

Abstract
Shank proteins are major scaffolds of the postsynaptic density of excitatory synapses. Mutations in SHANK genes are associated with autism and intellectual disability. The effects of missense mutations on Shank3 function, and therefore the pathomechanisms are unclear. Several missense mutations in SHANK3 affect the N-terminal region, consisting of the Shank/ProSAP N-terminal (SPN) domain and a set of Ankyrin (Ank) repeats. Here we identify a novel SHANK3 missense mutation (p.L270M) in the Ankyrin repeats in patients with an ADHD-like phenotype. We functionally analysed this and a series of other mutations, using biochemical and biophysical techniques. We observe two major effects: (1) a loss of binding to δ-catenin (e.g. in the p.L270M variant), and (2) interference with the intramolecular interaction between N-terminal SPN domain and the Ank repeats. This also interferes with binding to the α-subunit of the calcium-/calmodulin dependent kinase II (αCaMKII), and appears to be associated with a more severe neurodevelopmental pathology.
Subject terms: Biochemistry, Cell biology, Genetics, Neuroscience, Molecular medicine, Neurology, Neurological disorders
Introduction
Neurodevelopmental disorders are frequently caused by mutations in genes coding for proteins which contribute to the formation and function of synapses. This has led to the term “synaptopathies”1,2. Genetic alterations affect both pre- and postsynaptic components. One prominent family of genes, which has early on been implicated in the etiology of autism, intellectual disability and schizophrenia is the SHANK family of genes, coding for SHANK 1–32–4. Loss of one copy of the SHANK3 gene is observed in 22q13 deletion syndrome/Phelan McDermid syndrome; in addition to loss-of-function mutations, also numerous missense mutations have been described4–6. Shank proteins are multidomain scaffold proteins of the postsynaptic density of excitatory, glutamatergic synapses7. They connect, through numerous protein interactions, the glutamate receptors of the synapse with the F-actin based cytoskeleton of the dendritic spine8. In detail, the central PDZ domain of Shank proteins binds to SAPAP/GKAP family members, which links Shank to the PSD-95/NMDA receptor complex9,10. Multimerisation of the C-terminal SAM domains of Shank2 and Shank3 is believed to be essential for PSD formation11,12. A long proline rich region between these two domains provides anchoring points for cytoskeleton-associated proteins such as IRSp53, cortactin, Abi1, as well as Homer, a scaffold for metabotropic glutamate receptors10,13–15. Somewhat surprisingly, only in one case a missense mutation in SHANK genes was found in patients affecting these “core” motifs and domains of Shank proteins (i.e. the Abi1 binding site in Shank316). Instead, missense mutations in the C-terminal half of Shank proteins lead to alterations outside of known interaction motifs, making it difficult to assess the functional relevance of these mutations17.
The N-terminal region of Shank3 has emerged as a hotspot for missense mutations6. This region includes the Shank/ProSAP N-terminal domain (SPN) and a set of seven ankyrin repeats (Ank). Recent structural work has shown that the SPN domain is folded as a ubiquitin like (Ubl) domain, similar to Ras association domains, and high affinity binding of active, GTP-bound variants of Ras and Rap to the SPN domain of Shank3 has been demonstrated18,19. The mutations R12C and L68P in the SPN domain which were found in autistic patients disrupt G-protein binding18.
The Ank repeats interact with postsynaptic partners such as α-Fodrin and δ-catenin20,21, as well as Sharpin (the postsynaptic localisation of which has been unclear)22. In addition, the SPN domain is in close intramolecular contact to the Ank domain, to the effect that the SPN domain interferes with the access of some known interaction partners of the Ank repeats, namely α-Fodrin and Sharpin18,23. Furthermore, recent biochemical work demonstrated an interaction of the inactive, non-phosphorylated form of αCaMKII with the loop between SPN and Ank, partially covering the Ras binding site24. The functional relevance for most of these interactions for the synaptic function of Shank proteins remains largely unclear. More importantly, it remains to be clarified how individual missense mutations in the N-terminal part of Shank3 lead to neurodevelopmental disorders, such as autism and schizophrenia.
Here we perform a functional analysis of missense mutations affecting the N-terminal SPN and Ank domains of Shank3. Mutations are taken from patients presenting with attention deficit/hyperactivity disorder (ADHD) and autistic phenotypes, from the ClinVar database and from the published literature. Our experiments identify two major aspects of ASD-associated mutations: interference with the tight association of SPN and Ank domains, and reduced binding to δ-catenin.
Materials and methods
Genetic analyses
Whole exome sequencing (WES) was performed as a duo-analysis comparing data from the proband and his mother. DNA was extracted from EDTA-stabilised peripheral blood lymphocytes and subjected to exome capture using NimbleGen SeqCap EZ MedExome (Roche), followed by sequencing on an Illumina NovaSeq platform to a mean coverage of minimum 300×, with 95% of targeted bases covered with a minimum coverage of 30×. Raw reads were aligned using the Burrows-Wheeler Alignment tool v. 0.7.15 and the GATK (Genome Analysis Toolkit) Best Practice pipeline v. 3.8-0 was used for variant calling25. Annotation and filtering of variants was performed using VarSeq 2.2.0 (Golden Helix). The sequence variant was confirmed by bidirectional Sanger sequencing according to standard procedures, and Sanger sequencing was also used for segregation analysis. Written informed consent for publication of these data has been obtained from all individuals or their legal guardians presented in this study. Work with patients in the Institute for Human Genetics follows protocols approved by the Ethics Committee of the Hamburg Chamber of Physicians (approval number PV3802). All methods were performed in accordance with the relevant guidelines and regulations.
DNA constructs
Bacterial expression constructs coding for His6/SUMO-tagged fusion proteins of rat Shank3 (residues 1–348, SPN − Ank) were generated in pET-SUMO (Thermo Fisher Scientific) as described before18. Constructs coding for N-terminal (residues 1–339, SPN − Ank; residues 75–339, 95–339; longer and shorter Ank only construct) rat Shank3 with a C-terminal mRFP-tag were generated in pmRFP-N2 (Clontech). A construct coding for N-terminally GFP-tagged full-length rat Shank3 in the pHAGE vector was obtained from Alex Shcheglovitov (Univ. of Utah, Salt Lake City, UT)18,26. For live FRET imaging, rat Shank3 N-terminal constructs (WT, R12C, N52R, L68P and P141A) coding for amino acids 1–339 of Shank3 were used, including an N-terminal GFP and a C-terminal mCherry sequence. Missense mutations were introduced by site-directed mutagenesis using the Quik-Change II site-directed mutagenesis kit (Agilent) and mutagenic oligonucleotides obtained from Sigma-Aldrich. Construct identity was confirmed by Sanger sequencing. A construct coding for HA-tagged HRas G12V was obtained from Georg Rosenberger (UKE Hamburg, Germany). cDNA coding for Rap1a G12V was cloned into the pEGFP-C1 vector (Clontech). An expression vector coding for mouse δ-catenin carrying an N-terminal GFP-tag in pEGFP-C1 was obtained from K. Kosik (Univ. of California, Santa Barbara, CA). Constructs coding for the C-terminal part of α-Fodrin fused to an N-terminal EGFP-tag20 and full length αCaMKII in a modified pcDNA3 vector coding for an N-terminal T7-tag27 have been described before.
Cell culture and transient transfection
Human embryonic kidney (HEK) 293 T cells were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum and 1% Penicillin/Streptomycin. A maximum of 20 cell passages were used. Transient transfection of 293 T cells was performed using TurboFect Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer's instructions.
Cell lysis and immunoprecipitation
Cell lysis was performed using immunoprecipitation (IP) buffer (50 mM Tris pH 8, 120 mM NaCl, 0.5% NP40, 1 mM EDTA). Immunoprecipitation was performed using RFP-trap beads (Chromotek). Precipitates were washed five times in IP buffer; both input and precipitate samples were then processed for SDS-PAGE and Western blotting.
