

Abstract
Obesity and metabolic diseases, such as insulin resistance and type 2 diabetes (T2D), are associated with metastatic breast cancer in post-menopausal women. Here, we investigated the critical cellular and molecular factors behind this link. We found that primary human adipocytes shed extracellular vesicles—specifically exosomes—that induced the expression of genes associated with epithelial-to-mesenchymal transition (EMT) and cancer stem-like cell (CSC) traits in cocultured breast cancer cell lines. Transcription of these genes was further increased in cells exposed to exosomes shed from T2D patient-derived adipocytes or insulin-resistant adipocytes and required the epigenetic reader proteins BRD2 and BRD4 in recipient cells. The thrombospondin family protein TSP5, which is associated with cancer, was more abundant in exosomes from T2D or insulin-resistant adipocytes and partially contributed to EMT in recipient cells. Bioinformatic analysis of breast cancer patient tissue showed that greater co-expression of COMP (which encodes TSP5) and BRD2 or BRD3 correlated with poorer prognosis, specifically decreased distant metastasis-free survival. Our findings reveal a mechanism of exosome-mediated crosstalk between metabolically abnormal adipocytes and breast cancer cells that may promote tumor aggressiveness in T2D patients.
INTRODUCTION
Obesity, insulin resistance and Type 2 diabetes (T2D) are risk factors for breast cancer in post-menopausal women (1). The metabolic and inflammatory complications of obesity are implicated in carcinogenesis and progression of estrogen receptor (ER)-positive breast cancer (2). Population studies also establish that obesity-driven T2D associates with incidence (3), progression and mortality (4) of ER-negative breast cancer. However, the cellular and molecular pathways that mediate breast cancer incidence, progression and metastasis are still not fully delineated. In sporadic breast cancer, well known genes that control proliferation, cell cycle, signal transduction, genome stability and other pathways (genes such as MYC, CCND1, ERBB2, TP53 and CDH1) accumulate mutations and contribute in a multistep fashion to expansion of the malignant clone, immune evasion, cell survival, tissue invasion and metastasis. The tumor microenvironment (TME) plays a critical role to promote cancer progression; however, the adipocytes, endothelial cells, immune infiltrates and other somatic cells of the breast typically do not harbor any mutations. Thus, DNA mutational databases are insufficient to understand most TME mechanisms. Co-morbid immunological or metabolic diseases alter the function of non-mutated cells systemically and in the breast TME, which creates opportunities to explore why tumors may progress in one person but not another, despite a similar mutational landscape in the malignant clones. Here, we focused on differences in the TME of obesity that might reveal novel mechanisms that promote breast cancer progression.
Adipocytes are by mass the preponderant non-malignant cell type in the TME of breast cancer. Yet, compared to immune infiltrates, adipocytes are disproportionately understudied as modifiers of cancer progression. Adipocytes function as an active endocrine tissue, releasing adipokines [such as interleukin-6, tumor necrosis factor (TNF)-α, leptin and adiponectin (5)] that play critical roles in tumor cell proliferation, as well as matrix metalloproteinases (MMPs) (6) that are important for tumor invasiveness. Adipocytes also release lipid that nearby breast tumor cells adapt for fuel by fatty acid oxidation, becoming more aggressive upon this metabolic reprogramming (7).
As the obesity epidemic continues to deepen worldwide, understanding the nature and function of adipocyte intercellular communication is increasing in importance. Extracellular vesicles and a subtype thereof called exosomes have gained attention as facilitators of adipocyte crosstalk with nearby tissues, potentially including malignant or pre-malignant breast epithelial cells (2,7). Adipocyte dysfunction is a long-appreciated feature of obesity-associated metabolic diseases, including insulin resistance, glucose intolerance, and T2D. Here, we investigated whether the altered secretome of metabolically dysfunctional adipocytes, including changes to the adipocyte exosome profile, may promote breast cancer development and progression. Our investigation focused on novel exosome crosstalk between metabolically abnormal adipocytes and breast cancer cells.
RESULTS
Adipocyte-tumor cell co-culture as a microenvironment model
To investigate the role of adipocytes in breast cancer progression, we first used a transwell system to co-culture the human ER-positive cell line MCF7 with human adipocytes that had been differentiated from primary pre-adipocytes surgically obtained from cancer-free, female patients with or without T2D undergoing bariatric surgery. We observed that the expression of key genes that are important for epithelial-to-mesenchymal transition (EMT), a transcriptional signature associated with tumor progression (8; including SNAI1, SNAI2, VIM, CDH2 and TWIST1; and MMPs including MMP3 and MMP9) were increased upon co-culture (Fig. 1A and data file S1). Fold-changes in EMT gene expression were greater if the adipocytes had first been rendered insulin resistant (IR) by overnight exposure to low-dose TNF-α (9). Ingenuity Pathway Analysis (IPA) of differentially expressed genes (Fig. 1B and data file S2) indicated that IR-converted adipocytes (Fig. 1C) more strongly induced EMT pathways in co-cultured MCF7 cells than did insulin-sensitive (IS) adipocytes, as compared to MCF7 controls alone (without co-culture). To explore how crosstalk within the TME induces such transcriptional signatures and complex cellular phenotypes, we focused on the induction of EMT genes. This approach enabled us to explore how novel crosstalk in the TME induces transcriptional signatures and more complex cellular phenotypes than just growth, such as morphology, migration and stemness.
Fig. 1. Human adipocytes induce transcription of EMT genes in a co-cultured ER+ human breast cancer cell model.
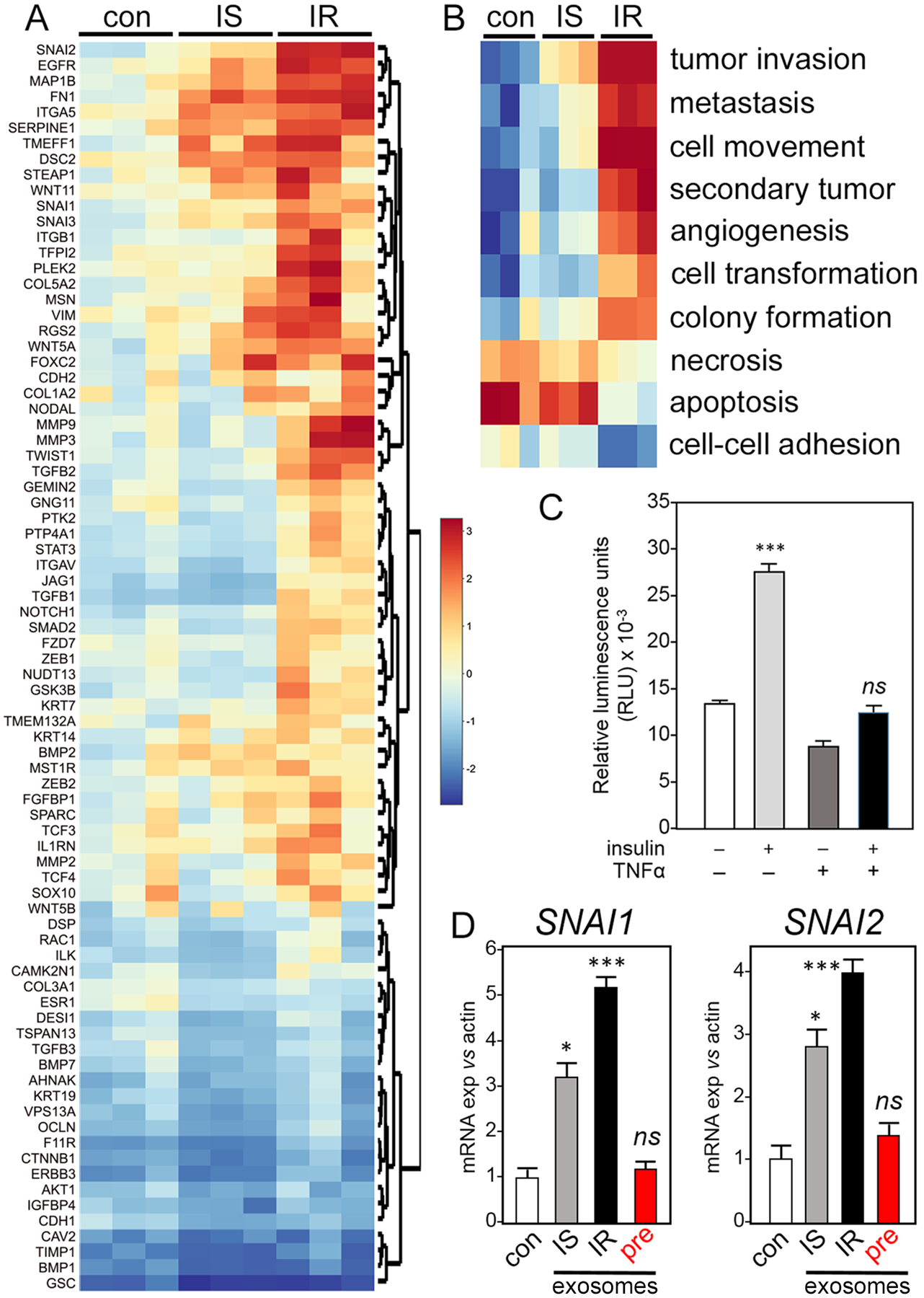
(A and B) MCF7 cells were co-cultured for 5 days with insulin-sensitive (IS) adipocytes that were differentiated from human primary pre-adipocytes, or ex vivo-induced insulin-resistant (IR) adipocytes from the same source, and compared to control MCF7 alone (con). Expression of selected EMT genes was analyzed by commercial PCR array (A) and Ingenuity pathway analysis (IPA; B). Data are from 3 independent experiments. (C) Fluorimetric glucose uptake assay in primary adipocytes after 24 hours treatment with recombinant human 250 pM TNF-α,10 nM insulin, both, or neither to assess insulin sensitivity status. Data are mean ± SEM from 3 independent experiments. (D) Expression of SNAI1 and SNAI2 in MCF7 cells cultured with exosomes purified from insulin-sensitive (IS) or insulin-resistant (IR) adipocyte–conditioned media or with exosomes purified from conditioned media of matched, undifferentiated pre-adipocyte control cells (pre) compared to MCF7 cells alone (con). Fold-change in expression of selected mRNAs encoding Snail (SNAI1) and Slug (SNAI2) was measured by RT-PCR relative to RNA encoding β-actin (ACTB). Data are mean ± SEM from 3 independent experiments; * P <0.05, *** P <0.005, and ns not significant by unpaired two-tailed t-test (C and D).
