

ABSTRACT
Increased usage of daptomycin (DAP) for methicillin-resistant Staphylococcus aureus (MRSA) infections has led to emergence of DAP-resistant (DAP-R) strains, resulting in treatment failures. DAP-fosfomycin (Fosfo) combinations are synergistically active against MRSA, although the mechanism(s) of this interaction is not fully understood. The current study explored four unique but likely interrelated activities of DAP-Fosfo combinations: (i) synergistic killing, (ii) prevention of evolution of DAP-R, (iii) resensitization of already DAP-R subpopulations to a DAP-susceptible (DAP-S) phenotype, and (iv) perturbations of specific cell envelope phenotypes known to correlate with DAP-R in MRSA. Using an isogenic DAP-S (CB1483)/DAP-R (CB185) clinical MRSA strain pair, we demonstrated that combinations of DAP plus Fosfo (DAP+Fosfo) (i) enhanced killing of both strains in vitro and ex vivo, (ii) increased target tissue clearances of the DAP-R strain in an in vivo model of experimental infective endocarditis (IE), (iii) prevented emergence of DAP-R in the DAP-S parental strain both in vitro and ex vivo, and (iv) resensitized the DAP-R strain to a DAP-S phenotype ex vivo. Phenotypically, following exposure to sub-MIC Fosfo, the DAP-S/DAP-R strain pair exhibited distinct modifications in (i) net positive surface charge (P < 0.05), (ii) quantity (P < 0.0001) and localization of cell membrane cardiolipin (CL), (iii) DAP surface binding, and (iv) membrane fluidity (P < 0.05). Furthermore, preconditioning this strain pair to DAP with or without Fosfo (DAP+/−Fosfo) sensitized these organisms to killing by the human host defense peptide LL37. These data underscore the notion that DAP-Fosfo combinations can impact MRSA clearances within multiple microenvironments, likely based on specific phenotypic adaptations.
KEYWORDS: MRSA, Staphylococcus, combination, daptomycin, fosfomycin
INTRODUCTION
Staphylococcus aureus is a major nosocomial and community pathogen and a leading cause of endovascular infections (e.g., infective endocarditis [IE]) (1). Daptomycin (DAP) has become a key standard-of-care antibiotic for the treatment of methicillin-resistant S. aureus (MRSA), especially in patients with complicated and/or recalcitrant infections, as well as in cases of vancomycin treatment failures (2). However, in vivo DAP resistance (DAP-R) emerges clinically, resulting in treatment failures (1–5). The most well-known genetic mutations associated with DAP-R in S. aureus are single point mutations in several specific regions of the multipeptide resistance factor (mprF) open reading frame (ORF), with or without concomitant point mutations in the yyc operon (6). The mprF operon primarily mediates the relative positive surface charge in S. aureus via lysinylation of cell membrane (CM) phosphatidylglycerol (PG), forming lysyl-PG (L-PG) (7). DAP-R in S. aureus mutants has also been linked with the histidine kinase of the system (YycG). Various mutations in distinct locations of the yyc operon have been reported, although the exact mechanism leading to DAP-R is not well understood. Of note, DAP-R has been well chronicled for complicated, deep-seated infections such as osteomyelitis and IE, where factors such as high inocula, reduced metabolic activity, and altered antibiotic penetration profiles each play a contributory role (3).
The optimal treatment for DAP-R MRSA strains is not known. Several investigations have examined newer combination therapy approaches for MRSA bacteremia, focusing mainly on DAP–β-lactam regimens (e.g., DAP-ceftaroline) (8, 9). Combinations of DAP plus Fosfo (DAP+Fosfo) have been recently highlighted as another potential salutary strategy for DAP-R strains. Such regimens exhibit profound bactericidal synergy in vitro against MRSA and yield synergistic activity in experimental IE (10). Fosfo, like β-lactams, targets cell wall peptidoglycan synthesis, but through inhibition of an earlier biosynthetic step (i.e., blockade of UDP-N-acetylglucosamine enolpyruvyl transferase [MurA]) (10). The combination of DAP-Fosfo was recently examined in a small (n = 81 patients), randomized, open-label, multicenter Spanish clinical trial of MRSA bacteremia (5). Although limited by including relatively few patients with complicated bacteremia (e.g., only 9 patients [11%] with IE), this combination yielded (i) improved clinical outcomes at end of treatment, (ii) reduced early in-hospital mortality, (iii) lowered rates of microbiologic failure, and (iv) prevention of complicated bacteremia (5). These interesting in vivo outcomes provided a strong impetus to understand the mechanisms underlying the interactive efficacies of DAP-Fosfo combinations.
The objective of the present study was to utilize a clinically derived, isogenic DAP-susceptible (DAP-S)/DAP-R MRSA strain pair to evaluate the potential of DAP-Fosfo combinations versus MRSA, in three specific facets: (i) synergistic killing, (ii) prevention of DAP-R development, and (iii) resensitization of DAP-R to a DAP-S phenotype. In addition, we correlated these effects with specific cell envelope phenotypic modifications in vitro, which have been noted to be perturbed previously among DAP-R MRSA isolates (6).
Note that for a more facile presentation, we have used the terminology “DAP-R” instead of “DAP nonsusceptibility.”
RESULTS
MICs.
The previously published DAP MICs of the CB1483/CB185 DAP-S/DAP-R MRSA strain pair (11) were reconfirmed. The DAP-S and DAP-R MRSA strains each exhibited Fosfo MICs of 8 mg/L, indicating Fosfo-S (Table 1).
TABLE 1.
MICs and genotypes of studied DAP-S/DAP-R (CB1483/CB185) MRSA strains
| Strain | DAP MICa (mg/L) | Fosfo MIC (mg/L) | mpr SNPa | yyc SNPa |
|---|---|---|---|---|
| CB1483 (DAP-S) | 0.25 | 8 | — | — |
| CB185 (DAP-R) | 4 | 8 | L826Fb | None |
Data in this table have been previously published (11). —, none (no mutation detected).
Mutation in putative mprF L-PG synthase domain.
In vitro time-kill curve analysis.
DAP+Fosfo was bactericidal (>3-log10 CFU/mL reduction) and substantially enhanced killing of both strains versus that obtained with either drug alone (P < 0.05); this combination was synergistic for the DAP-R strain (Fig. 1).
FIG 1.

Sublethal DAP+/−Fosfo (1× MIC of each antibiotic) killing of DAP-S/DAP-R MRSA strains in vitro. Arrows indicate the enhanced bactericidal synergistic killing impact of combination of DAP and Fosfo versus those of the single antibiotics.
Resistance prevention and resensitization during in vitro serial passaging.
DAP+Fosfo was able to prevent emergence of DAP-R in the DAP-S parental strain (Fig. 2A), but it did not resensitize the DAP-R variant strain in vitro to DAP-S. However, the initial DAP MIC of the DAP-R strain did not further increase during DAP+Fosfo passage, while the MIC doubled over 10 days of DAP-alone passage (Fig. 2B).