SDS-PAGE and Western blot
Proteins were denatured in 1 × Laemmli sample buffer (63 mM Tris/HCl, pH 6.8; 10% glycerol; 1.5% SDS; 0.1 M Dithiothreitol; and 0.01% bromophenol blue) at 95 °C, separated on 10% SDS-PAGE at 100–180 V and transferred to nitrocellulose membrane in transfer buffer (25 mM Tris; 192 mM glycine; 0.05% SDS; and 20% methanol) at 100 V for 100 min using a MINI PROTEAN II™ system (Bio-Rad). Membranes were blocked with 5% milk powder/TBS-T (Tris buffered saline; 10 mM Tris/HCl pH 8; 150 mM NaCl, with 0.05% Tween 20) and incubated with the indicated primary antibodies overnight at 4 °C followed by HRP-linked secondary antibodies at room temperature for 1 h. After washing the membranes with TBS-T, chemiluminescence was detected using WesternBright chemoluminescence substrate (Biozym, Hess. Oldendorf, Germany). Membranes were scanned using a ChemiDoc™ MP Imaging System (Bio-Rad) and images were processed and analysed using Image Lab Software (Bio-Rad). Exposure times were set to maximise the signal, but avoid saturation. For all blots a background subtraction was performed using Image Lab.
Animals
For preparing primary neuronal cultures, brain tissue was isolated from Rattus norvegicus embryos. Pregnant rats (Envigo; 4–5 months old) were sacrificed on day E18 of pregnancy using CO2 anaesthesia, followed by decapitation. Neurons were prepared from all embryos present, regardless of gender (12–16 embryos). All animal experiments were approved by, and conducted in accordance with, the guidelines of the Animal Welfare Committee of the University Medical Center (Hamburg, Germany) under permission number Org766. All animal experiments were performed in compliance with ARRIVE guidelines.
Hippocampal neuron culture and transfection
Primary hippocampal neurons were isolated from E18 rat embryos. The hippocampal tissue was dissected, and hippocampal neurons were extracted by enzymatic digestion with trypsin, followed by mechanical dissociation. Cells were grown in Neurobasal medium supplemented with 2% B27, 1% Glutamax and 1% Penicillin/Streptomycin. Neurons were transfected on DIV7 using the calcium phosphate method. The complete Neurobasal medium was collected from wells one hour before transfection and replaced with transfection medium (Minimal Essential Medium + Glutamax). 5 µg plasmid DNA was mixed with 10 µL 2.5 M CaCl2 and topped up to 100 µL with ddH2O. An equal amount of 2 × Hepes buffered salt solution (HBS pH 7.05; 274 mM NaCl; 10 mM KCl; 1.4 mM Na2HPO4; 15 mM d-glucose; 42 mM Hepes) was added dropwise to the reaction tube under continuous vortexing. The reaction was incubated at room temperature for 30 min and then divided between the wells of the cell culture plate. After a 2 h incubation with the transfection mixture, the cells were washed seven times with 1 × Hank’s Balanced Salt Solution (HBSS). After the final wash, the previously collected Neurobasal medium was added back to the cells.
Immunocytochemistry
Neurons (DIV14; 7 days after transfection) were fixed with 4% paraformaldehyde, 4% sucrose in PBS and permeabilised with 0.1% Triton X-100 in PBS for 5 min at room temperature. After blocking (10% horse serum in PBS) for 1 h at room temperature, neurons were incubated with corresponding antibodies overnight followed by 1 h of incubation with Alexa Fluor antibodies. The coverslips were mounted onto glass microscopic slides using ProLong™ Diamond Antifade mounting medium (Thermo Fisher Scientific).
Live FRET (Förster resonance energy transfer) imaging
293 T cells were transfected with FRET constructs and imaged live in Hank’s balanced salt solution (HBSS; without phenol red) on the following day for 1 min. The FRET coefficient was calculated point by point in the image by making the following operation between the acceptor channel (mCherry) and the donor channel (GFP):
FRET coefficient values range between 0 and 1. To display them as 8-bit images, these values were rescaled ranging between 0 and 255. The value 1 (255) represents maximum FRET efficiency while 0 is equivalent to no detectable FRET.
Expression and purification of recombinant protein
Shank3 protein (N-terminus: residues 1–348; Ank repeats: 99–348; WT and P141A mutant) as a fusion with a His6-SUMO-tag was expressed from pET-SUMO vectors (Champion pET SUMO Protein Expression System, Invitrogen) in the BL21*(DE3) E. coli strain. Cultures were grown in LB media supplemented with antibiotics. Cells were grown at 37 °C to an optical density at 600 nm of 0.6–0.7 and cooled to 18 °C before inducing with 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) overnight. Cells were harvested by centrifugation at 12,000×g, resuspended in low imidazole buffer (10 mM imidazole, 500 mM NaCl, 20 mM Na2HPO4, pH 7.4 with 10% glycerol v/v), frozen and stored at − 20 °C. Cells were lysed through treatment with lysozyme (0.2 mg/mL) in the presence of protease inhibitor cocktail and 2 mM dithiothreitol (DTT) (2 mM) for 30 min before sonication on a Soniprep 150 Plus Disintegrator (MSE). After centrifugation at 60,000×g the supernatant was used for purification using nickel-affinity chromatography on a HisTrap HP column (GE Healthcare), using an isocratic elution of low to high imidazole buffer (500 mM imidazole, 500 mM NaCl, 20 mM Na2HPO4, pH 7.4). Eluted protein was then cleaved using recombinant SUMO protease, followed by overnight dialysis. The His-SUMO-tag was removed by a reverse pass on the HisTrap HP column. Proteins were exchanged into 1 M NaCl, 20 mM Tris, 2 mM DTT pH 7.4. For NMR measurements, proteins were produced in BL21 STAR (DE3) cultured in 2xM9 minimal medium containing 1 g/l 15N-labeled NH4Cl, and purified as described previously18.
Differential scanning fluorimetry (DSF)
Proteins were diluted to 5 µM in varying salt conditions ranging from 0.125 to 1 M NaCl, 20 mM Tris pH 7.5. Samples were added to wells in a MicroAmp Fast 96 well reaction plate (Applied Biosystems), along with a 1:1000 dilution of Sypro Orange Protein Gel Stain (Invitrogen) and a 1:10 dilution of 200 μM Tris pH 7.5. Thermal stability was then measured on a StepOnePlusTm Real-Time PCR System (Applied Biosystems) over a temperature range of 25–95 °C. Results were analysed using StepOneTm melting software (Applied Biosystems). Smoothed melting curve data were taken and the normalised mean fluorescence for each sample was plotted to generate a melting transition curve. The transition curve was isolated and a Boltzmann statistical analysis was done to give a Tm for each sample.
NMR spectroscopy
NMR spectra were collected on Bruker Neo 800 MHz spectrometers equipped with TCI CryoProbe. Experiments were performed at 298 K in the buffer contained 20 mM Tris pH 7.5, 1 M NaCl and 0.5 mM TCEP (tris-carboxyethyl-phosphine) with 5% (v/v) 2H2O. Spectra were processed with TopSpin (Bruker).
Isothermal titration calorimetry (ITC)
ITC experiments were performed using an MicroCal PEAQ-ITC (Malvern Panalytical). ITC titrations were performed in the buffer contained 20 mM Tris pH 7.5, 1 M NaCl and 0.5 mM TCEP at 25 °C. Data were integrated and fitted to a single-site-binding equation using PEAQ-ITC software (Malvern Panalytical).
Microscopy
Confocal images were acquired with Leica SP5/SP8 confocal microscopes using a 63× objective. Quantitative analysis for neuron images was performed using ImageJ. Three independent experiments were performed for all neuron data. Primary dendrites were counted at a ring within 10 μm distance from the soma. For counting Shank3 clusters, dendritic fragments beginning at a minimum of 20 µm radial distance from the soma were analysed. For counting postsynaptic Shank3 clusters, only Shank3 clusters distinctly colocalising with PSD-95 were quantified. The counting was performed using the Multi-Point tool of ImageJ. The evaluation of the fluorescence resonance energy transfer was performed using Bitplane Imaris software.