We suspected that adipocyte-origin exosomes; 10 were conveying signals to tumor cells. TME exosomes (11) have been implicated in EMT (12), supporting a rationale to purify exosomes from IS or IR adipocyte conditioned media, challenge MCF7 cultures and test function on a target set of EMT genes (Fig. 1D, fig. S1A). Consistent with EMT, exosomes attenuated the expression of genes that encode characteristic epithelial markers, like E-cadherin (CDH1) (fig. S1A). Exosomes from undifferentiated, pre-adipocyte controls did not induce EMT genes (Fig. 1D). Several gene expression signatures associated with other important pathways, such as cell cycle or cytokine signaling, were relatively unperturbed in tumor cells by co-culture with adipocytes (fig. S1B and data file S7). Exosome size distributions were similar between exosomes of IS and IR adipocyte origin (fig. S1C). It has been suggested that, in obesity, breast adipocytes promote breast cancer progression through metabolic and inflammatory mediators (13), but mechanistic detail has been lacking. Here, we propose mechanisms that depend on adipocyte-origin exosomes. Furthermore, these exosomes convey more dangerous signals if the adipocytes are insulin resistant or obtained from patients with obesity-associated metabolic complications. These insights are consistent with data (13) that the contributions of obesity and metabolic syndrome to risk of breast cancer progression are different and resolvable.
Adipocyte exosomes encode metabolic status
In post-menopausal women with obesity, adipokines and adipocyte-induced estrogen production (14), pro-inflammatory, CD68+ adipose tissue macrophages in “crown-like structures” (15), and adipocyte-released free fatty acids (7) have each been implicated in ER+ breast tumor progression. The association of obesity-driven T2D with elevated risk for progression of ER-negative breast cancer (3,4), suggests novel TME crosstalk drives tumor progression independent of tumor hormone status. Thus, we next compared exosomes purified from primary T2D-derived adipocytes (isolated as pre-adipocyte progenitors from patients and differentiated ex vivo) to exosomes purified from adipocytes derived from non-diabetic controls matched by age, sex and BMI (table S1). Consistent with exosome results from IR adipocytes, exosomes from T2D adipocytes induced the expression of genes associated with pathways linked to breast cancer aggressiveness, such as invasion and migration (Fig. 2A) and EMT (fig. S2A), compared to non-diabetic (ND) exosomes (data files S3 and S8). Immunofluorescence imaging confirmed differential regulation of critical EMT proteins (Fig. 2B). Similar results were obtained for genes and their inferred pathways associated with cancer cell stemness (16) (Fig. 2C, fig. S2B, and data files S4 and S9). Like exosomes from IS and IR adipocytes, exosome size distributions were similar between exosomes from ND and T2D adipocytes (fig. S2C). Individual transcripts were validated by RT-PCR (Fig. 2, D and E). T2D adipocyte exosomes attenuated gene expression that is associated with cell death pathways (Fig. 2AC) and downregulated those encoding epithelial markers (Fig. 2F). Thus, the data suggest that patient metabolic status reprograms breast cancer cell gene expression—and possibly consequential aggressiveness—through adipocyte exosomes.
Fig. 2. Exosomes from T2D patient-derived adipocytes induce greater EMT- and CSC-associated gene expression in breast cancer cell models than exosomes from non-diabetic donor-derived adipocytes.
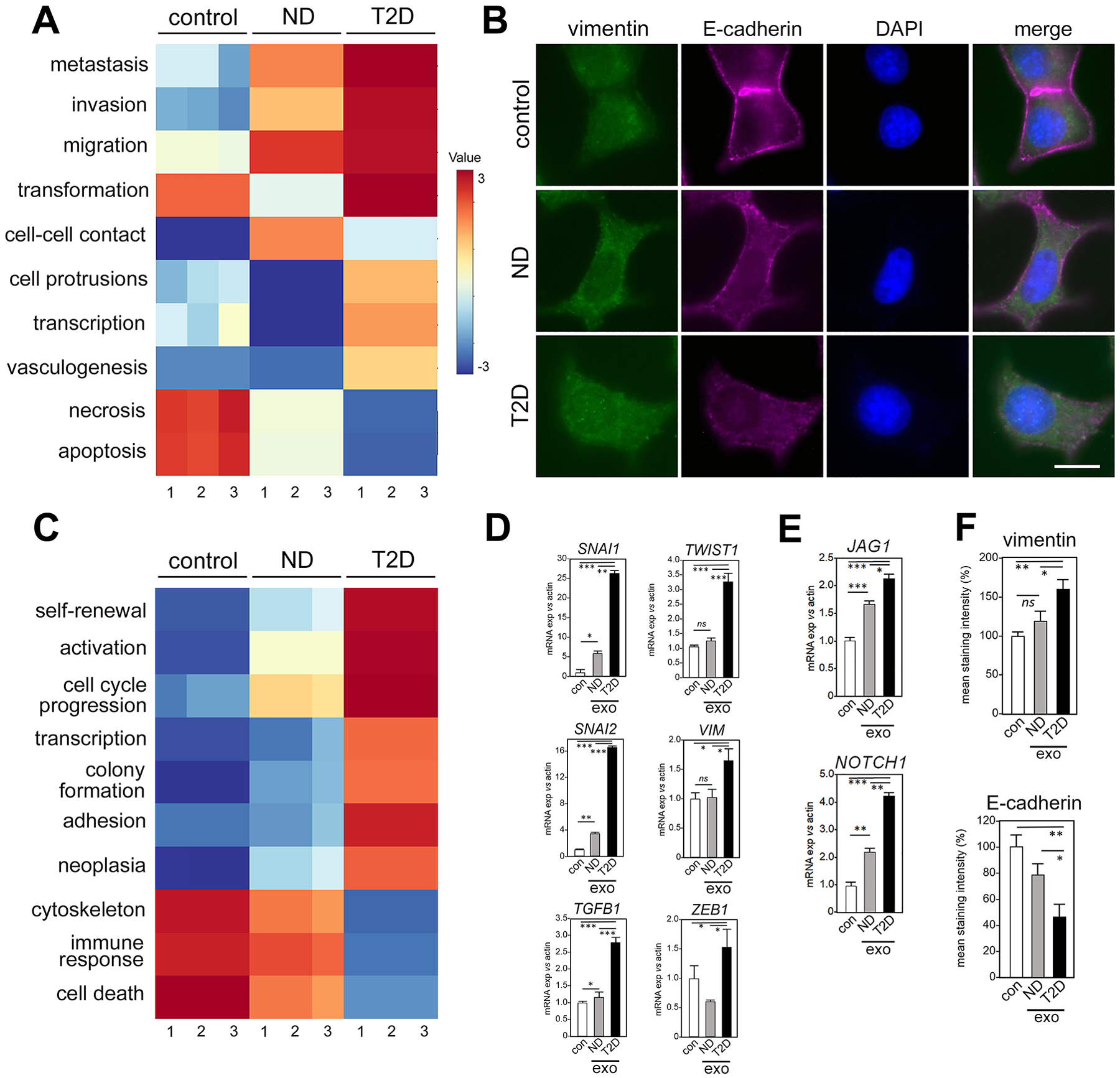
(A) Human primary subcutaneous pre-adipocytes from T2D and non-diabetic (ND) patients were differentiated and exosomes were isolated from conditioned media. MCF7 cells were treated with exosomes for 5 days and IPA was performed on EMT gene expression array data (fig. S2A, data file S3). Data are IPA-generated Z scores from N=3 independent cultures (fig. S2A and data file S3). Results are compared to vehicle control. (B) Immunofluorescence staining for vimentin and E-cadherin in MCF7 cells cultured with exosomes from ND or T2D adipocyte conditioned media, compared to control (vehicle-treated). DAPI, nuclear counterstain. Scale bar, 10 μm. For each of N=3 independent experiments, 25 images were examined by immunofluorescence. One representative image is shown, out of 25 images collected for each of the three experimental conditions with three replicates. (C) Pathways associated with cancer stem-like cell (CSCs) [N=3, as in (A)] were tested for induction by exosomes purified from adipocyte conditioned media of ND or T2D patients, and compared to vehicle control. Corresponding data in fig. S2B and data file S4. (D and E) Individual EMT genes from (A) and CSC genes from (C), respectively, were validated with PCR TaqMan probes. Data are means ± SEM from 3 independent experiments. (F) Quantitation of immunofluorescence signals in (B), as means ± SEM. Expression in each exosome experimental (exo) purified from either ND or T2D adipocyte-conditioned media was compared to vehicle control (con). * P <0.05, ** P <0.01, *** P <0.005, and ns not significant by unpaired, two-tailed t-test.
