

Abstract
Mitochondria are central to the metabolic circuitry that generates superoxide radicals/anions (O2•−) as a by-product of oxygen metabolism. By regulating superoxide levels, manganese superoxide dismutase plays important roles in numerous biochemical and molecular events essential for the survival of aerobic life. In this study, we used MitoParaquat (mPQ) to generate mitochondria-specific O2•− and stable isotope-resolved metabolomics tracing in primary human epidermal keratinocytes to investigate how O2•− generated in mitochondria regulates gene expression. The results reveal that isocitrate is blocked from conversion to α-ketoglutarate and that acetyl-coenzyme A (CoA) accumulates, which is consistent with a reduction in oxygen consumption rate and inactivation of isocitrate dehydrogenase (IDH) activity. Since acetyl-CoA is linked to histone acetylation and gene regulation, we determined the effect of mPQ on histone acetylation. The results demonstrate an increase in histone H3 acetylation at lysines 9 and 14. Suppression of IDH increased histone acetylation, providing a direct link between metabolism and epigenetic alterations. The activity of histone acetyltransferase p300 increased after mPQ treatment, which is consistent with histone acetylation. Importantly, mPQ selectively increased the nuclear levels and activity of the oxidative stress-sensitive nuclear factor erythroid 2-related factor 2. Together, the results establish a new paradigm that recognizes O2•− as an initiator of metabolic reprogramming that activates epigenetic regulation of gene transcription in response to mitochondrial dysfunction.
Keywords: mitochondria, superoxide, epigenetics, TCA cycle, metabolism, transcription
Graphical Abstract
Introduction
Mitochondria are the primary source of superoxide anion radical (O2•−) formation. It has been reported that about 1% to 2% of the consumed oxygen for respiration leaks as O2•− and, during normal respiration, intramitochondrial superoxide concentration can reach 10−11 – 10−12 M [1]. Thus, aerobic organisms are continuously exposed to high levels of O2•−, which can cause micro- and macromoleculer damage. O2•− also can serve as a signaling molecule that regulates many life and death events. In the mitochondrial matrix, O2•− superoxide radicals are enzymatically converted to hydrogen peroxide by manganese superoxide dismutase (MnSOD). A failure in this generation and removal relationship provokes mitochondrial dysfunction, which contributes to the pathogenesis of a wide variety of diseases, including neurodegenerative diseases and metabolic disorders such as diabetes, obesity, cardiovascular disease, and cancer [2–6]. Mitochondria play a central role in energy metabolism, generate ATP by oxidative phosphorylation, and serve as a hub for coordinating fatty acid and glucose metabolism. Mitochondrial functions and metabolic pathways including the tricarboxylic acid (TCA) cycle and oxidative phosphorylation are known to be protected by the guardian MnSOD [7,8]. Metabolic reprogramming resulting from altered enzymatic activities of the TCA cycle plays an important role in whether metabolic adaptations occur or pathological disorders (including cancer) progress [9,10].
It has been demonstrated that mutation of NADP+-dependent isocitrate dehydrogenase 1/2 (IDH1/2) leads to metabolic reprogramming in glioblastoma, which produces D-2-hydroxyglutarate (2-HG), thereby acquiring a function that regulates epigenetics [11–16]. In humans, IDH exists in three isoforms, which use different cofactors and occur in different cellular locations. IDH1 and IDH2 use NADP+ whereas IDH3 utilizes NAD+ as a cofactor. IDH1 localizes in the cytosol, IDH2 localizes in both cytosol and mitochondria, and IDH3 localizes mainly in mitochondria. IDH3 catalyzes the decarboxylation of isocitrate to α-ketoglutarate (α-KG) and thus supports TCA cycle activity.
Obstruction of the TCA cycle can result in the accumulation of TCA cycle intermediates or by-products that can modify epigenetic marks and in turn regulate gene expression [17]. Histone modification is a widely studied epigenetic mark. Specific histone modifications positively regulate gene expression and activate gene transcription. However, histone modification can also be repressive, which silences gene expression. Histone acetylation and methylation are well-studied epigenetic events [18,19] that are associated with increase or decrease of gene expression [20–22]. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are the enzymes responsible for adding and removing acetyl groups to and from histones, respectively [23–25]. P300/CBP is a HAT that also functions as a transcription coactivator and is closely associated with Pol II during trancription activation. In contrast, HDACs, including sirtuin family members, deacetylate histones to repress gene transcription [26].
The current study investigated the role of mitochondrially generated superoxides in the reprogramming of mitochondrial metabolism that leads to epigenetic modifications and activation of gene transcription. We show that mitochondrial superoxide blocks TCA cycle progression by inactivating IDH, which leads to cellular accumulation of acetyl-coenzyme A (CoA), increase of histone acetylation, and in turn increases in the levels of transcriptional activity mediated by nuclear factor erythroid 2-related factor 2 (NRF2).
