

Abstract
We report the structural revision via synthesis of the abietane diterpenoid plebeianiol A. The synthesis was accomplished via a short and convergent sequence that featured our previously established cobalt-catalyzed hydrogen-atom-transfer-induced radical bicyclization. We further connected plebeianiol A as the likely biogenetic precursor to another, previously reported ether-bridged abietane. Finally, we demonstrated that the key cyclization event is efficient with the A-ring diol protected with two different cyclic acetal protecting groups or in unprotected form.
Graphical Abstract

Species in the Salvia genus are well-known to produce structurally diverse abietane diterpenoids.1,2 Extracts of these plants are used in traditional Chinese medicine to treat bacterial infections and a variety of inflammatory disease indications.1–3 Common phenolic abietanes, such as carnosic acid (1, Figure 1) and carnosol (2), exhibit significant antioxidant activities,4 surely owing to their catechol function. They also demonstrate anti-inflammatory activities.5,6 There is significant ongoing interest in both of these compounds and analogues in the development of anticancer chemotherapeutics.7,8 Owing to their high abundance, some of these polycyclic diterpenoids have found application in real-world problems. For instance, rosemary extracts containing carnosic acid and carnosol are commonly used as preservatives for perishable produce,9 whereas ferruginol (3), which also has antitumor activity, serves as a biomarker in resin fossil analysis.10
Figure 1.

a. Select abietane diterpenoids and their applications. b. Proposed structures of plebeianiol A and structure of a biogenetically related abietane
In 2015, Liang, Wu, and coworkers disclosed a phytochemical study of Salvia plebeia R. Br. wherein a novel, highly oxidized abietane, plebeianiol A was isolated and assigned structure 4.11 These authors showed that plebeianiol A has radical-scavenging properties, and inhibits the production of reactive oxygen species and nitric oxide. Based on 2D NMR experiments, the authors proposed a structure with the canonical octahydrophenanthrene abietane substructure, bearing hydroxyl groups in positions C2, C3, C20, and C11, which are often found oxidized in abietane compounds.2 In an unusual twist, the authors proposed the presence of an isopropyl group on the B ring at C7, and a phenolic hydroxyl group in the location typical of the abietane isopropyl group, at C13. While the authors noted that this proposal was unusual given the typical structures of abietane natural products (see 1–3), and the co-isolation of plebeianiol A with carnosol and other “normal” abietanes, they nonetheless proposed a highly unusual biogenesis of this compound.11–13 Additionally, the absolute configuration at C10 was designated as R even though all documented aromatic abietanes have been assigned S configurations at this position. On these grounds and after careful re-examination of the NMR spectral data disclosed for plebeianiol A, we were led to conclude that the structure had likely been misassigned and might in fact be 5, with typical features of an abietane diterpenoid. Liang and coworkers had previously isolated a related abietane (6) from Ajuga forrestii (in the same Lamiaceae family as Salvia species), to which 5 is likely a biosynthetic precursor.13
Our group recently disclosed total syntheses of related oxidized abietane diterpenoids14 via cobalt-catalyzed hydrogen-atom-transfer-induced radical bicyclizations15 that were inspired by the cationic biogenetic cyclizations known to produce abietanes and related diterpenoids.12a We surmised that we could avail ourselves of a similar approach to access the proposed revised structure of plebeianiol A (5) and to thereby correct its structure. Furthermore, we anticipated accessing 6 via a further oxidative cyclization of 5, thus connecting these compounds via synthesis. In this disclosure, we describe our successes in these endeavors, as well as the efficiency of the hydrogen-atom-transfer from a metal hydride (MHAT) based method for bicyclization of substrates with the proto-A-ring diol protected as an acetonide, a butanedione diacetal (BDA), and in unprotected form.
On the basis of our previous work, we aimed to use the cobalt-catalyzed, MHAT-induced bicyclization to access the tricyclic core of 5 and 6 (Scheme 1). The corresponding polyene precursor 8 would arise from suitably protected fragments 9 and 1014 via Horner–Wadsworth–Emmons alkenylation. The key architecture-building bicyclization event would feature a more oxidized A-ring than in any of our previous studies.
Scheme 1.