Exosomes from IR adipocytes promote greater EMT and morphological changes in cancer cells, compared to IS
Characteristic morphological changes also accompany transcription and protein expression differences in breast tumor cells upon EMT (17), which we measured by image analysis of exosome-treated human cellular models (Fig. 3A). We observed increased cellular perimeter, elongation and reduced circularity upon treatment with exosomes derived from T2D adipocytes compared to ND controls (Fig. 3B). Transwell experiments with MDA-MB-231 cells (18), a model for human triple negative breast cancer, confirmed that T2D exosomes promote cell migration (Fig. 3, C and D). To identify candidate exosome proteins that could account for functional differences between T2D and ND adipocytes, we performed proteomics analysis by mass spectrometry (Fig. 3E). The full set of peptides identified by mass spectrometry (table S2 and data file S11) was analyzed by IPA. Results showed that pathways associated with tumor cell aggressiveness (motility, invasion and angiogenesis) were increased in IR exosome payloads compared to IS (fig. S3 and data file S10), whereas pathways associated with cell death were decreased, consistent with data described above (Figs. 1 and 2). We previously established that critical pathways that promote breast cancer aggressiveness and EMT are controlled by the somatic BET (bromodomain and extraterminal domain) protein family (18,19), although only BRD4, and not BRD2 or BRD3, is required for cellular migration in breast cancer (18,20) and prostate cancer models (21). Therefore, we tested the BET dependence of exosome induction of selected EMT genes in MCF7 cells. Upon siRNA knockdown of BRD2 and BRD4, but not BRD3, the exosomes purified from ND or T2D adipocytes lost their ability to increase SNAI1 and SNAI2 (Fig. 3F). Thus, exosome signaling to these EMT target genes requires the BET proteins BRD2 and BRD4 as essential effectors.
Fig. 3. Exosomes from adipocytes of T2D patients induce morphological, transcriptional and migration phenotypes characteristic of EMT in human breast cancer cells, compared to exosomes from adipocytes of ND patients.
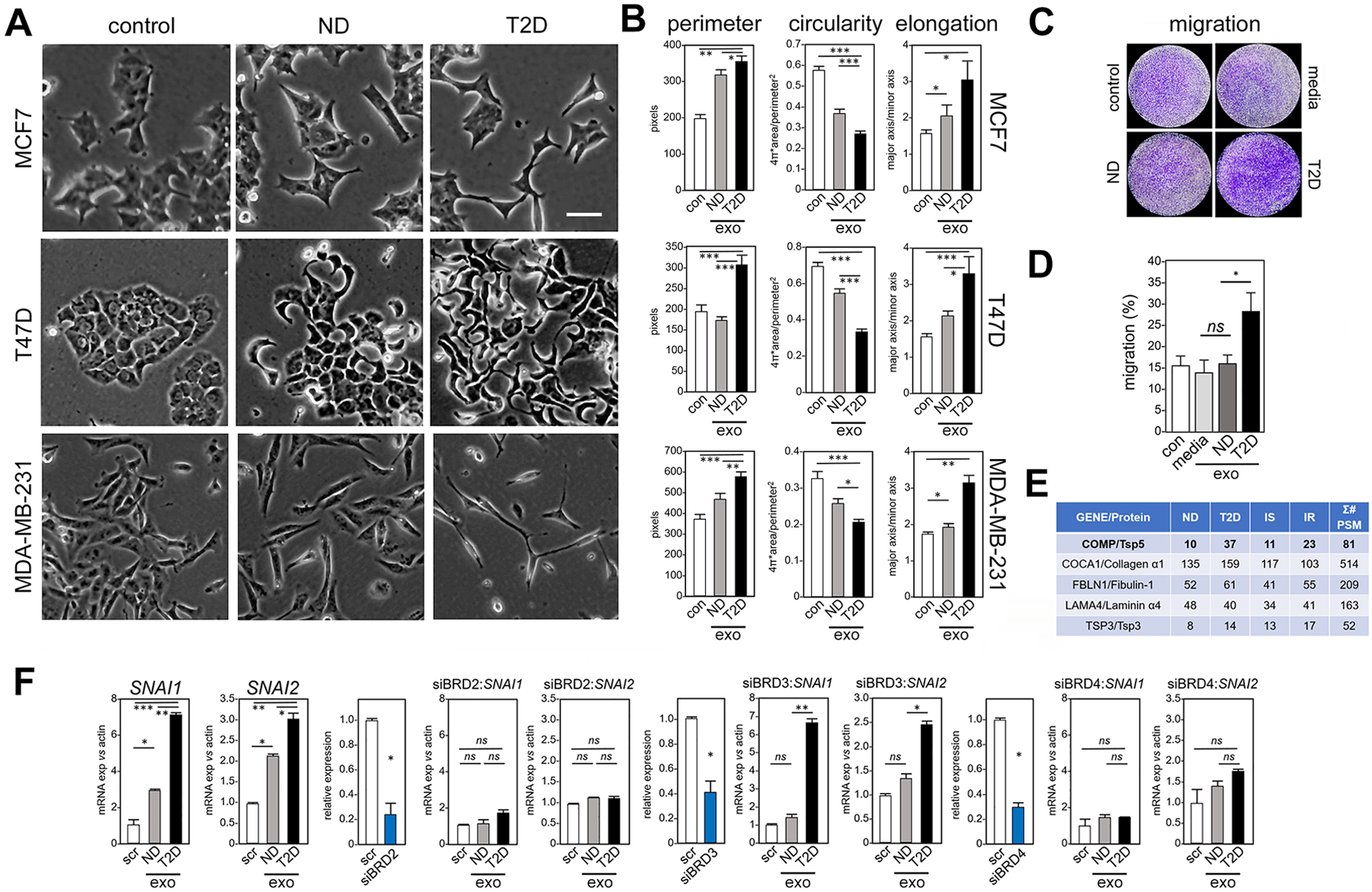
(A) Treatment for 5 days of three human breast cancer cell lines (MCF7, T47D, MDA-MB-231) with exosomes from adipocytes of T2D patients compared to ND patients or to untreated control (con). Morphology by light microscopy. Scale bar, 30 μm. One representative image is shown, out of 25 images collected for each of the three experimental conditions with three replicates. (B) Cellular perimeter, circularity and elongation (a parameter that is converse to circularity) were measured in each model after exposure to exosomes (exo) from adipocytes of ND patients and T2D patients, and compared to vehicle control (con). Morphological analysis was conducted using ImageJ. N=3 independent experiments, n = 25 cells each. * P <0.05, ** P <0.01, *** P <0.001 by unpaired, two-tailed t-test. (C) Migration of MDA-MB-231 breast cancer cells was measured in a 6-hour transwell assay after exposure to exosomes from conditioned media of adipocytes of ND patients and T2D patients, compared to control exosomes isolated from media without adipocyte culture, and vehicle control.. Cells that reached the distal side of the 8-μm pore membrane were visualized by crystal violet stain and microscopy. (D) Quantitation of (C). Data are from N=3 independent experiments. * P <0.05 and ns not significant by unpaired, two-tailed t-test compared to vehicle control. (E) Unbiased proteomic analysis of human adipocyte exosomes by LC-MS/MS. Σ# PSM (number of Peptide-Spectrum Matches) reports the sum of occurrences of unique peptides for each protein. (F) Exosomes from either T2D or ND adipocytes (exo) were tested for ability to induce transcription of SNAI and SNAI2 in MCF7 cells, in the context of siRNA knockdown of BRD2, BRD3 or BRD4. Gene expression is relative to β-actin and compared to scrambled siRNA control (scr). Control experiments without knockdown constructs are shown at far left, followed in order by knockdown of each BET gene. Positive controls for knockdown efficiency (blue bar) are shown before each experiment, with relative expression compared to scrambled control (scr). Data are from N=3 independent experiments. * P <0.05, ** P <0.01, *** P <0.001, and ns not significant by unpaired, two-tailed t-test compared to scrambled control.
We then tested whether the same functional relationships held for an entirely different system: murine 4T1 cells (22), a model for highly metastatic, triple-negative breast cancers (23). We used murine 3T3-L1 pre-adipocytes differentiated to mature adipocytes (24) as the source of exosomes, then treated mature adipocytes with murine TNF-α to induce insulin resistance as above. As in the human system, exosomes from mouse IR adipocytes induced expression of mesenchymal proteins and reduced epithelial proteins in 4T1 cells, compared to untreated control or exosomes from IS adipocytes (fig. S4A). As before, exosomes from IR adipocytes induced EMT genes (fig. S4B) and greater migration (fig. S4CD) in 4T1 cells than did exosomes from IS adipocytes. As we previously showed that BRD4 is a critical regulator of cellular migration in several human breast cancer cell models, including MCF7, SUM149PT and MDA-MB-231 (18), we used the BRD4-selective PROTAC degrader MZ-1 (25), which ablates prostate cancer cell migration (21), to inhibit 4T1 cell migration. As expected, MZ-1 effectively ablated 4T1 migration provoked by treatment with exosomes from IR adipocytes (fig. S4D).