Methods
Reagents
Anti-histone H3 acetyl-lysine 9 (H3K9Ac) (Cat. No. 9649), anti-H3K14Ac (Cat. No. 7627), anti-H3K27Ac (Cat. No. 8173), anti-H4(K5, K8, K12)Ac (Cat. No. 13944), anti-NRF2 (Cat. No. 33649), and anti-HDAC1 (Cat. No. 34589) antibodies were obtained from Cell Signaling Technology (Danvers, MA). Anti-p300 (sc-48343) monoclonal antibody and anti-NF-κB antibodies (Cat. No. p50 sc-7178, p65 sc-372) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-β-actin (Cat. No. SAB2100037) antibody from Sigma (St. Louis, MO). HAT/p300-inhibitor (C646) (Cat. No. SML0002) was purchased from Millipore (Temecula, CA). IDH (Cat. No. K756), HAT (Cat. No. K334), and NRF2 activity assay kit (Cat. No. E4337) were purchased from Biovision (Milpitas, CA).
Cell culture and treatments
The human primary neonatal epidermal keratinocytes (HEKn, Cat. No. C-001–5C) were obtained from Invitrogen Life Sciences (Carlsbad, CA). The JB6 mouse epidermal cells, originally obtained from Dr. Nancy H. Colburn of the National Cancer Institute (Rockville, MD), were maintained as described previously [27,28]. All cells were kept in a 5% CO2 incubator at 37°C in optimum growth in media consisting of either MEM supplemented with 10% fetal bovine serum (Hyclone Inc., Logan, UT), 1% (w/v) L-glutamine, and 1% P/S antibiotics (Invitrogen), or Epilife medium supplemented with S7 (Invitrogen) for primary cells. Cells were treated with MitoParaquat (mPQ; a gift from Professor Michael Murphy, Cambridge University, UK) to generate O2•− in mitochondria.
Detection and quantification of reactive oxygen species by fluorescence microscopy and flow cytometry
MitoSOX Red (ThermoScientific, Cat. No. M36008) is a highly selective indicator of mitochondrial superoxide in live cells. We employed MitoSOX Red to estimate the intracellular levels of O2•−. Briefly, HEKn or JB6 cells were treated with 5 μM MitoSOX Red for 10 min at 37°C and rinsed 3 times with 1× phosphate-buffered saline, and the MitoSOX Red fluorescence was detected in live cells by fluorescence microscopy (model IX71,Tokyo, Japan). MitoSOX Red was also used to measure superoxide levels by flow cytometry.
Isolation of nuclear and cytosolic fractions
Both nuclear and cytosolic fractions were isolated from HEKn cells or JB6 cells as described previously [29]. Briefly, a subconfluent monolayer of cells was collected and then resuspended in buffer containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride, and a cocktail of protease inhibitors. The cytoplasmic fractions were separated by centrifugation at 17,000 × g at 4°C for 30 s. Subsequently, the nuclear pellets were resuspended in buffer containing 20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 35% glycerol, 0.5 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride, and protease inhibitors, and the nuclear proteins were collected by centrifugation at 14,000 × g at 4°C for 2 min.
Measurement of oxygen consumption rate
Oxygen consumption rate (OCR) was determined using the Extracellular Flux XF assay (Seahorse-Bioscience, North Billerica, MA). The OCR experiments were performed by sequentially adding pyruvate, oligomycin, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP), and rotenone to cells.
Western blotting
Western blotting was used to analyze proteins as described previously [29]. Briefly, total proteins were subjected to 10% or 12% SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. Membranes, after blocking with 5% bovine serum albumin, were probed with specific primary antibody (dilution range 500- to 5,000-fold) followed by secondary antibody (dilution range 2,000- to 6,000-fold) to detect specific proteins. Protein bands were visualized using the enhanced chemiluminescence detection system (ECL®, Amersham Biosciences Corp., Piscataway, NJ). Densitometric analysis was performed for quantification of protein band intensity with ImageJ software (NIH).
Histone acetyltransferase activity
HAT activity in isolated nuclear fractions was measured fluorometrically using a HAT activity assay kit (Biovision, Cat. No. K33–100) according to the manufacturer’s protocol. Briefly, nuclear proteins were combined with acetyl-CoA and H3 peptide as substrates, and incubated for 60 min at 25°C. The HAT enzyme generates acetylated H3 peptide and CoA-SH. The CoA-SH reacts with the developer to produce fluoroscence products that were detected by fluorimetry with excitation at 535 nm and emission at 587 nm. All the data were collected in kinetic mode.
NRF2 DNA binding activity
NRF2 DNA binding activity in the nuclear extract was determined based on the binding capacity of NRF2 to an antioxidant response element consensus sequence using an ELISA kit (Active motif, Cat. No. 50296) according to the manufacturer’s protocol. Briefly, isolated nuclear protein was incubated with immobilized consensus DNA in a 96-well plate along with binding buffer. The non-specific binding of DNA to protein was counteracted by using herring sperm DNA or mutant oligonucleotide. After three washes, the DNA-protein complex was detected colorimetrically at 450 nm.
Enzymatic assay
IDH1/2 activity was determined by a colorometric assay using NADP+ or NAD+ as a co-factor and isocitrate as substrate following the manufacturer’s protocol (Biovision, Cat. No. K756).
Estimation of metabolites
The cellular levels of citrate, isocitrate, and acetyl-CoA were measured according to the assay manufacturer’s instructions (Biovision).
