998). With the epidemic of obesity in the United States and to a lesser extent in other wealthy nations, a better understanding of the weight regulatory system would be of enormous public health importance. It has long been known that the hypothalamus is the brain region most critical for appetite control. Lesions of the medial hypothalamus in rats and other animals lead to remarkable obesity, and lesions of the lateral hypothalamus produce profound anorexia (Levitt & Teitelbaum 1975). If peptides that are synthesized only in the hypothalamus could be found, they might be useful in appetite control. It would furthermore be reasonable to hope that receptors for peptides produced in this region would be ideal targets for pharmacological manipulation.
Sutcliffe’s group used the technique of subtractive RNA hybridization to detect mRNAs synthesized in the hypothalamus, but not in the cerebellum. They reasoned that the cerebellum is not known to be strongly involved in food intake regulation. Therefore, a comparison of hypothalamic and cerebellar mRNAs might reveal peptides having functions related to appetite control.
They discovered a hypothalamus-specific mRNA that encodes a substance they named preprohypocretin, the precursor of two peptides that they named hypocretin-1 and hypocretin-2, from their hypothalamic location and their structural similarities to the gut hormone secretin. They mapped the locations of the peptides and concluded that they were restricted to neuronal cell bodies in the hypothalamus and their projections. Both peptides are found in the same neurons. The work was led by Luis De Lecea and Thomas Kilduff (De Lecea et al. 1998).
At about the same time that Sutcliffe’s group was looking for hypothalamic peptides, Yanagisawa’s group in Texas was looking for ligands for a number of orphan receptors. “Orphan receptors” are encoded by cDNA sequences that resemble those of known receptors, in this case G protein–coupled receptors; the first step to discovering the function of such receptors is to identify their ligands. They screened ligands for a receptor that they eventually named orexin receptor-1. They found two peptides in hypothalamic extracts, orexin-A and orexin-B, that strongly activated cell lines carrying this orphan receptor. Noting that orexin-B had a much weaker effect than orexin-A, they hypothesized that a second receptor existed for these peptides. A search of the genomic database led to the identification of a second receptor (orexin receptor-2) encoded by a gene with substantial sequence similarity to that of the orexin receptor-1 gene. It was found that orexin receptor-1 responded relatively selectively to orexin-A, whereas orexin receptor-2 responded to both orexin-A and orexin-B. No additional receptors for orexin have so far been identified.
Aware of the hypothalamic role in food intake regulation, they administered the peptides to rats by injection into the lateral ventricle, so that the peptides would circulate throughout the ventricular system within the brain. They found that eating was increased and because of this finding named the peptides “orexins,” from the Greek word for appetite. The paper, whose lead author was Takeshi Sakurai, appeared less than a month after the paper from de Lecea et al. (Sakurai et al. 1998). Soon after the papers appeared, it became apparent that Hcrt-1 and -2 were identical to orexin-A and -B. In the current review we will use the term hypocretin (Hcrt) for the peptides (Hcrt-1 = orexin-A, Hcrt-2 = orexin-B).
As we will see, it is doubtful that the discovered peptides are critically important in appetite regulation. However, there is little doubt that the Hcrt peptides are vitally important in behavioral regulation, and that the integrity of the Hcrt system has important implications for proper neurological functioning.
STRUCTURE OF THE PEPTIDES
Hcrt-1 is a peptide that consists of 33 amino acids. The peptide bends back upon itself and is held in this position by disulfide bonds. Hcrt-2 is a 28 amino acid peptide that exists as a linear chain without any disulfide bonds (Sakurai et al. 1998). Both peptides are generated from preprohypocretin.
ANATOMY OF THE HCRT SYSTEM
Hcrt cell somas in the brain are concentrated in the hypothalamus. Although many papers have described them as being restricted to the “lateral hypothalamus,” this description is not entirely accurate. The fornix is usually considered the dividing line between lateral and medial hypothalamus. Many Hcrt cells are located medial to the fornix. This is true in rats and is also apparent in humans, where a dense cluster of Hcrt cells is located in the dorsomedial hypothalamic nucleus, medial to the fornix (Figure 1) (Thannickal et al. 2000). In addition to the hypothalamic grouping of cells staining for both Hcrt-1 and -2, it has been reported that small groups of neurons containing Hcrt-2, but not Hcrt-1, exist in the lateral division of the central nucleus of the amygdala, in the anterior lateral subnucleus of the bed nucleus of the stria terminalis immediately adjacent to the internal capsule, and in an area just ventral to the lateral ventricle (Ciriello et al. 2003), as well as in olfactory neurons (Caillol et al. 2003). Prior anatomical studies of the Hcrt system had not observed Hcrt cells in these areas (Nambu et al. 1999, Peyron et al. 1998).
Figure 1.
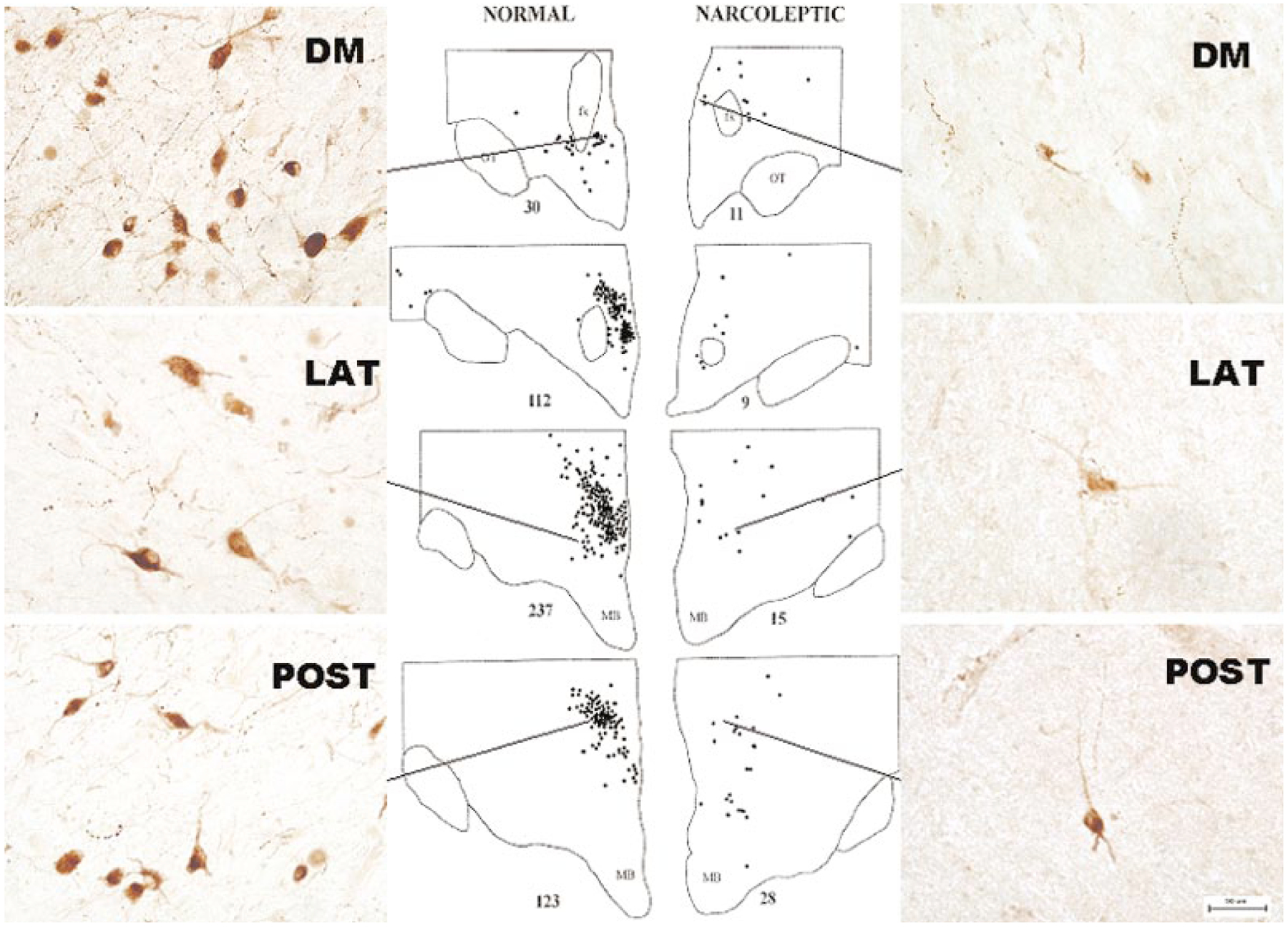
Distribution of Hcrt-labeled cells in normal and narcoleptic humans. (Left) Normal human brain has approximately 70,000 Hcrt cells located in the hypothalamus, both medial and lateral to the fornix. (Right) Narcoleptic human brain has an 85%–95% loss of Hcrt cells. Normal Hcrt cell morphology is visible in surviving Hcrt cells in narcoleptics. DM, dorsomedial; LAT, lateral; POST, posterior hypothalamic nuclei. Calibration 50 μm. (Redrawn from Thannickal et al. 2000)
Although most Hcrt somas are in the hypothalamus, these cells project axons widely throughout the brain. Heavily innervated regions include the locus coeruleus and raphe nuclei of the brainstem, the cholinergic neurons of the brainstem and forebrain, the histaminergic cells of the posterior hypothalamus and other hypothalamic nuclei (Figure 2). Less densely innervated regions include the dorsal and ventral horns of the spinal cord, brainstem motor nuclei, limbic regions of the brainstem, and the neocortex (Fung et al. 2001, Nambu et al. 1999, Peyron et al. 1998, van den Pol 1999).
Figure 2.