FIG 2.

DAP MICs of DAP-S/DAP-R strains serially passaged in DAP+Fosfo versus DAP alone in vitro. (A) DAP+Fosfo prevented selection of DAP-R; (B) DAP+Fosfo did not resensitize DAP-R CB185 to the DAP-S phenotype but prevented further MIC increases.
Impacts of DAP-Fosfo combinations in the ex vivo IE model.
Paralleling our in vitro studies described above, ex vivo-simulated endocardial vegetation (SEV) modeling demonstrated enhanced bactericidal impacts of DAP+Fosfo versus both the DAP-S and DAP-R strains (Fig. 3). In the parental DAP-S strain, both DAP and Fosfo alone were bactericidal over the 96-h exposure period, while only Fosfo alone demonstrated bactericidal activity in the DAP-R variant. In both strains, the combination of DAP+Fosfo provided rapid and sustained early bactericidal activity (at 8 h or less), sterilizing SEVs over the 24- to 96-h exposure period.
FIG 3.

Fosfo facilitates synergistic killing significantly with DAP against DAP-S/DAP-R MRSA strains in an ex vivo IE model within simulated IE vegetations. Simulated model doses were DAP at 6 mg/kg every 24 h and/or Fosfo at 4 g every 8 h. *, P ≤ 0.05 for DAP+Fosfo versus the single antibiotics in both the DAP-S and DAP-R strains. Arrows highlight the increased bactericidal synergistic killing impact of combination of DAP and Fosfo versus those of the single antibiotics.
We further analyzed the SEV-derived isolates from the ex vivo model. For the DAP-S parental strain treated with DAP alone, DAP-R subpopulations were detected at the 72- to 96-h exposure points (MICs = 4 mg/L); this correlated with bacterial regrowth noted at those time points. Treatment with DAP+Fosfo prevented emergence of DAP-R subpopulations (DAP MIC = 0.5 mg/L). In contrast, the combination DAP+Fosfo rapidly sterilized SEVs in both strains; therefore, resistance or further MIC increases did not emerge with this combination ex vivo.
In vivo IE model.
As shown in Table 2, none of the DAP monotherapy regimens reduced MRSA bioburdens in the three target tissues (versus untreated controls). Fosfo alone, as well as lower doses of DAP (2 to 6 mg/kg of body weight) in combination with Fosfo, yielded modest but significant reductions in MRSA bioburdens in these organs (versus those in untreated controls). Further, the combination of higher-dose DAP (10 mg/kg) with Fosfo significantly reduced MRSA bioburdens in all tissues, with organ sterilizations in the majority of animals (Table 2).
TABLE 2.
Treatment of DAP-R CB185 with DAP+Fosfoa
| Treatment group (5 rabbits/group) | Vegetation count (mean CFU/g ± SD) | Kidney count (mean CFU/g ± SD) | Spleen count (mean CFU/g ± SD) |
|---|---|---|---|
| Untreated | 8.28 ± 0.6 | 6.49 ± 0.5 | 6.55 ± 0.3 |
| DAP alone (2 mg) | 8.80 ± 0.2 | 7.88 ± 0.5 | 6.74 ± 0.3 |
| DAP alone (6 mg) | 8.62 ± 0.6 | 7.86 ± 0.6 | 6.80 ± 0.4 |
| DAP alone (10 mg) | 8.99 ± 0.4 | 7.81 ± 0.1 | 6.57 ± 0.9 |
| Fosfo alone (300 mg) | 5.95 ± 1.3 * | 3.53 ± 0.7 * | 3.65 ± 1.0 * |
| DAP (2 mg) + Fosfo (300 mg) | 6.84 ± 1.1 α | 2.71 ± 1.2 α | 2.61 ± 0.8 α |
| DAP (6 mg) + Fosfo (300 mg) | 7.44 ± 0.2 β | 3.70 ± 1.2 β | 3.05 ± 1.0 β |
| DAP (10 mg) + Fosfo (300 mg) | 1.98 ± 1.2 $ + | 1.07 ± 0.4 $ ++ | 0.87 ± 0.1 $ +++ |
*, P < 0.01 versus untreated control, DAP alone (2 to 10 mg); β, P < 0.05 versus untreated control, DAP alone (2 to 10mg); α, P < 0.01 versus untreated control, DAP alone (2 to 10 mg), Fosfo alone (300 mg), and DAP (2 mg) plus Fosfo (300 mg); $, P < 0.001 versus untreated control, DAP alone (2 to 10 mg), Fosfo alone (300 mg), and DAP (6 mg) plus Fosfo (300 mg); +, 3/5 animals sterile; ++, 4/5 animals sterile; +++, 5/5 animals sterile.
The DAP-R MRSA isolated from the cardiac vegetations of animals treated with DAP alone maintained their baseline in vitro DAP MICs. In addition, in animals receiving Fosfo alone, vegetation isolates evolved Fosfo-R subpopulations (MICs increasing from 8 mg/L to >32 mg/L). In contrast, vegetation isolates from animals treated with any of the DAP+Fosfo combination regimens maintained Fosfo susceptibility in vitro. Thus, although Fosfo alone was modestly active in reducing target organ MRSA bioburdens, this was at the expense of rapid emergence of Fosfo-R subpopulations. DAP resensitization was not seen in any treatment group.
LL-37 susceptibility.
DAP-R MRSA commonly exerts cross-resistance to variety of host defense peptides (HDPs), including mammalian neutrophils and platelets (12). To evaluate the impact of DAP with or without Fosfo (DAP+/−Fosfo) on HDP susceptibilities, LL-37 susceptibility profiles of our isogenic DAP-S/DAP-R strain pair were determined following overnight growth in these antibiotics at sublethal concentrations. As expected, the DAP-R CB185 strain exhibited higher baseline survival to LL-37 than the parental DAP-S strain (Fig. 4). Fosfo (with or without DAP) preconditioning of both strains rendered them more susceptible to killing by LL-37.
FIG 4.

Exposure to DAP, Fosfo, and a combination of DAP and Fosfo at 0.5× MIC to DAP-S (CB1483) and DAP-R (CB185) MRSA strains impacts HDP (i.e., LL-37) susceptibility.
Cell envelope phenotypes.
(i) Surface charge.
Sub-MIC exposures of both DAP-S and DAP-R cells to Fosfo significantly decreased net positive surface charge (P < 0.0001) versus that of untreated cells (Fig. 5). These data are consistent with enhancement in the cell envelope content of the negatively charged phospholipid (PL) species (cardiolipin [CL]) in Fosfo-exposed cells (see below).
FIG 5.