Compared to those from insulin-sensitive adipocytes, exosomes from insulin-resistant adipocytes carry more TSP5, which induces several EMT genes
Both ND and T2D types of exosomes contained proteins of known importance for the TME. The top differentially represented hit was thrombospondin-5 (TSP5, encoded by COMP; Fig. 3E), which is associated with cancer progression (26,27), suggesting that TSP5 is critical for exosome effector function. To test the role of TSP5, we transduced human primary pre-adipocytes with lentivirus overexpressing TSP5, then differentiated the cells into adipocytes and purified exosomes from conditioned media. We first confirmed increased expression of TSP5 mRNA in the adipocytes, and increased protein loading into exosomes using the exosome marker CD63 as a control (Fig. 4A), then tested EMT gene responses in MCF7 cell readouts. As expected, TSP5-enriched exosomes significantly increased transcription of ZEB1 and SNAI2 readout genes compared to control (Fig. 4B). To prove that exosomes deliver TSP5 to MCF7 cells, we loaded synthetic, cationic lipid vesicles with recombinant human TSP5 and identified them with fluorescently labeled ovalbumin. Immunofluorescence showed that TSP5-loaded exosomes indeed delivered their payload to MCF7 cells to induce vimentin expression (Fig. 4C). Additionally, exosomes released from TSP5 lentivirus-transduced, primary adipocytes (ABM-007) induced expected transcriptional changes by EMT array, whereas control exosomes from the same adipocytes transduced with lentivirus vector control did not (Fig. 4D and data file S5). Here, MMP9 was the top ranked gene. Consistent with previous results, pathway analysis TSP5-loaded exosomes increased signaling associated with invasion, migration and metastasis, and decreased pathways associated with cell death (Fig. 4E and data file S6). We also transfected MDA-MB-231 cells with plasmids for overexpression of recombinant, V5 epitope-tagged TSP5 and confirmed by immunofluorescence that the epithelial marker E-cadherin was decreased (fig. S5A) and the mesenchymal marker vimentin was increased (fig. S5B) Additionally, we used flow cytometry of CD24 and CD44 surface markers (28) to show that forced expression of TSP5 shifted non-malignant breast epithelial MCF10A cells toward a more mesenchymal-like (CD44hi/CD24lo) phenotype (fig. S6, A and B). These results confirm that forced expression of TSP5 is necessary and sufficient to induce EMT-like phenotypic changes in human breast cancer cell models, without ruling out the possibility that native exosomes released by adipocytes harbor as-yet uncharacterized proteins, enzymes, RNAs or metabolites that provoke similar shifts (7).
Fig. 4. TSP5 protein drives expression of EMT phenotypes.
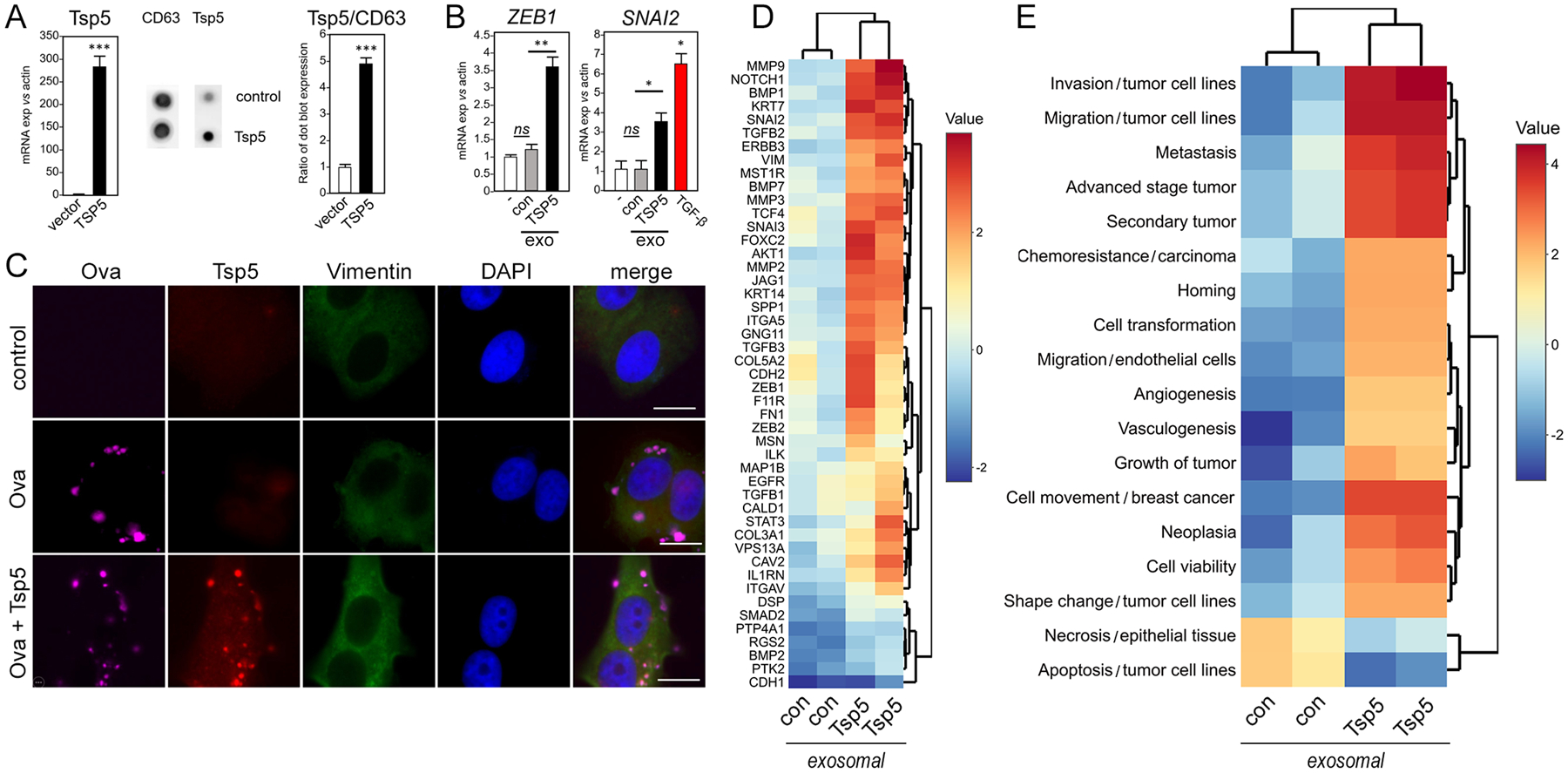
Human pre-adipocytes were transduced with TSP5-expressing lentivirus or lentivirus vector control and differentiated to mature adipocytes as above. (A) RT-PCR of adipocyte total RNA was used to compare gene expression in TSP5-transduced adipocytes to vector control. Exosomes werepurified from conditioned media of transduced adipocytes and characterized by Elispot for expression of TSP5 protein and exosomal surface marker CD63. Exosomal TSP5 expression is reported as ratio with CD63. Data are from N=3 independent experiments; *** P <0.001 by unpaired, two-tailed t-test. (B) Upon addition to MCF7 cells, exosomes (exo) from adipocytes transduced with TSP5 lentivirus were compared to exosomes from adipocytes transduced with lentivirus vector control (con) for ability to induce transcription of EMT genes ZEB1 and SNAI2. Induction of SNAI2 by human recombinant transforming growth factor (TGF)-β is shown as a positive control for a pro-EMT response (red bar). Expression in each experimental was compared to vehicle control (−) using unpaired, two-tailed t-test. Data are from N=3 independent experiments. * P <0.05, ** P <0.01, and ns not significant by unpaired, two-tailed t-test. (C) MCF7 cells were treated for 3 days with synthetic cationic vesicles loaded with recombinant human TSP5, then vimentin expression was measured by immunofluorescence imaging as a measure of EMT induction. Anti-TSP5 antibody confirmed delivery of protein by the synthetic vesicles. As a control for loading and delivery, AlexaFluor 647-labeled ovalbumin (Ova) was also incorporated into the vesicles. For each of N=3 independent experiments, 25 images were examined by immunofluorescence. One representative image is shown, out of 25 images collected for each of the three experimental conditions with three independent replicates. Scale bar, 10 μm. (D) Heatmap of EMT gene expression by PCR array. MCF7 cells were treated for 5 days with exosomes purified from conditioned media of human primary adipocytes that had been transduced with either lentivirus overexpressing TSP5 or lentivirus vector control. Color scale bar shows expression range (data file S5). (E) IPA analysis of (D) generated Z scores of pathways and functions (data file S6).
As a negative control, we knocked down Tsp5 in 3T3-L1 pre-adipocytes with shRNA and confirmed that knockdown ablated the ability of exosomes purified from mature adipocytes to induce several EMT genes in 4T1 cells (fig. S7), including SNAI1, NOTCH1, JAG1, ZEB1 and MMP3 (fig. S8, A and B). Exosomes from the TSP5 knockdown adipocytes no longer induce canonical cancer pathways upregulated by IR exosomes (ILK, ERK/MAPK, HGF and actin cytoskeleton signaling; fig. S8C). Other IR exosome-induced genes (such as SERPINE1 and MAP1B) were TSP5-independent (fig. S8B), as were other canonical cancer pathways (such as FAT10 cancer signaling, TGF-β signaling, and STAT3 signaling; fig. S8C).
Co-expression of genes encoding TSP5 and BET proteins associates with decreased distant-metastasis-free survival in breast cancer cohorts
Finally, we examined distant metastasis-free survival (DMFS) in cohorts of breast cancer patients with high or low expression of COMP, the gene encoding TSP5, co-expressed with high or low levels of each of the three BET bromodomain genes BRD2, BRD3 or BRD4 (Fig. 5). As expected, higher co-expression of COMP and a BET gene was associated with reduced DMFS, with the strongest effect for BRD2. Specifically, we observed a statistically significant, 35% increased risk of distant metastasis over 25 years for high co-expression of BRD2 and COMP, and a 19% increased risk for high co-expression of BRD3 and COMP. There was a trend for reduced DMFS for high co-expression of BRD4 and COMP, but the difference did not reach statistical significance for 332 patients per group. These findings reaffirm the importance of testing the functions of all three somatic BET genes (19) in obesity-related cancer, not just BRD4, which is commonly assumed to be the sole important player (29,30).