Antibodies
The following primary antibodies were used: mouse anti-GFP (Covance MMS-118P; RRID:AB_291290; WB: 1:3000), mouse anti-HA-7 (Merck H9658; RRID:AB_260092; WB 1:20,000) mouse anti-T7-tag (Merck 69522; RRID:AB_11211744; WB 1:10,000) and mouse anti-PSD-95 (Thermo Fisher Scientific MA1-046, RRID:AB_2092361; ICC: 1:500); rat anti-RFP (Chromotek 5F8; RRID:AB_2336064; WB: 1:1000); chicken anti-MAP2 (Antibodies-Online; RRID:AB_10786841; ICC: 1:1000). HRP-labeled goat secondary antibodies were from Jackson ImmunoResearch (RRID:AB_2338133; RRID:AB_10015289) and used for WB at 1:2500 dilution. For ICC, Alexa 633 goat anti-mouse IgG (Invitrogen A21050; RRID:AB_2535718) and Alexa 405 goat anti-chk IgG (abcam ab175675; RRID:AB_2819980) were used at 1:1000 dilution.
Protein stability prediction
Protein stability analysis was performed in silico using the PoPMuSiC program28 available at soft.dezyme.com.
Evaluation of data
All data are presented as mean ± SD. No statistical method was used to determine sample size. Sample size was based on experience with the relevant type of experiment18,21,23,27, and with the objective to minimise the number of animals killed. No randomisation methods were used. No test for outliers was conducted on the data; no data points were excluded. Statistical significance was determined using Prism8 software (GraphPad, San Diego, CA) and analysed by Student’s t-test or one-way ANOVA with post hoc Dunnett’s test.
Ethics approval
All mouse and rat experiments were approved by, and conducted in accordance with, the guidelines of the Animal Welfare Committee of the University Medical Center (Hamburg, Germany) under permission numbers Org_766 and Org_1018. The study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Human involvement and consent for publication
Written informed consent for participation in this study, and publication of these data has been obtained from all individuals or their legal guardians presented in this study. Work with patients in the Institute for Human Genetics follows protocols approved by the Ethics Committee of the Hamburg Chamber of Physicians (approval number PV3802).
Results
We identified a family with several individuals affected by behavioral issues (Fig. 1a). The proband, a 12 years old boy, had learning disability, outward reacting behavior, and demand avoidance, and fulfilled the criteria for oppositional defiant disorder. The mother had learning disability, ADHD and an emotional instable personality borderline disorder. The maternal uncle had an outward reacting behavior, and was diagnosed with ADHD as a young adult. He has since his teenage years had periodical substance abuse problems, and has been to prison twice. All three are overweight. Information about the phenotype of the maternal grandmother was very limited, but she was hot-tempered when she was younger. She is not overweight. A more detailed clinical description can be found in the “Supplemental information” for this manuscript.
Figure 1.

(a) Pedigree of the family with the SHANK3 missense variant c.808C>A, p.L270M. ± denotes presence/absence of the variant. (b + c) Domain structure of Shank3 and 3D-structure of the Shank3 N-terminus (model based on the crystal structure of the Shank3 N-terminus (5G4X)18. The positions of the N-terminal missense mutations examined in this study are indicated.
Upon duo exome sequencing of the proband and his mother, we identified a heterozygous missense mutation in the SHANK3 gene (NM_033517.1:c.808C>A, p.Leu270Met). No other candidate variants were detected. The variant was shown to be present also in the maternal uncle and the maternal grandmother by Sanger sequencing (“Supplemental information”, Fig. S1).
To clarify the pathogenicity of this variant in SHANK3 we began to compare the functional effects of this mutation with those of other variants in SHANK3 which are associated with autism spectrum disorders (ASD), intellectual disability (ID) or other disorders such as epilepsy. To do so, we performed a systematic analysis of this mutation and other patient missense mutations in SHANK3 affecting the N-terminal region of the Shank3 protein which had not been functionally analysed before (Fig. 1b,c). We analysed all missense mutations found in the ClinVar database which altered residues in the N-terminal SPN and Ank domains, and which were absent from the gnomAD database of 125,748 exome sequences and 15,708 whole-genome sequences29. This included the P141A mutation found in an autistic individual which had been previously been published, but not functionally analysed30. One variant (I245T) which was listed in ClinVar was present many times in gnomAD and was therefore excluded.
An initial bioinformatic analysis of the effect of the mutations on protein stability was performed based on the crystal structure of the Shank3 N-terminus (5G4X18). All mutations had a negative impact on thermodynamic stability of the protein, represented as positive ΔΔG values in Table 1. The P141A, G250C and Y272C mutations led to the highest ΔΔG values, whereas T337S had the smallest effect on protein stability.
Table 1.
Overview of SHANK3 variants analysed in this study.
| Variant, domain | Source | Phenotype | Interpretation | Inheritance | Solvent accessibility (%) | ΔΔG (kcal/mol) |
|---|---|---|---|---|---|---|
| D69G SPN | ClinVar | Autism | Unknown significance | De novo | 55.77 | 0.64 |
| Q106P linker | ClinVar | Phelan McDermid syndromea | Unknown significance | Not provided | 50.96 | 1.28 |
| P141A Ank | Ref30 | Developmental delay, seizures, autism | Pathogenic | De novo | 2.21 | 1.33 |
| G250C Ank | ClinVar | Not provided | Likely pathogenic | Not provided | 24.18 | 1.73 |
| Q256H Ank | ClinVar | Not provided | Likely pathogenic | Not provided | 5.98 | 0.87 |
| L270M Ank | This study | Learning difficulties; oppositional behavioral disorder; ADHD–like behavior | Unknown significance | Inherited | 3.01 | 1.11 |
| Y272C Ank | ClinVar | Phelan McDermid syndromea | Unknown significance | Not provided | 27.8 | 1.48 |
| Q317R Ank | ClinVar | Not provided | Unknown significance | Not provided | 24.48 | 0.53 |
| T337S Ank | ClinVar | 22q13 deletion syndromea | Unknown significance | De novo | 52.02 | 0.11 |
| N52R SPN (no patient mutation | Ref32 | – | – | – | 0.00 | 1.06 |
| L68P SPN (already studied before) | Ref39 | Autism | Pathogenic | Inherited | 5.71 | 1.47 |
Solvent accessibility of the residue at this position and free energy changes (ΔΔG values), indicating altered stability, are calculated using PoPMuSiC software. Note that the original publication for the ΔΔG calculation lists a maximum value of ± 4 kcal/mol for destabilising/stabilising variants28.
aseveral entries in ClinVar are listed as “Phelan-McDermid syndrome” or under the alternative name “22q13 deletion syndrome”, though there is no deletion involved but a missense mutation, as indicated.
For biochemical analyses of Shank3 interactions, mutations were introduced into an expression construct coding for the SPN and Ank domains (residues 1–339 of Shank3) in fusion with mRFP. The C-terminal residue 339 precisely aligns with the C-terminal end of the Ank repeats; previous work indicated the presence of a nuclear localization sequence between residues 339–37631. As we did not want a possible nuclear localization to interfere with binding experiments with cytosolic partners, we chose this rather short fragment for our assays. In addition, previous work had shown that presence or absence of a C-terminal extension to the Ank repeats did not alter stability of the Ank repeats, or their ability to interact with several known binding partners23. WT and mutant constructs, or a control vector coding for mRFP alone, were coexpressed in 293 T cells with Shank3 interaction partners of interest. Cells were lysed, and mRFP-tagged proteins were immunoprecipitated using the RFP-trap matrix. Input and precipitate samples were analysed for the presence of Shank3 and its interaction partners by Western blotting.