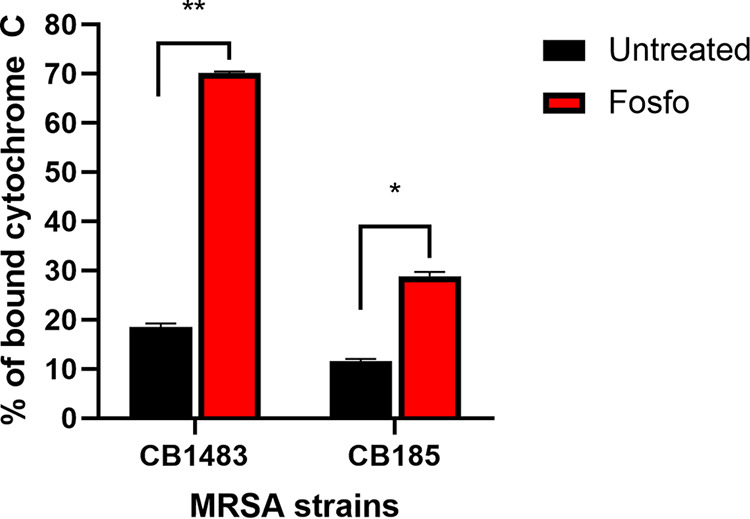
Surface charge of DAP-S CB1483/DAP-R CB185 S. aureus strains following exposure to 0.5× Fosfo MIC. *, P ≤ 0.05, and **, P ≤ 0.01, versus Fosfo-untreated strains. More residual cytochrome c present in the supernatant equates to a more positively charged surface.
(ii) DAP binding.
Overall, the degrees of BODIPY-DAP binding in the DAP-S/R strain pair were similar. With the addition of Fosfo (4 mg/L) to both strains, DAP binding to the cell envelope was enhanced in both strains (Fig. 6), resulting in an ∼3-log10 increase in total cell fluorescence in Fosfo-treated versus untreated cells (P < 0.001). Figure 6B displays confocal images with notable regions of focal BODIPY-DAP binding (versus more circumferential, comprehensive binding) in cells. This was particularly evident at the septal divisome in dividing cells.
FIG 6.

(A) Quantification of BODIPY-DAP binding; (B) BODIPY-DAP binding localization by confocal microscopy in the DAP-S/DAP-R strain pair with Fosfo treatment. *, P < 0.001 in Fosfo-treated versus untreated organisms. Yellow arrows indicate areas of high BODIPY-DAP binding.
(iii) Quantification and localization of anionic PLs (e.g., CL).
Following exposure to sub-MIC Fosfo (4 mg/L), the DAP-S/DAP-R strain pair exhibited significantly increased overall amounts of cell envelope CL (Fig. 6A). Compared to that of untreated strains, N-acrylamide orange (NAO) fluorescence was 2.8 times greater with Fosfo treatment in the DAP-S strain (P < 0.001) and 4.7 times greater in the DAP-R strain (P < 0.001). Further, using confocal microscopy, Fosfo-treated cells showed both overall increases in NAO fluorescence and apparent delocalization of CL into the cell envelope (versus untreated cells) (Fig. 7). For example, NAO binding was concentrated to one area of the cell in the untreated strains, while Fosfo treatment resulted in NAO distribution around the entire cell circumference while still retaining high intensity at these focal regions (Fig. 7B, green arrows).
FIG 7.
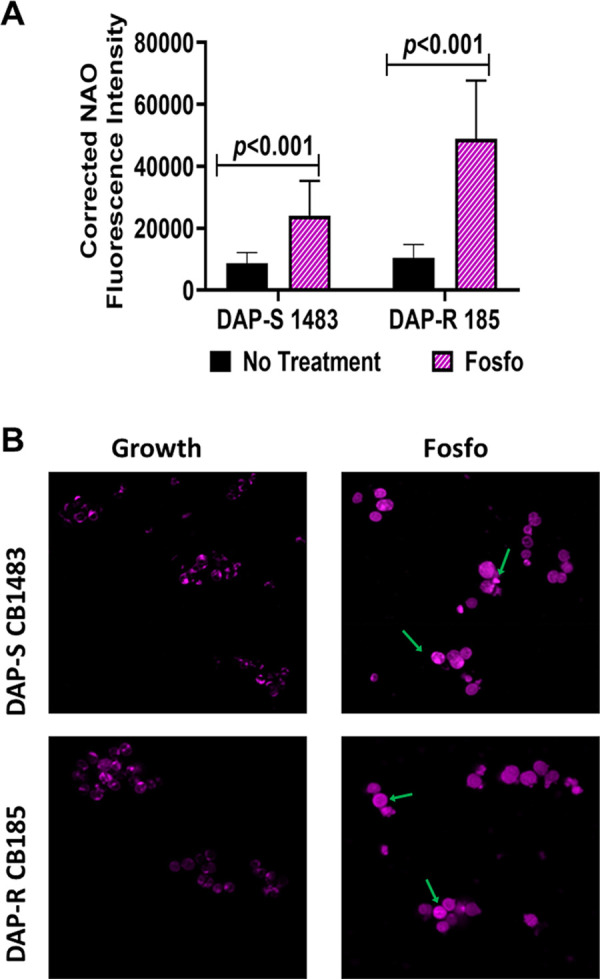
NAO fluorescence intensity (A) and NAO localization by microscopy (B) of DAP-S/DAP-R strain pair following subinhibitory Fosfo exposure. Green arrows indicate areas of high NAO localization.
(iv) CM order (fluidity/rigidity).
We have previously shown that for S. aureus, enterococci, and viridans group streptococci that develop DAP-R, notable changes in CM order occur as an “adaptive response” to DAP exposures (13–20). Interestingly, both the DAP-S and DAP-R strains demonstrated a significantly more fluid CM than that of untreated controls (P < 0.001) when exposed to sub-MIC Fosfo (0.5× MIC) (Fig. 8).
FIG 8.
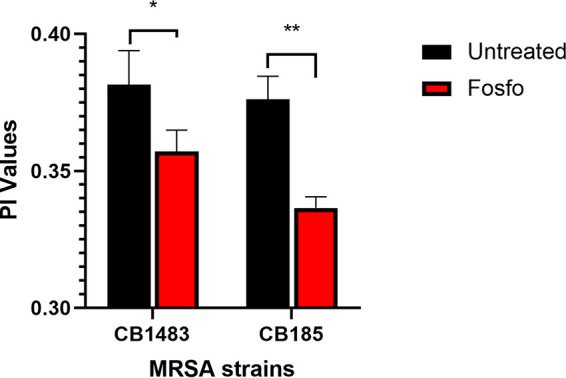
CM fluidity of DAP-S CB1483/DAP-R CB185 S. aureus strains following exposure to 0.5× Fosfo MIC. *, P ≤ 0.05, and **, P ≤ 0.001, versus Fosfo-untreated strains. Lower PI values indicate higher CM fluidity.
DISCUSSION
In the current study, we assessed the interrelatedness of bactericidal synergy, resistance prevention, and resensitization with DAP+Fosfo combinations, focusing mainly on the DAP-R phenotype. Several interesting themes emerged from this investigation.