NMR analysis
HEKn cells were treated with mPQ or DMSO as a vehicle control and incubated in neurobasal media supplemented with 6 mM 13C6-glucose, 1% fetal bovine serum, and 1% growth factor for 24 h. Medium was collected for NMR analysis and the cells were washed with ice-cold 1× phosphate-buffered saline followed by extraction of polar metabolites for analysis by NMR and ion chromatography coupled with ultra high-resolution Fourier transform mass spectrometry (IC-UHR-FTMS), as described previously [30]. Isotopomers were quantified as absolute (normalized against protein content) and fractional enrichment, as previously described [31–33].
Ion Chromatography-Ultra High-Resolution Fourier Transform Mass Spectrometric analysis
Polar extracts were reconstituted in 50 μL ultrapure deionized water (EMD Millipore) and spectra recorded as described previously [34]. Peak areas were integrated and exported to Excel via the Thermo TraceFinder (version 3.3) software package. Peak areas were corrected for natural abundance distributions for each isotopologue, and fractional enrichment of each metabolite was calculated based on the equation 13C amount/(13C+12C) amount.
Statistical analysis
Data were analyzed using one-way analysis (ANOVA) of variance for multiple-group comparisons, and Student’s t-test for two-group comparisons. For multiple-group statistics, Bonferroni’s post-test for multiple comparisons was used to determine statistical significance.
Results
mPQ generates mitochondrial superoxide
mPQ is a paraquat compound chemically linked with triphenylphosphonium, which produces superoxide anion radicals exclusively in mitochondria by accepting electrons from complex 1. The production of superoxide is caused by the paraquat moiety, not by the presence of the triphenylphosphonium ion [35]. To confirm the generation of superoxide radicals in mitochondria of primary human keratinocytes, we treated the HEKn cells with mPQ for 24 h. Mitochondrial O2•− was assessed by measuring MitoSOX Red fluorescence using fluorescence microscopy and flow cytometry. The results show that mPQ treatment significantly increased the MitoSOX Red flourescence in HEKn cells (Fig. 1A and B). Similarly, MitoSOX Red fluorescence was significantly increased in JB6 cells following mPQ treatment (Supplementary Fig. 1). These results agree with a previous report [35] and confirm that treatment with mPQ increases superoxide production in the mitochondria of keratinocytes under the conditions used in this study.
Figure 1.

mPQ generates superoxide radicals in mitochondria and impairs mitochondrial function in primary human keratinocytes. (A) Microscopic view of MitoSOX Red following mPQ treatment. (B) Quantitative determination of fluorescence using flow cytometry after mPQ treatment. (C) Measurement of OCR and determination of oxidative phosphorylation activity using Agilent Seahorse XF analysis of HEKn cells following the treatment of mPQ. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: *, p ≤ 0.05 and **, p ≤ 0.01.
mPQ alters mitochondrial function
To investigate how mitochondria adapt their function in response to O2•− generated in mitochondria, we treated HEKn cells with mPQ and used the Agilent Seahorse XF Analyzer to determined the OCR. The result (Fig. 1C) shows that the OCR was remarkably decreased, with a significant decrease in basal respiration, maximum respiration, spare capacity, and ATP-linked energy production. These results suggest that mitochondrial superoxide radical impairs mitochondrial oxidative phosphorylation.
Mitochondrial superoxide alters metabolic activity
Mitochondrial oxidative phosphorylation is tightly linked to TCA cycle activity. To identify metabolic changes that occur in mitochondria following mPQ-induced superoxide production, we profiled cellular metabolic activities using the stable isotope-resolved metabolomics (SIRM) approach. HEKn cells were cultured with normal cultured media until 70% confluent. Cells were then treated with either DMSO as vehicle control or mPQ at 5 μM or 10 μM concentration and cultured for 24 h in 13C6-glucose containing neurobasal medium. The labeled metabolites that were derived from 13C6-glucose were detected using NMR and IC-UHR-FTMS analyses. Fig. 2A illustrates the expected 13C labeling patterns for metabolites of glycolysis and the TCA cycle. We saw that 13C3-pyruvate and 13C3-lactate levels increased following mPQ treatment (Fig. 2B and C), which suggests cellular glucose uptake and increased utilization of labeled glucose by glycolysis. Although the lactate levels remain unchanged in the media following mPQ treatment for 24 h (Supplementary Fig. 2), we did find higher levels of cellular lactate synthesis in the mPQ-treated cells compared to the controls (Fig. 2C). This result is consistent with the observed reduction of OCR by mPQ treatment. Next, we measured the 13C labeling patterns of the metabolites in the TCA cycle, which includes citrate (Fig. 2D), isocitrate (Fig. 2E), α-KG (Fig. 2F), succinate (Fig. 2G), fumarate (Fig. 2H), and malate (Fig. 2I). We saw that the 13C2 and 13C5 isotopologues of citrate and isocitrate levels increased significantly while the downstream product, the 13C2 isotopologue of α-KG, was depleted. This points to a block at the IDH step in response to mPQ treatment. However, this block did not lead to depletion of the subsequent 13C products of the TCA cycle, which includes 13C-succinate, fumarate, and malate. We noted that mPQ reversed the abundance of 13C2 and 13C3 isotopologues in malate/fumarate and citrate/isocitrate/α-KG. The 13C2 isotopologues are generated from the forward pyruvate dehydrogenase initiated TCA cycle reactions (tracked by •) while the 13C3 isotopologues can be produced from pyruvate carboxylation and the reversal of the second half of the TCA cycle reactions (tracked by •) (Fig. 2A). Thus, this result suggests that the activation of pyruvate carboxylase-mediated anaplerosis compensates for the block at IDH. Interestingly, the levels of unlabeled (C0) isotopologues of succinate, fumarate, and malate increased in response to mPQ, indicating the contribution of non-glucose source(s) such as glutamine to this buildup.