Approach to the proposed revised structure of plebeianiol A
Our synthesis began with the ozonolysis of commercially available 4-pentenenitrile (11) and subsequent Wittig reaction of the in-situ generated aldehyde to give 12 (Scheme 2). The acrylate ester was subjected to Sharpless asymmetric dihydroxylation,16 which produced a diol with 97:3 er as measured by analysis of the corresponding Mosher esters (see SI for details). This highly polar diol proved exceedingly difficult to purify via silica gel chromatography and we therefore subjected the crude material to protection as the 2,3-butanedione-derived cyclic diacetal (BDA).17 Treatment of the crude diol with p-TsOH and 2,3-butanedione cleanly afforded the protected intermediate 13. Importantly, this circumvented the handling issues associated with the diol and provided a stable intermediate upon which to install the isopropenyl substituent. Owing to the differences in electrophilicity of the methyl ester and nitrile, selective nucleophilic methylation was achieved providing the tertiary alcohol. Several conditions were examined for elimination of the resulting tertiary alcohol (e.g., SOCl2, MsCl) but we found that the Burgess reagent was the most efficient for the formation of the requisite 1,1-disubstituted alkene of 14. Next, nitrile-stabilized carbanion formation and reaction with diethyl chlorophosphate afforded the desired cyanophosphonate 15 as an inconsequential mixture of diastereomers.18 Convergent coupling of 15 with orthoformate-protected catechol/aldehyde 1614 proceeded smoothly in 80% yield and with preference for the desired Z-alkene (>10:1 dr), thus permitting access to ample quantities of polyene cyclization precursor 17.
Scheme 2.

Total synthesis of plebeianiol A (5) and its oxidized cyclic ether congener 6. [(DHQD)2PHAL: hydroquinidine 1,4-phthalazinediyl diether; Burgess reagent: methyl N-(triethylammoniumsulfonyl)carbamate; LDA: lithium diisopropylamide; KHMDS: potassium bis(trimethylsilyl)amide; DTBP: 2,6-di-t-butylpyridine; TMDSO: 1,1,3,3-tetramethyldisiloxane; DIBAL-H: diisobutylaluminum hydride.]
Prior to evaluating the cyclization of 17, we had engaged in a model study to determine whether or not the use of the BDA protecting group was necessary for successful bicyclization. One potential advantage of this protecting group is the rigid chair-like configuration that might help enforce a reactive conformation for cyclization, and we therefore chose it initially over the more-often-used acetonide. Additionally, previous work had indicated that while unprotected carbinols are tolerated under the cyclization conditions, free alcohols at C3 were associated with only moderate levels of diastereoselectivity (~ 3:1),14 and we were thus unable to predict the outcome of a free C2/C3 diol. To probe this question, we prepared slightly simpler model substrates 21a–c (Figure 2) containing a dimethyl resorcinol terminating group in place of the protected catechol needed for the total synthesis. We found that the model BDA substrate 21a cyclized in excellent yield (84%) providing the product as a single diastereoisomer (>20:1 dr), confirming our predictions of its proclivity for cyclization. We were pleased to learn that the unprotected diol (21b) also reacted smoothly to provide the tricyclic product in 74% yield and again as a single diastereoisomer. These results are in line with our previous studies showing that these radical polyene cyclizations can be performed on highly oxygenated substrates, even in unprotected form.. Finally, acetonide 21c also cyclized efficiently (82%) and completely stereoselectively, indicating that the identity of the protecting group does not strongly influence the key reaction. The configurations of each of the three cyclized products (22a-c) were confirmed by X-ray crystallographic analysis.19 We note that our ultimate choice of the BDA protecting group was informed by its robustness and ease of handling during early stages of our cyclization substrate synthesis.
Figure 2.