In a first set of experiments, we analysed binding to the small G-proteins Rap1 (expressed as a GFP-tagged, constitutively active G12V mutant) and HRas (as HA-tagged, active G12V variant). Both have been shown to interact with the SPN domain of Shank proteins in their active, GTP bound forms18. Here, both Rap1a and HRas were efficiently coimmunoprecipitated from lysates of cells expressing WT and all variant forms of the Shank3 N-terminus; the L68P variant which had previously been shown to eliminate G-protein binding was used here as a non-binding control18. A quantitative analysis showed that there were no significant differences between mutants, suggesting that none of the mutations analysed here has any effect on small G-protein binding (Fig. 2).
Figure 2.
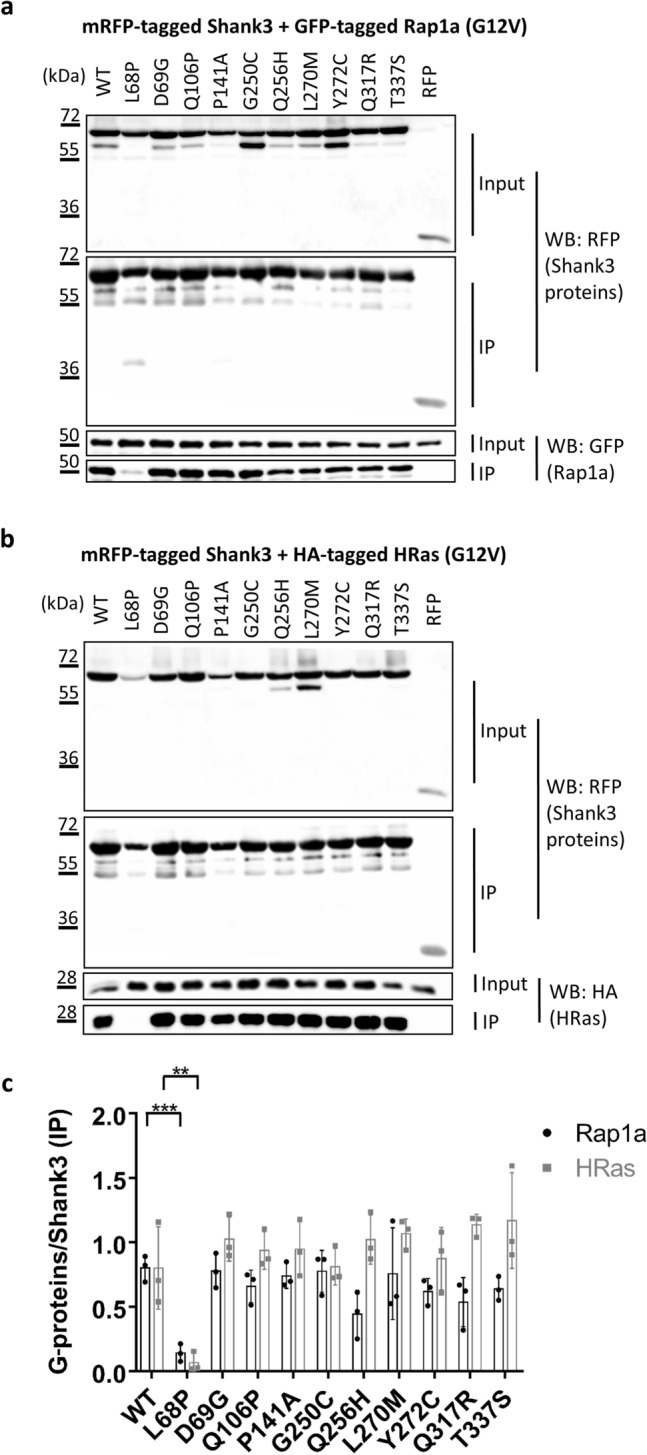
RFP-tagged WT and mutant variants of the Shank3 N-terminus (SPN + Ank domains), or mRFP alone, were coexpressed in 293 T cells with active (G12V-mutant) variants of the small G-proteins Rap1a (GFP-tagged; a) or HRas (HA-tagged, b). After cell lysis, RFP-tagged proteins were immunoprecipitated using the mRFP-trap matrix. Input and precipitate samples were analysed by Western blotting using epitope-specific antibodies. (c) Quantitative analysis of the data shown in (a) and (b). Signal intensities in IP samples for G-proteins were divided by IP signals for mRFP-Shank3 variants. **,***Significantly different from WT, p < 0.01, p < 0.001; data from three independent experiments; ANOVA, followed by Dunnett’s multiple comparisons test.
In the next step we determined binding to δ-catenin, which we have recently identified as interaction partner of the N-terminus of Shank321. Binding to δ-catenin was not affected by the D69G mutation in the SPN domain, but was significantly reduced by six mutations in the Ank repeats (e.g. L270M) (Fig. 3), consistent with our previous observation that the Ank repeats of Shank3 constitute the binding interface for δ-catenin21.
Figure 3.

(a) RFP-tagged WT and mutant variants of the Shank3 N-terminus (SPN + Ank domains), or mRFP alone, were coexpressed in 293 T cells with GFP-tagged δ-catenin. After cell lysis, RFP-tagged proteins were immunoprecipitated using the mRFP-trap matrix. Input and precipitate samples were analysed by Western blotting using mRFP- (upper panels) and GFP-specific antibodies (lower panels). (b) Quantitative analysis. Signal intensities in IP samples for δ-catenin were divided by IP signals for mRFP-Shank3 variants. *,**,***Significantly different from WT, p < 0.05, 0.01, p < 0.001, respectively; data from three independent experiments; ANOVA, followed by Dunnett’s multiple comparisons test.
Similarly, previous work had shown that the cytoskeletal protein α-Fodrin binds to the Ank repeats of Shank proteins20. However, our structural and biochemical studies had shown that the SPN domain, through its intramolecular interaction, blocks access of α-Fodrin to the Ank repeats18,23. Our current experiments supported this idea as binding of α-Fodrin to the WT N-terminus of Shank3 appeared to be rather weak. However, one of the mutations tested, P141A, strongly increased the binding of α-Fodrin to Shank3 (Fig. 4). This indicated that this mutation might open up the tight interaction between SPN and Ank repeats, allowing for better access of α-Fodrin to its binding site on the Ank repeats. In agreement, we observed that a residue at this position exhibits very low solvent accessibility (Table 1).
Figure 4.

(a) RFP-tagged WT and mutant variants of the Shank3 N-terminus (SPN + Ank domains), or mRFP alone, were coexpressed in 293 T cells with a GFP-tagged C-terminal fragment of α-Fodrin20,23. After cell lysis, RFP-tagged proteins were immunoprecipitated using the mRFP-trap matrix. Input and precipitate samples were analysed by Western blotting using mRFP- (upper panels) and GFP-specific antibodies (lower panels). (b) Quantitative analysis. Signal intensities in IP samples for α-Fodrin were divided by IP signals for mRFP-Shank3 variants. ****Significantly different from WT, p < 0.0001; data from three independent experiments; ANOVA, followed by Dunnett’s multiple comparisons test.