Figure 2.

Metabolic reprogramming in primary HEKn cells. SIRM analysis was performed to measure the responses of 13C6-glucose-derived TCA cycle metabolites following treatment with mPQ for 24 h. (A) Schematic diagram shows the atom-resolved metabolic network connecting glycolysis with the TCA cycle. (B-I) The levels of the 13C-labeled isotopologues of TCA cycle metabolites in cell extracts, quantified by IC-UHR-FTMS. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: *, p ≤ 0.05 and **, p ≤ 0.01.
mPQ suppresses IDH activity
Since mPQ treatment attenuated isocitrate to α-KG conversion, we then investigated this effect on selected mitochondrial enzyme activity in cell lysates. The data in Fig. 3A show that IDH1/2 activity decreased significantly following mPQ treatment. This decrease was not due to altered amounts of protein (Supplementary Fig. 3). More importantly, the activity of IDH3, a mitochondrially localized enzyme, was significantly blocked, falling with as low as 1 μM of mPQ treatment (Fig. 3B). This result suggests that IDHs are a target of superoxide radicals. We also assayed for citrate, isocitrate, and acetyl-CoA in the same lysates. The results show that citrate, isocitrate, and acetyl-CoA levels were significantly increased following mPQ treatment (Fig. 3C, D, and E). These findings are consistent with the SIRM data, and further suggest that mitochondrial superoxide inactivates IDH enzyme activity, which leads to accumulation of citrate and acetyl-CoA.
Figure 3.

Measurement of selected metabolites and metabolic enzymes using biochemical assays in mouse skin epidermal (JB6) cells. (A) IDH1/2 activity was measured using isocitrate as substrate and NADP+ as cofactor and (B) IDH3 activity was measured using NAD+ as cofactor. (C-E) The cellular levels of citrate, isocitrate, and acetyl-CoA were measured using an assay kit following treatment with mPQ. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: *, p ≤ 0.05 and **, p ≤ 0.01.
Mitochondrially generated superoxide mediates epigenetic modification
Both citrate and acetyl-CoA are known regulators of epigenetic modification of histones. Our data show an increase of cellular citrate and acetyl-CoA levels with mPQ treatment compared to vehicle-treated control cells. We hypothesized that the increased acetyl-CoA level is associated with altered histone acetylation. To test this hypothesis, we first examined histone H3 acetylation in HEKn cells following treatment with mPQ. Since K9, K14, and K27 are the main lysine redues in histone H3, they were examined for acetylation status using western blot analysis coupled with acetylation-specific antibodies. These results demonstrate that histone H3 acetylation at K9 and K14 was significantly increased both in total cell lysate and isolated nuclear extract (Fig. 4A and 4B). By contrast, histone H4 acetylation at residues K5, K8, and K12 was not altered following mPQ treatment compared to vehicle-treated control cells (Fig. 4A and 4B). In addition, the level of histone H3 acetylation at K27 detected in the nuclear extract remain unchanged, demonstrating that histone H3 was selectively modified upon exposure to superoxide radical (Fig. 4A and B). These results suggest that mitochondrial superoxide induces selective epigenetic modification, in part by histone H3 acetylation.
Figure 4.

mPQ-induced epigenetic modification. (A) Western blot analyses of histone marks in cell extract and isolated nucleus, and (B) quantification of western blot data. (C) Histone acetyltransferase/p300 activity was measured in isolated nucleus, as described in Materials and Methods, and C646 was used as a p300-specific inhibitor. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: *, p ≤ 0.05 and **, p ≤ 0.01.
To determine which HAT is responsible for histone 3 acetylation in these experiments, we examined the levels of HAT p300 protein by western blotting. As shown in Fig. 4A and 4B, the p300 level was significantly increased following treatment with mPQ as compared to vehicle-treated control cells. However, HDAC1, a histone deacetylase, and DNMT3A, a methyltransferase, were not altered following treatment with mPQ compared to vehicle-treated controls (Fig. 4 A and B). We then measured HAT activity using a fluorescence-based assay. Consistent with histone acetylation and p300 protein levels, the overall HAT activity significantly increased following mPQ treatment compared to vehicle-treated cells (Fig. 4C). These results suggest that mPQ treatment leads to p300 activation and the increase in histone H3 acetylation.
Role of IDH in epigenetic modification of histone
We hypothesized that the mitochondrial superoxide-mediated inactivation of IDH may be associated with metabolic reprogramming and epigenetic modification. To determine the relationship, we used the small interfering (si) RNA approach to suppress IDH protein expression and to determine the effect on histone H3 acetylation. The results show that IDH2 and IDH3 siRNA suppressed IDH2 and IDH3 protein levels, respectively (Fig. 5A and B) and enzyme activity (Fig. 5C and D). The suppression of IDH2 and IDH3 led to a significant increase of histone H3 acetylation at K9, compared to the scrambled siRNA-treated control cells (Fig. 5B). These data support a direct link between inactvation of IDH enzymes and epigenetic modification of histones.