Fig. 5. Kaplan–Meier curves of distant metastasis–free survival of breast cancer patients, calculated from TCGA data.
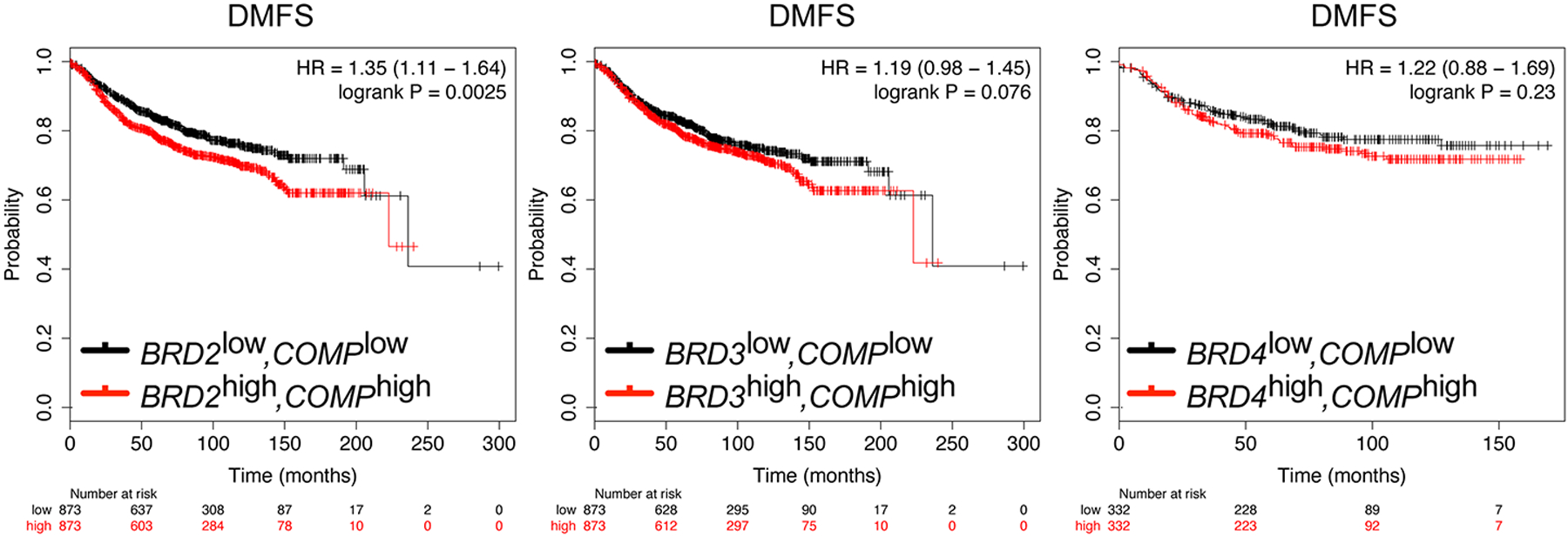
The level of expression of COMP, the gene encoding TSP5, was assessed for association with distant metastasis-free survival (DMFS) in breast cancer patients in co-expression with each of the three somatic BET bromodomain genes BRD2, BRD3 or BRD4. Expression groups of equal size were characterized as high (red) or low (black) for each gene, and joint probabilities were computed for 873 patients per group. Survival times are shown in months; hazard ratios (HR) and confidence intervals (in brackets) are shown for each pair of genes.
DISCUSSION
Population studies over twenty years have convincingly implicated cardiometabolic risk factors in incidence and progression of obesity-driven cancers, including breast cancers (31–33). Numerous hormones, metabolites, cytokines and tissue structural properties that change in concert with obesity, in both humans and animal models, have gained attention as potential mechanisms that link obesity and cancer. However, identification of the most important causal elements has been difficult. Seeking to leverage ex vivo and in vitro models, many investigators continue to test murine adipocytes against human breast cancer cell lines, in co-culture, conditioned media or organoid experiments, where species differences may complicate interpretation. Furthermore, the few reports that use human adipocytes often derive preadipocyte progenitors from normal volunteers and ignore insulin resistance. We determined that human breast cancer cell lines provide a useful ex vivo readout to assay factors produced by human primary adipocytes. We considered the metabolism of the adipocyte as the independent variable. High-throughput, cellular proliferation assays are widely used for discovery of novel biochemical activators or inhibitors, yet these techniques yield limited information. We focused instead on pathway analysis of transcriptionally induced EMT genes, which requires 3–5 days, rather than simpler, overnight proliferation assays. This approach enabled discovery of novel pathways important for tumor progression that might be missed with a more convenient assay.
Although systemic metabolism of the patient clearly plays a major role in the risk of breast cancer progression (34), the adipocyte contribution to the local signaling in the TME demands further study. As obesity worsens, adipose tissue develops insulin resistance (35,36), which provoked our overall hypothesis that insulin resistance reprograms exosome payloads (10). IPA shows gene regulation pathways associated with tumor cell aggressiveness (invasiveness, migration, angiogenesis) are upregulated by insulin-resistant cell-derived exosome payloads compared to those from insulin-sensitive cells. Angiogenesis pathways are also coupled to hypoxia in adipose tissue in obesity (37–39), and the increased expression of such genes as NOTCH1, SNAI1, SNAI2, SERPINE1, COL1A2, EGFR and those associated with the HIF1α pathway, as shown here, suggest that insulin-resistant and T2D-derived adipocyte exosomes may mediate differences in the breast TME of patients with metabolic disease, promoting tumor vascularization as the malignant cell clones undergo expansion. However, breast tumor cell invasiveness and migration mechanisms appear uniquely to require NOTCH1 and BRD4 signaling (18).
Together, our results implicate exosome proteins from IR adipocytes in tumor aggressiveness associated with EMT and cancer stem cell signatures. Results are borne out in breast cancer patient cohorts with long term follow-up (Fig. 5). Our conclusions are consistent with an extensive clinical and population literature that has implicated patient metabolism in risk of breast cancer incidence and progression. Furthermore, adipocyte-origin, circulating exosomes might be suitable as non-invasive, liquid biopsy biomarkers (40) to assist clinical decision making for breast cancer patients at risk for progression. Our insights into exosome communication among adipocytes and tumor models of divergent hormone status suggest that patient metabolism influences progression risk across breast cancer subtypes, through microenvironment mechanisms independent of hormone signaling.
MATERIALS AND METHODS
Cell lines
Cell lines, including MCF7 (HTB-22), MDA-MB-231 (HTB-26), T47D (HTB-133) and 3T3-L1 (CL-173), were obtained from the American Type Culture Collection (ATCC). MCF10A cells were gifted by Dr. Sam Thiagalingam, and 4T1 cells by Dr. Dennis Jones, both of Boston University Medical Center. Human primary pre-adipocytes were obtained and differentiated in Boston University’s Adipose Tissue Biology and Nutrient Metabolism Core.
Reagents
Unless otherwise specified, chemicals and biochemicals were from Sigma-Aldrich. Differentiation reagents for conversion of murine 3T3-L1 fibroblasts to mature adipocytes were dexamethasone (D8893-1MG), 3-isobutyl-1-methylxanthine (IBMX, I7018) and insulin (I0516).
Reagents used in experiments for transfer of exogenous TSP5 to target cells were recombinant COMP/TSP5 (SRP6457), Pierce™ Protein Transfection Reagent (89850, Thermo Scientific), ovalbumin, and Alexa Fluor™ 647 conjugate (O34784, Thermo Scientific). Paraformaldehyde solution (AAJ19943K2, Thermo Scientific) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, FluoroPure grade; D21490, Thermo Scientific) were used to fix and stain the nuclei.
Gene expression analysis by RT-PCR was performed using TaqMan™ master mix (Thermo Fisher, 4369510). The human gene probes were: SNAI1 (Hs00195591_m1), SNAI2 (Hs00950344_m1), CDH1 (Hs01023895_m1), COMP (Hs00164359_m1), ACTB (Hs00357333_g1). VIM (Hs00958111_m1), ZEB1 (Hs01566408_m1), TWIST1 (Hs01675818_s1), JAG1 (Hs01070032_m1), NOTCH1 (Hs01062014_m1), TGFB1 (Hs00998133_m1), BRD2 (Hs01121986_g1), BRD3 (Hs00978972_m1) and BRD4 (Hs04188087_m1).
The mouse gene probes were: Snai1 (Mm00441533_g1), Vim (Mm01333430_m1), Cdh1 (Mm01247357_m1), Comp (Mm00489490_m1), Twist1 (Mm00442036_m1) and Actb (Mm02619580_g1), Zeb1 (Mm00495564_m1) and Epcam (Mm00493214_m1).
Antibodies
Antibodies to CD63 (ab216130), TSP5 (ab74524), V5 tag (SV5-Pk1) (ab27671) and vimentin (monoclonal ab8978) were purchased from Abcam. Antibodies to E-cadherin (4A2, mouse mAb 14472), vimentin (D21H3, rabbit mAb 5741) and N-cadherin (D4R1H, rabbit mAb 13116) were obtained from Cell Signaling Technology. Filamentous actin (F-actin) was stained using either Alexa Fluor™ Phalloidin probes 488nm (A12379), 568nm (A12380) or Plus 647 (A30107) from Thermo Fisher. Mouse or rabbit Alexa Fluor Plus secondary antibodies were purchased from Thermo Fisher and used for immunofluorescence imaging.