This result prompted us to ask whether mutations might affect the intramolecular interaction between SPN and Ank domains. As a first approach for this, we coexpressed the GFP-tagged SPN domain with the full length N-terminus (SPN + Ank), reasoning that any mutation that would open up the SPN − Ank fold would allow for easier access of the exogenous SPN domain to the Ank repeats. Here we used the N52R mutation as a positive control. The change of Asn52 to Arg in the SPN domain has been designed based on the 3D-structure, with the intention to weaken the affinity of the SPN to the Ank domain. Indeed we have shown before that it truly opens up the SPN − Ank interaction32. Upon coexpression of WT and mutant forms of the complete N-terminus with the GFP-tagged SPN domain, only the N52R positive control induced a significant increase in binding of the SPN domain to the N-terminus (Fig. 5a,b). However, we realised that an opening of the SPN − Ank fold which would be induced by a loss of affinity for the SPN domain in the Ank repeats might not be detected in this experiment. To overcome this problem, we directly analysed the interaction between Ank repeats and the SPN domain by generating an mRFP-tagged construct coding for the Ank repeats alone, which was coexpressed together with the GFP-tagged SPN domain. Here, we observed that the P141A mutation induced a strong loss of interaction with the SPN domain (Fig. 5c,d). This observation clearly supported the idea that this mutation interferes with the SPN/Ank interaction and thereby allows for improved binding of α-Fodrin to the Ank repeats. Some of these experiments were repeated with a shorter Ank construct (residues 95–339); here we again observed that the P141A variant disrupted binding to the exogenous SPN domain (Supplementary Fig. S2; also see below).
Figure 5.

(a) RFP-tagged WT and mutant variants of the Shank3 N-terminus (SPN + Ank domains), or mRFP alone, were coexpressed in 293 T cells with the GFP-tagged SPN domain of Shank3. The N52R mutant, which was designed based on the 3D structure of Shank3 to disrupt the SPN − Ank contact, was included as a positive control here. After cell lysis, RFP-tagged proteins were immunoprecipitated using the mRFP-trap matrix. Input and precipitate samples were analysed by Western blotting using mRFP- (upper panels) and GFP-specific antibodies (lower panels). (b) Quantitative analysis. Signal intensities in IP samples for GFP-SPN were divided by IP signals for mRFP-Shank3 variants. ****Significantly different from WT, p < 0.0001; ANOVA, followed by Dunnett’s multiple comparisons test. (c) The experiment was repeated as in a, however using a Shank3 construct coding for Ank repeats only. d. Quantitative analysis performed as in (b). ***Significantly different from WT, p < 0.001; data from three independent experiments; ANOVA, followed by Dunnett’s multiple comparisons test.
For further structural analysis, we made use of a bacterial expression construct which had been used before for successful expression of the SPN + Ank repeats (residues 1–348); in addition, a shorter fragment for expression of only the Ank repeats was generated (residues 99–348). Pro141 and its surrounding sequence are highly conserved from the single Drosophila Shank protein, to all mammalian Shank proteins (Supplementary Fig. S3). We wondered whether the P141A variant might unfold the Ank repeats, or rather act locally in the binding site of the SPN domain on the Ank repeats. We recorded 1H,15N-HSQC NMR spectra of WT and P141A Ank repeats uniformly labelled with 15N. These are very sensitive to changes in the protein fold; spectra were very similar for the WT and P141A Ank domains, as the majority of the signals had identical chemical shifts, and only a small number of signals changes their positions (Fig. 6a). Thus, the mutation causes only local changes in the protein structure in the vicinity of the mutation.
Figure 6.

Effect of P141A mutation on Shank3 Ank domain. (a) Superposition of the 1H,15N-HSQC NMR spectra of WT Ank (blue) and P141A Ank (red) domains of Shank3 measured at concentrations of 0.25 mM in the buffer containing 20 mM Tris pH 7.5, 1 M NaCl and 0.5 mM TCEP. Circles highlight the main differences between the spectra. (b) Structure of the SPN − Ank fragment (PDB ID 5G4X) showing position of P141 (red) at the interface between SPN and Ank domains. (c + d) ITC titration and isotherm for the interactions between isolated SPN and WT Ank (c) and P141A Ank (d) domains of Shank3. Solid line in c represents fitting to the single-site-binding model with a Kd of 19 µM and ΔH = -3.7 kcal/mol. No binding was detected for the P141A mutant (d). ITC titrations were performed at 25 °C with 25 µM of SPN in the cell and 400 µM of WT Ank or P141A Ank in the syringe. The buffer contained 20 mM Tris pH 7.5, 1 M NaCl and 0.5 mM TCEP.
In the structure of the SPN − Ank fragment P141 residue is located in the loop between the helices of the first ankyrin repeat that makes extensive contacts with the linker region between SPN and Ank domain (Fig. 6b). The rigid ring structure of proline creates a kink in the protein backbone that allows a close packing of the linker to the ankyrin repeat. This packing is likely to be perturbed by the alanine mutation, leading to the disruption of the interaction between SPN and Ank. In the ITC experiments, when SPN was titrated into the solution of WT Ank domain, we detected heat release corresponding to exothermic binding with a Kd of ~ 20 μM and ΔH of ~ − 3.7 kcal/mol. In contrast, no interaction was detected for the titration of SPN to P141A Ank, the small heat change in the course of the ITC titration caused by the residual buffer mismatch (Fig. 6c,d). This provides direct experimental support for the interaction between WT SPN and Ank domains, and disruption of this interaction by the P141A mutation.
We used several biophysical methods to verify that the closed conformation of the SPN − Ank tandem is indeed opened up by some mutations found in patients. In the 3D structure of the Shank3 N-terminal domains, the N- and C-termini of the protein are located rather close to each other, indicating that FRET might be an ideal technique to monitor conformational changes. We generated FRET reporter constructs by fusing EGFP to the N-terminus, and mCherry to the C-terminus of the Shank3 fragment (residues 1–339, as before). After transfection in 293 T cells, the WT fragment indeed showed high FRET efficiency, supporting the notion that both fluorophores are in close proximity to each other (Fig. 7a). We analysed the P141A mutation, together with two controls for which we were confident that they would disrupt the intramolecular contact between SPN and Ank domains: N52R (see above, and32) and L68P, a patient mutation for which we have shown that it disrupts the folding of the SPN domain18,23,33. We also included, in a separate experiment, a mutant which is not involved in the interface between SPN and Ank: R12C. We observed a significant decrease in FRET efficiency for P141A, similar to N52R and L68P, confirming that the P141A mutation opens up the SPN − Ank fold (Fig. 7a,c). In contrast, R12C had no effect, in agreement with its location in the Ras binding site, pointing away from the SPN − Ank interface (Fig. 7b,d).
Figure 7.

(a) 293 T cells transfected with Shank3 N-terminal FRET constructs coding for a fusion of N-terminal GFP, followed by WT or mutant Shank3 (residues 1–339, SPN + Ank), and C-terminal mCherry, were subjected to live cell imaging using a FRET imaging program (scale bar 50 µm). The FRET intensity was compared after 1 min. The color-coded FRET signal intensity (left) represents a low FRET signal (FRET = 0) with the dark blue color and a very high FRET signal FRET = 1 with the dark red color. (b) Repeat of the experiments shown in (a) with the R12C mutant (which is not in the interface between SPN and Ank domains). (c) Quantitative evaluation of cells shown in (a). (d) Quantitative evaluation of cells shown in (b). ****p < 0.0001; ANOVA, followed by Dunnett’s multiple comparisons test; ns: non-significant; t-test. Analysis of n = 15–17 cells (a, c) or n = 9–10 cells (b, d) from three independent experiments.
In a second approach, we performed differential scanning fluorimetry, which measures the thermal unfolding of the protein. Protein samples were subjected to increasing temperatures, causing an unfolding event in the protein. The SYPRO Orange dye binds to hydrophobic regions exposed as the protein unfolds, and changes in the fluorescence are monitored as the temperature increases to generate the melting curves. The Tm calculated is the temperature at which the concentration of the folded state of the protein equals the concentration of the unfolded state of the protein. The P141A protein showed a decrease in Tm of six degrees, compared to that of the WT 1–348 Shank3, indicating a substantial reduction in stability of the mutant protein compared to WT at 0.5 M NaCl conditions (Fig. 8).
Figure 8.

DSF analysis of stability of Shank3 constructs (1–348 WT and 1–348 P141A) in 0.5 M NaCl, 20 mM Tris, 2 mM DTT, pH 7.5. Fluorescence based on SYPRO-Orange dye. Mean normalised data plotted showing melt curves (a) and Tm values calculated from V50 Boltzman calculation from mean fluorescence data (b); ****Significantly different from WT, p < 0.0001; data from three independent experiments; t-Test.