Figure 5.

Effect of IDH suppression on epigenetic marks in JB6 cells. (A) Suppression of IDH2/3 by treating JB6 cells with respective siRNA. Proteins were then detected by western blot analysis using antibodies and quantified in (B). (C-D) Activity of isocitrate dehydrogenase following suppression of IDH2/3 using siRNA. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: **, p ≤ 0.01.
mPQ-induced expression of redox regulator NRF2
Next, we asked whether mPQ-induced epigenetic modifications are associated with gene transcription. We assessed the level of redox-sensitive transcription factors in the isolated nuclear extract. Both NF-κB and NRF2 are known oxidation-sensitive transcription factors, and upon activation, both transcription factors localize to the nucleus. Our results show that NRF2 protein levels significantly increased in the nuclear extract following treatment with mPQ as compared to the vehicle-treated control cells (Fig. 6A and B). However, the p50 and p65 levels, which are the components of the NF-κB transcription complex, were not altered after mPQ treatment (Fig. 6A and B). Consistent with the changes of NRF2 protein levels, the NRF2 DNA-binding activity was significantly increased after mPQ treatment (Fig. 6C), suggesting that the NRF2 transcription factor was activated by mitochondria-derived superoxide radical.
Figure 6.

Activation of transcription factors by mPQ treatment. (A) Western blot analyses showed increased nuclear levels of NRF2 but not NF-κB subunits (p50 and p65) in JB6 cells in response to mPQ treatment for 24 h. (B) Quantification of western blot data. (C) NRF2 binding to target DNA in the nucleus following mPQ treatment. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: **, p ≤ 0.01.
NRF2 activation leads to transcription of its target genes
Because NRF2 DNA binding activity was increased by mPQ treatment (Fig. 6C), we tested whether the increased NRF2 activity led to increased expression of its target genes such as MnSOD. Real-time PCR data show that MnSOD mRNA levels significantly increased following treatment with mPQ compared to vehicle-treated control cells (Fig. 7A). Consistent with the changes in MnSOD mRNA levels, following mPQ treatment, MnSOD protein levels also significantly increased in the total cell lysate and isolated cytosolic fraction (Fig. 7B). These results demonstrate that mPQ induces NRF2 activity, leading to enhanced expression of NRF2 target genes.
Figure 7.

mPQ activates gene transcription. (A) Relative levels of MnSOD mRNA in JB6 cells as measured by real-time PCR following mPQ treatment for 24 h. (B) Western blot analysis of whole cell extracts (WC) as well as cytosolic (Cyt) or nuclear (NE) fractions were performed using MnSOD antibody. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: **, p ≤ 0.01.
Discussion
Mitochondria are the major producers of superoxide, and the superoxide anion radical is the substrate for MnSOD, which plays dual stimulatory and inhibitory roles in regulating cancer initiation and progression [36,37]. In normal cells, superoxide is very tightly regulated, but elevated levels of superoxide in cancer cells damage protein, DNA, and other macromolecular structures. Consequently, cancer cells undergo major metabolic alterations in response to mitochondrial injury [38,39]. Here, we have identified how superoxide radicals in mitochondria modulate metabolic pathways leading to alterations in epigenetic marks and gene transcription.
Redox cyclers such as mPQ generate superoxide in the mitochondrial matrix and suppress mitochondrial oxidative phosphorylation (Fig. 1). Reduced oxidative phosphorylation and increased glycolytic activity were also detected in UVB-mediated skin carcinogenesis [40], suggesting that superoxide generated in mitochondria can lead to cancer development. Here, we found that when excess superoxide is present in mitochondria, energy production by oxidative phosphorylation is compromised because the TCA cycle is disrupted at the IDH2 and IDH3 steps. The activities of these two mitochondrially localized enzymes were attenuated without altering their protein levels (Fig. 3A, 3B, Supplementary Fig. 3), suggesting inactivation of enzyme activity. Our results also show that HEKn cells adapted to this disruption by enhancing anaplerotic pyruvate carboxylation and reversing the reactions in the latter half of the TCA cycle. However, the disruption led to the buildup of citrate, isocitrate, and acetyl CoA. This isocitrate buildup presumably resulted, in part, from the blockage of mitochondrial IDH2/3 activity.
IDH can be inactivated by superoxide via reaction with the cysteine residues in the active site of the IDH enzyme protein [41]. When the effect of superoxide on the activities of purified IDH enzymes was measured using a xanthine-xanthine oxidase-mediated superoxide-generating system in vitro, the results support that the enzyme activity of IDH was inactivated by superoxide (Supplementary Fig. 4). It has also been shown that Cys128 and Cys216 of IDH are targets of oxidative modification and undergo homo-oligomeric complex formation leading to inactivation of enzyme activity [41]. These results support our findings that inactivation of the mitochondrial IDH enzymes suppresses isocitrate to α-KG conversion in the TCA cycle while causing the build-up of citrate and acetyl CoA upon mPQ treatment. Our data also suggest that IDH activity is controlled in a redox-dependent manner.