Cyclization experiments on model systems 21a–c
We next exposed 17 to our cobalt bicyclization to establish the tricyclic core of plebeianiol A (18, Scheme 2). This reaction proceeded with good efficiency (78% yield) when the reaction was buffered with 2,6-di-tert-butylpyridine (DTBP) to preserve the orthoformate protecting group. Reduction of the neopentylic nitrile with DIBAL-H provided the aldehyde which could be further reduced with sodium borohydride in a second step to give the desired carbinol 19. With all of the carbons at the proper oxidation states, we then removed both protecting groups using aqueous methanolic HCl, which afforded 5 in quantitative yield (11 steps LLS, ~13% overall yield). Spectroscopic analysis of this pentaol were in good accord with the 1H and 13C NMR data reported for plebeianiol A and originally assigned to structure 4.11 Further support for the structural revision was obtained by conversion of plebeianiol A to a related abietane natural product13 by oxidation of the catechol with molecular oxygen (presumably generating intermediate 20) followed by cycloisomerization to 6.20 The 1H and 13C NMR spectral data of this bridged ether product were also in good agreement with those previously published.13,19
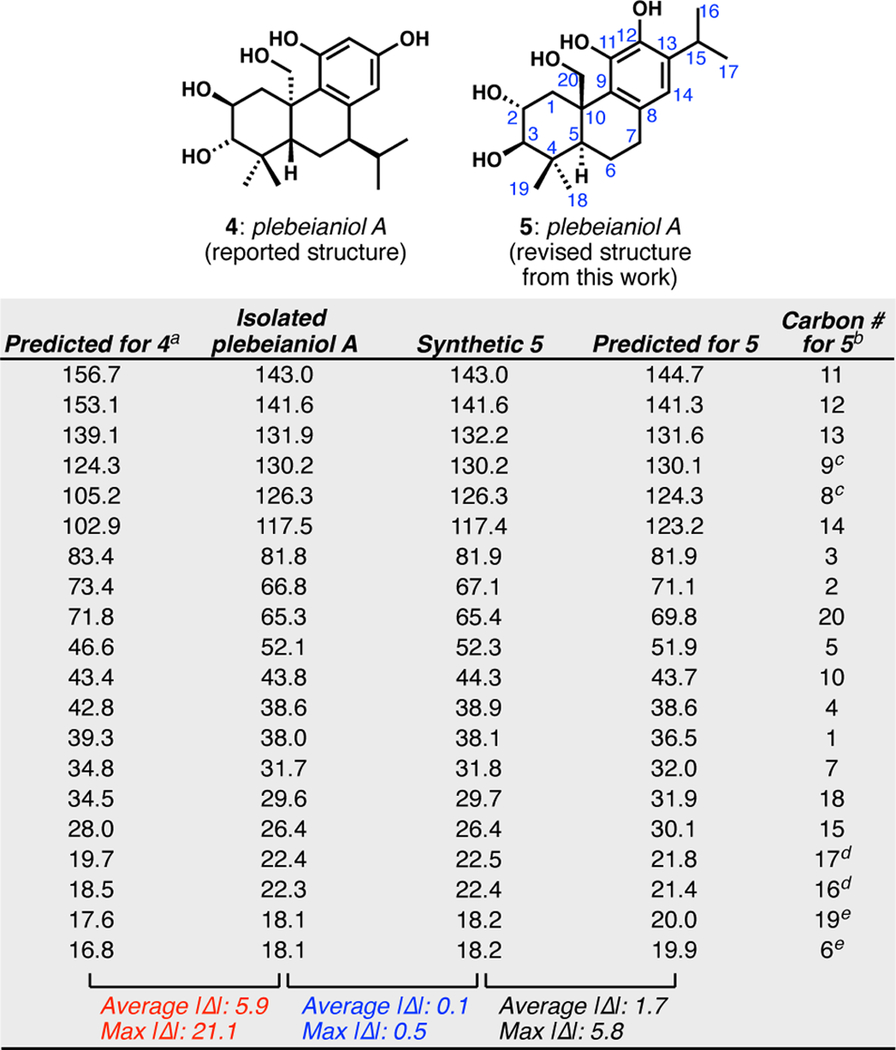
In parallel with our synthesis efforts, we calculated 13C NMR data for structures 4 and 5 to corroborate our proposed structural reassignment of plebeianiol A (Table 1). Reasoning that the fundamental differences in connectivity between 4 and 5 should manifest in the 13C spectrum of plebeianiol A, we adapted the predictive method described by Rychnovsky21–23 to calculate 13C chemical shifts for both structures (Table 1). As we anticipated, the predicted 13C shifts obtained for 4 deviate substantially from experimental 13C data previously reported for isolated plebeianiol A.11 With an absolute average deviation of 5.9 ppm and with four carbon atoms deviating by more than 10 ppm (in one instance by 21 ppm), these computational data bolstered our hypothesis that the initially reported structure 4 was misassigned. Conversely, the calculated chemical shifts for 5 are in good agreement with our experimental data for synthetic 5, as evidenced by a low absolute average deviation of 1.7 ppm and with no atoms deviating by more than 6 ppm. Direct comparison of the 13C data for isolated plebeianiol A 4 and synthetic plebeianiol A 5 reveals near congruence between the two data sets, strongly suggesting that the correct structure for plebeianiol A is in fact 5. Additional evidence is obtained by comparison of the two-dimensional NMR spectra (COSY, HMQC, and HMBC; see SI) as well as specific rotation data (+25.8 reported for 4, +26.2 for 5). The latter suggests that the absolute configuration of plebeianiol A (5) now fits with the observed configurations of all other abietane natural products (the α-disposition of the C20 carbon in 4 has not been observed in this class of molecules2). Taken together, we conclude that the originally proposed structure of plebeianiol A (4) was incorrect, and this work combining chemical synthesis with computational methods necessitates a revision of the structure of plebeianiol A to 5.