Finally, we turned our attention to the α-subunit of the calcium/calmodulin dependent kinase II (αCaMKII). In contrast to the aforementioned interaction partners, this protein, in its autoinhibited inactive form, does not specifically interact with one of the two clearly defined domains (SPN and Ank) but rather binds to the linker connecting the two domains and to the SPN domain24. Upon coexpression and coprecipitation of the WT Shank3 fragment with this protein, we observed strong and specific binding (Fig. 9).
Figure 9.
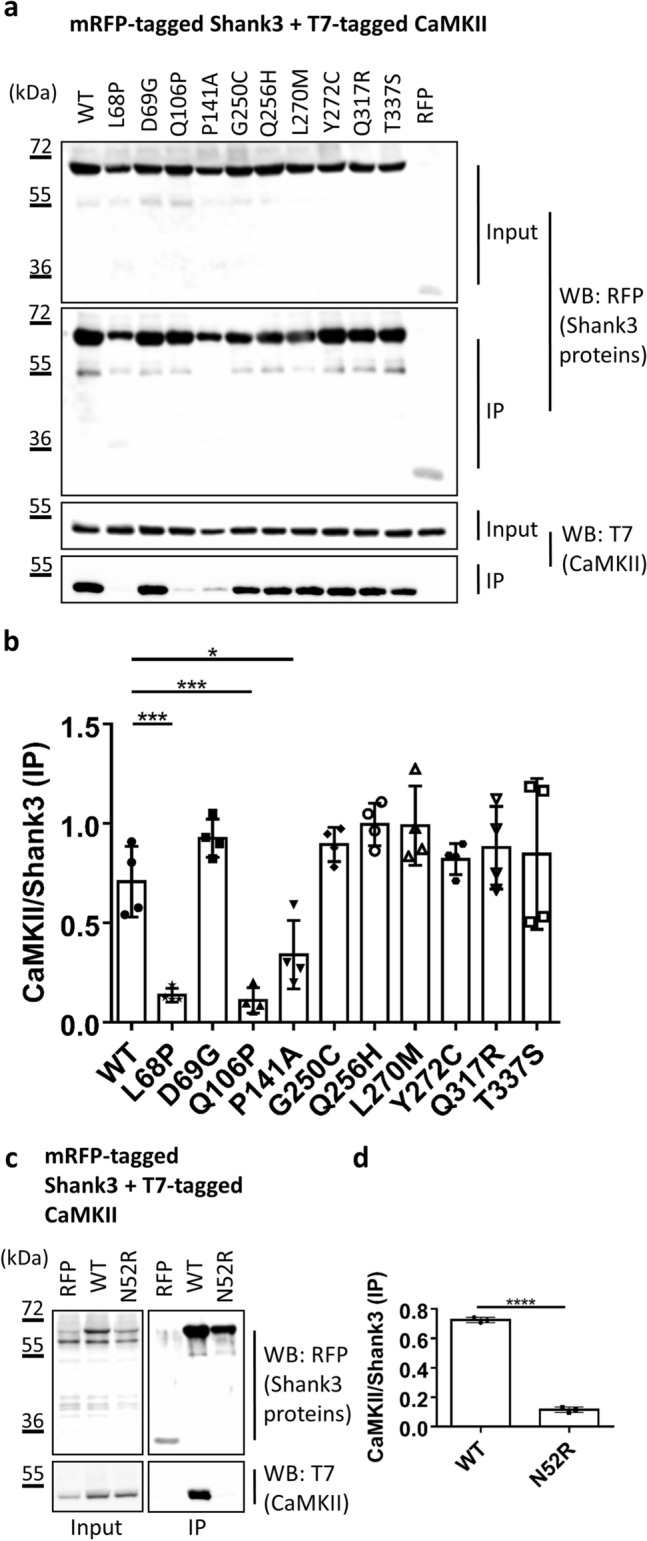
(a) RFP-tagged WT and mutant variants of the Shank3 N-terminus (SPN + Ank domains), or mRFP alone, were coexpressed in 293 T cells with T7-tagged αCaMKII. After cell lysis, RFP-tagged proteins were immunoprecipitated using the mRFP-trap matrix. Input and precipitate samples were analysed by Western blotting using mRFP- (upper panels) and T7-specific antibodies (lower panels). (b) Quantitative analysis. Signal intensities in IP samples for T7-αCaMKII were divided by IP signals for mRFP-Shank3 variants. *,**,***Significantly different from WT, p < 0.05, 0.01, p < 0.001, respectively; data from four independent experiments; ANOVA, followed by Dunnett’s multiple comparisons test. (c, d) The assay as in (a, b) was repeated for the N52R mutation in Shank3. ****Significantly different from WT, p < 0.0001; data from three independent experiments; t-Test.
This was reduced by the L68P mutation (unfolding the SPN domain, thus supporting the relevance of the SPN surface for interaction with αCaMKII). In addition, both Q106P and P141A showed significantly impaired binding to the αCaMKII. For Q106P, this may be explained by the close proximity of Q106 to the linker region and the actual αCaMKII binding site on Shank3. In the original publication on this interaction24, mutation of Ile102, which is in close proximity to Q106, also interfered strongly with this interaction. On the other hand, P141 is not localised in the linker region but is part of the interface between Ank and SPN domains. Our previous data (see Figs. 5, 6, 7) suggest that the primary effect of P141A is to disrupt the SPN − Ank intramolecular contact. We speculated that P141A affects αCaMKII binding by pushing SPN and Ank away from each other, thereby altering the conformation of the linker region which appears to act as a hinge between SPN and Ank repeats. To validate this hypothesis, we assessed the mutation which had been specifically designed to affect the SPN − Ank interface, namely the N52R mutation, for its effect on αCaMKII binding. Indeed, we observed that this mutation, which also alters a residue in the interface between Ank and SPN domains, destroyed binding of the αCaMKII.
In the next step we analysed the potential relevance of our findings for synaptic function; here we focused on the P141A and L270M mutants. GFP-tagged full-length Shank3 (WT and mutants) was expressed in primary cultured hippocampal neurons, and transfected cells were stained and analysed for the distribution of MAP2 (as a dendritic marker), PSD-95 (as a postsynaptic marker) and GFP-Shank3 (Fig. 10a,b). Both WT and mutant proteins were efficiently targeted to postsynaptic sites, as the GFP-Shank3 signal colocalised extensively with PSD-95; also, we observed the Shank3- and PSD-95 positive, presumably postsynaptic clusters mostly in some distance from the (MAP2-labelled) dendritic shaft, indicating a localisation on dendritic spines. The number of total Shank3 clusters was not affected by any of the mutations (Fig. 10c). However, a clear difference between WT and P141 mutant was observed with respect to the number of GFP-Shank3 clusters colocalising with PSD-95, as we found that this was significantly reduced for neurons expressing the P141A mutant (Fig. 10d). No difference was observed for the L270M variant. Furthermore, neuronal morphology was not affected by either variant as the number of primary dendrites did not change, regardless of the construct used (Fig. 10e).
Figure 10.

Primary hippocampal neurons expressing GFP-Shank3 (WT or P141A, L270M mutants) were fixed and stained for the postsynaptic marker PSD-95 and the dendritic marker MAP2 (gray). The expressed Shank3 variants were detected using the GFP fluorescence. (a) Overview images of neurons (scale bar 20 μm). (b) Boxed dendritic areas were magnified (scale bar 10 μm). GFP-Shank3 colocalises extensively with PSD-95 in presumably postsynaptic clusters, which are found in a small distance from the (MAP2-positive) main dendrite. (c) Quantitative evaluation of the total number of Shank3 clusters per 20 μm dendrite. ns: non-significant; analysis of 45 dendrites of n = 15 neurons from three independent experiments; t-Test. (d) Quantitative evaluation of the number of postsynaptic Shank3 clusters (Shank3 clusters colocalising with PSD-95) per 20 µm dendrite. *Significantly different from WT, p < 0.05; analysis of 45 dendrites of n = 15 neurons from three independent experiments; t-Test. (e) Quantitative evaluation of the number of primary dendrites per neuron. ns: non-significant; analysis of n = 15 neurons from three independent experiments; t-Test.