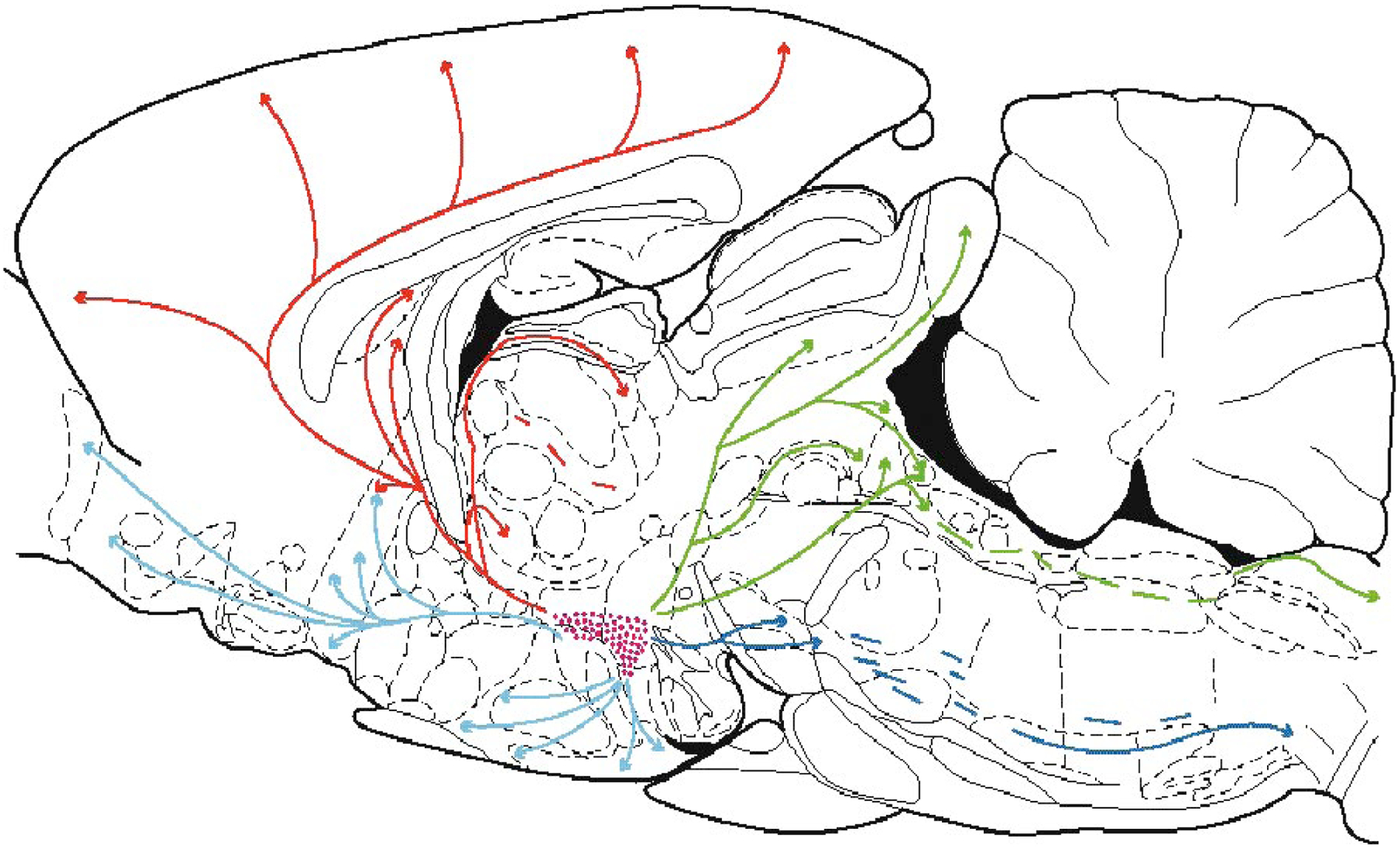
Locations of Hcrt cells in the hypothalamus in the rat and their ascending and descending projections. Some descending Hcrt axons terminate in the dorsal and ventral horns of the spinal cord. (From Peyron et al. 1998)
Brain Hcrt receptors are distributed throughout the innervated regions, but the distributions of mRNAs for Hcrt receptor-1 and -2 are strikingly different. Receptor-1 is the predominant receptor type in cingulate cortex, anterior olfactory nucleus, prefrontal and infralimbic cortex, bed nucleus of stria terminalis, anterior hypothalamus, and locus coeruleus. Receptor-2 is the predominant type in the medial septal nucleus, nucleus of the diagonal band, hippocampal CA3 field, arcuate nucleus of the hypothalamus, lateral hypothalamus, and tuberomammillary nucleus. Both receptors are present in substantial numbers in other regions (Marcus et al. 2001, Trivedi et al. 1998).
Most Hcrt cells in the hypothalamus also contain the peptide dynorphin (Chou et al. 2001). However, the vast majority of dynorphin containing neurons is located outside the hypothalamus. Neuronal activity-regulated pentraxin (Narp), a secreted neuronal protein implicated in regulating synapse formation and produced by an “immediate early gene” (one of a group of genes that is rapidly expressed in neurons when their metabolic activity is increased), is found in virtually all Hcrt cells. Narp is not present in non-Hcrt hypothalamic cells, although it is present in some non-Hcrt cells of the hippocampal mossy fiber pathway, projections of the habenula to the interpeduncular nucleus, and neurons of the vestibular system (Reti et al. 2002). It is likely that Narp is involved in the signaling functions of Hcrt neurons. Some evidence suggests that Hcrt cells may also contain glutamate as a cotransmitter (Abrahamson & Moore 2001); however, this work needs to be confirmed with studies of the distribution of a specific glutamate reuptake transporter, since glutamate is used in cellular metabolism and its presence alone does not prove that it is being used as a transmitter. Hcrt also has an important role in regulating amino acid release from non-Hcrt-containing cells, as will be discussed below.
Hcrt neurons exist outside the brain. Some neurons in the intestines and in the pancreas display Hcrt- and Hcrt receptor-like immunoreactivity, as do vagal and spinal primary afferent neurons (Kirchgessner 2002). Hcrt mRNA has been reported in the testis (Johren et al. 2001). Hcrt receptors have been reported in the adrenal glands, particularly in male rats (Johren et al. 2001). Receptors have also been identified in the pituitary and median eminence (Date et al. 2000).
SYNAPTIC MECHANISMS UNDERLYING HCRT ACTION
Hypocretin’s regulation of the release of other neurotransmitters appears to underlie many of its effects. These effects can be most readily investigated in brain slices maintained in a bath into which transmitter agonists and antagonists and chemicals that block action potentials can be applied. The disadvantage of this in vitro approach is that levels of transmitter agonists and other conditions in the living animal will undoubtedly differ from those created in vitro. Thus, a combination of in vitro studies and investigations of intact animals is essential for gaining a full understanding of how the peptides act.
Hcrt neurons have widely projecting axons innervating all the monoaminergic brain systems. Serotonergic and noradrenergic systems have descending connections strongly linked to movement and muscle tone control (Jacobs 1991, Kiyashchenko et al. 2001, Lai et al. 2001, Wu et al. 1999), as well as ascending projections to forebrain regions involved in sensory integration (Aston-Jones et al. 1994, Jacobs 1987). Histamine neurons are strongly involved in forebrain alerting (Lin et al. 1989). Dopamine neurons are involved in both alerting and reward (Dehaene & Changeux 2000, Rye & Jankovic 2002).
Hcrt cells also strongly innervate the major cholinergic cells in the brainstem (Burlet et al. 2002) and basal forebrain (Eggermann et al. 2003). These cells are known to play a central role in the cortical EEG activation that characterizes waking (Jones 2000). Therefore, the Hcrt system can serve to coordinate the activity of these arousal systems with motor activity.
Hcrt is excitatory at many of its projection sites. This is true for neurons of the noradrenergic locus coeruleus (Bourgin et al. 2000, Hagan et al. 1999, Horvath et al. 1999, Ivanov & Aston-Jones 2000, van den Pol et al. 2002), serotonergic dorsal raphe (Liu et al. 2002), histaminergic tuberomammillary (Bayer et al. 2001), cholinergic brainstem (Burlet et al. 2002), and cholinergic basal forebrain nuclei (Eggermann et al. 2003). Hcrt directly excites many dopaminergic neurons in the midbrain ventral tegmental region (Korotkova et al. 2003). Hcrt excites GABAergic neurons in the pars reticulata of the substantia nigra (Korotkova et al. 2003), neurons adjacent to the dopaminergic neurons supplying forebrain and brainstem regions. GABAergic neurons projecting from the septal nucleus to the hippocampus are also excited by Hcrt (Wu et al. 2002a). An important clue to its arousing actions is the report that Hcrt excites neurons of the nonspecific centromedian thalamic nucleus, which is known to be involved in generalized cortical arousal, but does not excite neurons of the specific thalamic sensory relays, which relay sensory information to the cortex. This centromedian excitation is mediated by type 2 Hcrt receptors (Bayer et al. 2002). Hcrt also increases sympathetic outflow from the medulla (Dun et al. 2000).
Cells in lamina 1 and 2 of the dorsal horn of the spinal cord, involved in the processing of somatosensory information, are excited by Hcrt, but most are also indirectly inhibited by Hcrt via glycine released by adjacent Hcrt responsive cells (Grudt et al. 2002). Similarly, other cells are affected both directly and indirectly by Hcrt. For example, serotonergic cells simultaneously receive direct Hcrt excitation and indirect GABAergic inhibition (Liu et al. 2002), and a similar pattern may occur with locus coeruleus cells (Kiyashchenko et al. 2002). Hcrt application onto vagal motoneurons has been reported both to depolarize (excite) these cells and simultaneously to inhibit them by increasing the number of inhibitory postsynaptic potentials.
This pattern of direct excitation accompanied by indirect inhibition or excitation suggests that the net effect of Hcrt in this situation is largely dependent on the inputs into the cells mediating the indirect effects. For example if Hcrt is directly exciting a postsynaptic cell and simultaneously exciting a GABAergic cell that inhibits the same postsynaptic cell, the inhibitory effect of Hcrt will be greatly enhanced if other excitatory inputs into the GABAergic cell are active. Conversely, the inhibitory effects will be blocked if the GABAergic cell is itself inhibited. In this manner, the indirect pathways utilized by Hcrt provide great flexibility for regulation of the effects of Hcrt. Like an electronic amplifier, this kind of circuit can amplify relatively weak inputs to produce a larger effect in postsynaptic cells.
Some critical sleep-related cell groups are not activated either directly or indirectly by Hcrt. For example, GABAergic sleep active neurons do not respond to Hcrt in the slice preparation (Eggermann et al. 2003). These neurons are thought to have a role in triggering sleep. Hcrt administration to the nucleus accumbens enhances inhibitory GABAergic currents but does not affect glycine action and it decreases NMDA glutamate receptor-induced currents (Martin et al. 2002). The nucleus accumbens is heavily innervated by dopaminergic cells and has a high density of Hcrt receptor-2.
Surprisingly, Hcrt neurons do not respond directly to Hcrt in vitro. Typically, neurons are inhibited when they are exposed to the transmitter they release, presumably as a means of feedback control. Li et al. (2002) showed that application of Hcrt to hypothalamic slices produced excitation of Hcrt cells. However, this effect was an indirect one. Blockade of action potentials within the slice with tetrodotoxin prevented the response, as did blockade of glutamatergic transmission. It was concluded that Hcrt excites the population of Hcrt neurons indirectly by increasing the activity of glutamatergic neurons adjacent to Hcrt neurons. This study also showed that norepinephrine and serotonin potently inhibit Hcrt neurons. Thus, one can identify an inhibitory feedback circuit onto Hcrt neurons mediated by serotonin and norepinephrine: Hcrt neurons directly excite norepinephrine and serotonin neurons and these neurons in turn project back onto Hcrt neurons, producing direct inhibition that counters the self excitation mediated by local glutamatergic neurons. Acetylcholine and histamine have little effect on Hcrt neurons; however, according to the study by Li et al. Hcrt has potent excitatory effects on histamine and acetylcholine-containing cells. Figure 3 summarizes some of the main synaptic connections described or hypothesized for Hcrt cells (Kiyashchenko et al. 2002; Li et al. 2002; Nitz & Siegel 1997a,b).
Figure 3.