First, DAP+Fosfo combinations substantially enhanced early in vitro killing of both DAP-S/DAP-R strains compared to that with either drug alone. Further, the combination appeared to prevent DAP MIC increases. In the DAP-S strains, the addition of Fosfo prevented the emergence of DAP-R. It also prevented further MIC increases in the DAP-R strain, although not resensitizing DAP-R subpopulations to a DAP-S phenotype in vitro. Each of these outcomes could be beneficial clinically, especially bactericidal synergy, against DAP-R MRSA, for which treatment options are quite limited. Moreover, combining DAP+Fosfo in treating DAP-S strains to prevent selection of DAP-R, and thus retain DAP activity, is a desirable clinical outcome.
Second, similar to the in vitro studies, the ex vivo SEV modeling also demonstrated increased bactericidal impacts of DAP + Fosfo against both the DAP-S and DAP-R strains. In addition, treatment of the DAP-S strain with this combination prevented emergence of DAP-R; further, the DAP-R strain treated with DAP+Fosfo was resensitized to DAP-S. The last outcome differed from the in vitro results described above. Moreover, within SEVs ex vivo, these combinations resensitized the DAP-R strain. We posit that the presence of a number of antibacterial host factors within SEVs (e.g., defense peptides, inflammatory cells, and platelets) may well amplify the collective impacts of either or both antibiotics in this combination.
Third, in parallel to the ex vivo parameters described above, the combination of subtherapeutic human-equivalent dosages of DAP and Fosfo was active in vivo at reducing MRSA counts in both kidneys and spleen (but not vegetations) in the IE model compared to those of untreated controls. Interestingly, a more human-mimicking dosage of DAP (10 mg/kg in rabbits = 6 mg/kg in humans [21]) in combination with Fosfo significantly reduced MRSA counts in all three target tissues, with organ sterilizations in the majority of animals. As opposed to the ex vivo outcomes described above, DAP+Fosfo did not resensitize DAP-R subpopulations within cardiac vegetations. This may relate to a number of factors, including (i) high initial inocula, (ii) growth phase and compositional differences between simulated ex vivo versus in vivo vegetations, (iii) DAP-Fosfo dose and pharmacokinetic/pharmacodynamic (PK-PD) differences in the two scenarios, (iv) impacts of blood flow and shear forces on microbial physiology, (v) distinct host defense factors in vivo, and (vi) differential bacterial gene expression profiles occurring ex vivo versus in vivo.
Fourth, to understand if enhanced susceptibility to host defense peptides induced by exposures to DAP+/−Fosfo could contribute to net in vivo efficacy of this drug combination, we studied the LL-37 susceptibility profiles of our isogenic DAP-S/DAP-R strain pair following DAP-Fosfo preconditioning. Of interest, both the DAP-S and DAP-R strains exhibited augmented susceptibility to LL-37 following such preconditioning (compared to DAP alone); this salutary impact was mainly driven by Fosfo preexposure. To our knowledge, this is the first study of the ability of Fosfo to synergize with LL-37 against MRSA.
Emergence of DAP-R in MRSA has been linked to a cadre of well-established cell envelope modifications (13–16, 22). Thus, prior studies demonstrated increased positive cell surface charge in DAP-R versus DAP-S MRSA (correlating with gain-in-function mutations in mprF and dysregulation of dltABCD expression) (13–16, 18), with repulsion of the positively charged DAP-calcium complex (18). Thus, we sought to analyze how Fosfo might influence these metrics. Both DAP-S/DAP-R MRSA strains exposed to Fosfo exhibited less positively charged surfaces, with an associated enhancement in DAP binding. In theory, these changes could explain, at least in part, enhanced DAP efficacy in the presence of Fosfo exposures.
The relationship between DAP activity and overall content of the major anionic cell envelope phospholipid species, CL and PG, has been well chronicled (13–16, 18). PG and CL are both essential for DAP’s initial interaction with target CMs (23). Moreover, the combined PG-CL “microdomains” are felt to serve as negatively charged CM “docking sites” for calcium-DAP and other cationic molecules for initial interaction with the CM (24). In addition, divisome CL localization is critical to DAP accumulation at this key site of its mechanism of action (18, 19). To further understand how Fosfo affects CM CL content and localization, the DAP-S/DAP-R strain pair was evaluated for quantitative and distribution perturbations in NAO binding. Fosfo significantly increased overall content of CL, as well as its accumulation at the septal divisome. This correlates with the observed relative decrease in positive surface charge. Furthermore, following Fosfo exposures, the DAP-S/DAP-R strain pair showed prominent mislocalization of CL away from the division septum. Such mislocalization of CL has been postulated to underpin DAP-R in enterococci (25). In counterbalance, CL-rich CMs become more susceptible to bending and stretching in terms of dividing, because of inadequate lateral interaction with other PLs. Thus, Fosfo-induced mislocalization of CL might actually improve DAP’s insertion and CM pore formation, increasing its bactericidal activity.
An optimum CM order exists for the initial bacterial surface interactions of many cationic peptides, including calcium-complexed DAP (13–16, 18, 19). We have previously shown that extremes of CM order (more rigid or more fluid versus respective DAP-S parental strains) occurred among most DAP-R strains derived in vitro, in vivo, or clinically (13–16). In the current strain pair, Fosfo induced significantly more fluid membranes versus those of untreated control cells. The precise mechanistic linkage(s) of enhanced CM fluidity elicited by Fosfo with amplification of DAP’s bactericidal activities, i.e., whether this impact relates to modifications of DAP binding, oligomerization, and/or CM insertion, remains to be defined.
We recognize several key limitations of our study. Only one DAP-S/DAP-R MRSA strain pair was studied, and therefore, strain-to-strain variability may exist beyond our study. Also, comprehensive analyses of the genetic basis for the enhancement effects of Fosfo on DAP activities in vitro, ex vivo, and in vivo were not carried out. In addition, future studies should employ more human-equivalent DAP dosing regimens, both ex vivo and in vivo, to encompass current treatment strategies (i.e., higher-dose DAP [26] and variable-dose Fosfo [26]). Furthermore, it would be informative to examine the ability of Fosfo (+/−DAP) preexposures to sensitize MRSA to a cadre of HDPs beyond LL-37. Lastly, we limited our cell envelope modification analytics to MRSA exposed to Fosfo alone. Follow-up studies need to also examine the impacts of the exposures to DAP alone and DAP+Fosfo on the same metrics. Many of these studies are currently in progress in our labs.
In conclusion, our studies have provided some key insights into a potential mechanistic basis for enhanced efficacies of DAP+Fosfo versus MRSA. This combination could be an alternative strategy to circumvent the emergence of resistance, as well as a method of enhancing the activity of each of the individual antibiotic agents. Moreover, these events could plausibly affect the capacity of DAP+Fosfo to impact bactericidal synergy, prevention of DAP-R, and/or resensitization of DAP-R subpopulations. These data underscore that signature cell envelope phenotypic modifications likely underpin, at least in part, the mechanistic bases of DAP-Fosfo combinations. These investigations provide justification for exploring the genotypic bases of these phenotypic correlates the effect of DAP+Fosfo. In addition, similar strategies might be helpful in establishing a novel platform for evaluating other combination regimens against MRSA and other Gram-positive pathogens (i.e., a mechanistic dissection of synergy versus prevention of resistance versus resensitization versus signature envelope adaptations).