Discussion
Mutations in SHANK3 have been mostly associated with autism and intellectual disability syndromes17; we report here a family with the p.L270M variant, where carriers of this variant exhibit a somewhat divergent phenotypic spectrum. Most remarkable is the spectrum of challenging behavior, including ADHD and oppositional behavioral disorder. ADHD has previously been described associated with the SHANK2 gene34, and with a duplication as well as a loss-of-function/frameshift variant in SHANK335,36. However, ADHD has not been associated with missense variants in SHANK3. Learning disabilities were reported in three of the four family members, while the maternal grandmother reported not having had problems in school. In addition, we observed obesity in three of four carriers of this variant. The fact that this variant segregates with the phenotype of the behavioral issues suggests that it might likely be causative for at least this phenotype.
To prove pathogenicity we performed a number of functional assays. For this, we included further variants in the N-terminal part of Shank3, which had so far not been functionally studied, including several mutations which had been deposited in the ClinVar database with only limited phenotypical description. We aimed for a thorough biochemical analysis of the effects of mutations affecting the N-terminal parts of the Shank3 protein. For this we used a standardised assay of coexpression/co-immunoprecipitation of various known interaction partners of Shank3, with an mRFP-tagged construct which contains the SPN and Ank repeat domains. We used this very “reduced” construct because we wanted to study the isolated effects of N-terminal mutations. It should be kept in mind that some interaction partners make more than one contact to the full length Shank3 protein; thus, the α-subunit of CaMKII which was studied here binds to the linker between SPN domain and Ank repeats in its inactive, non-phosphorylated form24. In addition, it binds to a short sequence segment within the proline rich region in its active form37. Thus, any isolated effect of mutations such as the Q106P mutation might have been lost in the context of the full-length protein.
The effects we observed may be divided into two categories (which are not mutually exclusive; see Table 2): several mutations had a negative impact on the interaction with δ-catenin. As we had identified the Ank repeats of Shank3 as the domain binding to δ-catenin, it is conceivable that the mutations alter the interface between δ-catenin and Shank3 and thereby reduce binding. A reduction in δ-catenin interaction was the only effect observed with the L270M mutation; Leu270 is somewhat buried in the hydrophobic core of the Ank repeats. It does not appear to be an unfolding mutation, as most aspects of the Shank3 N-terminal interactions remain intact. We speculate that it slightly alters the surface of the Ank repeats in a way which is incompatible with δ-catenin binding. It may be argued that loss of δ-catenin binding only may be associated with the somewhat milder phenotype exerted by this mutation. However, such a conclusion would require analysis of more patients/variants. Interestingly, loss of function mutations in CTNND2, which encodes δ-catenin, have been found to be associated with an ADHD phenotype in several patients38.
Table 2.
Summary of the functional analysis of the examined SHANK3 variants.

Another set of mutations interfered with the closed conformation between SPN domain and Ank repeats, and/or the binding of the α-subunit of the CaMKII to the linker between both domains. One intriguing finding we made here is that these two aspects are intricately linked to each other. Thus, we observed that, upon disruption of the SPN − Ank contact using N52R or P141A mutations, we also lose binding of αCaMKII (see model in Fig. 11). Loss of αCaMKII was also observed with the R12C and L68P mutations which have been more widely studied. R12, similar to Q106 analysed here, appears to be in direct contact with αCaMKII24, whereas the L68P mutation leads to a local unfolding of the SPN domain18,23,33. As loss of αCaMKII binding appears to be a more common result of SHANK3 missense mutations, one might ask what the physiological relevance of this interaction may be. Currently, we speculate that Shank3 acts as a negative regulator of the αCaMKII signalling pathway which is prominently involved in synaptic plasticity. One prediction here would be that Shank3 deficiency (or the presence of mutants R12C, L68P or P141A) would lead to a higher basal activity of αCaMKII due to loss of the inhibiting effect of the Shank3 N-terminus.
Figure 11.

Model of open/closed transition of the N-terminal domains of the Shank3 proteins. (a) In the closed conformation, a shallow cavity is formed between the SPN domain, the linker between SPN and Ank, and the N-terminal end of the helix beginning with Q106. Bound αCaMKII covers a large part of this interface through interactions with both the SPN and the linker sequence, including and up to Q106. (b) Upon disruption of the SPN − Ank interaction, the geometry of the αCaMKII binding site is altered and αCaMKII cannot bind anymore. Model based on the crystal structure of the Shank3 N-terminus (5G4X)18 and models of the Shank3/αCaMKII complex shown in Ref.24.
Importantly, it appears that the p.P141A variant of SHANK3 has the most dramatic effect on the function of the protein, as it disrupts the intramolecular interaction between SPN and Ank, and interferes with αCaMKII and δ-catenin binding. Our biophysical analyses, using NMR, FRET and DSF techniques, support this conclusion. They also show that the separation of domains destabilises the protein, as we recorded a lower melting point of the Shank3 N-terminus in the DSF assay. Importantly, the P141A variant reduces the capacity of Shank3 to support the formation of postsynaptic clusters in cultured hippocampal neurons. The total number of GFP-Shank3 clusters is similar for WT Shank3 and the P141A variant; however, the number of clusters colocalising with PSD-95 (presumably postsynaptic clusters) is reduced for this variant, indicating a tendency to cluster spontaneously on dendrites. No such effect was seen for the L270M variant. Thus, P141A and L270M may stand for two opposite ends of the mutational spectrum in this part of the Shank3 protein. P141A is associated with multiple disruptions of N-terminal interactions; this mutation was not inherited but occurred de novo in the patient and causes a rather severe phenotype with intellectual disability and autism. On the other hand, L270M affected only one of the interactions tested here; this variant was inherited and causes a comparatively mild phenotype involving ADHD and learning difficulties.
Supplementary Information
Acknowledgements
We thank UKE microscopy imaging facility (UMIF) for providing confocal microscopes. We thank the patients and their family for participation. The authors thank Hans-Hinrich Hönck (UKE Hamburg) for excellent technical assistance.