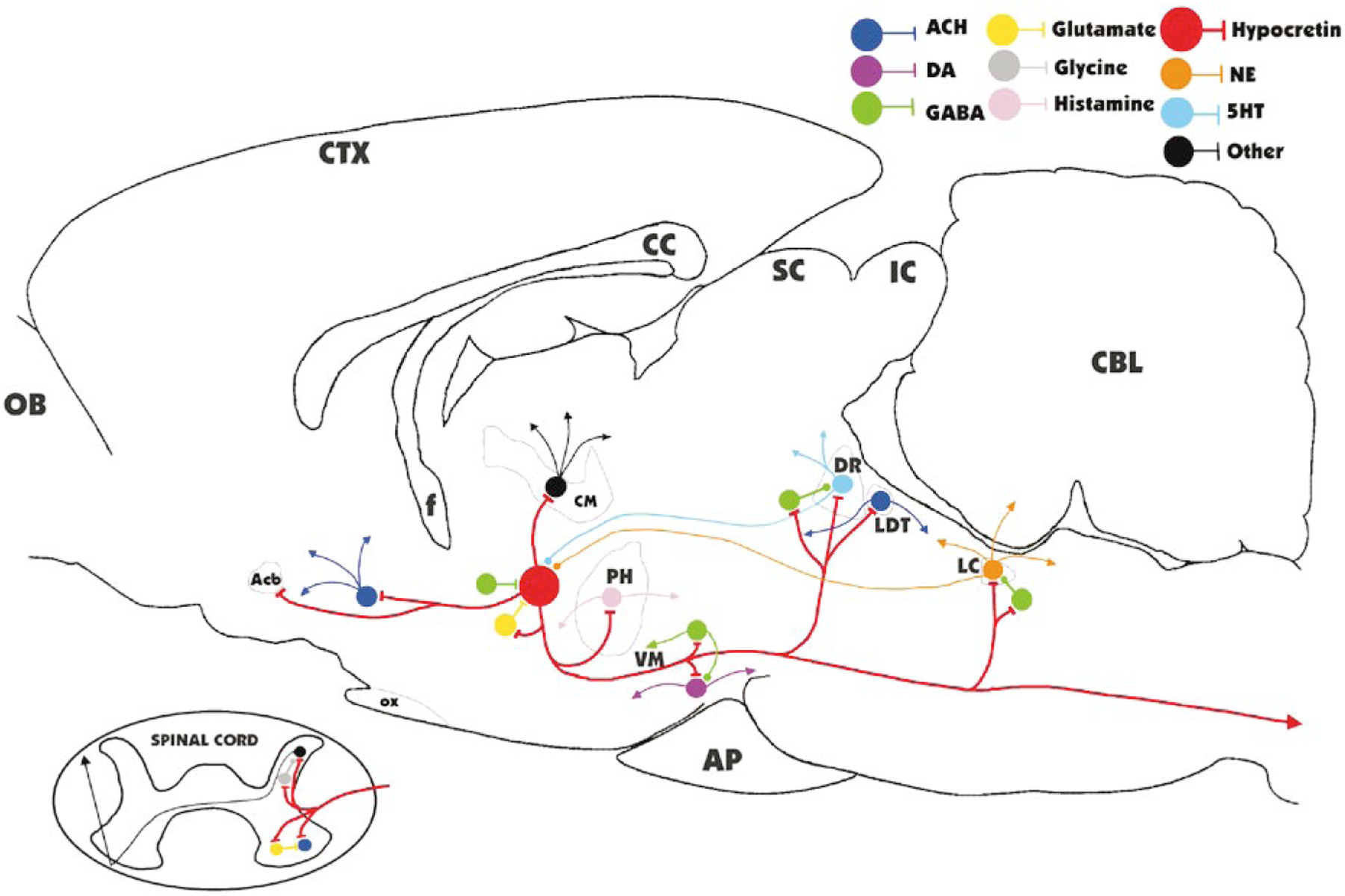
Major identified synaptic interactions of Hcrt neurons. Lines terminated by perpendicular lines denote excitation; circular terminations indicate inhibition. ACH, acetylcholine; DA, dopamine; NE, norepinephrine; 5HT, serotonin; OB, olfactory bulb; Acb, nucleus accumbens; f, fornix; OX, optic chiasm; CM, centromedian nucleus of the thalamus; PH, posterior hypothalamus; VM, ventral midbrain; AP, anterior pituitary; SC, superior colliculus; IC, inferior colliculus; DR, dorsal raphe; LDT, laterodorsal tegmental and pedunculopontine; LC, locus coeruleus; CBL, cerebellum.
The synaptic effects documented in in vitro work can explain aspects of the pathology associated with Hcrt cell or receptor loss, as occurs in narcolepsy. The potent excitation of histamine cells by Hcrt (Eriksson et al. 2001, Yamanaka et al. 2002), largely mediated by Hcrt receptor-2, may be one element in the arousing effect of Hcrt administration (Hagan et al. 1999, Ivanov & Aston-Jones 2000). The sleepiness seen in Hcrt receptor-2 mutant narcoleptic dogs (Siegel 1999), the reduced levels of histamine in these canine and in human narcoleptics (Nishino et al. 2001, 2002), and the soporific effects of antihistamines (Welch et al. 2003) support this conclusion, as does the major role of histamine in mediating the arousing effects of Hcrt infusion into the lateral ventricles (Huang et al. 2001). Histamine blockers attenuate the Hcrt-induced increase in waking, but even high doses do not completely block the arousing response (Yamanaka et al. 2002), however, which suggests that other systems might also be involved. Indeed, the noradrenergic, cholinergic, and dopaminergic systems are also strongly excited by Hcrt, and it is likely that it is this coordinated activation, rather than the release of histamine alone, that is mediating Hcrt’s arousing effects (Figure 3). Locomotion induced by injection of Hcrt into the lateral ventricle was blocked by dopamine D2 receptor antagonists (Nakamura et al. 2000, Sakurai et al. 1998), demonstrating that dopaminergic pathways play an important role in behavioral arousal due to Hcrt administration.
HCRT AND AMINO ACIDS
Van den Pol et al. (1998) and Follwell & Ferguson (2002) presented evidence from work in hypothalamic brain slices that Hcrt administration causes glutamate release, and under some conditions GABA release as well. John et al. (2001, 2003) measured changes in central glutamate release after intravenous administration of Hcrt in anesthetized rats. Intravenous administration caused a sustained (>50 min) and marked (>60%) increase in glutamate release in the amygdala, a region with a moderate concentration of Hcrt receptors, but no such change in the cerebellar cortex, a region without substantial numbers of Hcrt receptors (Figure 4) (John et al. 2001, 2003). This release is calcium dependent, which indicates that the glutamate is released from synaptic vesicles (John et al. 2003). Similar increases in glutamate release have been seen in unanesthetized animals following systemic Hcrt administration (Kodama & Kimura 2002). Intravenous infusion of Hcrt increases behavioral activity and decreases REM sleep (John et al. 2000), effects also seen with central administration. Therefore, the intravenous effects are likely to be mediated, in part, by Hcrt-induced glutamate release.
Figure 4.
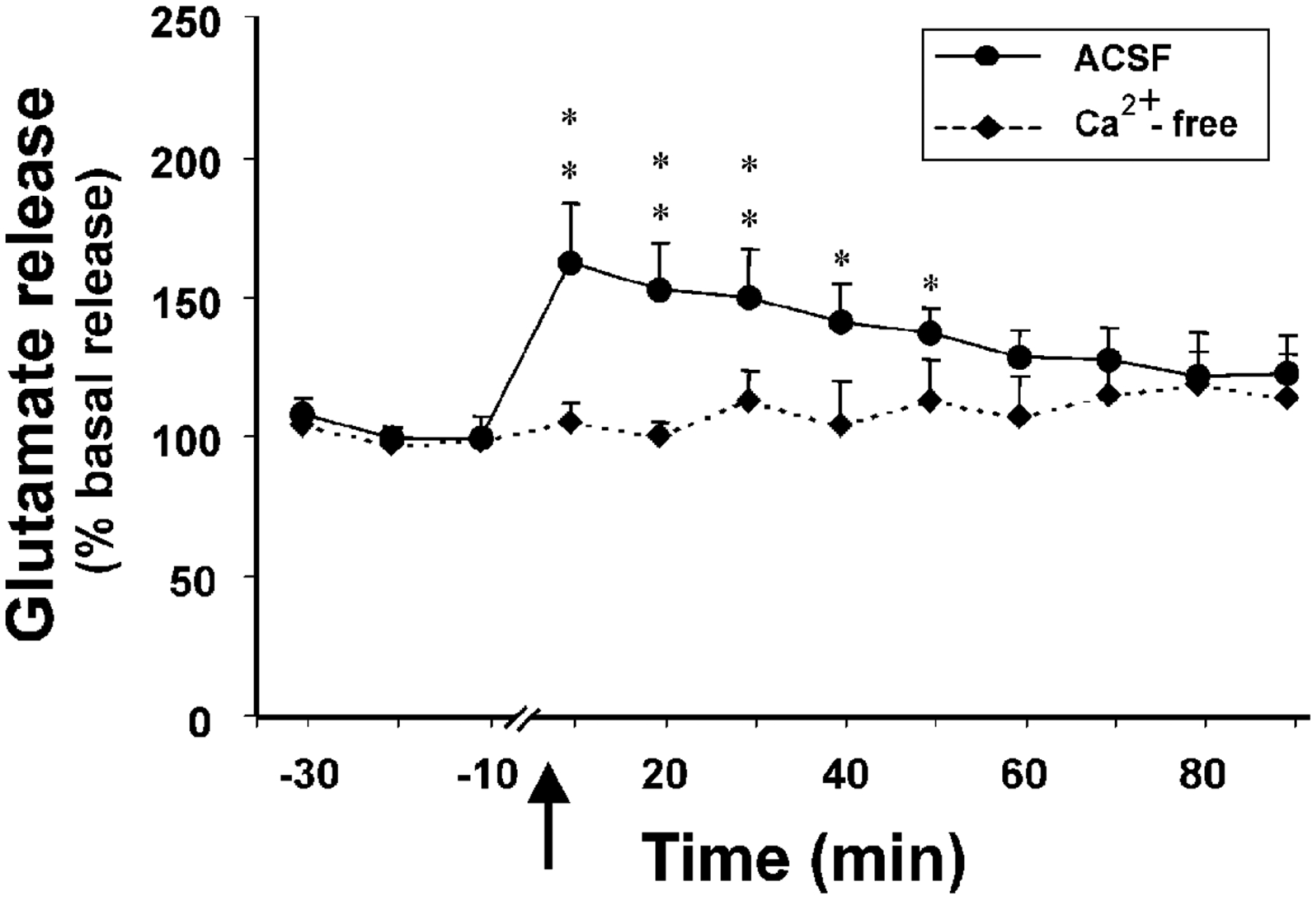
Effect of intravenous injection of Hcrt on central glutamate release in the amygdala, a Hcrt-innervated region. Intravenous Hcrt injection produces a marked, calcium-dependent increase in glutamate release. No such increase occurs in the cerebellar cortex, a region without substantial Hcrt innervation. This demonstrates that Hcrt effectively crosses the blood-brain barrier and produces a marked release of glutamate in Hcrt-innervated regions. (From John et al. 2003)
Hcrt neurons innervate motoneuron pools (Fung et al. 2001). When Hcrt was microinjected into the trigeminal motor nucleus, ipsilateral masseter muscle tone increased in a dose-dependent manner. Hcrt application into the hypoglossal motor nucleus also increased genioglossus muscle activity. However, pretreatment with D-AP5, an N-methyl-D-aspartic acid (NMDA) glutamate receptor antagonist, abolished the excitatory response of Hcrt application onto both groups of motoneurons (Figure 5) (Peever et al. 2003). These studies demonstrate that Hcrt’s effects on these motoneurons are mediated by the release of glutamate, the most ubiquitous excitatory brain neurotransmitter.
Figure 5.