Differentiation of 3T3-L1 pre-adipocytes
3T3-L1 pre-adipocytes were cultured in Dulbecco’s modified eagle medium (DMEM) containing 4.5 g/L glucose, L-glutamine and sodium pyruvate) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, 10 μg/mL streptomycin (all from Corning) until confluence, then incubated in the same medium for an additional 2 days. Differentiation was induced by addition of 1 μM dexamethasone, 0.5 mM IBMX, and 1.67 μM insulin for 3 days, at which time the medium was replaced with growth medium containing 0.41 μM insulin. After differentiation, cells were treated with 1 nM recombinant mouse TNF-α (Abcam, ab9740) for 24 hours to induce insulin resistance.
Differentiation of primary human adipocytes
The Adipocyte Core of the Boston Nutrition Obesity Research Center (BNORC) (P30DK046200) provided the pre-adipocytes. Samples were de-identified and not linked to any protected information. All subjects provided informed consent for participation. The protocol was approved by Institutional Review Board of Boston University Medical Center. To obtain pre-adipocytes, subcutaneous fat tissue was obtained by BNORC from subjects undergoing bariatric surgery. Adipose stromal cells (ASC) were isolated as previously described (36). Briefly, minced tissue was treated with collagenase solution (1mg/mL HBSS) (Type 1, Worthington Biochemical, Lakewood NJ) at 37°C, shaken at 100 rpm for 2 hours. The digested tissue was filtered through a 250 μm mesh (Component Supply, Inc. Smithville Hwy, Sparta, TN). Cells in the flow through were centrifuged at 500 × g for 10 min at room temperature. The red blood cells in the cell pellets were lysed (0.154 mM NH4Cl, 10 mM KH2PO4 and 0.1 mM EDTA, pH 7.3). Then, washed cells were plated using alpha MEM Media (Gibco Thermo Fisher Scientific, Waltham MA) with 10% FBS (Gemini Bio Products, West Sacramento, CA), 100 units/mL penicillin, 10 μg/mL streptomycin (Pen/Strep) (Corning, Corning NY). Differentiation of ASCs to adipocytes was performed in serum-free media using chemicals and reagents purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Complete differentiation media (CDM) was made in DMEM/F12 (GIBCO) with 25 mM NaHCO3 and Pen/Strep containing 33 μM D-(+)-biotin, 17 μM pantothenate, 10 μg/mL transferrin, 100 nM dexamethasone, 100 nM insulin, 1 μM rosiglitazone (Calbiochem), 2 nM 3,3′,5-triiodo-L-thyronine (T3) and 0.5 mM IBMX. Differentiation was initiated two days post-confluence and CDM was replenished every 2–3 days. After 7 days, CDM was removed and the cells were fed with maintenance media: DMEM/F12 (GIBCO) with 25 mM NaHCO3 and Pen/Strep containing 33 μM D-(+)-biotin, 17 μM pantothenate, 10 μg/mL transferrin, 10 nM dexamethasone and 10 nM insulin.
Negative control exosomes from undifferentiated adipose stromal cells
As a rigorous control to confirm the mature adipocyte origin of the biologically active exosomes, we used the 2-dimensional culture of primary ASCs from which adipocytes are differentiated by the standard cocktail (above) and omitted a key chemical required for differentiation: rosiglitazone. In this case, without the consequent activation of PPARγ, mature adipocytes do not form, yet pre-adipocyte fibroblasts, endothelial cells and any other ACS cells remain represented in the culture. Conditioned media from this pre-adipocyte culture, where differentiation was blocked, was used as the source of exosomes, which were purified and added to the MCF7 cultures.
Induction of insulin resistance
Treatment of mature, fully differentiated, primary adipocytes ex vivo with low concentrations (250 pM) of the pro-inflammatory cytokine tumor necrosis factor alpha (TNF-α) is well established to ablate insulin-sensitive glucose transport and induce phosphorylation of Akt and other molecular changes. Human primary adipocytes were treated with 250 pM recombinant human TNF-α (Abcam, ab9642), and 3T3-L1 adipocytes were treated with 1 nM recombinant mouse TNF-α (Abcam, ab9740), overnight to induce insulin resistance.
Exosome isolation
The conditioned media of both ND or T2D adipocytes yielded between 8.0 × 108 and 1 × 109 exosomes per mL, from which we purified and concentrated exosomes as follows. Conditioned media (typically 10 mL) was centrifuged at 300 × g for 10 min in a 15 mL conical tube to remove cells and debris. The supernatant was transferred to a new 15 mL conical tube and further centrifuged at 16,000 × g for 30 min to remove additional debris. This supernatant was then transferred to an Amicon Ultra-15 (Millipore-Sigma; REF-UFC910008) 100K centrifugal filter and centrifuged at 4,500 × g for 15 min to concentrate the material to 1 mL final volume. To precipitate exosomes, 1 mL of this concentrated, conditioned media was added to Exo-spin Buffer reagent (Cell Guidance Systems; # EX06) in a 2:1 ratio (sample:buffer; v:v) and incubated overnight at 4°C. The sample was then centrifuged at 16,000 × g for 1 hour at 4°C to pellet the exosomes. The supernatant was discarded and the pellet was re-suspended in 100 μL PBS (Cell Guidance System; LOT0619; EX-P10) for further separation of exosomes from other exosomes using Exo-spin columns. These spin columns work on the principle of size exclusion chromatography with separation of particles based on diameter. The columns are packed with spherical beads, with a 30 nm pore size. The space between the pores is such that the eluate contains exosomes with a size range of 30 nm-200 nm. The Exo-spin (Cell Guidance System; LOT0720) column was equilibrated for 15 min at room temperature before use. The column was calibrated twice with 250 μL PBS and centrifuged at 50 × g for 10 sec. Then, 100 μL of re-suspended total exosome sample was added to the top of the column, which was then centrifuged at 50 × g for 60 sec and the flow through was discarded. The column was then transferred to a new 1.5 mL centrifuge tube, whereupon 200 μL PBS was applied to the column, which was then centrifuged at 50 × g for 60 sec to elute 200 μL of final, pure exosomes for downstream functional assays.
Exosomes were also purified using a Total isolation kit (Thermo Fisher Scientific) and purified by size exclusion chromatography on a qEV Original column, using an Automatic Fraction Collector (AFC, serial number: V1-0395, IZON) and identified for similar functional outcomes in downstream assays. The size distribution and concentration of exosomes were always determined before each biological experiment using a NanoSight NS300 system (Malvern Panalytical). Day-to-day variation in exosome yields across adipocyte cultures was <10%, and TSP5 knockdown or overexpression adipocytes yielded exosome numbers within this range. For each experiment, exosome counts were always normalized after NanoSight quantitation such that equal numbers were added to cell wells in each experiment and across replicates.
Glucose transport assay
To measure glucose uptake in cells, the Glucose Uptake-Glo™ Assay (J1341, Promega) was used as outlined in the manufacturer’s protocol. This is a plate-based, bioluminescent method for measuring glucose uptake by living cells, based on the detection of 2-deoxyglucose-6-phosphate (2DG6P).
Cellular symmetry analysis
ImageJ was used to analyze cell morphological parameters. Perimeter was defined as the length of the outside boundary of a cell. Circularity was calculated as 4π × area/perimeter2 wherein a value of 1.0 indicates a perfect circle. As the value approaches 0.0, the parameter indicates an increasingly elongated shape. Cell elongation was measured as aspect ratio:major axis/minor axis.
Exogenous TSP5 transfer to cells
Recombinant TSP5 (Sigma-Aldrich, SRP6457) was loaded into target cells using Pierce Protein Transfection Reagent Kit (Thermo Scientific, 89850). Reagent protein complexes attach to negatively charged cell surfaces, and either can directly fuse with the membrane to deliver the captured protein into the cell or undergo endocytosis and then fuse with the endosome, to release the captured protein into the cytoplasm. The Pierce™ Reagent was vortexed for 10 to 20 seconds at top speed before each use. For a 6-well plate or 33 mm dishes, 10 μL of reagent was pipetted. The solvent was evaporated by placing the microcentrifuge tubes containing the reagent under a laminar flow hood for 2 hours. The dried reagent was hydrated with the diluted solution of recombinant protein and carrier (TSP5, 100 ng + AlexaFluor-647-labelled ovalbumin, 5 μg) in 50 to 100 μL PBS. Then, the solution was mixed briefly by pipetting up and down 3 to 5 times, incubated at room temperature for 5 min and vortexed for 3 to 5 seconds at low to medium speed. Serum-free medium was added to the reagent/protein complex to bring the final delivery volume up to 1 mL. The final delivery mix was transferred onto PBS-washed cells, incubated for 3 to 4 hours at 37°C, then one volume of 20% serum-containing medium was added directly to the well or dish to quench the reaction.
TSP5 knockdown and overexpression
All lentiviral particles were obtained from Sigma-Aldrich. To deplete TSP5, human primary pre-adipocytes (from subject ID: ABM- 007) were transduced with pLKO.1-COMP shRNA (TRCN0000056075) and non-target control (SHC001V) lentiviral particles. To overexpress TSP5, human primary pre-adipocytes were transduced with pLX_317-COMP (TRCN0000470297) and control vector (ORFBFPV) lentiviral particles. Human primary pre-adipocytes were selected using growth media containing puromycin (0.5 μg/mL). To knockdown Tsp5 in 3T3-L1 mouse pre-adipocytes, mouse-specific pLKO.1-COMP shRNA (TRCN0000066166) and non-target control (SHC001V) lentiviral particles were used. Cells were selected using growth media containing puromycin (1 μg/mL). After selection in each case, the transduced pre-adipocytes were differentiated to mature adipocytes as described above.