Abbreviations
- Abi-1
Abl interactor 1
- ADHD
Attention deficit hyperactivity disorder
- Ank
Ankyrin repeats
- ANOVA
Analysis of variance
- ASD
Autism spectrum disorder
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- cDNA
Complementary deoxyribonucleic acid
- chk
Chicken
- DAPI
4′,6-Diamidino-2-phenylindole
- DIV
Days in vitro
- DSF
Differential scanning fluorimetry
- DTT
Dithiothreitol
- EDTA
Ethylenediaminetetraacetic acid
- EGFP
Enhanced green fluorescent protein
- FRET
Förster energy resonance transfer
- GFP
Green fluorescent protein
- GKAP
Guanylate kinase-associated protein
- GTP
Guanosine triphosphate
- HA
Human influenza hemagglutinin
- HBS
Hepes buffered salt solution
- HBSS
Hank’s balanced salt solution
- HEK
Human embryonic kidney
- HRP
Horseradish peroxidase
- ICC
Immunocytochemistry
- ID
Intellectual disability
- IgG
Immunoglobulin G
- IP
Immunoprecipitation
- IRSp53
Insulin receptor substrate 53
- MAP2
Microtubule-associated protein 2
- mRFP
Monomeric red fluorescent protein
- ns
Not significant
- PAGE
Polyacrylamide gel electrophoresis
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PDZ
PSD-95/DLG/ZO1
- PSD
Post-synaptic density
- PSD-95
Postsynaptic density protein 95
- SAM
Sterile alpha motif
- SAPAP
SAP90/PSD-95-associated protein
- SD
Standard deviation
- SDS
Sodium dodecyl sulfate
- Shank
SH3 and multiple ankyrin repeats
- SPN
Shank3/ProSAP N-terminal
- SUMO
Small Ubiquitin-like Modifier
- TBS-T
Tris-buffered saline-Tween 20
- Ubl
Ubiquitin like
- WES
Whole exom sequencing
- WB
Western blot
- WT
Wild type
Author contributions
D.W., E.W., D.T., F.H.N., M.K., M.J.L. performed experiments; T.M.O. analysed patient data; A.V.F., I.B. and C.R.F. analysed data; I.B. and H.-J.K. conceived the project; D.W. and H.-J.K. wrote the manuscript; F.H.N. and H.-J.K. applied for funding. All authors read and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. Work in our laboratory was supported by Deutscher Akademischer Austauschdienst (DAAD; to F.H.N.), a postdoctoral fellowship from Forschungsförderungsfonds Medizin at UKE Hamburg (to F.H.N.), the Biotechnology and Biological Sciences Research Council (BBSRC) DTP fellowship (to E.W), the region of Southern Denmark (to C.F.) and a grant from Deutsche Forschungsgemeinschaft (DFG; KR 1321/9-1; to H.-J.K.). DAAD, UKE Hamburg, BBSRC, the region of Southern Denmark and DFG did not play a role in the design of the study, in the collection, analysis, and interpretation of data and in writing the manuscript.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files]. The raw next-generation sequencing data that support the findings in affected individuals cannot be made publicly available for reasons of patient confidentiality. Qualified researchers may apply for access to these data, pending institutional review board approval.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-04723-5.
References
- 1.Grant SG. Synaptopathies: Diseases of the synaptome. Curr. Opin. Neurobiol. 2012;22:522–529. doi: 10.1016/j.conb.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 2011;21:594–603. doi: 10.1016/j.tcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Durand CM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonaglia MC, et al. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 2006;43:822–828. doi: 10.1136/jmg.2005.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid Syndrome) Mol. Syndromol. 2011 doi: 10.1159/000334260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassani Nia F, Kreienkamp HJ. Functional relevance of missense mutations affecting the N-terminal part of Shank3 found in Autistic Patients. Front. Mol. Neurosci. 2018;11:268. doi: 10.3389/fnmol.2018.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheng M, Kim E. The Shank family of scaffold proteins. J. Cell Sci. 2000;113:1851. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- 8.Kreienkamp HJ. Scaffolding proteins at the postsynaptic density: Shank as the architectural framework. Handbook Exp. Pharmacol. 2008 doi: 10.1007/978-3-540-72843-6_15. [DOI] [PubMed] [Google Scholar]
- 9.Naisbitt S, et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/S0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 10.Tu JC, et al. Coupling of mGluR/homer and PSD-95 complexes by the shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/S0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 11.Baron MK, et al. An architectural framework that may lie at the core of the postsynaptic density. Science. 2006;311:531. doi: 10.1126/science.1118995. [DOI] [PubMed] [Google Scholar]
- 12.Grabrucker AM, et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011;30:569–581. doi: 10.1038/emboj.2010.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soltau M, Richter D, Kreienkamp H-J. The insulin receptor substrate IRSp53 links postsynaptic shank1 to the small G-protein cdc42. Mol. Cell. Neurosci. 2002;21:575–583. doi: 10.1006/mcne.2002.1201. [DOI] [PubMed] [Google Scholar]
- 14.Proepper C, et al. Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J. 2007;26:1397–1409. doi: 10.1038/sj.emboj.7601569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du Y, Weed SA, Xiong W-C, Marshall TD, Parsons JT. Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol. Cell. Biol. 1998;18:5838. doi: 10.1128/MCB.18.10.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, et al. An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol. Psychiatry. 2020;25:2534–2555. doi: 10.1038/s41380-018-0324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leblond CS, et al. Meta-analysis of SHANK mutations in autism spectrum disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014;10:e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lilja J, et al. SHANK proteins limit integrin activation by directly interacting with Rap1 and R-Ras. Nat. Cell Biol. 2017;19:292–305. doi: 10.1038/ncb3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Q, Hosokawa T, Zeng M, Hayashi Y, Zhang M. Shank3 binds to and stabilizes the active form of Rap1 and HRas GTPases via Its NTD-ANK tandem with distinct mechanisms. Structure. 2020;28:290–300.e294. doi: 10.1016/j.str.2019.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Bockers TM, et al. Synaptic scaffolding proteins in rat brain. Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein alpha-fodrin. J. Biol. Chem. 2001;276:40104–40112. doi: 10.1074/jbc.M102454200. [DOI] [PubMed] [Google Scholar]
- 21.Hassani Nia F, et al. Targeting of delta-catenin to postsynaptic sites through interaction with the Shank3 N-terminus. Mol. Autism. 2020;11:85. doi: 10.1186/s13229-020-00385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim S, et al. Sharpin, a novel postsynaptic density protein that directly interacts with the shank family of proteins. Mol. Cell Neurosci. 2001;17:385–397. doi: 10.1006/mcne.2000.0940. [DOI] [PubMed] [Google Scholar]
- 23.Mameza MG, et al. SHANK3 gene mutations associated with autism facilitate ligand binding to the Shank3 ankyrin repeat region. J. Biol. Chem. 2013;288:26697–26708. doi: 10.1074/jbc.M112.424747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai Q, et al. CaMKIIalpha-driven, phosphatase-checked postsynaptic plasticity via phase separation. Cell Res. 2021;31:37–51. doi: 10.1038/s41422-020-00439-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013;43:111011–111033. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shcheglovitov A, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quitsch A, Berhorster K, Liew CW, Richter D, Kreienkamp HJ. Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J. Neurosci. 2005;25:479–487. doi: 10.1523/JNEUROSCI.2699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011;12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boccuto L, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 2013;21:310–316. doi: 10.1038/ejhg.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassani Nia F, Woike D, Kloth K, Kortum F, Kreienkamp HJ. Truncating mutations in SHANK3 associated with global developmental delay interfere with nuclear beta-catenin signaling. J. Neurochem. 2020 doi: 10.1111/jnc.15014. [DOI] [PubMed] [Google Scholar]
- 32.Salomaa SI, et al. SHANK3 conformation regulates direct actin binding and crosstalk with Rap1 signaling. Curr. Biol. 2021 doi: 10.1016/j.cub.2021.09.022. [DOI] [PubMed] [Google Scholar]
- 33.Bucher M, et al. Autism associated SHANK3 missense point mutations impact conformational fluctuations and protein turnover at synapses. bioRxiv. 2021 doi: 10.1101/2020.12.31.424970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma SL, et al. Genetic overlap between attention deficit/hyperactivity disorder and autism spectrum disorder in SHANK2 gene. Front. Neurosci. 2021;15:649588. doi: 10.3389/fnins.2021.649588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han K, et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503:72. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanani F, Stud D, Balasubramanian M. SHANK3 variant as a cause of nonsyndromal autism in an 11-year-old boy and a review of published literature. Clin. Dysmorphol. 2018;27:113–115. doi: 10.1097/MCD.0000000000000232. [DOI] [PubMed] [Google Scholar]
- 37.Perfitt TL, et al. Neuronal L-Type calcium channel signaling to the nucleus requires a novel CaMKIIalpha–Shank3 interaction. J. Neurosci. 2020;40:2000–2014. doi: 10.1523/JNEUROSCI.0893-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adegbola A, et al. Disruption of CTNND2, encoding delta-catenin, causes a penetrant attention deficit disorder and myopia. HGG Adv. 2020 doi: 10.1016/j.xhgg.2020.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gauthier J, et al. Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its supplementary information files]. The raw next-generation sequencing data that support the findings in affected individuals cannot be made publicly available for reasons of patient confidentiality. Qualified researchers may apply for access to these data, pending institutional review board approval.