Table 1.
Comparison of 13C NMR chemical shifts calculated for structures 4 and 5, as well as those obtained by isolation and synthesis of plebeianiol A, reveal that its correct structure is 5.
|
For the comparison of the predicted shifts for 4 with the data reported by Liang, Wu, and co-workers,11 the resonances are simply listed in order of decreasing chemical shift because there was no way to use abietane numbering for the incorrect structure.
We have assigned the resonances to carbons atoms in 5, which are in the right-hand column. Please see the SI for more details.
Analysis of the HMBC spectrum was ambiguous with respect to the resonances for C8 and C9, but the predicted shifts strongly imply the assignments shown.
We were unable to unambiguously assign these methyl carbon resonances on the isopropyl group.
These resonances were exactly coincident.
Supplementary Material
Acknowledgments
We thank the Overman lab, and Dr. Yuriy Slutskyy in particular, for providing detailed instructions for calculation of 13C NMR chemical shifts.
Funding Sources
This work was funded by the NIH through grant GM-129264, and D.V. was supported by an NSF Graduate Research Fellowship.
Footnotes
Supporting Information
Supporting Information including experimental procedures, characterization data, NMR spectra for all new compounds, X-ray crystallographic data, and methods for 13C NMR shift calculations, is available free of charge on the ACS Publications website.
REFERENCES
- (1).Wu Y-B; Ni Z-Y; Shi Q-W; Dong M; Kiyota H; Gu Y-C; Cong B Constituents from Salvia Species and Their Biological Activities. Chem. Rev 2012, 112, 5967–6026. 10.1021/cr200058f [DOI] [PubMed] [Google Scholar]
- (2).González MA Aromatic Abietane Diterpenoids: Their Biological Activity and Synthesis. Nat. Prod. Rep 2015, 32, 684–704. 10.1039/C4NP00110A [DOI] [PubMed] [Google Scholar]
- (3).Li M; Li Q; Zhang C; Zhang N; Cui Z; Huang L; Xiao P An Ethnopharmacological Investigation of Medicinal Salvia Plants (Lamiaceae) in China. Acta Pharm. Sin. B 2013, 3, 273–280. 10.1016/j.apsb.2013.06.001 [DOI] [Google Scholar]
- (4).Inatani R; Nakatani N; Fuwa H Antioxidative Effect of the Constituents of Rosemary (Rosmarinus Officinalis L.) and Their Derivatives. Agric. Biol. Chem 1983, 47, 521–528. 10.1080/00021369.1983.10865682 [DOI] [Google Scholar]
- (5).Poeckel D; Greiner C; Verhoff M; Rau O; Tausch L; Hörnig C; Steinhilber D; Schubert-Zsilavecz M; Werz O Carnosic Acid and Carnosol Potently Inhibit Human 5-Lipoxygenase and Suppress pro-Inflammatory Responses of Stimulated Human Polymorphonuclear Leukocytes. Biochem. Pharmacol 2008, 76, 91–97. 10.1016/j.bcp.2008.04.013 [DOI] [PubMed] [Google Scholar]
- (6).Maione F; Cantone V; Pace S; Chini MG; Bisio A; Romussi G; Pieretti S; Werz O; Koeberle A; Mascolo N; Bifulco G Anti-Inflammatory and Analgesic Activity of Carnosol and Carnosic Acid in Vivo and in Vitro and in Silico Analysis of Their Target Interactions. Br. J. Pharmacol 2017, 174, 1497–1508. 10.1111/bph.13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Han S; Li X; Xia Y; Yu Z; Cai N; Malwal SR; Han X; Oldfield E; Zhang Y Farnesyl Pyrophosphate as a Target for Drug Development: Discovery of Natural-Product-Derived Inhibitors and Their Activity in Pancreatic Cancer Cells. J. Med. Chem 2019, 62, 10867–10896. 10.1021/acs.jmedchem.9b01405 [DOI] [PubMed] [Google Scholar]
- (8).O’Neill EJ; Den Hartogh DJ; Azizi K; Tsiani E Anticancer Properties of Carnosol: A Summary of In Vitro and In Vivo Evidence. Antioxidants 2020, 9, 961. 10.3390/antiox9100961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Etter SC Rosmarinus Officinalis as an Antioxidant. J. Herbs Spices Med. Plants 2005, 11, 121–159. 10.1300/J044v11n01_05 [DOI] [Google Scholar]