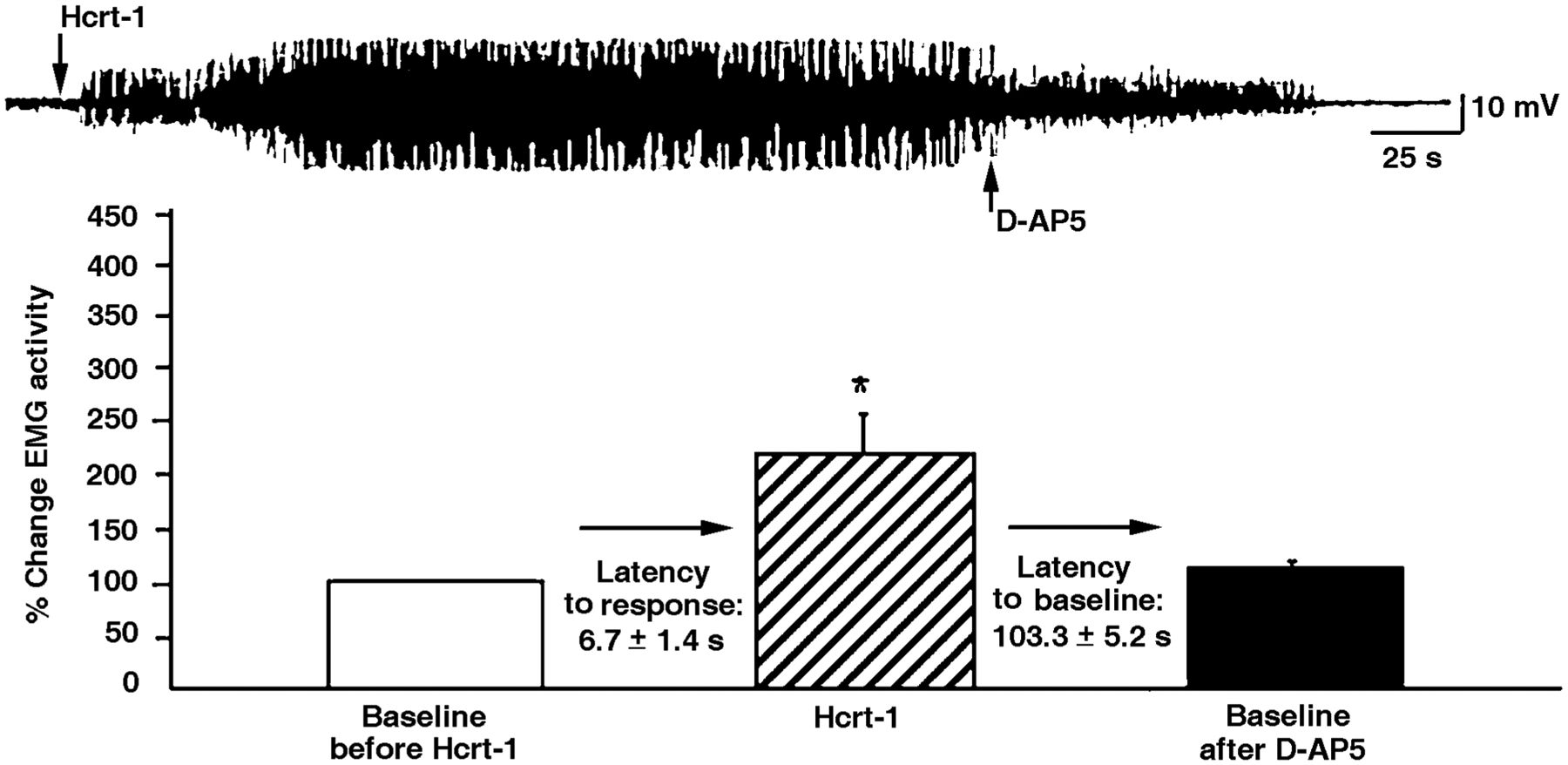
Interactions of Hcrt with glutamate after microinjection of Hcrt into motor nuclei. Hcrt microinjections potently excited trigeminal motoneurons, but this effect was completely blocked by the glutamate NMDA receptor antagonist AP-5. Glutamate is a major mediator of Hcrt effects in motor nuclei and in other regions of the brain. (From Peever et al. 2003)
HCRT’S ROLE IN NARCOLEPSY AND OTHER DISORDERS
After identifying the Hcrt peptides in rats, Yanagisawa’s group created preprohypocretin knockout mice in which the peptides were not generated. Careful observation of the behavior of these animals revealed unusual, abrupt onset periods of inactivity. After ruling out various causes, they concluded that they had created narcoleptic mice that were experiencing either attacks of sleep or cataplexy (Chemelli et al. 1999). At the same time, Lin et al. (1999) were investigating the genetics of canine narcolepsy. A genetic form of canine narcolepsy had been described in Doberman pinschers and Labrador retrievers (Baker & Dement 1985), and these dogs were bred and studied (Mitler et al. 1976; Siegel et al. 1991, 1999). Lin et al. found that this form of canine narcolepsy was caused by a mutation in the gene for Hcrt receptor-2.
Narcolepsy is a chronic disorder characterized by two types of symptoms (Siegel 1999, 2000a). The most debilitating symptom is usually chronic, overwhelming sleepiness. A narcoleptic person goes through the day as sleepy as a nonnarcoleptic person who has not slept for two or more days. The result is sleep in inappropriate and embarrassing situations and an inability to perform up to the individual’s intellectual capacity due to the difficulty of sustaining alertness. Excessive daytime sleepiness also occurs in sleep apnea and other disorders and therefore is not by itself diagnostic of narcolepsy. A second symptom experienced by most narcoleptics is loss of muscle tone during waking, as is seen in cataplexy or sleep paralysis. Cataplexy is most commonly triggered by the patient’s laughter, but can also be triggered by other sudden onset emotions such as anger, embarrassment, or by athletic exertion, or it may have no identified cause. During cataplexy, although unable to move to a greater or lesser extent, the patient is alert, aware of the environment, and understands and remembers what is going on. A similar loss of muscle tone called sleep paralysis frequently occurs at sleep onset or offset in narcoleptics, with awareness of the environment maintained during periods of motor paralysis lasting from seconds to minutes. Many narcoleptics also have hypnagogic hallucinations, often frightening hallucinations that incorporate elements of the environment. Narcoleptics have a very short sleep latency and tend to transition quickly from waking into REM sleep. Normal individuals who have been sleep deprived or nonnarcoleptics with other forms of sleep pathology can have equally short sleep latencies, but unlike narcoleptics they generally pass through an initial non-REM sleep period that lasts at least 5 minutes, at sleep onset. The hypnagogic hallucinations, cataplexy, sleep paralysis, and short REM sleep latency of narcoleptics have been explained as an “escape” of REM sleep components into the waking state, since in REM sleep, the skeletal motoneurons are inhibited and hallucinatory mentation is the norm. Narcolepsy is not uncommon, affecting more than 1 in 2000 individuals, an incidence approximately one half to one third that of multiple sclerosis and about one fourth that of Parkinson’s disease.
The discoveries that mutations in the preprohypocretin or Hcrt receptor genes can cause narcolepsy in animals suggested that human narcolepsy might be caused by a similar pathology of the Hcrt system. However, human narcolepsy is clearly not genetically determined in the way that canine genetic narcolepsy is. Most human narcoleptics do not have parents or siblings with diagnosed narcolepsy, and most pairs of identical twins in which narcolepsy has been identified in at least one twin are discordant for the disease (Aldrich 1998, Guilleminault & Anognos 2000, Hublin et al. 1994). However, Honda and colleagues (Honda et al. 1984) showed that 85% to 95% of human narcoleptics share a particular human leucocytic antigen (HLA) genotype that is found in about 25% of the overall population. Subsequent work determined that this HLA type is DQB1*0602 (Olerup et al. 1990). The proteins encoded by genes in the HLA region are involved in presenting antigens to the immune system, and many autoimmune disorders are linked to one or more HLA haplotypes. Because of the strong HLA linkage present in narcolepsy, it was suspected that this disorder might also be an autoimmune disease. In narcoleptic dogs, we have seen degenerative changes in forebrain limbic regions, including many of the major projection regions of Hcrt neurons, which suggests that a degenerative process contributes to canine genetic narcolepsy (Siegel et al. 1999). However, no evidence of abnormal immune function had been detected in human narcoleptics and no target for immunological attack had been identified at the time we began our studies. Could the Hcrt neurons be targeted in narcoleptic humans?
Because narcolepsy is never a direct cause of death, brain tissue from human narcoleptics is rarely saved for investigation. Thannickal et al. (2000) had acquired several brains of human narcoleptics from Dr. Michael Aldrich and other sources, but they had been in fixative for 4 to 12 years. Thannickal et al. (2000) first tested immunohistochemical staining techniques on control tissue that had been in fix for equally long periods, and discovered that Hcrt neurons could not be stained in this “overfixed” tissue by conventional means, although they could easily be stained in freshly fixed human brains. Using antigen retrieval techniques, the overfixed tissue was stained, and it was discovered that narcoleptics had lost 85% to 95% of their Hcrt neurons (Figure 1). Surviving Hcrt cells were found in all narcoleptics, which suggests that residual Hcrt function may modulate symptoms and that strategies to maximize the impact of surviving Hcrt cells could be useful in therapies for narcolepsy. A further finding was glial proliferation (gliosis) in brain regions that normally contain Hcrt cells and in regions to which Hcrt neurons projected. Glial cells proliferate during inflammatory reactions, which suggests that such a reaction may have accompanied cell loss. A paper by Peyron et al. published at the same time also reported Hcrt depletion in narcolepsy (Peyron et al. 2000). Significant differences between the two papers were Peyron et al.’s report of no gliosis, no surviving Hcrt cells in narcoleptics, and much lower numbers of Hcrt cells in normal humans (20,000 versus the 70,000 calculated by Thannickal et al.). These discrepancies remain to be resolved, but they may be due to differences in the sensitivities of the Hcrt identification and glial staining techniques used. In work done subsequent to their initial publication, Thannickal et al. stained relatively fresh narcoleptic and normal tissue, and continued to see surviving Hcrt cells in all narcoleptic brains, a consistent pattern of gliosis in narcoleptics, and a count of 60,000 to 80,000 Hcrt neurons in normal human brains (Thannickal et al. 2000, 2003).
An examination of the pattern of gliosis in narcoleptics revealed that it is not confined to regions normally containing Hcrt cell somas, but rather is present throughout the projection fields of Hcrt axons. The density of gliosis is most strongly correlated with the density of Hcrt receptor-2, rather than the density of Hcrt receptor-1 or the density of Hcrt cell somas. This finding suggests that an inflammatory process accompanies Hcrt cell loss and that this process is intensified in regions with Hcrt receptor-2, perhaps with retrograde loss of the cell somas. The identity of an antigen, if any, against which an autoimmune response might be triggered in human narcolepsy remains unknown (Thannickal et al. 2003).
Further confirmation that Hcrt cell loss is sufficient to cause narcolepsy came from work with mice and rats. Hara and colleagues (Hara et al. 2001) created a transgenic mouse that carried a cytotoxic “ataxin-3” gene fragment driven by a functional preprohypocretin promoter. These animals lost all identifiable Hcrt cells postnatally and developed symptoms of narcolepsy in parallel with cell loss. Gerashchenko et al. (2001) injected Hcrt linked to the toxin saporin into Hcrt cell regions. The toxin caused the death of Hcrt cells and adjacent cells containing the Hcrt receptor. It also caused symptoms of sleepiness resembling narcolepsy, but not cataplexy.