MATERIALS AND METHODS
Bacterial strains.
We utilized a well-characterized clinical bloodstream isogenic MRSA DAP-S/DAP-R strain pair (CB1483/CB185) (11). CB1483 was isolated before DAP therapy, while the DAP-R strain CB185 was isolated after initiation of DAP (11). Genetic relatedness (isogeneity) of the studied DAP-S/DAP-R MRSA strain pair was confirmed by genotypic profiling (i.e., clonal complex, agr, spa, and SCCmec typing) (Table 1) (11). Sequencing of the mprF and yycG genes of DAP-R CB185 revealed a single-point mutation (L826F) in one of the classic mprF “hot spots” in the N-terminal synthase domain, but not in yycG (Table 1). It should be noted that mprF single-nucleotide polymorphisms (SNPs) are the most common mutations induced in DAP-R strains (11, 13–15).
MICs.
DAP analytical powder was obtained from Merck & Co., Inc. (Whitehouse Station, NJ). Fosfo clinical powder was commercially purchased (Sigma). DAP MIC testing was performed by CLSI-recommended broth microdilution, with 50 mg calcium/L and 12.5 mg magnesium/L added to Mueller-Hilton broth (MHB; Difco, Detroit, MI). DAP MICs were also determined by standard Etest (bioMérieux) on Mueller-Hinton agar (MHA) plates (Difco). The generally agreed-upon breakpoint distinguishing DAP-S and DAP-R MRSA is ≥2 mg/L (39). Susceptibility to Fosfo was determined by broth microdilution MICs (range, 0.25 to 256 mg/L) using the above-mentioned cation-adjusted MHB supplemented with 25 μg glucose-6-phosphate/ml (Sigma-Aldrich Chemical Co., St. Louis, MO). Initial bacterial densities of 1 × 105 CFU/well (standardized from a McFarland unit of 0.5) were used, and incubations were done at 37°C. The lowest antibiotic concentration that prevented visible growth represented the MIC.
There is no formal CLSI breakpoint available to denote Fosfo resistance; however, the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines consider S. aureus strains having Fosfo MICs of <32 mg/L “susceptible” (27). S. aureus ATCC 29213 was used as a reference strain for both DAP and Fosfo MIC quality controls.
In vitro time-kill curves.
The MRSA strain pair was cultured aerobically overnight in brain heart infusion (BHI) broth (with 50 mg/L CaCl2 supplementation for DAP and 25 μg glucose-6-phosphate/ml supplementation for Fosfo) (10, 27). For bactericidal and synergistic assessments, time-kill assays were performed, utilizing MICs of DAP+/−Fosfo as defined above (8, 28). A standard initial inoculum of ∼1 × 106 CFU/mL was used to better approximate in vivo bacterial densities in IE. The extent of killing from 0 to 8 h was determined by quantitative culture of surviving CFU (log10 CFU per milliliter) on MHA plates incubated at 37°C for 16 to 20 h (10, 28). Extensive pilot studies revealed that the greatest bactericidal impacts of DAP-Fosfo combinations occurred over the first 8 h. At least three experimental runs were performed on separate days.
This method determined (i) synergy (≥2-log10 CFU/mL reduction for the combination versus the most active single agent), (ii) additive effect (<2- to ≥1-unit decrease in log10 CFU per milliliter), (iii) indifference (<1-unit decrease to ≥1-unit increase in log10 CFU per milliliter), or (iv) antagonism (≥2-log10 CFU/mL increase) (28). A “bactericidal effect” was defined as at least a 99.9% (≥3-log10) CFU reduction from the initial bacterial inoculum, while a “bacteriostatic effect” was defined as a less than 99.9% (<3-log10) CFU reduction (28).
Serial in vitro passage experiments.
Serial passage experiments with CB1483 and CB185 in DAP (with and without Fosfo) were performed daily for 10 total days to examine for both emergence of DAP-R and resensitization to a DAP-S phenotype.
Initial DAP concentrations of 0.5× MIC were used (0.125 mg/L for CB1483 and 2 mg/L for CB185), and then concentrations were increased daily 2-fold. If growth did not occur with this passage concentration, the previous concentration allowing growth was continued and passaging was repeated again. DAP concentrations did not exceed 4 mg/L, to minimize DAP MIC escalations beyond the range commonly reported for clinical DAP-R S. aureus (2 to 4 mg/L) (10, 11, 15). The initial concentration of Fosfo used for passage experiments was 4 mg/L (0.5× MIC for both strains) and was held constant with each passage round (10, 28).
All passage reaction mixtures were incubated at 37°C, with a flask-to-medium volume ratio of 7:1, using agitation at 225 rpm and passaged daily by inoculating 20 μL into 2 mL fresh medium. After each day’s passage, samples were obtained and stored at −80°C for further in vitro assays. The proportion of MRSA strains evolving a DAP-R phenotype was determined by parallel plating onto MHA, with or without DAP at 2 and 4 mg/L. Any colonies growing on antibiotic screening plates were also formally evaluated for DAP MICs by both broth microdilution and Etest (11, 13, 15, 16). The MIC stability of DAP-R strains that emerged during the 10-day passage period was determined by passaging the resulting resistant strains for 5 days in antibiotic-free media (17, 28), with repeat of DAP MIC determinations as described above (11, 13, 15, 16).
Ex vivo-simulated IE vegetation model.
The same in vitro metrics as defined above were evaluated in parallel using an ex vivo model of simulated endocardial vegetations. This model features a fluid phase (i.e., continuously replenished fresh media), as well as a tissue microenvironment phase comprised of infected artificial vegetations (simulated endocardial vegetations [SEVs]). The human component-derived SEVs (consisting primarily of human fibrinogen/fibronectin/fibrin/platelets) were prepared as described elsewhere (29–31). This model mirrors experimental and human IE in terms of vegetation bacterial burden, infection progression, and antibiotic PK-PD. In this ex vivo model, we employed computer-designed and programmed PK-PD drug administrations of DAP and Fosfo (alone and in combination) which simulated the following human-equivalent dosage regimens: DAP at 6 mg/kg every 24 h and/or Fosfo at 4 g every 8 h. The efficacy of DAP+/−Fosfo in synergy, preventing evolution of DAP-R and/or resensitizing the DAP-R strain to a DAP-S phenotype, was assessed over a 96-h ex vivo exposure period, allowing adequate time for DAP-R to emerge (30, 31).