Several of the mitochondrial TCA cycle metabolites are linked to epigenetic modification, including 2-HG, citrate, and acetyl-CoA [42]. 2-HG is produced from a mutated IDH1/2 gene and regulates the epigenome via DNA demethylation [13–16]. The epigenome is sensitive to the availability of substrates for chromatin-modifying enzymes, which renders histone modifications very sensitive to cellular metabolism. For example, the global level of histone acetylation is dependent on the relative abundance of acetyl-CoA. Our findings show that inactivation of IDH by superoxides and cellular accumulation of acetyl-CoA led to altered histone acetylation (Figs. 4 and 5). Although a very small amount of acetyl-CoA can be produced in the cytosol and nucleus, the majority of acetyl-CoA is produced in the mitochondria. During TCA cycle progression, the mitochondria use several processes to produce acetyl-CoA, including the conversion of pyruvate to acetyl-CoA, β-oxidation of fatty acids, amino acid metabolism, and ketone body formation [43,44]. Furthermore, mitochondria export citrate to the cytosol and the nucleus, where citrate converts to acetyl-CoA by the ATP-citrate lyase reaction [45]. Our results demonstrate that an increase in cellular citrate/acetyl-CoA enhanced histone H3 acetylation (Fig. 4), which can be mediated by inactivation of mitochondrial IDH enzymes (Fig. 3). This finding is supported by our observation that the suppression mitochondrial IDH2/3 expression using siRNA led to increased H3 acetylation (Fig. 5), which establishes a link between the IDH inactivation and the histone acetylation. Consistently, the activity of HAT p300 was found to be increased after mPQ treatment, and the p300 activity was attenuated by pretreatment with its specific inhibitor, C646 (Fig. 4C). Altogether, these results demonstrate that epigenetic alterations were initiated by mitochondrially generated superoxides.
We further show that mPQ selectively increased the nuclear level and transcription activity of NRF2, an oxidative stress-sensitive transcription factor (Fig. 6), which in turn led to increased expression of its target gene, MnSOD (Fig. 7). It has been reported that NRF2 DNA binding activity and expression of its target gene, heme oxygenase, are increased upon histone acetylation (46). Consistent with this finding, it has also been shown that inhibition of histone deacetylation increases the NRF2 DNA binding activity and its target gene expression (47). These reports support our findings that mitochondrial superoxide mediates epigenetic alterations and NRF2 activation, which are depicted in Figure 8.
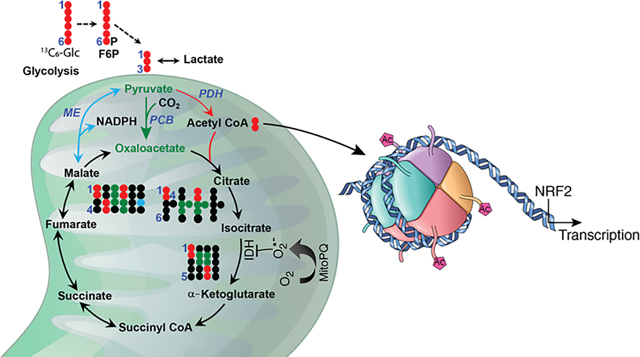
Figure 8.

Summary ilustration of the the role of mitochondrial superoxide radical in metabolic reprogramming, epigenetic modification, and gene transcription. The schematic diagram shows mPQ-generated superoxide in the mitochondrial matrix and its consequent effect on TCA cycle enzymes, metabolite formation, histone acetylation, and NRF2- mediated gene transcription.
Supplementary Material
Supplementary Figure 1. mPQ generates superoxide radicals in mitochondria of JB6 cells. (A) MitoSOX Red fluorescence-tagged cells were analyzed by flow cytometry following mPQ treatment for 24 h. (B) Relative levels of MitoSOX Red fluorescence. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: **, p ≤ 0.01.
Supplementary Figure 2. The levels of lactate in the media were measured using HSQCAD NMR analysis following mPQ treatment. The change in media 13C-3-lactate concentration over time was assessed by comparison with the zero-time point. Data points are presented as the mean +/− SD of 3 to 4 individual samples.
Supplementary Figure 3. mPQ has no effect on IDH protein expression. (A) Western blot analysis of IDH protein expression in JB6 cells. (B) Quantification of western blot data. Data points are presented as the mean +/− SD of 3 to 4 individual samples.
Supplementary Figure 4. Superoxide inactivates IDH enzyme activity in vitro. Activity of IDH was measured spectrophotometrically using isocitrate as substrate. Pure IDH enzyme was incubated with substrate and NADPH for 30 min in the absence or presence of xanthine/xanthine oxidase (X/XO). The progression of the reaction was measured spectrophotometrically at a wavelength of 340 nm in kinetic mode.
Highlights.
mPQ increases mitochondrial O2•− generation and suppresses mitochondrial functions.
Mitochondrial O2•− blocks TCA cycle progression by inactivating IDH.
Mitochondrial O2•− is an initiator of metabolic reprogramming that activates epigenetic regulation of gene transcription in response to mitochondrial dysfunction
Acknowledgements
This work was supported by NIH grants R01 CA214638 and P20 GM121327 to Dr. Daret K. St. Clair and in part by NIH grant 1U24DK097215–01A1 to Teresa W. M. Fan. The authors would like to thank Mr. Michael Alstott for performing the OCR experiments using the Redox Metabolism Shared Resource Facilities funded by a Markey Cancer Center support grant (P30 CA177558). Finally, we thank Professor Michael Murphy, Cambridge University, UK, for his generous gift of mPQ.