- (10).Popova M; Trusheva B; Cutajar S; Antonova D; Mifsud D; Farrugia C; Bankova V Identification of the Plant Origin of the Botanical Biomarkers of Mediterranean Type Propolis. Nat. Prod. Commun 2012, 7, 569–570. 10.1177/1934578X1200700505 [DOI] [PubMed] [Google Scholar]
- (11).Zhang B-B; He B-Q; Sun J-B; Zeng B; Shi X-J; Zhou Y; Niu Y; Nie S-Q; Feng F; Liang Y; Wu F-H Diterpenoids from Salvia Plebeia R. Br. and Their Antioxidant and Anti-Inflammatory Activities. Molecules 2015, 20, 14879–14888. 10.3390/molecules200814879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).The biosynthesis of abietane diterpenoids has been studied extensively. See, for example:; (a) Ignea C; Athanasakoglou A; Ioannou E; Georgantea P; Trikka FA; Loupassaki S; Roussis V; Makris AM; Kampranis SC Carnosic Acid Biosynthesis Elucidated by a Synthetic Biology Platform. Proc. Natl. Acad. Sci 2016, 113, 3681–3686. 10.1073/pnas.1523787113. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scheler U; Brandt W; Porzel A; Rothe K; Manzano D; Božić D; Papaefthimiou D; Balcke GU; Henning A; Lohse S; Marillonnet S; Kanellis AK; Ferrer A; Tissier A Elucidation of the Biosynthesis of Carnosic Acid and Its Reconstitution in Yeast. Nat. Commun 2016, 7, 12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xiong Y; Qu W; Sun J; Wang M; Liang J Eudesmane Sesquiterpenoid Lactones and Abietane Diterpenoids from Ajuga Forrestii Diels. Phytochem. Lett 2013, 6, 457–460. 10.1016/j.phytol.2013.05.017 [DOI] [Google Scholar]
- (14).Vrubliauskas D; Gross BM; Vanderwal CD Stereocontrolled Radical Bicyclizations of Oxygenated Precursors Enable Short Syntheses of Oxidized Abietane Diterpenoids. J. Am. Chem. Soc 2021, 143, 2944–2952. 10.1021/jacs.0c13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vrubliauskas D; Vanderwal CD Cobalt-Catalyzed Hydrogen-Atom Transfer Induces Bicyclizations That Tolerate Electron-Rich and Electron-Deficient Intermediate Alkenes. Angew. Chem. Int. Ed 2020, 59, 6115–6121. 10.1002/anie.202000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kolb HC; VanNieuwenhze MS; Sharpless KB Catalytic Asymmetric Dihydroxylation. Chem. Rev 1994, 94, 2483–2547. 10.1021/cr00032a009 [DOI] [Google Scholar]
- (17).Ley SV; Baeschlin DK; Dixon DJ; Foster AC; Ince SJ; Priepke HWM Reynolds DJ 1,2-Diacetals: A New Opportunity for Organic Synthesis. Chem. Rev 2001, 101, 53–80. 10.1021/cr990101j [DOI] [PubMed] [Google Scholar]
- (18). Unexpectedly, this phosphonation reaction was rather diastereoselective, ranging from about 4:1 to 10:1 diastereomeric ratio. We have no explanation for this apparent long-range stereoinduction.
- (19). Please see the Supporting Information for details.
- (20).Majetich G; Zou G Total Synthesis of (−)-Barbatusol, (+)-Demethylsalvicanol, (−)-Brussonol, and (+)-Grandione. Org. Lett 2008, 10, 81–83. 10.1021/ol701800d [DOI] [PubMed] [Google Scholar]
- (21).Rychnovsky SD Predicting NMR Spectra by Computational Methods: Structure Revision of Hexacyclinol. Org. Lett 2006, 8, 2895–2898. 10.1021/ol0611346 [DOI] [PubMed] [Google Scholar]
- (22).Cimino P; Gomez-Paloma L; Duca D; Riccio R; Bifulco G Comparison of Different Theory Models and Basis Sets in the Calculation of 13C NMR Chemical Shifts of Natural Products. Magn. Reson. Chem 2004, 42, S26–S33. 10.1002/mrc.1410 [DOI] [PubMed] [Google Scholar]
- (23).Lodewyk MW; Siebert MW; Tantillo DJ Computational Preduction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev 2012, 112, 1839–1862. 10.1021/cr200106v [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.