Pretherapy, and at daily time points, two SEVs each were removed from the chambers, weighed, homogenized, diluted in cold saline, plated onto calcium-salt-supplemented MHA plates, and then incubated at 35°C for 24 h. Surviving colonies were quantified, and those in each DAP-Fosfo treatment exposure group were calculated as mean log10 CFU per gram (±standard deviation [SD]). Models were performed in duplicate for each antibiotic regimen (monotherapy versus combination therapy), thereby providing four SEVs per time point per drug exposure for PD activity assessments. Synergy, additivity, indifference, or antagonism in this model was determined using the same definitions as outlined above for the combination versus the most active single agent at the end of therapy. To evaluate DAP-R emergence or resensitization, homogenized SEVs were parallel plated and cultured on MHA containing 50 mg CaCl2/L and 4 mg DAP/L for the DAP-S CB1483 strain and 4 and 8 mg/L for the DAP-R CB185 strain. Surviving colonies on antibiotic-free agar and DAP-containing agar were evaluated for DAP MIC by broth microdilution and Etest as described above for confirmation of MIC.
In vivo IE model.
In-parallel in vivo experiments are critical to confirm translatability of the observed activity from the in vitro and ex vivo studies described above. In addition, we focused on validating the potential capacity of DAP-Fosfo combinations to synergistically kill, as well as to resensitize DAP-R subpopulations in various target tissues to a DAP-S phenotype. For this purpose, the well-characterized rabbit aortic valve IE model was employed as described before (21, 32). Briefly, in aortic catheterized rabbits, an intravenous (i.v.) inoculation of the DAP-R CB185 MRSA strain was carried out at a 95% infective dose (ID95) inoculum established in prior IE studies (∼5 × 105 CFU i.v.) (33). This inoculum achieves reliable colonization of sterile cardiac vegetations, as well as consistent hematogenous seeding to other key target organs (kidneys and spleen).
Our IE studies utilized three distinct DAP dose regimens (2, 6, or 10 mg/kg i.v., given once daily for 4 days) to encompass a range of DAP exposures in rabbits, from sublethal (2 and 6 mg/kg) to lethal (10 mg/kg). This dose range was designed to allow an adequate “window” to disclose potential DAP-Fosfo synergy in vivo. For Fosfo, dose regimens of 300 mg/kg were employed; these regimens generate sublethal Fosfo human-like PK-PD in animals (34).
Animals were sacrificed at least 12 h after the last antibiotic doses to minimize antibiotic carryover (based on the respective half-lives of these agents in rabbits [data not shown]). Cardiac vegetations, kidneys, and spleens were sterilely excised, weighed, quantitatively cultured, and parallel plated onto antibiotic-free agar or agar supplemented with DAP as described above or Fosfo (at 32 mg/L in MHA plus 25 mg glucose-6-phosphate/L). Colonies from organ cultures growing on DAP-containing and Fosfo-containing plates were subjected to MIC determinations by broth microdilution. These studies defined the potential impact of DAP-Fosfo on emergence of DAP-R or resensitization of DAP-R MRSA to DAP-S.
LL-37 susceptibility.
LL-37 is a linear cathelicidin host defense peptide found in mammalian neutrophils and epithelium (12). We performed a 2-h LL-37 bactericidal assay in minimal liquid nutrient medium, potassium phosphate buffer (PPB; pH 7.4), and 0.25% brain heart infusion (BD, Sparks, MD) as detailed before (35). Both strains were preconditioned by overnight growth in DAP+/−Fosfo (each agent at 0.5× MIC). We used LL-37 at either 10 or 20 mg/L for the 2-h killing assay against the DAP-S/DAP-R MRSA strains. These LL-37 concentrations reflected those employed in previous investigations of HDP-S. aureus interactions (36). LL-37 was purchased from Peptide International, Louisville, KY. A final inoculum of 103 CFU/mL was incubated at 37°C in the presence or absence of LL-37 for 2 h, after which samples were collected for quantitative culture to assess the degree of killing. Final data were expressed as mean percent surviving CFU per milliliter (±SD). A minimum of three experiments were carried out on different days.
Phenotypic profiling.
Potential perturbations of specific cell envelope phenotypes previously associated with DAP-R were assessed for impacts of DAP+/−Fosfo in the strain set (13–15, 18, 19). For all assays described below, strains were exposed to sub-MIC levels of DAP and/or Fosfo. Outcome data for the isolates exposed to DAP+/−Fosfo were compared to data for the respective original strain grown in media alone. A minimum of three independent experiments were carried out for all phenotypic assays, using sublethal concentrations (sub-MICs) of DAP and/or Fosfo.
(i) Surface charge.
The relative positive cell surface charge was measured with the cytochrome c binding assay as described before (13–15, 18, 19). In this assay, more residual cytochrome c present in the supernatant equates to a more positively charged surface (13–15, 18, 19). Data are presented as means (±SDs) of bound cytochrome c.
(ii) DAP binding.
Binding and localization of DAP to MRSA whole cells were assessed by confocal fluorescence imaging using a BODIPY-DAP binding assay (19, 37). Cells were preconditioned with 4 mg/L Fosfo (0.5× MIC for both CB1483 and CB185) overnight and then exposed to 16 mg/L BODIPY-DAP (19, 37). As the reported potency of BODIPY-tagged DAP is roughly 4-fold lower than that of unbound DAP, the exposure concentrations were roughly 4-fold higher than the average free human serum level of 3.6 mg/L (19, 37).
(iii) Quantification of anionic PLs.
Cardiolipin (CL) is the principal anionic phospholipid (PL) in the cell membrane (CM) of MRSA; we used an anionic phospholipid-specific dye (N-acrylamide orange [NAO])-based spectrofluorometric assay as a quantitative readout for CL content. As negative controls for this NAO binding assay, we employed CL synthase knockout mutants, N315 Δcls1 and N315 Δcls2 (18). CB1483 and CB185 were first exposed to 4 mg/L Fosfo (0.5× MIC) overnight. Then, 1.0 × 107 CFU/mL of each MRSA strain was exposed to 20 μM NAO and incubated at 4°C for 20 min. NAO fluorescence intensity was measured using spectrofluorometry (excitation wavelength = 525 nm; emission wavelength = 640 nm).
(iv) Anionic PL localization.
Localization of anionic PLs (e.g., CL-rich domains) was visualized using NAO staining as described before (18). Fluorescence imaging was performed with stimulated emission depletion microscopy (STED) (18). Cells were grown to exponential phase at 37°C in a shaking incubator in LB medium overnight in the presence or absence of 4 mg/L Fosfo. NAO was then added at 20 μM for 1 h at room temperature. Cells were washed and resuspended in phosphate-buffered saline (PBS). The cells were concentrated 20-fold at the last step, and 3 μL was placed on a glass slide. Slides were set with ProLong diamond antifade mountant and a no. 1.5 glass coverslip. Images were collected using a Leica SP8 3X STED superresolution confocal microscope using the green fluorescent protein (GFP) standard filter set (495-nm excitation and 510- to 579-nm emission, with 592-nm depletion) to visualize NAO. The previously described knockout controls validated NAO binding specificity (see Fig. S1 in the supplemental material) (18, 38). The relative NAO content and distribution were compared between the Fosfo-treated vs untreated cells of the DAP-S/DAP-R MRSA pair.