Abbreviations:
- 2-HG
D-2-hydroxyglutarate
- α-KG
α-ketoglutarate
- CoA
coenzyme A
- H3K
histone 3 lysine residue
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HEKn
human primary neonatal epidermal keratinocytes
- IDH
isocitrate dehydrogenase
- mPQ
MitoParaquat
- MnSOD
manganese superoxide dismutase
- NRF2
nuclear factor erythroid 2-related factor 2
- OCR
oxygen consumption rate
- TCA
tricarboxylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Giulivi C, Boveris A, Cadenas E, The steady-state concentrations of oxygen radicals in mitochondria. Reactive Oxygen species in Biological Systems, chapter3 (1999) 77–102. [Google Scholar]
- [2].Oberley LW, Buettner GR, Role of superoxide dismutase in cancer: a review, Cancer Res. 39 (1979) 1141–1149. [PubMed] [Google Scholar]
- [3].Kowluru RA, Atasi L, Ho YS, Role of mitochondrial superoxide dismutase in the development of diabetes retinopathy, Invest Opthalmol Vis Sci. 47 (2006) 1594–1599. [DOI] [PubMed] [Google Scholar]
- [4].Leuner B et al. , RAGE influences obesity in mice. Effects of the presence of RAGE on weight gain, AGE accumulation, and insulin levels in mice on a high fat diet, Z Gerontol Geriatr. 45 (2012) 102–108. [DOI] [PubMed] [Google Scholar]
- [5].Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DKDK, The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice, J Clin Invest. 98 (1996) 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].St. Clair DK, Oberley LW, Manganese superoxide dismutase expression in human cancer cells: a possible role of mRNA processing, Free Radic. Res. Commun. 12–13 (1991) 771–778. [DOI] [PubMed] [Google Scholar]
- [7].Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK, Manganese superoxide dismutase: guardian of the powerhouse, Int. J Mol. Sci. 12 (2011) 7114–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yetkin-Arik B, et al. , The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis, Sci. Rep. 9 (2019) 12608. doi: 10.1038/s41598-019-48676-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hanahan D, Weinberg RA, The next generation, Cell 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [10].Pavlova MN, Thompson CB, The emerging hallmarks of cancer metabolism, Cell Metabol. 23 (2016) 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yan H, et al. , IDH1 and IDH2 mutations in gliomas, N Engl. J Med. 360 (2009) 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nobusawa S, Watanabe T, Kleihues P, Ohgaki H, Molecular structure and predictive factor of secondary glioblastoma, Clin. Can Res. 15 (2009) 6002–6007. [DOI] [PubMed] [Google Scholar]
- [13].Xu W, et al. , Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of a-ketoglutarate-dependent dioxygenases, Cancer Cell 19 (2011) 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chowdhury R, et al. , The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases, EMBO Reports.12 (2011) 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kohli RM, Zhang Y, TET enzymes, TDG and the dynamics of DNA demethylation, Nature 502 (2013) 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schaap FG, French PJ, Bovee JB, Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors, Adv. Anat. Pathol. 20 (2013) 32–38. [DOI] [PubMed] [Google Scholar]
- [17].Xu W, Wang F, Yu Z, Xin F, Epigenetics and cellular metabolism, Genetics & Epigenetics, 8 (2016) 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Menzies KJ, Zhang H, Katsyuba E, Auwerx J, Protein acetylation in metabolism-metabolites and cofactors, Nat. Rev. Endocrinol. 12 (2016) 43–60. [DOI] [PubMed] [Google Scholar]
- [19].Teperino R, Schoonjans K, Auwerx J, Histone methyl transferases and demethylases; can they link metabolism and transcription?, Cell Metabol. 12 (2010) 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Margueron R, Reinberg D, Chromatin structure and inheritance of epigenetic information, Nat. Rev. Genet 11 (2010) 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zenter GE, Henikoff S, Regulation of nucleosome dynamics by histone modifications, Nat. Struct. Mol Biol. 20 (2013) 259–266. [DOI] [PubMed] [Google Scholar]
- [22].Allis CD, Jenuwein T, The molecular hallmarks of epigenetic control, Nat. Rev. Genet. 17 (2016) 487–500. [DOI] [PubMed] [Google Scholar]
- [23].Seto E, Yoshida M, Erasers of histone acetylation: the histone deacetylase enzymes, Cold Spring Harbor Perspect. Biol. 6 (2014) doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee KK, Workman JL, Histone acetyltransferase complexes: one size does not fit all, Nat. Rev. Mol. Cell Biol. 8 (2007) 284–295. [DOI] [PubMed] [Google Scholar]
- [25].Goodman RH, Smolik S, CBP/p300 in cell growth, transformation and development, Genes Dev. 14 (2000) 1553–1577. [PubMed] [Google Scholar]
- [26].Hassig CA, et al. , A role for histone deacetylase activity in HDAC1-mediated transcriptional repression, Proc. Natl. Acad. Sci. USA. 95 (1998) 3519–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Colburn NH, Former DF, Nelson KA, Yuspa SH, Tumor promoter induces anchorage independent irreversibly, Nature 281 (1979) 589–591. [DOI] [PubMed] [Google Scholar]