The combination of direct excitatory and indirect inhibitory effects seen in dorsal raphe, locus coeruleus, and other regions (Figure 3) may underlie the dynamic aspects of the deficits caused by Hcrt cell loss. Narcolepsy is characterized by an instability of motor control and sleep-waking states. Narcoleptics inappropriately switch from having normal muscle tone to being partially or completely atonic when strong emotions are triggered. Conversely, their normal suppression of muscle tone in REM sleep tends to be interrupted by periods without muscle tone suppression (termed REM sleep behavior disorder) (Schenck & Mahowald 1992). Their waking rapidly switches into sleep, but they are unable to sleep for long periods because their non-REM sleep is frequently interrupted by waking (Guilleminault & Anognos 2000; Siegel 1999, 2000a,b; Thannickal et al. 2000). I hypothesize that the simultaneous inhibition and excitation mediated by Hcrt cells stabilizes control of the membranes of postsynaptic sleep- and motor-controlling neurons, in a way that would be impossible with only excitation or inhibition. “Push-pull” control produces stability. This is analogous to holding a large object with two hands rather than one. The loss of this mechanism with Hcrt cell death creates instability. Such instability in muscle tone and arousal control systems leads to the instability of motor and sleep-wake state control that characterizes narcolepsy.
The link between Hcrt and narcolepsy led to research into the possible role of Hcrt in other neurological disorders. A quick way of detecting Hcrt abnormalities is by measuring Hcrt levels in the cerebrospinal fluid; these levels were found to be undetectably low in human narcolepsy (Nishino et al. 2000). This abnormality seems relatively specific to narcolepsy, or to damage to the hypothalamic region in which the Hcrt cells are located. No obvious Hcrt abnormality was found in Alzheimer’s disease (Thannickal et al. 2000), despite the massive brain degeneration seen in this disorder, or in Parkinson’s disease (Ripley et al. 2001). Hcrt levels in schizophrenics did not differ from those in controls (Nishino et al. 2002). Levels in some cases of Guillain-Barré syndrome, a syndrome in which the hypothalamus can be damaged, were abnormally low (Ripley et al. 2001), as they are in myotonic dystrophy (Martinez-Rodriguez et al. 2003). In restless legs syndrome, whose sufferers cannot tolerate long periods of quiescence and must move to relieve uncomfortable sensations in their legs, Hcrt levels are elevated (Allen et al. 2002).
NORMAL BEHAVIORAL ROLE OF HCRT
The demonstrated link between the Hcrt system and the sleep disorder narcolepsy suggests that the release of Hcrt varies with sleep-waking state, but what is the nature of this variation? The direct excitatory connections with norepinephrine, serotonin, and histamine neurons suggest that Hcrt may drive these neurons and have the same sleep cycle discharge pattern shown by these cells groups. These three cell groups are all maximally active in active waking, decrease discharge in quiet waking, decrease discharge further in slow wave sleep, and are completely inactive in REM sleep (Siegel 2000b). On the other hand, Hcrt cells also strongly innervate and excite forebrain and brainstem cholinergic cells. These cholinergic cells are maximally active in waking, decrease discharge in quiet waking and non-REM sleep, but increase discharge in REM sleep to rates equal to or exceeding those in active waking (Siegel 2000b). Finally, dopaminergic cells are also strongly driven by Hcrt and have little or no change in activity over the sleep cycle (Shouse et al. 2000, Miller et al. 1983). Thus, the pattern of Hcrt release over the sleep cycle cannot easily be predicted by the activity of Hcrt target cell populations, all of which receive a variety of non-Hcrt inputs as well.
An initial recording study found cells in the Hcrt cell region of the rat hypothalamus with a variety of sleep cycle discharge patterns. Some were active in waking and off in REM sleep, others were active in both waking and REM sleep, and still others showed no significant change in discharge across the sleep cycle (Alam et al. 2002). However, this study did not determine which, if any, of these neuronal types contained Hcrt. The anatomical clustering of Hcrt cells, their generally similar and widespread projections, and their physiological uniformity (Li et al. 2002) suggest that the Hcrt cell population, like other widely projecting cell groups (Siegel 2000b), might show a relatively uniform pattern of sleep cycle discharge. But what is their discharge profile?
This question was addressed by Kiyashchenko et al. (2002) by using in vivo microdialysis in conjunction with the development of an extremely sensitive assay that allowed measurement of Hcrt in the 10 μl fluid samples that could be collected within 5-minute sleep periods (Kiyashchenko et al. 2002). The results are shown in Figure 6. It was discovered that Hcrt levels were at approximately equal levels in active waking and REM sleep and were greatly reduced in non-REM sleep and quiet waking. Furthermore, Hcrt levels were significantly and substantially higher in active waking than in alert, quiet waking.
Figure 6.
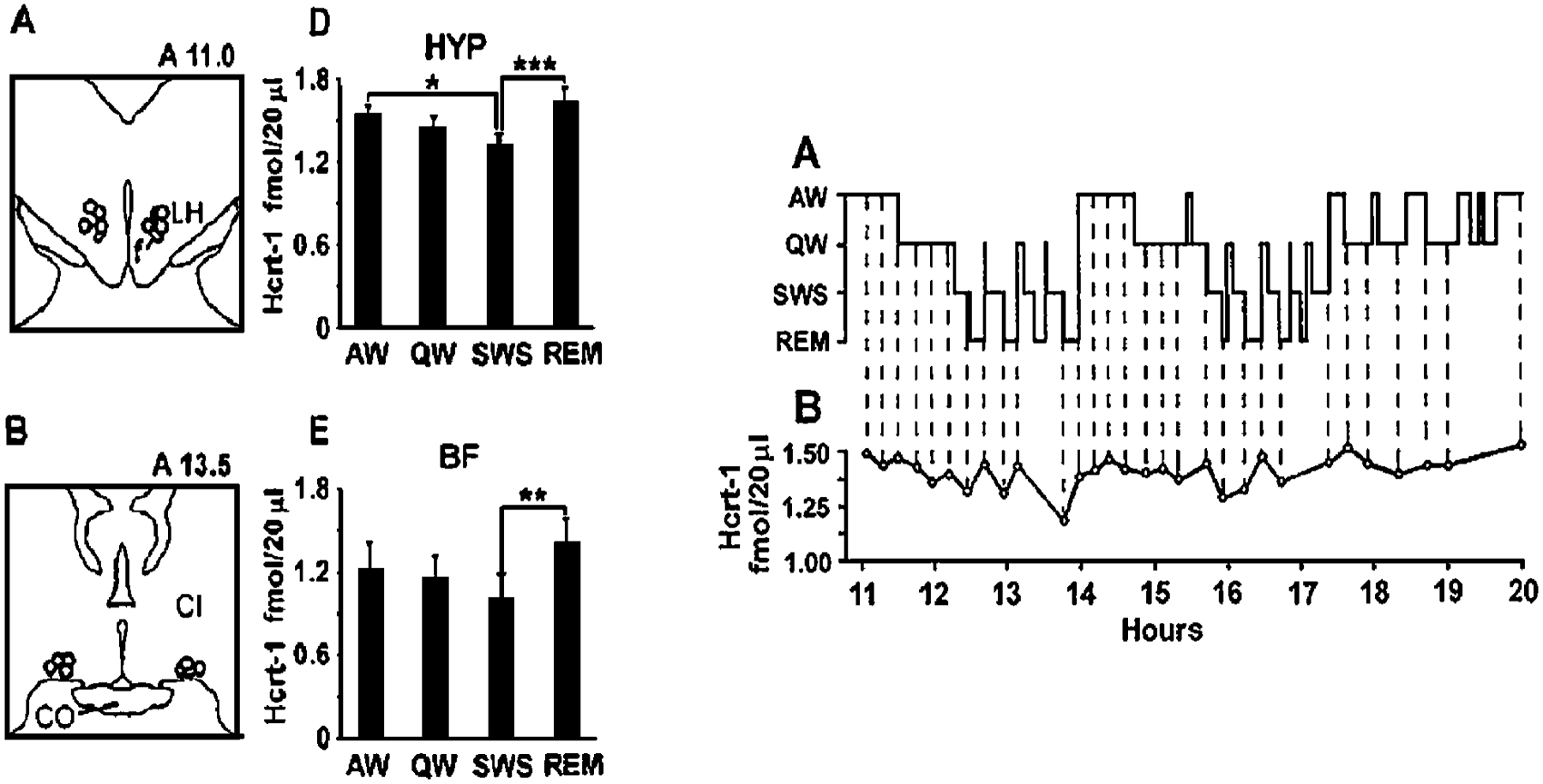
Sleep cycle release of Hcrt. (Left) Hcrt release is maximal in active waking and REM sleep, states with high levels of activity in central motor systems, and is significantly lower in non-REM sleep. Regions that were sampled are indicated in the drawing. CI, internal capsule; HYP, lateral hypothalamus; BF, basal forebrain. (Right) Time course of Hcrt release in successive 5-minute Hcrt samples across the sleep cycle. Maximal values are seen in waking and REM sleep. (From Kiyashchenko et al. 2002)
The roles of movement, sleep, and feeding variables in Hcrt release were compared (Wu et al. 2002b). The name “orexin” refers to the hypothesized role of Hcrt in inducing food consumption. Much of the behavioral Hcrt literature has dealt with the effects of Hcrt infusion on feeding. However, the infusion of very large amounts of Hcrt into the lateral hypothalamus, the hypothalamic region known to be involved in feeding, might induce changes in appetite by exciting lateral hypothalamic cells that would not normally be exposed to such high levels of Hcrt. In a similar manner, injection of Hcrt into the brain’s ventricles might cause an abnormal pattern of excitation that would increase food intake, even if Hcrt normally had little role in food intake regulation. Therefore, we attempted to determine the effects of food deprivation and food intake on Hcrt release in normal dogs, thus taking advantage of the relative ease in larger animals of extracting sufficient quantities of cerebrospinal fluid for accurate Hcrt assay. It was found that food deprivation for as long as 48 hours did not increase Hcrt levels (Figure 7, top). Feeding at the end of this period also did not produce any change in Hcrt levels. This lack of variation in release in relation to feeding and food deprivation contrasts with the changes shown by other peptides implicated in feeding (reviewed in Wu et al. 2002b).
Figure 7.