(v) CM fluidity.
CM fluidity was assessed by the orientation of the fluorescent probe 1,6-diphenyl-hexa-1,3,5-triene (DPH) within the CM by polarizing spectrofluorometry (excitation and emission wavelengths of 360 and 426 nm, respectively) as described before, to provide a quantitative measure of CM order (13–15, 18, 19). Polarization index (PI) values were generated; the lower the PI values, the higher the CM fluidity.
Statistical analyses.
Chi-square analyses were used for proportional data between treatment groups (e.g., tissue sterilization rates); Student’s t test was used for continuous data (e.g., bacterial densities in SEVs and within in vivo target organs, presented as mean log10 CFU per gram of tissue ± SD), as well as for all phenotypic assays. P values of ≤0.05 were considered significant.
ACKNOWLEDGMENTS
This research was supported by grants from NIH/NIAID: R01-AI132627 (to W.E.R.) and R01-AI130056 (to A.S.B.). N.N.M. was supported by The Lundquist Institute at Harbor-UCLA intramural research grant 532345-01-00.
We thank Sabrina Farah at The Lundquist Institute for excellent technical assistance and the University of Wisconsin Madison Optical Imaging Core for access and training on Leica SP8 3X STED superresolution confocal microscope.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Roch M, Gagetti P, Davis J, Ceriana P, Errecalde L, Corso A, Rosato AE. 2017. Daptomycin resistance in clinical MRSA strains is associated with a high biological fitness cost. Front Microbiol 8:2303. 10.3389/fmicb.2017.02303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rose WE, Fallon M, Moran JJ, Vanderloo JP. 2012. Vancomycin tolerance in methicillin-resistant Staphylococcus aureus: influence of vancomycin, daptomycin, and telavancin on differential resistance gene expression. Antimicrob Agents Chemother 56:4422–4427. 10.1128/AAC.00676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marty FM, Yeh WW, Wennersten CB, Venkataraman L, Albano E, Alyea EP, Gold HS, Baden LR, Pillai SK. 2006. Emergence of a clinical daptomycin-resistant Staphylococcus aureus isolate during treatment of methicillin-resistant Staphylococcus aureus bacteremia and osteomyelitis. J Clin Microbiol 44:595–597. 10.1128/JCM.44.2.595-597.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakoulas G, Rose W, Rybak MJ, Pillai S, Alder J, Moellering RC, Jr, Eliopoulos GM. 2008. Evaluation of endocarditis caused by methicillin-susceptible Staphylococcus aureus developing nonsusceptibility to daptomycin. J Clin Microbiol 46:220–224. 10.1128/JCM.00660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pujol M, Miró J-M, Shaw E, Aguado J-M, San-Juan R, Puig-Asensio M, Pigrau C, Calbo E, Montejo M, Rodriguez-Álvarez R, Garcia-Pais M-J, Pintado V, Escudero-Sánchez R, Lopez-Contreras J, Morata L, Montero M, Andrés M, Pasquau J, Arenas M-D-M, Padilla B, Murillas J, Jover-Sáenz A, López-Cortes L-E, García-Pardo G, Gasch O, Videla S, Hereu P, Tebé C, Pallarès N, Sanllorente M, Domínguez M-Á, Càmara J, Ferrer A, Padullés A, Cuervo G, Carratalà J, MRSA Bacteremia (BACSARM) Trial Investigators. 2021. Daptomycin plus fosfomycin versus daptomycin alone for methicillin-resistant Staphylococcus aureus bacteremia and endocarditis: a randomized clinical trial. Clin Infect Dis 72:1517–1525. 10.1093/cid/ciaa1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277:139–158. 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oku Y, Kurokawa K, Ichihashi N, Sekimizu K. 2004. Characterization of the Staphylococcus aureus mprF gene, involved in lysinylation of phosphatidylglycerol. Microbiology (Reading) 150:45–51. 10.1099/mic.0.26706-0. [DOI] [PubMed] [Google Scholar]
- 8.Geriak M, Haddad F, Rizvi K, Rose W, Kullar R, LaPlante K, Yu M, Vasina L, Ouellette K, Zervos M, Nizet V, Sakoulas G. 2019. Clinical data on daptomycin plus ceftaroline versus standard of care monotherapy in the treatment of methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob Agents Chemother 63:e02483-18. 10.1128/AAC.02483-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCreary EK, Kullar R, Geriak M, Zasowski EJ, Rizvi K, Schulz LT, Ouellette K, Vasina L, Haddad F, Rybak MJ, Zervos MJ, Sakoulas G, Rose WE. 2020. Multicenter cohort of patients with methicillin-resistant Staphylococcus aureus bacteremia receiving daptomycin plus ceftaroline compared with other MRSA treatments. Open Forum Infect Dis 7:ofz538. 10.1093/ofid/ofz538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reed JM, Gardner SG, Mishra NN, Bayer AS, Somerville GA. 2019. Metabolic interventions for the prevention and treatment of daptomycin non-susceptibility in Staphylococcus aureus. J Antimicrob Chemother 74:2274–2283. 10.1093/jac/dkz194. [DOI] [PubMed] [Google Scholar]
- 11.Mishra NN, McKinnell J, Yeaman MR, Rubio A, Nast CC, Chen L, Kreiswirth BN, Bayer AS. 2011. In vitro cross-resistance to daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 55:4012–4018. 10.1128/AAC.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanetti M. 2004. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 75:39–48. 10.1189/jlb.0403147. [DOI] [PubMed] [Google Scholar]
- 13.Mishra NN, Rubio A, Nast CC, Bayer AS. 2012. Differential adaptations of methicillin-resistant Staphylococcus aureus to serial in vitro passage in daptomycin: evolution of daptomycin resistance and role of membrane carotenoid content and fluidity. Int J Microbiol: 2012:1–6. 10.1155/2012/683450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani S, Cafiso V, Bertuccio T, Yeaman MR, Nast CC, Yang SJ. 2014. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One 9:e107426. 10.1371/journal.pone.0107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones T, Yeaman MR, Sakoulas G, Yang SJ, Proctor RA, Sahl HG, Schrenzel J, Xiong YQ, Bayer AS. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemother 52:269–278. 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra NN, Yang SJ, Sawa A, Rubio A, Nast CC, Yeaman MR, Bayer AS. 2009. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 53:2312–2318. 10.1128/AAC.01682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra NN, Tran TT, Seepersaud R, Garcia-de-la-Maria C, Faull K, Yoon A, Proctor R, Miro JM, Rybak MJ, Bayer AS, Arias CA, Sullam PM. 2017. Perturbations of phosphatidate cytidylyltransferase (CdsA) mediate daptomycin resistance in Streptococcus mitis/oralis by a novel mechanism. Antimicrob Agents Chemother 61:e02435-16. 10.1128/AAC.02435-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lew C, Mishra NN, Bayer AS, Rose WE. 2021. β-Lactam-induced cell envelope adaptations, not solely enhanced daptomycin binding, underlie daptomycin-beta-lactam synergy in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 65:e00356-21. 