Knockdown by siRNA transfection
Cells were transfected with 25 nmol/L of listed siRNAs for 72 hours with Lipofectamine™ RNAiMAX Transfection Reagent (13778075, Thermo Fisher). Human Bromodomain and ExtraTerminal (BET) and non-targeting (scramble) SMARTpool siRNAs were purchased from Dharmacon. The pools in each case were comprised of five independent siRNAs. Catalog numbers are as follows: siBRD2 (L-004935-00-0005), siBRD3 (L-004936-00-0005) and siBRD4 (L-004937-00-0005).
Immunofluorescence (IF) imaging
For IF imaging 100K cells were cultured in 27 mm glass bottom dishes (Thermo Fisher, 150682). For staining, cells were washed with DPBS including MgCl2 and CaCl2, then fixed with 4% paraformaldehyde (PFA) for 10 min. Then, cell membranes were permeabilized with Triton-X 100, 0.5% for 15 min. Cellular binding sites were blocked with bovine serum albumin (BSA), 2% (w:v) for 45 min. Then fixed, permeabilized and blocked cells were stained with primary antibodies (1:100 dilution, v:v) for 2 hours, washed 5 times, then stained with IF conjugated secondary antibodies (1:200 dilution, v:v) and phalloidin (1:1000 dilution, v:v) and DAPI counterstain. Finally, images were captured using a Nikon Deconvolution Wide-Field Epifluorescence System at the Boston University Cellular Imaging Core. Images were analyzed using ImageJ, with differences in means evaluated by comparison of a minimum of 25 individual, representative cells for each condition. Images were assembled in Adobe Photoshop Version 21.1.2. IF stain of proteins with primarily plasma membrane localization (such as E-cadherin) was quantified by selecting the plasma membrane and comparing across samples using the same method. Other proteins that have cytoplasmic or cell-wide distribution were quantified by selecting the whole cell. For TSP5 exosome delivery experiments (Fig 4), we found that the extent of staining of AlexaFluor 647-ovalbumin carrier protein was not changed by the presence or absence of TSP5, from which we concluded that TSP5 does not alter the ability of exosomes to bind to or merge with the target cells
Proteomics analysis (Nanospray LC-MS/MS analysis)
Exosome proteomics analysis was performed by Poochon Scientific LLC (Frederick, MD) using liquid chromatography with tandem mass spectrometry (LC-MS/MS).
The protein of the exosome samples was denatured by 1% SDS and heated at 95°C, followed by digestion with trypsin (Pierce Trypsin Protease, MS grade; #90057). In brief, the denatured protein was reduced with dithiothreitol at 56°C for 45 min, followed by alkylation with iodoacetamide for 30 min at room temperature in the dark. Alkylated proteins were then precipitated by 80% acetone, followed by trypsin digestion at 37°C for 16 hours. The digested peptide mixture was then concentrated and desalted using C18 Zip-tip (ZTC18S960, Millipore). Reconstituted, desalted peptides were dissolved in 20 μL of 0.1% formic acid (Formic Acid Optima LC/MS (A11–50), Fisher Scientific) in LC-MS/MS grade water. Peptides (12 μL) were analyzed by 110 min LC-MS/MS run.
The LC-MS/MS analysis of samples was performed using an Orbitrap Exploris 240 Mass Spectrometer and a Dionex UltiMate 3000 RSLCnano System (both from Thermo Scientific). The Orbitrap Exploris 240 instrument was operated in the data dependent mode to automatically switch between full scan MS and MS/MS acquisition. Peptide mixture from each sample was loaded onto a peptide trap cartridge at a flow rate of 5 μL/min. The trapped peptides were eluted onto a reversed-phase EasySpray C18 column (Thermo) using a linear gradient of acetonitrile (3–36%) in 0.1% formic acid. The elution duration was 110 min at a flow rate of 0.3 μL/min. Eluted peptides from the EasySpray column were ionized and sprayed into the mass spectrometer, using a Nano-EasySpray Ion Source (Thermo) under the following settings: spray voltage, 1.6 kV, capillary temperature, 275°C. The 15 most intense multiply charged ions (z ≥ 2) were sequentially isolated and fragmented in the octopole collision cell by higher-energy collisional dissociation (HCD) using normalized HCD collision energy 30 with an AGC target 1×105 and a maximum injection time of 200 ms at 17,500 resolution. The isolation window was set to 2. The dynamic exclusion was set to 20 s. Charge state screening was enabled to reject unassigned and 1+ and 7+ or higher charge state ions.
The raw data file acquired from each sample was searched against UniProtKB human protein sequences database (20,547 entries, downloaded on April 20, 2020) and target protein sequences were searched using the Proteome Discoverer 2.4 software (Thermo) based on the SEQUEST algorithm. Carbamidomethylation (+57.021 Da) of cysteines was fixed modification, and Oxidation Met and Deamidation Q/N-deamidated (+0.98402 Da) were set as dynamic modifications. The minimum peptide length was specified to be five amino acids. The precursor mass tolerance was set to 15 ppm, whereas fragment mass tolerance was set to 0.05 Da. The maximum false peptide discovery rate was specified as 0.01. The resulting Proteome Discoverer Report contains all assembled proteins with peptides sequences and peptide spectrum match counts (PSM#).
PCR array
RNA was isolated from cells using RNeasy Plus Mini Kit (74136, Qiagen). From each sample, 1 μg of RNA was used to prepare 20 μL cDNA using QuantiTect Reverse Transcription Kit (Qiagen, 205313). Human RT2 Profiler™ PCR Array, including Epithelial to Mesenchymal Transition (EMT) (PAHS-090Z) and Cancer Stem Cell (PAHS-176ZC) arrays, were purchased from Qiagen. Each cDNA sample (102 μL comprised of 1 μg RNA) was mixed with 1,350 μL RT2 SYBR Green ROX qPCR Mastermix (Qiagen, 330522) and 1,248 μL RNAse-free water. A total volume of 2,700 μL was mixed by vortexing and transferred to a reservoir. Then, an 8 channel pipettor was used to take aliquots of reaction mix from the reservoir, to distribute 25 uL into each well of the array. PCR reactions were performed and results were analyzed using a 7500 Fast Real-Time PCR instrument.
Gene expression analysis
All Ct values of the genes were normalized to the respective ACTB gene (delta Ct). Then the ΔCt of each gene was subtracted from the control gene ΔCt (delta.delta Ct). For the control groups, delta.delta Ct was calculated using this formula: ΔΔCt = ΔCt (C1 or C2 or C3) – ΔCt (Control average). Then, fold-change was calculated using 2^-ΔΔCt, (2 to the power of negative ΔΔCt). Next, the Z score was calculated based on this formula: Z score = (x-mean)/SD, where X is the fold-change. Hierarchical clustering analyses were performed using BioVinci Software (Bioturing, San Diego, CA, USA). Heatmaps were generated by applying clustering on rows (either: genes, pathways or diseases) using the Euclidean distance metrics and complete linkage criterion.
Ingenuity pathway analysis
To predict disease and function, and downstream pathways, data were analyzed through the use of Ingenuity Pathway Analysis, IPA (QIAGEN Inc.). https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). Data were uploaded to our IPA account through software. Core analysis was conducted, then comparison analysis was performed on all the conditions of each experiment, including replicates. The p-value cutoff (log10) was set to 1.3, which corresponds to p-value of 0.05. The measurement activation Z-score range was −3.5 to +3.5. After running each IPA analysis, a representative heatmap of proteomics signatures was generated.
Flow cytometry analysis
Single cell suspensions were washed after collection and stained in ice-cold Ca+2/Mg+2- free PBS with a viability dye (Zombie NIR, BioLegend) for 20 min at 4°C in the dark. Cell suspensions were then washed twice with ice-cold flow cytometry buffer (Ca+2/Mg+2- free PBS, supplemented with 2% FBS and 2 mM EDTA). Cell suspensions were then stained extracellularly with the appropriate antibodies for 25 min at 4°C, in the dark. CD324 (E-cadherin, clone 67A4) was conjugated to PerCP/Cy5.5 (BioLegend), CD44 (clone BJ18) was conjugated to APC (BioLegend) and CD24 (clone ML5) was conjugated to FITC (BD Biosciences). All cell suspensions were washed twice in ice-cold flow cytometry buffer prior to analysis. Unstained cells and single-stained controls were used to calculate flow cytometry compensation. Data acquisition (typically 1 million events) was performed on a BD LSRII instrument at the Boston University Flow Cytometry Core Facility. Data analysis was carried out using FlowJo Software (version 10.6.1, Tree Star).
Migration assay
MDA-MB-231 cells were cultured in DMEM media (25 mM glucose + 10% FBS + 1% antibiotic supplementation) for 3 days with adipocyte-derived exosomes from non-diabetic patients (ND group), Type 2 Diabetic patients (T2D group) and fresh media (exosome control) or PBS (control) for three days. Cells were then switched to serum-free media for three hours and subsequently plated in 24-well, 8-micron pore size Transwell plates (Thermo Fisher) for 6 hours. The cells were plated in the upper well of the transwell inserts with serum free media and the bottom well was filled with DMEM complete media to serve as a chemoattractant. Cells that stayed in the upper side of the membrane and did not migrate by the end of the assay were removed with a cotton swab, and the cells that migrated were fixed with ice-cold methanol for 5 minutes at −20°C. After fixation, cells were then stained with 1% crystal violet (v:v) in 2% ethanol for 10 minutes at room temperature. Images were captured by an EVOS XL Core digital inverted microscope. The percentage of migration and invasion was determined by first calculating the sum of the area of total migrated/invaded cells on the entire membrane with ImageJ software (National Institutes of Health, Bethesda, MD), and then converted to relative percent migration/invasion by comparing each condition to the control condition.