- [28].Dhar SK, St Clair DK, Nucleophosmin blocks mitochondrial localization of p53 and apoptosis, J Biol. Chem. 284 (2009) 16409–16418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dhar SK, Xu Y, Chen Y, St Clair DK, Specificity protein 1-dependent p53 mediated suppression of manganese superoxide dismutase gene expression, J Biol. Chem. 2006;281 (2006) 21698–21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fan TW, et al. , Distinctly perturbed metabolic networks underlie differential tumor tissue damages induced by immune modulator beta-glucan in a two-case ex vivo non-small-cell lung cancer study, Cold Spring Harb Mol Case Study 2(4) (2016) doi: 10.1101/mcs.a000893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fan TW, Lane AN, Structure-based profiling of metabolites and isotopomers by NMR, Prog Nucl Magn Reson Spectrosc. 52 (2008) 69–117. [Google Scholar]
- [32].Fan TW, Lane AN, NMR-based stable isotope resolved metabolomics in systems biochemistry, J Biomol NMR 49 (2011) 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lane AN, Fan TW, Higashi RM, Isotopomer-based metabolomic analysis by NMR and mass spectrometry, Methods Cell Biol. 84 (2008) 541–588. [DOI] [PubMed] [Google Scholar]
- [34].Moseley HN, Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry, BMC Bioinformatics 11 (2011) 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Robb EL, et al. , Selective superoxide generation within mitochondria by targeted redox cycler mitoparaquat, Free Rad. Biol. Med. 89 (2015) 883–894. [DOI] [PubMed] [Google Scholar]
- [36].Dhar SK, Tangpong J, Chaiswing L, Oberley TD, St Clair DK, Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages, Cancer Res. 71 (2011) 6684–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dhar SK, St Clair DK, Manganese superoxide dismutase regulation and cancer, Free Radic. Biol. Med. 52 (2012) 2209–2222. [DOI] [PubMed] [Google Scholar]
- [38].Ward PS, Thompson CB, Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate, Cancer Cell 21 (2012) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cairns RA, Harris I, McCracken S, Mak TW, Cancer cell metabolism, Cold Spring Harb Symp Quant Biol. 76 (2011) 299–311. [DOI] [PubMed] [Google Scholar]
- [40].Dhar SK, Batinic-Haberle I, St Clair DK, UVB-induced inactivation of manganese-containing superoxide dismutase promotes mitophagy via ROS-mediated mTORC2 pathway activation, J Biol. Chem. 294 (2019) 6831–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yoshida K, Hisabori T, Mitochondrial isocitrate dehydrogenase is inactivated upon oxidation and reactivated by thioredoxin-dependent reduction in Arabidopsis, Frontiers Enviro. Sci. 2 (2014) doi: 10.3389/fenvs.2014.00038. [DOI] [Google Scholar]
- [42].Matilainen O, Quiros PM, Auwerx J, Mitochondria and Epigenetics-Crosstalk in homeostasis and stress, Trends Cell Biol. 27 (2017) 453–463. [DOI] [PubMed] [Google Scholar]
- [43].Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G, Acetyl coenzyme A: a central metabolite and second messenger, Cell Metabol. 21 (2015) 805–821. [DOI] [PubMed] [Google Scholar]
- [44].Corbet C, Feron O, Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling, Biochem. Biophys. Acta-Rev. Cancer 1868 (2017) 7–15. [DOI] [PubMed] [Google Scholar]
- [45].Carrer A, Wellen KE, Metabolism and epigenetics: a link cancer cell exploit, Curr. Opin. Biotechnol. 34 (2015) 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang Z, Guo Z, Zhan Y, Li H, Wu S, Role of histone acetylation in activation of nuclear factor erythroid 2-related factor 2/heme oxygenase 1pathway by manganese chloride, Toxicol. and Appl. Pharmacol. 336 (2017) 94–100. [DOI] [PubMed] [Google Scholar]
- [47].Cai D, Yin S, Jiang Q, Cao W, Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis, Arthritis Res. Ther. 17 (2015) 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. mPQ generates superoxide radicals in mitochondria of JB6 cells. (A) MitoSOX Red fluorescence-tagged cells were analyzed by flow cytometry following mPQ treatment for 24 h. (B) Relative levels of MitoSOX Red fluorescence. Data points are presented as the mean +/− SD of three individual samples. Statistical significance is indicated by asterisks: **, p ≤ 0.01.
Supplementary Figure 2. The levels of lactate in the media were measured using HSQCAD NMR analysis following mPQ treatment. The change in media 13C-3-lactate concentration over time was assessed by comparison with the zero-time point. Data points are presented as the mean +/− SD of 3 to 4 individual samples.
Supplementary Figure 3. mPQ has no effect on IDH protein expression. (A) Western blot analysis of IDH protein expression in JB6 cells. (B) Quantification of western blot data. Data points are presented as the mean +/− SD of 3 to 4 individual samples.
Supplementary Figure 4. Superoxide inactivates IDH enzyme activity in vitro. Activity of IDH was measured spectrophotometrically using isocitrate as substrate. Pure IDH enzyme was incubated with substrate and NADPH for 30 min in the absence or presence of xanthine/xanthine oxidase (X/XO). The progression of the reaction was measured spectrophotometrically at a wavelength of 340 nm in kinetic mode.