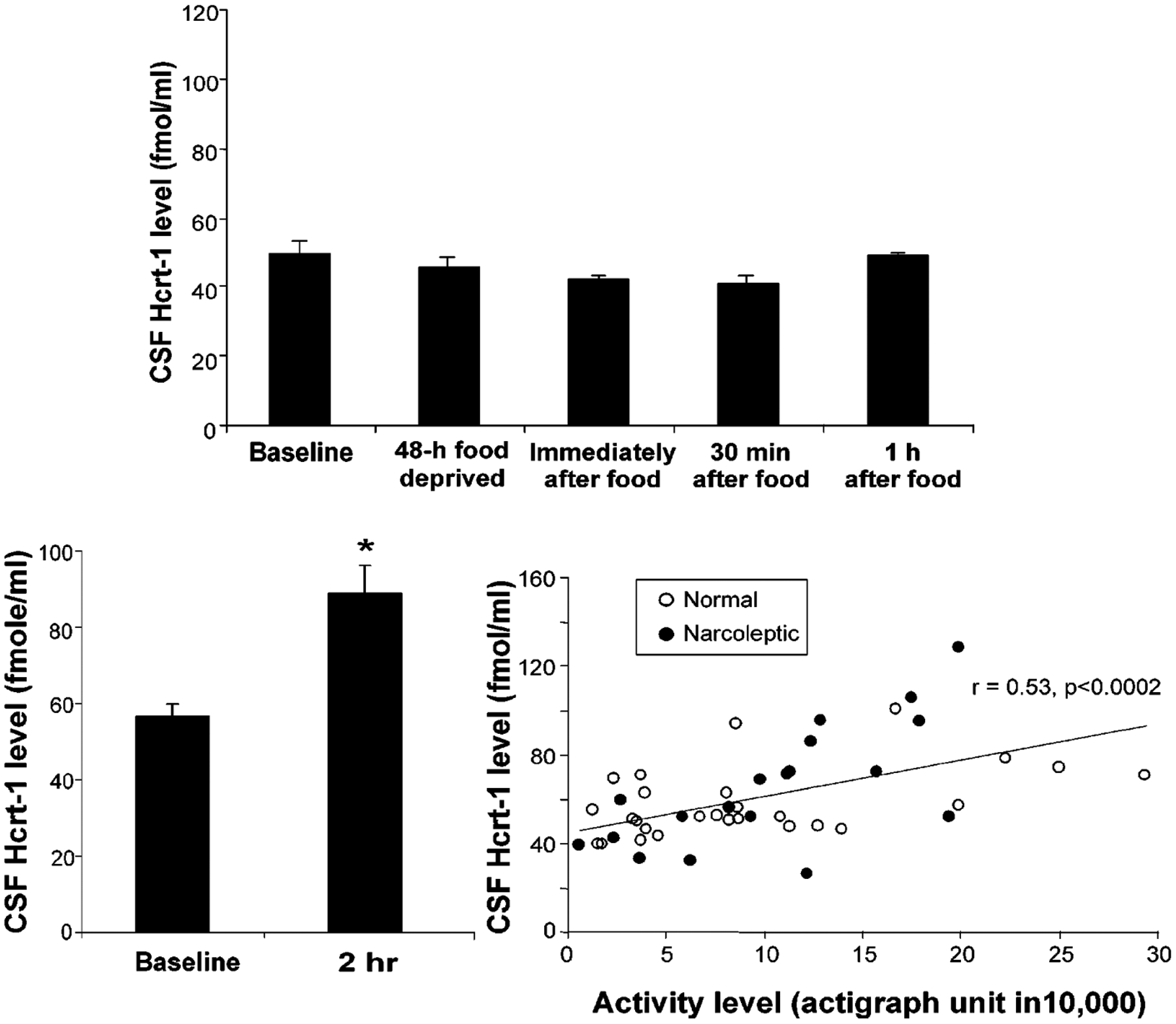
Behavioral studies of Hcrt release after food deprivation, feeding, and exercise. (Top) Effect of 48-hour food deprivation, feeding, and exercise on Hcrt release in normal dogs. In contrast to the changes in other putative feeding-related peptides, food deprivation does not increase Hcrt levels in cerebrospinal fluid. Feeding after deprivation also does not alter Hcrt levels. (Bottom left) Two-hour period of exercise produces marked increase in Hcrt levels. (Bottom right) Strong positive correlation between motor activity during exercise as measured by actigraph and levels of Hcrt in cerebrospinal fluid. (From Wu et al. 2002b)
The hypothesis that an essential, underlying correlate of Hcrt release is motor activity can explain the reported effects of Hcrt system manipulation on food intake. Although injection of high levels of Hcrt directly into the hypothalamus has been reported to induce food consumption in some studies, other studies have reported that although 24-hour infusion increased feeding during the normally inactive daytime period, it had no effect on overall food consumption (Kotz et al. 2002, Yamanaka et al. 1999). The effects of Hcrt administration on food consumption are weak compared to those of neuropeptide Y, an established feeding-related peptide (Edwards et al. 1999, Ida et al. 1999). Hcrt infusion produces increased motor activity, even in the absence of food intake (Ida et al. 1999; John et al. 2000, 2003; Yamanaka et al. 2003) but, as discussed above, food intake or food deprivation in the absence of increased movement does not produce any increase in Hcrt level (Wu et al. 2002b). Hcrt may also increase overall metabolic rate to support motor activity (Lubkin & Stricker-Krongrad 1998). Increased body temperature is a well-known correlate of increased motor activity.
Hcrt knockout animals, which do not produce the Hcrt peptides, are not emaciated, contrary to what would be predicted by the Hcrt-feeding hypothesis. Although they eat somewhat less, their weight is normal. This is consistent with the idea that they are moving less and hence consuming less energy. Ataxin mutant animals, which lose their Hcrt cells postnatally, eat less than normal mice but become extremely obese (Hara et al. 2001). Likewise, human narcoleptics, who have lost their Hcrt cells, eat less than control groups yet have a significantly higher body mass index (Schuld et al. 2000). The increased body weight of human narcoleptics and ataxin mutants may be due to the loss of the Hcrt cells themselves, as compared to the loss of the ability of these cells to generate the peptides in Hcrt “knockout” animals. Alternatively, the differences between the ataxin mutant and Hcrt knockout animals may be a consequence of the differing genetic backgrounds of the animals. The relatively small weight elevation seen in human narcoleptics may be attributable to a reduction in motor activity consequent to the loss of Hcrt activation of motor systems.
We next deprived dogs of sleep for 24 hours by walking them when they became inactive. This produced a near doubling of Hcrt levels in their cerebrospinal fluid. Because this deprivation technique induced greater levels of physical activity, we also studied the effect of activity alone. We found that as little as 2 hours of active waking produced a doubling of Hcrt levels relative to levels in the same dogs during quiet waking periods (Figure 7, lower left). The levels of Hcrt seen in individual dogs were highly correlated with their levels of motor activity as measured with activity monitors (Figure 7, lower right). A similar conclusion was reached in a study in which immunoreactivity for Fos (a protein generated by an “immediate early” gene) was used as a measure of Hcrt cell activity (Torterolo et al. 2003).
One likely mediator of the effects of Hcrt on muscle tone is the locus coeruleus. Hcrt neurons strongly innervate the locus coeruleus (Nambu et al. 1999, Peyron et al. 1998). Brainstem-induced muscle tone suppression is linked to a cessation of norepinephrine release (Lai et al. 2001). Cessation of activity in noradrenergic cells of the locus coeruleus precedes and accompanies cataplexy in narcoleptic dogs (Wu et al. 1999). Microinjection of Hcrt into the locus coeruleus and certain other brain regions increases muscle tone (Figure 8) (Kiyashchenko et al. 2001, Lai et al. 2001, Mileykovskiy et al. 2002). These findings fit with anatomical studies that have identified the noradrenergic locus coeruleus as the largest extrahypothalamic projection of the Hcrt system (Peyron et al. 1998) and with our behavioral studies. The loss of this and other motor facilitatory Hcrt projections in narcoleptics can explain the loss of muscle tone in cataplexy and sleep paralysis. Sudden, strong emotions can produce muscle weakness in normal individuals. We get “weak in the knees” when we hear bad news. We “double over with laughter” when we hear or tell a good joke. But normal individuals do not fall to the ground at these times. Presumably, this is because, along with the weakness induced by circuits activated during these emotions, there is a counteracting activation of systems that excite motor systems. Hcrt is one of these systems. In its absence, as in narcolepsy, these same emotions can result in a complete loss of muscle tone for periods of seconds to minutes.
Figure 8.
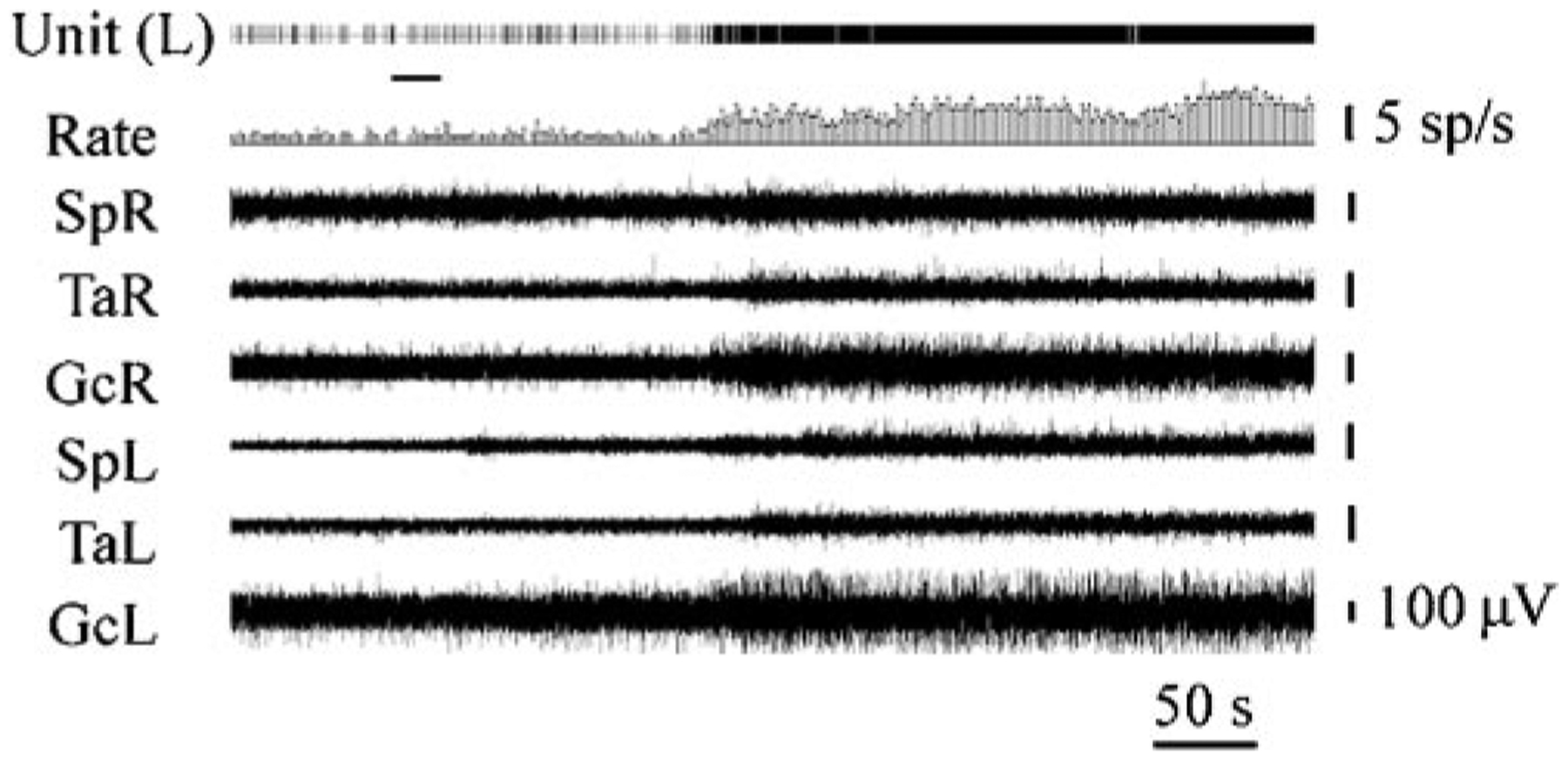
Effect of Hcrt microinjection adjacent to the locus coeruleus. This figure shows that Hcrt microinjection into the locus coeruleus raises muscle tone ipsilateral to the injection. The loss of this excitatory effect of Hcrt contributes to cataplexy, the sudden loss of muscle tone experienced by most narcoleptics. Locus coeruleus activity ceases during cataplexy (Wu et al. 1999). SpR, SpL: electromyogram (EMG) of muscle splenius (right and left side); TaR, TaL: EMG of tibialis anterior muscle (right and left side); GcR, GcL: EMG of gastrocnemius muscle (right and left side). (From Kiyashchenko et al. 2001)
CONCLUSIONS
We have found that a major correlate of Hcrt release in waking is motor activity. The high level of release of Hcrt in REM sleep can also be seen as a correlate of motor activation, because REM sleep is a state in which central motor systems are highly active (Siegel 1979, Siegel & Tomaszewski 1983, Siegel et al. 1983), even though the expression of this internal motor activity is blocked at the motoneuron level (Chase & Morales 2000, Kodama et al. 2003, Lai et al. 2001, Wu et al. 1999).