Kaplan-Meier analysis
To investigate the correlation between distant metastasis-free survival (DMFS) and BRD2, BRD3, BRD4 and COMP expression, we utilized data from 1803 (BRD2 and BRD3) and 664 (BRD4) patients from the TCGA repository (www.kmplot.com; list reference shown below). The normalized expression values of the RNAseq ID 214911 (BRD2), 212547 (BRD3), 226054 (BRD4) and 205713 (COMP) were used. For each gene, the mean expression was selected and cutoff values were determined by median expression. To assess the potential additive effects of the genes, samples with “low” BRD2, BRD3 or BRD4 expression and “low” COMP expression were combined into separate, individual cohorts and were compared to cohorts with “high” BRD2, BRD3 or BRD4 and “high” COMP expression. Kaplan-Meier plots were drawn by comparing the “low” and “high” cohorts to estimate the duration of survival. Hazard Ratios (HR) and p values are shown for distant metastasis-free survival (DMFS) (37).
Statistical analysis
Statistical analyses for PCR experiments and in vitro cell culture assays were typically performed using unpaired, two-tailed Student’s t-test. Biological and biochemical replicates were performed in at least triplicate and data are presented as means. Error bars represent standard error of measurement. Cellular morphology measurements were performed with at least 25 representative cells at the same magnification, in areas of equal culture density. The following symbols were used to indicate significant differences: ns P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.005, and **** P < 0.001.
Biohazards
All the experiments executed for this study were conducted in accordance with NIH guidelines, under the oversight of the Boston University Institutional Biosafety Committee and with approval. Universal precautions were always observed for human primary tissue or cell lines.
Supplementary Material
Acknowledgments:
We thank Alessio Ciulli for providing MZ-1 and helpful consultation, Sam Thiagalingam for the MI and MIII derivatives of the MCF10A cell line, and Dennis Jones for the 4T1 cells. We thank Guillaume Andrieu, Kunlin Huang, Pablo Llevenes, Conor Ross, Alexa Stathis, Hoàng Thắng and former members of the Denis lab, as well as Caroline Apovian, John Berk, Kimberly Bertrand, Anand Devaiah, Ruben Dries, Andrew Emili, Rachel Flynn, Neil Ganem, Raghuveera Goel, Naomi Ko, Stefano Monti, Valentina Perissi and Paola Sebastiani for thoughtful suggestions and discussion. We thank the Boston University-Boston Medical Center Flow Cytometry and Cellular Imaging Core Facilities for technical assistance.
Funding:
This work was supported by the Cancer Systems Biology Consortium of the National Cancer Institute (GVD: U01CA182898, U01CA243004, R01CA222170) and the National Institute of Diabetes and Digestive and Kidney Diseases (SRF: R01DK117161; P30DK046200 to Adipose Biology and Nutrient Metabolism Core of Boston Nutrition and Obesity Research Center).
Competing interests:
G.V.D., N.J and T.M. are inventors on U.S. patent 63/171,689 to use exosomes as a cancer diagnostic. The other authors declare that they have no conflict of interest. The funding agency played no role in the preparation of the manuscript or the decision to publish. Results and interpretation reported here do not necessarily represent the views of the NIH.
Footnotes
Data and materials availability:
All data are available in the main text or the supplementary materials. The RNA array data are deposited to GEO (https://www.ncbi.nlm.nih.gov/geo) under accession ID GSE186432. The mass spectrometry data are deposited to PRIDE (http://www.ebi.ac.uk/pride) under accession ID PXD029369.
References and Notes
- 1.Vona-Davis L, Rose DP, Type 2 diabetes and obesity metabolic interactions: common factors for breast cancer risk and novel approaches to prevention and therapy. Curr. Diabetes Rev 8, 116–130 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Quail DF, Dannenberg AJ, The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol 15, 139–154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer JR, Castro-Webb N, Bertrand K, Bethea TN, Denis GV, Type II diabetes and incidence of estrogen receptor negative breast cancer in African American women. Cancer Res. 77, 6462–6469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charlot M, Castro-Webb N, Bethea TN, Bertrand K, Boggs DA, Denis GV, Adams-Campbell LL, Rosenberg L, Palmer JR, Diabetes and breast cancer mortality in Black women. Cancer Causes Control 28, 61–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fantuzzi G, Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol 115, 911–919 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van Obberghen E, Tartare-Deckert S, Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J. Biol. Chem 278, 11888–11896 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Wang YY, Attané C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, et al. , Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2, e87489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J, Weinberg RA, Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Stephens JM, Lee J, Pilch PF, Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J. Biol. Chem 272, 971–976 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Quan M, Kuang S, Exosomal secretion of adipose tissue during various physiological states. Pharm. Res 37, 221–234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalluri R, The biology and function of exosomes in cancer. J. Clin. Invest 126, 1208–1215 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vella LJ, The emerging role of exosomes in epithelial-mesenchymal-transition in cancer. Front. Oncol 4, 361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA, Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J. Clin. Oncol 34, 4270–4276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown KA, Iyengar NM, Zhou XK, Gucalp A, Subbaramaiah K, Wang H, Giri DD, Morrow M, et al. , Menopause is a determinant of breast aromatase expression and its associations with BMI, inflammation and systemic markers. J. Clin. Endocrinol. Metab 102, 1692–1701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyengar NM, Zhou XK, Gucalp A, Morris PG, Howe LR, Giri DD, Morrow M, Wang H, et al. , Systemic correlates of white adipose tissue inflammation in early-stage breast cancer, Clin. Cancer Res 22, 2283–2289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A, Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3, e2888 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB, An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 7, e1002218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrieu G, Tran AH, Strissel KJ, Denis GV, BRD4 regulates breast cancer dissemination through Jagged1/Notch1 signaling. Cancer Res. 76, 6555–6567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrieu GP, Denis GV, BET proteins exhibit transcriptional and functional opposition in the epithelial-to-mesenchymal transition. Mol. Cancer Res 16, 580–586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu L, Chen Z, Lin X, et al. , Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ 27, 255–268 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shafran JS, Andrieu GP, Györffy B, Denis GV, BRD4 regulates metastatic potential of castration-resistant prostate cancer through AHNAK. Mol. Cancer Res 17, 1627–1638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur P, Nagaraja GM, Zheng H, Gizachew D, Galukande M, Krishnan S, Asea A, A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer 12, 120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pulaski BA, Ostrand-Rosenberg S, Mouse 4T1 breast tumor model. Curr. Protoc. Immunol 20, 20.2.1–20.2.16 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Wang F, Liu H, Blanton WP, Belkina A, Lebrasseur NK, Denis GV, Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem. J 425, 71–83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zengerle M, Chan KH, Ciulli A, Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol 10, 1770–1777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Englund E, Bartoschek M, Reitsma B, Jacobsson L, Escudero-Esparza A, Orimo A, et al. , Cartilage oligomeric matrix protein contributes to the development and metastasis of breast cancer. Oncogene 35, 5585–5596 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Papadakos KS, Bartoschek M, Rodriguez C, Gialeli C, Jin SB, Lendahl U, et al. , Cartilage Oligomeric Matrix Protein initiates cancer stem cells through activation of Jagged1-Notch3 signaling. Matrix Biol. 81, 107–121 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Liu S, Cong Y, Wang D, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports 2, 78–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belkina AC, Denis GV, BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 12, 465–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrieu G, Belkina AC, Denis GV, Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol 19, 45–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calle EE, Kaaks R, Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 4, 579–591 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Bosco JL, Palmer JR, Boggs DA, Hatch EE, Rosenberg L, Cardiometabolic factors and breast cancer risk in U.S. black women. Breast Cancer Res Treat. 134, 1247–1256 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bandera EV, Chandran U, Hong CC, Troester MA, Bethea TN, Adams-Campbell LL, Haiman CA, Park SY, Olshan AF, Ambrosone CB, Palmer JR, Rosenberg L, Obesity, body fat distribution, and risk of breast cancer subtypes in African American women participating in the AMBER Consortium. Breast Cancer Res Treat. 150, 655–666 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C, The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 8, 57622–57641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H, Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 112, 1821–1830 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B, Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 17, 4–12 (2006). [PubMed] [Google Scholar]
- 37.Christiaens V, Lijnen HR, Angiogenesis and development of adipose tissue. Mol Cell Endocrinol. 318, 2–9 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Corvera S, Gealekman O, Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim Biophys Acta. 1842, 463–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engin A, Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: Hypoxia hypothesis. Adv Exp Med Biol. 960, 305–326 (2017). [DOI] [PubMed] [Google Scholar]
- 40.LeBleu VS, Kalluri R, Exosomes as a multicomponent biomarker platform in cancer. Trends Cancer 6, 767–774 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Lee MJ, Fried SK, Optimal protocol for the differentiation and metabolic analysis of human adipose stromal cells. Methods Enzymol. 538, 49–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z, An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res Treatment 123, 725–731 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. The RNA array data are deposited to GEO (https://www.ncbi.nlm.nih.gov/geo) under accession ID GSE186432. The mass spectrometry data are deposited to PRIDE (http://www.ebi.ac.uk/pride) under accession ID PXD029369.