10.1128/AAC.00356-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra NN, Bayer AS, Baines SL, Hayes AS, Howden BP, Lapitan CK, Lew C, Rose WE. 2021. Cell membrane adaptations mediate beta-lactam-induced resensitization of daptomycin-resistant (DAP-R) Staphylococcus aureus in vitro. Microorganisms 9:1028. 10.3390/microorganisms9051028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishra NN, Bayer AS, Tran TT, Shamoo Y, Mileykovskaya E, Dowhan W, Guan Z, Arias CA. 2012. Daptomycin resistance in enterococci is associated with distinct alterations of cell membrane phospholipid content. PLoS One 7:e43958. 10.1371/journal.pone.0043958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kennedy S, Chambers HF. 1989. Daptomycin (LY146032) for prevention and treatment of experimental aortic valve endocarditis in rabbits. Antimicrob Agents Chemother 33:1522–1525. 10.1128/AAC.33.9.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muraih JK, Pearson A, Silverman J, Palmer M. 2011. Oligomerization of daptomycin on membranes. Biochim Biophys Acta 1808:1154–1160. 10.1016/j.bbamem.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Zhang T, Taylor SD, Palmer M, Duhamel J. 2016. Membrane binding and oligomerization of the lipopeptide A54145 studied by pyrene fluorescence. Biophys J 111:1267–1277. 10.1016/j.bpj.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilelee E, Pokorny A, Yeaman MR, Bayer AS. 2010. Lysyl-phosphatidylglycerol attenuates membrane perturbation rather than surface association of the cationic antimicrobial peptide 6W-RP-1 in a model membrane system: implications for daptomycin resistance. Antimicrob Agents Chemother 54:4476–4479. 10.1128/AAC.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran TT, Panesso D, Mishra NN, Mileykovskaya E, Guan Z, Munita JM, Reyes J, Diaz L, Weinstock GM, Murray BE, Shamoo Y, Dowhan W, Bayer AS, Arias CA. 2013. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. mBio 4:e00281-13. 10.1128/mBio.00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miro JM, Entenza JM, Del Rio A, Velasco M, Castaneda X, Garcia de la Maria C, Giddey M, Armero Y, Pericas JM, Cervera C, Mestres CA, Almela M, Falces C, Marco F, Moreillon P, Moreno A, Hospital Clinic Experimental Endocarditis Study Group. 2012. High-dose daptomycin plus fosfomycin is safe and effective in treating methicillin-susceptible and methicillin-resistant Staphylococcus aureus endocarditis. Antimicrob Agents Chemother 56:4511–4515. 10.1128/AAC.06449-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu CL, Liu CY, Huang YT, Liao CH, Teng LJ, Turnidge JD, Hsueh PR. 2011. Antimicrobial susceptibilities of commonly encountered bacterial isolates to fosfomycin determined by agar dilution and disk diffusion methods. Antimicrob Agents Chemother 55:4295–4301. 10.1128/AAC.00349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zapata B, Alvarez DN, Farah S, Garcia-de-la-Maria C, Miro JM, Sakoulas G, Bayer AS, Mishra NN. 2018. Prevention of high-level daptomycin-resistance emergence in vitro in Streptococcus mitis-oralis by using combination antimicrobial strategies. Curr Microbiol 75:1062–1067. 10.1007/s00284-018-1491-3. [DOI] [PubMed] [Google Scholar]
- 29.Werth BJ, Barber KE, Ireland CE, Rybak MJ. 2014. Evaluation of ceftaroline, vancomycin, daptomycin, or ceftaroline plus daptomycin against daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus in an in vitro pharmacokinetic/pharmacodynamic model of simulated endocardial vegetations. Antimicrob Agents Chemother 58:3177–3181. 10.1128/AAC.00088-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose WE, Leonard SN, Rybak MJ. 2008. Evaluation of daptomycin pharmacodynamics and resistance at various dosage regimens against Staphylococcus aureus isolates with reduced susceptibilities to daptomycin in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 52:3061–3067. 10.1128/AAC.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rose WE, Leonard SN, Sakoulas G, Kaatz GW, Zervos MJ, Sheth A, Carpenter CF, Rybak MJ. 2008. daptomycin activity against Staphylococcus aureus following vancomycin exposure in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 52:831–836. 10.1128/AAC.00869-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chambers HF, Basuino L, Diep BA, Steenbergen J, Zhang S, Tattevin P, Alder J. 2009. Relationship between susceptibility to daptomycin in vitro and activity in vivo in a rabbit model of aortic valve endocarditis. Antimicrob Agents Chemother 53:1463–1467. 10.1128/AAC.01307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hady WA, Bayer AS, Xiong YQ. 2012. Experimental endocarditis model of methicillin resistant Staphylococcus aureus (MRSA) in rat. J Vis Exp 2012(64):e3863. 10.3791/3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ortiz Zacarias NV, Dijkmans AC, Burggraaf J, Mouton JW, Wilms EB, van Nieuwkoop C, Touw DJ, Kamerling IMC, Stevens J. 2018. Fosfomycin as a potential therapy for the treatment of systemic infections: a population pharmacokinetic model to simulate multiple dosing regimens. Pharmacol Res Perspect 6:e00378. 10.1002/prp2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakoulas G, Bayer AS, Pogliano J, Tsuji BT, Yang SJ, Mishra NN, Nizet V, Yeaman MR, Moise PA. 2012. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 56:838–844. 10.1128/AAC.05551-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, Dhand A, Pogliano J, Yeaman MR, Hensler ME, Bayer AS, Nizet V. 2014. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med (Berl) 92:139–149. 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenson RE, Baines SL, Howden BP, Mishra NN, Farah S, Lew C, Berti AD, Shukla SK, Bayer AS, Rose WE. 2020. Prolonged exposure to β-lactam antibiotics reestablishes susceptibility of daptomycin-nonsusceptible Staphylococcus aureus to daptomycin. Antimicrob Agents Chemother 64:e00890-20. 10.1128/AAC.00890-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai M, Ohniwa RL, Kato Y, Takeshita SL, Ohta T, Saito S, Hayashi H, Morikawa K. 2011. Staphylococcus aureus requires cardiolipin for survival under conditions of high salinity. BMC Microbiol 11:13. 10.1186/1471-2180-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clinical and Laboratory Standards Institute. 2020. Performance standards for antimicrobial susceptibility testing. Approved standard M100-ED20. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Download AAC.01649-21-s0001.pdf, PDF file, 0.2 MB (176.6KB, pdf)