When animals are motorically active, they must be alert in order to execute movements properly, carry out consummatory acts, and integrate sensory inputs. Thus, an activation of sensory and related forebrain cognitive systems is necessary during periods of activity.
Our finding that Hcrt levels in alert, immobile waking are not significantly higher than the levels seen in non-REM sleep argues against a major role for Hcrt in nonmotor-associated attentional processes in animals. Given the explanatory power of the motor activity hypothesis, it becomes incumbent upon those who hypothesize a relation to physiological changes independent of motor activity to quantify or control for the motor activity elicited by their experimental manipulations.
Current data do not indicate the precise nature of the role of Hcrt in motor activity. However, clues to the likely dynamics of Hcrt release can be derived from the symptoms seen in hypocretin-deficient or hypocretin-receptor mutant animals and humans (i.e., narcoleptics). As pointed out above, narcoleptics have two main symptoms, excessive sleepiness and cataplexy. The excessive sleepiness is a tonic symptom, expressed throughout the day, whereas cataplexy is expressed episodically.
A tonic role of Hcrt is hypothesized to be the activation of monoaminergic, cholinergic, and amino acid neurotransmitter systems during periods of maintained muscle tone. The release of Hcrt at these times is hypothesized to occur continuously, through the spontaneous discharge of these cells. In REM sleep, the tonic effects of Hcrt on nonadrenergic and serotonergic cell groups are blocked by indirect Hcrt facilitation of GABA release onto these same cells (Kiyashchenko et al. 2002). In waking, the tonic activity of Hcrt cells extends the duration of waking periods, blocking the onset of sleep.
Phasic release of Hcrt is hypothesized to counter the effects of emotion on muscle tone. Sudden strong emotions of certain types may produce a disfacilitation of motoneurons as a result of the inactivation of monoaminergic and other brainstem cell groups with descending projections to motoneurons. This disfacilitation could be combined with active inhibition mediated by glycine and GABA release onto motoneurons.
In the normal animal, this disfacilitation/inhibition could be countered by a burst of Hcrt activity, facilitating the activity of motoneurons and thereby maintaining muscle tone. Even in normal animals, sudden, very strong emotions will still reveal a motor inhibition, as in the weakness produced by strong laughter or anger. However, the counteracting effect of Hcrt would prevent any complete loss of muscle tone.
Cataplexy does not occur with equal frequency during all types of movements. Rather, it accompanies particular kinds of emotionally charged behaviors. In narcoleptic dogs, the consummatory behavior required to consume desirable, novel food triggers cataplexy, while consumption of the food that is ordinarily available does not. Vigorous play with the narcoleptic dog, for example by pulling on a towel the dog is holding in its mouth, throwing a toy to the dog, and other such pleasurable, motorically engaging activities, will also trigger cataplexy. In contrast, startling the animal or bringing it into an unfamiliar room will not trigger cataplexy, although the situation is highly alerting. In humans, as mentioned above, laughter, anger, or other sudden-onset emotions are the typical triggers of cataplexy. On the other hand, high levels of attention or even pain that is highly alerting are not typical triggers. These observations suggest that Hcrt release is linked to the sudden onset of certain kinds of emotions whose precise quality undoubtedly varies among species just as it does to a much lesser extent among patients with narcolepsy.
In summary, it is hypothesized that Hcrt cells maintain some tonic discharge during periods of waking motor activity. This discharge would be phasically increased during the sorts of sudden, strong emotions that trigger cataplexy and this release would both act to maintain muscle tone phasically and contribute to the tonic maintenance of alertness. The targets of Hcrt release, as indicated in Figure 3, are systems related to motor facilitation and systems related to forebrain alerting. Thus, Hcrt release would act to coordinate motor activity and arousal.
This extrapolation from the phenomena of excessive sleepiness and cataplexy in narcoleptic animals to the normal function of Hcrt release needs to be tested in normal animals. This can best be done by characterizing the activity pattern of identified Hcrt neurons during a range of behaviors.
Our work in dogs, in which Hcrt administration has been shown to reverse the deficits of narcolepsy (John et al. 2000), offers hope that Hcrt administration will be an effective treatment for this disorder. Manipulation of the Hcrt system may also be useful in other behavioral disorders.
ACKNOWLEDGMENTS
Supported by the Medical Research Service of the Department of Veterans Affairs, NS14610, NS64109, MH64109, and HL060246.
LITERATURE CITED
- Abrahamson EE, Moore RY. 2001. The posterior hypothalamic area: chemoarchitecture and afferent connections. Brain Res. 889:1–22 [DOI] [PubMed] [Google Scholar]
- Alam MN, Gong H, Alam T, Jaganath R, McGinty D, Szymusiak R. 2002. Sleep-waking discharge patterns of neurons recorded in the rat perifornical lateral hypothalamic area. J. Physiol 538:619–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich MS. 1998. Diagnostic aspects of narcolepsy. Neurology 50:S2–7 [DOI] [PubMed] [Google Scholar]
- Allen RP, Mignot E, Ripley B, Nishino S, Earley CJ. 2002. Increased CSF hypocretin-1 (orexin-A) in restless legs syndrome. Neurology 59:639–41 [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Rajkowski J, Kubiak P, Alexinsky T. 1994. Locus coeruleus neurons in monkey are selectively activated by attended cues in a vigilance task. J. Neurosci 14:4467–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Dement WC. 1985. Canine narcolepsy-cataplexy syndrome: evidence for an inherited monoaminergic-cholinergic imbalance. In Brain Mechanisms of Sleep, ed. McGinty DJ, Drucker-Colin R, Morrison A, Parmeggiani PL, pp. 199–234. New York: Raven [Google Scholar]
- Bayer L, Eggermann E, Saint-Mleux B, Machard D, Jones BE, et al. 2002. Selective action of orexin (hypocretin) on nonspecific thalamocortical projection neurons. J. Neurosci 22:7835–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer L, Eggermann E, Serafin M, Saint-Mleux B, Machard D, et al. 2001. Orexins (hypocretins) directly excite tuberomammillary neurons. Eur. J. Neurosci 14:1571–75 [DOI] [PubMed] [Google Scholar]
- Bourgin P, Huitron-Resendiz S, Spier AD, Fabre V, Morte B, et al. 2000. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J. Neurosci 20:7760–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlet S, Tyler CJ, Leonard CS. 2002. Direct and indirect excitation of laterodorsal tegmental neurons by hypocretin/orexin peptides: implications for wakefulness and narcolepsy. J. Neurosci 22:2862–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillol M, Aioun J, Baly C, Persuy MA, Salesse R. 2003. Localization of orexins and their receptors in the rat olfactory system: possible modulation of olfactory perception by a neuropeptide synthetized centrally or locally. Brain Res. 960:48–61 [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR. 2000. Control of motoneurons during sleep. See Kryger et al. 2000, pp. 155–68 [Google Scholar]
- Chemelli RM, Willie JT, Sinton C, Elmquist J, Scammell T, et al. 1999. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98:437–51 [DOI] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, et al. 2001. Orexin (hypocretin) neurons contain dynorphin. J. Neurosci 21:RC168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello J, Rosas-Arellano MP, Solano-Flores LP, de Oliveira CV. 2003. Identification of neurons containing orexin-B (hypocretin-2) immunoreactivity in limbic structures. Brain Res. 967:123–31 [DOI] [PubMed] [Google Scholar]
- Date Y, Mondal MS, Matsukura S, Ueta Y, Yamashita H, et al. 2000. Distribution of orexin/hypocretin in the rat median eminence and pituitary. Brain Res. Mol. Brain Res 76:1–6 [DOI] [PubMed] [Google Scholar]
- Dehaene S, Changeux JP. 2000. Reward-dependent learning in neuronal networks for planning and decision making. Prog. Brain Res 126:217–29 [DOI] [PubMed] [Google Scholar]
- De Lecea L, Kilduff T, Peyron C, Gao XB, Foye PE, et al. 1998. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. USA 95:322–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass AB, Shipley JE, Haines RF, Scholten RC. 1993. Schizophrenia, narcolepsy, and HLA-DR15, DQ6. Biol. Psychiatry 34:773–80 [DOI] [PubMed] [Google Scholar]
- Dun NJ, Le Dun S, Chen CT, Hwang LL, Kwok EH, Chang JK. 2000. Orexins: a role in medullary sympathetic outflow. Regul. Pept 96:65–70 [DOI] [PubMed] [Google Scholar]
- Edwards CM, Abusnana S, Sunter D, Murphy KG, Ghatei MA, Bloom SR. 1999. The effect of the orexins on food intake: comparison with neuropeptide Y, melanin-concentrating hormone and galanin. J. Endocrinol 160:R7–12 [DOI] [PubMed] [Google Scholar]
- Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, et al. 2003. Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience 108:177–81 [DOI] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. 2001. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J. Neurosci 21:9273–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follwell MJ, Ferguson AV. 2002. Cellular mechanisms of orexin actions on paraventricular nucleus neurones in rat hypothalamus. J. Physiol 545:855–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung SJ, Yamuy J, Sampogna S, Morales FR, Chase MH. 2001. Hypocretin (orexin) input to trigeminal and hypoglossal motoneurons in the cat: a double-labeling immunohistochemical study. Brain Res. 903:257–62 [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Kohls MD, Greco M, Waleh NS, Salin-Pascual R, et al. 2001. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J. Neurosci 21:7273–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt TJ, van den Pol AN, Perl ER. 2002. Hypocretin-2 (orexin-B) modulation of superficial dorsal horn activity in rat. J. Physiol 538:517–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilleminault C, Anognos A. 2000. Narcolepsy. See Kryger et al. 2000, pp. 676–86 [Google Scholar]
- Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, et al. 1999. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc. Natl. Acad. Sci. USA 96:10911–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, et al. 2001. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30:345–54 [DOI] [PubMed] [Google Scholar]
- Honda Y, Doi Y, Juji T, Satake M. 1984. Narcolepsy and HLA: positive DR2 as a prerequisite for the development of narcolepsy. Folia Psychiatr. Neurol Jpn 38:360 [Google Scholar]
- Horvath TL, Peyron C, Diano S, Ivanov A, Aston-Jones G, et al. 1999. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J. Comp. Neurol 415:145–59 [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, et al. 2001. Arousal effect of orexin A depends on activation of the histaminergic system. Proc. Natl. Acad. Sci. USA 98:9965–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hublin C, Kaprio J, Partinen M, Koskenvuo M, Heikkila K, et al. 1994. The prevalence of narcolepsy: an epidemiological study of the Finnish twin cohort. Ann. Neurol 35:709–16 [DOI] [PubMed] [Google Scholar]
- Ida T, Nakahara K, Katayama T, Murakami N, Nakazato M. 1999. Effect of lateral cerebroventricular injection of the appetite-stimulating neuropeptide, orexin and neuropeptide Y, on the various behavioral activities of rats. Brain Res. 821:526–29 [DOI] [PubMed] [Google Scholar]
- Ivanov A, Aston-Jones G. 2000. Hypocretin/orexin depolarizes and decreases potassium conductance in locus coeruleus neurons. NeuroReport 11:1755–58 [DOI] [PubMed] [Google Scholar]
- Jacobs BL. 1987. Brain monoaminergic unit activity in behaving animals. In Progress in Psychobiology and Physiological Psychology, ed. Epstein AN, Morrison AR, pp. 171–206. New York: Academic [Google Scholar]
- Jacobs BL. 1991. Serotonin and behavior: emphasis on motor control. J. Clin. Psychiatry 52(Suppl.):17–23 [PubMed] [Google Scholar]
- John J, Wu MF, Kodama T, Siegel JM. 2003. Intravenously administered hypocretin-1 alters brain amino acid release: an in vivo microdialysis study in rats. J. Physiol 548.2:557–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, Wu MF, Kodama T, Siegel JM. 2001. Hypocretin-1 (orexin-A) produced changes in glutamate and GABA release: an in vivo microdialysis study. Sleep 24:A20 [Google Scholar]
- John J, Wu MF, Siegel JM. 2000. Systemic administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res. 3:23–28. http://www.sro.org/2000/John/23/ [PMC free article] [PubMed] [Google Scholar]
- Johren O, Neidert SJ, Kummer M, Dendorfer A, Dominiak P. 2001. Prepro-orexin and orexin receptor mRNAs are differentially expressed in peripheral tissues of male and female rats. Endocrinology 142:3324–31 [DOI] [PubMed] [Google Scholar]
- Jones BE. 2000. Basic mechanisms of sleep-wake states. See Kryger et al. 2000, pp. 134–54 [Google Scholar]
- Kirchgessner AL. 2002. Orexins in the braingut axis. Endocr. Rev 23:1–15 [DOI] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Lai YY, Siegel JM. 2001. Increased and decreased muscle tone with orexin (hypocretin) microinjections in the locus coeruleus and pontine inhibitory area. J. Neurophysiol 85:2008–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Maidment N, Lam HA, Wu MF, et al. 2002. Release of hypocretin (orexin) during waking and sleep states. J. Neurosci 22:5282–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Kimura M. 2002. Arousal effects of orexin-A correlate with GLU release from the locus coeruleus in rats. Peptides 23:1673–81 [DOI] [PubMed] [Google Scholar]
- Kodama T, Lai YY, Siegel JM. 2003. Changes in inhibitory amino acid release linked to pontine-induced atonia: an in vivo microdialysis study. J. Neurosci 23:1548–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE. 2003. Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J. Neurosci 23:7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotz CM, Teske JA, Levine JA, Wang C. 2002. Feeding and activity induced by orexin A in the lateral hypothalamus in rats. Regul. Pept 104:27–32 [DOI] [PubMed] [Google Scholar]
- Kryger MH, Roth T, Dement WC, eds. 2000. Principles and Practice of Sleep Medicine. Philadelphia: Saunders [Google Scholar]
- Lai YY, Kodama T, Siegel JM. 2001. Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J. Neurosci 21:7384–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Kodama T, Siegel JM. 2001. Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J. Neurosci 21:7384–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt DR, Teitelbaum P. 1975. Somnolence, akinesia, and sensory activation of motivated behavior in the lateral hypothalamic syndrome. Proc. Natl. Acad. Sci. USA 72:2819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN. 2002. Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron 36:1169–81 [DOI] [PubMed] [Google Scholar]
- Lin JS, Sakai K, Vanni-Mercier G, Jouvet M. 1989. A critical role of the posterior hypothalamus in the mechanisms of wakefulness determined by microinjection of muscimol in freely moving cats. Brain Res. 479:225–40 [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, et al. 1999. The REM sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor gene. Cell 98:365–76 [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN, Aghajanian GK. 2002. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J. Neurosci 22:9453–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubkin M, Stricker-Krongrad A. 1998. Independent feeding and metabolic actions of orexins in mice. Biochem. Biophys. Res. Commun 253:241–45 [DOI] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, et al. 2001. Differential expression of orexin receptors 1 and 2 in the rat brain. J. Comp. Neurol 435:6–25 [DOI] [PubMed] [Google Scholar]
- Martin G, Fabre V, Siggins GR, De Lecea L. 2002. Interaction of the hypocretins with neurotransmitters in the nucleus accumbens. Regul. Pept 104:111–17 [DOI] [PubMed] [Google Scholar]
- Martinez-Rodriguez JE, Lin L, Iranzo A, Genis D, Marti MJ, et al. 2003. Decreased hypocretin-1 (orexin-A) levels in the cerebrospinal fluid of patients with myotonic dystrophy and excessive daytime sleepiness. Sleep 26:287–90 [DOI] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. 2002. Muscle tone facilitation and inhibition after orexin-a (hypocretin-1) microinjections into the medial medulla. J. Neurophysiol 87:2480–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Farber J, Gatz P, Roffwarg H, German DC. 1983. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and waking in the rat. Brain Res. 273:133–41 [DOI] [PubMed] [Google Scholar]
- Mitler MM, Soave O, Dement WC. 1976. Narcolepsy in seven dogs. J. Am. Vet. Med. Assoc 168:1036–38 [PubMed] [Google Scholar]
- Nakamura T, Uramura K, Nambu T, Yada T, Goto K, et al. 2000. Orexin-induced hyperlocomotion and stereotypy are mediated by the dopaminergic system. Brain Res. 873:181–87 [DOI] [PubMed] [Google Scholar]
- Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. 1999. Distribution of orexin neurons in the adult rat brain. Brain Res. 827:243–60 [DOI] [PubMed] [Google Scholar]
- Nishino S, Fujiki N, Ripley B, Sakurai E, Kato M, et al. 2001. Decreased brain histamine content in hypocretin/orexin receptor-2 mutated narcoleptic dogs. Neurosci. Lett 3(3):125–28 [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Mignot E, Benson KL, Zarcone VP. 2002. CSF hypocretin-1 levels in schizophrenics and controls: relationship to sleep architecture. Psychiatry Res. 110:1–7 [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. 2000. Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355:39–41 [DOI] [PubMed] [Google Scholar]
- Nishino SS, Sakurai E, Nevisimalova S, Vankova J, Yoshida Y, et al. 2002. CSF histamine content is decreased in hypocretin-deficient human narcolepsy. Sleep 25:A476 [Google Scholar]
- Nitz D, Siegel JM. 1997a. GABA release in the dorsal raphe nucleus: role in the control of REM sleep. Am. J. Physiol. Regul. Integr. Comp. Physiol 273:R451–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitz D, Siegel JM. 1997b. GABA release in the cat locus coeruleus as a function of the sleep/wake state. Neuroscience 78:795–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olerup O, Schaffer M, Hillert J, Sachs C. 1990. The narcolepsy-associated DRw15, DQw6, Dw2 haplotype has no unique HLA-DQA or -DQB restriction fragments and does not extend to the HLA-DP subregion. Immunogenetics 32:41–44 [DOI] [PubMed] [Google Scholar]
- Passouant P 1976. The history of narcolepsy. In Narcolepsy, ed. Guilleminault C, Dement WC, Passouant P, pp. 3–14. New York: Spectrum [Google Scholar]
- Peever JH, Lai YY, Siegel JM. 2003. Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J. Neurophysiol 89:2591–600 [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, et al. 2000. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med 6:991–97 [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, et al. 1998. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J. Neurosci 18:9996–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reti IM, Minor LB, Baraban JM. 2002. Prominent expression of Narp in central vestibular pathways: selective effect of labyrinth ablation. Eur. J. Neurosci 16:1949–58 [DOI] [PubMed] [Google Scholar]
- Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, et al. 2001. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology 57:2253–58 [DOI] [PubMed] [Google Scholar]
- Rye DB, Jankovic J. 2002. Emerging views of dopamine in modulating sleep/wake state from an unlikely source: PD. Neurology 58:341–46 [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, et al. 1998. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–85 [DOI] [PubMed] [Google Scholar]
- Schenck CH, Mahowald MW. 1992. Motor dyscontrol in narcolepsy: rapid-eye-movement (REM) sleep without atonia and REM sleep behavior disorder. Ann. Neurol 32:3–10 [DOI] [PubMed] [Google Scholar]
- Schuld A, Hebebrand J, Geller F, Pollmacher T. 2000. Increased body-mass index in patients with narcolepsy. Lancet 355:1274–75 [DOI] [PubMed] [Google Scholar]
- Shouse MN, Staba RJ, Saquib SF, Farber PR. 2000. Monoamines and sleep: microdialysis findings in pons and amygdala. Brain Res. 860:181–89 [DOI] [PubMed] [Google Scholar]
- Siegel JM. 1979. Behavioral functions of the reticular formation. Brain Res. Rev 1:69–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM. 1999. Narcolepsy: a key role for hypocretins (orexins). Cell 98:409–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM. 2000a. Narcolepsy. Sci. Am 282:76–8111056991 [Google Scholar]
- Siegel JM. 2000b. Brainstem mechanisms generating REM sleep. See Kryger et al. 2000, pp. 112–33 [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer H, Paul R, Shiromani P, et al. 1991. Neuronal activity in narcolepsy: identification of cataplexy related cells in the medial medulla. Science 262:1315–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Gulyani S, Ouyang S, Wu MF, et al. 1999. Neuronal degeneration in canine narcolepsy. J. Neurosci 19:248–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Tomaszewski KS. 1983. Behavioral organization of reticular formation: studies in the unrestrained cat. I. Cells related to axial, limb, eye, and other movements. J. Neurophysiol 50:696–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Tomaszewski KS, Wheeler RL. 1983. Behavioral organization of reticular formation: studies in the unrestrained cat: II. Cells related to facial movements. J. Neurophysiol 50:717–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, et al. 2000. Reduced number of hypocretin neurons in human narcolepsy. Neuron 27:469–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Siegel JM, Nienhuis R, Moore RY. 2003. Pattern of hypocretin (orexin) soma and axon loss, and gliosis, in human narcolepsy. Brain Pathol. 13:340–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torterolo P, Yamuy J, Sampogna S, Morales FR, Chase MH. 2003. Hypocretinergic neurons are primarily involved in activation of the somatomotor system. Sleep 26:25–28 [PubMed] [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. 1998. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. 438:71–75 [DOI] [PubMed] [Google Scholar]
- van den Pol AN. 1999. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. J. Neurosci 19:3171–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB. 1998. Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J. Neurosci 18:7962–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Ghosh PK, Liu RJ, Li Y, Aghajanian GK, Gao XB. 2002. Hypocretin (orexin) enhances neuron activity and cell synchrony in developing mouse GFP-expressing locus coeruleus. J. Physiol 541:169–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MJ, Meltzer EO, Simons FE. 2003. H1-antihistamines and the central nervous system. Clin. Allergy Immunol 17:337–88 [PubMed] [Google Scholar]
- Wu M, Zhang ZM, Leranth C, Xu CQ, van den Pol AN, Alreja M. 2002a. Hypocretin increases impulse flow in the septohippocampal GABAergic pathway: implications for arousal via a mechanism of hippocampal disinhibition. J. Neurosci 22:7754–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, Gulyani S, Yao E, Mignot E, Phan B, Siegel JM. 1999. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience 91:1389–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, John J, Maidment N, Lam HA, Siegel JM. 2002b. Hypocretin release in normal and narcoleptic dogs after food and sleep deprivation, eating, and movement. Am. J. Physiol. Regul. Integr. Comp. Physiol 283:R1079–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, et al. 2003. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38:701–13 [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Sakurai T, Katsumoto T, Yanagisawa M, Goto K. 1999. Chronic intracere-broventricular administration of orexin-a to rats increases food intake in daytime, but has no effect on body weight. Brain Res. 849:248–52 [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Tsujino N, Funahashi H, Honda K, Guan JL, et al. 2002. Orexins activate histaminergic neurons via the orexin 2 receptor. Biochem. Biophys. Res. Commun 290:1237–45 [DOI] [PubMed] [Google Scholar]